Apoptosis Detection: PARP-1 Cleavage Analysis by Flow Cytometry vs. Western Blot
This article provides a comprehensive guide for researchers and drug development professionals on detecting apoptosis through PARP-1 cleavage, a key biochemical hallmark of programmed cell death.
Apoptosis Detection: PARP-1 Cleavage Analysis by Flow Cytometry vs. Western Blot
Abstract
This article provides a comprehensive guide for researchers and drug development professionals on detecting apoptosis through PARP-1 cleavage, a key biochemical hallmark of programmed cell death. We explore the foundational biology of PARP-1 and its cleavage fragments, detail step-by-step methodologies for both flow cytometric and western blot analysis, address common troubleshooting scenarios, and present a direct comparison of these techniques for validation purposes. By synthesizing methodological insights with comparative advantages, this resource aims to empower scientists in selecting and optimizing the most appropriate technique for their specific research context, from basic discovery to preclinical drug screening.
PARP-1 Cleavage: The Biochemical Hallmark of Apoptosis
The Biological Role of PARP-1 in DNA Repair and Cell Death
Poly(ADP-ribose) polymerase-1 (PARP-1) is a critical nuclear enzyme that functions as a primary DNA damage sensor and facilitator of DNA repair processes [1]. Beyond its repair functions, PARP-1 plays a paradoxical role in cell fate determination—its hyperactivation triggers a distinct form of programmed cell death known as parthanatos [1] [2]. This dual nature makes PARP-1 a protein of intense interest in both basic research and therapeutic development, particularly in cancer biology and neurodegenerative diseases [2] [3].
The detection of PARP-1 cleavage fragments serves as a crucial biomarker in apoptosis research, with flow cytometry and western blotting emerging as complementary techniques for its analysis [4] [5]. This application note delineates the biological context of PARP-1 in DNA damage response and cell death pathways, provides experimental protocols for its detection, and offers resources to facilitate research in this domain.
Biological Functions of PARP-1
Molecular Structure and Domains
PARP-1 is a multi-domain protein with specialized functional regions that coordinate its response to DNA damage [6]:
- Zinc finger domains (Zn1 and Zn2): Specifically recognize DNA strand breaks and form a dimeric module for DNA damage sensing
- Zinc finger domain (Zn3): Mediates interdomain contacts and regulates chromatin structure
- BRCT domain: Binds intact DNA without activating catalytic activity and enables rapid nuclear movements through a "monkey-bar mechanism"
- WGR domain: Bridges nucleosomes and aligns broken DNA for ligation, while signaling DNA break recognition to the catalytic domain
- PARP alpha-helical domain (HD): Maintains autoinhibition in the absence of activation signals; damaged DNA binding relieves this inhibition
PARP-1 in DNA Damage Repair
PARP-1 serves as a first responder in multiple DNA repair pathways, maintaining genomic stability through several mechanisms:
Base Excision Repair/Single-Strand Break Repair (BER/SSBR): PARP-1 rapidly binds to single-strand breaks (SSBs) induced by oxidative stress, alkylating agents, or irradiation, leading to its activation and poly(ADP-ribose) (PAR) synthesis [3]. This PARylation event recruits essential repair proteins including the scaffold protein XRCC1, which subsequently assembles with DNA Polβ, ligases, and other core factors to complete the repair process [1] [3].
Double-Strand Break (DSB) Repair: PARP-1 contributes to both homologous recombination (HR) and non-homologous end joining (NHEJ) pathways [1]. It facilitates HR by promoting the recruitment of the MRN complex (MRE11, RAD50, NBS1) and BRCA1 to damage sites [1]. In alternative NHEJ, PARP-1 binds to DSB sites when Ku70/Ku80 are absent, recruiting MRE11 to resect DNA ends that are subsequently joined using microhomology sequences [1].
DNA Replication: PARP-1 plays a significant role in DNA replication by recognizing unligated Okazaki fragments and controlling replication fork velocity [1] [3]. It acts as a sensor of replication stress and protects stalled replication forks from degradation by promoting the recruitment of MRE11 and RAD51 [3].
Table 1: PARP-1 Functions in DNA Damage Response and Repair
| Function | Mechanism | Key Interacting Proteins |
|---|---|---|
| SSB Repair | Recognizes SSBs, catalyzes PAR synthesis, recruits repair machinery | XRCC1, DNA Polβ, LIG1/3, PNKP [3] |
| DSB Repair (HR) | Promotes recruitment of HR proteins to damage sites | MRN complex, BRCA1, ATM [1] |
| DSB Repair (NHEJ) | Facilitates error-prone alternative NHEJ in absence of Ku proteins | MRE11, POLθ, LIG3 [1] |
| Replication | Recognizes unligated Okazaki fragments, protects stalled forks | MRE11, RAD51, RECQ1 [1] [3] |
| Chromatin Remodeling | PARylates histones, promoting chromatin decompaction at damage sites | Histones H2B, H3 [6] |
PARP-1 in Cell Death Pathways
PARP-1 activation exhibits a dose-dependent effect on cell fate. Moderate activation promotes DNA repair and cell survival, while excessive activation triggers cell death through multiple mechanisms:
Parthanatos: This distinct caspase-independent programmed necrosis occurs following severe DNA damage and PARP-1 hyperactivation [1] [2]. Key features include nuclear shrinkage, chromatin condensation, and large-scale DNA fragmentation (15-50 kb fragments) [1]. The process involves PAR-mediated deadly crosstalk between the nucleus and mitochondria, triggering apoptosis-inducing factor (AIF) release from mitochondria [1] [2]. AIF then recruits macrophage migration inhibitory factor (MIF), a 3' exonuclease, to the nucleus where it cleaves genomic DNA [1].
Energy Depletion-Mediated Cell Death: PARP-1 hyperactivation consumes large amounts of NAD+, leading to ATP depletion and eventual necrotic cell death [2] [7]. As a key cofactor in mitochondrial energy production, NAD+ depletion disrupts cellular energy homeostasis [2].
Apoptosis: PARP-1 serves as a cleavage substrate for caspases-3 and -7 during apoptosis, generating characteristic 24 kDa and 89 kDa fragments [4] [6] [5]. Recent evidence suggests these cleavage fragments may actively participate in cell death execution rather than merely serving as apoptotic markers [4] [5] [8].
PARP-1 Cleavage as an Apoptosis Marker
Cleavage Mechanism and Biological Significance
PARP-1 cleavage during apoptosis occurs primarily at the DEVD214 site by activated caspases-3 and -7, generating two fragments: a 24 kDa N-terminal fragment containing the DNA-binding domain and an 89 kDa C-terminal fragment containing the catalytic domain [4] [6] [5]. This cleavage event serves as a well-established biochemical marker of apoptosis, with several functional consequences:
Inhibition of DNA Repair: The 24 kDa fragment retains DNA-binding capability but lacks catalytic activity, potentially acting as a dominant-negative inhibitor that blocks DNA repair and conserves cellular energy during apoptosis [4] [8].
Regulation of Gene Expression: PARP-1 cleavage fragments can influence inflammatory responses by modulating NF-κB activity [4] [5]. The 89 kDa fragment has been shown to enhance NF-κB-dependent transcription of pro-inflammatory genes such as iNOS and COX-2 [4].
Novel Signaling Functions: Recent research indicates that the 89 kDa truncated PARP-1 (tPARP1) translocates to the cytoplasm during apoptosis and can ADP-ribosylate RNA Polymerase III, facilitating IFN-β production and enhancing apoptosis in response to cytosolic DNA [8].
Detection Methods: Flow Cytometry vs. Western Blot
Table 2: Comparison of PARP-1 Cleavage Detection Methods
| Parameter | Flow Cytometry | Western Blot |
|---|---|---|
| Sample Type | Single-cell suspensions | Tissue lysates, cell extracts |
| Cellular Resolution | Single-cell level | Population average |
| Information Obtained | Cleavage quantification, correlation with other markers | Fragment size confirmation, cleavage efficiency |
| Multiplexing Capability | High (with antibody panels) | Limited (typically 2-3 targets) |
| Throughput | High (hundreds to thousands of samples) | Moderate |
| Sensitivity | High (depends on antibody quality) | Moderate to High |
| Key Reagents | PARP-1 cleavage-specific antibodies, viability dyes, caspase substrates | PARP-1 antibodies recognizing full-length and cleaved fragments |
| Quantification | Percentage of positive cells | Band intensity ratio (cleaved/full-length) |
Experimental Protocols
Protocol: Detection of PARP-1 Cleavage by Western Blot
This protocol enables specific detection of PARP-1 cleavage fragments during apoptosis, suitable for both adherent and suspension cells.
Materials:
- Cell lysis buffer: RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) supplemented with protease inhibitors (including caspase inhibitors)
- Electrophoresis system: SDS-PAGE setup with 4-12% Bis-Tris gradient gels
- Transfer apparatus: Wet or semi-dry transfer system
- Primary antibodies: Anti-PARP-1 antibody recognizing full-length protein (116 kDa) and cleaved fragments (89 kDa), and caspase-3 antibody (optional validation)
- Secondary antibodies: HRP-conjugated anti-rabbit or anti-mouse IgG
- Detection system: Chemiluminescent substrate compatible with HRP
- Induction agents: Staurosporine (1-2 μM) or other apoptosis inducers as positive control
Procedure:
- Induce Apoptosis: Treat cells with apoptosis inducer for appropriate duration (typically 4-24 hours). Include untreated controls and caspase inhibitor controls (e.g., Z-VAD-FMK, 20-50 μM) if investigating caspase dependence.
- Harvest Cells: Collect both adherent and floating cells to capture all apoptotic populations. Wash twice with cold PBS.
- Prepare Lysates: Lyse cells in RIPA buffer (100-200 μL per 10^6 cells) on ice for 30 minutes. Centrifuge at 14,000 × g for 15 minutes at 4°C. Collect supernatant.
- Quantify Protein: Determine protein concentration using BCA or Bradford assay. Adjust samples to equal concentrations with lysis buffer.
- SDS-PAGE: Load 20-40 μg protein per lane. Include pre-stained molecular weight markers. Run gel at 120-150 V until dye front reaches bottom.
- Transfer: Transfer proteins to PVDF or nitrocellulose membrane using appropriate transfer system (1-2 hours at 100 V or overnight at 30 V).
- Blocking: Block membrane with 5% non-fat milk or BSA in TBST for 1 hour at room temperature.
- Primary Antibody Incubation: Incubate with anti-PARP-1 antibody (dilution per manufacturer's recommendation) in blocking buffer overnight at 4°C.
- Washing: Wash membrane 3× for 10 minutes each with TBST.
- Secondary Antibody Incubation: Incubate with HRP-conjugated secondary antibody (1:2000-1:5000) in blocking buffer for 1 hour at room temperature.
- Detection: Develop using chemiluminescent substrate according to manufacturer's instructions. Image with digital imaging system or X-ray film.
Expected Results: Apoptotic samples will show decreased full-length PARP-1 (116 kDa) and appearance of the 89 kDa cleavage fragment. The 24 kDa fragment is typically not detected in standard western blots due to poor transfer or antibody epitope location.
Protocol: Detection of PARP-1 Cleavage by Flow Cytometry
This protocol enables quantitative analysis of PARP-1 cleavage at the single-cell level with multiparameter capability.
Materials:
- Fixation buffer: 4% paraformaldehyde in PBS or commercial fixation buffers
- Permeabilization buffer: 0.1-0.5% Triton X-100 in PBS or commercial permeabilization buffers
- Staining buffer: PBS with 1% BSA or FBS
- Primary antibody: Anti-cleaved PARP-1 (Asp214) antibody or similar cleavage-specific antibody
- Secondary antibody: Fluorochrome-conjugated secondary antibody (if using indirect detection)
- Alternative: Fluorochrome-conjugated anti-cleaved PARP-1 antibody for direct detection
- Counterstains: Propidium iodide (1 μg/mL), 7-AAD, or DAPI for DNA content/cell cycle analysis
- Optional: Annexin V conjugates, active caspase-3 antibodies for multiparameter apoptosis analysis
- Flow cytometer with appropriate laser and filter configurations
Procedure:
- Induce Apoptosis: Treat cells with apoptosis inducer as described in Section 4.1.
- Harvest Cells: Collect both adherent (using gentle trypsinization or cell scraping) and floating cells. Wash twice with cold PBS.
- Fix Cells: Resuspend cell pellet in 4% paraformaldehyde and incubate for 20 minutes at room temperature. Wash twice with staining buffer.
- Permeabilize Cells: Resuspend cells in permeabilization buffer and incubate for 10 minutes on ice. Wash twice with staining buffer.
- Stain with Primary Antibody: Resuspend cells in staining buffer containing anti-cleaved PARP-1 antibody (dilution per manufacturer's recommendation). Incubate for 1 hour at room temperature or overnight at 4°C.
- Wash: Wash cells twice with staining buffer.
- Stain with Secondary Antibody (if using indirect detection): Resuspend cells in staining buffer containing fluorochrome-conjugated secondary antibody. Incubate for 30-45 minutes at room temperature in the dark.
- Wash: Wash cells twice with staining buffer.
- Counterstain (optional): Resuspend cells in staining buffer containing DNA dye (e.g., DAPI) for cell cycle analysis or viability dye for dead cell exclusion.
- Acquire Data: Analyze samples on flow cytometer within 24 hours. Use appropriate single-color controls for compensation.
Data Analysis:
- Gate on intact cells using forward and side scatter properties
- Exclude dead cells using viability dyes if needed
- Analyze cleaved PARP-1 fluorescence intensity in relevant channel
- For multiparameter analysis, create bivariate plots to correlate PARP-1 cleavage with other apoptosis markers (Annexin V, caspase activation)
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for PARP-1 Research
| Reagent Category | Specific Examples | Research Application |
|---|---|---|
| PARP-1 Antibodies | Anti-PARP-1 (full-length), Anti-cleaved PARP-1 (Asp214) | Western blot, flow cytometry, immunofluorescence [4] [6] |
| PARP Inhibitors | Olaparib (IC50 = 1.49 nM), PJ-34, 3-aminobenzamide | Functional studies, therapeutic applications [1] [2] [9] |
| Apoptosis Inducers | Staurosporine, MNNG (alkylating agent), Ionizing radiation | Induce PARP-1 cleavage and activation [1] [10] |
| Caspase Inhibitors | Z-VAD-FMK (pan-caspase inhibitor), DEVD-CHO (caspase-3 inhibitor) | Determine caspase dependence of PARP-1 cleavage [4] [5] |
| Detection Kits | Colorimetric/chemiluminescent substrates, apoptosis detection kits | Measure PARP-1 cleavage and enzymatic activity [4] |
| Cell Lines | PARP-1-deficient cells, BRCA-mutated cancer lines | Study PARP-1 function and synthetic lethality [3] |
Research Applications and Therapeutic Implications
PARP-1 in Cancer Therapy
PARP inhibitors have emerged as powerful targeted therapies for cancers with deficient DNA repair mechanisms, particularly those with BRCA1/2 mutations [1] [3]. The concept of synthetic lethality exploits the simultaneous disruption of PARP-mediated repair and homologous recombination in BRCA-deficient cells, leading to selective cancer cell death [3]. Several PARP inhibitors have received FDA approval, including olaparib, rucaparib, and talazoparib [2] [3].
Next-generation PARP-1 selective inhibitors are being developed to minimize toxicity associated with PARP-2 inhibition, particularly hematological adverse effects [3]. These selective inhibitors maintain efficacy in BRCA-mutated cancers while offering improved safety profiles [3].
PARP-1 in Neurodegenerative Diseases
In neurological contexts, PARP-1 overactivation contributes to the pathogenesis of various neurodegenerative diseases including Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis [2]. PARP-1 activation in neurons promotes pathological processes such as protein aggregation, neuroinflammation, mitochondrial dysfunction, and blood-brain barrier damage [2]. Consequently, PARP inhibition represents a promising therapeutic strategy for neuroprotection [2] [7].
Table 4: Quantitative Data on PARP-1 Inhibitors
| Inhibitor | IC50 for PARP-1 | Clinical Status | Primary Applications |
|---|---|---|---|
| Olaparib | 1.49 nM [9] | FDA-approved | Ovarian cancer, breast cancer, prostate cancer [2] [3] |
| PJ-34 | Not specified | Preclinical research | Experimental models of stroke, neurodegeneration [2] [10] |
| 3-aminobenzamide | Not specified | Preclinical research | Basic research, prototype inhibitor [2] |
| Compound 4a (novel hybrid) | 2.01 nM [9] | Preclinical development | Targeted liver cancer therapy (dual EGFR/PARP-1 inhibition) [9] |
| Compound 4f (novel hybrid) | 18.4 nM [9] | Preclinical development | Targeted liver cancer therapy (dual EGFR/PARP-1 inhibition) [9] |
PARP-1 represents a critical nexus in cellular fate decisions, balancing DNA repair and cell death functions through its multifaceted roles in DNA damage response. The detection of PARP-1 cleavage provides a valuable biomarker for apoptosis research, with both western blot and flow cytometry offering complementary approaches for its analysis. Ongoing research continues to reveal novel functions of PARP-1 and its cleavage fragments, particularly in inflammatory signaling and cytoplasmic activities during apoptosis. The development of increasingly selective PARP-1 inhibitors holds promise for enhanced therapeutic applications in oncology and neurodegenerative diseases, highlighting the continued importance of this protein in both basic research and clinical translation.
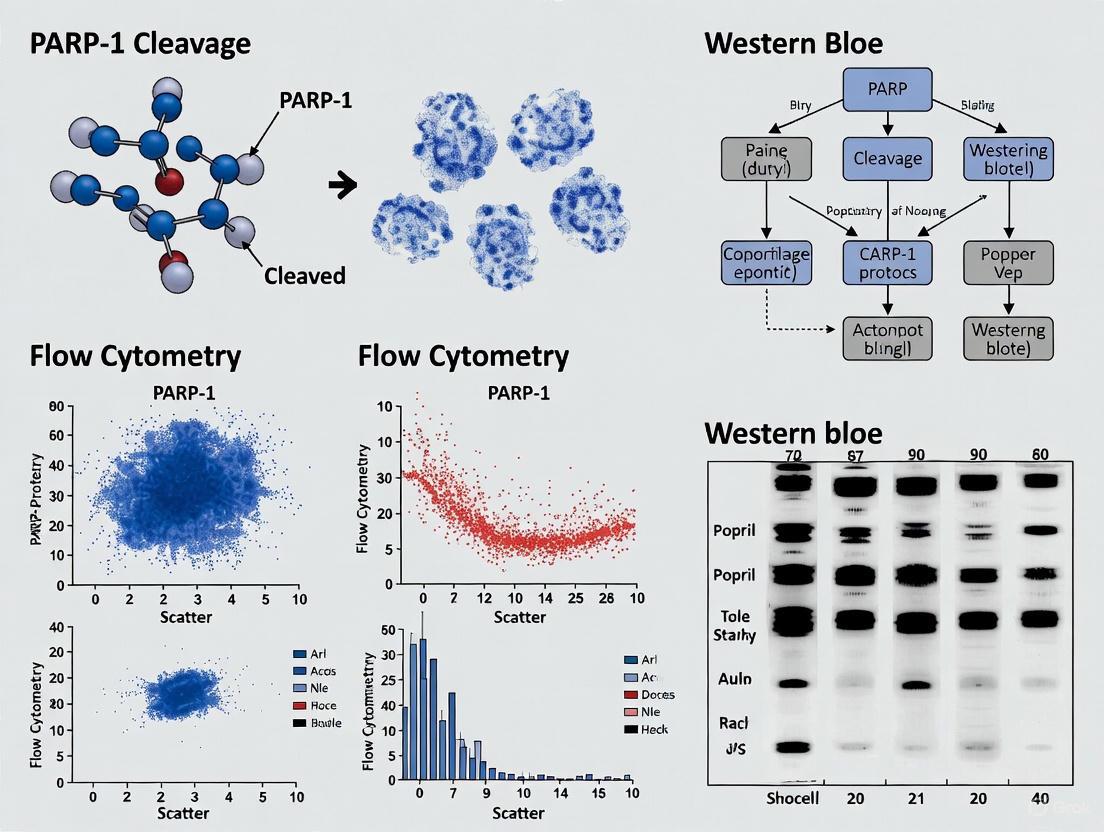
Poly(ADP-ribose) polymerase-1 (PARP-1) is a 116 kDa nuclear enzyme that plays a central role in the cellular response to DNA damage, primarily by detecting DNA strand breaks and initiating base excision repair [11]. Beyond its DNA repair functions, PARP-1 participates in various cellular processes, including transcription regulation, inflammation, and cell death signaling [11] [4]. A critical event in cell death pathways is the caspase-mediated cleavage of PARP-1, which serves as a well-established biochemical hallmark of apoptosis [12] [11]. This proteolytic cleavage occurs at a specific aspartic acid residue (Asp214) within the nuclear localization signal, generating two characteristic fragments: a 24 kDa DNA-binding fragment and an 89 kDa catalytic fragment [12] [4]. These signature fragments not only indicate apoptosis but also execute distinct functional roles in the cell death process. This application note details the mechanisms and detection methodologies for these PARP-1 cleavage fragments, providing researchers with practical protocols for apoptosis detection in the context of drug development and basic research.
Biological Significance of PARP-1 Cleavage Fragments
Functional Roles of the 24 kDa and 89 kDa Fragments
Caspase-mediated cleavage of PARP-1 represents a decisive step in the commitment to apoptosis, serving to suppress DNA repair and facilitate cellular disassembly [12]. The generation of the 24 kDa and 89 kDa fragments leads to the functional segregation of PARP-1's domains, with each fragment acquiring distinct roles in the apoptosis process (Table 1).
Table 1: Characteristics and Functions of PARP-1 Cleavage Fragments
| Fragment | Domains Contained | Localization After Cleavage | Primary Functions |
|---|---|---|---|
| 24 kDa | DNA-binding domain (with two zinc finger motifs), Nuclear Localization Signal (NLS) | Retained in nucleus [13] | Irreversibly binds to DNA strand breaks [11]; Acts as trans-dominant inhibitor of DNA repair [13] [11] |
| 89 kDa | Automodification domain, Catalytic domain (with BRCT and WGR domains) | Translocates to cytoplasm [13] [14] | Serves as PAR carrier to cytoplasm [13] [14]; Binds AIF to promote parthanatos [13]; Interacts with and ADP-ribosylates RNA Pol III [8] |
The 24 kDa fragment contains the DNA-binding domain with two zinc finger motifs and the nuclear localization signal [11]. After cleavage, this fragment remains in the nucleus where it irreversibly binds to DNA strand breaks [11]. This binding serves a critical function in apoptosis by acting as a trans-dominant inhibitor of DNA repair processes, preventing the recruitment of additional DNA repair machinery to damaged sites and thereby facilitating the apoptotic process [13] [11].
The 89 kDa fragment, containing the automodification and catalytic domains, translocates from the nucleus to the cytoplasm following cleavage [13] [14]. Recent research has revealed multifaceted roles for this fragment beyond the simple inactivation of PARP-1. It functions as a carrier for poly(ADP-ribose) (PAR) polymers, transporting them to the cytoplasm where they can bind to apoptosis-inducing factor (AIF) [13] [14]. This PAR-AIF interaction facilitates AIF release from mitochondria and its subsequent translocation to the nucleus, where it contributes to caspase-independent DNA fragmentation [13] [14]. This pathway represents a crucial intersection between caspase-dependent apoptosis and AIF-mediated parthanatos.
Additionally, the 89 kDa fragment has been shown to interact with the RNA polymerase III (Pol III) complex in the cytoplasm during poly(dA-dT)-stimulated apoptosis [8]. The BRCT domain of the 89 kDa fragment mediates this interaction, leading to ADP-ribosylation of Pol III and subsequent facilitation of interferon-beta (IFN-β) production, thereby connecting PARP-1 cleavage to innate immune responses during apoptosis [8].
PARP-1 Cleavage in Different Cell Death Pathways
While caspase-mediated cleavage generating the 24 kDa and 89 kDa fragments is characteristic of apoptosis, PARP-1 is also cleaved by other proteases in different cell death contexts. During necrosis, lysosomal proteases such as cathepsins B and G cleave PARP-1, producing a predominant 50 kDa fragment, which is not inhibited by broad-spectrum caspase inhibitors like zVAD-fmk [15]. This distinct cleavage pattern provides researchers with a valuable tool for differentiating between apoptotic and necrotic cell death.
The functional consequences of PARP-1 cleavage also vary significantly depending on the cellular context and death stimulus. Studies using uncleavable PARP-1 mutants (PARP-1UNCL) and individual fragment expression have demonstrated that the 24 kDa fragment confers protection against oxygen/glucose deprivation (OGD) in neuronal models, while expression of the 89 kDa fragment is cytotoxic [4]. These findings suggest that PARP-1 cleavage fragments may regulate cellular viability and inflammatory responses in opposing ways during ischemic stress [4].
Diagram: PARP-1 Cleavage and Fragment Signaling Pathways. This diagram illustrates the caspase-mediated cleavage of full-length PARP-1 (116 kDa) into 24 kDa and 89 kDa fragments and their distinct downstream signaling pathways that promote cell death.
Detection Methods and Comparative Analysis
Western Blot Detection of PARP-1 Cleavage
Western blot analysis remains the gold standard for detecting PARP-1 cleavage fragments due to its ability to provide direct molecular weight confirmation and clear differentiation between full-length PARP-1 and its cleavage products.
Table 2: Key Reagents for PARP-1 Cleavage Detection by Western Blot
| Reagent | Specification | Application | Key Features |
|---|---|---|---|
| Anti-Cleaved PARP (Asp214) Antibody [12] | Rabbit monoclonal, recognizes 89 kDa fragment | Western Blot (1:1000 dilution) | Specific for caspase-cleaved fragment; does not recognize full-length PARP-1 |
| PARP-1 Primary Antibody | Rabbit polyclonal, various vendors | Western Blot | Detects both full-length (116 kDa) and cleaved (89 kDa) PARP-1 |
| Secondary Antibody | HRP-conjugated anti-rabbit | Western Blot | For chemiluminescent detection |
| Cell Lysis Buffer | RIPA buffer with protease inhibitors | Sample preparation | Preserves protein integrity and post-translational modifications |
Protocol: Western Blot Detection of PARP-1 Cleavage Fragments
Sample Preparation:
- Harvest cells and wash with cold PBS [16].
- Lyse cells in RIPA buffer supplemented with protease inhibitors (e.g., PMSF) and caspase inhibitors (if measuring basal levels).
- Centrifuge at 12,000 × g for 15 minutes at 4°C to remove insoluble material.
- Determine protein concentration using a standard assay (e.g., BCA assay).
Gel Electrophoresis:
- Load 20-50 μg of total protein per lane on 4-12% Bis-Tris polyacrylamide gels.
- Run at constant voltage (120-150V) until appropriate separation is achieved.
- Transfer proteins to PVDF or nitrocellulose membranes using standard transfer protocols.
Immunoblotting:
- Block membranes with 5% non-fat dry milk in TBST for 1 hour at room temperature.
- Incubate with primary antibody (e.g., Cleaved PARP (Asp214) Antibody #9541 at 1:1000 dilution [12]) in blocking buffer overnight at 4°C.
- Wash membrane 3 times for 10 minutes each with TBST.
- Incubate with HRP-conjugated secondary antibody (1:2000-1:5000) in blocking buffer for 1 hour at room temperature.
- Wash membrane 3 times for 10 minutes each with TBST.
Detection:
- Develop blots using enhanced chemiluminescence (ECL) substrate.
- Image using a digital imaging system capable of detecting chemiluminescent signals.
- Strip and re-probe membrane with loading control antibodies (e.g., β-actin, GAPDH) for normalization.
Data Interpretation: Apoptotic samples will show the characteristic 89 kDa cleavage fragment, while non-apoptotic samples will display only the full-length 116 kDa PARP-1 band. Quantitative analysis can be performed by densitometry to calculate the ratio of cleaved to full-length PARP-1.
Flow Cytometry-Based Apoptosis Detection
Flow cytometry offers a high-throughput alternative for detecting apoptosis in cell populations, typically using Annexin V/propidium iodide (PI) staining as a complementary method to assess phosphatidylserine externalization and membrane integrity.
Protocol: Annexin V/Propidium Iodide Staining for Flow Cytometry [17] [16]
Sample Preparation:
Staining:
- Add 5 μL of fluorochrome-conjugated Annexin V to 100 μL of cell suspension [17].
- Incubate for 10-15 minutes at room temperature, protected from light [17].
- Add 2 mL of 1X Binding Buffer and centrifuge at 400-600 × g for 5 minutes at room temperature. Discard supernatant [17].
- Resuspend cells in 200 μL of 1X Binding Buffer.
- Add 5 μL of propidium iodide (PI) staining solution just before analysis [17] [16].
Flow Cytometry Analysis:
- Analyze samples within 4 hours of staining using a flow cytometer equipped with appropriate lasers and filters for the fluorochromes used [17].
- Collect a minimum of 10,000 events per sample.
- Use unstained cells, Annexin V-only stained cells, and PI-only stained cells as controls for compensation and gating [16].
Data Interpretation:
- Viable cells: Annexin V negative, PI negative
- Early apoptotic cells: Annexin V positive, PI negative
- Late apoptotic/necrotic cells: Annexin V positive, PI positive [16]
Diagram: Annexin V/PI Staining Workflow. This diagram outlines the key steps in the Annexin V/propidium iodide staining protocol for flow cytometry-based apoptosis detection, highlighting critical steps and potential pitfalls.
Caspase Activity Assays
Caspase-Glo 3/7 assays provide a luminescent method for specifically measuring the activities of executioner caspases (caspase-3 and -7) that directly cleave PARP-1.
Protocol: Caspase-Glo 3/7 Assay [18]
Sample Preparation:
- Culture cells in white-walled multiwell plates suitable for luminescence reading.
- Include positive control (cells treated with known apoptosis inducer) and negative control (untreated cells).
Assay Procedure:
- Equilibrate Caspase-Glo 3/7 reagent and cell plates to room temperature.
- Add equal volume of Caspase-Glo 3/7 reagent to each well (e.g., 100 μL reagent to 100 μL cells in medium).
- Mix contents gently using a plate shaker for 30 seconds.
- Incubate at room temperature for 1-3 hours to allow signal development.
- Measure luminescence using a plate-reading luminometer.
Data Analysis:
- Normalize luminescence readings to protein content or cell number.
- Calculate fold-increase in caspase activity compared to untreated controls.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for PARP-1 Cleavage and Apoptosis Detection
| Category | Specific Product/Kit | Application | Key Features |
|---|---|---|---|
| PARP-1 Cleavage Detection | Cleaved PARP (Asp214) Antibody #9541 [12] | Western Blot | Specifically detects 89 kDa fragment; validated for human and mouse samples |
| Flow Cytometry Apoptosis Detection | Annexin V Apoptosis Detection Kits [17] | Flow Cytometry | Multiple fluorochrome options; includes Annexin V and viability dye |
| Caspase Activity Assay | Caspase-Glo 3/7 Assay [18] | Luminescent plate assay | Homogeneous "add-mix-measure" format; high sensitivity |
| PARP Inhibition | PJ34, ABT-888 (PARP inhibitors) [13] | Functional studies | Tool compounds for investigating PARP-1 function in cell death |
| Caspase Inhibition | zVAD-fmk (pan-caspase inhibitor) [13] | Control experiments | Confirms caspase-dependent nature of PARP-1 cleavage |
Method Selection Guide
The choice between Western blot, flow cytometry, and caspase activity assays depends on the specific research question, sample type, and required throughput (Table 4).
Table 4: Comparative Analysis of Apoptosis Detection Methods
| Parameter | Western Blot | Flow Cytometry | Caspase Activity Assay |
|---|---|---|---|
| Information Obtained | Direct visualization of PARP-1 cleavage fragments; molecular weight confirmation | Apoptosis staging (early vs. late); population heterogeneity | Quantitative caspase-3/7 activity; high throughput |
| Sensitivity | High (with good antibodies) | Moderate to high | High |
| Throughput | Low to moderate | High | Very high |
| Sample Requirements | 20-50 μg protein per sample | 1-5 × 10^5 cells per sample | Cell lysates or direct culture |
| Key Advantages | Direct detection of PARP-1 cleavage; semi-quantitative | Single-cell analysis; multiparameter capabilities | Simple protocol; suitable for screening |
| Limitations | No single-cell information; requires protein extraction | Indirect measure of PARP-1 cleavage | Does not directly measure PARP-1 cleavage |
For comprehensive apoptosis analysis, researchers often combine multiple methods—for example, using flow cytometry for initial screening and population analysis, followed by Western blot to confirm PARP-1 cleavage in specific cell populations of interest.
Troubleshooting and Technical Considerations
Common Challenges and Solutions
Incomplete PARP-1 Cleavage Detection:
High Background in Western Blot:
- Potential cause: Non-specific antibody binding or insufficient blocking.
- Solution: Optimize blocking conditions (e.g., use 5% BSA instead of milk) and increase wash stringency.
Poor Annexin V Staining:
- Potential cause: Use of buffers containing EDTA or other calcium chelators.
- Solution: Ensure all buffers are calcium-containing and EDTA-free [17].
Variable Caspase Activity Results:
- Potential cause: Incorrect cell numbers or incubation times.
- Solution: Optimize cell density and perform time course experiments to determine optimal signal window.
Experimental Design Considerations
When designing experiments to investigate PARP-1 cleavage:
- Include appropriate positive controls (e.g., cells treated with 1 μM staurosporine for 4-6 hours [13]) and negative controls (e.g., cells pre-treated with zVAD-fmk caspase inhibitor [13]).
- Consider temporal dynamics—PARP-1 cleavage occurs relatively early in apoptosis, so time course experiments are essential for capturing the complete picture.
- Account for cell-type specific differences in PARP-1 expression and cleavage kinetics.
- For functional studies, consider using PARP-1 inhibitors (e.g., PJ34) [13] or caspase inhibitors to validate mechanism.
The detection of caspase-mediated PARP-1 cleavage fragments provides researchers with a powerful tool for investigating apoptotic pathways in both basic research and drug development contexts. The 24 kDa and 89 kDa fragments not only serve as biomarkers of apoptosis but also execute distinct biological functions that contribute to the cell death process. Western blot analysis remains the most specific method for directly detecting these fragments, while flow cytometry and caspase activity assays offer complementary approaches for higher-throughput screening and population analysis. The protocols and guidelines presented in this application note provide researchers with a comprehensive framework for implementing these detection methods in their apoptosis research, with particular relevance to cancer biology, neurobiology, and inflammatory disease research where PARP-1 mediated cell death pathways play crucial pathological roles.
PARP-1 Cleavage as a Universal Apoptosis Marker Across Cell Types
Poly(ADP-ribose) polymerase-1 (PARP-1) is a nuclear enzyme with well-established roles in DNA damage repair and cellular homeostasis. During apoptosis, PARP-1 serves as a primary substrate for executioner caspases (primarily caspase-3 and -7), which cleave the 116 kDa full-length protein into characteristic 24 kDa and 89 kDa fragments [11]. This cleavage event disables PARP-1's DNA repair capabilities, facilitating the dismantling of the nucleus and serving as a recognized biochemical hallmark of apoptotic cell death [8] [11]. The 89 kDa fragment, containing the catalytic domain, translocates to the cytoplasm, while the 24 kDa DNA-binding domain fragment remains nuclear and acts as a trans-dominant inhibitor of DNA repair [11]. This proteolytic cleavage has established PARP-1 as a universal marker for detecting apoptosis across diverse cell types and experimental conditions.
Biological Significance of PARP-1 Cleavage
The cleavage of PARP-1 is not merely a bystander event but an active contributor to the apoptotic process. The 24 kDa fragment irreversibly binds to DNA strand breaks, inhibiting the recruitment of DNA repair complexes and conserving cellular ATP pools that would otherwise be depleted by PARP-1 overactivation [11]. Recent research has revealed that the 89 kDa truncated PARP-1 (tPARP1) acquires novel biological functions, including the ability to recognize and mono-ADP-ribosylate the RNA polymerase III (Pol III) complex in the cytosol [8]. This tPARP1-mediated modification facilitates IFN-β production and enhances apoptosis during innate immune responses to pathogenic DNA [8]. The functional divergence of PARP-1 fragments underscores the critical role of PARP-1 cleavage in both extinguishing DNA repair and actively promoting cell death execution.
Table 1: PARP-1 Fragments Generated During Apoptosis and Their Functions
| Fragment | Molecular Weight | Domains Contained | Cellular Localization | Biological Function |
|---|---|---|---|---|
| Full-length PARP-1 | 116 kDa | Two zinc finger motifs, BRCT domain, WGR domain, Catalytic domain | Nucleus | DNA damage repair, transcriptional regulation |
| N-terminal fragment | 24 kDa | Two zinc finger motifs (DNA-binding domain) | Nucleus | Irreversibly binds DNA breaks, inhibits DNA repair |
| C-terminal fragment | 89 kDa | Third zinc finger, BRCT domain, WGR domain, Catalytic domain | Cytoplasm | Novel functions including Pol III ADP-ribosylation, promotes apoptosis |
Detection Methodologies: Flow Cytometry vs. Western Blot
Flow Cytometry for PARP-1 Cleavage Detection
Flow cytometry enables rapid, quantitative analysis of PARP-1 cleavage at the single-cell level, particularly when using antibodies specific for the cleaved forms. This approach allows for simultaneous multiparametric analysis of other apoptosis markers.
Detailed Protocol for Flow Cytometric Detection:
- Cell Preparation: Collect cells (approximately 1×10^6 cells/mL) and wash twice with cold phosphate-buffered saline (PBS).
- Fixation and Permeabilization: Treat cells with Cytofix/Cytoperm solution for 20 minutes at 4°C to preserve intracellular epitopes.
- Intracellular Staining: Incubate cells with saturating amounts of primary antibody specific for cleaved PARP-1 (Asp214) for 45 minutes at 4°C.
- Secondary Detection: For indirect detection, incubate with fluorochrome-conjugated secondary antibody (e.g., AlexaFluor 488) for 30 minutes at 4°C, protected from light.
- Data Acquisition and Analysis: Analyze stained cells using a flow cytometer. Report cleaved PARP-1 levels as Mean Fluorescent Intensity (MFI) or percentage of positive cells [19].
Western Blot for PARP-1 Cleavage Detection
Western blot provides definitive molecular weight confirmation of PARP-1 cleavage fragments and can detect both full-length and cleaved forms simultaneously.
Detailed Protocol for Western Blot Detection:
- Protein Extraction: Lyse cells in RIPA buffer supplemented with protease and phosphatase inhibitors. Quantify protein concentration using a BCA assay.
- Gel Electrophoresis: Separate 20-50 μg of total protein on 7.5-10% SDS-polyacrylamide gels at 100-120 V for 1-2 hours.
- Membrane Transfer: Transfer proteins to PVDF membranes using wet or semi-dry transfer systems.
- Immunoblotting: Block membranes with 5% non-fat milk for 1 hour. Incubate with primary antibodies against PARP-1 (detecting both full-length and 89 kDa fragment) or cleaved PARP-1 (specific to the 89 kDa fragment) overnight at 4°C.
- Detection: Incubate with HRP-conjugated secondary antibodies for 1 hour at room temperature. Develop using enhanced chemiluminescence substrate and visualize with a digital imaging system [20] [21].
Table 2: Comparison of Flow Cytometry and Western Blot for PARP-1 Cleavage Detection
| Parameter | Flow Cytometry | Western Blot |
|---|---|---|
| Sample Throughput | High (can analyze thousands of cells per second) | Low to medium (typically 10-20 samples per gel) |
| Cellular Resolution | Single-cell level analysis | Population average |
| Molecular Specificity | Lower (requires cleavage-specific antibodies) | Higher (confirms fragment size) |
| Multiplexing Capacity | High (can combine with other apoptosis markers) | Limited (typically 2-3 targets per membrane) |
| Quantitative Accuracy | Excellent for relative quantification | Good with proper normalization |
| Equipment Requirements | Flow cytometer | Gel electrophoresis and transfer systems |
| Protocol Duration | 4-6 hours | 24-48 hours (including overnight incubation) |
| Key Applications | Drug screening, heterogeneous populations, kinetic studies | Mechanism confirmation, fragment characterization |
PARP-1 Cleavage in Regulated Cell Death Pathways
PARP-1 cleavage traditionally serves as an apoptosis marker, but its role in other regulated cell death (RCD) pathways is increasingly recognized. PARP-1 hyperactivation can trigger parthanatos, a caspase-independent programmed cell death pathway characterized by excessive poly(ADP-ribose) (PAR) polymer formation [22]. In acute myeloid leukemia (AML), a standard frontline drug combination of cytarabine and idarubicin induces distinct features of parthanatos in primary cell samples, with parthanatos-positive patient groups showing a 3-fold improvement in survival rates (HR = 0.28-0.37, p = 0.002-0.046) [22]. This demonstrates the clinical relevance of PARP-1-mediated cell death beyond classical apoptosis. Furthermore, recent evidence indicates that ferroptosis inducers like RSL3 can trigger PARP-1 cleavage through caspase-dependent pathways while simultaneously suppressing full-length PARP-1 expression via inhibition of METTL3-mediated m6A RNA modification [20]. This dual mechanism represents a novel convergence point between ferroptotic and apoptotic signaling.
Diagram 1: PARP-1 Cleavage Signaling Pathway in Apoptosis. This diagram illustrates the sequential process from apoptotic stimuli to functional outcomes of PARP-1 cleavage.
Research Reagent Solutions for PARP-1 Cleavage Studies
Table 3: Essential Reagents for PARP-1 Cleavage Detection
| Reagent/Category | Specific Examples | Function/Application | Considerations |
|---|---|---|---|
| PARP-1 Antibodies | Anti-PARP-1 (full-length), Anti-cleaved PARP-1 (Asp214) | Detection of total and cleaved PARP-1 in WB and FC | Clone specificity; species reactivity; application validation |
| Caspase Inhibitors | Z-VAD-FMK (pan-caspase inhibitor) | Inhibition of PARP-1 cleavage to confirm caspase-dependence | Cell permeability; concentration optimization (typically 20-50 µM) |
| Apoptosis Inducers | RSL3, Chemotherapeutic agents (e.g., cytarabine/idarubicin) | Positive controls for PARP-1 cleavage experiments | Mechanism-specific (intrinsic vs. extrinsic pathways) |
| Flow Cytometry Reagents | Cytofix/Cytoperm solution, Fluorochrome-conjugated secondary antibodies | Cell fixation, permeabilization, and detection | Permeabilization optimization; antibody titration required |
| Western Blot Reagents | SDS-PAGE gels, PVDF membranes, ECL substrate | Protein separation, transfer, and detection | Transfer efficiency; antibody validation required |
| Cell Death Assays | Annexin V/7-AAD, MTT, LDH release assays | Complementary apoptosis/viability assessment | Multiplexing capacity with PARP-1 detection |
Experimental Workflows for PARP-1 Cleavage Analysis
Diagram 2: Experimental Workflow for PARP-1 Cleavage Detection. This diagram outlines the parallel pathways for flow cytometry and Western blot analysis of PARP-1 cleavage.
PARP-1 cleavage remains a robust, universal marker for apoptosis detection across diverse cell types, with significant implications for both basic research and drug development. The complementary application of flow cytometry and Western blot methodologies provides comprehensive insights into apoptotic progression, from single-cell quantification to molecular confirmation of cleavage events. Furthermore, the emerging roles of PARP-1 fragments in novel biological processes, such as innate immune activation through Pol III modification, highlight the expanding significance of this proteolytic event beyond its traditional status as a cell death marker. As research continues to elucidate the complex interplay between different regulated cell death pathways, PARP-1 cleavage analysis will remain an essential component in the molecular toolkit for cell death research and therapeutic development.
For decades, the cleavage of poly(ADP-ribose) polymerase 1 (PARP-1) has been recognized as a definitive biochemical hallmark of apoptosis, serving as a reliable marker for researchers employing flow cytometry and western blot techniques [4] [23]. This proteolytic event, mediated by executioner caspases-3 and -7, generates characteristic 24-kDa and 89-kDa fragments [4] [24]. However, emerging research reveals that these fragments are not merely inert byproducts of cell death but possess distinct and active biological functions that significantly influence cellular fate [4]. This Application Note reframes PARP-1 cleavage within a broader thesis of apoptosis detection, moving beyond its utility as a simple marker to explore its functional significance in cell death pathways and its implications for basic research and drug development.
The Functional Paradigm of PARP-1 Cleavage Fragments
The traditional view holds that PARP-1 cleavage serves to inactivate DNA repair processes during apoptosis, thereby conserving cellular energy for the orderly dismantling of the cell [23]. Contemporary studies, however, demonstrate that the resulting fragments actively regulate cellular viability and inflammatory responses in opposing ways [4].
Table 1: Biological Functions of PARP-1 Cleavage Fragments
| Fragment | Size | Primary Origin | Key Biological Functions | Impact on Cell Fate |
|---|---|---|---|---|
| PARP-1 24kDa | 24 kDa | Caspase-3/7 cleavage at DEVD214 [4] [24] | Irreversibly binds DNA breaks [20]; Regulates inflammatory response via NF-κB [4] | Pro-apoptotic [20]; Cytoprotective in specific contexts [4] |
| PARP-1 89kDa | 89 kDa | Caspase-3/7 cleavage at DEVD214 [4] [24] | Translocates to cytoplasm; induces caspase-mediated DNA fragmentation [20] | Cytotoxic [4]; Pro-apoptotic [20] |
| Full-length PARP-1 | 113 kDa | N/A | DNA damage repair; NAD+-consuming catalytic activity [24] [25] | Cell survival; overactivation leads to energy depletion-induced necrosis [24] |
The biological outcomes of PARP-1 cleavage are highly context-dependent, influenced by the extent of DNA damage and the cellular microenvironment. Research indicates that expression of the uncleavable PARP-1 (PARP-1UNCL) or the 24-kDa fragment can confer protection from ischemic damage in neuronal models, whereas the 89-kDa fragment is consistently cytotoxic [4]. This functional divergence is partly mediated through differential regulation of the NF-κB pathway, where the 89-kDa fragment increases NF-κB activity and the expression of pro-inflammatory proteins like iNOS and COX-2, while the 24-kDa fragment and PARP-1UNCL have the opposite effect [4].
Quantitative Assessment of Cleavage in Apoptosis Research
The detection of PARP-1 cleavage provides a quantifiable metric for apoptosis in both flow cytometry and western blot applications. The following table summarizes key quantitative findings from recent studies that utilize PARP-1 cleavage as a central readout.
Table 2: Quantitative Data on PARP-1 Cleavage in Experimental Models
| Inducer/Context | Cell Line/Model | Key Cleavage-Related Findings | Detection Method | Citation |
|---|---|---|---|---|
| RSL3 (Ferroptosis Inducer) | Various cancer cells (e.g., MHCC97H, Kuramochi) | Triggers caspase-3-dependent PARP-1 cleavage; also suppresses full-length PARP-1 via m6A mRNA modification [20] | Western Blot, RT-qPCR [20] | [20] |
| Sclerohumin D (Compound 4) | MIA PaCa-2 (Pancreatic Cancer) | Suppresses Bcl-2/Bcl-xL; triggers caspase-3 activation leading to PARP-1 cleavage [26] | Western Blot [26] | [26] |
| Particulate Matter (PM) | HBE Δα BKCa (Bronchial Epithelial) | Induced PARP1-dependent apoptosis, confirmed by cleavage fragment detection [27] | Flow Cytometry, Western Blot [27] | [27] |
| Oxygen/Glucose Deprivation (OGD) | SH-SY5Y (Neuroblastoma), Primary Rat Neurons | PARP-1UNCL and PARP-124 are cytoprotective; PARP-189 is toxic [4] | Viability Assays, Immunoblotting [4] | [4] |
Detailed Experimental Protocols
This section provides detailed methodologies for key experiments analyzing PARP-1 cleavage, designed for replication in a research setting.
Protocol 1: Detecting PARP-1 Cleavage via Western Blotting
This protocol is adapted from mechanistic studies on novel apoptogens [20] [26].
Key Reagents & Materials:
- Cell Line: MIA PaCa-2 pancreatic cancer cells [26] (or other relevant line).
- Treatment: Sclerohumin D (or other inducer like RSL3 [20] or standardized Particulate Matter [27]).
- Lysis Buffer: RIPA buffer supplemented with protease and phosphatase inhibitors.
- Antibodies: Primary antibodies against PARP-1 (to detect full-length and fragments), Caspase-3 (full-length and cleaved), Bcl-2, Bcl-xL [26]; and corresponding HRP-conjugated secondary antibodies.
- Other: SDS-PAGE gel, PVDF membrane, chemiluminescence detection kit.
Procedure:
- Cell Seeding and Treatment: Seed cells in 6-well plates and allow to adhere overnight. Treat cells with the desired apoptogen (e.g., Sclerohumin D at its IC50 concentration of ~1.35-41.70 μM [26]) for a time course (e.g., 24, 48 hours).
- Protein Extraction: Harvest cells, wash with PBS, and lyse in ice-cold RIPA buffer. Centrifuge at 14,000 × g for 15 minutes at 4°C. Collect the supernatant and determine protein concentration using a BCA assay.
- Gel Electrophoresis and Transfer: Load 20-40 μg of protein per lane onto an SDS-PAGE gel (8-12% gradient recommended). Separate proteins by electrophoresis and transfer to a PVDF membrane.
- Immunoblotting: Block the membrane with 5% non-fat milk in TBST for 1 hour. Incubate with primary antibodies (e.g., anti-PARP-1 at 1:1000 dilution [20]) overnight at 4°C. Wash the membrane and incubate with HRP-conjugated secondary antibody for 1 hour at room temperature.
- Detection: Develop blots using a chemiluminescence substrate and image with a digital imaging system. The cleavage of PARP-1 is indicated by the disappearance of the 113 kDa full-length band and the appearance of the 89 kDa fragment [20] [26].
Protocol 2: Assessing Apoptosis via Flow Cytometry with Cleaved PARP-1
This protocol is adapted from studies on environmental toxicology and DNA damage response [28] [27].
Key Reagents & Materials:
- Cell Line: Human Bronchial Epithelial (HBE) cells or similar [27].
- Treatment: Standardized Particulate Matter (PM) (e.g., 30-100 μg/ml) [27].
- Staining Reagents: FITC-conjugated anti-cleaved PARP-1 (Asp214) antibody (e.g., clone F21-852 [28]), PE-conjugated anti-active Caspase-3 antibody, cell fixation/permeabilization kit (e.g., Cytofix/Cytoperm [28]).
- Other: Flow cytometer with appropriate lasers and filters.
Procedure:
- Cell Treatment and Harvest: Treat cells with the genotoxic agent (e.g., PM at 50 μg/ml for 24 hours [27]). Harvest both adherent and floating cells by gentle trypsinization and combine.
- Fixation and Permeabilization: Wash cells twice with cold PBS. Fix and permeabilize cells using a commercial kit (e.g., incubate in Cytofix/Cytoperm solution for 20 minutes [28]).
- Intracellular Staining: Wash cells with perm/wash buffer. Incubate cells with saturating amounts of FITC-anti-cleaved PARP-1 and/or PE-anti-active Caspase-3 antibodies for 45 minutes at 4°C in the dark.
- Analysis: Wash cells and resuspend in flow cytometry buffer. Analyze on a flow cytometer. Cleaved PARP-1 positive cells are identified in the FITC channel, and the percentage of positive cells within the live cell gate is calculated [28] [27]. This allows for quantitation of the apoptotic population.
Visualization of Signaling Pathways
The following diagrams illustrate the key signaling pathways involving PARP-1 cleavage, as discussed in this note.
Diagram 1: PARP-1 Cleavage in Cell Fate Decisions. This diagram illustrates how different stresses lead to PARP-1 activation and cleavage. The resulting fragments (24 kDa and 89 kDa) execute distinct pro-apoptotic functions, with the 89 kDa fragment also promoting a pro-inflammatory response via NF-κB activation.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Studying PARP-1 Cleavage and Function
| Reagent / Material | Specific Example / Catalog Number | Primary Function in Research | Experimental Application |
|---|---|---|---|
| Anti-PARP-1 Antibody | Abcam ab81299 [20] | Detects full-length and cleavage fragments of PARP-1 | Western Blot, Immunofluorescence |
| Anti-Cleaved PARP-1 (Asp214) Antibody | BD Biosciences FITC-conjugated, clone F21-852 [28] | Specifically detects the caspase-cleaved form of PARP-1 | Flow Cytometry |
| Caspase Inhibitor | Z-VAD-FMK [20] | Pan-caspase inhibitor; blocks PARP-1 cleavage | Validation of caspase-dependent apoptosis |
| PARP Inhibitor | Olaparib, PJ34 [20] [25] | Inhibits PARP-1 catalytic activity; used to study non-cleavage functions | Studying synthetic lethality, differentiation |
| Ferroptosis Inducer | RSL3 [20] | Induces ferroptosis and caspase-mediated PARP-1 cleavage | Studying crosstalk between cell death pathways |
| Standardized Particulate Matter | SRM-2786 (NIST) [27] | Genotoxic agent inducing DNA damage and PARP-1 cleavage | Environmental toxicology, DNA damage response studies |
Concluding Remarks
The cleavage of PARP-1 represents a critical juncture in cell fate, transitioning from a DNA damage sensor to an executioner of cell death through its bioactive fragments. For the researcher, moving beyond a binary view of PARP-1 cleavage—as merely present or absent—to a more nuanced understanding of the functional consequences of its fragments enriches the interpretation of both flow cytometry and western blot data. This expanded paradigm opens new avenues for therapeutic intervention, particularly in diseases like cancer and neurodegeneration, where modulating specific fragment functions could alter pathological outcomes.
Differentiating Apoptotic Cleavage from Necrotic PARP-1 Processing
Poly(ADP-ribose) polymerase-1 (PARP-1) is a nuclear enzyme with multifaceted roles in cellular homeostasis, DNA repair, and cell death signaling. As a prominent substrate for various proteases, PARP-1 cleavage serves as a crucial biomarker for identifying specific cell death pathways. The detection of characteristic cleavage fragments provides researchers with valuable insights into whether cells are undergoing apoptosis or necrosis, which has significant implications for understanding disease mechanisms and developing therapeutic strategies. This application note details the experimental approaches for differentiating apoptotic from necrotic PARP-1 processing within the context of apoptosis detection methodologies, focusing on flow cytometry and western blot applications.
The fundamental difference between these processes lies in the proteases involved and the resulting cleavage fragments. Apoptotic cleavage is primarily mediated by caspases, producing signature fragments of 24 kDa and 89 kDa, while necrotic processing involves lysosomal proteases such as cathepsins, generating a distinct 50 kDa fragment [15] [11]. Understanding these differences enables researchers to accurately interpret cell death mechanisms in experimental models.
PARP-1 Cleavage Signatures in Apoptosis vs. Necrosis
Molecular Signatures and Protease Specificity
PARP-1 exhibits distinct cleavage patterns depending on the cell death pathway activated. The following table summarizes the key characteristics of PARP-1 processing in apoptosis versus necrosis:
Table 1: Characteristic PARP-1 Cleavage Patterns in Apoptosis and Necrosis
| Feature | Apoptotic Cleavage | Necrotic Cleavage |
|---|---|---|
| Primary Proteases | Caspases-3 and -7 [11] | Cathepsins B and G [15] |
| Characteristic Fragments | 24 kDa (DBD) + 89 kDa (CD+AMD) [4] [11] | 50 kDa fragment [15] |
| Catalytic Activity | Inactivated [11] | Not fully characterized |
| DNA Binding | 24 kDa fragment binds irreversibly to DNA [11] | Not fully characterized |
| Inhibitor Sensitivity | zVAD-fmk sensitive [15] | zVAD-fmk insensitive [15] |
| Associated Pathways | Caspase-dependent apoptosis [29] [11] | PARP-1-mediated necrosis [30] |
Biological Consequences of PARP-1 Cleavage
The functional outcomes of PARP-1 cleavage differ significantly between apoptosis and necrosis. In apoptosis, caspase-mediated cleavage generates a 24 kDa DNA-binding domain (DBD) fragment that remains tightly bound to DNA strand breaks, acting as a trans-dominant inhibitor of DNA repair by blocking access of repair enzymes to damaged sites [11]. This irreversible binding conserves cellular ATP pools and facilitates the apoptotic process. Simultaneously, the 89 kDa fragment containing the catalytic and automodification domains is liberated from the nucleus to the cytosol with greatly reduced DNA binding capacity [11].
In necrosis, the 50 kDa fragment resulting from lysosomal protease activity represents a fundamentally different processing mechanism. This cleavage pattern occurs independently of caspase activation and is associated with alternative cell death pathways involving PARP-1 hyperactivation [15] [30]. The functional consequences of this necrotic cleavage are less well characterized but represent an active area of investigation.
Experimental Detection Methodologies
Flow Cytometry for PARP-1 Cleavage Detection
Flow cytometry offers a powerful approach for detecting PARP-1 cleavage in individual cells, allowing for multiparametric analysis of cell death pathways while preserving cellular heterogeneity.
Protocol: Multiplexed Flow Cytometric Detection of Cell Death Pathways
Sample Preparation:
- Collect and wash cells in cold PBS
- For suspension cells: Use directly after treatment
- For adherent cells: Harvest using gentle enzymatic or mechanical dissociation
Cell Staining Procedure:
- Viability Staining: Resuspend cell pellet (~1×10⁶ cells) in Zombie NIR fixable viability dye (1:1000 dilution in PBS) and incubate for 15 minutes at room temperature, protected from light [31]
- Fixation: Wash cells twice with PBS, then resuspend in fixation Solution A (Caltag) for 15 minutes at room temperature [31]
- Permeabilization: Wash cells twice with PBS, then permeabilize with 0.25% Triton X-100 for 15 minutes at room temperature [31]
- Intracellular Staining: Incubate cells with antibody cocktail for 20 minutes at room temperature:
- Analysis: Wash cells and resuspend in 400 µL PBS for acquisition on a flow cytometer (e.g., ACEA Bioscience Novocyte 3000) [31]
Gating Strategy and Data Interpretation:
- Gate single cells using FSC-A vs FSC-H
- Identify viable (Zombie NIR-negative) and non-viable (Zombie NIR-positive) populations
- Analyze cleaved PARP-1 expression in conjunction with caspase-3 and RIP3:
- Apoptotic cells: Caspase-3+/cleaved PARP-1+/RIP3-
- Necroptotic cells: RIP3+/cleaved PARP-1+/caspase-3-
- Late apoptotic/necrotic: Zombie NIR+/cleaved PARP-1+ [31]
Table 2: Key Reagents for Flow Cytometric Detection of PARP-1 Cleavage
| Reagent | Specificity | Application | Reference |
|---|---|---|---|
| Anti-cleaved PARP-1 (Asp214) | Cleaved PARP-1 at aspartate 214 | Detection of apoptotic PARP-1 cleavage | [28] [19] |
| Anti-poly(ADP-ribose) (PAR) | Poly(ADP-ribose) polymers | Detection of PARP-1 activation | [28] [19] |
| Anti-active caspase-3 | Activated caspase-3 | Apoptosis marker | [31] |
| Zombie NIR fixable dye | Cell viability | Distinguishing live/dead cells | [31] |
| RIP3 antibody | RIP3 kinase | Necroptosis marker | [31] |
Western Blot Analysis of PARP-1 Cleavage
Western blotting provides complementary information to flow cytometry, allowing visualization of specific PARP-1 cleavage fragments and distinction between apoptotic and necrotic patterns.
Protocol: Western Blot Detection of PARP-1 Cleavage Fragments
Sample Preparation and Protein Extraction:
- Lyse cells in RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) supplemented with protease and phosphatase inhibitors
- Incubate on ice for 30 minutes with occasional vortexing
- Centrifuge at 14,000 × g for 15 minutes at 4°C
- Collect supernatant and quantify protein concentration using BCA assay
Electrophoresis and Immunoblotting:
- Separate 20-30 μg of protein by SDS-PAGE (8-12% gradient gel) at 100-120V for 1-2 hours
- Transfer to PVDF membrane using wet or semi-dry transfer system
- Block membrane with 5% non-fat milk in TBST for 1 hour at room temperature
- Incubate with primary antibodies diluted in blocking buffer overnight at 4°C:
- Anti-PARP-1 antibody (1:1000) to detect full-length (113 kDa) and fragments
- Anti-cleaved PARP-1 (Asp214) (1:1000) for apoptotic cleavage [29]
- Anti-β-actin (1:5000) or anti-GAPDH (1:5000) as loading control
- Wash membrane 3× with TBST, 10 minutes each
- Incubate with appropriate HRP-conjugated secondary antibody (1:5000) for 1 hour at room temperature
- Develop using enhanced chemiluminescence substrate and image with digital imaging system
Fragment Identification:
- Full-length PARP-1: 113 kDa
- Apoptotic fragments: 89 kDa and 24 kDa [4] [11]
- Necrotic fragment: 50 kDa [15]
Signaling Pathways in PARP-1-Mediated Cell Death
The following diagrams illustrate the key signaling pathways involved in PARP-1 cleavage during apoptosis and necrosis, highlighting the crucial differences in protease activation and fragment generation.
Diagram 1: PARP-1 Cleavage Pathways in Apoptosis vs. Necrosis
Integrated Experimental Workflow
The following diagram outlines a comprehensive experimental approach for differentiating apoptotic and necrotic PARP-1 cleavage using complementary techniques:
Diagram 2: Integrated Workflow for PARP-1 Cleavage Analysis
Research Reagent Solutions
The following table compiles essential reagents for studying PARP-1 cleavage in cell death pathways:
Table 3: Essential Research Reagents for PARP-1 Cleavage Studies
| Reagent Category | Specific Products | Application Purpose | Experimental Considerations |
|---|---|---|---|
| PARP-1 Antibodies | Anti-PARP-1 (full length), Anti-cleaved PARP-1 (Asp214), Anti-PAR antibody [28] [19] | Detecting PARP-1 expression, cleavage, and activity | Validate for specific applications (WB, FC, IF); check species reactivity |
| Protease Inhibitors | zVAD-fmk (caspase inhibitor), Necrostatin-1 (RIP1 inhibitor), CA-074 (cathepsin B inhibitor) [15] [30] | Pathway inhibition studies | Use appropriate concentrations; confirm specificity with multiple inhibitors |
| Cell Death Inducers | Staurosporine, H₂O₂, MNNG, β-Lapachone [15] [32] [30] | Inducing specific cell death pathways | Titrate for optimal response; include both apoptotic and necrotic inducers |
| Detection Systems | HRP-conjugated secondaries, fluorescently-labeled antibodies, ECL substrates [29] [31] | Signal detection and visualization | Match detection method to instrument capabilities; optimize signal-to-noise |
| Validation Tools | siRNA against PARP-1, caspase-3 knockout cells, PARP-1 deficient MEFs [4] [30] | Specificity controls and pathway validation | Include appropriate wild-type controls; confirm knockdown/knockout efficiency |
Differentiating apoptotic cleavage from necrotic PARP-1 processing requires complementary experimental approaches that leverage the specific characteristics of each cleavage pattern. Flow cytometry offers the advantage of single-cell analysis and multiparametric assessment of cell death pathways, while western blotting provides definitive fragment identification and clear distinction between the 89/24 kDa apoptotic fragments and the 50 kDa necrotic fragment.
The protocols and methodologies detailed in this application note provide researchers with a comprehensive framework for accurately identifying PARP-1 cleavage patterns in experimental systems. This differentiation is crucial for understanding cell death mechanisms in various pathological contexts, including neurodegenerative diseases, cancer biology, and toxicological assessments, ultimately supporting drug development efforts targeting specific cell death pathways.
Step-by-Step Protocols: From Cell Lysis to Data Acquisition
Within the broader investigation of apoptosis detection methodologies, Western blotting remains a foundational technique for identifying specific biochemical events, such as the cleavage of Poly (ADP-ribose) polymerase-1 (PARP-1). This protein is a well-established marker of apoptosis, and its detection via Western blot provides a key point of comparison for other techniques like flow cytometry. This protocol details a standardized method for preparing cell lysates, performing SDS-PAGE, and immunodetecting both full-length and cleaved PARP-1, enabling reliable assessment of apoptotic activity in research and drug development.
Background: PARP-1 as a Key Apoptosis Marker
PARP-1 is a 116 kDa nuclear enzyme that functions as a primary responder to DNA damage, playing a critical role in DNA repair pathways [33] [34]. During the execution phase of apoptosis, caspase-3, and other effector caspases, cleave PARP-1 at the Asp214-Gly215 bond [33] [35]. This proteolytic event separates the 24 kDa DNA-binding domain from the 89 kDa catalytic domain, resulting in the inactivation of PARP-1's DNA repair function and facilitating cellular disassembly [33] [20]. The appearance of the 89 kDa fragment is thus a definitive biochemical hallmark of caspase-mediated apoptosis. Research continues to elucidate the complex role of PARP-1 in cell fate, including its functions in novel forms of regulated cell death, such as its involvement in the crosstalk between ferroptosis and apoptosis [20], and its interactions with key immune signaling molecules like STING in response to severe DNA damage [10].
Research Reagent Solutions
The following table catalogues essential reagents for the successful detection of PARP-1 and its cleaved form in Western blot experiments.
Table 1: Key Reagents for PARP-1 Western Blot Analysis
| Reagent | Specific Product Example | Function in the Protocol |
|---|---|---|
| Anti-Cleaved PARP (Asp214) Antibody | Cleaved PARP (Asp214) (D64E10) XP Rabbit mAb #5625 [33] | Specifically detects the 89 kDa caspase-cleaved fragment of PARP-1; does not recognize full-length PARP-1. |
| Anti-PARP Antibody | PARP Antibody #9542 [34] | Detects both full-length PARP-1 (116 kDa) and the large cleaved fragment (89 kDa). |
| Cell Lysis Buffer | RIPA Lysis Buffer [36] [37] | Efficiently extracts total cellular protein, including membrane-bound and nuclear proteins like PARP-1. |
| Protease Inhibitor Cocktail | Halt Protease and Phosphatase Inhibitor Cocktail [36] | Prevents proteolytic degradation of target proteins, including PARP-1 and its cleavage fragments, during lysate preparation. |
| Chemiluminescent Substrate | SuperSignal West Pico PLUS Chemiluminescent Substrate [38] | Provides high-sensitivity detection for horseradish peroxidase (HRP)-conjugated secondary antibodies. |
Detailed Experimental Protocol
Stage 1: Cell Lysis and Sample Preparation
Proper cell lysis is critical for the accurate detection of PARP-1, a nuclear protein. The following procedure ensures complete protein extraction while maintaining integrity.
Materials:
- Ice-cold Phosphate-Buffered Saline (PBS)
- RIPA Lysis Buffer [36] [37]
- Protease Inhibitor Cocktail (add to lysis buffer immediately before use) [36] [39]
- BCA or Bradford Protein Assay Kit [36] [37]
- 4X LDS Sample Buffer [36]
- Sample Reducing Agent (e.g., Dithiothreitol (DTT)) [37]
Procedure:
- Prepare Lysis Buffer: Add protease inhibitor cocktail to ice-cold RIPA buffer (e.g., 10 µL of inhibitor per 1 mL of buffer) [36].
- Wash and Harvest Cells:
- Lyse Cells: Add ice-cold lysis buffer to the cell pellet or dish (~100-200 µL for a 6-well plate, ~1 mL per 10⁷ cells) [36] [37]. Incubate on ice for 10-15 minutes with periodic agitation [37].
- Clarify Lysate: Transfer the lysate to a microcentrifuge tube and centrifuge at 14,000-17,000 x g for 15 minutes at 4°C [36] [39]. Carefully transfer the supernatant (containing the soluble proteins) to a new tube and discard the pellet.
- Determine Protein Concentration: Use a BCA or Bradford assay to determine the protein concentration of each sample, following the kit manufacturer's instructions [36] [37]. The BCA assay is often preferred for RIPA buffer lysates as it is less affected by detergents [36].
- Prepare Samples for Electrophoresis: Dilute lysates with 4X LDS sample buffer and reducing agent (e.g., DTT) to a final 1X concentration [36]. Heat the samples at 70°C for 10 minutes to denature proteins [36]. At this stage, samples can be stored at -20°C or loaded directly onto a gel.
Stage 2: SDS-PAGE and Protein Transfer
This stage separates proteins by molecular weight and transfers them to a membrane for immunodetection.
Materials:
- Precast SDS-PAGE Gel (e.g., 4-12% Bis-Tris gradient gel) [37]
- SDS-PAGE Running Buffer (e.g., MOPS or MES for Bis-Tris gels) [37]
- Transfer Buffer [38]
- Nitrocellulose or PVDF Membrane [38]
- Methanol (for PVDF membrane activation)
Procedure:
- Set Up Gel Electrophoresis:
- Assemble the gel electrophoresis unit and fill the chamber with running buffer.
- Load 10-40 µg of total protein per well [37]. Include a pre-stained protein molecular weight ladder in one lane.
- Run the gel at a constant voltage (e.g., 120-150V) until the dye front nears the bottom, following the gel manufacturer's recommendations.
- Prepare for Protein Transfer:
- Transfer Proteins: Using a wet or semi-dry transfer apparatus, transfer proteins from the gel to the membrane according to the system's instructions. A common wet transfer condition is 100V for 60-90 minutes on ice [38].
Stage 3: Antibody Incubation and Detection
This stage uses specific antibodies to detect PARP-1 and its cleaved fragment.
Materials:
- Blocking Buffer (e.g., 5% non-fat dry milk or commercial blocking buffers) [38]
- Wash Buffer (TBST or PBST: TBS or PBS with 0.05% Tween 20) [38]
- Primary Antibody (see Table 1 for options)
- HRP-conjugated Secondary Antibody [38]
- Chemiluminescent HRP Substrate [38]
Procedure:
- Block Membrane: Incubate the membrane in a sufficient volume of blocking buffer for 30-60 minutes at room temperature with agitation [38].
- Incubate with Primary Antibody:
- Dilute the primary antibody in blocking buffer as per the manufacturer's datasheet. For example:
- Incubate the membrane with the primary antibody solution for 1 hour at room temperature or overnight at 2-8°C with agitation [38].
- Wash Membrane: Wash the membrane 3 times for 10 minutes each with wash buffer (TBST/PBST) to remove unbound antibody [38].
- Incubate with Secondary Antibody:
- Dilute the HRP-conjugated secondary antibody (e.g., Goat Anti-Rabbit) in wash buffer (typical dilutions range from 1:20,000 to 1:100,000) [38].
- Incubate the membrane for 1 hour at room temperature with agitation.
- Wash Membrane: Wash the membrane 6 times for 5 minutes each with wash buffer to thoroughly remove any unbound secondary antibody [38].
- Detect Signal:
- Mix the chemiluminescent substrate components as per the manufacturer's instructions.
- Incubate the membrane with the substrate working solution for 1-5 minutes.
- Drain excess reagent and image the blot using a digital imager or X-ray film [38].
Data Interpretation and Expected Results
A successful Western blot for apoptosis detection will show distinct bands corresponding to the molecular weights of full-length and cleaved PARP-1.
- Non-apoptotic cells: A single, dominant band at 116 kDa (full-length PARP-1).
- Apoptotic cells: A dominant band at 89 kDa (cleaved PARP-1 fragment) and a corresponding decrease in the intensity of the 116 kDa band [33] [34] [35].
The PARP Antibody #9542 will detect both forms, providing a complete picture, while the Cleaved PARP (Asp214) Antibody #5625 is specific for the 89 kDa fragment, offering high specificity for apoptosis confirmation [33] [34].
Experimental Workflow Diagram
The following diagram illustrates the key stages of the Western blot protocol for PARP-1 detection.
This detailed application note provides a robust Western blot protocol for the specific detection of PARP-1 cleavage, a cornerstone method for confirming apoptosis in cellular models. When framed within a broader thesis comparing flow cytometry and Western blotting, this protocol highlights Western blot's unique strength in providing direct, size-based molecular evidence of a key apoptotic event. The quantitative data on protein loading and molecular weight, combined with the high specificity of modern antibodies, makes this technique an indispensable tool for researchers and drug development professionals validating apoptotic pathways.
Poly (ADP-ribose) polymerase-1 (PARP-1) is a nuclear enzyme with a well-established role in the detection and repair of DNA single-strand breaks via the base excision repair pathway [40] [11]. Beyond its DNA repair functions, PARP-1 is a critical substrate for several cell-death proteases and is widely recognized as a biochemical hallmark of apoptosis. During programmed cell death, PARP-1 is cleaved by activated caspases at specific sites, most notably the Asp214 residue located within its nuclear localization signal in the DNA-binding domain [4] [11]. This proteolytic cleavage event terminates the DNA repair capacity of PARP-1 and facilitates the disassembly of the cell, serving as a reliable and early marker for apoptosis detection in research and drug development. Distinguishing between the full-length and cleaved forms of PARP-1 provides crucial information about cellular commitment to death pathways, making it an essential readout in cancer research, neurodegenerative disease studies, and therapeutic efficacy assessments [29] [11].
Biological Significance of PARP-1 Cleavage
The Cleavage Event and Fragment Generation
PARP-1 cleavage occurs primarily via caspase-3 and caspase-7 at the conserved sequence DEVD214↓G, generating two signature fragments: a 24 kDa DNA-binding domain (DBD) fragment and an 89 kDa catalytic fragment [11]. The 24 kDa fragment contains two zinc finger motifs that allow it to bind irreversibly to DNA strand breaks, acting as a trans-dominant inhibitor of DNA repair by blocking access to damaged DNA for other repair enzymes [11] [20]. Concurrently, the 89 kDa fragment, which contains the automodification domain and catalytic site, is liberated from the nucleus into the cytoplasm [11]. Research indicates that these fragments are not merely inert byproducts of cleavage but may actively regulate cellular viability and inflammatory responses in opposing ways [4].
Table: PARP-1 Proteolytic Fragments and Their Properties
| Fragment Size | Domains Contained | Cellular Localization After Cleavage | Functional Consequences |
|---|---|---|---|
| 24 kDa | DNA-binding domain (zinc fingers) | Retained in nucleus | Irreversibly binds DNA breaks, inhibits DNA repair |
| 89 kDa | Automodification domain, Catalytic domain | Liberated to cytoplasm | May directly induce caspase-mediated DNA fragmentation |
Functional Consequences in Cell Death and Inflammation
The cleavage of PARP-1 serves multiple physiological purposes during apoptosis. First, it inactivates the DNA repair function of PARP-1, preventing wasteful energy consumption (NAD+ and ATP depletion) in a doomed cell and ensuring efficient apoptotic progression [11]. Second, the generated fragments may actively participate in cell death regulation. Studies demonstrate that the 24 kDa fragment confers protection from ischemic damage in neuronal models, while the 89 kDa fragment exhibits cytotoxic properties and enhances pro-inflammatory NF-κB activity [4]. This functional divergence highlights the importance of specifically detecting these individual fragments rather than simply assessing total PARP-1 levels. Furthermore, PARP-1 cleavage is considered a hallmark of apoptosis, with its detection providing a reliable indicator of caspase activation and commitment to programmed cell death [29] [11].
Antibody-Based Detection Methods
Key Antibody Characteristics and Targets
The specific detection of full-length versus cleaved PARP-1 relies on antibodies with well-defined epitope recognition. The mouse monoclonal antibody PARP-1 (F-2) (sc-8007) serves as an excellent example, raised against amino acids 764-1014 mapping at the C-terminus of PARP of human origin [40]. This antibody recognizes both the full-length PARP-1 and the C-terminal 89 kDa cleavage fragment, making it particularly valuable for apoptosis detection. For research focusing specifically on the caspase cleavage event, antibodies targeting the neo-epitope created by cleavage at Asp214 are essential. The anti-cleaved PARP-1 (Asp214) antibody (clone F21-852) is specifically designed for this purpose and is effectively used in flow cytometric assays to distinguish apoptotic populations [28].
Table: Key Antibodies for PARP-1 Detection
| Antibody Target | Clone/Name | Recognized Forms | Primary Applications | Key Features |
|---|---|---|---|---|
| C-terminus (aa 764-1014) | PARP-1 (F-2) | Full-length & 89 kDa fragment | WB, IP, IF, IHC(P), ELISA | Mouse monoclonal IgG2a; detects both intact and cleaved C-terminal fragment |
| Cleavage site (Asp214) | F21-852 | Cleaved form only | Flow cytometry, IF | FITC-conjugated; specific for apoptosis-associated cleavage neo-epitope |
| Poly(ADP-ribose) polymer | 10H | PAR polymers | Flow cytometry, WB | Detects PARP activity; mouse monoclonal |
Method Comparison: Western Blot vs. Flow Cytometry
The choice between western blot and flow cytometry for PARP-1 cleavage detection depends on the research question, sample type, and required throughput.
Western Blotting provides high specificity for detecting the precise molecular weights of PARP-1 fragments (116 kDa full-length, 89 kDa cleavage product), allowing researchers to confirm the specific cleavage pattern associated with apoptosis [29]. This method enables the simultaneous assessment of multiple apoptosis markers (caspases, Bcl-2 family proteins) in the same sample, providing a comprehensive view of cell death pathways. However, western blotting is semi-quantitative at best, requires more cells, and lacks single-cell resolution.
Flow Cytometry offers rapid, quantitative analysis of PARP-1 cleavage at the single-cell level within heterogeneous populations, allowing researchers to identify subpopulations of apoptotic cells and correlate PARP-1 cleavage with other cellular markers [28]. The technique is suitable for high-throughput screening and can be combined with cell surface immunophenotyping. However, flow cytometry typically requires specific antibody conjugations and provides less direct information about fragment sizes compared to western blot.
Detailed Experimental Protocols
Western Blot Protocol for PARP-1 Cleavage Detection
Sample Preparation and Protein Extraction
Begin by lysing cells in RIPA or similar lysis buffer supplemented with protease and phosphatase inhibitors. For adherent cells, wash with cold PBS and scrape directly into lysis buffer. For tissue samples, homogenize thoroughly in lysis buffer. Centrifuge lysates at 15,000 × g for 15 minutes at 4°C to remove insoluble material. Quantify protein concentration using a BCA or Bradford assay, ensuring equal loading across samples [29].
Gel Electrophoresis and Transfer
Load 20-50 μg of total protein per lane onto 7.5-10% SDS-polyacrylamide gels, which provide optimal resolution for distinguishing full-length PARP-1 (116 kDa) from its major cleavage fragment (89 kDa). Include pre-stained molecular weight markers for accurate size determination. After separation, transfer proteins to PVDF or nitrocellulose membranes using wet or semi-dry transfer systems. Confirm successful transfer with Ponceau S staining if desired [29].
Immunodetection
Block membranes with 5% non-fat dry milk or BSA in TBST for 1 hour at room temperature. Incubate with primary antibodies diluted in blocking buffer overnight at 4°C. Key antibodies include:
- PARP-1 (F-2) (1:1000) for detecting both full-length and the 89 kDa C-terminal fragment [40]
- Cleaved PARP (Asp214) for apoptosis-specific detection
- Caspase-3 antibodies to corroborate apoptosis induction
- β-actin or GAPDH as loading controls
After TBST washes, incubate with appropriate HRP-conjugated secondary antibodies (e.g., m-IgG Fc BP-HRP or m-IgG2a BP-HRP for PARP-1 (F-2)) for 1-2 hours at room temperature [40]. Develop using enhanced chemiluminescence substrate and image with a digital imaging system.
Analysis and Interpretation
Quantify band intensities using densitometry software such as ImageJ. Calculate the ratio of cleaved PARP-1 (89 kDa) to full-length PARP-1 (116 kDa) to assess the extent of apoptosis. Normalize signals to loading controls to account for variations in protein loading. The appearance of the 89 kDa fragment with corresponding decrease in full-length PARP-1 indicates apoptotic activity [29].
Flow Cytometry Protocol for Cleaved PARP-1 Detection
Cell Preparation and Staining
Harvest cells and wash twice with cold PBS. Fix cells with Cytofix/Cytoperm solution for 20 minutes at 4°C, then permeabilize with Perm/Wash buffer for an additional 20 minutes to allow intracellular antibody access [28]. Incubate cells with saturating amounts of FITC-conjugated anti-cleaved PARP-1 (Asp214) antibody (clone F21-852) for 45 minutes at 4°C in the dark. Include isotype control antibodies to establish background fluorescence.
Data Acquisition and Analysis
After staining, wash cells twice with Perm/Wash buffer and resuspend in flow cytometry staining buffer. Acquire data using a flow cytometer equipped with a 488 nm laser and appropriate filter for FITC detection (typically 530/30 nm bandpass filter). Analyze a minimum of 10,000 events per sample. Use forward versus side scatter gating to exclude debris and focus on the intact cell population. Determine the percentage of cleaved PARP-1 positive cells by comparing fluorescence intensity to the isotype control [28].
Research Reagent Solutions
Table: Essential Research Reagents for PARP-1 Detection
| Reagent Category | Specific Examples | Research Applications | Key Considerations |
|---|---|---|---|
| Primary Antibodies | PARP-1 (F-2) (sc-8007) [40] | Detects full-length and C-terminal fragment in WB, IP, IF | Mouse monoclonal; target C-terminus (aa 764-1014) |
| Anti-cleaved PARP (Asp214) (F21-852) [28] | Specific detection of apoptotic cells in flow cytometry | FITC-conjugated; recognizes caspase cleavage neo-epitope | |
| Secondary Detection | m-IgG Fc BP-HRP, m-IgG2a BP-HRP [40] | Secondary detection for WB with PARP-1 (F-2) | Optimized for specific mouse IgG subclasses |
| Assay Kits | Cytofix/Cytoperm Fixation/Permeabilisation Kit [28] | Intracellular staining for flow cytometry | Critical for antibody access to nuclear PARP-1 |
| Apoptosis Western Blot Cocktail (ab136812) [29] | Simultaneous detection of multiple apoptosis markers | Includes caspases, PARP, actin controls | |
| PARP Inhibitors | ABT-888 (Veliparib), Olaparib [41] [28] | Experimental controls for PARP activity studies | Catalytic inhibitors; useful for validating specificity |
Troubleshooting and Technical Considerations
Common Challenges and Solutions
Incomplete or No PARP-1 Cleavage Detection: Ensure apoptosis has been adequately induced in your experimental system. Include positive controls (e.g., cells treated with known apoptosis inducers like staurosporine) to validate your detection system. Optimize antibody concentrations and incubation times, as under- or over-staining can lead to weak signals or high background.
High Background in Western Blot: Increase blocking time or try different blocking agents (BSA vs. non-fat milk). Optimize washing stringency and antibody concentrations. For flow cytometry, ensure proper titration of antibodies and use of isotype controls to set appropriate gates [29] [28].
Multiple Bands in Western Blot: PARP-1 can be cleaved by proteases other than caspases (calpains, cathepsins, granzymes, MMPs), generating fragments of different sizes (50-55 kDa, 40-42 kDa, 35-36 kDa) [11]. Use antibodies targeting specific cleavage sites (e.g., Asp214 for caspases) to distinguish between apoptosis and other cell death pathways. Ensure fresh protease inhibitors are included in lysis buffers.
Method-Specific Optimization
For Western Blot: Ensure proper protein separation by using fresh electrophoresis buffers and appropriate gel percentages. Validate antibody specificity by including lysates from PARP-1 knockout cells if available. For quantification, ensure signals are within the linear range of detection by testing different exposure times [29].
For Flow Cytometry: Optimize fixation and permeabilization conditions, as over-permeabilization can damage cellular structures and increase background. Include single-stained controls for compensation when performing multiparameter panels. Consider including viability dyes to exclude false-positive signals from dead cells [28].
The detection of PARP-1 cleavage, particularly at the Asp214 site, remains a cornerstone method for apoptosis assessment in biomedical research. The selection of appropriate antibodies, particularly those specific for either the C-terminal region (e.g., PARP-1 (F-2)) or the caspase cleavage neo-epitope (e.g., anti-Asp214), combined with properly executed western blot or flow cytometry protocols, provides researchers with robust tools for quantifying programmed cell death. As research continues to reveal the complex roles of PARP-1 fragments in cell death, inflammation, and disease pathogenesis [4] [11], these detection methods will remain essential for advancing our understanding of cell death mechanisms and developing novel therapeutic strategies.
Within the broader context of apoptosis detection methodologies, the cleavage of poly(ADP-ribose) polymerase-1 (PARP-1) serves as a critical biochemical hallmark of programmed cell death. During the execution phase of apoptosis, activated caspase-3 and caspase-7 cleave the 113-kDa PARP-1 protein into characteristic fragments of 24 kDa and 89 kDa [11]. This proteolytic event represents a definitive commitment to apoptosis, as the 24-kDa DNA-binding fragment irreversibly binds to DNA strand breaks, inhibiting DNA repair and facilitating cellular dismantling [11]. The detection and quantification of these cleavage fragments provide researchers with a powerful tool for investigating apoptotic pathways in various experimental models, from basic research to drug development.
The choice between flow cytometry and Western blot for PARP-1 cleavage detection represents a fundamental methodological consideration, each offering distinct advantages and limitations. Flow cytometry enables single-cell analysis of PARP-1 cleavage while maintaining cellular heterogeneity, allowing for the simultaneous assessment of multiple parameters including cell surface markers, viability, and intracellular targets within the same cell [42] [19]. Conversely, Western blot provides population-average data that lacks cellular resolution but offers definitive fragment size confirmation. This application note details optimized protocols for flow cytometric detection of intracellular antigens, with particular emphasis on PARP-1 cleavage detection within the context of apoptosis research.
Technical Comparison: Flow Cytometry vs. Western Blot for PARP-1 Detection
Table 1: Methodological Comparison for PARP-1 Cleavage Detection
| Parameter | Flow Cytometry | Western Blot |
|---|---|---|
| Cellular Resolution | Single-cell level analysis maintained [19] | Population average; cellular context lost |
| Multiparametric Capability | High (cell surface markers, viability, phospho-signaling) [42] [43] | Limited typically to target protein and loading control |
| Throughput | Higher (96-well plate format possible) [42] | Lower (typically 10-20 samples per gel) |
| Semi-Quantification | Excellent (based on fluorescence intensity) [19] | Good (based on band density) |
| Fragment Sizing | Indirect (via antibody specificity) [19] | Direct (based on molecular weight) [11] |
| Required Cell Number | 5×10⁵ to 1×10⁶ cells [44] | Typically 1-5×10⁶ cells per sample |
| Key Application | Phenotyping apoptotic populations, signaling studies | Definitive fragment identification, cleavage efficiency |
Flow Cytometry Protocol for Intracellular Antigen Detection
The successful detection of intracellular proteins such as cleaved PARP-1 requires careful optimization of fixation and permeabilization conditions to preserve epitope integrity while allowing antibody access. The following protocols have been validated for detection of intracellular antigens including PARP-1 cleavage fragments [19].
Surface and Intracellular Staining for Apoptosis Detection
This protocol enables simultaneous analysis of cell surface markers and intracellular PARP-1 cleavage fragments, allowing for the identification of specific apoptotic cell populations within heterogeneous samples.
Materials Required:
- 12×75 mm round-bottom test tubes or 96-well U-bottom plates
- Intracellular Fixation & Permeabilization Buffer Set (e.g., cat. no. 88-8824) [42]
- Flow Cytometry Staining Buffer (PBS with 0.5-1% BSA) [42] [44]
- Fluorochrome-conjugated antibodies against cell surface markers and cleaved PARP-1
- Fixable Viability Dye (e.g., eFluor series) [42]
- Permeabilization Buffer (10X) [42]
Experimental Procedure:
Prepare single-cell suspension (1×10⁶ to 1×10⁷ cells/mL) in Flow Cytometry Staining Buffer [42] [43].
(Optional) Viability Staining: Resuspend cell pellet in Fixable Viability Dye diluted in PBS. Incubate for 10-30 minutes at 4°C in the dark. Wash with 2 mL of Flow Cytometry Staining Buffer [42] [43].
Cell Surface Staining: Resuspend cell pellet in 100 µL of Flow Cytometry Staining Buffer containing titrated antibodies against cell surface markers. Incubate for 20-60 minutes at 4°C in the dark. Wash with 2 mL of Flow Cytometry Staining Buffer [42].
Fixation: After the last wash, resuspend the cell pellet in 100 µL (for tubes) or 200 µL (for 96-well plates) of IC Fixation Buffer. Vortex gently and incubate for 20-60 minutes at room temperature, protected from light [42].
Permeabilization:
- Add 2 mL (tubes) or 200 µL (plate) of 1X Permeabilization Buffer and centrifuge at 400-600× g for 5 minutes. Discard supernatant.
- Repeat the wash step with Permeabilization Buffer [42].
Intracellular Staining: Resuspend fixed and permeabilized cells in 100 µL of 1X Permeabilization Buffer containing the recommended amount of antibody against cleaved PARP-1 (e.g., clone F21-852 specific for Asp214) [19]. Incubate for 20-60 minutes at room temperature, protected from light.
Washing: Add 2 mL of 1X Permeabilization Buffer, centrifuge, and discard supernatant. Repeat this wash step once [42].
Acquisition: Resuspend stained cells in an appropriate volume of Flow Cytometry Staining Buffer (typically 200-400 µL) and analyze by flow cytometry immediately or store at 4°C for analysis within 24 hours [42] [44].
Fixation and Permeabilization Methods
The choice of fixation and permeabilization method significantly impacts antibody binding and signal quality. The following table summarizes common approaches optimized for different intracellular targets.
Table 2: Fixation and Permeabilization Methods for Intracellular Staining
| Method | Fixative | Permeabilization Agent | Primary Application | Key Considerations |
|---|---|---|---|---|
| Aldehyde-Detergent | 1-4% Paraformaldehyde (PFA) [43] [44] | Triton X-100 (0.1-0.5%) [43] [44] | Cytoplasmic proteins, cytokines [42] | Preserves protein structure; compatible with protein fluorophores [45] |
| Transcription Factor Buffer Set | Foxp3 Fixation/Permeabilization Concentrate [42] | Combined in working solution [42] | Nuclear antigens, transcription factors [42] | One-step fixation/permeabilization; optimized for nuclear targets |
| Methanol-Based | 4% PFA or standalone [46] [45] | Ice-cold Methanol (90-100%) [43] [46] | Phosphoproteins (e.g., STATs), nuclear antigens [46] [45] | Denatures protein fluorophores (PE, APC); can unmask some epitopes [46] |
| Saponin-Based | 4% PFA [45] | Saponin (0.1-0.5%) [43] [45] | Cytosolic antigens, labile epitopes [43] | Reversible permeabilization; requires saponin in all buffers [45] |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for PARP-1 Cleavage Detection
| Reagent | Function | Example Products |
|---|---|---|
| Intracellular Fixation Buffer | Stabilizes cellular structures and antigens by cross-linking; halts biological processes. | Intracellular Fixation Buffer (cat. no. 88-8824) [42]; 4% Formaldehyde, Methanol-Free (#47746) [44] |
| Permeabilization Buffer | Dissolves membrane lipids to create pores for antibody access to intracellular compartments. | Permeabilization Buffer (10X) [42]; Cell Permeabilization Buffer (#39487) with Triton X-100 [44] |
| Fixable Viability Dyes | Distinguishes live from dead cells based on covalent binding to amine groups in non-viable cells, critical for excluding false positives. | eFluor 450, 506, 660, 780 [42] |
| Fc Receptor Blocking Reagent | Reduces non-specific antibody binding by blocking Fc receptors on immune cells. | Normal Mouse/Rat Serum [42]; Purified anti-CD16/CD32 [43] |
| Anti-Cleaved PARP Antibody | Specifically detects the caspase-generated neo-epitope of PARP-1 (e.g., at Asp214), providing a direct marker of apoptosis. | FITC anti-cleaved PARP-1 (Asp214) (Clone F21-852) [19] |
| Flow Cytometry Staining Buffer | Provides a protein-rich environment for antibody dilutions and washes to minimize non-specific background staining. | Flow Cytometry Staining Buffer (#00-4222) [42]; PBS with 0.5% BSA [44] |
Workflow and Signaling Pathway Visualization
Experimental Workflow for PARP-1 Cleavage Detection
PARP-1 Cleavage in Apoptotic Signaling Pathway
Flow cytometric detection of PARP-1 cleavage fragments represents a powerful methodology for apoptosis research, particularly when combined with cell surface phenotyping to identify specific responding populations within complex samples. The protocols detailed in this application note, utilizing optimized fixation and permeabilization conditions, provide researchers with robust tools for investigating cell death mechanisms in both basic research and drug development contexts. When compared with Western blot analysis, flow cytometry offers superior cellular resolution and multiparametric capabilities, making it ideally suited for screening applications and heterogeneous sample analysis, while Western blot remains the gold standard for definitive fragment identification.
The detection of apoptosis is a cornerstone of research in cell biology, cancer therapeutics, and drug development. While Western blotting has been a traditional method for identifying key apoptotic events, such as the cleavage of Poly(ADP-ribose) polymerase-1 (PARP-1), it lacks the ability to provide multidimensional analysis at the single-cell level. Multiparametric flow cytometry overcomes this limitation by enabling the simultaneous detection of multiple apoptotic characteristics within a heterogeneous cell population. This application note details a protocol for integrating the analysis of PARP-1 cleavage—a hallmark mid-to-late apoptotic event—with the detection of phosphatidylserine externalization (Annexin V binding) and caspase-3 activation. This tri-parametric approach provides a powerful tool for delineating the complex and sequential progression of cell death, offering significant advantages over single-parameter assays for mechanistic studies and high-throughput drug screening [47] [48].
Background and Significance
The Apoptotic Cascade and Key Biomarkers
Apoptosis is executed via a cascade of proteolytic enzymes known as caspases. Caspase-3 is a key effector caspase that, upon activation, cleaves a multitude of cellular substrates, including PARP-1 [29]. The cleavage of PARP-1 at the DEVD214 motif by caspase-3 (and caspase-7) serves as a definitive biochemical marker of apoptosis, generating signature fragments of 24 kDa and 89 kDa [4] [8]. This event inactivates PARP-1's DNA repair function, facilitating the dismantling of the cell. Prior to PARP-1 cleavage, cells undergoing apoptosis externalize phosphatidylserine (PS) from the inner to the outer leaflet of the plasma membrane, a phenomenon that can be detected by the binding of fluorescently conjugated Annexin V. The combination of these markers allows researchers to capture a continuum of apoptotic stages: from early (caspase activation and PS exposure) to intermediate/late stages (PARP-1 cleavage and loss of membrane integrity) [47] [48].
Flow Cytometry vs. Western Blot in Apoptosis Detection
The choice between flow cytometry and Western blotting for apoptosis detection depends on the research question. The following table summarizes their core distinctions:
Table 1: Comparison of Apoptosis Detection by Flow Cytometry and Western Blot
| Feature | Multiparametric Flow Cytometry | Western Blot |
|---|---|---|
| Analysis Level | Single-cell | Bulk population |
| Multiparameter Capacity | High (3+ markers simultaneously) | Low (typically 1-2 markers per blot) |
| Information Obtained | Heterogeneity, quantification of subpopulations | Presence/absence and cleavage status of protein |
| Throughput | High | Medium to Low |
| Primary Output | Percentage of cells in specific death stages | Band intensity indicating protein cleavage |
| Key Apoptosis Markers | PARP-1 cleavage, caspase activation, PS exposure, membrane integrity | PARP-1 cleavage, caspase activation |
The primary advantage of flow cytometry is its ability to identify and quantify distinct cell subpopulations—such as those that are Annexin V positive but still have an intact membrane (early apoptotic) versus those that are positive for both Annexin V and a viability dye (late apoptotic/necrotic)—all while simultaneously measuring caspase activity or intracellular PARP-1 cleavage [47] [29]. This is invaluable for understanding the dynamics of cell death in response to experimental treatments.
Experimental Design and Workflow
This protocol is designed for the tri-parametric analysis of apoptosis using a flow cytometer equipped with a 488 nm laser and a red (633-640 nm) laser.
Assay Principle and Logical Workflow
The assay logic progresses from early to late apoptotic events, enabling the classification of cells into distinct stages based on the combination of markers.
Gating Strategy and Data Interpretation
The following diagram illustrates the sequential gating strategy for data analysis, from selecting single cells to the final identification of apoptotic subpopulations based on the three key parameters.
Research Reagent Solutions
A successful multiparametric assay depends on the careful selection and combination of reagents. The table below lists essential materials and their functions.
Table 2: Key Research Reagents for Multiparametric Apoptosis Analysis
| Reagent | Specificity/Function | Key Characteristics | Example Products |
|---|---|---|---|
| Anti-PARP-1 (Cleaved Form) Antibody | Detects the 89 kDa caspase-derived fragment of PARP-1 | Must be validated for flow cytometry; confirms executioner caspase activity. | Multiple commercial suppliers |
| Fluorogenic Caspase Substrate | Measures caspase-3/7 activity | Cell-permeable, becomes fluorescent upon cleavage. Early apoptotic marker. | PhiPhiLux G1D2 [47], FAM-DEVD-FMK (FLICA) [49] [50], CellEvent Caspase-3/7 |
| Annexin V Conjugate | Binds externalized Phosphatidylserine (PS) | Marks early apoptosis. Requires calcium in buffer. | Annexin V-PE, Annexin V-APC [48] |
| DNA Binding Dye / Viability Probe | Assesses plasma membrane integrity | Distinguishes late apoptotic/necrotic cells (permeable) from early apoptotic/viable cells (impermeable). | Propidium Iodide (PI) [47] [48], 7-AAD [47] [48] |
| Wash Buffer | Cell washing and staining | Must contain Ca²⁺ for Annexin V binding. | Dulbecco's PBS with calcium/magnesium and 2% FBS [48] |
Detailed Experimental Protocol
Sample Preparation and Staining
This procedure is optimized for suspension cells but can be adapted for adherent cells.
- Induce Apoptosis & Harvest Cells: Treat cells with the apoptotic stimulus of choice (e.g., RSL3 [20]). Include untreated and positive control (e.g., Staurosporine-treated) samples. Harvest cells, wash once with cold PBS, and resuspend at 1-5 x 10^6 cells/mL in complete medium or wash buffer.
- Stain for Active Caspases:
- For FLICA (FAM-DEVD-FMK): Dilute the FLICA reagent according to the manufacturer's instructions. Add the diluted FLICA to the cell suspension and incubate for 45-60 minutes at 37°C in the dark [49] [50]. FLICA is cell-permeable and covalently binds to active caspases.
- For PhiPhiLux G1D2: Dilute the substrate and incubate with cells for 45-60 minutes at 37°C. Note that PhiPhiLux is not immobilized and requires prompt analysis post-staining [47].
- Wash Cells: Pellet cells and wash twice with 1X Apoptosis Wash Buffer to remove unbound reagent.
- Stain with Annexin V and Viability Dye: Resuspend the cell pellet in Annexin V Binding Buffer. Add the appropriate volume of fluorescently conjugated Annexin V (e.g., Annexin V-APC) and a DNA dye like 7-AAD or PI. Incubate for 15-20 minutes at room temperature in the dark.
- Fix Cells (Optional): If required for downstream analysis or biosafety, cells can be fixed after FLICA staining (but not PhiPhiLux) using a mild paraformaldehyde solution (e.g., 1-2%). Fixation must be performed after the caspase staining and wash steps, but can be done before or after the Annexin V/viability dye staining [47] [50].
- Acquire Data by Flow Cytometry: Analyze samples promptly on a flow cytometer. Keep samples on ice in the dark until acquisition.
Flow Cytometry Configuration
Configure your cytometer's lasers and detectors according to the fluorochromes used. A common configuration is:
- 488 nm laser: Detect FLICA (FITC/FAM channel ~530 nm) and PI/7-AAD (PI: ~617 nm; 7-AAD: ~670 nm).
- 633 nm red laser: Detect Annexin V-APC (~660 nm).
Adjust PMT voltages using single-stained and unstained controls, and apply compensation to correct for spectral overlap.
Data Analysis and Interpretation
The power of this multiparametric approach lies in the stratification of cells into distinct stages of health and death. The table below provides a guide for interpreting the complex phenotype of each population.
Table 3: Phenotypic Interpretation of Multiparametric Apoptosis Assay Results
| Caspase-3/7 (FLICA) | Annexin V | Viability Dye (e.g., 7-AAD) | PARP-1 Cleavage | Cell Status Interpretation |
|---|---|---|---|---|
| - | - | - | - | Viable, healthy cells. No apoptosis. |
| + | - | - | - | Early apoptosis: Caspase pathway activated, but PS not yet externalized. |
| + | + | - | +/- | Early/Mid apoptosis: PS externalized, caspases active. PARP-1 cleavage may begin. |
| + | + | + | + | Late apoptosis: Loss of membrane integrity, caspases active, PARP-1 cleaved. |
| - | + | + | - | Necrotic cells or late apoptotic secondary necrotic cells. |
Application in Research
This robust protocol has wide-ranging applications, particularly in translational research. It is extensively used in cancer research to evaluate the efficacy and mechanism of action of novel chemotherapeutic agents, such as the ferroptosis inducer RSL3, which has been shown to promote PARP-1 cleavage and apoptosis [20]. In drug development, it serves as a powerful tool for high-throughput screening of pro-apoptotic compounds and for assessing the emergence of therapy resistance by monitoring evasive cell death pathways. Furthermore, the ability to detect PARP-1 cleavage at the single-cell level is crucial for studying heterogeneous cell populations, such as stem cells or tumor cells, where a bulk assay like Western blotting might miss critical subpopulations that survive treatment.
Troubleshooting Guide
- High Background in Caspase Signal: Ensure thorough washing after FLICA/PhiPhiLux incubation. Titrate the reagent to determine the optimal concentration for your cell type.
- Low Annexin V Signal: Verify that the binding buffer contains calcium. Ensure the cells are not over-fixed before Annexin V staining.
- Poor Resolution of Populations: Check compensation settings using single-stained controls. Confirm antibody/reagent titrations. Use a viability dye with appropriate permeability (7-AAD can be preferable to PI for better discrimination of early apoptotic cells) [48].
- Discrepancy with Western Blot Data: Remember that flow cytometry is more sensitive in detecting small subpopulations of dying cells that may be masked in a bulk Western blot analysis.
The proteolytic cleavage of Poly(ADP-ribose) polymerase-1 (PARP-1) is a well-established biochemical hallmark of apoptosis, occurring early in the cell death cascade. Caspase-mediated cleavage, primarily by caspase-3, separates PARP-1 (113 kDa) into two characteristic fragments: a 24 kDa N-terminal fragment and an 89 kDa C-terminal fragment [4] [51]. The 89 kDa fragment, which contains the catalytic domain, loses its DNA-binding capacity due to the separation from the N-terminal DNA-binding domain, leading to inactivation of its DNA repair function and facilitating cellular disassembly [8] [51]. This specific cleavage event serves as a critical biomarker for distinguishing apoptosis from other forms of cell death, making its accurate detection paramount in diverse research areas from oncology to neuroscience. The selection of an appropriate detection method—whether flow cytometry or Western blot—fundamentally shapes the type and quality of data obtained, with each technique offering distinct advantages for specific research contexts.
Technical Comparison: Flow Cytometry vs. Western Blot
The choice between flow cytometry and Western blot for detecting PARP-1 cleavage hinges on the specific research question, particularly the required data dimensionality (population statistics vs. molecular detail) and experimental workflow.
Table 1: Core Technical Comparison for PARP-1 Cleavage Detection
| Feature | Flow Cytometry | Western Blot |
|---|---|---|
| Data Output | Single-cell resolution, population heterogeneity | Bulk population analysis, no cellular heterogeneity |
| Information Gained | Percentage of cleaved PARP-1 positive cells; correlation with other markers (e.g., active Caspase-3) [28] | Molecular weight confirmation (24 kDa and/or 89 kDa fragments) [51] |
| Throughput | Higher | Lower |
| Cell Requirement | ~1x10^6 cells per sample [28] | Varies, typically more than flow cytometry |
| Key Reagent | Anti-cleaved PARP-1 antibody (e.g., Clone 4G4C8) [51] | Anti-cleaved PARP-1 or PAR-specific antibody [28] |
| Multiplexing | High (with intracellular staining for active Caspase-3, etc.) [28] | Moderate (by stripping/re-probing or multiplex fluorescent detection) |
Table 2: Method Selection Guide Based on Research Goals
| Research Goal | Recommended Method | Rationale |
|---|---|---|
| Quantifying apoptotic population frequency | Flow Cytometry | Provides direct, quantitative counts of cells positive for PARP-1 cleavage within a heterogeneous sample [28]. |
| Confirming specific cleavage fragment identity | Western Blot | Superior for verifying the molecular weights of the 89 kDa and 24 kDa fragments, confirming the specific apoptotic signature [51]. |
| Correlating PARP-1 cleavage with other intracellular events | Flow Cytometry | Ideal for co-staining with markers like active Caspase-3 or phospho-proteins to study signaling relationships at a single-cell level [28]. |
| Analyzing samples with limited cell numbers | Flow Cytometry | More efficient for obtaining population data from smaller cell samples, as in primary patient samples [22]. |
| Detecting PARP activation (PAR formation) without cleavage | Flow Cytometry or Western Blot | Both can use anti-PAR antibodies (e.g., clone 10H) to detect PARylation, an indicator of PARP-1 activation in contexts like parthanatos [28] [22]. |
Detailed Experimental Protocols
Protocol A: Flow Cytometric Detection of Cleaved PARP-1
This protocol is optimized for the quantitative analysis of PARP-1 cleavage at the single-cell level and is amenable to multiplexing.
Key Reagent Solutions:
- Antibody: Cleaved PARP-1 Monoclonal Antibody (e.g., Clone 4G4C8, reactive to human, mouse, rat) [51].
- Critical Buffers: Cytofix/Cytoperm Fixation/Permeabilization Solution Kit [28].
- Staining Buffer: PBS supplemented with a low concentration of detergent (e.g., 0.5% BSA, 0.1% saponin) for intracellular antibody staining.
Step-by-Step Workflow:
- Cell Preparation and Stimulation: Harvest and wash cells in cold PBS. Induce apoptosis using appropriate stimuli (e.g., 1 μM Staurosporine for 3 hours [51]).
- Fixation and Permeabilization: Resuspend cell pellet (~1x10^6 cells) in 100 μL of PBS. Add Cytofix/Cytoperm solution and incubate for 20 minutes at 4°C to fix and make intracellular epitopes accessible [28].
- Intracellular Staining: Wash cells twice with Perm/Wash Buffer. Incubate cells with saturating amounts of anti-cleaved PARP-1 antibody (recommended: 0.40 μg per 10^6 cells in a 100 μL suspension) for 45 minutes at 4°C [51].
- Secondary Staining (If required): For unconjugated primary antibodies, wash cells and incubate with a fluorochrome-conjugated secondary antibody (e.g., AlexaFluor 488) for 30 minutes at 4°C in the dark.
- Data Acquisition: Resuspend cells in PBS and analyze immediately on a flow cytometer. Collect a minimum of 10,000 events per sample. Use fluorescence minus one (FMO) controls to set positive gates accurately.
Protocol B: Western Blot Detection of PARP-1 Cleavage
This protocol provides definitive confirmation of PARP-1 cleavage through visualization of the specific proteolytic fragments.
Key Reagent Solutions:
- Antibodies: Anti-cleaved PARP-1 antibody (Clone 4G4C8, for the 89 kDa fragment) [51]. Antibodies against full-length PARP-1 are also needed for comparison.
- Lysis Buffer: RIPA buffer supplemented with protease and phosphatase inhibitors.
- Gel System: Standard SDS-PAGE setup (8-12% gels are suitable for resolving the 113 kDa full-length and 89 kDa cleaved PARP-1).
Step-by-Step Workflow:
- Protein Extraction: Lyse cell pellets in ice-cold RIPA buffer for 30 minutes. Centrifuge at >12,000 x g for 15 minutes at 4°C to clear insoluble debris.
- Protein Quantification and Denaturation: Determine protein concentration using a Bradford or BCA assay. Denature equal amounts of protein (20-40 μg) in Laemmli buffer at 95°C for 5 minutes.
- Gel Electrophoresis and Transfer: Load and separate denatured proteins via SDS-PAGE. Transfer proteins from the gel to a nitrocellulose or PVDF membrane.
- Immunoblotting: Block the membrane with 5% non-fat milk in TBST for 1 hour. Incubate with primary antibody (e.g., Anti-cleaved PARP1 at 1:5000-1:50000 dilution [51]) overnight at 4°C. Wash and incubate with an appropriate HRP-conjugated secondary antibody.
- Detection: Develop blots using enhanced chemiluminescence (ECL) substrate and image with a digital imager. The cleaved 89 kDa fragment and full-length 113 kDa PARP-1 should be clearly visible.
Signaling Pathways and Experimental Workflows
The decision to use flow cytometry or Western blot is rooted in the biological context of PARP-1's role in cell death. The following diagrams map the signaling pathways and experimental workflows.
Diagram 1: PARP-1 Cleavage in Apoptosis. This diagram illustrates the core signaling pathway where apoptotic stimuli trigger caspase activation, leading to PARP-1 cleavage. The resulting fragments contribute to cell death through DNA repair inhibition and other fragment-specific effects [4] [8] [52].
Diagram 2: PARP-1 Cleavage Detection Workflow. This workflow chart outlines the critical decision points in the experimental process, guiding researchers toward the most appropriate method based on whether their primary need is population quantification or molecular confirmation of PARP-1 cleavage.
The Scientist's Toolkit: Essential Research Reagents
Successful detection of PARP-1 cleavage relies on a core set of validated reagents and tools.
Table 3: Key Research Reagent Solutions for PARP-1 Cleavage Analysis
| Reagent / Tool | Specific Example / Model | Function & Application Note |
|---|---|---|
| Anti-Cleaved PARP-1 Antibody | Mouse Monoclonal (4G4C8) [51] | Specifically recognizes the 89 kDa cleavage fragment; validated for WB, IHC, IF/ICC, and Flow Cytometry (Intra). |
| Anti-PAR Antibody | Mouse Monoclonal (clone 10H) [28] | Detects poly(ADP-ribose) polymers; used to measure PARP-1 activation in models of parthanatos or inflammation [28] [22]. |
| PARP Inhibitor (Control) | ABT-888 (Veliparib) [28] | A potent PARP inhibitor used as a control to suppress PAR formation and confirm the specificity of PARP-1-related signals. |
| Apoptosis Inducer | Staurosporine [51] | A broad-spectrum kinase inducer used as a positive control to trigger apoptosis and PARP-1 cleavage in experimental systems. |
| Flow Cytometry Assay Kit | Cytofix/Cytoperm Fixation/Permeabilization Kit [28] | Enables intracellular staining for flow cytometry by making the cleaved PARP-1 epitope accessible to antibodies while preserving cell structure. |
Solving Common Problems and Enhancing Assay Performance
In the study of programmed cell death, the detection of apoptosis remains a cornerstone of cellular and molecular biology research, particularly in drug development where quantifying cell death mechanisms is essential. Among the various biomarkers for apoptosis, cleavage of Poly(ADP-ribose) polymerase-1 (PARP-1) by executioner caspases stands as a definitive biochemical hallmark [8]. During apoptosis, caspase-3 cleaves the 116 kDa full-length PARP-1 into characteristic 24 kDa and 89 kDa fragments, a transition that serves as a critical indicator of apoptotic progression [20] [8]. While flow cytometry offers rapid quantification of apoptosis in cell populations, Western blotting provides unparalleled specificity in confirming the molecular events through direct visualization of PARP-1 cleavage fragments [20].
However, achieving reliable detection of these fragments presents significant technical challenges. The signal-to-noise ratio must be optimized to distinguish specific cleavage products from non-specific bands, and the potential for high background can obscure critical results, particularly when detecting low-abundance cleavage fragments [53] [54]. This application note addresses these challenges within the context of apoptosis research, providing optimized protocols and troubleshooting guides to ensure accurate, reproducible detection of PARP-1 cleavage for the research and drug development communities.
Understanding PARP-1 Cleavage in Apoptosis Signaling
The cleavage of PARP-1 represents a committed step in the apoptotic pathway, with the resulting fragments exerting distinct biological functions. The 89 kDa truncated PARP-1 (tPARP1) translocates from the nucleus to the cytoplasm, where recent research indicates it recognizes the RNA polymerase III (Pol III) complex and facilitates innate immune responses during apoptosis [8]. Meanwhile, the 24 kDa fragment remains nuclear and may act as a dominant-negative by occupying DNA damage sites [8]. This cleavage event not only inactivates PARP-1's DNA repair functions but also generates fragments with novel biological activities that promote apoptotic progression [55].
The following diagram illustrates the PARP-1 cleavage pathway during apoptosis and the subsequent cellular events:
Diagram Title: PARP-1 Cleavage Pathway During Apoptosis
Key Challenges in Western Blot Optimization
Non-Specific Bands and Background Interference
In the context of PARP-1 cleavage detection, non-specific bands present a particular challenge as they may be mistaken for the characteristic 24 kDa and 89 kDa fragments, leading to false interpretation of apoptosis. The table below summarizes the primary causes and targeted solutions for these issues:
Table 1: Troubleshooting Non-Specific Bands and High Background in Western Blotting
| Problem | Primary Causes | Recommended Solutions | Considerations for PARP-1 Detection |
|---|---|---|---|
| Non-specific Bands | Low antibody specificity [54]; Protein degradation [53]; Post-translational modifications [53] | Titrate primary antibody [54]; Include protease inhibitors [56]; Perform secondary-only control [53] | PARP-1 has multiple isoforms and modification states that may produce additional bands [20] |
| High Background | Insufficient blocking [57]; Excessive antibody concentration [53]; Inadequate washing [53] | Optimize blocking buffer [53]; Reduce antibody concentration [57]; Increase wash frequency/duration [53] | BSA is preferred over milk for phosphoprotein detection [56] |
| Weak/No Signal | Failed transfer [56]; Antibody degradation [56]; Insufficient antigen [53] | Verify transfer with Ponceau S [56]; Use fresh aliquots of antibodies [56]; Load positive control [53] | PARP-1 cleavage fragments may be low abundance; ensure sufficient protein load |
| Smearing Bands | Protein degradation [57]; Overloading [53]; Improper transfer [53] | Use fresh protease inhibitors [57]; Reduce protein load [53]; Optimize transfer conditions [53] | Apoptotic samples have active proteases; use stronger protease inhibitors |
Signal-to-Noise Optimization Framework
The following workflow provides a systematic approach for optimizing signal-to-noise ratio in Western blotting, particularly relevant for detecting PARP-1 cleavage fragments:
Diagram Title: Signal-to-Noise Optimization Workflow
Experimental Protocols for Apoptosis Detection
Optimized Western Blot Protocol for PARP-1 Cleavage Detection
Sample Preparation
- Prepare cell lysates using RIPA buffer supplemented with fresh protease inhibitor cocktail (essential for preventing degradation of PARP-1 fragments) [56]
- Include phosphatase inhibitors if detecting post-translationally modified forms of PARP-1
- Protein quantification: Use BCA assay to ensure equal loading [20]
- For apoptosis induction: Treat cells with appropriate apoptotic agents (e.g., RSL3 for ferroptosis-apoptosis crosstalk [20] or other inducers)
Gel Electrophoresis and Transfer
- Use 4-20% gradient gels for optimal resolution of full-length PARP-1 (116 kDa) and cleavage fragments (89 kDa and 24 kDa)
- Load 20-50 μg of total protein per lane for whole cell lysates [53]
- Include molecular weight markers and positive control (apoptotic cell lysate) [53]
- Transfer to PVDF membrane using wet transfer system at 100V for 1 hour [58]
Blocking and Antibody Incubation
- Block with 5% BSA in TBST for 1 hour at room temperature (BSA preferred over milk for potential phospho-epitopes) [56]
- Primary antibody incubation: Anti-PARP-1 antibody (diluted in blocking buffer), incubate overnight at 4°C with gentle agitation [54]
- Wash: 3 × 10 minutes with TBST
- Secondary antibody incubation: HRP-conjugated secondary (species appropriate), 1:10,000 dilution in blocking buffer, 1 hour at room temperature [56]
- Wash: 5 × 5 minutes with TBST [53]
Detection and Analysis
- Develop with enhanced chemiluminescence (ECL) substrate [53]
- Image using appropriate system (e.g., iBright Imaging System [59])
- For quantitative analysis: Ensure exposures are within linear dynamic range [59]
Stripping and Reprobing Protocol for Loading Controls
To confirm equal loading across lanes, stripping and reprobing for housekeeping proteins is often necessary. The following protocol is optimized for PVDF membranes:
Table 2: Stripping Buffer Compositions and Applications
| Stripping Method | Buffer Composition | Incubation Conditions | Best For | Efficiency |
|---|---|---|---|---|
| Mild Stripping | 1.5% glycine, 0.1% SDS, 1% Tween-20, pH 2.2 [58] | 10-20 min, room temperature [58] | High-abundance targets; Fragile epitopes | Preserves >90% antigen [58] |
| Stringent Stripping | 62.5 mM Tris-HCl, 2% SDS, 0.8% β-mercaptoethanol, pH 6.8 [58] | 30 min, 50°C [58] | Strong antibody-antigen interactions | May reduce antigen quantity |
Procedure
- After initial detection, rinse membrane in water to remove residual ECL substrate [58]
- Incubate membrane with mild stripping buffer (15-20 minutes, room temperature with agitation) [58]
- Wash membrane 3 × 5 minutes with TBST [58]
- Test stripping efficiency: Re-incubate with secondary antibody and ECL substrate - no signal should be detected [58]
- If signal remains, repeat with stringent stripping buffer [58]
- Re-block membrane and reprobe with loading control antibody (e.g., GAPDH, β-actin) [59]
Quantitative Western Blot Normalization Strategies
Accurate quantification of PARP-1 cleavage requires proper normalization to account for loading variations. The field is increasingly moving toward total protein normalization (TPN) as the gold standard, as it addresses limitations of traditional housekeeping protein approaches [59].
Diagram Title: Western Blot Normalization Strategy Comparison
Implementation of Total Protein Normalization
For quantitative analysis of PARP-1 cleavage, implement TPN using the following protocol:
- After transfer, stain membrane with No-Stain Protein Labeling Reagent or similar total protein stain [59]
- Image total protein before blocking using appropriate channels (e.g., 700 nm channel for near-infrared) [59]
- Process through standard immunoblotting procedure for PARP-1 detection
- Calculate ratio of PARP-1 signal (full-length or cleaved) to total protein in each lane
- Express results as normalized band intensity relative to control samples
This approach is particularly valuable for apoptosis studies where traditional housekeeping proteins may be proteolyzed or whose expression may change during cell death [59].
Research Reagent Solutions
The following table details essential reagents and their optimal applications for PARP-1 cleavage detection and general Western blot optimization:
Table 3: Essential Research Reagents for Apoptosis Detection by Western Blot
| Reagent Category | Specific Products | Optimal Application | Technical Notes |
|---|---|---|---|
| Blocking Buffers | Non-fat dry milk; BSA; AzureChemi Blot Blocking Buffer [54] | Milk: general use; BSA: phosphoproteins; AzureBuffer: low background [56] | BSA recommended for PARP-1 detection due to potential phosphorylation [56] |
| Membranes | PVDF; Nitrocellulose [53] | PVDF: multiple reprobing; Nitrocellulose: lower background [58] [57] | PVDF preferred for stripping/reprobing protocols [58] |
| Detection Systems | Enhanced chemiluminescence (ECL); Fluorescent secondaries [59] | ECL: general use; Fluorescent: multiplexing [59] | ECL sufficient for PARP-1 cleavage detection |
| Stripping Buffers | Restore PLUS Western Blot Stripping Buffer; Mild stripping buffer [58] | Gentle stripping for fragile epitopes; Stringent for strong interactions [58] | Always start with mild conditions [58] |
| Normalization Reagents | No-Stain Protein Labeling Reagent; Total protein stains [59] | Total protein normalization (TPN) [59] | Required for publication in top journals [59] |
| PARP-1 Antibodies | Cleavage-specific antibodies; Full-length recognizing [20] | Apoptosis quantification; Cleavage fragment detection | Validate with positive control (apoptotic lysate) |
Optimizing Western blotting for apoptosis detection through PARP-1 cleavage analysis requires meticulous attention to both technical execution and appropriate controls. The key recommendations for researchers in this field include:
- Implement total protein normalization as the preferred quantification method to account for potential variations in housekeeping proteins during apoptosis [59]
- Always include appropriate controls - positive control (apoptotic cell lysate), secondary antibody-only control, and molecular weight markers [53]
- Validate antibody specificity for the specific PARP-1 fragments of interest, particularly when investigating novel cell death inducers like RSL3 [20]
- Follow journal guidelines for image presentation including avoidance of excessive cropping and appropriate processing without manipulation [59]
By adhering to these optimized protocols and troubleshooting approaches, researchers can achieve reliable, reproducible detection of PARP-1 cleavage, enabling accurate assessment of apoptotic pathways in both basic research and drug development contexts.
Detecting apoptosis through the analysis of Poly(ADP-ribose) polymerase-1 (PARP-1) cleavage represents a crucial methodology in cell death research and drug development. While Western blotting has long served as the gold standard for confirming PARP-1 cleavage into its characteristic 89 kDa fragment [60], flow cytometry offers unparalleled advantages for single-cell analysis and rapid quantification of heterogeneous cell populations. However, the technical pitfalls of autofluorescence and improper compensation can severely compromise data accuracy, potentially leading to false conclusions about therapeutic efficacy or toxicity mechanisms. This application note details structured protocols to identify, mitigate, and correct these pervasive issues, specifically within the context of PARP-1 cleavage detection in apoptosis research.
Understanding the Technical Pitfalls
The Autofluorescence Problem in PARP-1 Analysis
Autofluorescence (AF) presents a particularly challenging issue in flow cytometry applications, as it arises from intrinsic cellular fluorophores such as NADPH, flavins, and lipofuscin, creating a background signal that can obscure specific fluorescence detection. This background varies significantly between cell types; for instance, macrophages and monocytes exhibit substantially higher AF than lymphocytes [61]. Critically, cellular treatments common in apoptosis research—including drug exposure, fixation, and permeabilization procedures required for intracellular PARP-1 staining—can markedly increase autofluorescence [61]. This elevated background directly reduces the signal-to-noise ratio for detecting cleaved PARP-1, potentially obscuring genuine apoptotic populations and leading to underestimation of treatment effects.
Compensation Artifacts in Multiparametric Panels
Compensation errors represent another fundamental challenge, arising from the unavoidable spectral overlap between fluorophores used in multiparametric flow cytometry. When uncorrected, this spillover causes fluorophore signals to be detected in "inappropriate" detectors, generating false-positive populations and distorting data interpretation [62]. The problem intensifies when detecting cleaved PARP-1, as this typically requires intracellular staining with permeabilization, which alters cellular light scattering and fluorescence properties [19] [61]. Furthermore, tandem dyes commonly used in multicolor panels are susceptible to degradation, which alters their spectral signatures and invalidates compensation calculations [63] [62]. These artifacts can create the illusion of distinct cell populations or mask true biological changes in PARP-1 cleavage patterns, directly impacting conclusions about drug-induced apoptosis.
Table 1: Quantitative Impact of Technical Pitfalls on PARP-1 Cleavage Detection
| Technical Issue | Effect on Signal Detection | Impact on Apoptosis Quantification | Common Experimental Manifestations |
|---|---|---|---|
| Autofluorescence | Increased background in all channels; Reduced signal-to-noise ratio [61] | Underestimation of cleaved PARP-1+ populations; Reduced statistical significance | False-negative populations; Compression of positive signals toward background |
| Under-compensation | Artificial signal spread into adjacent detectors [62] | False double-positive populations; Overestimation of apoptosis in gated populations | Diagonal streaking in biparametric plots; Non-alignment with axis in control samples |
| Over-compensation | Over-subtraction of genuine signal [63] | Artificial negative populations; Loss of true cleaved PARP-1+ events | "Over-compensated" populations appearing in negative quadrant |
| Tandem Dye Breakdown | Altered spectral signature invalidates compensation [63] | Unpredictable false positives/negatives dependent on degradation level | Multiple peaks in single-color controls; Non-round negative populations |
Experimental Protocols for Pitfall Mitigation
Protocol 1: Assessment and Minimization of Autofluorescence
Principle: Characterize and account for cell-type-specific autofluorescence signatures before designing PARP-1 detection panels.
Materials:
- Untreated control cells (identical to experimental conditions)
- Viability dye (e.g., Propidium Iodide or LIVE/DEAD fixable dyes)
- Fixation/Permeabilization reagents (e.g., Cytofix/Cytoperm kit [19])
- Flow cytometer with appropriate laser and detector configuration
Procedure:
- Prepare Unstained Controls: Harvest untreated cells and divide into two aliquots. Process one aliquot identical to experimental conditions (including fixation/permeabilization if used for intracellular PARP-1 staining). Keep the second aliquot untreated as a baseline control [61].
- Analyze AF Signature: Acquire data from both samples on your flow cytometer. Use the same voltages and gains planned for your PARP-1 detection experiment.
- Identify Problematic Detectors: Create density plots of all fluorescence channels. Identify detectors with significant AF, typically appearing as a diagonal spike in violet laser-excited channels (e.g., BV510, BV605) [61].
- Implement AF Management Strategy:
- Compensation Approach: Designate an empty channel in a high-AF region (e.g., BUV496 or PerCP-Cy5.5) as an "AF dump channel." Use unstained controls to calculate and subtract this AF signal from other detectors during analysis [61].
- Gating Approach: Include a marker for highly autofluorescent cells (e.g., CD11b for myeloid cells) in your panel and gate these populations out during analysis [61].
- Validate with Experimental Samples: Process a small set of experimental samples (including apoptosis-induced positive controls) to confirm that AF management does not exclude genuine cleaved PARP-1 positive populations.
Protocol 2: Optimization of Single-Color Controls for Accurate Compensation
Principle: Establish robust reference controls that precisely define the spectral signature of each fluorophore used in PARP-1 detection panels.
Materials:
- Compensation beads (e.g., CompBeads [62]) or cell-based controls
- Antibody conjugates identical to those used in experimental panels
- PARP-1 inhibitor (e.g., ABT-888/veliparib [19]) for specificity controls
- Apoptosis inducer (e.g., staurosporine [60] or doxorubicin [19]) for positive controls
Procedure:
- Prepare Single-Color Controls: For each fluorophore conjugate in your panel (including cleaved PARP-1 antibody), prepare either:
- Bead-based controls: Use anti-mouse or anti-rat compensation beads stained with each individual antibody conjugate [62].
- Cell-based controls: Use cells with known marker expression for surface antigens. For cleaved PARP-1, use apoptosis-induced cells (e.g., 1μM staurosporine for 3 hours [60]) and unstained cells as negative control.
- Apply the "Five Rules" for Reference Controls [63]:
- Bright is Better: Ensure positive signals in controls are as bright or brighter than experimental samples.
- Like-with-Like: Match autofluorescence between positive and negative populations (e.g., for viability dyes, use heat-killed cells split into stained and unstained aliquots).
- Matched Fluorophore: Use identical fluorophores (not similar colors) between controls and experiments.
- Same Tandem Lot: Use antibodies from identical manufacturing lots to control for tandem dye variability.
- Identical Conditions: Expose controls to the same fixation, permeabilization, and staining buffers as experimental samples.
- Validate Control Quality: Check that each single-color control shows a single, tight positive population without multiple peaks or excessive spread [63].
- Establish Compensation Settings: Use the single-color controls to calculate compensation matrices in your flow cytometry software.
- Verify with Dual-Color Controls: Fine-tune compensation using cells stained with two fluorophores that identify mutually exclusive populations or by mixing single-stained controls [62].
Protocol 3: Integrated Workflow for PARP-1 Cleavage Detection with Built-In Controls
Principle: Implement a comprehensive staining and analysis protocol that incorporates autofluorescence and compensation controls directly into the PARP-1 cleavage detection workflow.
Materials:
- Cells treated with experimental conditions (e.g., chemotherapeutic agents [64] [22])
- Annexin V-FITC (for early apoptosis detection [65])
- Propidium Iodide (for viability assessment [65])
- Anti-cleaved PARP-1-APC antibody (clone 4G4C8 [60])
- CD11b-BV510 or similar (for autofluorescent population identification [61])
- Fixation/Permeabilization kit (e.g., Cytofix/Cytoperm [19])
- Flow cytometer with blue (488nm) and red (640nm) lasers
Procedure:
- Induce Apoptosis: Treat cells with experimental compounds (e.g., 5μM cisplatin for SW620 cells [64]) for appropriate duration.
- Harvest and Stain Surface Markers: Collect cells by gentle centrifugation. Resuspend in staining buffer and add Annexin V-FITC and CD11b-BV510. Incubate 15-20 minutes at room temperature in the dark.
- Viability Staining: Add Propidium Iodide (1μg/mL final concentration) immediately before acquisition of live cells. Alternatively, use fixable viability dyes for fixed samples.
- Fix and Permeabilize: Wash cells twice with PBS, then fix and permeabilize using Cytofix/Cytoperm solution according to manufacturer's instructions [19]. Note that fixation increases autofluorescence, so maintain consistent timing [61].
- Intracellular Staining: Wash cells with Perm/Wash buffer, then incubate with anti-cleaved PARP-1-APC antibody (0.4μg per 10^6 cells in 100μL [60]) for 45 minutes at 4°C.
- Acquire Data: Resuspend cells in staining buffer and acquire data on flow cytometer using compensation settings established in Protocol 2.
- Analysis Strategy:
- Gate on Singlets: Exclude doublets using FSC-H vs FSC-A.
- Exclude Autofluorescent Cells: Gate out CD11b-BV510 bright cells to reduce AF interference [61].
- Identify Viable, Apoptotic, and Dead Populations: Use Annexin V/PI staining to distinguish viable (Annexin V-/PI-), early apoptotic (Annexin V+/PI-), and late apoptotic/necrotic (Annexin V+/PI+) populations [65].
- Analyze PARP-1 Cleavage: Examine cleaved PARP-1-APC expression within each apoptotic population.
Diagram 1: Experimental workflow for PARP-1 cleavage detection with integrated controls. The dashed lines indicate where control samples are prepared in parallel with experimental samples.
Table 2: Research Reagent Solutions for PARP-1 Cleavage Detection
| Reagent / Resource | Specific Function | Application Notes | Validation Criteria |
|---|---|---|---|
| Anti-Cleaved PARP-1 Antibody (clone 4G4C8 [60]) | Specific detection of apoptosis-associated PARP-1 fragment (89 kDa) | Works in WB, IHC, IF/ICC, FC (Intra); Reacts with human, mouse, rat samples | Shows increased signal in staurosporine-treated positive controls [60] |
| PARP Inhibitor (ABT-888/Veliparib [19]) | Specific inhibition of PARP activity; control for PARP-related effects | Use at 1μM final concentration; validates specificity of PARP-1 activation | Reduces PAR levels in LPS-stimulated PBMCs [19] |
| Annexin V / PI Apoptosis Detection [65] | Differentiation of viable, early apoptotic, and late apoptotic/necrotic cells | Essential for contextualizing PARP-1 cleavage within apoptosis progression | Clear population separation in drug-treated cells (e.g., doxorubicin) [65] |
| Compensation Beads (e.g., CompBeads [62]) | Consistent single-color controls for compensation setup | Critical for tandem dyes (PE-Cy7, APC-Cy7); reduces cell-based variability | Tight, single positive populations without multiple peaks [63] |
| Fixation/Permeabilization Kit (e.g., Cytofix/Cytoperm [19]) | Enables intracellular cleaved PARP-1 antibody access | Optimize incubation time (e.g., 20 min [19]); increases autofluorescence | Maintains cell integrity while allowing antibody penetration |
Data Interpretation and Validation Strategies
Correlation with Western Blot Analysis
Given the thesis context comparing flow cytometry with Western blot for PARP-1 cleavage detection, implementing correlation studies is essential. While flow cytometry provides single-cell resolution and population heterogeneity data, Western blotting confirms the specific molecular weight of PARP-1 cleavage fragments (89 kDa) [60]. For method validation, parallel samples should be analyzed using both techniques. As demonstrated in drug toxicity studies, Western blotting can detect cleaved PARP-1 and caspase-3 in the same samples analyzed by flow cytometry, providing orthogonal validation of apoptotic induction [64]. This correlation is particularly important when establishing new flow cytometry panels or when investigating novel cell types where autofluorescence and staining conditions may require optimization.
Recognizing and Troubleshooting Data Artifacts
Even with careful experimental design, artifacts may still appear in flow cytometry data. Recognizing these patterns is essential for accurate interpretation:
- "Swooping" Populations: Curved distributions in biparametric plots indicate unmixing errors, often from poor single-color controls [63].
- Non-Round Negative Populations: Asymmetrical negative distributions suggest compensation problems that can create false positives [63].
- Unexpected CD25+ Populations: Apparent novel immune cell populations may actually be autofluorescent contaminants, as seen with macrophage AF in T-cell gates [61].
When these artifacts are identified, revisit single-color controls, ensure consistent sample processing, and consider implementing autofluorescence extraction or exclusion strategies as detailed in Protocols 1 and 2.
Diagram 2: Troubleshooting guide for identifying and addressing common flow cytometry artifacts in PARP-1 analysis.
Accurate detection of PARP-1 cleavage via flow cytometry requires meticulous attention to technical details surrounding autofluorescence and compensation. The protocols outlined herein provide a systematic approach to overcome these challenges, enabling researchers to confidently apply this powerful technology to apoptosis research and drug development. By implementing robust control strategies, understanding the sources and manifestations of technical artifacts, and validating flow cytometry data with orthogonal methods like Western blotting, researchers can generate reliable, reproducible data that accurately reflects biological reality. Through this rigorous approach, flow cytometry remains an indispensable tool for advancing our understanding of cell death mechanisms and evaluating therapeutic interventions.
The detection of apoptosis through Poly(ADP-ribose) polymerase-1 (PARP-1) cleavage serves as a cornerstone in cellular death research, providing critical insights for cancer biology, neurotoxicology, and therapeutic development. During apoptosis, executioner caspases-3 and -7 cleave the 116-kDa PARP-1 protein into characteristic 24-kDa and 89-kDa fragments, a process considered a biochemical hallmark of programmed cell death [13] [11]. The 24-kDa fragment contains the DNA-binding domain, while the 89-kDa fragment comprises the automodification and catalytic domains [13]. This cleavage event inactivates PARP-1's DNA repair function, facilitating cellular dismantling while preventing energy depletion through uncontrolled PARP activation [11].
The reliability of detecting these cleavage fragments hinges overwhelmingly on meticulous sample preparation that preserves both protein epitopes for immunodetection and enzyme activities for functional assays. Inadequate preparation can obscure critical findings, leading to false negatives or inaccurate quantification. This application note details optimized protocols for sample preparation tailored to two principal detection methodologies—western blotting and flow cytometry—within the broader context of apoptosis research. We emphasize procedures that maintain sample integrity from collection through analysis, enabling researchers to confidently capture these transient apoptotic signatures.
PARP-1 Cleavage Biology and Detection Significance
Caspase-Mediated Cleavage and Fragment Fate
PARP-1 cleavage occurs at a specific DEVD216↓G217 motif located between its DNA-binding domain and automodification domain [11]. This proteolytic event yields two primary fragments: a 24-kDa N-terminal fragment that remains tightly bound to DNA damage sites, acting as a trans-dominant inhibitor of DNA repair, and an 89-kDa C-terminal fragment that translocates to the cytoplasm [13]. Recent research has revealed that the 89-kDa fragment can carry poly(ADP-ribose) (PAR) polymers to the cytoplasm, where they facilitate apoptosis-inducing factor (AIF) release from mitochondria, creating a bridge between caspase-dependent apoptosis and parthanatos, a caspase-independent programmed cell death pathway [13].
Methodological Selection Criteria
Choosing between western blot and flow cytometry depends on specific experimental questions and resource availability. The table below compares their fundamental characteristics for apoptosis detection via PARP-1 cleavage.
Table 1: Comparison of Western Blot and Flow Cytometry for PARP-1 Cleavage Detection
| Feature | Western Blot | Flow Cytometry |
|---|---|---|
| Key Readout | Separation and visualization of full-length (116-kDa) and cleaved (89-kDa, 24-kDa) fragments | Presence of cleaved PARP epitopes or caspase activity at single-cell level |
| Sample Type | Lysates from cell populations or homogenized tissues | Single-cell suspensions |
| Throughput | Medium (typically 10-20 samples per gel) | High (thousands of cells per second) |
| Information Context | Population-average protein profile | Heterogeneity analysis within cell populations |
| Multiplexing Potential | Low to medium (sequential probing for different targets) | High (simultaneous measurement of multiple parameters) |
| Key Advantage | Direct confirmation of specific fragment sizes | Correlation of PARP cleavage with other apoptotic markers (e.g., phosphatidylserine exposure) |
Critical Sample Preparation Protocols
Sample Preparation for Western Blot Analysis
Western blotting remains the gold standard for directly visualizing the distinct PARP-1 cleavage fragments, confirming the specific molecular weight shifts that unequivocally demonstrate apoptosis.
Cell Lysis and Protein Extraction
Reagents Needed:
- RIPA Lysis Buffer: Provides effective extraction of nuclear and cytoplasmic proteins.
- Protease Inhibitor Cocktail: Essential to prevent post-lysis degradation of PARP fragments.
- Phosphatase Inhibitors: Preserve phosphorylation states if studying regulatory modifications.
- Nuclease-free Water: Prevents nucleic acid contamination that can affect protein quantification.
- Protein Assay Kit (e.g., BCA or Bradford): For accurate protein quantification.
Protocol Steps:
- Rapid Harvesting and Washing: Pellet cells by centrifugation (300 × g, 5 min at 4°C). Wash once with ice-cold phosphate-buffered saline (PBS). Critical: Perform all steps at 4°C to halt enzymatic activity.
- Lysis: Aspirate PBS completely and add appropriate volume of ice-cold RIPA buffer supplemented with fresh protease inhibitors (e.g., 1 mM PMSF, 1 μg/mL leupeptin, 1 μg/mL aprotinin). Use approximately 100-200 μL lysis buffer per 1 × 10⁶ cells.
- Incubation: Incubate samples on ice for 30 minutes with occasional gentle vortexing to ensure complete lysis.
- Clarification: Centrifuge lysates at 12,000 × g for 15 minutes at 4°C to pellet insoluble debris, including nuclei.
- Protein Quantification: Transfer supernatant to a fresh tube and determine protein concentration using a BCA or Bradford assay. Critical: Normalize all samples to the same concentration (e.g., 1-2 μg/μL) using lysis buffer to ensure equal loading.
- Sample Preparation for SDS-PAGE: Mix normalized lysates with 4× Laemmli sample buffer. Do not boil PARP-1 samples, as excessive heat can cause protein aggregation. Instead, heat at 65-70°C for 10-15 minutes.
- Storage: Aliquot and store prepared samples at -80°C. Avoid repeated freeze-thaw cycles.
Gel Electrophoresis and Normalization
For optimal resolution of PARP-1 fragments, use 4-12% or 4-15% gradient polyacrylamide gels, which provide superior separation of the 116-kDa full-length protein from the 89-kDa fragment compared to fixed-percentage gels. Include pre-stained protein molecular weight markers on every gel. For normalization, Total Protein Normalization (TPN) is now recommended over housekeeping proteins (e.g., GAPDH, β-actin) by major journals, as housekeeping protein expression can vary significantly with experimental conditions [59]. TNP can be achieved with total protein stains or fluorogenic labeling methods performed post-transfer [59].
Sample Preparation for Flow Cytometry
Flow cytometry enables multiparametric analysis of PARP-1 cleavage at the single-cell level, allowing researchers to correlate cleavage events with other apoptotic markers and identify heterogeneous responses within cell populations.
Cell Harvesting and Fixation
Reagents Needed:
- Annexin V Binding Buffer: For assays detecting phosphatidylserine exposure.
- Permeabilization Buffer (e.g., saponin-based or detergent-based).
- Intracellular Staining Permeabilization Wash Buffer (e.g., BD Cytofix/Cytoperm).
- Fluorochrome-conjugated Antibodies against cleaved PARP (e.g., specific to the 89-kDa fragment).
- Viability Dye (e.g., 7-AAD, DAPI, or fixable viability stains).
Protocol Steps:
- Gentle Harvesting: For adherent cells, use gentle, non-enzymatic dissociation methods such as cell scraping or EDTA-based solutions to preserve membrane integrity. Avoid trypsin, which can cleave surface proteins.
- Viability Staining (for exclusion): Resuspend cell pellet in Annexin V binding buffer containing a viability dye (e.g., 7-AAD) to distinguish early apoptotic cells (Annexin V+/7-AAD-) from late apoptotic/necrotic cells (Annexin V+/7-AAD+).
- Fixation: Add an equal volume of 4%-8% paraformaldehyde (PFA) in PBS to the cell suspension for a final concentration of 2%-4%. Incubate for 10-20 minutes at room temperature. Critical: Avoid over-fixation (>30 minutes), which can mask epitopes.
- Permeabilization: Pellet cells (400 × g, 5 min) and thoroughly resuspend in ice-cold permeabilization buffer (e.g., 90% methanol, 0.1% Triton X-100 in PBS, or commercial perm buffers). Incubate for 30 minutes on ice or at -20°C for methanol-based permeabilization.
- Washing: Pellet cells and wash twice with flow cytometry staining buffer (PBS with 1% BSA).
Intracellular Staining for Cleaved PARP
- Antibody Staining: Resuspend fixed and permeabilized cells in staining buffer containing a titrated amount of fluorochrome-conjugated anti-cleaved PARP antibody. Include isotype control antibodies for accurate gating.
- Incubation: Protect samples from light and incubate for 30-60 minutes at room temperature or overnight at 4°C for enhanced signal-to-noise ratio with some antibodies.
- Washing: Pellet cells and wash twice with staining buffer to remove unbound antibody.
- Resuspension and Analysis: Resuspend cells in an appropriate volume of staining buffer or PBS for immediate acquisition on a flow cytometer. If analysis cannot be performed immediately, resuspend cells in staining buffer containing 1% PFA and store at 4°C for up to 24-48 hours.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for PARP-1 Cleavage Studies
| Reagent Category | Specific Examples | Function in Protocol |
|---|---|---|
| PARP-1 Antibodies | Anti-PARP-1 (full length), Anti-cleaved PARP-1 (89 kDa), Anti-cleaved PARP-1 (24 kDa) | Detection of full-length and specific cleavage fragments in western blot (WB) and flow cytometry (FC) |
| Caspase Assays | Fluorogenic caspase-3/7 substrates (e.g., DEVD-AFC), Active caspase-3 antibodies | Functional confirmation of apoptosis execution; multiplexing with PARP cleavage in FC |
| Viability Indicators | 7-AAD, Propidium Iodide, DRAQ7, Fixable Viability Dyes (e.g., BV421) | Distinguish apoptotic from necrotic cells; exclude dead cells in FC analysis |
| Membrane Asymmetry Probes | Fluorochrome-conjugated Annexin V (FITC, PE, BV421) | Detect phosphatidylserine exposure as an early apoptotic marker in FC |
| Cell Lysis Reagents | RIPA Buffer, NP-40 Buffer, Commercial Lysis Kits | Efficient extraction of nuclear and cytoplasmic proteins while preserving protein integrity for WB |
| Protein Normalization Tools | No-Stain Protein Labeling Reagent, Fluorescent Total Protein Stains | Accurate loading control for WB via Total Protein Normalization [59] |
| Fixation/Permeabilization Kits | BD Cytofix/Cytoperm, FoxP3 Staining Buffer Set, Methanol | Preserve cellular structure and allow antibody access to intracellular epitopes for FC |
Visualization of PARP-1 Cleavage Pathway and Detection
PARP-1 Cleavage in Apoptosis Signaling
Experimental Workflow for PARP Cleavage Detection
Troubleshooting and Quality Control
Common Pitfalls and Solutions
Problem: Absence or weak signal of PARP-1 fragments in western blot despite apoptotic induction. Solution: Verify caspase activity using fluorogenic substrates. Ensure lysis buffer contains fresh protease inhibitors to prevent fragment degradation. Avoid over-boiling samples before SDS-PAGE.
Problem: High background in flow cytometry. Solution: Titrate all antibodies carefully. Include proper isotype and unstained controls. Increase wash steps after antibody incubation. Ensure permeabilization is complete but not excessive.
Problem: Inconsistent results between technical replicates. Solution: Standardize cell counting methods. Pre-aliquot lysis buffers with inhibitors to minimize variation. Use consistent timing for all processing steps.
Validation Strategies
Corroborate PARP-1 cleavage data with complementary apoptotic markers. In flow cytometry, combine cleaved PARP detection with Annexin V staining and viability dyes. In western blotting, probe for other caspase substrates (e.g., cleaved caspase-3). Employ positive controls (e.g., cells treated with staurosporine or actinomycin D) to confirm assay performance [13]. For quantitative western blotting, adhere to journal guidelines requiring total protein normalization and provide uncropped blots with molecular weight markers [59].
By implementing these sample preparation protocols with careful attention to critical steps that preserve epitopes and enzyme activities, researchers can reliably detect PARP-1 cleavage as a definitive marker of apoptosis, advancing research in cell death mechanisms and therapeutic development.
Antibody Validation and Specificity Controls for Accurate Interpretation
Poly (ADP-ribose) polymerase-1 (PARP-1) is a 113 kDa nuclear enzyme that plays a critical role in DNA repair and maintenance of genomic integrity [66] [67]. During the early stages of apoptosis, PARP-1 becomes a primary target for cleavage by caspase proteases, particularly caspase-3 and caspase-7 [68] [66]. This cleavage occurs at the highly conserved aspartic acid residue 214 (within the DEVD sequence), separating the PARP-1 molecule into two characteristic fragments: a 24 kDa DNA-binding fragment and an 89 kDa catalytic fragment [69] [66]. The detection of these cleavage fragments, especially the 89 kDa fragment, has become a well-established biochemical marker for identifying apoptotic cells in various research contexts, from basic cancer biology to drug development screening [69] [67].
The significance of PARP-1 cleavage extends beyond merely serving as an apoptosis indicator. Research indicates that the cleavage fragments themselves may play active roles in regulating cell death processes. While the 24 kDa fragment may act as a trans-dominant inhibitor of intact PARP-1, the 89 kDa fragment has been associated with enhanced cytotoxicity in some models [4]. This complexity underscores the importance of accurate detection and interpretation of PARP-1 cleavage patterns, which depends heavily on antibody specificity and appropriate validation controls. The choice between flow cytometry and Western blot detection methods further influences the experimental design and interpretation of results, each offering distinct advantages for different research applications in apoptosis detection.
PARP-1 Cleavage Signatures Across Cell Death Pathways
Protease-Specific Cleavage Patterns
PARP-1 serves as a substrate for multiple proteases activated in different cell death pathways, each generating distinctive cleavage signatures. While caspase-mediated cleavage during apoptosis produces the characteristic 89 kDa fragment, other proteases create different PARP-1 fragments that can indicate alternative cell death mechanisms [67]. During necrosis, PARP-1 is processed into a major 50 kDa fragment through the action of lysosomal proteases such as cathepsins B and G, a process not inhibited by broad-spectrum caspase inhibitors like zVAD-fmk [15]. Calpains, granzymes, and matrix metalloproteinases (MMPs) can also cleave PARP-1, generating fragments ranging from 42-89 kDa [67] [70]. This protease-specific cleavage creates unique "signature fragments" that serve as biomarkers for identifying specific cell death programs operating in particular pathological conditions [67].
The biological consequences of these different cleavage events extend beyond simply inactivating PARP-1. Research suggests that PARP-1 cleavage fragments may actively regulate cellular viability and inflammatory responses in opposing ways [4]. For instance, in models of oxygen/glucose deprivation (an in vitro ischemia model), expression of the 24 kDa fragment was cytoprotective, while expression of the 89 kDa fragment was cytotoxic [4]. These findings highlight the importance of not only detecting PARP-1 cleavage but also accurately identifying the specific fragments present, as they may have distinct functional implications in cell death pathways.
PARP-1 Cleavage in Apoptosis Signaling
The following diagram illustrates the central role of PARP-1 cleavage in the apoptosis signaling pathway and its detection:
Key Validation Parameters for Cleaved PARP Antibodies
Establishing Antibody Specificity
The foundation of accurate PARP-1 cleavage detection rests on demonstrating antibody specificity through rigorous validation. Knockout (KO) validated antibodies represent the gold standard for confirming specificity, as they should show no signal in PARP-1 knockout cell lines [71]. For example, the Anti-Cleaved PARP1 antibody [E51] (ab32064) demonstrates complete loss of signal at the expected molecular weight in PARP-1 knockout A549 and HAP1 cell lines, while showing clear detection of the ~25-30 kDa fragment in wild-type cells treated with apoptosis inducers like staurosporine [71]. Similarly, Cell Signaling Technology's Cleaved PARP (Asp214) Antibody #9541 is confirmed to detect only the 89 kDa fragment produced by caspase cleavage and does not recognize full-length PARP-1 or other PARP isoforms [69].
Appropriate biological controls are equally crucial for validation. This includes comparing untreated cells with cells treated with known apoptosis inducers such as camptothecin or staurosporine [71] [66]. Camptothecin, a topoisomerase I inhibitor, induces apoptosis in a dose-dependent manner and serves as a positive control for cleaved PARP detection in protocols [66]. The use of caspase inhibitors provides additional validation by preventing PARP-1 cleavage and subsequent antibody detection, further confirming specificity for the caspase-cleaved form [28]. Antibodies should also be tested across multiple cell lines and species to establish cross-reactivity profiles, with many validated antibodies showing reactivity in human, mouse, and rat samples [71] [69] [70].
Method-Specific Validation Considerations
Validation requirements differ significantly between flow cytometry and Western blot applications. For flow cytometry, key parameters include optimal fixation and permeabilization conditions to allow antibody access to intracellular epitopes without destroying antigenicity [28] [66]. The BD Pharmingen PE Mouse Anti-Cleaved PARP (Asp214) protocol specifies using Cytofix/Cytoperm solution with a 20-minute incubation on ice, followed by Perm/Wash buffer washes [66]. Titration experiments are essential to determine the optimal antibody concentration that provides maximal signal-to-noise ratio, with recommended volumes typically around 20 μl per test of 1×10^6 cells [66].
For Western blot applications, validation must confirm the antibody detects the correct molecular weight fragment (typically 89 kDa for caspase-cleaved PARP-1, though some antibodies detect smaller fragments around 25-30 kDa) [71] [69]. Membrane blocking conditions and antibody dilution buffers can significantly impact results, with 5% non-fat dry milk/TBST being commonly used [71]. The inclusion of loading controls such as GAPDH or alpha-tubulin is essential for normalizing protein loading and enabling quantitative comparisons [71]. For both applications, appropriate isotype controls and secondary antibody-only controls are necessary to distinguish specific signal from background.
Research Reagent Solutions for PARP-1 Cleavage Detection
Table 1: Essential Reagents for Cleaved PARP Detection
| Reagent Category | Specific Examples | Function & Importance |
|---|---|---|
| Primary Antibodies | Anti-Cleaved PARP1 [E51] (ab32064) [71]Cleaved PARP (Asp214) Antibody #9541 [69]PE Mouse Anti-Cleaved PARP (Asp214) (Clone F21-852) [66]Cleaved PARP1 Monoclonal (60555-1-Ig) [70] | Detect specific caspase-cleaved fragments of PARP-1; clone and host species determine application compatibility |
| Apoptosis Inducers | Camptothecin (4-6 μM) [66]Staurosporine (0.1-3 μM) [71] | Positive controls that induce apoptosis and PARP-1 cleavage; essential for validation experiments |
| Cell Lines | Jurkat (human T-cell leukemia) [66]A549 (human lung carcinoma) [71]HAP1 (haploid human cell line) [71] | Well-characterized models for apoptosis studies; Jurkat cells particularly responsive to camptothecin |
| Fixation/Permeabilization Kits | Cytofix/Cytoperm Fixation/Permeabilization Kit [66] | Critical for intracellular staining in flow cytometry; preserves cell structure while allowing antibody access |
| Validation Tools | PARP-1 knockout cell lines [71]Caspase inhibitors (e.g., zVAD-fmk) [15] | Confirm antibody specificity; knockout lines provide definitive negative controls |
Comparative Analysis of Commercial Cleaved PARP Antibodies
Table 2: Commercial Cleaved PARP Antibody Comparison
| Antibody Product | Host/Clonality | Applications | Reactivity | Specific Fragment Detected | Key Validation Data |
|---|---|---|---|---|---|
| Anti-Cleaved PARP1 [E51] (ab32064) [71] | Rabbit monoclonal | WB, IHC-P | Human, Mouse, Rat | ~25-30 kDa fragment | KO validated in A549 and HAP1 cells; 402 publications |
| Cleaved PARP (Asp214) #9541 [69] | Rabbit polyclonal | WB, Simple Western | Human, Mouse | 89 kDa fragment | Does not recognize full-length PARP-1; peptide affinity purified |
| PE Mouse Anti-Cleaved PARP (Asp214) (552933) [66] | Mouse monoclonal (F21-852) | Flow Cytometry | Human, Mouse | 89 kDa fragment | Does not react with intact PARP-1; optimized for intracellular staining |
| Cleaved PARP1 (60555-1-Ig) [70] | Mouse monoclonal (4G4C8) | WB, IHC, IF/ICC, FC, ELISA | Human, Mouse, Rat | 89 kDa fragment | Detects only cleaved form, not full-length; multiple applications |
Experimental Protocols for Detection Methods
Flow Cytometry Protocol for Cleaved PARP Detection
The following workflow outlines the key steps for detecting cleaved PARP-1 using flow cytometry:
Step-by-Step Protocol:
- Induce Apoptosis: Treat approximately 1×10^6 proliferating Jurkat cells with camptothecin at a final concentration of 4-6 μM. Include an untreated control aliquot. Incubate for 4 hours at 37°C [66].
- Cell Harvesting: Wash cells twice with cold PBS, then resuspend in Cytofix/Cytoperm solution at a concentration of 2×10^6 cells/ml [66].
- Fixation/Permeabilization: Incubate cells for 20 minutes on ice. Pellet cells by centrifugation and carefully aspirate the Cytofix/Cytoperm solution [66].
- Wash Steps: Wash cells twice at room temperature with 0.5 ml Perm/Wash buffer per 1×10^6 cells, discarding supernatants after each wash [66].
- Antibody Staining: Resuspend cells in Perm/Wash buffer at 10×10^6/ml. Aliquot 1×10^6 cells per 100-μl test. Add 20 μl antibody per test and incubate for 30 minutes at room temperature, protected from light [66].
- Final Wash: Wash each test in 1.0 ml Perm/Wash Buffer, discard supernatant, and resuspend in 0.5 ml Perm/Wash Buffer for flow cytometry analysis [66].
Western Blot Protocol for Cleaved PARP Detection
Sample Preparation and Electrophoresis:
- Cell Lysis: Prepare whole cell lysates from treated and untreated cells using RIPA buffer supplemented with protease and phosphatase inhibitors. Maintain samples on ice throughout preparation.
- Protein Quantification: Determine protein concentration using a standard assay such as BCA to ensure equal loading across gels.
- Gel Electrophoresis: Load 20-30 μg of protein per lane on an appropriate SDS-PAGE gel (8-12% acrylamide). Include molecular weight markers and both positive (apoptosis-induced) and negative (untreated) controls [71].
- Protein Transfer: Transfer proteins from gel to nitrocellulose or PVDF membrane using standard wet or semi-dry transfer systems.
Membrane Blocking and Antibody Incubation:
- Blocking: Block membrane in 5% non-fat dry milk in TBST (Tris-buffered saline with 0.1% Tween-20) for 1 hour at room temperature with gentle agitation [71].
- Primary Antibody Incubation: Incubate membrane with cleaved PARP antibody diluted in 5% non-fat dry milk/TBST. For ab32064, use 1:10,000 dilution; for #9541, use 1:1,000 dilution. Incubate overnight at 4°C with gentle agitation [71] [69].
- Wash Steps: Wash membrane four times in TBST for 5-10 minutes each wash.
- Secondary Antibody Incubation: Incubate with appropriate HRP-conjugated or fluorescently-labeled secondary antibody diluted in blocking buffer (typically 1:10,000 to 1:20,000) for 1 hour at room temperature [71].
- Final Washes: Wash membrane four times in TBST before detection.
Detection and Analysis:
- Signal Detection: Use enhanced chemiluminescence (ECL) for HRP-based detection or direct imaging for fluorescent secondary antibodies.
- Membrane Stripping and Reprobing: If analyzing multiple proteins from the same membrane, strip and reprobe with loading control antibodies such as anti-GAPDH (1:20,000) or anti-alpha-tubulin (1:20,000) [71].
- Data Interpretation: The cleaved PARP-1 fragment typically appears at ~89 kDa for most antibodies, though some detect smaller fragments (~25-30 kDa). Compare signal intensity between treated and untreated samples, normalized to loading controls.
Troubleshooting and Data Interpretation
Common Technical Challenges and Solutions
Weak or No Signal may result from insufficient apoptosis induction, suboptimal antibody concentration, or improper fixation/permeabilization. To address this, first verify apoptosis induction using alternative methods such as Annexin V staining or caspase activity assays. Titrate the primary antibody to determine optimal concentration, and confirm that fixation/permeabilization conditions are appropriate for the specific cell type being analyzed [71] [66]. For Western blot, ensure adequate protein transfer by verifying transfer efficiency with reversible stains.
High Background or Non-Specific Staining often stems from insufficient blocking, over-fixation, or antibody concentration that is too high. Extend blocking time to at least 1 hour, optimize fixation duration, and titrate both primary and secondary antibodies to find the optimal signal-to-noise ratio [71]. Include appropriate controls such as isotype-matched antibodies and secondary antibody-only controls to distinguish specific from non-specific signal.
Inconsistent Results Between Experiments frequently arises from cell passage number effects, variations in apoptosis induction efficiency, or lot-to-lot antibody variability. Use low-passage cells whenever possible, standardize apoptosis induction conditions carefully, and validate new antibody lots against previous lots using standardized control samples. For flow cytometry, ensure consistent cell processing and instrument calibration between experiments.
Critical Controls for Accurate Interpretation
Appropriate controls are essential for validating cleaved PARP detection experiments. Biological controls should include untreated cells, cells treated with known apoptosis inducers (e.g., camptothecin, staurosporine), and cells treated with caspase inhibitors prior to apoptosis induction [28] [66]. Technical controls for flow cytometry include isotype-matched antibodies and secondary antibody-only controls to assess non-specific binding [66]. For Western blot, PARP-1 knockout cell lines provide definitive negative controls to confirm antibody specificity [71]. Loading controls such as GAPDH or alpha-tubulin are essential for normalizing Western blot data and assessing sample quality [71]. When establishing methods, include positive control cell lines known to show robust PARP-1 cleavage, such as camptothecin-treated Jurkat cells [66].
Quantitative Considerations and Pitfalls
While cleaved PARP detection is widely used as an apoptosis marker, several caveats warrant consideration. The appearance of cleaved PARP fragments is not exclusively associated with apoptosis, as other cell death pathways can generate different PARP-1 cleavage products [15] [67]. The temporal expression of cleaved PARP varies by cell type and apoptosis inducer, potentially making it less sensitive for detecting very early or late apoptosis. Additionally, certain cell types may express alternative PARP isoforms that could cross-react with some antibodies. Quantitative comparisons should be made within linear detection ranges, particularly for Western blot, where signal saturation can distort results. For flow cytometry, proper gating strategies that exclude debris and aggregated cells are essential for accurate quantification of cleaved PARP-positive populations.
Accurate data normalization is a foundational step in protein analysis techniques such as western blotting and flow cytometry. It ensures that observed differences in target protein signals reflect true biological changes rather than technical artifacts arising from uneven sample loading, pipetting inaccuracies, or transfer inconsistencies. Within the specific context of apoptosis detection, monitoring the cleavage of Poly(ADP-ribose) polymerase-1 (PARP-1) serves as a well-established biomarker. The transition of full-length PARP1 (113 kDa) to its characteristic 89 kDa cleavage fragment is a definitive indicator of caspase-mediated apoptosis [4] [72]. Validating this key molecular event relies heavily on robust normalization strategies to generate reliable, interpretable, and reproducible data. This application note details these strategies, framed within a research thesis comparing apoptosis detection via PARP-1 cleavage using flow cytometry and western blot.
The Critical Role of Housekeeping Proteins and Their Limitations
Housekeeping proteins (HKPs) are ubiquitously expressed proteins presumed to maintain consistent levels across different tissues and experimental conditions. They are traditionally used as internal controls to normalize the expression levels of target proteins.
- Common Housekeeping Proteins: The most frequently used HKPs include β-actin, Glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and β-tubulin [73] [74]. These proteins are chosen for their roles in essential cellular structures and processes.
- Inherent Challenges and Variability: A critical assumption underlying the use of HKPs is their stable expression. However, substantial evidence demonstrates that this stability cannot be universally relied upon. The table below summarizes documented variations in common HKPs under various conditions [73]:
| Housekeeping Protein | Documented Variations and Contexts |
|---|---|
| β-actin | ↑ in spinal cord injury; ↓ in Alzheimer's disease; ↓ in steatosis and alcoholic hepatitis; variable in kidney tumors; altered with cell density; unreliable at very high/low protein loads [73]. |
| GAPDH | ↓ in Alzheimer's disease brain; ↑ in kidney tumor tissue; ↓ in steatosis and alcoholic hepatitis; reduced by VHL transfection; variable with cell confluence; unreliable across a range of protein loads [73]. |
| β-tubulin | Regional decreases/increases in schizophrenia brain; unstable in adipose tissue; no significant change after spinal cord injury [73]. |
This variability indicates that using an unstable HKP for normalization can lead to the misinterpretation of data, generating both false-positive and false-negative results [73] [74]. Therefore, the choice of an HKP requires empirical validation for each specific experimental system, including the cell type, treatment, and disease context being studied.
Normalization in Western Blotting for Apoptosis Detection
Western blotting is a cornerstone technique for visualizing PARP-1 cleavage, providing direct molecular weight confirmation of the full-length and cleaved forms.
Standard Protocol: Detecting PARP-1 Cleavage via Western Blot
Objective: To reliably detect and quantify the cleavage of PARP-1 as a marker of apoptosis in cell lysates.
Materials:
- Primary Antibodies: Anti-PARP1 antibody (detects full-length and cleaved forms) and/or anti-cleaved PARP1 (Asp214) specific antibody [72]. Anti-β-actin (or other validated HKP) for loading control.
- Secondary Antibodies: HRP-conjugated antibodies specific to the host species of the primary antibodies.
- Lysis Buffer: RIPA buffer supplemented with protease and phosphatase inhibitors.
- Other Reagents: SDS-PAGE gels, PVDF or nitrocellulose membranes, ECL or similar chemiluminescent substrate.
Methodology:
- Sample Preparation: Harvest control and treated cells. Lyse cells using ice-cold RIPA buffer with inhibitors. Quantify total protein concentration for each sample using a colorimetric assay (e.g., BCA or Bradford).
- Gel Electrophoresis: Load an equal mass of total protein (e.g., 20-30 µg) for each sample onto an SDS-PAGE gel. Include a pre-stained protein ladder. Resolve proteins by electrophoresis.
- Protein Transfer: Transfer proteins from the gel to a membrane using a standard wet or semi-dry transfer system.
- Immunoblotting:
- Blocking: Incubate the membrane in 5% non-fat milk or BSA in TBST for 1 hour at room temperature.
- Primary Antibody Incubation: Incubate membrane with primary antibodies (e.g., anti-PARP1 and anti-β-actin) diluted in blocking buffer, overnight at 4°C.
- Washing: Wash membrane 3 times for 5-10 minutes each with TBST.
- Secondary Antibody Incubation: Incubate membrane with appropriate HRP-conjugated secondary antibodies for 1 hour at room temperature.
- Washing: Repeat washing step as above.
- Signal Detection: Incubate membrane with chemiluminescent substrate and image using a digital imager. Ensure that band intensities for both target and HKP are within the linear range of detection and are not saturated [73] [75].
- Densitometric Analysis and Normalization:
- Quantify the band intensity for full-length PARP-1, cleaved PARP-1 (89 kDa fragment), and the HKP (e.g., β-actin) using analysis software.
- Calculate the normalized intensity for each PARP-1 form:
Normalized Intensity = (Intensity of PARP-1 Band) / (Intensity of HKP Band). - The ratio of cleaved PARP-1 to full-length PARP-1, often further normalized to the control sample, provides a quantitative measure of apoptosis.
Alternative Strategy: Total Protein Normalization (TPN)
Given the limitations of single HKPs, Total Protein Normalization (TPN) is a highly reliable alternative. TPN uses the total protein signal in each lane as the loading control, circumventing issues related to the variability of a single protein [75].
- Implementation: After transfer, the membrane can be stained with Ponceau S or more sensitive fluorescent total protein stains (e.g., SYPRO Ruby). "Stain-Free" technology, which uses trihalo compounds to label proteins directly in the gel prior to transfer, is another robust option [73] [75].
- Normalization Calculation: The intensity of the PARP-1 band is normalized to the total protein signal from the entire lane or a relevant section of the lane.
Normalization in Flow Cytometry for Apoptosis Detection
Flow cytometry offers a high-throughput, multi-parametric approach to analyze PARP-1 cleavage at the single-cell level, allowing for the examination of heterogeneous cell populations.
Standard Protocol: Detecting PARP-1 Cleavage via Flow Cytometry
Objective: To quantify intracellular levels of PAR and/or cleaved PARP-1 in specific leukocyte subpopulations.
Materials:
- Primary Antibodies: Anti-PAR antibody (e.g., clone 10H) [28], Anti-cleaved PARP-1 (Asp214) antibody (conjugated to a fluorophore, e.g., FITC) [72].
- Surface Marker Antibodies: e.g., anti-CD45 (pan-leukocyte, PE-conjugated), anti-CD14 (macrophages, PerCP-conjugated) [28].
- Staining Reagents: Fixation/Permeabilization buffer kit (e.g., Cytofix/Cytoperm), Flow cytometry staining buffer (PBS with BSA).
Methodology:
- Cell Preparation and Stimulation: Collect cells (e.g., peripheral blood mononuclear cells - PBMCs, or purified milk somatic cells). Treat cells with apoptosis inducer (e.g., LPS, staurosporine, poly(dA-dT)) [28] [8].
- Cell Surface Staining: Resuspend cell pellet in flow cytometry buffer and incubate with conjugated surface marker antibodies for 30 minutes in the dark at 4°C. Wash cells to remove unbound antibody.
- Fixation and Permeabilization: Fix and permeabilize cells using a commercial kit (e.g., BD Cytofix/Cytoperm) to allow intracellular antibody access. An optimal incubation time of 20 minutes is recommended [28].
- Intracellular Staining: Incubate cells with antibodies against PAR or cleaved PARP-1 for 45 minutes at 4°C in the dark. If using an unconjugated primary antibody, include a subsequent step with a fluorophore-conjugated secondary antibody.
- Data Acquisition and Analysis:
- Acquire data on a flow cytometer.
- First, gate on the live cell population based on forward and side scatter.
- Within the live gate, identify specific cell subsets (e.g., lymphocytes, macrophages) using the surface markers.
- For each gated population, analyze the fluorescence intensity of the PAR or cleaved PARP-1 channel.
- Report data as Mean Fluorescence Intensity (MFI) or the percentage of positive cells for the PAR/cleaved PARP-1 signal.
Normalization Strategies in Flow Cytometry
- Internal Biological Controls: The most effective normalization leverages internal controls within the same sample. For example, in a mixed population of cells, the signal from unstimulated cells or a specific unresponsive cell subset can serve as an internal negative control. This is evident in mastitis research, where PAR levels in lymphocytes and macrophages from uninfected cows provide a baseline for comparison to infected samples [28].
- Fluorescence Minus One (FMO) Controls: FMO controls are essential for accurately setting positive gates, especially for low-abundance targets like cleaved PARP-1. An FMO control contains all fluorophore-conjugated antibodies except the one of interest.
- Standardized Beads: For longitudinal studies, fluorescent calibration beads can be used to monitor and correct for day-to-day instrument variability, ensuring consistent MFI measurements over time.
Comparative Analysis: Western Blot vs. Flow Cytometry
The choice between western blot and flow cytometry for detecting PARP-1 cleavage depends on the specific research question. The table below outlines a comparative analysis of the two techniques:
| Parameter | Western Blot | Flow Cytometry |
|---|---|---|
| Primary Readout | Separation by molecular weight; visual confirmation of cleavage fragments (89 kDa) [72]. | Fluorescence intensity at the single-cell level. |
| Normalization Method | Housekeeping Protein (e.g., β-actin) or Total Protein Normalization [75]. | Internal cell population controls, FMO controls, MFI comparison [28]. |
| Key Advantages | Directly confirms proteolytic cleavage; semi-quantitative; relatively accessible equipment. | High-throughput; single-cell resolution in mixed populations; multi-parametric analysis. |
| Key Limitations | Bulk population measurement; cannot assess heterogeneity; more prone to loading artifacts. | Does not directly show molecular weight; requires specific instrumentation and expertise. |
| Ideal Context | Initial validation of PARP-1 cleavage; when protein size confirmation is critical. | Analyzing apoptosis in specific cell subtypes within a complex mixture (e.g., immune cells). |
The Scientist's Toolkit: Essential Reagents
| Research Reagent | Function and Application |
|---|---|
| Anti-Cleaved PARP-1 (Asp214) | Primary antibody specifically recognizing the caspase-cleaved 89 kDa fragment of PARP1; used for specific apoptosis detection in WB, IF, and FC [72]. |
| Anti-PAR Antibody (e.g., clone 10H) | Detects poly(ADP-ribose) polymers, indicating PARP-1 enzymatic activity; used in flow cytometry to monitor early PARP-1 activation in inflammation and apoptosis [28]. |
| Pan-Lymphocyte/Macrophage Markers (e.g., CD45, CD14) | Conjugated antibodies used in flow cytometry to identify and gate on specific leukocyte subpopulations for cell-type-specific analysis of PARP-1 cleavage [28]. |
| Fixation/Permeabilization Kit | Essential reagent for flow cytometry protocols detecting intracellular targets like PAR or cleaved PARP-1; enables antibody access to nuclear antigens [28]. |
| PARP Inhibitor (e.g., ABT-888/Veliparib) | Small molecule inhibitor used as a control to confirm the specificity of PAR detection in assays, verifying that the signal is dependent on PARP enzymatic activity [28]. |
Connecting PARP-1 Cleavage to Apoptotic Signaling
The cleavage of PARP-1 is a pivotal event in the execution phase of apoptosis. During apoptosis, caspases-3 and -7 are activated and cleave PARP-1 at the DEVD214 site. This cleavage generates two fragments: a 24 kDa fragment that may act as a dominant-negative inhibitor of DNA repair, and an 89 kDa fragment (tPARP1) [4] [8]. While the full-length PARP-1 is involved in DNA repair, its cleavage inactivates this function and facilitates cellular disassembly. Recent research has revealed that the 89 kDa truncated PARP1 (tPARP1) translocates to the cytoplasm, where it can mono-ADP-ribosylate the RNA Polymerase III (Pol III) complex. This modification enhances the innate immune response by promoting IFN-β production, creating a link between apoptosis and anti-viral defense mechanisms [8]. Furthermore, studies using non-cleavable PARP-1 mutants (PARP-1UNCL) have demonstrated their protective effect against ischemic challenge, while the expression of the 89 kDa fragment (PARP-189) is cytotoxic, underscoring the critical and opposing roles of PARP-1 and its cleavage products in determining cell fate [4] [55].
Technique Comparison: Choosing the Right Tool for Your Research
Within cell biology and pre-clinical drug discovery, the detection of apoptosis is a critical endpoint for evaluating treatment efficacy and understanding disease mechanisms. A cornerstone methodological question involves selecting the most appropriate technique for identifying this form of programmed cell death. This application note provides a direct comparative analysis of two fundamental techniques—flow cytometry and western blotting—for the detection of apoptosis via the measurement of Poly (ADP-ribose) polymerase-1 (PARP-1) cleavage. Cleavage of PARP-1 by executioner caspases is a well-established hallmark of apoptosis, generating signature 89 kDa and 24 kDa fragments that serve as definitive biochemical markers of this cell death pathway [11] [76]. We frame this comparison within the context of a broader research thesis, providing detailed protocols and quantitative data to guide researchers and drug development professionals in their experimental design.
The Biological Rationale: PARP-1 Cleavage as an Apoptosis Biomarker
PARP-1 is a nuclear enzyme with a pivotal role in DNA repair and genomic integrity. During the early stages of apoptosis, caspase-3 and caspase-7 are activated and cleave PARP-1 at a specific aspartic acid residue (Asp214) within its DNA-binding domain [11] [76]. This proteolytic event results in the separation of the 24 kDa DNA-binding domain fragment from the 89 kDa catalytic domain fragment. The biological consequence is the inactivation of PARP-1's DNA repair function, which prevents cellular energy depletion and facilitates the orderly dismantling of the cell [11]. The appearance of the 89 kDa fragment, and the concomitant disappearance of the full-length 113 kDa PARP-1, is thus a widely accepted and specific indicator of apoptosis.
The following diagram illustrates the process of PARP-1 cleavage during apoptosis and its subsequent detection:
Comparative Technique Analysis: Flow Cytometry vs. Western Blot
The choice between flow cytometry and western blotting for detecting PARP-1 cleavage hinges on the specific experimental requirements, including the need for quantification, single-cell analysis, and throughput. The table below summarizes the core capabilities of each technique.
Table 1: Direct Comparison of Flow Cytometry and Western Blot for PARP-1 Cleavage Detection
| Analytical Parameter | Flow Cytometry | Western Blot |
|---|---|---|
| Sensitivity | High (can detect low-abundance targets in single cells) [28] | Moderate (limited by transfer efficiency and antibody affinity) |
| Specificity | High (achieved via intracellular staining with specific antibodies and gating on cellular subpopulations) [28] | High (achieved via molecular weight separation and specific antibodies) [76] |
| Quantitative Capability | High. Provides robust, statistical data (e.g., % positive cells) from large cell numbers (>10,000 events) [28]. | Semi-Quantitative. Relies on densitometry to compare band intensity ratios (cleaved/full-length) [4]. |
| Key Advantage | Single-cell analysis within heterogeneous samples; ability to multiplex with other markers (e.g., active Caspase-3) [28]. | Direct visualization of specific cleavage fragments (e.g., 89 kDa); confirms proteolytic event. |
| Primary Limitation | Does not directly visualize the molecular weight of the target fragment. | Lacks single-cell resolution; results represent a population average. |
| Throughput | Higher (suitable for multi-sample screening) | Lower (more labor-intensive and time-consuming) |
| Sample Requirement | Can be performed with a lower cell number (~1x10^6 cells) [28]. | Typically requires a higher cell number for protein extraction. |
Detailed Experimental Protocols
Protocol A: Detection of Cleaved PARP-1 by Flow Cytometry
This protocol is adapted from methods used to evaluate PAR content in bovine milk leukocyte subpopulations, demonstrating its application in complex cellular mixtures [28].
Research Reagent Solutions & Key Materials:
- Antibody: Mouse anti-PAR monoclonal antibody (e.g., clone 10H) [28] or anti-cleaved PARP-1 antibody (e.g., clone 194C1439) [76]. The latter specifically recognizes the caspase-cleaved epitope.
- Fixation/Permeabilization Solution: Cytofix/Cytoperm solution or similar commercial kit (e.g., Foxp3/Transcription Factor Staining Buffer Set) [28] [77].
- Secondary Antibody: Goat anti-mouse IgG conjugated to a fluorophore (e.g., AlexaFluor 488) [28].
- Flow Cytometer: Equipped with appropriate lasers and filters for your fluorophore.
Workflow Diagram for Flow Cytometry Protocol:
Step-by-Step Methodology:
- Cell Preparation: Harvest cells and wash twice with cold PBS. Count and adjust concentration to approximately 1 × 10^6 cells per 100 µL of staining buffer [28] [77].
- Fixation and Permeabilization: Resuspend the cell pellet in Fixation/Permeabilization solution. Incubate for 20 minutes at 4°C to fix the cells and make the intracellular epitopes accessible. Wash twice with a permeabilization buffer [28].
- Intracellular Staining: Resuspend the cell pellet in permeabilization buffer containing a saturating concentration of the primary antibody against cleaved PARP-1. Incubate for 45 minutes at 4°C. Include an isotype control for background subtraction.
- Secondary Staining (if required): Wash cells twice to remove unbound antibody. Resuspend in buffer containing the fluorophore-conjugated secondary antibody. Incubate for 30 minutes at 4°C in the dark.
- Data Acquisition and Analysis: Wash cells and resuspend in flow cytometry staining buffer. Acquire data on a flow cytometer, collecting a minimum of 10,000 events per sample. Analyze the data by gating on the single-cell population and then determining the percentage of cells positive for cleaved PARP-1 fluorescence.
Protocol B: Detection of Cleaved PARP-1 by Western Blot
This is a standard protocol for confirming PARP-1 cleavage, leveraging the molecular weight shift as a key identifier [76].
Research Reagent Solutions & Key Materials:
- Lysis Buffer: RIPA buffer or NP-40 buffer supplemented with protease and phosphatase inhibitors.
- Antibody: Mouse anti-cleaved PARP-1 antibody (e.g., clone 194C1439) which is specifically recommended for detection of the cleaved product of PARP-1 [76].
- Gel Electrophoresis System: For SDS-PAGE separation of proteins.
- Membrane: PVDF or nitrocellulose for protein transfer.
Workflow Diagram for Western Blot Protocol:
Step-by-Step Methodology:
- Protein Extraction: Lyse cells in RIPA buffer on ice for 30 minutes. Centrifuge at high speed to pellet debris and collect the supernatant containing the soluble protein. Quantify the protein concentration using a standard assay (e.g., BCA).
- Gel Electrophoresis: Load an equal amount of protein (20-50 µg) per lane on an SDS-polyacrylamide gel (e.g., 8-10%). Run the gel to separate proteins by molecular weight.
- Protein Transfer: Transfer the separated proteins from the gel onto a PVDF or nitrocellulose membrane using a wet or semi-dry transfer system.
- Blocking and Antibody Probing: Block the membrane with 5% non-fat milk in TBST for 1 hour at room temperature to prevent non-specific binding. Incubate the membrane with the primary antibody against cleaved PARP-1 (diluted in blocking buffer) overnight at 4°C. Wash the membrane and incubate with an HRP-conjugated secondary antibody for 1 hour at room temperature.
- Detection and Quantification: Detect the bound antibodies using a chemiluminescent substrate and image the membrane. The cleaved PARP-1 fragment will appear as a band at 89 kDa, while the full-length protein is at 113 kDa. Use densitometry software to quantify the band intensities and calculate the ratio of cleaved to full-length PARP-1.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for PARP-1 Cleavage Detection
| Reagent | Function in the Experiment | Example Product & Specification |
|---|---|---|
| Anti-Cleaved PARP-1 Antibody | Specifically binds to the caspase-generated neo-epitope on the 89 kDa fragment, enabling specific detection. | PARP1 Antibody (194C1439); mouse monoclonal IgG2b; recommended for WB and IP [76]. |
| Fixation/Permeabilization Kit | Preserves cell structure while dissolving membranes, allowing antibodies to access intracellular targets. | Cytofix/Cytoperm Solution Kit; Foxp3/Transcription Factor Staining Buffer Set [28] [77]. |
| Fluorophore-Conjugated Secondary Antibody | Binds to the primary antibody and emits a fluorescent signal for detection in flow cytometry. | Goat anti-mouse IgG AlexaFluor 488 conjugate [28]. |
| HRP-Conjugated Secondary Antibody | Binds to the primary antibody and catalyzes a chemiluminescent reaction for detection in western blot. | Mouse IgG2b Binding Protein (m-IgG2b BP-HRP) [76]. |
| PARP Inhibitor (Control) | Serves as a positive control for PARP-related assays and can help induce or modulate cell death pathways. | ABT-888 (Veliparib); used at 1 µM final concentration [28]. |
The decision between flow cytometry and western blot is not a matter of which is superior, but which is most appropriate for the research question.
- Flow cytometry is the unequivocal choice for high-throughput, quantitative screening of apoptosis in heterogeneous cell populations, such as in mixed cell cultures or primary patient samples, where understanding the response of specific cell subtypes is crucial [28]. Its ability to be multiplexed with other markers like active Caspase-3 provides a multi-parametric view of cell death.
- Western blotting remains the gold standard for confirmatory analysis, providing definitive proof of the proteolytic cleavage event by visualizing the specific molecular weight fragments. It is indispensable for initial assay validation and for studies where the primary goal is to confirm that apoptosis has occurred via the canonical caspase pathway.
For a comprehensive research thesis, an integrated approach is highly recommended. Initial screening and quantification of apoptotic responses can be efficiently conducted using flow cytometry, while key findings are subsequently validated and confirmed using western blot analysis. This synergistic use of both techniques provides both robust statistical power and definitive molecular confirmation.
The detection of apoptosis through the cleavage of Poly(ADP-ribose) polymerase-1 (PARP-1) is a cornerstone event in cellular biology, cancer research, and drug development. The 89 kDa cleavage fragment generated by caspase-3 activity serves as a definitive biochemical marker of programmed cell death. The choice of detection methodology—single-cell analysis (e.g., flow cytometry) or population-level analysis (e.g., Western blot)—critically impacts the throughput, statistical power, and biological insights of a study. This application note details the quantitative advantages and limitations of each approach, providing structured protocols and data to guide researchers in selecting the optimal strategy for their experimental needs, particularly within the context of drug discovery and validation.
Technology Comparison: Single-Cell vs. Population-Level Analysis
The following table summarizes the core characteristics of flow cytometry (a single-cell analysis technique) and Western blot (a population-average technique) for detecting PARP-1 cleavage.
Table 1: Comparison of Flow Cytometry and Western Blot for Apoptosis Detection
| Feature | Flow Cytometry (Single-Cell) | Western Blot (Population Average) |
|---|---|---|
| Throughput | High (can analyze >10,000 cells per second) [78] | Low (manual processing, limited samples per gel) |
| Statistical Power | High (analysis of large, discrete cell numbers enables robust statistical testing of heterogeneous populations) [78] | Low (result is an average from a lysate of millions of cells) |
| Multiplexing Capability | High (simultaneous detection of cleaved PARP, active caspase-3, and cell surface markers) [28] | Low (typically probes for one or two targets per membrane) |
| Detection of Rare Cell Populations | Excellent (can identify and quantify small subpopulations) [78] | Poor (rare cell signals are diluted and masked by the majority) |
| Information on Cellular Heterogeneity | Directly reveals cell-to-cell variation within a sample [78] | Masks cellular heterogeneity; provides a population average |
| Key Advantage | Uncovering heterogeneity and identifying rare cells. | Confirmation of cleavage fragment size; established, accessible protocol. |
| Primary Limitation | Cannot confirm the precise molecular weight of the detected protein. | Lack of cellular resolution and inability to detect heterogeneity. |
Detailed Experimental Protocols
Protocol A: Multiparameter Flow Cytometry for Cleaved PARP-1
This protocol is adapted from studies detecting PARP in leukocyte subpopulations and apoptotic cells [28] [79].
3.1.1 Workflow Diagram
The following diagram outlines the key steps in the flow cytometry protocol for detecting cleaved PARP-1:
3.1.2 Step-by-Step Procedure
- Cell Preparation: Harvest approximately 1x10^6 cells per sample. Wash cells twice with cold PBS.
- Fixation and Permeabilization: Resuspend the cell pellet in 100 µL of PBS. Add the recommended volume of a commercial fixation/permeabilization solution (e.g., Cytofix/Cytoperm Kit). Incubate for 20 minutes in the dark at 4°C [28].
- Intracellular Staining: Wash cells twice with a permeabilization/wash buffer. Incubate cells with saturating amounts of a primary antibody specific for the cleaved form of PARP-1 (e.g., Asp214) for 45 minutes at 4°C [28].
- Secondary Staining (if using a non-conjugated primary antibody): Wash cells and incubate with a fluorophore-conjugated secondary antibody (e.g., AlexaFluor 488) for 30 minutes at 4°C in the dark.
- Multiplexing (Optional): To correlate PARP-1 cleavage with caspase activation, include a fluorophore-conjugated antibody against active Caspase-3 (e.g., PE-conjugated) during the intracellular staining step [28].
- Data Acquisition and Analysis: Resuspend cells in wash buffer and acquire data immediately on a flow cytometer. Analyze data using software such as FlowJo or Kaluza. Set gates based on untreated or isotype-control stained cells to determine the positive population for cleaved PARP-1.
Protocol B: Western Blot for PARP-1 Cleavage Fragments
This protocol is standard for confirming PARP-1 cleavage, as used in studies involving mithramycin and etoposide [79].
3.2.1 Workflow Diagram
The following diagram illustrates the Western blot protocol for detecting PARP-1 cleavage:
3.2.2 Step-by-Step Procedure
- Protein Extraction: Lyse harvested cells in RIPA buffer supplemented with protease and phosphatase inhibitors. Incubate on ice for 30 minutes, then centrifuge at 14,000 x g for 15 minutes to collect the supernatant [79].
- Protein Quantification: Determine protein concentration using a BCA assay. Dilute samples with Laemmli buffer to an equal concentration.
- Gel Electrophoresis and Transfer: Load 20-30 µg of protein per well on an 8-12% SDS-polyacrylamide gel. Separate proteins by electrophoresis, then transfer to a PVDF membrane.
- Immunoblotting: Block the membrane with 5% non-fat milk in TBST for 1 hour. Incubate with primary antibodies against PARP-1 (to detect both full-length and cleaved fragments) and a loading control (e.g., β-actin) overnight at 4°C [79].
- Detection: Wash the membrane and incubate with HRP-conjugated secondary antibodies for 1 hour at room temperature. Detect bands using a chemiluminescent substrate and image with a digital system.
- Analysis: The intact PARP-1 appears at ~116 kDa, while the key apoptotic cleavage fragment appears at ~89 kDa [79]. Densitometry analysis can be used to quantify the ratio of cleaved to full-length PARP-1.
The Scientist's Toolkit: Key Research Reagents
The following table lists essential reagents and their applications for studying PARP-1 in apoptosis.
Table 2: Key Reagent Solutions for PARP-1 Cleavage Analysis
| Reagent / Solution | Function / Application | Example |
|---|---|---|
| Anti-cleaved PARP-1 Antibody | Specific detection of the caspase-generated 89 kDa fragment in flow cytometry and Western blot. | FITC-conjugated anti-cleaved PARP-1 (Asp214) [28] |
| Anti-PARP-1 Antibody | Detects both full-length and cleaved PARP-1; essential for Western blot confirmation. | PARP-1 antibody for Western blot [79] |
| Fixation/Permeabilization Kit | Enables intracellular staining for flow cytometry by making internal epitopes accessible to antibodies. | Cytofix/Cytoperm Solution Kit [28] |
| Anti-active Caspase-3 Antibody | Multiplexing reagent to confirm the apoptotic pathway and correlate with PARP-1 cleavage. | PE-conjugated anti-active Caspase-3 [28] |
| PARP Inhibitors (PARPi) | Positive controls for inducing PARP-1 cleavage and studying synthetic lethality in BRCA-deficient models. | BMN 673 (Talazoparib), ABT-888 (Veliparib) [28] [80] |
| Chemotherapeutic Agents | Inducers of DNA damage and apoptosis, leading to PARP-1 cleavage. Useful for assay validation. | Etoposide, Doxorubicin [28] [79] |
Apoptosis Signaling and PARP-1 Cleavage Pathway
The central role of PARP-1 cleavage in the execution of apoptosis is summarized in the pathway below:
Poly(ADP-ribose) polymerase 1 (PARP-1) is a nuclear enzyme with well-characterized roles in DNA damage repair and the maintenance of genomic integrity. During the execution phase of apoptosis, caspases-3 and -7 cleave PARP-1 at the DEVD214 site, generating two characteristic fragments: a 24-kDa DNA-binding domain fragment and an 89-kDa catalytic domain fragment [4]. This cleavage event serves as a critical biochemical hallmark of apoptosis, as it inactivates DNA repair functions and facilitates the dismantling of the cell. The detection of cleaved PARP-1, particularly the 89-kDa fragment, has become a established method for identifying apoptotic cells in diverse research contexts, from cancer drug development to studies of neuronal cell death [4] [81]. This application note details methodologies for integrating PARP-1 cleavage detection with other apoptotic markers using flow cytometry and western blotting, enabling robust, multiplexed assessment of cell death in research and drug discovery.
PARP-1 in Apoptotic Signaling Pathways
PARP-1 cleavage occupies a decisive position in the apoptotic cascade, typically occurring downstream of caspase-3 activation. The functional consequences of this cleavage are twofold. First, the generation of the 89-kDa fragment is believed to produce a dominant-negative inhibitor of DNA repair, ensuring the irreversibility of the cell death process. Second, research indicates that the cleavage fragments themselves may actively propagate pro-apoptotic signals; the 89-kDa fragment has been demonstrated to be cytotoxic, while the 24-kDa fragment can bind irreversibly to DNA breaks, further preventing DNA repair [20] [4]. Beyond its role as a caspase substrate, PARP-1 also influences cell fate through its interaction with key signaling pathways, including the NF-κB-mediated inflammatory response [4] [55]. Furthermore, recent studies elucidate that inducers of ferroptosis, such as RSL3, can trigger apoptosis through PARP1, involving both caspase-dependent cleavage and METTL3-mediated translational suppression of PARP1, highlighting its role as a nexus for crosstalk between different cell death pathways [20]. The following diagram illustrates the central role of PARP-1 cleavage within the broader context of apoptotic signaling.
Comparative Analysis of Detection Methodologies
The choice between flow cytometry and western blotting for detecting PARP-1 cleavage depends on the specific research question, as each technique offers distinct advantages and limitations concerning throughput, quantitative capability, and single-cell resolution.
Table 1: Comparison of Flow Cytometry and Western Blot for Apoptosis Detection
| Feature | Flow Cytometry | Western Blot |
|---|---|---|
| Primary Readout | Percentage of cells with cleaved PARP-1 (at single-cell level) | Presence/absence of cleaved PARP-1 band in cell population lysate |
| Throughput | High (can analyze 10,000+ cells/sample) | Low to moderate |
| Multiplexing Potential | High (with intracellular staining panels) | Moderate (by stripping/reprobing membranes) |
| Quantification | Semi-quantitative (Mean Fluorescence Intensity) | Semi-quantitative (Band Densitometry) |
| Key Advantage | Ability to correlate cleavage with other markers (e.g., cell cycle, surface markers) on a per-cell basis | Direct visualization of fragment size; well-established, trusted technique |
| Key Limitation | Requires specific antibody validated for intracellular staining and fixation | Loses single-cell and heterogeneous population information |
Key Experimental Findings and Data
Recent studies have quantified PARP-1 cleavage in various experimental models, providing reference data for assay development and validation.
Table 2: Quantitative Data on PARP-1 Cleavage from Key Studies
| Induction Method/Context | Cell Model | Detection Method | Key Findings & Quantitative Data |
|---|---|---|---|
| Staurosporine (1 μM, 4h) | HeLa Cells [81] | Western Blot | Clear detection of the 89 kDa cleaved PARP1 fragment; used for antibody validation. |
| Staurosporine (1 μM, 4h) | HL-60 Cells [81] | Flow Cytometry | Demonstrated a significant increase in the population positive for cleaved PARP1. |
| Oxygen/Glucose Deprivation (OGD) | SH-SY5Y & Primary Rat Neurons [4] [55] | Viability Assays | Expression of cleaved 89 kDa fragment (PARP-1$_{89}$) was cytotoxic, reducing cell viability. |
| Cadmium Acetate (5 μM) | NRK-52E & rPT Cells [82] | Western Blot | PARP-1 overexpression and cleavage contributed to parthanatos, a PARP-1-dependent cell death. |
| RSL3 (Ferroptosis Inducer) | Various Cancer Cells [20] | Western Blot, RT-qPCR | Induced PARP1 cleavage via caspase-3 and reduced full-length PARP1 via m6A-mediated translational suppression. |
| LPS (1 μg/mL, 1h & 16h) | Bovine PBMCs [19] | Flow Cytometry | PAR levels (MFI) increased significantly after 1h; Cleaved PARP-1+ cells appeared after 16h, indicating late apoptosis. |
Integrated Experimental Protocols
This section provides detailed protocols for the simultaneous detection of PARP-1 cleavage alongside other apoptotic markers, enabling a comprehensive analysis of cell death.
Multiplexed Flow Cytometry Protocol for Apoptosis
This protocol allows for the concurrent analysis of cleaved PARP-1, active caspase-3, and cell cycle status within a single sample, providing a multi-parametric view of apoptosis.
Key Research Reagent Solutions:
- Anti-Cleaved PARP-1 (Asp214) Antibody [clone F21-852]: FITC-conjugated antibody validated for flow cytometry to specifically detect the apoptotic 89 kDa fragment [19].
- PE-conjugated Anti-Active Caspase-3 Antibody [clone C92-605]: Detects the active form of executioner caspase-3, a direct mediator of PARP-1 cleavage [19].
- 7-Aminoactinomycin D (7-AAD): A fluorescent DNA intercalator used to exclude late apoptotic/necrotic cells and for cell cycle analysis.
- Annexin V Binding Buffer: Essential for maintaining calcium-dependent phosphatidylserine binding by Annexin V conjugates.
- Cytofix/Cytoperm Fixation/Permeabilization Solution: Required for intracellular staining of cleaved PARP-1 and active caspase-3.
Procedure:
- Cell Stimulation & Harvest: Induce apoptosis in cells (e.g., with 1 μM Staurosporine for 4 hours). Harvest both adherent and suspension cells, washing once with cold PBS.
- Annexin V Staining (Optional Live Cell Marker): Resuspend cell pellet in 100 μL of 1X Annexin V Binding Buffer. Add 5 μL of Annexin V conjugate (e.g., APC) and incubate for 15 minutes in the dark at room temperature. Add 400 μL of binding buffer and proceed immediately to the next step without washing [81].
- Fixation and Permeabilization: Pellet cells and thoroughly resuspend in 250 μL of Cytofix/Cytoperm solution. Incubate for 20 minutes on ice [19].
- Intracellular Staining: Wash cells twice with 1X Perm/Wash Buffer. Resuspend the cell pellet in 50-100 μL of Perm/Wash Buffer containing predetermined optimal concentrations of FITC anti-cleaved PARP-1 and PE anti-active Caspase-3 antibodies. Incubate for 45 minutes at 4°C in the dark.
- DNA Staining (for Cell Cycle): After two final washes, resuspend cells in Perm/Wash Buffer containing 20 μg/mL 7-AAD and 100 μg/mL RNase A. Incubate for 30 minutes at room temperature in the dark [81].
- Flow Cytometric Analysis: Analyze samples on a flow cytometer equipped with appropriate lasers and filters. Collect a minimum of 10,000 events per sample. Use fluorescence minus one (FMO) controls to establish gating boundaries.
Data Interpretation:
- Early Apoptotic: Annexin V+/ cleaved PARP-1-/ active Caspase-3±
- Mid Apoptotic: Annexin V+/ cleaved PARP-1+/ active Caspase-3+
- Late Apoptotic: Annexin V+/ cleaved PARP-1+/ 7-AAD+
- Necrotic: Annexin V-/ 7-AAD+
Sequential Western Blot Protocol for Multiplexing
This protocol enables the sequential detection of multiple apoptotic proteins from a single membrane, maximizing data output from limited samples.
Key Research Reagent Solutions:
- Anti-Cleaved PARP1 Antibody [clone 4B5BD2, ab110315]: A recombinant monoclonal antibody highly specific for the 89 kDa fragment generated by cleavage at Asp214; does not recognize full-length PARP1 [81].
- Anti-Caspase-3 Antibody: Detects both full-length (35 kDa) and cleaved (17/19 kDa) fragments of caspase-3.
- Anti-γH2AX (Ser139) Antibody: Marker for DNA double-strand breaks, useful for confirming genotoxic stress [20].
- HRP-conjugated Secondary Antibodies: For chemiluminescent detection.
- Stripping Buffer: A mild, acidic solution (e.g., Glycine-HCl, SDS-containing buffer) to remove primary and secondary antibodies without stripping the transferred proteins.
Procedure:
- Sample Preparation: Lyse control and treated cells in RIPA buffer supplemented with protease and phosphatase inhibitors. Determine protein concentration using a BCA assay.
- Gel Electrophoresis and Transfer: Load 20-40 μg of total protein per lane onto a 4-12% Bis-Tris polyacrylamide gel. Separate proteins via SDS-PAGE and transfer to a nitrocellulose or PVDF membrane.
- Primary Antibody Incubation (Round 1): Block the membrane with 5% non-fat milk in TBST for 1 hour. Incubate with a primary antibody cocktail targeting proteins of distinct molecular weights (e.g., Cleaved PARP1 [89 kDa], Caspase-3 [17/19 kDa], and a loading control like GAPDH [37 kDa]) overnight at 4°C [82] [81].
- Detection (Round 1): Incubate with appropriate HRP-conjugated secondary antibodies for 1-2 hours at room temperature. Develop using enhanced chemiluminescence (ECL) reagent and image.
- Membrane Stripping: Wash the membrane thoroughly. Incubate with gentle agitation in stripping buffer (e.g., 0.2 M Glycine, 0.1% SDS, 1% Tween 20, pH 2.2) for 10-20 minutes. Wash extensively with TBST.
- Primary Antibody Incubation (Round 2): Re-block the membrane for 30 minutes. Probe with the next set of primary antibodies (e.g., γH2AX [17 kDa], PARP1 [113 kDa full-length]) overnight at 4°C [20].
- Detection (Round 2): Repeat the secondary antibody incubation and ECL detection steps.
Data Interpretation:
- Apoptotic induction is confirmed by the appearance of the 89 kDa cleaved PARP1 band and the 17/19 kDa cleaved caspase-3 bands, concurrent with the diminution of the full-length PARP1 (113 kDa) and pro-caspase-3 (35 kDa) signals.
- γH2AX upregulation indicates the presence of DNA damage, which may be the initiating factor for apoptosis.
The workflow for this multiplexed western blot approach is summarized below.
The integration of PARP-1 cleavage detection with other apoptotic markers significantly enhances the resolution and reliability of cell death analysis. Flow cytometry offers unparalleled power for multiparametric, single-cell analysis, ideal for heterogeneous populations and high-throughput screening. In contrast, western blotting provides definitive confirmation of protein cleavage events based on molecular weight. The choice of technique and the specific multiplexing strategy should be guided by the experimental goals. The protocols and data outlined herein provide a robust framework for researchers to systematically dissect apoptotic pathways, which is fundamental for advancing our understanding of cell death mechanisms in disease and therapy.
The detection of apoptosis, a programmed and controlled form of cell death, is essential for both basic biological research and pharmaceutical development. Poly(ADP-ribose) polymerase-1 (PARP-1) cleavage has emerged as a definitive biochemical marker of apoptosis, as it is a preferred substrate for executioner caspases-3 and -7 [11]. During apoptosis, these caspases cleave the 116-kDa full-length PARP-1 at the DEVD214 site, generating signature fragments of 24 kDa and 89 kDa [4] [11]. The 89 kDa truncated PARP-1 (tPARP-1) loses its nuclear localization signal and translocates to the cytoplasm, while the 24 kDa DNA-binding domain fragment remains in the nucleus [8]. The choice between flow cytometry and western blotting to detect this cleavage event is not merely technical but fundamentally shapes the type of biological questions a researcher can address. This application note provides detailed protocols and frameworks for implementing these techniques in two distinct contexts: high-throughput drug screening and targeted mechanistic studies.
Biological Significance of PARP-1 Cleavage
From DNA Repair to Apoptosis Execution
PARP-1 is a nuclear enzyme with a primary role in the detection and repair of DNA single-strand breaks via the poly(ADP-ribosyl)ation (PARylation) pathway [4] [83]. However, upon initiation of apoptosis and activation of the caspase cascade, PARP-1 is cleaved, which serves two key purposes. First, it inactivates the DNA repair function of PARP-1, preventing futile DNA repair efforts and facilitating the apoptotic process [11]. Second, recent evidence indicates that the cleavage fragments themselves may have active biological roles. The 24-kDa fragment acts as a trans-dominant inhibitor of intact PARP-1 by irreversibly binding to DNA strand breaks, while the 89-kDa tPARP-1 has been shown to translocate to the cytoplasm and mediate ADP-ribosylation of RNA Polymerase III, potentially influencing innate immune responses during apoptosis [11] [8].
PARP-1 in Cell Death and Inflammation Signaling
Beyond its role as a caspase substrate, PARP-1 influences cell fate through its function as a cofactor for transcription factors like NF-κB. Research demonstrates that different PARP-1 cleavage products differentially regulate the inflammatory response. Expression of the 89-kDa fragment increases NF-κB activity and expression of pro-inflammatory proteins like iNOS and COX-2, while the uncleavable PARP-1 (PARP-1UNCL) and the 24-kDa fragment have the opposite effect, reducing inflammatory mediators and increasing expression of the anti-apoptotic protein Bcl-xL [4]. This complex interplay positions PARP-1 cleavage as a critical node connecting cell death execution to inflammatory signaling, a consideration vital for interpreting experimental results in both drug screening and mechanistic contexts.
The following diagram illustrates the core apoptosis signaling pathway leading to PARP-1 cleavage and the divergent biological outcomes influenced by its fragments.
Figure 1: Apoptosis Signaling Pathway and PARP-1 Cleavage Outcomes. The diagram illustrates key apoptotic stimuli leading to caspase activation and PARP-1 cleavage, generating fragments with distinct biological activities that influence cell death and inflammatory processes [4] [11] [8].
Method Selection Guide: Flow Cytometry vs. Western Blot
The decision between flow cytometry and western blotting hinges on the specific experimental goals, throughput requirements, and information needs. The table below provides a structured comparison to guide method selection.
Table 1: Comparative Analysis of Flow Cytometry and Western Blot for PARP-1 Cleavage Detection
| Parameter | Flow Cytometry | Western Blotting |
|---|---|---|
| Primary Application | High-throughput screening, population heterogeneity analysis [19] | Mechanistic studies, fragment characterization, pathway mapping [29] |
| Throughput | High (96/384-well plates, thousands of samples) | Low to medium (typically 10-20 samples per gel) |
| Information Type | Single-cell, multi-parametric data on protein level/localization [84] | Bulk population data, molecular weight confirmation [29] |
| Key Readout | Percentage of PARP-1 positive cells, fluorescence intensity shift [19] | Band presence/intensity (full-length vs. cleaved fragments) [29] |
| Detected Fragment | Often the 89 kDa fragment (using C-terminal antibodies) [19] | Both 24 kDa and 89 kDa fragments (using domain-specific antibodies) [4] [11] |
| Cell State Context | Yes (via light scatter: FSC/SSC for size/granularity) [84] | No (lysate-based, no morphological context) |
| Multiplexing Potential | High (with caspase-3, annexin V, viability markers) [84] [19] | Limited (typically 2-3 targets with stripping/re-probing) |
| Data Output | Population statistics (e.g., % positive cells, MFI) [84] [19] | Semi-quantitative band intensity (e.g., cleaved/full-length ratio) [29] |
Workflow I: Flow Cytometry for Drug Screening
Flow cytometry offers unparalleled advantages for drug discovery applications where rapid assessment of compound efficacy across thousands of samples is required. Its ability to quantify PARP-1 cleavage at the single-cell level within a heterogeneous population provides rich datasets for prioritizing lead compounds.
Detailed Protocol: PARP-1 Cleavage Detection by Flow Cytometry
Step 1: Cell Preparation and Treatment
- Plate cells in 96-well or 384-well U-bottom plates suitable for centrifugation. Common models include SH-SY5Y neuroblastoma cells or primary cortical neurons for neuronal apoptosis studies [4].
- Treat cells with experimental compounds (e.g., chemotherapeutic agents, targeted therapeutics) for defined time periods. Include controls: untreated cells (negative control) and cells treated with a known apoptosis inducer like 1 μM doxorubicin or staurosporine (positive control) [19].
Step 2: Cell Harvesting and Staining
- Harvest cells by gentle centrifugation (300 × g for 5 minutes) and wash with cold PBS.
- Fix and permeabilize cells using the Cytofix/Cytoperm solution (BD Biosciences) for 20 minutes at 4°C [19]. This critical step allows antibody access to intracellular epitopes.
- Incubate with primary antibody against PARP-1 (specific for the 89 kDa cleavage fragment, e.g., anti-cleaved PARP-1 Asp214) for 45 minutes at 4°C [19].
- Wash and incubate with a fluorochrome-conjugated secondary antibody (e.g., AlexaFluor 488) for 30 minutes at 4°C in the dark.
Step 3: Data Acquisition and Analysis
- Resuspend cells in flow cytometry buffer and acquire data on a flow cytometer capable of detecting the chosen fluorochrome.
- Use forward scatter (FSC) vs. side scatter (SSC) dot plots to gate on intact single cells, excluding debris and aggregates [84].
- Analyze the fluorescence intensity of the PARP-1 cleavage marker. The percentage of cells positive for cleaved PARP-1 is calculated by comparing to the isotype control or untreated cells [84] [19].
- For enhanced data richness, simultaneously stain with Annexin V and a viability dye like propidium iodide (PI) to distinguish early apoptotic (Annexin V+/PI-), late apoptotic (Annexin V+/PI+), and necrotic (Annexin V-/PI+) cells [8].
Workflow Diagram: Flow Cytometry for Drug Screening
The following diagram outlines the key steps in the flow cytometry workflow, highlighting its suitability for parallel processing and multi-parametric analysis.
Figure 2: Flow Cytometry Workflow for PARP-1 Cleavage Detection. The protocol emphasizes high-throughput processing and the potential for multiplexed analysis to provide context on cell death stages [84] [19].
Workflow II: Western Blot for Mechanistic Studies
Western blotting remains the gold standard for confirming the specific proteolytic cleavage of PARP-1, providing unambiguous evidence of apoptosis through molecular weight verification of the characteristic 24 kDa and 89 kDa fragments.
Detailed Protocol: PARP-1 Cleavage Detection by Western Blot
Step 1: Cell Lysis and Protein Quantification
- Lyse treated cells in RIPA buffer supplemented with protease and phosphatase inhibitors to preserve post-translational modifications.
- Centrifuge lysates at 12,000 × g for 15 minutes at 4°C to remove insoluble material.
- Quantify protein concentration using a Bradford or BCA assay. Normalize all samples to the same concentration (e.g., 1-2 μg/μL) with lysis buffer [29].
Step 2: Gel Electrophoresis and Transfer
- Load 20-40 μg of total protein per well on a 4-12% Bis-Tris polyacrylamide gel. A 4-20% gradient gel can also provide optimal resolution for separating the 116 kDa full-length and 89 kDa cleaved PARP-1.
- Include a pre-stained protein molecular weight ladder for accurate size determination.
- Run gel electrophoresis at constant voltage (120-150V) until the dye front reaches the bottom.
- Transfer proteins from the gel to a PVDF or nitrocellulose membrane using a wet or semi-dry transfer system.
Step 3: Immunoblotting and Detection
- Block the membrane with 5% non-fat milk or BSA in TBST for 1 hour at room temperature to prevent non-specific antibody binding.
- Incubate with primary antibodies overnight at 4°C. For comprehensive analysis, use a combination of:
- Anti-PARP-1 antibody (recognizing both full-length and cleaved fragments)
- Anti-cleaved PARP-1 antibody (specific to the 89 kDa fragment neo-epitope) [29]
- Anti-caspase-3 antibody (to show correlated activation of the apoptotic executor)
- Wash membrane thoroughly (3 × 5 minutes with TBST) and incubate with an appropriate HRP-conjugated secondary antibody for 1 hour at room temperature.
- Develop the blot using enhanced chemiluminescence (ECL) substrate and image with a digital imaging system.
Step 4: Data Analysis and Normalization
- Quantify band intensity using densitometry software such as ImageJ [29].
- Calculate the ratio of cleaved PARP-1 (89 kDa fragment) to full-length PARP-1 (116 kDa) to normalize for total PARP-1 expression levels.
- Normalize data to a housekeeping protein (e.g., β-actin, GAPDH) to account for loading variations [29].
- For mechanistic insights, re-probe the membrane for other apoptosis-related proteins like Bcl-2, Bax, or phosphorylated signaling molecules.
Workflow Diagram: Western Blot for Mechanistic Studies
The following diagram outlines the western blot workflow, emphasizing its sequential nature and the key steps that enable precise molecular characterization.
Figure 3: Western Blot Workflow for PARP-1 Cleavage Detection. The protocol highlights steps that ensure specific detection and quantification of PARP-1 cleavage fragments, providing molecular weight confirmation essential for mechanistic validation [29].
The Scientist's Toolkit: Essential Research Reagents
Successful detection of PARP-1 cleavage requires carefully selected reagents and controls. The table below details essential materials and their specific functions in apoptosis detection assays.
Table 2: Key Research Reagent Solutions for PARP-1 Cleavage Studies
| Reagent Category | Specific Examples | Function & Application Notes |
|---|---|---|
| Cell Models | SH-SY5Y neuroblastoma cells, Primary cortical neurons [4] | Validated models for neuronal apoptosis; primary cells provide physiological relevance. |
| Apoptosis Inducers | Doxorubicin (1 μM), Staurosporine, Etoposide phosphate (VP-16) [11] [19] | Positive controls for inducing caspase-dependent apoptosis and PARP-1 cleavage. |
| PARP-1 Antibodies (Flow Cytometry) | Anti-cleaved PARP-1 (Asp214) [19] | Specifically detects the 89 kDa fragment generated by caspase cleavage; essential for intracellular staining. |
| PARP-1 Antibodies (Western Blot) | Anti-PARP-1 (full-length + cleaved), Anti-cleaved PARP-1 (89 kDa), Anti-24 kDa fragment [4] [29] [11] | Confirm complete cleavage pattern; domain-specific antibodies verify fragment identity. |
| Apoptosis Antibody Cocktails | Pro/p17-caspase-3 + Cleaved PARP1 + Actin [29] | Pre-mixed multiplex antibodies for efficient detection of multiple apoptotic markers in a single blot. |
| Caspase Inhibitors | Z-VAD-FMK (pan-caspase inhibitor) [11] | Negative control to confirm caspase-dependence of observed PARP-1 cleavage. |
| Flow Cytometry Reagents | Cytofix/Cytoperm Kit, Annexin V Binding Buffer, Propidium Iodide [19] | Enable cell fixation/permeabilization and multiparametric analysis of cell death stages. |
| Detection Enzymes | HRP-conjugated secondary antibodies, ECL substrate [29] | Generate measurable signal for western blot detection; choice impacts sensitivity and dynamic range. |
Data Interpretation and Analysis
Flow Cytometry Data Analysis
Flow cytometry data should be analyzed systematically. Begin by gating on intact single cells using FSC-A vs. SSC-A to exclude debris and doublets [84]. The resulting population can then be plotted on a histogram to visualize the fluorescence intensity of the cleaved PARP-1 stain. A clear rightward shift in the histogram compared to the isotype control indicates the presence of cleaved PARP-1. The percentage of positive cells is determined by setting a threshold based on the negative control, typically exceeding 95% of control cells. Data can be reported as both the percentage of positive cells and the Mean Fluorescence Intensity (MFI), which provides information about the extent of cleavage per cell [19]. When multiplexed with Annexin V and PI, cells in early apoptosis will typically be Annexin V+/PI- and may show intermediate levels of PARP-1 cleavage, while late apoptotic cells (Annexin V+/PI+) typically exhibit high levels of cleavage.
Western Blot Data Analysis
Western blot analysis provides semi-quantitative data on PARP-1 cleavage. A successful apoptotic experiment will show a decrease in the 116 kDa full-length PARP-1 band intensity with a corresponding increase in the 89 kDa cleavage product band [29] [11]. In some cases, the 24 kDa fragment may also be detectable with specific antibodies. Band intensities should be quantified by densitometry. The key metric is the ratio of cleaved PARP-1 to full-length PARP-1, which normalizes for total PARP-1 expression and provides a clear indicator of apoptotic progression. This ratio should then be normalized to a loading control (e.g., β-actin or GAPDH) to account for any variations in sample loading [29]. For mechanistic studies, correlate PARP-1 cleavage with caspase-3 activation (appearance of the p17/p19 cleaved fragments) and changes in regulators like Bcl-2 family proteins.
Application to Research Objectives
Drug Screening Implementation
In drug discovery, flow cytometry enables high-throughput compound screening to identify novel apoptosis-inducing therapeutics. Implementation involves:
- Automation: Utilize liquid handling robots for cell plating, compound addition, and staining procedures in 384-well plates.
- Validation: Establish baseline PARP-1 cleavage levels in control cells and determine the Z'-factor for assay quality assessment.
- Multiplexing: Combine cleaved PARP-1 detection with viability markers (e.g., propidium iodide) to calculate specific apoptosis, distinguishing genuine programmed cell death from general toxicity.
- Dose-Response: Screen compounds across a concentration range (e.g., 1 nM - 10 μM) to establish potency (EC50 values) for lead prioritization [19].
Mechanistic Studies Implementation
For mechanistic investigations, western blotting provides the molecular specificity required to:
- Confirm Apoptotic Mechanism: Verify PARP-1 cleavage fragment sizes to distinguish apoptosis from other cell death pathways [11].
- Map Signaling Pathways: Analyze temporal relationships between PARP-1 cleavage and activation of upstream/downstream factors (e.g., caspase-8, -9, Bid, AIF).
- Investigate Fragment Functions: Study the distinct biological activities of the 24 kDa and 89 kDa fragments using overexpression constructs (PARP-124, PARP-189) [4].
- Explore Non-Apoptotic Roles: Investigate emerging functions of tPARP-1, such as its role in cytoplasmic RNA Pol III modification and innate immune activation [8].
Troubleshooting Common Challenges
Low Signal in Flow Cytometry: Optimize antibody titration and permeabilization duration (20 minutes recommended) [19]. Include a positive control (doxorubicin-treated cells) to validate the assay.
High Background in Western Blot: Increase blocking time, optimize antibody concentrations, and extend wash durations. Ensure sufficient protein normalization during sample preparation.
Incomplete Cleavage Pattern: Verify apoptosis induction efficiency and timing. Some cell types or death stimuli may produce partial cleavage. Include a robust positive control.
Discordant Results Between Techniques: Consider biological differences—flow cytometry may detect early cleavage in a subpopulation, while western blot requires substantial cleavage across the population for detection.
The detection of apoptosis is a critical endpoint in many biological and pharmacological studies, particularly in cancer research and drug development. Among the various molecular markers of apoptosis, the cleavage of Poly(ADP-ribose) polymerase 1 (PARP1) stands out as a well-established and reliable indicator of caspase-dependent programmed cell death. PARP1, a 116 kDa nuclear enzyme involved in DNA repair, is cleaved by executioner caspases (primarily caspase-3) during apoptosis into characteristic fragments of approximately 89 kDa and 24 kDa [85] [86] [87]. This cleavage event serves as a definitive marker of irreversible commitment to the apoptotic pathway.
While flow cytometry offers rapid, quantitative analysis of PARP1 cleavage at the single-cell level within heterogeneous populations, Western blotting provides complementary verification of specific proteolytic fragment size and identity. This application note details a correlative approach that leverages the strengths of both techniques to generate validated, high-quality data on apoptotic progression, with specific focus on methodological considerations for detecting PARP1 cleavage.
PARP1 Cleavage as an Apoptosis Marker
Biological Significance and Mechanism
In response to minor DNA damage, PARP1 is activated and initiates DNA repair pathways through poly(ADP-ribosyl)ation (PARylation) of target proteins [88] [19]. However, during apoptosis, caspase-3 and caspase-7 cleave PARP1 at the conserved Asp214-Gly215 site, separating the N-terminal DNA-binding domains (24 kDa) from the C-terminal catalytic domain (89 kDa) [86] [89]. This cleavage inactivates PARP1's DNA repair function, preventing futile energy consumption and facilitating cellular disassembly.
Recent research has revealed that the cleavage fragments themselves may have regulatory roles. The N-terminal fragment (ZnF1–2PARP1) remains bound to DNA breaks and can trans-dominantly inhibit DNA repair by competing with full-length PARP1 and PARP2 [89]. The C-terminal catalytic fragment (PARP1ΔZnF1–2) retains basal enzymatic activity but is incapable of DNA-dependent stimulation [89].
Technical Comparison of Detection Methods
Table 1: Comparison of Flow Cytometry and Western Blot for Detecting PARP1 Cleavage
| Parameter | Flow Cytometry | Western Blot |
|---|---|---|
| Primary Output | Percentage of cells with cleaved PARP1 | Molecular weight confirmation of fragments |
| Quantification | Single-cell, statistical analysis of populations | Semi-quantitative, bulk population analysis |
| Sensitivity | High (can detect rare cells) | Moderate (requires sufficient protein load) |
| Spatial Context | Lost (cells are permeabilized) | Lost (tissue/cells are homogenized) |
| Key Advantage | Multiparametric analysis, cell sorting capability | Fragment size validation, specificity confirmation |
| Typical Antibody Target | Intracellular epitope near cleavage site (e.g., Asp214) | Full-length and/or cleaved fragments |
| Sample Throughput | High (rapid analysis of thousands of cells) | Low to moderate (gel electrophoresis required) |
| Recommended Application | Screening, kinetic studies, heterogeneous populations | Validation, specificity confirmation, fragment analysis |
Integrated Experimental Workflow
The following diagram illustrates the correlative experimental workflow for validating flow cytometry data with Western blot analysis:
Detailed Methodologies
Flow Cytometry Protocol for Detecting Cleaved PARP1
This protocol is adapted from published methodologies using intracellular staining for cleaved PARP1 followed by flow cytometric analysis [19] [81].
Materials Required:
- Antibody: Anti-Cleaved PARP1 (Asp214) monoclonal antibody (e.g., Clone 4B5BD2) [81]
- Cells: Treated and untreated control cells (e.g., HeLa, HL-60)
- Induction Agent: Staurosporine (1 μM, 4 hours) or other apoptosis inducer [81]
- Buffers: Flow cytometry staining buffer, fixation/permeabilization solution
- Equipment: Flow cytometer with 488 nm laser and appropriate filter sets
Step-by-Step Procedure:
Apoptosis Induction and Cell Harvesting
- Treat cells with apoptosis-inducing agent (e.g., 1 μM staurosporine for 4 hours) alongside untreated controls.
- Harvest cells by gentle trypsinization or scraping, followed by centrifugation at 300 × g for 5 minutes.
- Wash cells once with cold PBS and adjust concentration to 1 × 10^6 cells/mL in PBS.
Cell Fixation and Permeabilization
- Fix cells with 4% paraformaldehyde for 20 minutes at room temperature.
- Centrifuge and resuspend in permeabilization buffer (e.g., 0.1% Triton X-100 in PBS) for 15 minutes on ice.
- Note: Optimization may be required for different cell types; bovine cells required 20 minutes permeabilization in published studies [19].
Antibody Staining
- Incubate cells with anti-cleaved PARP1 antibody (1.0 μg/mL recommended for clone 4B5BD2) [81] for 2 hours at room temperature or overnight at 4°C.
- Include appropriate controls: unstained cells, isotype control, and secondary antibody-only control.
- Wash cells twice with staining buffer to remove unbound antibody.
Data Acquisition and Analysis
- Analyze samples using a flow cytometer with capability for detecting FITC or Alexa Fluor 488.
- Collect a minimum of 10,000 events per sample.
- Set gates based on untreated and isotype controls to determine positive population for cleaved PARP1.
- Analyze data using flow cytometry analysis software to determine percentage of cleaved PARP1-positive cells.
Western Blot Protocol for Validating PARP1 Cleavage
This protocol provides a complementary method to confirm the presence of specific PARP1 cleavage fragments detected in flow cytometry.
Materials Required:
- Antibodies:
- Cell Lysis: RIPA buffer or IP lysis buffer with protease inhibitors [88]
- Gel Electrophoresis: SDS-PAGE system, PVDF or nitrocellulose membrane
- Detection: ECL or similar chemiluminescent substrate
Step-by-Step Procedure:
Protein Extraction and Quantification
- Lyse cells in IP lysis buffer (50 mM Tris-HCl pH 7.4, 1% Triton X-100, 1% NP-40, 150 mM NaCl, 1 mM EDTA) containing protease inhibitor cocktail [88].
- Incubate on ice for 30 minutes, then centrifuge at 13,500 rpm for 20 minutes at 4°C.
- Collect supernatant and quantify protein concentration using Bradford or BCA assay.
- Prepare samples with 40-50 μg total protein per lane in SDS loading buffer [88].
Gel Electrophoresis and Transfer
- Separate proteins by SDS-PAGE using 8-12% gradient gels.
- Transfer to PVDF membrane using standard wet or semi-dry transfer methods.
- Confirm transfer by Ponceau S staining if desired.
Antibody Detection and Development
- Block membrane with 5% non-fat milk or BSA in TBST for 1 hour at room temperature.
- Incubate with primary antibodies diluted in blocking buffer:
- Incubate overnight at 4°C with gentle agitation.
- Wash membrane 3× with TBST, 10 minutes each.
- Incubate with appropriate HRP-conjugated secondary antibody (1:2000-1:10000) for 1 hour at room temperature.
- Develop using enhanced chemiluminescence substrate and image with digital imaging system.
Method Validation and Correlation
Specificity Controls and Antibody Validation
Rigorous validation is essential when employing correlative approaches. Key considerations include:
- Antibody Specificity: Ensure antibodies are validated for the specific application. For example, the cleaved PARP1 antibody #5625 specifically recognizes the 89 kDa fragment produced by caspase cleavage but does not recognize full-length PARP1 [86].
- Genetic Controls: Use PARP1 knockout cell lines (e.g., HAP1 PARP1 KO) to confirm antibody specificity, as demonstrated for antibody ab110315 [81].
- Orthogonal Validation: Employ multiple antibodies targeting different epitopes. For instance, use both N-terminal-specific cleaved PARP1 antibodies and C-terminal antibodies that recognize both full-length and cleaved PARP1 [87] [90].
- Apoptosis Induction Controls: Include both positive (staurosporine-treated) and negative (untreated) controls in every experiment.
Table 2: Key Research Reagent Solutions for PARP1 Cleavage Detection
| Reagent | Specific Example | Application | Key Feature | Validation Evidence |
|---|---|---|---|---|
| Anti-Cleaved PARP1 (Asp214) | Clone D64E10 (CST #5625) [86] | WB, IHC, IF, FC | Specific for 89 kDa fragment only | Does not recognize full-length PARP1 [86] |
| Anti-PARP1 | CST #9542 [85] | WB | Detects full-length and 89 kDa fragment | Specific for PARP1, not other isoforms [85] |
| Anti-Cleaved PARP1 | Clone 4B5BD2 (ab110315) [81] | FC, WB, ICC | Recombinant monoclonal | KO-validated; specific for Asp214 cleavage site [81] |
| Anti-PARP1 | 13371-1-AP (Proteintech) [90] | WB, IHC, IF, IP | C-terminal region | Detects full-length and 89 kDa fragment [90] |
| Anti-Cleaved PARP1 | ab4830 [87] | WB | Polyclonal | Specific for 85 kDa fragment; multiple publications [87] |
Data Interpretation and Correlation
The following diagram illustrates the molecular events of PARP1 cleavage and the corresponding detection approaches:
Interpreting Correlative Data:
- Strong Correlation: When flow cytometry shows a high percentage of cleaved PARP1-positive cells and Western blot confirms strong 89 kDa band intensity, this validates both the quantification and specificity of detection.
- Discordant Results: If flow cytometry indicates cleaved PARP1-positive cells but Western blot shows weak or absent 89 kDa band, consider:
- Sensitivity differences between techniques
- Potential degradation of protein samples
- Antibody specificity issues
- Subcellular localization changes not detected by Western blot
The correlative approach of using Western blot to validate flow cytometry data for PARP1 cleavage detection provides a robust framework for apoptosis assessment. This dual-method strategy leverages the quantitative power of flow cytometry with the specificity and fragment confirmation capability of Western blotting. By implementing the detailed protocols and validation strategies outlined in this application note, researchers can generate high-quality, reproducible data on apoptotic progression, essential for reliable research outcomes in basic science and drug development contexts.
The integration of these techniques is particularly valuable in preclinical drug screening, where verifying the mechanism of drug-induced apoptosis through PARP1 cleavage can provide critical insights for therapeutic development, especially in cancer research where PARP1 inhibitors are increasingly employed [88] [91].
Conclusion
The detection of PARP-1 cleavage remains a cornerstone of apoptosis research, with both western blot and flow cytometry offering distinct and complementary advantages. Western blot provides definitive fragment identification and is ideal for initial validation, while flow cytometry enables high-throughput, multiparametric single-cell analysis crucial for heterogeneous cell populations. The choice between these techniques should be guided by the specific research objectives, required throughput, and need for multiplexing. Future directions include the development of more specific antibodies, standardized protocols for complex samples like tissue extracts, and the integration of PARP-1 cleavage data with other omics technologies to build a more comprehensive understanding of cell death pathways in disease and therapy.
References
- 1. - PARP and its associated nucleases in 1 damage response - PMC DNA [https://pmc.ncbi.nlm.nih.gov/articles/PMC6764844/]
- 2. Roles and therapeutic potential of PARP-1 in ... [https://www.sciencedirect.com/science/article/abs/pii/S0006295225006380]
- 3. Targeting PARP1: A Promising Approach for Next- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12162764/]
- 4. Poly(ADP-ribose) polymerase-1 and its cleavage products ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4013233/]
- 5. Inflammasome-Activated Caspase 7 Cleaves PARP1 to ... [https://www.sciencedirect.com/science/article/pii/S109727651200175X]
- 6. | Abcam PARP 1 [https://www.abcam.com/en-us/targets/parp1/412710]
- 7. Poly(ADP-ribose) polymerase-1 protects neurons against ... [https://www.nature.com/articles/4402117]
- 8. Truncated PARP1 mediates ADP-ribosylation of RNA ... [https://www.nature.com/articles/s41421-021-00355-1]
- 9. Potent EGFR/ PARP - 1 inhibition by spirooxindole-triazole hybrids for... [https://pubs.rsc.org/en/content/articlelanding/2025/ra/d4ra05966b]
- 10. STING directly interacts with PAR to promote apoptosis ... [https://www.nature.com/articles/s41418-025-01457-z]
- 11. PARP-1 cleavage fragments: signatures of cell-death ... [https://biosignaling.biomedcentral.com/articles/10.1186/1478-811X-8-31]
- 12. Cleaved PARP (Asp214) Antibody #9541 [https://www.cellsignal.com/products/primary-antibodies/cleaved-parp-asp214-antibody/9541?srsltid=AfmBOor0_BZzKZpxmg4RXaaP4k7o38Weg6ayekHe8lV6MnBUtDcFO8iP]
- 13. The 89-kDa PARP1 cleavage fragment serves as a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7948984/]
- 14. The 89-kDa PARP1 cleavage fragment serves as a ... - PubMed [https://pubmed.ncbi.nlm.nih.gov/33168626/]
- 15. Characterization of the necrotic cleavage of poly(ADP- ... [https://www.nature.com/articles/4400851]
- 16. Protocol for Apoptosis Assay by Flow Cytometry Using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4943750/]
- 17. Annexin V Staining Protocol for Flow Cytometry [https://www.thermofisher.com/us/en/home/references/protocols/cell-and-tissue-analysis/protocols/annexin-v-staining-flow-cytometry.html]
- 18. Caspase-Glo® 3/7 Assay Protocol [https://www.promega.com/resources/protocols/technical-bulletins/101/caspase-glo-37-assay-protocol/]
- 19. New Insights into the Significance of PARP-1 Activation [https://www.mdpi.com/2073-4409/10/3/599]
- 20. RSL3 promotes PARP1 apoptotic functions by distinct ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12492750/]
- 21. Correction: Carnosol Induces ROS-Mediated Beclin1 ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0337572]
- 22. Article PARP-1 improves leukemia outcomes by inducing ... [https://www.sciencedirect.com/science/article/pii/S2666379123003580]
- 23. Caspases as master regulators of programmed cell death [https://www.nature.com/articles/s12276-025-01470-9]
- 24. Biological function, mediate cell death pathway and their ... [https://www.frontiersin.org/journals/nutrition/articles/10.3389/fnut.2022.1093939/full]
- 25. Role of poly(ADP-ribose) polymerase-1 in regulating ... [https://www.nature.com/articles/s41598-022-25405-w]
- 26. Unusual diterpenoids featuring a 4/9-fused ring system ... [https://www.sciencedirect.com/science/article/abs/pii/S0045206825011356]
- 27. BKCa channel as a novel regulator of cellular DNA ... [https://www.nature.com/articles/s41598-025-03824-9]
- 28. New Insights into the Significance of PARP-1 Activation [https://pmc.ncbi.nlm.nih.gov/articles/PMC8001672/]
- 29. guide | Abcam Apoptosis western blot [https://www.abcam.com/en-us/knowledge-center/western-blot/western-blot-analysis-of-apoptosis]
- 30. PARP1-mediated necrosis is dependent on parallel JNK ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4179488/]
- 31. Simultaneous polychromatic flow of multiple... cytometric detection [https://link.springer.com/article/10.1007/s10495-019-01528-w]
- 32. Evaluation of poly(ADP-ribose) polymerase cleavage ... [https://www.sciencedirect.com/science/article/pii/S0015028208005621]
- 33. Cleaved PARP (Asp214) (D64E10) XP® Rabbit mAb | Cell Signaling Technology [https://www.cellsignal.com/products/primary-antibodies/cleaved-parp-asp214-d64e10-xp-rabbit-mab/5625]
- 34. PARP Antibody | Cell Signaling Technology [https://www.cellsignal.com/products/primary-antibodies/parp-antibody/9542]
- 35. Purified Mouse Anti-Cleaved PARP (Asp214) [https://www.bdbiosciences.com/en-us/products/reagents/western-blotting-and-molecular-reagents/western-blot-reagents/purified-mouse-anti-cleaved-parp-asp214.552596]
- 36. Western Blot Sample Preparation Protocol [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-gel-electrophoresis-information/western-blot-protocols/western-blot-sample-preparation.html]
- 37. Western blot protocol [https://www.abcam.com/en-us/technical-resources/protocols/western-blot]
- 38. Western Blot Protocols and Recipes [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-gel-electrophoresis-information/western-blot-protocols.html]
- 39. Lysate preparation protocol for western blotting [https://www.abcam.com/en-us/technical-resources/protocols/lysate-preparation-for-western-blot]
- 40. Antibody (F-2) | SCBT - Santa Cruz Biotechnology PARP 1 [https://www.scbt.com/p/parp-1-antibody-f-2]
- 41. Differential trapping of PARP1 and PARP2 by clinical PARP ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3528345/]
- 42. Staining Intracellular Antigens for Flow Cytometry [https://www.thermofisher.com/us/en/home/references/protocols/cell-and-tissue-analysis/protocols/staining-intracellular-antigens-flow-cytometry.html]
- 43. Flow Cytometry Protocol | Abcam [https://www.abcam.com/en-us/technical-resources/protocols/flow-cytometry-for-intracellular-and-extracellular-targets]
- 44. Flow Cytometry, Triton X-100 Permeabilization Protocol [https://www.cellsignal.com/learn-and-support/protocols/protocol-flow-triton-x100?srsltid=AfmBOorc8675dWqVR3urwMHSgrXLLiITva2D3h5oWHQClQVfO8zitWEz]
- 45. Intracellular Flow Cytometry Staining Protocol [https://www.ptglab.com/news/blog/intracellular-flow-cytometry-staining-protocol/?srsltid=AfmBOorxZqRRGovpZj8OXBb9c5IK0NZCpiW9WTQqMSUqyEX9DjsPMGi-]
- 46. Flow Cytometry Protocol with Alcohol Permeabilization [https://www.rndsystems.com/resources/protocols/flow-cytometry-protocol-staining-intracellular-molecules-using-alcohol]
- 47. Multiparametric Analysis of Apoptosis by Flow Cytometry [https://pmc.ncbi.nlm.nih.gov/articles/PMC8063493/]
- 48. Multiparametric Analysis of Apoptosis by Flow Cytometry ... [https://www.creative-diagnostics.com/multiparametric-analysis-of-apoptosis-by-flow-cytometry-protocol.htm]
- 49. Green Fluorescent FAM-FLICA® Caspase-3/7 Assay | 94 [https://www.antibodiesinc.com/products/fam-flica-caspase-3-7-assay-kit-93?srsltid=AfmBOoqw_RCsffHCEalNZJ4-MXyzSvwSYd0_PJT87NXR_NajVWzk9L38]
- 50. Vybrant™ FLICA Caspase Apoptosis Assay Kits for flow ... [https://www.thermofisher.com/order/catalog/product/V35118]
- 51. Cleaved PARP1 antibody (60555-1-Ig) [https://www.ptglab.com/products/Cleaved-PARP1-Antibody-60555-1-Ig.htm?srsltid=AfmBOorJBDqBGgb4W_2xPwdfMIqpnpOhfzbo8QO6oAf8pyqzQhMVo6W0]
- 52. Cleavage of Poly(ADP-ribose) Polymerase by Interleukin ... [https://www.sciencedirect.com/science/article/pii/S0021925818518913]
- 53. Griffin:Western Blot Optimization [https://openwetware.org/wiki/Griffin:Western_Blot_Optimization]
- 54. Western Blot Troubleshooting: Non-specific bands [https://azurebiosystems.com/blog/western-blot-troubleshooting-what-to-do-about-non-specific-bands/]
- 55. Poly(ADP-ribose) polymerase-1 and its cleavage products ... [https://www.sciencedirect.com/science/article/pii/S0167488913004242]
- 56. The 8 Western Blot Failures and How to Prevent Them (2025 ... [https://wildtypeone.substack.com/p/the-8-western-blot-failures-and-how?utm_campaign=post&triedRedirect=true]
- 57. A Scientist's Guide to Conquering High Background in ... - clyte [https://www.clyte.tech/post/guide-to-conquering-high-background-in-western-blotting?srsltid=AfmBOoo50rxwV0qQbB6taPT-6JfGu0UYTgMid6xrB1M2Q6AkAg0VLSXu]
- 58. Stripping and Reprobing Western Blots [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/stripping-reprobing-western-blots.html]
- 59. 2024 Guide to Quantitative Western Blot Publication [https://www.thermofisher.com/blog/life-in-the-lab/2023-guide-to-quantitative-western-blot-publication/]
- 60. Cleaved PARP1 antibody (60555-1-Ig) [https://www.ptglab.com/products/Cleaved-PARP1-Antibody-60555-1-Ig.htm?srsltid=AfmBOopYN2RVmONVi-qxmI8ieqGs2cPFCzX0FOHrPp1o-2sJPblgipmM]
- 61. Autofluorescence in conventional flow cytometry [https://www.colibri-cytometry.com/post/autofluorescence-in-conventional-flow-cytometry]
- 62. Setting Compensation Multicolor Flow [https://www.bdbiosciences.com/en-us/resources/protocols/setting-compensation-multicolor-flow]
- 63. Spectral Unmixing in Flow Cytometry: 7 Top Tips for Success [https://bitesizebio.com/70438/spectral-unmixing-in-flow-cytometry/]
- 64. Drug toxicity assessment: cell proliferation versus cell death [https://www.nature.com/articles/s41420-022-01207-x]
- 65. Flow cytometry-based quantitative analysis of cellular ... [https://www.sciencedirect.com/science/article/pii/S2405844024096968]
- 66. BD Pharmingen™ PE Mouse Anti-Cleaved PARP (Asp214) [https://www.bdbiosciences.com/en-us/products/reagents/flow-cytometry-reagents/research-reagents/single-color-antibodies-ruo/pe-mouse-anti-cleaved-parp-asp214.552933]
- 67. PARP-1 cleavage fragments: signatures of cell-death ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3022541/]
- 68. Cleavage of Automodified Poly(ADP-ribose) Polymerase ... [https://www.sciencedirect.com/science/article/pii/S0021925819520610]
- 69. Cleaved PARP (Asp214) Antibody #9541 [https://www.cellsignal.com/products/primary-antibodies/cleaved-parp-asp214-antibody/9541?srsltid=AfmBOoos1GXu_RNhOrM1z7RmuDGNWF3Cd3gRX9MFGMFhegY_duMbcb-Y]
- 70. Cleaved PARP1 antibody (60555-1-Ig) [https://www.ptglab.com/products/Cleaved-PARP1-Antibody-60555-1-Ig.htm?srsltid=AfmBOooeCtBlNQHRh3i8YtL2XQUA-ooRMKC256hdPB0zYUnFohNoPy1V]
- 71. Anti-Cleaved PARP1 antibody [E51] (ab32064) [https://www.abcam.com/en-us/products/primary-antibodies/cleaved-parp1-antibody-e51-ab32064]
- 72. Cleaved PARP1 antibody (60555-1-Ig) [https://www.ptglab.com/products/Cleaved-PARP1-Antibody-60555-1-Ig.htm?srsltid=AfmBOoofbRvYw-IWMIfzcwPWjcVOOHu7uvdR48s4KFDzKkyrsbnlizvw]
- 73. re-visiting housekeeping proteins as internal reference ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3614345/]
- 74. Re-visiting housekeeping proteins as internal reference ... [https://www.sciencedirect.com/science/article/abs/pii/S0024320513001197]
- 75. How To Calculate Western Blot Normalization - TotalLab [https://totallab.com/resources/how-to-calculate-western-blot-normalization/]
- 76. Antibody (194C1439) | SCBT - Santa Cruz Biotechnology PARP 1 [https://www.scbt.com/p/cleaved-parp-1-antibody-194c1439]
- 77. PARP1 Characterization as a Potential Biomarker for BCR [https://pmc.ncbi.nlm.nih.gov/articles/PMC10705790/]
- 78. Single Cell Analysis – Advantages, Challenges, and ... [https://www.technologynetworks.com/drug-discovery/blog/single-cell-analysis-advantages-challenges-and-applications-322768]
- 79. Evaluation of Mithramycin in Combination with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12468201/]
- 80. Poly(ADP-ribose) polymerase-1 antagonizes DNA ... [https://www.nature.com/articles/s41467-019-10741-9]
- 81. Anti-Cleaved PARP1 antibody [4B5BD2] (ab110315) [https://www.abcam.com/en-us/products/primary-antibodies/cleaved-parp1-antibody-4b5bd2-ab110315]
- 82. PARP-1 overexpression contributes to Cadmium-induced ... [https://www.nature.com/articles/s41598-017-04555-2]
- 83. The contribution of PARP1, PARP2 and poly(ADP-ribosyl) ... [https://www.nature.com/articles/s41598-021-84351-1]
- 84. How to Interpret Flow Cytometry Data [https://www.fortislife.com/resources/antibody-resources/how-to-interpret-flow-cytometry-data]
- 85. PARP Antibody #9542 [https://www.cellsignal.com/products/primary-antibodies/parp-antibody/9542?srsltid=AfmBOoqXcIOtvkd917sVLfTaXnU5ZRS8FAAZlKT0bLASFrSh_NQuuXJ6]
- 86. Cleaved PARP (Asp214) (D64E10) Rabbit Monoclonal ... [https://www.cellsignal.com/products/primary-antibodies/cleaved-parp-asp214-d64e10-rabbit-monoclonal-antibody/5625?srsltid=AfmBOopshqlHz_R3GTo_1ufVTjloWSlzTjYoWEkmdFOTbEFKc0qIJ4MJ]
- 87. Anti-Cleaved PARP1 antibody (ab4830) [https://www.abcam.com/en-us/products/primary-antibodies/cleaved-parp1-antibody-ab4830]
- 88. The deubiquitination-PARylation positive feedback loop of ... [https://www.nature.com/articles/s41388-025-03428-7]
- 89. Regulatory apoptotic fragment of PARP1 complements ... [https://www.sciencedirect.com/science/article/abs/pii/S1568786423001477]
- 90. PARP1 antibody (13371-1-AP) [https://www.ptglab.com/products/PARP1-Antibody-13371-1-AP.htm?srsltid=AfmBOoqyF-H0K05Egs6wmv81vOqH3aeeaKERR4h_YnOMrV0tbwM4w2ua]
- 91. Comprehensive interrogation of synthetic lethality in the ... [https://www.nature.com/articles/s41586-025-08815-4]