Apoptotic Pathways Decoded: Death Receptor vs. Mitochondrial Signaling in Disease and Therapy
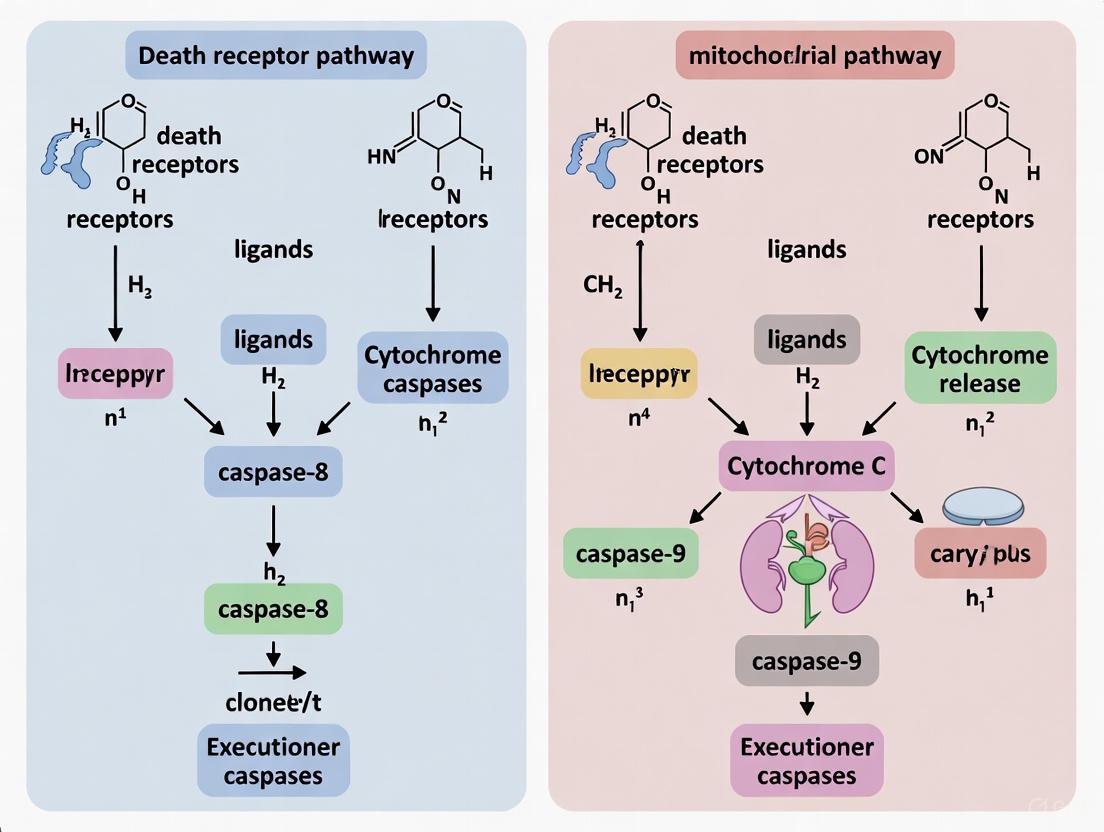
This article provides a comprehensive analysis of the two principal apoptotic pathways: the death receptor (extrinsic) and mitochondrial (intrinsic) pathways.
Apoptotic Pathways Decoded: Death Receptor vs. Mitochondrial Signaling in Disease and Therapy
Abstract
This article provides a comprehensive analysis of the two principal apoptotic pathways: the death receptor (extrinsic) and mitochondrial (intrinsic) pathways. Tailored for researchers and drug development professionals, it details the core mechanisms, key molecular players (e.g., caspases, Bcl-2 family, FADD), and critical regulatory nodes. The scope extends from foundational biology and established research methodologies to the translational application of this knowledge in developing targeted cancer therapeutics, such as BH3 mimetics and DR5 agonists. It further explores the pervasive crosstalk between these pathways and their integration with non-apoptotic cell death processes like necroptosis and pyroptosis, offering a strategic framework for overcoming treatment resistance and designing novel combination therapies.
Core Mechanisms: Deconstructing the Death Receptor and Mitochondrial Apoptotic Pathways
Apoptosis, or programmed cell death, is a highly regulated process essential for maintaining tissue homeostasis, eliminating damaged cells, and ensuring proper embryonic development [1] [2]. The precise initiation and execution of apoptosis occur through two primary signaling pathways: the extrinsic pathway, triggered by external death ligands, and the intrinsic pathway, activated by internal cellular stress [3]. While distinct in their initiation, these pathways converge on a common execution phase mediated by caspase proteases. Understanding the mechanistic details of these triggers is fundamental for apoptosis research and the development of novel therapeutics for cancer, neurodegenerative disorders, and autoimmune diseases [1] [4]. This guide provides a technical overview of these initiation mechanisms, framed within the broader context of death receptor pathway versus mitochondrial pathway research.
The Extrinsic Pathway: Death Receptor-Mediated Initiation
The extrinsic pathway, or death receptor pathway, begins outside the cell in response to signals from the immune system or other cellular neighbors [3]. It is characterized by a ligand-receptor interaction that rapidly initiates the apoptotic cascade.
Core Mechanism and Key Components
This pathway is activated when specific extracellular death ligands bind to their corresponding transmembrane death receptors, which belong to the Tumor Necrosis Factor (TNF) receptor superfamily [1] [3]. Key ligand-receptor pairs include FasL/Fas, TNF-α/TNFR1, and Apo2L (TRAIL)/DR4 or DR5 [3] [2]. Upon ligand binding, the receptors trimerize and recruit intracellular adapter proteins, such as FADD (Fas-Associated protein with Death Domain), via shared death domains (DD) [3]. FADD then recruits procaspase-8 (and in some cases, procaspase-10) via death effector domains (DED), forming a multi-protein complex known as the Death-Inducing Signaling Complex (DISC) [1] [5].
Table 1: Key Components of the Extrinsic Apoptotic Pathway
| Component Type | Example Molecules | Function |
|---|---|---|
| Death Ligands | FasL, TNF-α, TRAIL (Apo2L) | Extracellular signal that binds and activates death receptors [6] [3]. |
| Death Receptors | Fas (CD95), TNFR1, DR4, DR5 | Transmembrane receptors that transduce the death signal into the cell [3] [2]. |
| Adaptor Proteins | FADD, TRADD | Bridge the death receptor and initiator caspases to form the DISC [3]. |
| Initiator Caspase | Caspase-8, Caspase-10 | Autocatalytically activated at the DISC; initiates the protease cascade [1] [5]. |
Within the DISC, procaspase-8 molecules are brought into close proximity, leading to their autocatalytic activation [5]. Activated caspase-8 then propagates the death signal by cleaving and activating downstream executioner caspases, primarily caspase-3, -6, and -7, which proceed to dismantle the cell by cleaving hundreds of cellular substrates [1] [6].
Type I and Type II Cells
Research has revealed a critical nuance in the extrinsic pathway, leading to the classification of cells as Type I or Type II, which determines how the death signal is amplified [5] [7].
- Type I Cells: In these cells, the amount of active caspase-8 generated at the DISC is sufficient to directly and robustly activate executioner caspases like caspase-3, leading to rapid apoptosis without the need for mitochondrial amplification [5] [7].
- Type II Cells: In these cells, the initial caspase-8 signal is weaker. To achieve effective apoptosis, the signal must be amplified through the intrinsic mitochondrial pathway. This is achieved when caspase-8 cleaves the BH3-only protein BID into its active truncated form, tBID [5] [7]. tBID translocates to the mitochondria, where it promotes the activation of BAX and BAK, leading to Mitochondrial Outer Membrane Permeabilization (MOMP) and the release of cytochrome c, thus engaging the intrinsic pathway [5] [7]. The cellular levels of inhibitors like c-FLIP (which competes with caspase-8 at the DISC) and XIAP (which inhibits caspases) are key determinants of whether a cell behaves as Type I or Type II [5] [7].
Diagram 1: The Extrinsic Apoptosis Pathway and Type I/Type II Cell Distinction.
The Intrinsic Pathway: Mitochondrial-Mediated Initiation
The intrinsic pathway, also known as the mitochondrial pathway, is activated in response to diverse internal cellular stresses, acting as a guardian to eliminate damaged cells [3].
Core Mechanism and Key Components
This pathway is initiated by non-receptor-mediated stimuli, including DNA damage, oxidative stress, growth factor deprivation, radiation, and cytotoxic drugs [6] [3]. These stresses are sensed and integrated by key regulators, most notably the tumor suppressor protein p53 [3]. p53 acts as a transcription factor to upregulate pro-apoptotic members of the Bcl-2 family, such as PUMA, Noxa, and Bax [1] [3].
The decisive event in the intrinsic pathway is Mitochondrial Outer Membrane Permeabilization (MOMP) [1]. This process is tightly controlled by the balance between pro-apoptotic and anti-apoptotic members of the Bcl-2 protein family. Anti-apoptotic proteins (e.g., Bcl-2, Bcl-xL) preserve mitochondrial integrity, while pro-apoptotic effectors (BAX, BAK) are responsible for pore formation [1] [3]. Upon activation by stress signals, "activator" BH3-only proteins (like BIM) and "sensitizer" BH3-only proteins (like Bad, Noxa) tip the balance by neutralizing anti-apoptotic proteins and directly activating BAX/BAK [6]. Oligomerization of BAX and BAK at the mitochondrial outer membrane leads to MOMP, causing the release of several mitochondrial intermembrane space proteins into the cytosol [1] [3].
Table 2: Key Components of the Intrinsic Apoptotic Pathway
| Component Type | Example Molecules | Function |
|---|---|---|
| Initiation Stimuli | DNA damage, Oxidative stress, Radiation | Internal distress signals that activate the pathway [3]. |
| Key Regulator | p53 | Stress sensor and transcription factor that induces pro-apoptotic genes [3]. |
| Anti-apoptotic Bcl-2 | Bcl-2, Bcl-xL, Mcl-1 | Preserve mitochondrial membrane integrity [1] [6]. |
| Pro-apoptotic Bcl-2 | BAX, BAK | Form pores in the mitochondrial outer membrane (MOMP) [1] [3]. |
| BH3-only Proteins | BIM, Bid, Bad, PUMA, Noxa | Regulate BAX/BAK activity; sentinels for cellular stress [6] [3]. |
| Mitochondrial Factors | Cytochrome c, SMAC/DIABLO, AIF | Released upon MOMP; activate caspases and other death mechanisms [1] [3]. |
The release of cytochrome c is a pivotal step. In the cytosol, cytochrome c binds to Apaf-1, which in the presence of dATP/ATP, oligomerizes to form the apoptosome [3] [2]. The apoptosome recruits and activates procaspase-9. Activated caspase-9 then cleaves and activates the executioner caspase-3, committing the cell to death [3]. Other released proteins, such as SMAC/DIABLO, promote apoptosis by antagonizing Inhibitor of Apoptosis Proteins (IAPs), which normally suppress caspase activity [1] [3].
Diagram 2: The Intrinsic Apoptosis Pathway Triggered by Cellular Stress.
Experimental Methodologies for Pathway Analysis
Dissecting the specific apoptotic pathway involved in a biological context or drug response requires a combination of well-established assays.
Discriminating Between Pathways
- Extrinsic Pathway Assays: A core method involves immunoprecipitation of the DISC to detect the formation of the complex containing the death receptor, FADD, and caspase-8 following ligand stimulation [3]. Caspase-8 activity can be measured using fluorogenic substrates or cleavage-specific antibodies in Western blotting. Flow cytometry using fluorescently labeled death ligands (e.g., FasL) can also quantify receptor binding and internalization.
- Intrinsic Pathway Assays: The hallmark of the intrinsic pathway is MOMP. This can be assessed by measuring the release of cytochrome c from isolated mitochondria or in permeabilized cells via Western blotting or immunofluorescence [3]. Changes in mitochondrial membrane potential (ΔΨm) can be monitored using fluorescent dyes like JC-1 or TMRM. Additionally, the oligomerization status of BAX and BAK can be analyzed by cross-linking experiments and Blue Native PAGE.
- Common Downstream Assays: Activation of executioner caspases (caspase-3/7) is a convergent point and is easily measurable using commercial activity kits or antibodies against cleaved caspase-3. Annexin V staining is a gold standard for detecting phosphatidylserine externalization, an early apoptotic event [8]. DNA fragmentation, a late apoptotic event, can be detected by TUNEL assay or DNA laddering.
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents for Apoptosis Pathway Research
| Reagent / Assay | Function / Application | Experimental Example |
|---|---|---|
| Recombinant Death Ligands (e.g., FasL, TRAIL) | To specifically activate the extrinsic pathway in vitro. | Treat cultured cells to induce DISC formation and caspase-8 activation [3]. |
| Annexin V Apoptosis Detection Kits | Detect phosphatidylserine exposure on the cell surface (early apoptosis) via flow cytometry. | Distinguish between viable, early apoptotic, and late apoptotic/necrotic cells after a drug treatment [8]. |
| Caspase Activity Assays (Fluorometric/Colorimetric) | Quantify the enzymatic activity of specific caspases (e.g., 8, 9, 3/7). | Determine if cell death is caspase-8 (extrinsic) or caspase-9 (intrinsic) dependent [1]. |
| Mitochondrial Membrane Potential Dyes (e.g., JC-1, TMRM) | Assess the loss of ΔΨm, a key event in intrinsic apoptosis. | Confirm mitochondrial involvement in a stress-induced cell death model [3]. |
| Cytochrome c Release Assays | Monitor the translocation of cytochrome c from mitochondria to cytosol. | Validate the occurrence of MOMP after treatment with a DNA-damaging agent [3]. |
| BH3 Mimetics (e.g., ABT-263/Navitoclax) | Small molecule inhibitors that bind and neutralize anti-apoptotic Bcl-2 proteins. | Sensitize cancer cells to intrinsic apoptosis; test Bcl-2 family dependency [1]. |
| Caspase Inhibitors (e.g., z-VAD-fmk (pan-caspase), z-IETD-fmk (caspase-8)) | Pharmacologically inhibit caspase activity to determine caspase-dependency of death. | Confirm if a cell death phenotype is apoptotic [1]. |
The extrinsic and intrinsic pathways of apoptosis represent two sophisticated molecular machines for initiating cell death, one looking outward for immune commands, the other inward for signs of distress. While this review has delineated their distinct triggers and initial signaling events, it is crucial to recognize the extensive crosstalk between them, most elegantly exemplified by the caspase-8-mediated cleavage of BID in Type II cells, which effectively couples the extrinsic pathway to the intrinsic amplification loop [6] [5] [7].
From a therapeutic perspective, this mechanistic understanding is being leveraged to develop targeted therapies. For instance, TRAIL receptor agonists and BH3 mimetics are designed to specifically activate the extrinsic and intrinsic pathways, respectively, in cancer cells [1]. The growing knowledge of the complex interplay between different cell death modalities, including apoptosis, necroptosis, and ferroptosis, further expands the arsenal of potential strategies to tackle diseases characterized by aberrant cell survival [6] [4]. Therefore, continued rigorous investigation into the initiation triggers of apoptosis remains a cornerstone of translational cell death research.
The death receptor pathway represents a critical mechanism of apoptosis initiation, triggered by extracellular signals and culminating in the controlled demise of the cell. This in-depth technical guide examines the molecular orchestration of death receptor signaling, with particular focus on the events from ligand binding through death-inducing signaling complex (DISC) formation and subsequent caspase-8 activation. As research continues to elucidate the intricate balance between cellular survival and death, understanding the precise regulation of this pathway provides crucial insights for therapeutic interventions in cancer, autoimmune disorders, and neurodegenerative diseases. This review contrasts the death receptor pathway with the mitochondrial apoptosis pathway, highlighting key distinctions in initiation mechanisms, regulatory components, and cellular contexts where each predominates.
Apoptosis, or programmed cell death, is a fundamental process essential for development, tissue homeostasis, and immune function. Two principal pathways mediate apoptosis: the intrinsic (mitochondrial) pathway and the extrinsic (death receptor) pathway. While the mitochondrial pathway responds to internal cellular damage signals such as DNA damage, oxidative stress, and endoplasmic reticulum stress, the death receptor pathway is activated by extracellular ligands binding to cell surface receptors [9] [10]. This death receptor pathway enables rapid transmission of apoptotic signals without requiring mitochondrial amplification in certain cellular contexts, though crosstalk between these pathways significantly expands the regulatory landscape of apoptotic cell death [11].
The death receptor pathway exhibits remarkable specificity and efficiency in signal transduction, with complete apoptotic commitment occurring within minutes to hours of receptor engagement in sensitive cells. This pathway plays particularly important roles in immune system regulation, including immune privilege maintenance, lymphocyte homeostasis, and cytotoxic T-cell mediated killing of infected or transformed cells [10] [12]. Dysregulation of death receptor signaling contributes to numerous pathological conditions, including cancer immune evasion, autoimmune disorders, and neurodegenerative diseases, highlighting its therapeutic significance [13].
Molecular Components of the Death Receptor Pathway
Death Receptors and Their Ligands
Death receptors constitute a specialized subset of the tumor necrosis factor (TNF) receptor superfamily characterized by a conserved cytoplasmic region of approximately 80 amino acids termed the "death domain" (DD) [10] [13]. This protein interaction module is essential for initiating the apoptotic signal transduction cascade. Eight members of the death receptor family have been characterized to date, with the most extensively studied being Fas (CD95/APO-1), TNFR1 (DR1), TRAILR1 (DR4), and TRAILR2 (DR5) [13].
Table 1: Major Death Receptors and Their Corresponding Ligands
| Death Receptor | Alternative Names | Ligand | Primary Functions |
|---|---|---|---|
| Fas | CD95, APO-1, DR2 | FasL (CD95-L) | Immune regulation, lymphocyte homeostasis |
| TNFR1 | DR1, CD120a, p55/p60 | TNF-α | Inflammation, survival/apoptosis balance |
| TRAILR1 | DR4, APO-2 | TRAIL (Apo-2L) | Immune surveillance, tumor cell elimination |
| TRAILR2 | DR5, KILLER, TRICK2 | TRAIL (Apo-2L) | Immune surveillance, tumor cell elimination |
| DR3 | APO-3, LARD, TRAMP, WSL1 | TWEAK/TL1A | T-cell costimulation, inflammation |
| DR6 | - | APP (amyloid precursor protein) | Neuronal regulation, controversial role |
| EDAR | - | Ectodysplasin-A | Ectodermal development |
| NGFR | p75NTR | Nerve Growth Factor | Neuronal apoptosis, development |
These receptors typically exist as pre-assembled trimers on the cell surface prior to ligand binding, contrary to earlier models suggesting ligand-induced trimerization [10]. The death ligands, including FasL, TNF-α, and TRAIL, are homotrimeric type II transmembrane proteins that belong to the TNF superfamily. Upon binding to their cognate receptors, these ligands induce conformational changes that enable higher-order clustering of receptor trimers and subsequent recruitment of intracellular adapter proteins [10].
Intracellular Adapter Proteins and Signaling Complexes
The death domain serves as a critical protein interaction module that recruits intracellular adapter proteins following receptor activation. The primary adapter protein for Fas, TRAILR1, and TRAILR2 is FADD (Fas-Associated Death Domain protein), which contains both a death domain (for receptor interaction) and a death effector domain (DED) for engaging downstream signaling components [10] [12].
The complex formed at activated Fas, TRAILR1, or TRAILR2 is termed the death-inducing signaling complex (DISC), which serves as the central apoptotic signaling platform in the death receptor pathway [12] [13]. In contrast, TNFR1 engages a more complex signaling mechanism, initially recruiting the adapter protein TRADD (TNFR-Associated Death Domain protein), which then serves as a platform for assembling alternative signaling complexes that can activate either apoptotic or inflammatory pathways [13].
DISC Formation and Molecular Architecture
Sequential Assembly of the DISC
The DISC forms within seconds of death receptor engagement and represents the biochemical core of the death receptor pathway. The assembly follows a precise sequence of molecular interactions:
- Receptor Activation: Death ligands bind to their cognate pre-trimerized receptors, inducing conformational changes that expose the intracellular death domains [10].
- FADD Recruitment: The exposed death domains recruit FADD through homologous death domain interactions, forming a two-layer structure with interfaces at three different binding surfaces [10].
- Caspase-8 Recruitment: FADD exposes its death effector domain (DED), which recruits procaspase-8 through homologous DED interactions [10] [12].
The minimal functional unit for DISC formation requires at least two receptor trimers to be brought together, enabling the formation of higher-order complexes necessary for signal amplification [10]. This assembly creates a filamentous structure that facilitates caspase-8 activation through induced proximity.
Diagram 1: Sequential Assembly of the Death-Inducing Signaling Complex (DISC)
Caspase-8 Activation Mechanism
Caspase-8 activation at the DISC occurs through an induced proximity model that involves several sequential steps:
- Filament Formation: Caspase-8 contains two N-terminal death effector domains (DED1 and DED2). DED1 binds to FADD, which exposes DED2, enabling it to bind to DED1 of another caspase-8 molecule. This creates an extended filament of caspase-8 monomers [10].
- Dimerization and Autocatalysis: The close proximity of caspase-8 molecules in the filament facilitates dimerization and interchain autocatalytic cleavage, which stabilizes the active enzyme form [10] [12].
- Executioner Caspase Activation: Active caspase-8 dimers then cleave and activate downstream effector caspases, including caspase-3, -6, and -7, which orchestrate the proteolytic events responsible for apoptotic morphology [9] [10].
The regulation of caspase-8 activation is precisely controlled by cellular FLICE-inhibitory protein (c-FLIP), which exists in multiple isoforms that can either inhibit or promote caspase-8 activation depending on cellular context and expression levels [12]. When FLIP is present at the DISC, it can form heterodimers with caspase-8 that exhibit restricted substrate specificity, potentially leading to survival signaling rather than apoptosis [10] [12].
Key Regulatory Mechanisms at the DISC
c-FLIP-Mediated Regulation
The cellular FLICE-inhibitory protein (c-FLIP) represents a critical regulatory checkpoint in death receptor signaling. c-FLIP exists in three principal isoforms: FLIPLong (FLIPL), FLIPShort (FLIPS), and FLIPR [12]. These isoforms contain DED domains that enable them to compete with caspase-8 for binding to FADD at the DISC.
Table 2: c-FLIP Isoforms and Their Functions in DISC Regulation
| Isoform | Structure | Function | Effect on Apoptosis |
|---|---|---|---|
| FLIPLong (FLIPL) | Contains two DEDs and catalytically inactive caspase-like domain | Forms heterodimers with caspase-8; limited proteolytic activity | Can promote or inhibit depending on expression level |
| FLIPShort (FLIPS) | Contains two DEDs only, no caspase-like domain | Blocks caspase-8 filament formation | Potent inhibition of apoptosis |
| Viral FLIP (v-FLIP) | Similar to FLIPS, encoded by viruses | Competes with caspase-8 for FADD binding | Strong inhibition of apoptosis |
The regulatory function of c-FLIP is concentration-dependent. At high expression levels, c-FLIP predominantly inhibits caspase-8 activation by preventing filament formation. However, at lower expression levels, FLIPL can form proteolytically active heterodimers with caspase-8 that cleave a restricted set of substrates, potentially promoting cell survival or non-apoptotic functions [10] [12].
Quantitative Aspects of Life/Death Decisions
The decision between survival and apoptosis in response to death receptor engagement is governed by quantitative parameters at the DISC. Key factors influencing this decision include:
- Receptor Expression Levels: Cells expressing high levels of death receptors are generally more sensitive to apoptosis induction [12].
- Caspase-8 to FLIP Ratio: The relative abundance of caspase-8 versus c-FLIP determines signaling outcome, with high FLIP levels favoring survival [12].
- DISC Assembly Kinetics: The rate and extent of DISC formation influence signal strength, with rapid, robust DISC assembly favoring apoptosis [12].
Studies using quantitative biochemical approaches have revealed that low-level death receptor stimulation often leads to survival signaling through limited caspase-8 activation, while higher stimulation thresholds trigger full apoptotic commitment [12]. This threshold behavior provides a mechanism for discriminating between subtle regulatory signals and definitive death signals in physiological contexts.
Type I and Type II Cell Signaling Paradigms
The functional outcome of death receptor engagement varies between cell types, leading to the classification of Type I and Type II cells based on their signaling characteristics:
Type I Cells
In Type I cells, robust DISC formation generates sufficient active caspase-8 to directly activate executioner caspases, leading to rapid apoptosis independent of mitochondrial amplification [11]. This pathway is characterized by:
- Efficient DISC formation and caspase-8 activation
- Minimal dependence on mitochondrial amplification
- Resistance to Bcl-2 overexpression
- Typically observed in certain lymphocytes and transformed cell lines
Type II Cells
In Type II cells, limited DISC formation generates insufficient active caspase-8 to directly activate executioner caspases, requiring mitochondrial amplification through Bid cleavage [11]. This pathway features:
- Less efficient DISC formation
- Critical dependence on mitochondrial amplification
- Sensitivity to inhibition by Bcl-2 family proteins
- Prevalent in hepatocytes, pancreatic β-cells, and many primary cell types
The molecular basis for this distinction involves multiple factors, including the efficiency of DISC formation, the relative expression of XIAP (which inhibits caspase-3 and -9), and the availability of Bid for cleavage by caspase-8 [11]. In Type II cells, caspase-8 cleaves Bid to generate truncated Bid (tBid), which translocates to mitochondria and promotes cytochrome c release through activation of Bax/Bak, thereby engaging the mitochondrial pathway [11].
Diagram 2: Type I and Type II Apoptosis Signaling Pathways
Experimental Methodologies for Death Receptor Research
DISC Immunoprecipitation and Analysis
The biochemical characterization of DISC composition and dynamics represents a fundamental methodology in death receptor research. The standard protocol involves:
- Cell Stimulation: Treat cells with death receptor-specific ligands (e.g., FasL, TRAIL) or receptor-crosslinking antibodies for varying time periods.
- Cell Lysis: Use mild non-ionic detergents (e.g., 1% Triton X-100 or CHAPS) in physiological salt conditions to preserve protein complexes.
- Immunoprecipitation: Incubate lysates with antibodies specific to the death receptor extracellular domain or tagged receptor constructs.
- Complex Analysis: Resolve immunoprecipitates by SDS-PAGE and analyze by Western blotting for DISC components (FADD, caspase-8, c-FLIP).
Quantitative analysis of DISC composition can be achieved through densitometric scanning of Western blots or, more precisely, through stable isotope labeling with amino acids in cell culture (SILAC) combined with mass spectrometry [12]. This approach enables precise determination of stoichiometric relationships between DISC components under different experimental conditions.
Caspase-8 Activation Assays
Multiple complementary approaches are employed to assess caspase-8 activation:
- Enzyme Activity Assays: Fluorogenic substrates (e.g., IETD-AFC) that release fluorescent products upon cleavage by caspase-8.
- Western Blot Analysis: Detection of caspase-8 cleavage fragments (p43/41 and p18) as indicators of activation.
- Flow Cytometry: Using fluorescently-labeled inhibitors of caspases (FLICA) to detect active enzyme in intact cells.
- FRET-Based Reporters: Genetically-encoded biosensors that undergo fluorescence resonance energy transfer changes upon caspase-8-mediated cleavage.
Table 3: Essential Research Reagents for Death Receptor Pathway Investigation
| Reagent Category | Specific Examples | Research Application | Key Functions |
|---|---|---|---|
| Death Receptor Agonists | Recombinant FasL, TRAIL, Anti-Fas antibodies (CH11) | Pathway activation | Induce receptor clustering and DISC formation |
| Death Receptor Antagonists | Soluble decoy receptors, neutralizing antibodies | Pathway inhibition | Block ligand-receptor interaction |
| Caspase Inhibitors | z-IETD-fmk (caspase-8 inhibitor), z-VAD-fmk (pan-caspase) | Mechanistic studies | Determine caspase-dependent effects |
| Detection Antibodies | Anti-FADD, anti-caspase-8, anti-FLIP, anti-CD95 | Western blot, IP, flow cytometry | Identify and quantify pathway components |
| Cell Lines | Jurkat (Type I), Huh7 (Type II), BJAB transfectants | Model systems | Provide cellular context for signaling studies |
| Gene Manipulation Tools | siRNA/shRNA for knockdown, overexpression vectors | Functional studies | Modulate expression of specific components |
Death Receptor Pathway in Therapeutic Applications
Understanding the molecular intricacies of death receptor signaling has enabled several therapeutic approaches, particularly in oncology:
Agonistic Antibodies and Recombinant Ligands
Agonistic antibodies targeting death receptors (particularly TRAIL-R1 and TRAIL-R2) and recombinant TRAIL have been developed to selectively induce apoptosis in transformed cells. While showing promise in preclinical models, clinical efficacy has been limited by variable expression of death receptors and regulatory proteins in tumors [13].
Sensitization Strategies
Combination approaches that sensitize tumor cells to death receptor-mediated apoptosis represent an active area of investigation. These include:
- Chemotherapeutic agents that upregulate death receptor expression
- Proteasome inhibitors that reduce FLIP levels
- HDAC inhibitors that modulate multiple components of the apoptotic machinery
- SMAC mimetics that antagonize IAP-mediated caspase inhibition
The differential expression of death receptors and regulatory molecules between normal and malignant cells provides a theoretical basis for selective tumor targeting, though successful clinical translation requires deeper understanding of pathway regulation in specific tumor contexts [13].
Comparative Analysis: Death Receptor vs. Mitochondrial Pathways
The death receptor and mitochondrial pathways represent two distinct but interconnected mechanisms for initiating apoptosis. Key comparative aspects include:
Initiation Mechanisms
- Death Receptor Pathway: Initiated by extracellular ligands binding to cell surface receptors; enables rapid, directed cell elimination in response to external cues.
- Mitochondrial Pathway: Activated by intracellular damage signals (DNA damage, oxidative stress, ER stress); functions as a cellular damage sensor.
Amplification Mechanisms
- Death Receptor Pathway: Signal amplification occurs through caspase-8 activation at the DISC; in Type II cells, further amplification via mitochondrial engagement.
- Mitochondrial Pathway: Signal amplification through cytochrome c-mediated apoptosome formation and caspase-9 activation.
Regulatory Networks
- Death Receptor Pathway: Primarily regulated by c-FLIP at the level of DISC composition; receptor expression levels; decoy receptors.
- Mitochondrial Pathway: Governed by Bcl-2 family protein interactions controlling mitochondrial outer membrane permeabilization; IAP proteins.
Physiological Contexts
- Death Receptor Pathway: Critical for immune system homeostasis; elimination of infected or damaged cells by cytotoxic lymphocytes; immune privilege maintenance.
- Mitochondrial Pathway: Essential for developmental remodeling; response to cellular stress; elimination of genetically compromised cells.
Therapeutic manipulation of these pathways continues to present opportunities for drug development, with death receptor pathway targeting offering potential for immune-mediated killing of tumor cells, while mitochondrial pathway targeting may enhance cellular sensitivity to conventional DNA-damaging agents [9] [11] [13].
The death receptor pathway represents a sophisticated molecular machinery for translating extracellular signals into controlled cellular dismantling. From initial ligand-receptor interactions through DISC formation and caspase-8 activation, this pathway exemplifies the precision of apoptotic signaling networks. The regulatory mechanisms governing this pathway, particularly the balance between caspase-8 and c-FLIP at the DISC and the distinction between Type I and Type II signaling paradigms, highlight the complex cellular logic underlying life/death decisions.
Ongoing research continues to reveal novel aspects of death receptor biology, including non-apoptotic functions in proliferation, inflammation, and differentiation. The integration of quantitative approaches with structural biology and single-cell analysis promises to further elucidate the dynamic regulation of this pathway. As our understanding deepens, so too will opportunities for therapeutic intervention in the numerous pathological conditions characterized by dysregulated apoptosis.
The mitochondrial pathway of apoptosis, also known as the intrinsic pathway, represents a fundamental cellular process essential for development and tissue homeostasis in multicellular organisms [14] [15]. This genetically regulated cell death pathway functions as a critical counterbalance to cell proliferation, ensuring maintenance of physiological equilibrium in adult tissues [1]. Dysregulation of this pathway underpins numerous pathological conditions; insufficient apoptosis can lead to autoimmune disorders and cancer, whereas excessive cell death contributes to debilitating degenerative diseases affecting the heart and nervous system [14]. The mitochondrial pathway is primarily controlled by the B-cell lymphoma 2 (BCL-2) protein family, which integrates diverse cellular stress signals to determine cellular fate [16]. These proteins ultimately decide whether a cell commits to death by controlling the pivotal event in intrinsic apoptosis: mitochondrial outer membrane permeabilization (MOMP) [14]. Following MOMP, proteins normally confined to the mitochondrial intermembrane space, including cytochrome c, are released into the cytosol, triggering the activation of caspases that execute the orderly dismantling of the cell [15] [1].
Within the broader context of apoptosis research, the mitochondrial pathway stands alongside the death receptor (extrinsic) pathway as one of the two principal suicide programs [17] [15]. While the extrinsic pathway is initiated by ligand binding to death receptors at the cell surface, the intrinsic pathway is activated in response to internal derangements including DNA damage, growth factor withdrawal, oxidative stress, and oncogene activation [14] [1]. Although these pathways operate largely independently, significant cross-talk exists between them, particularly in certain cell types designated as "type II cells" where the extrinsic pathway requires mitochondrial amplification to effectively execute cell death [15]. This integration occurs through caspase-8-mediated cleavage of the BH3-only protein BID, generating truncated BID (tBID) which translocates to mitochondria to activate the core apoptotic machinery [15]. The mitochondrial pathway's centrality in responding to internal cellular damage has made it a prime target for therapeutic intervention, particularly in oncology where restoring apoptotic sensitivity in cancer cells represents a promising treatment strategy [16].
The BCL-2 Protein Family: Architects of Cell Fate
The BCL-2 protein family constitutes an intricate regulatory network that governs mitochondrial outer membrane permeabilization (MOMP), the commitment point in intrinsic apoptosis [14] [15]. These globular, α-helical proteins share sequence homology within conserved regions known as BCL-2 homology (BH) domains and can be functionally categorized into three distinct subgroups based on their structure and apoptotic function [16] [18].
Anti-apoptotic proteins, including BCL-2 itself, BCL-XL, BCL-w, MCL-1, BFL-1, and BCL-B, contain four BH domains (BH1-4) and serve as crucial survival factors that protect cells from apoptotic stimuli [14] [16]. Their canonical function involves embedding in the outer mitochondrial membrane via a C-terminal transmembrane domain, where they prevent MOMP by sequestering pro-apoptotic family members [14] [18]. Multi-domain pro-apoptotic effector proteins, principally BAX and BAK (and to a lesser extent BOK), contain BH1-3 domains and directly execute MOMP [14]. In healthy cells, BAX resides largely in the cytosol or loosely associates with mitochondria, while BAK is constitutively integrated into the outer mitochondrial membrane [14]. Upon activation, both proteins undergo conformational changes, oligomerize, and form pores in the mitochondrial outer membrane [14]. BH3-only proteins (BID, BIM, BAD, PUMA, NOXA, BIK, BMF, and HRK) function as specialized sentinels that monitor cellular well-being [14] [16]. They are activated through diverse mechanisms including transcriptional upregulation, post-translational modifications, and subcellular relocalization in response to specific stress signals [14].
Table 1: The BCL-2 Protein Family: Classification and Function
| Category | Representative Members | BH Domains | Function | Regulation |
|---|---|---|---|---|
| Anti-apoptotic | BCL-2, BCL-XL, MCL-1, BCL-w | BH1-4 | Prevent MOMP by sequestering pro-apoptotic members | Overexpressed in cancers; inhibited by BH3-only proteins |
| Pro-apoptotic Effectors | BAX, BAK, BOK | BH1-3 | Directly mediate MOMP through oligomerization | Activated by direct activator BH3-only proteins |
| BH3-only Proteins | BID, BIM, PUMA, BAD, NOXA | BH3 only | Sense cellular stress and initiate apoptosis | Activated transcriptionally (PUMA, NOXA) or post-translationally (BID cleavage, BAD dephosphorylation) |
The "interaction model" between these family members revolves around the hydrophobic groove formed by the BH1-3 domains of anti-apoptotic proteins, which serves as the main binding site for the BH3 domains of pro-apoptotic partners [16]. Anti-apoptotic proteins preserve mitochondrial integrity by sequestering either activated BH3-only proteins or monomeric forms of BAX and BAK, thereby preventing their oligomerization [14]. In response to cellular stress, specific BH3-only proteins are activated and either directly engage and activate BAX/BAK ("direct activation" model) or neutralize anti-apoptotic proteins, thereby displacing their inhibition of BAX/BAK ("displacement" model) [14] [16]. The critical balance between these competing pro-survival and pro-death factions determines whether a cell succumbs to apoptosis or survives.
Mitochondrial Outer Membrane Permeabilization (MOMP): The Point of No Return
Mitochondrial outer membrane permeabilization (MOMP) represents the decisive commitment point in the intrinsic apoptotic pathway, a nearly irreversible step that seals cellular fate [15]. This process is characterized by the formation of pores in the mitochondrial outer membrane through which proteins normally confined to the intermembrane space escape into the cytosol [14]. While the outer mitochondrial membrane is typically permeable to molecules under 5 kDa, MOMP creates pores sufficiently large to permit the passage of proteins exceeding 100 kDa [15]. The execution of MOMP absolutely requires the multidomain pro-apoptotic effector proteins BAX and BAK, as demonstrated by the profound resistance of BAX/BAK double-knockout cells to diverse death stimuli including staurosporine, UV radiation, growth factor deprivation, and endoplasmic reticulum stress [14].
The process of BAX/BAK activation and pore formation involves several sequential steps. For BAX, activation involves translocation from the cytosol to the mitochondrial outer membrane, where it inserts via its C-terminal transmembrane domain [14]. Both BAX and BAK undergo N-terminal conformational changes during activation, exposing epitopes that can be detected by specific antibodies such as the 6A7 antibody for BAX [14]. Following activation, these proteins form higher-order homo-oligomers that can be visualized through western blotting after chemical crosslinking [14]. These oligomers subsequently participate in forming proteolipid pores in the mitochondrial outer membrane, though the precise architecture of these pores remains an active research area [14] [15]. The duration of MOMP for individual mitochondria is remarkably brief, occurring within seconds, but the asynchronous engagement of MOMP across all mitochondria within a cell typically spans approximately five minutes [15]. High-resolution imaging techniques have revealed that MOMP propagation within single cells can occur in a wave-like pattern, potentially facilitated by endoplasmic reticulum calcium channels, though the precise mechanistic link to BCL-2 family proteins remains incompletely understood [15].
While MOMP has traditionally been considered a binary, all-or-nothing event at the cellular level, recent observations challenge this dogma with the description of "partial MOMP" phenomena. Incomplete MOMP (iMOMP) occurs when most but not all mitochondria within a cell undergo permeabilization, with cell survival dependent on the absence or inhibition of caspase activity [15]. Minority MOMP (miniMOMP) describes the scenario where only a small fraction of mitochondria experience MOMP following sublethal stress, resulting in limited caspase activation that falls below the threshold for cell death execution [15]. Rather than triggering apoptosis, miniMOMP can induce DNA damage and non-apoptotic caspase signaling that may promote oncogenic transformation, representing a potentially deleterious outcome of failed apoptotic execution [15].
Table 2: MOMP Variants and Their Consequences
| MOMP Type | Mitochondria Affected | Caspase Activation | Cell Fate | Potential Consequences |
|---|---|---|---|---|
| Complete MOMP | All mitochondria | Robust, sustained | Apoptosis | Ordered cell dismantling and clearance |
| Incomplete MOMP (iMOMP) | Majority (but not all) | Variable, often limited | Survival (if caspases inhibited) | Potential repopulation by intact mitochondria |
| Minority MOMP (miniMOMP) | Small minority | Sublethal levels | Survival with signaling | DNA damage, oncogenic transformation |
Cytochrome c Release and Caspase Activation
The permeabilization of the mitochondrial outer membrane during MOMP enables the efflux of several pro-apoptotic proteins from the intermembrane space into the cytosol, with cytochrome c representing the most critical of these factors in committing the cell to apoptosis [15]. Cytochrome c is a heme protein normally embedded in the mitochondrial inner membrane space, where it serves an essential function in the electron transport chain and cellular respiration [1]. Upon release into the cytosol, cytochrome c initiates the formation of the apoptosome, a multi-protein complex that serves as the activation platform for caspase-9 [16] [1]. The apoptosome assembly involves cytochrome c binding to Apaf-1 (apoptotic protease-activating factor 1) in the presence of ATP/dATP, triggering Apaf-1 oligomerization into a wheel-like complex that recruits and activates procaspase-9 [1].
Once activated, caspase-9 cleaves and activates the executioner caspases, primarily caspase-3 and caspase-7, which then proceed to systematically dismantle the cell by cleaving over 1,000 cellular substrates [15] [1]. These caspase targets include proteins involved in DNA repair (such as PARP), structural proteins (including nuclear lamins and cytoskeletal components), and cell cycle regulators, ultimately producing the characteristic morphological hallmarks of apoptosis: chromatin condensation (pyknosis), nuclear fragmentation (karyorrhexis), cell shrinkage, membrane blebbing, and formation of apoptotic bodies [15] [1]. In addition to cytochrome c, MOMP results in the simultaneous release of other pro-apoptotic mitochondrial proteins including second mitochondria-derived activator of caspase (SMAC, also known as DIABLO) and Omi/HtrA2 [14] [15]. These proteins promote apoptosis by neutralizing inhibitor of apoptosis proteins (IAPs), which would otherwise suppress caspase activity, thereby further ensuring the irreversible commitment to cell death [15] [1].
Experimental Approaches: BH3 Profiling and Beyond
The development of BH3 profiling has provided researchers with a powerful functional assay to quantitatively measure how close a cell is to the apoptotic threshold, effectively interrogating the "primed" state of mitochondria for apoptosis [14]. This technique uses synthetic peptides corresponding to the BH3 domains of various BH3-only proteins to apply standardized death signals to isolated mitochondria or permeabilized cells, then measures the resulting mitochondrial outer membrane permeabilization [14]. The pattern of response to different BH3 peptides reveals the specific anti-apoptotic proteins upon which a cell depends for survival and can identify distinct classes of apoptotic blocks utilized by cancer cells to evade cell death [14].
The BH3 profiling protocol involves several key steps. First, mitochondria are isolated from the cell line or patient sample of interest [14]. These mitochondria are then incubated with individual BH3 domain peptides derived from different BH3-only proteins (such as BIM, BID, BAD, PUMA, and NOXA), each with distinct binding specificities for various anti-apoptotic BCL-2 family members [14]. Finally, mitochondrial outer membrane permeabilization is measured, typically by assessing the loss of mitochondrial membrane potential or the release of cytochrome c [14]. The resulting pattern of which peptides induce MOMP provides a functional readout of the anti-apoptotic dependencies within the tested sample [14].
BH3 profiling can distinguish between three fundamental classes of apoptotic block in cancer cells. A Class A block indicates relatively low levels of functional activator BH3-only proteins, where only direct activator peptides (BID and BIM) cause MOMP while sensitizer peptides do not [14]. A Class B block reflects the absence or non-functionality of BAX and/or BAK, in which case none of the BH3 peptides induce MOMP as these effector proteins are essential for pore formation [14]. A Class C block identifies cells whose survival depends on specific anti-apoptotic proteins (BCL-2, BCL-XL, or MCL-1) that are "primed" with BH3-only protein activators or activated BAX/BAK; in this case, the pattern of which sensitizer BH3 peptides cause MOMP reveals which anti-apoptotic proteins are primarily responsible for maintaining survival [14].
Table 3: Research Reagent Solutions for Mitochondrial Apoptosis Studies
| Reagent Category | Specific Examples | Research Application | Key Features |
|---|---|---|---|
| BH3-Mimetic Compounds | ABT-737, ABT-263 (Navitoclax), ABT-199 (Venetoclax), Obatoclax | Experimental therapeutic targeting of anti-apoptotic BCL-2 proteins | Small molecules that bind hydrophobic groove of specific anti-apoptotic proteins; research tools and clinical candidates |
| BCL-2 Family Antibodies | Conformation-specific antibodies (e.g., 6A7 for active BAX) | Detection of protein localization, expression, and activation status | Western blot, immunofluorescence, and immunoprecipitation applications; specific for activated conformations |
| BH3 Profiling Peptides | Synthetic peptides from BIM, BID, BAD, PUMA, NOXA BH3 domains | Functional assessment of mitochondrial priming | 20-amino acid sequences; distinct binding patterns to different anti-apoptotic family members |
| Caspase Activity Assays | Fluorogenic or chromogenic caspase substrates (e.g., DEVD-afc) | Measurement of caspase activation downstream of MOMP | Quantitative assessment of apoptotic progression; specific for different caspase family members |
| Mitochondrial Dyes | TMRE, JC-1, MitoTracker | Assessment of mitochondrial membrane potential and integrity | Flow cytometry and fluorescence microscopy applications; indicators of MOMP |
Therapeutic Targeting and Clinical Translation
The intricate molecular regulation of the mitochondrial apoptotic pathway has made it an attractive target for therapeutic intervention, particularly in oncology where cancer cells frequently exploit overexpression of anti-apoptotic BCL-2 family members to evade cell death [16]. The development of BH3-mimetic drugs represents a paradigm of successful translational research, with these small molecules designed to structurally mimic the BH3 domain of pro-apoptotic proteins and competitively inhibit anti-apoptotic BCL-2 family members [16]. The first breakthrough in this field came with ABT-737, a potent inhibitor of BCL-2, BCL-XL, and BCL-w discovered through nuclear magnetic resonance (NMR)-based screening and structure-based design [16]. Its orally available successor, ABT-263 (navitoclax), progressed to clinical trials but revealed dose-limiting thrombocytopenia due to BCL-XL inhibition in platelets [16]. This toxicity prompted the development of the highly selective BCL-2 inhibitor ABT-199 (venetoclax), which demonstrated remarkable efficacy in hematologic malignancies with manageable toxicities and received FDA approval in 2016 [16].
Following the clinical success of venetoclax, several chemically similar BCL-2 inhibitors including sonrotoclax and lisaftoclax are currently under clinical evaluation, both as monotherapies and in rational combination regimens [16]. However, the development of BH3-mimetics targeting BCL-XL or MCL-1 has proven more challenging due to on-target toxicities; BCL-XL inhibition causes profound thrombocytopenia, while MCL-1 inhibition has been associated with cardiac toxicity, complications that have limited clinical development of these agents [16]. Novel targeting approaches are emerging to overcome these limitations, including proteolysis targeting chimeras (PROTACs) that selectively degrade target proteins, antibody-drug conjugates (ADCs) for tumor-specific drug delivery, and compounds targeting the BH4 domain of BCL-2 [16]. These innovative strategies to achieve tumor-specific BCL-XL or MCL-1 inhibition could prove transformational for many cancer subtypes, particularly solid tumors that often depend on these alternative anti-apoptotic proteins rather than BCL-2 alone [16].
Table 4: Clinically Developed BH3-Mimetic Compounds
| Compound | Molecular Targets | Clinical Status | Key Applications | Limitations |
|---|---|---|---|---|
| ABT-737 | BCL-2, BCL-XL, BCL-w | Preclinical research tool | Laboratory studies of apoptosis mechanisms | Not orally bioavailable |
| Navitoclax (ABT-263) | BCL-2, BCL-XL, BCL-w | Phase I/II clinical trials | NHL, CLL, SCLC | Dose-limiting thrombocytopenia from BCL-XL inhibition |
| Venetoclax (ABT-199) | BCL-2 (selective) | FDA/EMA approved (2016) | CLL, AML | Limited efficacy in solid tumors |
| Obatoclax (GX15-070) | Pan-BCL-2 inhibitor | Phase I/II trials | Hematological malignancies, NSCLC | Lower potency and specificity |
| AT-101 (R-(-)-gossypol) | Multiple BCL-2 family | Phase II trials | Various cancers | Limited specificity |
Beyond direct cancer therapy, understanding mitochondrial apoptosis has implications for treating other pathological conditions. In neurodegenerative diseases where excessive apoptosis contributes to neuronal loss, strategies to enhance anti-apoptotic signaling or inhibit pro-apoptotic factors may provide neuroprotection [1]. Conversely, in autoimmune disorders, promoting apoptosis in autoreactive immune cells could restore immune tolerance [1]. The ongoing refinement of BH3 profiling as a biomarker strategy may enable better prediction of chemotherapy sensitivity and patient stratification for BH3-mimetic therapies [15]. As our knowledge of the BCL-2 family continues to evolve, incorporating non-canonical functions, subcellular localization dynamics, and interactions with non-apoptotic cellular pathways, new therapeutic opportunities will undoubtedly emerge for manipulating this fundamental cell death pathway in human health and disease [18].
Apoptosis, or programmed cell death, is a fundamental process for maintaining tissue homeostasis and eliminating damaged or unwanted cells. Research has long focused on two primary initiating pathways: the extrinsic (death receptor) pathway and the intrinsic (mitochondrial) pathway [1]. While these pathways originate from different cellular locations and stimuli, they converge with remarkable precision at a critical juncture: the activation of executioner caspases. This convergence represents one of the most crucial regulatory points in cellular fate, where disparate death signals become channeled into a common execution mechanism [19] [20].
The execution phase of apoptosis is characterized by a series of stereotypic morphological changes: cell shrinkage, chromatin condensation, DNA fragmentation, and eventual disintegration into apoptotic bodies [21] [1]. These changes are primarily mediated by a family of cysteine proteases called caspases, which cleave their substrates at specific aspartic acid residues [22] [23]. This whitepaper examines the molecular machinery of this converging point, detailing how both apoptotic pathways activate the caspase cascade that executes the cell's demise, with particular focus on implications for therapeutic intervention.
Molecular Machinery of Apoptosis
The Caspase Protease Family
Caspases (cysteine-aspartic proteases) are the principal effectors of apoptotic cell death. These enzymes are synthesized as inactive zymogens (pro-caspases) that require proteolytic cleavage for activation [23]. The caspase family can be functionally categorized based on their position in the apoptotic cascade:
- Initiator Caspases (caspase-2, -8, -9, -10): Characterized by long prodomains that facilitate interaction with adaptor molecules. They initiate the apoptotic cascade but have limited substrate specificity [22] [19].
- Executioner Caspases (caspase-3, -6, -7): Feature short prodomains and are activated by initiator caspases. They cleave numerous cellular substrates to systematically dismantle the cell [22] [23].
- Inflammatory Caspases (caspase-1, -4, -5, -11): Primarily involved in inflammation and pyroptosis rather than apoptosis [22] [23].
All caspases share a common structural feature: they cleave their substrates after aspartic acid residues, a specificity unique among proteases [21] [23]. The active enzyme is typically a heterotetramer composed of two large and two small subunits, forming two catalytic sites per molecule [21].
The Extrinsic (Death Receptor) Pathway
The extrinsic pathway is initiated by extracellular signals binding to death receptors on the cell surface. These receptors belong to the tumor necrosis factor (TNF) receptor superfamily and include Fas (CD95), TNFR1, and TRAIL receptors [10] [3].
Key Molecular Events:
- Receptor Ligand Binding: Trimeric death ligands (e.g., FasL, TNF-α, TRAIL) bind to their cognate receptors, inducing receptor trimerization and conformational changes [10].
- DISC Formation: The intracellular death domains (DD) of activated receptors recruit adaptor proteins such as FADD (Fas-associated death domain) through homotypic DD interactions [10] [20]. FADD then recruits procaspase-8 via death effector domain (DED) interactions, forming the Death-Inducing Signaling Complex (DISC) [10] [3].
- Caspase-8 Activation: Within the DISC, caspase-8 molecules are brought into close proximity, enabling their auto-proteolytic activation [10] [23]. In some contexts, caspase-10 may be activated similarly [10].
The activated caspase-8 then propagates the death signal, serving as the bridge to the execution phase.
The Intrinsic (Mitochondrial) Pathway
The intrinsic pathway is triggered by intracellular stress signals, including DNA damage, oxidative stress, and growth factor withdrawal [3] [19]. This pathway is critically regulated by the Bcl-2 family of proteins, which govern mitochondrial outer membrane permeabilization (MOMP) [1] [19].
Key Molecular Events:
- BCL-2 Family Regulation: Cellular stress disrupts the balance between pro-apoptotic (e.g., Bax, Bak, Bid, Bad) and anti-apoptotic (e.g., Bcl-2, Bcl-xL) Bcl-2 family members [3] [19].
- Mitochondrial Outer Membrane Permeabilization (MOMP): Pro-apoptotic proteins Bax and Bak oligomerize to form pores in the mitochondrial outer membrane, facilitating the release of apoptogenic factors including cytochrome c, SMAC/DIABLO, and Omi/HtrA2 into the cytosol [1] [19].
- Apoptosome Formation: Cytochrome c binds to Apaf-1 in the presence of dATP/ATP, inducing oligomerization into a wheel-like complex called the apoptosome [19] [20].
- Caspase-9 Activation: The apoptosome recruits and activates procaspase-9, which remains bound to the complex while catalytically active [19] [20].
The Converging Point: Executioner Caspase Activation
Molecular Integration Mechanisms
Both apoptotic pathways converge at the point of executioner caspase activation, primarily through the proteolytic activation of caspase-3 and caspase-7 [19]. The molecular events at this convergence point can be summarized as follows:
Table 1: Caspase Activation in Apoptotic Pathways
| Pathway Component | Extrinsic Pathway | Intrinsic Pathway |
|---|---|---|
| Activation Trigger | Death receptor ligation | Cellular stress |
| Initiator Caspase | Caspase-8, -10 | Caspase-9 |
| Activation Complex | DISC (Death-Inducing Signaling Complex) | Apoptosome |
| Key Adaptor | FADD | Apaf-1 |
| Primary Executioner | Caspase-3, -7 | Caspase-3, -7 |
| Cross-Talk Mediator | Bid cleavage | N/A |
The convergence is not merely parallel but involves significant cross-talk between the pathways. In many cell types (classified as Type II cells), caspase-8 activated via the extrinsic pathway cleaves the BH3-only protein Bid to generate truncated Bid (tBid) [3] [20]. tBid translocates to mitochondria where it activates Bax/Bak, thereby engaging the mitochondrial amplification loop [3] [20]. This cross-talk ensures that even weakly initiated death signals can be amplified through mitochondrial involvement.
The Execution Phase Cascade
Once activated, executioner caspases initiate a proteolytic cascade that systematically dismantles cellular structures through limited proteolysis of key substrates [23] [19]. The major events include:
- Nuclear Demolition: Caspase-3 activates caspase-activated DNase (CAD) by cleaving its inhibitor ICAD. CAD then mediates internucleosomal DNA fragmentation, a hallmark of apoptosis [3] [19]. Additionally, caspases cleave nuclear structural proteins like lamins, facilitating nuclear shrinkage and fragmentation [21].
- Cytoskeletal Disassembly: Caspases cleave numerous cytoskeletal components including actin, fodrin, and gelsolin, leading to loss of cell shape and membrane blebbing [21] [19].
- Cell Surface Changes: Caspase-mediated cleavage of focal adhesion kinases facilitates cell detachment, while phospholipid scramblases externalize phosphatidylserine, providing an "eat me" signal for phagocytic cells [19].
- Organelle Disruption: Caspases cleave proteins involved in Golgi organization and protein translation, effectively halting cellular operations [21].
This controlled demolition occurs with remarkable precision, preventing the release of inflammatory cellular contents – a key distinction from necrotic cell death [1].
Experimental Analysis of Caspase Activation
Key Methodologies and Workflows
Research into apoptotic signaling relies on a multifaceted experimental approach to dissect the complex molecular interactions. The following diagram illustrates a generalized workflow for analyzing caspase activation in apoptosis research:
Research Reagent Solutions
The following table details essential research tools for investigating caspase activation and apoptotic pathways:
Table 2: Essential Research Reagents for Apoptosis Studies
| Reagent/Category | Specific Examples | Research Application |
|---|---|---|
| Caspase Activity Assays | Fluorogenic substrates (DEVD-AFC, IETD-AMC), Luminescent caspase-Glo assays | Quantification of caspase activation kinetics using specific peptide sequences cleaved by different caspases [24]. |
| Antibodies for Apoptosis | Phospho-specific antibodies, Cleaved caspase-3 (Asp175), Cleaved PARP (Asp214) | Detection of caspase activation and substrate cleavage via Western blot, immunofluorescence, and flow cytometry [3]. |
| Pathway-Specific Modulators | FasL/TRAIL (extrinsic pathway inducers), ABT-263 (Bcl-2 inhibitor), SMAC mimetics | Selective activation or inhibition of specific apoptotic pathways to dissect molecular mechanisms [1]. |
| Cell Viability/Versus Death Assays | Annexin V/propidium iodide staining, MTT/WST-1 assays, LDH release assays | Discrimination between apoptotic, necrotic, and viable cell populations [24]. |
| Live-Cell Analysis Tools | Fluorescent caspase reporters (SCAT1, Caspase-3/7 Green Detection Reagent), Mitochondrial membrane potential dyes (JC-1, TMRM) | Real-time monitoring of caspase activation and mitochondrial events in live cells [24]. |
Advanced Technical Approaches
Specialized techniques have been developed to dissect the precise molecular interactions in caspase activation:
DISC and Apoptosome Analysis: Immunoprecipitation of the Death-Inducing Signaling Complex (DISC) using anti-Fas or anti-caspase-8 antibodies followed by Western blotting for associated proteins (FADD, caspase-8) enables direct examination of this critical activation complex [10]. Similarly, the apoptosome can be studied through gel filtration chromatography and co-immunoprecipitation of Apaf-1 and caspase-9 [20].
Caspase Substrate Profiling: Advanced proteomic approaches, including mass spectrometry analysis of cleaved cellular proteins, help identify novel caspase substrates and map the proteolytic landscape during apoptosis execution [23].
Pathway Interplay and Regulatory Networks
The relationship between the extrinsic and intrinsic pathways is not merely convergent but involves complex regulatory interactions. The following diagram illustrates these relationships and the central role of caspase activation:
Critical regulatory mechanisms fine-tune this convergence point:
- Cellular Context Determination: Cells are classified as Type I or Type II based on their apoptotic signaling. Type I cells (e.g., thymocytes) demonstrate strong caspase-8 activation at the DISC sufficient to directly activate executioner caspases without mitochondrial amplification. Type II cells (e.g., hepatocytes) require mitochondrial amplification through Bid cleavage and cytochrome c release [20].
- Inhibitor of Apoptosis Proteins (IAPs): IAP family proteins, including XIAP, directly bind and inhibit active caspases, particularly caspase-3, -7, and -9. This inhibition is counteracted by mitochondrial proteins like SMAC/DIABLO and Omi/HtrA2, which are released during intrinsic apoptosis and displace IAPs from caspases [19] [20].
- FLIP Regulation: Cellular FLIP (FLICE-inhibitory protein) proteins regulate the extrinsic pathway by competing with caspase-8 for binding to FADD. High levels of FLIP can inhibit death receptor-mediated apoptosis, while specific splice forms may even promote caspase-8 activation in certain contexts [10].
Therapeutic Implications and Research Applications
Apoptosis Targeting in Disease Treatment
Dysregulation of apoptosis is a hallmark of numerous diseases, making the convergence point of caspase activation an attractive therapeutic target:
Cancer Therapeutics: Many cancers exhibit evasion of apoptosis through upregulation of anti-apoptotic proteins (e.g., Bcl-2, IAPs) or defects in death receptor signaling [1] [25]. Therapeutic strategies include:
- BH3 mimetics (e.g., Venetoclax) that inhibit anti-apoptotic Bcl-2 proteins to promote mitochondrial apoptosis [1] [25].
- SMAC mimetics that antagonize IAPs to enhance caspase activation [1].
- Agonistic death receptor antibodies that directly activate the extrinsic pathway [10] [25].
Neurodegenerative Disorders: Excessive apoptosis contributes to neuronal loss in conditions like Alzheimer's disease, Parkinson's disease, and Huntington's disease [1] [22]. Caspase inhibition has shown neuroprotective effects in preclinical models, though clinical translation remains challenging due to efficacy and specificity concerns [22].
Research and Diagnostic Applications
The quantitative analysis of apoptosis has significant research and clinical applications:
Table 3: Apoptosis Assay Market by Application (2025 Projections)
| Application Area | Market Share | Key Drivers |
|---|---|---|
| Oncology Research | 40.5% | High cancer prevalence and need for therapy response assessment [25]. |
| Neurodegenerative Disease Research | Significant growth segment | Elucidation of apoptotic mechanisms in neuronal death [25]. |
| Toxicology & Drug Safety | Growing application | Regulatory requirements for apoptosis assessment in preclinical screening [24]. |
| Autoimmune Disease Research | Established segment | Dysregulated apoptosis in immune cell homeostasis [1]. |
The North American apoptosis assay market is projected to grow from USD 2.7 billion in 2024 to USD 6.1 billion by 2034, reflecting the expanding research and clinical applications of apoptosis analysis [24].
The convergence of the extrinsic and intrinsic apoptotic pathways at the point of executioner caspase activation represents a critical regulatory node in cell fate determination. This converging point integrates diverse death signals into a coordinated execution program that systematically dismantles cellular structures while minimizing inflammatory consequences. The molecular machinery at this junction – including caspase activation complexes, regulatory proteins, and feedback mechanisms – offers multiple therapeutic intervention points for diseases characterized by dysregulated apoptosis.
Continued research into the precise mechanisms of caspase activation and the nuanced interplay between apoptotic pathways will undoubtedly yield new insights into cellular homeostasis and novel therapeutic strategies for cancer, neurodegenerative disorders, and other conditions where apoptosis goes awry. The development of more specific caspase modulators and advanced detection methodologies will further enhance both our fundamental understanding and clinical manipulation of this essential biological process.
Apoptosis, or programmed cell death, is a genetically regulated process essential for maintaining tissue homeostasis and eliminating damaged or superfluous cells in multicellular organisms [26] [27]. Dysregulation of apoptotic pathways is a hallmark of numerous diseases, including cancer, autoimmune disorders, and neurodegenerative conditions, making its molecular players prime targets for therapeutic intervention [16] [28]. The execution of apoptosis is primarily orchestrated through two core, interconnected signaling pathways: the extrinsic (death receptor) pathway and the intrinsic (mitochondrial) pathway [6] [26]. Both pathways converge on the activation of a cascade of proteolytic enzymes known as caspases, which dismantle the cell in a controlled manner [29]. This technical guide provides a detailed examination of the key molecular components—death receptors, BCL-2 family proteins, and caspases—that govern these processes, framed within the context of comparative apoptosis research. A thorough understanding of these mechanisms is foundational for developing novel targeted therapies, such as the successful BCL-2 inhibitor Venetoclax, which has transformed treatment for specific hematologic malignancies [16] [30].
The Extrinsic Pathway: Death Receptors and Initiator Caspases
The extrinsic pathway is initiated by extracellular death signals transduced through specific cell surface receptors [6] [26].
Core Death Receptors and Ligands
The extrinsic pathway is activated by the binding of extracellular death ligands to their corresponding transmembrane death receptors, members of the tumor necrosis factor (TNF) receptor superfamily [26]. Key receptor-ligand pairs include Fas (CD95) with FasL (Fas Ligand), TNF Receptor 1 (TNFR1) with TNF-α, and Death Receptors 4 and 5 (DR4/DR5) with TNF-Related Apoptosis-Inducing Ligand (TRAIL) [29] [26]. This ligand-receptor interaction triggers the intracellular assembly of a multi-protein complex known as the Death-Inducing Signaling Complex (DISC) [6].
DISC Formation and Caspase Activation
The core event in the extrinsic pathway is the formation of the DISC. Upon ligand binding, the intracellular death domains (DD) of the receptor recruit adaptor proteins, such as FADD (Fas-Associated Death Domain protein), which in turn binds initiator caspases, primarily caspase-8 and caspase-10, via death effector domains (DED) [29] [26]. This recruitment leads to the dimerization and auto-proteolytic activation of these initiator caspases within the DISC [26]. Once activated, caspase-8 and -10 directly cleave and activate downstream effector caspases, such as caspase-3, -6, and -7, which then execute the apoptotic program by degrading over 600 cellular substrates, leading to the characteristic morphological changes of apoptosis, including cell shrinkage, chromatin condensation, and DNA fragmentation [6] [26].
Table 1: Key Death Receptors, Ligands, and Primary Functions
| Death Receptor | Ligand | Key Adaptor Protein | Initiator Caspase | Primary Functions/Notes |
|---|---|---|---|---|
| Fas (CD95) | FasL | FADD | Caspase-8, -10 | Critical for immune cell regulation and elimination of auto-reactive cells [29] [26] |
| TNF-R1 | TNF-α | TRADD / FADD | Caspase-8 | Has a dual role; can promote cell survival or inflammation in addition to apoptosis [26] |
| DR4/DR5 | TRAIL | FADD | Caspase-8, -10 | Being explored for cancer therapy due to potential for selective apoptosis in cancer cells [26] |
The following diagram illustrates the sequence of events in the extrinsic apoptosis pathway:
The Intrinsic Pathway: BCL-2 Proteins and Mitochondrial Permeabilization
The intrinsic pathway integrates internal cellular stress signals, such as DNA damage, growth factor withdrawal, and oxidative stress, and is critically regulated by the BCL-2 protein family at the mitochondrial level [16] [14].
The Tripartite BCL-2 Family
The BCL-2 family consists of about 20 proteins that can be structurally and functionally categorized into three subgroups based on their BH (BCL-2 Homology) domains and their role in apoptosis [16] [27]:
- Anti-apoptotic Proteins: These include BCL-2, BCL-XL, MCL-1, BCL-W, and BFL-1/A1. They contain four BH domains (BH1-BH4) and act as guardians of mitochondrial integrity by sequestering pro-apoptotic members [16] [14].
- Multi-Domain Pro-apoptotic Effectors: This group includes BAX and BAK, which contain BH1-3 domains. They are the direct executors of mitochondrial outer membrane permeabilization (MOMP) [14].
- BH3-only Proteins: These are sentinels for cellular stress and include activators (e.g., BIM, BID, PUMA) and sensitizers (e.g., BAD, NOXA, HRK). They share only the BH3 domain and initiate apoptosis by either directly activating BAX/BAK or neutralizing anti-apoptotic proteins [16] [14].
Regulation of Mitochondrial Outer Membrane Permeabilization (MOMP)
The pivotal event in the intrinsic pathway is MOMP. In response to cellular stress, activated BH3-only proteins displace the activators BIM and BID from anti-apoptotic proteins or directly engage BAX and BAK [16] [14]. This triggers a conformational change in BAX and BAK, leading to their homo-oligomerization and integration into the outer mitochondrial membrane, forming pores [14]. MOMP results in the irreversible release of pro-apoptotic factors from the mitochondrial intermembrane space into the cytosol, most notably cytochrome c [16] [14]. Cytochrome c then binds to APAF-1, forming the "apoptosome" complex, which recruits and activates the initiator caspase-9. Caspase-9, in turn, activates the effector caspases-3, -6, and -7, culminating in cell death [6] [26].
Table 2: The BCL-2 Protein Family: Classification, Members, and Key Roles
| Subfamily | BH Domains | Key Members | Primary Mechanism of Action |
|---|---|---|---|
| Anti-apoptotic | BH1-BH4 | BCL-2, BCL-XL, MCL-1, BCL-W, BFL-1 | Preserve mitochondrial integrity by binding and neutralizing pro-apoptotic BH3-only proteins and activated BAX/BAK [16] [27] |
| Pro-apoptotic Effectors | BH1-BH3 | BAX, BAK, BOK | Upon activation, homo-oligomerize to form pores in the mitochondrial outer membrane, causing MOMP [14] |
| BH3-only Proteins | BH3 only | Activators: BIM, BID, PUMASensitizers: BAD, NOXA, HRK, BIK, BMF | Activators: Directly induce BAX/BAK activation.Sensitizers: Displace activators from anti-apoptotic proteins, indirectly promoting BAX/BAK activation [16] [14] |
The interplay between these proteins and the key step of MOMP is summarized below:
Caspases: The Executioners of Apoptosis
Caspases (cysteine-aspartic proteases) are a family of endoproteases that are synthesized as inactive zymogens and become activated by proteolytic cleavage at specific aspartic residues [29] [26]. They are the central executioners of both apoptotic pathways.
Classification and Substrate Specificity
Caspases are classified based on their role in the apoptotic cascade:
- Initiator Caspases (Caspase-8, -9, -10): These have long pro-domains containing protein-protein interaction motifs (DED or CARD) that allow them to be recruited to and activated within large signaling complexes like the DISC (caspase-8, -10) or the apoptosome (caspase-9) [29] [26].
- Effector Caspases (Caspase-3, -6, -7): These have short pro-domains and are activated by initiator caspases. They are responsible for the systematic cleavage of key cellular proteins, such as PARP, nuclear lamins, and cytoskeletal proteins, leading to the characteristic biochemical and morphological hallmarks of apoptosis [29] [26].
Table 3: Caspase Classification, Activation, and Key Functions
| Caspase | Type | Activation Complex/Pathway | Primary Functions / Key Substrates |
|---|---|---|---|
| Caspase-8 | Initiator | DISC (Extrinsic) | Initiates the extrinsic pathway; cleaves and activates effector caspases; cleaves BID to tBID, linking to intrinsic pathway [6] [26] |
| Caspase-9 | Initiator | Apoptosome (Intrinsic) | Initiates the intrinsic pathway; activated by cytochrome c/APAF-1; cleaves and activates effector caspases [26] |
| Caspase-3 | Effector | Activated by initiator caspases | Key executioner caspase; cleaves PARP, ICAD/DFF45, leading to DNA fragmentation [29] [26] |
| Caspase-6 | Effector | Activated by initiator caspases | Cleaves nuclear lamins and other substrates [26] |
| Caspase-7 | Effector | Activated by initiator caspases | Cooperates with caspase-3 in substrate proteolysis [26] |
Pathway Crosstalk and Integration
The extrinsic and intrinsic pathways are not isolated; they communicate and amplify the death signal through key molecular links. The most well-established connection is the cleavage of the BH3-only protein BID by caspase-8 (active in the extrinsic pathway) into its truncated, potent form, tBID [6] [14]. tBID then translocates to the mitochondria, where it potently activates BAX and BAK, thereby engaging the intrinsic pathway and amplifying the apoptotic signal through a mitochondrial feedback loop [14]. This crosstalk ensures that even a weak extrinsic signal can be robustly amplified, leading to irreversible cell commitment to death.
Experimental Approaches and Research Tools
The study of apoptosis and its key molecular players relies on a suite of sophisticated biochemical, cellular, and functional assays.
Key Methodologies for Apoptosis Research
- BH3 Profiling: This functional assay measures the mitochondrial priming state—how close a cell is to the apoptotic threshold. Isolated mitochondria or permeabilized cells are exposed to synthetic BH3 peptides, and the degree of MOMP is measured, often by cytochrome c release or changes in membrane potential. The pattern of response reveals which anti-apoptotic proteins a cancer cell is dependent on for survival, predicting sensitivity to specific BH3-mimetic drugs [14] [30].
- NIADS v2 (Non-Invasive Apoptosis Detection Sensor v2): A advanced bioluminescence-based assay that allows for rapid, sensitive, and real-time detection of caspase-3 activity, a key marker of apoptosis execution. It is particularly useful for high-throughput screening of drug efficacy and has been applied in studies of drug resistance, such as in Imatinib-resistant leukemia cells [31].
- Co-immunoprecipitation (Co-IP) and Western Blotting: These classic techniques are used to study protein-protein interactions within the BCL-2 family, such as the binding between anti-apoptotic BCL-2 and pro-apoptotic BIM, which is a hallmark of a primed cell [30].
- PRIMABs (Conformation-Specific Antibodies): A novel diagnostic platform using antibodies that specifically recognize complexes between anti-apoptotic proteins (e.g., BCL-2, MCL-1) and the pro-apoptotic BIM. This allows for direct measurement of the functional priming state in patient samples and can detect the disruption of these complexes by BH3-mimetic drugs, serving as a predictive biomarker for treatment response [30].
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Reagents and Tools for Apoptosis Research
| Research Tool / Reagent | Category | Key Function / Application |
|---|---|---|
| Recombinant Death Ligands (e.g., FasL, TRAIL) | Protein | To experimentally activate the extrinsic apoptosis pathway in cell cultures [26] |
| BH3 Peptides (e.g., BIM, BAD, HRK, NOXA) | Synthetic Peptide | Used in BH3 profiling to determine mitochondrial dependencies and identify "primed" cells [14] [30] |
| BH3-Mimetic Drugs (e.g., Venetoclax, ABT-737) | Small Molecule Inhibitor | Tool compounds and approved drugs that selectively inhibit anti-apoptotic BCL-2 proteins to induce apoptosis in cancer cells [16] [14] |
| Conformation-Specific Antibodies (e.g., PRIMABs) | Antibody | Detect specific, functional protein-protein interactions (e.g., BCL-2:BIM complex) for predictive diagnostics [30] |
| Caspase Activity Kits (e.g., NIADS v2) | Bioluminescence/Fluorescence Assay | Quantify the activity of key caspases (like caspase-3) to measure apoptosis induction in real-time [31] |
| Pan-Caspase Inhibitors (e.g., zVAD-fmk) | Small Molecule Inhibitor | To broadly inhibit caspase activity and confirm the caspase-dependent nature of cell death in experiments [26] |
The workflow for a key functional assay, BH3 profiling, is detailed below:
The molecular machinery of apoptosis—comprising death receptors, BCL-2 family proteins, and caspases—forms an intricate and highly regulated network that determines cellular life-and-death decisions. The extrinsic pathway responds to external cues, while the intrinsic pathway integrates internal damage signals, with both converging on caspase activation. The critical regulation of the intrinsic pathway by the BCL-2 family at the mitochondria presents a powerful therapeutic node, as successfully exploited by BH3-mimetics like Venetoclax. Ongoing research continues to refine our understanding of the complex crosstalk between these pathways, the development of resistance mechanisms, and the role of non-apoptotic functions of these proteins. For researchers and drug development professionals, the advancing toolkit—from functional assays like BH3 profiling and NIADS to novel reagents like conformation-specific antibodies—is enabling more precise diagnostic and therapeutic strategies, ultimately paving the way for more effective, personalized treatments for cancer and other diseases characterized by apoptotic dysregulation.
From Bench to Bedside: Research Tools and Therapeutic Targeting of Apoptosis
Apoptosis, or programmed cell death, is a fundamental process essential for development, tissue homeostasis, and disease pathogenesis. Research has largely focused on two principal initiation pathways: the death receptor (extrinsic) pathway and the mitochondrial (intrinsic) pathway. The death receptor pathway is triggered by extracellular ligands binding to cell surface receptors of the tumor necrosis factor (TNF) family, leading to the rapid assembly of a death-inducing signaling complex (DISC) and activation of initiator caspases [10]. In contrast, the mitochondrial pathway is initiated by cellular stress, leading to mitochondrial outer membrane permeabilization (MOMP) and the release of pro-apoptotic factors such as cytochrome c [32] [33]. While distinct in their initiation, these pathways converge on the activation of executioner caspases that orchestrate the controlled dismantling of the cell.
Advanced imaging technologies, particularly single-molecule imaging and correlative microscopy, are revolutionizing our understanding of these apoptotic pathways. They enable researchers to visualize the dynamic molecular interactions and structural changes in real-time and with unprecedented resolution. This technical guide explores how these methodologies are being applied to dissect the intricate mechanisms of apoptosis, providing detailed experimental protocols and resources for the research community.
Correlative Light and Electron Microscopy (CLEM) in Apoptosis
Principles and Workflow
Correlative Light and Electron Microscopy (CLEM) is a powerful methodology that integrates the live-cell dynamic imaging capabilities of fluorescence microscopy with the high-resolution ultrastructural detail provided by electron microscopy. This combination allows researchers to first observe a dynamic process like apoptosis in living cells and then fix and examine the very same cells at nanometer resolution to see the structural consequences of the observed events [34].
The general workflow for a CLEM study of apoptosis involves several key stages, as visualized below:
Figure 1: The Correlative Light and Electron Microscopy (CLEM) Workflow for Apoptosis Research
Detailed Protocol: Tracking Mitochondrial Apoptosis via CLEM
The following protocol, adapted from studies on HeLa cells, outlines a specific application of CLEM to study the mitochondrial pathway of apoptosis [34]:
Cell Culture and Labeling:
- Use HeLa cells stably transfected with a tetracysteine-tagged cytochrome c construct (Cyt. c-4CYS).
- Grow cells on MatTek petri dishes with etched grids to facilitate relocation.
- Induce apoptosis using 100 μM etoposide for 12-18 hours.
- Label the Cyt. c-4CYS with 250 nM FlAsH reagent.
- Simultaneously stain mitochondria with 50 nM TMRE to monitor membrane potential (ΔΨm).
Confocal Microscopy and Staging:
- Acquire time-lapse images using a confocal microscope (e.g., Leica TCS SP2).
- Excite FlAsH with a 488 nm laser and detect emission between 497-553 nm.
- Excite TMRE with a 543 nm laser and detect emission between 555-620 nm.
- Define apoptotic stages based on fluorescence patterns:
- Stage 1: Punctate FlAsH (cytochrome c not released) and punctate TMRE (ΔΨm maintained).
- Stage 2: Diffuse FlAsH (cytochrome c released) but punctate TMRE (ΔΨm maintained).
- Stage 3: Diffuse FlAsH (cytochrome c released) and loss of TMRE signal (ΔΨm lost).
Sample Preparation for EM:
- Fix cells immediately after imaging with primary fixative (2% paraformaldehyde, 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.4) for 1 hour on ice.
- Wash 3x with ice-cold 0.1 M sodium cacodylate buffer with 3 μM calcium chloride.
- Post-fix with 1% osmium tetroxide, 0.8% potassium ferrocyanide, and 3 μM calcium chloride in 0.1 M sodium cacodylate for 60 minutes on ice.
- Wash with distilled water and perform en bloc staining with 2% uranyl acetate.
- Dehydrate through an ethanol series and embed in Durcupan resin.
Electron Tomography:
- Relocate the identical cells of interest using the etched grid.
- Prepare 250-300 nm semi-thick sections.
- Acquire tilt series images typically from ±60° at 1° increments.
- Reconstruct 3D tomograms for detailed ultrastructural analysis.
Key Insights into Mitochondrial Remodeling
Application of this CLEM protocol has yielded critical insights into the mitochondrial pathway. Research has revealed that the formation of large holes in the mitochondrial outer membrane—essential for cytochrome c release—is driven by the coordinated action of Bax protein clusters and profound inner membrane remodeling [33]. The unfolding of the inner membrane cristae appears to exert physical pressure on the outer membrane, promoting its rupture and the subsequent release of intermembrane space proteins into the cytosol to activate the caspase cascade [33].
Single-Molecule Imaging and Fluorescent Reporters
Real-Time Caspase Activity Monitoring
Single-molecule and live-cell fluorescence imaging techniques allow for the real-time observation of caspase activation, a key event in both death receptor and mitochondrial pathways. A state-of-the-art reporter system utilizes a split-GFP design, where the GFP molecule is divided into two fragments tethered by a flexible linker containing a caspase-3/7-specific DEVD cleavage motif [35].
The mechanism of this sophisticated reporter system is illustrated below:
Figure 2: Mechanism of a Split-GFP Caspase Reporter
- Protocol: Using Stable Caspase Reporter Cell Lines [35]:
- Generate stable cell lines (e.g., via lentiviral transduction) expressing the ZipGFP-based caspase-3/7 reporter and a constitutive mCherry marker.
- Plate cells in 2D or 3D culture systems (e.g., spheroids or patient-derived organoids).
- Induce apoptosis with chosen stimuli (e.g., 100 nM carfilzomib or 50 μM oxaliplatin).
- Perform live-cell imaging using a confocal microscope equipped with environmental control (37°C, 5% CO₂).
- Excite GFP at 488 nm and mCherry at 587 nm.
- Quantify GFP fluorescence intensity over time, normalizing to mCherry signal to account for cell presence.
- For caspase specificity validation, include controls with pan-caspase inhibitor (e.g., 50 μM zVAD-FMK).
Advanced Single-Molecule Probe Design
Beyond reporter cells, novel fluorescent probes are being engineered for detecting specific apoptotic markers. A recently developed mitochondria-targeted probe (MIC) can independently detect sulfur dioxide (SO₂) and DNA simultaneously [36]. During apoptosis, the probe detects increased SO₂ levels, which are associated with reduced mitochondrial membrane potential and activation of the caspase-3 cascade. The apoptotic process also triggers changes in mitochondrial membrane permeability, causing the probe to release and translocate to the nucleus, where it emits red fluorescence upon binding DNA [36]. This dual-channel monitoring provides a sophisticated tool for dissecting the role of specific molecules in apoptosis.
Bright-to-Dark Apoptosis Reporters
An alternative to the dark-to-bright reporter strategy is the bright-to-dark system, where an existing fluorescent protein is inactivated by caspase cleavage. This approach involves mutagenesis-based insertion of a caspase-3 cleavage motif (DEVD) directly into the green fluorescent protein (GFP) [37]. Upon caspase-3 activation during apoptosis, the engineered GFP is cleaved and loses fluorescence. Studies suggest this bright-to-dark system may offer greater sensitivity compared to dark-to-bright reporters, and it functions across various model systems, including different mammalian cell types and other species [37].
Successful implementation of these advanced imaging techniques requires specialized reagents and tools. The table below summarizes key resources for apoptosis imaging research.
Table 1: Essential Research Reagent Solutions for Apoptosis Imaging
| Reagent/Tool | Function/Application | Example Use Case |
|---|---|---|
| Caspase-3/7 Reporter Cells | Real-time visualization of executioner caspase activation | Stable cell lines expressing ZipGFP-DEVD for live-cell apoptosis tracking [35] |
| Tetracysteine Tagged Proteins | Specific protein tracking with biarsenical dyes | Cyt. c-4CYS for monitoring cytochrome c localization via FlAsH/ReAsH staining [34] |
| Mitochondrial Membrane Potential Dyes | Assessment of mitochondrial health and function | TMRE staining to monitor ΔΨm loss during apoptosis [34] |
| Caspase Inhibitors | Specific pathway inhibition for mechanistic studies | zVAD-FMK (pan-caspase inhibitor) to confirm caspase-dependent processes [35] |
| MIC Probe | Simultaneous detection of mitochondrial SO₂ and nuclear DNA | Investigating SO₂-mediated apoptotic mechanisms via dual-channel fluorescence [36] |
| Annexin V Assay Kits | Detection of phosphatidylserine externalization | Endpoint validation of apoptosis by flow cytometry [35] [24] |
The market for apoptosis assays is robust and growing, with the North American market projected to reach USD 6.1 billion by 2034, driven by technological advancements and increased research in chronic diseases [24]. Leading suppliers include Thermo Fisher Scientific, Danaher, and Merck, which offer comprehensive portfolios of reagents, assay kits, and instrumentation [24].
Pathway-Specific Insights from Advanced Imaging
Visualizing the Death Receptor Pathway
Advanced imaging has refined our understanding of the death receptor pathway. For the CD95 receptor, ligand binding induces conformational changes that allow the intracellular death domain (DD) to recruit the adapter protein FADD via DD-DD interactions [10]. FADD then recruits procaspase-8 through death effector domain (DED) interactions, leading to caspase-8 oligomerization and activation [10]. This complex, known as the death-inducing signaling complex (DISC), forms within seconds of receptor ligation.
Single-molecule techniques have revealed that the activation mechanism involves caspase-8 filament formation, a process regulated by cellular FLIP proteins [10]. The ability to visualize these molecular assemblies in real-time has been crucial for understanding the precise regulation of this rapid apoptotic pathway.
Elucidating the Mitochondrial Pathway
In the mitochondrial pathway, cutting-edge microscopy has been instrumental in revealing the molecular mechanism of Bax-mediated outer membrane permeabilization. Correlative light and electron microscopy studies have shown that Bax does not merely form pores but assembles into large clusters that cooperate with inner membrane remodeling to create large ruptures [33]. The unfolding of the inner membrane cristae appears to exert mechanical pressure on the outer membrane, facilitating the formation of these openings and the subsequent release of cytochrome c and other pro-apoptotic factors [33].
Comparative Analysis of Imaging Platforms
Selecting the appropriate imaging technology is crucial for experimental success. The table below compares key platforms used in advanced apoptosis research.
Table 2: Technical Comparison of Apoptosis Imaging Platforms
| Imaging Platform | Key Strengths | Limitations | Typical Resolution | Applications in Apoptosis |
|---|---|---|---|---|
| Confocal Live-Cell Imaging | Real-time dynamics, specific molecular labeling, 3D capability | Photobleaching, limited resolution (~200 nm laterally) | ~200 nm lateral, ~500 nm axial | Caspase activation kinetics, mitochondrial potential changes, protein translocation [34] [35] |
| Electron Tomography | Ultrastructural detail, 3D reconstruction of organelles | Requires fixation, not applicable to live cells, technically demanding | ~2-5 nm | Mitochondrial membrane remodeling, Bax cluster morphology, cristae structure [32] [33] |
| Correlative CLEM | Combines dynamic and structural information, targeted high-resolution imaging | Complex workflow, potential sample preparation artifacts | Light microscopy resolution for live-cell, EM resolution for ultrastructure | Linking cytochrome c release to specific mitochondrial structural changes [34] [33] |
| Super-Resolution Microscopy | Beyond diffraction limit, molecular localization precision | Often slower acquisition, specialized equipment | ~20 nm lateral | Molecular organization in DISC, Bax oligomerization patterns |
The integration of single-molecule imaging and correlative microscopy is fundamentally advancing apoptosis research. These technologies have transitioned from merely documenting apoptotic endpoints to illuminating the dynamic molecular and structural changes that define both the death receptor and mitochondrial pathways. Future developments will likely focus on increasing the multiplexing capacity of reporters, improving the temporal resolution of CLEM workflows, and integrating artificial intelligence for automated image analysis and prediction of cell fate decisions [24].
These advanced techniques are not only refining our basic understanding of cell death but also creating new opportunities in drug discovery and therapeutic monitoring. As these tools become more accessible and integrated into standard research workflows, they will undoubtedly continue to reveal new layers of complexity in the regulation of apoptosis, potentially identifying novel therapeutic targets for diseases ranging from cancer to neurodegenerative disorders.
The intrinsic apoptotic pathway, also known as the mitochondrial pathway, represents a critically regulated process of programmed cell death essential for maintaining tissue homeostasis and eliminating damaged cells. This pathway is initiated by intracellular stressors—including DNA damage, oncogene activation, and hypoxia—which converge at the mitochondria to determine cellular fate [3]. The B-cell lymphoma 2 (BCL-2) protein family functions as the fundamental regulator of this pathway, acting as a tripartite apoptotic switch that balances cellular survival and death signals [16]. The discovery that cancer cells frequently exploit the anti-apoptotic members of this family to evade programmed cell death has positioned the intrinsic pathway as a compelling target for therapeutic intervention [38]. The development of BCL-2 inhibitors, particularly venetoclax, represents a paradigm shift in cancer therapy, demonstrating that directly targeting the apoptotic machinery can yield profound clinical benefits for patients with hematological malignancies [39] [40].
The BCL-2 protein family comprises approximately 20 proteins that share BCL-2 homology (BH) domains [16]. These proteins are structurally and functionally categorized into three distinct subgroups: (1) multi-domain anti-apoptotic proteins (BCL-2, BCL-XL, MCL-1, BCL-W, BFL-1, and BCL-B) that promote cellular survival; (2) multi-domain pro-apoptotic effector proteins (BAK, BAX, and BOK) that execute mitochondrial outer membrane permeabilization (MOMP); and (3) BH3-only pro-apoptotic proteins (BID, BIM, BAD, PUMA, NOXA, and others) that sense cellular stress and initiate the apoptotic cascade [16] [38] [6]. The precise interplay between these competing factions determines whether a cell will survive or undergo apoptosis, making the regulatory dynamics of this protein family a critical focal point for drug development.
Molecular Mechanisms of the Intrinsic Pathway
Regulation by the BCL-2 Protein Family
The intrinsic pathway is tightly regulated through protein-protein interactions among the three subgroups of the BCL-2 family. In healthy cells, anti-apoptotic proteins such as BCL-2 and BCL-XL bind and sequester the pro-apoptotic effectors BAX and BAK, preventing their activation and maintaining mitochondrial integrity [38] [40]. When cells experience internal stress signals—such as DNA damage or growth factor deprivation—the BH3-only proteins become transcriptionally upregulated or post-translationally activated [6]. These activated BH3-only proteins then bind to the anti-apoptotic proteins, displacing the sequestered BAX and BAK molecules [41]. The freed BAX and BAK undergo conformational changes, oligomerize, and integrate into the mitochondrial outer membrane, forming pores that trigger MOMP [16] [3].
This permeabilization represents the point of no return in the intrinsic pathway, as it leads to the release of several mitochondrial intermembrane space proteins into the cytosol, including cytochrome c, SMAC/DIABLO, and Omi/HTRA2 [3]. Cytochrome c binds to APAF-1 (apoptotic protease activating factor-1) in the presence of dATP/ATP, forming a complex known as the apoptosome, which recruits and activates procaspase-9 [42] [6]. The initiator caspase-9 then cleaves and activates the executioner caspases-3, -6, and -7, which systematically dismantle the cell through proteolytic cleavage of key structural and regulatory proteins, culminating in the characteristic morphological changes of apoptosis, including chromatin condensation, DNA fragmentation, and formation of apoptotic bodies [41] [6].
Pathway Visualization
The following diagram illustrates the key molecular events in the intrinsic apoptotic pathway and the mechanism of BCL-2 inhibition:
Venetoclax: Mechanism of Action and First-Generation Clinical Success
Venetoclax as a BH3 Mimetic
Venetoclax (ABT-199) represents the first highly selective BCL-2 inhibitor approved for clinical use, belonging to a class of compounds known as BH3 mimetics [16]. This orally administered small molecule was specifically designed to bind with high affinity to the hydrophobic groove of the BCL-2 protein, effectively mimicking the action of native BH3-only proteins [40]. By occupying this critical interaction site, venetoclax displaces pro-apoptotic proteins such as BIM that are normally sequestered by BCL-2, thereby allowing these activated proteins to trigger BAX/BAK-mediated mitochondrial outer membrane permeabilization [40]. The selectivity profile of venetoclax for BCL-2 over BCL-XL is particularly important from a therapeutic perspective, as it minimizes the thrombocytopenia associated with broader BCL-2 family inhibition—a dose-limiting toxicity observed with earlier agents like navitoclax [16] [40].
The molecular interactions between venetoclax and BCL-2 have been extensively characterized through structural studies. Venetoclax binds into the hydrophobic groove on the BCL-2 surface formed by the BH1, BH2, and BH3 domains, utilizing four hydrophobic pockets (P1-P4) within this binding groove [16]. This specific binding mode enables venetoclax to effectively neutralize BCL-2's anti-apoptotic function while sparing other anti-apoptotic family members, contributing to its favorable therapeutic index compared to earlier generation inhibitors [40]. The drug's mechanism capitalizes on the concept of "primed apoptosis" in cancer cells, wherein malignant cells exist in a state of heightened readiness for apoptosis, dependent on BCL-2 for continued survival. By specifically inhibiting BCL-2, venetoclax tips this balance toward cell death in susceptible tumors [39].
Clinical Applications and Efficacy
Venetoclax has demonstrated remarkable efficacy across various hematological malignancies, fundamentally changing treatment paradigms for several conditions. The following table summarizes key clinical applications and efficacy data for venetoclax-based therapies:
Table 1: Clinical Efficacy of Venetoclax in Hematological Malignancies
| Disease | Combination Therapy | Patient Population | Efficacy Outcomes | References |
|---|---|---|---|---|
| Acute Myeloid Leukemia (AML) | Azacitidine or Decitabine | Older/unfit for intensive chemotherapy | Significant improvement in response rates and survival; new standard of care | [39] |
| Chronic Lymphocytic Leukemia (CLL) | Anti-CD20 monoclonal antibodies | Treatment-naïve and relapsed/refractory | High overall response rates (up to 85%); prolonged progression-free survival | [40] |
| Multiple Myeloma (MM) | Dexamethasone + proteasome inhibitors/BiTEs | Relapsed/refractory, particularly t(11;14) | Promising activity, especially in t(11;14) subtype | [40] |
| Myelodysplastic Syndromes (MDS) | Azacitidine | Higher-risk MDS | Encouraging response rates in ongoing trials | [43] |
The efficacy of venetoclax is particularly pronounced in acute myeloid leukemia (AML), where its combination with hypomethylating agents (azacitidine or decitabine) has become a standard of care for older patients or those unfit for intensive chemotherapy [39]. This combination leverages synergistic mechanisms—the hypomethylating agents presumably sensitize leukemia cells to venetoclax by altering the expression of BCL-2 family members and other apoptotic regulators [39] [43]. In chronic lymphocytic leukemia (CLL), venetoclax has demonstrated exceptional activity, with studies reporting overall response rates as high as 84% in some patient populations, significantly improving progression-free survival compared to standard chemoimmunotherapy regimens [40].
Next-Generation BCL-2 Inhibitors and Novel Targeting Strategies
Overcoming Venetoclax Resistance
Despite the remarkable success of venetoclax, the development of acquired resistance remains a significant clinical challenge [39]. Resistance mechanisms are multifactorial and include: (1) BCL-2 mutations (e.g., F104L and F104C) that reduce venetoclax binding affinity without compromising BCL-2's anti-apoptotic function; (2) compensatory upregulation of alternative anti-apoptotic proteins, particularly MCL-1 and BCL-XL; and (3) transcriptional reprogramming mediated by factors such as NF-κB that establish co-dependencies on multiple anti-apoptotic proteins [38] [44]. Single-cell transcriptomic analyses of venetoclax-resistant chronic lymphocytic leukemia (CLL) cells have revealed the emergence of distinct subpopulations characterized by concurrent expression of BCL-2 with MCL-1 or by switching dependency to BFL-1, particularly in TP53-mutant cases [44].
To address these resistance mechanisms, several next-generation BCL-2 inhibitors and novel therapeutic approaches are under development. Lisaftoclax (APG-2575) is an investigational BCL-2 inhibitor that has demonstrated promising activity in patients with venetoclax-refractory myeloid malignancies [43]. In a global Phase Ib/II study presented at ASCO 2025, lisaftoclax in combination with azacitidine achieved an overall response rate of 31.8% in venetoclax-refractory patients with relapsed/refractory AML, with responses observed even in high-risk populations characterized by TP53 mutations and complex karyotypes [43]. This represents the first clinical evidence of a BCL-2 inhibitor overcoming resistance to venetoclax, suggesting potential structural or functional distinctions between these agents despite targeting the same protein [43].
Innovative Therapeutic Platforms
Beyond conventional small-molecule inhibitors, several innovative platforms are being explored to target the BCL-2 family more effectively:
PROTAC-Based Degraders: Proteolysis-Targeting Chimeras (PROTACs) represent a groundbreaking approach to overcome resistance mediated by BCL-2 mutations. These heterobifunctional molecules simultaneously bind to the target protein (BCL-2) and an E3 ubiquitin ligase, facilitating ubiquitination and subsequent proteasomal degradation of the target [16]. A novel BCL-2/BCL-XL dual-efficacy degrader has demonstrated potent activity against venetoclax-resistant CLL cells, including those harboring BCL-2 mutations, while showing improved platelet safety compared to traditional BCL-XL inhibitors [44]. This technology fundamentally differs from inhibition by permanently removing the target protein from the cellular environment.
Selective BCL-XL and MCL-1 Inhibitors: While selective BCL-XL and MCL-1 inhibitors have faced developmental challenges due to on-target toxicities (thrombocytopenia for BCL-XL inhibitors and cardiac toxicity for MCL-1 inhibitors), novel delivery strategies—including antibody-drug conjugates (ADCs) and tissue-specific activation approaches—are being explored to improve their therapeutic indices [16]. These agents hold particular promise for tumors dependent on these alternative anti-apoptotic proteins.
BH4 Domain-Targeting Therapies: Emerging research is exploring the therapeutic potential of targeting the BH4 domain of BCL-2, which is critical for its anti-apoptotic function and involved in interactions with regulatory proteins outside the immediate BCL-2 family [16]. This approach may provide an alternative strategy for disrupting BCL-2 function in resistant malignancies.
The following table compares key next-generation agents in development:
Table 2: Next-Generation BCL-2 Family-Targeting Therapies
| Therapeutic Agent | Target | Mechanism | Development Status | Key Features | |
|---|---|---|---|---|---|
| Lisaftoclax (APG-2575) | BCL-2 | Inhibition | Phase III (NDA accepted for CLL/SLL in China) | Activity in venetoclax-refractory patients | [43] |
| BCL-2/BCL-XL PROTAC | BCL-2, BCL-XL | Degradation | Preclinical | Overcomes mutation-based resistance; improved platelet safety | [44] |
| Sonrotoclax | BCL-2 | Inhibition | Clinical Evaluation | Similar target profile with potential pharmacokinetic differences | [16] |
| Novel MCL-1 Inhibitors | MCL-1 | Inhibition | Clinical Development | Addresses MCL-1-mediated resistance; toxicity challenges | [16] |
Experimental Approaches for Studying the Intrinsic Pathway and BCL-2 Inhibition
Core Methodologies and Assays
Research on the intrinsic apoptotic pathway and therapeutic targeting relies on a suite of well-established experimental techniques that enable investigators to assess apoptotic commitment, mitochondrial integrity, and caspase activation. The following experimental workflow outlines key methodologies used in this field:
Key Methodologies:
BH3 Profiling: This functional assay evaluates mitochondrial priming by measuring the mitochondrial response to synthetic BH3 peptides. It can identify which anti-apoptotic proteins a particular cancer cell depends on for survival, enabling more precise patient stratification and therapy selection [44]. The assay measures cytochrome c release or mitochondrial membrane depolarization after exposure to specific BH3 domain peptides.
Annexin V/Propidium Iodide (PI) Staining: A flow cytometry-based method that detects phosphatidylserine externalization on the outer leaflet of the plasma membrane, an early event in apoptosis. Viable cells are Annexin V-negative/PI-negative; early apoptotic cells are Annexin V-positive/PI-negative; and late apoptotic/necrotic cells are double-positive [41].
Caspase Activation Assays: These include Western blot analysis of caspase cleavage (e.g., procaspase-3 to cleaved caspase-3) and PARP cleavage, as well as fluorometric or colorimetric assays using caspase-specific substrates (e.g., DEVD for caspase-3, IETD for caspase-8, LEHD for caspase-9) [41] [42].
TUNEL Assay: Terminal deoxynucleotidyl transferase dUTP nick end labeling detects DNA fragmentation, a hallmark of late-stage apoptosis, by labeling the 3'-OH ends of fragmented DNA with modified nucleotides [41]. This assay is particularly useful in tissue sections and can be combined with caspase staining for more comprehensive analysis.
Mitochondrial Membrane Potential Assessment: Using fluorescent dyes such as TMRE (tetramethylrhodamine ethyl ester) or JC-1 that accumulate in healthy mitochondria based on membrane potential. Apoptotic cells show decreased fluorescence due to loss of mitochondrial membrane potential during early apoptosis [41].
Cytochrome c Release Assays: Performed using subcellular fractionation followed by Western blotting or immunofluorescence microscopy to visualize the translocation of cytochrome c from mitochondria to the cytosol following MOMP [42] [3].
Cell-Free Caspase Activation Assays: These utilize cell lysates (S16 extracts) supplemented with cytochrome c and dATP/ATP to reconstitute apoptosome formation and caspase activation in a controlled system, allowing researchers to study specific components of the intrinsic pathway without confounding cellular processes [42].
Research Reagent Solutions
Table 3: Essential Research Reagents for Studying Intrinsic Apoptosis
| Reagent Category | Specific Examples | Research Application | Key Features | |
|---|---|---|---|---|
| Viability & Early Apoptosis | Annexin V-FITC/PI kits; TMRE | Flow cytometry detection of phosphatidylserine exposure; mitochondrial membrane potential | Distinguishes early vs. late apoptosis; compatible with multiple detection platforms | [41] |
| Late Apoptosis Detection | TUNEL Assay Kits | Fluorescence or IHC detection of DNA fragmentation | Specific for apoptotic DNA cleavage; applicable to cells, tissue sections, and paraffin-embedded samples | [41] |
| Antibody-Based Detection | Anti-cleaved caspase-3; Anti-PARP; Anti-cytochrome c; Anti-BCL-2 family proteins | Western blot, immunofluorescence, IHC for apoptosis markers | Confirms specific protein activation/cleavage; allows subcellular localization | [41] [42] |
| BCL-2 Family Protein Analysis | Pro- and anti-apoptotic BCL-2 family antibody sampler kits | Comprehensive profiling of BCL-2 family proteins by Western | Enables simultaneous assessment of multiple pathway components; ideal for mechanism studies | [41] |
| Functional Assays | BH3 peptides; Granzyme B | BH3 profiling; induction of apoptosis in cell-free systems | Measures mitochondrial priming; identifies BCL-2 family dependencies | [42] [44] |
| Cell-Free Systems | S16 cell-free extracts from tissues | Study of cytochrome c/dATP-induced caspase activation | Enables pathway dissection without intact cellular compartments; useful for studying age- and tissue-specific effects | [42] |
The targeting of the intrinsic apoptotic pathway through BCL-2 inhibition represents a landmark achievement in cancer therapeutics, validating the deliberate reactivation of programmed cell death as a viable treatment strategy. Venetoclax has demonstrated transformative efficacy across multiple hematologic malignancies, particularly in acute myeloid leukemia and chronic lymphocytic leukemia, establishing a new paradigm for targeting the fundamental mechanisms of cancer cell survival [39] [40]. However, the emergence of resistance mechanisms—including BCL-2 mutations, compensatory upregulation of alternative anti-apoptotic proteins, and transcriptional reprogramming—highlights the remarkable adaptability of cancer cells and underscores the need for continued therapeutic innovation [38] [44].
The future of intrinsic pathway targeting lies in combination strategies and next-generation agents that can overcome or prevent resistance. Promising approaches include the rational combination of BCL-2 inhibitors with other targeted agents, the development of dual-specificity inhibitors and degraders capable of addressing compensatory survival pathways, and the implementation of functional assays like BH3 profiling to guide personalized treatment selection [43] [44]. As these advanced therapeutic modalities progress through clinical development, they hold the potential to expand the benefits of intrinsic pathway targeting to broader patient populations, including those with solid tumors, and to ultimately improve outcomes for patients with refractory hematologic malignancies. The continued elucidation of apoptotic regulation and the development of increasingly sophisticated targeting strategies ensure that the intrinsic pathway will remain a fertile ground for therapeutic innovation in the coming years.
The tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) pathway, specifically through its death receptor 5 (DR5), represents a promising therapeutic avenue in oncology due to its unique ability to selectively induce apoptosis in malignant cells while sparing normal tissues. This whitepaper provides a comprehensive technical analysis of strategies to reactivate the extrinsic apoptotic pathway in cancer, focusing on TRAIL analogs and DR5 agonists. Within the broader context of apoptosis research, which contrasts the extrinsic death receptor pathway with the intrinsic mitochondrial pathway, we examine the molecular mechanisms, current therapeutic agents, resistance challenges, and emerging combination strategies. The content is structured to serve researchers, scientists, and drug development professionals with detailed experimental methodologies, quantitative data comparisons, and essential research tools for advancing this field.
Apoptosis, a genetically programmed cell death process essential for maintaining tissue homeostasis, proceeds primarily through two distinct yet interconnected pathways: the extrinsic (death receptor) pathway and the intrinsic (mitochondrial) pathway [1] [3].
The extrinsic pathway initiates when extracellular death ligands bind to cell surface death receptors belonging to the tumor necrosis factor (TNF) receptor superfamily. This binding triggers receptor oligomerization and formation of the death-inducing signaling complex (DISC), which activates initiator caspases (primarily caspase-8) that subsequently activate executioner caspases (caspase-3, -6, and -7) [41] [3]. In contrast, the intrinsic pathway activates in response to intracellular stress signals such as DNA damage, oxidative stress, or growth factor deprivation. These signals cause mitochondrial outer membrane permeabilization (MOMP), leading to cytochrome c release and formation of the apoptosome, which activates caspase-9 and subsequently the executioner caspases [45] [3].
The TRAIL-DR5 axis represents a particularly promising extrinsic pathway for cancer therapeutic intervention because DR5 demonstrates the highest affinity for TRAIL under physiological conditions and selectively induces apoptosis in transformed cells while minimally affecting normal cells [46] [47]. This selectivity forms the rationale for developing TRAIL analogs and DR5 agonists as potential cancer therapeutics with favorable safety profiles.
DR5 Biology and Expression in Malignancy
Structural and Functional Characteristics
DR5 (also known as TRAIL-R2, TNFRSF10B, CD262, Apo2, Killer/Ly98, TRICK2A, and TRICKB) is a type I transmembrane protein consisting of a signal peptide, an extracellular domain, a transmembrane domain, and an intracellular death domain [46] [47]. The full-length DR5 cDNA is 1,146 bp, encoding 381 amino acids, with gene transcription occurring at 8q21.3 [46]. While DR5 shares significant homology with DR4 in the cysteine-rich domain and death domain, their distribution and physiological functions differ substantially [47].
A critical distinction lies in their expression patterns: DR4 is distributed and highly expressed in many immune-related tissues and some specific tumor types, while DR5 is widely distributed in normal tissue cells at very low levels but highly expressed across many different cancer types [46] [47]. This differential expression profile, combined with DR5's highest affinity for TRAIL at the optimal human body temperature of 37°C, positions it as a superior target for tumor-selective therapy [46].
DR5 Expression in Normal and Tumor Tissues
DR5 is expressed at low levels across various normal human tissues, including the heart, lungs, thymus, liver, kidneys, colon, small intestine, ovaries, prostate, testes, and skeletal muscles [46] [47]. In contrast, DR5 is significantly upregulated in numerous malignancies, including:
- Carcinomas: Breast, endometrial, cervical, ovarian, pancreatic, hepatocellular, and rectal cancers [46]
- Sarcomas: Bone sarcomas (Ewing's sarcoma, osteosarcoma, and chondrosarcoma) and soft tissue sarcomas [46] [47]
- Hematological malignancies: Multiple myeloma and other blood cancers [46]
This upregulated expression in malignant tissues, combined with minimal expression in normal tissues, provides the fundamental therapeutic window for DR5-targeted therapies.
Mechanisms of DR5 Upregulation
Multiple molecular mechanisms regulate DR5 expression, offering opportunities for therapeutic intervention through combinatorial approaches:
- Transcription Factors: CHOP, which may form heterodimers with C/EBPb on the DR5 promoter [46] [47]
- Stress Response Pathways: ERK activation leads to ATF4 activation, promoting CHOP induction and subsequent DR5 expression [46]
- Tumor Suppressors: p53 directly transactivates the DR5 gene [46] [47]
- Kinase Signaling: JNK activates CHOP by binding to the AP-1 site in the CHOP promoter [46]
- Proliferation Regulators: Sp1 binds to TATA-minor promoter regions of the DR5 gene for basal transcription [46]
- Inflammatory Pathways: NF-κB p65 subunit increases DR5 expression by binding to the first intronic region of the DR5 gene [46]
- Repressors: YY1 negatively regulates DR5 transcription by binding to putative DNA binding sites in the DR5 promoter [46]
Table 1: Key Regulators of DR5 Expression
| Regulator | Mechanism of Action | Therapeutic Implication |
|---|---|---|
| CHOP | Forms heterodimers with C/EBP proteins on DR5 promoter | ER stress inducers can upregulate DR5 |
| p53 | Directly transactivates DR5 gene | DNA-damaging agents can enhance DR5 expression |
| ERK | Leads to ATF4 activation promoting CHOP induction | Kinase pathway modulation affects DR5 expression |
| Sp1 | Binds TATA-minor promoter for basal transcription | Fundamental for constitutive and inducible expression |
| NF-κB | p65 subunit binds first intronic region of DR5 gene | Inflammatory signals can modulate DR5 levels |
| YY1 | Binds DR5 promoter to negatively regulate transcription | Inhibition may enhance DR5 expression |
Therapeutic Agents: TRAIL Analogs and DR5 Agonists
First-Generation TRAIL Receptor Agonists
Initial therapeutic development focused on recombinant soluble TRAIL and receptor-agonsitic antibodies. However, these first-generation agonists demonstrated limited antitumor activity in clinical trials due to several limitations, including poor pharmacokinetics, insufficient receptor clustering, and inherent resistance mechanisms [48].
Advanced TRAIL Variants and Fusion Proteins
Novel protein engineering approaches have yielded more potent TRAIL-based therapeutics with enhanced anti-tumor properties. For instance, the multitarget recombinant fusion protein SRH-DR5-B-p48 incorporates a DR5-selective TRAIL variant (DR5-B) with synthetic peptides (SRH and p48) that antagonize VEGFR2 and FGFR1 receptors, respectively [48]. This design enables simultaneous induction of tumor cell apoptosis via DR5 and suppression of angiogenesis in the tumor microenvironment [48].
Molecular dynamics analyses confirm that the SRH and p48 peptides form non-specific temporary contacts with the DR5-B domain while maintaining high affinity for their respective targets (nanomolar dissociation constants for VEGFR2 and FGFR1) [48]. This fusion protein demonstrates superior tumor-killing activity across various cancer types and effectively destroys tumor-like structures in 3D cell models while inhibiting FGF2-mediated fibroblast proliferation [48].
Clinical-Stage DR5 Agonists
Several DR5-targeted agonists have advanced to clinical development with promising results:
- INBRX-109: A DR5 agonist that showed encouraging antitumor activity and a favorable safety profile in patients with unresectable/metastatic chondrosarcoma in a Phase I study [46] [47]
- Drozitumab: A human monoclonal agonistic antibody against DR5 evaluated as a novel therapeutic for bone and soft tissue sarcomas [46]
Table 2: Selected TRAIL Analogs and DR5 Agonists in Development
| Agent | Type | Key Features | Development Status |
|---|---|---|---|
| Recombinant TRAIL | Soluble ligand | First-generation agonist; limited clinical efficacy | Early clinical trials |
| SRH-DR5-B-p48 | Fusion protein | DR5-selective variant with anti-angiogenic peptides; multitargeted | Preclinical |
| INBRX-109 | DR5 agonist | Encouraging activity in chondrosarcoma; favorable safety | Phase I |
| Drozitumab | Agonistic antibody | Human monoclonal antibody; evaluated in sarcomas | Preclinical/Clinical |
Resistance Mechanisms and Combination Strategies
The Dual Nature of DR5 Signaling
A critical challenge in DR5-targeted therapy is the receptor's capacity to activate both pro-apoptotic and pro-survival signaling pathways. Research reveals that in the context of DR4–DR5–DcR2 hetero-oligomeric complexes, a single death receptor (DR5) can assemble composite plasma membrane-proximal platforms that propagate TRAIL signaling to both death and survival pathways in clonal cancer cell populations [49].
This dual signaling leads to "fractional survival" in response to TRAIL treatment, wherein only a portion of the initial cellular population dies while the surviving fraction develops resistance [49]. Surprisingly, key apoptotic proteins including FADD and procaspase-8 participate in transducing non-apoptotic signals in response to TRAIL, creating a complex signaling landscape [49].
Beyond insufficient death receptor activation, multiple molecular mechanisms contribute to resistance against TRAIL and DR5 agonists:
- Upregulation of Anti-Apoptotic Proteins: Overexpression of Bcl-2, Bcl-xL, and other anti-apoptotic Bcl-2 family members prevents mitochondrial amplification of the death signal [45]
- DISC Modulation: Elevated cellular FLICE-inhibitory protein (c-FLIP) levels compete with caspase-8 for binding to FADD, inhibiting initiator caspase activation [45] [49]
- Inhibitor of Apoptosis Proteins (IAPs): XIAP, cIAP1, and cIAP2 directly inhibit caspase activity and promote cell survival [45]
- Decoy Receptor Expression: DcR1 and DcR2 compete for TRAIL binding without transmitting death signals, acting as molecular sinks [46] [49]
Rational Combination Therapies
Strategic combination approaches can overcome resistance by simultaneously targeting multiple components of the apoptotic machinery:
- DR5 Agonists + Bcl-2 Inhibitors: Co-targeting extrinsic and intrinsic pathways circumvents mitochondrial blockade of apoptosis [45]
- DR5 Agonists + ER Stress Inducers: Compounds that enhance CHOP-mediated DR5 upregulation sensitize cells to TRAIL-induced apoptosis [46]
- DR5 Agonists + Kinase Inhibitors: Inhibition of pro-survival kinases (ERK, Akt, p38) enhances apoptotic signaling [49]
- DR5 Agonists + HDAC Inhibitors: Histone deacetylase inhibitors can upregulate DR5 expression while modulating other apoptotic regulators [46]
Experimental Approaches and Research Tools
Key Methodologies for DR5 Pathway Analysis
Death Receptor Activation and DISC Analysis
- Immunoprecipitation/Western Blotting: To detect DR5 expression and DISC composition (FADD, caspase-8, c-FLIP) [49]
- Flow Cytometry: To quantify surface DR5 expression and analyze apoptosis via Annexin V/PI staining [50]
- Co-Immunoprecipitation: To characterize TRAIL receptor complexes and their associated proteins [49]
Apoptosis Detection Methods
- TUNEL Assay: Detects DNA fragmentation during late-stage apoptosis [41]
- Caspase Activity Assays: Fluorometric or colorimetric substrates to measure caspase-3, -8, and -9 activation [41]
- Mitochondrial Membrane Potential Assessment: TMRE or JC-1 staining to detect MOMP [41]
- Western Blotting for Apoptotic Markers: Cleaved caspase-3, cleaved PARP, and cytochrome c release [41] [50]
Cell Viability and Death Assays
- Cell Counting Kit-8 (CCK-8): Measures metabolic activity as a viability indicator [50]
- Clonogenic Survival Assays: Assess long-term reproductive cell death after treatment [46]
- Live/Dead Staining: Differential dye uptake to distinguish viable, apoptotic, and necrotic cells [41]
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for DR5 Pathway Investigation
| Reagent/Category | Specific Examples | Research Application |
|---|---|---|
| Recombinant Proteins | Human TRAIL, sDR5-Fc, SRH-DR5-B-p48 | Ligand-receptor studies; apoptosis induction; antagonist studies [48] [50] |
| Agonistic Antibodies | Anti-DR5 agonistic antibodies (e.g., drozitumab) | Receptor activation; apoptosis induction [46] |
| Apoptosis Detection Kits | Annexin V-FITC/PI, TUNEL, Caspase-3/8/9 activity assays | Quantification and staging of apoptotic cell death [41] [50] |
| DR5 Detection Reagents | Anti-DR5 antibodies for flow cytometry, IHC, WB | Receptor expression analysis; tissue localization [46] [50] |
| Pathway Inhibitors | zVAD-fmk (pan-caspase inhibitor), c-FLIP inhibitors | Mechanism studies; pathway validation [49] |
| Cell Lines | Cancer cell lines with varying DR5 expression | In vitro efficacy models; mechanism studies [46] [49] |
Signaling Pathway Visualization
TRAIL-DR5 Apoptotic Signaling Pathway
Experimental Workflow for DR5 Therapeutic Evaluation
Reactivation of the extrinsic apoptotic pathway through TRAIL analogs and DR5 agonists continues to represent a promising strategy for selective cancer therapy. The unique advantage of this approach lies in the differential expression of DR5 in malignant versus normal tissues, providing a potential therapeutic window. However, the complexity of DR5 signaling—with its capacity to activate both death and survival pathways—demands sophisticated therapeutic designs and strategic combination approaches.
Future directions in this field include the development of more effective multivalent DR5 agonists with enhanced receptor clustering capabilities, dual-targeting agents that simultaneously engage DR5 while blocking pro-survival pathways, and personalized medicine approaches based on DR5 expression profiles and apoptotic competency of individual tumors. As our understanding of the intricate balance between death receptor and mitochondrial pathways deepens, more effective therapeutic strategies will emerge to overcome resistance and fully harness the apoptosis-inducing potential of the TRAIL-DR5 system for cancer treatment.
Apoptosis, or programmed cell death, is a critical process for maintaining tissue homeostasis and is executed primarily via two core signaling pathways: the extrinsic (death receptor) pathway and the intrinsic (mitochondrial) pathway [1]. The extrinsic pathway is initiated by the binding of extracellular death ligands (e.g., TNFα, FasL, TRAIL) to their corresponding cell-surface death receptors, leading to the assembly of the Death-Inducing Signaling Complex (DISC) and activation of caspase-8 [6] [1]. Conversely, the intrinsic pathway is activated by internal cellular stresses, such as DNA damage or oxidative stress, culminating in mitochondrial outer membrane permeabilization (MOMP), release of cytochrome c, formation of the apoptosome, and activation of caspase-9 [6] [1]. Both pathways converge on the activation of executioner caspases (e.g., caspase-3 and -7), which dismantle the cell [1].
A hallmark of cancer is the ability of malignant cells to evade apoptosis, a resistance often mediated by the Inhibitor of Apoptosis (IAP) proteins [51] [52]. IAPs, such as XIAP, cIAP1, and cIAP2, are frequently overexpressed in human cancers and block cell death by directly inhibiting caspase activity and modulating key survival signaling pathways, notably NF-κB [53] [52]. This review delineates the role of SMAC mimetics, a class of IAP antagonists, in combating this inhibition, with a specific focus on their mechanism at the intersection of the extrinsic and intrinsic apoptotic pathways.
IAP Proteins: Key Regulators of Apoptosis and Survival
IAP proteins are defined by the presence of one to three baculovirus IAP repeat (BIR) domains, which facilitate protein-protein interactions [51]. Their anti-apoptotic functions are multifaceted:
- XIAP is the most potent direct caspase inhibitor, binding to and suppressing the activity of caspase-3, -7, and -9 [52].
- cIAP1 and cIAP2 do not directly inhibit caspases as potently but function as E3 ubiquitin ligases. They are crucial regulators of the canonical and non-canonical NF-κB pathways, promoting cell survival and proliferation [53] [52]. They also form part of the TNF receptor signaling complex, where they modulate cell survival decisions [53].
The endogenous IAP antagonist, SMAC (Second Mitochondria-derived Activator of Caspases), is released from the mitochondria during intrinsic apoptosis [54]. Upon its release, SMAC binds to IAP proteins via its N-terminal AVPI motif, displacing them from caspases and thereby promoting apoptosis [54].
SMAC Mimetics: Mechanism of Action as IAP Antagonists
SMAC mimetics are small molecules designed to replicate the AVPI binding motif of the native SMAC protein [55]. Their primary mechanism is to antagonize IAP proteins, thereby re-sensitizing cancer cells to apoptosis. The pharmacological action is twofold, critically engaging both the intrinsic and extrinsic apoptotic pathways:
Neutralization of XIAP and Promotion of Caspase Activity
By occupying the BIR domains of XIAP, SMAC mimetics sterically hinder XIAP from binding to and inhibiting caspases. This relieves the suppression of both caspase-9 (from the intrinsic pathway) and the executioner caspases-3 and -7, facilitating the apoptotic cascade [56].
Induction of cIAP1/2 Degradation and TNFα-Mediated Cell Death
A more rapid and profound effect of SMAC mimetics is the targeted degradation of cIAP1 and cIAP2 [53]. The binding of a SMAC mimetic induces a conformational change in cIAPs, stimulating their auto-ubiquitination and leading to proteasomal degradation within minutes of treatment [53]. The loss of cIAPs has two major consequences:
- Activation of NF-κB Signaling: Degradation of cIAPs leads to the stabilization of the kinase NIK, activating both the canonical and non-canonical NF-κB pathways [53]. This activation can induce the transcription and secretion of TNFα in sensitive cancer cells.
- Formation of a Pro-Death Complex: In the absence of cIAPs, the TNFα secreted in an autocrine manner binds to TNFR1 and promotes the formation of a complex containing RIPK1 and FADD, which activates caspase-8 [53] [57].
This elegant mechanism reveals that SMAC mimetics, while targeting intracellular inhibitors, ultimately kill cancer cells primarily by activating the extrinsic apoptotic pathway in a TNFα-dependent manner [53] [57]. The following diagram illustrates this key signaling cascade.
Quantitative Profile of Clinical-Stage SMAC Mimetics
Continuous research has yielded several potent SMAC mimetics, which can be monovalent or bivalent, and are currently in various stages of clinical development [55] [56]. The table below summarizes key candidates.
Table 1: Selected SMAC Mimetics in Clinical Development
| Compound Name | Chemical Type | Primary IAP Targets | Key Preclinical/Clinical Findings | Clinical Status |
|---|---|---|---|---|
| Birinapant (TL32711) | Bivalent | cIAP1, XIAP | Suppresses cIAP1 & XIAP; promises antitumor activity as single agent or in combination [54]. | Phase II [54] |
| UC-112 | Novel Monovalent | XIAP, Survivin | Potent single-agent activity; inhibits tumor growth in A375 melanoma xenograft model; overcomes P-gp mediated multidrug resistance [54]. | Preclinical |
| AZD5582 | Not Specified | XIAP, cIAP1 | Potent activator of caspase-3/7 and caspase-9; induces TNFα-dependent apoptosis [54]. | Preclinical/Clinical |
| LCL161 | Not Specified | cIAP1 | Demonstrates efficacy in xenograft models; well-tolerated in clinical trials [56]. | Phase II |
Experimental Protocols for Evaluating SMAC Mimetics
To elucidate the mechanism and efficacy of SMAC mimetics, a suite of standardized experimental methodologies is employed. The workflow below outlines a generalized cascade for validation.
In Vitro Binding and Target Engagement
- Fluorescence Polarization (FP) / Surface Plasmon Resonance (SPR): These assays quantify the binding affinity of SMAC mimetics to purified BIR domains of IAPs (e.g., XIAP-BIR3) [54]. FP involves a fluorescently labeled AVPI peptide competing with the test compound for binding to the IAP protein. A decrease in polarization indicates displacement and successful binding of the mimetic [54].
- Pull-Down Assays: Cell lysates from cancer lines treated with SMAC mimetics are incubated with immobilized mimetics. Bound proteins are eluted and analyzed via immunoblotting to confirm direct engagement with intended targets (XIAP, cIAP1, cIAP2) and associated proteins like TRAF1/2 [53].
Cellular Viability and Apoptosis Mechanisms
- Cell Viability Assays (e.g., MTT, CellTiter-Glo): Cancer cell lines are treated with a dose range of SMAC mimetics, alone or in combination with chemotherapeutics or death receptor ligands. Viability is measured after 48-72 hours to determine IC₅₀ values [57] [54].
- Analysis of cIAP Degradation: Cells are treated with SMAC mimetics for short periods (30 minutes to 4 hours). Lysates are subjected to immunoblotting with antibodies against cIAP1 and cIAP2. Rapid loss of signal confirms the compound's ability to induce proteasomal degradation. This can be blocked by pre-treatment with proteasome inhibitors like MG132 [53].
- Caspase Activation Assays: Caspase activity is measured using fluorogenic substrates specific for caspase-3/7, -8, or -9. Increased fluorescence indicates apoptosis induction. To distinguish pathway dependence, assays are repeated in the presence of specific caspase inhibitors (e.g., Z-VAD-fmk for pan-caspase, crmA for caspase-8) [53] [57].
- TNFα Dependency and Autocrine Signaling:
- TNFα Neutralization: Cells are treated with SMAC mimetics alongside a neutralizing TNFα antibody. Inhibition of cell death confirms the role of autocrine TNFα signaling [53] [57].
- siRNA Knockdown: Knockdown of TNFα, TNFR1, or key signaling components like RIPK1 via siRNA is performed. Resistance to SMAC mimetic-induced death in knocked-down cells validates the specificity of the pathway [53] [57].
In Vivo Efficacy Models
- Xenograft Mouse Models: Immunodeficient mice are implanted with human cancer cells. Once tumors are established, animals are treated with vehicle control, SMAC mimetic, standard chemotherapy, or their combination. Tumor volume is measured regularly, and at endpoint, tumors are harvested for immunohistochemical analysis of IAP levels (e.g., survivin), caspase-3 cleavage, and markers of proliferation [57] [54].
The Scientist's Toolkit: Essential Research Reagents
The following table catalogues critical reagents used in the experimental evaluation of SMAC mimetics.
Table 2: Key Reagents for SMAC Mimetics Research
| Reagent / Tool | Function / Mechanism | Example Usage |
|---|---|---|
| SMAC Mimetics | Antagonize IAPs by mimicking AVPI motif; induce cIAP degradation. | Birinapant, LCL161, UC-112 used as investigational compounds [54] [56]. |
| Proteasome Inhibitor | Blocks proteasomal protein degradation. | MG132 used to confirm SMAC mimetic-induced cIAP degradation is proteasome-dependent [53]. |
| Caspase Inhibitors | Irreversibly inhibits caspase activity to define pathway dependence. | Z-VAD-fmk (pan-caspase); crmA (caspase-8) used to confirm caspase-dependent apoptosis [53]. |
| TNFα Neutralizing Antibody | Binds and neutralizes secreted TNFα. | Validates the role of autocrine TNFα in SMAC mimetic-induced cell death [53] [57]. |
| siRNA / shRNA | Silences expression of specific target genes. | Knockdown of TNFα, TNFR1, or RIPK1 to confirm their essential role in the cell death mechanism [53] [57]. |
| Caspase Activity Assay Kits | Fluorometric or colorimetric detection of caspase cleavage activity. | Quantify activation of caspase-8, -9, and -3/7 following SMAC mimetic treatment [54]. |
Combination Therapies and Overcoming Resistance
The efficacy of SMAC mimetics is significantly enhanced in combination therapies, which help overcome inherent resistance mechanisms in cancer cells.
- With Conventional Chemotherapy: SMAC mimetics synergize with genotoxic agents (e.g., 5-fluorouracil, etoposide) by enhancing an autocrine TNFα feedback loop, shifting the cellular response towards RIP1-dependent extrinsic apoptosis, even when the chemotherapy initially triggers the intrinsic pathway [57].
- With Death Receptor Agonists (e.g., TRAIL): SMAC mimetics sensitize cancer cells to TRAIL-induced apoptosis by simultaneously relieving caspase inhibition (via XIAP neutralization) and promoting caspase-8 activation [56].
- To Overcome Multidrug Resistance (MDR): Some novel SMAC mimetics, such as UC-112, are not substrates for the P-glycoprotein (P-gp) drug efflux pump, allowing them to retain potency in MDR cancer cells where other drugs fail [54].
SMAC mimetics represent a paradigm-shifting class of targeted therapeutics that effectively combat the inhibition of apoptosis by IAP proteins. Their mechanism masterfully exploits the crosstalk between the intrinsic and extrinsic apoptotic pathways, ultimately triggering a TNFα-mediated, caspase-8 dependent suicide signal in cancer cells. While clinical development is ongoing, the strategic combination of SMAC mimetics with conventional therapies holds immense promise for overcoming drug resistance and improving outcomes in cancer treatment. Future research will continue to refine these compounds, identify predictive biomarkers for patient selection, and optimize combination regimens to fully realize their potential in clinical oncology.
Apoptosis, or programmed cell death, is a critical biological process for maintaining cellular homeostasis, and its dysregulation is a hallmark of cancer. The induction of apoptosis in target cancer cells is a primary therapeutic goal for many cancer therapies [58]. Two principal signaling pathways initiate apoptosis: the extrinsic (death receptor) pathway and the intrinsic (mitochondrial) pathway. The extrinsic pathway is activated by external ligands such as tumor necrosis factor (TNF)-α or Fas ligand (FasL) binding to their respective death receptors on the cell surface. This interaction leads to the formation of the death-inducing signaling complex (DISC), which activates initiator caspases-8 and -10, ultimately leading to the activation of effector caspases-3, -6, and -7 and cell death [6] [7]. Conversely, the intrinsic pathway is activated by internal cellular disturbances, including oxidative stress, DNA damage, and mitochondrial damage. This pathway is controlled by the BCL-2 family of proteins and results in mitochondrial outer membrane permeabilization (MOMP), cytochrome c release, formation of the apoptosome, and activation of caspase-9, which then activates the same effector caspases [6] [59].
A critical concept in apoptosis regulation is the classification of cells as Type I or Type II. In Type I cells, sufficient amounts of activated caspase-8 from the DISC can directly activate effector caspase-3, leading to apoptosis without mitochondrial involvement. In Type II cells, the death signal requires amplification through the mitochondrial pathway. Here, caspase-8 cleaves the BH3-only protein BID to generate truncated BID (tBID), which activates BAX and BAK to induce MOMP and cytochrome c release [7]. The reasons for these differences are not fully understood but may involve varying expression levels of inhibitors such as XIAP (X-linked inhibitor of apoptosis protein), which can block caspase activity and necessitate mitochondrial amplification to overcome its inhibition [7]. Understanding which pathway dominates in a patient's tumor, and the associated biomarker profile, is fundamental to developing biomarker-driven strategies for apoptotic therapies.
Key Biomarkers of Apoptotic Pathways
Biomarkers are objectively measured characteristics that indicate normal or pathogenic biological processes or pharmacological responses. Their qualification is a lengthy process requiring retrospective and prospective clinical trials, but they are essential for confirming a drug hits its intended target (proof of mechanism) and achieves the anticipated outcome (proof of concept) [60]. In the context of apoptosis, biomarkers can be proteins, nucleic acids, or other molecules detected in tissues or biological fluids like serum [60]. Ideal biomarkers should be specific, accurately quantifiable, rapid to measure, robust, validatable, and correlate with disease burden and treatment effects [60].
The following tables summarize key biomarkers for the intrinsic and extrinsic apoptotic pathways, along with caspase-independent mediators, and their clinical significance as demonstrated in recent studies.
Table 1: Key Molecular Biomarkers of Apoptotic Pathways
| Biomarker | Pathway | Function | Measurement Method |
|---|---|---|---|
| Caspase-8 [59] | Extrinsic | Initiator caspase; activated by DISC. Cleaves and activates executioner caspases or BID. | IHC (cleaved form), ELISA |
| Caspase-3 [59] | Executioner (Both) | Primary executioner caspase; responsible for DNA fragmentation and cell dismantling. | IHC (cleaved form), ELISA |
| BID [7] | Crosstalk | BH3-only protein; cleaved by caspase-8 to tBID, which links extrinsic to intrinsic pathway. | IHC, Western Blot |
| BCL-2 [59] | Intrinsic | Anti-apoptotic protein; prevents cytochrome c release by controlling BAX/BAK. | IHC, ELISA |
| BAX/BAK [59] | Intrinsic | Pro-apoptotic effectors; oligomerize to cause MOMP and cytochrome c release. | IHC, ELISA |
| Cytochrome c [59] | Intrinsic | Mitochondrial protein; released upon MOMP, forms apoptosome with Apaf-1 to activate caspase-9. | ELISA, IHC |
| AIF1 (AIFM1) [59] | Caspase-Independent | Apoptosis-inducing factor; upon release, translocates to nucleus and induces DNA fragmentation. | IHC, ELISA |
| SMAC/DIABLO [7] | Regulation | Mitochondrial protein; released with cytochrome c, neutralizes IAP inhibition of caspases. | ELISA |
Table 2: Clinical Prognostic Value of Apoptosis-Related Biomarkers in TNBC (2025 Study)
| Biomarker | Prognostic Value in TNBC | Impact on Overall Survival (OS) | Subgroup Analysis |
|---|---|---|---|
| AIF1 (Protein) [59] | Favorable | Significantly enhanced OS (HR=0.40, p=0.0033) | Greater OS benefit in chemotherapy-treated patients (HR=0.36, p=0.0072) |
| AIFM1 (mRNA) [59] | Favorable | Significantly enhanced OS (HR=0.48, p=0.018) | - |
| Caspase-3 (Protein) [59] | Favorable | Significant OS advantage | Particularly in chemotherapy-treated patients |
| BCL-2 (Protein) [59] | Favorable | Significant OS advantage | - |
Experimental Protocols for Biomarker Analysis
Validated experimental protocols are crucial for the accurate detection and quantification of apoptotic biomarkers in clinical and research settings. Below are detailed methodologies for key techniques cited in recent research.
Immunohistochemistry (IHC) for Protein Biomarkers in TNBC
A 2025 study on triple-negative breast cancer (TNBC) utilized IHC to quantify the expression of apoptosis-related proteins in a clinical cohort of 103 cases [59].
Protocol:
- Tissue Preparation: Formalin-fixed, paraffin-embedded (FFPE) primary TNBC tissue sections are cut to 4-5 μm thickness.
- Deparaffinization and Rehydration: Sections are deparaffinized in xylene and rehydrated through a graded series of ethanol to water.
- Antigen Retrieval: Heat-induced epitope retrieval (HIER) is performed using a citrate-based or EDTA-based buffer (pH 6.0 or 9.0) in a pressure cooker or water bath to unmask target epitopes.
- Endogenous Peroxidase Blocking: Incubate sections with 3% hydrogen peroxide to quench endogenous peroxidase activity.
- Blocking: Apply a protein block (e.g., normal serum or BSA) to reduce non-specific antibody binding.
- Primary Antibody Incubation: Incubate sections with validated primary antibodies against the target proteins (e.g., AIF1, cleaved-caspase-3, BCL-2) at optimized dilutions and conditions (overnight at 4°C). Include positive and negative controls.
- Secondary Antibody Incubation: Apply a biotinylated secondary antibody followed by streptavidin-horseradish peroxidase (HRP) complex. Alternatively, use a polymer-based HRP system.
- Chromogen Development: Visualize antibody binding using 3,3'-Diaminobenzidine (DAB) as a chromogen, which produces a brown precipitate.
- Counterstaining and Mounting: Counterstain with hematoxylin to visualize nuclei, dehydrate, clear, and mount with a permanent mounting medium.
- Scoring and Analysis: Two independent pathologists, blinded to clinical data, score the stained tissues. For cytoplasmic markers like AIF1 and cleaved-caspase-3, an immunoreactive score (e.g., H-score) that considers both staining intensity (0-3) and the percentage of positive tumor cells is used. Data are then correlated with clinical outcomes such as overall survival (OS) and recurrence-free survival (RFS) using statistical methods like Kaplan-Meier analysis [59].
Real-Time Live-Cell Imaging for Discriminating Apoptosis and Necrosis
This sensitive confirmatory assay allows for the real-time discrimination of apoptosis and necrosis at a single-cell level using genetically encoded fluorescent probes [58].
Protocol:
- Cell Line Engineering:
- Generate a stable cell line (e.g., U251 neuroblastoma) expressing two fluorescent probes:
- A FRET-based caspase sensor (e.g., ECFP-DEVD-EYFP), where ECFP (donor) and EYFP (acceptor) are linked by a caspase-3/7 cleavage sequence (DEVD). Upon caspase activation, cleavage separates the fluorophores, resulting in a loss of FRET, measurable as a change in the ECFP/EYFP emission ratio.
- A non-soluble fluorescent protein targeted to an organelle, such as DsRed localized to the mitochondria (Mito-DsRed), which serves as a marker for cell integrity.
- Generate a stable cell line (e.g., U251 neuroblastoma) expressing two fluorescent probes:
- Experimental Setup:
- Plate the stable cells on glass-bottom dishes suitable for live-cell imaging.
- Treat cells with the agent of interest (e.g., chemotherapeutic drugs like doxorubicin, or necrosis inducers like H₂O₂).
- Mount the dish on a live-cell imaging system (wide-field, confocal, or high-throughput imager) with environmental control (37°C, 5% CO₂).
- Image Acquisition:
- Acquire time-lapse images at regular intervals (e.g., every 15-45 minutes) over 24-48 hours.
- For each time point, capture images for ECFP, EYFP, and Mito-DsRed fluorescence channels.
- Data Analysis and Cell Fate Determination:
- Live Cells: Retain both the FRET probe (no ratio change) and Mito-DsRed fluorescence.
- Apoptotic Cells: Exhibit a loss of FRET (increase in ECFP/EYFP ratio) indicating caspase activation, while retaining Mito-DsRed fluorescence.
- Necrotic Cells: Lose the soluble FRET probe due to membrane permeabilization (no ECFP/EYFP fluorescence) but retain the organelle-targeted Mito-DsRed fluorescence for a prolonged period. This occurs without a preceding FRET ratio change in primary necrosis, or after caspase activation in secondary necrosis [58].
Visualization of Apoptotic Signaling Pathways
Diagram 1: Extrinsic vs. Intrinsic Apoptosis Pathways
Diagram 2: Experimental Workflow for Apoptosis vs. Necrosis
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Apoptosis Biomarker Analysis
| Reagent / Assay | Function / Target | Application in Research |
|---|---|---|
| Phosphatidylserine (PS) Detection (Annexin V) [60] | Binds to externalized PS on the outer leaflet of the plasma membrane, an early apoptosis marker. | Flow Cytometry, Fluorescence Microscopy. Often used with viability dyes (e.g., PI) to distinguish early apoptosis from necrosis. |
| Caspase Activity Assays [60] | Detect activated caspases using fluorogenic or chromogenic substrates (e.g., DEVD-pNA). | ELISA, Flow Cytometry, Spectrophotometry. Provides proof of mechanism for caspase-inducing therapies. |
| BCL-2 Family Antibodies [6] [59] | Detect pro- and anti-apoptotic proteins (e.g., BCL-2, BAX, BID) in tissue or cell lysates. | Immunohistochemistry (IHC), Western Blot, ELISA. Used for prognostic stratification and monitoring target engagement. |
| Mitochondrial Membrane Potential Dyes (JC-1, TMRM) | Detect loss of mitochondrial membrane potential (ΔΨm), an early event in the intrinsic pathway. | Flow Cytometry, Fluorescence Microscopy. Indicator of mitochondrial integrity and early apoptosis. |
| FRET-Based Caspase Biosensor [58] | Genetically encoded probe (e.g., ECFP-DEVD-EYFP) that loses FRET upon caspase cleavage. | Real-Time Live-Cell Imaging, HTS. Enables kinetic, single-cell analysis of caspase activation and discrimination from necrosis. |
| ELISA for Soluble Biomarkers [60] | Quantify proteins released during apoptosis (e.g., cytochrome c, nucleosomal DNA, CK18) in serum/plasma. | Serological Analysis. Allows for minimally invasive, serial monitoring of treatment response and disease burden. |
| ApoEV Isolation & Analysis Kits [61] | Isolate and characterize apoptotic extracellular vesicles (ApoEVs) based on size and surface markers (e.g., PS). | Flow Cytometry, Proteomics, Genomic Analysis. Emerging tool for non-invasive biomarker discovery and liquid biopsy. |
Navigating Resistance and Pathway Plasticity in Apoptotic Therapies
Common Mechanisms of Therapy Resistance in Cancer Cells
Therapy resistance represents a fundamental challenge in clinical oncology, directly leading to treatment failure in a majority of patients with advanced disease. This whitepaper synthesizes current mechanistic understanding of resistance, framing it within the critical interplay between the Death Receptor (extrinsic) and Mitochondrial (intrinsic) apoptotic pathways. We delineate how genetic, epigenetic, and metabolic adaptations converge to disable apoptotic signaling, enabling cancer cell survival. The analysis extends to emerging concepts of mitochondrial plasticity, cancer stem cells (CSCs), and integrated cell death pathways like PANoptosis. A detailed examination of experimental methodologies provides a resource for interrogating these mechanisms, while advanced therapeutic strategies targeting apoptotic pathways are evaluated for their potential to overcome resistance and improve patient outcomes.
Cancer drug resistance is a pervasive clinical obstacle, accounting for up to 90% of chemotherapy failures [62] [63]. This resistance manifests as either intrinsic (pre-existing) or acquired (developed during treatment), ultimately leading to disease recurrence and progression [62]. The efficacy of most anticancer therapies, including chemotherapy, radiation, and targeted agents, depends on ultimately triggering apoptosis in malignant cells [45]. Consequently, a tumor's ability to evade programmed cell death is a cornerstone of therapeutic resistance.
The two primary apoptotic pathways—the extrinsic (Death Receptor) pathway and the intrinsic (Mitochondrial) pathway—serve as critical arbiters of cell fate under therapeutic stress [45] [10]. The delicate balance between these pathways is frequently dysregulated in cancer, with tumors exploiting a myriad of mechanisms to suppress apoptosis. This whitepaper dissects the molecular foundations of resistance rooted in the failure of these pathways, exploring the interplay between genetic mutations, metabolic reprogramming, and microenvironmental adaptation. By framing resistance through the lens of apoptotic signaling, we aim to provide a structured framework for developing novel, effective therapeutic interventions.
Molecular Mechanisms of Resistance in Apoptotic Pathways
Resistance via the Death Receptor Pathway
The extrinsic apoptotic pathway initiates when extracellular ligands such as Fas-L (CD95-L), TRAIL, or TNF-α bind to their cognate death receptors (e.g., CD95, DR4/DR5, TNFR1) [10] [64]. Ligand binding induces receptor trimerization and recruitment of the adaptor protein FADD (Fas-Associated Death Domain protein), which then recruits and activates procaspase-8 to form the Death-Inducing Signaling Complex (DISC) [10]. Active caspase-8 then propagates the death signal by directly cleaving and activating executioner caspases-3, -6, and -7 [64].
Key Resistance Mechanisms:
- Inhibitory Protein Expression: A primary mechanism of resistance is the overexpression of regulatory proteins like c-FLIP (FLICE-like inhibitory protein) [10]. c-FLIP bears structural homology to caspase-8 but lacks catalytic activity. By binding to FADD within the DISC, it competitively inhibits caspase-8 recruitment and activation, effectively shutting down the extrinsic trigger [10] [64].
- Receptor Downregulation: Many tumors exhibit reduced surface expression of death receptors like CD95 or TRAIL receptors, limiting the initiation of the death signal [10].
- DISC Modulation: Some herpes viruses and mammalian cells express short isoforms of FLIP (FLIPS and v-FLIP) that potently inhibit caspase-8 filament formation and activation within the DISC [10].
Table 1: Key Resistance Mechanisms in the Death Receptor Pathway
| Mechanism | Molecular Component | Functional Consequence |
|---|---|---|
| DISC Inhibition | c-FLIP overexpression | Competes with procaspase-8 binding to FADD, preventing initiation of the caspase cascade [10]. |
| Reduced Signal Initiation | Death Receptor downregulation (e.g., CD95, DR5) | Limits the cell's ability to receive and transduce external pro-apoptotic signals [10]. |
| Caspase-8 Mutation | Inactivating mutations in CASP8 gene | Directly ablates the core enzymatic activator of the extrinsic pathway [10]. |
Resistance via the Mitochondrial Pathway
The intrinsic pathway is regulated by the Bcl-2 family of proteins and integrates diverse intracellular stress signals, including DNA damage, oxidative stress, and oncogene activation [45] [65]. The pivotal event is Mitochondrial Outer Membrane Permeabilization (MOMP), controlled by the balance between pro-apoptotic (e.g., Bax, Bak, Bid) and anti-apoptotic (e.g., Bcl-2, Bcl-xL, Mcl-1) proteins [45] [66]. MOMP leads to cytochrome c release, apoptosome formation with Apaf-1 and caspase-9, and subsequent activation of executioner caspases [45].
Key Resistance Mechanisms:
- Anti-apoptotic Protein Overexpression: Many cancers overexpress anti-apoptotic Bcl-2 family proteins like Bcl-2 and Bcl-xL, which sequester pro-apoptotic proteins like Bax and Bak, preventing MOMP and cytochrome c release [45] [65]. This is a common mechanism of resistance to chemotherapeutics that act through the intrinsic pathway.
- Impaired Balance of Bcl-2 Proteins: Dysregulation of BH3-only proteins, which act as sentinels for cellular stress, can prevent the activation of Bax/Bak, making cells insensitive to internal damage signals [45].
- Altered Mitochondrial Metabolism: Cancer stem cells (CSCs) and resistant cells often undergo metabolic reprogramming, enhancing oxidative phosphorylation (OXPHOS) and mitochondrial fitness to resist stress-induced apoptosis [67] [65]. They also upregulate mitochondrial quality control mechanisms like mitophagy to remove damaged mitochondria and maintain survival [67].
Table 2: Key Resistance Mechanisms in the Mitochondrial Pathway
| Mechanism | Molecular Component | Functional Consequence |
|---|---|---|
| Blocked MOMP | Bcl-2/Bcl-xL overexpression | Sequesters pro-apoptotic effectors (Bax, Bak) and BH3-only proteins, preventing pore formation in the OMM [45] [65]. |
| Metabolic Reprogramming | Enhanced OXPHOS, Mitophagy | Increases energy production and removes damaged mitochondria, allowing cells to withstand metabolic and therapeutic stress [67] [65]. |
| Reduced Pro-apoptotic Signal | p53 mutation, Loss of Bax/Bak | Ablates critical sensors and effectors required for initiating the mitochondrial pathway in response to damage [45]. |
Convergent and Integrated Resistance Mechanisms
Cancer cells deploy multifaceted defenses that simultaneously impair both apoptotic pathways and involve broader adaptive processes.
- Caspase Inhibition: Executioner caspases represent a convergent point for both pathways. Overexpression of Inhibitor of Apoptosis Proteins (IAPs), particularly XIAP, directly binds and inhibits caspases-3, -7, and -9, blocking the final execution phase of apoptosis [45] [64].
- Cancer Stem Cells (CSCs): CSCs are a subpopulation with enhanced resistance capabilities. They exhibit upregulated DNA repair, metabolic plasticity, and increased expression of drug efflux pumps like P-glycoprotein, which reduces intracellular drug concentrations [67] [63].
- Enhanced DNA Repair: Resistance to DNA-damaging agents is frequently mediated by enhanced DNA repair pathways. For instance, upregulation of Homologous Recombination Repair (HRR) genes, including BRCA1/2 reversion mutations, and increased Nucleotide Excision Repair (NER) activity via ERCC1 overexpression are common in resistant tumors [63].
- Tumor Microenvironment (TME): The TME contributes physically and biochemically. Dense fibrotic stroma in cancers like pancreatic ductal adenocarcinoma can form a physical barrier to drug delivery [62]. Interactions with cancer-associated fibroblasts (CAFs) and immune cells within the TME provide pro-survival signals that counteract therapy-induced apoptosis [62] [63].
Advanced Concepts and Emerging Mechanisms
PANoptosis and Mitochondrial Orchestration
PANoptosis is a novel, unified concept of inflammatory programmed cell death that integrates components from apoptosis, necroptosis, and pyroptosis [66]. Mitochondria serve as the central hub for this pathway, coordinating crosstalk between these different death modalities. Key integrative events include:
- MOMP as a Shared Node: MOMP releases not only cytochrome c (for apoptosis) but also mitochondrial DNA (mtDNA) and ROS, which can activate inflammasomes and amplify pyroptotic and necroptotic signaling [66].
- Signal Convergence: Proteins like caspase-8, a key initiator of apoptosis, also play regulatory roles in suppressing necroptosis, demonstrating the intricate interplay within the PANoptosis network [66]. Therapeutic evasion can therefore occur through dysregulation of this broader death network, not just classical apoptosis.
Metabolic Adaptation and a Novel Cell Death Pathway
Metabolic stress and innate immune activation can combine to trigger a newly discovered cell death pathway named mitoxyperilysis [68]. This pathway is characterized by:
- Mitochondrial Perilysis: Damaged mitochondria persistently localize near the plasma membrane, where they produce reactive oxygen species (ROS) that cause oxidative damage and eventual membrane rupture [68].
- Therapeutic Potential: In mouse models, combining innate immune activation (via a bacterial component) with fasting (metabolic stress) synergistically induced mitoxyperilysis, leading to significant tumor regression, whereas neither stimulus alone was effective [68]. This discovery highlights how combination therapies targeting multiple stress pathways can overcome resistance by engaging non-canonical cell death mechanisms.
Experimental Toolkit for Investigating Resistance
Core Methodologies and Workflows
A multi-faceted approach is essential for dissecting the complex mechanisms of therapy resistance.
1. Profiling Apoptotic Competence
- Method: Immunoblotting and Immunohistochemistry to assess the protein expression levels of key apoptotic regulators (e.g., Bcl-2, Bcl-xL, Bax, Bak, c-FLIP, procaspases, cleaved caspases) in treated vs. untreated resistant and sensitive cell lines or patient samples [45] [64].
- Workflow: Cells/tissues -> Protein extraction -> SDS-PAGE -> Transfer to membrane -> Blocking -> Incubation with primary antibodies (e.g., anti-Bcl-2, anti-cleaved Caspase-3) -> Incubation with HRP-conjugated secondary antibodies -> Chemiluminescent detection [64].
2. Functional Assessment of Mitochondrial Integrity
- Method: Flow Cytometry using fluorescent probes to measure critical parameters of mitochondrial health, including:
- Mitochondrial Membrane Potential (ΔΨm): Using JC-1 or TMRM dyes. Collapse of ΔΨm is an early event in intrinsic apoptosis.
- Mitochondrial ROS Production: Using MitoSOX Red.
- MOMP and Cytochrome c Release: By combining mitochondrial staining with immunofluorescence for cytochrome c localization [45] [65].
- Workflow: Culture cells -> Induce treatment -> Harvest and stain with fluorescent probes -> Analyze by flow cytometry or confocal microscopy [65].
3. Validating Death Receptor Pathway Function
- Method: DISC Immunoprecipitation and Caspase-8 Activation Assay. This methodology directly probes the initiation of the extrinsic pathway [10].
- Workflow: Treat cells with a death receptor ligand (e.g., TRAIL, Fas-L) -> Lyse cells -> Immunoprecipitate the death receptor complex (DISC) using an antibody against the receptor (e.g., anti-Fas) -> Analyze the immunoprecipitate by Western blot for co-precipitated FADD, caspase-8, and c-FLIP. Caspase-8 activity can be measured separately using fluorogenic substrates [10].
4. Inducing and Quantifying a Novel Cell Death Pathway (Mitoxyperilysis)
- Method: Combined Metabolic and Immune Stress Protocol, followed by microscopy and viability assays [68].
- Workflow:
- Step 1 - Metabolic Stress: Subject cancer cells to nutrient deprivation (e.g., using low-glucose media or fasting in vivo) or treat with an mTOR inhibitor [68].
- Step 2 - Innate Immune Activation: Co-treat with a Pathogen-Associated Molecular Pattern (PAMP), such as a bacterial lipopolysaccharide (LPS), to activate innate immune signaling [68].
- Step 3 - Visualization: Use live-cell microscopy with MitoTracker (for mitochondria) and membrane-impermeant dyes (e.g., Propidium Iodide) to visualize mitochondrial positioning at the plasma membrane and subsequent cell lysis [68].
- Step 4 - Viability Assay: Quantify cell death using assays like LDH release, which measures plasma membrane integrity [68].
The following diagram illustrates the logical workflow for designing experiments to probe resistant mechanisms, incorporating classical and emerging pathways:
Essential Research Reagents
Table 3: Key Reagents for Investigating Therapy Resistance
| Reagent / Tool | Category | Primary Function in Research |
|---|---|---|
| Recombinant TRAIL/Fas-L | Death Receptor Ligand | To directly activate the extrinsic apoptotic pathway and assess cellular sensitivity and DISC formation [10]. |
| ABT-263 (Navitoclax) | BH3 Mimetic / Small Molecule Inhibitor | Binds and inhibits Bcl-2, Bcl-xL, and Bcl-w, disrupting their anti-apoptotic function and promoting MOMP; used to probe mitochondrial priming [45] [65]. |
| JC-1 Dye | Fluorescent Probe | A potentiometric dye that aggregates in healthy mitochondria (red fluorescence) and remains monomeric in depolarized mitochondria (green fluorescence), allowing ratiometric measurement of ΔΨm by flow cytometry [65]. |
| mTOR Inhibitors (e.g., Rapamycin) | Small Molecule Inhibitor | Used to model nutrient stress and inhibit anabolic signaling; critical for studying metabolic stress-induced death pathways like mitoxyperilysis [68]. |
| c-FLIP Inhibitors / siRNA | Genetic & Pharmacological Tool | To knock down or inhibit the c-FLIP protein, thereby relieving its inhibition on the DISC and sensitizing cells to Death Receptor-mediated apoptosis [10] [64]. |
| Z-VAD-FMK | Pan-Caspase Inhibitor | An irreversible broad-spectrum caspase inhibitor. Used to determine whether cell death is caspase-dependent (apoptosis) or caspase-independent [66]. |
Therapeutic Strategies to Overcome Resistance
The intricate understanding of resistance mechanisms has catalyzed the development of targeted therapeutic strategies.
- BH3 Mimetics: Drugs like venetoclax (ABT-199, Bcl-2 selective) directly target and inhibit anti-apoptotic Bcl-2 proteins, displacing pro-apoptotic partners to trigger MOMP and apoptosis. They are particularly effective in hematological malignancies and are being explored in solid tumors [45] [65].
- Death Receptor Agonists: Recombinant TRAIL and agonistic antibodies against DR4/DR5 are designed to selectively activate the extrinsic pathway in cancer cells. Clinical success has been limited by inherent resistance, often mediated by high c-FLIP levels, necessitating combination therapies [10].
- SMAC Mimetics: These small molecules antagonize IAPs like XIAP, relieving the inhibition on caspases and promoting apoptosis. They can synergize with other agents, including TRAIL receptor agonists and conventional chemotherapy [45].
- Combination with Nanotechnology: Nanoparticles can overcome pharmacokinetic limitations of natural compounds (phytochemicals) and targeted agents. They enhance tumor-specific delivery, improve solubility, minimize side effects, and can be engineered to co-deliver multiple drugs to attack several resistance pathways simultaneously [45] [64].
- Targeting Mitochondrial Metabolism: Inhibitors of oxidative phosphorylation (e.g., IACS-010759) or mitochondrial translation are being investigated to target the metabolic vulnerabilities of CSCs and OXPHOS-dependent resistant cells [67] [65].
- Exploiting Integrated Cell Death: Engaging multiple nodes of the PANoptosis network or combining innate immune activators with metabolic stressors (as in mitoxyperilysis) represents a frontier for overcoming resistance by making cell death inescapable [66] [68].
The evasion of apoptosis through the dysregulation of the Death Receptor and Mitochondrial pathways is a hallmark of therapy-resistant cancer. The molecular mechanisms are complex, spanning from genetic mutations and inhibitory protein expression to metabolic adaptation and ecological niche remodeling. Moving forward, overcoming this challenge requires a multifaceted strategy: the continued development of targeted agents like BH3 mimetics and SMAC mimetics; the intelligent use of combination therapies that attack both apoptosis pathways and parallel survival signals; and the clinical translation of emerging concepts such as PANoptosis and metabolic stress-induced death. By systematically targeting the vulnerabilities within the cell death signaling network, the next generation of cancer therapies holds the promise of restoring apoptosis and defeating drug resistance.
The therapeutic targeting of apoptosis, a form of programmed cell death, represents a cornerstone in the modern oncological arsenal. Cell death occurs primarily through two major pathways: the extrinsic (death receptor) pathway, initiated by extracellular ligands binding to cell surface death receptors, and the intrinsic (mitochondrial) pathway, activated by internal cellular stress signals and governed by the BCL-2 protein family at the mitochondrial membrane [6] [51]. While these pathways are distinct in their initiation, they converge on the activation of executioner caspases that dismantle the cell. A critical point of crosstalk is the caspase-8-mediated cleavage of the pro-apoptotic protein BID to its active form, tBID, which amplifies the death signal by engaging the mitochondrial pathway [5].
Despite the logical appeal of inducing apoptosis in cancer cells, the clinical success of single-agent therapies has been limited by the pervasive development of resistance. Tumor cells exploit a multitude of mechanisms to evade cell death, including overexpression of anti-apoptotic proteins (e.g., BCL-2, MCL-1, XIAP), mutations in death receptors or caspases, and impaired death-inducing signaling complex (DISC) formation [51]. Overcoming this resistance requires a sophisticated, rational approach that leverages our deepening understanding of apoptotic signaling networks. This guide details the current strategies—encompassing second-generation targeted agents and rationally designed combinations—that are being deployed to reinstate effective cell death in resistant malignancies, with a specific focus on the interplay between the death receptor and mitochondrial pathways.
Resistance Mechanisms in Apoptosis Pathways
Resistance in the Death Receptor Pathway
The extrinsic pathway is triggered by ligands such as TRAIL (TNF-related apoptosis-inducing ligand) binding to death receptors DR4 and DR5. Resistance arises through multiple mechanisms:
- Downregulation of Death Receptors: Many cancer cells, including colorectal cancer cells, exhibit decreased DR4/5 expression or function due to defective p53, epigenetic changes, or mutations [51].
- Decoy Receptor Overexpression: Tumor cells can overexpress non-signaling decoy receptors (DcR1/2) that compete with DR4/5 for TRAIL binding, effectively sequestering the ligand and preventing death signaling [51].
- DISC Inhibition: High levels of the cellular FLICE-inhibitory protein (c-FLIP) can compete with caspase-8 for binding to FADD at the DISC, preventing the initiation of the caspase cascade [51].
Resistance in the Mitochondrial Pathway
The intrinsic pathway is critically regulated by the balance between pro- and anti-apoptotic members of the BCL-2 family. Key resistance mechanisms include:
- Overexpression of Anti-Apoptotic BCL-2 Proteins: Malignancies often overexpress proteins like BCL-2, BCL-XL, and MCL-1, which sequester pro-apoptotic activators (like BIM) and effectors (BAX/BAK), preventing mitochondrial outer membrane permeabilization (MOMP) [16] [51].
- Impaired Pro-Apoptotic Signaling: Loss of function in apoptotic effectors like BAX and BAK, or insufficient release of cytochrome c, can halt the apoptotic process [51].
- Upregulation of Inhibitor of Apoptosis Proteins (IAPs): Proteins like XIAP directly bind and inhibit caspases-9, -3, and -7, effectively blocking the execution phase of apoptosis [51].
Second-Generation Therapeutic Agents
The limitations of first-generation apoptosis-inducing therapies have driven the development of more potent, selective, and stable second-generation agents.
Table 1: Second-Generation Agents Targeting Apoptosis Pathways
| Agent Name | Target | Mechanism of Action | Key Advancement / Feature | Development Status / Indication |
|---|---|---|---|---|
| Sonrotoclax & Lisaftoclax [16] | BCL-2 | BH3-mimetic; inhibits BCL-2 | Next-generation BCL-2 inhibitors with improved profiles; chemically similar to venetoclax. | Under clinical evaluation |
| TLY012 [51] | DR4/5 | PEGylated recombinant human TRAIL (rhTRAIL) | Prolonged half-life (12-18 hours); marked activity against fibrotic cells. | Orphan drug designation for systemic sclerosis (2019) |
| Eftozanermin alfa (ABBV-621) [51] | DR4/5 | TRAIL receptor agonist fusion protein | Engineered to induce higher-order clustering of DR trimers for a stronger apoptotic signal. | In clinical studies |
| ONC201 [51] | DR5-inducer | Small molecule inducer of DR5 and integrated stress response | Can overcome TRAIL resistance; shows synergy with TLY012 in pancreatic cancer models. | In clinical studies |
| KT-253 [69] | MDM2 | Novel MDM2 degrader (Homo-PROTAC) | Promotes degradation of MDM2; effective regardless of p53 status. | Orphan drug designation for Acute Myeloid Leukemia (AML) |
| AOH1996 [69] | PCNA | First-in-class, small-molecule PCNA inhibitor | Selectively targets cancer-associated PCNA isoforms; disrupts DNA replication/repair. | Preclinical/Early Clinical |
Advanced BH3-Mimetics and Protein Degraders
The success of the first-generation BCL-2 inhibitor venetoclax validated BH3-mimetics as a powerful therapeutic class [16] [51]. However, resistance can emerge through upregulation of other anti-apoptotic proteins like MCL-1 or BCL-XL. Second-generation BH3-mimetics such as sonrotoclax and lisaftoclax are under active investigation to refine efficacy and safety [16]. Beyond inhibition, novel strategies like proteolysis targeting chimeras (PROTACs) are being employed to eliminate, rather than just inhibit, target proteins. KT-253, an MDM2 degrader, represents this innovative approach. By degrading MDM2, it potently reactivates the p53 tumor suppressor pathway, a key upstream regulator of the intrinsic pathway and the potent pro-apoptotic protein PUMA [70] [69].
Enhanced Death Receptor Agonists
First-generation TRAIL receptor agonists suffered from poor pharmacokinetics and an inability to efficiently cluster receptors. TLY012, a PEGylated version of rhTRAIL, addresses the short half-life of earlier compounds, demonstrating a significantly prolonged circulation time and enhanced anti-tumor activity in preclinical models [51]. Another agent, eftozanermin alfa (ABBV-621), is a engineered fusion protein designed to overcome the weak signaling of earlier agonists by inducing high-order clustering of DR4 and DR5, thereby delivering a more potent apoptotic trigger [51].
Rational Combination Therapies
Monotheracies often fail due to the redundancy and crosstalk within cell death pathways. Rational combinations are essential to overcome resistance.
Table 2: Rational Combination Therapy Strategies to Overcome Resistance
| Combination Strategy | Mechanistic Rationale | Example Agents | Targeted Resistance Mechanism |
|---|---|---|---|
| BH3-mimetic + BCL-XL/MCL-1 inhibitor | Block multiple anti-apoptotic proteins simultaneously to prevent compensatory upregulation. | Venetoclax + MCL-1 inhibitor | Upregulation of MCL-1 or BCL-XL following BCL-2 inhibition. |
| DR Agonist + IAP Antagonist | DR agonist activates extrinsic pathway; IAP antagonist blocks XIAP to relieve caspase inhibition. | Eftozanermin alfa + SMAC mimetic | High IAP expression that blocks caspase activity downstream of death receptors. |
| DR Agonist + BH3-mimetic | DR agonist initiates caspase-8 activation; BH3-mimetic lowers mitochondrial threshold for apoptosis, favoring type II apoptosis. | TLY012 + Venetoclax | Low efficiency of BID cleavage and MOMP in type II cells. |
| Apoptosis Inducer + Immune Checkpoint Inhibitor | Apoptotic cell death can enhance tumor immunogenicity and T-cell infiltration. | TLY012 + anti-PD-1 | Immunosuppressive tumor microenvironment. |
| ONC201 + TLY012 | ONC201 upregulates DR5 and stress pathways, sensitizing cells to TRAIL-induced apoptosis. | ONC201 + TLY012 | Overexpression of IAPs (cIAP-1, XIAP, survivin) and cFLIP. |
Vertical Combinations: Targeting Multiple Nodes in a Pathway
- Synergizing Extrinsic and Intrinsic Pathways: In so-called "type II" cells, robust apoptosis requires the caspase-8/BID-mediated amplification of the death signal through the mitochondrial pathway [5]. Combining a death receptor agonist like TLY012 with a BH3-mimetic like venetoclax simultaneously activates the initial extrinsic trigger and lowers the mitochondrial threshold for apoptosis, effectively forcing the cell to commit to death. This combination has shown synergistic apoptosis in pancreatic cancer models, a cancer type notorious for its resistance [51].
- Dual Targeting of Anti-Apoptotic BCL-2 Family: Cancer cells reliant on BCL-2 can shift dependence to MCL-1 upon venetoclax treatment. Co-administering BCL-2 and MCL-1 inhibitors prevents this adaptive resistance, leading to profound and synergistic tumor cell killing in preclinical models [16].
Horizontal Combinations: Targeting Parallel Resistance Pathways
- Counteracting IAP-Mediated Inhibition: Even with effective MOMP, IAPs like XIAP can block caspase activity. Combining a DR agonist or a BH3-mimetic with a SMAC mimetic (which antagonizes IAPs) ensures that the caspase cascade proceeds unimpeded [51].
- Engaging the Immune System: Inducing apoptosis with agents like TLY012 can promote the infiltration of CD8+ T cells into the tumor microenvironment. Combining it with an immune checkpoint inhibitor like anti-PD-1 creates a positive feedback loop, where apoptosis stimulates an anti-tumor immune response that is further amplified by checkpoint blockade [51].
Experimental Protocols for Evaluating Combination Therapies
In Vitro Assessment of Apoptotic Synergy
Objective: To determine the synergistic potential of two agents in inducing apoptosis in a cancer cell line model. Materials:
- Cancer cell line (e.g., pancreatic adenocarcinoma cell line resistant to TRAIL).
- Test agents (e.g., TLY012 and ONC201).
- Annexin V-FITC / Propidium Iodide (PI) Apoptosis Detection Kit (e.g., from Merck or Bio-Rad) [8] [71].
- Flow cytometer.
- Cell culture reagents and equipment.
Methodology:
- Cell Seeding and Treatment: Seed cells in a 96-well plate. After 24 hours, treat with a matrix of concentrations for each drug alone and in combination (e.g., using a fixed-ratio design).
- Incubation and Harvest: Incubate cells for 16-48 hours. Harvest adherent and floating cells by gentle trypsinization and combine.
- Staining: Wash cells with PBS and resuspend in Annexin V binding buffer. Add Annexin V-FITC and PI according to the kit protocol. Incubate for 15 minutes in the dark.
- Flow Cytometry Analysis: Analyze samples on a flow cytometer within 1 hour. Collect a minimum of 10,000 events per sample.
- Data Analysis: Identify cell populations:
- Viable cells: Annexin V-/PI-
- Early Apoptotic: Annexin V+/PI-
- Late Apoptotic/Necrotic: Annexin V+/PI+ Calculate the total apoptosis percentage (Early + Late). Use software (e.g., CompuSyn) to calculate the Combination Index (CI) to quantify synergy (CI < 1 indicates synergy) [51].
Ex Vivo Analysis of Mitochondrial Involvement
Objective: To confirm engagement of the mitochondrial pathway and MOMP in treated cells. Materials:
- Treated cells from Protocol 5.1.
- Antibodies for Western Blot: anti-caspase-8, anti-BID, anti-tBID, anti-cytochrome c, anti-caspase-9, anti-caspase-3, anti-PARP [71].
- Mitochondrial and cytosolic fractionation kit.
- Equipment for Western blotting.
Methodology:
- Cell Fractionation: After treatment, fractionate cells into mitochondrial and cytosolic fractions using a commercial kit.
- Western Blotting: Resolve proteins from both fractions and whole-cell lysates by SDS-PAGE. Transfer to membranes and probe with relevant antibodies.
- Key Readouts:
- BID Cleavage: Appearance of tBID in whole-cell lysates indicates caspase-8 activation.
- Cytochrome c Release: Translocation of cytochrome c from the mitochondrial fraction to the cytosolic fraction confirms MOMP.
- Caspase Cascade: Sequential cleavage/activation of caspase-9, caspase-3, and its substrate PARP confirms execution of apoptosis [5] [51].
Visualization of Signaling Pathways and Experimental Workflows
Apoptosis Signaling and Therapeutic Targeting
High-Throughput Apoptosis Screening Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for Apoptosis Mechanism and Therapeutic Evaluation
| Reagent / Assay Type | Specific Example | Function / Application in Research |
|---|---|---|
| Annexin V Assays | Annexin V-FITC / PI Kit (Merck) [8] | Gold-standard for detecting phosphatidylserine externalization (early apoptosis) and membrane integrity. |
| Caspase Assays | Fluorometric/Luminescent Caspase-3/7, -8, -9 Activity Kits [71] | Quantify activation of initiator and executioner caspases to delineate pathway engagement. |
| Mitochondrial Assays | JC-1, TMRM dyes for membrane potential; Cytochrome c Antibodies [71] | Assess mitochondrial health and confirm MOMP, a key commitment step in intrinsic apoptosis. |
| DNA Fragmentation Assays | TUNEL Assay [71] | Detect late-stage apoptotic cells by labeling DNA strand breaks. |
| Flow Cytometry Panels | Annexin V StarBright Conjugates (Bio-Rad) [71] | Enable multiplexed, high-resolution analysis of apoptotic markers in complex cell populations. |
| Antibodies for Western Blot | Anti-cleaved Caspase-3, -8, -9; anti-PARP; anti-BID/tBID; anti-BCL-2 family proteins [71] | Confirm proteolytic cleavage events and protein expression changes central to apoptotic signaling. |
The challenge of overcoming resistance in apoptosis-targeted cancer therapy is being met with an increasingly sophisticated toolkit. The era of single-agent therapy is giving way to a new paradigm defined by second-generation agents with improved properties and, more importantly, rational combination strategies informed by a deep understanding of the crosstalk and redundancy between the death receptor and mitochondrial pathways. The convergence of novel protein degraders, enhanced biologic agonists, and selective small molecules allows for a multi-pronged attack on the core machinery of cell survival.
Future progress will hinge on several key areas: the development of robust predictive biomarkers to identify patients most likely to benefit from specific combinations; the exploration of non-canonical cell death pathways like necroptosis and ferroptosis, and their integration with apoptotic signaling in phenomena like PANoptosis [66]; and the refinement of drug delivery platforms to mitigate on-target toxicities, particularly for agents targeting BCL-XL and MCL-1 [16]. As our molecular maps of apoptotic resistance become more detailed, so too will our ability to design precision combination therapies that effectively and durable induce cancer cell death.
Cell death pathways are fundamental to maintaining tissue homeostasis, and their dysregulation is a hallmark of cancer. The intricate balance between the death receptor (extrinsic) pathway and the mitochondrial (intrinsic) pathway of apoptosis represents a critical axis in cellular survival decisions. While the death receptor pathway initiates apoptosis through external signal engagement, the mitochondrial pathway responds to internal cellular damage. However, tumor cells exhibit remarkable death pathway plasticity—the ability to shift between different regulated cell death (RCD) mechanisms when a primary pathway is compromised. This plasticity represents a significant therapeutic challenge, as tumors can develop resistance by switching from apoptosis to alternative death mechanisms such as necroptosis and ferroptosis. Understanding the molecular cross-talk and regulatory networks governing these transitions is essential for developing effective cancer therapies that can overcome treatment resistance [72] [73].
The concept of death pathway plasticity extends beyond mere alternative pathway activation. It encompasses a sophisticated cellular adaptation strategy where tumors rewire their death signaling networks in response to therapeutic pressure. This review examines the molecular mechanisms underlying this plasticity, with particular focus on how tumors shift between apoptosis, necroptosis, and ferroptosis. We explore the key regulatory nodes that control these transitions, provide quantitative comparisons of the distinct pathways, and present experimental approaches for investigating these processes in cancer models [6] [72].
Molecular Mechanisms of Core Cell Death Pathways
Apoptosis: The Death Receptor and Mitochondrial Pathways
Apoptosis is a highly regulated process characterized by specific morphological features including cell shrinkage, chromatin condensation, and formation of apoptotic bodies. It is mediated by caspases, a family of cysteine-aspartic proteases that cleave specific cellular targets, leading to orderly cellular dismantling [73].
The extrinsic (death receptor) pathway initiates when death ligands such as Fas ligand (FasL), tumor necrosis factor (TNF), or TNF-related apoptosis-inducing ligand (TRAIL) bind to their corresponding death receptors on the cell surface. This binding triggers formation of the death-inducing signaling complex (DISC), where adapter proteins FADD or TRADD recruit and activate initiator caspase-8. Active caspase-8 then directly cleaves and activates executioner caspases-3 and -7, culminating in apoptosis [6] [73].
The intrinsic (mitochondrial) pathway activates in response to intracellular stressors including DNA damage, oxidative stress, and growth factor deprivation. These signals trigger mitochondrial outer membrane permeabilization (MOMP), controlled by the BCL-2 protein family. The BCL-2 family includes anti-apoptotic members (BCL-2, BCL-XL, MCL-1), pro-apoptotic effectors (BAX, BAK), and BH3-only sensors (BID, BIM, PUMA). MOMP enables cytochrome c release from mitochondria, which then binds APAF-1 to form the apoptosome complex, activating caspase-9 and subsequently executioner caspases [6] [74] [26].
The classification of cells as Type I or Type II reflects the interplay between these pathways. In Type I cells, sufficient caspase-8 activation at the DISC directly triggers executioner caspases. In Type II cells, caspase-8-mediated BID cleavage to tBID is required to amplify the death signal through mitochondrial involvement [7].
Necroptosis: A Backup Death Pathway
Necroptosis represents a caspase-independent, inflammatory form of cell death that can be activated when apoptosis is blocked. Morphologically, it features cell swelling, plasma membrane rupture, and release of damage-associated molecular patterns (DAMPs) that trigger immune responses [75] [73].
The pathway typically initiates through death receptors like TNFR1 when caspase-8 activity is inhibited. Under these conditions, RIPK1 and RIPK3 form a heterodimeric complex via their RHIM domains, leading to RIPK3 autophosphorylation. Activated RIPK3 then phosphorylates MLKL, causing its oligomerization and translocation to the plasma membrane. The pMLKL oligomers form pores that disrupt membrane integrity, leading to cytoplasmic leakage and inflammatory responses [75] [73].
Ferroptosis: An Iron-Dependent Death Mechanism
Ferroptosis is characterized by iron-dependent accumulation of lipid peroxides leading to oxidative membrane damage. This process differs morphologically and biochemically from apoptosis and necroptosis, featuring reduced mitochondrial cristae and outer membrane rupture [76] [72].
The core mechanism involves the glutathione-dependent antioxidant defense system. Depletion of glutathione or inhibition of glutathione peroxidase 4 (GPX4), which normally reduces lipid hydroperoxides, leads to lethal lipid peroxide accumulation. This process is driven by iron metabolism and polyunsaturated fatty acid peroxidation, making it genetically and biochemically distinct from other death forms [76] [72].
Mechanisms of Death Pathway Plasticity
Molecular Switches and Cross-Talk Between Pathways
Death pathway plasticity is governed by intricate molecular cross-talk that enables tumors to adapt to therapeutic pressure. Key regulatory nodes act as molecular switches that determine cell fate decisions between apoptosis, necroptosis, and ferroptosis.
Caspase-8 serves as a critical switch between apoptosis and necroptosis. In conditions of active caspase-8, cells undergo apoptosis, while caspase-8 inhibition redirects death signaling toward necroptosis through RIPK1-RIPK3-MLKL activation [77] [7]. This switch is particularly relevant in development, as demonstrated in RIPK3/Caspase-8 double knockout mice, which show a 12.6% increase in total cell count in the telencephalon compared to wild-type mice, highlighting the coordinated role of these pathways in regulating cell numbers [77].
The BCL-2 family proteins regulate cross-talk between apoptosis and other death mechanisms. Anti-apoptotic BCL-2 members not only control MOMP but also influence metabolic pathways that affect ferroptosis sensitivity. Additionally, cardiolipin/caspase-8/BID platforms at mitochondrial membranes integrate signals from multiple death pathways, demonstrating the organelle's central role in death decision networks [7].
Metabolic reprogramming provides another layer of regulation, with oncometabolites and energy sensors like AMPK and mTOR influencing death pathway selection. For instance, inhibition of glycolysis can sensitize cancer cells to death receptor-mediated apoptosis by activating AMPK and reducing MCL-1 expression [75].
Tumor Microenvironment and Death Pathway Adaptation
The tumor microenvironment (TME) significantly influences death pathway plasticity through various components including cancer-associated fibroblasts, immune cells, extracellular matrix, and metabolic factors. Hypoxic regions within tumors often show increased resistance to apoptosis but may become vulnerable to ferroptosis due to altered iron metabolism and oxidative stress responses [72].
Oxidative stress levels in the TME can determine death pathway activation. Moderate oxidative stress may trigger apoptosis, while severe oxidative damage can induce ferroptosis through overwhelming of cellular antioxidant systems. The presence of extracellular metabolites from apoptotic cells can also influence the metabolic programs of surrounding cells, potentially altering their death pathway preferences [75] [72].
Table 1: Key Molecular Regulators of Death Pathway Plasticity
| Regulator | Primary Function | Pathway Cross-Talk | Therapeutic Targeting |
|---|---|---|---|
| Caspase-8 | Initiator caspase in extrinsic apoptosis | Switches between apoptosis and necroptosis; cleaves BID to engage mitochondrial pathway | Combination therapies with caspase inhibitors |
| BID/tBID | BH3-only protein connecting death receptors to mitochondria | Integrates extrinsic apoptosis with intrinsic pathway; regulated by cardiolipin platform | BH3 mimetics to modulate mitochondrial priming |
| RIPK1/RIPK3 | Serine-threonine kinases in necroptosis signaling | Activated when caspase-8 inhibited; alternative to apoptosis | RIPK1 inhibitors in development |
| GPX4 | Selenoprotein reducing lipid hydroperoxides | Central regulator of ferroptosis; inhibition induces iron-dependent death | GPX4 inhibitors (e.g., RSL3) |
| BCL-2 Family | Controls mitochondrial outer membrane permeabilization | Determines apoptotic threshold; influences metabolic pathways | BH3 mimetics (venetoclax) in clinical use |
| MLKL | Executor of necroptosis through membrane disruption | Phosphorylated by RIPK3; forms membrane pores | MLKL inhibitors in preclinical development |
Quantitative Analysis of Death Pathway Characteristics
Understanding the quantitative differences between cell death pathways provides insights into their distinct biological roles and therapeutic potential.
Table 2: Comparative Analysis of Cell Death Pathways in Cancer
| Characteristic | Apoptosis | Necroptosis | Ferroptosis |
|---|---|---|---|
| Morphological Features | Cell shrinkage, chromatin condensation, apoptotic bodies | Cell swelling, organelle enlargement, membrane rupture | Reduced mitochondrial cristae, outer membrane rupture |
| Key Initiators | Death receptors, DNA damage, growth factor withdrawal | Death receptors with caspase inhibition, TLR signaling | Glutathione depletion, GPX4 inhibition, iron overload |
| Main Executors | Caspases-3/7/8 | pMLKL oligomers | Lipid peroxides |
| Energy Requirement | ATP-dependent | ATP-independent | ATP-independent |
| Immunogenicity | Generally low (immunologically silent) | High (releases DAMPs) | Moderate to high (releases DAMPs) |
| Metabolic Regulators | Glucose metabolism, AMPK/mTOR signaling | Glucose metabolism, ROS | Iron metabolism, glutathione synthesis, lipid metabolism |
| Primary Assays | Caspase activation, phosphatidylserine exposure, DNA fragmentation | MLKL phosphorylation/oligomerization, membrane integrity loss | Lipid peroxidation, glutathione depletion, mitochondrial shrinkage |
Data derived from multiple sources indicate that during development, the percentage of cleaved caspase-3 positive (CC3+) cells increases by approximately 203% from E13 to P4 in the mouse telencephalon, while cells with compromised membrane integrity (Cisplatin+) increase by about 130% over the same period, reflecting the temporal dynamics of different death mechanisms [77].
Experimental Approaches for Studying Death Pathway Plasticity
Methodologies for Pathway Detection and Quantification
Single-cell mass cytometry (CyTOF) enables simultaneous quantification of multiple cell death markers across diverse cell populations. This approach allows researchers to identify distinct cell death populations based on marker combinations: CC3+Cisplatin− cells (early apoptosis), CC3−Cisplatin+ cells (non-apoptotic death), and CC3+Cisplatin+ cells (later-stage apoptotic/non-apoptotic death) [77].
Metabolic profiling approaches help elucidate connections between metabolism and death pathways. Techniques include extracellular flux analysis to measure mitochondrial function, LC-MS-based metabolomics to track metabolic intermediates, and seahorse analysis for real-time monitoring of energy metabolism [75].
Genetic perturbation models are essential for establishing causal relationships. The use of RIPK3/Caspase-8 double knockout mice provides a powerful system for dissecting the contributions of extrinsic apoptosis and necroptosis, circumventing the embryonic lethality of Caspase-8 single knockouts while revealing the coordinated regulation of these pathways [77].
Research Reagent Solutions
Table 3: Essential Research Reagents for Death Pathway Investigation
| Reagent/Category | Specific Examples | Primary Function | Application Context |
|---|---|---|---|
| Caspase Inhibitors | zVAD-fmk, Q-VD-OPh | Pan-caspase inhibitors | Inducing necroptosis by blocking apoptotic pathway |
| BH3 Mimetics | Venetoclax (ABT-199), Navitoclax (ABT-263) | Inhibit anti-apoptotic BCL-2 proteins | Restoring apoptotic sensitivity in cancer cells |
| RIPK1 Inhibitors | Necrostatin-1, GSK'872 | Inhibit RIPK1 kinase activity | Blocking necroptosis initiation |
| Ferroptosis Inducers | Erastin, RSL3, FIN56 | Inhibit system Xc− or GPX4 | Inducing ferroptosis in apoptosis-resistant cells |
| Metabolic Modulators | 2-DG, Oligomycin, Etomoxir | Inhibit glycolysis, OXPHOS, or fatty acid oxidation | Studying metabolic regulation of death pathways |
| Death Receptor Agonists | Recombinant TRAIL, FasL antibodies | Activate death receptor pathway | Studying extrinsic apoptosis initiation |
| Lipid Peroxidation Reporters | C11-BODIPY⁵⁸¹/⁵⁹¹, Liperfluo | Detect lipid peroxidation in live cells | Quantifying ferroptosis activation |
| Viability Dyes | Cisplatin-based dyes, Propidium Iodide | Assess membrane integrity | Distinguishing apoptotic vs. necrotic death |
Signaling Pathway Diagrams
Diagram Title: Death Pathway Cross-Talk and Molecular Switches
This diagram illustrates the molecular interconnections between apoptosis, necroptosis, and ferroptosis pathways. Key switching points include caspase-8, which determines apoptosis versus necroptosis commitment, and metabolic inputs that influence ferroptosis sensitivity. The integration of death receptor and mitochondrial pathways through BID/tBID represents a critical node for signal amplification in Type II cells.
Diagram Title: Experimental Workflow for Death Pathway Plasticity
This workflow outlines a systematic approach for investigating death pathway plasticity in response to therapeutic challenges. The protocol emphasizes parallel assessment of multiple death mechanisms using complementary techniques, enabling researchers to identify pathway switching events that underlie treatment resistance.
The plasticity of cell death pathways represents both a challenge and opportunity for cancer therapy. Understanding how tumors shift between apoptosis, necroptosis, and ferroptosis provides a roadmap for rational combination therapies that can overcome resistance by simultaneously targeting multiple death mechanisms or blocking escape routes.
Therapeutic strategies exploiting death pathway plasticity include: BH3 mimetics to lower the apoptotic threshold; SMAC mimetics to counteract IAP-mediated caspase inhibition; ferroptosis inducers for apoptosis-resistant tumors; and necroptosis promotion in caspase-compromised environments [72] [73]. The emerging concept of "metal death codes"—where specific transition metals dictate death pathway activation—offers additional therapeutic avenues for manipulating cell fate decisions [76].
Future research directions should focus on: biomarker development to predict death pathway dependencies in individual tumors; temporal mapping of death pathway dynamics during treatment; and microenvironmental modulation to create contexts that favor specific death mechanisms. The integration of death pathway modulation with immunotherapy approaches represents a particularly promising frontier, as different death modalities vary in their ability to stimulate anti-tumor immunity [73].
In conclusion, death pathway plasticity underscores the remarkable adaptability of cancer cells, but also reveals novel vulnerabilities. By understanding the molecular switches that govern transitions between apoptosis, necroptosis, and ferroptosis, researchers can develop more effective therapeutic strategies that anticipate and counter resistance mechanisms, ultimately improving outcomes for cancer patients.
The Impact of the Tumor Microenvironment on Apoptotic Signaling
Apoptosis, or programmed cell death, is a fundamental process for maintaining tissue homeostasis by eliminating damaged or unwanted cells, and it represents a primary mechanism through which cytotoxic therapies kill cancer cells [78]. This highly regulated form of cell death proceeds via two principal signaling pathways: the extrinsic (death receptor) pathway and the intrinsic (mitochondrial) pathway [79] [80]. While the core components of these pathways have been extensively characterized, their function cannot be understood in isolation from their physiological context. The Tumor Microenvironment (TME)—comprising stromal cells, immune cells, the extracellular matrix, and signaling molecules—creates a dynamic ecosystem that profoundly influences apoptotic signaling [72]. This review examines how the TME modulates both the death receptor and mitochondrial pathways of apoptosis, exploring the complex interplay that determines cell fate and shapes therapeutic responses in cancer. We integrate current molecular insights with experimental methodologies and emerging therapeutic strategies that target apoptosis within the TME.
Core Apoptotic Signaling Pathways
The Death Receptor Pathway (Extrinsic Apoptosis)
The extrinsic apoptosis pathway is initiated by the binding of death ligands to their cognate cell surface receptors. Key death receptor ligands include TNFα, FAS ligand, and TRAIL, which engage TNFR1, FAS, and DR4/DR5 receptors, respectively [81] [80]. Upon ligand binding, these receptors recruit adapter proteins such as FADD and TRADD, forming a Death-Inducing Signaling Complex (DISC) [81] [78]. The DISC recruits and activates initiator caspase-8 (and in some cases caspase-10), which then cleaves and activates executioner caspases-3, -6, and -7, leading to the proteolytic dismantling of the cell [81] [80]. In some cellular contexts, activated caspase-8 cleaves the pro-apoptotic Bcl-2 family protein BID to its truncated form (tBID), which translocates to mitochondria and amplifies the apoptotic signal through the intrinsic pathway [82].
The Mitochondrial Pathway (Intrinsic Apoptosis)
The intrinsic apoptosis pathway is triggered by intracellular stress signals, including DNA damage, oxidative stress, and oncogene activation [79] [80]. These stresses tip the balance of Bcl-2 family proteins in favor of pro-apoptotic members. The executioners BAX and BAK undergo activation and oligomerization, leading to Mitochondrial Outer Membrane Permeabilization (MOMP) [79] [80]. MOMP allows the release of mitochondrial intermembrane space proteins into the cytosol, most critically cytochrome c [81]. Cytochrome c, together with APAF1 and procaspase-9, forms the apoptosome, a multi-protein complex that activates caspase-9, which in turn activates the executioner caspases [79] [80]. Other factors released during MOMP, such as SMAC/Diablo and AIF, further promote cell death by counteracting Inhibitor of Apoptosis Proteins (IAPs) or contributing to caspase-independent apoptosis [79] [82].
Table 1: Core Components of Apoptotic Pathways
| Pathway Element | Key Components | Primary Function |
|---|---|---|
| Death Receptors | TNFR1, FAS, DR4, DR5 | Transduce extracellular death signals |
| Adaptor Proteins | FADD, TRADD | Form the DISC complex |
| Initiator Caspases | Caspase-8, Caspase-10 (Extrinsic); Caspase-9 (Intrinsic) | Initiate caspase activation cascade |
| Bcl-2 Family | BAX, BAK (Pro-apoptotic); Bcl-2, Bcl-xL (Anti-apoptotic) | Regulates Mitochondrial Outer Membrane Permeabilization (MOMP) |
| Mitochondrial Factors | Cytochrome c, SMAC/Diablo, AIF | Promote apoptosome formation & inhibit IAPs |
| Executioner Caspases | Caspase-3, Caspase-6, Caspase-7 | Execute apoptotic program via substrate cleavage |
Pathway Crosstalk and Integration
The extrinsic and intrinsic pathways are not isolated but exhibit significant crosstalk, primarily through caspase-8-mediated BID cleavage [82]. The "cross-talk" molecule tBID provides a critical link, allowing death receptor signaling to amplify apoptosis through the mitochondrial pathway, especially in contexts where caspase-8 activation alone is insufficient [80] [82]. This integration is regulated by the cellular context and the TME, which can determine the dominance of one pathway over the other or their synergistic interaction.
The following diagram illustrates the core components and crosstalk between these two pathways:
Diagram 1: Core Apoptotic Signaling Pathways. The extrinsic (yellow) and intrinsic (blue) pathways converge on a common execution phase (red). Crosstalk occurs via tBID (green).
TME-Mediated Regulation of Apoptosis
The TME is not a passive bystander but an active regulator of apoptotic signaling, creating barriers that protect tumor cells from therapy-induced death while also presenting opportunities for novel interventions.
Immune Cells and Soluble Mediators
Immune cells within the TME constitute a double-edged sword in apoptotic regulation. Cytotoxic T lymphocytes and Natural Killer (NK) cells can directly induce apoptosis in tumor cells via the extrinsic pathway by expressing FAS ligand or secreting perforin and granzymes [83] [78]. However, the TME often promotes the recruitment and polarization of immunosuppressive cells. Macrophages polarized to an M2 phenotype and myeloid-derived suppressor cells secrete anti-inflammatory cytokines like IL-10 and TGF-β, which can suppress death receptor expression and promote resistance to apoptosis [82]. Furthermore, the execution of apoptosis itself can shape the immune landscape. For instance, apoptotic cells have been shown to recruit platelets via externalized phosphatidylserine, forming protective clots around circulating tumor cells and paradoxically enhancing metastatic survival [84].
Hypoxia, Metabolic Adaptation, and Oxidative Stress
Hypoxia, a hallmark of most solid tumors, profoundly reprograms cellular metabolism and inhibits apoptosis. It stabilizes HIF-1α, which transcriptionally upregulates anti-apoptotic proteins like Bcl-2 and MCL-1 while simultaneously downregulating pro-apoptotic Bcl-2 family members [72]. This shift in the Bcl-2 family balance raises the threshold for MOMP, rendering cells resistant to intrinsic apoptosis. Hypoxia also alters glucose metabolism, favoring aerobic glycolysis (the Warburg effect), which can suppress mitochondrial apoptosis by maintaining mitochondrial membrane potential and reducing ROS production [79]. However, the TME is metabolically heterogeneous, and nutrient starvation in poorly vascularized regions can also induce autophagy, a process that can either suppress or promote apoptosis depending on context and intensity [80] [82].
Dysregulated Cell Death Crosstalk in the TME
The TME fosters death pathway plasticity, whereby inhibition of one form of regulated cell death (RCD) can lead to the upregulation of another as a compensatory mechanism [81] [72]. For example, when caspase-8 is inhibited—a key node for extrinsic apoptosis—cells may switch to RIPK1/RIPK3/MLKL-dependent necroptosis [81] [82]. Similarly, resistance to apoptosis can be overcome by inducing ferroptosis, an iron-dependent, lipid peroxidation-driven cell death [81] [82]. The specific composition of the TME, including factors like nutrient availability, immune cell infiltration, and cytokine profiles, dictates which alternative death pathways are available, presenting both a challenge for therapy and an opportunity for combinatorial approaches.
Table 2: TME Factors Influencing Apoptotic Signaling
| TME Factor | Impact on Apoptosis | Proposed Mechanism |
|---|---|---|
| Hypoxia | Inhibits Intrinsic Pathway | HIF-1α mediated upregulation of Bcl-2, MCL-1; downregulation of pro-apoptotic proteins |
| M2 Macrophages | Suppresses Extrinsic Pathway | Secretion of IL-10, TGF-β leading to death receptor downregulation |
| Lactate / Acidity | Promotes Survival | Inhibition of caspase activity; activation of autophagy |
| Reactive Oxygen Species (ROS) | Dual Pro-/Anti-apoptotic Role | Low levels promote survival signaling; high levels induce MOMP |
| Cancer-Associated Fibroblasts (CAFs) | Confers Therapy Resistance | Secretion of survival factors; remodeling of extracellular matrix |
Experimental Analysis of Apoptosis in the TME
A Representative Workflow for Investigating Apoptosis
The following diagram outlines a generalized experimental workflow for studying apoptotic signaling and its modulation by the TME, integrating key methodologies discussed across multiple studies [85] [84] [82]:
Diagram 2: Experimental Workflow for Apoptosis Research. A generalized pipeline from model establishment to functional validation.
Detailed Methodologies for Key Assays
Cell Viability and Cytotoxicity (MTT Assay): The MTT assay is a cornerstone for initial screening of therapeutic efficacy. Cells are plated and treated with the agent of interest (e.g., Resveratrol, Temozolomide) across a range of concentrations and time points [85]. Following treatment, MTT reagent (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) is added and incubated, allowing viable cells to reduce it to purple formazan crystals. The crystals are dissolved in DMSO, and the absorbance is measured spectrophotometrically at 490 nm. Viability is calculated as a percentage relative to the vehicle-treated control [85].
Apoptosis Quantification (Annexin V/Propidium Iodide Staining): This flow cytometry-based assay distinguishes between viable, early apoptotic, late apoptotic, and necrotic cells. After treatment, cells are stained with Annexin V AlexaFluor 488, which binds to phosphatidylserine (PS) externalized on the outer leaflet of the plasma membrane in early apoptosis, and Propidium Iodide (PI), which penetrates cells with compromised membrane integrity (late apoptosis/necrosis) [85] [84]. The populations are quantified as follows: Annexin V-/PI- (viable), Annexin V+/PI- (early apoptotic), Annexin V+/PI+ (late apoptotic), and Annexin V-/PI+ (necrotic).
Gene Expression Analysis (RT-qPCR): To investigate molecular mechanisms, RNA is extracted from treated cells and reverse transcribed to cDNA. Quantitative PCR is performed using primers specific for genes of interest (e.g., p21, p27, p53, Bcl-2, Bax). Expression levels are normalized to housekeeping genes (e.g., GAPDH, β-actin), and the relative fold change is calculated using the 2^(-ΔΔCt) method [85].
Synergy Evaluation (Combination Index): For drug combination studies (e.g., Resveratrol + Temozolomide), the nature of the interaction (synergistic, additive, antagonistic) is determined by calculating the Combination Index (CI) using the Chou-Talalay method [85]. The formula is: CI = (D1/Dx1) + (D2/Dx2) where D1 and D2 are the doses of each drug in combination required to achieve a specific effect (e.g., 50% inhibition), and Dx1 and Dx2 are the doses of each drug alone required to achieve the same effect. A CI < 1 indicates synergy, CI = 1 additivity, and CI > 1 antagonism [85].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Apoptosis and TME Research
| Reagent / Assay | Function / Application | Example from Literature |
|---|---|---|
| Temozolomide (TMZ) | DNA alkylating chemotherapeutic agent; induces intrinsic apoptosis. | Used in GBM studies to investigate resistance mechanisms [85]. |
| Resveratrol | Natural polyphenol; can induce cell cycle arrest and apoptosis via p53/p21 pathway. | Shown to upregulate p27 and p53 in GBM cells [85]. |
| Annexin V Kits | Detect phosphatidylserine externalization for early apoptosis identification. | Used with Tali cytometer to quantify apoptosis in U118 GBM cells [85] [84]. |
| Z-VAD-FMK | Pan-caspase inhibitor; used to block apoptotic execution and investigate alternative death pathways. | Used to demonstrate caspase-dependency of cell death [81]. |
| Necrostatin-1 (Nec-1) | RIPK1 inhibitor; specifically blocks necroptosis. | Used to dissect the contribution of necroptosis vs. apoptosis [80] [82]. |
| Erastin | System xc- inhibitor; inducer of ferroptosis. | Used to study ferroptosis as an alternative death pathway when apoptosis is blocked [81] [82]. |
| SMAC Mimetics | Small molecules that antagonize IAPs; sensitize cells to apoptosis. | In clinical development to overcome apoptosis resistance [78] [72]. |
Therapeutic Implications and Future Directions
Targeting Apoptosis by Modulating the TME
The understanding that the TME suppresses apoptotic signaling has led to novel therapeutic strategies aimed at "re-sensitizing" tumor cells to death.
- Overcoming Immune Suppression: Combining immune checkpoint inhibitors (e.g., anti-PD-1/PD-L1) with agents that induce immunogenic cell death (ICD) can convert an immunologically "cold" TME into a "hot" one. ICD inducers, such as certain chemotherapeutics, radiotherapy, and oncolytic viruses, cause dying tumor cells to release damage-associated molecular patterns (DAMPs), which promote dendritic cell maturation and antigen presentation, leading to robust T-cell-mediated killing primarily via the extrinsic pathway [82].
- Directly Targeting Apoptotic Machinery: BH3 mimetics (e.g., Venetoclax) are small molecules that inhibit anti-apoptotic Bcl-2 proteins, thereby lowering the threshold for MOMP and promoting intrinsic apoptosis [72]. These are particularly promising for tumors dependent on specific Bcl-2 family members for survival. Similarly, SMAC mimetics promote apoptosis by antagonizing IAPs, thereby unleashing caspase activity [78] [72].
- Exploiting Death Pathway Plasticity: When tumors develop resistance to apoptosis, inducing alternative, non-apoptotic forms of RCD can be an effective workaround. For instance, ferroptosis inducers (e.g., Erastin, RSL3) or strategies to enhance necroptosis are being explored to eliminate apoptosis-resistant cell populations [81] [82] [72].
Quantitative Modeling and Personalized Medicine
Given the complexity of signaling networks in the TME, mathematical models of intracellular and intercellular signaling are becoming invaluable tools. These models can integrate quantitative data on pathway interactions to generate predictive insights into tumor behavior and response to combination therapies, helping to prioritize the most promising therapeutic strategies for experimental validation [86]. Furthermore, high-throughput sequencing technologies, including single-cell RNA sequencing, are unraveling the heterogeneity of the TME and its impact on cell death pathways. This enables the identification of biomarkers that can predict susceptibility to specific apoptosis-inducing therapies, paving the way for truly personalized treatment regimens [80] [72].
The impact of the Tumor Microenvironment on apoptotic signaling is profound and multifaceted. It modulates both the death receptor and mitochondrial pathways through a complex network of cellular interactions, soluble factors, and metabolic conditions. This regulation often promotes therapy resistance but also reveals a remarkable plasticity in cell death signaling. The future of effective cancer therapy lies in moving beyond a tumor-cell-centric view to embrace a TME-informed perspective. By understanding and targeting the intricate crosstalk between the TME and apoptotic pathways, researchers and clinicians can develop more effective combinatorial strategies that overcome resistance and rewire the TME to favor tumor elimination.
The precise induction of apoptosis in specific cell populations represents a cornerstone of modern therapeutics, particularly in oncology and the treatment of inflammatory diseases. The fundamental challenge in leveraging programmed cell death for clinical benefit lies in the delicate balance between achieving sufficient efficacy against target cells while minimizing off-target toxicity. This balance defines the therapeutic window—the dosage range where a drug is effective without causing unacceptable adverse effects. Central to this challenge are two principal apoptotic signaling cascades: the extrinsic (death receptor) pathway and the intrinsic (mitochondrial) pathway. These pathways exhibit distinct regulatory mechanisms, amplification potential, and crosstalk with other cell death modalities, creating both opportunities and obstacles for therapeutic intervention. A deep understanding of their operational dynamics, points of convergence, and contextual activation is paramount for designing targeted strategies that maximize therapeutic benefit while sparing healthy tissues.
The death receptor pathway initiates at the cell surface through ligand-receptor interactions, typically manifesting rapid activation kinetics, while the mitochondrial pathway integrates intracellular damage signals, serving as an amplification mechanism that often determines commitment to cell death. Emerging research reveals a surprisingly high degree of redundancy and crosstalk between these core apoptotic pathways and other cell death mechanisms, including necroptosis, pyroptosis, and ferroptosis [7] [6]. This intricate network of cell death signaling introduces both challenges and opportunities for therapeutic optimization, as modulation of one pathway may influence others in unexpected ways. Furthermore, the classification of cells as type I or type II based on their reliance on mitochondrial amplification highlights fundamental biological differences that must be considered for effective treatment strategies [7]. This technical guide examines the molecular machinery, experimental approaches, and therapeutic targeting strategies for these two fundamental apoptosis pathways within the critical framework of therapeutic window optimization.
Molecular Mechanisms of Apoptosis Pathways
The Extrinsic (Death Receptor) Pathway
The extrinsic apoptosis pathway initiates when extracellular death ligands, such as Fas ligand (FasL) or Tumor Necrosis Factor (TNF)-related apoptosis-inducing ligand (TRAIL), bind to their cognate death receptors on the cell surface [6]. This interaction induces receptor multimerization and recruitment of intracellular adaptor proteins including FADD/MORT1, which in turn recruits initiator caspases (primarily caspase-8 and caspase-10) to form the Death-Inducing Signaling Complex (DISC) [7]. At the DISC, caspase-8 undergoes dimerization-induced activation through conformational change and autoproteolytic processing [7] [87]. The activated caspase-8 is then released into the cytosol where it directly cleaves and activates effector caspases-3 and -7, executing the final stages of apoptotic cell death [7] [6].
Cells exhibit significant heterogeneity in their requirement for mitochondrial amplification of the death receptor signal. In type I cells, high levels of caspase-8 activation at the DISC are sufficient to directly process effector caspases and induce apoptosis without mitochondrial involvement [7]. This direct activation pathway typically demonstrates faster kinetics and may be more resistant to anti-apoptotic regulation. The classification between type I and type II cells is not merely academic; it has profound implications for therapeutic targeting and resistance mechanisms, particularly in cancer treatment where tumor cells may exploit these differential signaling architectures to evade death receptor-targeted therapies.
The Intrinsic (Mitochondrial) Pathway
The intrinsic apoptosis pathway functions as a critical integrator of intracellular stress signals, including DNA damage, oxidative stress, and metabolic disturbances [6]. These stressors activate the tumor suppressor p53, which transcriptionally upregulates pro-apoptotic BH3-only proteins such as PUMA, and directly activates the pro-apoptotic effectors BAX and BAK [6]. The core event in mitochondrial apoptosis is Mitochondrial Outer Membrane Permeabilization (MOMP), a process tightly regulated by the BCL-2 protein family [7] [88]. BAX and BAK, when activated, undergo conformational changes and oligomerize to form pores in the mitochondrial outer membrane, leading to the release of cytochrome c and other pro-apoptotic proteins from the intermembrane space [7] [6].
Once in the cytosol, cytochrome c binds to Apaf-1, forming the apoptosome complex which recruits and activates caspase-9 [7] [6]. This initiator caspase then processes and activates the effector caspases-3 and -7, culminating in apoptotic cell death. Simultaneously, SMAC/DIABLO and HtrA2/Omi are released from mitochondria and counteract the inhibitory effects of XIAP (X-linked Inhibitor of Apoptosis Protein) on caspases, thereby promoting apoptosis execution [7] [87]. The mitochondrial pathway thus serves as a critical amplification step, especially important in type II cells where death receptor signaling alone is insufficient for full apoptosis commitment.
Key Regulatory Nodes and Pathway Integration
The BCL-2 protein family constitutes a critical regulatory network governing MOMP and serves as a primary integration point between extrinsic and intrinsic pathways. This family includes anti-apoptotic members (BCL-2, BCL-xL, MCL-1), pro-apoptotic effector proteins (BAX, BAK), and BH3-only proteins that function as sensors and initiators of apoptosis [7] [6]. The "BH3-only" protein BID provides a crucial molecular connection between the pathways; it is cleaved by caspase-8 to generate truncated tBID, which then translocates to mitochondria where it activates BAX/BAK and promotes MOMP [7]. This caspase-8-mediated BID cleavage represents the primary mechanism for cross-talk between death receptor and mitochondrial pathways in type II cells.
Another critical regulatory node is XIAP, which potently inhibits the enzymatic activity of caspase-3, -7, and -9 [7] [87]. Cells with high XIAP levels typically require mitochondrial amplification to overcome this inhibition through SMAC/DIABLO release, classifying them as type II cells [7]. The mitochondrial pathway is further regulated by complex structural features of mitochondria themselves, particularly the Mitochondrial Contact Site and Cristae Organizing System (MICOS), which controls cristae architecture and the availability of cytochrome c for release during apoptosis [7]. These multi-layered regulatory mechanisms create numerous potential intervention points for therapeutic modulation of apoptosis signaling.
Table 1: Core Components of Extrinsic and Intrinsic Apoptosis Pathways
| Component | Extrinsic Pathway Role | Intrinsic Pathway Role | Therapeutic Significance |
|---|---|---|---|
| Initiator Caspases | Caspase-8, -10 (activated at DISC) | Caspase-9 (activated at apoptosome) | Differential activation kinetics and regulation |
| Effector Caspases | Caspase-3, -6, -7 (directly activated in type I cells) | Caspase-3, -6, -7 (activated after MOMP) | Common execution machinery; primary drug targets |
| BCL-2 Family | Limited role (except BID cleavage) | Central regulators of MOMP | Prime targets for small molecule inhibitors (venetoclax, etc.) |
| Regulatory Proteins | c-FLIP (DISC regulation), XIAP (caspase inhibition) | SMAC/DIABLO (XIAP antagonism), BID (cross-talk) | Determinants of type I/II classification; resistance mechanisms |
| Amplification Mechanism | Not required in type I cells | MOMP essential for type II cells | Critical for therapeutic efficacy in resistant cells |
Quantitative Dynamics and Heterogeneity in Apoptosis Signaling
Single-cell analysis techniques have revealed substantial heterogeneity in apoptosis signaling dynamics within nominally homogeneous cell populations [87]. This variability presents a significant challenge for achieving predictable therapeutic responses. Live-cell imaging studies using FRET-based caspase activity reporters have demonstrated a prolonged and variable delay between death receptor engagement and the commitment to apoptosis, often lasting several hours [87] [58]. During this pre-commitment phase, initiator caspases (e.g., caspase-8) can be actively signaling while effector caspases (e.g., caspase-3) remain restrained by regulatory mechanisms.
Quantitative modeling combined with experimental perturbation has identified XIAP and proteasome-dependent degradation of effector caspases as critical mechanisms maintaining this pre-commitment state [87]. When these restraining mechanisms are compromised, cells can enter an indeterminate state of partial cell death execution, potentially leading to genomic instability through incomplete caspase activation and sublethal damage [87]. This phenomenon has important implications for therapeutic window optimization, as suboptimal dosing may promote survival of damaged cells with malignant potential rather than achieving complete elimination.
The dynamics of MOMP represent a key control point in apoptosis commitment. Once a certain threshold of MOMP is reached, caspase activation becomes rapid and irreversible, creating a bistable switch from life to death [87]. The timing of this switch varies considerably between individual cells, influenced by the expression levels of BCL-2 family proteins, IAPs, and other regulatory factors. Understanding and quantifying this heterogeneity is essential for predicting therapeutic response and designing dosing strategies that maximize target cell elimination while minimizing damage to healthy tissues.
Table 2: Quantitative Parameters of Apoptosis Signaling from Live-Cell Studies
| Parameter | Experimental System | Measurement | Implications for Therapeutic Window |
|---|---|---|---|
| Pre-MOMP Delay | HeLa cells + TRAIL/CHX [87] | Highly variable (2-8 hours) | Determines time to efficacy; influences dosing schedule |
| Caspase Activation Threshold | FRET reporters + mathematical modeling [87] | ~20% effector caspase activity for commitment | Defines minimum effective concentration for apoptosis-inducing drugs |
| Type I vs. Type II Response | Multiple cell lines + death receptor ligands [7] | Mitochondrial amplification required in ~70% of cell types | Informs combination therapy strategies for different cancers |
| Secondary Necrosis Transition | Caspase sensor + Mito-DsRed [58] | 45 min - 3 hours after caspase activation | Affects inflammatory consequences of treatment |
| Heterogeneity Index | Single-cell tracking + TRAIL [87] | Coefficient of variation >50% in time to death | Predicts fractional killing requiring repeated dosing or combination therapy |
Experimental Approaches for Apoptosis Analysis
Live-Cell Imaging and Single-Cell Analysis
Advanced live-cell imaging techniques enable real-time tracking of apoptosis signaling dynamics in individual cells, providing crucial insights that are obscured in population-level analyses. Genetically encoded FRET-based reporters allow specific monitoring of initiator and effector caspase activation simultaneously with MOMP [87] [58]. A typical experimental setup involves stable expression of caspase reporter proteins—such as IC-RP (initiator caspase reporter with IETD cleavage sequence) and EC-RP (effector caspase reporter with DEVDR sequence)—together with mitochondrial markers like IMS-RP (intermembrane space targeted red fluorescent protein) [87]. This multi-parameter approach enables discrimination between apoptotic cells (showing FRET ratio changes while retaining mitochondrial fluorescence) and necrotic cells (losing FRET probe without ratio change while retaining mitochondrial fluorescence) [58].
For high-throughput applications, this live-cell imaging approach can be adapted to automated microscopy platforms, allowing quantification of apoptosis and necrosis dynamics across multiple experimental conditions in parallel [58]. The single-cell resolution provided by these methods is particularly valuable for characterizing heterogeneous responses to therapy and identifying subpopulations with differential sensitivity. This heterogeneity directly impacts therapeutic window, as resistant subpopulations may require alternative targeting strategies or combination approaches to achieve complete elimination.
Biochemical and Molecular Assessment Techniques
Traditional biochemical methods remain essential for validating apoptosis signaling events and providing complementary information to live-cell approaches. Western blot analysis of caspase substrate cleavage (e.g., PARP-1 cleavage by caspases-3 and -7) provides evidence of effector caspase activation, while detection of cytochrome c release from mitochondria confirms MOMP [89]. DNA fragmentation analysis through agarose gel electrophoresis (revealing the characteristic "DNA ladder" pattern) or TUNEL assay offers additional confirmation of apoptotic progression [89].
Flow cytometry-based approaches using Annexin V/propidium iodide staining enable quantitative assessment of apoptosis at the population level, distinguishing early apoptotic (Annexin V+/PI-), late apoptotic (Annexin V+/PI+), and necrotic (Annexin V-/PI+) cells [58]. However, this method has limitations in discriminating primary necrosis from secondary necrosis following apoptosis, highlighting the importance of complementary caspase activity assays [58]. For comprehensive analysis, these biochemical methods should be integrated with functional assessments in a time-resolved manner to establish the sequence of apoptotic events—from caspase activation and chromatin condensation to DNA fragmentation and changes in membrane permeability [89].
Targeted Perturbation Strategies
Elucidating the functional roles of specific apoptosis regulators requires targeted perturbation approaches combined with phenotypic assessment. RNA interference (RNAi) enables selective depletion of specific pathway components (e.g., caspase-8, BID, XIAP), revealing their necessity for apoptosis execution in different contexts [87]. Conversely, overexpression of anti-apoptotic proteins (e.g., BCL-2, BCL-xL, c-FLIP) can identify sufficient mechanisms for resistance [87]. Small molecule inhibitors targeting specific apoptosis regulators—including caspase inhibitors (Z-VAD-FMK), BCL-2 family inhibitors (venetoclax), and IAP antagonists—provide pharmacologically relevant tools for pathway dissection and therapeutic exploration [87] [6].
When combining these perturbation strategies with the quantitative readouts described above, researchers can construct detailed maps of apoptosis regulatory networks and identify critical nodes that determine life-death decisions. This systems-level understanding is fundamental to optimizing therapeutic windows, as it reveals which pathway components offer the greatest leverage for achieving selective target cell elimination while sparing healthy tissues.
Research Reagent Solutions
Table 3: Essential Research Reagents for Apoptosis Pathway Investigation
| Reagent Category | Specific Examples | Experimental Function | Application Context |
|---|---|---|---|
| Live-Cell Reporters | IC-RP (IETD sequence), EC-RP (DEVDR sequence), IMS-RP [87] | Real-time monitoring of caspase activation and MOMP dynamics | Single-cell analysis of apoptosis heterogeneity and kinetics |
| Caspase Substrates | Fluorogenic DEVD-AMC (caspase-3/7), IETD-AFC (caspase-8) [58] | Quantitative enzyme activity measurements in cell lysates | Biochemical validation of caspase activation |
| Antibodies | Anti-PARP-1 (cleaved form), anti-cytochrome c, anti-active caspase-3 [89] | Detection of specific apoptosis markers by western blot or immunofluorescence | Endpoint confirmation of apoptosis signaling events |
| Inducers/Inhibitors | TRAIL (receptor agonist), ABT-263 (BCL-2 inhibitor), Z-VAD-FMK (pan-caspase inhibitor) [87] [6] | Pathway perturbation to establish causal relationships | Mechanistic studies and combination therapy screening |
| Viability Assays | Annexin V/PI staining, MTT/XTT assays, ATP-based luminescence [58] [89] | Assessment of cell death and metabolic activity | Population-level efficacy quantification |
Pathway Visualization and Computational Modeling
Computational models of apoptosis signaling provide a framework for integrating quantitative data and generating testable predictions about pathway behavior under different conditions. Mathematical models based on ordinary differential equations can capture the core logic of caspase activation, MOMP regulation, and feedback loops [87]. These models have revealed design principles of apoptosis signaling, including threshold responses, bistability, and noise-filtering mechanisms. When parameterized with experimental data, such models can predict how variations in specific protein concentrations (e.g., BCL-2 family members or IAPs) influence apoptosis sensitivity, potentially explaining differential responses between normal and malignant cells.
Visualization of apoptosis pathways and their interconnections is essential for comprehending the complex network topology that governs cell fate decisions. The following diagrams, generated using Graphviz DOT language with a standardized color palette, depict key signaling relationships and experimental workflows.
Diagram 1: Apoptosis Signaling Pathways and Cross-Talk. This diagram illustrates the core components of extrinsic (blue) and intrinsic (red) apoptosis pathways, their convergence on effector caspases, and key regulatory nodes (green). The cross-talk through BID cleavage (yellow) represents a critical integration point determining type I/II cell behavior.
Diagram 2: Live-Cell Apoptosis Analysis Workflow. This diagram outlines the experimental workflow for discriminating apoptosis and necrosis using FRET-based caspase sensors and mitochondrial markers, enabling quantitative single-cell analysis of cell death dynamics.
Therapeutic Targeting Strategies and Window Optimization
Direct Pathway Targeting Approaches
Therapeutic targeting of apoptosis pathways has yielded several clinically successful agents, particularly in oncology. Death receptor pathway activation using recombinant TRAIL or agonistic antibodies against DR4/DR5 receptors initially showed promise for selectively inducing apoptosis in malignant cells [87]. However, many tumors demonstrate inherent or acquired resistance through multiple mechanisms, including decoy receptor expression, high c-FLIP levels, or insufficient caspase-8 activation [7] [87]. The recognition of type I/II differentiation provides a framework for understanding these resistance patterns and developing rational combination strategies.
BCL-2 family inhibition represents the most successful approach for targeting the intrinsic pathway, with venetoclax (ABT-199) achieving notable clinical success in hematological malignancies [6]. These BH3 mimetics function by displacing pro-apoptotic proteins from their anti-apoptotic counterparts, effectively priming cells for apoptosis. Therapeutic window optimization for BCL-2 inhibitors requires careful consideration of tissue-specific dependencies on different anti-apoptotic family members (BCL-2, BCL-xL, MCL-1), as inhibition of BCL-xL can cause dose-limiting thrombocytopenia due to platelet dependency on this protein for survival.
Biomarker-Driven Patient Stratification
Optimizing therapeutic windows for apoptosis-targeting agents increasingly relies on biomarker-driven patient selection to identify those most likely to benefit. For BCL-2 inhibitors, BCL-2 expression levels and BH3 profiling can predict sensitivity [6]. For death receptor agonists, the classification of tumors as type I or type II based on their requirement for mitochondrial amplification can inform treatment selection, with type I tumors expected to demonstrate greater sensitivity to single-agent therapy [7]. Additionally, assessment of caspase-8 expression and mutation status, FLIP levels, and XIAP expression can provide further stratification markers for predicting response to death receptor-targeted therapies.
The development of functional biomarkers that capture the dynamic state of apoptosis signaling networks offers particular promise for therapeutic window optimization. BH3 profiling measures mitochondrial priming—the proximity to the apoptosis threshold—and has demonstrated predictive value for chemotherapy response [6]. Similarly, live-cell imaging approaches using patient-derived cells could potentially provide dynamic biomarkers of apoptosis competence, enabling more precise matching of targeted agents with susceptible tumors.
Rational Combination Strategies
Overcoming the limitations of single-agent apoptosis induction requires rational combination strategies that simultaneously target multiple regulatory nodes. For death receptor agonists in type II tumors, combination with BCL-2 family inhibitors can overcome the mitochondrial amplification requirement and sensitize resistant cells [7] [87]. Similarly, IAP antagonists can lower the threshold for apoptosis by relieving caspase inhibition, particularly in combination with agents that induce SMAC/DIABLO release [87].
The integration of apoptosis-targeting agents with conventional chemotherapy, radiation, or other targeted therapies represents another important combination approach. These combinations must be carefully optimized to maximize synergistic killing of malignant cells while minimizing overlapping toxicities to normal tissues. Sequential dosing strategies informed by quantitative modeling of apoptosis dynamics may further enhance therapeutic windows by exploiting temporal aspects of pathway regulation and feedback loops [87]. As our understanding of apoptosis crosstalk with other cell death modalities deepens, additional combination opportunities will likely emerge, particularly with agents targeting necroptosis, ferroptosis, or autophagy pathways [7] [6].
Integrated Cell Death: Crosstalk, Validation, and Comparative Analysis of Pathways
The extrinsic (death receptor-mediated) and intrinsic (mitochondria-mediated) pathways represent two core apoptotic signaling routes. For decades, a critical biological question has persisted: how do these seemingly distinct pathways communicate to ensure an integrated cellular response to diverse death stimuli? This whitepaper examines the established body of evidence identifying the BH3-only protein tBID as the essential molecular link enabling this cross-talk. We explore the biochemical mechanism of tBID generation by caspase-8, its translocation to mitochondria, and its direct role in activating effector proteins BAX and BAK to induce mitochondrial outer membrane permeabilization (MOMP). The discussion is framed within the context of the type I/type II cell paradigm, which explains the differential dependence of cells on mitochondrial amplification of death receptor signals. This synthesis provides a foundational understanding for researchers investigating apoptotic mechanisms and developing therapeutics that target cell death pathways.
Apoptotic cell suicide can be initiated by a plethora of stimuli that generally feed into one of two known cell death signaling pathways [90]. The extrinsic pathway transduces signals of extracellular 'death ligands' belonging to the TNF superfamily (e.g., FasL, TRAIL). Binding of these ligands to preassembled receptor complexes triggers the activation of caspase-8 through the adapter molecule FADD (Fas-associated death domain), forming the Death-Inducing Signaling Complex (DISC) [90]. Conversely, the intrinsic pathway feeds cell death signals through the mitochondrion, which acts as a generic damage sensor and monitor of metabolic status. With the assistance of cytochrome c, cell death is initiated by the formation of a macromolecular complex (the apoptosome), which utilizes APAF-1 to mediate the activation of caspase-9 [90].
Once the activation of initiator caspases occurs by either of these two routes, the pathways converge on the effector caspases (caspase-3 and caspase-7), which are the proteolytic engines of cell death [90]. The discovery of tBID resolved a significant controversy in apoptosis research, explaining why the extrinsic apoptotic pathway was insensitive to Bcl-2 overexpression in some cells but sensitive in others [90]. The identification of Bid as the 'go-between,' transmitting signals from the extrinsic to the intrinsic pathway, provided a molecular basis for the cross-talk between the two pathways [90].
The Type I/Type II Cell Distinction: A Context for tBID Function
The functional necessity for tBID is best understood through the type I/type II cell paradigm, which emerged to explain contradictory observations regarding the sensitivity of death receptor signaling to mitochondrial inhibition [90].
- Type I Cells: In these cells, stimulation of death receptors (e.g., CD95) generates large amounts of active caspase-8 at the DISC. This robust activation allows for the direct cleavage and activation of effector caspases (e.g., caspase-3) without requiring mitochondrial amplification [11] [5]. Consequently, apoptosis in type I cells is largely resistant to overexpression of the mitochondrial anti-apoptotic protein Bcl-2 [90].
- Type II Cells: In contrast, type II cells exhibit slower DISC assembly and generate low levels of active caspase-8 [91]. These levels are insufficient to directly activate effector caspases, particularly in the presence of inhibitors like XIAP [5]. Therefore, type II cells must engage the intrinsic (mitochondrial) pathway to amplify the death signal. This dependency makes them sensitive to Bcl-2 overexpression [90]. Hepatocytes represent a classic example of type II cells, as evidenced by the protection from Fas-induced liver failure afforded by Bcl-2 overexpression or Bid deficiency [90].
Table 1: Characteristics of Type I and Type II Cells in Death Receptor-Mediated Apoptosis
| Feature | Type I Cells | Type II Cells |
|---|---|---|
| DISC Formation | Robust, high levels | Weak, low levels |
| Caspase-8 Activation | High | Low |
| Mitochondrial Dependency | Not required | Essential |
| Effect of Bcl-2 | Insensitive | Sensitive |
| BID Cleavage | Occurs but not essential | Critical for apoptosis |
| Key Determinant | High DISC formation | XIAP levels; need for signal amplification [90] [91] [5] |
The Molecular Mechanism of tBID Generation and Action
From Caspase-8 Activation to BID Cleavage
The journey of tBID begins at the DISC, where caspase-8 is activated. Caspase-8 typically exists as a monomer in the cytosol, but upon DISC formation, it undergoes dimerization, which induces a conformational change that exposes its active site [91]. This dimerization is sufficient for activation, though full proteolytic activity requires self-cleavage [91]. The activated caspase-8 then proteolytically cleaves its substrates, one of the most critical being the cytosolic, inactive protein BID.
BID is cleaved by caspase-8 at a specific aspartate residue, generating a 15 kDa C-terminal fragment known as truncated BID (tBID) and a 7 kDa N-terminal fragment (p7) [92]. This cleavage represents the first committed step in bridging the two apoptotic pathways.
The Mitochondrial Platform: Cardiolipin, Caspase-8, and BID
Recent research has illuminated that the process is more spatially organized than previously thought. Evidence indicates that active caspase-8 itself can translocate to and insert into the mitochondrial outer membrane [91]. This localization is dependent on the initial cleavage of caspase-8 into the p43-p10 heterodimer [91]. Furthermore, a native complex containing caspase-8 and BID has been identified on the mitochondrial membrane [91]. This complex forms on a platform enriched with the mitochondrial phospholipid cardiolipin [93] [5].
Within this cardiolipin-rich platform, caspase-8 directly cleaves mitochondrial-associated BID to generate tBID [91] [5]. This compartmentalization ensures that even low levels of active caspase-8 can efficiently generate tBID precisely where it is needed—on the mitochondrial surface [91].
tBID-Mediated Activation of BAX/BAK and MOMP
The final and decisive action of tBID is to trigger mitochondrial outer membrane permeabilization (MOMP). tBID, now stably associated with the mitochondrial membrane, adopts an extended structure with six α-helices, all of which interact with the membrane [92]. Critically, this includes its BH3 domain, which is contained within helix α3 [92].
The membrane-associated tBID functions as a membrane-targeted death ligand that activates the pro-apoptotic effector proteins BAK and BAX [94]. The current "embedded together" model posits that tBID, via its BH3 domain, engages with BAK or BAX, inducing their allosteric activation and intramembranous oligomerization [92] [94]. These oligomers form pores in the mitochondrial outer membrane through which cytochrome c and other apoptogenic factors are released into the cytosol [94]. Once in the cytosol, cytochrome c initiates the formation of the apoptosome, leading to the activation of caspase-9 and the downstream effector caspases, thereby committing the cell to death [5].
Table 2: Key Molecular Events in tBID-Mediated Cross-Talk
| Step | Key Players | Molecular Action | Functional Outcome |
|---|---|---|---|
| Initiation | Death Receptor, FADD, Pro-caspase-8 | Formation of the DISC and caspase-8 activation. | Triggering of the extrinsic pathway. |
| Coupling | Active Caspase-8, BID | Caspase-8 cleaves cytosolic BID. | Generation of tBID fragment. |
| Translocation & Complex Formation | tBID, Caspase-8, Cardiolipin | tBID and caspase-8 translocate to mitochondria and form a complex on cardiolipin platforms. | Spatial coordination of the death signal at the MOM. |
| Execution | tBID, BAX, BAK | tBID activates and oligomerizes BAX/BAK. | Pore formation in the MOM (MOMP). |
| Amplification | Cytochrome c, Apaf-1, Caspase-9 | Cytochrome c release leads to apoptosome formation. | Activation of the intrinsic pathway and effector caspases. |
Experimental Insights: Key Methodologies and Reagents
The model of tBID function is supported by a multitude of experimental approaches. Below is a summary of key methodologies and the critical reagents that have been instrumental in dissecting this pathway.
Detailed Experimental Protocol: Assessing Caspase-8 and tBID on Mitochondria
Objective: To investigate the formation of the native caspase-8/BID complex on mitochondria and its functional consequence in type II cells.
Methodology Summary (based on [91]):
- Cell Model: Use a well-established type II cell line, such as HeLa (human cervical adenocarcinoma) cells.
- Gene Silencing: Stably silence endogenous caspase-8 expression using a short hairpin RNA (shRNA) system. A control cell line should be generated with a non-targeting shRNA (shCont).
- Caspase-8 Mutagenesis: Create a series of caspase-8 point mutants (e.g., C360S, DM1, DM2) tagged with GFP to study the impact of processing and catalytic activity on mitochondrial localization and function.
- Stimulation & Fractionation:
- Treat both wild-type and mutant-expressing cells with a death receptor agonist (e.g., anti-Fas antibody or TRAIL).
- At various time points post-stimulation, lyse cells and perform subcellular fractionation via differential centrifugation to isolate a pure mitochondrial fraction.
- Analysis:
- Immunoblotting: Analyze mitochondrial fractions for the presence of caspase-8 (full-length and cleaved fragments), full-length BID, and tBID. Compare with cytosolic fractions.
- Co-Immunoprecipitation (Co-IP): Use antibodies against caspase-8 or BID to immunoprecipitate the complex from the mitochondrial fraction. Immunoblot for the other partner to confirm direct interaction.
- Confocal Microscopy: Image cells expressing caspase-8-GFP mutants co-stained with a mitotracker dye (e.g., Mito-dsRed2) to visually confirm mitochondrial localization.
- Viability Assay: Measure apoptosis in response to death receptor stimulation in the presence of the different caspase-8 mutants (e.g., by flow cytometry for Annexin V/propidium iodide staining).
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Studying tBID-Mediated Cross-Talk
| Research Reagent | Function in Experimental Design |
|---|---|
| Recombinant Death Ligands (e.g., FasL, TRAIL) | To specifically activate the extrinsic apoptosis pathway in vitro. |
| Agonistic Anti-Fas Antibodies (e.g., clone CH11) | A common tool to stimulate the Fas receptor and initiate DISC formation. |
| Caspase-8 shRNA/siRNA | To genetically knock down caspase-8 and validate its essential role in the pathway. |
| Bid Knockout Mice | In vivo models to demonstrate the physiological role of BID; Bid-/- mice are resistant to Fas-induced hepatocellular apoptosis [90]. |
| Caspase-8 Point Mutants (e.g., C360S, DM1) | To dissect the roles of caspase-8 catalytic activity and proteolytic processing in mitochondrial translocation and BID cleavage [91]. |
| Cardiolipin-Containing Liposomes | Artificial membrane systems (e.g., Giant Unilamellar Vesicles) to study the biophysical interactions between caspase-8, BID, and the mitochondrial membrane [93] [5]. |
| LPPG Micelles | Membrane-mimetic micelles used in NMR studies to determine the structure of membrane-associated tBID [92]. |
| Anti-BID / Anti-tBID Antibodies | Critical for detecting full-length BID and the cleaved tBID fragment in western blotting and immunofluorescence. |
| Anti-Cytochrome c Antibodies | To monitor the release of cytochrome c from mitochondria, a key functional readout for MOMP. |
The role of tBID as the critical link between the extrinsic and intrinsic apoptotic pathways is a cornerstone of modern cell death biology. The mechanism—from its generation by caspase-8 at the DISC or on mitochondrial membranes, to its direct activation of BAX/BAK—provides a coherent molecular explanation for the cross-talk that ensures an effective cellular suicide response in type II cells.
Future research continues to uncover new layers of regulation. The discovery of the mitochondrial cardiolipin platform as an activation site for caspase-8 and BID adds significant complexity to the model [93] [5]. Furthermore, emerging evidence suggests that the proteins involved in this cross-talk, including caspase-8 and BID, are entangled in multiple other cell death pathways, such as necroptosis and ferroptosis [93] [77]. This highlights a surprising degree of redundancy and communication between different cell death modalities. For drug development professionals, a deep understanding of the tBID bridge is crucial. It presents a node for therapeutic intervention—strategies to modulate BID activation or function could potentially sensitize resistant type II cancer cells to death receptor-targeted therapies or protect healthy cells in degenerative diseases. As our knowledge of this pivotal connection deepens, so too will our ability to harness it for therapeutic benefit.
For decades, apoptosis was considered the principal form of regulated cell death, with research largely focused on two core pathways: the extrinsic death receptor pathway and the intrinsic mitochondrial pathway. The death receptor pathway initiates at the plasma membrane through ligand binding to receptors like Fas (CD95) and TNF receptor, leading to the formation of the death-inducing signaling complex (DISC) and activation of caspase-8 [10] [95]. In contrast, the mitochondrial pathway responds to intracellular stress signals, culminating in mitochondrial outer membrane permeabilization (MOMP), cytochrome c release, and activation of caspase-9 via the apoptosome [95] [96]. While these apoptotic pathways remain fundamental to our understanding of development and homeostasis, recent research has revealed a more complex landscape of regulated cell death (RCD) pathways that extend beyond apoptosis [74].
The discovery of non-apoptotic programmed cell death forms—particularly necroptosis, pyroptosis, and ferroptosis—has fundamentally transformed our understanding of cellular demise [97] [98]. Unlike apoptosis, which is generally immunologically silent, these lytic forms of cell death amplify inflammatory responses through the release of damage-associated molecular patterns (DAMPs) and pro-inflammatory cytokines [95]. Furthermore, emerging evidence demonstrates extensive crosstalk among these pathways, leading to the conceptualization of PANoptosis—a unified inflammatory cell death pathway incorporating components from pyroptosis, apoptosis, and necroptosis that cannot be accounted for by any single pathway alone [95] [99]. This evolving understanding is particularly relevant in disease contexts, including perioperative neurocognitive disorders (PNDs), where anesthetic exposure has been shown to activate multiple cell death pathways simultaneously [97] [98].
This review synthesizes current knowledge of non-apoptotic cell death pathways, their molecular mechanisms, and their intricate interactions, with particular emphasis on their relevance to apoptosis research and therapeutic development.
Molecular Mechanisms of Non-Apoptotic Cell Death Pathways
Pyroptosis: Inflammatory Programmed Cell Death
Pyroptosis represents a lytic form of programmed cell death characterized by pore formation in the plasma membrane, cell swelling, and release of pro-inflammatory cytokines [95]. This pathway serves as a crucial defense mechanism against intracellular pathogens and is increasingly recognized for its role in sterile inflammation and disease.
Key Molecular Components:
- Inflammasome Activation: Multiprotein complexes including NLRP3, AIM2, and NLRC4 that serve as molecular platforms for caspase activation [95].
- Inflammatory Caspases: Caspase-1 (activated by inflammasomes) and caspase-4/5 (human) or caspase-11 (mouse) that directly cleave gasdermin family proteins [95].
- Gasdermin Family: Gasdermin D (GSDMD) is the primary executioner protein; its N-terminal domain forms plasma membrane pores upon cleavage [95].
- Pro-inflammatory Cytokine Release: Mature IL-1β and IL-18 are processed and released through gasdermin pores [95].
The pyroptotic cascade initiates with inflammasome assembly in response to pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs). Inflammasomes recruit and activate caspase-1, which subsequently cleaves pro-IL-1β and pro-IL-18 into their active forms while simultaneously cleaving GSDMD [95]. The N-terminal fragments of GSDMD oligomerize and form pores in the plasma membrane, leading to osmotic cell lysis and release of inflammatory mediators [95]. Recent evidence indicates that caspase-3 and caspase-8 can also cleave gasdermin E (GSDME), providing an alternative route to pyroptosis and highlighting the interconnectivity between different cell death pathways [95].
Necroptosis: Regulated Necrosis
Necroptosis represents a caspase-independent form of regulated cell death with morphological features of necrosis that is triggered by death receptor ligands, Toll-like receptor ligands, or viral infection [97] [95]. This pathway serves as a backup cell death mechanism when apoptotic signaling is compromised.
Core Signaling Machinery:
- Receptor-Interacting Protein Kinases: RIPK1 and RIPK3 form the necrosome complex through RIP homotypic interaction motif (RHIM) domain interactions [97].
- Mixed Lineage Kinase Domain-Like Protein: MLKL serves as the executioner protein; upon phosphorylation by RIPK3, MLKL oligomerizes and translocates to the plasma membrane, causing membrane disruption [97] [95].
- Death Receptors: TNFR1, CD95, and TRAIL receptors can initiate necroptotic signaling when caspase-8 activity is inhibited [97].
The necroptosis pathway initiates with death receptor ligation under conditions of caspase inhibition, leading to RIPK1 activation. Activated RIPK1 recruits and phosphorylates RIPK3 through RHIM domain interactions, forming the necrosome complex [97]. RIPK3 then phosphorylates MLKL, inducing a conformational change that enables MLKL oligomerization and translocation to the inner leaflet of the plasma membrane [95]. MLKL oligomers disrupt membrane integrity, resulting in ionic imbalance, cellular swelling, and release of cellular contents, including DAMPs such as HMGB1 and ATP, which propagate inflammatory responses [97].
Ferroptosis: Iron-Dependent Cell Death
Ferroptosis is characterized by iron-dependent accumulation of lipid peroxides that ultimately lead to oxidative membrane damage and cell death [97] [95]. This pathway is morphologically, biochemically, and genetically distinct from other forms of cell death.
Key Regulatory Mechanisms:
- Glutathione Peroxidase 4: GPX4 serves as the primary defense enzyme that converts lipid hydroperoxides to nontoxic lipid alcohols using glutathione as a cofactor [95].
- System Xc- Antiporter: The cystine/glutamate antiporter imports cystine for glutathione synthesis; inhibition depletes glutathione and sensitizes cells to ferroptosis [95].
- Lipid Peroxidation: Polyunsaturated fatty acids in membrane lipids are highly susceptible to peroxidation in the presence of free iron and reactive oxygen species [95].
- Iron Metabolism: Intracellular iron levels directly influence ferroptosis sensitivity through Fenton chemistry that generates reactive oxygen species [95].
Ferroptosis can be triggered through two primary mechanisms: direct inhibition of GPX4 or depletion of glutathione by impairing system Xc- function [95]. The resulting accumulation of lipid peroxides disrupts membrane integrity, leading to cell death that can be inhibited by iron chelators and lipophilic antioxidants but not by caspase inhibitors or necroptosis inhibitors [95].
Table 1: Key Characteristics of Non-Apoptotic Cell Death Pathways
| Feature | Pyroptosis | Necroptosis | Ferroptosis |
|---|---|---|---|
| Inducers | Pathogen infection, DAMPs | Death receptor ligation, caspase inhibition | GPX4 inhibition, glutathione depletion |
| Key Executors | Gasdermin D, caspase-1 | MLKL, RIPK1, RIPK3 | Lipid peroxides, iron |
| Morphology | Cell swelling, membrane pore formation | Organelle swelling, plasma membrane rupture | Mitochondrial shrinkage, normal nucleus |
| Immune Response | Strongly inflammatory | Inflammatory | Immunogenic |
| Key Inhibitors | Caspase inhibitors, gasdermin inhibitors | Necrostatin-1, MLKL inhibitors | Iron chelators, antioxidants |
PANoptosis: Integration of Cell Death Pathways
The PANoptosis Concept
PANoptosis represents a unified inflammatory cell death pathway that integrates components from pyroptosis, apoptosis, and necroptosis [95] [99]. This concept emerged from observations that multiple RCD pathways can be simultaneously activated by specific triggers and that combined loss of key molecules from all three pathways—but not individual pathway deficiencies—prevents cell death in these contexts [95].
Defining Features of PANoptosis:
- Activation by Specific Triggers: Bacterial and viral infections, sterile insults, and cytokine combinations can induce PANoptosis [95] [99].
- Molecular Complex Formation: The PANoptosome serves as a molecular scaffold for contemporaneous engagement of key molecules from multiple RCD pathways [99].
- Inflammatory Nature: PANoptosis results in substantial inflammatory responses due to lytic cell death and cytokine release [95].
- Resistance to Single Pathway Inhibition: Only combined inhibition of multiple pathways blocks PANoptosis, explaining why targeting individual pathways often shows limited efficacy [95].
The PANoptosome complex provides the structural basis for PANoptosis, serving as an integrated molecular platform that recruits and activates components from pyroptosis, apoptosis, and necroptosis [99]. Several PANoptosomes have been identified, including the Z-DNA binding protein 1 (ZBP1) PANoptosome and the NLRP12 PANoptosome, which form in response to specific infectious or inflammatory stimuli [99].
Molecular Crosstalk in PANoptosis
Extensive molecular crosstalk enables the integration of distinct RCD pathways into PANoptosis. Key molecular players facilitate this interconnectivity:
Caspase-8 as a Molecular Switch: Caspase-8 occupies a critical position in cell death regulation, functioning as a switch between apoptosis, necroptosis, and pyroptosis [95] [99]. Active caspase-8 promotes apoptosis through direct cleavage of executioner caspases while simultaneously suppressing necroptosis by cleaving RIPK1 and RIPK3 [95]. When caspase-8 activity is inhibited, the cell shifts toward necroptosis. Additionally, caspase-8 can contribute to pyroptosis by cleaving gasdermin proteins and can participate in inflammasome activation under certain conditions [95].
Inflammasome Platforms as Integration Hubs: Inflammasomes, particularly the NLRP3 inflammasome, serve as platforms for PANoptosome assembly [99]. These complexes can recruit not only caspase-1 but also caspase-8, RIPK1, RIPK3, and FADD, thereby enabling coordinated activation of pyroptosis, apoptosis, and necroptosis [99]. For instance, the NLRP3 inflammasome can activate caspase-8 alongside caspase-1, bridging pyroptotic and apoptotic signaling [99].
ZBP1 as a PANoptosis Trigger: ZBP1 has emerged as a critical sensor of viral infection that can initiate PANoptosis by recruiting multiple cell death components [99]. Upon detecting viral nucleic acids, ZBP1 nucleates a PANoptosome containing caspase-8, caspase-6, RIPK1, RIPK3, NLRP3, ASC, and caspase-1, leading to simultaneous activation of pyroptosis, apoptosis, and necroptosis [99].
Diagram 1: PANoptosis Signaling Integration. Multiple triggers activate sensor proteins that nucleate the PANoptosome complex, leading to simultaneous activation of pyroptosis, apoptosis, and necroptosis.
Comparative Analysis of Cell Death Pathways
Morphological and Functional Distinctions
The different RCD pathways exhibit distinct morphological features and functional consequences that reflect their unique molecular mechanisms and physiological roles.
Table 2: Morphological and Functional Comparison of Cell Death Pathways
| Characteristic | Apoptosis | Pyroptosis | Necroptosis | Ferroptosis | PANoptosis |
|---|---|---|---|---|---|
| Nuclear Changes | Condensation, fragmentation | Condensation | Minimal | Normal | Variable, combined features |
| Mitochondrial Changes | Cytochrome c release | Swelling | Swelling | Shrinkage, membrane density | Variable, combined features |
| Plasma Membrane | Blebbing, intact | Pore formation, rupture | Rupture | Rupture | Rupture with pore formation |
| Inflammatory Response | None (immunologically silent) | Strong | Moderate | Immunogenic | Strong |
| Caspase Dependence | Dependent | Dependent (caspase-1/4/5/11) | Independent | Independent | Partial dependence |
| Key Regulators | Caspases, Bcl-2 family | Gasdermins, inflammasomes | RIPK1, RIPK3, MLKL | GPX4, system Xc- | ZBP1, PANoptosome components |
Pathway Interconnections and Crosstalk
The traditional view of cell death pathways as linear, independent cascades has been replaced by a more nuanced understanding of extensive crosstalk and redundancy. Several molecular nodes facilitate communication between different RCD pathways:
Mitochondrial Integration: Mitochondria serve as central hubs for crosstalk between apoptosis, necroptosis, and ferroptosis [95] [100]. In the intrinsic apoptosis pathway, mitochondrial outer membrane permeabilization (MOMP) represents a commitment step that leads to cytochrome c release and caspase activation [95] [96]. Similarly, mitochondria play crucial roles in necroptosis by producing reactive oxygen species that enhance necrosome formation and in ferroptosis through their role in iron metabolism and lipid peroxidation [95].
Caspase Functional Versatility: Certain caspases demonstrate remarkable functional plasticity, participating in multiple cell death pathways. While caspase-8 is traditionally associated with extrinsic apoptosis, it also regulates necroptosis and can contribute to pyroptosis under specific conditions [95] [99]. Similarly, caspase-3, the primary executioner caspase in apoptosis, can cleave GSDME to induce pyroptosis when activated [95].
Transcriptional Coordination: Common transcription factors, particularly NF-κB and p53, coordinately regulate multiple cell death pathways [95]. NF-κB activation generally promotes cell survival but can also enhance expression of components from multiple RCD pathways, while p53 can transcriptionally activate genes involved in apoptosis, ferroptosis, and necroptosis [95].
Diagram 2: Molecular Crosstalk Between Cell Death Pathways. Caspase-8 serves as a critical molecular switch between apoptosis, necroptosis, and pyroptosis, while mitochondria integrate signals from multiple pathways.
Research Methodologies and Experimental Approaches
Pathway-Specific Assessment Techniques
Accurate characterization of cell death modalities requires multidisciplinary approaches that assess morphological, biochemical, and functional parameters.
Morphological Assessment:
- Electron Microscopy: Provides ultrastructural details essential for distinguishing cell death modalities. Apoptotic cells show chromatin condensation and membrane blebbing; pyroptotic and necroptotic cells display swelling and membrane discontinuities; ferroptotic cells exhibit mitochondrial shrinkage with normal nuclei [95].
- Time-Lapse Microscopy: Enables real-time observation of cell death dynamics, including membrane permeability changes and cell swelling [95].
Biochemical and Molecular Techniques:
- Western Blotting: Detects cleavage and activation of pathway-specific markers including caspases (apoptosis), GSDMD/GSDME (pyroptosis), phosphorylated MLKL (necroptosis), and GPX4 degradation (ferroptosis) [97] [95].
- ELISA and Luminex: Quantify released cytokines and DAMPs to characterize inflammatory responses associated with lytic cell death [97].
- Flow Cytometry: Multiparametric analysis using viability dyes, caspase activity probes, and specific antibodies enables discrimination of cell death pathways in heterogeneous populations [95].
Functional Assays:
- Pharmacological Inhibition: Pathway-specific inhibitors help dissect contributions of individual death mechanisms [97] [95].
- Genetic Manipulation: CRISPR/Cas9-mediated gene knockout or RNA interference against key regulators provides definitive evidence for pathway involvement [95] [99].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Studying Non-Apoptotic Cell Death Pathways
| Reagent Category | Specific Examples | Primary Application | Mechanism of Action |
|---|---|---|---|
| Pyroptosis Inhibitors | VX-765, Ac-YVAD-cmk | Caspase-1 inhibition | Blocks inflammatory caspase activity |
| Necroptosis Inhibitors | Necrostatin-1, GSK'872, Necrosulfonamide | RIPK1 and MLKL inhibition | Prevents necrosome formation and MLKL membrane translocation |
| Ferroptosis Inhibitors | Ferrostatin-1, Liproxstatin-1, Deferoxamine | Lipid antioxidant and iron chelation | Scavenges lipid radicals and reduces iron availability |
| PANoptosis Modulators | CRID3, NSA | PANoptosome disruption | Interferes with inflammasome and PANoptosome assembly |
| Pathway Activators | Nigericin, TSZ, Erastin, RSL3 | Inducing specific cell death forms | Activates inflammasomes, death receptors, or inhibits GPX4 |
| Detection Reagents Anti-cleaved caspase-3, anti-pMLKL, anti-GSDMD, C11-BODIPY | Pathway activity assessment | Identifies activated executors and lipid peroxidation |
Experimental Workflow for Cell Death Characterization
A systematic approach is essential for accurately identifying cell death mechanisms, particularly when multiple pathways may be activated simultaneously.
Diagram 3: Experimental Workflow for Cell Death Characterization. A stepwise approach combining viability assessment, morphological analysis, molecular marker detection, and functional validation enables accurate identification of cell death mechanisms.
Therapeutic Implications and Future Directions
Targeting Cell Death Pathways in Disease
The therapeutic manipulation of non-apoptotic cell death pathways holds significant promise for numerous disease areas, particularly in oncology, neurodegenerative disorders, and inflammatory conditions.
Cancer Therapeutics:
- Ferroptosis Inducers: Show particular promise for treating apoptosis-resistant malignancies [95]. GPX4 inhibitors (e.g., RSL3) and system Xc- inhibitors (e.g., erastin, sorafenib) can trigger ferroptosis in tumor cells [95].
- Necroptosis Activators: Second mitochondrial-derived activator of caspase (SMAC) mimetics promote necroptosis in apoptosis-resistant cancer cells by degrading cellular inhibitor of apoptosis proteins (cIAPs) [95].
- PANoptosis-Based Approaches: Combination therapies that simultaneously engage multiple cell death pathways may overcome treatment resistance common in advanced cancers [99].
Inflammatory and Neurodegenerative Disorders:
- Pyroptosis Inhibition: Caspase-1 and GSDMD inhibitors show potential for treating autoinflammatory diseases, including familial Mediterranean fever and cryopyrin-associated periodic syndrome [95].
- Necroptosis Inhibition: RIPK1 inhibitors (e.g., necrostatin-1) demonstrate efficacy in animal models of neurodegenerative diseases, including amyotrophic lateral sclerosis and Alzheimer's disease [97].
- Multi-Target Strategies: Given the pathway crosstalk, multi-target therapies that simultaneously modulate several RCD pathways may offer enhanced efficacy for complex disorders [97] [99].
Emerging Research Frontiers
Several emerging areas represent particularly promising directions for future research:
Temporal-Spatial Regulation of Cell Death: Advanced imaging and biosensor technologies are enabling real-time visualization of cell death initiation and propagation within tissues [95]. Understanding how cell death pathways are spatially organized and temporally coordinated will provide crucial insights into their physiological regulation.
Cell Death in the Tumor Microenvironment: The interplay between different cell death modalities in the tumor microenvironment significantly influences anticancer immunity [95] [74]. Ferroptosis and necroptosis can enhance antitumor immunity through immunogenic cell death, while certain forms of apoptosis may promote immune tolerance [95].
Metabolic Regulation of Cell Death: Cellular metabolism profoundly influences susceptibility to different cell death pathways [95]. Glucose metabolism regulates apoptosis, iron metabolism controls ferroptosis, and energy metabolism affects necroptosis execution [95]. Understanding these connections may enable metabolic interventions to modulate cell death.
PANoptosis in Host Defense: The role of PANoptosis in antimicrobial defense represents an exciting research frontier [95] [99]. Several pathogens, including influenza A virus, herpes simplex virus, and Francisella novicida, induce PANoptosis, suggesting this integrated cell death pathway serves as an evolutionarily conserved mechanism for combating infection [99].
The landscape of programmed cell death has expanded dramatically beyond the traditional boundaries of apoptosis. The discovery of pyroptosis, necroptosis, and ferroptosis, along with the emerging concept of PANoptosis, has revealed unprecedented complexity in how cells orchestrate their own demise. Rather than operating in isolation, these pathways form an integrated network with extensive molecular crosstalk and redundancy. This interconnectedness provides cells with flexibility in responding to diverse stresses but also presents challenges for therapeutic intervention. The future of cell death research lies in understanding the precise contextual regulation of these pathways and developing sophisticated strategies to manipulate them for therapeutic benefit. As our knowledge continues to evolve, so too will our ability to harness these fundamental biological processes to treat human disease.
Apoptosis, or programmed cell death, is a fundamental biological process essential for maintaining tissue homeostasis and eliminating damaged or unwanted cells. The two principal routes to apoptotic cell death are the Death Receptor (extrinsic) Pathway and the Mitochondrial (intrinsic) Pathway [26]. While these pathways are initiated by distinct stimuli and involve unique upstream signaling events, they converge on the activation of a common set of executioner caspases, illustrating a core principle of cellular biology: the interplay between functional redundancy and specificity [101] [72].
The death receptor pathway is typically activated by extracellular ligands binding to cell surface receptors of the tumor necrosis factor (TNF) receptor superfamily, such as CD95 (Fas) or TRAIL receptors [10]. In contrast, the mitochondrial pathway is initiated in response to internal cellular stresses, including DNA damage, oxidative stress, and endoplasmic reticulum (ER) stress, leading to mitochondrial outer membrane permeabilization (MOMP) [96] [26]. Understanding the nuanced crosstalk, regulatory checkpoints, and unique outputs of these pathways is not only critical for fundamental cell biology but also for developing targeted therapies, particularly in cancer, where apoptotic evasion is a hallmark [72].
This whitepaper provides a comparative analysis of the death receptor and mitochondrial apoptosis pathways. It details their molecular mechanisms, highlights key experimental methodologies for their study, and discusses the implications of their functional redundancy and specificity for drug discovery.
The following diagram illustrates the core components and sequence of events for the two major apoptotic pathways, highlighting points of convergence and crosstalk.
Diagram 1: Core Apoptotic Signaling Pathways.
The Death Receptor (Extrinsic) Pathway
The death receptor pathway is characterized by its rapid, direct activation of caspase cascades. The process begins with the binding of a trimeric death ligand (e.g., FasL, TRAIL) to its cognate death receptor, which exists as a pre-assembled trimer on the cell surface [10]. This binding induces a conformational change that recruits the adapter protein FADD (FAS-associated death domain protein) via homophilic death domain (DD) interactions. FADD then recruits procaspase-8 via death effector domain (DED) interactions, forming the Death-Inducing Signaling Complex (DISC) [10].
Within the DISC, caspase-8 undergoes proximity-induced dimerization and auto-proteolytic activation [10] [101]. A key regulatory element at this stage is the protein c-FLIP, which can bind to FADD and caspase-8. Depending on its isoform, c-FLIP can either inhibit caspase-8 activation or form a active heterodimer that cleaves a limited set of substrates, thereby modulating the apoptotic signal [10]. In some cell types (designated Type I cells), active caspase-8 is sufficient to directly cleave and activate the executioner caspases-3 and -7. In other cells (Type II cells), the signal is amplified through the mitochondrial pathway via caspase-8-mediated cleavage of the Bcl-2 family protein Bid into its truncated form (tBid). tBid then translocates to the mitochondria, activating the intrinsic pathway [10] [26].
The Mitochondrial (Intrinsic) Pathway
The mitochondrial pathway integrates a wide array of intracellular damage signals. Key inducers include DNA damage, oxidative stress, ER stress, and metabolic perturbations [102] [26]. The central event in this pathway is Mitochondrial Outer Membrane Permeabilization (MOMP), a point of no return that is tightly regulated by the Bcl-2 protein family [96] [26].
The Bcl-2 family comprises both pro-apoptotic (e.g., Bax, Bak, Bid, Bim) and anti-apoptotic (e.g., Bcl-2, Bcl-xL, Mcl-1) members. In response to stress signals, activated pro-apoptotic "effector" proteins Bax and Bak oligomerize on the mitochondrial outer membrane, forming pores that cause MOMP [26]. This leads to the release of several mitochondrial intermembrane space proteins, most critically cytochrome c. In the cytosol, cytochrome c binds to the protein Apaf-1, triggering its oligomerization into a wheel-like signaling platform called the apoptosome. The apoptosome recruits and activates procaspase-9, which then cleaves and activates the executioner caspases-3 and -7 [101] [26]. Other proteins released during MOMP, such as Smac/DIABLO, promote apoptosis by neutralizing Inhibitor of Apoptosis Proteins (IAPs), which would otherwise inhibit active caspases [72].
Comparative Analysis: Redundancy and Specificity
The following table summarizes the key characteristics that distinguish these two pathways, as well as their points of convergence.
Table 1: Comparative Analysis of Apoptotic Pathways
| Feature | Death Receptor Pathway | Mitochondrial Pathway |
|---|---|---|
| Primary Initiators | Extracellular death ligands (FasL, TRAIL) [10] | Intracellular stress (DNA damage, oxidative stress, ER stress) [102] [26] |
| Key Regulatory Complexes | Death-Inducing Signaling Complex (DISC) [10] | Apoptosome [26] |
| Key Initiator Caspase | Caspase-8 (and -10 in humans) [10] [101] | Caspase-9 [101] [26] |
| Key Regulatory Proteins | FADD, c-FLIP [10] | Bcl-2 Family, IAPs [72] [26] |
| Central Signaling Event | Caspase-8 activation at the DISC [10] | Mitochondrial Outer Membrane Permeabilization (MOMP) [26] |
| Primary Output | Caspase activation; in Type II cells, amplification via Bid cleavage [10] | Cytochrome c release; caspase activation [26] |
| Functional Redundancy | Convergence on executioner caspase-3/7 activation [101] [26] | Convergence on executioner caspase-3/7 activation [101] [26] |
| Crosstalk Mechanism | Caspase-8 cleavage of Bid (tBid) links to mitochondrial amplification [10] [26] | ER stress can induce DR5 expression and caspase-8 activation [10] |
Redundancy is evident in the pathways' convergence on the same executioner phase. Both pathways ultimately lead to the proteolytic activation of caspase-3 and -7, which then orchestrate the systematic dismantling of the cell by cleaving hundreds of cellular substrates, resulting in the characteristic morphological changes of apoptosis [101] [72].
Specificity is maintained through their unique initiation, regulation, and amplification mechanisms. The death receptor pathway provides a fast, direct response to extracellular commands, particularly from the immune system. The mitochondrial pathway acts as a sensitive integrator of the cell's internal health, with the Bcl-2 family acting as a crucial gatekeeper. The concept of "Type I" and "Type II" cells further exemplifies context-specificity, determining whether the death receptor pathway operates independently or requires mitochondrial amplification [10].
Crosstalk between the pathways is a critical feature ensuring robust cell death execution. The primary link is the caspase-8-mediated cleavage of Bid to generate tBid, which engages the mitochondrial pathway, thereby creating a positive feedback loop that amplifies the initial death signal [10] [26]. Furthermore, intrinsic stressors like those causing ER stress can transcriptionally upregulate death receptors like DR5 and even promote their ligand-independent activation, demonstrating bidirectional communication [10].
Experimental Analysis of Apoptotic Pathways
A comprehensive analysis of these pathways requires a multi-faceted experimental approach. The workflow below outlines a protocol for dissecting pathway-specific contributions in a cellular model.
Diagram 2: Experimental Workflow for Pathway Analysis.
Detailed Experimental Protocols
4.1.1 Inducing and Measuring Apoptosis via the Death Receptor Pathway
- Stimulation: Treat cells with a death receptor agonist. A common method is to use an anti-Fas antibody (e.g., CH11) to activate the CD95 receptor or recombinant TRAIL to engage the TRAIL receptors. A typical protocol involves treating cells (e.g., RLE-6TN rat lung epithelial cells) with the agonist at a predetermined concentration (e.g., 100-500 ng/mL) for 6-24 hours [103].
- Viability and Caspase Readout:
- Cell Viability: Use a Cell Counting Kit-8 (CCK-8). After treatment, add CCK-8 solution (10 µL/well in a 96-well plate) and incubate for 2 hours. Measure absorbance at 450 nm with a reference wavelength of 650 nm. Viability is calculated relative to untreated controls [103].
- Caspase-8 Activity: Use a luminescent caspase-8 assay. Lyse treated cells and incubate lysates with a caspase-8 substrate (e.g., Ac-IETD-pNA or a luminescent counterpart). Caspase-8 cleavage of the substrate generates a signal quantifiable with a plate reader.
4.1.2 Inducing and Measuring Apoptosis via the Mitochondrial Pathway
- Stimulation: Treat cells with an intrinsic stressor. For example, to model smoking-induced lung injury, prepare Cigarette Smoke Extract (CSE) by bubbling smoke from one unfiltered cigarette through 10 mL of PBS, then filter through a 0.22 µm filter. Treat RLE-6TN cells with 2-10% CSE for 24 hours [103]. Alternatively, use chemotherapeutics like etoposide (20-50 µM) to induce DNA damage.
- Mitochondrial and Apoptotic Readouts:
- Mitochondrial Membrane Potential (MMP): Use the JC-1 dye. Harvest treated cells and incubate with 10 µg/mL JC-1 for 20 minutes at 37°C. Analyze by fluorescence microscopy or flow cytometry. A decrease in the red/green fluorescence ratio indicates MMP loss, a hallmark of MOMP [103] [102].
- Oxidative Stress: Use MitoSOX Red dye (5 µM) to specifically detect mitochondrial superoxide. Incubate cells for 30 minutes at 37°C and analyze by flow cytometry [102].
- Western Blot for Intrinsic Pathway Markers: Lyse cells in RIPA buffer. Separate 30 µg of protein by SDS-PAGE, transfer to a PVDF membrane, and probe for key proteins such as:
- Cleaved Caspase-9 and Cleaved Caspase-3 as markers of activation.
- BAX/BCL-2 Ratio: Pro-apoptotic BAX and anti-apoptotic BCL-2 levels indicate the pro-apoptotic balance [103].
- Cytochrome c release can be detected by fractionating cells into cytosolic and mitochondrial fractions before Western blotting.
4.1.3 Pathway Modulation and Specificity Testing
- Genetic Knockdown: To confirm the specific role of a pathway, use siRNA to knock down key adapters like FADD (for the extrinsic pathway) or effectors like Bax/Bak (for the intrinsic pathway). Transfect cells with siRNA 48-72 hours before apoptotic stimulation and assess the impact on cell death readouts [103].
- Pharmacological Inhibition: Use caspase-specific inhibitors to dissect pathway contributions. For example, pre-treat cells with Z-IETD-FMK (a caspase-8 inhibitor) to block the extrinsic pathway, or Z-LEHD-FMK (a caspase-9 inhibitor) to block the intrinsic pathway, before applying pathway-specific stimuli [72].
The Scientist's Toolkit: Key Research Reagents
Table 2: Essential Reagents for Apoptosis Research
| Reagent / Assay | Function / Application | Specific Example |
|---|---|---|
| Recombinant Death Ligands | Activate the extrinsic pathway by binding death receptors. | Recombinant TRAIL, Anti-Fas Antibody (CH11) [10] |
| Intrinsic Pathway Inducers | Induce cellular stress to trigger the mitochondrial pathway. | Cigarette Smoke Extract (CSE), Etoposide, UV Radiation [103] |
| Caspase Activity Assays | Quantify the enzymatic activity of initiator and executioner caspases. | Luminescent Caspase-Glo 8/9/3 Assays [72] |
| Mitochondrial Dyes | Assess mitochondrial health and function. | JC-1 (MMP), MitoSOX Red (mitochondrial ROS) [103] [102] |
| Selective Caspase Inhibitors | Chemically block specific caspases to determine their necessity. | Z-IETD-FMK (Caspase-8), Z-LEHD-FMK (Caspase-9) [72] |
| siRNA/shRNA | Genetically knock down specific proteins to validate their role. | siRNA against FADD, BAX, BAK, or Caspase-9 [103] |
| Western Blot Antibodies | Detect protein expression, cleavage, and post-translational modifications. | Antibodies for BAX, BCL-2, Cleaved Caspases, Cytochrome c [103] [26] |
| Cell Viability Assays | Measure overall cell health and survival post-treatment. | CCK-8, MTT, CellTiter-Glo [103] |
The functional redundancy and specificity of apoptotic pathways have profound implications for drug development, especially in oncology. Cancer cells often exploit these regulatory mechanisms to evade cell death [72]. Therapeutic strategies are being developed to specifically target one pathway or exploit their crosstalk.
- Targeting the Death Receptor Pathway: Agonistic antibodies against DR5 (e.g., HexaBody DR5/DR5) and Conatumumab are designed to directly activate the extrinsic pathway in cancer cells and are under clinical investigation [72] [26].
- Targeting the Mitochondrial Pathway: "BH3 mimetics" like Venetoclax (ABT-199) are small molecules that inhibit anti-apoptotic Bcl-2, thereby promoting MOMP and activating the intrinsic pathway. These have shown remarkable success in treating certain hematological cancers [72] [26].
- Combination Therapies: Given the crosstalk and plasticity of death pathways, combining agents that target both extrinsic and intrinsic pathways is a rational strategy to overcome resistance. For instance, combining a DR5 agonist with a BH3 mimetic may synergistically induce robust apoptosis in tumors that would otherwise be resistant to either agent alone [72].
In conclusion, the death receptor and mitochondrial apoptotic pathways exemplify how biological systems achieve both robust output and nuanced control through the principles of redundancy and specificity. A deep understanding of their distinct mechanisms, points of convergence, and context-dependent crosstalk is indispensable for unraveling disease pathogenesis and designing the next generation of targeted, effective therapeutics.
Apoptosis, or programmed cell death, is a fundamental cellular process critical for maintaining tissue homeostasis and eliminating damaged cells. In the context of drug discovery, particularly for cancer and neurodegenerative diseases, the targeted modulation of apoptosis pathways represents a promising therapeutic strategy. The two principal apoptosis pathways—the extrinsic (death receptor) pathway and the intrinsic (mitochondrial) pathway—converge on a common execution phase but are initiated through distinct molecular mechanisms [1] [6]. The death receptor pathway is triggered by extracellular ligands binding to cell surface receptors, while the mitochondrial pathway is activated by intracellular stress signals originating within the cell [6]. A comprehensive understanding of both pathways, including their unique triggers, key molecular components, and points of crosstalk, is essential for developing targeted therapies. This guide provides an in-depth technical overview of the experimental models and validation methodologies used to investigate these critical pathways in apoptosis research, with emphasis on translating in vitro findings to in vivo preclinical systems.
Molecular Mechanisms of Apoptosis Pathways
The Extrinsic (Death Receptor) Pathway
The extrinsic pathway initiates when extracellular death ligands, such as Tumor Necrosis Factor (TNF-α) or Fas ligand (FasL), bind to their corresponding transmembrane death receptors (e.g., Fas, TNFR1) [1] [6]. This ligand-receptor interaction triggers the assembly of a multi-protein complex known as the Death-Inducing Signaling Complex (DISC). The DISC serves as a platform for the recruitment and activation of initiator caspases, primarily caspase-8 and caspase-10 [6]. Once activated, these initiator caspases directly cleave and activate executioner caspases (caspase-3, -6, and -7), initiating the proteolytic cascade that leads to characteristic apoptotic morphological changes, including cell shrinkage, chromatin condensation, and DNA fragmentation [1].
The Intrinsic (Mitochondrial) Pathway
The intrinsic pathway is activated in response to internal cellular distress signals, such as DNA damage, oxidative stress, or growth factor withdrawal [1] [6]. These stimuli cause a disruption in the balance between pro-apoptotic (e.g., Bax, Bak, Bid, PUMA) and anti-apoptotic (e.g., Bcl-2, Bcl-xL) proteins from the Bcl-2 family [1]. The ensuing dominance of pro-apoptotic factors leads to Mitochondrial Outer Membrane Permeabilization (MOMP), a critical event resulting in the release of several mitochondrial intermembrane space proteins into the cytosol [1]. Among these, cytochrome c forms the apoptosome complex with Apaf-1 and procaspase-9, leading to the activation of the initiator caspase-9, which then activates the executioner caspases [6]. Other released factors, such as Smac/DIABLO, promote apoptosis by neutralizing Inhibitor of Apoptosis Proteins (IAPs) [1].
Pathway Crosstalk and Integration
The extrinsic and intrinsic pathways are not entirely separate entities; significant crosstalk exists between them. The key integrating molecule is Bid, a pro-apoptotic protein from the Bcl-2 family. In certain cell types, activated caspase-8 from the extrinsic pathway cleaves Bid into its truncated, active form (tBid). tBid then translocates to the mitochondria, where it promotes MOMP and engages the intrinsic pathway, thereby amplifying the apoptotic signal [6]. This crosstalk ensures a robust and committed cellular response to death signals.
Table 1: Key Components of Apoptosis Pathways
| Pathway Component | Extrinsic Pathway | Intrinsic Pathway |
|---|---|---|
| Primary Activators | Extracellular death ligands (TNF-α, FasL) [6] | Intracellular stress (DNA damage, oxidative stress) [6] |
| Key Initiation Complex | Death-Inducing Signaling Complex (DISC) [6] | Apoptosome [6] |
| Key Regulatory Proteins | Death Receptors (Fas, TNFR1) [6] | Bcl-2 Family Proteins (Bax, Bak, Bcl-2, Bcl-xL) [1] |
| Initiator Caspases | Caspase-8, Caspase-10 [6] | Caspase-9 [6] |
| Critical Molecular Events | Receptor ligation, DISC formation [6] | MOMP, Cytochrome c release [1] |
| Executioner Caspases | Caspase-3, -6, -7 (activated by initiator caspases) [1] [6] |
The following diagram illustrates the sequence of molecular events in both apoptosis pathways and their crosstalk:
In Vitro Validation Models
In vitro systems provide a controlled environment for the initial dissection of apoptosis pathways and screening of potential modulators. They offer advantages of scalability, reproducibility, and the ability to manipulate specific molecular targets.
Cell-Based Assays for Apoptosis Detection
A suite of assays is available to detect and quantify apoptotic events in cell cultures, each targeting specific biochemical or morphological hallmarks of apoptosis.
Table 2: Common In Vitro Apoptosis Assays
| Assay Type | Target/Principle | Key Reagents & Kits | Detection Method |
|---|---|---|---|
| Phosphatidylserine Exposure | Annexin V binding to externalized PS [8] | Annexin V-FITC/PI kits (e.g., Merck APOAF) [8] | Flow Cytometry, Fluorescence Microscopy |
| Caspase Activation | Cleavage of caspase-specific substrates [104] | Z-DEVD-aminoluciferin (Caspase-3/7), Fluorogenic substrates, FLICA probes [104] | Luminescence, Fluorescence, Flow Cytometry |
| Mitochondrial Membrane Potential (ΔΨm) | Loss of ΔΨm as early apoptosis event | JC-1 dye, TMRM, TMRE | Fluorescence Shift (JC-1: red to green) |
| DNA Fragmentation | Cleavage of DNA into oligonucleosomal fragments | TUNEL assay kits | Fluorescence Microscopy, Flow Cytometry |
| Western Blot Analysis | Protein cleavage (e.g., PARP, Caspases) or expression changes (e.g., Bcl-2, Bax) | Antibodies against target proteins | Chemiluminescence |
Functional Studies with Genetic Manipulation
To establish causal relationships and delineate pathway-specific functions, genetic manipulation is indispensable.
- Gene Knockdown/Knockout: Technologies such as RNA interference (siRNA, shRNA) or CRISPR-Cas9 are used to silence or knock out specific genes in the apoptosis pathways (e.g., death receptors, Bcl-2 family members, caspases) [1]. For instance, knockdown of INHBB, a mitochondria-associated programmed cell death gene, was shown to suppress the proliferation and migration of colorectal cancer cells in vitro, validating its functional role [105].
- Gene Overexpression: Conversely, plasmids or viral vectors can be used to overexpress anti-apoptotic genes (e.g., Bcl-2) to study their protective effects or pro-apoptotic genes to induce cell death [1].
In Vivo Preclinical Validation Models
Validation in live animal models is a critical step to confirm the physiological relevance of in vitro findings and assess therapeutic efficacy and potential toxicity in a complex organism.
Animal Models of Human Disease
- Xenograft Models: These are widely used in cancer research. Immunodeficient mice are subcutaneously or orthotopically implanted with human cancer cells. The ability of a drug to induce apoptosis and inhibit tumor growth can be monitored. For example, the efficacy of topotecan in inducing apoptosis in human ovarian tumor xenografts correlated with its antitumor efficacy in vivo [106].
- Genetically Engineered Mouse Models (GEMMs): These models involve mice with genetically altered genes relevant to apoptosis pathways (e.g., p53 knockout, Bcl-2 transgenic). They are valuable for studying the role of specific genes in tumorigenesis and treatment response in an intact immune system [106].
In Vivo Apoptosis Imaging
Non-invasive imaging allows for the real-time monitoring of apoptosis in the same animal over time, which is crucial for longitudinal studies and assessing therapeutic response.
- Bioluminescence Imaging: A prominent approach uses a caspase-3/7-specific substrate like Z-DEVD-aminoluciferin. Upon caspase activation in apoptotic cells engineered to express luciferase, the substrate is cleaved, releasing aminoluciferin and producing a measurable luminescent signal. This method has been successfully used to image early-stage apoptosis in tumor xenograft models after treatment with chemotherapeutic agents [104].
- Radionuclide and MRI Imaging: Other strategies involve radiolabeled annexin V analogs for Positron Emission Tomography (PET) or magnetic resonance contrast agents that detect apoptotic cells.
The following workflow summarizes a multi-faceted validation strategy integrating bioinformatics, in vitro, and in vivo approaches:
Integrated Workflow: From Biomarker Discovery to Functional Validation
Modern apoptosis research often employs an integrated workflow that leverages large-scale data analysis to identify candidate targets, which are subsequently validated through iterative in vitro and in vivo experiments. This approach is exemplified by recent studies exploring mitochondrial and programmed cell death-related genes in various diseases [107] [108] [105].
- Bioinformatic Identification of Candidate Genes: Analysis of public transcriptomic datasets (e.g., from GEO, TCGA) is performed to identify Differentially Expressed Genes (DEGs) between disease and control samples. These DEGs are then intersected with curated lists of known Mitochondrial-Related Genes (MRGs) and Programmed Cell Death-Related Genes (PCD-RGs) to pinpoint candidate genes of interest [107] [105]. For instance, this method identified NDUFA1 and COX7C as key mitochondrial-related biomarkers in obsessive-compulsive disorder, and S100A9, S100A8, and BCL2A1 in multiple organ dysfunction syndrome [107] [108].
- In Vitro Functional Validation: The expression and functional role of candidate genes are confirmed in relevant cell lines.
- Expression Validation: Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR) and Western Blotting are used to verify the mRNA and protein expression levels of the candidate genes in disease-relevant cell lines or patient samples [107] [105].
- Functional Assays: Loss-of-function (e.g., siRNA) and gain-of-function experiments are conducted to assess the impact of the candidate gene on apoptosis and other cellular phenotypes like proliferation and migration. For example, downregulation of INHBB in colorectal cancer cells was shown to suppress tumorigenesis in vitro [105].
- In Vivo Preclinical Confirmation: Findings are ultimately validated in animal models.
- Therapeutic Efficacy: The impact of modulating the target (genetically or pharmacologically) on disease progression and apoptosis induction is evaluated [106].
- Non-invasive Monitoring: Imaging techniques, such as bioluminescence imaging with Z-DEVD-aminoluciferin, can be applied to monitor apoptosis activation in real-time following treatment [104].
The Scientist's Toolkit: Essential Reagents and Materials
Successful apoptosis research relies on a collection of specialized reagents, assays, and technologies.
Table 3: Key Research Reagent Solutions for Apoptosis Studies
| Category | Specific Examples | Primary Function in Apoptosis Research |
|---|---|---|
| Assay Kits & Reagents | Annexin V-FITC/PI Apoptosis Detection Kit [8] | Detects phosphatidylserine exposure and membrane integrity to distinguish early/late apoptosis. |
| Z-DEVD-aminoluciferin [104] | Caspase-3/7 substrate for bioluminescent detection of caspase activity in vitro and in vivo. | |
| Fluorochrome-Labeled Inhibitors of Caspases (FLICA) [104] | Cell-permeable probes that bind active caspases for flow cytometric detection. | |
| Cell Culture Models | Immortalized Cell Lines (e.g., HCT116, SW620) [105] | Provide a consistent, renewable source of cells for mechanistic studies and drug screening. |
| Primary Cells | Offer a more physiologically relevant model, though with limited lifespan. | |
| Genetic Tools | siRNA/shRNA Libraries [1] | For transient or stable gene knockdown to study gene function. |
| CRISPR-Cas9 Systems | For precise gene knockout or editing [1]. | |
| Overexpression Plasmids/Viral Vectors | For studying the effects of gene upregulation. | |
| Antibodies | Anti-PARP, Anti-Cleaved Caspase-3, Anti-Bcl-2, Anti-Bax, Anti-Cytochrome c | For Western blot, immunohistochemistry, and immunofluorescence to detect protein levels, cleavage, and localization. |
| In Vivo Imaging | In vivo Imaging System (IVIS) | Platform for detecting bioluminescent and fluorescent signals in live animals [104]. |
| Z-DEVD-aminoluciferin | Caspase-3/7 substrate for in vivo bioluminescence apoptosis imaging [104]. |
A rigorous, multi-stage validation process that progresses from in vitro systems to in vivo preclinical models is paramount for advancing our understanding of apoptosis pathways and translating this knowledge into novel therapeutics. The initial mechanistic insights gained from reductionist cell culture models must be confirmed in the complex, physiologically relevant context of a living organism. The continuous development of sophisticated tools—such as high-throughput flow cytometry, CRISPR-based genetic editing, sensitive in vivo imaging probes, and integrated bioinformatic analyses—empowers researchers to dissect the intricacies of the death receptor and mitochondrial pathways with unprecedented precision. This systematic approach to validation is the cornerstone of successful drug development in the field of apoptosis research.
Therapeutic Implications of Pathway Integration for Complex Disease Treatment
Programmed cell death, or apoptosis, is a fundamental biological process essential for maintaining tissue homeostasis, eliminating damaged cells, and ensuring proper embryonic development [1] [26]. The sophisticated molecular machinery governing apoptosis proceeds primarily through two well-characterized signaling pathways: the extrinsic (death receptor) pathway and the intrinsic (mitochondrial) pathway [1] [26]. While these pathways were historically studied in isolation, contemporary research reveals extensive crosstalk and redundancy between them, creating a complex regulatory network that determines cellular fate [93] [72]. This interconnectivity presents both challenges and opportunities for therapeutic intervention, particularly in complex diseases like cancer, where apoptosis is frequently dysregulated.
The evasion of apoptosis is a recognized hallmark of cancer, enabling uncontrolled proliferation and tumor survival [72] [109]. Cancer cells develop resistance to apoptotic cell death through various mechanisms, including the downregulation of pro-apoptotic factors, overexpression of anti-apoptotic proteins such as Bcl-2 and IAP family members, and mutation of key regulators like p53 [72] [52]. Overcoming this resistance requires a deep understanding of the molecular architecture of apoptotic signaling and the points of convergence between different cell death pathways. This whitepaper examines the therapeutic implications of integrating knowledge about the death receptor and mitochondrial apoptosis pathways, with a focus on developing innovative strategies to treat complex diseases, particularly resistant cancers, by exploiting the inherent redundancy and crosstalk within cell death networks.
Molecular Mechanisms of Apoptotic Pathways
The Extrinsic (Death Receptor) Pathway
The extrinsic pathway is initiated by the binding of specific death ligands to their corresponding cell surface death receptors. Key death ligands include Tumor Necrosis Factor (TNF)-α, Fas ligand (FasL), and TNF-Related Apoptosis-Inducing Ligand (TRAIL) [1] [26]. These interactions trigger receptor trimerization and the recruitment of adapter proteins such as FADD (Fas-Associated protein with Death Domain) to form the Death-Inducing Signaling Complex (DISC) [72]. The DISC serves as a platform for the auto-activation of initiator caspase-8, which then directly cleaves and activates executioner caspases-3, -6, and -7, culminating in the characteristic morphological changes of apoptosis, including DNA fragmentation, membrane blebbing, and phagocytic clearance [1] [26].
The Intrinsic (Mitochondrial) Pathway
The intrinsic pathway is activated in response to internal cellular stressors, such as DNA damage, oxidative stress, and growth factor withdrawal [1] [109]. These signals provoke mitochondrial outer membrane permeabilization (MOMP), a pivotal event controlled by the balanced interactions between pro-apoptotic (e.g., Bax, Bak, Bid, Bim) and anti-apoptotic (e.g., Bcl-2, Bcl-xL, Mcl-1) members of the Bcl-2 protein family [88] [109]. MOMP facilitates the release of several apoptogenic factors from the mitochondrial intermembrane space into the cytosol, including cytochrome c, SMAC/DIABLO, and Omi/HtrA2 [1] [109]. Cytochrome c, in concert with Apaf-1 and dATP/ATP, forms the apoptosome, which activates caspase-9. SMAC/DIABLO and Omi/HtrA2 promote apoptosis by neutralizing Inhibitor of Apoptosis Proteins (IAPs), thereby relieving their inhibition on caspases [52] [109].
Critical Integration Points and Crosstalk
The extrinsic and intrinsic pathways are not isolated but are molecularly entangled, with Bid serving as a critical node for signal integration [93]. In scenarios where caspase-8 activation alone is insufficient to fully execute apoptosis (particularly in "Type II" cells), this initiator caspase cleaves the pro-apoptotic Bcl-2 family protein Bid to generate its truncated, active form, tBid [93] [109]. tBid translocates to the mitochondria, where it promotes Bax/Bak oligomerization and MOMP, thereby amplifying the death signal through the intrinsic pathway [93] [88]. Recent research has highlighted the importance of a cardiolipin/caspase-8/BID platform on the outer mitochondrial membrane, which facilitates the localized generation of tBid and its subsequent action on mitochondrial membranes [93].
Furthermore, mitochondria act as central hubs integrating signals from various cell death modalities, including apoptosis, necroptosis, and ferroptosis [93] [88]. The Mitochondrial Contact Site and Cristae Organizing System (MICOS) complex is emerging as a critical determinant of mitochondrial membrane architecture and physiology, influencing the cell's commitment to death [93].
Diagram 1: Integrated Apoptotic Signaling Network. This diagram illustrates the molecular crosstalk between the extrinsic and intrinsic apoptotic pathways, highlighting critical integration points like Bid and the mitochondrial cardiolipin platform.
Therapeutic Targeting of Apoptosis in Cancer
The strategic reactivation of apoptotic pathways in malignant cells represents a cornerstone of modern oncology drug development. The integrated view of cell death signaling has fostered several mechanistically distinct therapeutic classes.
Table 1: Therapeutic Agents Targeting Apoptotic Pathways
| Therapeutic Class | Target/Mechanism | Representative Agents | Clinical/Preclinical Status |
|---|---|---|---|
| BH3 Mimetics | Inhibit anti-apoptotic Bcl-2 proteins (Bcl-2, Bcl-xL, Mcl-1) | Navitoclax, Venetoclax | Approved/Clinical Trials [72] [109] |
| SMAC Mimetics | Antagonize IAP proteins, promoting caspase activation | Birinapant, LCL161 | Clinical Trials [72] [52] |
| Death Receptor Agonists | Activate extrinsic pathway via TRAIL or DR4/5 | ABBV-621, MEDI3039, Hexa-Body DR5/DR5 | Clinical Trials [1] [26] |
| p53-MDM2 Interaction Inhibitors | Stabilize p53 by disrupting its degradation | Idasanutlin, Nutlin-3 | Clinical Trials [72] |
| Multi-Kinase Inhibitors | Induce mitochondrial apoptosis via kinase inhibition | Meriolin derivatives | Preclinical [110] |
Overcoming Resistance via Pathway Crosstalk
A significant therapeutic challenge is death pathway plasticity, wherein cancer cells develop resistance to a specific pro-apoptotic stimulus by shifting their dependence to alternative survival pathways [72]. This underscores the limitation of monotherapies and the rational basis for combination treatments. For instance, the efficacy of Death Receptor agonists can be limited in cells with high thresholds for mitochondrial apoptosis. Combining TRAIL receptor agonists with SMAC mimetics or BH3 mimetics can synergistically overcome this resistance by simultaneously engaging the extrinsic pathway and disabling the intrinsic inhibitory blocks [72] [52].
Novel agents like meriolin derivatives demonstrate the potential of this integrated approach. These multi-kinase inhibitors can activate the mitochondrial apoptosis pathway with remarkable potency and rapid kinetics, even in the presence of overexpressed anti-apoptotic Bcl-2—a common resistance mechanism in cancer cells [110]. Furthermore, they have shown efficacy in models of imatinib-resistant chronic myeloid leukemia and cisplatin-resistant carcinomas, highlighting their ability to bypass major clinical resistance mechanisms [110].
Experimental Analysis of Apoptotic Signaling
Methodologies for Assessing Pathway Activity
Rigorous experimental protocols are essential for dissecting the complex interplay between apoptotic pathways and evaluating therapeutic efficacy.
Protocol 1: Analysis of Mitochondrial Apoptosis Activation
- Objective: To assess the early events of the intrinsic apoptosis pathway, including MOMP and caspase activation.
- Procedure:
- Treatment: Expose cells (e.g., Jurkat leukemia or Ramos lymphoma cells) to the apoptotic stimulus (e.g., 50-200 nM Meriolin derivatives, 1 µM Staurosporine as positive control) for 1-8 hours [110].
- Mitochondrial Membrane Potential (ΔΨm): Harvest cells and stain with potentiometric dyes like JC-1 or TMRM. Analyze by flow cytometry. A collapse in ΔΨm is an early indicator of mitochondrial dysfunction [110].
- Protein Release: Isolate cytosolic fractions via digitonin permeabilization or differential centrifugation. Detect the release of mitochondrial proteins such as Cytochrome c and SMAC into the cytosol by Western blotting [110] [109].
- Caspase Activity: Measure the catalytic activity of executioner caspases (e.g., caspase-3/7) using fluorogenic substrates (e.g., Ac-DEVD-AMC). Monitor fluorescence kinetics over time [110].
- PARP Cleavage: Analyze the cleavage of poly(ADP-ribose) polymerase (PARP), a classic caspase-3 substrate, by Western blotting as a downstream marker of apoptosis execution [110].
Protocol 2: Differentiating Extrinsic and Intrinsic Pathway Contribution
- Objective: To determine the relative contribution of death receptor versus mitochondrial signaling in apoptosis induction.
- Procedure:
- Genetic/Pharmacological Inhibition: Pre-treat cells with specific inhibitors:
- Pan-caspase inhibitor: Q-VD-OPh (20 µM) to confirm caspase-dependent death [110].
- Caspase-8 inhibitor: Z-IETD-FMK to probe the extrinsic pathway.
- Caspase-9 inhibitor: Z-LEHD-FMK to probe the intrinsic pathway.
- Disc Complex Immunoprecipitation: For death receptor signaling, lyse cells and immunoprecipitate the DISC using antibodies against Fas or FADD. Analyze for the recruitment and processing of caspase-8 by Western blotting [72].
- Bid Cleavage Analysis: Detect the cleavage of full-length Bid to its active truncated form (tBid) by Western blotting. This serves as a direct molecular marker of crosstalk from the extrinsic to the intrinsic pathway [93] [109].
- Viability Assay: Measure cell viability 24-48 hours post-treatment using assays like MTT or Annexin V/propidium iodide staining by flow cytometry to quantify the protective effect of each inhibitor [110].
- Genetic/Pharmacological Inhibition: Pre-treat cells with specific inhibitors:
The Scientist's Toolkit: Key Research Reagents
Table 2: Essential Reagents for Apoptosis Research
| Reagent Category | Specific Examples | Research Application |
|---|---|---|
| Caspase Inhibitors | Q-VD-OPh (pan-caspase), Z-IETD-FMK (caspase-8), Z-LEHD-FMK (caspase-9) | Determining caspase dependency and delineating specific pathway contributions [110]. |
| Death Receptor Ligands | Recombinant TRAIL, Fas Agonistic Antibodies | Specific activation of the extrinsic apoptosis pathway [1] [109]. |
| Mitochondrial Dyes | JC-1, TMRM, MitoTracker | Assessing mitochondrial membrane potential (ΔΨm) and integrity [110]. |
| Apoptosis Inducers | Staurosporine (broad), Meriolin derivatives (kinase/mitochondrial), Etoposide (DNA damage) | Positive controls and mechanistic studies of intrinsic pathway activation [110]. |
| Antibodies for WB | Anti-Cytochrome c, Anti-SMAC/DIABLO, Anti-cleaved PARP, Anti-caspase-8, -9, -3, Anti-Bid/tBid | Detecting protein localization, cleavage, and activation as hallmarks of apoptosis [93] [110] [109]. |
| Flow Cytometry Assays | Annexin V/PI Apoptosis Detection Kit, APO-BrdU TUNEL Assay | Quantifying phosphatidylserine externalization (early apoptosis) and DNA fragmentation (late apoptosis), respectively [110]. |
Diagram 2: Experimental Workflow for Apoptosis Analysis. A generalized protocol for the comprehensive assessment of apoptotic signaling in response to therapeutic agents.
The traditional binary view of the death receptor versus mitochondrial apoptosis pathways is evolving into a more nuanced understanding of a highly integrated and plastic cell death network. The therapeutic implications of this paradigm shift are profound. Success in treating complex, multifactorial diseases like cancer will increasingly depend on strategies that co-opt this natural redundancy and crosstalk, rather than targeting isolated components.
Future directions will focus on rational combination therapies that simultaneously engage multiple nodes within the cell death network to preempt or overcome resistance [72] [109]. This includes combinations of BH3 mimetics with SMAC mimetics, death receptor agonists, or conventional chemotherapeutics. Furthermore, the integration of apoptotic inducers with immunotherapies represents a promising frontier, as certain forms of therapy-induced apoptosis can stimulate immunogenic cell death, enhancing anti-tumor immunity [109]. The application of advanced techniques, such as single-molecule imaging and multi-correlative microscopy, will continue to refine our understanding of dynamic processes like MOMP at the nanoscale, revealing new vulnerabilities for therapeutic exploitation [88]. Ultimately, leveraging the integrated nature of cell death signaling paves the way for more adaptive, effective, and personalized treatment regimens for patients with cancer and other complex diseases characterized by apoptotic dysregulation.
Conclusion
The death receptor and mitochondrial pathways, while initiated by distinct signals, are deeply interconnected components of a sophisticated cell death network. The convergence on caspase activation and the pivotal crosstalk mediated by molecules like tBID underscore a high degree of redundancy and integration, which is further expanded by interactions with non-apoptotic death pathways like PANoptosis. For clinical translation, this integrated view is paramount. The success of BH3 mimetics validates targeting the intrinsic pathway, but overcoming resistance requires a multifaceted approach. Future directions must focus on exploiting pathway crosstalk through rational drug combinations, developing biomarkers to predict therapeutic response, and creating next-generation agents that can modulate the broader cell death network to restore apoptosis in refractory cancers and other diseases characterized by apoptotic dysregulation.
References
- 1. Apoptosis: A Comprehensive Overview of Signaling Pathways ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11592877/]
- 2. Apoptosis [https://en.wikipedia.org/wiki/Apoptosis]
- 3. Intrinsic and Extrinsic Pathways of Apoptosis [https://www.thermofisher.com/us/en/home/life-science/antibodies/antibodies-learning-center/antibodies-resource-library/cell-signaling-pathways/cellular-apoptosis-pathway.html]
- 4. Mechanism of RCD and the Role of Different Death ... [https://www.mdpi.com/2227-9059/13/8/1880]
- 5. Multiple entanglements of different cell death pathways, in ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2025.1667611/full]
- 6. Insights on the crosstalk among different cell death ... [https://www.nature.com/articles/s41420-025-02328-9]
- 7. Multiple entanglements of different cell death pathways, in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12644007/]
- 8. Apoptosis Assay Market Size Report, 2025 – 2034 [https://www.gminsights.com/industry-analysis/apoptosis-assay-market]
- 9. Frontiers | Regulation of apoptosis and interaction with cartilage... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2025.1571448/full]
- 10. The Death of Receptor - PMC Pathway Apoptosis [https://pmc.ncbi.nlm.nih.gov/articles/PMC8805650/]
- 11. Caught in the act between death receptors and mitochondria [https://www.sciencedirect.com/science/article/pii/S0167488911000346]
- 12. Regulation of CD95/Fas signaling at the DISC [https://www.nature.com/articles/cdd2011155]
- 13. Death Receptor Pathway [https://www.thermofisher.com/us/en/home/life-science/antibodies/antibodies-learning-center/antibodies-resource-library/cell-signaling-pathways/apoptosis-death-receptors.html]
- 14. Control of mitochondrial apoptosis by the Bcl-2 family - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2714431/]
- 15. MOMP, cell suicide as a BCL-2 family business - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5729535/]
- 16. The BCL2 family: from apoptosis mechanisms to new ... [https://www.nature.com/articles/s41392-025-02176-0]
- 17. Molecular steps of death receptor and mitochondrial ... [https://pubmed.ncbi.nlm.nih.gov/11758823/]
- 18. Subcellular Localization and Dynamics of the Bcl-2 Family ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2018.00013/full]
- 19. Apoptosis Signaling Pathway: Stages, Types and Key ... [https://geneglobe.qiagen.com/us/knowledge/pathways/apoptosis-signaling]
- 20. Death Receptor Signals to Mitochondria - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC2941887/]
- 21. Caspases: the executioners of apoptosis - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC1218630/]
- 22. Caspases as master regulators of programmed cell death [https://www.nature.com/articles/s12276-025-01470-9]
- 23. Caspase [https://en.wikipedia.org/wiki/Caspase]
- 24. North America Apoptosis Assay Market Trends 2025–2034 [https://www.gminsights.com/industry-analysis/north-america-apoptosis-assay-market]
- 25. Apoptosis Market Size, Share and Opportunities, 2025-2032 [https://www.coherentmarketinsights.com/industry-reports/apoptosis-market]
- 26. Apoptosis: A Comprehensive Overview of Signaling ... [https://www.mdpi.com/2073-4409/13/22/1838]
- 27. The role of BCL-2 family proteins in regulating apoptosis ... [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2022.985363/full]
- 28. Mitochondria-associated programmed cell death as a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10474116/]
- 29. Current concepts in apoptosis: The physiological suicide ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6275981/]
- 30. Development of a Novel Biomarker Platform for Profiling ... [https://www.mdpi.com/2072-6694/17/11/1852]
- 31. Unveiling IL6R and MYC as Targeting Biomarkers in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11011921/]
- 32. Correlated light and electron microscopy illuminates the ... [https://www.sciencedirect.com/science/article/pii/S0005272808001606]
- 33. Cutting-edge microscopy reveals how apoptosis starts in ... [https://www2.mrc-lmb.cam.ac.uk/cutting-edge-microscopy-reveals-how-apoptosis-starts-in-the-mitochondria/]
- 34. Correlated Light and Electron Microscopy/ ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2730195/]
- 35. Integrated real-time imaging of executioner caspase ... [https://www.nature.com/articles/s41420-025-02662-y]
- 36. A Novel Single-Molecule Fluorescence Lifetime Probe for ... [https://pubmed.ncbi.nlm.nih.gov/40708524/]
- 37. Designing an apoptosis reporter by mutagenesis-based ... [https://www.sciencedirect.com/science/article/pii/S2090123225004795]
- 38. The role of BCL-2 family proteins in regulating apoptosis and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9597512/]
- 39. Venetoclax and new BCL-2 inhibitors in acute myeloid ... [https://pubmed.ncbi.nlm.nih.gov/41143709/]
- 40. Mechanisms of action of the BCL-2 inhibitor venetoclax in ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2023.1291920/full]
- 41. Mechanisms of Cell Death: Apoptosis - CST Blog [https://blog.cellsignal.com/mechanisms-of-cell-death-apoptosis]
- 42. Differential regulation of the intrinsic pathway of apoptosis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1847410/]
- 43. Live from ASCO 2025 [https://www.ascentage.com/live-from-asco-2025-ascentage-pharma-presents-clinical-data-on-bcl-2-inhibitor-lisaftoclax-in-venetoclax-refractory-patients-in-oral-report/]
- 44. Overcoming Venetoclax Resistance in CLL: A Novel BCL2 ... [https://mediamedic.co/overcoming-venetoclax-resistance-in-cll-a-novel-bcl2-bcl-xl-dual-efficacy-degrader-reshapes-the-therapeutic-landscape%E4%B8%A8eha-2025/]
- 45. Overcoming cancer drug resistance: insights into Apoptotic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12583045/]
- 46. Frontiers | Advances in the study of death receptor 5 [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1549808/full]
- 47. Advances in the study of death receptor 5 - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC11936945/]
- 48. Novel Fusion Protein Based on DR -Specific 5 Variant with... TRAIL [https://link.springer.com/article/10.1134/S0006297925602096]
- 49. Dual role of DR5 in death and survival signaling leads to ... [https://www.nature.com/articles/cddis2017423]
- 50. Blocking TRAIL-DR5 signaling pathway with soluble death ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2023.1171293/full]
- 51. Targeting apoptotic pathways for cancer therapy - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11245162/]
- 52. Targeting the inhibitors of apoptosis proteins (IAPs) to ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1562167/full]
- 53. Smac mimetics and TNFα: a dangerous liaison? - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3184251/]
- 54. Discovery of Novel Second Mitochondria-Derived Activator ... [https://www.sciencedirect.com/science/article/abs/pii/S0022356524272748]
- 55. IAP Proteins Antagonist: An Introduction and Chemistry of ... [https://pubmed.ncbi.nlm.nih.gov/29532750/]
- 56. Review Smac mimetics as IAP antagonists [https://www.sciencedirect.com/science/article/abs/pii/S1084952114003279]
- 57. Smac mimetics increase cancer cell response to ... [https://www.nature.com/articles/cdd201044]
- 58. A quantitative real-time approach for discriminating ... [https://www.nature.com/articles/cddiscovery2016101]
- 59. Prognostic Potential of Apoptosis-Related Biomarker ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12346514/]
- 60. Biomarkers of apoptosis - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2538762/]
- 61. Apoptotic extracellular vesicles: emerging biomarkers for ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1619456/full]
- 62. Drug resistance in cancer: molecular mechanisms and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12623568/]
- 63. Multilevel Mechanisms of Cancer Drug Resistance [https://www.mdpi.com/1422-0067/25/22/12402]
- 64. Overcoming cancer drug resistance: insights into Apoptotic ... [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2025.1677380/full]
- 65. Mitochondrial metabolism and cancer therapeutic innovation [https://www.nature.com/articles/s41392-025-02311-x]
- 66. Mitochondrial orchestration of PANoptosis - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC12540656/]
- 67. Cancer stem cells: mitochondria signalling pathway and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12226654/]
- 68. Inflammation and metabolic stress combine to drive a new ... [https://www.stjude.org/media-resources/news-releases/2025-medicine-science-news/inflammation-and-metabolic-stress-combine-to-drive-new-cell-death-pathway-mitoxyperilysis.html]
- 69. Targeting MDM2, RAS, and PCNA for cancer ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1663766/full]
- 70. Harnessing PUMA's lethal potential: BCL-2 family ... [https://www.sciencedirect.com/science/article/pii/S246829422500111X]
- 71. Apoptosis Testing Market Size, Share, Growth [https://market.us/report/apoptosis-testing-market/]
- 72. Reactivating apoptotic pathways in cancer: A review of ... [https://www.sciencedirect.com/science/article/abs/pii/S0014299925007198]
- 73. Targeting regulated cell death: Apoptosis, necroptosis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11921819/]
- 74. Comprehensive landscape of cell death mechanisms [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2025.1611055/full]
- 75. Cell death and cancer: Metabolic interconnections - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12187904/]
- 76. Navigating Transition Metal-Dependent Cell Death [https://pubmed.ncbi.nlm.nih.gov/41178545/]
- 77. Extrinsic apoptosis and necroptosis in telencephalic ... [https://www.nature.com/articles/s41418-025-01594-5]
- 78. and cancer Apoptosis | Abcam signaling pathway [https://www.abcam.com/en-us/technical-resources/pathways/apoptosis-and-cancer-signaling-pathway]
- 79. Mitochondria and tumorigenesis: Molecular basis and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12508591/]
- 80. Cell death in tumor microenvironment: an insight for ... [https://www.nature.com/articles/s41420-025-02376-1]
- 81. Specific signaling mediated programmed pathways in... cell death [https://pmc.ncbi.nlm.nih.gov/articles/PMC12084487/]
- 82. Therapeutic targeting of cell death-immune crosstalk in cancer ... [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-025-02491-8]
- 83. in Cell Immune Apoptosis and Its... Tumor Microenvironment [https://www.frontiersin.org/research-topics/62976/cell-apoptosis-in-tumor-immune-microenvironment-and-its-therapeutic-potential/magazine]
- 84. Apoptotic cells promote circulating tumor cell survival and ... [https://www.nature.com/articles/s42003-025-08541-7]
- 85. Resveratrol and temozolomide induce apoptosis and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12333572/]
- 86. Quantitative modeling to understand cell signaling in the ... [https://www.sciencedirect.com/science/article/pii/S2452310021000305]
- 87. Quantitative analysis of pathways controlling extrinsic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2858979/]
- 88. Mitochondrial dynamics and pore formation in regulated ... [https://www.sciencedirect.com/science/article/pii/S0968000425002191]
- 89. Apoptosis: A Four-Week Laboratory Investigation for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC256977/]
- 90. Cross-Talk in Cell Death Signaling - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2195865/]
- 91. BID is cleaved by caspase-8 within a native complex on the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3132005/]
- 92. Structural Insights of tBid, the Caspase-8-activated Bid, and Its ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3861634/]
- 93. Multiple entanglements of different cell death pathways, in ... [https://pubmed.ncbi.nlm.nih.gov/41306288/?utm_source=FeedFetcher&utm_medium=rss&utm_campaign=None&utm_content=1VSjW0JqT_v3o4ef_i7fczOFyon91YoRD2Oh6DG1jbIZYnq94z&fc=None&ff=20251127070858&v=2.18.0.post22+67771e2]
- 94. Pro-apoptotic cascade activates BID, which oligomerizes ... [https://www.nature.com/articles/4400783]
- 95. Regulated cell death pathways and their roles in ... [https://www.nature.com/articles/s12276-023-01069-y]
- 96. Living with death: the evolution of the mitochondrial ... [https://www.nature.com/articles/cdd200865]
- 97. Beyond apoptosis: Exploring necroptosis, ferroptosis, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12512153/]
- 98. Beyond apoptosis: Exploring necroptosis, ferroptosis, and ... [https://pubmed.ncbi.nlm.nih.gov/41078395/]
- 99. From pyroptosis, apoptosis and necroptosis to PANoptosis [https://www.sciencedirect.com/science/article/pii/S2001037021003287]
- 100. Death receptor signals to mitochondria - PubMed - NIH [https://pubmed.ncbi.nlm.nih.gov/15640619/]
- 101. structural and molecular mechanisms and functions in cell ... [https://www.nature.com/articles/s41421-025-00791-3]
- 102. Mitochondrial, metabolic and bioenergetic adaptations ... [https://www.nature.com/articles/s41419-025-07596-y]
- 103. FASN regulates CSE-induced apoptosis, oxidative stress and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12486463/]
- 104. imaging of early stage In by measuring real-time... vivo apoptosis [https://link.springer.com/article/10.1007/s10495-010-0553-1]
- 105. A Novel Mitochondria‐Associated Programmed Cell Death ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12648302/]
- 106. as a determinant of tumor sensitivity to topotecan in human... Apoptosis [https://pubmed.ncbi.nlm.nih.gov/9815771/]
- 107. Identification and experimental validation of biomarkers ... [https://www.nature.com/articles/s41598-025-17606-w]
- 108. Identification and validation of key genes related to apoptosis ... [https://eurjmedres.biomedcentral.com/articles/10.1186/s40001-025-03292-x]
- 109. Impact of Complex Apoptotic Signaling Pathways ... - MDPI [https://www.mdpi.com/2072-6694/16/5/984]
- 110. Novel meriolin derivatives activate the mitochondrial ... [https://www.nature.com/articles/s41420-024-01901-y]