A Researcher's Guide to Validating Intrinsic Apoptosis Activation: From Bench to Biomarkers
This guide provides a comprehensive framework for researchers, scientists, and drug development professionals to accurately detect and validate the activation of the intrinsic apoptosis pathway.
A Researcher's Guide to Validating Intrinsic Apoptosis Activation: From Bench to Biomarkers
Abstract
This guide provides a comprehensive framework for researchers, scientists, and drug development professionals to accurately detect and validate the activation of the intrinsic apoptosis pathway. It covers the foundational biology of the mitochondrial pathway, details established and emerging methodological approaches, offers solutions for common troubleshooting scenarios, and outlines a strategic multi-parametric validation workflow. By synthesizing current scientific literature and technological advancements, this article serves as a critical resource for ensuring robust and reproducible analysis of intrinsic apoptosis in both basic research and therapeutic development contexts.
Understanding the Intrinsic Apoptosis Pathway: Core Mechanisms and Key Biomarkers
Defining the Mitochondrial Pathway of Apoptosis
The mitochondrial pathway of apoptosis, or the intrinsic apoptotic pathway, is a precisely regulated cellular process essential for maintaining tissue homeostasis and eliminating damaged cells. This pathway is primarily controlled by the BCL-2 protein family, which integrates diverse cellular stress signals to determine cell fate [1] [2]. When activated, this pathway leads to mitochondrial outer membrane permeabilization (MOMP), triggering the release of cytochrome c and other pro-apoptotic factors that ultimately execute programmed cell death [3] [2]. The critical importance of this pathway is evident in various pathological conditions, particularly cancer, where its deregulation contributes to tumor development and therapy resistance [1]. This guide provides a comprehensive comparison of current methodologies for validating intrinsic apoptosis pathway activation, offering researchers a framework for selecting appropriate experimental approaches based on their specific research needs.
Core Mechanisms of the Mitochondrial Apoptosis Pathway
The BCL-2 Protein Family: Regulators of Cell Fate
The BCL-2 protein family serves as the central regulatory unit of the intrinsic apoptosis pathway, comprising both pro-apoptotic and anti-apoptotic members that interact to control MOMP [1] [2]. This family can be divided into three functional subgroups:
- Anti-apoptotic proteins (BCL-2, BCL-XL, MCL-1, BCL-w, A1/Bfl-1) that preserve mitochondrial integrity and prevent cytochrome c release
- Multi-domain pro-apoptotic proteins (BAX, BAK) that directly mediate MOMP
- BH3-only proteins (BID, BIM, PUMA, BAD, NOXA) that sense cellular stress and initiate the apoptotic cascade [1] [2]
These proteins engage in complex interactions that ultimately determine whether a cell survives or undergoes apoptosis. The balance between these competing factions is influenced by various cellular stress signals, including DNA damage, growth factor deprivation, and oxidative stress [1].
Mitochondrial Outer Membrane Permeabilization: The Point of No Return
MOMP represents the critical commitment point in the intrinsic apoptosis pathway. This process is primarily mediated by the activation and oligomerization of BAX and BAK, which form pores in the mitochondrial outer membrane [2] [4]. These pores facilitate the release of cytochrome c and other pro-apoptotic factors from the mitochondrial intermembrane space into the cytosol [3].
Once in the cytosol, cytochrome c binds to Apaf-1, triggering the formation of the apoptosome complex, which then activates caspase-9 [3]. Activated caspase-9 subsequently cleaves and activates effector caspases-3 and -7, initiating the execution phase of apoptosis [1] [2]. Recent research has revealed that MOMP involves a two-step process requiring both cristae junction opening and BAX/BAK pore formation, with cardiolipin oxidation playing an important regulatory role [3].
Table 1: Key Components of the Mitochondrial Apoptosis Pathway
| Component Category | Key Elements | Primary Function |
|---|---|---|
| Anti-apoptotic BCL-2 Proteins | BCL-2, BCL-XL, MCL-1, BCL-w, A1/Bfl-1 | Neutralize pro-apoptotic members; maintain mitochondrial integrity [1] [2] |
| Pro-apoptotic Effectors | BAX, BAK | Form pores in mitochondrial outer membrane; execute MOMP [1] [2] |
| BH3-only Proteins | BIM, PUMA, BID, BAD, NOXA | Sense apoptotic stimuli; activate BAX/BAK or inhibit anti-apoptotic members [1] |
| Mitochondrial Factors | Cytochrome c, SMAC/DIABLO, AIF | Released after MOMP; activate caspases and promote cell death [1] [3] |
| Downstream Signaling | Apaf-1, Caspase-9, Caspases-3/7 | Form apoptosome; execute apoptotic program [3] |
Comparative Analysis of Validation Methodologies
Flow Cytometry: Multiparameter Single-Cell Analysis
Flow cytometry offers a powerful approach for simultaneously assessing multiple parameters of apoptosis in individual cells. A recently developed integrated protocol enables comprehensive analysis of eight different cellular parameters from a single sample, providing a detailed view of the cellular state [5].
Experimental Protocol: Integrated Flow Cytometry Workflow
- Cell Preparation: Harvest approximately 0.5 million cells per experimental condition
- Staining Procedure: Combine multiple stains including:
- Annexin V/PI: Distinguishes viable (Annexin V-/PI-), early apoptotic (Annexin V+/PI-), late apoptotic (Annexin V+/PI+), and necrotic cells (Annexin V-/PI+)
- JC-1: Measures mitochondrial membrane potential (ΔΨm) – depolarization indicated by shift from red (aggregates) to green (monomers) fluorescence
- BrdU/PI: Assesses cell cycle progression and DNA synthesis
- CellTrace Violet: Tracks proliferation rates and cell generations [5]
- Data Acquisition: Analyze samples using a flow cytometer with appropriate laser configurations and filters
- Analysis: Use specialized software to quantify population distributions for each parameter
This multiparametric approach enables researchers to correlate mitochondrial depolarization with other apoptotic markers and cell cycle status, providing mechanistic insight into apoptosis activation [5].
Fluorescence Microscopy: Spatial and Temporal Resolution
Fluorescence microscopy techniques provide valuable spatial and temporal information about mitochondrial dynamics during apoptosis, allowing researchers to monitor changes in real-time within living cells [6] [7].
Experimental Protocol: Live-Cell Imaging of Mitochondrial Metrics
- Cell Line Preparation: Utilize cells expressing fluorescent reporters (e.g., COX8-EGFP for mitochondria, ACTIN-mCherry as internal control) [7]
- Image Acquisition:
- Widefield Microscopy: Cost-effective for imaging ΔΨm, NAD(P)H, and FAD autofluorescence
- Confocal Microscopy: Enhanced resolution through pinhole elimination of out-of-focus light
- FLIM (Fluorescence Lifetime Imaging): Measures nanosecond-scale fluorescence decay, providing sensitivity to microenvironment changes [6]
- Parameters Measured:
- ΔΨm: Using potentiometric dyes (JC-1, TMRM) or FRET-based sensors
- NAD(P)H/FAD: Autofluorescence imaging of metabolic cofactors
- ROS Production: Using fluorescent probes (DHR, DCFDA) [6]
- Mitochondrial Morphology: Tracking fusion/fission dynamics
- Mitochondrial-Cellular Protein Ratio: Quantitative analysis of mitochondrial biogenesis [7]
This approach enables direct visualization of mitochondrial events during apoptosis, including the timing and pattern of MOMP, mitochondrial networking, and interactions with other organelles [6] [7].
Computational Modeling: Systems-Level Analysis
Computational approaches provide a systems-level understanding of the complex regulatory networks controlling intrinsic apoptosis, offering predictive capabilities that complement experimental methods [8].
Methodological Framework: Kinetic Systems Modeling
- Network Definition: Identify key components and interactions based on experimental literature
- Model Formulation: Develop ordinary differential equations or stochastic models describing:
- BCL-2 family protein interactions
- MOMP activation kinetics
- Caspase activation cascades [8]
- Parameter Estimation: Use quantitative experimental data to constrain model parameters
- Model Validation: Test predictions against experimental observations not used in model building
- Simulation Analysis: Investigate systems-level properties including:
- Bistability and switch-like behavior in MOMP
- Robustness to parameter variation
- Signal integration across multiple stress pathways [8]
These models have provided mechanistic insight into how rapid, irreversible MOMP emerges from complex protein interactions and have helped reconcile competing theories of BH3-only protein function [8].
Diagram 1: The mitochondrial pathway of apoptosis integrates stress signals to commit to cell death via BCL-2 family proteins.
Table 2: Comparison of Apoptosis Validation Methodologies
| Methodology | Key Parameters Measured | Temporal Resolution | Spatial Information | Throughput | Key Advantages |
|---|---|---|---|---|---|
| Flow Cytometry | ΔΨm, phosphatidylserine exposure, membrane integrity, cell cycle, proliferation [5] | Single time point (multiple time points possible) | No | High (10,000+ cells/ sample) | Multiparametric, quantitative, statistically robust population data [5] |
| Fluorescence Microscopy | ΔΨm, mitochondrial morphology, metabolite levels (NADH, FAD), ROS, organelle interactions [6] [7] | Real-time (seconds to minutes) | Yes (subcellular) | Medium | Spatiotemporal dynamics, live-cell imaging, subcellular localization [6] |
| Computational Modeling | System dynamics, network properties, MOMP kinetics, prediction of drug responses [8] | Continuous (simulated time) | Network topology | Theoretical | Predictive power, mechanistic insight, in silico experimentation [8] |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Mitochondrial Apoptosis Studies
| Reagent Category | Specific Examples | Research Application | Mechanistic Insight |
|---|---|---|---|
| BCL-2 Family Inhibitors (BH3-mimetics) | Venetoclax (ABT-199), Navitoclax (ABT-263), ABT-737 [2] | Selective targeting of anti-apoptotic BCL-2 proteins; induce apoptosis in cancer cells [1] [2] | Probe dependencies on specific anti-apoptotic members; therapeutic development [2] |
| Fluorescent Mitochondrial Dyes | JC-1, TMRM, TMRE, Rhod-123 [6] [5] | Measure mitochondrial membrane potential (ΔΨm); indicator of early apoptosis [6] [5] | Mitochondrial depolarization precedes caspase activation [5] |
| Apoptosis Detection Reagents | Annexin V, Propidium Iodide (PI), caspase substrates/ inhibitors [5] | Distinguish apoptotic stages; measure caspase activity [5] | Phosphatidylserine externalization (early apoptosis); membrane permeabilization (late apoptosis) [5] |
| Metabolic Probes | DCFDA (ROS), MitoSOX (mitochondrial superoxide), NAO (cardiolipin) [6] [3] | Detect reactive oxygen species; monitor cardiolipin oxidation [6] [3] | ROS promotes cardiolipin oxidation and cytochrome c release [3] |
| Genetic Reporters | COX8-EGFP (mitochondria), ACTIN-mCherry (normalization) [7] | Live-cell tracking of mitochondrial mass and biogenesis [7] | Monitor mitochondrial dynamics in real-time; quantitative analysis of mitochondrial changes [7] |
Discussion and Research Applications
Integration of Methodological Approaches
Effective validation of intrinsic apoptosis pathway activation typically requires a combination of the methodologies described above. For instance, flow cytometry provides robust quantitative data on population responses, while live-cell imaging offers temporal and spatial context for key events such as MOMP [5] [7]. Computational models can then integrate these experimental data to generate testable hypotheses about system behavior and identify critical control points in the pathway [8].
A particularly powerful approach involves using BH3-mimetics as experimental tools to probe the "priming" of cells for apoptosis. Compounds such as venetoclax (BCL-2 selective) or A-1331852 (BCL-XL selective) can identify which anti-apoptotic proteins a particular cell type depends on for survival, providing functional insight into BCL-2 family interactions [2].
Technical Considerations and Challenges
Each methodology presents specific technical considerations. Flow cytometry requires careful compensation between fluorescent channels and appropriate controls for autofluorescence [5]. Fluorescence microscopy must account for potential phototoxicity during live-cell imaging, particularly when using high-intensity illumination [6]. Computational models require rigorous parameter estimation and validation against experimental data to ensure biological relevance [8].
A common challenge across all methods is the heterogeneity of apoptotic responses within cell populations. Single-cell techniques like flow cytometry and live-cell imaging are particularly valuable for characterizing this heterogeneity and identifying subpopulations with distinct apoptotic sensitivities [5] [7].
Future Directions
Recent advances in apoptosis research include the development of novel targeting approaches such as proteolysis targeting chimeras (PROTACs) for BCL-XL or MCL1, which may achieve tumor-specific apoptosis induction with reduced toxicity [2]. High-content imaging approaches combining multiple fluorescent biosensors are enabling more comprehensive profiling of apoptotic signaling dynamics [6]. Additionally, computational models are increasingly being applied to predict therapy responses and identify rational combination therapies [8].
As our understanding of the mitochondrial apoptosis pathway continues to evolve, the integration of complementary validation methodologies will remain essential for translating basic mechanistic insights into therapeutic advances for cancer and other diseases characterized by apoptotic dysregulation.
Diagram 2: Integrated experimental workflow for validating intrinsic apoptosis combines multiple methods for comprehensive analysis.
The BCL-2 protein family constitutes a critical regulatory circuit that governs mitochondrial outer membrane permeabilization (MOMP), the decisive event in the intrinsic apoptosis pathway [2] [9]. This pathway is activated in response to cellular stress signals, including DNA damage, growth factor deprivation, and oncogenic stress [10]. The founding member, BCL-2, was first identified in 1984 as the gene involved in the t(14;18) chromosomal translocation found in follicular lymphoma, representing the first example of an oncogene that promotes cancer by blocking cell death rather than stimulating proliferation [2] [11]. The BCL-2 family employs an intricate network of protein-protein interactions to maintain tissue homeostasis by balancing pro- and anti-apoptotic signals, with dysregulation of this system contributing to numerous diseases, including cancer, neurodegenerative disorders, and autoimmune conditions [12] [2] [9]. For researchers validating intrinsic apoptosis activation, understanding these interactions provides fundamental insights into cellular fate decisions and offers therapeutic targets for modulating pathological cell survival or death.
Structural and Functional Classification of BCL-2 Family Proteins
The BCL-2 family consists of approximately 20 proteins that share BCL-2 homology (BH) domains and are categorized into three functional subgroups based on their structure and role in apoptosis regulation [2] [13].
Multi-Domain Anti-Apoptotic Proteins
Anti-apoptotic members, including BCL-2, BCL-XL, BCL-w, MCL-1, BCL2A1, and BCL-B, typically contain four BH domains (BH1-BH4) [2] [13]. These proteins preserve mitochondrial integrity by preventing MOMP and subsequent cytochrome c release [2]. They feature a characteristic hydrophobic groove on their surface formed by BH1, BH2, and BH3 domains, which serves as the primary interaction site for binding pro-apoptotic family members [2] [13].
Multi-Domain Pro-Apoptotic Proteins
Pro-apoptotic effectors BAK, BAX, and BOK contain three BH domains (BH1-BH3) and directly execute MOMP [12] [2]. In healthy cells, BAX resides inactive in the cytoplasm, while BAK is embedded in the mitochondrial membrane. Upon activation, both proteins undergo conformational changes, oligomerize, and form pores in the mitochondrial outer membrane, enabling cytochrome c release [2] [14] [9].
BH3-Only Pro-Apoptotic Proteins
The BH3-only proteins, including BIM, BID, BAD, BIK, NOXA, PUMA, BMF, and HRK, function as cellular sentinels that sense stress signals and initiate apoptosis [2] [11]. They share only the BH3 domain, which is necessary and sufficient for their killing activity [13]. These proteins act through two mechanisms: directly activating BAX/BAK or neutralizing anti-apoptotic proteins by occupying their hydrophobic grooves [2].
Table 1: Classification of Principal BCL-2 Family Proteins
| Protein Class | Representative Members | BH Domains | Primary Function | Subcellular Localization |
|---|---|---|---|---|
| Anti-apoptotic | BCL-2, BCL-XL, MCL-1, BCL-w | BH1-BH4 | Inhibit MOMP, block cytochrome c release | Mitochondria, ER |
| Pro-apoptotic (multi-domain) | BAX, BAK, BOK | BH1-BH3 | Execute MOMP, form mitochondrial pores | BAX: cytosol→mitochondria; BAK: mitochondria |
| Pro-apoptotic (BH3-only) | BIM, BID, BAD, PUMA, NOXA | BH3 only | Sense stress, inhibit anti-apoptotic or activate pro-apoptotic | Various (cytosol, cytoskeleton, mitochondria) |
Molecular Mechanisms of Apoptotic Regulation
The BCL-2 Protein Interaction Network
The intrinsic apoptosis pathway is regulated through a complex interaction network among BCL-2 family members. Anti-apoptotic proteins preserve mitochondrial integrity by sequestering activated BH3-only proteins and directly inhibiting BAX and BAK [2] [14]. In response to cellular stress, upregulated or activated BH3-only proteins either directly activate BAX/BAK or function as sensitizers by neutralizing anti-apoptotic proteins, thereby displacing bound activators [2]. Once activated, BAX and BAK oligomerize to form proteolipid pores in the mitochondrial outer membrane, leading to MOMP and the release of cytochrome c and other apoptogenic factors [2] [9]. Cytochrome c then facilitates the formation of the apoptosome, which activates caspase-9 and the downstream caspase cascade, ultimately executing cell death [2] [10].
Diagram 1: BCL-2 Protein Network Regulating Intrinsic Apoptosis
Non-Canonical Functions of BCL-2 Proteins
Beyond their established role in apoptosis regulation, BCL-2 family proteins participate in diverse physiological processes, including autophagy, calcium homeostasis, mitochondrial dynamics, and neuronal function [12] [11]. BCL-2 inhibits autophagy by binding to the BH3 domain of Beclin 1, a critical autophagy initiator, thereby suppressing autophagosome formation [15]. During metabolic stress, post-translational modifications such as phosphorylation disrupt BCL-2/Beclin 1 interaction, enabling autophagy activation [15]. At the endoplasmic reticulum (ER), BCL-2 proteins modulate calcium signaling by regulating calcium release from ER stores to mitochondria, influencing both energy production and cell death initiation [2] [15] [11]. These non-apoptotic functions highlight the multifaceted nature of BCL-2 proteins in cellular homeostasis and stress adaptation.
Experimental Approaches for Validating Intrinsic Apoptosis Activation
Assessing BCL-2 Protein Interactions and Conformational Changes
Co-Immunoprecipitation (Co-IP) for Protein-Protein Interactions
Objective: To detect and quantify interactions between pro- and anti-apoptotic BCL-2 family proteins in response to apoptotic stimuli.
Protocol:
- Cell Lysis: Harvest treated and control cells using mild non-ionic detergent lysis buffer (e.g., 1% CHAPS or NP-40) to preserve protein complexes. Include protease and phosphatase inhibitors.
- Antibody Incubation: Incubate cell lysates with antibody against your target protein (e.g., anti-BCL-2, anti-BCL-XL, or anti-BAX) overnight at 4°C with gentle rotation.
- Bead Capture: Add protein A/G agarose beads and incubate for 2-4 hours at 4°C to immobilize immune complexes.
- Washing: Pellet beads and wash 3-5 times with lysis buffer to remove non-specifically bound proteins.
- Elution and Analysis: Elute bound proteins with SDS-PAGE sample buffer, separate by electrophoresis, and transfer to PVDF membrane. Detect interacting partners using specific antibodies (e.g., detect BIM bound to BCL-2).
Interpretation: Increased binding of BH3-only proteins to anti-apoptotic members indicates apoptotic initiation. Displacement interactions can be quantified by comparing immunoprecipitation efficiency across treatment conditions.
BH3 Profiling to Measure Mitochondrial Priming
Objective: To functionally assess cellular proximity to the apoptotic threshold by measuring mitochondrial sensitivity to BH3 peptides.
Protocol:
- Mitochondrial Isolation: Permeabilize cells with digitonin or isolate mitochondria from treated and control cells.
- BH3 Peptide Exposure: Incubate with synthetic BH3-only domain peptides (e.g., BIM, BAD, NOXA, HRK) at varying concentrations.
- MOMP Detection: Measure cytochrome c release by ELISA or western blot, or monitor mitochondrial membrane depolarization using JC-1 or TMRM fluorescent dyes.
- Quantification: Calculate the percentage of cytochrome c release or degree of membrane depolarization for each BH3 peptide.
Interpretation: Cells with high "mitochondrial priming" undergo MOMP with minimal BH3 stimulation. Distinct response patterns to specific BH3 peptides indicate dependence on particular anti-apoptotic proteins (e.g., BAD sensitivity indicates BCL-2/BCL-XL dependence).
Measuring Mitochondrial Outer Membrane Permeabilization
Cytochrome c Release Assay
Objective: To directly quantify cytochrome c release from mitochondria, the definitive marker of MOMP.
Protocol:
- Subcellular Fractionation: Separate mitochondrial and cytosolic fractions from treated cells using differential centrifugation.
- Western Blot Analysis: Detect cytochrome c in both fractions using specific antibodies. COX IV or VDAC serve as mitochondrial fraction markers, while β-tubulin or GAPDH mark cytosolic fractions.
- Quantification: Normalize cytochrome c signals to fraction markers and calculate the cytosolic:mitochondrial ratio across conditions.
Interpretation: Increased cytochrome c in cytosolic fractions indicates MOMP has occurred. This method provides direct biochemical evidence of intrinsic pathway activation.
BAX/BAK Conformational Change Detection
Objective: To monitor activation status of BAX and BAK using conformation-specific antibodies.
Protocol:
- Cell Staining: Fix and permeabilize cells, then incubate with antibodies that specifically recognize active conformations of BAX (clone 6A7) or BAK (clone Ab-1).
- Flow Cytometry Analysis: Quantify the percentage of cells with active BAX/BAK using flow cytometry.
- Confocal Microscopy: For spatial analysis, perform immunofluorescence and image by confocal microscopy to visualize mitochondrial localization of active BAX/BAK.
Interpretation: Increased active BAX/BAK correlates with apoptosis induction. Mitochondrial translocation of BAX provides additional evidence of activation.
Table 2: Key Methodologies for Validating BCL-2 Family-Mediated Apoptosis
| Method | Measured Parameters | Information Gained | Advantages | Limitations |
|---|---|---|---|---|
| Co-Immunoprecipitation | Protein-protein interactions between BCL-2 family members | Direct evidence of binding events; identifies specific complexes | Measures native interactions; can detect displacement | May not capture transient interactions; requires specific antibodies |
| BH3 Profiling | Mitochondrial depolarization or cytochrome c release in response to BH3 peptides | Functional assessment of apoptotic priming; identifies anti-apoptotic dependencies | Predictive of therapeutic response; highly quantitative | Requires specialized peptides; technically challenging |
| Cytochrome c Release Assay | Cytochrome c localization (mitochondrial vs. cytosolic) | Direct measurement of MOMP execution | Definitive marker of intrinsic pathway activation | Requires subcellular fractionation; may miss early events |
| Conformation-Specific BAX/BAK Flow Cytometry | Active BAX/BAK conformation | Early detection of pro-apoptotic effector activation | Single-cell resolution; quantitative | Does not directly measure downstream events |
Diagram 2: Experimental Workflow for Validating Intrinsic Apoptosis Activation
Therapeutic Targeting of BCL-2 Proteins in Human Diseases
BH3-Mimetic Drugs: From Basic Research to Clinical Application
The structural characterization of BH3 domain interactions with anti-apoptotic BCL-2 proteins enabled rational design of BH3-mimetic compounds that occupy the hydrophobic groove, thereby neutralizing anti-apoptotic function and promoting apoptosis [2]. Venetoclax (ABT-199), the first FDA-approved selective BCL-2 inhibitor, demonstrates remarkable efficacy in hematologic malignancies, particularly chronic lymphocytic leukemia (CLL) and acute myeloid leukemia (AML) [2] [14]. Its development followed earlier inhibitors ABT-737 and navitoclax (ABT-263), which targeted both BCL-2 and BCL-XL but exhibited dose-limiting thrombocytopenia due to BCL-XL inhibition in platelets [2] [14]. Subsequent BH3-mimetics targeting other anti-apoptotic members, including BCL-XL and MCL-1 inhibitors, have faced greater developmental challenges due to on-target toxicities—thrombocytopenia for BCL-XL inhibitors and cardiac toxicity for MCL-1 inhibitors [2]. Novel approaches such as proteolysis targeting chimeras (PROTACs) and antibody-drug conjugates (ADCs) are being explored to achieve tumor-specific inhibition of these targets [2].
Resistance Mechanisms and Combination Strategies
Resistance to BH3-mimetics emerges through various mechanisms, including upregulation of alternative anti-apoptotic proteins (e.g., MCL-1 or BCL-XL elevation following BCL-2 inhibition), mutations in BCL-2 that reduce drug binding, and changes in the expression of BH3-only proteins [2] [14]. Functional BH3 profiling can identify these adaptive dependencies and guide rational combination therapies. For instance, venetoclax combined with MCL-1 inhibitors demonstrates synergistic killing in multiple myeloma and AML models, while combinations with standard chemotherapeutics or targeted agents often overcome primary resistance [2] [14]. In multiple myeloma, venetoclax monotherapy achieved an 84% overall response rate, with combination regimens showing further improvement in clinical outcomes [14].
Table 3: Clinically Advanced BH3-Mimetics Targeting BCL-2 Family Proteins
| Therapeutic Agent | Primary Target | Clinical Stage | Key Indications | Notable Toxicities |
|---|---|---|---|---|
| Venetoclax (ABT-199) | BCL-2 | FDA-approved | CLL, AML, NHL | Tumor lysis syndrome, neutropenia |
| Navitoclax (ABT-263) | BCL-2, BCL-XL, BCL-w | Phase I/II | CLL, SCLC, NHL | Thrombocytopenia (dose-limiting) |
| Sonrotoclax (BGB-11417) | BCL-2 | Phase I/II | CLL, NHL, WM | Lower thrombocytopenia risk |
| Lisaftoclax (APG-2575) | BCL-2 | Phase I/II | CLL, AML, MM | Well-tolerated profile |
| BCL-XL specific agents | BCL-XL | Preclinical/Early clinical | Solid tumors | Thrombocytopenia (mitigated by PROTACs/ADCs) |
| MCL-1 inhibitors (S64315) | MCL-1 | Early clinical | AML, MM, Lymphoma | Cardiac toxicity |
Research Reagent Solutions for BCL-2 Family Studies
The expanding apoptosis assay market, projected to grow from USD 2.7 billion in 2024 to USD 6.1 billion by 2034, reflects increasing research focus on cell death mechanisms [16]. Several companies provide essential tools for studying BCL-2 family proteins and apoptosis:
- Thermo Fisher Scientific (26.5% market share) offers comprehensive solutions including Annexin V-FITC apoptosis detection kits, antibodies for BCL-2 family proteins, mitochondrial membrane potential dyes (JC-1, TMRM), and cytochrome c detection assays [16].
- Beckman Coulter (Danaher) provides flow cytometry systems with automated apoptosis analysis modules, caspase activity assays, and high-content screening platforms for multiparametric cell death analysis [16].
- Merck (Sigma-Aldrich) supplies validated antibodies for BCL-2 family members, BH3 peptides for profiling, caspase inhibitors, and specialized reagents for mitochondrial isolation and analysis [16].
- Bio-Rad Laboratories offers automated Western blotting systems and reagents for protein interaction studies, while Becton, Dickinson and Company provides flow cytometry instrumentation and analysis software optimized for apoptosis marker detection [16].
Emerging technologies include AI-powered platforms for automated gating and image analysis, 3D cell culture-compatible apoptosis assays, and high-throughput screening systems that integrate multiple apoptosis parameters for comprehensive pathway assessment [16].
The BCL-2 protein family constitutes a critical regulatory node determining cellular fate through an intricate balance of pro- and anti-apoptotic signals. For researchers validating intrinsic apoptosis activation, multiparametric assessment spanning protein interactions, mitochondrial priming, effector activation, and downstream events provides the most comprehensive insight into pathway status. The development and clinical success of BH3-mimetics like venetoclax exemplify how fundamental understanding of BCL-2 family biology can translate into effective therapeutics. However, challenges remain in targeting specific anti-apoptotic members without on-target toxicities and overcoming resistance mechanisms. Future directions include developing more selective inhibitors, advancing predictive biomarkers through BH3 profiling, and designing rational combination therapies that leverage the complex interplay between BCL-2 family members to achieve selective cancer cell elimination while sparing normal tissues.
The intrinsic apoptosis pathway is a tightly regulated cell death program essential for development, tissue homeostasis, and eliminating damaged cells. A pivotal event in this pathway is Mitochondrial Outer Membrane Permeabilization (MOMP), which enables the release of pro-apoptotic proteins from the mitochondrial intermembrane space into the cytosol [17]. This process is primarily regulated by Bcl-2 family proteins, with Bax and Bak serving as essential gatekeepers. Upon activation, these proteins form pores in the outer mitochondrial membrane, leading to the release of key factors including cytochrome c and Smac/DIABLO [18] [17]. Cytochrome c initiates apoptosome formation and caspase activation, while Smac/DIABLO counteracts Inhibitor of Apoptosis Proteins (IAPs), thereby promoting cell death [18] [19]. This guide provides a comparative analysis of these molecular events and presents validated experimental approaches for researchers studying intrinsic apoptosis activation in drug discovery and basic research.
Comparative Analysis of Key Apoptotic Molecules
Understanding the distinct characteristics, functions, and regulatory mechanisms of cytochrome c and Smac/DIABLO is fundamental for designing appropriate experimental strategies to monitor apoptosis activation.
Table 1: Key Characteristics and Functions of Cytochrome c and Smac/DIABLO
| Feature | Cytochrome c | Smac/DIABLO |
|---|---|---|
| Primary Function | Activates caspase cascade via apoptosome formation [17] | Neutralizes IAP proteins (e.g., XIAP), relieving caspase inhibition [18] [19] |
| Localization | Mitochondrial intermembrane/intercristae spaces, associated with cardiolipin [17] | Mitochondrial intermembrane space [19] |
| Release Mechanism | Bax/Bak-dependent pore formation; can occur via Bax/Bak-independent mechanisms [20] [17] | Primarily through Bax/Bak-dependent pores; regulated by Bcl-2 [18] [19] |
| Downstream Effect | Binds to Apaf-1 to form apoptosome, activating caspase-9 [17] | Binds to XIAP, displacing it from caspases-9 and -3 [18] |
| Regulation | Modulated by Bcl-2 family proteins (Bcl-2, Bcl-xl, Bax, Bak) [18] | Release is inhibited by Bcl-2 overexpression [19] |
Table 2: Regulatory Proteins and Experimental Modulations
| Protein Target | Pro-apoptotic Effect | Anti-apoptotic Effect | Experimental Modulations |
|---|---|---|---|
| Bax/Bak | Forms pores in mitochondrial outer membrane [18] | Deficiency confers resistance to apoptotic stimuli [18] | Use Bax-/- Bak-/- DKO MEFs to study requirement [20] |
| Bcl-2/Bcl-xl | - | Inhibits cytochrome c and Smac release [18] [19] | Overexpression blocks MOMP [18] |
| XIAP | - | Directly inhibits caspases-3, -7, and -9 [18] | Smac mimetics or gene knockdown to relieve inhibition [18] |
| Caspases | Execute apoptosis via proteolytic cleavage [19] | Inhibition blocks apoptotic morphology [19] | z-VAD-fmk (pan-caspase inhibitor) to test caspase dependence [21] |
Experimental Approaches for Validating Apoptosis Activation
Assessing Mitochondrial Membrane Permeabilization
The release of intermembrane space proteins is a definitive marker for MOMP. Two complementary approaches are recommended:
Subcellular Fractionation and Immunoblotting This biochemical method involves separating mitochondrial and cytosolic fractions following apoptotic stimulation. Cells are gently lysed with digitonin, which permeabilizes the plasma membrane but leaves mitochondria intact. Subsequent centrifugation yields a heavy membrane (mitochondrial) fraction and a cytosolic fraction [19]. Immunoblotting for cytochrome c and Smac/DIABLO confirms their translocation. Loss from the mitochondrial fraction with concurrent appearance in the cytosolic fraction indicates MOMP. This method provides population-level data and is highly quantitative when combined with densitometry.
Single-Cell Live Imaging of Protein Release For kinetic analysis in individual living cells, researchers can engineer cells to express fluorescent protein tags (e.g., GFP, YFP) fused to cytochrome c or Smac/DIABLO [21]. Upon induction of apoptosis (e.g., with UV irradiation), time-lapse confocal microscopy can monitor the release of these proteins from mitochondria into the cytosol, which appears as a transition from a punctate mitochondrial pattern to a diffuse cellular distribution. This technique revealed that cytochrome c and Smac/DIABLO release during UV-induced apoptosis occurs within the same time window, coinciding with mitochondrial membrane potential depolarization [21].
Functional Validation Using Genetic and Pharmacological Tools
Confirming the functional requirement of specific components strengthens validation studies.
- Bax/Bak Dependency: Utilize Bax/Bak double-knockout (DKO) mouse embryonic fibroblasts (MEFs). Resistance to apoptosis in DKO cells, compared to wild-type cells, confirms the essential gateway function of Bax/Bak for a given stimulus [20]. Reconstitution experiments (re-introducing Bax or Bak) can rescue the apoptotic phenotype [18].
- Caspase Dependency: To determine if a process is caspase-dependent, pre-treat cells with a broad-spectrum caspase inhibitor like z-VAD-fmk. If cytochrome c release is inhibited, it suggests a caspase-amplification loop is involved. In contrast, cytochrome c release is often caspase-independent, while Smac/DIABLO release can be caspase-dependent in some contexts [19] [21].
- IAP Antagonism: To demonstrate Smac/DIABLO's functional role, cells can be transfected with a cytosolic, active form of Smac. In Bax-deficient cells, which are resistant to TRAIL, this expression is sufficient to reconstitute TRAIL sensitivity by overcoming XIAP-mediated caspase inhibition [18].
The following diagram illustrates the interconnected experimental workflow for validating key events in intrinsic apoptosis.
Research Reagent Solutions
A successful apoptosis validation study requires a toolkit of reliable reagents. The table below lists essential materials and their applications, as evidenced by the cited research.
Table 3: Essential Research Reagents for Apoptosis Pathway Validation
| Reagent / Tool | Primary Function / Application | Example Use in Research |
|---|---|---|
| Bax-/- / Bak-/- DKO MEFs | Determines the essential gateway role of Bax/Bak for specific death stimuli [20] | Testing resistance to TRAIL or other agents [18] |
| Bcl-2 Overexpression System | Inhibits MOMP, blocking cytochrome c and Smac release [18] [19] | Validating mitochondrial pathway involvement [18] |
| Pan-Caspase Inhibitor (z-VAD-fmk) | Distinguishes caspase-dependent and independent events [19] [21] | Testing if protein release requires caspase activity [19] |
| Subcellular Fractionation (Digitonin) | Separates cytosolic and mitochondrial fractions [19] | Detecting cytochrome c/Smac translocation via immunoblotting [19] |
| Fluorescent Protein Tags (GFP/YFP) | Labels proteins for live-cell imaging of release kinetics [21] | Real-time tracking of Cytochrome c/Smac release in single cells [21] |
| Recombinant Active Smac | Antagonizes XIAP to promote caspase activity [18] | Reconstituting apoptosis in resistant (e.g., Bax-deficient) cells [18] |
| Serine Protease Inhibitors (AEBSF, TPCK) | Inhibits Bax/Bak-independent cytochrome c release [20] | Investigating alternative death mechanisms [20] |
Integrated Signaling Pathway
The following diagram integrates the molecular events, regulatory proteins, and experimental tools into a cohesive visual representation of the intrinsic apoptosis pathway and its key validation points.
The Apoptosome Formation and Caspase-9 Activation Cascade
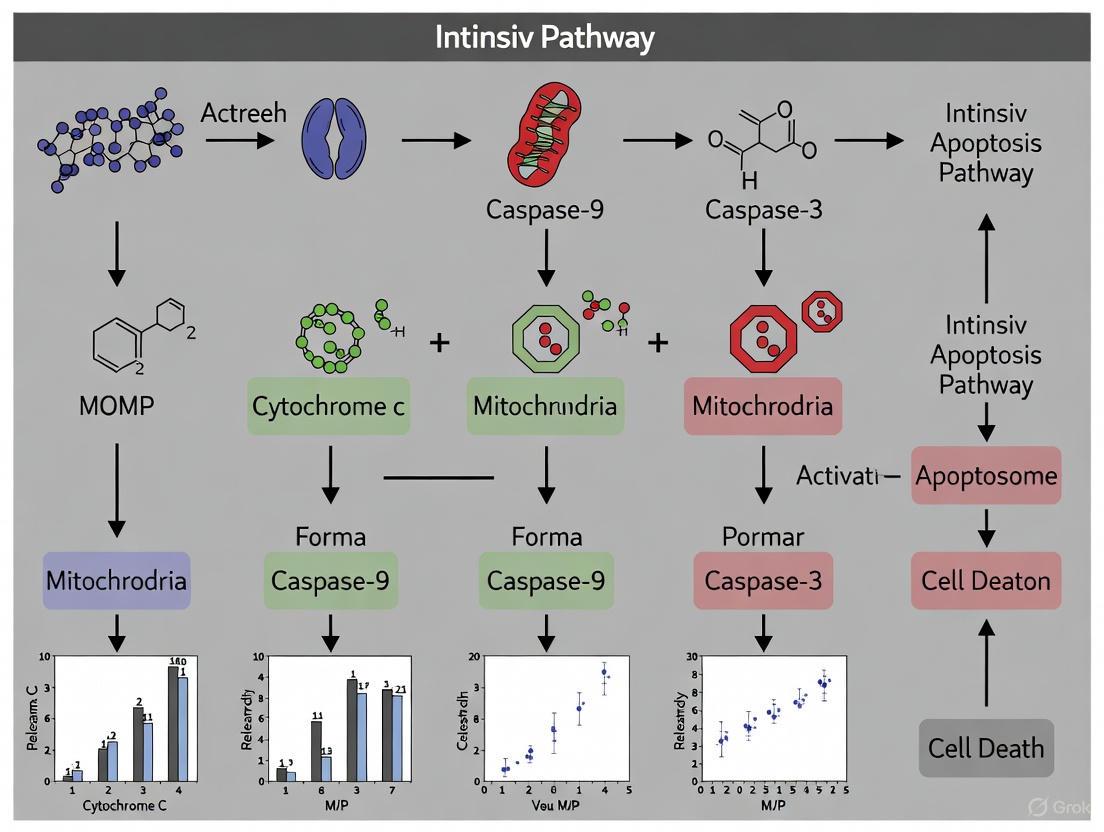
The intrinsic apoptotic pathway, a cornerstone of programmed cell death, is critical for maintaining cellular homeostasis and eliminating damaged cells. The apoptosome stands as the central signaling hub of this pathway, a large protein complex whose formation is triggered by internal cellular stressors such as DNA damage, oxidative stress, or chemotherapeutic agents [22] [23]. The core event in intrinsic apoptosis is the mitochondrial outer membrane permeabilization (MOMP), which leads to the release of cytochrome c from the mitochondrial intermembrane space into the cytosol [24] [25]. Once in the cytosol, cytochrome c, in the presence of (d)ATP, binds to the protein Apoptotic Protease-Activating Factor 1 (Apaf-1), inducing a conformational change that promotes Apaf-1 oligomerization into a wheel-like structure with sevenfold symmetry—the apoptosome [22] [24] [25]. This heptameric scaffold then recruits and activates the initiator caspase, caspase-9, which subsequently triggers a proteolytic cascade leading to controlled cellular disassembly [22] [23] [24]. Given its pivotal role in deciding cell fate, validating the activation of this pathway is a fundamental objective in cancer research, drug development, and the study of degenerative diseases. This guide provides a comparative analysis of the core mechanisms and the experimental methodologies essential for this validation.
Core Activation Mechanism: Dimerization vs. Allosteric Regulation
The precise mechanism by which the apoptosome activates caspase-9 has been the subject of intense scientific debate, primarily between two models: the "induced proximity" (dimerization) model and the "holoenzyme" (allosteric) model. A summary of their characteristics is provided in Table 1.
Table 1: Comparison of Caspase-9 Activation Models
| Feature | Induced Proximity / Dimerization Model | Holoenzyme / Allosteric Model |
|---|---|---|
| Core Principle | Apoptosome serves as a platform to concentrate caspase-9 monomers, facilitating homodimerization and trans-activation [22] [26]. | Apoptosome induces conformational changes within a monomeric caspase-9, allosterically activating it without requiring dimerization [24]. |
| Role of Apoptosome | Platform to increase local concentration of caspase-9 [22]. | Allosteric activator that directly modifies caspase-9 conformation [24]. |
| Key Evidence | Activation of caspase-9 by kosmotropic salts; functional replacement of caspase-9 recruitment domain with that of caspase-8 [26]. | Sustained catalytic activity of caspase-9 is dependent on continued binding to the apoptosome [22]. |
| Status of Bound Caspase-9 | Can form homodimers on the apoptosome platform [25]. | Primarily monomeric until substrate binding induces dimerization [27] [24]. |
Recent research has begun to reconcile these models. A landmark 2023 study using methyl-TROSY NMR spectroscopy revealed that the protease domain of caspase-9 remains predominantly monomeric while tethered to the apoptosome. However, the apoptosome "primes" caspase-9, enabling it to undergo rapid and extensive dimerization specifically upon substrate binding [27] [24]. This mechanism adds a crucial layer of regulation, ensuring caspase-9 is only fully active when its downstream targets are available. Furthermore, studies show the apoptosome can also facilitate the formation of caspase-9/Apaf-1 heterodimers, which may represent a distinct, highly active form of the enzyme [25].
The following diagram illustrates this integrated activation pathway and the associated validation experiments detailed in subsequent sections.
Comparative Experimental Data for Pathway Validation
Validating apoptosome-mediated caspase-9 activation requires a multifaceted approach. The table below summarizes quantitative and qualitative data from key experimental methodologies, providing a benchmark for interpreting results.
Table 2: Key Assays for Validating Apoptosome Formation and Caspase-9 Activation
| Experimental Method | Key Readout / Parameter | Typical Results & Data Interpretation | Context from Search Results |
|---|---|---|---|
| In Vitro Apoptosome Reconstitution + Activity Assay | Caspase-9 activity (Vmax/Km) using fluorogenic substrate (e.g., LEHD-amc). | ProC9-TM (uncleavable mutant): Higher Vmax, sustained activity [25].C9-p35/p12 (cleaved form): Lower Vmax, activity decays (molecular timer) [25] [22]. | Demonstrates functional consequences of caspase-9 binding and processing [22] [25]. |
| Size-Exclusion Chromatography with Multi-Angle Light Scattering (SEC-MALS) | Molecular weight (MW) and oligomeric state of proteins/complexes in solution. | Caspase-9 PD (no substrate): ~30 kDa (Monomer) [24].Caspase-9 PD + Z-LEHD-fmk: ~60 kDa (Dimer) [24].ProC9-TM (40 µM): Can form dimers; C9-p35/p12 does not [25]. | Directly probes the oligomeric state critical for the activation mechanism [24] [25]. |
| Methyl-TROSY NMR Spectroscopy | Atomic-level dynamics and conformation of proteins within large complexes. | Caspase-9 PD is monomeric on apoptosome; dimerizes only upon substrate addition [27] [24]. | Provided pivotal evidence for the substrate-induced dimerization model [27] [24]. |
| Crosslinking + Immunoblotting | Detection of specific protein-protein interactions and complexes. | Proof of caspase-9 homodimerization within the apoptosome [25]. | Direct biochemical evidence for proximity-induced dimerization [25]. |
Detailed Experimental Protocols
This section outlines core protocols for reconstituting the apoptosome and assessing its activity, which are fundamental for validating pathway activation.
Protocol 1: Reconstitution of the Apoptosome and Analysis of Caspase-9 Activation
This protocol is adapted from methodologies used to demonstrate caspase-9 dimerization and allosteric regulation [24] [26] [25].
Workflow Diagram:
Materials:
- Recombinant Proteins: Human Apaf-1, cytochrome c, and caspase-9 (wild-type and mutants like non-cleavable ProC9-TM [25]).
- Buffers: Reconstitution Buffer (e.g., 20 mM HEPES-KOH pH 7.5, 100 mM NaCl, 5 mM MgCl₂, 1 mM DTT).
- Cofactor: 1 mM dATP.
- Activity Assay Reagents: Fluorogenic caspase-9 substrate (e.g., Ac-LEHD-amc or Ac-LEHD-afc) and reaction buffer.
Step-by-Step Method:
- Apoptosome Reconstitution: Combine purified Apaf-1 (e.g., 1 µM) with a molar excess of cytochrome c (e.g., 5 µM) and 1 mM dATP in reconstitution buffer. Incubate the mixture at 30°C for 30-60 minutes to allow for complex assembly [25].
- Caspase-9 Recruitment: Add purified procaspase-9 (or its variants) to the reconstituted apoptosome and incubate further to allow binding and activation.
- Activity Measurement: Dilute an aliquot of the activation mixture into a plate containing reaction buffer with the LEHD-amc substrate (e.g., 100 µM). Monitor the increase in fluorescence (excitation/~380 nm, emission/~460 nm) over time using a plate reader. Compare the initial rates of hydrolysis (V₀) between different caspase-9 variants (e.g., ProC9-TM vs. C9-p35/p12) to assess the impact of processing on activity [25].
- SEC-MALS Analysis: To directly determine the oligomeric state, inject the apoptosome-caspase-9 complex onto a size-exclusion column coupled to MALS and refractive index detectors. This will confirm the formation of the high molecular weight apoptosome (~1.1 MDa) and can provide information on the stoichiometry of bound caspase-9 [24] [25].
Protocol 2: Investigating Activation via NMR Spectroscopy
This advanced protocol provides unique insights into the dynamics of caspase-9 activation, as demonstrated in recent studies [27] [24].
Workflow Diagram:
Materials:
- Isotope-Labeled Protein: Uniformly deuterated caspase-9 with protonated, ¹³C-labeled methyl groups on isoleucine, leucine, and valine residues.
- NMR Instrumentation: High-field NMR spectrometer (e.g., 600 MHz or higher) equipped with a cryogenic probe.
- Apoptosome Mimic or Native Complex: Either the native Apaf-1 apoptosome or an engineered mimic (e.g., based on a heptameric proteasome scaffold) [24].
- Substrate/Inhibitor: The tetrapeptide inhibitor Z-LEHD-fmk, which acts as a substrate mimic and traps the active conformation [24].
Step-by-Step Method:
- Sample Preparation: Reconstitute the apoptosome complex with the isotopically labeled caspase-9 in a suitable NMR buffer.
- NMR Data Acquisition: Collect methyl-TROSY NMR spectra of the caspase-9/apoptosome complex. The quality of the spectra will confirm whether the protease domain is flexibly tethered to the large scaffold.
- Substrate Titration: Titrate the substrate mimic Z-LEHD-fmk into the sample and acquire subsequent NMR spectra.
- Data Interpretation: Spectral changes, such as line broadening or chemical shift perturbations, upon substrate addition indicate a change in the oligomeric state and dynamics—specifically, the transition of the caspase-9 protease domain from a monomeric to a dimeric state [24].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Apoptosome and Caspase-9 Research
| Reagent / Material | Function / Utility | Example Use Case |
|---|---|---|
| Recombinant Apaf-1 | Core scaffold protein for in vitro reconstitution of the apoptosome. | Required for building the functional activation platform from purified components [25]. |
| Caspase-9 Mutants (e.g., ProC9-TM, F404D) | Mechanistic probes. ProC9-TM (non-cleavable) tests processing effects. F404D (dimerization-deficient) tests dimerization role [25]. | Comparing activity of ProC9-TM vs wild-type reveals the molecular timer function of autoprocessing [22] [25]. |
| Fluorogenic Substrate (Ac-LEHD-amc/afc) | Quantitative measurement of caspase-9 enzymatic activity. | Used in in vitro activity assays to determine kinetic parameters (Vmax, Km) of apoptosome-bound caspase-9 [25]. |
| Caspase-9 Inhibitor (Z-LEHD-fmk) | Irreversibly inhibits and traps caspase-9 in its active conformation. Used as a substrate mimic. | Essential for stabilizing the caspase-9 dimer for structural studies like SEC-MALS and NMR [24] [25]. |
| Antibodies (Anti-Caspase-9, Anti-cleaved-Caspase-9) | Detect expression, recruitment, and activation status in cell lysates or tissues. | Immunoblotting to confirm caspase-9 activation in a disease model (e.g., pulmonary fibrosis) [28]. |
| Kosmotropic Salts (e.g., Ammonium Citrate) | Induce protein dimerization in solution by excluding volume and enhancing hydrophobic interactions. | Enforces dimerization of caspase-9 independently of the apoptosome, testing the induced proximity model [26] [25]. |
The current model of caspase-9 activation is nuanced, integrating elements of both dimerization and allosteric regulation. The apoptosome functions as a regulated activation platform that recruits and primes caspase-9 monomers, which then undergo substrate-induced dimerization to achieve full catalytic power [27] [24]. This mechanism ensures tight control over the initiation of cell death. To conclusively validate the activation of the intrinsic pathway, a combination of biochemical, biophysical, and cellular assays is required. Researchers should correlate data from in vitro activity assays, which show enhanced and sustained activity with non-cleavable caspase-9, with structural data from SEC-MALS or NMR that confirm the expected oligomeric states. Furthermore, using dimerization-deficient mutants serves as a critical negative control. Mastering these techniques and their interpretation is essential for accurately screening novel chemotherapeutic agents, understanding disease mechanisms involving dysregulated apoptosis, and advancing drug development in oncology and neurodegeneration.
Morphological Hallmarks of Intrinsic Apoptosis
The intrinsic apoptosis pathway is a genetically programmed form of cell death essential for development, tissue homeostasis, and disease prevention. For researchers and drug development professionals, validating its activation is crucial in areas ranging from cancer therapy development to toxicological assessment. Unlike accidental cell death, intrinsic apoptosis is a tightly regulated process characterized by specific morphological and biochemical events that occur in a predictable sequence [29] [30]. This guide provides a comprehensive comparison of the key morphological hallmarks and the experimental methodologies used to detect them, offering a practical framework for confirming pathway activation in research settings.
Core Morphological Hallmarks of Intrinsic Apoptosis
The intrinsic apoptosis pathway, also known as the mitochondrial pathway, presents a stereotypical sequence of morphological changes that distinguish it from other cell death modalities such as necrosis. These hallmarks represent critical checkpoints for validation [30].
Table 1: Core Morphological Hallmarks of Intrinsic Apoptosis
| Morphological Hallmark | Description | Biological Significance | Contrast with Necrosis |
|---|---|---|---|
| Cell Shrinkage | Reduction in cell volume and density, disintegration of cell junctions, and detachment from extracellular matrix [29] [30]. | One of the earliest and most ubiquitous events, driven by ion efflux and water loss [30]. | Necrotic cells undergo swelling (oncosis) and only lyse at late stages [31]. |
| Chromatin Condensation | Nuclear chromatin condenses into compact, sharply delineated masses, often in a crescent shape at the nuclear periphery [29] [30]. | Indicates irreversible commitment to death; results from caspase-mediated degradation of nuclear proteins [30]. | Necrosis features karyolysis (nuclear dissolution) without organized, dense chromatin aggregates [30]. |
| Mitochondrial Outer Membrane Permeabilization (MOMP) | The outer mitochondrial membrane becomes permeable, leading to the release of pro-apoptotic proteins like cytochrome c [29] [3]. | The "point of no return" for intrinsic apoptosis; triggers caspase activation cascade [8] [32]. | Not a defined feature of necrosis; instead, mitochondria often appear swollen and degraded [29]. |
| Membrane Blebbing | The plasma membrane forms dynamic, outward protrusions (blebs) due to cytoskeletal disruption and caspase-mediated ROCK-I activation [30] [32]. | A hallmark of the execution phase; facilitates the formation of apoptotic bodies [30]. | Not a typical feature; necrotic membranes may rupture but do not systematically bleb [29]. |
| Formation of Apoptotic Bodies | The cell fragments into small, sealed membrane vesicles containing intact organelles and condensed nuclear fragments [29] [30]. | "Packages" the dying cell for efficient clearance by phagocytes, preventing inflammatory responses [30]. | Necrosis leads to membrane rupture and disorganized leakage of cellular contents, provoking inflammation [29] [31]. |
Advanced Imaging Techniques for Morphological Assessment
Accurate validation requires imaging techniques capable of resolving these subcellular morphological changes. The choice of methodology balances resolution, label requirements, and the ability to monitor dynamics.
Table 2: Comparison of Imaging Techniques for Apoptosis Morphology
| Technique | Principle | Key Advantages for Apoptosis Research | Key Limitations |
|---|---|---|---|
| Fluorescence Microscopy | Uses fluorescent dyes (e.g., Hoechst) or antibodies to label specific cellular components [30]. | High specificity for nuclear morphology (condensation, fragmentation); widely accessible; allows multiplexing with biochemical markers (e.g., caspase activation) [33] [30]. | Requires staining, which can be invasive and cause photobleaching; provides primarily 2D information [31]. |
| Transmission Electron Microscopy (TEM) | Images ultrathin cell sections with a beam of electrons to reveal ultrastructural details [30]. | Gold standard for morphology; reveals definitive features like MOMP, chromatin crescents, and organelle integrity with nanometer resolution [30]. | Requires sample fixation (not live-cell); labor-intensive; low throughput; expensive [30] [31]. |
| Full-Field Optical Coherence Tomography (FF-OCT) | A label-free interferometric technique that captures high-resolution 3D tomography of living cells [31]. | Non-invasive, label-free, and real-time visualization of dynamics like membrane blebbing and cell shrinkage in 3D; no photobleaching [31]. | Lower resolution than TEM; limited molecular specificity unless combined with other probes [31]. |
| Quantitative Phase Microscopy (QPM) | Measures phase shifts in transmitted light to map cell density and morphology without labels [31]. | Label-free and non-invasive; quantitative analysis of cell shrinkage and dry mass distribution [31]. | Can struggle with low contrast internal structures; complex data processing; typically 2D [31]. |
Visualizing the Process: Intrinsic Apoptosis Signaling Pathway
The following diagram illustrates the key molecular events of the intrinsic apoptosis pathway, from the initial stress signal to the execution of morphological changes.
Experimental Protocols for Validation
Combining multiple methods provides the most robust validation of intrinsic apoptosis. Below are detailed protocols for key experiments.
Protocol 1: Morphological Assessment via Fluorescence Microscopy
This is a widely used method for identifying classic nuclear hallmarks [30].
- Key Reagents: Hoechst 33342 or DAPI stain; cell culture with appropriate apoptotic inducer (e.g., 5 μmol/L doxorubicin [31]); paraformaldehyde (4%) for fixation; permeabilization buffer (e.g., 0.1% Triton X-100) [30].
- Procedure:
- Induction and Fixation: Treat cells with the apoptotic stimulus for the desired time. Wash cells with PBS and fix with 4% paraformaldehyde for 15 minutes at room temperature.
- Permeabilization and Staining: Permeabilize cells with 0.1% Triton X-100 in PBS for 10 minutes. Stain with Hoechst 33342 (e.g., 1 μg/mL) for 10-15 minutes, protected from light.
- Imaging and Analysis: Wash slides and mount for microscopy. Image using a UV filter set. Apoptotic nuclei are identified by intensely stained, condensed chromatin, often marginated at the nuclear periphery, and nuclear fragmentation [30].
Protocol 2: Real-Time, Label-Free Dynamics with FF-OCT
This protocol leverages FF-OCT to monitor morphological changes in living cells without labels [31].
- Key Reagents: Live cell culture (e.g., HeLa cells); apoptotic inducer (e.g., 5 μmol/L doxorubicin); culture chamber for microscopy [31].
- Procedure:
- System Setup: Use a custom-built time-domain FF-OCT system with a broadband halogen light source and high-NA water immersion objectives for sub-micrometer resolution [31].
- Image Acquisition: Initiate time-lapse imaging immediately after adding the apoptotic inducer. Acquire en face (x-y) cross-sectional images and z-stacks at regular intervals (e.g., every 20 minutes) for several hours.
- Analysis: Reconstruct 3D surface topography. Identify and track the progression of key events: initial cell contraction, subsequent membrane blebbing, and the formation of echinoid spines and filopodia reorganization [31].
Protocol 3: Correlative Assay Combining Morphology and Biochemistry
This protocol combines morphological analysis with a key biochemical marker of apoptosis.
- Key Reagents: reagents for fluorescence microscopy; primary antibody against cleaved caspase-3; fluorescently-labeled secondary antibody; Annexin V-FITC conjugate to detect phosphatidylserine (PS) externalization [29] [33].
- Procedure:
- Live-Cell Staining for PS Exposure: Incubate live cells with Annexin V-FITC in a binding buffer for 15 minutes. Rinse and acquire fluorescence images to detect green fluorescence on the cell membrane, an early marker of apoptosis.
- Fixation and Staining for Caspase-3: Fix and permeabilize the same cells. Incubate with an anti-cleaved-caspase-3 antibody, followed by a fluorescent secondary antibody (e.g., red).
- Correlative Analysis: Image the cells. Co-localization of condensed/fragmented nuclei (by Hoechst), PS externalization (Annexin V), and cleaved caspase-3 signal provides multi-parametric, definitive evidence of intrinsic apoptosis execution [29] [33].
Visualizing the Workflow: Experimental Validation Strategy
The following diagram outlines a logical workflow for validating intrinsic apoptosis by integrating multiple experimental approaches.
The Scientist's Toolkit: Key Research Reagents
A curated list of essential reagents for studying the morphological hallmarks of intrinsic apoptosis is provided below.
Table 3: Essential Research Reagents for Intrinsic Apoptosis Studies
| Reagent / Tool | Function / Target | Key Application in Validation |
|---|---|---|
| Hoechst 33342 / DAPI | Cell-permeable DNA dyes that bind AT-rich regions [30]. | Staining of nuclei to identify chromatin condensation and nuclear fragmentation by fluorescence microscopy [30]. |
| Anti-Cleaved Caspase-3 Antibody | Antibody specific to the activated, cleaved form of caspase-3 [29] [33]. | Immunofluorescence or Western blot detection of effector caspase activation, a key downstream biochemical event [29] [33]. |
| Annexin V Conjugates | Protein that binds phosphatidylserine (PS) when exposed on the outer leaflet of the plasma membrane [29]. | Flow cytometry or microscopy to detect an early marker of apoptosis (PS externalization), often used with viability dyes to exclude necrotic cells [29]. |
| OptoBAX System | Optogenetic construct (Cry2-CIB-BAX) for light-controlled BAX activation [33] [34]. | Precise, temporal initiation of MOMP to study the direct consequences of intrinsic pathway activation on cell morphology in real time [33] [34]. |
| Doxorubicin | Anthracycline chemotherapeutic agent that intercalates into DNA, causing double-strand breaks [31]. | A well-characterized chemical inducer of the intrinsic apoptosis pathway, activating p53 and generating ROS [31]. |
| Z-VAD(OMe)-FMK | Broad-spectrum, cell-permeable caspase inhibitor [33]. | A control tool to confirm the caspase-dependence of the observed cell death morphology [33]. |
Validating the activation of the intrinsic apoptosis pathway relies on the definitive identification of its characteristic morphological hallmarks, primarily cell shrinkage, chromatin condensation, and membrane blebbing. No single assay is sufficient; confidence is achieved through a multi-parametric approach that correlates these distinct morphological features with key biochemical events like caspase-3 cleavage. The choice of imaging technique—from standard fluorescence microscopy to advanced label-free methods like FF-OCT—depends on the specific research needs regarding throughput, resolution, and the ability to monitor dynamics. By applying the compared methods and reagents detailed in this guide, researchers can robustly validate intrinsic apoptosis activation in both basic research and drug discovery contexts.
A Practical Toolkit: Assays and Techniques for Detecting Intrinsic Apoptosis
Assessing Mitochondrial Membrane Potential (ΔΨm) with JC-1 and TMRM
The intrinsic apoptosis pathway is a precisely regulated mechanism of programmed cell death, and one of its defining early events is the disruption of mitochondrial integrity. A critical change during this activation is the collapse of the mitochondrial membrane potential (ΔΨm), an electrochemical gradient across the inner mitochondrial membrane that is essential for energy production and serves as a key indicator of mitochondrial health [35] [36]. Accurately measuring this depolarization is therefore fundamental for validating the initiation of the intrinsic apoptotic cascade. Among the most widely used tools for this purpose are the fluorescent dyes JC-1 and Tetramethylrhodamine Methyl Ester (TMRM). This guide provides a detailed comparison of these two dyes, equipping researchers with the knowledge to select the appropriate probe and implement optimal protocols for detecting apoptosis in their experimental systems.
Dye Comparison: Mechanism, Performance, and Data Interpretation
JC-1 and TMRM operate on distinct photophysical principles, leading to different strengths and considerations for application in apoptosis detection.
JC-1: Ratiometric Measurement via J-Aggregates
JC-1 is a unique ratiometric dye that exhibits potential-dependent accumulation within mitochondria. Its key feature is the formation of two distinct fluorescent species:
- At low ΔΨm (depolarized): JC-1 exists in a monomeric state, emitting green fluorescence (emission peak ~529 nm).
- At high ΔΨm (polarized): JC-1 accumulates sufficiently to form J-aggregates, which emit red fluorescence (emission peak ~590 nm) [35] [37].
The quantitative ratio of red-to-green fluorescence is directly dependent on the ΔΨm. Consequently, during the early stages of apoptosis, a decrease in this ratio signals mitochondrial depolarization. This internal ratio control makes JC-1 less sensitive to variables like mitochondrial density, dye concentration, and cell size, which can be a significant advantage [35].
TMRM: Intensity-Based Measurement
TMRM is a cationic, lipophilic dye that accumulates in the mitochondrial matrix in a Nernstian manner, driven by the negative potential inside. It functions as a single-wavelength, intensity-based probe.
- In healthy, polarized mitochondria, TMRM is concentrated within the organelles, yielding bright fluorescence.
- Upon depolarization, the dye diffuses out into the cytoplasm, leading to a dimming of the mitochondrial signal and a concomitant increase in general cytoplasmic fluorescence [38] [39] [40].
A critical methodological consideration for TMRM is the use of sub-quenching concentrations, where the fluorescence intensity remains proportional to the dye concentration and, by extension, the ΔΨm. High, quenching concentrations must be avoided, as they distort this relationship [40].
Table 1: Key Characteristics of JC-1 and TMRM
| Feature | JC-1 | TMRM |
|---|---|---|
| Measurement Type | Ratiometric (Red/Green) | Intensity-based |
| Signal Change with Depolarization | ↓ Red/Green Ratio | ↓ Fluorescence Intensity |
| Excitation/Emission (Monomer/Aggregate) | 514/529 nm (Monomer), 514/590 nm (J-Aggregate) [35] | ~548/573 nm [37] |
| Advantages | - Internal calibration via ratio- Less sensitive to dye loading & cell size- Clear visual distinction | - Simpler setup and analysis- Suitable for kinetic live-cell imaging- Lower inhibition of electron transport chain [37] |
| Disadvantages | - More complex data analysis- J-aggregate formation can be influenced by factors beyond ΔΨm [41] | - Signal depends on exact dye loading & cell volume- Requires careful control of concentration to avoid quenching |
Table 2: Performance in Experimental Applications
| Application | JC-1 | TMRM |
|---|---|---|
| Flow Cytometry | Excellent for identifying distinct cell populations based on red/green ratio [35] | Well-suited, but requires careful gating based on intensity shifts [38] |
| Fluorescence Microscopy | Provides a color-coded map of mitochondrial health [35] | Excellent for high-resolution imaging and tracking dynamics over time [38] [40] |
| Kinetic Studies | Less ideal due to slow response to rapid potential changes | Excellent for real-time monitoring of ΔΨm flickering or oscillations [39] |
| Sensitivity to ΔΨm Loss | High; pronounced color shift from red to green | High; clear loss of intense punctate staining [38] |
Experimental Protocols for Apoptosis Detection
JC-1 Staining Protocol for Flow Cytometry
This protocol is optimized for detecting apoptosis-induced ΔΨm loss in cell populations.
- Cell Preparation: Harvest and wash cells in phosphate-buffered saline (PBS). Adjust cell concentration to 1-5 x 10^6 cells/mL in pre-warmed serum-free medium or PBS.
- Staining: Incubate cells with 2-5 μM JC-1 for 15-30 minutes at 37°C in the dark [35]. Note: The optimal concentration and time should be determined empirically for each cell type.
- Washing: Centrifuge cells and gently resuspend in fresh pre-warmed buffer to remove excess dye.
- Analysis: Analyze cells immediately on a flow cytometer equipped with a 488 nm laser. Collect green monomer fluorescence using a ~530/30 nm bandpass filter (FITC channel) and red J-aggregate fluorescence using a ~585/42 nm bandpass filter (PE channel).
- Data Interpretation: Plot the red (PE) vs. green (FITC) fluorescence. A healthy population will show high red and low green signal, while apoptotic cells will exhibit decreased red and increased green fluorescence. The results can be quantified as the ratio of geometric mean fluorescence intensities (PE/FITC) [35].
TMRM Staining Protocol for Live-Cell Imaging
This protocol is designed for quantifying ΔΨm in adherent cells using microscopy.
- Cell Preparation: Plate cells on glass-bottom culture dishes and allow them to adhere under normal growth conditions.
- Dye Loading: Replace the culture medium with a pre-warmed, dye-containing solution with 10-50 nM TMRM. Incubate for 15-30 minutes at 37°C in the dark to allow for dye equilibration [38] [40].
- Image Acquisition: For live-cell imaging, maintain the TMRM at a low concentration (e.g., 5-10 nM) in the imaging medium to prevent dye loss. Use a microscope with a Texas Red or Cy3 filter set. Take care to use low illumination intensity to avoid phototoxicity and dye bleaching.
- Controls: Include a control well treated with a depolarizing agent like FCCP (1-10 μM) or CCCP (50-100 μM) for 10 minutes prior to imaging to confirm the specificity of the signal [38].
- Data Analysis: Quantify the fluorescence intensity of individual mitochondria or regions of interest (ROIs) over time. A drop in intensity indicates depolarization. Normalize the fluorescence to the initial baseline value (F/F₀) for kinetic analysis.
Integration into the Intrinsic Apoptosis Pathway
The loss of ΔΨm is not merely a biomarker but a pivotal event in the intrinsic apoptosis pathway, and its detection with JC-1 or TMRM serves as a key validation point. The diagram below illustrates how this measurement fits into the broader apoptotic cascade.
Diagram 1: The role of ΔΨm loss in the intrinsic apoptosis pathway. Detection of ΔΨm collapse with JC-1 or TMRM validates the critical step between mitochondrial outer membrane permeabilization and the release of pro-apoptotic factors.
As visualized, the drop in ΔΨm typically coincides with or immediately precedes the opening of the mitochondrial permeability transition pore (MPTP) or other forms of outer membrane permeeabilization, leading to the release of cytochrome c and other intermembrane space proteins [35]. This irreversible step commits the cell to the caspase-dependent execution phase of apoptosis. Therefore, quantitatively measuring this depolarization event provides critical evidence for the activation of the intrinsic pathway.
The Scientist's Toolkit: Essential Reagents for Validation
A robust validation of apoptosis requires a multi-parameter approach. Beyond ΔΨm dyes, the following reagents are essential for correlative analysis.
Table 3: Key Research Reagents for Apoptosis Pathway Validation
| Reagent | Function in Apoptosis Research |
|---|---|
| JC-1 | Ratiometric fluorescent dye to detect loss of ΔΨm, an early marker of intrinsic apoptosis activation [35]. |
| TMRM/TMRE | Intensity-based fluorescent dyes for quantifying ΔΨm; ideal for kinetic imaging studies with minimal respiratory chain impact [38] [37]. |
| Annexin V | Binds to phosphatidylserine (PS) externalized on the cell surface, a mid-stage marker of apoptosis. Often used with propidium iodide (PI) to distinguish early apoptotic (Annexin V+/PI-) from late apoptotic/necrotic (Annexin V+/PI+) cells [42]. |
| Propidium Iodide | A membrane-impermeant DNA dye that identifies dead cells with compromised plasma membrane integrity. Used to exclude necrotic cells or label late-stage apoptotic cells [42]. |
| Caspase Inhibitors & Substrates | Fluorogenic caspase substrates (e.g., for caspase-3) directly measure the activity of key executioner enzymes. Inhibitors (e.g., Z-VAD-FMK) are used to confirm caspase-dependent apoptosis. |
| Mitochondrial Uncouplers | Chemicals like FCCP/CCCP that completely dissipate ΔΨm by shuttling protons across the inner membrane. Served as essential positive controls for ΔΨm staining experiments [38] [35]. |
Both JC-1 and TMRM are powerful and reliable tools for assessing mitochondrial membrane potential in the context of apoptosis research. The choice between them hinges on the specific experimental requirements. JC-1 is often superior for endpoint assays where its ratiometric property provides robust, semi-quantitative data that is less susceptible to technical artifacts, making it excellent for flow cytometry and confirming depolarization in heterogeneous cell populations. TMRM, conversely, is the preferred choice for detailed kinetic studies and high-resolution live-cell imaging, where its single-wavelength behavior and minimal impact on mitochondrial function allow for real-time observation of ΔΨm dynamics. For the most compelling validation of intrinsic apoptosis activation, scientists should consider integrating these ΔΨm measurements with other complementary assays, such as Annexin V staining and caspase activity tests, to build a comprehensive picture of the cell death process.
Within the intricate cascade of the intrinsic apoptosis pathway, the release of cytochrome c from the mitochondrial intermembrane space into the cytosol represents a decisive, point-of-no-return event. [43] [44] This process, primarily regulated by the BCL-2 protein family and triggered by cellular stress, culminates in the formation of the apoptosome and the activation of executioner caspases. [45] [46] [23] For researchers and drug development professionals, the accurate detection of cytochrome c release is therefore paramount for validating the induction of intrinsic apoptosis, especially when screening novel anti-cancer therapeutics designed to reactivate this programmed cell death in malignant cells. [45] [46] Among the plethora of available biochemical techniques, subcellular fractionation and immunofluorescence (IF) have emerged as two cornerstone methodologies. This guide provides a detailed, objective comparison of these techniques, complete with experimental protocols and data, to inform your choice of assay for validating intrinsic apoptosis pathway activation.
The Central Role of Cytochrome c in Apoptosis
The intrinsic apoptosis pathway is a tightly regulated mechanism essential for eliminating damaged or unwanted cells. Cytochrome c, a component of the mitochondrial electron transport chain, plays a dual role in cellular fate. Under normal physiological conditions, it is confined to the mitochondrial intermembrane space and is indispensable for aerobic ATP synthesis. [44] Upon receiving a potent apoptotic stimulus, such as DNA damage or photothermal stress, the mitochondrial outer membrane becomes permeabilized (MOMP). This event is controlled by the equilibrium of pro- and anti-apoptotic BCL-2 family proteins. [45] [44] The permeabilization allows cytochrome c to escape into the cytosol, where it binds to the adaptor protein APAF1. This binding triggers APAF1 oligomerization into a wheel-like signaling platform known as the apoptosome, which then recruits and activates the initiator caspase, caspase-9. [44] [23] Active caspase-9 subsequently cleaves and activates effector caspases-3 and -7, leading to the systematic proteolysis of cellular components and apoptotic cell death. [46] [23] Consequently, detecting the translocation of cytochrome c from the mitochondria to the cytosol serves as a definitive functional readout for the activation of this pathway.
The following diagram illustrates the key steps of the intrinsic apoptosis pathway, culminating in the critical event detected by the methods discussed in this guide.
Methodologies and Experimental Protocols
This section provides detailed, step-by-step protocols for subcellular fractionation and immunofluorescence, two widely used methods for detecting cytochrome c release.
Subcellular Fractionation Protocol
The subcellular fractionation protocol, often referred to as the "Lyse-and-Wash" (L&W) method, is designed to biochemically separate the cytoplasmic contents from the nucleus, allowing for independent analysis of cytochrome c localization. [47] [48] The following workflow outlines the key stages of this process.
Detailed Step-by-Step Protocol [47] [48]:
Cell Harvesting and Lysis:
- Harvest approximately 1-2 x 10^6 cells (e.g., HeLa or Caov-4) by gentle scraping or trypsinization, and pellet them by centrifugation.
- Crucially, perform all subsequent steps on ice or at 4°C.
- Resuspend the cell pellet in 1 mL of a hypotonic lysis solution containing 0.1% NP-40.
- Incubate the suspension on ice for 3 minutes to allow for plasma membrane disruption.
Separation of Cytoplasmic Fraction:
- Centrifuge the lysed cell suspension at 1,000 rcf for 5 minutes at 4°C.
- Carefully transfer the supernatant to a new, pre-chilled microcentrifuge tube. This is the cytoplasmic fraction.
- For a cleaner fraction, re-centrifuge this supernatant at 15,000 rcf for 3 minutes to pellet any residual debris or organelle contamination.
Nuclear Washing and Purification:
- The pellet from the first centrifugation contains the nuclei but is often contaminated with cytoplasmic organelles like the endoplasmic reticulum that remain attached to the outer nuclear membrane. [48]
- Resuspend this nuclear pellet in 1 mL of an isotonic buffer containing a higher concentration of 0.3% NP-40.
- Incubate on ice for several minutes and then pellet the nuclei again by centrifugation at 1,000 rcf for 5 minutes.
- Discard the supernatant. The resulting pellet is the purified nuclear fraction.
Downstream Analysis:
- Both the cytoplasmic and nuclear fractions can now be analyzed by Western blotting.
- Probe the blots with an antibody against cytochrome c.
- In healthy, non-apoptotic cells, cytochrome c should be detected only in the heavy mitochondrial fraction (often removed during the first debris spin) and be absent from the final cytoplasmic fraction. Upon apoptosis induction, a clear signal for cytochrome c will appear in the cytoplasmic fraction. [47]
- Essential Validation: Always confirm the purity of your fractions using specific markers:
Immunofluorescence Protocol
Immunofluorescence allows for the visualization of cytochrome c redistribution within the fixed cell, preserving valuable spatial context. The following workflow outlines the key stages of this protocol.
Detailed Step-by-Step Protocol [49] [50]:
Cell Preparation and Fixation:
- Grow cells on sterile glass coverslips placed in a culture dish until they reach 50-70% confluency.
- Treat cells with an apoptotic inducer (e.g., cisplatin, staurosporine) and include an untreated control.
- Aspirate the medium and wash the cells gently with phosphate-buffered saline (PBS).
- Fix the cells by incubating with a suitable fixative (e.g., 4% paraformaldehyde in PBS) for 15 minutes at room temperature.
- Wash the fixed cells three times with PBS for 5 minutes each.
Permeabilization and Blocking:
- Permeabilize the cells to allow antibody access by incubating with PBS containing 0.1% Triton X-100 (or NP-40) for 5 minutes at room temperature. [49]
- Wash the cells three times with PBS.
- To prevent non-specific antibody binding, incubate the coverslips in a blocking buffer (PBS with 0.1% Tween-20 and 5% serum from the host species of the secondary antibody) for 1-2 hours in a humidified chamber. [49]
Antibody Incubation:
- Prepare the primary antibody (e.g., anti-cytochrome c) diluted in the blocking buffer. A typical starting dilution is 1:200. [49]
- Apply 100 µL of the antibody solution to the coverslip and incubate overnight at 4°C in a humidified chamber.
- The next day, wash the coverslips three times for 10 minutes each with PBS/0.1% Tween 20 to remove unbound primary antibody.
- Prepare the fluorophore-conjugated secondary antibody (e.g., Alexa Fluor 488) diluted 1:500 in PBS. [49]
- Apply 100 µL of the secondary antibody solution to the coverslip and incubate for 1-2 hours at room temperature, protected from light.
- Perform a final series of washes (three times for 5 minutes each with PBS), keeping the samples protected from light.
Mounting and Imaging:
- Mount the coverslips onto glass slides using a compatible anti-fade mounting medium.
- Seal the edges with nail polish if necessary.
- Image the cells using a fluorescence or confocal microscope.
- Interpretation: In non-apoptotic cells, cytochrome c will display a punctate, filamentous pattern, colocalizing with mitochondrial markers. Upon apoptosis induction, this pattern is lost and replaced by a diffuse, cytoplasmic staining, signifying its release. [43] [44]
Objective Comparison of Techniques
The choice between subcellular fractionation and immunofluorescence depends heavily on the specific research question, required data output, and available resources. The table below summarizes the core characteristics of each method for a direct, objective comparison.
Table 1: Method Comparison at a Glance
| Feature | Subcellular Fractionation | Immunofluorescence (IF) |
|---|---|---|
| Core Principle | Biochemical separation of cellular compartments followed by Western blot. | In situ antibody-based detection and visualization via microscopy. |
| Spatial Resolution | Compartment-level (Cytoplasm vs. Nucleus). | Single-cell and sub-cellular (can show punctate vs. diffuse patterns). [43] |
| Data Output | Quantitative or semi-quantitative data on protein levels across fractions. | Qualitative and semi-quantitative spatial data. |
| Key Advantage | Objectivity and specificity; direct confirmation via fraction purity markers; suitable for downstream activity assays. [47] | Visual confirmation with morphological context; ability to perform multiplexing with other markers. [49] |
| Main Limitation | Loses single-cell and spatial information; potential for cross-contamination between fractions. [48] | Lower throughput; semi-quantitative; relies on antibody specificity and image interpretation. [49] |
| Throughput | Medium to High (can process multiple samples in parallel). | Low to Medium (time-consuming imaging and analysis). |
| Best Suited For | Validating release in bulk cell populations; when quantitative data is required. | Confirming heterogeneity of response within a population; assessing correlation with other cellular events. |
Supporting Experimental Data from Literature:
- Fractionation Validation: A study comparing fractionation methods demonstrated that the L&W protocol using NP-40 effectively isolated pure nuclear fractions, free from cytoplasmic contaminants like GAPDH and ERp29, and mitochondrial cytochrome c. This purity is critical for unambiguous interpretation of Western blot results. [48]
- High-Resolution IF: Advanced forms of IF, such as Surface-Enhanced Raman Spectroscopy (SERS), have been used to achieve spatial profiling of cytochrome c release at the single-cell level. This technique can map the distribution of cytochrome c around single apoptotic cells, revealing isotropic release patterns and enabling the study of variability between different cell lines. [43]
The Scientist's Toolkit: Essential Research Reagents
Successful execution of these protocols hinges on the quality and appropriate use of key reagents. The following table lists essential materials and their functions.
Table 2: Essential Research Reagents and Resources
| Reagent / Resource | Function / Application | Critical Considerations |
|---|---|---|
| Anti-Cytochrome c Antibody | Primary antibody for detection in both WB and IF. | Validate for specific applications (WB vs. IF). Confirm species reactivity. |
| NP-40 / Triton X-100 | Non-ionic detergent for cell membrane permeabilization (IF) and lysis (Fractionation). | Concentration is critical; optimize for different cell lines to avoid nuclear damage. [47] [48] |
| Protease Inhibitor Cocktail | Added to lysis and fractionation buffers to prevent protein degradation. | Essential for preserving the integrity of proteins like cytochrome c and caspases during processing. |
| Primary Antibody (e.g., GAPDH, Lamin B1) | Western blot markers for validating fraction purity. [47] | GAPDH for cytoplasm; Lamin B1 or PARP for nucleus. Confirmation of pure fractions is non-negotiable. |
| Fluorophore-Conjugated Secondary Antibody | For detection of primary antibody in IF. | Choose a fluorophore compatible with your microscope's lasers and filter sets. Protect from light. [49] |
| Coverslips & Mounting Medium | Support for cell growth during IF and preservation of fluorescence post-staining. | Use #1.5 thickness for high-resolution microscopy. Use anti-fade mounting medium. |
Both subcellular fractionation and immunofluorescence are robust and validated methods for detecting cytochrome c release, each offering distinct advantages. Subcellular fractionation coupled with Western blotting provides a population-averaged, biochemical confirmation that is highly objective and quantifiable, making it ideal for initial screening and validation studies in drug discovery. [47] [48] In contrast, immunofluorescence offers unparalleled spatial resolution at the single-cell level, allowing researchers to visualize the heterogeneity of apoptotic response within a population and correlate cytochrome c release with other morphological changes. [43] [49]
The optimal choice is context-dependent. For definitive, quantitative confirmation of intrinsic pathway activation, subcellular fractionation is the gold standard. When investigating the dynamics of release, heterogeneity, or requiring co-localization data, immunofluorescence is the superior technique. In many cases, these methods are not mutually exclusive but are used complementarily to provide a comprehensive validation of apoptosis, strengthening the conclusions drawn from any single experimental approach.
Measuring Bax/Bak Oligomerization and BCL-2 Family Interactions
Within the intrinsic apoptosis pathway, the oligomerization of BAX and BAK represents the decisive step that commits a cell to death. These pro-apoptotic effectors undergo major conformational changes to form pores in the mitochondrial outer membrane (MOM), leading to cytochrome c release and caspase activation [51] [52]. For researchers validating intrinsic apoptosis activation, accurately measuring BAX/BAK oligomerization and their interactions with regulatory BCL-2 family proteins is essential. This guide objectively compares key methodological approaches, providing supporting experimental data and protocols to inform your experimental design.
Key Techniques for Studying BAX/BAK Activation
The following techniques provide complementary insights into BAX/BAK conformation, interactions, and oligomerization states.
Table 1: Comparison of Core Methodologies for Studying BAX/BAK Oligomerization
| Method | Key Applications | Key Findings Enabled | Temporal Resolution | Spatial Context |
|---|---|---|---|---|
| Bimolecular Fluorescence Complementation (BiFC) | Protein-protein interactions in living cells [53] | Direct Bim-Bax interaction in cytosol prior to mitochondrial translocation [53] | Minutes to hours | Preserved in living cells |
| Computational Protein Design | High-affinity binder design; structure-function analysis [54] | BAK binders with 60 nM affinity; induction of BAK unfolding [54] | N/A (structural snapshots) | Atomic resolution |
| Super-Resolution Microscopy | Direct oligomer visualization; pore architecture [55] | BAK forms smaller structures faster than BAX; co-assembly into mixed pores [55] | Seconds to minutes | Mitochondrial membrane |
| Size Exclusion Chromatography (SEC) & Crosslinking | Oligomer separation and stabilization [56] | BCL-w dimers dissociate BAX oligomers [56] | Minutes | Solution-based |
Experimental Protocols for Key Assays
BiFC Analysis in Living Cells
The BiFC assay captures transient protein interactions in living cells by expressing target proteins fused to complementary fragments of a fluorescent protein [53].
Detailed Protocol:
- Construct Generation: Subclone cDNAs for BCL-2 family proteins (Bax, Bak, Bim, PUMA, Noxa, etc.) into BiFC plasmids containing Venus fragments (VN173 or VC155) [53].
- Cell Culture and Transfection: Culture adherent cells (e.g., HeLa) and transfect at 50-80% confluence using lipid-based transfection reagents. Adjust DNA amounts to achieve near-endogenous expression levels to avoid artificial complementation [53].
- Apoptosis Inhibition: Add 50 μM Z-VAD-fmk to prevent caspase-mediated cell death when expressing pro-apoptotic pairs [53].
- Image Acquisition and Analysis: After 24 hours, analyze BiFC complex formation by confocal or time-lapse fluorescence microscopy. Co-stain with MitoTracker Red to assess mitochondrial localization [53].
- Controls: Include untransfected cells and single-transfected controls for background fluorescence assessment.
Super-Resolution Imaging of BAX/BAK Oligomers
This technique enables visualization of apoptotic pore formation dynamics directly at the MOM [55].
Detailed Protocol:
- Cell Preparation: Plate cells on imaging-optimized dishes and induce apoptosis using appropriate stimuli (e.g., UV irradiation, staurosporine).
- Immunostaining: At defined apoptosis timepoints, fix cells and permeabilize. Incubate with primary antibodies against BAX and BAK, followed by fluorophore-conjugated secondary antibodies validated for super-resolution microscopy [55].
- Image Acquisition: Use STORM or PALM super-resolution systems to acquire images with 20-30 nm resolution.
- Quantitative Analysis: Apply specialized software to quantify oligomer size, distribution, and composition. BAK oligomers typically appear faster and smaller than BAX structures [55].
SEC-MALS for Oligomer Characterization
Size exclusion chromatography with multi-angle light scattering (SEC-MALS) determines absolute molecular weights of protein complexes in solution [56].
Detailed Protocol:
- Protein Preparation: Express and purify recombinant BAX or BAK. Treat monomeric BAX with Fos-12 detergent to induce oligomer formation [56].
- Chromatography: Inject samples onto an analytical SEC column equilibrated with appropriate buffer.
- Light Scattering Detection: Use inline MALS and refractive index detectors to determine absolute molecular weights without size standards.
- Data Analysis: Calculate molecular weights and oligomeric states using instrument software. BAX oligomers typically exhibit molecular weights ~150 kDa [56].
Apoptotic Pathway and Experimental Framework
The intrinsic apoptosis pathway involves a tightly regulated sequence of BCL-2 family protein interactions culminating in BAX/BAK-mediated mitochondrial outer membrane permeabilization (MOMP).
Diagram Title: BAX/BAK Activation in Intrinsic Apoptosis
Research Reagent Solutions
This table catalogs essential reagents and tools for studying BAX/BAK interactions and oligomerization.
Table 2: Essential Research Reagents for BAX/BAK Studies
| Reagent Category | Specific Examples | Research Application | Key Features |
|---|---|---|---|
| Expression Plasmids | pBiFC-VN173/VC155 [53] | Protein interaction studies | Venus fluorescent protein fragments for BiFC |
| BAX/BAK Binders | BAK-CDP02, BAX-CDP01 [54] | Modulating BAX/BAK activity | Computationally designed binders with nanomolar affinity |
| Cell Lines | HeLa (ATCC) [53] | Cellular localization studies | Well-characterized for apoptosis research |
| Apoptosis Inducers | Staurosporine, UV irradiation [55] | Inducing intrinsic apoptosis pathway | Activate BAX/BAK through different upstream signals |
| Detection Antibodies | Anti-BAX (6A7, conformational) [52] | Detecting activation-specific epitopes | Recognizes exposed N-terminal in active BAX |
| BH3 Peptides | BIM BH3, BID BH3 [54] [52] | In vitro activation studies | Directly activate BAX/BAK in biochemical assays |
Technical Considerations for Experimental Design
When designing studies to measure BAX/BAK oligomerization, several technical factors significantly impact data interpretation:
Cellular Context Matters: BAX primarily localizes to the cytosol and translocates to mitochondria during apoptosis, while BAK is constitutively mitochondrial [52]. This fundamental difference affects experimental approaches for each protein.
Dynamic Equilibrium Considerations: BAX exists in a dynamic equilibrium between cytosol and mitochondria, maintained through retrotranslocation by anti-apoptotic proteins like BCL-xL [52]. Disrupting this equilibrium during cell lysis can artificially alter oligomerization states.
Membrane Environment Effects: BAX/BAK oligomerization and function depend on specific membrane lipid compositions [56]. Pure lipid systems provide control but may lack native complexity.
Oligomer Heterogeneity: BAX and BAK form structurally distinct oligomers with different kinetics - BAK oligomerizes faster into smaller structures than BAX [55]. Mixed oligomers containing both proteins also occur, complicating attribution of specific functions.
Selecting the appropriate methodology for measuring BAX/BAK oligomerization requires careful consideration of the specific research question. BiFC offers unparalleled insight into dynamic interactions in living cells [53], while super-resolution microscopy directly visualizes oligomer architecture [55]. Biochemical approaches like SEC-MALS provide quantitative size characterization [56], and computational design enables precise mechanistic dissection through engineered binders [54]. For comprehensive validation of intrinsic apoptosis activation, combining multiple complementary techniques provides the most robust experimental evidence, as each method captures different aspects of the complex BAX/BAK activation cascade.
Quantifying Caspase-9 and Caspase-3/7 Activity with Fluorogenic Assays
The validation of intrinsic apoptosis pathway activation is a cornerstone of research in cell biology, cancer research, and drug development. Apoptosis, or programmed cell death, is an essential process for maintaining tissue homeostasis and eliminating damaged cells [57] [58]. The intrinsic apoptosis pathway, also known as the mitochondrial pathway, is initiated in response to cellular stress signals such as DNA damage, oxidative stress, or growth factor deprivation [57] [59]. This pathway is characterized by mitochondrial outer membrane permeabilization, leading to the release of cytochrome c into the cytosol [57]. Cytochrome c then binds to apoptotic protease-activating factor 1 (Apaf-1), forming a complex called the apoptosome, which is indispensable for the activation of the initiator caspase, caspase-9 [60] [57].
Caspases, a family of cysteine-aspartic proteases, are the central executioners of apoptosis [57]. They are synthesized as inactive zymogens and must undergo proteolytic cleavage to become activated [57]. Caspase-9 is a prototypical initiator caspase that, once activated by the Apaf-1 apoptosome, cleaves and activates the effector caspases-3 and -7 [60] [57]. These effector caspases then proceed to cleave numerous cellular substrates, including poly (ADP-ribose) polymerase (PARP), leading to the characteristic morphological changes of apoptosis, such as cell shrinkage, chromatin condensation, and DNA fragmentation [61] [62]. The critical positioning of caspase-9 and caspase-3/7 in the apoptotic cascade makes them prime targets for quantifying intrinsic apoptosis activation. Fluorogenic assays, which utilize synthetic substrates that emit fluorescence upon cleavage, provide sensitive and quantitative means to measure the enzymatic activity of these caspases, offering direct functional readouts of apoptosis progression [63] [64].
Fluorogenic Assays: Principles and Key Reagents
Fluorogenic assays for caspase activity leverage the specific proteolytic activity of caspases. These assays use synthetic peptides containing a caspase-specific recognition sequence conjugated to a fluorophore [61] [64]. The fluorophore is typically quenched when the peptide is intact. Upon cleavage by the target caspase, the fluorophore is released, resulting in a measurable increase in fluorescence intensity that is proportional to caspase activity [61].
The core component of these assays is the substrate, which consists of two essential parts: the peptide sequence that confers specificity and the fluorophore that enables detection. The four-amino-acid peptide sequence is designed based on the known cleavage preferences of different caspases. For caspase-9, the preferred sequence is LEHD, while for the executioner caspases-3 and -7, it is DEVD [65] [64]. This difference in sequence specificity allows for the discriminative measurement of initiator and effector caspase activities.
Commonly used fluorophores include 7-amino-4-methylcoumarin (AMC), aminomethylcoumarin (AFC), and rhodamine 110 (R110) [61] [64]. The choice of fluorophore influences the assay's sensitivity and compatibility with instrumentation. For instance, R110-based substrates can provide a higher signal amplitude because the fluorophore is conjugated to two peptide chains, and both must be cleaved to generate the maximum fluorescent signal [61]. More recent advancements have also led to the development of luminogenic substrates, where caspase cleavage releases aminoluciferin, which is then used by luciferase to generate a luminescent signal. These luminescent assays are reported to be 20-50 times more sensitive than their fluorogenic counterparts and are highly amenable to high-throughput screening in 1536-well plate formats [61].
Table 1: Key Fluorogenic Substrates for Caspase-9 and Caspase-3/7
| Caspase Target | Preferred Substrate Sequence | Common Fluorophores | Notes on Specificity |
|---|---|---|---|
| Caspase-9 | LEHD | AMC, AFC | The primary substrate for the initiator caspase-9 [65] [64]. |
| Caspase-3/7 | DEVD | AMC, AFC, R110 | The canonical substrate for executioner caspases-3 and -7 [61] [64]. Can also be cleaved by other caspases like caspase-8 and -10 [64]. |
Quantitative Comparison of Caspase-9 and Caspase-3/7 Activities
A direct comparison of caspase-9 and caspase-3/7 activities reveals critical functional differences that are rooted in their distinct biological roles and activation mechanisms. While both are key mediators of apoptosis, their kinetic parameters and responses to activation complexes differ significantly.
Biochemical studies have shown that the activity of caspase-9 is profoundly enhanced upon incorporation into the Apaf-1 apoptosome, forming the caspase-9 holoenzyme (C9Holo) [60] [65]. Interestingly, a engineered, dimeric form of caspase-9 (LZ-C9) exhibits higher activity than the C9Holo against the optimal synthetic peptide substrate LEHD-AFC [65]. This suggests that dimerization is a key mechanism for the catalytic activation of caspase-9. However, the physiological relevance of this finding is limited because the C9Holo demonstrates a much higher affinity (lower Km) for its natural substrate, procaspase-3, compared to LZ-C9 [65]. This indicates that the apoptosome not only promotes caspase-9 dimerization but also allosterically enhances its ability to recognize and process its downstream effector, procaspase-3. This enhancement is crucial for efficient procaspase-3 activation at physiological concentrations [65].
In contrast, caspase-3 is an executioner caspase that is activated downstream of caspase-9. Its activity is often used as a definitive marker for cells committed to apoptosis [61]. The activation of caspase-3/7 is a convergent point in both the intrinsic and extrinsic apoptotic pathways, making DEVDase activity a widely used indicator of apoptosis. Assays measuring DEVD cleavage are typically highly sensitive, with luminescent versions capable of detecting activity in small cell numbers in high-density plate formats [61].
Table 2: Kinetic and Functional Comparison of Caspase-9 and Caspase-3/7
| Parameter | Caspase-9 (Holoenzyme) | Caspase-3/7 |
|---|---|---|
| Primary Role | Initiator Caspase | Executioner Caspase |
| Activation Trigger | Binding to the Apaf-1 apoptosome [60] | Cleavage by initiator caspases (e.g., caspase-9) [57] |
| Optimal Synthetic Substrate | LEHD-AFC [65] [64] | DEVD-AMC/AFC/R110 [61] [64] |
| Key Physiological Substrate | Procaspase-3 [65] | PARP, DFF45/ICAD, and other cellular proteins [61] |
| Affinity for Physiological Substrate | High affinity (Low Km) for procaspase-3 [65] | N/A |
| Interpretation of High Activity | Indicates initiation of intrinsic apoptosis | Indicates irreversible commitment to apoptotic execution [61] |
Experimental Protocols for Fluorometric Caspase Activity Assays
This section provides detailed methodologies for measuring caspase activity via fluorometric assays, covering both population-based and single-cell approaches using flow cytometry.
Population-Based Fluorometric Assay
This protocol utilizes a spectrofluorometer to measure the average caspase activity in a population of cells [63].
- Cell Lysis and Sample Preparation: After experimental treatments, harvest cells (e.g., 1-2 x 10^6 cells) by trypsinization and centrifugation. Wash the cell pellet with cold phosphate-buffered saline (PBS). Lyse the cells in a suitable ice-cold lysis buffer for 30 minutes. Clarify the lysate by centrifugation at high speed (e.g., 10,000 x g) for 10 minutes at 4°C. Transfer the supernatant to a new tube and perform protein quantification to normalize caspase activity.
- Reaction Setup: In a suitable container (e.g., a microcuvette or a well in a black-walled microplate), combine cell lysate (containing 50-100 μg of total protein), reaction buffer, and the fluorogenic substrate (e.g., DEVD-AFC for caspase-3/7 or LEHD-AFC for caspase-9) at a final concentration of 10-50 μM. Include a negative control with lysate from untreated cells and a blank with lysis buffer instead of lysate.
- Incubation and Measurement: Incubate the reaction mixture at 37°C for 1-2 hours. Protect the plate from light. Measure the fluorescence at the appropriate wavelengths for the liberated fluorophore (e.g., excitation ~400 nm and emission ~505 nm for AFC) using a spectrofluorometer or a multi-mode plate reader.
- Data Analysis: Subtract the fluorescence of the blank from all samples. Normalize the fluorescence values to the total protein concentration in the lysate. Express caspase activity as fold-change relative to the untreated control.
Single-Cell Assay Using Flow Cytometry
This protocol allows for the detection of caspase activity at the single-cell level, revealing heterogeneity within a cell population [63].
- Cell Staining: After treatment, harvest adherent cells using a gentle cell dissociation reagent to preserve membrane integrity. Wash the cells once with cold PBS. Resuspend the cell pellet (approximately 1 x 10^6 cells) in culture medium or a suitable buffer containing the cell-permeable fluorogenic substrate (e.g., DEVD-AFC for caspase-3/7) at a recommended working concentration. Vortex gently and incubate for 30-60 minutes at 37°C in the dark.
- Data Acquisition: After incubation, wash the cells with cold PBS to remove excess substrate. Resuspend the cells in ice-cold PBS or a buffer suitable for flow cytometry. Analyze the cells immediately using a flow cytometer. Excite the fluorophore with the appropriate laser (e.g., a 488-nm laser for AFC) and detect emission with a standard FITC/GFP filter set (e.g., 530/30 nm).
- Data Analysis: Use flow cytometry software to analyze the data. Gate on the live cell population based on forward and side scatter. The fluorescence intensity in the relevant channel corresponds to caspase activity. The percentage of cells with high caspase activity (an indicator of apoptosis) can be quantified.
The Scientist's Toolkit: Essential Research Reagents
A successful caspase activity assay relies on a set of key reagents, each serving a specific function in the experimental workflow.
Table 3: Essential Reagents for Fluorogenic Caspase Assays
| Reagent / Tool | Function / Description |
|---|---|
| Fluorogenic Substrates (e.g., DEVD-AFC, LEHD-AMC) | Synthetic peptides that release a fluorescent molecule upon cleavage by specific caspases; the core of the activity measurement [61] [64]. |
| Caspase Lysis Buffer | A detergent-based buffer used to lyse cells and extract active caspases while preserving their enzymatic activity. |
| Caspase Inhibitors (e.g., Z-VAD-FMK, Ac-DEVD-CHO) | Irreversible (FMK) or reversible (CHO) inhibitors used as negative controls to confirm that substrate cleavage is caspase-specific [62]. |
| Annexin V Conjugates | A protein that binds to phosphatidylserine exposed on the outer leaflet of the plasma membrane during early apoptosis; used for multiplexing and validating apoptosis [61] [62] [59]. |
| MitoView Blue / TMRE | Fluorescent dyes used to assess mitochondrial membrane potential (ΔΨm), a key event in the intrinsic apoptosis pathway [62] [59]. |
| Antibodies for Western Blot (vs. Cleaved Caspase-3, PARP) | Used for orthogonal validation of caspase activation and apoptosis through the detection of specific cleavage events [62] [58]. |
Visualizing the Pathway and Workflow
The following diagrams illustrate the logical sequence of the intrinsic apoptosis pathway and the standard experimental workflow for a fluorogenic caspase assay.
Diagram 1: The intrinsic apoptosis pathway leads to caspase-9 and caspase-3/7 activation, which can be measured with specific fluorogenic substrates.
Diagram 2: The core workflow for fluorogenic caspase assays, showing the parallel paths for population-based and single-cell analysis.
Fluorogenic assays for caspase-9 and caspase-3/7 provide distinct yet complementary windows into the activation of the intrinsic apoptosis pathway. Caspase-9 activity, best measured with the LEHD substrate, reports on the initial triggering of the pathway and the formation of the functional apoptosome complex. Its activity is uniquely regulated by the apoptosome, which enhances its affinity for its physiological substrate, procaspase-3. In contrast, caspase-3/7 activity, detected with the DEVD substrate, serves as a robust marker of irreversible commitment to apoptotic execution. The choice between population-based and single-cell assays depends on the research question, with the former offering quantitative simplicity and the latter revealing cellular heterogeneity. By understanding the principles, kinetics, and appropriate methodologies outlined in this guide, researchers can effectively apply these powerful tools to validate intrinsic apoptosis activation in diverse experimental contexts, from basic research to high-throughput drug discovery.
Apoptosis, or programmed cell death, is a fundamental biological process crucial for maintaining cellular homeostasis, embryonic development, and immune system regulation [66]. This highly controlled, energy-dependent process eliminates unwanted or damaged cells without triggering an inflammatory response, distinguishing it from necrotic cell death [50]. The intrinsic apoptosis pathway, also known as the mitochondrial pathway, becomes activated in response to cellular stress signals such as DNA damage, oxidative stress, or growth factor withdrawal [50]. This activation leads to characteristic biochemical hallmarks that serve as detectable markers for researchers, with two of the most significant being phosphatidylserine externalization and DNA fragmentation [67] [68].
The translocation of phosphatidylserine (PS) from the inner to the outer leaflet of the plasma membrane represents an early event in apoptosis, occurring before the loss of membrane integrity [67]. This externalized PS serves as an "eat-me" signal to phagocytes for the clearance of dying cells [66]. Subsequently, during the later stages of apoptosis, endonucleases are activated that cleave nuclear DNA at internucleosomal regions, producing DNA fragments of approximately 180-200 base pairs, a process known as DNA fragmentation [68] [69]. These specific biochemical events form the basis for the two primary detection methods compared in this guide: Annexin V/Propidium Iodide (PI) staining for PS externalization and TUNEL (Terminal deoxynucleotidyl transferase dUTP nick end labeling) for DNA fragmentation.
Understanding the temporal sequence of these biochemical hallmarks is essential for validating intrinsic apoptosis pathway activation. This guide provides a comprehensive comparison of these two fundamental techniques, enabling researchers to select the most appropriate method for their specific experimental needs in drug development and mechanistic studies.
Principle and Mechanism of Annexin V/PI Staining
Biochemical Basis
The Annexin V/PI staining method leverages the calcium-dependent binding properties of Annexin V to phosphatidylserine (PS), a phospholipid that maintains strict asymmetric distribution in the plasma membrane of viable cells [67] [66]. In healthy cells, PS is predominantly confined to the inner (cytoplasmic) leaflet of the plasma membrane through the activity of ATP-dependent translocases [66]. During early apoptosis, this membrane asymmetry collapses due to the inactivation of translocases and activation of scramblases, resulting in the rapid externalization of PS to the outer membrane leaflet [67]. Annexin V, a 35-36 kDa cellular protein, exhibits high affinity for PS in the presence of calcium ions (Ca²⁺), making it an ideal probe for detecting this early apoptotic event [67].
The standard Annexin V protocol incorporates propidium iodide (PI) as a viability dye to distinguish between early apoptotic cells and those in later stages of cell death [67] [66]. PI is a membrane-impermeant DNA intercalating dye that is excluded from viable and early apoptotic cells with intact plasma membranes [66]. When membrane integrity becomes compromised in late apoptosis and necrosis, PI enters the cell and binds to DNA, emitting red fluorescence upon excitation [67]. This dual-staining approach enables researchers to differentiate between four distinct cell populations: viable (Annexin V⁻/PI⁻), early apoptotic (Annexin V⁺/PI⁻), late apoptotic (Annexin V⁺/PI⁺), and necrotic (Annexin V⁻/PI⁺) cells [66] [70].
Detection Workflow and Pathway
The diagram below illustrates the key detection stages in the Annexin V/PI assay and how they correspond to different phases of intrinsic apoptosis:
Principle and Mechanism of TUNEL Assay
Biochemical Basis
The TUNEL (Terminal deoxynucleotidyl transferase dUTP nick end labeling) assay detects a later biochemical hallmark of apoptosis: DNA fragmentation [68] [69]. During the execution phase of apoptosis, endonucleases are activated that cleave DNA at the linker regions between nucleosomes, generating abundant double-stranded and single-stranded DNA breaks [68]. These breaks contain 3'-hydroxyl (3'-OH) termini that serve as substrates for terminal deoxynucleotidyl transferase (TdT), an enzyme that catalyzes the template-independent addition of deoxynucleotides to 3'-OH ends of DNA fragments [69].
The standard TUNEL assay utilizes TdT to incorporate labeled deoxynucleotides (typically dUTP modified with fluorophores, biotin, or bromine) to the 3'-OH ends of fragmented DNA [68] [69]. Early TUNEL methodologies employed direct labeling with fluorescein-dUTP or indirect labeling with biotin-dUTP followed by streptavidin-enzyme conjugates [68]. More advanced approaches now incorporate alkyne-modified nucleotides like EdUTP, detected via copper-catalyzed azide-alkyne cycloaddition ("click" chemistry), which offers enhanced specificity and signal-to-noise ratio [69]. The Click-iT Plus TUNEL assay further optimizes copper concentration to preserve fluorescent protein signals and maintain compatibility with phalloidin staining, enabling multiplexed applications [69].
Detection Workflow and Pathway
The diagram below illustrates the key detection stages in the TUNEL assay and how they correspond to DNA fragmentation during intrinsic apoptosis:
Comparative Analysis: Annexin V/PI vs. TUNEL Assay
Technical Comparison Table
The following table provides a comprehensive comparison of the key technical aspects and performance characteristics between Annexin V/PI staining and TUNEL assay:
| Parameter | Annexin V/PI Staining | TUNEL Assay |
|---|---|---|
| Detection Principle | Calcium-dependent binding to externalized phosphatidylserine [67] | Enzyme-mediated (TdT) labeling of 3'-OH DNA ends [68] |
| Primary Detection Target | Loss of membrane asymmetry [66] | DNA fragmentation [69] |
| Apoptosis Stage Detected | Early to mid-stage (before membrane integrity loss) [67] | Mid to late stage (after DNA cleavage) [69] |
| Cell Processing Requirements | Can be performed on live, unfixed cells [70] | Requires cell fixation and permeabilization [69] |
| Compatibility with Flow Cytometry | Excellent - standard application [67] [66] | Good - requires optimized protocols [71] [69] |
| Compatibility with Microscopy | Good - for adherent cells and smears [67] | Excellent - particularly for tissue sections [69] |
| Multiplexing Capability | Limited due to calcium dependence and live cell requirement [67] | High - compatible with IHC, IF, and other staining methods [69] |
| Assay Duration | Rapid (~20-30 minutes incubation) [66] [70] | Longer protocol (2+ hours including fixation) [69] |
| Specificity for Apoptosis | May detect other forms of PS-exposing cell death (e.g., necroptosis) [67] | May detect non-apoptotic DNA damage if not properly controlled [68] |
| Key Limitations | Sensitivity to calcium concentration; reversible binding; requires immediate analysis [67] | Potential for false positives from necrosis or mechanical damage; more complex protocol [68] |
Temporal Detection Profile in Intrinsic Apoptosis
A critical consideration when selecting an apoptosis detection method is understanding where each assay functions within the temporal sequence of intrinsic apoptosis activation. The following table compares the detection profiles:
| Apoptosis Phase | Key Biochemical Events | Annexin V/PI Detection | TUNEL Assay Detection |
|---|---|---|---|
| Initiation | Mitochondrial outer membrane permeabilization; Cytochrome c release [50] | Not detectable | Not detectable |
| Early Execution | Phosphatidylserine externalization; Caspase activation [67] [50] | Strong detection (Annexin V+/PI-) | Not detectable or weak |
| Mid Execution | Membrane blebbing; Cell shrinkage [50] | Detectable (Annexin V+/PI-) | Beginning of detection |
| Late Execution | DNA fragmentation; Chromatin condensation [68] [50] | Detectable (Annexin V+/PI+) | Strong detection |
| Termination | Formation of apoptotic bodies; Phagocytosis [50] | Not typically analyzed | Detectable |
This temporal profile highlights the complementary nature of these techniques, with Annexin V/PI staining detecting earlier events and TUNEL identifying later-stage apoptosis.
Detailed Experimental Protocols
Annexin V/PI Staining Protocol for Flow Cytometry
Materials Required:
- Annexin V-FITC conjugate (e.g., Bio-Techne #NBP2-29373) [70]
- Propidium Iodide solution (50 µg/mL) [66]
- 1X Binding Buffer: 10 mM HEPES (pH 7.4), 140 mM NaCl, 2.5 mM CaCl₂ [66] [70]
- Phosphate-buffered saline (PBS), cold
- Flow cytometer with 488 nm excitation and filters for FITC (530 nm) and PI (617 nm) [67]
Step-by-Step Procedure:
Cell Preparation and Harvesting:
- For adherent cells: gently detach using non-enzymatic methods (e.g., EDTA) or mild trypsinization followed by serum-containing media to inactivate trypsin [67]. Avoid harsh detachment that may damage membrane integrity.
- For suspension cells: collect directly by centrifugation.
- Wash cells twice with cold PBS by centrifuging at 300 × g for 5 minutes at room temperature [66].
Cell Staining:
Incubation:
Flow Cytometry Analysis:
- Analyze samples immediately (within 1 hour) using flow cytometry [70].
- Use unstained cells and single-stained controls (Annexin V-only and PI-only) for compensation and gating [66].
- Set up quadrant gates based on controls: viable cells (Annexin V⁻/PI⁻), early apoptotic (Annexin V⁺/PI⁻), late apoptotic/necrotic (Annexin V⁺/PI⁺) [66].
Critical Considerations:
- Maintain calcium concentration in binding buffer as Annexin V binding is Ca²⁺-dependent [67].
- Process controls and experimental samples in parallel to ensure consistency.
- Analyze samples promptly as apoptosis may progress during extended storage.
- For adherent cells, include a wash step with serum-containing media before Annexin V binding to neutralize any residual trypsin activity [67].
TUNEL Assay Protocol for Cultured Cells
Materials Required:
- Click-iT TUNEL Alexa Fluor Imaging Assay (e.g., Thermo Fisher Scientific) [69]
- Phosphate-buffered saline (PBS)
- Fixation solution (4% formaldehyde in PBS)
- Permeabilization solution (0.25% Triton X-100 in PBS)
- DNase I (for positive control)
- Hoechst 33342 or DAPI for nuclear counterstaining
Step-by-Step Procedure:
Cell Preparation and Fixation:
- Culture cells on appropriate chamber slides or coverslips.
- After treatment, carefully aspirate media and wash cells gently with PBS.
- Fix cells with 4% formaldehyde in PBS for 15 minutes at room temperature.
- Wash twice with PBS for 5 minutes each.
Permeabilization:
- Permeabilize cells with 0.25% Triton X-100 in PBS for 20 minutes at room temperature [69].
- Wash twice with PBS for 5 minutes each.
TUNEL Reaction:
- Prepare TUNEL reaction mixture according to manufacturer's instructions.
- For Click-iT TUNEL assay: add EdUTP using TdT enzyme, then detect with appropriate azide dye [69].
- Apply reaction mixture to samples and incubate in a humidified chamber for 60 minutes at 37°C protected from light.
Detection and Counterstaining:
- Wash cells three times with PBS containing 1% BSA.
- For colorimetric detection: apply streptavidin-peroxidase conjugate followed by DAB substrate [69].
- For fluorescence detection: apply appropriate detection reagents (e.g., Alexa Fluor azide).
- Counterstain nuclei with Hoechst 33342 or DAPI for 5 minutes.
- Wash and mount slides with appropriate mounting medium.
Microscopy and Analysis:
- Visualize using fluorescence or brightfield microscopy.
- Include appropriate controls: untreated negative control, DNase I-treated positive control, and no-TdT enzyme negative control.
- Quantify TUNEL-positive cells by counting stained nuclei relative to total nuclei.
Critical Considerations:
- Optimize fixation and permeabilization conditions to preserve morphology while allowing reagent access.
- Include rigorous controls to distinguish apoptosis from non-specific DNA damage.
- For flow cytometry applications, use the APO-BrdU TUNEL assay with BrdU incorporation and Alexa Fluor 488-labeled anti-BrdU antibody [69].
Research Reagent Solutions
The following table outlines essential reagents and kits for implementing these apoptosis detection methods:
| Reagent/Kits | Supplier Examples | Primary Function | Key Applications |
|---|---|---|---|
| Annexin V-FITC Apoptosis Detection Kits | Abcam (ab14085), Bio-Techne (NBP2-29373) [67] [70] | Detection of phosphatidylserine externalization | Flow cytometry, fluorescence microscopy |
| Propidium Iodide Solution | Sigma-Aldrich, Thermo Fisher Scientific [66] | Viability dye for membrane integrity assessment | Discrimination of late apoptotic/necrotic cells |
| Click-iT TUNEL Assay Kits | Thermo Fisher Scientific [69] | Detection of DNA fragmentation via click chemistry | Fluorescence microscopy, HCS |
| APO-BrdU TUNEL Assay | Thermo Fisher Scientific [69] | Flow cytometry-based DNA fragmentation detection | Quantitative analysis of apoptotic cells |
| Annexin V Binding Buffer | Multiple suppliers [67] [66] | Provides optimal calcium-dependent binding conditions | Maintenance of Annexin V-PS binding |
| Terminal Deoxynucleotidyl Transferase (TdT) | Multiple suppliers [68] [69] | Enzyme for labeling DNA breaks in TUNEL assay | Catalyzes nucleotide addition to 3'-OH ends |
Application Data and Validation in Intrinsic Apoptosis Research
Comparative Sensitivity in Experimental Models
Research comparing multiple apoptosis detection techniques has revealed important insights into the sensitivity and appropriate application of Annexin V/PI staining versus TUNEL assay. A comparative study on murine immortalized astrocytes treated with urine from multiple sclerosis patients demonstrated that both propidium iodide labeling (by flow cytometry) and TUNEL assay were well-suited for detecting apoptosis in adherent cellular models [72]. Notably, this investigation observed that phosphatidylserine externalization (detected by Annexin V) and DNA fragmentation (detected by TUNEL) occurred concomitantly after induction of apoptosis in their experimental system [72].
The market analysis for apoptosis assays indicates growing technological advancements, with flow cytometry-based methods representing a significant segment valued at USD 4.9 billion in 2022 and estimated to register over 8.4% CAGR between 2023 and 2032 [73]. This growth reflects the continued importance of these detection methods in both basic research and drug development applications.
Validation in Disease Models and Drug Development
Both Annexin V/PI staining and TUNEL assays play critical roles in validating intrinsic apoptosis pathway activation across various disease models:
In cancer research, these assays are extensively used to evaluate chemotherapeutic efficacy by measuring treatment-induced apoptosis [67] [73]. The Annexin V/PI assay is particularly valuable for high-throughput screening of drug candidates, providing rapid quantification of early apoptotic populations [67]. Meanwhile, TUNEL assays offer definitive confirmation of late-stage apoptosis in tissue sections, making them invaluable for preclinical studies of novel therapeutics [69].
In neurodegenerative disease research, where apoptosis contributes significantly to disease pathology, TUNEL assays have been instrumental in identifying apoptotic neurons in post-mortem tissue [66]. The ability to visualize DNA fragmentation in situ provides crucial spatial information about cell death patterns in affected brain regions.
For immunological applications, particularly in studying Autoimmune Lymphoproliferative Syndrome (ALPS), Fas-mediated apoptosis assays represent a specialized application of these principles in clinical diagnostics [74]. While distinct from the intrinsic pathway, these assays share methodological similarities and highlight the importance of validated apoptosis detection in clinical contexts.
The comparative analysis of Annexin V/PI staining and TUNEL assay reveals two complementary techniques for validating intrinsic apoptosis pathway activation, each with distinct advantages and applications. Annexin V/PI staining offers rapid, quantitative detection of early apoptotic events through phosphatidylserine externalization, making it ideal for flow cytometry-based screening and kinetic studies [67] [66]. In contrast, the TUNEL assay provides specific identification of later apoptotic stages through DNA fragmentation detection, with particular strength in microscopy-based applications and tissue analysis [68] [69].
The selection between these methods should be guided by specific research objectives, experimental timeline, and required throughput. For drug discovery and high-throughput screening where early detection is paramount, Annexin V/PI staining represents the preferred approach. For pathological confirmation and tissue-based studies where definitive apoptosis identification is required, TUNEL assay offers superior specificity. In many research scenarios, employing both techniques in a complementary manner provides the most comprehensive validation of intrinsic apoptosis pathway activation, capturing both early membrane changes and late nuclear events in the apoptotic cascade.
As apoptosis research continues to evolve, technological advancements in both methodologies are enhancing their sensitivity, specificity, and compatibility with multiplexed approaches. The development of click chemistry-based TUNEL assays and improved Annexin V conjugates demonstrates the ongoing refinement of these essential tools for cell death research [69]. By understanding the principles, applications, and limitations of each technique, researchers can make informed decisions to optimally validate intrinsic apoptosis activation in their experimental systems.
Overcoming Experimental Challenges: Pitfalls and Optimization Strategies
Addressing False Positives and Necroptosis Interference
In the field of cell death research, accurately interpreting data for intrinsic apoptosis pathway activation is complicated by the potential for false positives and interference from other cell death mechanisms, particularly necroptosis. Both pathways can be initiated by similar stimuli and share upstream signaling components, yet they diverge into morphologically and biochemically distinct processes. This guide provides a structured framework for researchers to validate intrinsic apoptosis activation through specific experimental design, targeted reagent selection, and strategic pathway inhibition to ensure data accuracy in drug development and mechanistic studies.
Key Distinctions: Intrinsic Apoptosis vs. Necroptosis
The intrinsic apoptosis and necroptosis pathways, while occasionally initiated by overlapping cellular stresses, are characterized by unique molecular executors and defining biochemical events. The table below outlines the core differentiating features.
Table 1: Fundamental Characteristics of Intrinsic Apoptosis and Necroptosis
| Feature | Intrinsic Apoptosis | Necroptosis |
|---|---|---|
| Key Initiators | DNA damage, oxidative stress, ER stress [75] [76] | TNF-α, TLR ligands, viral infection [77] [78] |
| Core Regulators | Bcl-2 family proteins (Bax/Bak, Bid, Bim) [75] [8] | RIPK1, RIPK3, MLKL [77] |
| Critical Executioner | Caspase-3 & Caspase-7 [45] [75] | Phosphorylated MLKL [77] |
| Mitochondrial Involvement | Cytochrome c release, MOMP [75] [8] | Not required for execution; can occur downstream of some inducers |
| Morphological Hallmarks | Cell shrinkage, chromatin condensation, apoptotic bodies [75] | Cellular swelling, plasma membrane rupture [77] |
| Immunogenicity | Generally non-immunogenic/anti-inflammatory [45] | Highly immunogenic/pro-inflammatory [78] |
Validating Key Pathway Components: Experimental Design & Reagents
Definitive validation requires a multi-parametric approach that confirms the activation of apoptosis-specific components and the absence of necroptosis markers. The following experimental strategies are critical for generating conclusive data.
Caspase Activation Assessment
Caspase-3 is a primary executioner protease in apoptosis and serves as a key activation marker.
- Experimental Protocol (Western Blotting): Lyse cells and perform SDS-PAGE followed by immunoblotting. Use antibodies that detect both the full-length (inactive, ~35 kDa) and cleaved (active, ~17/19 kDa) forms of caspase-3. The appearance of the cleaved fragments is a definitive indicator of activation [45] [79].
- Data Interpretation: The presence of cleaved caspase-3 fragments, alongside a decrease in the full-length protein, strongly indicates apoptotic execution. This cleavage is a downstream consequence of initiator caspases (caspase-9 in intrinsic pathway) and is a reliable metric [75] [80].
Necroptosis Executioner Analysis
Phosphorylated MLKL is the definitive marker for necroptosis execution.
- Experimental Protocol (Immunofluorescence/Phospho-Western Blotting): For immunofluorescence, fix and permeabilize cells, then stain with an antibody specific for phosphorylated MLKL (e.g., at Thr-357/Ser-358). Counterstain with a nuclear dye (DAPI) and image. Phosphorylated MLKL translocates and forms oligomeric pores on the plasma membrane, visible as distinct punctate staining [77]. Phospho-specific Western blotting can also confirm MLKL phosphorylation.
- Data Interpretation: The presence of phosphorylated MLKL, especially its localization to the plasma membrane, is a non-apoptotic signal and confirms necroptosis induction. Its absence, in the context of cell death, helps rule out necroptotic interference [77].
Pharmacological Inhibition for Pathway Specificity
Using specific inhibitors is a powerful tool to functionally dissect the contributing pathways.
- Pan-Caspase Inhibitor (e.g., Z-VAD-FMK): This cell-permeable compound irreversibly binds to the catalytic site of most caspases. If Z-VAD-FMK fails to inhibit cell death, it suggests a non-apoptotic, caspase-independent mechanism is involved [45] [76].
- Necroptosis Inhibitor (e.g., Necrostatin-1): Necrostatin-1 is a specific allosteric inhibitor of RIPK1 kinase activity. If cell death is suppressed by Necrostatin-1, it confirms a significant necroptotic component [77] [78].
- Experimental Protocol: Pre-treat cells with the inhibitor for 1-2 hours before applying the apoptotic or necroptotic stimulus. Measure cell viability (e.g., by ATP-based assays, dye exclusion) and cell death markers (e.g., caspase-3 cleavage, MLKL phosphorylation) after an appropriate incubation period (e.g., 24 hours).
Table 2: Research Reagent Solutions for Cell Death Pathway Analysis
| Reagent / Tool | Function / Target | Key Application in Validation |
|---|---|---|
| Z-VAD-FMK (Pan-caspase Inhibitor) | Irreversibly inhibits caspase enzymatic activity [45] [76] | Functionally confirms caspase-dependence of cell death; its inability to block death suggests alternative pathways. |
| Necrostatin-1 (Necroptosis Inhibitor) | Specifically inhibits RIPK1 kinase activity [77] [78] | Functionally confirms RIPK1-dependent necroptosis; used to rule out necroptotic interference. |
| Anti-Cleaved Caspase-3 Antibody | Detects the activated (cleaved) form of caspase-3 [45] [79] | A definitive biochemical marker for apoptosis execution via Western Blot or IF. |
| Anti-Phospho-MLKL Antibody | Detects the active, phosphorylated form of MLKL [77] | A definitive biochemical marker for necroptosis execution via Western Blot or IF. |
| BH3 Mimetics (e.g., Venetoclax, S63845) | Small molecules that inhibit anti-apoptotic proteins (Bcl-2, Mcl-1) [45] [79] | Directly activates the intrinsic apoptosis pathway; used as a positive control or combination agent. |
Integrated Validation Workflow
A robust validation strategy involves parallel experiments to confirm apoptosis while actively ruling out necroptosis. The diagram below illustrates this multi-pronged experimental workflow.
Integrated experimental workflow for validating intrinsic apoptosis and excluding necroptosis.
Molecular Crosstalk and Signaling Pathways
Understanding the molecular decision points between apoptosis and necroptosis is fundamental to experimental design. The intrinsic apoptosis pathway is primarily regulated by the Bcl-2 protein family, which controls mitochondrial outer membrane permeabilization (MOMP), leading to caspase activation [75] [8]. Necroptosis, in contrast, is a caspase-independent pathway mediated by the RIPK1-RIPK3-MLKL axis [77]. Caspase-8 activity serves as a critical molecular switch; when active, it cleaves and inactivates RIPK1 and RIPK3, promoting apoptosis and suppressing necroptosis. Inhibition of caspase-8, however, can shift the signaling balance towards necroptosis [78] [76]. The following diagram illustrates these pathways and their key interactions.
Molecular pathways of intrinsic apoptosis and necroptosis, highlighting key components, inhibitors, and the regulatory role of caspase-8.
Optimizing Assay Conditions for Different Cell Types and Tissues
Validating the activation of the intrinsic apoptosis pathway is a critical requirement in diverse fields of biological research, from fundamental cell biology to pre-clinical drug discovery. This pathway, initiated by intracellular stressors and regulated by the BCL-2 protein family, leads to mitochondrial outer membrane permeabilization (MOMP), caspase activation, and controlled cellular demise [3] [8]. The accurate distinction of this programmed cell death from other forms, such as necrosis, is essential for correctly interpreting experimental outcomes, particularly when evaluating the efficacy and mechanism of action of novel chemotherapeutic agents [81] [82]. This guide provides a comparative analysis of modern methodologies, offering structured experimental data and protocols to assist researchers in selecting and optimizing the most appropriate assays for their specific cell types and tissue contexts.
Core Technologies for Detecting Intrinsic Apoptosis
The intrinsic apoptosis pathway is characterized by a well-defined sequence of molecular events. Cellular stress signals lead to the activation of pro-apoptotic BH3-only proteins, which in turn activate the effector proteins BAX and BAK. These proteins oligomerize to form pores in the mitochondrial outer membrane, a process known as MOMP. This permits the release of cytochrome c into the cytosol, where it binds to APAF-1 and triggers the formation of the apoptosome, ultimately leading to the activation of caspase-9 and the downstream effector caspases-3 and -7 [3] [8]. The following diagram illustrates this key signaling pathway.
Comparative Platform Analysis for Spatial Apoptosis Detection
Advanced spatial transcriptomics platforms enable researchers to profile gene expression within the native tissue architecture, providing powerful tools for visualizing apoptotic signaling in complex tissue microenvironments, such as tumors.
Performance Comparison of Spatial Transcriptomics Platforms
A 2025 benchmark study systematically compared the performance of three major commercial imaging-based spatial transcriptomics platforms—CosMx, MERFISH, and Xenium—using formalin-fixed paraffin-embedded (FFPE) lung adenocarcinoma and pleural mesothelioma samples [83]. The table below summarizes key performance metrics from this study.
Table 1: Platform comparison for spatial analysis of FFPE tumor samples [83]
| Platform | Panel Size (Genes) | Transcripts per Cell (Mean) | Unique Genes per Cell (Mean) | Key Strengths | Notable Limitations |
|---|---|---|---|---|---|
| CosMx | 1,000-plex | Highest (p < 2.2e-16) | Highest (p < 2.2e-16) | Highest transcript capture sensitivity | Limited field of view; some key markers (CD3D, FOXP3) expressed at level of negative controls in older samples |
| MERFISH | 500-plex | Variable (lower in older samples) | Variable (lower in older samples) | Whole-tissue coverage | Lack of negative control probes in panel |
| Xenium (Unimodal) | 339-plex | Intermediate | Intermediate | Whole-tissue coverage; reliable negative control separation | Lower transcript counts than CosMx |
| Xenium (Multimodal) | 339-plex | Lower than unimodal | Lower than unimodal | Multimodal segmentation capabilities | Lowest transcript counts among platforms |
Experimental Protocol: Spatial Transcriptomics with FFPE Tissues
Methodology: The comparative study used serial 5 μm sections of FFPE surgically resected lung adenocarcinoma and pleural mesothelioma samples assembled in tissue microarrays (TMAs) [83].
- Sample Preparation: FFPE blocks were sectioned following standard pathological protocols.
- Platform Processing: Serial TMA sections were submitted to the respective companies (NanoString, Vizgen, 10x Genomics) to run their standard single-cell imaging-based ST assays.
- Data Analysis: Performance was evaluated by comparing metrics like transcripts per cell, gene counts per cell, and signal-to-background using negative control probes. Cell segmentation and phenotyping accuracy were assessed against pathologists' evaluations of multiplex immunofluorescence and H&E-stained serial sections [83].
Single-Cell Functional Assays for Apoptosis Dynamics
Beyond spatial mapping, quantifying the dynamics of apoptosis execution at the single-cell level is crucial for validating pathway activation.
Comparison of Live-Cell Apoptosis Detection Assays
Different live-cell assays provide unique insights into the kinetics and modality of cell death, each with distinct advantages.
Table 2: Functional assays for detecting apoptosis and distinguishing from necrosis
| Assay Technology | Key Readout | Key Advantage | Throughput Potential | Reference |
|---|---|---|---|---|
| FRET-based Caspase Sensor | Caspase activation (FRET loss) & cell permeability | Confirmatory, real-time distinction of apoptosis vs. primary necrosis at single-cell level | Adaptable to HTS imaging platforms | [81] |
| Quantitative Phase Imaging (QPI) | Cell density (pg/pixel) & Cell Dynamic Score (CDS) | Label-free, non-invasive; monitors subtle morphological dynamics | Suitable for time-lapse analysis of population dynamics | [82] |
| Annexin V/PI Staining | Phosphatidylserine exposure & membrane integrity | Widely established, accessible | High (flow cytometry) | [81] |
Experimental Protocols for Functional Assays
A. FRET-based Caspase Sensor Assay
This protocol enables real-time discrimination between apoptosis and necrosis using a genetically encoded FRET-based caspase sensor [81].
- Cell Line Development: Generate stable cell lines expressing a FRET-based caspase sensor (e.g., ECFP-DEVD-EYFP) and a stable non-soluble marker, such as Mito-DsRed, targeted to mitochondria.
- Real-Time Imaging: Plate cells and treat with apoptosis inducers (e.g., 0.5 μM staurosporine, 0.1 μM doxorubicin). Perform time-lapse imaging using a fluorescence microscope equipped with appropriate filters for ECFP, EYFP, and DsRed.
- Data Interpretation:
- Apoptotic cells show loss of FRET (increase in ECFP/EYFP ratio) while retaining mitochondrial DsRed fluorescence.
- Necrotic cells show sudden loss of both ECFP and EYFP fluorescence (without FRET loss) but retain Mito-DsRed signal.
- Live cells show intact FRET probe and Mito-DsRed fluorescence [81].
The following workflow outlines the key steps and decision points in this assay.
B. Quantitative Phase Imaging (QPI) Assay
This label-free method distinguishes cell death subroutines based on morphological and dynamic features [82].
- Cell Preparation: Plate cells (e.g., DU145, LNCaP, PNT1A) and induce death using agents like staurosporine, doxorubicin, or black phosphorus. Include controls with caspase inhibitor (e.g., 10 μM z-VAD-FMK).
- Time-Lapse QPI: Use a quantitative phase microscope (e.g., Q-PHASE) to acquire time-lapse images, maintaining standard culture conditions (37°C, 5% CO₂).
- Feature Extraction: Analyze QPI micrographs to extract parameters such as cell density (in pg/pixel) and Cell Dynamic Score (CDS), which measures average intensity change of cell pixels.
- Classification: Apply machine learning (e.g., LSTM neural network) to classify cell death modality. Caspase 3,7-dependent death typically shows different density and CDS dynamics compared to caspase-independent lytic death [82].
The Scientist's Toolkit: Key Research Reagent Solutions
Selecting the appropriate reagents and tools is fundamental for successfully validating intrinsic apoptosis.
Table 3: Essential reagents and tools for intrinsic apoptosis research
| Tool / Reagent | Function | Example Use Case |
|---|---|---|
| Spatial Transcriptomics Panels | Profiling expression of hundreds of genes in situ | Mapping immune and tumor cell interactions in FFPE tumor sections [83] |
| FRET-based Caspase Sensor | Real-time visualization of caspase activation | Distinguishing apoptotic and necrotic death in single cells after drug treatment [81] |
| Quantitative Phase Microscope | Label-free imaging of cell mass and morphology | Tracking morphological dynamics of cell death without fluorescent labels [82] |
| BCL-2 Family Modulators | Specific activators or inhibitors of pathway nodes | Testing dependence of cell death on specific BCL-2 family proteins (e.g., BH3 mimetics) [8] |
| Caspase Inhibitors (z-VAD-FMK) | Pan-caspase inhibitor | Confirming caspase-dependent nature of cell death [82] |
Choosing the optimal assay for validating intrinsic apoptosis requires careful consideration of the research question, sample type, and required throughput. For profiling apoptotic signaling within the complex architecture of intact tissues, such as FFPE tumor samples, spatial transcriptomics platforms are unparalleled. CosMx offers high sensitivity, while Xenium provides robust whole-tissue coverage. For kinetic analysis of cell death dynamics at the single-cell level, FRET-based biosensors provide a confirmatory, real-time method to unambiguously distinguish apoptosis from necrosis, whereas QPI offers a powerful label-free alternative. By leveraging the complementary strengths of these technologies and following the standardized protocols outlined herein, researchers can robustly validate intrinsic apoptosis pathway activation across diverse experimental conditions.
In the investigation of the intrinsic apoptosis pathway, caspases stand as central executioners. These cysteine-dependent proteases are synthesized as inactive zymogens and become activated through proteolytic cleavage, leading to the dismantling of the cell. The use of caspase inhibitors, particularly pan-caspase inhibitors like Z-VAD-FMK (benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone), has become a cornerstone for validating the involvement of caspases in programmed cell death. However, the pharmacological profile of these tools is complex, and their application requires rigorous experimental design to confirm specificity and draw accurate biological conclusions. This guide objectively compares the performance of Z-VAD-FMK with other caspase inhibitors, providing supporting experimental data and methodologies essential for researchers, scientists, and drug development professionals.
Caspase Inhibitor Mechanisms and Specificity Profiles
Caspase inhibitors function by targeting the active site cysteine residue, often through reversible or irreversible covalent bonds. The specificity and spectrum of inhibition vary significantly between compounds.
Table 1: Comparison of Common Caspase Inhibitors
| Inhibitor Name | Primary Target(s) | Spectrum of Action | Key Characteristics | Reported Experimental Concentrations |
|---|---|---|---|---|
| Z-VAD-FMK | Multiple caspases | Pan-caspase inhibitor | Irreversible binding; cell-permeable; can have off-target effects at high concentrations. | 1-100 μM (context-dependent) [84] [85] |
| Boc-D-FMK | Multiple caspases | Broad-spectrum | Similar to Z-VAD-FMK but showed no pro-apoptotic effect in neutrophils [84]. | 1-100 μM [84] |
| Q-VD-OPh | Multiple caspases | Pan-caspase inhibitor | Irreversible binding; highly potent; reduced cytotoxicity in vivo even at high concentrations (up to 500-1000 μM) [86]. | Up to 500-1000 μM in vivo [86] |
| Z-IETD-FMK | Caspase-8 | Selective inhibitor | Irreversible binding; targets the extrinsic apoptosis initiator caspase. | 1-100 μM [84] |
| Ac-LEHD-cmk | Caspase-9 | Selective inhibitor | Targets the intrinsic apoptosis initiator caspase; showed no effect on TNFα-induced apoptosis in neutrophils [84]. | 1-100 μM [84] |
| VX-765 (Belnacasan) | Caspase-1 | Selective inhibitor | Reversible inhibitor; developed for inflammatory diseases; clinical trials terminated due to liver toxicity [86]. | N/A (clinical compound) |
The Z-VAD-FMK Specificity Paradox
A critical consideration for researchers is that the effects of Z-VAD-FMK can be compound-specific and not universally generalizable to all broad-spectrum inhibitors. In a study on neutrophil apoptosis, while Z-VAD-FMK at concentrations >100 μM enhanced TNFα-stimulated cell death, lower concentrations (1-30 μM) effectively inhibited it. In contrast, Boc-D-fmk, another broad-spectrum inhibitor, caused only concentration-dependent inhibition of apoptosis without any augmentation, underscoring that pro-apoptotic effects are not a class-wide phenomenon [84]. This highlights the necessity of dose-response experiments and the use of multiple, structurally distinct inhibitors to confirm caspase dependency.
Experimental Protocols for Validating Caspase Inhibition
To confidently attribute an observed phenotype to caspase inhibition, a multi-faceted experimental approach is required. The following protocols outline key methodologies.
Protocol: Validating Inhibitor Efficacy and Specificity in Vitro
This protocol is designed to test the functional activity of a caspase inhibitor in a cell-based model of intrinsic apoptosis.
- 1. Induction of Apoptosis: Treat cells with a known intrinsic apoptotic stimulus (e.g., etoposide at 50 μg/ml, UV irradiation, or serum starvation) [85].
- 2. Inhibitor Treatment: Pre-treat cells (30 minutes prior to apoptosis induction) with a range of Z-VAD-FMK concentrations (e.g., from 1 μM to 100 μM). Include a control group with Boc-D-fmk or Q-VD-OPh for comparison [84] [86].
- 3. Assessment of Apoptotic Markers:
- Cell Viability: Measure using assays like WST-1 or CCK-8 after 24-48 hours [85] [87].
- Caspase-3/7 Activity: Use a fluorometric or colorimetric substrate assay (e.g., DEVD-pNA). Effective inhibition should significantly reduce substrate cleavage.
- Western Blot Analysis: Probe for key apoptosis indicators:
- Cleaved Caspase-3: Reduction or absence confirms inhibitor efficacy.
- Cleaved PARP: Reduction of the 89 kD cleavage fragment indicates suppression of executioner caspase activity [85].
- BCL-2 Family Proteins: Monitor changes in Bax/Bcl-2 ratio as an upstream event.
- 4. Specificity Confirmation: Use selective inhibitors for initiator caspases (e.g., Ac-LEHD-cmk for caspase-9). In the intrinsic pathway, caspase-9 inhibition should mirror the protective effect of Z-VAD-FMK, while caspase-8 inhibition (Z-IETD-fmk) may not [84].
Protocol: Differentiating Apoptosis from Necroptosis
Caspase inhibition can, under certain conditions, shift the mode of cell death from apoptosis to necroptosis, a programmed form of necrosis.
- 1. Experimental Setup: Stimulate cells with TNFα in the presence of Z-VAD-FMK (e.g., 20-80 μM) to simultaneously induce apoptosis and inhibit caspases [87].
- 2. Cell Death Analysis:
- Flow Cytometry: Use annexin V and propidium iodide (PI) staining. Apoptotic cells are typically annexin V+/PI- (early) or annexin V+/PI+ (late), while necroptotic cells may be annexin V-/PI+ [85].
- LDH Release Assay: Measure lactate dehydrogenase release into the culture medium, a hallmark of plasma membrane rupture in necroptosis.
- 3. Pathway Inhibition: Co-treat with a specific necroptosis inhibitor, such as Necrostatin-1 (an RIPK1 inhibitor). If cell death is rescued, it confirms a shift to necroptosis [87].
- 4. Molecular Confirmation: Perform Western blot analysis for phosphorylated MLKL, the executioner of necroptosis [88].
The diagram below illustrates the cell fate decision between apoptosis and necroptosis upon caspase inhibition.
The Scientist's Toolkit: Key Research Reagent Solutions
A selection of essential reagents for studying caspase-mediated apoptosis is presented in the table below.
Table 2: Essential Reagents for Apoptosis Research
| Reagent Name | Function/Description | Key Application in Research |
|---|---|---|
| Z-VAD-FMK | Irreversible, cell-permeable pan-caspase inhibitor. | Primary tool for initial validation of caspase involvement in cell death [85] [87]. |
| Q-VD-OPh | Irreversible, cell-permeable pan-caspase inhibitor with lower reported cellular toxicity. | Preferred for in vivo studies and long-term experiments where Z-VAD-FMK toxicity is a concern [86]. |
| Selective Inhibitors (e.g., Z-IETD-FMK, Ac-LEHD-cmk) | Target specific initiator caspases. | Determining the contribution of extrinsic (caspase-8) vs. intrinsic (caspase-9) pathways [84]. |
| Annexin V / Propidium Iodide (PI) | Fluorescent probes for phosphatidylserine exposure (early apoptosis) and membrane integrity (necrosis). | Standard flow cytometry method to quantify and differentiate modes of cell death [85]. |
| Caspase Activity Assay Kits (e.g., DEVD-pNA) | Fluorogenic or colorimetric substrates for specific caspases (e.g., DEVD for caspases-3/7). | Directly measuring the enzymatic activity of caspases in cell lysates [85]. |
| Antibodies: Cleaved Caspase-3, Cleaved PARP | Detect activated (cleaved) forms of key apoptotic proteins. | Western blot confirmation of apoptosis execution and inhibitor efficacy [85]. |
Data Interpretation and Common Pitfalls
Interpreting data from caspase inhibition experiments requires caution to avoid common pitfalls.
- Incomplete Inhibition: Assess caspase activity directly, rather than relying solely on cell viability. Incomplete inhibition can lead to residual caspase activity and misinterpretation of results.
- Off-Target Effects: As demonstrated in neutrophil studies, high concentrations of Z-VAD-FMK can produce compound-specific, pro-apoptotic effects unrelated to its caspase-inhibitory function [84]. This underscores the critical need for multiple, complementary inhibitors.
- Alternative Death Pathways: Inhibition of caspases may not rescue cell viability if alternative death pathways like necroptosis [87] or pyroptosis [88] are activated. The experimental protocol for differentiating necroptosis is essential in such cases.
- Non-Apoptotic Roles of Caspases: Emerging evidence indicates caspases regulate other processes like cellular differentiation and proliferation. Inhibiting them may affect these non-apoptotic functions, complicating phenotype interpretation [86].
Z-VAD-FMK remains an invaluable, though complex, tool for confirming the activation of caspase-dependent apoptosis. Its use must be strategically complemented with selective inhibitors, careful dose-response analyses, and assays that detect alternative cell death pathways. The selection of an appropriate control inhibitor, such as Boc-D-fmk or Q-VD-OPh, is crucial for verifying the specificity of observed effects. By employing the detailed experimental protocols and comparative data outlined in this guide, researchers can robustly validate the role of caspases in the intrinsic apoptosis pathway and advance the development of targeted therapeutic strategies.
Interpreting the Bax/Bcl-2 Ratio as a Critical Tipping Point
In the intrinsic apoptosis pathway, the precise balance between pro-apoptotic and anti-apoptotic proteins determines cellular life-or-death decisions. The Bax/Bcl-2 expression ratio serves as a critical tipping point in this regulatory mechanism, functioning as a molecular rheostat that controls mitochondrial outer membrane permeabilization and subsequent caspase activation. This comprehensive review examines the quantitative assessment of the Bax/Bcl-2 ratio as a biomarker for predicting therapeutic response across various cancer types, explores the underlying molecular mechanisms, and details standardized experimental approaches for its measurement. Evidence from melanoma, breast cancer, cervical cancer, and other malignancies demonstrates that a high Bax/Bcl-2 ratio characteristic of sensitive cells, while a low ratio is associated with resistance to apoptosis-inducing stimuli. Understanding and measuring this critical balance provides researchers with a powerful tool for validating intrinsic apoptosis pathway activation in both basic research and clinical applications.
The B-cell lymphoma-2 (Bcl-2) family proteins are fundamental regulators of the intrinsic apoptosis pathway, which is activated in response to cellular stress, DNA damage, and various anticancer therapies [89]. These proteins are characterized by Bcl-2 homology (BH) domains and functionally divided into three subgroups: (1) anti-apoptotic proteins (Bcl-2, Bcl-XL, Bcl-w, Mcl-1); (2) pro-apoptotic multi-domain proteins (Bax, Bak, Bok); and (3) BH3-only proteins (Bid, Bim, Bad, Puma, Noxa) [90] [89]. The rheostat model of apoptosis regulation proposes that cell fate is determined by the delicate balance between counteracting pro-apoptotic and anti-apoptotic BCL-2 family proteins [91].
At the center of this regulatory system lies the Bax/Bcl-2 ratio, which integrates apoptotic signals to determine cellular susceptibility to death stimuli. As Raisova et al. demonstrated, "a low Bax/Bcl-2 ratio was characteristic for resistant cells and a high Bax/Bcl-2 ratio was characteristic for sensitive cells" in human melanoma cells exposed to CD95/Fas-mediated apoptosis [92]. This ratio determines the susceptibility of cells to undergo mitochondrial outer membrane permeabilization (MOMP), the critical commitment point in intrinsic apoptosis [3]. When the Bax/Bcl-2 ratio favors Bax oligomerization, it triggers cytochrome c release from mitochondria, leading to apoptosome formation and caspase activation [3] [93]. Conversely, when Bcl-2 predominates, it preserves mitochondrial integrity and promotes cell survival [90].
Molecular Mechanisms: How the Ratio Controls Apoptotic Commitment
Regulation of Mitochondrial Outer Membrane Permeabilization (MOMP)
The Bax/Bcl-2 ratio primarily governs the intrinsic apoptosis pathway at the mitochondrial level by regulating MOMP. Upon activation by apoptotic signals, Bax undergoes conformational changes and translocates from the cytoplasm to the mitochondrial outer membrane, where it oligomerizes to form pores [3]. These Bax/Bak pores facilitate the release of cytochrome c and other pro-apoptotic factors from the mitochondrial intermembrane space into the cytoplasm [3] [89].
Anti-apoptotic Bcl-2 counteracts this process through multiple mechanisms: (1) direct binding to Bax via its BH3 domain, preventing Bax activation and oligomerization; (2) sequestration of BH3-only proteins that would otherwise activate Bax; and (3) potential regulation of mitochondrial dynamics and cristae structure [3] [90]. The relative abundance of these opposing forces creates the critical tipping point that determines whether a cell will survive or undergo apoptosis.
Table 1: Bcl-2 Family Proteins and Their Functions in Apoptosis Regulation
| Protein Type | Representative Members | Key Functions | Domain Structure |
|---|---|---|---|
| Anti-apoptotic | Bcl-2, Bcl-XL, Bcl-w, Mcl-1 | Inhibit MOMP, bind and neutralize pro-apoptotic members | BH4, BH3, BH1, BH2 |
| Pro-apoptotic effector | Bax, Bak, Bok | Form mitochondrial pores, mediate cytochrome c release | BH3, BH1, BH2 |
| BH3-only | Bid, Bim, Bad, Puma, Noxa | Initiate apoptosis by antagonizing anti-apoptotic members | BH3 only |
From Cytochrome c Release to Caspase Activation
Once cytochrome c is released into the cytoplasm due to Bax-mediated MOMP, it initiates apoptosome formation by binding to apoptotic protease-activating factor-1 (Apaf-1) in an ATP-dependent manner [3]. This complex then recruits and activates procaspase-9, which subsequently cleaves and activates executioner caspases-3 and -7, leading to the systematic dismantling of the cell [3] [93].
The integrity of this pathway relies heavily on the Bax/Bcl-2 balance. As demonstrated in Peyronie's disease research, Bax overexpression with minimal Bcl-2 expression correlated with strong caspase-3 immunostaining and positive TUNEL assay results, confirming apoptosis activation through the intrinsic pathway [93].
Diagram 1: Intrinsic Apoptosis Pathway Regulation by Bax/Bcl-2 Ratio. A high Bax/Bcl-2 ratio promotes mitochondrial outer membrane permeabilization, triggering the caspase activation cascade.
Quantitative Evidence: The Bax/Bcl-2 Ratio as a Predictive Biomarker
Cancer Therapy Response Prediction
Substantial clinical evidence supports the use of the Bax/Bcl-2 ratio as a predictive biomarker for therapeutic response across various cancer types:
Radiotherapy Response in Breast Cancer: A 2018 study examining breast cancer patients undergoing radiotherapy demonstrated that Bax expression and the Bax/Bcl-2 ratio significantly increased following irradiation with 1 and 2 Gy doses (P<0.001 and P<0.0001, respectively) [94]. Most notably, a significant correlation was observed between the dose-response curve slope (as an in vitro radiosensitivity index) and acute skin toxicity score following irradiation (as a clinical radiosensitivity index) [94]. This finding suggests that the Bax/Bcl-2 ratio determined before radiation therapy could serve as a biomarker to identify radiosensitive individuals.
Melanoma Cell Apoptosis Susceptibility: Seminal research on human melanoma cells established that the Bax/Bcl-2 ratio directly determines susceptibility to CD95/Fas-mediated apoptosis [92]. The study examined 11 melanoma cell populations and found a clear relationship between the Bax/Bcl-2 ratio and apoptotic susceptibility. Additionally, the ratio correlated with sensitivity to N-acetylsphingosine, another cell death inducer [92]. This regulatory function was confirmed through Bcl-2 overexpression experiments, which abolished apoptosis triggered by both apoptotic stimuli.
Cervical Cancer Mutational Profiles: Recent genomic analysis of HPV-associated squamous cell carcinoma of the cervix revealed distinct mutation patterns in both Bcl-2 and Bax genes [91]. Bcl-2 gene mutations primarily showed transition mutations (100%), with nucleotide changes of A>G (50%), C>T (33.33%), and G>A>T (16.67%) [91]. In contrast, BAX gene mutations included Indel (50%), Transversion (33.4%), and Transition (16.6%) mutations [91]. These mutational differences likely alter the functional Bax/Bcl-2 ratio and contribute to apoptosis resistance in cervical cancer.
Table 2: Bax/Bcl-2 Ratio as Predictive Biomarker Across Cancer Types
| Cancer Type | Therapeutic Context | Bax/Bcl-2 Ratio Correlation with Outcomes | Study Reference |
|---|---|---|---|
| Melanoma | CD95/Fas-mediated apoptosis | High ratio = sensitive cells; Low ratio = resistant cells | [92] |
| Breast Cancer | Radiotherapy response | Significant increase post-irradiation; correlates with skin toxicity | [94] [95] |
| Cervical Cancer | HPV-associated squamous cell carcinoma | Distinct mutation patterns affecting ratio | [91] |
| Various Cancers | Chemotherapy resistance | Low ratio associated with poor clinical response | [90] |
Experimental Models and Protective Interventions
Beyond human cancers, the Bax/Bcl-2 ratio has been validated as a sensitive biomarker in experimental models:
UV-B Radiation Protection: A 2024 study on Rattus norvegicus lens epithelial cells demonstrated that UV-B radiation significantly affected the expression of Bax, Bcl-2, and their ratio [96]. More importantly, both anti-UV-B glasses and contact lenses provided similar protection to the lens epithelium, as evidenced by normalized Bax/Bcl-2 ratios in protected groups compared to significantly altered ratios in unprotected exposure [96]. This research established that the Bax/Bcl-2 ratio can serve as a quantitative biomarker for comparing the efficacy of protective interventions against apoptotic stimuli.
Peyronie's Disease Fibrosis: Research on fibrotic plaques in Peyronie's disease revealed Bax overexpression with little to no Bcl-2 immunostaining in fibroblasts and myofibroblasts of plaques from patients [93]. This altered ratio was associated with strong caspase-3 immunostaining and positive TUNEL assay results, confirming activation of the intrinsic apoptosis pathway in this non-malignant condition [93].
Experimental Approaches: Methodologies for Ratio Quantification
Standardized Workflow for Bax/Bcl-2 Ratio Determination
Accurate measurement of the Bax/Bcl-2 ratio requires careful experimental design and appropriate methodological choices. The following workflow represents a standardized approach for ratio determination:
Diagram 2: Experimental Workflow for Bax/Bcl-2 Ratio Determination. Multiple methodological approaches can be employed for quantification at protein or mRNA levels.
Key Methodological Platforms
Protein-Level Quantification
Western Blot Analysis: Quantitative Western blotting was used in the seminal melanoma study to assign Bax/Bcl-2 ratios across 11 melanoma cell populations [92]. This method provides information about protein size and specificity through molecular weight detection. The standard protocol includes: protein extraction from cells or tissues, quantification by BCA or Bradford assay, SDS-PAGE separation, transfer to PVDF or nitrocellulose membranes, blocking with 5% non-fat milk or BSA, incubation with primary antibodies (anti-Bax and anti-Bcl-2), washing, incubation with HRP-conjugated secondary antibodies, and chemiluminescent detection. Normalization to housekeeping proteins (e.g., β-actin, GAPDH) is essential for accurate ratio calculation.
Immunohistochemistry (IHC): IHC enables localization of Bax and Bcl-2 expression within tissue architecture, as demonstrated in Peyronie's disease research [93]. Standard IHC protocol includes: tissue fixation in 10% neutral buffered formalin, paraffin embedding, sectioning at 5μm thickness, antigen retrieval using citrate buffer (pH 6) in microwave oven, peroxidase quenching with 3% H₂O₂, blocking with normal serum, incubation with primary antibodies (typically overnight at 4°C), detection using biotinylated secondary antibodies and streptavidin-HRP, DAB chromogen development, and hematoxylin counterstaining. Semiquantitative analysis involves scoring intensity (0-4) and proportion of immunopositive cells (<5% to >75%) [93].
mRNA-Level Quantification
Reverse Transcription Quantitative PCR (RT-qPCR): This method was employed in breast cancer radiotherapy studies to measure Bax and Bcl-2 expression in peripheral blood mononuclear cells [94]. The standard protocol includes: RNA extraction using TriPure or similar reagents, DNAse treatment, cDNA synthesis using reverse transcriptase with oligo(dT) or random hexamers, quantitative PCR with gene-specific primers, and normalization to reference genes (e.g., GAPDH, β-actin). The comparative Ct (ΔΔCt) method is typically used for relative quantification of expression ratios.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for Bax/Bcl-2 Ratio Analysis
| Reagent/Category | Specific Examples | Application Notes | Quality Control |
|---|---|---|---|
| Primary Antibodies | Anti-Bax (polyclonal, Dako), Anti-Bcl-2 (monoclonal, Dako) | IHC, Western blot; species-specific | Validate with positive controls (basal cell carcinoma specimens) [93] |
| RNA Extraction Kits | TriPure reagent (Roche) | Maintain RNA integrity for RT-qPCR | Assess by agarose gel electrophoresis [94] |
| PCR Components | GoTaq colorless reaction buffer, MgCl₂, primers specific for Bax/Bcl-2 | Optimize annealing temperatures | Include no-template controls |
| IHC Detection | LSAB+System-HRP (Dako), DAB substrate kit (Vector Labs) | Optimize developing time to prevent background | Include negative controls (normal serum instead of primary antibody) [93] |
| Cell Culture Media | RPMI 1640 (GIBCO) with FBS, penicillin/streptomycin | Maintain PBMCs for in vitro irradiation studies | Test for mycoplasma contamination |
Discussion: Implications for Research and Therapeutic Development
The Bax/Bcl-2 ratio represents a quantifiable molecular tipping point with significant implications for basic research and clinical applications. As a critical determinant of cellular susceptibility to apoptosis, it provides researchers with a reliable parameter for validating intrinsic pathway activation across multiple contexts.
In drug development, measuring the Bax/Bcl-2 ratio offers a powerful approach for screening pro-apoptotic compounds and identifying potential resistance mechanisms. The development of BH3 mimetics, such as the Bcl-2 inhibitor Venetoclax, highlights the therapeutic potential of targeting this regulatory system [89]. However, emerging mutations in the BCL-2 gene (e.g., F104L and F104C) that reduce drug binding affinity without altering affinity for Bax and BIM represent a challenge that requires further investigation [89].
For personalized medicine, the Bax/Bcl-2 ratio shows promise as a predictive biomarker for tailoring cancer therapies. The correlation between pre-treatment ratios and radiotherapy response in breast cancer patients suggests potential for identifying individuals who would benefit from treatment intensification or alternative approaches [94]. Similarly, in cervical cancer, the distinct mutation patterns in BAX and Bcl-2 genes may inform prognosis and treatment selection [91].
From a methodological perspective, standardization of Bax/Bcl-2 ratio measurement protocols is essential for comparing results across studies. While various techniques provide valuable data, consistency in sample processing, normalization methods, and quantification approaches would enhance reproducibility and clinical translation.
The Bax/Bcl-2 expression ratio serves as a critical tipping point in the regulation of intrinsic apoptosis, functioning as a molecular rheostat that determines cellular fate in response to diverse stimuli. Robust experimental evidence from cancer research, therapeutic response studies, and experimental models confirms that this ratio provides a reliable biomarker for apoptosis susceptibility. Through standardized methodologies including Western blot, immunohistochemistry, and RT-qPCR, researchers can accurately quantify this decisive parameter to validate intrinsic pathway activation. As drug development increasingly targets Bcl-2 family interactions, and personalized medicine seeks predictive biomarkers for treatment response, the Bax/Bcl-2 ratio remains an essential measurement for both basic research and clinical applications.
Navigating Technical Limitations of Flow Cytometry and Imaging Assays
Validating the activation of the intrinsic apoptosis pathway is a fundamental requirement in biomedical research, spanning drug discovery, toxicology, and basic cell biology. This pathway, initiated by cellular stress signals that trigger mitochondrial outer membrane permeabilization (MOMP), leads to the release of cytochrome c and subsequent caspase activation. Researchers primarily rely on flow cytometry (FCM) and various imaging assays to detect these molecular events, yet each platform presents distinct technical limitations that can compromise data accuracy and interpretation.
This guide provides an objective comparison of these technologies, supported by experimental data, to enable researchers to select appropriate methodologies, implement optimized protocols, and accurately validate intrinsic apoptosis activation in their experimental systems.
Technology Platform Comparison: Capabilities and Limitations
The choice between conventional flow cytometry, imaging flow cytometry (IFC), and live-cell imaging platforms involves significant trade-offs between throughput, spatial resolution, and temporal resolution.
Table 1: Technical Comparison of Apoptosis Analysis Platforms
| Platform | Throughput | Spatial Resolution | Temporal Resolution | Key Advantages | Primary Limitations |
|---|---|---|---|---|---|
| Conventional Flow Cytometry | High (up to 70,000 cells/sec) [5] | None (no imaging) | Single time-point (end-point) | High-throughput, multiparametric quantification (up to 8 parameters from one sample) [5] | No morphological context; relies on population-level statistics [97] |
| Imaging Flow Cytometry (IFC) | Medium-High (up to 10,000 cells/sec) [97] | High (sub-micron, ~780 nm) [98] | Single time-point (end-point) | Combines high-throughput with high-resolution morphological imaging [97] | Higher cost; complex data analysis; requires specialized expertise [97] |
| Live-Cell Imaging (e.g., Incucyte) | Low (limited fields of view) | Medium (diffraction-limited) | High (real-time, kinetic) | Enables kinetic quantification of apoptosis in real-time [99] | Lower throughput; potential phototoxicity; limited multiplexing depth [99] |
Advanced IFC systems have pushed throughput boundaries, with one system demonstrating real-time analysis exceeding 1,000,000 events per second while maintaining sub-micron resolution [98]. However, this generates massive data volumes, requiring sophisticated processing pipelines. Conversely, live-cell imaging systems like the Incucyte address the temporal limitation of endpoint assays by allowing continuous, non-invasive monitoring of the same population over time, which is crucial for capturing the dynamic sequence of apoptotic events [99].
Performance Comparison in Apoptosis Detection
Direct methodological comparisons reveal critical differences in sensitivity, specificity, and practical performance between flow cytometry and imaging assays.
Table 2: Experimental Performance Data: Flow Cytometry vs. Imaging Assays
| Assay Type | Detection Method | Sensitivity | Specificity | Key Experimental Findings | Reference/Context |
|---|---|---|---|---|---|
| Viability (FM vs. FCM) | FM: FDA/PI; FCM: Hoechst, DiIC1, Annexin V, PI | FM: 9-10% viability; FCM: 0.2-0.7% viability (under high cytotoxic stress) [100] | Strong correlation (r=0.94) between methods [100] | FCM showed superior precision and ability to distinguish apoptosis from necrosis under high cytotoxic stress [100] | Cytotoxicity of Bioglass 45S5 on SAOS-2 cells [100] |
| Autoantibody Detection (CBA vs. FACS) | Live Cell-Based Assay (CBA) vs. FACS | CBA: 100%; FACS: 87% [101] | 100% for both methods [101] | FACS showed high inter-assay variation; CBA deemed more reliable for clinical detection of NMDAR antibodies [101] | Detection of NMDAR antibodies in patient serum [101] |
| T-cell Response (FC-ICS vs. QF) | Flow Cytometry Intracellular Cytokine Staining (FC-ICS) vs. QuantiFERON (QF) | FC-ICS detected more positive results (134 vs. 120) [102] | Significantly discordant qualitative results (P<0.001) [102] | FC-ICS demonstrated greater sensitivity, urging caution when comparing immune responses across platforms [102] | Detection of SARS-CoV-2-S-reactive T cells post-vaccination [102] |
A comparative study of cell viability assessment highlighted flow cytometry's advantage in precision, especially under high cytotoxic stress. While both fluorescence microscopy (FM) and FCM showed a strong correlation (r=0.94), FCM provided more precise viability measurements (0.2-0.7%) compared to FM (9-10%) when assessing bioactive glass cytotoxicity, and could further distinguish early and late apoptosis from necrosis [100]. This underscores FCM's superior resolution for dissecting complex cell death mechanisms.
Experimental Protocols for Validating Intrinsic Apoptosis
A comprehensive understanding of intrinsic apoptosis activation requires a multiparametric approach. The following protocols can be implemented individually or integrated into a unified workflow.
Integrated Multiparametric Flow Cytometry Protocol
This protocol enables the assessment of key apoptotic parameters—mitochondrial membrane potential, apoptosis induction, and cell cycle status—from a single sample [5].
Step-by-Step Workflow:
Cell Staining:
- Proliferation Tracking: Incubate cells with CellTrace Violet (1-5 µM) for 20 minutes at 37°C. Quench with complete medium [5].
- DNA Synthesis Labeling: Add BrdU (10 µM final concentration) for 30-60 minutes at 37°C to pulse-label S-phase cells [5].
- Mitochondrial Staining: Incubate with JC-1 dye (2 µM) for 30 minutes at 37°C, protected from light [5].
- Apoptosis Staining: Wash cells and resuspend in Annexin V Binding Buffer. Add Annexin V-APC and propidium iodide (PI) [5].
Fixation and Permeabilization: Fix cells with 70% ethanol at -20°C for 1 hour. Permeabilize and denature DNA for BrdU detection using HCl and Triton X-100 [5].
BrdU Staining: Stain cells with anti-BrdU-FITC antibody for 30 minutes at room temperature [5].
DNA Staining: Resuspend cells in PI/RNase A solution and incubate for 30 minutes at 37°C [5].
Flow Cytometry Analysis: Acquire data on a flow cytometer equipped with lasers for violet (405 nm), blue (488 nm), and red (633 nm) excitation. Analyze a minimum of 10,000 events per sample [5].
Integrated Multiparametric Flow Cytometry Workflow
Live-Cell Imaging Apoptosis Assay Protocol
This protocol uses the Incucyte system for real-time, kinetic quantification of apoptosis [99].
Step-by-Step Workflow:
Cell Seeding: Seed adherent cells (e.g., 2,000-5,000 cells per well) in a 96-well or 384-well plate and allow to adhere for 18-24 hours [99].
Treatment and Dye Addition: Treat cells with experimental compounds. Simultaneously, add Incucyte Caspase-3/7 Green Dye (or Annexin V Dye) directly to the medium at a 1:1000 dilution. Mix gently by pipetting [99]. Note: No wash steps are required.
Real-Time Data Acquisition: Place the plate in the Incucyte Live-Cell Analysis System. Acquire both phase-contrast and fluorescent images (e.g., 20X magnification) from multiple fields per well at regular intervals (e.g., every 2-6 hours) for the duration of the experiment [99].
Image and Data Analysis: Use integrated software to automatically segment and quantify fluorescent objects (apoptotic cells). Correlate fluorescence with morphological changes visible in phase-contrast images, such as membrane blebbing and cell shrinkage [99].
The Intrinsic Apoptosis Pathway: A Visual Guide
The intrinsic (mitochondrial) pathway is coordinated through a series of key molecular events. The following diagram maps this process and highlights the specific steps that can be measured using the protocols and reagents described in this guide.
Intrinsic Apoptosis Pathway & Detection Points
Research Reagent Solutions
Selecting appropriate reagents is critical for successful apoptosis detection. The following table summarizes key solutions used in the featured protocols.
Table 3: Essential Reagents for Apoptosis Pathway Analysis
| Reagent | Detection Target | Function & Application | Platform |
|---|---|---|---|
| JC-1 | Mitochondrial Membrane Potential (ΔΨm) | Fluorescent dye that shifts emission from red (high ΔΨm) to green (low ΔΨm) upon depolarization [5]. Marker for early intrinsic apoptosis. | Flow Cytometry, Fluorescence Microscopy |
| Annexin V (FITC/APC) | Phosphatidylserine (PS) Externalization | Binds to PS on the outer leaflet of the plasma membrane, a hallmark of early apoptosis. Used with viability dyes (PI) to distinguish stages of cell death [103] [5]. | Flow Cytometry, Imaging Flow Cytometry, Live-Cell Imaging |
| Caspase-3/7 Dyes (FLICA, Incucyte) | Caspase-3/7 Enzyme Activity | Cell-permeable substrates that become fluorescent upon cleavage by active caspases, indicating irreversible commitment to apoptosis [99]. | Flow Cytometry, Live-Cell Imaging |
| Propidium Iodide (PI) | Plasma Membrane Integrity / DNA Content | DNA-binding dye excluded by live cells. Used with Annexin V to identify late apoptotic/necrotic cells, or in fixed samples for cell cycle analysis (sub-G1 peak) [104] [103] [5]. | Flow Cytometry |
| BrdU | DNA Synthesis / S-Phase Cells | Thymidine analog incorporated during DNA replication. Detected with specific antibodies to assess cell proliferation and cell cycle dynamics [5]. | Flow Cytometry |
| CellTrace Violet | Cell Proliferation & Generations | Fluorescent cytoplasmic dye that dilutes evenly with each cell division, allowing tracking of proliferation rates and generations [5]. | Flow Cytometry |
Flow cytometry and imaging assays offer complementary strengths for validating intrinsic apoptosis. Flow cytometry provides unmatched statistical power for multiparametric analysis at the population level, while imaging assays deliver crucial morphological context and temporal dynamics. The choice between these platforms is not a matter of superiority but of strategic alignment with research goals. By understanding their specific limitations—such as FCM's lack of spatial information and conventional microscopy's lower throughput—researchers can implement the robust, cross-validated methodologies necessary for conclusive validation of intrinsic apoptosis pathway activation.
Ensuring Robust Results: A Multi-Parametric Validation and Cross-Comparison Framework
In the investigation of complex biological processes like the intrinsic apoptosis pathway, reliance on a single measurement provides an incomplete and potentially misleading picture. The intrinsic pathway is characterized by a cascade of interconnected cellular events, including mitochondrial outer membrane permeabilization (MOMP), redox state changes, and caspase activation. This article makes the case for multi-parametric analysis—the simultaneous measurement of multiple parameters in the same biological sample—as an essential approach for robust validation in cell death research. Supported by comparative data and detailed protocols, we demonstrate how this methodology provides a comprehensive, systems-level understanding that is unattainable with single-assay approaches.
The Critical Limitations of Single-Parameter Assays
Single-parameter assays, while simple and cost-effective, are fraught with limitations that can compromise research conclusions.
- Inability to Capture Heterogeneity: Cell populations are inherently heterogeneous. A single-parameter measurement, such as a bulk caspase activity assay, can mask distinct subpopulations of cells at different stages of apoptosis [105].
- High Risk of False Negatives/Positives: A process may be active without significantly altering the single parameter being measured. For instance, early apoptotic events may occur without immediate changes in plasma membrane integrity, leading to false negatives in assays based solely on viability dyes [103].
- Lack of Mechanistic Insight: A single data point offers no information on the temporal or causal relationships between key events. Understanding whether mitochondrial fragmentation precedes or follows Bax activation is critical for elucidating apoptotic regulation, a relationship that cannot be decoded from sequential single assays [105].
The evidence is clear: "Comparisons between tests in clinical trials derived cohorts provide consistent evidence that combining test results generally improves prognostic value" [106]. This principle, well-established in clinical diagnostics, is equally applicable to fundamental biological research.
Multi-Parametric Methodologies in Action
Live-Cell Multiparametric Microscopy
This technique allows for the real-time tracking of multiple fluorescent biomarkers in individual living cells, revealing the dynamics of apoptosis.
Experimental Protocol:
- Cell Preparation: Stable expression of a FRET-based caspase-3 sensor (e.g., mKate2-DEVD-iRFP) is required. The sensor exhibits a change in fluorescence lifetime upon caspase-3 cleavage [107].
- Data Acquisition: Use Two-Photon Excitation Fluorescence Lifetime Imaging Microscopy (FLIM) to acquire time-lapse images. Simultaneously measure:
- Caspase-3 Activation: Track the fluorescence lifetime of the mKate2 donor.
- Redox State: Image the autofluorescence of metabolic cofactors NAD(P)H and FAD to calculate the FAD/NAD(P)H redox ratio.
- Reactive Oxygen Species (ROS): Use a fluorescent ROS-sensitive dye.
- Data Analysis: Correlate the timing of ROS accumulation, shifts in redox ratio, and caspase-3 activation on a per-cell basis [107].
The diagram below illustrates the workflow and the key parameters measured in this live-cell multiparametric assay.
Flow Cytometry with Annexin V/Propidium Iodide and Protein Labeling
Flow cytometry is a powerful high-throughput platform for multi-parametric analysis of apoptosis in cell populations.
Experimental Protocol:
- Cell Staining: Harvest cells and resuspend in binding buffer. Dual-stain with:
- Annexin V-FITC: Binds to phosphatidylserine (PS) exposed on the outer leaflet of the plasma membrane during early apoptosis.
- Propidium Iodide (PI): A membrane-impermeant dye that enters cells in late apoptosis and necrosis.
- APC-conjugated Antibodies: For simultaneous detection of specific protein markers (e.g., CD44) [103].
- Data Acquisition and Gating: Analyze samples using a flow cytometer with appropriate filters and compensation controls. The gating strategy typically identifies four populations:
- Viable cells: Annexin V⁻ / PI⁻
- Early Apoptotic cells: Annexin V⁺ / PI⁻
- Late Apoptotic cells: Annexin V⁺ / PI⁺
- Necrotic cells: Annexin V⁻ / PI⁺ (if present)
- Analysis: Quantify the percentage of cells in each quadrant and measure the mean fluorescence intensity of the APC channel (specific protein marker) within each gated population to track protein expression changes from viable to apoptotic states [103].
High-Content Analysis of Mitochondrial Morphology and Function
This approach combines automated microscopy with computational analysis to quantify complex morphological phenotypes and functional parameters at a single-cell level.
Experimental Protocol:
- Cell Staining and Imaging: Label mitochondria (e.g., with Mito-GFP) and incubate with potentiometric dyes (e.g., TMRM) to measure mitochondrial membrane potential (ΔΨm). Expose cells to apoptotic stimuli and acquire high-resolution images [105].
- Automated Image Analysis: Use software like CellProfiler to perform:
- Image Preprocessing: Improve signal-to-noise ratio.
- Segmentation: Identify individual cells and mitochondria.
- Feature Extraction: Calculate hundreds of morphological features (e.g., size, shape, texture, connectivity) for each cell [105].
- Machine Learning Classification: Train a Random Forest classifier to automatically categorize mitochondrial morphology into classes such as networked, fragmented, or swollen based on the extracted features [105].
- Data Integration: Integrate morphological classifications with functional data (e.g., ΔΨm, Bax activation) using data-driven modeling (e.g., fuzzy logic) to explore non-linear relationships between morphology and apoptosis [105].
Comparative Data: Single vs. Multi-Parametric Insights
The following tables summarize key experimental findings that highlight the superior information gained from a multi-parametric approach.
Table 1: Contrasting Insights from Single vs. Multi-Parametric Assays in Apoptosis Research
| Research Question | Single-Parameter Assay Insight | Multi-Parametric Assay Insight | Key Reference |
|---|---|---|---|
| How does cisplatin induce cell death? | Activation of caspase-3 (a single downstream event). | Cisplatin induces a generalized oxidative shift (increased FAD/NAD(P)H ratio) in both apoptotic and non-apoptotic cells, suggesting a stress response independent of caspase commitment. | [107] |
| What is the role of mitochondrial morphology? | Correlation of overall fragmented state with apoptosis. | A pre-fragmented mitochondrial state can be protective by limiting cell-to-cell apoptosis propagation, revealing a paradoxical role. | [105] |
| How does a drug affect a specific protein? | Change in total protein expression level in a whole population. | Tracking CD44 protein expression separately in viable, early, and late apoptotic cells reveals stage-specific regulation. | [103] |
Table 2: Quantitative Results from a Multiparametric Live-Cell FLIM Study [107]
| Apoptotic Stimulus | Caspase-3 Activation (Fluorescence Lifetime, ns) | Redox Ratio FAD/NAD(P)H | Correlation between ROS and Caspase-3 |
|---|---|---|---|
| Staurosporine (5 µM) | Increased from 1.53 to 1.86 ns at 0.5 h | Increased from ~1.1 to ~2.6 | Strong correlation observed |
| Cisplatin (2.2 µM) | Increased to 1.88 ns in 75% of cells by 0.5 h | Increased from ~1.1 to ~2.8 | ROS accumulation correlated with caspase-3 activation |
| Hydrogen Peroxide (1 mM) | Increased to 1.90 ns at 4 h (77% cells) | Gradually increased from ~0.9 to ~1.3 | Strong correlation observed |
The Scientist's Toolkit: Essential Reagents for Multi-Parametric Analysis
Table 3: Key Research Reagent Solutions for Apoptosis Validation
| Reagent / Assay | Function / Target | Key Application in Multi-Parametric Analysis | |
|---|---|---|---|
| FRET-based Caspase Sensor (e.g., mKate2-DEVD-iRFP) | Caspase-3/7 activity | Allows kinetic tracking of caspase activation in live cells via FLIM, without interference from other fluorescent probes. | [107] |
| Annexin V (FITC conjugate) | Phosphatidylserine (PS) exposure | Marker for early apoptosis. Used in combination with PI and antibody conjugates (e.g., APC) in flow cytometry. | [103] |
| Propidium Iodide (PI) | DNA in membrane-compromised cells | Distinguishes late apoptotic/necrotic cells (PI-positive) from early apoptotic cells (PI-negative). | [103] |
| TMRM / TMRE | Mitochondrial membrane potential (ΔΨm) | Potentiometric dye to assess mitochondrial health and function. A loss of signal indicates depolarization. | [105] |
| NAD(P)H & FAD Autofluorescence | Cellular metabolic redox state | The FLIM lifetime and intensity ratio (FAD/NAD(P)H) serve as non-invasive metabolic indicators. | [107] |
| CellProfiler Software | Image analysis and feature extraction | Enables automated, high-throughput quantification of morphological features from thousands of cells. | [105] |
| Random Forest Classifier | Machine learning classification | Automatically classifies complex morphological phenotypes (e.g., networked/fragmented mitochondria) from extracted features. | [105] |
A Logical Framework for Multi-Parametric Analysis
The following diagram maps the logical relationships between key components of the intrinsic apoptosis pathway, illustrating the interconnected system that multi-parametric analysis seeks to measure.
The validation of intrinsic apoptosis pathway activation demands a move beyond simplistic, single-parameter assays. As demonstrated, multi-parametric analysis through techniques like live-cell microscopy, advanced flow cytometry, and high-content imaging provides a comprehensive, mechanistic, and clinically relevant understanding of cell death. It reveals critical heterogeneities, establishes causal and temporal relationships, and builds a robust framework for evaluating therapeutic efficacy and resistance. For researchers and drug developers committed to rigorous science, a multi-parametric strategy is not just an option—it is an imperative.
Correlating Biochemical, Morphological, and Functional Readouts
Validating the activation of the intrinsic apoptosis pathway is a cornerstone of biomedical research, particularly in cancer biology and drug discovery. This pathway, initiated by cellular stress signals such as DNA damage or growth factor deprivation, is characterized by mitochondrial outer membrane permeabilization (MOMP), leading to the release of cytochrome c and the activation of executioner caspases [29]. Given the complexity and transient nature of this process, relying on a single analytical method provides an incomplete picture. A multi-parametric approach that correlates biochemical, morphological, and functional readouts is essential for a definitive assessment of apoptotic commitment. This guide objectively compares the performance of current technologies and assays, providing the experimental data and protocols necessary to triangulate intrinsic apoptosis activation confidently.
Morphological Hallmarks of Apoptosis
The initiation of the intrinsic apoptotic cascade precipitates a series of distinctive morphological changes. These cellular alterations serve as the first line of evidence for apoptosis and can be visualized using various imaging techniques.
- Cell Shrinkage and Condensation: The cell undergoes a reduction in volume and its cytoplasm becomes denser [108] [109].
- Membrane Blebbing: The plasma membrane displays dynamic, outward blebbing events [110] [29].
- Nuclear Fragmentation: The nucleus exhibits chromatin condensation (pyknosis), followed by nuclear membrane budding and fragmentation into discrete apoptotic bodies [108] [109] [29]. This is a consequence of caspase-activated DNase (CAD) activity.
- Formation of Apoptotic Bodies: The cell dismantles into small, membrane-bound vesicles containing intact organelles and nuclear fragments, which are rapidly phagocytosed by neighboring cells without inciting inflammation [109] [29].
Advanced analytical approaches are now leveraging these morphological features. For instance, artificial intelligence (AI) and machine learning models have been developed to detect early apoptotic cells based on quantifiable textural changes in nuclear chromatin, such as alterations in Gray-Level Entropy Matrix (GLEM) Entropy and Run-Length Matrix (RLM) features [111].
Biochemical Assays for Apoptosis Detection
Biochemical assays provide specific, measurable readouts of key molecular events in the apoptotic pathway. The table below compares the most commonly used biochemical assays.
Table 1: Comparison of Key Biochemical Assays for Apoptosis Detection
| Assay Target | Detection Method | Key Feature | Stage of Apoptosis | Throughput | Key Performance Data |
|---|---|---|---|---|---|
| Caspase-3/7 Activity | Fluorogenic/Luminogenic substrates (e.g., DEVD-AMC, DEVD-aminoluciferin) [61] | Irreversible commitment to cell death; direct marker of executioner phase [99] | Mid to Late | High (HTS amenable) | Luminogenic assays are 20-50x more sensitive than fluorogenic versions [61]. |
| Phosphatidylserine (PS) Exposure | Annexin V binding (conjugated to fluorophores or luciferase subunits) [99] [61] | "Eat-me" signal on the outer leaflet of the plasma membrane [112] | Early | Medium-High (with no-wash protocols) | Amenable to multiplexing with caspase-3/7 or cytotoxicity dyes [99]. |
| DNA Fragmentation | TUNEL Assay [110] [109] | Detects DNA strand breaks in situ | Late | Low (manual, multi-step) | Prone to false positives from non-apoptotic DNA damage; requires careful standardization [109]. |
| Mitochondrial Membrane Potential (ΔΨm) | Cationic dyes (e.g., TMRE, JC-1) [110] | Loss of ΔΨm is an early event in intrinsic pathway [110] | Early-Mid | Medium | Decreased fluorescence intensity correlates with dysfunctional mitochondria [110]. |
Experimental Protocols for Key Biochemical Assays
1. Caspase-3/7 Activity Assay (Luminometric, HTS Protocol)
- Principle: Upon cleavage by caspase-3/7, a DEVD-aminoluciferin substrate is released and consumed by firefly luciferase, generating a luminescent signal proportional to caspase activity [61].
- Protocol: a. Cell Seeding: Plate adherent or suspension cells in opaque-walled, white microplates (96-, 384-, or 1536-well format). b. Treatment: Apply apoptotic inducers (e.g., chemotherapeutics like Camptothecin or Cisplatin) for a designated time course. c. Assay Reagent Addition: Add an equal volume of Caspase-Glo 3/7 Reagent directly to each well. The lytic nature of the reagent lyses the cells, exposing caspases to the substrate. d. Incubation and Reading: Mix plates on an orbital shaker and incubate at room temperature for 30-60 minutes. Measure luminescence using a plate-reading luminometer [61].
- Validation Data: This assay is highly robust against DMSO concentrations up to 10% and has been validated in ultra-HTS campaigns screening over 325,000 compounds [61].
2. Annexin V Staining (No-wash, Live-Cell Kinetic Protocol)
- Principle: Recombinant Annexin V conjugated to a bright, photostable fluorophore (e.g., Cyanine dye) binds with high affinity to externally exposed PS [99].
- Protocol: a. Cell Preparation: Seed cells in a standard microplate. b. Dye Addition: Add Incucyte Annexin V Dye directly to the culture medium at the recommended concentration (mix-and-read, no wash steps). c. Treatment and Imaging: Treat cells with apoptotic inducers. Place the plate in a live-cell imaging system (e.g., Incucyte) for automated, kinetic imaging. d. Quantification: Use integrated software to automatically segment and quantify Annexin V-positive fluorescent objects over time [99].
- Validation Data: This protocol has been used to generate pharmacological dose-response curves and kinetic apoptosis profiles for compounds like Camptothecin in A549 cancer cells, demonstrating concentration-dependent effects over 72 hours [99].
Integrated Analysis: Multiplexing and Live-Cell Imaging
The most powerful validation comes from correlating multiple readouts simultaneously in the same sample. Modern platforms enable this integrated analysis.
Table 2: Research Reagent Solutions for Multiplexed Apoptosis Analysis
| Reagent / Solution | Function | Application in Apoptosis Research |
|---|---|---|
| Incucyte Annexin V Dyes | Fluorescently labels exposed phosphatidylserine | Kinetic, no-wash measurement of early apoptosis in live cells [99]. |
| Incucyte Caspase-3/7 Dyes | Cell-permeable, non-fluorescent substrates cleaved to release DNA-binding fluorophore | Real-time measurement of executioner caspase activation in live cells [99]. |
| Incucyte Nuclight Reagents | Lentiviral reagents for constitutive nuclear labeling (e.g., with H2B-GFP or H2B-RFP) | Multiplexing with apoptosis dyes to simultaneously track cell proliferation, confluence, and death [99]. |
| ZipGFP-based Caspase Reporter | Genetically encoded, stable biosensor for caspase-3/7 activity | Real-time, single-cell tracking of apoptosis dynamics in 2D and 3D cultures; irreversible fluorescence upon DEVD cleavage marks apoptotic events [113]. |
| Caspase-Glo 3/7 Assay | Lytic, luminogenic reagent for caspase-3/7 activity | Highly sensitive, endpoint or kinetic measurement of caspase activity in HTS formats [61]. |
Workflow for Multiplexed Kinetic Analysis: A study demonstrated the power of multiplexing by treating HT-1080 fibrosarcoma cells (stably labeled with Nuclight NIR for nuclear tracking) with a serial dilution of Camptothecin in the presence of a Caspase-3/7 Green dye. The integrated analysis allowed for the simultaneous quantification of two key parameters over 48 hours: a concentration-dependent decrease in NIR nuclear count (a functional readout of anti-proliferative effect and cell loss) and a corresponding increase in green caspase-3/7 signal (a biochemical readout of apoptotic induction) [99]. This multi-parametric approach provides a comprehensive view of compound efficacy.
Signaling Pathway and Experimental Workflow
Diagram 1: Intrinsic Apoptosis Pathway & Correlated Readouts.
Diagram 2: Experimental Workflow for Multiplexed Validation.
Validating intrinsic apoptosis requires moving beyond single-parameter snapshots to a dynamic, multi-faceted view of cell death. As demonstrated, technologies like live-cell analysis systems and stable fluorescent reporter cells enable researchers to correlate the biochemical commitment of caspase activation with the ensuing morphological changes and functional outcomes like proliferation inhibition in real time. The experimental data and protocols provided here underscore that a synergistic approach—leveraging biochemical precision, morphological context, and functional relevance—is paramount for generating robust, publication-quality data on intrinsic apoptosis activation, ultimately accelerating the pace of discovery in basic research and therapeutic development.
Distinguishing Intrinsic from Extrinsic Apoptosis and Other RCD Forms
Apoptosis, a form of programmed cell death (PCD), is an evolutionarily conserved, energy-dependent process crucial for embryonic development, tissue homeostasis, and the elimination of damaged or dangerous cells [114] [115]. This genetically programmed cell suicide mechanism is characterized by distinct morphological changes, including cell shrinkage, chromatin condensation, membrane blebbing, and nuclear fragmentation, culminating in the formation of apoptotic bodies that are swiftly phagocytosed without inducing inflammation [114] [116]. The precise regulation of apoptotic pathways is fundamental to cellular health, with dysregulation contributing to various pathologies, including cancer, autoimmune disorders, and neurodegenerative diseases [115] [116].
For researchers and drug development professionals, distinguishing between the two primary apoptotic pathways—intrinsic and extrinsic—and other forms of regulated cell death (RCD) is paramount for accurate experimental interpretation and therapeutic development. The intrinsic (mitochondrial) pathway responds to internal cellular stressors, while the extrinsic (death receptor) pathway initiates from external death signals [10] [117]. Both pathways converge on the activation of caspases, a family of cysteine proteases that execute the dismantling of the cell [118]. This guide provides a comprehensive comparison of these pathways, detailed experimental methodologies for their validation, and essential tools for researchers focused on elucidating the mechanisms of intrinsic apoptosis activation.
Comparative Analysis of Apoptotic Pathways
Defining Features and Key Distinctions
Apoptosis is primarily executed through two initiating pathways that possess distinct triggers and regulatory mechanisms but ultimately converge on a common execution phase.
Table 1: Fundamental Characteristics of Intrinsic and Extrinsic Apoptosis
| Feature | Intrinsic Apoptosis | Extrinsic Apoptosis |
|---|---|---|
| Initiating Stimulus | Internal cellular stress: DNA damage, hypoxia, oxidative stress, cytokine deprivation [10] [119] [114] | External ligand binding: Death ligands (e.g., FasL, TNF-α) from immune cells or other sources [117] [115] |
| Key Regulatory Proteins | Bcl-2 family proteins (BAX, BAK, Bcl-2, Bcl-xL) [119] [116] | Death Receptors (Fas, TNFR1), FADD, Caspase-8 [10] [117] |
| Central Signaling Hub | Mitochondria [119] [115] | Plasma Membrane Death-Inducing Signaling Complex (DISC) [10] [120] |
| Key Initiator Caspase | Caspase-9 [119] [114] | Caspase-8 [117] [120] |
| Biomarker Release | Cytochrome c, SMAC/DIABLO, AIF from mitochondria [10] [119] | No specific biomarker release; initiated by surface receptor clustering [117] |
| Core Signaling Complex | Apoptosome (APAF-1, Cytochrome c, Caspase-9) [119] [115] | DISC (Death Receptor, FADD, Caspase-8) [10] [120] |
| Primary Physiological Role | Eliminating damaged or stressed cells; maintaining tissue homeostasis [119] [118] | Immune surveillance; eliminating infected or abnormal cells [117] |
Molecular Mechanisms and Cross-Talk
The intrinsic and extrinsic pathways, while independently activated, are not entirely isolated and can interconnect through specific molecular mediators.
Figure 1: Integrated Apoptotic Signaling Pathways. The intrinsic (yellow/red) and extrinsic (green) pathways converge through caspase activation and the tBID cross-talk mechanism [10] [119] [115].
The intrinsic pathway is characterized by Mitochondrial Outer Membrane Permeabilization (MOMP), a pivotal event controlled by the balance of pro- and anti-apoptotic Bcl-2 family proteins [119]. Cellular stress stabilizes p53, which transcriptionally activates pro-apoptotic BH3-only proteins. These activators (like BIM and PUMA) neutralize anti-apoptotic proteins (like Bcl-2 and Bcl-xL) and directly activate the executioner proteins BAX and BAK. Oligomerized BAX/BAK form pores in the mitochondrial outer membrane, leading to MOMP and the release of cytochrome c into the cytosol [119] [116]. Cytochrome c then binds to APAF-1, forming the apoptosome, which recruits and activates caspase-9 [119] [115].
The extrinsic pathway is initiated by the binding of death ligands (e.g., FasL, TRAIL, TNF-α) to their cognate death receptors on the cell surface. This binding induces receptor trimerization and the recruitment of the adaptor protein FADD and procaspase-8, forming the Death-Inducing Signaling Complex (DISC). Within the DISC, caspase-8 undergoes autocatalytic activation [10] [117] [120].
Both pathways converge on the execution phase, where initiator caspases (8 or 9) cleave and activate effector caspases (3, 6, and 7). Caspase-3, the primary executioner, proteolytically cleaves key cellular substrates such as PARP, lamin A/C, and ICAD, leading to the characteristic morphological and biochemical hallmarks of apoptosis [114] [116]. The pathways are interconnected via caspase-8-mediated cleavage of the BH3-only protein BID to its active form, tBID, which translocates to mitochondria and amplifies the apoptotic signal by activating BAX/BAK, thereby engaging the intrinsic pathway [10] [119].
Distinguishing Apoptosis from Other Forms of Regulated Cell Death
While apoptosis is a well-defined PCD, cells can undergo other forms of regulated death with distinct mechanisms and morphological features.
Table 2: Key Differences Between Apoptosis and Other Regulated Cell Death (RCD) Forms
| Form of RCD | Key Initiators | Molecular Hallmarks | Morphological Features | Immunogenic Response |
|---|---|---|---|---|
| Apoptosis | Caspase-8, -9, -3; Bcl-2 family; Death Receptors [114] [116] | Caspase activation, phosphatidylserine externalization, DNA laddering [114] [116] | Cell shrinkage, membrane blebbing, apoptotic bodies [114] | Immunologically silent (non-inflammatory) [114] |
| Necroptosis | TNF-α, RIPK1, RIPK3, MLKL [114] | RIPK1/RIPK3 necrosome formation, MLKL phosphorylation/oligomerization [114] | Organelle swelling, plasma membrane rupture [114] | Pro-inflammatory (release of cellular contents) [114] |
| Pyroptosis | Caspase-1, -4, -5, -11; inflammasomes [114] [116] | Inflammasome activation, cleavage of gasdermin D, pore formation [114] | Cell swelling, large bubble formation, lysis [114] | Pro-inflammatory (release of IL-1β, IL-18) [114] |
| Ferroptosis | Glutathione depletion, GPX4 inhibition, iron accumulation [114] | Iron-dependent lipid peroxidation, ROS accumulation [114] | Mitochondrial shrinkage, loss of cristae [114] | Pro-inflammatory [114] |
The Scientist's Toolkit: Essential Reagents for Apoptosis Research
Table 3: Key Research Reagent Solutions for Apoptosis Validation
| Reagent / Assay | Primary Function / Target | Key Readout / Application |
|---|---|---|
| Annexin V / PI Staining [114] [116] | Binds externalized phosphatidylserine (PS); PI stains DNA in permeable cells. | Flow cytometry to distinguish early apoptotic (Annexin V+/PI-), late apoptotic (Annexin V+/PI+), and necrotic (Annexin V-/PI+) cells. |
| TUNEL Assay [114] [116] | Labels 3'-OH ends of fragmented DNA. | Microscopy or flow cytometry to detect late-stage apoptotic cells with DNA strand breaks. |
| Caspase Activity Assays [114] [116] | Fluorogenic substrates cleaved by active caspases. | Spectrofluorometry to measure activity of specific caspases (e.g., Casp-3, -8, -9). |
| Mitochondrial Dyes (JC-1, TMRE) [114] [116] | Accumulate in active mitochondria based on membrane potential (ΔΨm). | Loss of fluorescence indicates ΔΨm collapse, an early event in intrinsic apoptosis. |
| Antibodies for WB/IF/IHC | Detect full-length and cleaved proteins (e.g., Caspase-3, PARP, Cytochrome c). | Western Blot (WB), Immunofluorescence (IF), IHC to confirm protein cleavage/translocation. |
| BH3 Mimetics (e.g., Venetoclax) [116] | Small molecules that inhibit anti-apoptotic Bcl-2 proteins. | Induce intrinsic apoptosis; used as a tool compound and therapeutic agent. |
Experimental Protocols for Validating Intrinsic Apoptosis Activation
A multi-parametric approach is essential to conclusively demonstrate the activation of the intrinsic apoptotic pathway. The workflow below integrates several key assays.
Figure 2: Multi-Parametric Workflow for Validating Intrinsic Apoptosis. This integrated approach uses complementary techniques to provide conclusive evidence [114] [116].
Protocol 1: Annexin V/Propidium Iodide (PI) Staining for Flow Cytometry
This assay is a standard for detecting early and late apoptotic cells based on plasma membrane changes [114] [116].
- Cell Preparation: Harvest treated and control cells (both adherent and floating) by gentle trypsinization followed by centrifugation.
- Washing: Wash cells twice with cold PBS and resuspend in 1X Annexin V Binding Buffer at a density of 1-5 x 10^5 cells/mL.
- Staining: Add Annexin V-fluorochrome conjugate (e.g., FITC) and Propidium Iodide (PI) to a 100 μL cell suspension. Incubate for 15 minutes at room temperature in the dark.
- Analysis: Add 400 μL of Annexin V Binding Buffer to each tube and analyze by flow cytometry within 1 hour.
- Interpretation: Annexin V-/PI- (viable), Annexin V+/PI- (early apoptotic), Annexin V+/PI+ (late apoptotic/secondary necrotic), Annexin V-/PI+ (necrotic/primary necrotic) [116].
Protocol 2: Western Blot Analysis for Caspase Activation and Protein Cleavage
Western blotting provides direct evidence of proteolytic cleavage events central to apoptosis execution [114] [116].
- Protein Extraction: Lyse cells in RIPA buffer supplemented with protease and phosphatase inhibitors. Determine protein concentration.
- Gel Electrophoresis: Separate 20-50 μg of total protein per lane on 4-20% gradient SDS-PAGE gels.
- Transfer and Blocking: Transfer proteins to a PVDF membrane. Block the membrane with 5% non-fat milk in TBST for 1 hour.
- Antibody Probing: Incubate with primary antibodies overnight at 4°C. Key targets for intrinsic apoptosis include:
- Cleaved Caspase-9 (Activated form)
- Cleaved Caspase-3 (Activated executioner caspase)
- Cleaved PARP (A key substrate of caspase-3) [116]
- Bax / Bcl-2 (To assess pro-/anti-apoptotic protein balance)
- Detection: Incubate with HRP-conjugated secondary antibodies and visualize using enhanced chemiluminescence (ECL) substrate. The appearance of cleavage products confirms apoptotic signaling.
Protocol 3: Immunofluorescence Microscopy for Cytochrome c Release and BAX Translocation
This protocol visualizes the critical mitochondrial events of intrinsic apoptosis [116].
- Cell Seeding and Fixation: Seed cells on glass coverslips. After treatment, fix cells with 4% paraformaldehyde for 15 minutes at room temperature.
- Permeabilization and Blocking: Permeabilize cells with 0.1% Triton X-100 in PBS for 10 minutes. Block with 5% BSA in PBS for 1 hour.
- Staining: Incubate with primary antibodies against Cytochrome c and a mitochondrial marker (e.g., COX IV) or against BAX for 1-2 hours. After washing, incubate with appropriate fluorescent secondary antibodies (e.g., Alexa Fluor 488, 594).
- Mounting and Imaging: Mount coverslips with an anti-fade mounting medium containing DAPI to stain nuclei. Image using a confocal microscope.
- Interpretation: In healthy cells, cytochrome c displays a punctate pattern overlapping perfectly with the mitochondrial marker. Upon MOMP, cytochrome c shows a diffuse cytoplasmic pattern. Similarly, BAX translocates from the cytosol to mitochondria, appearing as punctate foci [116].
The precise distinction between intrinsic and extrinsic apoptosis, as well as other RCD forms, is a cornerstone of modern cell biology and translational research. As detailed in this guide, this requires a thorough understanding of the unique initiators, molecular regulators, and morphological outcomes of each pathway. The provided comparative tables, experimental workflows, and detailed protocols offer a framework for researchers to design robust validation strategies. Mastering these distinctions and techniques is essential for accurately modeling disease mechanisms, such as the evasion of apoptosis in cancer [120], and for developing targeted therapies that specifically modulate these critical cell death pathways to achieve therapeutic efficacy.
Integrating Western Blot, Flow Cytometry, and Live-Cell Imaging Data
Validating activation of the intrinsic apoptosis pathway is a cornerstone of biomedical research, particularly in oncology and drug development. This pathway, triggered by cellular stress, is characterized by mitochondrial outer membrane permeabilization, cytochrome c release, and caspase cascade activation. No single technique can fully capture this complexity; researchers must instead integrate data from complementary methodologies. Western blotting, flow cytometry, and live-cell imaging each provide unique and non-redundant insights into the apoptotic process. This guide objectively compares these technologies, providing experimental data and protocols to enable researchers to develop a comprehensive, multi-platform validation strategy for intrinsic apoptosis activation.
Technology Comparison for Apoptosis Analysis
The following table provides a systematic comparison of the three key technologies for analyzing intrinsic apoptosis pathway activation.
Table 1: Comparative Analysis of Key Apoptosis Detection Technologies
| Parameter | Western Blotting | Flow Cytometry | Live-Cell Imaging |
|---|---|---|---|
| Primary Readout | Protein presence, cleavage, and modification at population level [58] | Multiparametric protein expression and light scatter at single-cell level [121] [104] | Real-time spatial localization and morphological dynamics in live cells [122] |
| Sample Type | Lysed cell populations [58] | Single-cell suspensions [123] [124] | Adherent or suspended live cells [122] |
| Key Apoptosis Markers | Cleaved caspases (3, 9), cleaved PARP, Bcl-2 family proteins [58] | Phosphatidylserine exposure (Annexin V), ΔΨm loss, caspase activation, DNA fragmentation [104] | Mitochondrial transmembrane potential (ΔΨm) dynamics, membrane blebbing, nuclear condensation and fragmentation [104] [122] |
| Single-Cell Resolution | No [121] | Yes [125] [121] | Yes [122] |
| Temporal Resolution | End-point (snapshot) [58] | End-point or kinetic (with sampling) [104] | Real-time, continuous kinetic data [122] |
| Throughput | Low to medium [121] | High (thousands of cells/second) [125] [124] | Low to medium [122] |
| Quantification Capability | Semi-quantitative (densitometry) [58] | Highly quantitative (fluorescence intensity) [121] [124] | Quantitative with optimized algorithms (morphology, intensity, translocation) [122] [126] |
| Key Advantage | Specific detection of protein cleavage events (e.g., caspase-3, PARP) [58] | Statistical power from analyzing thousands of cells; identifies heterogeneous subpopulations [125] [121] | Reveals dynamic spatiotemporal processes without perturbing cellular context [122] |
| Main Limitation | Averages signal across entire population, missing rare events [125] [121] | Requires single-cell suspension, potentially perturbing physiology [122] | Limited availability of specific fluorescent probes compared to antibodies [122] |
Experimental Protocols for Apoptosis Validation
Western Blotting for Key Apoptotic Markers
Western blotting remains a fundamental method for confirming apoptosis through specific protein cleavages.
Detailed Protocol:
- Sample Preparation: Lyse 5×10⁵ to 10⁶ cells per sample in RIPA buffer containing protease and phosphatase inhibitors. Centrifuge to remove debris and quantify protein concentration [58].
- Gel Electrophoresis & Transfer: Load equal protein amounts (20-50 µg) for SDS-PAGE. Transfer proteins to a PVDF or nitrocellulose membrane [58].
- Antibody Incubation: Block membrane with 5% non-fat milk or BSA. Incubate with primary antibodies against key apoptotic markers:
- Cleaved Caspase-3 (key executioner caspase)
- Cleaved PARP (89 kDa fragment; hallmark of apoptosis)
- Cytochrome c (release from mitochondria)
- Bax/Bcl-2 (pro- vs. anti-apoptotic balance)
- Use β-actin or GAPDH as loading control [58].
- Detection & Analysis: Incubate with HRP-conjugated secondary antibody. Develop with chemiluminescent substrate and image. Use densitometry software (e.g., ImageJ) to quantify band intensities. Calculate cleaved-to-full-length ratios (e.g., cleaved PARP:full-length PARP) to assess activation extent [58].
Multiparametric Flow Cytometry for Single-Cell Analysis
Flow cytometry excels at detecting early and mid-stage apoptotic markers in heterogeneous cell populations.
Detailed Protocol:
- Sample Preparation: Harvest and wash cells. Use 0.5-1×10⁶ cells per sample in polystyrene tubes. For adherent cells, use gentle, non-enzymatic dissociation to preserve surface epitopes [123].
- Viability Staining: Resuspend cells in viability dye (e.g., 7-AAD, DAPI) according to manufacturer's instructions. Incubate on ice in the dark, wash, and retain supernatant [123].
- Annexin V Staining: Resuspend cell pellet in Annexin V Binding Buffer. Add Annexin V-FITC/APC and incubate for 15-20 minutes at room temperature in the dark. Add PI just before analysis to distinguish early apoptotic (Annexin V+/PI-) from late apoptotic/necrotic (Annexin V+/PI+) cells [104].
- Intracellular Staining (for active caspases): Fix cells with 1-4% PFA for 15-20 minutes on ice. Permeabilize with 0.1% Triton X-100 for 10-15 minutes at room temperature. Incubate with fluorochrome-labeled caspase inhibitors (FLICA) for 60 minutes at 37°C. Wash and analyze [104].
- Analysis: Acquire data on a flow cytometer, collecting at least 10,000 events per sample. Use fluorescence minus one (FMO) controls for accurate gating. Analyze data to quantify percentages of cells in various apoptotic stages [104] [124].
Live-Cell Imaging for Kinetic Analysis of Apoptosis
Live-cell imaging captures dynamic morphological changes and protein translocations in real time.
Detailed Protocol:
- Cell Preparation & Staining: Plate cells in optical-bottom 96-well plates. For mitochondrial potential (ΔΨm) assessment, load cells with 50-100 nM TMRM or TMRE in culture medium and incubate for 20-30 minutes at 37°C. For caspase activation, use cell-permeable FLICA probes or genetically encoded biosensors [104] [122].
- Image Acquisition: Use an environmentally controlled live-cell imaging system maintaining 37°C and 5% CO₂. Acquire images at regular intervals (e.g., every 5-15 minutes) over 24-48 hours. Use a 20x or 40x objective for sufficient resolution and field of view [122].
- Key Parameters to Quantify:
- Mitochondrial Depolarization: Loss of TMRM fluorescence intensity [104].
- Membrane Blebbing: Phase-contrast or differential interference contrast (DIC) imaging.
- Nuclear Fragmentation: Use DNA-binding dyes like Hoechst 33342 (validated for live cells).
- Biosensor Activation: e.g., FRET-based SCAT biosensor for caspase-3 activation [122].
- Image Analysis: Use image analysis software (e.g., ImageJ, commercial HCS software) with algorithms to segment individual cells and track morphological changes and fluorescence intensity/localization over time [122].
Visualizing the Experimental Workflow and Pathway
The following diagrams illustrate the core apoptosis pathway and the integrated experimental approach for its validation.
Diagram 1: Core intrinsic apoptosis pathway and associated detection methods. Key events include Mitochondrial Outer Membrane Permeabilization (MOMP) and caspase activation, which are detected by complementary techniques.
Diagram 2: Integrated experimental workflow for apoptosis validation. The convergent approach leverages parallel data streams from different technologies to build robust evidence for pathway activation.
Research Reagent Solutions for Apoptosis Detection
Successful validation requires high-quality, well-characterized reagents. The following table details essential tools for apoptosis research.
Table 2: Key Reagents for Apoptosis Pathway Analysis
| Reagent Category | Specific Examples | Primary Function in Apoptosis Detection | Key Application(s) |
|---|---|---|---|
| Antibodies for Western Blot | Anti-cleaved Caspase-3, Anti-cleaved PARP, Anti-Bax, Anti-Bcl-2 [58] | Detect specific protein cleavage events and expression level changes in apoptotic signaling [58] | Western Blot [58] |
| Fluorescent Probes for Flow Cytometry | Annexin V conjugates (FITC, APC), TMRM/TMRE, FLICA kits, PI/7-AAD [104] | Label phosphatidylserine exposure, measure mitochondrial potential, detect active caspases, and assess viability/ploidy [104] | Flow Cytometry [123] [104] |
| Live-Cell Dyes & Biosensors | TMRM, Hoechst 33342, CellEvent Caspase-3/7 reagents, SCAT FRET biosensor [122] [127] | Monitor kinetics of ΔΨm loss, nuclear fragmentation, and caspase activation in live, unperturbed cells [104] [122] | Live-Cell Imaging [122] |
| Viability Dyes | Propidium Iodide (PI), 7-AAD, Fixable Viability Dyes [123] [104] | Distinguish live, early apoptotic, late apoptotic, and necrotic cell populations by membrane integrity [123] | Flow Cytometry, Microscopy [123] [104] |
| Apoptosis Antibody Cocktails | Pre-mixed antibodies targeting cleaved caspases, PARP, and Bcl-2 family proteins [58] | Enable efficient, multiplexed detection of multiple apoptotic markers in a single assay, improving reproducibility [58] | Western Blot [58] |
Validating intrinsic apoptosis pathway activation requires a strategic, multi-faceted approach that leverages the distinct advantages of Western blotting, flow cytometry, and live-cell imaging. Western blotting provides definitive evidence of specific protein cleavages, flow cytometry offers unparalleled statistical power for analyzing heterogeneous populations at the single-cell level, and live-cell imaging captures the dynamic progression of cell death in its native context. By integrating experimental protocols and data from these complementary platforms—using the reagent solutions and workflows outlined in this guide—researchers can achieve a robust and comprehensive validation of intrinsic apoptosis, strengthening conclusions in fundamental research and accelerating the development of novel therapeutics.
Apoptosis, or programmed cell death, is a critical process in development and homeostasis, and its dysregulation is a hallmark of cancer [45]. The intrinsic apoptotic pathway, also known as the mitochondrial pathway, is regulated by BCL-2 family proteins and represents a key therapeutic target for cancer treatment [45]. Neuroblastoma is the most common extracranial solid tumor in children, accounting for approximately 8-10% of pediatric malignancies and 15% of childhood cancer-related deaths [128]. Despite intensive multimodal therapies, the overall survival rate for high-risk neuroblastoma remains low, creating an urgent need for more effective therapeutic strategies [128] [129].
Oxysterols, oxidized derivatives of cholesterol, have gained research interest due to their pro-apoptotic and anti-tumor properties [128]. Among these, 25-Hydroxycholesterol (25OHChol) has demonstrated cytotoxic effects in various cancer cell types [128] [130]. This case study systematically validates the induction of intrinsic apoptosis by 25OHChol in human neuroblastoma models, providing a framework for apoptosis pathway validation in cancer research.
Experimental Validation of 25OHChol-Induced Cytotoxicity
Concentration and Time-Dependent Viability Reduction
The initial validation of 25OHChol's effects on BE(2)-C human neuroblastoma cells demonstrated clear concentration-dependent and time-dependent cytotoxicity. Using the CCK-8 assay to assess cell viability, researchers observed that 25OHChol treatment resulted in a significant decline in viable cells, whereas treatment with cholesterol or other oxysterols (24sOHChol, 27OHChol) showed no noticeable effect at comparable concentrations [128].
Table 1: Viability of BE(2)-C Cells Following 25OHChol Treatment
| Concentration (µg/mL) | 24h Viability (%) | 48h Viability (%) | 72h Viability (%) |
|---|---|---|---|
| 0.5 | - | 92.1% | - |
| 1.0 | 87.1% | 58.1% | 50.6% |
| 2.0 | - | 40.7% | 38.2% |
After 48 hours of treatment with 1 µg/mL 25OHChol, cell viability decreased to approximately 50%, confirming its potent cytotoxic effects on neuroblastoma cells [128]. This concentration- and time-dependent response provides the foundational evidence necessary for subsequent apoptosis mechanism investigation.
Comparative Cytotoxicity Profile
In the same study, when BE(2)-C cells were treated with cholesterol or various oxysterols at concentrations of 0.5, 1, and 2 µg/mL for 24, 48, and 72 hours, only 25OHChol resulted in a significant reduction in viability (decreasing to 61.7%), while other treatments had no noticeable effect [128]. This specificity highlights 25OHChol's unique potency against neuroblastoma cells compared to other oxysterols.
Methodological Framework for Apoptosis Validation
Morphological Assessment of Apoptosis
Optical microscopy and DAPI staining served as initial methods to identify characteristic morphological changes associated with apoptosis. Following treatment with 25OHChol (1 µg/mL) for 48 hours, BE(2)-C cells exhibited significant cell shrinkage and loss of cell-cell adhesion under optical microscopy [128].
DAPI staining, which fluorescently labels nuclear material, revealed chromatin condensation and nuclear fragmentation - both hallmarks of apoptotic cell death [128]. These morphological changes provide the first visual evidence of apoptosis induction before proceeding to more specific molecular analyses.
Flow Cytometric Analysis with Annexin V/PI Staining
Annexin V/Propidium Iodide (PI) flow cytometry provides a quantitative measure of apoptosis progression by distinguishing between viable, early apoptotic, late apoptotic, and necrotic cells [128]. This method detects the externalization of phosphatidylserine - an event occurring in early apoptosis - using Annexin V binding, while PI identifies cells with compromised membrane integrity [16] [131].
Table 2: Apoptotic Cell Populations Following 25OHChol Treatment
| Treatment Group | Early Apoptotic Cells | Late Apoptotic Cells | Total Apoptotic Rate |
|---|---|---|---|
| Control | - | - | 6.82% |
| Chol | - | - | 6.74% |
| 24sOHChol | - | - | 9.86% |
| 25OHChol | - | - | 79.17% |
In the 25OHChol-treated group, the combined rate of early and late apoptosis reached 79.17%, significantly higher than control groups, confirming the critical role of 25OHChol in apoptosis induction in BE(2)-C cells [128].
Molecular Mechanism of Intrinsic Apoptosis Activation
BCL-2 Family Protein Dynamics
Western blot analysis of Bcl-2 family proteins provides crucial insight into the regulation of the intrinsic apoptotic pathway. Treatment with 25OHChol resulted in an increased Bax/Bcl-2 ratio, indicating a shift in the balance toward pro-apoptotic signaling [128] [130]. This protein ratio is a critical determinant of mitochondrial outer membrane permeabilization (MOMP), representing a key regulatory point in intrinsic apoptosis execution.
The intrinsic apoptosis pathway is primarily regulated by the BCL-2 protein family, which includes both anti-apoptotic (e.g., Bcl-2) and pro-apoptotic members (e.g., Bax) [45]. In most mammalian cells, these proteins control the intrinsic pathway by regulating mitochondrial membrane permeability [45]. The increased Bax/Bcl-2 ratio observed following 25OHChol treatment promotes mitochondrial membrane permeabilization, leading to cytochrome c release [128].
Mitochondrial Membrane Potential Reduction
Measurement of mitochondrial membrane potential (MMP) using flow cytometry with JC-1 or similar fluorescent dyes provides functional evidence of intrinsic pathway activation. 25OHChol treatment caused a significant reduction in MMP, indicating mitochondrial membrane permeabilization - a definitive step in intrinsic apoptosis initiation [128] [130]. This loss of MMP facilitates the release of pro-apoptotic factors, including cytochrome c, from the mitochondrial intermembrane space into the cytoplasm.
Caspase Activation Analysis
Caspase activity assays demonstrated increased caspase-9 and caspase-3/7 activity following 25OHChol treatment [128] [130]. Caspase-9 activation occurs following cytochrome c release and apoptosome formation, while caspase-3 and -7 function as executioner caspases that mediate the proteolytic cleavage of cellular targets [45].
The critical role of caspases in 25OHChol-induced apoptosis was further confirmed through inhibition experiments. Treatment with the pan-caspase inhibitor Z-VAD-FMK resulted in a dose-dependent increase in cell viability, establishing caspase activity as essential for 25OHChol-mediated cell death [128].
Research Reagent Solutions for Apoptosis Validation
Table 3: Essential Research Reagents for Apoptosis Validation
| Reagent/Assay | Application | Key Features |
|---|---|---|
| CCK-8 Assay | Cell viability assessment | Measures metabolic activity; concentration-dependent cytotoxicity [128] |
| Annexin V/PI Apoptosis Detection | Flow cytometry-based apoptosis quantification | Distinguishes between early/late apoptosis and necrosis [128] [16] |
| DAPI Staining | Nuclear morphology analysis | Visualizes chromatin condensation and nuclear fragmentation [128] |
| Western Blotting | Protein expression analysis | Detects Bcl-2 family proteins and caspase cleavage [128] |
| Caspase Activity Assays | Caspase activation measurement | Fluorometric or colorimetric detection of caspase-9, -3/7 activity [128] |
| Mitochondrial Membrane Potential Dyes | Mitochondrial function assessment | JC-1 or similar dyes detect loss of MMP [128] |
| Z-VAD-FMK | Caspase inhibition studies | Pan-caspase inhibitor to confirm caspase-dependent apoptosis [128] |
Comparative Analysis with Alternative Apoptosis-Inducing Strategies
Targeted Therapeutic Approaches in Neuroblastoma
Other experimental approaches for inducing apoptosis in neuroblastoma include:
BCL-2 Inhibition: MYCN-amplified neuroblastomas demonstrate sensitivity to the BCL-2 inhibitor ABT-199 (venetoclax), with this sensitivity occurring in part through low anti-apoptotic BCL-xL expression and high pro-apoptotic NOXA expression [132]. Combination with Aurora Kinase A inhibitor MLN8237 induces widespread apoptosis in MYCN-amplified neuroblastoma models [132].
JNK-Independent Chemotherapeutics: Research has identified approved oncology drugs capable of inducing apoptosis in a JNK-independent manner, which may overcome forms of nongenetic chemoresistance in neuroblastoma [133].
Novel Small Molecule Therapeutics: FLIX5, a recently identified small compound, exhibits broad cytotoxicity against neuroblastoma and medulloblastoma cells primarily by triggering apoptosis, potentially through targeting EPLIN (Epithelial Protein Lost In Neoplasm) [129].
STAT3 Inhibition: Napabucasin, a STAT3 inhibitor, has been found to induce apoptosis in SH-SY5Y neuroblastoma cells by modulating the Bcl-2/Bax signaling pathway and demonstrates synergistic effects when combined with doxorubicin [134].
Technical Comparison of Apoptosis Detection Methods
Table 4: Comparison of Apoptosis Detection Methodologies
| Methodology | Information Provided | Throughput | Key Advantages |
|---|---|---|---|
| CCK-8/MTT Viability Assays | Overall cell viability | High | Simple, quantitative, suitable for initial screening [128] |
| Annexin V/PI Flow Cytometry | Apoptosis staging and quantification | Medium | Distinguishes apoptosis stages, quantitative [128] |
| DAPI Nuclear Staining | Nuclear morphology changes | Low | Visual confirmation of apoptosis, simple [128] |
| Western Blot Analysis | Protein expression and cleavage | Low | Mechanistic insight, specific protein detection [128] |
| Caspase Activity Assays | Caspase activation kinetics | Medium | Functional enzyme activity, pathway-specific [128] |
| MMP Measurement | Mitochondrial membrane integrity | Medium | Direct assessment of intrinsic pathway activation [128] |
Integrated Experimental Workflow for Apoptosis Validation
This integrated workflow represents a systematic approach for validating intrinsic apoptosis pathway activation. The process begins with cytotoxicity assessment, progresses through morphological and flow cytometric analysis, investigates molecular mechanisms, and concludes with specific inhibition studies to confirm the apoptotic mechanism.
The validation of 25-hydroxycholesterol-induced apoptosis in BE(2)-C human neuroblastoma cells provides a comprehensive case study in intrinsic apoptosis pathway analysis. Through a multi-faceted experimental approach, 25OHChol demonstrates potent, concentration-dependent cytotoxicity mediated specifically through the mitochondrial apoptotic pathway, characterized by an increased Bax/Bcl-2 ratio, reduced mitochondrial membrane potential, and subsequent caspase-9 and caspase-3/7 activation.
This case study also illustrates a robust methodological framework for apoptosis validation that can be applied to other therapeutic candidates. The integration of viability assays, morphological analysis, flow cytometry, protein detection, and functional assays provides complementary evidence to conclusively establish apoptotic mechanisms. As research in apoptosis-targeting therapies continues to advance, particularly for challenging malignancies like high-risk neuroblastoma, such rigorous validation approaches will be essential for developing effective treatment strategies that overcome therapeutic resistance.
Conclusion
Accurately validating intrinsic apoptosis pathway activation requires a holistic, multi-faceted approach that integrates foundational knowledge with a suite of complementary techniques. Relying on a single biomarker or assay is insufficient to confirm this complex process. By adopting a rigorous, multi-parametric validation strategy—correlating key events like Bax/Bak activation, cytochrome c release, caspase-9 cleavage, and characteristic morphological changes—researchers can generate robust, reproducible data. The continued development of more sensitive and high-throughput detection technologies, combined with a deeper understanding of pathway crosstalk, will further refine our ability to modulate intrinsic apoptosis for therapeutic benefit, particularly in oncology and neurodegenerative disease research. This systematic framework empowers scientists to confidently elucidate mechanisms of cell death in basic research and advance the development of novel therapeutics that target this critical pathway.
References
- 1. Mitochondrial signaling in cell death via the Bcl-2 family - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2874116/]
- 2. The BCL2 family: from apoptosis mechanisms to new ... [https://www.nature.com/articles/s41392-025-02176-0]
- 3. Regulation of the Intrinsic Apoptosis Pathway by Reactive ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3717204/]
- 4. Endolysosomal Targeting of Mitochondria Is Integral to ... [https://www.sciencedirect.com/science/article/pii/S1534580720304032]
- 5. From proliferation to mitochondrial membrane potential ... [https://www.nature.com/articles/s41420-025-02391-2]
- 6. Fluorescence microscopy imaging of mitochondrial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10326048/]
- 7. In situ observation of mitochondrial biogenesis as the early ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8441175/]
- 8. From computational modelling of the intrinsic apoptosis ... [https://www.nature.com/articles/cddis201436]
- 9. Apoptosis: A Comprehensive Overview of Signaling Pathways ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11592877/]
- 10. Intrinsic and Extrinsic Pathways of Apoptosis [https://www.thermofisher.com/us/en/home/life-science/antibodies/antibodies-learning-center/antibodies-resource-library/cell-signaling-pathways/cellular-apoptosis-pathway.html]
- 11. Multiple Functions of BCL-2 Family Proteins - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3552500/]
- 12. Multifaceted Roles of Bcl - 2 : Family Roles in... Proteins Regulatory [https://pubmed.ncbi.nlm.nih.gov/41182644/]
- 13. Bcl-2 family [https://en.wikipedia.org/wiki/Bcl-2_family]
- 14. Mechanisms of action of the BCL-2 inhibitor venetoclax in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10657905/]
- 15. Molecular Medicine Reports [https://www.spandidos-publications.com/10.3892/mmr.2025.13537]
- 16. North America Apoptosis Assay Market Trends 2025–2034 [https://www.gminsights.com/industry-analysis/north-america-apoptosis-assay-market]
- 17. Mechanisms of cytochrome c release from mitochondria [https://www.nature.com/articles/4401950]
- 18. TRAIL-induced apoptosis requires Bax-dependent ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC155309/]
- 19. Apoptosis-associated release of Smac/DIABLO from ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC125329/]
- 20. A Bax/Bak-independent Mechanism of Cytochrome c ... [https://www.sciencedirect.com/science/article/pii/S0021925820649808]
- 21. Smac/DIABLO and cytochrome c are released from ... [https://link.springer.com/article/10.1007/s10495-005-0803-9]
- 22. Caspase-9: structure, mechanisms and clinical application [https://pmc.ncbi.nlm.nih.gov/articles/PMC5410359/]
- 23. Caspases as master regulators of programmed cell death [https://www.nature.com/articles/s12276-025-01470-9]
- 24. Activation of caspase-9 on the apoptosome as studied by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10743466/]
- 25. The Apaf-1 apoptosome induces formation of caspase-9 ... [https://www.nature.com/articles/ncomms13565]
- 26. The Apoptosome Activates Caspase-9 by Dimerization [https://www.sciencedirect.com/science/article/pii/S1097276506001730]
- 27. Activation of caspase-9 on the apoptosome as studied by ... [https://pubmed.ncbi.nlm.nih.gov/38085782/]
- 28. Caspase-9 activates β-catenin signaling to promote ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-025-07020-1]
- 29. Research progress on morphology and mechanism of ... [https://www.nature.com/articles/s41419-024-06712-8]
- 30. Morphological assessment of apoptosis [https://www.sciencedirect.com/science/article/abs/pii/S1046202307002150]
- 31. High-Resolution Imaging of Morphological Changes ... [https://www.mdpi.com/2079-6374/15/8/522]
- 32. Hallmarks and Significance of Apoptosis [https://encyclopedia.pub/entry/41155]
- 33. Imaging of morphological and biochemical hallmarks ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6851291/]
- 34. Imaging of morphological and biochemical hallmarks ... [https://www.sciencedirect.com/science/article/pii/S0021925820305330]
- 35. JC-1 Dye for Mitochondrial Membrane Potential [https://www.thermofisher.com/us/en/home/life-science/cell-analysis/cell-viability-and-regulation/apoptosis/mitochondria-function/jc-1-dye-mitochondrial-membrane-potential.html]
- 36. Measuring mitochondrial membrane potential | The EMBO ... [https://link.springer.com/article/10.1038/s44318-025-00632-9]
- 37. What is the difference between MitoPT TMRE and JC-1? [https://www.antibodiesinc.com/blogs/news/ict-answers-your-questions-5?srsltid=AfmBOorONisa9AkrGB1vm7We1hwPQFdQa6-kUp3cxru1GLelzuo6mV_y]
- 38. Performance of TMRM and Mitotrackers in mitochondrial ... [https://www.sciencedirect.com/science/article/pii/S0005272823000737]
- 39. Spontaneous Changes in Mitochondrial Membrane Potential ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6762873/]
- 40. Mitochondrial Membrane Potential in Axons Increases with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2597466/]
- 41. Characterization of the far-red fluorescent probe MitoView ... [https://www.frontiersin.org/journals/physiology/articles/10.3389/fphys.2023.1257739/full]
- 42. From proliferation to mitochondrial membrane potential ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11933298/]
- 43. In situ spatial profiling of cytochrome c release at the single ... [https://www.sciencedirect.com/science/article/abs/pii/S0956566325006943]
- 44. Diverse functions of cytochrome c in cell death and disease [https://pmc.ncbi.nlm.nih.gov/articles/PMC11043370/]
- 45. Targeting apoptotic pathways for cancer therapy - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11245162/]
- 46. Reactivating apoptotic pathways in cancer: A review of ... [https://www.sciencedirect.com/science/article/abs/pii/S0014299925007198]
- 47. Simple and Efficient Protocol for Subcellular Fractionation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8069826/]
- 48. Apoptosis regulation by subcellular relocation of caspases [https://www.nature.com/articles/s41598-018-30652-x]
- 49. Protocol for detection of caspases using immunofluorescence [https://www.abcam.com/en-us/technical-resources/protocols/immunofluorescence-protocol-to-detect-caspases]
- 50. Programmed cell death detection methods: a systematic ... [https://link.springer.com/article/10.1007/s10495-022-01735-y]
- 51. Building blocks of the apoptotic pore: how Bax and Bak are ... [https://www.nature.com/articles/cdd2013139]
- 52. BAX, BAK, and BOK: A Coming of Age for the BCL-2 Family ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7111251/]
- 53. Direct Interaction of Bax and Bak Proteins with Bcl-2 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3576097/]
- 54. Computational design of potent and selective binders of BAK ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12412652/]
- 55. The interplay between BAX and BAK tunes apoptotic pore ... [https://www.sciencedirect.com/science/article/pii/S1097276522000089]
- 56. Inhibition of oligomeric BAX by an anti-apoptotic dimer [https://www.sciencedirect.com/science/article/pii/S0092867425012425?dgcid=rss_sd_all]
- 57. A Comprehensive Exploration of Caspase Detection Methods [https://pmc.ncbi.nlm.nih.gov/articles/PMC11121653/]
- 58. Apoptosis western blot guide [https://www.abcam.com/en-us/knowledge-center/western-blot/western-blot-analysis-of-apoptosis]
- 59. Apoptosis Assay - an overview [https://www.sciencedirect.com/topics/pharmacology-toxicology-and-pharmaceutical-science/apoptosis-assay]
- 60. Mechanistic insights into caspase-9 activation by the structure ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5320974/]
- 61. Apoptosis Marker Assays for HTS - NCBI [https://www.ncbi.nlm.nih.gov/books/NBK572437/]
- 62. Triple Fluorescence staining to Evaluate Mechanism ... [https://www.nature.com/articles/s41598-018-31575-3]
- 63. Measuring Caspase Activity Using a Fluorometric Assay or ... [https://www.jove.com/t/64745/measuring-caspase-activity-using-fluorometric-assay-or-flow]
- 64. Fluorogenic substrates for caspase activity—Table 15.5 [https://www.thermofisher.com/us/en/home/references/molecular-probes-the-handbook/tables/fluorogenic-substrates-for-caspase-activity.html]
- 65. Article Caspase-9 Holoenzyme Is a Specific and Optimal ... [https://www.sciencedirect.com/science/article/pii/S1097276506002206]
- 66. Guide for Apoptosis Detection | Boster Bio Annexin V PI Staining [https://www.bosterbio.com/blog/post/methods/flow-cytometry-with-annexin-v-pi-staining]
- 67. assay Annexin for apoptosis | Abcam V staining protocol [https://www.abcam.com/en-us/technical-resources/protocols/annexin-v-for-apoptosis]
- 68. TUNEL assay [https://en.wikipedia.org/wiki/TUNEL_assay]
- 69. TUNEL Assays | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/cell-analysis/cell-viability-and-regulation/apoptosis/tunel-assays.html]
- 70. : Protocol and Annexin by V PI Staining Flow Cytometry [https://www.bio-techne.com/cn/resources/protocols-troubleshooting/annexin-v-and-pi-staining-flow-cytometry]
- 71. TUNEL assay-Standardized method for testing sperm DNA ... [https://pubmed.ncbi.nlm.nih.gov/32706440/]
- 72. Comparison of several techniques for the detection ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6495494/]
- 73. Apoptosis Assay Market Size Report, 2025 – 2034 [https://www.gminsights.com/industry-analysis/apoptosis-assay-market]
- 74. The Fas-mediated apoptosis assay: From concept to ... [https://www.sciencedirect.com/science/article/abs/pii/S0022175925000122]
- 75. Research progress on natural plant metabolites targeting ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1624569/full]
- 76. Insights on the crosstalk among different cell death ... [https://www.nature.com/articles/s41420-025-02328-9]
- 77. Development and validation of hierarchical signature for ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1470145/full]
- 78. Identification of the pyroptosis, apoptosis, and necroptosis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12211273/]
- 79. Targeting activation of intrinsic apoptotic pathway through ... [https://www.sciencedirect.com/science/article/abs/pii/S0304383525005312]
- 80. Mathematical modeling of apoptosis [https://biosignaling.biomedcentral.com/articles/10.1186/1478-811X-11-44]
- 81. A quantitative real-time approach for discriminating ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5253725/]
- 82. The Quantitative-Phase Dynamics of Apoptosis and Lytic ... [https://www.nature.com/articles/s41598-020-58474-w]
- 83. Comparison of imaging based single-cell resolution spatial ... [https://www.nature.com/articles/s41467-025-63414-1]
- 84. z-VAD-fmk augmentation of TNF alpha-stimulated ... [https://pubmed.ncbi.nlm.nih.gov/15572588/]
- 85. In vitro evaluation of the anti-apoptotic drug Z-VAD-FMK on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4615917/]
- 86. A long way to go: caspase inhibitors in clinical use [https://www.nature.com/articles/s41419-021-04240-3]
- 87. The Caspase Inhibitor Z-VAD-FMK Alleviates Endotoxic Shock ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6687755/]
- 88. Caspases as master regulators of programmed cell death [https://pmc.ncbi.nlm.nih.gov/articles/PMC12229594/]
- 89. The role of BCL-2 family proteins in regulating apoptosis ... [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2022.985363/full]
- 90. Targeting the Bcl-2 Family for Cancer Therapy - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3955095/]
- 91. Genomic analysis of BAX and Bcl - 2 gene mutations in human... [https://surgexppathol.biomedcentral.com/articles/10.1186/s42047-024-00153-5]
- 92. The Bax/Bcl-2 ratio determines the susceptibility of human ... [https://pubmed.ncbi.nlm.nih.gov/11511312/]
- 93. The Role of Intrinsic Pathway in Apoptosis Activation and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4147380/]
- 94. Bax/Bcl-2 expression ratio in prediction of response to breast ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5817177/]
- 95. / Bax - Bcl expression 2 in prediction of response to breast cancer... ratio [https://ijbms.mums.ac.ir/article_10207.html]
- 96. Expression of bax , Bcl - 2 , and Bax / Bcl - 2 of rattus norvegicus lens... ratio [https://scholar.unair.ac.id/en/publications/expression-of-bax-bcl-2-and-baxbcl-2-ratio-of-rattus-norvegicus-l]
- 97. Imaging flow cytometry: from high - resolution ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2025.1580749/full]
- 98. Imaging flow cytometry with a real-time throughput beyond ... [https://www.nature.com/articles/s41377-025-01754-9]
- 99. Kinetic quantification of apoptosis using Incucyte® live-cell ... [https://www.news-medical.net/whitepaper/20251027/Kinetic-quantification-of-apoptosis-using-Incucytec2ae-live-cell-imaging-assays.aspx]
- 100. Evaluating cell viability assessment techniques [https://pmc.ncbi.nlm.nih.gov/articles/PMC12492793/]
- 101. Comparison of Diagnostic Accuracy of Microscopy and Flow ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0122037]
- 102. Performance comparison of a flow cytometry immunoassay ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8853233/]
- 103. Flow cytometry-based quantitative analysis of cellular ... [https://www.sciencedirect.com/science/article/pii/S2405844024096968]
- 104. Flow cytometry-based apoptosis detection - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3863590/]
- 105. Multi-Parametric Analysis and Modeling of Relationships ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0028694]
- 106. Comparative survival analysis of multiparametric tests—when ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8266887/]
- 107. Insight into redox regulation of apoptosis in cancer cells ... [https://www.nature.com/articles/s41598-022-08509-1]
- 108. Morphologic and biochemical hallmarks of apoptosis [https://pubmed.ncbi.nlm.nih.gov/10728374/]
- 109. Morphologic criteria and detection of apoptosis [https://pubmed.ncbi.nlm.nih.gov/10412642/]
- 110. Seven Assays to Detect Apoptosis - CST Blog [https://blog.cellsignal.com/blog.cellsignal.com/cell-process-seven-assays-to-detect-apoptosis]
- 111. Advanced concept for identifying chemico-biological ... [https://www.sciencedirect.com/science/article/abs/pii/S0010482525015392]
- 112. Biomarkers of apoptosis - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2538762/]
- 113. Integrated real-time imaging of executioner caspase ... [https://www.nature.com/articles/s41420-025-02662-y]
- 114. Apoptosis - StatPearls - NCBI Bookshelf [https://www.ncbi.nlm.nih.gov/sites/books/NBK499821/]
- 115. Apoptosis: A Comprehensive Overview of Signaling ... [https://www.mdpi.com/2073-4409/13/22/1838]
- 116. Mechanisms of Cell Death: Apoptosis - CST Blog [https://blog.cellsignal.com/mechanisms-of-cell-death-apoptosis]
- 117. Biochemistry, Extrinsic Pathway of Apoptosis - StatPearls - NCBI [https://www.ncbi.nlm.nih.gov/books/NBK560811/]
- 118. The concept of intrinsic versus extrinsic apoptosis [https://pubmed.ncbi.nlm.nih.gov/35147165/]
- 119. Intrinsic Apoptosis - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/intrinsic-apoptosis]
- 120. Restoring apoptosis in breast cancer via peptide mediated ... [https://www.nature.com/articles/s41598-025-24772-4]
- 121. When should I use flow cytometry vs western blot? - CST Blog [https://blog.cellsignal.com/when-should-i-use-flow-cytometry-for-signaling-instead-of-western-blot]
- 122. A Quantitative Image Cytometry Technique for Time Series or ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0009955]
- 123. Flow Cytometry Protocol | Abcam [https://www.abcam.com/en-us/technical-resources/protocols/flow-cytometry-for-intracellular-and-extracellular-targets]
- 124. Review of Flow Cytometry as a Tool for Cell and Gene Therapy [https://pmc.ncbi.nlm.nih.gov/articles/PMC10872958/]
- 125. Imaging Flow Cytometry: Coping with Heterogeneity in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3524563/]
- 126. BD CellView™ Image Technology [https://www.bdbiosciences.com/en-us/learn/applications/cell-view-image-technology]
- 127. Cell Cycle Analysis Assays [https://www.thermofisher.com/us/en/home/life-science/cell-analysis/cell-viability-and-regulation/cell-cycle.html]
- 128. 25-Hydroxycholesterol Induces Intrinsic Apoptosis via ... [https://www.mdpi.com/1422-0067/26/16/8012]
- 129. Identification of a small molecule targeting EPLIN as ... [https://www.nature.com/articles/s41419-025-07876-7]
- 130. 25-Hydroxycholesterol Induces Intrinsic Apoptosis via ... [https://pubmed.ncbi.nlm.nih.gov/40869333/]
- 131. Apoptosis Assays Market Size, Share, Trends and Industry ... [https://www.consegicbusinessintelligence.com/apoptosis-assays-market]
- 132. Exploitation of the Apoptosis-Primed State of MYCN ... [https://pubmed.ncbi.nlm.nih.gov/26859456/]
- 133. Inclusion of JNK-independent drugs within multiagent ... [https://pubmed.ncbi.nlm.nih.gov/41313779/?utm_source=SimplePie&utm_medium=rss&utm_campaign=pubmed-2&utm_content=1bAXfGTh08tVk-e1kpwuqxoVfiUn_5olMN9ewCML2bdAMGPEOD&fc=20220524054416&ff=20251128223443&v=2.18.0.post22+67771e2]
- 134. Exploring apoptotic pathways in SH-SY5Y neuroblastoma ... [https://journals.viamedica.pl/folia_morphologica/article/view/103887]