A Strategic Guide to Eliminating Non-Specific Bands in PARP-1 Western Blotting
This article provides a comprehensive, step-by-step framework for researchers, scientists, and drug development professionals to troubleshoot and eliminate non-specific bands in PARP-1 Western blots.
A Strategic Guide to Eliminating Non-Specific Bands in PARP-1 Western Blotting
Abstract
This article provides a comprehensive, step-by-step framework for researchers, scientists, and drug development professionals to troubleshoot and eliminate non-specific bands in PARP-1 Western blots. Covering foundational knowledge of PARP-1 biology, optimized methodological protocols, systematic troubleshooting, and rigorous antibody validation, this guide synthesizes current best practices to ensure specific, reproducible, and publication-quality data for both basic research and therapeutic development applications.
Understanding PARP-1 Complexity: Why Multiple Bands Are Not Always Non-Specific
Technical Support Center
Troubleshooting Guides
FAQ: My PARP-1 Western blot shows multiple bands. What could be the cause?
Answer: Multiple bands in PARP-1 Western blots are a common challenge and can stem from various biological and technical factors. The table below summarizes the primary causes and their solutions.
| Cause | Evidence in Band Pattern | Recommended Solution |
|---|---|---|
| Apoptotic Cleavage [1] [2] | Appearance of an 89 kDa fragment alongside the full-length 116 kDa protein. | Include appropriate controls (e.g., apoptotic inducers); confirm with caspase inhibitors. |
| Necrotic Cleavage [2] | Appearance of a 50 kDa fragment; not inhibited by caspase inhibitors (zVAD-fmk). | Use necroptosis inhibitors; ensure healthy cell conditions to prevent accidental necrosis. |
| Protein Degradation [3] [1] | Multiple lower molecular weight bands or smearing below the main bands. | Use fresh protease inhibitors during lysate preparation; keep samples on ice [3]. |
| Antibody Cross-reactivity [3] [4] | Bands at unpredictable molecular weights unrelated to known PARP-1 fragments. | Optimize primary antibody concentration; switch to a monoclonal antibody [3]; use a validated antibody [1]. |
| Post-Translational Modifications [5] | Band smearing or shifts above the expected molecular weight. | Check literature for known PARP-1 PTMs; use specific enzymatic assays for confirmation. |
| Alternative Splice Variants [1] | Consistent bands at weights other than 116, 89, or 50 kDa. | Consult genomic databases; use isoform-specific antibodies if available. |
FAQ: How can I confirm that the multiple bands in my blot are specific to PARP-1?
Answer: The most definitive method to confirm antibody specificity is to use a genetic validation control.
- Knockdown/Knockout Validation: As demonstrated in one study, transfection of cells with PARP-1-targeting shRNA resulted in a loss of signal for all bands (both the full-length and lower molecular weight bands) in the Western blot. This confirms that the antibody is specifically detecting PARP-1 and its cleavage or degradation products [1].
- Protocol: Transfer your protein samples from a gel to a nitrocellulose membrane. Block the membrane with 5% skim milk for 1 hour. Incubate with your primary anti-PARP-1 antibody (diluted in blocking buffer) overnight at 4°C with gentle agitation. After washing, incubate with an HRP-conjugated secondary antibody for 1 hour at room temperature. Detect using a chemiluminescent substrate [6] [1].
Experimental Protocols
Detailed Protocol: Optimizing Antibody Incubation to Reduce Non-Specific Bands
The following methodology can help conserve valuable antibody reagents and reduce background.
- Membrane Preparation: After blocking, briefly immerse the membrane in TBST and then blot it thoroughly with a paper towel to absorb residual moisture [6].
- Minimal-Volume Application: Place the semi-dried membrane on a clean surface. Apply a small volume of the primary antibody working solution directly onto the membrane. For a mini-gel membrane, 20–150 µL may be sufficient [6].
- Create a Sheet Protector Unit: Gently overlay the membrane with a leaflet of a sheet protector. The downward pressure will help distribute the antibody solution as a thin, even layer across the entire membrane surface [6].
- Incubation: Incubate the sheet protector unit at room temperature. To prevent evaporation during longer incubations, place the unit on a wet paper towel and seal it inside a zipper bag [6].
- Washing and Detection: Proceed with standard washing steps and secondary antibody incubation as in a conventional protocol [6].
PARP-1 Cleavage Pathways and Experimental Workflow
The following diagram illustrates the key cellular processes that lead to the characteristic banding pattern of PARP-1 in Western blots.
PARP-1 Cleavage Pathways
The Scientist's Toolkit: Research Reagent Solutions
The table below lists essential materials and reagents used in PARP-1 research, particularly for Western blot analysis.
| Item | Function & Application |
|---|---|
| Validated Anti-PARP1 Antibody | Critical for specific detection of full-length and cleaved PARP-1. Look for antibodies validated for Western blotting, preferably with knockout/shRNA confirmation data [1]. |
| Caspase Inhibitors (e.g., zVAD-fmk) | Used to distinguish apoptotic cleavage. If PARP-1 cleavage is inhibited by zVAD-fmk, it is likely caspase-dependent [2]. |
| Protease Inhibitor Cocktails | Added to lysis buffers to prevent non-specific protein degradation during sample preparation, which can cause smearing or extra bands [3] [6]. |
| PARP Inhibitors (e.g., PJ34) | Used to study the enzymatic function of PARP-1. Inhibiting PARP-1 can affect the poly(ADP-ribosyl)ation of other transcription factors like Sp1 [5]. |
| Chemiluminescent Substrate | For signal detection after incubation with HRP-conjugated secondary antibodies [6] [1]. |
| Sheet Protector | A stationery item used to create a minimal-volume incubation chamber for antibodies, significantly reducing reagent consumption [6]. |
| Skim Milk or BSA | Common blocking agents used to cover non-specific binding sites on the membrane, reducing high background and non-specific bands [6] [4]. |
FAQ: Understanding Your PARP-1 Western Blot
What are the specific PARP-1 cleavage fragments and what do they indicate?
PARP-1 is cleaved by different proteases into specific signature fragments that serve as biomarkers for distinct cell death pathways. The table below summarizes the key fragments and their biological significance.
| Fragment Size | Protease Responsible | Cell Death Pathway | Domains Contained | Biological Consequence |
|---|---|---|---|---|
| 89 kDa & 24 kDa | Caspases-3 and -7 [7] | Apoptosis [7] | 89 kDa: Auto-Modification Domain (AMD) & Catalytic Domain (CD) [7]24 kDa: DNA-Binding Domain (DBD) [7] | 24 kDa fragment irreversibly binds DNA, inhibiting repair; hallmark of apoptosis [7]. |
| 89 kDa (poly-ADP-ribosylated) | Caspases-3 and -7 [8] | Apoptosis-to-Parthanatos Transition [8] | Catalytic Domain with attached PAR polymers [8] | Fragment translocates to cytoplasm, acts as PAR carrier, inducing AIF-mediated parthanatos [8]. |
| ~50 kDa | Cathepsins B and G (Lysosomal Proteases) [2] | Necrosis [2] | Not Specified | A hallmark of necrotic cell death; not inhibited by caspase inhibitors like zVAD-fmk [2]. |
The following diagram illustrates the protease-specific cleavage of PARP-1 and the fate of the resulting fragments in different cell death pathways.
Why am I seeing non-specific or unexpected bands in my PARP-1 western blot?
Unexpected bands can arise from several experimental and biological factors. The troubleshooting flowchart below guides you through the systematic identification and resolution of common issues.
Detailed Troubleshooting Protocols
1. Issue: Low Antibody Specificity or High Background
- Primary Cause: The primary antibody concentration is too high, leading to off-target binding, or the blocking step was incomplete [4] [9] [3].
- Recommended Protocol:
- Antibody Titration: Perform a dilution series of your primary antibody to find the optimal concentration. A higher dilution can reduce non-specific binding without significantly impacting the target signal [4] [3].
- Incubation Temperature: Perform the primary antibody incubation step at 4°C overnight. This decreases non-specific binding compared to room temperature incubations [4].
- Blocking Buffer: Switch from general blockers like milk or BSA to an engineered blocking buffer specifically designed to reduce non-specific interactions. If using milk or BSA, increase the concentration from 2% to 5% and ensure blocking is performed for at least 1 hour at room temperature or overnight at 4°C [4] [9].
- Washing Stringency: Increase the number and volume of washes after antibody incubations. Wash the membrane 4-5 times for 5 minutes each with gentle agitation, using a wash buffer containing 0.1% Tween-20 [9] [3].
2. Issue: Protein Degradation, Multimers, or Overloading
- Primary Cause: Protease activity in the sample creates lower molecular weight bands, or too much protein per lane causes "ghost bands" and smearing [9] [3].
- Recommended Protocol:
- Sample Preparation: Always prepare fresh lysis buffers and add a broad-spectrum protease inhibitor cocktail immediately before cell lysis to prevent protein degradation [3].
- Heat Denaturation: Ensure samples are properly boiled in Laemmli buffer for 5-10 minutes to disrupt protein complexes and multimers [3].
- Protein Load Optimization: Precisely quantify your protein samples. For membrane, nuclear, and total cell lysates, aim to load 20-30 µg of protein per lane. For purified proteins, 10-100 ng is typically sufficient [9] [3]. Reduce the load if you observe over-saturation.
How do Post-Translational Modifications (PTMs) like ADP-ribosylation affect PARP-1?
PARP-1 catalyzes poly(ADP-ribosyl)ation (PARylation), a key PTM in the DNA damage response. PARP-1 can modify itself (auto-modification) and other target proteins.
- Auto-modification: PARP-1 adds long chains of poly(ADP-ribose) (PAR) to itself on aspartate, glutamate, and serine residues [10] [11]. This auto-modification is crucial for its release from DNA damage sites, which facilitates repair and prevents replication stress [11].
- Detection Challenge: Aspartate/glutamate ADP-ribosylation is ester-linked and highly chemically labile. Standard sample preparation involving boiling can destroy this signal.
- Protocol for Preserving Labile PTMs: To detect labile Asp/Glu-ADPr, avoid boiling samples. Instead, lyse cells in a denaturing buffer containing 4% SDS at room temperature to inactivate enzymes while preserving the ester-linked modification [10].
The Scientist's Toolkit: Key Research Reagents
| Reagent / Material | Function / Role | Example & Notes |
|---|---|---|
| Caspase Inhibitor (e.g., zVAD-fmk) | To inhibit caspase-mediated PARP-1 cleavage and confirm the absence of apoptotic fragments [2]. | Useful for distinguishing apoptosis from other cleavage events. |
| PARP Inhibitor (e.g., PJ34) | To chemically inhibit PARP-1's enzymatic activity for studying its functional roles [5]. | Used to demonstrate PARP-1's role in regulating transcription factor Sp1 [5]. |
| Engineered Blocking Buffer | To effectively block the membrane and reduce non-specific antibody binding, minimizing background [4]. | Azure Chemi Blot Blocking Buffer; superior to milk/BSA for some targets [4]. |
| Protease Inhibitor Cocktail | To prevent non-specific proteolytic degradation of PARP-1 during sample preparation [3]. | Essential for obtaining clean, reproducible bands. |
| Anti-PARP-1 Antibody (Monoclonal) | To increase specificity and reduce non-specific bands compared to polyclonal antibodies [3]. | Critical for clear identification of specific fragments. |
| HRP-Coupled Substrate | For sensitive chemiluminescent detection of the target protein. | Use maximum sensitivity substrates (e.g., SuperSignal West Femto) for low-abundance targets [9]. |
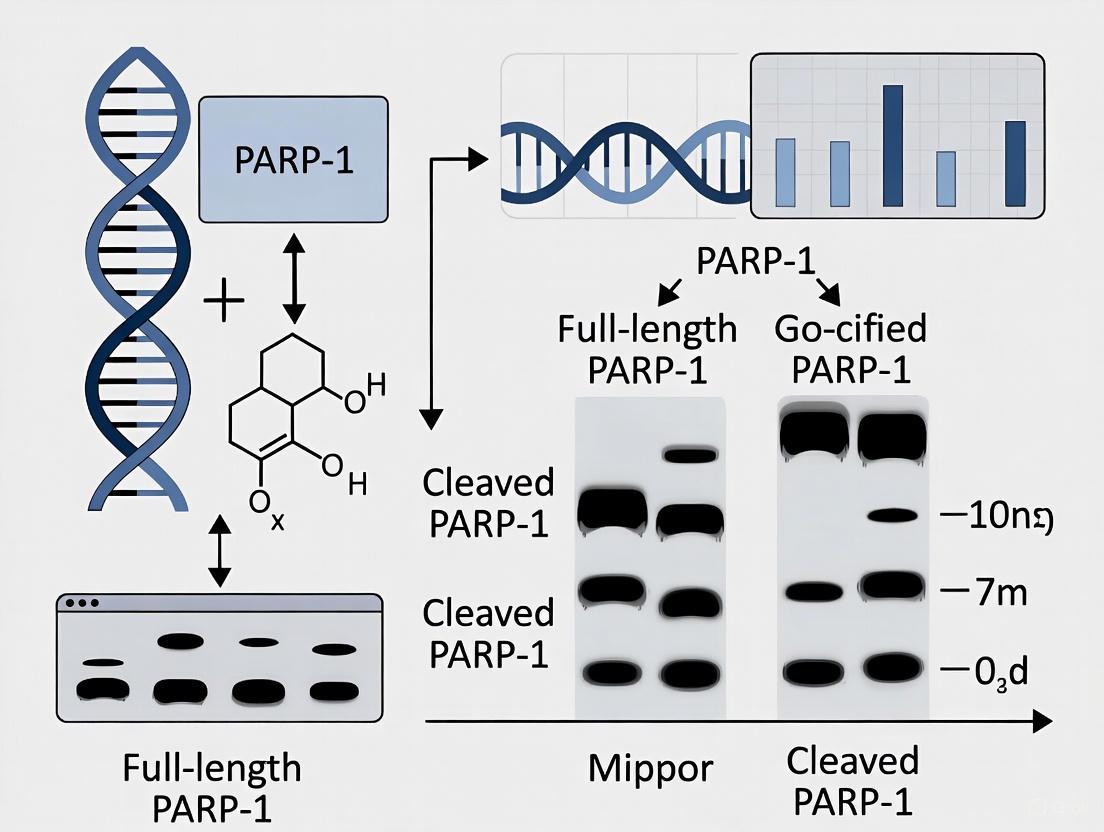
This case study focuses on the specific detection of full-length poly (ADP-ribose) polymerase 1 (PARP1) and its cleavage product using validated antibodies in western blotting. PARP1 is a 116 kDa nuclear enzyme crucial for DNA repair processes. During apoptosis, caspase-3 cleaves PARP1 into a definitive 89 kDa fragment, a well-established biochemical marker for programmed cell death [12] [13] [14]. Distinguishing these specific bands from non-specific signals is critical for accurate data interpretation in research and drug development.
The technical support content below provides troubleshooting guides and FAQs to help researchers reduce non-specific bands and optimize their PARP1 western blot experiments, directly supporting the broader thesis of improving experimental reliability in PARP-1 research.
Troubleshooting Guide: Non-Specific Bands in PARP1 Western Blot
Primary Troubleshooting Table
| Problem Area | Potential Cause | Recommended Solution | Underlying Principle |
|---|---|---|---|
| Antibody Issues | Primary antibody concentration too high [3] [9] | Titrate antibody; use recommended dilution (e.g., 1:1000 for CST #9542) [12] | Prevents antibody "promiscuity" and off-target binding [3]. |
| Polyclonal antibody cross-reactivity [3] | Use monoclonal antibodies or affinity-purified polyclonals [3]; validate via knockdown/knockout [1]. | Increases epitope specificity. | |
| Sample Quality | Protein degradation by proteases [3] | Use fresh protease inhibitors during sample preparation [3]. | Prevents appearance of lower molecular weight degradation bands. |
| Excessive protein loading [3] [9] | Load 20-30 µg for cell lysates; confirm concentration [3]. | Prevents "ghostbands" and over-saturation. | |
| Presence of protein multimers [3] | Boil samples in Laemmli buffer for 5-10 minutes pre-loading [3]. | Disrupts non-covalent protein aggregates. | |
| Blocking & Washing | Incomplete blocking [3] [4] | Increase blocking reagent (e.g., 2-5% BSA), extend blocking time, include 0.05% Tween-20 [3]. | Masks non-specific binding sites on the membrane. |
| Insufficient washing [3] | Perform 4-5 washes of 5 minutes each with 0.05-0.1% Tween-20 [3]. | Removes unbound antibodies and contaminants. |
PARP1-Specific Band Interpretation Guide
| Observed Band (kDa) | Specific Band? | Explanation & Validation Tips |
|---|---|---|
| 116 | Yes | Specific full-length PARP1 [12] [13]. Expected primary band in healthy cells. |
| 89 | Yes | Specific cleavage product of apoptosis [12] [14]. Appears with the 116 kDa band during early apoptosis. |
| ~24 | Yes (but often unseen) | Small N-terminal fragment; often runs off gel or is not detected by C-terminal antibodies [13]. |
| Bands between 40-89 kDa | Potentially Non-Specific | May result from degradation (if below 89 kDa) [3] or alternative cleavage by other proteases (e.g., calpains, cathepsins) [13]. Validate via knockdown [1]. |
| Bands above 116 kDa | Potentially Non-Specific | Could be protein multimers [3] or non-specific binding. Ensure sample is properly boiled and reduced. |
Experimental Protocols for Validation
Protocol 1: Validating Antibody Specificity via Knockdown
This protocol is considered a gold standard for confirming antibody specificity [1].
Key Materials:
- Validated PARP1 Antibody: e.g., Proteintech 13371-1-AP [13] or CST #9542 [12].
- shRNA for PARP1: Designed to target the PARP1 mRNA sequence.
- Control shRNA: Non-targeting scrambled sequence.
- Transfection Reagent: Suitable for the cell line used (e.g., Lipofectamine RNAiMAX).
- Standard Western Blotting reagents.
Expected Outcome: A specific antibody will show a significant reduction or complete loss of signal at both 116 kDa and 89 kDa in the knockdown lane compared to the control, confirming that all observed bands are specific to PARP1 [1].
Protocol 2: Inducing and Detecting PARP1 Cleavage
This protocol outlines how to reliably generate the 89 kDa cleavage fragment for antibody validation.
Procedure:
- Cell Treatment: Treat cells (e.g., Jurkat or HeLa) with a known apoptosis inducer.
- Sample Preparation: Lyse cells in RIPA buffer supplemented with fresh protease inhibitors [3]. Determine protein concentration.
- Western Blotting: Load 20-30 µg of protein per lane. Follow standard SDS-PAGE and transfer protocols.
- Detection: Incubate with primary antibody (e.g., Anti-Cleaved PARP1 antibody ab4830 at 1:1000 dilution) [14] and then with an appropriate HRP-conjugated secondary antibody.
Expected Outcome: Untreated cells show a dominant band at 116 kDa. Apoptotic cells show a strong band at 89 kDa, often with a corresponding decrease in the 116 kDa band intensity [14].
Frequently Asked Questions (FAQs)
Q1: My antibody data sheet shows a single clean band, but I see multiple bands in my experiment. Does this mean my antibody is non-specific? Not necessarily. Multiple bands can be due to specific biological reasons, especially for PARP1. The 89 kDa band is a valid cleavage product [14]. Other bands could result from protein degradation, alternative splicing, or post-translational modifications [1]. Validation in your specific experimental system using a knockdown control is crucial [1].
Q2: Why should I use a monoclonal antibody over a polyclonal antibody for PARP1 detection? Polyclonal antibodies, by nature, are a mixture that can bind to multiple epitopes, which sometimes leads to non-specific binding [3]. Monoclonal antibodies recognize a single epitope, offering higher specificity and lower chances of off-target binding, which helps reduce non-specific bands [3].
Q3: I see a band at the correct molecular weight, but my background is high. What should I do? High background is often a separate issue from non-specific bands but can obscure results. To reduce background:
- Decrease antibody concentration, especially the primary antibody [9] [4].
- Increase the concentration of your blocking reagent (e.g., BSA to 5%) or switch to an engineered blocking buffer [3] [4].
- Increase the number and duration of washes with TBST (0.05-0.1% Tween-20) after antibody incubations [3] [9].
- Ensure the membrane does not dry out at any step [9].
Research Reagent Solutions
This table lists key reagents essential for successful PARP1 western blotting, as cited in the literature.
| Reagent | Example Product / Specification | Function in Experiment |
|---|---|---|
| Validated PARP1 Antibody | Cell Signaling Technology #9542 [12] | Detects endogenous full-length (116 kDa) and cleaved (89 kDa) PARP1. |
| Antibody for Cleaved PARP1 | Abcam ab4830 [14] | Specifically detects the 85-89 kDa caspase-cleaved fragment of PARP1. |
| Broad-Specificity PARP1 Antibody | Proteintech 13371-1-AP [13] | A polyclonal antibody suitable for detecting PARP1 in WB, IHC, IP. |
| Positive Control Lysate | Etoposide-treated Jurkat cell lysate [14] | Provides a control sample known to contain the 89 kDa cleaved PARP1 fragment. |
| Protease Inhibitor Cocktail | Added fresh to lysis buffer [3] | Prevents proteolytic degradation of PARP1 during sample preparation. |
| Blocking Buffer | 5% BSA in TBST [3] | Blocks non-specific binding sites on the membrane to reduce background. |
PARP1 Cleavage and Detection Workflow
The following diagram summarizes the biological process of PARP1 cleavage during apoptosis and the subsequent experimental detection via western blotting.
In PARP-1 Western blot research, distinguishing true biological complexity from technical artifacts is fundamental to accurate data interpretation. Non-specific bands can lead researchers down incorrect experimental paths, wasting precious time and resources. This technical support guide provides targeted troubleshooting advice and FAQs to help you identify the root causes of Western blot anomalies specifically in PARP-1 research, enabling more reliable conclusions about this critical DNA damage response protein.
FAQs: Addressing PARP-1 Western Blot Challenges
Q1: My PARP-1 Western blot shows multiple bands at unexpected molecular weights. How can I determine if these represent biological variants or technical artifacts?
Multiple bands in PARP-1 blots can originate from both biological and technical sources. To systematically diagnose the issue, follow this decision pathway:
The most common biological explanations for multiple PARP-1 bands include:
- Post-translational modifications: PARP-1 undergoes extensive ADP-ribosylation, ubiquitylation, and acetylation, which can alter its apparent molecular weight [15] [16]. Recent research has identified specific modification sites on PARP-1, including serine ADP-ribosylation that can be targeted by ester-linked ubiquitylation [16].
- Protein isoforms and splice variants: Check literature databases for known PARP-1 variants and their expected molecular weights [17].
- Multimer formation: PARP-1 can form complexes with itself and other proteins. Boiling samples in Laemmli buffer for 5-10 minutes can disrupt these multimers [3].
Technical causes often include:
- Protein degradation: Use fresh protease inhibitors and maintain samples on ice during preparation [3] [17].
- Antibody issues: Polyclonal antibodies are particularly prone to non-specific binding. Consider switching to monoclonal antibodies or titrating your antibody concentration [3] [4].
Q2: What specific controls should I include when optimizing PARP-1 Western blots?
Proper controls are essential for validating PARP-1 Western blot results. This table summarizes the critical controls and their purposes:
| Control Type | Purpose | Recommended Implementation for PARP-1 |
|---|---|---|
| Positive Control | Verifies detection system functionality | Use PARP-1 overexpressing cell lines or cells treated with DNA damaging agents (e.g., H₂O₂) to induce PARP-1 expression [15] [18] |
| Negative Control | Checks for non-specific antibody binding | Use PARP-1 knockout cells or siRNA-mediated knockdown [19] |
| Loading Control | Ensures equal protein loading across lanes | Use housekeeping proteins (e.g., GAPDH, actin) from the same subcellular compartment as PARP-1 (nuclear) |
| Molecular Weight Marker | Identifies protein size accurately | Use pre-stained markers spanning 50-150 kDa (PARP-1 is ~116 kDa) |
Q3: The background in my PARP-1 blots is consistently high. What optimization strategies should I prioritize?
High background obscures results and complicates interpretation. Implement these specific solutions:
- Enhance blocking conditions: Increase blocking buffer concentration from 2% to 5% BSA, extend blocking incubation times, or switch to an engineered blocking buffer specifically designed to reduce non-specific binding [3] [4]. Prepare primary antibody in blocking buffer rather than wash buffer.
- Optimize antibody concentrations: High antibody concentrations are a common cause of background. Titrate both primary and secondary antibodies to find the optimal dilution that maintains signal while reducing background [4]. Incubate primary antibody at 4°C overnight rather than at room temperature.
- Increase washing efficiency: Wash membranes with gentle agitation using sufficient buffer volume to completely cover the membrane. Increase to 4-5 washes of 5 minutes each and consider increasing Tween-20 concentration to 0.1% in wash buffers [3].
- Verify antibody specificity: For PARP-1 studies, ensure your antibody is validated for Western blotting and recognizes denatured protein. Run a secondary antibody-only control to identify background caused by secondary antibody non-specific binding [17].
Research Reagent Solutions for PARP-1 Studies
This table outlines essential reagents and their specific applications in PARP-1 Western blot research:
| Reagent Category | Specific Examples | Application in PARP-1 Research |
|---|---|---|
| Protease Inhibitors | PMSF, complete protease inhibitor cocktails | Prevent PARP-1 degradation during sample preparation [3] [17] |
| Phosphatase Inhibitors | Sodium fluoride, beta-glycerophosphate | Preserve phosphorylation states of PARP-1 |
| PARP Inhibitors | AG14361, Olaparib analogs | Control for PARP-1 enzymatic activity in experimental treatments [18] |
| DNA Damage Agents | H₂O₂, camptothecin, hydroxyurea | Induce PARP-1 activation and recruitment to DNA damage sites [15] [16] |
| Blocking Buffers | BSA, non-fat milk, engineered blocking buffers | Reduce non-specific antibody binding [4] |
| Detection Systems | Chemiluminescent, fluorescent substrates | Optimize for sensitivity needed to detect PARP-1 at endogenous levels |
Advanced PARP-1 Specific Considerations
PARP-1 presents unique challenges due to its complex biology and modification status:
DNA Damage Response Dynamics PARP-1 rapidly responds to DNA damage (within ~1 second of DNA break occurrence) and undergoes rapid auto-ADP-ribosylation [20]. This dynamic modification state means that the banding pattern observed can vary significantly depending on the DNA damage status of your cells. When studying PARP-1 in DNA damage contexts, include appropriate damage-inducing controls and carefully track time post-treatment.
MAR vs PAR Modifications Recent advances have distinguished between mono(ADP-ribose) (MAR) and poly(ADP-ribose) (PAR) modifications on PARP-1, which have different biological consequences and detection requirements [20]. MARylation may be more common than previously thought, even after DNA damage. Ensure you're using detection reagents that can differentiate between these forms if relevant to your research question.
Interaction Networks PARP-1 functions within complex protein networks, interacting with partners like MARVELD1, which stabilizes PARP-1 and enhances NAA50-mediated acetylation [15]. These interactions can affect PARP-1 migration on Western blots. When observing unexpected banding patterns, consider potential interacting proteins that might be co-migrating or affecting PARP-1 modifications.
Experimental Protocol: Validating PARP-1 Specific Bands
To confirm that observed bands represent genuine PARP-1 signals rather than artifacts, implement this verification protocol:
Knockdown/Knockout Validation: Use siRNA or CRISPR to reduce or eliminate PARP-1 expression. Compare band patterns between control and PARP-1 depleted cells. Bands that disappear in knockout samples are likely specific [19].
Protease Protection Test: Treat samples with specific proteases (e.g., trypsin) in a time-course experiment. Genuine PARP-1 fragments should show predictable degradation patterns, while non-specific bands may degrade differently.
Antibody Validation with Blocking Peptides: Pre-incubate the primary antibody with the immunizing peptide (if available). Specific bands should be significantly reduced or eliminated, while non-specific bands will remain [17].
Cross-Verification with Alternative Antibodies: Repeat the blot with antibodies targeting different PARP-1 epitopes or domains. True PARP-1 bands should be detected by multiple validated antibodies.
Modification-Specific Treatments: Use enzymatic treatments (e.g., phosphatases, glycosidases) or specific conditions that remove known PARP-1 modifications to determine if band shifts correspond to specific PTMs.
By systematically implementing these troubleshooting approaches and validation protocols, researchers can significantly improve the reliability of their PARP-1 Western blot data and make more confident conclusions about this complex and highly modified protein.
Optimized Western Blot Protocol for PARP-1: From Sample Prep to Detection
In PARP-1 research, the integrity of your experimental data is fundamentally determined by the quality of your sample preparation. The core challenge researchers face is a dual problem: preventing the proteolytic degradation of the PARP-1 protein itself, while simultaneously ensuring that the sample preparation process does not inadvertently activate PARP-1. PARP-1 is a highly abundant and sensitive DNA damage sensor that is easily activated by minor nucleic acid perturbations during cell lysis [21]. This activation triggers its auto-modification (autoPARylation), leading to a characteristic smear or non-specific bands on a western blot, which can obscure the clear detection of the full-length protein at 113 kDa and its cleavage fragments [22] [23]. This article provides a targeted troubleshooting guide to navigate these critical steps, framed within the broader thesis of obtaining clean, specific, and interpretable PARP-1 western blot data.
FAQs and Troubleshooting Guides
What causes smears or high molecular weight bands in my PARP-1 western blot?
Answer: Smears or high molecular weight bands are typically the result of PARP-1 activation and subsequent autoPARylation during sample preparation. When PARP-1 binds to DNA nicks or breaks present in your lysate, it catalyzes the addition of long, branched chains of poly(ADP-ribose) (PAR) onto itself. This post-translational modification increases the protein's apparent molecular weight, creating a smear on a gel [11] [21] [24].
- Primary Cause: The presence of damaged DNA (e.g., from overly vigorous pipetting, sonication, or nuclease activity) during cell lysis activates PARP-1.
- Solution: Implement gentle lysis techniques and include PARP inhibitors in your lysis buffer to prevent the catalytic activity that leads to PAR chain formation.
How can I prevent PARP-1 activation during sample preparation?
Answer: Preventing activation is key to avoiding non-specific bands.
- Use Gentle Lysis Methods: Avoid sonication or excessive vortexing of lysates. Use a mild detergent-based lysis buffer and allow lysis to proceed on ice for 30-60 minutes with occasional gentle pipetting [25].
- Include PARP Inhibitors in Lysis Buffer: Supplement your standard lysis buffer with a potent PARP catalytic inhibitor. This prevents the initiation of ADP-ribosylation as soon as the cell membrane is compromised.
- Benzonase Nuclease Treatment: A highly effective strategy is to add Benzonase to your lysis buffer. This enzyme degrades all forms of DNA and RNA, removing the essential trigger for PARP-1 activation [21]. This step is crucial for studies focusing on PARP1's non-canonical roles involving RNA interactions [26].
How do I prevent the proteolytic degradation of PARP-1?
Answer: Proteolysis results in the appearance of lower molecular weight bands and the loss of the strong 113 kDa signal.
- Work Quickly and Keep Samples Cold: Perform all steps on ice or at 4°C using pre-chilled buffers.
- Use Comprehensive Protease Inhibitors: Employ a broad-spectrum protease inhibitor cocktail (e.g., targeting serine, cysteine, and metalloproteases) in your lysis buffer. Avoid repeated freeze-thaw cycles of lysates.
- Optimize Sample Boiling: While boiling is standard for SDS-PAGE, note that the ADP-ribosylation modification can be heat-labile [22]. The optimal balance between denaturation and preserving the protein modification must be determined empirically. Always use a fresh, reducing Laemmli buffer.
My experimental design involves inducing DNA damage. How can I distinguish specific PARylation from preparation artifacts?
Answer: This is a critical distinction for accurate data interpretation.
- Include a PARP Inhibitor Control: For every experiment, prepare a parallel sample treated with a high concentration of a PARP inhibitor (e.g., Olaparib, Rucaparib) during the lysis and throughout sample preparation. Any PARylation signal absent in this control can be confidently attributed to bona fide biological activation that occurred in your cells prior to lysis, rather than an artifact of the preparation process.
- Validate with Activation Markers: Use antibodies specific for PAR chains to confirm the presence of PARylation in your experimental samples, but again, compare to the inhibitor-controlled lysate.
Experimental Protocols for Optimal Sample Preparation
Standardized Protocol for Preventing Proteolysis and PARP-1 Activation
This protocol is designed for adherent cells and should be adapted as needed for specific experimental contexts.
Reagents Needed:
- Lysis Buffer Base: 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% Triton X-100, 1% NP-40, 1 mM EDTA.
- Protease Inhibitor Cocktail (e.g., tablets or liquid mix).
- PARP Catalytic Inhibitor (e.g., Olaparib or Rucaparib, prepare a 10 mM stock in DMSO).
- Benzonase Nuclease (optional, but highly recommended).
- 2X Laemmli Sample Buffer with β-mercaptoethanol.
Procedure:
- Pre-chill all buffers and equipment on ice.
- Prepare Complete Lysis Buffer fresh: To the Lysis Buffer Base, add 1X Protease Inhibitor Cocktail and 10-20 µM PARP Inhibitor. If using, also add 25 U/mL Benzonase.
- Aspirate culture media from adherent cells and wash once with ice-cold PBS.
- Add cold lysis buffer directly to the culture dish (e.g., 100 µL per 10 cm²).
- Incubate on ice for 30 minutes. Gently rock the plate every 10 minutes to ensure even coverage.
- Scrape the cells and transfer the lysate to a pre-chilled microcentrifuge tube.
- Clarify the lysate by centrifugation at 13,500 rpm for 20 minutes at 4°C.
- Transfer the supernatant (cleared lysate) to a new tube.
- Protein Quantification & Denaturation: Mix an aliquot of the lysate with an equal volume of 2X Laemmli buffer. Do not boil at 95°C as a first choice. Instead, heat at 70-80°C for 10-15 minutes to denature while better preserving heat-labile modifications [22].
- Proceed with SDS-PAGE and Western blotting.
Protocol Validation with Controls
To ensure your protocol is working, always run the following controls on your western blot:
- Positive Control for PARP-1 Expression: A whole-cell extract from a known PARP-1-expressing cell line (e.g., HeLa, HEK-293T) [23].
- Negative Control for Specificity: A lysate from PARP1-knockout or shRNA-treated cells [23].
- Inhibition Control: A sample prepared with the PARP inhibitor in the lysis buffer to confirm the absence of preparation-induced smearing.
Data Presentation: Troubleshooting PARP-1 Western Blots
The table below summarizes common issues, their causes, and solutions to achieve a clean western blot.
Table: Troubleshooting Guide for PARP-1 Western Blotting
| Observed Issue | Potential Cause | Recommended Solution |
|---|---|---|
| Smear above 113 kDa | PARP-1 autoPARylation due to activation by damaged DNA during lysis. | Add a PARP inhibitor (e.g., 10 µM Olaparib) and/or 25 U/mL Benzonase to the lysis buffer. |
| Multiple non-specific bands | Proteolytic degradation of the PARP-1 protein. | Use fresh, comprehensive protease inhibitors; keep samples consistently cold; avoid freeze-thaw cycles. |
| Loss of signal / Weak band | Over-boiling; protein not transferring efficiently; antibody issues. | Reduce denaturation temperature to 70-80°C; validate transfer efficiency with protein stain; check antibody dilution and specificity using a KO control [23]. |
| Band at ~89 kDa | Caspase-mediated cleavage of PARP-1, an indicator of apoptosis. | This is often a biological result. Confirm with other apoptosis assays. Ensure inhibitors of apoptosis are not required for your experimental design. |
Signaling Pathways and Experimental Workflows
The following diagram illustrates the critical decision points during sample preparation that determine the quality of your PARP-1 western blot data, and how they connect to the broader goal of reducing non-specific bands.
The Scientist's Toolkit: Research Reagent Solutions
Table: Essential Reagents for PARP-1 Sample Preparation and Analysis
| Reagent / Tool | Function / Application | Key Consideration |
|---|---|---|
| PARP Catalytic Inhibitors (e.g., Olaparib, Rucaparib, Talazoparib) | Inhibits PARP enzymatic activity. Added to lysis buffer to prevent autoPARylation during preparation. | Distinguish from PARP1-degrading PROTACs (e.g., 180055 [27]), which remove the protein entirely, as their use in sample prep is not standard. |
| Benzonase Nuclease | Degrades all forms of DNA and RNA (single/double-stranded, linear/circular). Removes the primary activator of PARP-1. | Crucial for preventing activation, especially when studying PARP1 in contexts involving RNA or chromatin binding [26] [21]. |
| Anti-PARP1 Antibody (ab227244) | Detects PARP1 in Western Blot (WB), Immunoprecipitation (IP), Immunofluorescence (IF), etc. [23] | Validated for a clean 113 kDa band. Use a KO lysate control to confirm specificity and rule out non-specific bands. |
| Anti-PAR Antibody | Specifically detects poly(ADP-ribose) chains. Used to confirm PARylation in assays. | Essential for validating that a smear is due to PARylation. Use in conjunction with PARP inhibitor controls. |
| Protease Inhibitor Cocktail | Broad-spectrum inhibition of serine, cysteine, and metalloproteases to prevent PARP-1 degradation. | Must be added fresh to lysis buffer. Avoid EDTA-free cocktails if your purification or assay requires divalent cations. |
FAQs on Blocking Buffer Selection and Troubleshooting
1. Why is the choice of blocking buffer so critical for detecting PARP1 by western blot?
The primary purpose of a blocking buffer is to saturate all unoccupied protein-binding sites on the membrane after transfer. This prevents detection antibodies from binding non-specifically, which causes high background noise and non-specific bands that can obscure your true results. For a protein like PARP1, which has multiple isoforms and can be subject to post-translational modifications, a clean background is essential for accurate interpretation. An inappropriate blocking agent can mask your target signal or increase non-specific binding, complicating data analysis [28] [29].
2. When should I use BSA over non-fat dry milk for PARP1 blots?
BSA is generally the preferred choice for PARP1 immunoblotting, especially when investigating DNA damage and repair pathways. This is because PARP1 is heavily regulated by phosphorylation and other modifications. Non-fat dry milk contains phosphoproteins and phosphatases that can interfere with the detection of phosphorylated proteins and may lead to high background or degraded signal. BSA, being a single, purified protein, lacks these interfering substances and is more compatible with phospho-specific antibodies often used in PARP1 functional studies [30] [28] [29].
3. What are engineered blocking buffers, and what advantages do they offer?
Engineered blocking buffers are specialized, often proprietary formulations designed to provide superior and consistent blocking. They are typically based on a single, highly purified protein (such as casein) or other polymers. Their advantages include:
- Higher Specificity: Reduced chance of cross-reaction with assay components compared to mixed-protein blockers like milk [29].
- Optimized Performance: Formulated to minimize background while maximizing target signal, which can be crucial for detecting low-abundance proteins [4] [29].
- Faster Blocking: Some can effectively block membranes in as little as 10-15 minutes [29].
- Compatibility: They are often serum- and biotin-free, making them ideal for a wide range of detection systems, including fluorescent western blotting [29].
4. I'm seeing multiple non-specific bands in my PARP1 blot. How can my blocking strategy help?
Multiple non-specific bands are often a symptom of incomplete blocking or low antibody specificity. Your blocking strategy can directly address this:
- Increase Blocking Buffer Concentration or Time: Try boosting the concentration of your blocking agent (e.g., from 2% to 5%) or extending the blocking incubation time [3] [28].
- Switch Blocking Agents: If using milk, switch to BSA or an engineered buffer to rule out interference from milk proteins [4].
- Incorporate Detergent: Add Tween-20 to a final concentration of 0.05%-0.1% to your blocking and wash buffers to further reduce weak, non-specific interactions [3] [28].
- Optimize Antibody Concentration: A too-high antibody concentration is a common cause of non-specific bands. Dilute your primary antibody further and consider incubating at 4°C to increase binding specificity [3] [9] [4].
Blocking Buffer Comparison and Selection Guide
The table below summarizes key characteristics of common blocking buffers to guide your selection.
| Blocking Buffer | Best For | Advantages | Disadvantages | Considerations for PARP1 Research |
|---|---|---|---|---|
| Non-Fat Dry Milk | General purpose, low-cost applications [31] [29] | Inexpensive; provides more complete blocking for some targets [31] [29] | Contains phosphoproteins and phosphatases that can interfere with phospho-specific detection; contains biotin [30] [29] | Not recommended for studies involving PARP1 phosphorylation or biotin-streptavidin systems. |
| Bovine Serum Albumin (BSA) | Detecting phosphorylated proteins; biotin-streptavidin systems [28] [29] | Lacks phosphoproteins and phosphatases; compatible with a wide range of detection systems [30] [31] | Can be more expensive than milk; may provide weaker blocking, leading to higher background for some antibodies [31] [29] | The default recommended choice for most PARP1 applications, especially in DNA damage signaling contexts. |
| Engineered (Casein-based) | High-sensitivity applications; minimizing non-specific bands [29] | Single-protein source reduces cross-reactivity; high-performance replacement for milk [29] | More expensive than traditional, homemade blockers [29] | Ideal for troubleshooting high background or when detecting PARP1 isoforms and cleavage products. |
| Specialty Commercial Buffers | Optimizing new systems; fluorescent western blotting; high background situations [4] [29] | Often serum- and biotin-free; fast blocking (10-15 min); low fluorescence [29] | Highest cost; proprietary formulations [29] | Useful for advanced applications like multiplex fluorescent detection of PARP1 and its interaction partners. |
Troubleshooting Guide: Non-Specific Bands and High Background
Problem: High Background Throughout the Membrane
- Possible Cause: Incomplete blocking of the membrane.
- Solution: Increase the concentration of your blocking agent to 5% and ensure you are blocking for at least 1 hour at room temperature with gentle agitation. For persistent background, try blocking overnight at 4°C [9] [28].
- Possible Cause: Antibody concentration is too high.
- Solution: Titrate your primary and secondary antibodies. A higher dilution often reduces background significantly. Start with the manufacturer's recommended dilution and increase it sequentially [9] [32] [4].
- Possible Cause: Insufficient washing.
- Solution: Perform 4-5 washes for 5 minutes each with TBST (Tris-Buffered Saline with 0.1% Tween-20) after both primary and secondary antibody incubations. Ensure sufficient buffer volume and gentle agitation [3] [32].
Problem: Multiple Non-Specific Bands
- Possible Cause: Low specificity of the primary antibody.
- Solution: Incubate the primary antibody at 4°C instead of room temperature to promote specific binding. If possible, use an antibody that has been validated for western blotting [9] [4].
- Possible Cause: Protein degradation or modification.
- Solution: Prepare fresh lysates with adequate protease inhibitors to prevent PARP1 degradation, which can create lower molecular weight bands [3] [9]. Be aware that PARP1 can undergo cleavage and form multimers, which may appear as additional bands [3].
Problem: Weak or No Target Signal
- Possible Cause: The blocking buffer is masking the antigen.
- Solution: Switch to a different blocking agent. If using milk, which can sometimes mask antigens, try BSA or an engineered buffer to see if the signal improves [28] [29].
- Possible Cause: Over-blocking the membrane.
- Solution: Reduce the concentration of the blocking agent or the incubation time [28].
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function | Example Use Case |
|---|---|---|
| Blocker BSA | A purified BSA formulation for blocking; ideal for phosphoprotein detection and biotin-free systems. | The standard choice for PARP1 blots, especially when studying its phosphorylation status in DNA damage repair [29]. |
| Blocker Casein | A purified casein solution; a high-performance, single-protein alternative to non-fat dry milk. | Useful for reducing non-specific bands when detecting PARP1, offering a cleaner background than milk [29]. |
| StartingBlock Blocking Buffer | A proprietary, single-protein, serum-free buffer that blocks quickly and is compatible with many systems. | Excellent for troubleshooting high background issues or when setting up a new PARP1 detection protocol [29]. |
| Blocker FL Fluorescent Blocking Buffer | A detergent-free, low-fluorescence buffer optimized for fluorescent western blot detection. | Essential for multiplex fluorescent experiments detecting PARP1 and other DNA repair proteins simultaneously [29]. |
| Tween-20 Detergent | A non-ionic detergent added to buffers (0.05-0.1%) to reduce non-specific antibody binding. | A critical additive to all wash buffers and often to blocking buffers to minimize background in PARP1 blots [3] [28]. |
Experimental Workflow for Blocking Buffer Optimization
The following diagram outlines a systematic workflow to optimize your blocking conditions for the cleanest PARP1 detection.
Key Experimental Protocols
Protocol: Standard Blocking Procedure with BSA
This is a reliable starting point for PARP1 western blotting.
- Preparation: Following transfer, place the membrane in a clean container.
- Blocking Buffer: Prepare a 5% (w/v) solution of BSA in TBST (Tris-Buffered Saline with 0.1% Tween-20). Filter the buffer if necessary to remove particulates.
- Incubation: Completely submerge the membrane in the blocking buffer. Incubate for 1 hour at room temperature with gentle rocking on an orbital shaker.
- Washing: Proceed to antibody incubation. No need to wash the membrane after blocking if the antibody is diluted in a compatible buffer [28].
Protocol: Troubleshooting High Background with Enhanced Blocking
If you are consistently getting high background, use this enhanced protocol.
- Increase Blocking Time/Temperature: Block the membrane for 1 hour at room temperature, then continue incubation overnight at 4°C.
- Use Antibody Diluent with Blocker: Dilute your primary antibody in a fresh batch of your blocking buffer (e.g., 5% BSA in TBST) instead of plain TBST.
- Enhanced Washing: After primary and secondary antibody incubations, wash the membrane 5 times for 5 minutes each with a large volume of TBST (0.1% Tween-20) under constant agitation [3] [9].
Frequently Asked Questions: Optimizing PARP-1 Western Blots
Why am I seeing multiple non-specific bands in my PARP-1 western blot? Multiple non-specific bands are a common challenge. For PARP-1, which has a predicted molecular weight of approximately 113-116 kDa, extra bands can arise from several sources [33]. The most frequent causes are:
- Antibody Concentration Too High: Excessive primary antibody concentration is a primary cause of off-target binding [3] [4].
- Antibody Type: Polyclonal antibodies, by nature, can be "promiscuous" and bind to similar epitopes on other proteins. Shifting to a monoclonal antibody can significantly improve specificity [3].
- Protein Degradation or Cleavage: PARP-1 is a known target for cleavage by caspases during apoptosis, producing a classic 89 kDa fragment [33] [34]. Other proteases can also generate cleavage products ranging from 42-89 kDa [33]. Incomplete inhibition of these proteases during sample preparation can lead to smearing or lower molecular weight bands.
- Incomplete Blocking or Washing: Insufficient blocking of the membrane or inadequate washing steps can leave "junk" that antibodies bind to non-specifically [3].
What does the appearance of an ~89 kDa band indicate? A band at approximately 89 kDa is a well-characterized cleavage fragment of PARP-1 and is a key biomarker of apoptosis [33]. This fragment is generated when caspases cleave the full-length 116 kDa protein. Its presence often indicates that a portion of your cells are undergoing programmed cell death. Specific antibodies, like the Anti-Cleaved PARP1 [SP276] (ab225715), are designed to detect this fragment [34].
How can I confirm that my primary band is truly PARP-1? The best practice is to include a PARP-1 knockout cell lysate as a control. In a valid experiment, the primary band at ~116 kDa should be present in wild-type cell lysates but absent in the knockout lysate, confirming the antibody's specificity [35] [34]. Many cited antibodies, such as [EPR18461] (ab191217) and 13371-1-AP, are KO-validated for this purpose [33] [35].
Optimization Parameters for PARP-1 Antibody Incubation
The table below summarizes key variable parameters you can optimize during the primary antibody incubation step to enhance specificity and signal.
Table 1: Antibody Incubation Optimization Guide
| Parameter | Typical Starting Range for PARP-1 | Optimization Purpose & Tips |
|---|---|---|
| Dilution Factor | 1:500 to 1:10,000 [33] [35] | Decreasing the antibody concentration is one of the most effective ways to reduce non-specific binding. If you see high background or extra bands, try a higher dilution (e.g., from 1:1000 to 1:2000) [3] [4]. |
| Incubation Time | Overnight (~16 hours) | A longer incubation at a higher dilution can improve the signal-to-noise ratio. Ensure consistency for reproducible results. |
| Incubation Temperature | 4°C [4] | Incubating at 4°C is highly recommended as it decreases non-specific binding of the antibody compared to room temperature [4]. |
| Antibody Diluent | 2-5% BSA or non-fat dry milk in TBST | Preparing the primary antibody in your blocking buffer can help reduce non-specific binding. For difficult blots, try boosting the blocking reagent concentration or adding 0.05% Tween-20 [3]. |
Detailed Troubleshooting Protocols
Protocol 1: Troubleshooting Non-Specific Bands
If your blot shows multiple bands, follow this systematic troubleshooting workflow.
Materials:
- Freshly prepared lysis buffer with protease inhibitors
- Fresh aliquots of primary and secondary antibodies
- Blocking buffer (e.g., 5% BSA or non-fat dry milk)
- Wash buffer (TBST with 0.1% Tween-20)
Step-by-Step Method:
- Titrate the Primary Antibody: Start with the highest dilution recommended on the datasheet. For example, the PARP1 Polyclonal Antibody (13371-1-AP) has a wide range of 1:1000–1:8000 [33]. Test a range of dilutions (e.g., 1:1000, 1:2000, 1:5000) on the same membrane if using a multi-well apparatus.
- Incubate at 4°C: Perform the primary antibody incubation step at 4°C to minimize off-target binding [4].
- Optimize Blocking: Block the membrane for at least 1 hour at room temperature (or longer at 4°C) using a sufficient concentration (e.g., 5%) of your blocking reagent [3].
- Increase Wash Stringency: After primary antibody incubation, wash the membrane 4-5 times for 5 minutes each with gentle agitation, using TBST containing 0.1% Tween-20 [3].
- Validate Results: Always include a positive control (a lysate known to express PARP-1) and, if possible, a negative control (PARP-1 knockout lysate) to confirm the specificity of the observed bands [35].
Protocol 2: Detecting PARP-1 Cleavage (Apoptosis Assay)
This protocol is optimized for detecting the full-length PARP-1 (116 kDa) and its cleavage fragment (89 kDa).
Materials:
- Apoptosis-inducer (e.g., Staurosporine)
- Cell lysis buffer (e.g., RIPA or 1% SDS hot lysis buffer)
- Primary antibody specific for full-length and cleaved PARP-1 (e.g., ab191217 or 13371-1-AP) [33] [35]
- Secondary antibody (HRP-conjugated), diluted 1:50,000 [35]
Step-by-Step Method:
- Induce Apoptosis: Treat cells with an apoptosis-inducing agent (e.g., 1µM Staurosporine for 3-4 hours) [35] [34].
- Prepare Lysates: Lyse cells promptly. Using a hot lysis method (e.g., 1% SDS) can help inactivate proteases and preserve the protein state [35].
- Western Blot: Load 20-30 µg of protein per well and separate by SDS-PAGE.
- Antibody Incubation:
- Detection: Develop the blot using a chemiluminescent substrate. Expect to see the full-length band at 113-116 kDa and the cleavage fragment at 89 kDa.
The Scientist's Toolkit: Key Research Reagents
Table 2: Essential Reagents for PARP-1 Western Blotting
| Reagent | Function & Application | Example from Literature |
|---|---|---|
| PARP1 Monoclonal Antibody [EPR18461] | Highly specific monoclonal antibody for detecting full-length and cleaved PARP1 in WB, IHC-P, and ICC/IF. KO-validated. | Used at 1:1000 dilution in 5% NFDM/TBST for WB in HeLa cell lysates [35]. |
| PARP1 Polyclonal Antibody (13371-1-AP) | A polyclonal option for various applications including WB, IP, and ICC. Recognizes the C-terminal region (full-length and cleavage). | Recommended WB dilution range: 1:1000-1:8000; tested in HeLa, Jurkat, and THP-1 cells [33]. |
| Anti-Cleaved PARP1 [SP276] | Recombinant monoclonal antibody designed to specifically detect the cleaved form of PARP1, a key apoptosis marker. | Used at 1/100 dilution for WB to detect the ~27 kDa cleaved fragment in staurosporine-treated cells [34]. |
| Protease Inhibitor Cocktail | Prevents proteolytic degradation of PARP-1 during sample preparation, reducing smearing and lower MW bands. | Critical for preparing samples to avoid artifacts from protein "nibbling" [3]. |
| Laemmli Buffer | SDS-PAGE sample buffer; boiling samples in it for 5-10 minutes can disrupt protein multimers that cause higher MW bands [3]. | Used to dissociate PARP-1 dimers or trimers that may form [3]. |
FAQs on Minimizing Background and Non-Specific Bands
What are the primary causes of high background in chemiluminescent western blots?
High background is frequently caused by incomplete blocking of the membrane, excessive antibody concentrations, or insufficient washing [4] [9]. Using an incompatible blocking buffer, such as milk with an avidin-biotin system or phosphate-buffered saline (PBS) with alkaline phosphatase (AP)-conjugated antibodies, can also lead to high background signals [9].
How can I reduce non-specific bands when detecting PARP-1?
Non-specific bands in PARP-1 blots can often be resolved by optimizing antibody concentration, as too high a concentration of primary antibody is a common cause of off-target binding [4] [3]. Incubating the primary antibody at 4°C can help decrease non-specific binding [4]. Furthermore, ensure sample integrity by using fresh protease inhibitors to prevent protein degradation that creates lower molecular weight bands [3].
My signal is weak, even though I know my protein is present. What should I check?
Weak or no signal can result from several issues. First, confirm efficient transfer to the membrane by staining the gel post-transfer [9]. Check that your antibody concentrations are sufficient and that the antigen has not been degraded or masked by your blocking buffer [9]. Also, ensure your chemiluminescent substrate is not expired, and consider increasing the membrane's incubation time with substrate or the film exposure time [9].
Why is it important to choose the right blocking buffer, and what are the recommendations?
The blocking buffer covers unused binding sites on the membrane to prevent antibodies from adhering non-specifically. Incompatible blockers can cause high background or mask your target [9]. For general purposes, 5% BSA or an engineered blocking buffer is often effective [4] [3]. When detecting phosphoproteins, avoid milk or casein and use BSA in Tris-buffered saline (TBS) instead [9]. If using an AP-conjugated antibody, ensure your blocker is in TBS, not PBS [9].
Troubleshooting Guide: Background and Non-Specific Signals
The following table outlines common problems and their verified solutions to help you achieve cleaner blots.
| Problem | Possible Cause | Recommended Solution |
|---|---|---|
| High Background | Incomplete blocking [4] | Increase blocking reagent concentration (e.g., 2% to 5% BSA), extend blocking time, or switch to an engineered blocking buffer [4] [3]. |
| Excessive antibody concentration [4] [9] | Titrate both primary and secondary antibodies to find the optimal dilution. | |
| Insufficient washing [3] [9] | Increase wash number and volume; use wash buffer with 0.05-0.1% Tween-20 [3] [9]. | |
| Non-Specific Bands | Low antibody specificity [4] | Use a monoclonal antibody if possible; for polyclonals, try further purification [4] [3]. |
| Too much protein loaded [3] [9] | Reduce protein load per lane (e.g., 10-30 µg for cell lysates). | |
| Protein degradation [3] | Use fresh protease inhibitors during sample preparation. | |
| Protein multimers [3] | Boil samples in Laemmli buffer for 5-10 minutes to disrupt aggregates. | |
| Weak or No Signal | Inefficient transfer [9] | Stain gel post-transfer to assess efficiency; ensure proper membrane activation and stack orientation. |
| Antibody concentration too low [9] | Increase primary and/or secondary antibody concentration. | |
| Incompatible buffer [9] | Avoid sodium azide with HRP-conjugated antibodies; check blocker compatibility. |
Experimental Protocol: Optimizing Detection for PARP-1
This protocol provides a detailed methodology for detecting PARP-1 with minimal background, incorporating specific optimization steps.
Sample Preparation
- Homogenize tissue or cell samples in RIPA buffer (25 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) supplemented with a fresh protease inhibitor cocktail [36] [25].
- Centrifuge the homogenate at 20,000 x g for 20 minutes at 4°C to remove insoluble debris [36].
- Determine protein concentration using a BCA or Bradford assay, ensuring the standard curve has an R-squared value ≥ 0.99 for accuracy [36].
- Prepare samples by adding loading buffer. Heat at 70°C for 10 minutes instead of boiling to avoid proteolysis and aggregation, which is particularly useful for preserving PARP-1 integrity [9]. A standard load for detecting PARP-1 (∼116 kDa) is 15-30 µg of total cell lysate per lane [36] [3].
Electrophoresis and Transfer
- Use a 4-12% Bis-Tris gradient gel for optimal separation across a broad molecular weight range [36].
- For the running buffer, MES is preferred for better resolution of proteins between 3.5-160 kDa [36].
- Transfer proteins to a nitrocellulose or PVDF membrane. For the low molecular weight PARP-1 cleavage fragment (89 kDa), add 20% methanol to the transfer buffer to help bind proteins and prevent them from passing through the membrane [9] [8].
Blocking and Antibody Incubation
- Block the membrane for at least 1 hour at room temperature or overnight at 4°C using 5% BSA in TBST (Tris-Buffered Saline with 0.05% Tween-20) [3] [9]. BSA is recommended over milk for potential phospho-targets and generally lower background.
- Prepare primary antibody dilutions in the blocking buffer. A good starting dilution for anti-PARP-1 is 1:1,000 [37]. For problematic non-specific bands, further dilution or incubation at 4°C is highly recommended [4].
- Wash the membrane 4-5 times for 5 minutes each with ample TBST [3].
- Incubate with HRP-conjugated secondary antibody diluted in blocking buffer. A starting dilution of 1:100,000 is appropriate for high-sensitivity substrates [37]. Perform another series of thorough washes.
Chemiluminescent Detection and Imaging
- Select a substrate based on PARP-1 abundance. For endogenous levels, a high-sensitivity substrate like SuperSignal West Femto (mid-femtogram level) is recommended [37].
- Incubate the membrane with the substrate according to the manufacturer's instructions.
- Capture the signal using a CCD camera-based imager, which offers a larger dynamic range and quantitative analysis compared to film. If using film, multiple exposure times should be tested to avoid saturation [37] [9].
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function & Rationale |
|---|---|
| Engineered Blocking Buffer | Specifically formulated to reduce non-specific antibody binding without masking the target epitope, superior to milk or standard BSA in some cases [4]. |
| Protease Inhibitor Cocktail | Prevents protein degradation during sample preparation, which is a common source of lower molecular weight non-specific bands [3] [25]. |
| SuperSignal West Femto Substrate | Provides maximum sensitivity (low- to mid-femtogram level) for detecting low-abundance proteins or proteins like PARP-1 under sub-optimal conditions [37]. |
| HRP-conjugated Secondary Antibodies | The small size and high turnover rate of HRP make it the enzyme of choice for chemiluminescent detection, offering high sensitivity and a wide range of available substrates [37]. |
| PVDF Membrane | Offers high protein binding capacity and mechanical strength, ideal for reprobing. Must be activated in methanol before use [36] [9]. |
PARP-1 Western Blot Optimization Workflow
The diagram below outlines the key decision points in the optimization process to achieve a clean PARP-1 blot.
Chemiluminescent Substrate Selection Table
Selecting the appropriate substrate is critical for balancing sensitivity and background. The table below compares popular commercial substrates.
| Substrate | Sensitivity | Signal Duration | Recommended Primary Antibody Dilution* | Ideal Use Case |
|---|---|---|---|---|
| Pierce ECL | Low- to mid-picogram | 0.5–2 hours | 1:1,000 | Abundant target protein; cost-sensitive work [37]. |
| SuperSignal West Pico PLUS | Low-picogram to high-femtogram | 6–24 hours | 1:1,000 | Good balance of value and sensitivity for most blots [37]. |
| SuperSignal West Dura | Mid-femtogram | 24 hours | 1:5,000 | Less abundant targets; requires long signal duration [37]. |
| SuperSignal West Femto | Low- to mid-femtogram | 8 hours | 1:5,000 | Maximum sensitivity for very low abundance proteins [37]. |
| SuperSignal West Atto | Low femtogram- high attogram | 6 hours | 1:5,000 | Ultimate sensitivity with minimal optimization needed [37]. |
*Antibody dilutions are a starting point based on a 1 mg/mL antibody concentration and may require further optimization.
Systematic Troubleshooting of Non-Specific PARP-1 Bands and High Background
Troubleshooting Guides and FAQs
Why do non-specific bands appear on my PARP-1 western blot?
Non-specific bands are a common challenge in western blotting, often resulting from antibodies binding to off-target proteins or regions of the membrane itself. The two most prevalent causes are incomplete blocking and excessive antibody concentration [4] [3]. In the context of PARP-1 research, which is crucial in DNA repair and cell fate studies, these artifacts can obscure results and lead to incorrect conclusions about this important protein [5] [18].
How can I troubleshoot incomplete blocking?
Incomplete blocking occurs when the blocking buffer fails to cover all non-specific protein-binding sites on the membrane. This allows antibodies to bind to these sites, creating a high background or non-specific bands [4].
- Optimize Your Blocking Buffer: Standard blockers like milk or BSA may not be sufficient. Consider switching to an engineered, proprietary blocking buffer specifically designed to reduce non-specific binding without masking your target epitope [4].
- Increase Concentration and Time: Boost the concentration of your blocking reagent (e.g., from 2% to 5% BSA) and extend the blocking incubation time [3].
- Add Detergent: Incorporate Tween-20 to a final concentration of 0.05% into your blocking buffer to help minimize background [9] [3].
- Avoid Incompatible Blockers: If using an avidin-biotin detection system, do not use milk as a blocker because it contains biotin, which will cause high background. Similarly, when detecting phosphoproteins, avoid phosphate-based buffers and milk; instead, use BSA in Tris-buffered saline [9].
How can I troubleshoot excessive antibody concentration?
Using too high a concentration of primary or secondary antibody is a frequent mistake. Excess antibody will bind to lower-affinity, off-target sites, producing non-specific bands [4] [38].
- Perform an Antibody Titration: The most effective solution is to optimize the antibody concentration through a titration experiment. Test a range of dilutions to find the one that provides the strongest specific signal with the lowest background [38].
- Incubate at 4°C: Performing the primary antibody incubation step at 4°C can help decrease non-specific binding [4].
- Extend Incubation Time: If you decrease the antibody concentration and the signal becomes weak, you can offset this by increasing the incubation time to ensure sufficient binding to your target [4].
Are there other common causes for non-specific bands?
Yes. While blocking and antibody concentration are primary culprits, other factors can contribute to a messy blot.
- Antibody Specificity: Polyclonal antibodies, by nature, are a mixture that can bind to multiple epitopes and are more prone to non-specificity than monoclonal antibodies [3].
- Protein Overloading: Loading too much protein per lane can cause "ghost bands" or smearing. For cell lysates, aim for 20-30 µg per lane [3] [39].
- Protein Degradation: Protease activity can degrade your target protein, producing lower molecular weight bands. Always use fresh protease inhibitors during sample preparation [3].
- Insufficient Washing: Inadequate washing can leave unbound "junk" that interferes with detection. Ensure thorough washing with a buffer containing 0.05-0.1% Tween-20 with gentle agitation [9] [3].
Table 1: Optimized Reagent Concentrations for PARP-1 Western Blotting
| Reagent / Parameter | Recommended Concentration / Value | Troubleshooting Adjustment |
|---|---|---|
| Blocking Buffer (BSA) | 2-5% (w/v) in TBST [3] | Increase concentration for insufficient blocking. |
| Blocking Time | 1 hour at room temperature [9] | Extend to overnight at 4°C for persistent background [9]. |
| Primary Antibody | Manufacturer's recommended dilution | Decrease concentration for high background/non-specific bands [4] [9]. |
| Primary Antibody Incubation | Variable | Perform at 4°C to reduce non-specific binding [4]. |
| Tween-20 in Wash Buffer | 0.05% (v/v) [9] | Increase to 0.1% for high background [3]. |
| Wash Duration/Frequency | 3-5 washes for 5 minutes each [3] | Increase number or volume of washes for high background [9]. |
| Cell Lysate Load | 20-30 µg per lane [3] | Reduce load if non-specific bands persist. |
Experimental Protocols
Protocol 1: Standard Blocking and Antibody Incubation for Low-Background Blots
This protocol is a baseline for detecting PARP-1, a protein involved in critical nuclear functions like DNA repair and transcription [5].
- Transfer: Following SDS-PAGE, transfer proteins to a PVDF or nitrocellulose membrane using standard methods.
- Blocking: Incubate the membrane in 5% BSA in Tris-Buffered Saline with 0.1% Tween-20 (TBST) for 1 hour at room temperature with gentle agitation. For stubborn background, block overnight at 4°C [9] [3].
- Primary Antibody Incubation:
- Dilute the anti-PARP-1 primary antibody in the same 5% BSA/TBST blocking buffer.
- Incubate the membrane with the antibody solution for 1-2 hours at room temperature or overnight at 4°C with agitation.
- Critical Note: The optimal dilution must be determined by titration. A too-high concentration is a primary cause of non-specific bands [4] [38].
- Washing: Wash the membrane 4-5 times with TBST for 5 minutes per wash with vigorous agitation [3].
- Secondary Antibody Incubation:
- Dilute the HRP- or fluorochrome-conjugated secondary antibody in 5% BSA/TBST.
- Incubate for 1 hour at room temperature with agitation.
- Avoid over-concentration of the secondary antibody [9].
- Washing: Repeat Step 4.
- Detection: Proceed with chemiluminescent or fluorescent detection according to your system's instructions.
Protocol 2: Antibody Titration to Eliminate Non-Specific Bands
This is an essential optimization experiment when establishing a new antibody or troubleshooting.
- Prepare Identical Blots: Run and transfer your PARP-1 samples (e.g., a cell lysate known to express PARP-1) across multiple identical gel lanes. Cut the membrane into individual strips, each containing one PARP-1 lane.
- Block: Block all strips simultaneously using the standard protocol.
- Titrate Primary Antibody: Prepare a series of primary antibody dilutions in blocking buffer. For example, if the recommended dilution is 1:1000, test dilutions of 1:500, 1:1000, 1:2000, and 1:5000.
- Incubate and Wash: Incubate each strip with a different antibody dilution, then wash all strips identically.
- Incubate with Secondary Antibody: Apply the same, optimal dilution of secondary antibody to all strips.
- Wash and Detect: Wash all strips identically and develop them using the same detection conditions and exposure time.
- Analyze: Select the dilution that yields a strong specific PARP-1 band with the cleanest background. The signal-to-noise ratio is your key metric [38].
PARP-1 Regulation and Detection Diagram
PARP-1 Regulation & Detection Issues
This diagram illustrates the simplified regulatory feedback loop of PARP-1 gene expression, based on research showing that PARP-1 activity poly(ADP-ribosyl)ates the transcription factor Sp1, reducing its DNA-binding ability and thereby downregulating PARP-1 promoter activity [5]. The primary culprits of incomplete blocking and excessive antibody concentration are shown as factors that obscure this biology by creating non-specific bands.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Troubleshooting PARP-1 Western Blots
| Item | Function / Application in Troubleshooting |
|---|---|
| BSA (Bovine Serum Albumin) | A common protein used in blocking buffers (1-5% w/v) and antibody diluents to saturate non-specific binding sites on the membrane [40] [9]. |
| Engineered Blocking Buffers | Proprietary buffers designed to provide superior blocking for specific applications (e.g., chemiluminescent or fluorescent detection), often yielding better signal-to-noise ratios than standard milk or BSA [4]. |
| Tween-20 | A non-ionic detergent added (typically at 0.05-0.1%) to wash and blocking buffers to reduce hydrophobic interactions and minimize background [9] [3]. |
| Protease Inhibitor Cocktail | Added to lysis buffer during sample preparation to prevent protein degradation, which can cause aberrant lower molecular weight bands [3]. |
| HRP Conjugated Secondary Antibodies | Enzymatically-conjugated antibodies for chemiluminescent detection. Must be titrated to avoid high background [9]. |
| PARP Inhibitors (e.g., PJ34) | Small molecule inhibitors used in research to study the consequences of inhibiting PARP-1 enzymatic activity, which can affect the transcription of its own gene and other cellular processes [5] [18]. |
| Membrane Stripping Buffer | A buffer used to remove bound antibodies from a blot, allowing it to be re-probed. Use should be minimized as it can damage proteins on the membrane [9]. |
For researchers studying apoptosis, DNA damage repair, and related signaling pathways, PARP-1 serves as a critical biomarker. However, western blot analysis of PARP-1 is frequently complicated by the appearance of non-specific bands that can obscure results and lead to misinterpretation. This technical guide addresses the common challenges associated with PARP-1 antibody specificity and provides detailed, actionable protocols for troubleshooting these issues, with a focus on antibody titration and purification techniques essential for obtaining clean, interpretable data.
Understanding PARP-1 and Its Detection Challenges
PARP-1 (poly(ADP-ribose) polymerase 1) is a nuclear enzyme with a calculated molecular weight of approximately 113 kDa, though it often migrates at 116 kDa on western blots due to post-translational modifications. During apoptosis, caspase cleavage generates a characteristic 89 kDa fragment, which serves as a key indicator of programmed cell death [41] [42]. Common detection challenges include:
- Non-specific bands at unexpected molecular weights
- High background interference
- Multiple bands due to protein degradation, splice variants, or multimers
- Weak or absent target band signal
The diagram below illustrates the PARP-1 cleavage process and the relationship between its full-length and cleaved forms:
Frequently Asked Questions (FAQs)
Q1: What are the expected bands for PARP-1 in western blotting?
A1: A specific PARP-1 antibody should detect the full-length protein at approximately 116 kDa and the caspase-cleaved fragment at 89 kDa [41] [42]. Some antibodies may also detect additional cleavage products depending on the epitope recognized.
Q2: Why do I see multiple non-specific bands in my PARP-1 western blot?
A2: Non-specific bands can arise from several factors, including:
- Antibody concentration too high, causing off-target binding [4] [3]
- Incomplete blocking of the membrane, allowing antibodies to bind non-specifically [4]
- Protein degradation from insufficient protease inhibition during sample preparation [3]
- Protein multimers forming complexes that migrate at higher molecular weights [3]
- Cross-reactivity with related proteins or different PARP isoforms [41]
Q3: How can I confirm that my detected band is specifically PARP-1?
A3: The most reliable method is using a knockout validation approach, where the antibody is tested on PARP-1 knockout cell lines. Specific antibodies will show a band in wild-type cells but no signal in knockout cells [35] [43] [42]. Additionally, induction of apoptosis with agents like staurosporine should generate the characteristic 89 kDa cleavage fragment [35].
Q4: What is the recommended starting dilution for PARP-1 antibodies?
A4: Optimal dilution varies by antibody and application. The table below summarizes recommended dilutions from various manufacturers:
Table: PARP-1 Antibody Dilution Recommendations for Western Blotting
| Antibody Source | Clone/Product # | Recommended Dilution | Host & Clonality |
|---|---|---|---|
| Cell Signaling Technology | #9542 | 1:1000 | Rabbit Polyclonal |
| Abcam | EPR18461 (ab191217) | 1:1000 - 1:10000 | Rabbit Monoclonal |
| Abcam | E102 (ab32138) | 1:1000 | Rabbit Monoclonal |
| Proteintech | 66520-1-Ig | 1:5000 - 1:50000 | Mouse Monoclonal |
| Rockland | 200-401-x51 | 1:1000 | Rabbit Polyclonal |
Troubleshooting Guides
Problem: Non-Specific Bands in PARP-1 Western Blot
Potential Causes and Solutions:
Excessive Antibody Concentration
- Solution: Perform antibody titration to determine optimal concentration
- Protocol: Prepare serial dilutions of primary antibody (e.g., 1:500, 1:1000, 1:2000, 1:5000) and test on identical membrane strips with the same protein sample
Incomplete Blocking
- Solution: Optimize blocking conditions
- Protocol:
- Increase blocking buffer concentration from 2% to 5% BSA or non-fat dry milk
- Extend blocking time from 1 hour to 2 hours at room temperature or overnight at 4°C
- Consider using engineered blocking buffers specifically designed to reduce non-specific binding [4]
Protein Degradation
- Solution: Improve protein extraction and storage conditions
- Protocol:
- Always include fresh protease inhibitors in lysis buffers
- Keep samples on ice during extraction
- Aliquot and store at -80°C to prevent freeze-thaw cycles
- Verify protein integrity by running a fresh gel
Non-Specific Antibody Binding
- Solution: Adjust incubation conditions
- Protocol: Perform primary antibody incubation at 4°C instead of room temperature to decrease non-specific binding [4]
The following workflow illustrates the systematic approach to troubleshooting non-specific bands:
Step-by-Step Protocol: Antibody Titration for PARP-1
Materials Needed
- PARP-1 antibody (primary antibody)
- Appropriate positive control cell lysate (e.g., HeLa, untreated and staurosporine-treated)
- Membrane with transferred proteins
- Blocking buffer (5% BSA or non-fat dry milk in TBST)
- Wash buffer (TBST)
- Secondary antibody conjugated to HRP or fluorescent tag
- Detection reagents
Procedure
Prepare Membrane Strips
- Transfer protein from gel to membrane using standard protocol
- Cut membrane into strips, each containing the same protein samples and molecular weight markers
Blocking
- Incubate all membrane strips in blocking buffer for 2 hours at room temperature with gentle agitation
Primary Antibody Incubation
- Prepare different dilutions of PARP-1 antibody in blocking buffer
- Recommended dilution range: 1:500 to 1:50000, depending on manufacturer's suggestion
- Incubate each strip with a different antibody dilution overnight at 4°C with gentle agitation
Washing
- Wash all strips 4-5 times for 5 minutes each with TBST buffer containing 0.1% Tween-20
Secondary Antibody Incubation
- Incubate with appropriate secondary antibody at recommended dilution for 1 hour at room temperature
Detection
- Develop using preferred detection method (chemiluminescent, fluorescent, etc.)
- Compare results across dilutions to identify the concentration that provides the strongest specific signal with minimal background
Documentation
- Record the optimal dilution for future reference
The Scientist's Toolkit: Research Reagent Solutions
Table: Essential Reagents for PARP-1 Western Blot Optimization
| Reagent | Function | Specific Recommendations |
|---|---|---|
| Blocking Buffers | Reduce non-specific antibody binding | 5% non-fat dry milk/TBST or engineered blocking buffers [4] |
| Protease Inhibitors | Prevent PARP-1 degradation during extraction | Include in lysis buffer; use fresh aliquots |
| PARP-1 Antibodies | Detect target protein | Select KO-validated antibodies [35] [43] |
| Positive Controls | Verify antibody performance | HeLa cells ± staurosporine treatment [35] |
| Staurosporine | Induce apoptosis and PARP cleavage | 1μM for 4 hours to generate 89 kDa fragment [35] |
| Wash Buffers | Remove unbound antibodies | TBST with 0.1% Tween-20 for effective washing [3] |
Advanced Technical Notes
Antibody Purification Techniques
For polyclonal antibodies that continue to show non-specific binding despite optimization, consider additional purification steps:
- Affinity Purification: Isolate specific antibodies using protein A or antigen-affinity columns
- Pre-adsorption: Incubate antibody with cell lysates from knockout models or related tissues lacking the target protein to remove non-specific antibodies
Validation Methods
- Knockout Validation: Test antibody on isogenic wild-type and PARP-1 knockout cell lines to confirm specificity [35] [43]
- Genetic Silencing: Use siRNA or shRNA to knock down PARP-1 expression and verify loss of signal
- Competition Assays: Pre-incubate antibody with immunizing peptide to block specific binding
By implementing these systematic approaches to antibody titration, purification, and validation, researchers can significantly improve the specificity and reliability of their PARP-1 detection, leading to more accurate interpretation of experimental results in apoptosis and DNA damage repair studies.
Frequently Asked Questions (FAQs)
Q1: I see unexpected lower molecular weight bands in my PARP-1 western blot. Could this be due to protein degradation? Yes, protein degradation is a common cause of unexpected lower molecular weight bands. For PARP-1, which has a full-length size of 116 kDa, a prominent cleavage fragment appears at 89 kDa when caspases are activated during apoptosis [44]. However, nonspecific degradation by proteases can produce other random fragments. To prevent this, always add fresh protease inhibitors to your lysis buffer and keep samples on ice [3].
Q2: My blot has a high background and smeary bands. Could sample contaminants be the cause? Absolutely. Contaminants like genomic DNA can cause viscosity in your lysate, leading to smearing, distorted lanes, and poor protein resolution during electrophoresis [9]. High salt or detergent concentrations can also cause band spreading and widening of lanes [9].
Q3: How can I confirm if my PARP-1 sample has been degraded? A key indicator is the presence of a specific 89 kDa band, which is a known caspase cleavage fragment of PARP-1 [44]. Comparing your results with a control sample from cells induced to undergo apoptosis (e.g., with a DNA-damaging agent) can be very informative. Nonspecific degradation may appear as a smear or multiple lower molecular weight bands.
Q4: What are the best practices for preparing a clean protein sample for PARP-1 western blotting? To ensure a high-quality sample, follow these steps:
- Use Fresh Inhibitors: Always supplement your lysis buffer with a fresh cocktail of protease inhibitors immediately before use.
- Handle Gently: Keep samples cold and avoid excessive vortexing to prevent shearing.
- Manage DNA: Shear viscous genomic DNA by briefly sonicating or passing the lysate through a small-gauge needle [9].
- Control Salt/Detergent: Ensure your final sample buffer has a salt concentration below 100 mM and an appropriate SDS-to-nonionic detergent ratio [9].
Troubleshooting Guide: Key Problems and Solutions
The table below summarizes common sample-level issues and how to resolve them for a cleaner PARP-1 blot.
Table 1: Troubleshooting Sample-Level Problems in PARP-1 Western Blotting
| Problem | Possible Cause | Recommended Solutions |
|---|---|---|
| Lower MW bands (~89 kDa) | Caspase-mediated cleavage during apoptosis [44] | Intended biological signal; use as apoptosis marker. |
| Lower MW bands (smear or multiple bands) | Nonspecific protein degradation by proteases | Prepare fresh lysate with fresh protease inhibitors; keep samples on ice [3]. |
| High background, smeared bands | Genomic DNA contamination | Shear DNA by brief sonication or needle pass [9]. |
| Streaking, distorted lanes | High salt concentration in sample | Dialyze sample or dilute to <100 mM salt before loading [9]. |
| Widened lanes, streaking | High detergent concentration (e.g., Triton X-100) | Keep SDS-to-nonionic detergent ratio at 10:1 or greater; use detergent removal columns [9]. |
Experimental Protocols for Sample Integrity
Protocol 1: Preparing Cell Lysates to Minimize PARP-1 Degradation
This protocol is designed to preserve the integrity of PARP-1 and other nuclear proteins.
- Lysis Buffer Preparation: Prepare a fresh RIPA or NP-40 based lysis buffer. Immediately before use, add a commercial protease inhibitor cocktail. For PARP-1, which is involved in DNA repair, consider also adding PARP inhibitors if you wish to study its basal state.
- Cell Harvesting and Lysis:
- Wash cells with cold PBS.
- Lyse cells directly in the culture dish by adding cold lysis buffer (e.g., 500 µL for a 10 cm dish).
- Scrape cells and transfer the lysate to a cold microcentrifuge tube.
- DNA Shearing: If the lysate is viscous, sonicate on ice using a low-power setting (e.g., 3 pulses of 5-10 seconds each) or pass the lysate 5-10 times through a 25-gauge needle.
- Clarification: Centrifuge the lysate at >12,000 x g for 15 minutes at 4°C to pellet insoluble debris, including genomic DNA.
- Protein Quantification and Storage: Transfer the supernatant to a new tube. Determine protein concentration, prepare aliquots with Laemmli buffer, and store at -80°C. Avoid repeated freeze-thaw cycles.
Protocol 2: Verifying Sample Quality Pre-Electrophoresis
- Visual Check for Viscosity: After clarification, the lysate should pipette easily. A viscous, "stringy" consistency indicates significant residual DNA contamination.
- Salt/Detergent Concentration Check: Use a conductivity meter to check salt levels if you suspect issues. Alternatively, dialyze or desalt the sample if background problems persist.
- Load a Test Gel: Before running your main western blot, load a small amount of your sample (2-5 µg) on an SDS-PAGE gel and stain with Coomassie Blue. A clean sample will show sharp, well-defined bands without a heavy smear at the dye front or top of the gel.
Signaling Pathways and Workflows
The following diagram illustrates the relationship between sample preparation problems and their observable effects on a western blot, specifically for PARP-1.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Addressing PARP-1 Sample Preparation Issues
| Item | Function | Specific Example/Consideration |
|---|---|---|
| Protease Inhibitor Cocktail | Inhibits a broad spectrum of proteases to prevent nonspecific protein degradation. | Use EDTA-free cocktails if studying metal-dependent proteins. Always add fresh. |
| PARP-1 Antibody | Specifically detects full-length (116 kDa) and cleaved (89 kDa) PARP-1. | Antibody #9542 from Cell Signaling Technology is validated to detect both forms [44]. |
| Sonicator | Shears genomic DNA to reduce sample viscosity and prevent smearing. | Use a microtip sonicator with low-power, short bursts to avoid heating and foaming. |
| Dialysis Kit / Desalting Columns | Reduces high salt and detergent concentrations from samples. | Thermo Scientific Slide-A-Lyzer MINI Dialysis Devices or Pierce Protein Concentrators [9]. |
| BCA or Bradford Assay Kit | Accurately quantifies protein concentration to ensure optimal loading. | Avoid overloading; for cell lysates, aim for 20-30 µg per lane [9]. |
Step-by-Step Troubleshooting Flowchart for Common PARP-1 Blotting Issues
Frequently Asked Questions (FAQs)
Q1: Why do I see multiple bands or high molecular weight smears in my PARP-1 western blot? Multiple bands or smears are frequently caused by the post-translational modification of PARP-1, most notably poly(ADP-ribosyl)ation (PARylation), which significantly increases its apparent molecular weight [45] [11]. This extensive modification can create a characteristic "ladder" or smear pattern above the expected 116 kDa band. Non-specific antibody binding is another common culprit [4].
Q2: My PARP-1 blot has a high background. What is the likely cause? High background is often a result of incomplete blocking of the membrane or using a primary or secondary antibody concentration that is too high [4]. This allows antibodies to bind non-specifically to the membrane or to off-target proteins.
Q3: What are the key experimental controls for a valid PARP-1 western blot? To confidently interpret your results, include these essential controls:
- Positive Control: A whole cell lysate from cells treated with a DNA-damaging agent (e.g., hydrogen peroxide or MNNG) to induce PARP-1 activation and cleavage.
- Negative Control: A lysate from PARP-1 knockout cells [45] [5] or cells treated with a PARP inhibitor (e.g., PJ34 [5] or Olaparib [25]) to confirm antibody specificity.
- Loading Control: An antibody for a constitutively expressed protein like tubulin or actin to ensure equal loading [45].
Troubleshooting Guide: PARP-1 Specific Issues and Solutions
The following table outlines common issues, their potential causes, and targeted solutions to help you obtain clean and interpretable PARP-1 blots.
| Problem | Possible Cause | Recommended Solution |
|---|---|---|
| High molecular weight smears/ladders | PARP-1 auto-PARylation [45] [11] | Treat cells with a PARP inhibitor (e.g., 300 μM DHIQ [45]) prior to lysis to prevent modification. |
| Multiple non-specific bands | Low antibody specificity or over-concentration [4] | Titrate the primary antibody to find the optimal dilution; incubate at 4°C to reduce non-specific binding [4]. |
| High background | Incomplete blocking or high antibody concentration [4] | Use an engineered blocking buffer; ensure adequate blocking time; dilute primary and/or secondary antibodies. |
| Missing or weak signal | Over-activation of PARP-1 leading to NAD+ depletion and necrotic cell death [45] | Optimize treatment conditions with DNA-damaging agents; use early time points to avoid extensive PARP-1 activation and cell death. |
| Unexpected band at ~89 kDa | Caspase-mediated cleavage of PARP-1 during apoptosis | Include controls for apoptosis (e.g., serum starvation) and use caspase inhibitors to confirm. |
Detailed Experimental Protocols
Protocol 1: Preventing PARylation-Dependent Smearing
Objective: To eliminate high molecular weight smears caused by PARP-1 auto-modification.
Methodology:
- Cell Culture & Pre-treatment: Culture cells (e.g., Mouse Embryonic Fibroblasts) as described [45]. Pre-treat cells with a PARP inhibitor, such as 300 μM 1,5-isoquinolinediol (DHIQ), for 30 minutes prior to any DNA-damaging treatment [45].
- Cell Lysis: Lyse cells in radioimmune precipitation (RIPA) buffer (1% Sodium deoxycholate, 0.1% SDS, 1% Triton X-100, 10 mM Tris pH 8.0, 0.14 M NaCl) supplemented with protease inhibitor cocktail [45].
- Protein Quantification & Immunoblotting: Determine protein concentration using a BCA assay. Load 10-20 μg of protein per well on a 4-12% Bis-Tris gradient gel. Perform Western blotting using standard procedures with anti-PARP-1 and anti-PAR antibodies to confirm inhibition of PARylation [45].
Protocol 2: Optimizing Antibody Specificity
Objective: To reduce non-specific bands and high background.
Methodology:
- Antibody Titration: Prepare a series of primary anti-PARP-1 antibody dilutions (e.g., 1:500, 1:1000, 1:2000, 1:5000) in the recommended blocking buffer.
- Incubation Conditions: Incubate the membrane with the primary antibody at 4°C overnight. Lower temperatures decrease non-specific binding [4].
- Enhanced Blocking: If non-specific bands persist, switch from general blocking buffers like milk or BSA to an engineered blocking buffer specifically designed to reduce non-specific interactions [4].
- Validation: Always validate your antibody signal using a PARP-1 knockout cell line as a negative control [5].
PARP-1 Blotting Troubleshooting Workflow
The following diagram outlines a logical, step-by-step decision-making process to diagnose and resolve the most common PARP-1 western blot issues.
PARP-1 Biology and Antibody Interference
This diagram illustrates the key biological processes involving PARP-1 that can lead to common blotting artifacts, such as smearing and band shifts.
The Scientist's Toolkit: Research Reagent Solutions
The table below lists key reagents and their specific functions in PARP-1 research and troubleshooting.
| Reagent | Function/Application in PARP-1 Research |
|---|---|
| PARP Inhibitors (e.g., DHIQ, PJ34, Olaparib) | Catalytically inhibits PARP-1; used to prevent auto-PARylation in blots and to study PARP-1 function [45] [5] [25]. |
| DNA-damaging Agents (e.g., MNNG, H₂O₂) | Induces DNA strand breaks, leading to PARP-1 activation and cleavage; used as a positive control in experiments [45] [5]. |
| Anti-PAR Antibody | Detects poly(ADP-ribose) chains; essential for confirming PARP-1 activation and the efficacy of PARP inhibitors [45] [25]. |
| Engineered Blocking Buffer | Superior to milk or BSA for blocking membranes; reduces non-specific antibody binding and high background [4]. |
| PARP-1 Knockout Cell Lines | Critical negative control for verifying the specificity of anti-PARP-1 antibodies and for functional studies [45] [5]. |
Confirming Specificity: Gold-Standard Validation Techniques for PARP-1 Detection
In PARP-1 western blot research, the appearance of non-specific bands is a frequent challenge that can lead to misinterpretation of experimental results. These extraneous bands often arise from antibody cross-reactivity with unrelated proteins, degraded protein fragments, or splice variants sharing similar epitopes. For a protein like PARP-1, which has a known cleavage product of approximately 85 kDa during apoptosis, distinguishing specific signals from non-specific binding is crucial for accurate data interpretation. Genetic controls, particularly knockout (KO) and knockdown (KD) strategies, provide the most definitive method for verifying antibody specificity and ensuring that your observed bands truly represent your target protein.
FAQ: Understanding Genetic Controls for Antibody Validation
What are knockout (KO) and knockdown (KD) validation methods?
Knockout (KO) and knockdown (KD) validation are genetic approaches that confirm antibody specificity by comparing samples with and without the target protein expression. KO validation uses CRISPR-Cas9 or other gene-editing technologies to permanently disrupt the gene encoding the target protein, creating a cell line that completely lacks the protein of interest. KD validation utilizes RNA interference (RNAi) technology, such as siRNA or shRNA, to temporarily reduce target protein expression by degrading its mRNA [46] [47].
Why are genetic controls considered the gold standard for antibody validation?
Genetic controls are considered the gold standard because they provide the most direct evidence of antibody specificity. The International Working Group for Antibody Validation has identified genetic approaches as the first of five pillars for antibody validation due to the unambiguous readout they provide [47]. When an antibody shows a strong signal in wild-type cells that disappears or significantly diminishes in KO or KD cells, this provides conclusive evidence that the antibody is specifically recognizing the intended target protein and not cross-reacting with other proteins.
How do genetic controls help troubleshoot non-specific bands in PARP-1 western blots?
For PARP-1 research, genetic controls can definitively identify which bands are specific and which are non-specific. PARP-1 has a full-length molecular weight of approximately 113-116 kDa and a cleaved form of around 85 kDa during apoptosis. When using a KO control for PARP-1, any bands that remain present in the KO sample are confirmed to be non-specific and should not be interpreted as related to PARP-1 [14] [47]. This is particularly valuable for identifying degradation products, splice variants, or cross-reactive proteins that might be mistaken for specific signals.
Experimental Protocols: Implementing Genetic Controls
CRISPR-Cas9 Knockout Validation Protocol
Workflow Overview:
Detailed Methodology:
gRNA Design and Complex Formation: Design a single-guide RNA (sgRNA) targeting an early exon of the PARP1 gene to ensure disruption of the functional protein. Complex the sgRNA with Cas9 enzyme to form ribonucleoproteins (RNPs) [46].
Cell Transfection: Introduce the RNP complex into an appropriate cell line (e.g., HeLa, HEK293) using electroporation or lipid-based transfection methods.
Selection and Cloning: After transfection, use antibiotic selection or single-cell sorting to isolate clonal populations. Allow cells to expand for 2-3 weeks [46].
Knockout Validation: Confirm successful PARP1 knockout through:
- DNA sequencing of the targeted genomic region
- Functional assays demonstrating lack of PARP-1 activity
- Western blotting with a validated PARP-1 antibody
Western Blot Analysis: Prepare protein lysates from wild-type (WT) and PARP1 knockout (KO) cells. Load equal protein amounts (typically 20-30 μg) alongside a molecular weight marker. Perform western blotting using your PARP-1 antibody under standard conditions [46] [47].
Expected Results: A specific PARP-1 antibody will show bands at the expected molecular weights (full-length ~116 kDa, cleaved ~85 kDa) in WT cells that are completely absent in KO cells. Any bands remaining in the KO lane represent non-specific binding [47].
RNA Interference Knockdown Validation Protocol
Workflow Overview:
Detailed Methodology:
siRNA Design: Select validated siRNA sequences targeting PARP1 mRNA. Include appropriate controls: non-targeting scrambled siRNA and untreated cells [46].
Cell Transfection: Plate cells at 60-70% confluence and transfect with PARP1-specific siRNA using lipid-based transfection reagents optimized for your cell type.
Incubation: Allow 48-72 hours for knockdown efficiency, as PARP-1 protein turnover must be considered.
Protein Extraction and Western Blot: Prepare lysates from siRNA-treated and control cells. Measure protein concentrations precisely using BCA or Bradford assay. Load equal amounts (20-30 μg) for western blot analysis [46].
Normalization: Use total protein normalization or housekeeping proteins as loading controls, though note that housekeeping proteins may vary under experimental conditions [48].
Expected Results: Successful knockdown typically shows 70-90% reduction in PARP-1 signal compared to controls. The pattern of disappearing bands indicates specificity, while unchanged bands represent non-specific interactions [46].
Troubleshooting Guide: Addressing Common Challenges
How should I interpret different western blot outcomes with genetic controls?
What if my PARP-1 antibody shows a band in the knockout control?
If you observe bands in the knockout sample, this indicates non-specific antibody binding. Consider these troubleshooting steps:
Optimize Antibody Conditions: Titrate your antibody to find the optimal concentration. Too high antibody concentration often causes non-specific binding [4] [9] [3]. For PARP-1 antibodies, try testing a range of dilutions (e.g., 1:500 to 1:5000).
Enhance Blocking and Washing: Increase blocking time to at least 1 hour at room temperature or overnight at 4°C. Use engineered blocking buffers specifically designed to reduce non-specific binding rather than traditional milk or BSA [4]. Increase wash stringency by adding 0.1% Tween-20 and performing 4-5 washes for 5 minutes each with agitation [3].
Verify Knockout Efficiency: Confirm that your KO cells truly lack PARP-1 protein by using multiple validation methods, including sequencing, functional assays, and alternative antibodies known to be specific.
Consider Alternative Antibodies: If optimization fails, the antibody may have inherent cross-reactivity issues. Source antibodies that have been genetically validated for specificity [14] [47].
Why might there be only partial reduction of signal in my knockdown experiment?
Partial reduction of signal in KD experiments can occur due to:
Inefficient Knockdown: Optimize transfection conditions and try different siRNA sequences. Use a pool of multiple siRNAs targeting different regions of PARP1 mRNA.
Protein Stability: PARP-1 may have a long half-life. Extend the time between transfection and harvest to 72-96 hours.
Antibody Cross-reactivity: The antibody may be recognizing both PARP-1 and homologous proteins (e.g., PARP-2). In this case, the specific band should reduce while non-specific bands remain constant [47].
Research Reagent Solutions for Genetic Validation
Table: Essential Reagents for Genetic Control Experiments
| Reagent/Category | Specific Examples | Function & Application Notes |
|---|---|---|
| CRISPR-Cas9 System | sgRNA targeting PARP1, Cas9 enzyme | Creates permanent genetic knockout by disrupting PARP1 gene; use validated sgRNAs for efficiency |
| RNAi Reagents | PARP1-specific siRNA, shRNA vectors | Achieves temporary knockdown of PARP1; use pooled siRNAs for improved efficiency |
| Validation Antibodies | Anti-PARP1 antibodies with KO validation [14] | Confirm specificity; ensure antibodies show loss of signal in KO models |
| Cell Lines | Wild-type and PARP1 KO versions (e.g., HeLa, HEK293) | Provide positive and negative controls; essential for side-by-side comparison |
| Blocking Buffers | Azure Chemi Blot Blocking Buffer, SuperBlock T20 Buffer [4] [9] | Reduce non-specific binding; protein-free options available for cross-reactive antibodies |
| Detection System | Fluorescent secondary antibodies, HRP-conjugated antibodies | Enable signal detection; fluorescent systems offer better quantitation [36] |
| Loading Controls | No-Stain Protein Labeling Reagent, housekeeping proteins [48] | Normalize protein loading; total protein normalization preferred over housekeeping proteins |
Practical Application: Publication Guidelines
Leading journals now emphasize proper antibody validation in publications. When submitting western blot data:
- Provide Unprocessed Images: Nature Portfolio journals require submission of unprocessed original images of western blots as supplementary information [49].
- Document Genetic Controls: Clearly describe the KO or KD validation methods in your Methods section, including cell lines used and validation data.
- Avoid Excessive Processing: Do not use editing tools that obscure manipulations. Adjustments should be applied equally to the entire image [49].
- Use Proper Normalization: Implement total protein normalization rather than relying solely on housekeeping proteins, as the latter may vary under experimental conditions [48].
Implementing genetic controls through knockout and knockdown methods represents the most reliable approach for validating antibody specificity in PARP-1 western blot research. By incorporating these strategies into your experimental workflow, you can confidently distinguish specific PARP-1 signals from non-specific bands, leading to more accurate data interpretation and reproducible results. As the field moves toward higher standards of antibody validation, genetic controls will continue to play an essential role in ensuring scientific rigor, particularly in critical applications such as drug development where PARP-1 is a therapeutic target.
Frequently Asked Questions (FAQs) for PARP-1 Western Blot Troubleshooting
FAQ 1: I see multiple bands in my PARP-1 western blot. What are the most common causes?
Multiple bands are a frequent issue in PARP-1 immunoblotting. The causes can be biological or technical. Key culprits and their solutions are summarized below.
Table 1: Troubleshooting Non-Specific Bands in PARP-1 Western Blots
| Issue Category | Specific Problem | Proposed Solution |
|---|---|---|
| Biological Factors | Caspase-mediated cleavage producing the 89 kDa fragment [50] | Confirm identity using an antibody specific to the cleavage site; compare with apoptotic samples. |
| Protein degradation from protease activity [3] | Prepare fresh sample lysates with sufficient, fresh protease inhibitors. | |
| Presence of protein multimers (dimers, trimers) [3] | Boil samples in Laemmli buffer for 5-10 minutes pre-loading to disrupt aggregates. | |
| Technical Factors | Primary antibody concentration is too high [3] [4] | Titrate the antibody to find the optimal dilution (e.g., test 1:500 to 1:2000). |
| Incomplete blocking of the membrane [3] [4] | Increase blocking reagent concentration (e.g., to 5% BSA), extend blocking time, or use an engineered blocking buffer. | |
| Low antibody specificity (common with polyclonals) [3] [51] | Use a monoclonal or recombinant antibody validated for western blotting; pre-clear antibody. |
FAQ 2: My antibody datasheet shows a single clean band, but I get multiple non-specific bands in my experiment. How can I validate its specificity?
An antibody's performance is highly dependent on your specific assay context and sample type [51]. A vendor-validated antibody is a good start, but user validation is critical.
- Genetic Knockout (KO) Validation: The gold standard is to run your experiment alongside a lysate from cells where the PARP-1 gene has been knocked out. The disappearance of all bands in the KO lane confirms the antibody's specificity [51].
- Orthogonal Method Correlation:
- Immunocytochemistry/Immunofluorescence (ICC/IF): If your western blot shows bands at 116 kDa and 89 kDa, a corresponding ICC/IF experiment should show strong nuclear signal. Correlation with a second method strengthens your conclusion [51].
- Mass Spectrometry (MS): For definitive identification, excise the non-specific band from the gel and analyze it by MS. This directly identifies the proteins in the band, confirming whether they are PARP-1 isoforms, cleavage products, or entirely different off-target proteins [52].
FAQ 3: I have confirmed that my extra band is non-specific. What are the best procedural steps to eliminate it?
After confirming the band is non-specific, follow this optimization workflow.
FAQ 4: What are the key reagent solutions for successful PARP-1 detection?
Using well-validated reagents is the first line of defense against irreproducible results.
Table 2: Research Reagent Solutions for PARP-1 Detection
| Reagent | Function & Importance | Recommendation |
|---|---|---|
| Validated Primary Antibody | Binds specifically to PARP-1. Select antibodies validated for WB and, if possible, KO-confirmed. | PARP Antibody #9542 (CST) detects full-length (116 kDa) and cleaved (89 kDa) PARP1 [50]. |
| Recombinant Antibodies | Offers superior lot-to-lot consistency, reducing irreproducibility associated with polyclonal antibodies [51]. | Consider recombinant antibodies for long-term projects. |
| Engineered Blocking Buffer | Blocks nonspecific binding sites on the membrane. Protein-free options can reduce background [4]. | Use commercial blocking buffers designed for chemiluminescent or fluorescent western blotting. |
| Protease Inhibitors | Prevents proteolytic degradation of PARP-1 during lysate preparation, preventing lower MW bands [3]. | Always use fresh, broad-spectrum protease inhibitor cocktails in lysis buffers. |
| Positive Control Lysate | Verifies that your entire immunoblotting protocol is working correctly [51]. | Use a cell lysate known to express PARP-1 (e.g., HeLa, HEK293). |
Detailed Experimental Protocols
Protocol 1: Antibody Validation Using Genetic Controls
This protocol outlines the definitive method for confirming antibody specificity.
Sample Preparation:
Gel Electrophoresis and Western Blotting:
Immunodetection:
Interpretation: Specific bands are those present in the WT lane but absent in the KO lane. Any band remaining in the KO lane is non-specific.
Protocol 2: Correlative Analysis by ICC/IF and Mass Spectrometry
This protocol uses orthogonal methods to identify non-specific bands.
Part A: Immunocytochemistry/Immunofluorescence (ICC/IF)
Cell Culture and Fixation:
- Plate the same cells used for western blotting on glass coverslips.
- At ~80% confluency, fix cells with 4% formaldehyde for 15 minutes.
- Permeabilize with 0.1% Triton X-100 for 10 minutes.
Staining:
Imaging and Analysis: Image using a fluorescence microscope. PARP-1 typically shows strong nuclear localization. A clean, specific antibody should produce a clear nuclear pattern without cytoplasmic speckling or other diffuse staining.
Part B: Protein Identification by Mass Spectrometry
Band Excision:
- Run a preparative western blot with your sample. Do not transfer. Stain the gel with Coomassie Blue.
- Carefully excise the gel pieces corresponding to the non-specific band and a control band (e.g., the true PARP-1 band).
In-Gel Digestion:
- Destain the gel pieces, reduce with dithiothreitol, and alkylate with iodoacetamide.
- Digest the proteins within the gel with trypsin overnight at 37°C [52].
LC-MS/MS Analysis and Data Processing:
- Extract peptides and analyze by Liquid Chromatography-tandem Mass Spectrometry (LC-MS/MS) [52].
- Search the resulting mass spectra against a protein sequence database (e.g., Swiss-Prot) using search engines like Mascot or Sequest.
- The output will list all proteins identified in the excised band, providing definitive evidence of its composition [52].
FAQ: Understanding Your PARP-1 Western Blot
Why do I see multiple bands for PARP-1 in my western blot? Multiple bands can be expected due to PARP-1's biological characteristics. The full-length PARP-1 protein is approximately 116 kDa. A common lower band around 89 kDa represents a well-documented cleavage fragment generated by caspases during apoptosis [45]. Additional bands can also arise from splice variants, post-translational modifications (especially poly(ADP-ribosyl)ation), or protein degradation [3] [54].
How can I confirm that a lower band is the apoptotic cleavage fragment? Inducing apoptosis in a controlled experiment (e.g., using a DNA-damaging agent) and comparing the band pattern to untreated cells can serve as a confirmation. In apoptotic cells, the intensity of the 89 kDa fragment should increase while the full-length 116 kDa band may decrease [45]. The use of caspase inhibitors should prevent the appearance of this fragment.
Why might band patterns differ between cell lines? Different cell lines can have varying basal levels of apoptosis, different expression profiles of PARP-1 splice variants, or distinct cellular environments that influence post-translational modifications like poly(ADP-ribosyl)ation [3] [45]. Furthermore, high-passage cell lines can accumulate changes in their expression profiles, potentially altering the observed band pattern [3].
Systematic Troubleshooting Guide for Multi-band Patterns
When your western blot shows unexpected bands, follow this diagnostic table to identify potential causes and solutions.
| Potential Cause | Indicators | Recommended Solutions |
|---|---|---|
| Apoptotic Cleavage [45] | Band at ~89 kDa; increases with DNA damage or other apoptotic stimuli. | Use a positive control for apoptosis; include caspase inhibitors in experiments. |
| Protein Degradation [3] [54] | Smearing or multiple lower molecular weight bands in a non-specific pattern. | Always prepare samples on ice; use fresh, comprehensive protease inhibitor cocktails in lysis buffer. |
| Protein Multimers [3] [54] | Bands at roughly double (∼232 kDa) or triple the expected molecular weight. | Add fresh DTT or β-mercaptoethanol to sample buffer; boil samples for 5-10 minutes in Laemmli buffer. |
| Incomplete Denaturation [55] | Bands at unexpected high molecular weights. | Ensure sample buffer contains fresh reducing agent; use a chaotropic agent like urea if necessary. |
| Non-Specific Antibody Binding [3] [4] [56] | Bands at unpredictable molecular weights not consistent with known isoforms or fragments. | Titrate the primary antibody to find the optimal concentration; incubate at 4°C to increase specificity. |
| Post-Translational Modifications [54] | Band smearing or shifts in apparent molecular weight. | Check literature for known PARP-1 modifications; use enzymatic treatments (e.g., glycosidases). |
| Alternative Splicing [3] [54] | Consistent additional bands at specific molecular weights across experiments. | Review literature for known PARP-1 splice variants; confirm molecular weights match predicted sizes. |
| Overloaded Gel [3] [55] | High background, "ghost bands," or distorted band shapes. | Reduce the amount of total protein loaded (aim for 20-30 µg for cell lysates). |
Experimental Protocols for Verification
Protocol 1: Verifying Apoptotic Cleavage
This protocol is adapted from methods used to study PARP-1 cleavage during cell death [45].
- Cell Treatment: Treat cells with a DNA-alkylating agent such as MNNG (e.g., 0.5 mM for 15 minutes) or another apoptosis inducer. Include a control group of untreated cells.
- Sample Preparation:
- Lyse cells in RIPA buffer (1% sodium deoxycholate, 0.1% SDS, 1% Triton X-100, 10 mM Tris pH 8.0, 0.14 M NaCl) supplemented with a protease inhibitor cocktail.
- Determine protein concentration using a BCA assay.
- Prepare samples with Laemmli buffer containing fresh DTT or β-mercaptoethanol and boil for 5-10 minutes.
- Gel Electrophoresis: Load 10-20 µg of protein per well onto a 4-12% Bis-Tris gradient gel for optimal resolution of the 116 kDa and 89 kDa bands.
- Western Blotting: Proceed with standard western blotting procedures and probe with your anti-PARP-1 antibody.
Expected Outcome: The untreated control should show a dominant band at 116 kDa. The apoptosis-induced samples should show a marked increase in the 89 kDa cleavage fragment.
Protocol 2: Optimizing Antibody Specificity
This protocol helps determine if non-specific bands are due to antibody cross-reactivity.
- Checkerboard Titration:
- Prepare a dilution series of your primary PARP-1 antibody (e.g., 1:500, 1:1000, 1:2000, 1:5000) using the blocking buffer (e.g., 5% BSA or an engineered blocking buffer).
- Similarly, prepare a dilution series of your secondary antibody.
- Dot Blot Setup:
- Spot small, equal volumes of your cell lysate directly onto a nitrocellulose membrane and allow to dry.
- Incubate each spot with a different combination of primary and secondary antibody dilutions.
- Analysis: After detection, identify the combination that provides the strongest specific signal for the true PARP-1 band(s) with the lowest background and the fewest non-specific bands. Use this optimized concentration for future western blots [54] [4].
PARP-1 Biology and Band Interpretation
PARP-1 is a nuclear enzyme activated by DNA damage. Its primary function is to catalyze the transfer of ADP-ribose units from NAD+ to itself and other acceptor proteins, a process known as poly(ADP-ribosyl)ation. This modification can significantly increase the protein's apparent molecular weight, leading to smearing or shifts on a blot [45]. Furthermore, during apoptosis, caspases (primarily caspase-3) cleave PARP-1 between Asp214 and Gly215, separating the N-terminal DNA-binding domain (∼24 kDa) from the C-terminal catalytic domain (∼89 kDa). The 89 kDa fragment is the one typically detected in western blots when using antibodies raised against the C-terminal region [45].
The following diagram illustrates the relationship between cellular events and the bands observed on a PARP-1 western blot.
The Scientist's Toolkit: Research Reagent Solutions
The following table lists key reagents and their roles in achieving clean and interpretable PARP-1 western blots.
| Reagent | Function in PARP-1 Research | Key Considerations |
|---|---|---|
| Protease Inhibitors [57] | Prevents non-specific proteolytic degradation that creates artifactual low-MW bands. | Use a broad-spectrum cocktail including PMSF (for serine proteases) and inhibitors for caspases if studying apoptosis. |
| Phosphatase Inhibitors [57] | Prevents dephosphorylation, which can sometimes affect antibody binding or protein migration. | Include sodium orthovanadate (tyrosine phosphatases) and sodium fluoride (serine/threonine phosphatases). |
| Reducing Agents (DTT/BME) [54] [55] | Disrupts disulfide bonds, preventing multimer formation that appears as high-MW bands. | Must be added fresh to the sample buffer prior to boiling. |
| PARP-1 Specific Antibodies [58] | Detects PARP-1 protein. Monoclonal antibodies generally offer higher specificity. | Select antibodies validated for WB. Check if the epitope is within the 89kDa fragment to ensure cleavage detection. |
| Chemiluminescent Substrate | Enables detection of the HRP-conjugated secondary antibody. | Overly sensitive substrates can exaggerate background and non-specific bands. |
| Enhanced Blocking Buffer [4] [56] | Blocks non-specific binding sites on the membrane to reduce background and non-specific bands. | For phospho-specific antibodies, use BSA instead of milk. Engineered buffers can offer superior blocking. |
Comparative Analysis of Commercial PARP-1 Antibodies and Lot-to-Lot Variation
Poly(ADP-ribose) polymerase 1 (PARP-1) is a 116 kDa nuclear enzyme critical for DNA damage signaling, chromatin remodeling, and epigenetic regulation. When analyzing PARP-1 via Western blot, researchers frequently encounter multiple bands, which can represent specific biological phenomena or indicate antibody-related issues. The full-length 116 kDa PARP-1 protein can be cleaved by caspases during apoptosis to generate an 89 kDa fragment, and sometimes additional lower molecular weight bands may appear due to degradation or other post-translational modifications [1]. These multiple bands present a significant interpretation challenge that requires careful experimental validation to distinguish true biological signals from antibody-related artifacts, particularly given the issues of lot-to-lot variance (LTLV) that plague immunoassays [59].
Understanding Lot-to-Lot Variance (LTLV) in Immunoassays
Causes and Impact of LTLV
Lot-to-lot variance (LTLV), also known as batch variability, negatively affects assay accuracy, precision, and specificity, leading to considerable uncertainty in reported results [59]. In the context of PARP-1 research, this variability can significantly impact data interpretation and experimental reproducibility.
Primary causes of LTLV in PARP-1 antibodies include:
- Quality fluctuation in raw materials: Antibodies are biological reagents that are difficult to regulate consistently [59]
- Manufacturing process deviations: Changes in production can affect final antibody performance [59]
- Antibody aggregation: High-concentration antibodies, particularly IgG3, tend to form aggregates that can lead to high background and signal leap [59]
- Variations in labeling efficiency: Purity issues can negatively impact assay specificity, signal, and background [59]
Case Study: Recombinant vs. Hybridoma Antibodies
Recent experiments demonstrate that even antibodies with identical amino acid sequences can show significant performance differences when produced using different methods. When a monoclonal antibody was exchanged from hybridoma to recombinant production, researchers observed substantially lower sensitivity and maximal signals despite adequate SEC-HPLC purity (98.7%). Further analysis by CE-SDS revealed nearly 13% impurity in the recombinant antibody, causing reduced assay performance [59].
Table 1: Impact of Antibody Production Method on Assay Performance
| Parameter | Hybridoma Antibody | Recombinant Antibody | Percent Deviation |
|---|---|---|---|
| Max signals (RLU) | 493,180 | 412,901 | -19.4% |
| Background (RLU) | Information not provided | Information not provided | Information not provided |
Troubleshooting PARP-1 Western Blot Issues: FAQ
FAQ 1: Why does my PARP-1 Western blot show multiple bands?
Multiple bands on a PARP-1 Western blot may indicate either specific biological detection or antibody non-specificity. PARP-1 is known to undergo caspase cleavage during apoptosis, generating an 89 kDa fragment from the full-length 116 kDa protein [1]. Additional bands may represent:
- Specific cleavage products: The 89 kDa fragment is a validated cleavage product [1]
- Protein degradation: Bands below 116 kDa and 89 kDa may represent proteolytic fragments [1]
- Post-translational modifications: PARP-1 undergoes extensive modification including ADP-ribosylation [11]
- Alternative splicing isoforms: Multiple protein isoforms may derive from a single gene [1]
- Non-specific binding: Antibody may detect unrelated proteins with similar epitopes
Validation experiment: To confirm antibody specificity, perform PARP-1 knockdown using siRNA or shRNA. As shown in the figure below, successful knockdown should diminish all bands, confirming specificity [1].
FAQ 2: How can I minimize lot-to-lot variability in my PARP-1 experiments?
Strategies to minimize LTLV impact:
Source recombinant antibodies: Recombinant antibodies demonstrate better batch-to-batch consistency compared to traditional polyclonal or hybridoma-derived monoclonal antibodies [51]. Production via synthetic DNA expression vectors introduced into suitable expression systems removes traditional reliance on hybridoma cells and avoids "genetic drift" that can compromise performance [51].
Implement rigorous quality control: Regularly test and verify the purity, stability, aggregation, and activity of antibody reagents [59]. Key methods include:
- Size exclusion chromatography with high-performance liquid chromatography (SEC-HPLC)
- Capillary electrophoresis sodium dodecyl sulfate gel electrophoresis (CE-SDS)
- SDS-PAGE for purity assessment
Maintain consistent storage conditions: Antibodies should be aliquoted and stored according to manufacturer specifications to maintain stability.
Validate each new lot: Before adopting a new antibody lot, perform parallel testing with the previous lot using standardized control samples.
FAQ 3: What controls are essential for validating PARP-1 antibody specificity?
Essential controls for PARP-1 Western blot validation:
Table 2: Required Controls for PARP-1 Antibody Validation
| Control Type | Purpose | Recommended Material | Expected Result |
|---|---|---|---|
| Positive control | Confirm antibody functionality | Cell lines with known PARP-1 expression (e.g., HEK293) | Strong bands at 116 kDa |
| Knockdown/KO negative control | Verify specificity | PARP-1 siRNA/shRNA treated cells | Significant reduction of all bands |
| Cleavage induction control | Validate detection of cleaved forms | Apoptosis-induced cells (e.g., staurosporine treatment) | Additional band at 89 kDa |
| Molecular weight marker | Accurate size determination | Prestained protein ladder | Correct size assignment |
| Loading control | Normalize protein loading | Housekeeping proteins (GAPDH, β-actin, α-tubulin) | Equal loading across lanes |
FAQ 4: Why is my PARP-1 signal weak or absent?
Common causes and solutions for weak/absent PARP-1 signal:
- Incomplete transfer: Verify transfer efficiency by staining gel post-transfer or using reversible membrane stains [9]
- Antibody concentration too low: Titrate antibody to determine optimal concentration [9]
- Insufficient antigen present: Increase protein loading; PARP-1 may be cleaved/degraded [9]
- Antigen masked by blocking buffer: Try different blocking buffers (BSA vs. milk) [9]
- Improforme buffer conditions: Avoid sodium azide in HRP-based detection systems [9]
FAQ 5: How can I reduce background noise in PARP-1 Western blots?
Strategies to reduce high background:
- Optimize antibody concentration: Decrease concentration of primary and/or secondary antibody [9]
- Modify blocking conditions: Increase blocking time; use different blocking buffers; add 0.05% Tween 20 to blocking buffer [9]
- Increase washing stringency: Increase number and volume of washes; include Tween 20 in wash buffer [9]
- Avoid membrane drying: Keep membrane covered with liquid at all times [9]
Experimental Protocols for PARP-1 Antibody Validation
Protocol 1: Knockdown Validation for PARP-1 Antibody Specificity
Purpose: Confirm that all bands detected by PARP-1 antibody are specific through genetic knockdown.
Materials:
- PARP-1 siRNA or shRNA (validated sequence)
- Scrambled siRNA negative control
- Appropriate transfection reagent
- Cells with confirmed PARP-1 expression (e.g., HeLa, HEK293)
- Lysis buffer (RIPA or similar)
- Standard Western blot equipment and reagents
Procedure:
- Plate cells at appropriate density (typically 50-70% confluency)
- Transfect with PARP-1-targeting siRNA and scrambled control using manufacturer's protocol
- Incubate for 48-72 hours to allow protein knockdown
- Harvest cells and prepare lysates
- Perform Western blot with standardized protocol
- Compare band patterns between knockdown and control samples
Expected Results: Validated PARP-1 antibody will show significant reduction in all bands in knockdown samples compared to scrambled control [1].
Protocol 2: Cross-Lot Comparison Testing
Purpose: Evaluate consistency between different lots of PARP-1 antibodies.
Materials:
- Multiple lots of the same PARP-1 antibody
- Standardized control lysates (multiple cell lines recommended)
- Reference antibody (if available)
Procedure:
- Prepare identical sets of gels with standardized control lysates
- Run Western blots in parallel using identical conditions
- Use the same secondary antibody, detection reagents, and exposure times
- Compare band intensity, pattern, and background across lots
- Quantify signals using densitometry if available
Interpretation: Consistent bands across lots indicate good lot-to-lot consistency. Significant variations suggest problematic LTLV.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents for PARP-1 Research
| Reagent/Category | Specific Examples | Function/Purpose | Considerations |
|---|---|---|---|
| PARP-1 Antibodies | Monoclonal, polyclonal, recombinant | Detection of PARP-1 in various applications | Recombinant antibodies offer better lot consistency [51] |
| Validation Tools | siRNA, shRNA, knockout cells | Confirm antibody specificity | Essential for verifying multiple bands [1] |
| Positive Controls | PARP-1 expressing cell lines (HEK293, HeLa) | Assay performance verification | Should show expected band pattern [51] |
| Apoptosis Inducers | Staurosporine, other agonists | Generate cleaved PARP-1 positive control | Validates detection of 89 kDa fragment [1] |
| Detection Systems | HRP, fluorescent conjugates | Signal detection and visualization | Different sensitivities and dynamic ranges [9] |
Visual Guide to PARP-1 Band Interpretation
The following diagram illustrates the decision process for interpreting multiple bands in PARP-1 Western blots:
Effectively working with PARP-1 antibodies requires understanding both the biology of PARP-1 and the technical challenges of immunoassays. The presence of multiple bands can represent legitimate biological phenomena rather than antibody quality issues. Through rigorous validation, appropriate controls, and strategic reagent selection, researchers can generate reliable and reproducible PARP-1 data. Implementing the troubleshooting guides and validation protocols outlined in this technical support document will significantly enhance experimental outcomes in PARP-1 research.
Conclusion
Achieving a clean, specific PARP-1 Western blot requires an integrated strategy that acknowledges its complex biology while implementing rigorous technical controls. Success hinges on moving beyond simple protocol execution to a deeper understanding that multiple bands can reflect genuine biological events like caspase cleavage or post-translational modifications. By systematically applying the troubleshooting and validation frameworks outlined—from optimizing blocking conditions and antibody dilutions to employing essential genetic knockout controls—researchers can generate reliable, interpretable data. As PARP-1 remains a critical therapeutic target in oncology, these robust blotting practices are fundamental for advancing both basic molecular knowledge and the development of next-generation PARP inhibitors, ensuring that experimental findings are both accurate and reproducible.
References
- 1. Does a Western blot with multiple bands indicate antibody ... [https://www.genuinbiotech.com/post/antibody-non-specificity]
- 2. Characterization of the necrotic cleavage of poly(ADP- ... [https://www.nature.com/articles/4400851]
- 3. Western Blot troubleshooting: Non-Specific Bands - Blog [https://www.arp1.com/blog/post/western-blot-troubleshooting-non-specific-bands.html]
- 4. Western Blot Troubleshooting: Non-specific bands [https://azurebiosystems.com/blog/western-blot-troubleshooting-what-to-do-about-non-specific-bands/]
- 5. Regulation of poly(ADP-ribose) polymerase-1 (PARP-1) gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2175517/]
- 6. Sheet Protector Strategy for Western Blot to Reduce ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12462392/]
- 7. PARP-1 cleavage fragments: signatures of cell-death ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3022541/]
- 8. The 89-kDa PARP1 cleavage fragment serves as a ... [https://www.sciencedirect.com/science/article/pii/S0021925820000320]
- 9. Western Blot Troubleshooting [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-gel-electrophoresis-information/western-blot-troubleshooting.html]
- 10. Preserving ester-linked modifications reveals glutamate ... [https://www.nature.com/articles/s41467-024-48314-0]
- 11. PARP1 auto-modification promotes faithful Okazaki ... [https://www.sciencedirect.com/science/article/pii/S1097276525007476]
- 12. PARP Antibody #9542 [https://www.cellsignal.com/products/primary-antibodies/parp-antibody/9542?srsltid=AfmBOoo_8tZNJSvAZC3l-e6GcJzxjAC_yhwY1F9WX0umumeI7t42ek1G]
- 13. PARP1 antibody (13371-1-AP) [https://www.ptglab.com/products/PARP1-Antibody-13371-1-AP.htm?srsltid=AfmBOopxJHbOUmwA-DM6vSue7RF33naZvhKPC334oRvR19dm8zU47Ffw]
- 14. Anti-Cleaved PARP1 antibody (ab4830) [https://www.abcam.com/en-us/products/primary-antibodies/cleaved-parp1-antibody-ab4830]
- 15. The regulation loop of MARVELD1 interacting with PARP1 ... [https://www.nature.com/articles/s41418-023-01118-z]
- 16. Serine ADPr on histones and PARP1 is a cellular target of ... [https://www.nature.com/articles/s41589-025-01974-5]
- 17. Western Blotting Troubleshooting [https://www.southernbiotech.com/troubleshooting-tips-for-western-blotting/]
- 18. PARP-1 inhibition influences the oxidative stress response ... [https://www.sciencedirect.com/science/article/pii/S2213231716300222]
- 19. Common Troubleshooting Tips for Western Blot Results [https://www.bosterbio.com/blog/post/common-troubleshooting-tips-for-western-blot-results-part-1?srsltid=AfmBOorFWlqBjdrO0jiqqrzFFDNDtiW_GD340Qc4qYuyoW3aLCP592Hk]
- 20. PARPs and ADP-ribosylation: Deciphering the Complexity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10202152/]
- 21. The dynamics and Regulation of PARP1 and PARP2 in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12217778/]
- 22. Protein and RNA ADP-ribosylation detection is influenced ... [https://www.life-science-alliance.org/content/6/1/e202201455]
- 23. Anti-PARP1 antibody (ab227244) [https://www.abcam.com/en-us/products/primary-antibodies/parp1-antibody-ab227244]
- 24. PARP1-dependent DNA-protein crosslink repair [https://www.nature.com/articles/s41467-024-50912-x]
- 25. The deubiquitination-PARylation positive feedback loop of ... [https://www.nature.com/articles/s41388-025-03428-7]
- 26. Non-coding Y RNA fragments in a complex with YBX1 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12203914/]
- 27. Minimizing DNA trapping while maintaining activity ... [https://www.nature.com/articles/s41419-024-07277-2]
- 28. Western blot blocking: Best practices [https://www.abcam.com/en-us/knowledge-center/western-blot/western-blot-blocking-methods]
- 29. Blocking Buffers for Western Blot and ELISA [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/blocking-buffers-western-blot-elisa.html]
- 30. Milk or BSA? How to Choose a Blocking Protein for Western Blot [https://blog.cellsignal.com/tech-tips-video-milk-or-bsa-choosing-a-blocking-protein-for-western-blot]
- 31. Western Blot Blocking Buffer Optimization | Boster Bio [https://www.bosterbio.com/protocol-and-troubleshooting/western-blotting-optimization/blocking?srsltid=AfmBOoqNA2BVPm_RFeXDgJj5jasCHzHP0WZ3I5ve4IDlOWVt-JN6qwG7]
- 32. Help! Why do my Western blots look terrible? [https://azurebiosystems.com/blog/help-why-do-my-western-blots-look-terrible/]
- 33. PARP1 antibody (13371-1-AP) [https://www.ptglab.com/products/PARP1-Antibody-13371-1-AP.htm?srsltid=AfmBOooiA7nCdbNRY_0YNgUNOqS5rGr4fS5RQSzrys_uHjVpdOlwFmzy]
- 34. Anti-Cleaved PARP1 antibody [SP276] (ab225715) [https://www.abcam.com/en-us/products/primary-antibodies/cleaved-parp1-antibody-sp276-ab225715]
- 35. Anti-PARP1 antibody [EPR18461] (ab191217) [https://www.abcam.com/en-us/products/primary-antibodies/parp1-antibody-epr18461-ab191217]
- 36. A Guide to Modern Quantitative Fluorescent Western ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4354265/]
- 37. Chemiluminescent Western Blotting [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/chemiluminescent-western-blotting.html]
- 38. What causes non-specific antibody binding and how can it ... [https://www.cytometry.org/web/q_view.php?id=153&filter=Analysis%20Techniques]
- 39. Western Blot Troubleshooting Guide - TotalLab [https://totallab.com/resources/western-blot-troubleshooting-guide/]
- 40. Blocking Strategies for IHC [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/blocking-strategies-ihc.html]
- 41. PARP Antibody #9542 [https://www.cellsignal.com/products/primary-antibodies/parp-antibody/9542?srsltid=AfmBOopjdE52ruFVn1DLE4s6rSmPr0MpfKY5OLbcTD2pjGndFbEk-hUY]
- 42. PARP1 antibody (66520-1-Ig) [https://www.ptglab.com/products/PARP1-Antibody-66520-1-Ig.htm?srsltid=AfmBOorma25SQ3CEJiHRWz9-Yg2QjRTtMkpi6O6YZUhxXcfbAC_GCSfb]
- 43. Anti-PARP1 antibody [E102] (ab32138) [https://www.abcam.com/en-us/products/primary-antibodies/parp1-antibody-e102-ab32138]
- 44. PARP Antibody #9542 [https://www.cellsignal.com/products/primary-antibodies/parp-antibody/9542?srsltid=AfmBOoprPt9hyrLuiV1bVDfrtwmbfU873BIeBwoynooaOnsvuyzzoSsP]
- 45. Activation of Poly(ADP)-ribose Polymerase (PARP-1) ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3140953/]
- 46. Knockout and Knockdown Antibody Validation [https://www.thermofisher.com/us/en/home/life-science/antibodies/invitrogen-antibody-validation/knockout-knockdown-antibody-validation.html]
- 47. Knockout (KO) Validation [https://www.antibodies.com/primary-antibodies/knockout-validation]
- 48. 2024 Guide to Quantitative Western Blot Publication [https://www.thermofisher.com/blog/life-in-the-lab/2023-guide-to-quantitative-western-blot-publication/]
- 49. Image integrity and standards | Nature Portfolio [https://www.nature.com/nature-portfolio/editorial-policies/image-integrity]
- 50. PARP Antibody #9542 [https://www.cellsignal.com/products/primary-antibodies/parp-antibody/9542?srsltid=AfmBOoolyilYrotd5ngWW-RT_K5NGQyNSpg0vR3-nCeluETbWAeOoLtS]
- 51. Antibody validation for Western blot: By the user, for the user [https://pmc.ncbi.nlm.nih.gov/articles/PMC6983856/]
- 52. Investigation of PARP-1, PARP-2, and PARG interactomes by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2861645/]
- 53. Cytoplasmic ADP-ribosylation levels correlate with markers ... [https://www.nature.com/articles/s41379-021-00788-9]
- 54. Western Blot Troubleshooting [https://www.antibodies.com/applications/western-blotting/western-blot-troubleshooting]
- 55. Western blot troubleshooting guide! [https://www.jacksonimmuno.com/secondary-antibody-resource/technical-tips/western-blot-trouble-shooting/]
- 56. 5 Tips for Reducing Non-specific Signal on Western Blots [https://www.biossusa.com/blogs/news/5-tips-for-reducing-non-specific-signal-on-western-blots?srsltid=AfmBOopiJ_xQYwBuGKD5k9NECXyzPQ9dUtjkNrApVf9tjcJ8d81DQWFh]
- 57. Western Blot: The Complete Guide [https://www.antibodies.com/applications/western-blotting]
- 58. Anti-PARP1 Antibodies | Invitrogen [https://www.thermofisher.com/antibody/primary/target/parp1]
- 59. Lot-to-Lot Variance in Immunoassays—Causes, ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10252387/]