Beyond the Full-Length Enzyme: The Pro-Apoptotic Functions and Therapeutic Potential of Truncated PARP-1 Fragments
This review synthesizes current knowledge on the biological functions of truncated PARP-1 (tPARP1) fragments in cell death pathways.
Beyond the Full-Length Enzyme: The Pro-Apoptotic Functions and Therapeutic Potential of Truncated PARP-1 Fragments
Abstract
This review synthesizes current knowledge on the biological functions of truncated PARP-1 (tPARP1) fragments in cell death pathways. For researchers and drug development professionals, we explore the foundational mechanisms of PARP-1 cleavage by caspases and other proteases, detailing how the resulting fragments actively promote apoptosis rather than merely inactivating the parent enzyme. We examine methodological approaches for studying tPARP1 and its applications in overcoming PARP inhibitor resistance. The content also addresses challenges in targeting these pathways and provides comparative analysis of tPARP1 functions across different cell death modalities, offering a comprehensive resource for advancing therapeutic strategies in oncology and neurodegenerative diseases.
From DNA Repair to Death Signal: The Genesis and Structure of Truncated PARP-1
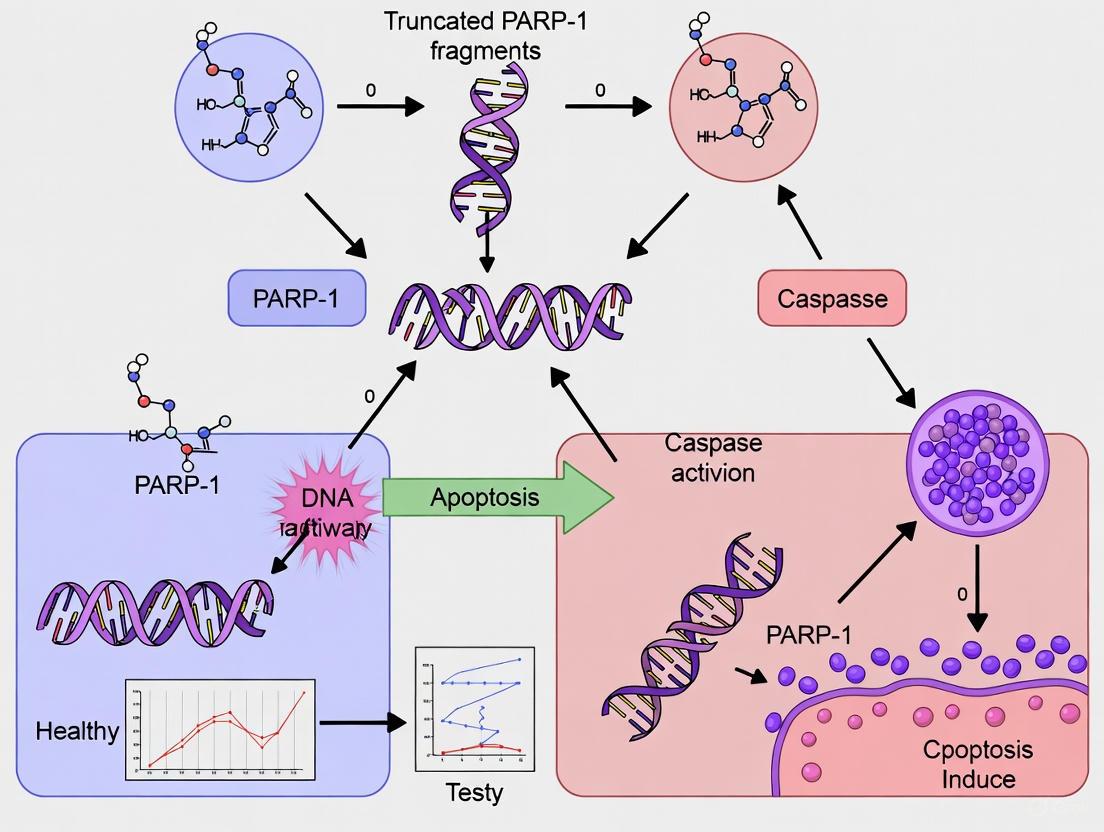
The cleavage of Poly (ADP-ribose) polymerase 1 (PARP1) by caspase-3 represents a definitive biochemical hallmark of apoptotic cell death. As the primary executioner caspase, caspase-3 specifically targets PARP1 at the aspartic acid residue 214 (D214), generating characteristic 24-kD and 89-kD fragments [1] [2]. This proteolytic event serves as a critical molecular switch that inactivates the DNA repair function of PARP1 and facilitates the dismantling of the cell during apoptosis. While historically viewed as merely an inactivation mechanism, emerging research reveals that the resulting truncated PARP1 (tPARP1) fragments possess novel biological activities that extend beyond their traditional roles. This technical guide examines the molecular mechanism, functional consequences, and experimental approaches for studying PARP1 cleavage, with particular emphasis on the emerging biological functions of tPARP1 fragments in cell death pathways.
Molecular Mechanism of PARP1 Cleavage by Caspase-3
Structural Domains of PARP1 and Cleavage Site
PARP1 is a 116-kDa nuclear enzyme composed of several functional domains that dictate its cellular functions. The modular structure includes:
- DNA-binding domain (DBD): Contains three zinc finger motifs (Zn1, Zn2, Zn3) that recognize DNA strand breaks [1] [3]
- Nuclear localization signal (NLS): Directs PARP1 to the nucleus [4]
- Caspase cleavage domain: Contains the DEVD/G sequence recognized by caspase-3 [3]
- BRCT domain: Facilitates protein-protein interactions [5]
- WGR domain: Regulates catalytic activity [3]
- Catalytic domain (CAT): Mediates poly(ADP-ribose) synthesis using NAD+ as substrate [1] [3]
Caspase-3 cleaves human PARP1 specifically between Asp214 and Gly215 within the DEVD/G sequence, separating the N-terminal DNA-binding domain (24 kDa) from the C-terminal automodification and catalytic domains (89 kDa) [2] [6]. This cleavage event is highly specific and serves as a recognized biomarker for apoptosis.
Caspase-3 Activation and Specificity
Caspase-3 exists as an inactive zymogen (caspase-3p32) that requires proteolytic activation through apoptotic signaling. The activation process involves:
- Proteolytic processing: Generation of p20 and p12 subunits that form the active enzyme [7]
- Extrinsic pathway activation: Death receptor signaling leads to caspase-8 activation, which directly processes caspase-3 [8]
- Intrinsic pathway activation: Mitochondrial cytochrome c release promotes apoptosome formation and caspase-9 activation, which subsequently activates caspase-3 [8]
Caspase-3 demonstrates exclusive specificity for the DEVD sequence in PARP1, with cleavage occurring efficiently at physiological enzyme concentrations [2] [9]. The structural basis for this specificity involves complementary interactions between the caspase-3 substrate-binding cleft and the PARP1 DEVD motif [10].
Table 1: PARP1 Fragments Generated by Caspase-3 Cleavage
| Fragment | Molecular Weight | Domains Contained | Cellular Localization | Primary Functions |
|---|---|---|---|---|
| N-terminal fragment | 24 kDa | Zinc fingers 1-2, NLS | Nuclear | Dominant-negative inhibitor of DNA repair, binds DNA breaks |
| C-terminal fragment (tPARP1) | 89 kDa | Zinc finger 3, BRCT, WGR, CAT | Cytosolic translocation | Novel signaling functions, RNA Pol III regulation, PAR carrier |
Biological Consequences of PARP1 Cleavage
Traditional View: Inactivation of DNA Repair
The conventional understanding of PARP1 cleavage centers on the irreversible inactivation of its DNA repair capabilities:
- The 24-kDa fragment retains the DNA-binding domain and nuclear localization signal, acting as a trans-dominant inhibitor by occupying DNA strand breaks and blocking access by intact DNA repair proteins [1] [4]
- The 89-kDa fragment loses nuclear retention capability due to separation from the NLS-containing domain, leading to its cytosolic translocation [4]
- ATP conservation results from preventing excessive PARP1 activation, which would deplete NAD+ and ATP pools through futile repair cycles [9]
This inactivation mechanism ensures that the apoptotic process proceeds without interference from DNA repair pathways that might attempt to rescue damaged cells.
Emerging Functions of Truncated PARP1 Fragments
Recent research has revealed novel biological activities associated with the tPARP1 fragments, particularly the 89-kDa C-terminal fragment:
Cytosolic Signaling Functions
The 89-kDa tPARP1 fragment translocates to the cytoplasm during apoptosis, where it engages in non-canonical signaling pathways:
- RNA Polymerase III Interaction: tPARP1 recognizes and mono-ADP-ribosylates the RNA polymerase III (Pol III) complex in the cytosol, facilitating IFN-β production during poly(dA-dT)-stimulated apoptosis [5]
- BRCT domain dependency: The interaction with Pol III requires an intact BRCT domain in tPARP1, with F473A mutation abolishing this binding capability [5]
- Immune activation: tPARP1-mediated ADP-ribosylation of Pol III enhances innate immune responses to foreign DNA, connecting apoptosis to antiviral defense mechanisms [5]
Role in Parthanatos
The 89-kDa fragment serves as a PAR carrier during specific cell death pathways:
- When PARP1 undergoes auto-poly(ADP-ribosyl)ation prior to caspase cleavage, the 89-kDa fragment retains covalently attached PAR polymers [4]
- These PAR polymers facilitate AIF (Apoptosis-Inducing Factor) release from mitochondria, triggering nuclear shrinkage and large-scale DNA fragmentation [4]
- This mechanism creates a molecular bridge between caspase-dependent apoptosis and PAR-dependent parthanatos [4]
Experimental Approaches for Studying PARP1 Cleavage
Detection Methodologies
Immunological Detection
Western blotting using cleavage-specific antibodies provides the most direct method for detecting PARP1 fragmentation:
- Antibody specificity: Anti-cleaved PARP1 (Asp214) antibodies specifically recognize the 89-kDa fragment without cross-reacting with full-length PARP1 or other isoforms [2] [6]
- Recommended dilutions:
- Cellular validation: Apoptotic induction with staurosporine (0.5-1 μM for 4-6 hours) or actinomycin D (1-5 μM for 6-12 hours) provides positive controls [4]
Functional Assays
- Caspase activity measurement: Fluorometric assays using DEVD-afc substrate (12.5 μM) detect caspase-3 activation with peak activity typically 30-60 minutes after apoptotic stimulation [7]
- Cellular localization studies: Immunofluorescence staining demonstrates nuclear-to-cytoplasmic translocation of the 89-kDa fragment following cleavage [4]
- Interaction studies: Co-immunoprecipitation assays validate tPARP1 interactions with protein complexes such as RNA Pol III [5]
Experimental Models for PARP1 Cleavage Research
Table 2: Experimental Models for Studying PARP1 Cleavage
| Model System | Induction Method | Key Readouts | Applications |
|---|---|---|---|
| HeLa cells | Staurosporine (0.5-1 μM, 4-6h) | 89-kDa fragment generation, PAR accumulation, AIF translocation | General apoptosis mechanisms, parthanatos cross-talk |
| 293T PARP1-/- | Poly(dA-dT) transfection (1-2 μg/mL, 6-8h) | tPARP1 and Pol III interaction, IFN-β production | Innate immune activation during apoptosis |
| Primary neurons | Glutamate excitotoxicity or oxygen-glucose deprivation | Caspase-3 activation, PARP1 cleavage, cell viability | Neurodegeneration models, cerebral ischemia |
| L929 fibroblasts | TNF-α (10-50 ng/mL) with caspase inhibition | PARP overactivation, necrosis-to-apoptosis switch | Cell death modality studies |
Technical Considerations and Controls
- Inhibition controls: Include caspase inhibitors (zVAD-fmk, 20-50 μM) and PARP inhibitors (PJ34, 10-20 μM) to validate specificity [4] [9]
- Time course experiments: PARP1 cleavage typically precedes DNA fragmentation, with detectable 89-kDa fragment appearing 1-3 hours after apoptotic stimulation [7]
- Non-cleavable PARP1 mutants: Express PARP1-D214N to distinguish cleavage-dependent and independent functions [5] [9]
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for PARP1 Cleavage Research
| Reagent Category | Specific Examples | Function/Application | Working Concentrations |
|---|---|---|---|
| Caspase-3 substrates | DEVD-afc, DEVD-fmk | Fluorometric activity measurement, inhibition | 12.5 μM substrate, 10-50 μM inhibitor |
| PARP1 antibodies | Cleaved PARP1 (Asp214) #9541, #5625 | Specific detection of 89-kDa fragment | WB: 1:1000, IF: 1:400, FC: 1:200-1:800 |
| Apoptosis inducers | Staurosporine, Actinomycin D, Etoposide | Experimental apoptosis induction | 0.5-1 μM, 1-5 μM, 10-50 μM respectively |
| PARP inhibitors | PJ34, ABT-888 | PARP catalytic activity inhibition | 10-20 μM, 1-10 μM respectively |
| Cell death modulators | zVAD-fmk (pan-caspase inhibitor) | Caspase activity blockade | 20-50 μM |
| Expression vectors | PARP1-D214N (non-cleavable mutant) | Cleavage-independent function analysis | Varies by transfection method |
The cleavage of PARP1 by caspase-3 at D214 represents a critical control point in cell fate decisions, generating 24-kD and 89-kD fragments with distinct biological activities. While the 24-kD fragment functions as a dominant-negative inhibitor of DNA repair, the 89-kD tPARP1 fragment exhibits novel signaling functions in the cytosol, including regulation of innate immune responses through RNA Pol III interaction and facilitation of parthanatos through PAR carrier activity. These emerging functions highlight the complex interplay between different cell death pathways and suggest new therapeutic targets for conditions involving dysregulated cell death, including cancer, neurodegenerative diseases, and ischemic injury. Continued investigation into the biological functions of tPARP1 fragments will undoubtedly yield additional insights into the sophisticated mechanisms controlling cellular survival and death.
During apoptosis, the full-length poly(ADP-ribose) polymerase 1 (PARP1) is cleaved by caspase-3 to generate a truncated fragment (tPARP1). This proteolytic event results in a fundamental reorganization of the protein's domain architecture, specifically removing the N-terminal zinc-finger domains responsible for DNA damage recognition while retaining the BRCT, WGR, and Catalytic domains. This structural transformation facilitates the translocation of tPARP1 from the nucleus to the cytoplasm and enables its interaction with novel binding partners, most notably the RNA polymerase III (Pol III) complex. This review comprehensively details the domain architecture of tPARP1, the functional consequences of this reorganization, and its emerging role in mediating cytosolic innate immune responses and cell death pathways, providing a critical framework for understanding its non-canonical functions beyond DNA repair.
Poly(ADP-ribose) polymerase 1 (PARP1) is a nuclear enzyme renowned for its role as a primary sensor of DNA damage. Upon activation by DNA strand breaks, it catalyzes the synthesis of poly(ADP-ribose) (PAR) chains onto itself and other nuclear proteins to initiate DNA repair pathways [11] [12]. A pivotal event in the execution phase of apoptosis is the caspase-mediated cleavage of PARP1. Caspase-3, a key effector caspase, primarily cleaves human PARP1 at aspartic acid 214 (D214), generating two distinct fragments: a 24 kDa N-terminal fragment and an 89 kDa C-terminal fragment known as truncated PARP1 (tPARP1) [5]. This cleavage event is considered a biochemical hallmark of apoptosis and fundamentally alters the protein's localization, interaction partners, and biological functions. The 24 kDa fragment, which contains the nuclear localization signal (NLS) and the first two zinc-finger domains, remains in the nucleus. In contrast, tPARP1, which loses the NLS, translocates from the nucleus to the cytoplasm, where it engages in novel, non-canonical functions that are distinct from its role in DNA repair [5]. The evolutionary conservation of PARP1 orthologs in lower organisms that naturally lack the N-terminal zinc fingers suggests that tPARP1 is not merely an inert byproduct of cleavage but possesses specific biological activities [5].
Comparative Domain Architecture: Full-Length PARP1 vs. tPARP1
The functional divergence between full-length PARP1 and tPARP1 is a direct consequence of their distinct structural compositions. The table below provides a detailed comparison of their domain architectures and the functional implications of these differences.
Table 1: Domain Architecture and Functional Comparison of Full-Length PARP1 and tPARP1
| Domain/Feature | Full-Length PARP1 (113 kDa) | tPARP1 (89 kDa) | Functional Consequence of Change |
|---|---|---|---|
| ZnF1 & ZnF2 | Present | Lost (cleaved off) | Loss of primary DNA break sensing; no longer occupies DNA ends in nucleus [5]. |
| ZnF3 | Present | Retained | Retained but functional role in tPARP1 context is less clear [5]. |
| BRCT Domain | Present | Retained | Becomes critical for novel protein-protein interactions (e.g., with Pol III complex) in the cytosol [5]. |
| WGR Domain | Present | Retained | May work in concert with the catalytic domain; potential role in nucleic acid binding or protein interactions [5] [3]. |
| Catalytic Domain (CAT) | Present | Retained | Retains enzymatic (ADP-ribosyl transferase) activity; can be activated in a DNA-damage-independent manner [5]. |
| Auto-modification Domain | Present (linkers around BRCT) | Partially Retained | Contains key PARylation sites; enables auto-modification and regulation [12]. |
| Nuclear Localization Signal (NLS) | Present | Lost (with ZnF1/ZnF2 fragment) | Relocates tPARP1 from the nucleus to the cytoplasm, accessing new substrates [5]. |
| Primary Localization | Nucleus | Cytoplasm | Enables interaction with cytosolic proteins and participation in innate immune signaling. |
| Primary Activator | DNA Strand Breaks | Unknown (possibly protein interactions) | Shifts the paradigm of PARP1 activation from a DNA-centric to a protein-centric mechanism. |
Detailed Analysis of Retained Domains in tPARP1
The BRCT Domain: From DNA Binding to Protein-Protein Interactions
In full-length PARP1, the BRCT domain has been recently identified as a DNA-binding domain that interacts with intact nucleosomal DNA, contributing to the "monkey-bar mechanism" that facilitates PARP1's rapid movement through chromatin [12]. However, in tPARP1, the BRCT domain undergoes a functional switch. It is repurposed for specific protein-protein interactions in the cytosol. Research has demonstrated that the BRCT domain of tPARP1 is both necessary and sufficient for recognizing and binding to the RNA polymerase III (Pol III) complex [5]. Mutational analysis, specifically a F473A mutation within the BRCT domain, disrupts its tertiary structure and abolishes this interaction, highlighting a critical and direct role for this domain in tPARP1's apoptotic function [5].
The WGR and Catalytic Domains: A Functional Unit
The WGR domain in full-length PARP1 is a key DNA-binding domain that works cooperatively with the zinc fingers and is essential for the DNA damage-induced allosteric activation of the catalytic domain [11] [12] [3]. In the activated state, the helical subdomain (HD) unfolds, allowing NAD+ access to the active site [11] [12]. While the precise mechanism of tPARP1 activation in the cytosol is not fully elucidated, it retains its catalytic capability. The WGR and Catalytic domains are retained in tPARP1, and it is hypothesized that they continue to function as a coordinated unit. The WGR domain may potentially interact with cytosolic nucleic acids (such as poly(dA-dT) used in experiments) or other proteins to transduce an activation signal to the catalytic domain, leading to PARylation activity independent of nuclear DNA damage [5].
Biological Functions and Signaling Pathways of tPARP1
tPARP1 in Innate Immune Activation and Apoptosis
The biological function of tPARP1 extends beyond the mere loss of its DNA repair capacity. Once in the cytoplasm, tPARP1 is positioned to participate in innate immune signaling pathways. Upon transfection of poly(dA-dT), which mimics pathogenic cytosolic DNA, cells undergo apoptosis and tPARP1 specifically interacts with the Pol III complex [5]. Pol III is known to transcribe foreign dsDNA into double-stranded RNA (dsRNA), which subsequently triggers type I interferon (IFN-β) production and apoptosis [5]. tPARP1 catalyzes the mono-ADP-ribosylation (MARylation) of Pol III, an event that facilitates its activation and the ensuing production of IFN-β [5]. This establishes a novel, non-canonical pathway where a cleaved fragment of a nuclear DNA repair enzyme directly modulates the cytosolic innate immune response.
Diagram 1: tPARP1 in Innate Immune Signaling and Apoptosis
USP10-PARP1 Positive Feedback Loop in DNA Repair
While not exclusive to tPARP1, recent research on the full-length protein reveals sophisticated regulatory mechanisms that may have implications for its truncated form. A deubiquitination-PARylation positive feedback loop between USP10 and PARP1 promotes DNA damage repair. Upon DNA damage and ROS generation, USP10 is recruited to deubiquitinate and stabilize PARP1 at the K418 site in an ATM-dependent manner. In turn, PARP1 PARylates USP10 at residues D634, D645, and E648, enhancing USP10's deubiquitination activity and creating a positive feedback loop that amplifies the DNA damage response [13]. This loop is highly relevant in breast cancer, where high PARP1 expression correlates with USP10 levels, and USP10 inhibition sensitizes cancer cells to PARP inhibitors [13]. The K418 ubiquitination site is located in the region retained in tPARP1, raising questions about whether similar regulatory mechanisms could influence tPARP1 stability or function in specific contexts.
Diagram 2: USP10-PARP1 Positive Feedback Loop
Experimental Approaches for Studying tPARP1
Key Methodologies and Workflows
Investigating the domain architecture and function of tPARP1 requires a combination of molecular biology, biochemistry, and cell-based assays. The following workflow outlines a standard experimental pipeline for validating tPARP1 interactions and functions.
Diagram 3: Experimental Workflow for tPARP1 Interaction and Function Studies
The Scientist's Toolkit: Essential Research Reagents
The table below catalogs key reagents and their applications for studying tPARP1, as derived from the cited methodologies.
Table 2: Research Reagent Solutions for tPARP1 Studies
| Reagent / Tool | Function / Application | Example Use in Context |
|---|---|---|
| PARP1-Deficient Cell Lines | Provides a clean background for expression of exogenous wild-type or mutant tPARP1 without interference from endogenous full-length PARP1. | Used to stably express SFB- or HA-tagged mtPARP1 (E988A) for affinity purification [5]. |
| Caspase-3 | The executioner protease that cleaves PARP1 to generate tPARP1. | Used in vitro to confirm cleavage or activated during apoptosis in cells. |
| Apoptosis Inducers (e.g., poly(dA-dT)) | Mimic pathogenic DNA to trigger the caspase cascade and subsequent PARP1 cleavage. | Transfected into cells to induce apoptosis and study tPARP1's cytosolic role [5]. |
| Tagged Constructs (SFB, HA, Myc) | Enable detection, immunoprecipitation, and purification of tPARP1 and its interactors. | SFB-tagged mtPARP1 used for tandem affinity purification; HA-tagged for Co-IP [5]. |
| Catalytic Mutant (E988A) | "Traps" substrates by forming a more stable enzyme-substrate complex, facilitating interaction partner identification. | Expressed in cells for unbiased proteomics to identify Pol III as a binding partner [5]. |
| Domain-Specific Mutants (e.g., F473A in BRCT) Disrupts specific domain function to establish its necessity in protein-protein interactions. | Used in Co-IP assays to demonstrate the essential role of the BRCT domain in binding Pol III subunits [5]. | |
| POLR3A, POLR3B, POLR3F Antibodies | Detect and immunoprecipitate specific subunits of the RNA Polymerase III complex. | Used in Co-IP and Western Blotting to confirm interaction with tPARP1 [5]. |
| PAR Antibody | Detects poly(ADP-ribose) chains, indicating PARP1/tPARP1 enzymatic activity. | Used in Western Blotting (in vitro and in cells) to confirm PARylation of Pol III [5]. |
| PARG/TARG1 Inhibitors | Block the degradation of PAR chains, stabilizing PARylation events for easier detection. | Could be used to augment detection of transient PARylation by tPARP1. |
The caspase-mediated cleavage of PARP1 and the subsequent generation of tPARP1 represent a fundamental shift in protein function driven by a dramatic alteration in domain architecture. The loss of the N-terminal zinc-finger domains disengages tPARP1 from its canonical role in nuclear DNA repair, while the retention of the BRCT, WGR, and Catalytic domains allows it to adopt a new function in the cytosol. The BRCT domain, in particular, is repurposed for a critical protein-protein interaction with the Pol III complex, enabling tPARP1 to modulate innate immune signaling and apoptosis through MARylation. This non-canonical pathway highlights the functional plasticity of PARP1 domains and adds a new layer of complexity to our understanding of cell death processes. From a therapeutic perspective, the distinct structure and function of tPARP1 present a unique opportunity. Targeting the specific interactions of tPARP1, such as its binding to Pol III via the BRCT domain, could lead to novel strategies for modulating immune responses or sensitizing cancer cells to apoptosis, potentially overcoming limitations associated with traditional catalytic PARP inhibitors. Future research should focus on elucidating the precise mechanism of tPARP1 activation in the cytosol and identifying other potential binding partners and substrates to fully unravel its biological significance in cell death and disease.
This review explores the evolutionary and functional significance of poly(ADP-ribose) polymerase 1 (PARP1) orthologs that naturally lack the N-terminal zinc finger domains. PARP1 is a critical DNA damage sensor and regulator of cell death pathways. While human PARP1 contains three zinc fingers (ZnF1, ZnF2, ZnF3) in its DNA-binding domain, bioinformatic analyses reveal that orthologs in several lower eukaryotes naturally lack the first two zinc fingers, resembling the truncated PARP1 (tPARP1) fragment generated during apoptosis in higher organisms. The conservation of this architecture through evolution suggests these truncated forms represent functional adaptations rather than merely degenerative states. This analysis, framed within the broader context of truncated PARP-1 fragments in cell death research, provides insights for developing targeted therapeutic strategies that exploit these natural structural variations.
PARP1 plays a dual role in cellular homeostasis, functioning in DNA damage repair under mild stress while promoting cell death pathways under severe damage. The protein's modular structure includes N-terminal zinc fingers for DNA binding, a central automodification domain (AMD), and a C-terminal catalytic domain (CAT). In humans, caspase-mediated cleavage during apoptosis generates truncated PARP1 (tPARP1) fragments, a process long considered a hallmark of programmed cell death [1].
Recent evolutionary analyses challenge this paradigm, revealing that the last common ancestor of extant eukaryotes encoded at least two PARP proteins, with one resembling human PARP1 and functioning in DNA damage response [14]. Surprisingly, PARP1 orthologs in several lower organisms naturally lack the first two N-terminal zinc finger motifs, mirroring the tPARP1 fragment generated during apoptosis in mammals [5]. This conservation across diverse lineages suggests positive selection for these truncated forms, indicating they represent functional adaptations with distinct biological roles rather than merely incomplete proteins.
This review synthesizes evidence from evolutionary biology, structural studies, and functional assays to examine the significance of naturally truncated PARP1 orthologs, their mechanisms of action, and their implications for understanding PARP1's role in cell death pathways.
Structural Organization of PARP1: Full-Length and Natural Variants
Domain Architecture of Canonical PARP1
Human PARP1 is a 1014-amino acid protein comprising three primary functional regions:
- DNA-binding domain (DBD): Contains three zinc finger motifs (ZnF1, ZnF2, ZnF3) and nuclear localization signals
- Automodification domain (AMD): Features a BRCT (Breast cancer-associated C-terminal) domain and WGR (Trp-Gly-Arg rich) domain
- Catalytic domain (CAT): Comprises helical subdomain (HD) and ADP-ribosyl transferase (ART) subdomain with NAD+ binding site [3] [15]
The zinc fingers exhibit specialized functions: ZnF1 and ZnF2 collaboratively recognize DNA strand breaks, while ZnF3 links structural domains to activate target proteins [16] [3].
Naturally Occurring Truncated PARP1 Orthologs
Evolutionary studies identify PARP1 orthologs in lower eukaryotes that naturally lack the first two zinc finger domains, resembling the apoptotic fragment of human PARP1. These naturally truncated forms contain:
- The third zinc finger (ZnF3)
- BRCT domain
- WGR domain
- C-terminal catalytic domain [5]
Table 1: Comparison of PARP1 Structural Variants Across Species
| Organism Category | Zinc Finger Composition | Structural Features | Functional Implications |
|---|---|---|---|
| Human (Full-length) | ZnF1, ZnF2, ZnF3 | Complete DNA-binding domain | Robust DNA damage recognition and repair |
| Human tPARP1 (Apoptotic) | ZnF3 only (lacks ZnF1-2) | Caspase-cleaved fragment | Altered function in cell death pathways |
| Lower Eukaryote Orthologs | ZnF3 only (naturally lacking ZnF1-2) | Evolutionary conserved truncation | Adapted DNA recognition mechanisms |
The conservation of this truncated architecture through evolution suggests these variants represent functional specialization rather than gene degradation [5] [14].
Evolutionary History of PARP1 Structural Diversity
Phylogenetic Distribution of PARP1 Variants
Comprehensive phylogenetic analyses of PARP genes across eukaryotic supergroups reveal:
- The ancestral eukaryote encoded at least two PARP proteins with different functions and activities
- One ancestral PARP was similar to human PARP1 and functioned in DNA damage response
- PARP1 genes are found in all eukaryotic supergroups, though some lineages have independently lost these genes
- The PARP superfamily can be subdivided into six clades, with two present in the last common eukaryotic ancestor [14]
Evolutionary Trajectory of Zinc Finger Domains
The evolutionary persistence of PARP1 variants lacking ZnF1-2 domains indicates positive selection for this structural configuration. Key findings include:
- Domain architecture comparisons show PARP1 orthologs in several lower organisms do not have the two N-terminal zinc fingers or even lack the third zinc finger motif
- This natural structural variation predates the evolutionary emergence of caspase-mediated apoptosis
- The conservation pattern suggests subfunctionalization or neofunctionalization of truncated PARP1 forms in specific biological contexts [5]
Table 2: Evolutionary Evidence for Naturally Truncated PARP1 Orthologs
| Evidence Type | Key Findings | Supporting References |
|---|---|---|
| Phylogenetic Analysis | PARP1 orthologs in lower eukaryotes naturally lack first two zinc fingers | [5] [14] |
| Domain Architecture | Similarity to apoptotic tPARP1 fragment in humans | [5] [15] |
| Functional Conservation | Retention of catalytic activity despite structural differences | [5] [14] |
Functional Mechanisms of Truncated PARP1 Orthologs
Altered DNA Recognition Mechanisms
While full-length PARP1 utilizes ZnF1 and ZnF2 as primary DNA damage sensors, truncated orthologs have evolved alternative recognition mechanisms:
- ZnF3-dependent binding: The third zinc finger maintains DNA binding capability, though with altered specificity
- WGR domain involvement: The WGR domain, which can interact with DNA, may play a heightened role in DNA recognition
- Collaborative domain interactions: Truncated forms likely rely on enhanced cooperation between remaining domains for DNA engagement [16] [15]
Structural studies reveal that ZnF1 and ZnF2 from separate PARP1 molecules form a strand-break recognition module that facilitates dimerization and trans-automodification, a mechanism necessarily altered in truncated variants [16].
Regulatory Functions in Cell Death Pathways
Naturally truncated PARP1 orthologs exhibit functional capabilities relevant to cell death regulation:
- Cytosolic localization: Unlike nuclear full-length PARP1, truncated forms may localize to cytoplasm
- Alternative binding partners: tPARP1 recognizes RNA polymerase III (Pol III) complex in cytosol during apoptosis
- Altered catalytic regulation: The basal activity of truncated PARP1 is inhibited by PAR, unlike DNA-stimulated full-length PARP1 [5] [15]
During poly(dA-dT)-stimulated apoptosis, tPARP1 mono-ADP-ribosylates RNA Pol III, facilitating IFN-β production and apoptosis, representing a distinct mechanism from canonical PARP1 function [5].
Diagram 1: Functional divergence of PARP1 variants in DNA damage response
Experimental Approaches for Studying Truncated PARP1 Orthologs
Structural Characterization Methods
X-ray Crystallography of PARP1-DNA Complexes
- Purpose: Determine atomic-level structure of PARP1 domains bound to DNA
- Methodology:
- Express and purify PARP1 DNA-binding domain (residues 5-202) in insect cells
- Co-crystallize with DNA molecules containing single-strand breaks
- Solve structure using molecular replacement and refine at 3.1Å resolution
- Key Findings: Revealed dimeric assembly where ZnF1 and ZnF2 from separate PARP1 molecules form strand-break recognition module [16]
Hybrid NMR/X-ray Crystallography for Complex Structures
- Purpose: Determine structure of complexes with weak binding or flexibility
- Methodology:
- Produce untagged, 15N-labelled or 15N,13C labelled protein versions
- Use multinuclear 2D and 3D NMR experiments for signal assignments
- Calculate structures using inter- and intra-molecular NOE-derived distance constraints
- Incorporate known crystal structure domains as fixed templates
- Applications: Suitable for characterizing flexible multi-domain proteins and their complexes [17]
Functional Assays for Truncated PARP1
In Vitro PARylation Assays
- Purpose: Assess PARylation activity of PARP1 variants
- Methodology:
- Incubate PARP1 variants with NAD+ substrate and DNA templates
- Detect PAR synthesis using SDS-PAGE or immunoblotting
- Measure kinetics under varying conditions (presence/absence of DNA, PAR inhibitors)
- Key Findings: PAR inhibits basal activity of PARP1ΔZnF1-2; ZnF1-2PARP1 complements PARP1ΔZnF1-2 for DNA-dependent activation [15]
Surface Plasmon Resonance (SPR) Binding Studies
- Purpose: Quantify protein-protein and protein-DNA interactions
- Methodology:
- Immobilize binding partners on sensor chips
- Pass analytes over surface and measure binding kinetics
- Determine equilibrium (KD) and kinetic (kon, koff) parameters
- Key Findings: tPARP1 binds VPS29 with KD of 2-3 µM; binding requires Zn++ coordination [17]
Co-immunoprecipitation (Co-IP) Assays
- Purpose: Identify protein-protein interactions in cellular contexts
- Methodology:
- Express tagged proteins (e.g., HA-tagged mtPARP1, myc-tagged Pol III subunits)
- Immunoprecipitate with tag-specific antibodies
- Detect co-precipitating partners by immunoblotting
- Key Findings: tPARP1 interacts with POLR3A, POLR3B, and POLR3F subunits of Pol III complex during apoptosis [5]
Diagram 2: Experimental workflow for characterizing truncated PARP1 orthologs
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Studying Truncated PARP1 Orthologs
| Reagent Category | Specific Examples | Research Applications | Key Functions |
|---|---|---|---|
| PARP1 Expression Constructs | PARP1ΔZnF1-2 (aa: 215-1014); ZnF1-2PARP1 (aa: 1-214) | Functional studies of apoptotic fragments | Express specific PARP1 domains and truncations |
| DNA Damage Probes | Oligonucleotides with single-strand breaks; poly(dA-dT) | Apoptosis induction; DNA binding assays | Activate PARP1; study DNA recognition mechanisms |
| Activity Assay Components | NAD+; anti-PAR antibodies; PARG inhibitors | PARylation assays; activity quantification | Measure catalytic activity and PAR synthesis |
| Interaction Partners | RNA Pol III subunits; VPS29; caspase enzymes | Protein-protein interaction studies | Identify novel binding partners and pathways |
| Structural Biology Tools | 15N/13C isotopic labeling; crystallization screens | NMR; X-ray crystallography | Determine atomic structures of complexes |
Research Implications and Future Directions
The evolutionary conservation of truncated PARP1 orthologs provides fundamental insights with significant research applications:
Implications for Drug Development
Understanding natural PARP1 variants offers strategic opportunities for therapeutic innovation:
- Selective inhibition: Truncated PARP1 forms may be differentially sensitive to PARP inhibitors, enabling selective targeting
- Alternative binding sites: The structural differences in truncated orthologs reveal alternative binding pockets for drug development
- Combination therapies: Leveraging the distinct functions of PARP1 variants could inform rational combination therapies
Future Research Priorities
Key unanswered questions present compelling research directions:
- Functional repertoire: Systematic characterization of biological processes regulated by truncated PARP1 orthologs across species
- Regulatory mechanisms: Elucidation of how naturally truncated forms are integrated into cellular signaling networks
- Pathological contexts: Investigation of whether human diseases involve the re-emergence of ancestral PARP1 functions
- Therapeutic exploitation: Development of strategies to selectively target different PARP1 structural forms for precision medicine
The evolutionary conservation of PARP1 orthologs that naturally lack N-terminal zinc fingers provides compelling evidence that these truncated forms represent functional adaptations rather than degenerative states. These natural variants mirror the apoptotic tPARP1 fragment in humans, suggesting deep evolutionary roots for PARP1's dual role in DNA repair and cell death regulation. The structural and functional insights from these orthologs reveal alternative mechanisms for DNA recognition, catalytic regulation, and pathway engagement that expand our understanding of PARP biology beyond the canonical model.
From a therapeutic perspective, these evolutionary insights highlight potential strategies for developing more selective PARP-targeted agents that exploit structural differences between full-length and truncated forms. Furthermore, understanding how naturally occurring PARP1 variants integrate into cell death pathways may reveal novel regulatory mechanisms that could be harnessed for controlling cell fate decisions in pathological contexts. As research continues to unravel the functional significance of these evolutionary adaptations, we anticipate new opportunities for leveraging this knowledge in drug development and therapeutic interventions.
Poly(ADP-ribose) polymerase 1 (PARP1) is a nuclear protein with well-established roles in DNA damage repair and maintenance of genomic integrity. Following activation by DNA strand breaks, PARP1 catalyzes the synthesis of poly(ADP-ribose) (PAR) chains on target proteins, facilitating the recruitment of DNA repair machinery [18] [4]. Beyond its nuclear functions, PARP1 plays a decisive role in cell fate decisions through the action of its proteolytic fragments. During apoptosis, PARP1 is a primary substrate for executioner caspases-3 and -7, which cleave the 116-kDa full-length protein into two major fragments: a 24-kDa DNA-binding fragment and an 89-kDa fragment (tPARP1) containing the automodification and catalytic domains [18] [4] [19]. This cleavage event was initially considered merely an apoptotic hallmark that inactivated DNA repair to facilitate cellular demise. However, emerging evidence demonstrates that the 89-kDa fragment undergoes active nuclear export and executes specific cytoplasmic functions that actively contribute to cell death pathways [18] [5] [20]. This whitepaper examines the mechanisms underlying the subcellular relocalization of the 89-kDa PARP1 fragment and its cytosolic functions, framing these processes within the broader context of truncated PARP1 fragments in cell death research.
Proteolytic Generation and Nuclear Export of the 89-kDa Fragment
Caspase-Mediated Cleavage and Fragment Characteristics
Caspase-mediated cleavage of PARP1 occurs at a specific aspartate residue (D214 in human PARP1) located within a nuclear localization signal (NLS) near the DNA-binding domain [4] [19]. This proteolytic event generates two distinct fragments with different properties and cellular destinations:
- The 24-kDa N-terminal fragment: Contains the DNA-binding domain with two zinc finger motifs and the NLS. This fragment remains tightly bound to DNA strand breaks in the nucleus, acting as a trans-dominant inhibitor of DNA repair by blocking access to DNA damage sites [4] [19].
- The 89-kDa C-terminal fragment (tPARP1): Comprises the automodification domain (with BRCT fold), the WGR domain, and the catalytic domain. This fragment loses its strong nuclear localization capability due to the cleavage within the NLS and becomes primed for nuclear export [4] [5].
Table 1: Characteristics of PARP1 Cleavage Fragments
| Feature | 24-kDa Fragment | 89-kDa Fragment (tPARP1) |
|---|---|---|
| Domains Contained | DNA-binding domain (ZnF1, ZnF2) | Automodification domain, WGR domain, Catalytic domain |
| Nuclear Localization Signal | Retained (with partial cleavage site) | Disrupted by cleavage |
| Primary Localization | Nuclear (bound to DNA lesions) | Cytoplasmic (after export) |
| Function | Dominant-negative inhibitor of DNA repair | PAR carrier; Cytoplasmic signaling activator |
Mechanisms of Nuclear Export and Cytosolic Translocation
The nuclear export of the 89-kDa fragment represents a critical step in its non-canonical functions. Research indicates that this process is facilitated by two potentially complementary mechanisms:
Passive Diffusion and Active Transport: Cleavage within the NLS disrupts the efficient nuclear import of the 89-kDa fragment, allowing its passive diffusion to the cytoplasm or active export via nuclear export signals [4]. Once in the cytoplasm, the fragment can execute its signaling functions.
Vesicle-Mediated Translocation: Recent evidence suggests an alternative pathway involving vesicular translocation. Studies in microglia show that PARP1 can translocate from the nucleus to the cytoplasm in vesicular structures upon inflammatory stimulation [21]. These PARP1-containing vesicles show colocalization with Lamin A/C, suggesting they might derive from the nuclear envelope through budding processes [21]. This vesicular transport mechanism may also apply to the 89-kDa fragment during specific cell death contexts.
Cytosolic Functions of the 89-kDa PARP1 Fragment
Once exported to the cytoplasm, the 89-kDa PARP1 fragment executes at least two distinct pro-death functions through different molecular interactions.
PAR Carrier for AIF-Mediated Death
The 89-kDa fragment serves as a vehicle for transporting nuclear PAR polymers to the cytoplasm, where they trigger mitochondrial apoptosis-inducing factor (AIF) release—a process bridging caspase-dependent apoptosis and PAR-mediated parthanatos [18] [4] [20].
Molecular Mechanism: In response to apoptotic stimuli like staurosporine or actinomycin D, PARP1 undergoes auto-poly(ADP-ribosyl)ation before caspase cleavage. The generated 89-kDa fragment retains covalently attached PAR polymers through this process. Once translocated to the cytoplasm, the PAR moieties on the fragment bind directly to AIF, which is anchored to the mitochondrial membrane [18] [4]. This binding facilitates AIF release from mitochondria and its subsequent translocation to the nucleus, where it collaborates with other factors to trigger large-scale DNA fragmentation and nuclear condensation [18] [4] [20].
Biological Significance: This pathway establishes a caspase-mediated interaction between classical apoptosis and PAR-mediated parthanatos, expanding understanding of programmed cell death mechanisms and suggesting new therapeutic targets for conditions where these pathways are dysregulated [18] [20].
Activation of Cytosolic Nucleic Acid Sensing
Beyond AIF-mediated death, the 89-kDa fragment regulates innate immune responses during apoptosis through interactions with the RNA polymerase III (Pol III) complex [5].
Molecular Mechanism: In the cytoplasm, the BRCT domain of the 89-kDa fragment directly interacts with subunits of the Pol III complex (POLR3A, POLR3B, and POLR3F). This interaction enables tPARP1 to catalyze mono-ADP-ribosylation of Pol III, enhancing its activity in transcribing double-stranded DNA (such as foreign pathogenic DNA) into double-stranded RNA [5]. This dsRNA production then stimulates type I interferon responses (IFN-β production) and amplifies apoptosis during cytoplasmic DNA sensing [5].
Evolutionary Context: The functional significance of this cleavage fragment is highlighted by evolutionary conservation—PARP1 orthologs in several lower organisms naturally lack the first two zinc finger motifs, resembling the 89-kDa fragment and suggesting conserved biological functions for this form of the protein [5].
Table 2: Cytosolic Functions of the 89-kDa PARP1 Fragment
| Function | Molecular Mechanism | Downstream Effect | Biological Context |
|---|---|---|---|
| PAR Carrier for AIF Release | Fragment-bound PAR binds mitochondrial AIF | AIF release and nuclear translocation; Large-scale DNA fragmentation | Staurosporine/actinomycin D-induced apoptosis [18] [4] |
| Activation of Cytosolic DNA Sensing | BRCT domain-mediated interaction with and ADP-ribosylation of Pol III complex | Enhanced IFN-β production; Amplification of apoptosis | Cytosolic DNA-induced apoptosis (e.g., pathogen infection) [5] |
Experimental Analysis of 89-kDa Fragment Relocalization and Function
Key Methodologies and Workflows
Investigating the subcellular relocalization and functions of the 89-kDa PARP1 fragment requires integrated experimental approaches. Below is a generalized workflow for studying fragment translocation and AIF-mediated death:
Research Reagent Solutions
Table 3: Essential Research Reagents for Studying 89-kDa PARP1 Fragment Biology
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Apoptosis Inducers | Staurosporine, Actinomycin D | Activate caspase cascade leading to PARP1 cleavage [18] [4] |
| PARP Inhibitors | PJ34, ABT-888 (Veliparib) | Inhibit PARP1 enzymatic activity and PAR formation; validate PAR-dependent mechanisms [4] [21] |
| Caspase Inhibitors | zVAD-fmk | Pan-caspase inhibitor; blocks PARP1 cleavage and downstream events [4] |
| Cell Lines | HeLa, PARP1-deficient 293T, PARP1(-/-) MEFs | Model systems for genetic and pharmacological studies [4] [5] |
| Antibodies | Anti-PARP1 (full-length and cleaved), Anti-PAR, Anti-AIF | Detect cleavage, PAR formation, and AIF translocation via Western blot, IF [18] [4] [21] |
| Expression Vectors | tPARP1 constructs, PARP1 shRNA | Mechanistic studies through overexpression or knockdown [4] [5] |
Detailed Experimental Protocol
Based on methodologies from key studies, below is a representative protocol for investigating 89-kDa fragment relocalization and AIF-mediated death:
Objective: To assess 89-kDa PARP1 fragment translocation and its functional consequences in response to apoptotic stimuli.
Procedure:
- Cell Culture and Treatment: Culture HeLa or other appropriate cells under standard conditions. Induce apoptosis using staurosporine (e.g., 0.5-1 μM) or actinomycin D (e.g., 0.5-1 μg/mL) for 2-6 hours. Include control groups with PARP inhibitors (PJ34, 10 μM) or caspase inhibitors (zVAD-fmk, 20 μM) added 1 hour before apoptosis inducers [4].
- Subcellular Fractionation: Harvest cells and separate nuclear and cytoplasmic fractions using commercial kits or differential centrifugation. Validate fraction purity using markers (e.g., Lamin A/C for nucleus, tubulin for cytoplasm) [4].
- Western Blot Analysis: Resolve proteins from whole cell lysates or fractions by SDS-PAGE. Transfer to membranes and immunoblot using:
- Immunofluorescence Microscopy: Plate cells on coverslips, treat as above, then fix and permeabilize. Co-stain with:
- Viability Assessment: Perform parallel experiments using MTT, ATP-based, or dye exclusion assays to correlate biochemical events with cell death [4].
The nuclear export of the caspase-generated 89-kDa PARP1 fragment and its subsequent cytoplasmic functions represent a significant expansion of PARP1 biology beyond its nuclear DNA repair roles. The 89-kDa fragment acts as a molecular link between different cell death pathways, serving as a PAR carrier for AIF-mediated death and as a regulator of cytosolic nucleic acid sensing. These processes underscore the functional importance of truncated PARP1 fragments in cell death research. Understanding these mechanisms provides deeper insights into physiological cell death control and pathological cell loss in conditions where these pathways are dysregulated, offering new avenues for therapeutic intervention in cancer, neurodegenerative diseases, and inflammatory disorders.
The 24-kD DNA-Binding Fragment as a Trans-Dominant Inhibitor of DNA Repair
Within the intricate landscape of cell death, the cleavage of poly(ADP-ribose) polymerase-1 (PARP-1) by executioner caspases is a established biochemical hallmark of apoptosis. This proteolytic event generates distinct fragments, the roles of which extend beyond mere inactivation of the parent protein. This whitepaper delves into the specific function of the 24-kilodalton (kD) DNA-binding fragment (DBD), characterizing its mechanism as a trans-dominant inhibitor of DNA repair. We synthesize evidence demonstrating that this fragment is not a passive byproduct but an active participant in the apoptotic cascade, ensuring the irreversibility of cell death by binding irreversibly to DNA strand breaks and blocking DNA repair pathways. The implications of this mechanism for cancer therapy and neurodegenerative diseases are profound, offering a framework for understanding cell fate decisions and informing novel therapeutic strategies.
PARP-1 is a critical nuclear enzyme involved in the base excision repair (BER) pathway, acting as a primary sensor of DNA single-strand breaks (SSBs) [1] [22]. Upon detecting DNA damage, PARP-1 becomes activated and catalyzes the synthesis of poly(ADP-ribose) (PAR) chains on itself and other nuclear proteins, facilitating the recruitment of DNA repair machinery [22] [23].
During the execution phase of apoptosis, caspase-3 and -7 cleave PARP-1 at a specific amino acid sequence (Asp214/Glu-Val-Asp-Gly), producing two signature fragments: an 89-kD catalytic fragment and a 24-kD DNA-binding fragment [1] [5]. While the 89-kD fragment, containing the auto-modification and catalytic domains, is liberated from the nucleus into the cytosol, the 24-kD fragment, which contains the first two zinc finger motifs and the nuclear localization signal, is retained in the nucleus [1] [24]. The generation of these fragments is a key event in a cell's commitment to death, and their functions are central to the broader biological role of truncated PARP-1 in cell death research.
Molecular Mechanism of the 24-kD Fragment
The 24-kD fragment exerts its trans-dominant inhibitory effect through a high-affinity, irreversible binding to DNA strand breaks. This action effectively blocks the DNA damage repair process at its earliest stage.
Structural Basis for DNA Binding
The 24-kD fragment comprises the N-terminal DNA-binding domain of PARP-1, which includes two zinc finger motifs (ZF1 and ZF2) with a CCHC ligand pattern that is highly unusual among zinc fingers [24]. Biophysical and structural studies have shown that these fingers are structurally independent in the absence of DNA and share a highly similar fold [24]. Crucially, recognition of DNA damage is primarily achieved by ZF2, which interacts much more strongly with nicked or gapped DNA ligands than ZF1 [24]. The fragment recognizes DNA single-strand breaks as a monomer and in a single orientation, forming a 1:1 monomeric complex with the damaged DNA [24].
Table 1: Key Domains of the 24-kD DNA-Binding Fragment
| Domain/Motif | Description | Functional Role in Trans-Dominant Inhibition |
|---|---|---|
| Zinc Finger 1 (ZF1) | First of two N-terminal zinc finger motifs. | Contributes to DNA damage recognition; essential for initial binding to DNA strand breaks [24]. |
| Zinc Finger 2 (ZF2) | Second zinc finger motif, structurally similar to ZF1. | Primary mediator of high-affinity binding to DNA single-strand breaks and gaps [24]. |
| Nuclear Localization Signal (NLS) | Signal sequence located between ZF2 and ZF3 in full-length PARP-1. | Ensures the 24-kD fragment is retained in the nucleus following cleavage, enabling its access to genomic DNA [1]. |
| Caspase Cleavage Site | Aspartate residue at position 214 (within the DEVD sequence). | Target site for caspase-3/7; generation of the fragment is absolutely dependent on this cleavage event [1] [5]. |
The Trans-Dominant Inhibition Model
The model of trans-dominant inhibition posits that the 24-kD fragment, by virtue of retaining the DNA-binding capability of the full-length protein but lacking the catalytic and auto-modification domains, acts as a competitive and irreversible blocker.
- Sequestration of DNA Breaks: The fragment binds to DNA strand breaks with high affinity, occupying the sites where active, full-length PARP-1 would normally dock to initiate repair [1] [24].
- Prevention of Repair Enzyme Recruitment: By occupying the damage site, the 24-kD fragment acts as a physical barrier, preventing the recruitment of other DNA repair enzymes, including the base excision repair (BER) machinery [1].
- Inhibition of Full-Length PARP-1: Even if full-length PARP-1 is present in the cell, the 24-kD fragment bound to DNA breaks prevents its activation, thereby halting any attempted repair and the associated consumption of cellular ATP pools [1]. This conservation of energy is thought to facilitate the efficient execution of apoptosis.
The following diagram illustrates the transition from normal DNA repair to the state of trans-dominant inhibition following PARP-1 cleavage during apoptosis.
Experimental Evidence and Key Data
The trans-dominant inhibitor model is supported by a body of experimental data from cellular and biochemical studies.
Key Experimental Findings
Research has consistently shown that the expression of the isolated 24-kD DNA-binding domain is sufficient to suppress DNA repair and sensitize cells to DNA-damaging agents. Key evidence includes:
- In Vivo Suppression: Overexpression of the 24-kD fragment in cells acts as a dominant-negative, suppressing the activation of endogenous full-length PARP-1 at DNA damage sites and attenuating DNA repair [1] [24].
- Irreversible Binding: The 24-kD fragment, unlike full-length PARP-1 which is released following auto-modification, remains persistently bound to DNA strand breaks. This irreversible binding is the cornerstone of its inhibitory function [1].
- Conservation of Energy: By blocking PARP-1 activation, the fragment prevents the catastrophic depletion of NAD+ and ATP that characterizes PARP-1 overactivation in other cell death pathways like parthanatos, thereby conserving cellular energy for the efficient execution of apoptosis [1] [25].
Table 2: Summary of Quantitative Data on PARP-1 and its 24-kD Fragment
| Parameter | Full-Length PARP-1 | 24-kD DNA-Binding Fragment | Experimental Context & Notes |
|---|---|---|---|
| Molecular Weight | 113 kD / 1014 amino acids [23] | 24 kD [1] | Determined by SDS-PAGE. |
| Cellular Copies | ~1-2 million copies per cell [1] | N/A | Fragment levels depend on apoptotic stimulus. |
| DNA Binding Affinity | High (nanomolar range), reversible after auto-PARylation [22] | High (nanomolar range), irreversible [1] [24] | Binds as a monomer to DNA single-strand breaks [24]. |
| Primary Functional Domains | DBD, AMD, CAT (WGR, BRCT) [1] | Zinc Fingers 1 & 2, NLS [24] | DBD=DNA-Binding Domain; AMD=Auto-Modification Domain; CAT=Catalytic Domain. |
Research Methods and Protocols
Studying the formation and function of the 24-kD fragment requires a combination of molecular biology, biochemistry, and cell-based techniques.
Detecting PARP-1 Cleavage
Protocol: Western Blot Analysis for PARP-1 Cleavage Fragments
- Cell Lysis and Protein Extraction: Harvest cells (apoptotically induced and controls) and lyse using RIPA buffer supplemented with protease inhibitors (e.g., PMSF, aprotinin) and a caspase inhibitor (e.g., Z-VAD-FMK) in control samples to prevent artifactual cleavage during processing.
- Protein Quantification: Determine protein concentration using a Bradford or BCA assay. Load equal amounts of protein (20-40 µg) onto a 4-12% Bis-Tris polyacrylamide gel for SDS-PAGE separation, which optimally resolves proteins in the 10-250 kD range.
- Electrophoresis and Transfer: Run the gel at constant voltage (120-150V) until adequate separation is achieved. Transfer proteins from the gel to a PVDF or nitrocellulose membrane using a wet or semi-dry transfer system.
- Immunoblotting:
- Block the membrane with 5% non-fat milk in TBST (Tris-Buffered Saline with Tween-20) for 1 hour at room temperature.
- Incubate with primary antibody overnight at 4°C. Critical Antibodies:
- Anti-PARP-1 antibody (C-terminal specific): Detects both full-length PARP-1 (~116 kD) and the 89-kD cleavage fragment.
- Anti-PARP-1 [Asp214] cleaved antibody: Specifically detects the 24-kD fragment generated by caspase cleavage.
- Wash the membrane 3 times for 5 minutes each with TBST.
- Incubate with an appropriate HRP-conjugated secondary antibody for 1 hour at room temperature. Wash again 3 times with TBST.
- Detection: Develop the blot using a chemiluminescent substrate and image with a digital imager. The appearance of the 89-kD and 24-kD bands, concomitant with the decrease of the full-length 116-kD band, is a definitive marker of caspase-mediated apoptosis.
Assessing DNA Repair Inhibition
Protocol: Immunofluorescence for DNA Damage Foci Co-localization
This protocol assesses whether the 24-kD fragment prevents the recruitment of repair factors to DNA damage sites.
- Cell Culture and Transfection: Culture cells on glass coverslips. Transfect with a plasmid expressing the isolated 24-kD DBD fragment (or a GFP-tagged version) or induce apoptosis.
- DNA Damage Induction: Treat cells with a DNA-damaging agent (e.g., H₂O₂ for oxidative stress, etoposide for topoisomerase II inhibition, or laser micro-irradiation for localized damage).
- Fixation and Permeabilization: At designated time points post-damage, fix cells with 4% paraformaldehyde for 15 minutes and permeabilize with 0.2% Triton X-100 in PBS for 10 minutes.
- Immunostaining:
- Block with 3% BSA in PBS for 30 minutes.
- Incubate with primary antibodies in blocking buffer for 1-2 hours. Key combinations include:
- Mouse anti-GFP (to detect transfected 24-kD-GFP).
- Rabbit anti-γH2AX (a marker for DNA double-strand breaks).
- Rabbit anti-XRCC1 (a key scaffold protein in SSB repair).
- Wash with PBS and incubate with fluorescently labeled secondary antibodies (e.g., Alexa Fluor 488 anti-mouse, Alexa Fluor 594 anti-rabbit) for 45 minutes in the dark. Counterstain nuclei with DAPI.
- Imaging and Analysis: Image cells using a confocal microscope. In control cells, XRCC1 and γH2AX should form distinct foci at sites of DNA damage. In cells expressing the 24-kD fragment, measure the reduction in co-localization of these repair factors with the 24-kD fragment itself, indicating successful blockade of repair machinery recruitment.
The following workflow summarizes the key experimental approaches for investigating the 24-kD fragment.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Studying the 24-kD PARP-1 Fragment
| Reagent / Assay | Function / Utility | Specific Example / Application |
|---|---|---|
| Anti-PARP-1 Cleavage Site Antibody | Specifically detects the 24-kD fragment generated by caspase-3 cleavage at Asp214. Essential for confirming apoptotic cleavage vs. other proteolytic events. | Rabbit anti-human PARP1 [Asp214] cleaved antibody; used in Western blotting and IF [1] [5]. |
| Caspase-3/7 Activity Assay | Quantifies the enzymatic activity of executioner caspases, providing context for PARP-1 cleavage. | Fluorometric or colorimetric assays based on cleavage of DEVD-pNA or DEVD-AFC substrates. |
| PARP-1 DNA-Binding Domain (DBD) Plasmid | Plasmid for expressing the isolated 24-kD fragment (or a tagged version) in cells. Critical for dominant-negative experiments. | GFP-tagged human PARP-1 DBD (aa 1-214) for transfection, tracking localization, and pull-down assays [24]. |
| DNA Damage Inducers | Agents used to create defined DNA lesions to probe the fragment's inhibitory function. | H₂O₂ (oxidative strand breaks), Etoposide (topoisomerase II-induced DSBs), Methyl methanesulfonate (MMS; alkylating agent) [1]. |
| Repair Protein Antibodies (e.g., XRCC1, γH2AX) | Markers for DNA damage and repair. Used to visualize repair foci and test if their formation is blocked by the 24-kD fragment. | Mouse anti-γH2AX (Ser139), Rabbit anti-XRCC1; used in immunofluorescence and Western blotting [1] [22]. |
Implications for Disease and Therapeutics
The functional role of the 24-kD PARP-1 fragment has significant implications across pathophysiology and drug discovery.
- Cancer Therapy: The irreversible DNA binding of the 24-kD fragment underscores the importance of PARP trapping as a mechanism for several PARP inhibitors (PARPi) used in oncology [22]. These inhibitors not only block PARP catalytic activity but also stabilize PARP-DNA complexes, mimicking the action of the 24-kD fragment and preventing DNA repair, which is synthetically lethal in BRCA-deficient cancers [26] [22].
- Neurodegenerative Diseases: In contrast, in contexts of extensive DNA damage such as in neurodegenerative diseases (Alzheimer's, Parkinson's), PARP-1 overactivation can lead to energy depletion and parthanatos [25]. Here, the caspase-mediated shutdown of PARP-1 via cleavage into the 24-kD and 89-kD fragments can be viewed as a protective mechanism to prevent necrotic cell death and inflammation [1] [25]. The balance between these cell death modalities is a key area of investigation.
The 24-kD DNA-binding fragment of PARP-1 is a critical molecular switch in cell fate determination. Its function as a trans-dominant inhibitor of DNA repair ensures that once a cell commits to apoptosis, the DNA repair machinery is decisively disengaged, promoting the irreversible execution of the cell death program. This mechanism bridges the gap between DNA damage sensing and the point of no return in apoptosis, highlighting the sophisticated regulatory networks that govern cellular life and death. Further research into modulating this pathway holds promise for enhancing the efficacy of cancer therapies and developing new interventions for neurodegenerative disorders.
Research Tools and Therapeutic Translation: Studying and Harnessing tPARP1 Functions
Poly(ADP-ribose) polymerase 1 (PARP1) is a nuclear enzyme crucial for DNA damage repair, transcriptional regulation, and the maintenance of genomic integrity. A pivotal event in its functional repertoire is its proteolytic cleavage during programmed cell death. This cleavage, primarily mediated by caspase-3 at the conserved aspartate residue 214 (within the DEVD sequence), serves as a well-established biochemical hallmark of apoptosis [27] [1]. The cleavage event generates two distinct fragments: a 24 kDa N-terminal DNA-binding domain (DBD) fragment and an 89 kDa C-terminal fragment, often referred to as truncated PARP1 (tPARP1) [27] [5]. For decades, the appearance of these fragments was viewed primarily as a marker of apoptosis, but emerging research reveals that the fragments, particularly tPARP1, are not merely inert byproducts but possess unique biological activities that actively regulate cell death pathways and inflammatory responses [27] [5] [28]. Consequently, antibodies capable of specifically detecting full-length PARP1, its cleavage fragments, and cleavage-site mutants have become indispensable tools for dissecting the complex roles of PARP1 in cell death, inflammation, and disease pathogenesis. This technical guide provides an in-depth overview of these critical research reagents and their applications.
PARP1 Cleavage Fragments: Signatures and Functions
Fragment Specification and Detection
The cleavage of PARP1 by caspases results in specific fragments with distinct domains and molecular weights, which can be resolved and identified using Western blotting with specific antibodies. The table below summarizes the key characteristics of these fragments.
Table 1: PARP1 Proteolytic Fragments and Their Characteristics
| Fragment Name | Molecular Weight | Domains Contained | Cellular Localization Post-Cleavage | Primary Function |
|---|---|---|---|---|
| Full-Length PARP1 | 113-116 kDa [29] [30] | DBD (Zn1, Zn2, Zn3), AMD, CAT [3] | Nucleus | DNA damage repair, transcription regulation |
| tPARP1 (89 kDa Fragment) | 85-89 kDa [27] [30] | Zn3, AMD, CAT (WGR, BRCT) [5] | Cytosol [5] | Mediates ADP-ribosylation of RNA Pol III, promotes IFN-β production and apoptosis [5] |
| 24 kDa Fragment | 24 kDa [27] | DBD (Zn1, Zn2) [1] | Nucleus [5] | Acts as a trans-dominant inhibitor of PARP1 by blocking DNA repair [1] |
Biological Functions of Cleavage Fragments
The biological significance of PARP1 cleavage extends far beyond inactivating the DNA repair function of the full-length protein.
- The 24 kDa Fragment: This N-terminal fragment, which contains the first two zinc finger motifs, remains tightly bound to DNA strand breaks. This binding acts as a trans-dominant inhibitor, preventing other DNA repair enzymes, including intact PARP1, from accessing and repairing damaged DNA, thereby facilitating the apoptotic process [1].
- The tPARP1 (89 kDa) Fragment: Once considered to lack significant function, recent studies have uncovered a novel and active role for tPARP1. Following cleavage, tPARP1 translocates to the cytosol [5]. Through its BRCT domain, it interacts with the RNA Polymerase III (Pol III) complex [5]. tPARP1 then mono-ADP-ribosylates Pol III, which enhances the transcription of foreign DNA (e.g., from pathogens) and facilitates the production of interferon-beta (IFN-β), thereby amplifying the innate immune response and apoptosis during cellular stress [5] [28].
- Functional Opposition of Fragments: Research indicates that these fragments can regulate cellular viability in opposing ways. Expression of the 24 kDa fragment or an uncleavable PARP1 mutant (PARP-1UNCL) conferred protection from ischemic damage in neuronal models, whereas expression of the 89 kDa tPARP1 fragment was cytotoxic [27].
The following diagram illustrates the PARP1 cleavage process and the subsequent functions of its fragments.
The Research Toolkit: Antibodies and Critical Reagents
The specific detection of PARP1 and its cleavage fragments relies on a suite of well-characterized antibodies and engineered molecular tools.
Antibodies for Detecting PARP1 and Its Cleavages
Antibodies used in PARP1 cleavage detection are often characterized by their specific recognition of epitopes in different domains, allowing for the differentiation between full-length and cleaved forms.
Table 2: Key Antibody Reagents for PARP1 Cleavage Research
| Antibody Clone / Name | Host & Isotype | Reactive Species | Applications | Key Feature / Epitope | Catalog Example & Details |
|---|---|---|---|---|---|
| Clone 123 | Mouse / IgG1 [31] | Human, Dog, Horse, Mouse, Rat [31] | WB, IHC, ICC/IF, IP [31] | Binds C-terminal region of human PARP1 [31] | Thermo Fisher (#436400) [31] |
| EPR18461 (ab191217) | Rabbit / IgG [29] | Human, Mouse, Rat [29] | WB, IHC, ICC/IF [29] | KO-validated; detects full-length (113 kDa) and cleaved (89 kDa) forms [29] | Abcam (ab191217) [29] |
| 1D7D4 (66520-1-PBS) | Mouse / IgG1 [30] | Human, Mouse, Rat [30] | WB, IHC, IF/ICC, IP, ELISA [30] | Binds N-terminal region (1-327 aa); detects full-length and cleavage fragments [30] | PTGLab (66520-1-PBS) [30] |
Cleavage-Site Mutants as Essential Research Tools
To investigate the functional consequences of PARP1 cleavage, researchers employ cleavage-site mutants and truncated constructs.
- Uncleavable PARP1 (PARP-1UNCL): This mutant, where the aspartate at the caspase cleavage site (D214) is mutated, cannot be cleaved by caspases. Studies using PARP-1UNCL have demonstrated its cytoprotective effects in models of ischemia and inflammation, highlighting the critical role of cleavage in promoting cell death [27].
- Expression Constructs for Fragments (PARP-124 and PARP-189): Plasmids or viral vectors allowing for the direct expression of the 24 kDa (PARP-124) or 89 kDa (PARP-189) fragments are used to study their individual functions. As noted, PARP-124 can be protective, while PARP-189 expression induces cytotoxicity and enhances NF-κB and iNOS activity [27].
Table 3: Engineered PARP1 Constructs for Functional Studies
| Construct Name | Description | Utility in Research | Key Experimental Finding |
|---|---|---|---|
| PARP-1UNCL | Uncleavable mutant (D214 mutation) [27] | To study the effect of blocking PARP1 cleavage | Confers protection from ischemic and inflammatory damage [27] |
| PARP-124 | Expression construct for the 24 kDa N-terminal fragment [27] | To study the function of the DNA-binding fragment alone | Mimics the cytoprotective effect of PARP-1UNCL [27] |
| PARP-189 (tPARP1) | Expression construct for the 89 kDa C-terminal fragment [27] [5] | To study the function of the truncated catalytic fragment alone | Is cytotoxic and enhances pro-inflammatory NF-κB signaling [27] |
Experimental Protocols for Detection and Functional Analysis
Standard Western Blotting Protocol for PARP1 Cleavage Detection
This protocol is adapted from methodologies cited across multiple sources [31] [27] [29].
Sample Preparation:
Gel Electrophoresis and Transfer:
- Load 10-30 µg of total protein per lane on a 4-12% Bis-Tris polyacrylamide gel [32].
- Perform electrophoresis and subsequently transfer proteins to a PVDF or nitrocellulose membrane.
Immunoblotting:
- Block the membrane with 5% non-fat dry milk (NFDM) in TBST for 1 hour at room temperature.
- Incubate with primary antibody (e.g., anti-PARP1 at 1:1,000 to 1:10,000 dilution [29]) in 1% BSA or 5% NFDM/TBST overnight at 4°C.
- Wash the membrane and incubate with an appropriate HRP-conjugated secondary antibody (e.g., 1:50,000 dilution [29]) for 1 hour at room temperature.
- Detect using enhanced chemiluminescence (ECL) reagents [32].
Expected Results: A successful blot will show a band at 113-116 kDa (full-length PARP1) and, in apoptotic samples, a band at 85-89 kDa (tPARP1) [29] [30]. The 24 kDa fragment is less commonly detected in standard Western blots but can be observed under optimized conditions.
Co-immunoprecipitation (Co-IP) to Identify tPARP1-Binding Partners
This protocol is based on the unbiased approach used to identify the interaction between tPARP1 and the RNA Pol III complex [5].
Cell Transfection and Treatment:
- Stably express a catalytically inactive, tagged tPARP1 mutant (e.g., SFB-tagged mtPARP1 with E988A mutation) in PARP1-deficient cells (e.g., 293T) to "trap" substrates.
- Induce apoptosis by transfecting with poly(dA-dT) to mimic pathogenic DNA [5].
Cell Lysis and Preparation:
- Lyse cells in a mild, non-denaturing lysis buffer (e.g., with 0.5% Triton X-100) to preserve protein interactions. Isolate the soluble fraction by centrifugation.
Affinity Purification:
- Incubate the cell lysate with streptavidin- and FLAG-conjugated beads for tandem affinity purification.
- Wash the beads extensively with lysis buffer to remove non-specifically bound proteins.
Elution and Analysis:
- Elute the bound protein complexes.
- Identify the interacting partners by mass spectrometric analysis [5].
- Validate specific interactions (e.g., between tPARP1 and POLR3A) by repeating the Co-IP with specific antibodies and immunoblotting.
Immunofluorescence (ICC/IF) for Localization Studies
This protocol is adapted from vendor datasheets and research articles [31] [29].
Cell Culture and Fixation:
- Culture cells (e.g., HeLa, A549) on glass coverslips until 70% confluent.
- Fix cells with 4% paraformaldehyde for 15 minutes at room temperature.
Permeabilization and Blocking:
Antibody Staining:
- Label cells with primary antibody (e.g., PARP1 Mouse Monoclonal at 1 µg/mL) in 1% BSA and incubate for 3 hours at room temperature [31].
- After washing, incubate with a fluorophore-conjugated secondary antibody (e.g., Alexa Fluor 488, at 1:1,000 dilution) for 1 hour [29].
- Counterstain the nucleus with DAPI.
Imaging and Analysis:
- Image using a confocal microscope. In healthy cells, PARP1 is predominantly nuclear. During apoptosis, the cleaved tPARP1 (89 kDa) can be detected in the cytosol, which can be visualized using specific antibodies [5].
Visualizing the Experimental Workflow
The following diagram outlines a logical workflow for a typical research project investigating PARP1 cleavage, integrating the reagents and methods described above.
The study of PARP1 cleavage has evolved from treating it as a simple apoptotic marker to understanding it as a critical regulatory mechanism that generates functionally active fragments with distinct roles in cell death and inflammation. The deployment of highly specific antibodies against different PARP1 domains and forms, coupled with the use of cleavage-site mutants and standardized experimental protocols, provides a powerful toolkit for researchers. These tools are essential for elucidating the nuanced biological functions of tPARP1 in health and disease, with significant implications for understanding cancer biology, neurodegenerative disorders, and the development of novel therapeutic strategies.
Poly (ADP-ribose) polymerase 1 (PARP1) is a nuclear enzyme with well-characterized roles in DNA damage repair and maintenance of genomic integrity. The proteolytic cleavage of PARP1 during programmed cell death represents a critical biochemical event that generates distinct truncated fragments (tPARP1) with potentially novel biological functions beyond their parental protein [1] [33]. During apoptosis, PARP1 is primarily cleaved by caspase-3 at aspartate 214, separating the 113-kDa full-length protein into two major fragments: a 24-kDa DNA-binding domain fragment that remains nuclear-localized, and an 89-kDa truncated PARP1 (tPARP1) encompassing the BRCT domain, WGR domain, and C-terminal catalytic domain that translocates to the cytoplasm [1] [34]. This cleavage event was historically viewed as merely an inactivation mechanism to conserve cellular ATP during apoptosis. However, emerging evidence suggests that tPARP1 fragments possess unique biological activities that actively contribute to cell death pathways and immune signaling [34] [33]. This technical guide explores the application of unbiased proteomics approaches to identify novel tPARP1 interactors, with particular emphasis on the groundbreaking discovery of RNA Polymerase III as a key binding partner and substrate.
PARP1 Domains and Cleavage Fragments
Structural Organization of PARP1
PARP1 exhibits a modular architecture consisting of several functionally specialized domains:
- DNA-binding domain (DBD): Contains three zinc finger motifs (Zn1, Zn2, Zn3) that recognize and bind to DNA strand breaks [1] [3]
- Automodification domain (AMD): Features a BRCT (BRCA1 C-terminal) domain that facilitates protein-protein interactions and serves as a target for auto-ADP-ribosylation [1]
- WGR domain: Connects the AMD to the catalytic domain and participates in DNA-dependent allosteric activation [34]
- Catalytic domain (CAT): Mediates poly(ADP-ribose) synthesis using NAD+ as substrate [1]
Cleavage Fragments and Their Properties
Caspase-mediated cleavage of PARP1 during apoptosis occurs within the DBD, generating two primary fragments with distinct cellular localizations and functions:
Table 1: PARP1 Cleavage Fragments and Their Characteristics
| Fragment | Molecular Weight | Domains Contained | Cellular Localization | Known Functions |
|---|---|---|---|---|
| 24-kDa DBD Fragment | 24 kDa | Zn1, Zn2, nuclear localization signal | Nuclear | Acts as trans-dominant inhibitor of PARP1; irreversibly binds damaged DNA [1] |
| 89-kDa tPARP1 Fragment | 89 kDa | Zn3, BRCT, WGR, CAT | Cytosolic translocation | Retains catalytic activity; mediates novel interactions including with Pol III [34] |
The following diagram illustrates the domain architecture of full-length PARP1 and its cleavage fragments:
Unbiased Proteomics Methodologies for tPARP1 Interactor Identification
Tandem Affinity Purification and Mass Spectrometry
The identification of novel tPARP1 interactors necessitates robust, unbiased proteomic approaches that can capture transient interactions while minimizing false positives. The following workflow outlines the key methodological steps:
Critical Experimental Considerations
Strategic Use of Catalytic Domain Mutants
A pivotal innovation in identifying genuine tPARP1 interactors involves the strategic mutation of the catalytic E988 residue to alanine (E988A). This mutation creates a substrate-trapping variant (mtPARP1) that stabilizes otherwise transient enzyme-substrate interactions by preventing ADP-ribose transfer and release [34]. This approach is particularly valuable for capturing interactions with proteins that undergo rapid ADP-ribosylation, as the modified proteins remain bound to the catalytically inactive tPARP1.
Complementary Trapping Strategies
To further validate identified interactors, complementary trapping approaches targeting ADP-ribosylation eraser enzymes can be employed:
- Mutant TARG1 (mTARG1): D125A mutation in TARG1, which normally removes mono-ADP-ribose modifications, creates a trapping variant for mono-ADP-ribosylated substrates [34]
- Mutant PARG (mPARG): E756A mutation in poly(ADP-ribose) glycohydrolase traps poly-ADP-ribosylated substrates [34]
Proteins identified through both mtPARP1 and mTARG1/mPARG approaches represent high-confidence candidates for genuine tPARP1 interactors and substrates.
Experimental Controls and Validation
Rigorous controls are essential for distinguishing specific interactors from non-specific background binding:
- Parallel experiments with full-length PARP1 containing the E988A mutation
- Competition assays with excess free NAD+ or PARP inhibitors
- Isotopic labeling strategies (SILAC, TMT) for quantitative comparison
- Orthogonal validation through co-immunoprecipitation and colocalization studies
Case Study: Identification of RNA Polymerase III as a Novel tPARP1 Interactor
Proteomic Discovery and Validation
The application of the aforementioned unbiased proteomics approach led to the seminal discovery that tPARP1 specifically interacts with the RNA Polymerase III (Pol III) complex in the cytosol during apoptosis [34]. This finding was particularly intriguing given Pol III's established role in transcribing foreign DNA during innate immune responses.
Key experimental evidence supporting this interaction includes:
- Mass spectrometry identification: POLR3A, POLR3B, and POLR3F (core subunits of Pol III) were consistently identified as common interactors with both mtPARP1 and mTARG1 [34]
- Co-immunoprecipitation confirmation: Interactions between tPARP1 and Pol III subunits were validated through conventional co-IP experiments in PARP1-deficient cells [34]
- Domain mapping: The BRCT domain of tPARP1 was identified as critical for mediating interactions with Pol III subunits, with the F473A mutation abolishing binding capacity [34]
- Apoptosis-dependent association: The tPARP1-Pol III interaction was enhanced during poly(dA-dT)-stimulated apoptosis, confirming the pathophysiological relevance [34]
Functional Consequences of the tPARP1-Pol III Interaction
The biological significance of this novel interaction extends to multiple cellular processes:
ADP-ribosylation of Pol III
tPARP1 catalyzes mono-ADP-ribosylation of Pol III subunits, which enhances the polymerase's transcriptional activity toward foreign DNA templates [34]. This modification creates a positive feedback loop that amplifies cytosolic DNA sensing and interferon-beta production during apoptotic processes triggered by pathogenic infections.
Enhancement of Innate Immune Signaling
The tPARP1-Pol III axis functions as an amplifier of innate immune responses by:
- Increasing transcription of foreign DNA into double-stranded RNA
- Enhancing RIG-I-mediated detection of viral pathogens
- Potentiating IFN-β production and apoptosis of infected cells [34]
Evolutionary Conservation Considerations
Interestingly, the domain architecture of tPARP1 (lacking the first two zinc fingers) resembles PARP1 orthologs in several lower organisms, suggesting that this truncated form may represent an evolutionarily conserved functional module rather than merely an inactivation byproduct [34].
Technical Protocols for tPARP1 Interactor Identification
Detailed Experimental Workflow
Construct Design and Preparation
- Plasmid design: Generate SFB (S-protein, Flag, Streptavidin-binding peptide) tagged constructs of wild-type tPARP1 and catalytic mutant mtPARP1 (E988A) in mammalian expression vectors
- Control constructs: Create parallel constructs for full-length PARP1 with equivalent mutations
- Domain mapping variants: Develop truncated tPARP1 constructs lacking specific domains (ΔBRCT, ΔWGR) for interaction domain identification
Cell Culture and Transfection
- Cell lines: Utilize PARP1-deficient 293T cells or appropriate cell models relevant to specific research questions
- Stable expression: Establish stable cell lines expressing SFB-tagged tPARP1 variants using puromycin selection
- Apoptosis induction: Implement appropriate apoptosis inducers (e.g., poly(dA-dT) transfection, staurosporine treatment) where physiologically relevant
Tandem Affinity Purification
- Cell lysis: Use mild lysis conditions (e.g., 50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% NP-40) with protease and PARP inhibitors
- Clearing: Centrifuge lysates at 13,500 × g for 20 minutes at 4°C
- First purification: Incubate with Streptavidin beads for 2 hours at 4°C
- Washing: Perform sequential washes with lysis buffer containing 500 mM NaCl and standard concentration
- Elution: Cleave with TEV protease or competitive elution with biotin
- Second purification: Incubate eluate with S-protein agarose beads
- Final elution: Boil in SDS-PAGE loading buffer or use low pH elution
Mass Spectrometry Analysis
- Protein digestion: Perform in-gel or in-solution tryptic digestion
- Peptide cleanup: Use C18 stage tips for desalting
- LC-MS/MS: Analyze on nanoUHPLC system coupled to high-resolution mass spectrometer (e.g., Orbitrap Q-Exactive)
- Database search: Process raw data with Proteome Discoverer against Swiss-Prot human database
- Filtering: Apply false discovery rate threshold of <1% and require at least one unique peptide
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagents for tPARP1 Interactor Studies
| Reagent/Category | Specific Examples | Function/Application | Technical Notes |
|---|---|---|---|
| Expression Vectors | SFB-tag, GFP-tag, HA-tag | Tagged protein expression | SFB enables tandem affinity purification [34] |
| Cell Lines | PARP1-deficient 293T, BRCA1-isogenic pairs | Background reduction, context-specific studies | Isogenic lines reveal pathway-specific interactions [35] |
| Affinity Resins | Streptavidin beads, Anti-Flag M2 agarose | Interaction complex purification | Sequential purification reduces non-specific binding [34] |
| Protease Inhibitors | PARP inhibitors (Olaparib, Rucaparib) | Stabilization of complexes | Used in competition controls [35] |
| Apoptosis Inducers | Poly(dA-dT), Etoposide, Staurosporine | Physiological context establishment | Poly(dA-dT) mimics pathogenic DNA [34] |
| Mass Spectrometry | TMT labeling, LC-MS/MS systems | Quantitative interactome profiling | Enables comparative analysis between conditions [35] |
| Validation Antibodies | Anti-PARP1 cleaved forms, Pol III subunits | Orthogonal verification | Specific antibodies confirm cleavage and interactions [34] |
Data Analysis and Bioinformatics Approaches
Proteomics Data Processing Pipeline
Effective analysis of proteomics data requires a structured bioinformatics approach:
- Raw data conversion: Transform raw mass spectrometry files to open formats (mzML)
- Database searching: Utilize search engines (MaxQuant, MS-GF+) against appropriate protein databases
- Quality filtering: Implement strict FDR thresholds (typically 1% at protein and peptide levels)
- Quantitative analysis: Apply normalization and statistical testing for significance
- Bioinformatic enrichment: Identify overrepresented pathways, domains, and cellular compartments
Interactor Prioritization Strategies
Not all identified proteins represent biologically relevant interactors. Effective prioritization requires multi-parameter assessment:
Table 3: Criteria for Prioritizing Potential tPARP1 Interactors
| Prioritization Criteria | High-Confidence Indicators | Exclusion Signals |
|---|---|---|
| Statistical Significance | Significant enrichment (p < 0.01), high peptide counts | Non-significant p-values, single peptide identification |
| Specificity | Enriched in tPARP1 vs full-length PARP1 pulldowns | Equal enrichment in both experimental conditions |
| Reproducibility | Identified across multiple biological replicates | Inconsistent detection across replicates |
| Technical Validation | Confirmed by co-IP, PLA, or other orthogonal methods | Failure in validation experiments |
| Biological Plausibility | Known roles in apoptosis, DNA sensing, or related pathways | No logical connection to tPARP1 biology |
| Evolutionary Conservation | Interactions conserved across multiple trapping approaches | Limited to single methodological approach |
Future Directions and Technical Innovations
Emerging Proteomics Technologies
The field of tPARP1 interactome research stands to benefit from several emerging proteomic technologies:
- Photoactivatable crosslinkers: Incorporation of azido groups or other photo-crosslinking moieties into ADP-ribose analogs enables covalent trapping of transient interactions [36]
- Chemically-defined ADP-ribose probes: Synthetic ADP-ribose polymers with defined lengths and structures facilitate identification of modification-specific readers [36]
- Integrated multi-omics approaches: Combination of interactome data with phosphoproteomics and ADP-ribosylome data provides systems-level insights [35]
- Spatial proteomics: Application of proximity-dependent labeling (BioID, APEX) to map tPARP1 interactions in specific cellular compartments
Biological Applications and Therapeutic Implications
The continued identification of novel tPARP1 interactors holds significant promise for multiple research domains:
- Cancer therapeutics: Understanding tPARP1 functions may inform improved PARP inhibitor strategies and combination therapies
- Innate immunity: Elucidation of tPARP1-Pol III signaling may reveal novel antiviral defense mechanisms
- Neurodegeneration: Characterizing tPARP1-mediated cell death pathways could identify targets for neuroprotective interventions
- Inflammatory diseases: Modulating tPARP1 interactions may offer therapeutic opportunities in autoinflammatory conditions
Unbiased proteomics approaches have revolutionized our understanding of tPARP1 biology, moving beyond the historical perception of PARP1 cleavage as merely an inactivation mechanism. The systematic application of tandem affinity purification, catalytic mutant trapping strategies, and advanced mass spectrometry has revealed a previously unrecognized functional network centered on tPARP1, with the identification of RNA Polymerase III as an interactor representing a paradigm shift in our understanding of apoptotic signaling. The continued refinement of these proteomic methodologies, coupled with innovative bioinformatic analysis and rigorous validation, promises to further elucidate the complex biological functions of tPARP1 fragments in cell death pathways and their implications for human health and disease.
Poly(ADP-ribose) polymerase 1 (PARP1) is a nuclear enzyme with well-established roles in DNA damage repair and maintenance of genomic integrity. Beyond its canonical functions, PARP1 serves as a critical substrate for proteolytic cleavage during various forms of programmed cell death. The specific cleavage of PARP1 by different proteases generates stable, characteristic fragments that serve as molecular signatures for distinct cell death pathways. These truncated PARP1 (tPARP1) fragments are not merely inert byproducts of proteolysis but possess unique biological activities that contribute to the progression and regulation of cell death mechanisms.
The diagnostic potential of tPARP1 fragments stems from the exquisite specificity of different proteases that target PARP1 during apoptosis, necrosis, and other cell death pathways. Caspases, calpains, granzymes, cathepsins, and matrix metalloproteinases each cleave PARP1 at specific recognition sites, generating fragments with characteristic molecular weights and biological functions. This comprehensive review examines the current understanding of tPARP1 fragments as biomarkers, their roles in cell death pathways, and the experimental approaches for their detection and quantification in research and diagnostic contexts.
PARP1 Cleavage Fragments as Protease-Specific Signatures
Signature Fragments Across Cell Death Pathways
Different proteases cleave PARP1 at specific sites, generating signature fragments that serve as molecular indicators of the specific cell death pathway activated.
Table 1: PARP1 Cleavage Fragments as Signatures of Specific Protease Activities
| Protease | Cleavage Fragments | Cell Death Pathway | Molecular Consequences |
|---|---|---|---|
| Caspase-3/7 | 24 kDa + 89 kDa | Apoptosis | 24 kDa fragment binds DNA irreversibly; 89 kDa fragment translocates to cytoplasm [1] [5] |
| Caspase-3 | 24 kDa + 89 kDa | Apoptosis (alternative functions) | 89 kDa fragment mediates ADP-ribosylation of RNA Pol III, facilitating IFN-β production [5] |
| Lysosomal Proteases (Cathepsins B, D, G) | ~50 kDa | Necrosis | Not inhibited by zVAD-fmk; mediated by lysosomal proteases released during necrosis [37] |
| Calpain | 55 kDa + 62 kDa | Excitotoxicity, Calcium-mediated death | Alternative cleavage pattern in specific pathological contexts [1] |
| Granzyme A | ~50 kDa | Immune-mediated cytotoxicity | Unique fragment generated during T-cell mediated killing [1] |
The caspase-generated 89-kDa tPARP1 fragment has been shown to translocate to the cytoplasm where it serves as a poly(ADP-ribose) (PAR) carrier, inducing apoptosis-inducing factor (AIF) release from mitochondria and facilitating AIF translocation to the nucleus [20]. This function establishes tPARP1 as an active participant in the parthanatos cell death pathway rather than merely a bystander effect.
Structural Determinants of PARP1 Cleavage
PARP1 contains several functionally distinct domains that determine its cleavage patterns:
- DNA-binding domain (DBD): Contains three zinc finger structures (Zn1, Zn2, Zn3) and nuclear localization signals [3]
- Auto-modification domain (AMD): Contains a BRCT domain involved in protein-protein interactions [3]
- Catalytic domain (CAT): Comprises the helical subdomain (HD) and ADP-ribosyl transferase (ART) subdomain with NAD+ binding site [3]
The caspase-3 cleavage site at aspartate 214 separates the DNA-binding domain from the catalytic domain, while necrotic cleavage occurs at distinct sites vulnerable to lysosomal proteases [5] [37].
Experimental Protocols for tPARP1 Detection and Analysis
Western Blot Analysis for tPARP1 Fragments
Protocol for Detection of PARP1 Cleavage Fragments:
- Cell Lysis: Prepare samples using RIPA lysis buffer supplemented with protease inhibitor cocktail (including caspase inhibitors if specifically analyzing necrotic cleavage) and phosphatase inhibitor cocktail [38] [39]
- Protein Quantification: Use BCA protein assay kit to normalize protein concentrations across samples [39]
- Gel Electrophoresis: Separate proteins using 4-12% Bis-Tris gradient gels under denaturing conditions
- Membrane Transfer: Transfer to PVDF membranes using standard western blot protocols
- Antibody Detection:
- Fragment Identification: Identify specific fragments by molecular weight comparison with standards and using cleavage-specific antibodies
Key Consideration: For distinguishing apoptotic versus necrotic cleavage, include caspase inhibitors (e.g., zVAD-fmk) in parallel samples – caspase inhibition prevents apoptotic cleavage but not necrotic cleavage [37].
Immunoprecipitation and Mass Spectrometry Analysis
Protocol for Identification of tPARP1-Interacting Partners:
- Cell Transfection: Stably express tagged tPARP1 (e.g., SFB-tagged) in PARP1-deficient cells [5]
- Induction of Apoptosis: Treat cells with apoptotic inducers (e.g., staurosporine, actinomycin D, or poly(dA-dT) to stimulate caspase-mediated PARP1 cleavage [5] [20]
- Cell Fractionation: Separate nuclear and cytoplasmic fractions to track tPARP1 translocation
- Tandem Affinity Purification: Purify tPARP1 and associated proteins using sequential anti-FLAG and anti-SBP immunoprecipitation [5]
- Mass Spectrometry Analysis: Identify interacting proteins by liquid chromatography-tandem mass spectrometry (LC-MS/MS)
- Validation: Confirm interactions through co-immunoprecipitation assays with candidate proteins
This approach identified RNA Polymerase III subunits as tPARP1 interaction partners, revealing a novel role for tPARP1 in innate immune response during apoptosis [5].
Signaling Pathways and Molecular Functions of tPARP1
Caspase-tPARP1 Pathway in Apoptosis
The 89-kDa tPARP1 fragment generated by caspase-3 cleavage translocates to the cytoplasm where its BRCT domain interacts with the RNA Polymerase III complex [5]. tPARP1 then catalyzes ADP-ribosylation of Pol III, enhancing its activity in transcribing foreign DNA and promoting IFN-β production, which amplifies the apoptotic response [5].
tPARP1 in Parthanatos
In parthanatos, the 89-kDa tPARP1 fragment serves as a cytoplasmic PAR carrier that binds to AIF and facilitates its translocation to the nucleus, where it induces large-scale DNA fragmentation and chromatin condensation [20]. This pathway represents a critical caspase-independent cell death mechanism with particular relevance in neurological diseases and other pathological conditions.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Research Reagents for tPARP1 Studies
| Reagent/Category | Specific Examples | Research Application | Mechanistic Insight |
|---|---|---|---|
| PARP Inhibitors | Olaparib, PJ-34, Nicotinamide, 3-aminobenzamide | Modulate PARP1 activity in experimental models | Establish causal relationship between PARP1 activation and cleavage patterns [40] [25] |
| Caspase Inhibitors | zVAD-fmk, DEVD-CHO | Distinguish caspase-dependent vs independent cleavage | Confirm apoptotic cleavage mechanism; isolate necrotic cleavage [37] |
| Apoptosis Inducers | Staurosporine, Actinomycin D, Etoposide, poly(dA-dT) | Activate caspase-mediated PARP1 cleavage | Study tPARP1 formation and translocation under controlled conditions [5] [20] |
| Necrosis Inducers | H2O2, Ethanol, HgCl2 | Activate lysosomal protease-mediated PARP1 cleavage | Investigate alternative cleavage mechanisms in non-apoptotic cell death [37] |
| PARP1 Cleavage-Specific Antibodies | Anti-89 kDa tPARP1, Anti-24 kDa fragment | Detect specific fragments in western blot, immunofluorescence | Identify specific cleavage patterns; determine subcellular localization [5] [20] |
| Cell Fractionation Kits | Nuclear/Cytoplasmic Fractionation kits | Track tPARP1 translocation following cleavage | Establish functional consequences of cleavage and cellular redistribution [20] |
Specialized Reagents for Advanced Applications
PARP1-Deficient Cell Lines: Engineered 293T cells with PARP1 knockout allow for clean background when expressing mutant forms of PARP1 to study cleavage-specific functions [5].
Non-Cleavable PARP1 Mutants: Expression vectors encoding PARP1 with mutated caspase cleavage sites (D214A) help establish the functional significance of PARP1 cleavage in apoptosis [5].
Metabolic Labeling Reagents: [$^{32}P]NAD+ for monitoring PARP1 enzymatic activity and PAR formation following cleavage [20].
Diagnostic Applications and Therapeutic Implications
The specific cleavage patterns of PARP1 have significant implications for diagnostic development and therapeutic monitoring:
Disease-Specific Cleavage Signatures
In neurodegenerative diseases including Parkinson's disease, Alzheimer's disease, and amyotrophic lateral sclerosis, PARP1 overactivation and cleavage contribute to disease pathology through multiple mechanisms, including parthanatos [41] [25]. Detection of specific tPARP1 fragments in cerebrospinal fluid or post-mortem tissue could serve as biomarkers for disease progression and cell death mechanisms.
In liver diseases, including non-alcoholic fatty liver disease (NAFLD), alcoholic liver disease, and hepatocellular carcinoma, PARP1 expression and cleavage patterns correlate with disease severity and therapeutic response [40]. The dual role of PARP1 in liver regeneration and cell death makes it a particularly interesting biomarker in hepatic pathologies.
Therapeutic Monitoring Applications
PARP inhibitors have shown promise in various malignancies, particularly in BRCA-mutant cancers [38] [39]. However, resistance to PARP inhibition remains a significant clinical challenge. Detection of alternative tPARP1 fragments in resistant tumors may indicate activation of bypass cell death pathways and inform combination therapy approaches.
Recent evidence demonstrates that RSL3, a ferroptosis inducer, promotes PARP1 apoptotic functions through distinct mechanisms, including caspase-dependent PARP1 cleavage and reduced full-length PARP1 via inhibition of METTL3-mediated m6A modification [39]. This suggests that tPARP1 fragments could serve as biomarkers for effective ferroptosis induction in resistant malignancies.
tPARP1 fragments represent specific biomarkers of protease activation in distinct cell death pathways. The characteristic cleavage patterns generated by caspases, lysosomal proteases, and other cell death-associated enzymes provide a molecular fingerprint that identifies the activated cell death mechanism in physiological and pathological contexts.
Future directions in tPARP1 research include the development of quantitative assays for specific tPARP1 fragments in clinical samples, the exploration of tPARP1 as a therapeutic target in its own right, and the investigation of cross-talk between different cell death pathways through simultaneous monitoring of multiple cleavage events. The continued refinement of detection methods and experimental approaches will further establish tPARP1 fragments as crucial biomarkers in cell death diagnostics and therapeutic development.
Poly (ADP-ribose) polymerase inhibitor (PARPi) resistance represents a critical challenge in oncology, particularly for BRCA-mutant cancers. While PARPi exploit synthetic lethality in homologous recombination (HR)-deficient cells, multiple resistance mechanisms including HR restoration, replication fork protection, and pharmacological alterations enable tumor survival. This review explores the emerging role of truncated PARP1 (tPARP1) fragments in cell death pathways as a potential therapeutic vulnerability to overcome PARPi resistance. We examine the molecular mechanisms of tPARP1-mediated apoptosis, detail experimental methodologies for investigating these pathways, and propose novel therapeutic strategies targeting tPARP1 biology to eradicate resistant malignancies.
PARP inhibitors have revolutionized cancer treatment through synthetic lethality in HR-deficient cancers, particularly those with BRCA1/2 mutations. However, both de novo and acquired resistance significantly limit their clinical efficacy, with over 40% of BRCA-deficient patients failing to respond [42]. The predominant resistance mechanisms center around restoration of DNA repair capacity—specifically, HR restoration through loss of non-homologous end joining (NHEJ) factors, BRCA reversion mutations, and replication fork stabilization [43]. Additionally, pharmacological alterations including increased drug efflux and restoration of PARylation contribute to treatment failure. This resistance landscape necessitates innovative approaches to target resistant malignancies beyond conventional PARP inhibition strategies.
Table 1: Major Mechanisms of PARPi Resistance and Their Frequency
| Mechanism Category | Specific Process | Key Effectors | Estimated Frequency in Resistant Cases |
|---|---|---|---|
| HR Restoration | Loss of NHEJ factors | 53BP1, REV7, SHLD complex | 25-40% [43] |
| BRCA reversion mutations | Restoration of BRCA1/2 function | 15-30% [42] | |
| Increased HR factor expression | RAD51, PALB2, BRCA1 | 10-20% [42] | |
| Replication Fork Dynamics | Fork stabilization | Reduced MRE11-mediated degradation | 20-35% [43] |
| Fork protection | Enhanced RAD51 loading | 15-25% [42] | |
| ssDNA Gap Suppression | Reduced gap accumulation | Altered Polθ activity | 10-20% [43] |
| Pharmacological Alterations | Increased drug efflux | P-glycoprotein upregulation | 10-15% [42] |
| PARP Activity | Restoration of PARylation | PARP1 mutations | 5-15% [42] |
The Biology of Truncated PARP1 Fragments in Cell Death
PARP1 Cleavage Fragments: Signatures of Cell Death Programs
PARP1 serves as a substrate for multiple proteases during various cell death programs, generating specific signature fragments with distinct biological activities [1]. Caspase-3 and -7 cleave PARP1 at aspartate 214 to produce 24-kDa DNA-binding and 89-kD catalytic fragments during apoptosis [5] [1]. This cleavage inactivates PARP1's DNA repair function while generating fragments with novel signaling capabilities. Other proteases, including calpains, cathepsins, granzymes, and matrix metalloproteinases, generate different PARP1 cleavage patterns characteristic of alternative cell death pathways such as necrosis and parthanatos [1].
The Dual Nature of the 89-kDa tPARP1 Fragment
The 89-kDa tPARP1 fragment exhibits context-dependent functions that may be exploitable in PARPi-resistant malignancies:
Cytosolic Translocation and Innate Immune Activation: During poly(dA-dT)-stimulated apoptosis, the 89-kDa tPARP1 translocates to the cytoplasm where it recognizes and mono-ADP-ribosylates the RNA polymerase III (Pol III) complex [5]. This modification facilitates IFN-β production and enhances apoptosis, representing a bridge between DNA damage and immune response activation.
PAR Carrier Function in Parthanatos: Recent research demonstrates that the caspase-cleaved 89-kDa fragment can serve as a carrier for poly(ADP-ribose) (PAR) polymers to the cytoplasm [20]. Once in the cytoplasm, PAR attached to tPARP1 facilitates apoptosis-inducing factor (AIF) release from mitochondria and subsequent nuclear translocation, amplifying cell death signaling.
Evolutionary Conservation: Interestingly, PARP1 orthologs in several lower organisms naturally lack the N-terminal zinc fingers present in full-length mammalian PARP1, suggesting that tPARP1 represents an evolutionarily conserved functional unit with specific biological roles beyond dominant-negative inhibition of DNA repair [5].
Experimental Analysis of tPARP1 Pathways
Methodologies for Investigating tPARP1- Mediated Apoptosis
Protein Affinity Purification and Mass Spectrometry
- Cell Line Preparation: Generate PARP1-deficient 293T cells stably expressing SFB-tagged catalytic mutant tPARP1 (E988A) to trap substrates [5]
- Complex Isolation: Perform tandem affinity purification from soluble fractions under native conditions
- Interactome Analysis: Identify binding partners via liquid chromatography-tandem mass spectrometry (LC-MS/MS)
- Validation: Confirm interactions through co-immunoprecipitation with Pol III subunits (POLR3A, POLR3B, POLR3F)
Apoptosis Induction and tPARP1 Functional Assays
- Stimulus Application: Transfert cells with poly(dA-dT) to mimic pathogenic DNA and induce caspase-mediated PARP1 cleavage [5]
- Apoptosis Quantification: Employ Annexin V-FITC/propidium iodide staining with FACS analysis
- Localization Studies: Perform subcellular fractionation and immunofluorescence to track tPARP1 translocation
- Functional Consequences: Assess IFN-β production via ELISA and Pol III ADP-ribosylation through in vitro modification assays
Table 2: Key Research Reagents for tPARP1 Investigation
| Reagent/Cell Line | Specific Application | Function/Utility | Key Features |
|---|---|---|---|
| PARP1-deficient 293T cells | Background for expression studies | Eliminates endogenous PARP1 interference | Enables clean interaction studies [5] |
| Catalytic mutant tPARP1 (E988A) | Substrate trapping | Binds but doesn't modify targets | Identifies transient interactions [5] |
| Poly(dA-dT) | Apoptosis induction | Mimics pathogenic DNA | Stimulates innate immune response [5] |
| BRCT domain mutants (e.g., F473A) | Interaction mapping | Disrupts BRCT domain function | Identifies BRCT-dependent interactions [5] |
| Caspase-3/7 inhibitors | Pathway dissection | Blocks PARP1 cleavage | Distinguishes caspase-dependent/independent death |
| PARG/TARG1 mutants | PAR metabolism studies | Traps ADP-ribosylated substrates | Identifies PARylation targets [5] |
Visualizing tPARP1-Mediated Apoptosis Signaling
Figure 1: tPARP1-Mediated Apoptosis Signaling in PARPi-Resistant Cells. The 89-kDa tPARP1 fragment generated during apoptosis translocates to the cytoplasm and engages multiple pro-death pathways through RNA Pol III interaction and AIF release.
Strategic Exploitation of tPARP1 in PARPi-Resistant Malignancies
Therapeutic Approaches to Leverage tPARP1 Biology
The unique properties of tPARP1 fragments suggest several strategic approaches to overcome PARPi resistance:
Caspase Activation Combinatorial Therapy: PARPi combined with agents that enhance caspase activation could promote tPARP1 generation in resistant cells. This approach leverages the dual role of caspase activation in both inhibiting DNA repair (through PARP1 cleavage) and activating tPARP1-mediated death signaling.
tPARP1 Stabilization Strategies: Developing small molecules that stabilize the 89-kDa fragment or enhance its cytosolic translocation could amplify its pro-death functions independent of PARP catalytic inhibition.
BRCT Domain-Targeted Therapeutics: The BRCT domain of tPARP1 is essential for its interaction with Pol III [5]. Compounds that modulate this interaction could potentiate the immune-activating aspect of tPARP1-mediated apoptosis.
PAR Polymer Metabolism Manipulation: Since tPARP1 functions as a PAR carrier, regulating PAR degradation through PARG inhibition could enhance tPARP1-mediated AIF release and cell death execution.
Experimental Framework for Therapeutic Development
Figure 2: Strategic Approaches to Overcome PARPi Resistance via tPARP1 Pathways. Multiple therapeutic strategies can bypass common resistance mechanisms by engaging tPARP1-mediated apoptosis.
The exploration of tPARP1 biology represents a paradigm shift in addressing PARPi resistance. Rather than focusing exclusively on preventing resistance mechanisms, leveraging the cell death signals encoded within PARP1 cleavage fragments offers a promising alternative approach. The 89-kDa tPARP1 fragment serves as a molecular nexus connecting DNA damage, innate immune activation, and mitochondrial death pathways—all of which can be exploited therapeutically.
Future research should prioritize the development of standardized assays for tPARP1 detection and functional assessment in clinical samples, the identification of biomarkers predicting responsiveness to tPARP1-targeting approaches, and the optimization of combination strategies that maximize tPARP1-mediated death while minimizing normal tissue toxicity. By shifting the therapeutic focus from PARP inhibition alone to the broader landscape of PARP1 biology, including its cleavage fragments, we may uncover novel vulnerabilities in treatment-resistant malignancies.
The cleavage of poly(ADP-ribose) polymerase 1 (PARP1) has long been recognized as a hallmark of apoptosis, serving as a surrogate marker for caspase activation. However, emerging research reveals that PARP1 fragmentation represents more than merely a cellular epiphenomenon; it constitutes a critical molecular switch that coordinates crosstalk between different cell death modalities [5] [1]. Within the context of ferroptosis—an iron-dependent form of regulated cell death characterized by lipid peroxidation—the role of PARP1 has remained particularly enigmatic until recent investigations illuminated its central positioning at the intersection of these pathways [44] [45].
This technical guide examines how the canonical ferroptosis inducer RSL3 orchestrates dual cell death outcomes through distinct mechanisms involving PARP1. As a glutathione peroxidase 4 (GPX4) inhibitor, RSL3 typically triggers ferroptosis through iron-dependent lipid peroxidation [46] [47]. Surprisingly, recent work demonstrates that RSL3 also engages apoptotic signaling through multifaceted regulation of PARP1, encompassing both proteolytic cleavage and translational suppression [44] [45]. These findings position truncated PARP1 (tPARP1) fragments not as mere bystanders in cell death, but as active mediators with specific biological functions that potentially bridge ferroptotic and apoptotic execution.
Molecular Mechanisms of Ferroptosis-Apoptosis Crosstalk
Distinct Morphological and Biochemical Features of Ferroptosis and Apoptosis
Ferroptosis and apoptosis represent distinct forms of regulated cell death with divergent morphological and biochemical characteristics. As delineated in Table 1, these pathways differ fundamentally in their execution mechanisms, though emerging evidence reveals significant crosstalk under specific cellular contexts [46] [47].
Table 1: Key Characteristics of Apoptosis and Ferroptosis
| Feature | Apoptosis | Ferroptosis |
|---|---|---|
| Morphological Characteristics | Cell membrane blebbing, cell rounding, chromatin condensation, nuclear fragmentation, apoptotic bodies | Normal nuclear size, absent chromatin condensation, small mitochondria with condensed membranes, reduced mitochondrial cristae |
| Biochemical Features | Caspase activation, oligonucleosomal DNA fragmentation | GPX4 inhibition, glutathione depletion, iron accumulation, lipid peroxidation |
| Key Regulators | Caspases, p53, Fas, Bcl-2 family | SLC7A11, GPX4, Nrf2, TFR1 |
| Common Inducers | FASL, UNC5B | Erastin, RSL3, sorafenib |
| Common Inhibitors | Z-VAD-FMK, XIAP | Ferrostatin-1, liproxstatin-1, deferoxamine |
PARP1 as a Molecular Integrator of Cell Death Signals
PARP1 serves as a critical node in cell death decision-making. This multifunctional nuclear enzyme contains three zinc finger DNA-binding domains at its N-terminus, a central automodification domain with a BRCT motif, and a C-terminal catalytic domain [48]. During apoptosis, caspase-3 cleaves PARP1 at the DEVD214 motif, generating two principal fragments: a 24-kD DNA-binding fragment and an 89-kD truncated PARP1 (tPARP1) fragment containing the automodification and catalytic domains [1] [48]. While the 24-kD fragment acts as a trans-dominant inhibitor of DNA repair by occupying DNA strand breaks, the biological function of the 89-kD tPARP1 has remained enigmatic until recent studies revealed its capacity to translocate to the cytoplasm and engage novel signaling partners [5].
RSL3-Induced PARP1 Regulation: Dual Mechanisms of Apoptotic Activation
Biphasic PARP1 Regulation During RSL3 Treatment
The ferroptosis inducer RSL3, known primarily as a GPX4 inhibitor, unexpectedly engages apoptotic signaling through two parallel mechanisms targeting PARP1, as summarized in Table 2.
Table 2: Dual Mechanisms of RSL3-Mediated PARP1 Regulation
| Mechanism | Molecular Process | Functional Consequences |
|---|---|---|
| Caspase-Dependent Cleavage | ROS-mediated caspase-3 activation cleaves PARP1 at D214 | Generates 89-kD tPARP1 fragment that translocates to cytoplasm and potentially engages novel substrates |
| Translational Suppression | Inhibition of METTL3-mediated m⁶A modification reduces PARP1 mRNA stability and translation | Decreases full-length PARP1 protein levels, impairing DNA damage repair and promoting genomic instability |
RSL3 triggers reactive oxygen species (ROS) production during ferroptosis initiation, which subsequently activates caspase-3. This caspase activation drives the proteolytic cleavage of PARP1, generating the characteristic 89-kD tPARP1 fragment [44] [45]. Simultaneously, RSL3 inhibits METTL3-mediated N⁶-methyladenosine (m⁶A) modification of PARP1 mRNA, reducing its stability and translational efficiency. This dual mechanism—combining post-translational cleavage and translational suppression—ensures comprehensive alteration of PARP1 function during RSL3 treatment [44].
Signaling Pathway Integration
The following diagram illustrates the coordinated mechanisms through which RSL3 modulates PARP1 to drive ferroptosis-apoptosis crosstalk:
Biological Functions of Truncated PARP1 Fragments
Non-Canonical Functions of Cleaved PARP1
The 89-kD tPARP1 fragment generated during apoptosis exhibits functions beyond its traditional role in DNA damage response. Following caspase-3 mediated cleavage and nuclear-to-cytoplasmic translocation, tPARP1 interacts with novel protein complexes, including the RNA polymerase III (Pol III) complex [5]. Through its BRCT domain, tPARP1 recognizes and mono-ADP-ribosylates Pol III subunits, enhancing their transcriptional activity and promoting interferon-beta (IFN-β) production during innate immune activation [5]. This function represents a paradigm shift in understanding tPARP1 as not merely an inactive cleavage product, but as a signaling molecule with distinct biological activities that potentially contribute to cell fate decisions.
Context-Dependent Cell Death Modulation
The interplay between ferroptotic and apoptotic signaling exhibits significant context dependency. As demonstrated in recent investigations, cells undergoing GPX4 inhibition display hybrid features of both ferroptosis and apoptosis, including transient membrane blebbing, submaximal cytochrome c release, and caspase activation alongside characteristic lipid peroxidation [47]. The specific cellular outcome depends on factors including the magnitude of oxidative stress, compensatory antioxidant capacities, and the relative expression of BCL-2 family proteins [47]. This complex interplay positions PARP1 cleavage fragments as potential determinants of final cell death execution, particularly under conditions of moderate ferroptotic stress where apoptotic signaling remains engaged.
Experimental Approaches for Studying RSL3-PARP1 Axis
Key Research Reagents and Methodologies
Table 3: Essential Research Reagents for Investigating RSL3-Mediated PARP1 Regulation
| Reagent/Category | Specific Examples | Research Application |
|---|---|---|
| Ferroptosis Inducers | RSL3, Erastin | Induction of ferroptosis via GPX4 inhibition or system Xc- blockade |
| Cell Death Inhibitors | Ferrostatin-1 (Fer-1), Liproxstatin-1 (Lip-1), Z-VAD-FMK | Distinguishing ferroptosis vs. apoptosis contribution |
| PARP Detection Reagents | Anti-PARP1 antibodies (cleavage-specific), PARP activity assays | Monitoring PARP1 cleavage and enzymatic activity |
| Oxidative Stress Probes | C11-BODIPY 581/591, DCFDA, MitoSOX | Quantifying lipid peroxidation and general ROS production |
| Apoptosis Assays | Annexin V/PI staining, caspase activity assays, cytochrome c release | Quantifying apoptotic activation |
| Gene Manipulation Tools | PARP1 siRNA/shRNA, CRISPR-Cas9 PARP1 knockout cells | Determining PARP1-specific functions |
Experimental Workflow for Mechanistic Studies
The diagram below outlines a comprehensive experimental approach for investigating RSL3-mediated PARP1 regulation:
Detailed Methodological Protocols
PARP1 Cleavage Detection by Western Blotting
Cell Treatment and Protein Extraction:
- Seed cells in 6-well plates (3×10⁵ cells/well) and incubate for 24 hours
- Treat cells with RSL3 (dose range: 0.1-10 µM) for 6-24 hours
- Include control groups with Fer-1 (1 µM) and/or Z-VAD-FMK (20 µM)
- Harvest cells and lyse in RIPA buffer with protease inhibitors
- Quantify protein concentration using BCA assay
Western Blot Procedure:
- Separate 20-30 µg protein by 4-12% Bis-Tris SDS-PAGE
- Transfer to PVDF membrane and block with 5% non-fat milk
- Incubate with primary antibodies: anti-PARP1 (1:1000), anti-cleaved PARP1 (1:1000), anti-β-actin (1:5000)
- Apply HRP-conjugated secondary antibodies (1:5000)
- Develop with ECL substrate and image using chemiluminescence detection
m⁶A Modification Analysis
m⁶A RNA Immunoprecipitation (MeRIP-qPCR):
- Extract total RNA using TRIzol reagent
- Fragment RNA to ~100 nucleotides by incubation at 94°C for 5 minutes
- Incubate RNA with anti-m⁶A antibody or normal IgG (negative control)
- Add protein A/G magnetic beads and incubate at 4°C for 2 hours
- Wash beads and elute m⁶A-containing RNA with m⁶A nucleotide solution
- Analyze PARP1 mRNA enrichment by qPCR with specific primers
Therapeutic Implications and Research Applications
Overcoming PARP Inhibitor Resistance
The dual cell death induction capability of RSL3 presents promising therapeutic opportunities, particularly in PARP inhibitor (PARPi)-resistant malignancies. Research demonstrates that RSL3 retains pro-apoptotic function in PARPi-resistant cells and effectively suppresses PARPi-resistant xenograft tumor growth in vivo [44] [45]. This effect stems from RSL3's capacity to bypass conventional resistance mechanisms by simultaneously targeting GPX4-mediated antioxidant defenses and PARP1 function through complementary mechanisms.
Research Translation and Drug Development
The RSL3-PARP1 axis offers multiple avenues for therapeutic exploration:
- Combination therapies: RSL3 with BH3-mimetics in apoptosis-refractory cancers
- Biomarker development: PARP1 cleavage fragments as indicators of treatment response
- Resistance mitigation: Targeting both ferroptotic and apoptotic pathways to prevent adaptive resistance
The molecular characterization of tPARP1 functions opens new possibilities for leveraging PARP1 cleavage as a therapeutic goal rather than merely a marker of cell death. The discovery that tPARP1 actively participates in cytoplasmic signaling events, including innate immune activation, suggests broader physiological roles beyond its established nuclear functions [5].
The investigation of RSL3 as an inducer of dual cell death via PARP1 fragments exemplifies the evolving understanding of cell death crosstalk in cancer biology. The molecular characterization of PARP1 cleavage fragments has transitioned from viewing them as mere biomarkers to recognizing them as active mediators with specific biological functions. This paradigm shift offers new therapeutic opportunities, particularly for malignancies resistant to conventional apoptosis-inducing agents. Future research directions should include comprehensive structural characterization of tPARP1 interactions, in vivo validation of tPARP1 signaling functions, and clinical translation of dual cell death induction strategies in therapy-resistant cancers.
Navigating Complexity: Challenges in Targeting tPARP1 Pathways and Resistance Mechanisms
Poly(ADP-ribose) polymerase-1 (PARP-1) serves as a critical molecular switch governing cellular fate in response to DNA damage. This nuclear enzyme integrates DNA repair signaling with cell death pathways through proteolytic cleavage events that determine whether a cell undergoes apoptosis or necrosis. During apoptosis, caspase-mediated cleavage generates signature PARP-1 fragments (89-kDa and 24-kDa) that inactivate DNA repair and facilitate programmed death, while during necrosis, lysosomal proteases produce distinct cleavage patterns (notably a 50-kDa fragment) leading to inflammatory cell death. The specific proteolytic fragments generated serve as molecular signatures that not only identify the cell death pathway activated but also execute distinct functions in death progression. This review examines the mechanisms and consequences of PARP-1 cleavage, detailing how different proteolytic fragments influence cellular fate decisions through their interactions with key death mediators. Understanding these processes provides critical insights for therapeutic interventions in cancer, neurodegenerative diseases, and other conditions characterized by dysregulated cell death.
PARP-1 is a nuclear enzyme with approximately 1-2 million copies per cell, accounting for approximately 85% of total cellular PARP activity [19]. This abundant protein contains several structurally and functionally distinct domains: a 46-kD DNA-binding domain (DBD) at the NH2 terminus containing two zinc finger motifs that facilitate tight binding to DNA damage sites; a 22-kD auto-modification domain (AMD) in the central region that functions as a target for covalent auto-modification; and a 54-kD catalytic domain (CD) at the carboxyl terminus that polymerizes linear or branched poly-ADP-ribose units from NAD+ onto target proteins [19]. A third zinc finger motif located between the second zinc finger motif and AMD plays an important role in inter-domain interactions and is vital for PARP-1 enzymatic action [19].
The AMD contains a BRCT fold, a motif found in many DNA repair proteins that facilitates protein-protein interactions and promotes the recruitment of DNA repair enzymes to sites of DNA damage [19]. Under physiological conditions, PARP-1 performs routine repair of DNA damage through poly(ADP-ribosyl)ation, which serves as a post-translational modification crucial for maintaining cellular homeostasis [19]. Beyond DNA repair, PARP-1 participates in diverse cellular functions including gene transcription, immune responses, inflammation, synaptic functions, and aging [19] [49].
PARP-1 Cleavage Fragments as Signatures of Specific Cell Death Programs
PARP-1 serves as a preferred substrate for multiple "suicidal" proteases, which cleave the enzyme at specific sites to generate signature fragments that serve as biomarkers for particular cell death pathways [19]. The proteolytic action of caspases, calpains, cathepsins, granzymes, and matrix metalloproteinases on PARP-1 produces distinct proteolytic fragments with different molecular weights and functions [19]. These PARP-1 signature fragments represent recognized biomarkers for specific patterns of protease activation in unique cell death programs.
Table 1: PARP-1 Cleavage Fragments in Different Cell Death Pathways
| Cell Death Pathway | Proteases Involved | Cleavage Fragments | Functional Consequences |
|---|---|---|---|
| Apoptosis | Caspases-3 and -7 | 89-kDa (AMD+CD) and 24-kDa (DBD) | Inactivation of DNA repair; conservation of ATP; facilitation of death program |
| Necrosis | Cathepsins B and G (lysosomal proteases) | 50-kDa major fragment | ATP depletion; inflammatory cell death |
| Parthanatos | Not applicable (PAR signaling) | PAR polymers attached to 89-kDa fragment | AIF-mediated caspase-independent death |
Caspase-Mediated Cleavage in Apoptosis
During apoptosis, caspases-3 and -7 cleave PARP-1 at the DEVD site (specifically between Asp214 and Gly215 in human PARP-1), resulting in the separation of the two zinc-finger DNA-binding motifs from the automodification and catalytic domains [19] [9]. This proteolytic cleavage generates two specific fragments: an 89-kD catalytic fragment (containing the AMD and catalytic domain) and a 24-kD DNA-binding fragment (containing the two zinc-finger motifs) [19]. The 24-kD cleaved fragment with its zinc-finger motifs is retained in the nucleus, where it irreversibly binds to damaged DNA and acts as a trans-dominant inhibitor of active PARP-1, thereby blocking DNA repair processes [19]. The 89-kD fragment has a greatly reduced DNA binding capacity and can be liberated from the nucleus into the cytosol [19].
Recent research has revealed a more complex role for the 89-kDa fragment, demonstrating that it can serve as a carrier for poly(ADP-ribose) (PAR) polymers to the cytoplasm [20] [18]. When PARP-1 undergoes auto-poly(ADP-ribosyl)ation prior to caspase cleavage, the 89-kDa fragment retains covalently attached PAR polymers during its translocation to the cytoplasm [20]. In the cytoplasm, these PAR polymers facilitate apoptosis-inducing factor (AIF) release from mitochondria, ultimately leading to AIF translocation to the nucleus and triggering caspase-independent cell death (parthanatos) [20] [18]. This mechanism represents a crucial intersection between apoptotic and parthanatos pathways.
Necrotic Cleavage by Lysosomal Proteases
In contrast to apoptotic cleavage, necrosis induces PARP-1 processing that generates a major fragment of 50 kDa [37] [50]. This necrotic cleavage event is not inhibited by zVAD-fmk, a broad-spectrum caspase inhibitor, indicating that caspases are not implicated in this process [37]. Instead, lysosomal proteases—particularly cathepsins B and G—released during necrotic cell death are responsible for PARP-1 cleavage during necrosis [37] [50].
Experimental evidence demonstrates that in vitro lysosomal proteolytic cleavage of affinity-purified PARP-1 produces fragments corresponding in apparent molecular weight to those found in Jurkat T cells treated with necrotic inducers like 0.1% H₂O₂, 10% EtOH, or 100 μM HgCl₂ [37]. The functional consequence of PARP-1 activation during necrosis is extensive consumption of NAD⁺ and, in efforts to resynthesize NAD⁺, massive ATP depletion, which shifts cell death toward necrosis [9]. This ATP depletion occurs because PARP-1 overactivation consumes large amounts of NAD⁺, and cellular efforts to resynthesize NAD⁺ deplete ATP stores [9].
PARP-1 Cleavage as a Molecular Switch Between Apoptosis and Necrosis
The cleavage of PARP-1 represents a critical molecular switch that directs cellular fate toward either apoptotic or necrotic death [9]. This switching function depends on the differential activation of specific proteases and the consequent generation of distinct PARP-1 fragments with unique biological activities.
The Energy Switch Hypothesis
The decision between apoptosis and necrosis is fundamentally regulated by cellular ATP levels [9]. Apoptosis is an energy-dependent process requiring ATP, whereas necrosis typically occurs under conditions of ATP depletion [9]. PARP-1 activation and cleavage patterns directly influence this energy status:
- In apoptosis, early caspase-mediated cleavage of PARP-1 prevents excessive NAD⁺ and ATP consumption, thereby maintaining sufficient energy stores for the orderly execution of the apoptotic program [9].
- In necrosis, the absence of PARP-1 cleavage allows continued PARP-1 activation, leading to severe NAD⁺ and ATP depletion that forces the cell into necrotic death [9].
Supporting this hypothesis, studies in L929 cells demonstrated that TNF-induced necrosis involves PARP activation leading to ATP depletion, whereas CD95-mediated apoptosis features caspase-mediated PARP-1 cleavage and maintained ATP levels [9]. Furthermore, caspase inhibitors potentiate TNF-induced necrosis by preventing PARP-1 cleavage, thereby allowing continued PARP-1 activation and ATP depletion [9].
Functional Consequences of PARP-1 Cleavage Fragments
The specific PARP-1 fragments generated during different cell death pathways execute distinct functions that influence the death process:
- The 24-kDa DBD fragment acts as a trans-dominant inhibitor of BER by irreversibly binding to DNA strand breaks, thereby preventing DNA repair and conserving cellular ATP pools [19].
- The 89-kDa fragment can serve as a cytoplasmic PAR carrier that facilitates AIF-mediated cell death, creating a link between caspase-dependent apoptosis and parthanatos [20].
- The 50-kDa necrotic fragment represents incomplete cleavage that may allow residual PARP-1 activity while disrupting normal DNA repair functions [37].
Table 2: Experimental Evidence for PARP-1 Cleavage in Cell Death Models
| Experimental Model | Inducing Stimulus | PARP-1 Fragments Observed | Key Findings | Research Reagents Used |
|---|---|---|---|---|
| Jurkat T cells | 0.1% H₂O₂, 10% EtOH, 100 μM HgCl₂ | 50-kDa | Necrotic cleavage inhibited by cathepsin inhibitors but not zVAD-fmk | zVAD-fmk (caspase inhibitor), cathepsin inhibitors |
| HL-60 cells | Etoposide phosphate (VP-16) | 89-kDa and 24-kDa | Caspase-3 mediated cleavage; influenced by PARP-1 auto-modification | Etoposide phosphate (VP-16) |
| L929 fibrosarcoma | TNF vs. anti-CD95 | 89-kDa and 24-kDa (apoptosis) | PARP cleavage acts as switch between apoptosis and necrosis | TNF, anti-CD95, zVAD, 3-aminobenzamide |
| Fibroblasts | PARP-1-D214N mutant | Uncleavable PARP-1 | Increased sensitivity to TNF-induced death | PARP-1-D214N mutant expression vector |
Research Reagent Solutions for PARP-1 Cleavage Studies
Investigating PARP-1 cleavage requires specific reagents and experimental approaches. The following toolkit provides essential resources for studying PARP-1 in cell death pathways:
Table 3: Research Reagent Solutions for PARP-1 Cleavage Studies
| Reagent Category | Specific Examples | Experimental Function | Application Context |
|---|---|---|---|
| Caspase Inhibitors | zVAD-fmk | Broad-spectrum caspase inhibitor; blocks apoptotic PARP-1 cleavage | Distinguishing caspase-dependent vs independent cleavage |
| PARP Inhibitors | 3-aminobenzamide (3AB) | Inhibits PARP enzymatic activity; prevents ATP depletion | Studying necrotic cell death and energy dynamics |
| Necrosis Inducers | H₂O₂ (0.1%), EtOH (10%), HgCl₂ (100 μM) | Induces necrotic cell death with characteristic PARP-1 cleavage | Generating 50-kDa PARP-1 fragment |
| Apoptosis Inducers | Staurosporine, Actinomycin D, Etoposide | Triggers caspase activation and apoptotic PARP-1 cleavage | Generating 89-kDa and 24-kDa PARP-1 fragments |
| Lysosomal Protease Inhibitors | Cathepsin B and G inhibitors | Blocks necrotic PARP-1 cleavage | Confirming lysosomal protease involvement in necrosis |
| PARP-1 Mutants | PARP-1-D214N (caspase-resistant) | Uncleavable PARP-1 mutant; enhances necrotic sensitivity | Studying functional consequences of PARP-1 cleavage |
Experimental Protocols for PARP-1 Cleavage Analysis
Detecting PARP-1 Cleavage by Western Blotting
Purpose: To identify and characterize PARP-1 cleavage fragments in different cell death models.
Procedure:
- Induce cell death using appropriate stimuli (e.g., 1μM staurosporine for apoptosis; 0.1% H₂O₂ for necrosis).
- Harvest cells at various timepoints post-treatment (typically 2-24 hours).
- Prepare whole cell extracts using RIPA buffer with protease inhibitors.
- Separate proteins by SDS-PAGE (8-12% gradient gels recommended).
- Transfer to PVDF or nitrocellulose membranes.
- Incubate with anti-PARP-1 antibodies (specific for different domains).
- Detect using enhanced chemiluminescence and compare fragment sizes to molecular weight standards.
Key Considerations: Use antibodies targeting different PARP-1 domains to confirm fragment identities. The 89-kDa fragment contains the catalytic domain, while the 24-kDa fragment contains the DNA-binding domain [19].
Distinguishing Apoptotic vs. Necrotic PARP-1 Cleavage
Purpose: To determine the proteases responsible for PARP-1 cleavage in specific cell death contexts.
Procedure:
- Pre-treat cells with either:
- 50μM zVAD-fmk (caspase inhibitor) for 1 hour before death induction
- Cathepsin inhibitors (e.g., CA-074 Me for cathepsin B) for necrotic models
- Induce cell death with appropriate stimuli.
- Analyze PARP-1 cleavage patterns by Western blotting.
- Compare fragment sizes: 89-kDa and 24-kDa indicate apoptotic cleavage; 50-kDa indicates necrotic cleavage [37] [50].
Interpretation: Caspase-dependent cleavage is inhibited by zVAD-fmk but not by cathepsin inhibitors, while necrotic cleavage shows the opposite inhibition pattern.
Subcellular Localization of PARP-1 Fragments
Purpose: To track the movement of PARP-1 fragments during cell death.
Procedure:
- Induce cell death in appropriate model system.
- Fractionate cells into nuclear and cytoplasmic components.
- Analyze PARP-1 fragments in each fraction by Western blotting.
- Use markers for different compartments (e.g., Lamin B for nucleus, GAPDH for cytoplasm).
Expected Results: In apoptosis, the 24-kDa fragment remains nuclear, while the 89-kDa fragment can translocate to the cytoplasm, particularly when poly(ADP-ribosyl)ated [20].
Signaling Pathway Visualizations
PARP-1 Cleavage in Apoptotic and Necrotic Pathways
Intersection of Apoptosis and Parthanatos via PARP-1 Fragments
Discussion and Research Implications
The role of PARP-1 cleavage as a molecular switch between apoptosis and necrosis has significant implications for understanding disease pathophysiology and developing therapeutic strategies. In cerebral ischemia, trauma, and excitotoxicity, PARP inhibition attenuates injury, demonstrating a central role of PARP-1 in these pathologies [19]. The recognition that specific PARP-1 fragments serve as signatures for particular cell death programs provides valuable biomarkers for diagnostic applications and therapeutic monitoring.
The functional significance of PARP-1 cleavage extends beyond simple inactivation of DNA repair. The 89-kDa fragment's role as a cytoplasmic PAR carrier reveals an elegant mechanism connecting caspase-dependent and independent death pathways [20] [18]. This intersection suggests potential therapeutic targets for conditions where both apoptosis and parthanatos contribute to cell loss, such as in neurodegenerative diseases and stroke.
From a therapeutic perspective, modulating PARP-1 cleavage represents a promising approach for various diseases. In cancer therapy, combining PARP inhibitors with standard treatments may enhance efficacy by preventing energy depletion and shifting cell death toward apoptosis [51]. For acute injuries such as myocardial infarction and cerebral ischemia, inhibiting PARP-1 activation may prevent necrotic cell death and inflammatory damage [9] [52].
Future research should focus on delineating the precise molecular interactions of different PARP-1 fragments with their binding partners and exploring the therapeutic potential of modulating these interactions in disease contexts. The development of more specific inhibitors targeting particular PARP-1 cleavage events or fragment functions may provide enhanced therapeutic benefits with reduced side effects.
PARP-1 cleavage serves as a critical molecular switch in cell fate decisions, directing cells toward either apoptotic or necrotic death through the generation of specific proteolytic fragments. The 89-kDa and 24-kDa fragments produced by caspases facilitate apoptotic death while conserving cellular energy, whereas the 50-kDa fragment generated by lysosomal proteases contributes to necrotic death through energy depletion. The recently discovered role of the 89-kDa fragment as a cytoplasmic PAR carrier that activates AIF-mediated parthanatos reveals an important connection between different cell death pathways. Understanding these mechanisms provides not only fundamental insights into cell death regulation but also promising therapeutic avenues for cancer, neurodegenerative diseases, and acute tissue injury where balanced cell death decisions are crucial for optimal outcomes.
Truncated Poly(ADP-ribose) polymerase-1 (tPARP1), generated through proteolytic cleavage of full-length PARP1, represents a critical molecular switch in cell death pathways with functions extending beyond its well-characterized role in DNA repair. This technical review examines the context-dependent nature of tPARP1, whose biological activities vary significantly based on cellular environment, tissue type, and the specific stressor triggering its formation. We synthesize emerging evidence demonstrating how tPARP1 fragments mediate divergent outcomes in apoptosis, inflammation, and innate immunity through tissue-specific and stressor-specific mechanisms. The comprehensive analysis of tPARP1 signaling networks, experimental methodologies, and reagent tools presented herein provides researchers with essential resources for advancing both fundamental understanding and therapeutic targeting of these proteolytic fragments in human disease.
PARP1, a nuclear enzyme central to DNA damage repair, undergoes proteolytic cleavage during various forms of cell death, generating distinct truncated fragments with unique biological activities. The canonical cleavage by caspase-3 during apoptosis produces 24 kDa and 89 kDa fragments [19] [27], but additional proteases including calpains, cathepsins, granzymes, and matrix metalloproteinases can generate different PARP1 fragments with potentially distinct functions [19]. While historically viewed as a mere marker of apoptosis, emerging research reveals that tPARP1 fragments are not merely inert byproducts but actively participate in diverse cellular processes in a context-dependent manner.
The functional divergence of tPARP1 stems from several factors: the specific cleavage site within PARP1's multi-domain structure, the cellular compartment where fragments localize, the tissue-specific expression of interaction partners, and the nature of the initial stress stimulus [19] [27]. For instance, the 89 kDa tPARP1 fragment translocates to the cytoplasm during apoptosis [5], where it gains access to novel substrates not available to nuclear full-length PARP1. This spatial redistribution enables context-specific functions that vary across tissue types and stress conditions.
This review systematically addresses the complex landscape of tPARP1 biology by examining how tissue context and stressor specificity shape its functional outcomes. We integrate structural, biochemical, and functional evidence to provide researchers with a comprehensive framework for investigating tPARP1 in diverse experimental and therapeutic contexts.
Structural Basis of PARP1 Cleavage and tPARP1 Generation
Domain Architecture of PARP1
PARP1 comprises three primary functional domains: an N-terminal DNA-binding domain (DBD) containing three zinc finger motifs (Zn1, Zn2, Zn3); a central auto-modification domain (AMD) with a BRCT (BRCA1 C-terminal) fold; and a C-terminal catalytic domain (CAT) responsible for ADP-ribosyl transferase activity [3]. The DBD mediates recognition of DNA strand breaks, while the AMD serves as a target for auto-ADP-ribosylation and contains protein-protein interaction interfaces. The CAT domain includes the NAD+-binding pocket that catalyzes poly(ADP-ribose) formation [53].
Table 1: Primary Domains of Full-Length PARP1
| Domain | Key Structural Features | Functional Role |
|---|---|---|
| DNA-binding Domain (DBD) | Three zinc finger motifs (Zn1, Zn2, Zn3); Nuclear localization signal (NLS); Caspase-3 cleavage site (DEVD214) | Recognizes and binds to DNA strand breaks; Contains caspase cleavage site |
| Auto-modification Domain (AMD) | BRCT fold; WGR domain | Mediates protein-protein interactions; Target for auto-ADP-ribosylation |
| Catalytic Domain (CAT) | Helical subdomain (HD); ADP-ribosyl transferase (ART) subdomain; NAD+ binding site | Catalyzes poly(ADP-ribose) formation; Binds PARP inhibitors |
Proteolytic Cleavage Patterns
Different proteases cleave PARP1 at specific sites, generating characteristic tPARP1 fragments with distinct domain compositions and biological activities:
- Caspase-3/7 cleavage: Occurs at aspartate 214 (DEVD214) within the DBD, separating the first two zinc fingers from the rest of the protein and producing 24 kDa (DBD-containing) and 89 kDa (AMD- and CAT-containing) fragments [19] [27].
- Calpain cleavage: Generates a 55 kDa fragment and additional smaller fragments with different biological activities [19].
- Other proteases (cathepsins, granzymes, MMPs): Produce distinct cleavage patterns that remain less characterized but contribute to the diversity of tPARP1 functions [19].
The 89 kDa tPARP1 fragment retains the BRCT domain, WGR domain, and catalytic domain but loses the nuclear localization signal and two zinc fingers, facilitating its cytoplasmic redistribution and altering its substrate specificity [5] [19].
Diagram 1: PARP1 cleavage by caspase-3/7 generates distinct fragments with different subcellular localizations.
Stressor-Specific tPARP1 Activation and Signaling
Apoptotic Stressors and Innate Immune Activation
During caspase-dependent apoptosis triggered by DNA-damaging agents or pathogenic stimuli, tPARP1 translocates to the cytoplasm where it interacts with novel substrates. A key mechanism involves the recognition of the RNA polymerase III (Pol III) complex by the BRCT domain of tPARP1 [5]. This interaction enables tPARP1 to mono-ADP-ribosylate Pol III, enhancing its activity in transcribing foreign DNA (such as poly(dA-dT)) into double-stranded RNA. This dsRNA then activates innate immune signaling pathways leading to IFN-β production and amplification of apoptotic signaling [5].
Table 2: Stressor-Specific tPARP1 Functions and Outcomes
| Stressor Type | Cleaving Protease | tPARP1 Fragment | Primary Function | Cellular Outcome |
|---|---|---|---|---|
| Apoptotic stimuli (DNA damage, pathogenic infection) | Caspase-3/7 | 89 kDa (cytosolic) | Mono-ADP-ribosylation of Pol III; Enhanced IFN-β production | Amplification of apoptosis; Innate immune activation |
| Excitotoxicity, ischemia-reperfusion | Calpain | 55 kDa and others | Altered gene regulation; Modulation of inflammatory response | Context-dependent cell death |
| Severe DNA damage | PARP1 overactivation | Not applicable (full-length hyperactivation) | NAD+ depletion; Energy crisis | Necrotic cell death |
| Ischemic challenge (OGD/ROG) | Caspase-3/7 | 24 kDa and 89 kDa | Regulation of NF-κB activity; Modulation of inflammatory genes | Altered cell survival/death balance |
This stressor-specific functionality demonstrates how tPARP1 can serve different roles based on the initial cellular insult. In the case of poly(dA-dT)-stimulated apoptosis, tPARP1 functions as a bridge between apoptotic and innate immune pathways, potentially contributing to clearance of infected cells [5].
Metabolic and Ischemic Stressors
Under conditions of oxygen/glucose deprivation (OGD) and restoration (ROG) modeling ischemia-reperfusion injury, tPARP1 fragments differentially influence cell survival and inflammatory responses. Expression of the 24 kDa fragment or an uncleavable PARP1 mutant conferred protection from OGD and OGD/ROG damage in neuronal models, while the 89 kDa fragment exhibited cytotoxic effects [27]. This differential effect occurred without significant changes in NAD+ levels or PAR formation, suggesting a PAR-independent mechanism.
Instead, the protective versus toxic effects correlated with modulation of NF-κB signaling and downstream inflammatory mediators. The cytotoxic 89 kDa fragment enhanced NF-κB activity and increased expression of pro-inflammatory proteins (iNOS, COX-2), while reducing anti-apoptotic Bcl-xL [27]. This demonstrates how the same cleavage event can produce fragments with opposing biological activities under metabolic stress conditions.
Diagram 2: Stressor-specific pathways leading to different tPARP1 functions and cellular outcomes.
Tissue-Specific Manifestations of tPARP1 Function
Neuronal Systems
In neuronal tissues, PARP1 cleavage and tPARP1 fragment formation have been extensively studied in cerebral ischemia, trauma, excitotoxicity, and neurodegenerative conditions [19] [27]. The balance between different tPARP1 fragments significantly influences neuronal survival versus death decisions. The 24 kDa fragment appears to exert neuroprotective effects in OGD models, while the 89 kDa fragment promotes inflammatory signaling and cytotoxicity [27].
PARP1 cleavage serves as a biomarker for specific protease activities in unique cell death programs in neurological contexts [19]. The presence of characteristic tPARP1 fragments can help distinguish between apoptotic, necrotic, and other forms of neuronal cell death, with potential diagnostic and therapeutic implications for conditions like Alzheimer's disease, Parkinson's disease, and traumatic brain injury [19].
Liver Pathology
In liver diseases, including alcoholic liver disease, non-alcoholic fatty liver disease, viral hepatitis, and hepatocellular carcinoma, PARP1 expression and cleavage play important roles in disease progression [40]. PARP1 knockout mice exhibit impaired liver regeneration and reduced hepatocyte proliferation, indicating a role for PARP1 (and potentially its cleavage products) in liver repair mechanisms [40]. The duality of PARP1 function in liver pathology—beneficial for regeneration but potentially harmful when overactivated—highlights the importance of understanding the specific contexts in which tPARP1 fragments exert their effects.
Cancer Systems
In breast cancer, PARP1 cleavage fragments may influence therapeutic responses to PARP inhibitors [54]. The molecular heterogeneity of breast cancer subtypes results in different patterns of PARP1 expression and potentially distinct cleavage dynamics in response to therapy [54]. The role of tPARP1 in regulating transcription factors like TFAP2A through ADP-ribosylation in a subtype-specific manner suggests tissue- and context-dependent functions in cancer systems [54].
Experimental Approaches for tPARP1 Research
Key Methodologies
Investigating tPARP1 functions requires specialized methodological approaches to detect fragments, characterize interactions, and determine functional consequences:
Detection and Quantification Methods:
- Western blotting using antibodies recognizing specific PARP1 epitopes can distinguish full-length PARP1 from cleavage fragments [27]. Antibodies specific for the 89 kDa fragment are particularly valuable for apoptosis assessment [5].
- Mass spectrometry-based approaches enable identification of ADP-ribosylation sites and interacting partners of tPARP1 fragments [54].
- Co-immunoprecipitation assays validate interactions between tPARP1 and proposed binding partners like the Pol III complex [5].
Functional Assessment Methods:
- Viability assays (MTT, Annexin V/PI staining) determine the consequences of tPARP1 expression under different stress conditions [27].
- Transcriptional reporter assays assess how tPARP1 fragments influence NF-κB and other transcription factor activities [27].
- Subcellular localization studies using immunofluorescence or fractionation track the redistribution of tPARP1 fragments during cell death [5] [19].
Table 3: Essential Research Reagents for tPARP1 Investigation
| Reagent Category | Specific Examples | Research Application | Key Features |
|---|---|---|---|
| PARP1 antibodies | Anti-PARP1 (cleavage-specific); Anti-PAR antibody | Detection of cleavage fragments; Assessment of PARP activity | Distinguish full-length vs. truncated PARP1; Recognize PAR polymers |
| Expression constructs | PARP1-UNCL (uncleavable); PARP-124; PARP-189 | Functional analysis of specific fragments | Enable isolated study of fragment functions |
| Protease inhibitors | Caspase inhibitors (Z-VAD-FMK); Calpain inhibitors | Determine protease specificity in cleavage | Identify proteases responsible for tPARP1 generation |
| PARP inhibitors | Olaparib; Talazoparib; PJ34 | Assess catalytic-dependent vs independent functions | Distinguish structural vs enzymatic roles of tPARP1 |
| Cell death inducers | Poly(dA-dT); Staurosporine; H2O2 | Activate specific cell death pathways with different tPARP1 outcomes | Trigger stressor-specific tPARP1 functions |
Protocol: Assessing tPARP1-Pol III Interactions During Apoptosis
This protocol outlines key methodology for investigating tPARP1 functions in cytosolic DNA-induced apoptosis, based on approaches described in [5]:
Induction of Apoptosis:
- Transfect cells with poly(dA-dT) (1-2 μg/mL) using appropriate transfection reagent to mimic cytosolic DNA from pathogens.
- Incubate for 6-24 hours depending on cell type.
- Include controls with empty vector or transfection reagent alone.
Validation of Apoptosis and PARP1 Cleavage:
- Harvest cells and prepare whole-cell lysates using RIPA buffer supplemented with protease inhibitors.
- Perform Western blotting using anti-PARP1 antibodies that recognize both full-length and cleaved PARP1.
- Confirm apoptosis by Annexin V/propidium iodide staining and flow cytometry.
Co-immunoprecipitation of tPARP1-Complexes:
- Prepare lysates from apoptotic cells in non-denaturing lysis buffer.
- Incubate lysates with anti-HA magnetic beads (for HA-tagged tPARP1) or anti-PARP1 antibodies.
- Wash beads extensively and elute bound proteins.
- Analyze eluates by Western blotting for POLR3A, POLR3B, and POLR3F (Pol III subunits).
Functional Assessment of Innate Immune Activation:
- Measure IFN-β production by ELISA or quantitative RT-PCR.
- Assess Pol III-mediated dsRNA production using specific antibodies or reporter systems.
- Evaluate the functional consequences of disrupting tPARP1-Pol III interactions using BRCT domain mutants (e.g., F473A).
The Scientist's Toolkit: Essential Research Reagents
Successful investigation of tPARP1 functions requires carefully selected research tools and reagents. The following table summarizes key resources based on methodologies cited in the literature:
Table 4: Research Reagent Solutions for tPARP1 Investigation
| Reagent Type | Specific Examples | Function/Application | Experimental Notes |
|---|---|---|---|
| PARP1 antibodies | Anti-PARP1 (Active Motif, 39559); Cleavage-specific antibodies | Detect full-length and truncated PARP1; Assess cleavage during apoptosis | Use antibodies that recognize different epitopes to distinguish fragments |
| Expression constructs | PARP1-UNCL (uncleavable mutant); PARP-124; PARP-189 | Express specific fragments; Determine functional contributions | Generate using site-directed mutagenesis (D214A for PARP1-UNCL) |
| PARP inhibitors | Olaparib (HY-10162); Talazoparib (HY-16106); PJ34 (ALX-270-289) | Inhibit catalytic activity; Distinguish enzymatic vs. scaffolding functions | Use at appropriate concentrations (e.g., Olaparib IC50 = 5 nM) |
| Protease inhibitors | Z-VAD-FMK (caspase inhibitor); Calpain inhibitors | Identify proteases responsible for PARP1 cleavage | Use in concentration ranges that selectively inhibit target proteases |
| Apoptosis inducers | Poly(dA-dT); Staurosporine; Etoposide | Activate caspase-dependent apoptosis; Generate tPARP1 fragments | Optimize concentration and duration for specific cell types |
| Tagging systems | SFB-tag (S-protein, FLAG, streptavidin-binding peptide); HA-tag | Facilitate affinity purification; Detect expressed proteins | SFB-tag enables tandem affinity purification for interaction studies |
The investigation of tPARP1 has evolved from viewing PARP1 cleavage as a simple cell death marker to understanding it as a mechanism generating functionally diverse fragments with context-dependent activities. The tissue-specific and stressor-specific functions of tPARP1 fragments represent an important layer of complexity in cell death regulation, with significant implications for pathogenesis and therapeutic development.
Key challenges moving forward include developing more specific tools to distinguish the functions of different tPARP1 fragments in complex biological systems, understanding how tissue-specific expression of interaction partners influences tPARP1 activities, and determining whether different tPARP1 fragments could serve as biomarkers for specific disease states or treatment responses. The potential for targeting specific tPARP1 functions therapeutically—either by inhibiting detrimental fragments or promoting protective ones—represents a promising avenue for future research, particularly in neurological disorders, cancer, and inflammatory conditions where PARP1 cleavage plays an established role in disease progression.
The cleavage of poly(ADP-ribose) polymerase 1 (PARP1) during apoptosis represents a fundamental paradox in cell biology. While historically considered a simple marker of caspase-mediated cell death, emerging research reveals that the resulting truncated PARP1 (tPARP1) fragments are not merely inactive debris but rather gain-of-function effectors that actively modulate cell death pathways. This whitepaper examines the biological significance of truncated PARP1 fragments within the broader context of redundant DNA repair and cell death mechanisms. The proteolytic processing of PARP1 by caspases and other proteases generates specific signature fragments that initiate unique signaling cascades, challenging the conventional view of PARP1 cleavage as merely an apoptotic bystander effect. Understanding how these fragments overcome compensatory cellular mechanisms provides critical insights for developing novel cancer therapeutic strategies that exploit inherent redundancies in biological systems.
PARP1 Structure and Cleavage Fragment Generation
Domain Architecture of Full-Length PARP1
PARP1 is a multifunctional enzyme comprising several structured domains that confer its DNA damage sensing and catalytic capabilities. The N-terminal DNA-binding domain (DBD) contains three zinc finger motifs (Zn1, Zn2, Zn3) that recognize DNA strand breaks [1] [3]. The Zn1 and Zn2 motifs specifically recognize DNA damage gaps by binding to the 5' and 3' ends respectively, distributed on both sides of DNA break sites, while Zn3 links the structural domains to activate the target protein [3]. Adjacent to the DBD is the nuclear localization signal (NLS) and a DEVD structural motif related to apoptosis recognition [3]. The central automodification domain (AMD) contains a BRCT (BRCA1 C-terminal) fold that mediates protein-protein interactions and serves as the primary target for PARP1 auto-ADP-ribosylation [1] [3]. The C-terminal catalytic domain (CAT) consists of the α-helical subdomain (HD) and ADP-ribosyl transferase (ART) subdomain, which contains the NAD+ binding site and PAR catalytic site essential for poly(ADP-ribose) formation [3].
Proteolytic Cleavage and Fragment Signatures
PARP1 serves as a preferred substrate for multiple "suicidal" proteases, with each protease family generating distinctive cleavage fragments that serve as biomarkers for specific cell death pathways [1]. Caspase-3 and -7 cleavage during apoptosis occurs primarily at aspartate 214 within the DEVD motif, producing a 24-kD N-terminal fragment containing two zinc-finger motifs and an 89-kD C-terminal fragment containing the third zinc finger, BRCT domain, WGR domain, and catalytic domain [5] [1]. This 89-kD truncated PARP1 (tPARP1) translocates from the nucleus to the cytoplasm, while the 24-kD fragment remains nuclear due to its retention of the NLS [5]. Calpain cleavage generates a 55-kD fragment, while granzyme A produces 50-kD and 62-kD fragments, and matrix metalloproteinases (MMPs) generate 55-kD, 40-kD, and 35-kD fragments, each associated with distinct pathological conditions [1].
Table 1: PARP1 Cleavage Fragments by Protease Family
| Protease | Cleavage Fragments | Primary Function | Cell Death Context |
|---|---|---|---|
| Caspase-3/7 | 24-kD + 89-kD (tPARP1) | Apoptosis execution; cytoplasmic signaling | Apoptosis |
| Calpain | 55-kD | Necrotic cell death | Ischemia/reperfusion injury |
| Granzyme A | 50-kD + 62-kD | Immune-mediated cell death | Cytotoxic T-cell response |
| MMPs | 55-kD, 40-kD, 35-kD | Tissue remodeling | Inflammatory conditions |
Compensatory Mechanisms in DNA Repair Pathways
DNA Repair Pathway Redundancy
Cells maintain genomic integrity through multiple complementary DNA repair pathways that exhibit significant functional redundancy. The base excision repair (BER) pathway serves as the primary mechanism for removing oxidative base damage, alkylation damage, and abasic sites, with PARP1 playing a central role in detecting and initiating repair of single-strand breaks (SSBs) [55]. Nucleotide excision repair (NER) provides backup for BER by processing bulky DNA lesions and certain types of oxidative damage, with demonstrated cross-pathway compensation observed in C. elegans models where loss of NTH-1 (BER) and XPA-1 (NER) produces a synthetic viability phenotype [56]. For double-strand breaks (DSBs), homologous recombination (HR) and non-homologous end joining (NHEJ) provide complementary error-free and error-prone repair mechanisms respectively, with PARP1 influencing both pathways through its roles in replication fork stabilization and alternative NHEJ regulation [55].
Transcriptional Compensation in Repair-Deficient Models
Studies in C. elegans reveal sophisticated transcriptional compensation mechanisms that maintain viability in DNA repair-deficient backgrounds. Loss of the base excision repair enzyme NTH-1 triggers upregulation of endogenous stress response genes and downregulation of insulin/IGF-1 signaling (ILS) pathway components, creating a pro-survival transcriptional profile that mirrors changes observed in NER-deficient models [56]. This compensatory transcriptomic shift involves 2074 differentially expressed probe sets in nth-1 mutants, including 84 genes downstream of the FOXO transcription factor DAF-16, though the response appears distinct from canonical DAF-16 activation [56]. Simultaneous disruption of both NTH-1 and XPA-1 generates a fundamentally different expression profile with downregulation of Aurora-B and Polo-like kinase 1 signaling networks, suggesting activation of alternative compensatory mechanisms when primary redundancy fails [56].
Biological Functions of Truncated PARP1 Fragments
Apoptosis-Specific Signaling by tPARP1
The 89-kD tPARP1 fragment generated by caspase cleavage exhibits gain-of-function properties that actively promote apoptotic signaling through novel mechanisms. Unlike full-length PARP1, tPARP1 translocates to the cytoplasm where it recognizes and binds the RNA polymerase III (Pol III) complex via its BRCT domain [5]. This interaction facilitates mono-ADP-ribosylation of Pol III subunits, enhancing Pol III-mediated transcription of foreign DNA and stimulating IFN-β production during poly(dA-dT)-stimulated apoptosis [5]. This mechanism represents a paradigm shift in understanding PARP1 cleavage, demonstrating that tPARP1 actively bridges DNA damage responses with innate immune activation during apoptosis. Expression of non-cleavable PARP1 mutants impairs these processes, confirming the biological significance of tPARP1-specific signaling [5].
Metabolic Regulation of Cell Fate Decisions
PARP1 cleavage fragments play a decisive role in directing cellular fate through metabolic regulation. Under conditions of moderate DNA damage, caspase-mediated PARP1 cleavage prevents NAD+ depletion, conserving cellular energy stores and facilitating the orderly process of apoptosis [3] [57]. In contrast, severe DNA damage causes PARP1 overactivation that depletes NAD+ and ATP, shifting cell death toward necrosis [3]. The 89-kD tPARP1 fragment exhibits altered enzymatic activity偏向 toward mono-ADP-ribosylation rather than poly-ADP-ribosylation, potentially modifying its impact on cellular energy metabolism [5]. This metabolic regulation creates a cell fate switch where the extent of DNA damage and PARP1 activation determines death modality, with tPARP1 generation representing an adaptive response that promotes apoptosis over necrosis when damage is moderate.
Caspase-Independent Cell Death Pathways
Beyond apoptosis, PARP1 fragments mediate caspase-independent cell death through mechanisms involving apoptosis-inducing factor (AIF). PARP1 overactivation triggers AIF release from mitochondria and translocation to the nucleus, where it induces chromatin condensation and large-scale DNA fragmentation (~50 kbp) [57]. This PARP1/AIF-mediated cell death pathway contributes significantly to ischemia/reperfusion injury and represents an important alternative death mechanism when caspase activation is impaired [57]. The specific PARP1 fragments involved in this pathway remain partially characterized, though the 89-kD tPARP1 likely participates given its retention of catalytic capacity and nuclear export capabilities.
Experimental Approaches and Methodologies
Identification of tPARP1-Interacting Partners
The discovery of tPARP1-Pol III interactions employed sophisticated protein affinity purification techniques. Researchers stably expressed catalytically inactive tPARP1 (E988A mutant) in PARP1-deficient 293T cells to "trap" transient interaction partners, followed by tandem affinity purification under native conditions [5]. To stabilize normally transient ADP-ribosylation events, parallel experiments expressed mutant versions of ADP-ribose removal enzymes (PARG E756A and TARG1 D125A) [5]. Interacting proteins were identified by mass spectrometry analysis, with the Pol III subunits POLR3A, POLR3B, and POLR3F emerging as common interactors with both mtPARP1 and mTARG1 [5]. Interaction validation included co-immunoprecipitation assays with hemagglutinin (HA)-tagged mtPARP1 and myc-tagged Pol III subunits in PARP1-deficient cells, confirming direct binding [5]. Domain mapping experiments using internal truncation mutants identified the BRCT domain as essential for Pol III interaction, with point mutation F473A ablating binding capacity [5].
Table 2: Essential Research Reagents for tPARP1 Studies
| Reagent/Solution | Specific Application | Function/Rationale |
|---|---|---|
| Catalytic E988A PARP1 mutant | Substrate trapping | Stabilizes transient enzyme-substrate interactions |
| Non-cleavable PARP1 mutants | Apoptosis signaling studies | Dissects cleavage-dependent and independent functions |
| PARP1-deficient 293T cells | Interaction studies | Eliminates confounding endogenous PARP1 |
| Poly(dA-dT) transfection | Apoptosis induction | Mimics pathogenic DNA; stimulates cytosolic DNA sensing |
| BRCT domain mutants (e.g., F473A) | Domain function mapping | Identifies critical interaction interfaces |
| Tandem affinity purification tags | Protein complex isolation | Enables high-specificity purification under native conditions |
Functional Assays for tPARP1 Activity
Poly(dA-dT)-stimulated apoptosis assays provide a robust system for evaluating tPARP1 function in physiological contexts. Transfection of poly(dA-dT) into cells mimics pathogenic DNA exposure, triggering caspase activation and PARP1 cleavage [5]. Apoptosis validation employs multiple complementary approaches: antibody-based detection of PARP1 cleavage fragments using both general PARP1 antibodies and tPARP1-specific reagents; Annexin V-FITC/propidium iodide staining with FACS analysis for quantitative apoptosis measurement; and morphological assessment of characteristic apoptotic changes [5]. For evaluating tPARP1 enzymatic activity, in vitro ADP-ribosylation assays with purified tPARP1 and Pol III components confirm direct modification capacity, while cellular ADP-ribosylation status during apoptosis can be monitored using ADP-ribose-specific antibodies [5].
Diagram 1: PARP1 Cleavage Fragment Signaling Pathways
Therapeutic Implications and Research Applications
PARP Inhibitor Development
Understanding PARP1 fragment biology has profound implications for targeted cancer therapy development. Current PARP inhibitors (PARPis) trap both PARP1 and PARP2 on DNA, causing synthetic lethality in BRCA-deficient cancers but exhibiting dose-limiting toxicities, particularly hematological effects linked to PARP2 inhibition [55]. Next-generation PARP1-selective inhibitors leverage structural differences between PARP1 and PARP2 to minimize toxicity while maintaining efficacy in homologous recombination-deficient tumors [55]. The discovery of tPARP1-specific functions suggests potential for therapeutics that modulate rather than completely inhibit PARP1 activity, potentially exploiting the distinct enzymatic properties of cleavage fragments for more selective intervention.
Biomarker and Diagnostic Applications
PARP1 cleavage fragments serve as specific biomarkers for different cell death modalities and therapeutic responses. The distinctive 24-kD and 89-kD caspase-generated fragments provide specific indicators of apoptotic activation, while alternative fragments generated by calpain, granzymes, or MMPs signal distinct death pathways operative in various pathological contexts [1]. Detection of cytoplasmic tPARP1 could specifically indicate apoptosis with innate immune activation, potentially serving as a biomarker for immunogenic cell death and response to DNA-damaging therapies [5]. The development of fragment-specific antibodies enables precise discrimination of cell death mechanisms in clinical samples, providing valuable diagnostic and prognostic information.
Diagram 2: Experimental Workflow for tPARP1 Interaction Mapping
The biological functions of truncated PARP1 fragments represent a compelling example of how compensatory mechanisms and redundancy in DNA repair and cell death pathways can be co-opted for specific biological outcomes. Rather than representing mere inactivation, PARP1 cleavage generates functionally distinct fragments that actively mediate apoptosis signaling through novel mechanisms, including cytosolic innate immune activation via Pol III interaction. This paradigm challenges traditional views of protease-mediated fragmentation as purely destructive and suggests similar gain-of-function roles for other cleaved signaling molecules. Future research should focus on identifying additional tPARP1 interaction partners, characterizing the structural basis for its altered enzymatic specificity, and exploring therapeutic opportunities for modulating tPARP1-specific functions in cancer and other diseases. The continued elucidation of how cells overcome compensatory mechanisms through regulated proteolysis will undoubtedly yield valuable insights for both basic biology and clinical applications.
Poly (ADP-ribose) polymerase 1 (PARP1) is a multifunctional enzyme traditionally recognized for its paramount role in the DNA damage response (DDR), particularly in the repair of DNA single-strand breaks via the base excision repair (BER) pathway [55] [58]. Its catalytic activity, triggered by DNA damage, results in the synthesis of poly(ADP-ribose) (PAR) chains on itself and other acceptor proteins, facilitating the recruitment of repair machinery and maintaining genomic integrity [59] [58]. However, during the execution of apoptosis, PARP1 undergoes caspase-mediated cleavage, a hallmark of programmed cell death. Caspase-3 primarily cleaves PARP1 at D214, generating a 24 kDa N-terminal fragment and an 89 kDa truncated PARP1 (tPARP1) [5]. While the 24 kDa fragment remains nuclear and may act as a dominant-negative by occupying DNA ends, the 89 kDa tPARP1, which contains the BRCT domain, WGR domain, and the C-terminal catalytic domain, translocates to the cytoplasm [5]. For years, this cleavage was viewed merely as a mechanism to inactivate PARP1's DNA repair function and prevent energy depletion. Recent research, however, has unveiled that tPARP1 is not merely an inactive relic but possesses its own pro-apoptotic biological functions, positioning it as a compelling therapeutic target [5] [39].
This whitepaper delves into the emerging biology of tPARP1, framing it within the context of cell death research and outlining a strategic framework for optimizing its induction in combination with PARP inhibitors (PARPi). We provide a detailed analysis of tPARP1's mechanisms, current combination therapy paradigms, validated experimental protocols for exploring this axis, and a toolkit of essential reagents, aiming to equip researchers with the knowledge to design novel and effective anticancer strategies.
The Biological Function of Truncated PARP1 in Cell Death
A Novel Role in Innate Immune Activation
The biological significance of tPARP1 has remained elusive until recently. An unbiased protein affinity purification study identified that tPARP1, upon translocation to the cytosol, specifically recognizes and interacts with the RNA polymerase III (Pol III) complex [5]. This interaction is mediated by the BRCT domain of tPARP1, and mutation of a key residue (F473A) in this domain disrupts the binding to Pol III subunits (POLR3A, POLR3B, POLR3F) [5].
Notably, tPARP1 catalyzes mono-ADP-ribosylation (MARylation) of the Pol III complex. This post-translational modification occurs during poly(dA-dT)-stimulated apoptosis, which mimics pathogenic DNA infection and triggers the innate immune response [5]. The tPARP1-mediated MARylation of Pol III facilitates the production of interferon-beta (IFN-β), a key cytokine in antiviral immunity, and enhances apoptosis [5]. In contrast, suppression of PARP1 or expression of a non-cleavable PARP1 mutant impairs these events. This reveals a previously unknown pathway wherein tPARP1, generated during apoptosis, actively participates in amplifying the innate immune response, potentially to eliminate infected or damaged cells more effectively.
Interplay with Ferroptosis and Apoptosis
The functional output of PARP1 cleavage is context-dependent and intersects with other cell death pathways. Research on the ferroptosis inducer RSL3 has shown that it promotes PARP1's apoptotic functions through distinct mechanisms [39]. During RSL3-induced ferroptosis, which is characterized by iron-dependent lipid peroxidation, two parallel apoptotic pathways are triggered:
- Caspase-Dependent Cleavage: RSL3 increases reactive oxygen species (ROS), leading to the activation of caspase-3, which subsequently cleaves PARP1 to generate the pro-apoptotic tPARP1 fragments [39].
- Epitranscriptomic Regulation of Full-Length PARP1: Simultaneously, RSL3 inhibits METTL3-mediated N6-methyladenosine (m6A) modification of PARP1 mRNA. This reduces PARP1 mRNA stability and translational efficiency, leading to decreased levels of full-length PARP1. The loss of full-length PARP1 exacerbates DNA damage and contributes to apoptosis [39].
This dual mechanism underscores that the depletion of full-length PARP1 and the generation of tPARP1 can cooperatively drive cell death. Importantly, RSL3 retains its pro-apoptotic efficacy even in PARPi-resistant cells, highlighting the potential of inducing tPARP1 to overcome therapeutic resistance [39].
The following diagram illustrates the sequence of tPARP1 generation and its role in immune activation during apoptosis.
Rationale for Combining tPARP1-Inducing Agents with PARP Inhibitors
The combination of tPARP1-inducing agents with PARPi is grounded in a multi-faceted mechanistic rationale designed to exploit distinct vulnerabilities in cancer cells and overcome resistance.
Overcoming PARPi Resistance
Resistance to PARPi monotherapy is a significant clinical challenge, often arising through mechanisms such as the restoration of homologous recombination (HR) repair [58]. By inducing tPARP1 formation via caspase-activating agents, this strategy introduces a parallel, irreversible pro-death signal that is independent of HR status. The tPARP1-mediated cytosolic immune signaling pathway can trigger cell death even in cells that have developed resistance to the synthetic lethal effects of PARPi [5] [39]. Furthermore, since tPARP1 lacks the N-terminal zinc fingers of full-length PARP1, its functions fall outside the classic PARP trapping and catalytic inhibition mechanisms, providing a novel angle of attack [5].
Exploiting Synergistic Cell Death Pathways
Many PARPi, such as olaparib, have been shown to downregulate SLC7A11, a component of the cystine/glutamate antiporter, in a p53-dependent manner. This impairs glutathione synthesis and sensitizes cells to ferroptosis [60]. Concurrently, ferroptosis inducers like RSL3 can enhance the cleavage of PARP1 and the generation of tPARP1 [39]. This creates a positive feedback loop where each agent potentiates the other's efficacy: PARPi primes the cell for ferroptosis, and the ferroptosis inducer accelerates the caspase-mediated generation of pro-apoptotic tPARP1. This crosstalk between apoptosis, ferroptosis, and tPARP1 signaling can lead to a more robust and sustained tumor cell kill.
Expanding the Therapeutic Window with Targeted Delivery and Scheduling
A major limitation of combining DNA-damaging agents with PARPi has been overlapping toxicity, particularly myelosuppression [26] [58]. Innovative approaches are now focused on mitigating these toxicities. A key strategy is tumor-targeted drug delivery. For instance, the nanoparticle topoisomerase I inhibitor CRLX101 exhibits prolonged circulation and preferential tumor accumulation, reducing systemic exposure [26].
Another critical strategy is gapped scheduling. Preclinical models informed a clinical trial design where olaparib dosing was initiated 48 hours after CRLX101 administration. This schedule allowed for adequate marrow recovery and enabled the delivery of higher, more effective doses of both agents, successfully widening the therapeutic window [26]. The following table summarizes key clinical evidence for combination strategies that can be leveraged to induce tPARP1.
Table 1: Clinical Evidence for Combination Strategies Pertinent to tPARP1 Induction
| Combination Therapy | Clinical Context | Key Findings & Relevance to tPARP1 | Source |
|---|---|---|---|
| CRLX101 (Nanoparticle TOP1i) + Olaparib (Gapped Schedule) | Advanced Solid Tumors (Phase I) | MTD: CRLX101 12 mg/m² q2w + Olaparib 250 mg BID (Days 3-13, 17-26). Elevated γH2AX (DNA damage) with combination. Tolerable safety profile. Rationale: TOP1i are potent inducers of DNA damage and apoptosis, potentially triggering PARP1 cleavage. | [26] |
| Talazoparib (PARPi) + Nedisertib (DNA-PKi) | In vitro model of BRCA-mutated Prostate Cancer | Additive decrease in cell proliferation and increased necrosis in BRCA-KO cells. Rationale: DNA-PKi impairs error-prone NHEJ, enhancing genomic chaos and apoptosis in HRD cells, potentially synergizing with PARPi and increasing tPARP1 generation. | [61] |
| Niraparib (PARPi) | Ovarian Cancer Preclinical Models | Induced ferroptosis via upregulation of fatty acid transporter CD36, independent of p53/BRCA status. Rationale: Connects PARP inhibition to ferroptosis, which can activate caspases and lead to PARP1 cleavage. | [60] |
Experimental Protocols for Investigating tPARP1 Function and Combination Effects
To empirically validate and optimize tPARP1-focused combination therapies, robust and detailed experimental methodologies are required. Below are key protocols adapted from recent studies.
Protocol: Validating tPARP1 and Pol III Interaction
- Objective: To confirm the physical interaction between caspase-generated tPARP1 and the RNA Pol III complex during apoptosis.
- Methodology:
- Cell Line & Transfection: Use PARP1-deficient 293T cells. Transfect with plasmids encoding:
- HA-tagged mutant tPARP1 (catalytic dead E988A to trap substrates).
- Myc-tagged Pol III subunits (POLR3A, POLR3B, POLR3F).
- A plasmid encoding caspase-3 for co-transfection experiments.
- Apoptosis Induction: 24-48 hours post-transfection, induce apoptosis by transfection with poly(dA-dT) (1 μg/mL for 6-8 hours) to mimic cytosolic DNA stress [5].
- Co-immunoprecipitation (Co-IP):
- Lyse cells in a mild, non-denaturing lysis buffer (e.g., NP-40 based).
- Incubate the soluble protein fraction with anti-HA affinity gel overnight at 4°C.
- Wash beads extensively to remove non-specifically bound proteins.
- Elute bound proteins and denature in Laemmli buffer.
- Detection: Analyze eluates and whole-cell lysates (inputs) by Western blotting.
- Probe for Myc-tag (Pol III subunits) to confirm interaction.
- Probe for HA-tag to verify tPARP1 pulldown.
- Use an antibody specific for the 89 kDa tPARP1 fragment to confirm cleavage.
- Cell Line & Transfection: Use PARP1-deficient 293T cells. Transfect with plasmids encoding:
- Key Controls: Include cells transfected with empty vector and cells transfected with full-length PARP1 (E988A mutant) to demonstrate the specificity of the tPARP1-Pol III interaction [5].
Protocol: Assessing Synergy in PARPi-Resistant Models
- Objective: To evaluate the cytotoxic synergy between a tPARP1-inducing agent (e.g., RSL3) and a PARPi in a PARPi-resistant cell line.
- Methodology:
- Cell Culture: Establish a PARPi-resistant cell line (e.g., by chronic, low-dose exposure to olaparib) or use a known resistant line. Maintain alongside the parental sensitive line.
- Drug Treatment:
- Treat cells with a matrix of concentrations for RSL3 (e.g., 0, 0.5, 1, 2 μM) and olaparib (e.g., 0, 5, 10, 20 μM) for 72 hours.
- Include control wells for DMSO (vehicle) and single agents.
- Viability Assay: Use the CellTiter-Glo 2.0 Luminescent assay to quantify ATP levels as a surrogate for cell viability, following the manufacturer's protocol [61].
- Synergy Analysis:
- Calculate the percentage of viability for each combination.
- Input the dose-response matrix into synergy analysis software such as SynergyFinder.
- Use the Zero Interaction Potency (ZIP) model to calculate synergy scores. A score >10 indicates synergy [61].
- Mechanistic Validation:
- Perform Western blotting on treated cells to detect PARP1 cleavage (using tPARP1-specific antibody) and caspase-3 activation.
- Measure lipid peroxidation (e.g., with C11-BODIPY 581/591 probe) to confirm ferroptosis induction.
- Expected Outcome: Synergistic cell death is anticipated in the resistant model, correlated with enhanced tPARP1 cleavage and lipid peroxidation, demonstrating the ability of the combination to bypass resistance mechanisms [39].
The Scientist's Toolkit: Essential Reagents and Models
To effectively research the tPARP1 combination therapy axis, the following tools and reagents are indispensable.
Table 2: Key Research Reagent Solutions for tPARP1 Studies
| Reagent / Model | Function / Application | Key Feature / Consideration |
|---|---|---|
| Non-Cleavable PARP1 Mutant | Control to distinguish functions of full-length PARP1 vs. tPARP1. | Mutated caspase cleavage site (D214A); expression impairs tPARP1-mediated IFN-β production and apoptosis [5]. |
| BRCT Domain Mutant (F473A) | Validates the specificity of tPARP1-Pol III interaction. | Disrupts the tertiary structure of the BRCT domain, abolishing binding to Pol III subunits [5]. |
| PARP1-Deficient Cell Lines | Isolate the phenotypic effects of re-introduced tPARP1. | CRISPR/Cas9-generated knockout cells (e.g., in 293T background) are ideal for transfection-based rescue experiments [5] [61]. |
| tPARP1-Specific Antibody | Specifically detects the 89 kDa apoptotic fragment. | Critical for distinguishing tPARP1 from full-length PARP1 in Western blot and immunofluorescence; avoids cross-reactivity. |
| Caspase-3 Activators | Induce apoptosis and generate endogenous tPARP1. | Poly(dA-dT) transfection (mimics cytosolic DNA) [5]; Chemotherapeutic agents (e.g., Topoisomerase inhibitors [26]); Ferroptosis inducers (e.g., RSL3 [39]). |
| PARP1 Degrader (e.g., NN3) | Alternative strategy to deplete full-length PARP1. | Induces ferroptosis in p53-positive, PARPi-resistant cells by downregulating SLC7A11, complementing tPARP1 induction strategies [60]. |
The discovery of tPARP1's non-canonical, pro-apoptotic role in the cytoplasm represents a paradigm shift in our understanding of PARP1 biology. Moving beyond its traditional nuclear confines, tPARP1 emerges as a critical mediator at the intersection of apoptosis and innate immunity. Strategically inducing its formation, particularly in concert with PARP inhibition, presents a powerful and rational approach to enhance anticancer efficacy and counter the pervasive challenge of drug resistance.
Future research must focus on delineating the precise structural determinants of tPARP1's catalytic activity towards Pol III and identifying other potential cytosolic substrates. The clinical translation of these strategies will heavily rely on the continued refinement of drug delivery systems, such as nanoparticles, and the optimization of scheduling regimens to maximize tumor cell kill while sparing normal tissues. By integrating the induction of tPARP1 into the next generation of combination therapies, researchers and drug developers can open a new front in the battle against cancer, leveraging the full complexity of cell death pathways for therapeutic gain.
The hyperactivation of poly(ADP-ribose) polymerase-1 (PARP-1) in response to genotoxic stress is a well-established trigger of cell death. For decades, the prevailing paradigm attributed this form of cell demise, known as parthanatos, to catastrophic NAD+ and ATP depletion resulting from attempts to sustain NAD+ resynthesis. However, recent research has unveiled a more nuanced and complex picture, implicating specific proteolytic fragments of PARP-1 and signaling-driven metabolic dysfunction beyond simple energetic failure. This whitepaper synthesizes current evidence on the metabolic consequences of PARP-1 activation, with a specific focus on the role of truncated PARP-1 (tPARP-1) fragments. We provide a detailed analysis of the mechanisms governing NAD+ and ATP depletion, present structured quantitative data and experimental methodologies, and discuss the implications for drug development in cancer and neurodegenerative diseases.
PARP-1 is a nuclear enzyme and a primary DNA damage sensor. Its domain architecture comprises an N-terminal DNA-binding domain (DBD) with two zinc fingers, a central auto-modification domain (AMD), and a C-terminal catalytic domain (CAT) [1] [3]. Upon detecting DNA strand breaks, PARP-1 becomes activated, cleaving NAD+ to synthesize poly(ADP-ribose) (PAR) chains on itself (auto-modification) and target proteins [1].
A critical event in PARP-1-mediated cell death is its proteolytic cleavage by various "suicidal" proteases, including caspases, calpains, and granzymes [1]. Caspase-3 and -7 cleavage, a hallmark of apoptosis, generates a specific 89-kD fragment (containing the AMD and CAT domains) and a 24-kD DBD fragment [1]. The 24-kD fragment irreversibly binds to damaged DNA, acting as a trans-dominant inhibitor of BER and conserving cellular ATP, thereby facilitating the apoptotic process [1]. The generation of these specific tPARP-1 fragments serves as a recognized biomarker for particular protease activities and cell death pathways [1]. Understanding the interplay between these fragments and cellular metabolism is crucial for dissecting the mechanistic underpinnings of parthanatos.
Established and Emerging Models of Metabolic Dysregulation
The Traditional "Suicide Hypothesis" and its Evolution
The classical "suicide hypothesis" posits that severe DNA damage leads to hyperactivation of PARP-1, causing a rapid depletion of the NAD+ pool [1] [62]. As NAD+ is essential for glycolysis and mitochondrial respiration, its loss impairs ATP generation. Furthermore, the cell's attempt to resynthesize NAD+ consumes significant ATP, creating a vicious cycle that culminates in bioenergetic collapse and necrotic cell death [62]. While this model is well-supported, recent evidence suggests it is not the sole mechanism.
The PAR-Dependent Inhibition of Glycolysis
A paradigm-shifting study demonstrated that bioenergetic collapse following PARP-1 activation is not solely dependent on NAD+ depletion [63]. Instead, the PAR polymer itself directly binds to and inhibits hexokinase (HK), the first and key regulatory enzyme of glycolysis [63]. This inhibition occurs prior to significant NAD+ depletion and leads to a profound defect in glycolytic flux, reducing ATP production and the availability of pyruvate for mitochondrial oxidation [63]. This mechanism re-frames the metabolic catastrophe as an active, signaling-driven process rather than a passive consequence of substrate consumption.
Table 1: Key Mechanisms in PARP-1-Mediated Metabolic Dysregulation
| Mechanism | Key Molecular Event | Primary Metabolic Consequence | Proposed Contribution to Cell Death |
|---|---|---|---|
| NAD+ Consumption (Suicide Hypothesis) [62] | PARP-1 hyperactivation consumes NAD+ for PAR synthesis. | Depletion of NAD+ pool. | Loss of cofactor for ATP production; energy failure. |
| PAR-Dependent Hexokinase Inhibition [63] | PAR polymer binds to and inhibits hexokinase. | Severe reduction in glycolytic flux and ATP. | Bioenergetic collapse independent of initial NAD+ drop. |
| Metabolic Shift to OxPhos [64] | PARP-1 activation triggers a cell-wide increase in protein-bound NADH. | Shift in metabolic preference from glycolysis to oxidative phosphorylation. | Survival response; OxPhos becomes critical for damaged cell viability. |
Context-Dependent Metabolic Shifts
The metabolic response to PARP-1 activation is not monolithic. Phasor-FLIM imaging has revealed that DNA damage induces a rapid, PARP-1-dependent increase in the protein-bound fraction of NADH, indicating a shift in metabolic balance toward oxidative phosphorylation (OxPhos) over glycolysis [64]. Inhibition of OxPhos, but not glycolysis, under these conditions sensitizes cells to parthanatos, suggesting this shift constitutes a pro-survival adaptation to DNA damage [64]. Furthermore, the energetic outcome is context-dependent; in glucose-deprived cells, PARP-1 hyperactivation can paradoxically trigger an adenylate kinase-dependent increase in ATP, challenging the invariant link between PARP-1 activity and ATP loss [62].
Figure 1: Signaling Pathways in PARP-1-Mediated Metabolic Collapse. This diagram illustrates the interplay between NAD+ depletion, PAR-dependent hexokinase inhibition, and metabolic shifts leading to parthanatos.
Quantitative Data on Metabolic Alterations
The following tables summarize key quantitative findings from seminal studies investigating the metabolic consequences of PARP-1 activation.
Table 2: Temporal Sequence of Metabolic Events Following PARP-1 Activation in Cortical Neurons (MNNG Treatment) [63]
| Time Post-MNNG | PAR Formation | Glycolytic Function (Lactate Production) | ATP Levels | NAD+ Levels | Mitochondrial OCR |
|---|---|---|---|---|---|
| 0-15 min | Detected immediately | Significantly reduced (~40-50%) | Significantly reduced | No appreciable change | Significantly reduced |
| 30-45 min | Present | Further reduced | Reduced further | No significant loss | n/d |
| 60 min | Present | Reduced (similar to 45 min) | Reduced | Significantly decreased | n/d |
| With PARP Inhibitor (DPQ) | Prevented | Completely preserved | Completely preserved | Preserved | Completely rescued |
Table 3: Metabolic Parameters in Response to Varied DNA Damage Doses (Laser Microirradiation) [64]
| Laser Input Power | DNA Damage Complexity | NADH Intensity | Bound NADH Fraction | Cell Viability at 24h |
|---|---|---|---|---|
| Low | Simple strand breaks | Reduced | Transient increase | Recovery & Proliferation |
| Medium | Moderate complexity | Reduced | Transient increase | >90% |
| High | Complex damage (SSBs, DSBs) | Significantly reduced | Significant & sustained increase (>12h) | >70% |
Detailed Experimental Protocols
To aid in the replication and design of studies in this field, we outline key methodologies used to generate the data discussed.
Protocol 1: Assessing Glycolytic and Mitochondrial Function via Seahorse Analyzer
This protocol is used to comprehensively analyze the real-time effects of PARP-1 activation on cellular bioenergetics [63].
- Cell Preparation: Plate primary mouse cortical neurons or other cell lines of interest (e.g., HeLa) in XF24 cell culture microplates.
- Treatment: Treat cells with a PARP-1 activator (e.g., 50 µM MNNG) for 15 minutes. Include controls with a PARP inhibitor (e.g., DPQ).
- Glycolytic Function (ECAR):
- Replace medium with Seahorse XF assay medium (without glucose, bicarbonate, or serum).
- Sequentially inject: a) 10 mM Glucose, b) 1 µM Oligomycin (ATP synthase inhibitor), c) 50 mM 2-Deoxyglucose (glycolysis inhibitor).
- Measure the Extracellular Acidification Rate (ECAR). Basal glycolysis is derived from the last rate measurement before oligomycin. Glycolytic capacity is the maximum rate after oligomycin. Glycolytic reserve is the difference between capacity and basal glycolysis.
- Mitochondrial Function (OCR):
- In a separate assay, sequentially inject: a) 1 µM Oligomycin, b) 1 µM FCCP (mitochondrial uncoupler), c) 1 µM Rotenone/Antimycin A (Complex I/III inhibitors).
- Measure the Oxygen Consumption Rate (OCR). Calculate basal OCR, ATP-linked OCR, maximal OCR, and spare respiratory capacity.
Protocol 2: Measuring NAD+ and ATP Levels
Standardized methods for quantifying nucleotide levels are critical [63] [62].
- Cell Lysis and Extraction:
- For NAD+ measurement, lyse cells in 1N HClO4, then neutralize with an equal volume of 1N KOH.
- For ATP measurement, lyse cells in a commercial lysis buffer (e.g., from an ATPlite kit).
- For AMP/ADP measurement, extract cells in 0.25N HCl and analyze via HPLC.
- Quantification:
- NAD+: Use an enzymatic cycling assay. Neutralized extract is mixed with Bicine buffer containing ethanol, MTT, phenazine ethosulfate, and alcohol dehydrogenase. NAD+ reduction is measured by absorbance at 550 nm.
- ATP: Use a commercial luminescence assay based on the luciferin-luciferase reaction. Luminescence is measured and correlated to an ATP standard curve.
- AMP/ADP: Analyze HCl extracts by HPLC with a reverse-phase column and UV detection at 260 nm.
Protocol 3: Monitoring Metabolic Dynamics via Phasor-FLIM
This advanced imaging technique allows for real-time, label-free monitoring of NADH metabolism in living cells [64].
- Cell Culture and Damage Induction: Culture cells (e.g., HeLa) on imaging dishes. Induce DNA damage using laser microirradiation with titrated input power to control damage dose and complexity.
- Image Acquisition: Use a multiphoton microscope to capture NADH autofluorescence. Acquire fluorescence lifetime images at high spatiotemporal resolution before and after damage induction.
- Phasor-FLIM Analysis: Process the fluorescence decay data using the fit-free phasor approach. This transforms the decay at each pixel into a graphical coordinate on a phasor plot.
- Data Interpretation: Identify clusters on the phasor plot corresponding to free (short lifetime) and protein-bound (long lifetime) NADH. The shift in the ratio of bound/free NADH over time indicates a metabolic shift toward OxPhos.
Figure 2: Experimental Workflow for PARP-1 Metabolic Studies. This workflow outlines the parallel approaches for assessing metabolic parameters.
The Scientist's Toolkit: Key Research Reagents
Table 4: Essential Reagents for Investigating PARP-1-Mediated Cell Death
| Reagent / Tool | Function / Specificity | Example Use Case |
|---|---|---|
| PARP Activators (e.g., MNNG, H₂O₂) | Induces DNA strand breaks, leading to PARP-1 hyperactivation. | Triggering parthanatos in cortical neurons or other cell models [63]. |
| PARP Inhibitors (e.g., DPQ, Olaparib) | Competitively or non-competitively inhibits PARP catalytic activity. | Determining PARP-dependence of observed metabolic and cell death phenotypes [63]. |
| FK866 | Highly specific inhibitor of NAMPT, depletes NAD+ independently of PARP. | Disentangling the effects of NAD+ depletion from PAR signaling [63]. |
| Seahorse XF Analyzer | Real-time measurement of ECAR and OCR in live cells. | Profiling glycolytic and mitochondrial function after PARP activation [63]. |
| Anti-PAR Antibody (e.g., 10H) | Detects poly(ADP-ribose) polymer formation by immunofluorescence/Western blot. | Visualizing and quantifying PARP-1 activation [63] [62]. |
| PARG Overexpression | Degrades PAR polymers. | Confirming the role of PAR (vs. NAD+ depletion) in metabolic inhibition [63]. |
| Caspase-3 Inhibitor (e.g., Z-DEVD-FMK) | Inhibits executioner caspase-3 activity. | Differentiating apoptotic (caspase-dependent) from parthanatos (caspase-independent) death pathways [1]. |
The study of metabolic consequences in tPARP-1-mediated cell death has evolved from a simple model of energy depletion to a sophisticated understanding of active signaling and metabolic adaptation. The cleavage of PARP-1 into specific fragments serves as both a biomarker and a functional switch in cell fate decisions. The discovery that PAR polymer directly inhibits hexokinase, and that cells can initiate a compensatory shift toward OxPhos, reveals multiple layers of regulation. These findings have profound implications for therapeutic strategies. Targeting PAR signaling or the specific tPARP-1 fragments, rather than solely aiming to preserve NAD+, may offer more precise interventions in conditions like stroke, neurodegeneration, and cancer. Future research should focus on further elucidating the structural basis of PAR-hexokinase interaction, the transcriptional and epigenetic rewiring driven by tPARP-1 fragments, and the development of novel inhibitors that can selectively modulate these specific pathways to achieve desired therapeutic outcomes.
Functional Verification and Cross-Pathway Analysis: Validating tPARP1 Roles in Cell Death
This technical guide provides a comprehensive analysis of the cleavage specificities of three proteases central to cell death pathways: caspases, calpains, and granzyme B. These enzymes exhibit distinct yet sometimes overlapping substrate preferences, which ultimately determine their functional consequences in cellular remodeling, apoptosis, and necrosis. Particular emphasis is placed on the cleavage of poly(ADP-ribose) polymerase 1 (PARP1) as a canonical marker, whose truncated fragments serve as key indicators of protease activity and mediate important biological functions. The systematic comparison of cleavage motifs, structural determinants of specificity, and experimental methodologies presented herein provides researchers with a framework for interrogating protease functions in cell death research and drug development.
Proteases are fundamental executioners of cell death, with caspases, calpains, and granzyme B representing three distinct families that coordinate cellular demise through specific substrate cleavage events. These enzymes recognize unique amino acid sequences and structural motifs in their substrates, thereby dictating the downstream consequences of their activation. Caspases (cysteine aspartyl proteases) primarily drive apoptosis and pyroptosis, with their activity characterized by cleavage after aspartic acid residues. Calpains (calcium-dependent cysteine proteases) participate in both apoptotic and necrotic death, exhibiting more diverse sequence preferences. Granzyme B, a serine protease delivered by cytotoxic lymphocytes, shares the caspase preference for aspartic acid but operates within a different structural context to eliminate target cells.
The biological significance of protease-mediated cleavage is exemplified by the processing of PARP1, a nuclear enzyme involved in DNA repair and other cellular processes. During apoptosis, caspase-3 cleaves PARP1 at a specific aspartic acid residue (DEVD↑G), generating truncated fragments that lose their DNA repair capacity but may gain new functions [1] [5]. Similarly, calpain-mediated cleavage of various substrates produces fragments that are rapidly degraded by the N-end rule pathway, representing another layer of post-proteolytic regulation [65]. Understanding these precise cleavage patterns is therefore essential for deciphering cell death mechanisms and developing targeted therapeutic interventions.
Protease Specificity Profiles and Structural Determinants
Caspase Family
Table 1: Caspase Specificity and Function
| Caspase | Primary Function | Optimal Cleavage Motif | Key Substrates | Biological Role |
|---|---|---|---|---|
| Caspase-3/7 | Executioner apoptosis | DEVD↓ | PARP1, ICAD, Caspase-6 | Primary apoptotic effectors |
| Caspase-8 | Initator apoptosis | LETD↓ | Caspase-3, Bid | Death receptor pathway |
| Caspase-9 | Initator apoptosis | LEHD↓ | Caspase-3/7 | Apoptotic protease activating factor-1 (Apaf-1) pathway |
| Caspase-1 | Inflammation | YVAD↓ | IL-1β, IL-18 | Pyroptosis, inflammasome activation |
Caspases are cysteine aspartyl proteases that cleave their substrates C-terminal to aspartic acid residues. Their substrate specificity is determined by at least four amino acids (P4-P1) N-terminal to the cleavage site, with executioner caspases-3 and -7 preferentially recognizing DEVD sequences [1] [66]. Caspases can be categorized as initiators (caspases-2, -8, -9, -10) that activate the executioners (caspases-3, -6, -7), which in turn cleave hundreds of cellular substrates to systematically dismantle the cell. Proteomic studies have revealed that individual caspases can have dozens to hundreds of cellular targets, with cleavage rates varying over 500-fold within each caspase's substrate cohort [66].
The biological roles of caspases extend beyond classical apoptosis to include non-apoptotic functions such as stem cell fate determination, spermatogenesis, and erythroid differentiation [66]. These non-lethal roles involve activation of apoptotic caspases in cells that do not die, raising fundamental questions about which substrates are cleaved when caspases induce non-lethal outcomes and how these differ from substrates cleaved in dying cells [67].
Calpain Family
Table 2: Calpain Specificity and Substrates
| Calpain Type | Activation | Cleavage Specificity | Key Substrates | Functional Consequences |
|---|---|---|---|---|
| Calpain-1 (μ-calpain) | Micromolar Ca²⁺ | Prefers hydrophobic residues at P1, lacks strict motif | c-Fos, IκBα, Ankrd2 | Fragment degradation via N-end rule |
| Calpain-2 (m-calpain) | Millimolar Ca²⁺ | Similar to calpain-1, context-dependent | Spectrin, ion channels, kinases | Cytoskeletal remodeling, signaling |
| Calpain-3 | Tissue-specific | Muscle-specific substrates | Titin, calpastatin | Muscle function, limb-girdle muscular dystrophy |
Calpains are Ca²⁺-dependent intracellular cysteine proteases that typically make a limited number of specific cuts in their substrates, often within unstructured loops [65]. Unlike caspases, calpains do not have a strict consensus sequence but often prefer hydrophobic residues at the P1 position. Calpain-mediated cleavages are controlled by Ca²⁺ levels, phosphorylation of calpains and/or their substrates, natural inhibitors like calpastatin, and regulated subcellular localization.
A critical functional aspect of calpain cleavage is that the generated C-terminal fragments frequently bear destabilizing N-terminal residues recognized by the Arg/N-end rule pathway, leading to their rapid degradation [65]. This connection positions calpains as upstream components of the N-end rule pathway, with the generated fragments serving as short-lived substrates. This regulatory mechanism mediates the remodeling of oligomeric complexes by eliminating protein fragments produced through calpain cleavage.
Granzyme B
Table 3: Granzyme B Specificity and Roles
| Feature | Description | Biological Significance |
|---|---|---|
| Classification | Serine protease, chymotrypsin-like | Caspase-like specificity with serine protease mechanism |
| Optimal Motif | IEPD↓ (preferred), also recognizes DEVD, VEID, VGPD | Extended substrate recognition (P4-P4') |
| Source | Cytotoxic T lymphocytes, NK cells, other immune and non-immune cells | Multiple cellular sources in pathological conditions |
| Inhibition | SERPINB9 (PI-9), viral inhibitors | Protection from accidental cell death, viral evasion |
Granzyme B is a serine protease with caspase-like specificity, preferentially cleaving after aspartic acid residues in target cells. Its specificity is primarily determined by an extended substrate-binding cleft that accommodates at least eight consecutive residues (P4-P4') [68] [69]. The structural basis for its Asp specificity involves an arginine residue (Arg226) located deep in the S1 pocket that forms a side-on interaction with the P1 aspartic acid side chain [70] [69].
Granzyme B can activate multiple cell death pathways by cleaving and activating executioner caspases (caspases-3 and -7) and initiator caspases (caspases-8 and -10), albeit with varying efficiency [68]. It also cleaves key apoptotic substrates directly, including Bid and ICAD, and can initiate mitochondrial permeabilization through cleavage of Mcl-1 and HAX1. Beyond its cytotoxic functions, granzyme B participates in extracellular matrix remodeling, cytokine processing, and inflammation, with elevated levels implicated in autoimmune diseases, cardiovascular diseases, and skin disorders [68] [69].
PARP1 Cleavage as a Central Signaling Node
PARP1 serves as a key integration point for multiple protease activities during cell death, with different cleavage patterns producing signature fragments that serve as biomarkers for specific protease activation.
Caspase-Mediated PARP1 Cleavage
During apoptosis, caspase-3 and -7 cleave PARP1 at the DEVD↑G site (located between amino acids 214 and 215 in human PARP1), generating two signature fragments: a 24-kDa DNA-binding domain (DBD) fragment and an 89-kDa fragment containing the automodification domain (AMD) and catalytic domain (CAT) [1]. The 24-kDa fragment remains tightly bound to DNA breaks, acting as a trans-dominant inhibitor of DNA repair by blocking access of additional DNA repair proteins to the damage site. The 89-kDa fragment translocates to the cytoplasm where it has been recently shown to recognize and ADP-ribosylate the RNA polymerase III (Pol III) complex, facilitating interferon-β production and enhancing apoptosis during innate immune responses [5].
Calpain-Mediated PARP1 Cleavage
Calpain cleaves PARP1 at distinct sites from caspases, producing different signature fragments. While the exact cleavage sites for calpain in PARP1 are less well-characterized than for caspases, they typically generate fragments of approximately 55-kDa and 62-kDa [1]. These calpain-generated PARP1 fragments are often targeted for rapid degradation by the N-end rule pathway due to their exposed N-terminal residues with destabilizing properties [65]. This connection between calpain cleavage and the N-end rule pathway represents an important mechanism for eliminating protein fragments produced during limited proteolysis.
Granzyme B-Mediated PARP1 Cleavage
Granzyme B cleaves PARP1 at the IEPD↑S site (located between amino acids 246 and 247 in the human protein), producing fragments of approximately 50-kDa and 64-kDa [1] [69]. This cleavage event contributes to the nuclear disassembly and apoptotic phenotype observed in target cells of cytotoxic lymphocytes. The ability of granzyme B to directly process PARP1, independent of caspase activation, provides a mechanism for inducing cell death even when caspases are inhibited by viral or cellular inhibitors.
Experimental Methodologies for Protease Analysis
Proteomic Approaches for Substrate Identification
Modern protease substrate identification relies heavily on mass spectrometry-based proteomic methods that provide global mapping of proteolytic events:
N-terminomics (TAILS and COFRADIC): These methods employ selective enrichment and identification of protein N-terminal peptides to comprehensively map protease cleavage sites on a proteome-wide scale. The process involves: (1) blocking native N-terminal with stable isotope labels; (2) protease treatment of cell extracts or live cells; (3) enrichment of newly generated N-terminal; and (4) LC-MS/MS analysis for identification [67] [66]. These approaches have identified hundreds to thousands of caspase cleavage events in apoptotic cells and revealed distinct substrate profiles in non-apoptotic contexts.
Quantitative MS-based Enzymology: This approach quantitatively measures the kinetics of substrate cleavage by incubating recombinant caspases with proteomes and monitoring the appearance of cleavage products over time. Julien et al. used this method to establish hierarchical substrate profiles for several caspases, revealing that each caspase has preferred substrates cleaved with rates varying over 500-fold [66].
Cleavage Site Prediction Tools
Table 4: Computational Tools for Cleavage Site Prediction
| Tool Name | Applicable Proteases | Methodology | Special Features |
|---|---|---|---|
| SitePrediction | Broad range | Frequency scoring, similarity matrices | PEST analysis, solvent accessibility, secondary structure |
| GraBCas | Granzyme B, caspases | Frequency-based scoring | Limited to specific proteases |
| CaSPredictor | Caspases | Additive frequency scoring | User-friendly interface |
| PoPS | Predefined protease set | Statistical modeling | PEST region prediction |
| PeptideCutter | Multiple proteases | Fixed consensus matching | Simple sequence scanning |
Several computational tools have been developed to predict potential protease cleavage sites in protein sequences. SitePrediction stands out for its user-friendly interface and incorporation of multiple extra features, including statistical evaluation of prediction quality, PEST sequence analysis, solvent accessibility predictions, and secondary structure predictions [71]. The reliability of these tools is often evaluated using ROC curve analysis, with tools like SitePrediction demonstrating high accuracy (AUC values of 0.995 for caspase-3 and 0.951 for calpain-2) [71].
The Scientist's Toolkit: Essential Research Reagents
Table 5: Essential Research Reagents for Protease Studies
| Reagent Category | Specific Examples | Research Application | Technical Notes |
|---|---|---|---|
| Activity Assays | DEVD-AFC (caspases), IEPD-AFC (GrB) | Fluorometric activity measurement | Use with appropriate controls and Z-VAD-fmk for specificity |
| Protease Inhibitors | Z-VAD-fmk (pan-caspase), Calpeptin (calpain), PI-9 (GrB inhibitor) | Pathway inhibition studies | Confirm specificity with complementary approaches |
| Antibodies | Anti-PARP1 (cleavage-specific), Anti-active caspase-3, Anti-granzyme B | Western blot, immunohistochemistry | Cleavage-specific antibodies are essential for apoptosis detection |
| Recombinant Proteases | Active caspase-3, calpain-1, granzyme B | In vitro cleavage assays | Requires optimization of buffer conditions (e.g., Ca²⁺ for calpains) |
| Cell Death Inducers | Staurosporine, etoposide, TNF-α+cycloheximide | Apoptosis induction models | Titrate for optimal caspase activation without secondary necrosis |
| Live Cell Reporters | FRET-based caspase substrates, Annexin V-FITC | Real-time apoptosis monitoring | Combine with viability dyes (PI) to distinguish early/late apoptosis |
Biological Context of Truncated PARP1 Fragments
The cleavage of PARP1 by different proteases generates truncated fragments with distinct biological activities that extend beyond the simple inactivation of DNA repair:
Caspase-Generated tPARP1 in Innate Immunity
Recent research has revealed that the 89-kDa tPARP1 fragment generated by caspase cleavage translocates to the cytoplasm where it recognizes and mono-ADP-ribosylates the RNA polymerase III (Pol III) complex [5]. This modification enhances Pol III-mediated transcription of foreign DNA, leading to increased IFN-β production and potentiation of apoptosis during antiviral responses. This discovery provides a biological rationale for the conservation of the PARP1 cleavage event across evolution and suggests that tPARP1 plays an active role in promoting cell death rather than simply inactivating DNA repair.
Calpain-Generated Fragments and the N-End Rule Pathway
Calpain-generated C-terminal fragments of various proteins, including transcriptional regulators, transmembrane ion channels, apoptosis controllers, kinases, and phosphatases, are short-lived substrates of the Arg/N-end rule pathway [65]. This pathway targets destabilizing N-terminal residues of protein substrates for ubiquitin-dependent degradation. The N-terminal residues of calpain-generated fragments are often conserved across evolution with respect to their destabilizing activity rather than their specific identity, suggesting selective pressure for rapid fragment turnover.
Differentiation-Specific Cleavage Events
Proteomic studies comparing caspase substrates in apoptotic versus differentiating cells have revealed that caspase activity in non-apoptotic contexts cleaves a specific subset of substrates distinct from those targeted during cell death [67]. In C2C12 myogenic differentiation, caspase-3 activity cleaves specific cytoskeletal proteins and regulators of muscle differentiation, facilitating the differentiation process without triggering cell death. This suggests that the functional consequences of protease activity are determined by both the specific substrates cleaved and the cellular context in which cleavage occurs.
Caspases, calpains, and granzyme B represent three distinct protease families with specialized roles in cellular remodeling and cell death. While they exhibit overlapping substrate specificities in some cases, each protease family possesses unique structural features and regulation that dictate its biological functions. The cleavage of PARP1 serves as an exemplary model for understanding how different proteases target the same protein at distinct sites to produce fragments with different biological activities. Future research in this field will likely focus on understanding the context-specific regulation of protease activity and the functional consequences of individual cleavage events within the global substrate pool. The continued development of proteomic methods and computational prediction tools will enhance our ability to dissect these complex proteolytic networks and identify novel therapeutic targets for diseases characterized by dysregulated cell death.
Poly(ADP-ribose) polymerase 1 (PARP1) is a nuclear enzyme with well-characterized roles in DNA repair and cell death signaling. During apoptosis, caspase-mediated cleavage of PARP1 is a established hallmark, generating specific fragments with potentially distinct biological activities. This technical guide examines the functional validation of non-cleavable PARP1 mutants as critical tools for delineating the contribution of PARP1 cleavage fragments to apoptotic efficiency. We present comprehensive experimental methodologies, quantitative data comparisons, and essential research tools for investigating how preventing PARP1 proteolysis influences cell death pathways. Within the broader context of truncated PARP1 fragment biology, this work provides researchers with validated approaches for determining whether non-cleavable PARP1 mutants attenuate or enhance apoptotic progression across different cellular models and death stimuli.
PARP1 is a multifunctional nuclear enzyme comprising several structured domains: three zinc finger motifs (ZnF1, ZnF2, ZnF3) that facilitate DNA damage recognition, a BRCT domain mediating protein-protein interactions, a WGR domain, and a C-terminal catalytic domain (CAT) responsible for ADP-ribosyl transferase activity [72] [73]. During apoptosis, PARP1 becomes a primary substrate for executioner caspases (particularly caspase-3), which cleave the protein at a specific aspartic acid residue (D214 in human PARP1) located between ZnF2 and ZnF3 [34] [19]. This proteolytic event generates two signature fragments: a 24-kDa DNA-binding fragment (DBD) containing the first two zinc fingers that remains nuclear, and an 89-kDa fragment (tPARP1) containing the third zinc finger, BRCT, WGR, and catalytic domains that translocates to the cytoplasm [34] [19].
The traditional interpretation of PARP1 cleavage posits that it serves to inactivate DNA repair capacity during apoptosis, thereby conserving cellular ATP pools for the execution of the death program [19]. However, emerging evidence suggests the cleavage fragments may possess gain-of-function activities that actively promote cell death signaling. For instance, recent research has demonstrated that the 89-kDa tPARP1 fragment can recognize and mono-ADP-ribosylate the RNA polymerase III (Pol III) complex in the cytoplasm during innate immune signaling, facilitating IFN-β production and apoptosis [34]. This finding challenges the purely passive model of PARP1 cleavage and suggests the fragments may actively participate in death signaling.
Non-cleavable PARP1 mutants, in which the caspase cleavage site has been modified through site-directed mutagenesis, represent powerful tools for functionally validating the biological significance of PARP1 proteolysis in apoptosis. This technical guide provides detailed methodologies and analytical frameworks for employing these mutants to dissect the functional relationship between PARP1 cleavage and apoptotic efficiency.
The Rationale for Non-Cleavable PARP1 Mutants in Apoptosis Research
Non-cleavable PARP1 mutants are constructed by mutating the caspase recognition sequence (DEVD214G in human PARP1) to render the protein resistant to caspase-mediated proteolysis [34]. These mutants enable researchers to address fundamental questions about PARP1 biology during cell death:
- Determining the active versus passive role of PARP1 cleavage in apoptotic progression
- Identifying specific signaling pathways dependent on PARP1 proteolysis
- Quantifying the contribution of PARP1 fragments to apoptotic efficiency versus full-length protein function
- Dissecting the temporal relationship between PARP1 cleavage and commitment to cell death
From an evolutionary perspective, the conservation of PARP1 cleavage sites across species suggests functional importance, while the existence of natural PARP1 orthologs in lower organisms that lack the N-terminal zinc fingers (structurally similar to the 89-kDa tPARP1 fragment) hints at potential biological activities for the cleavage products [34].
Table 1: PARP1 Cleavage Fragments and Their Documented Functions
| Fragment | Size | Domains Contained | Localization After Cleavage | Reported Functions |
|---|---|---|---|---|
| N-terminal fragment | 24 kDa | ZnF1, ZnF2 | Nuclear | Acts as dominant-negative by binding DNA damage sites; inhibits DNA repair [19] |
| C-terminal fragment (tPARP1) | 89 kDa | ZnF3, BRCT, WGR, CAT | Cytoplasmic | Binds and activates RNA Pol III; promotes IFN-β production; may have distinct substrate specificity [34] |
Experimental Strategies for Validating Non-Cleavable PARP1 Mutants
Molecular Engineering of Non-Cleavable PARP1 Mutants
The generation of non-cleavable PARP1 mutants requires precise molecular techniques to alter the caspase cleavage site without disrupting normal protein folding and function. The following protocol outlines the key steps:
Site-Directed Mutagenesis: Mutate the caspase-3 recognition sequence (DEVD214G) in human PARP1 to a non-cleavable sequence (e.g., DEVG214A or similar amino acid substitutions) using overlap extension PCR or commercial mutagenesis kits [34].
Vector Construction: Clone the mutated PARP1 sequence into appropriate mammalian expression vectors with suitable promoters (e.g., CMV) for transient or stable expression. Include tags (e.g., FLAG, HA, GFP) for detection and purification.
Control Constructs: Generate and parallelly test:
- Wild-type (WT) PARP1
- Catalytically inactive PARP1 (E988K mutation in the catalytic domain) [74]
- Non-cleavable and catalytically inactive double mutant
Protein Validation: Express constructs in PARP1-deficient cells (e.g., PARP1-/- MEFs or 293T cells) and verify:
- Protein expression levels by Western blotting
- Caspase resistance by in vitro cleavage assays
- Proper nuclear localization via immunofluorescence
- Enzymatic activity through PARylation assays [34]
Functional Assays for Apoptotic Efficiency
To quantitatively assess the impact of non-cleavable PARP1 mutants on apoptotic efficiency, implement the following experimental approaches:
Cell Death Induction and Analysis:
- Treat transfected cells with apoptosis-inducing agents:
Monitor apoptotic progression using multi-parameter assays:
- Phosphatidylserine externalization: Annexin V-FITC/PI staining with flow cytometry
- Caspase activation: Fluorogenic substrate assays (DEVD-AMC for caspase-3)
- Mitochondrial membrane potential: JC-1 or TMRM staining
- DNA fragmentation: TUNEL assay or DNA laddering
- Nuclear morphology: Hoechst 33342 staining for chromatin condensation
Time-course experiments: Analyze samples at multiple time points (0, 2, 4, 8, 16, 24 hours) post-treatment to capture kinetic differences in apoptotic progression.
Key Experimental Controls:
- Empty vector transfection controls
- Wild-type PARP1 reconstitution controls
- Caspase inhibition (Z-VAD-FMK, 20-50 μM) to confirm apoptosis-specific effects
- PARP inhibitor treatment (Olaparib, 1-10 μM) to dissect catalytic-dependent effects [75] [76]
Table 2: Quantitative Apoptosis Assessment Methods for PARP1 Cleavage Studies
| Method | Parameter Measured | Key Reagents | Data Output | Advantages |
|---|---|---|---|---|
| Annexin V/PI staining | Phosphatidylserine exposure; membrane integrity | Annexin V-FITC, Propidium Iodide | Flow cytometry quantification of early/late apoptotic and necrotic populations | Distinguishes apoptotic stages; high throughput |
| Caspase activity assay | Caspase-3/7 activation | DEVD-AMC or other fluorogenic substrates | Fluorescence intensity over time | Specific for executioner caspase activity; quantitative |
| TUNEL assay | DNA strand breaks | Terminal deoxynucleotidyl transferase, labeled nucleotides | Fluorescence microscopy or flow cytometry | Specific for late apoptosis; single-cell resolution |
| Western blot analysis | Cleavage of PARP1 and other caspase substrates | PARP1 antibodies (full-length and cleaved forms) | Band intensity quantification | Direct monitoring of PARP1 cleavage; multiplexing possible |
Signaling Pathways Involving PARP1 Cleavage in Cell Death
The functional impact of non-cleavable PARP1 mutants must be interpreted within the context of known PARP1-dependent cell death pathways. Research indicates that PARP1 cleavage fragments participate in multiple signaling cascades:
Cytoplasmic tPARP1 Signaling: The 89-kDa tPARP1 fragment translocates to the cytoplasm during apoptosis and can interact with the RNA polymerase III (Pol III) complex via its BRCT domain [34]. This interaction facilitates mono-ADP-ribosylation of Pol III subunits, enhancing IFN-β production and promoting apoptosis in response to cytosolic DNA sensing. Non-cleavable PARP1 prevents this cytoplasmic signaling function by retaining the full-length protein in the nucleus.
PARP1 Hyperactivation and Parthanatos: Under conditions of severe genotoxic stress, PARP1 becomes hyperactivated, leading to NAD+ and ATP depletion, which triggers a caspase-independent form of cell death called parthanatos [73]. Non-cleavable PARP1 mutants may potentially exacerbate this pathway by remaining active at DNA damage sites, though this hypothesis requires experimental validation.
Transcription Factor Regulation: PARP1 regulates numerous transcription factors including p53, NF-κB, and HIF1α through both enzymatic and non-enzymatic mechanisms [74]. Non-cleavable PARP1 mutants may differentially affect these regulatory interactions compared to wild-type PARP1 or its cleavage fragments.
Research Reagent Solutions for PARP1 Cleavage Studies
Table 3: Essential Research Tools for Investigating Non-Cleavable PARP1 Mutants
| Reagent Category | Specific Examples | Application/Function | Key Features |
|---|---|---|---|
| PARP1 Expression Constructs | Wild-type PARP1 cDNA; Non-cleavable PARP1 (DEVD→DEVG); Catalytically inactive (E988K) | Reconstitution studies in PARP1-deficient cells; structure-function analysis | Epitope-tagged versions for detection; mammalian expression vectors with selectable markers |
| PARP1-Deficient Cell Lines | PARP1-/- MEFs; PARP1 knockout 293T cells (generated via CRISPR/Cas9) | Background-free systems for expressing PARP1 mutants | Validated complete PARP1 deficiency; normal karyotype and growth characteristics |
| PARP1 Antibodies | Anti-PARP1 (full-length specific); Anti-cleaved PARP1 (Asp214); Anti-PARP1 (C-terminal) | Western blot, immunofluorescence, immunoprecipitation | Specific for different PARP1 forms; validated for particular applications |
| Apoptosis Inducers | Etoposide (50 μM); Staurosporine (1 μM); TNF-α/CHX (10 ng/mL/10 μg/mL); H₂O₂ (250 μM) | Induction of controlled apoptotic response | Different mechanisms of action; titratable potency |
| Caspase Inhibitors | Z-VAD-FMK (pan-caspase inhibitor); Z-DEVD-FMK (caspase-3 specific inhibitor) | Confirming caspase-dependent mechanisms | Cell-permeable; reversible or irreversible options |
| PARP Inhibitors | Olaparib (1-10 μM); Rucaparib (1-10 μM); DPQ (10-50 μM) [75] [76] [74] | Distinguishing catalytic-dependent vs independent functions | Clinical relevance; varying potency and specificity profiles |
| Detection Assays | Annexin V-FITC/PI kits; Caspase-3 fluorogenic substrates (DEVD-AMC); TUNEL assay kits | Quantitative apoptosis assessment | Commercial availability; standardized protocols |
Interpretation of Experimental Results and Technical Considerations
When evaluating the impact of non-cleavable PARP1 mutants on apoptotic efficiency, consider these critical analytical frameworks:
Differential Responses by Death Stimulus: The functional consequences of preventing PARP1 cleavage may vary significantly depending on the apoptotic stimulus. For example, non-cleavable PARP1 might show more pronounced effects in DNA damage-induced apoptosis compared to death receptor-mediated pathways. Always include multiple death inducers in experimental designs.
Catalytic-Independent Functions: PARP1 possesses both enzymatic and scaffolding functions. The WGR domain of PARP1, for instance, can promote the proteasomal degradation of HIPK2 independently of PARP1's catalytic activity [74]. Non-cleavable PARP1 mutants may therefore affect apoptotic signaling through both PARylation-dependent and -independent mechanisms.
Cell Type-Specific Effects: The impact of non-cleavable PARP1 on apoptotic efficiency may vary across cell types due to differences in:
- Expression levels of PARP1 and related family members
- Cellular metabolic states and NAD+ availability
- Baseline expression of apoptosis regulators
- Efficiency of cytoplasmic translocation mechanisms
Quantitative Assessment Criteria: Establish clear metrics for evaluating "apoptotic efficiency":
- Timing of commitment to irreversible cell death
- Percentage of population undergoing apoptosis at given time points
- Caspase activation kinetics
- Morphological progression through apoptotic stages
Table 4: Interpreting Non-Cleavable PARP1 Mutant Phenotypes in Apoptosis Assays
| Experimental Observation | Possible Interpretation | Follow-up Experiments |
|---|---|---|
| Delayed apoptosis with non-cleavable mutant | PARP1 cleavage fragments normally promote apoptotic progression | Test specific fragment activities; examine alternative death pathways |
| Accelerated apoptosis with non-cleavable mutant | Cleavage normally serves a protective function; fragments may inhibit pro-death signals | Assess NAD+/ATP depletion; examine parthanatos markers |
| No significant difference from wild-type PARP1 | PARP1 cleavage is not rate-limiting for apoptosis in this context | Verify mutant expression and cleavage resistance; test additional cell types |
| Stimulus-dependent effects | PARP1 cleavage has context-specific functions | Map signaling pathways specific to each stimulus |
Non-cleavable PARP1 mutants represent essential tools for functionally validating the significance of PARP1 proteolysis in apoptotic pathways. The experimental frameworks outlined in this technical guide provide researchers with robust methodologies for determining how preventing PARP1 cleavage impacts cell death efficiency across different biological contexts. As research continues to reveal novel functions for PARP1 cleavage fragments, particularly the cytoplasmic activities of the 89-kDa tPARP1 fragment, these approaches will remain critical for advancing our understanding of PARP1 biology in cell death decision-making. The integration of quantitative apoptosis assays with careful controls and appropriate interpretation frameworks will ensure meaningful conclusions about the functional relationship between PARP1 cleavage and apoptotic efficiency.
This whitepaper elucidates the novel biological function of the caspase-cleaved, 89 kDa truncated PARP1 (tPARP1) fragment in bridging apoptosis with innate immune activation. Canonically, PARP1 cleavage is a hallmark of apoptosis, yet the physiological role of the resulting fragments has been enigmatic. Recent research reveals that tPARP1 translocates to the cytosol, where it mono-ADP-ribosylates (MARylates) the RNA Polymerase III (Pol III) complex. This post-translational modification enhances the ability of Pol III to sense foreign cytosolic DNA, leading to elevated production of interferon-beta (IFN-β) and the amplification of the apoptotic response. This guide details the molecular mechanism, experimental methodologies, and key reagents essential for investigating this emerging pathway, providing a framework for its exploration in drug development and disease pathology.
The cleavage of poly(ADP-ribose) polymerase 1 (PARP1) by executioner caspases is a quintessential biomarker of apoptosis. The protease activity yields two primary fragments: a 24 kDa DNA-binding fragment and an 89 kDa truncated PARP1 (tPARP1) encompassing the BRCT domain, WGR domain, and the catalytic domain [34] [1]. For decades, the 24 kDa fragment was thought to be the primary functional product, acting as a dominant-negative inhibitor of DNA repair by occupying DNA strand breaks [1]. In contrast, the biological significance of the 89 kDa tPARP1 fragment remained largely unknown.
The discovery that tPARP1 is not a mere byproduct but an active contributor to cell death signaling represents a paradigm shift. This function is framed within a broader thesis that specific PARP1 cleavage fragments are not just markers but active mediators of distinct cell death programs, carrying unique "signatures" that influence pathological outcomes [1]. The tPARP1-mediated pathway exemplifies this principle, revealing a sophisticated cross-talk between the apoptotic machinery and the innate immune system, which may have profound implications for cancer therapy, antiviral defense, and inflammatory diseases.
Molecular Mechanism: From Nuclear Cleavage to Cytosolic Immune Signaling
The following diagram illustrates the core signaling pathway through which tPARP1 activates an innate immune response.
Figure 1: tPARP1-Mediated Innate Immune Activation Pathway. During apoptosis, caspase-3 cleaves PARP1, generating tPARP1, which translocates to the cytosol. Via its BRCT domain, tPARP1 binds and mono-ADP-ribosylates (MARylates) the RNA Pol III complex, stimulating IFN-β production and enhancing cell death.
Key Molecular Steps
- Caspase-3 Cleavage and Fragment Generation: During apoptosis, activated caspase-3 cleaves full-length PARP1 at aspartate residue 214 (D214), separating the N-terminal DNA-binding domain (containing two zinc fingers) from the C-terminal 89 kDa tPARP1 fragment [34] [77].
- Cytosolic Translocation: The 24 kDa fragment remains nuclear, while tPARP1, lacking a strong nuclear localization signal, relocates to the cytoplasm [34] [1]. This spatial separation is critical for its non-canonical function.
- Recognition of the RNA Pol III Complex: In the cytosol, tPARP1 specifically interacts with subunits of the RNA Pol III complex (POLR3A, POLR3B, POLR3F). This interaction is mediated by the BRCT domain within tPARP1. A point mutation (F473A) in the BRCT domain ablates this binding, confirming its essential role [34].
- ADP-ribosylation of Pol III: tPARP1 catalyzes the mono-ADP-ribosylation (MARylation) of the Pol III complex. This is a distinct activity from the poly-ADP-ribosylation (PARylation) PARP1 typically performs in the nucleus in response to DNA damage [34] [28].
- Activation of Innate Immunity and Apoptosis: MARylation enhances the activity of Pol III in transcribing cytosolic DNA (e.g., from pathogens or self-DNA under stress), producing RNA ligands that activate the RIG-I/MDA5 pathway. This leads to the induction of type I interferon, specifically IFN-β, which can further potentiate the apoptotic cascade [34].
Experimental Protocols & Data
This section outlines the key methodologies used to delineate the tPARP1-Pol III-IFN-β pathway.
Identification of tPARP1-Interacting Partners
Objective: To unbiasedly identify proteins that specifically interact with the truncated form of PARP1 (tPARP1) during apoptosis.
Workflow Diagram:
Figure 2: Workflow for Identifying tPARP1 Interactors.
Detailed Protocol:
- Cell Line Engineering: Generate PARP1-deficient 293T cells to eliminate background from endogenous PARP1.
- Construct Design: Stably express a tandem-affinity tagged (e.g., S-protein-Flag-Streptavidin binding peptide, or SFB) mutant tPARP1 (mtPARP1) where the catalytic glutamate E988 is mutated to alanine (E988A). This catalytic-dead mutant acts as a "substrate trap," stabilizing transient interactions with its substrates [34].
- Affinity Purification: Perform tandem affinity purification from the soluble fraction of cell lysates under native conditions to isolate the mtPARP1 protein complex.
- Mass Spectrometry (MS): Resolve the co-purified proteins by SDS-PAGE, digest them with trypsin, and analyze the resulting peptides by liquid chromatography-tandem mass spectrometry (LC-MS/MS).
- Data Analysis: Identify proteins from MS data and cross-reference them with interactors of catalytic mutants of the ADP-ribose erasers TARG1 (mTARG1) and PARG (mPARG). This clever triangulation strategy helps pinpoint genuine ADP-ribosylation substrates, identifying the Pol III subunits as common interactors [34].
Validating Functional Interactions during Apoptosis
Objective: To confirm the interaction between endogenous tPARP1 and Pol III in a physiological context of apoptosis.
Detailed Protocol:
- Apoptosis Induction: Transfect cells with poly(deoxyadenylic-deoxythymidylic) acid (poly(dA-dT)), a synthetic double-stranded DNA that mimics pathogenic DNA and stimulates the cytosolic DNA-sensing pathway, thereby inducing apoptosis [34].
- Apoptosis Confirmation:
- Western Blotting: Use antibodies specific for cleaved PARP1 to detect the generation of the 89 kDa tPARP1 fragment.
- Flow Cytometry: Stain cells with Annexin V-FITC and propidium iodide (PI) to quantify the percentage of apoptotic cells.
- Morphology Assessment: Observe characteristic apoptotic changes (e.g., cell shrinkage, membrane blebbing) via microscopy.
- Co-immunoprecipitation (Co-IP):
- Lyse cells and perform immunoprecipitation using an antibody against a Pol III subunit (e.g., POLR3A) or a tag on transfected tPARP1.
- Analyze the immunoprecipitated complex by western blotting to detect the presence of co-precipitated tPARP1.
Key Quantitative Data: The table below summarizes critical data from the referenced studies [34].
| Experimental Readout | Method Used | Key Finding / Quantitative Result |
|---|---|---|
| Apoptosis Induction | Annexin V/PI FACS | Transfection with poly(dA-dT) "remarkably increased" the population of apoptotic cells. |
| PARP1 Cleavage | Western Blot | Detection of the 89 kDa tPARP1 fragment using a cleavage-specific antibody. |
| tPARP1-Pol III Interaction | Co-IP + Western Blot | Confirmed physical interaction between tPARP1 and POLR3A, POLR3B, and POLR3F. |
| Domain Mapping | Co-IP with Truncation Mutants | Deletion or F473A point mutation of the BRCT domain "disrupted the interaction." |
| IFN-β Production | Not Specified (Likely ELISA/qPCR) | tPARP1 "facilitates IFN-β production"; suppressed by non-cleavable PARP1. |
Table 1: Summary of Key Experimental Evidence for the tPARP1-Pol III Pathway.
The Scientist's Toolkit: Essential Research Reagents
The following table catalogs crucial reagents and tools used to dissect this pathway, as derived from the primary research.
| Research Reagent | Function / Application in the Study | Key Experimental Insight |
|---|---|---|
| PARP1-deficient 293T Cells | Provides a clean genetic background to study exogenously expressed wild-type and mutant PARP1/tPARP1 without interference from the endogenous protein. | Essential for unbiased identification of tPARP1-specific interactors. |
| Catalytic-dead mtPARP1 (E988A) | Acts as a substrate trap by binding to but not modifying substrates, allowing for the stabilization and purification of transient enzyme-substrate complexes. | Enabled the identification of Pol III subunits as tPARP1 interactors via affinity purification-MS [34]. |
| Non-cleavable PARP1 Mutant | A PARP1 variant engineered to resist caspase-3 cleavage. Used to demonstrate the necessity of PARP1 cleavage for the downstream events. | Impairs Pol III ADP-ribosylation and IFN-β production during poly(dA-dT)-stimulated apoptosis [34] [77]. |
| Poly(dA-dT) | A synthetic double-stranded DNA used to transfect cells. Mimics cytosolic viral or self-DNA, activating the innate immune response and inducing apoptosis. | Key for experimentally stimulating the pathway that leads to tPARP1 generation and its cytosolic function [34]. |
| BRCT Domain Mutant (F473A) | A tPARP1 mutant with a disrupted protein-protein interaction interface in the BRCT domain. | Abolishes the interaction with Pol III subunits, confirming the BRCT domain is critical for this binding [34]. |
Table 2: Key Research Reagents for Investigating the tPARP1 Pathway.
Discussion and Research Implications
The elucidated pathway positions tPARP1 as a critical molecular switch that amplifies cell death by engaging the innate immune system. This has several significant implications:
- Amplification of Apoptotic Signaling: The pathway establishes a positive feedback loop where apoptosis generates tPARP1, which enhances IFN-β production, which in turn can further promote apoptosis. This ensures a robust and irreversible commitment to cell death, which is crucial for eliminating infected or damaged cells.
- Therapeutic Opportunities: In cancer therapy, enhancing this pathway could synergize with DNA-damaging agents or immunotherapies to improve tumor cell killing. Conversely, inhibiting this axis might be beneficial in pathological inflammatory conditions where excessive cell death and IFN production drive disease.
- Evolutionary Perspective: The finding is consistent with the observation that PARP1 orthologs in some lower eukaryotes naturally lack the N-terminal zinc fingers found in higher organisms, resembling the apoptotic tPARP1 fragment. This suggests that the primordial function of PARP1 may have been more closely aligned with its role in immune defense, which was later co-opted for DNA damage repair in complex organisms [34].
The discovery that truncated PARP1 mediates the ADP-ribosylation of RNA Pol III to stimulate IFN-β production fundamentally expands our understanding of PARP1 biology beyond DNA repair. It provides a definitive biological function for a major apoptotic cleavage fragment and reveals a sophisticated mechanism of cross-talk between two fundamental cellular processes: programmed cell death and innate immunity. This pathway underscores the broader thesis that proteolytic fragments of cell death regulators are not mere inert markers but active players with distinct signaling capabilities. Further research into this pathway will undoubtedly yield deeper insights into infection, cancer, and inflammation, opening new avenues for therapeutic intervention.
Within the complex landscape of cellular biology, regulated cell death (RCD) represents a critical process governing development, homeostasis, and disease pathogenesis. While apoptosis has been extensively characterized as a non-inflammatory programmed cell death, and necrosis was historically viewed as an accidental pathway, recent research has uncovered sophisticated molecular mechanisms underlying multiple RCD forms, including ferroptosis. These distinct death modalities employ unique signaling cascades and executioner mechanisms, yet demonstrate remarkable interconnectivity. Central to understanding these pathways is poly (ADP-ribose) polymerase-1 (PARP-1), a nuclear enzyme with multifaceted roles in DNA repair and cell fate decisions. The proteolytic cleavage of PARP-1 generates specific truncated fragments that serve as molecular signatures for particular cell death pathways, offering critical insights into cellular stress responses. This technical review provides an in-depth comparison of apoptosis, necrosis, and ferroptosis, with particular emphasis on the biological function of PARP-1 cleavage fragments as biomarkers and regulatory elements within these distinct cell death contexts, providing essential knowledge for researchers and drug development professionals working in oncotherapy and degenerative diseases.
Molecular Mechanisms of Cell Death Pathways
Apoptosis: The Programmed Cascade
Apoptosis represents a highly regulated, caspase-dependent form of cell death characterized by distinct morphological features including cell shrinkage, chromatin condensation, nuclear fragmentation, and formation of apoptotic bodies that are efficiently cleared by phagocytes without inducing inflammation [78] [79]. This death modality proceeds through two principal signaling pathways:
Intrinsic Pathway (Mitochondrial): Triggered by intracellular stressors such as DNA damage, oxidative stress, or growth factor deprivation, this pathway is regulated by BCL-2 family proteins which control mitochondrial outer membrane permeabilization (MOMP) [78] [79]. MOMP facilitates the release of cytochrome c into the cytosol, where it binds to APAF-1 and procaspase-9 to form the apoptosome complex, activating caspase-9 which subsequently activates executioner caspases-3 and -7 [78] [80].
Extrinsic Pathway (Death Receptor): Initiated through ligand binding to death receptors (e.g., Fas, TNFR) on the plasma membrane, leading to the formation of the death-inducing signaling complex (DISC) which activates caspase-8 [78]. Caspase-8 directly cleaves and activates executioner caspases-3 and -7, orchestrating the apoptotic program [78].
Both pathways converge on the activation of executioner caspases-3 and -7, which systematically cleave cellular substrates, including PARP-1, leading to the characteristic biochemical and morphological changes of apoptosis [78] [79].
Necrosis and Necroptosis: From Accidental to Regulated Death
Necrosis has traditionally been classified as an accidental, unregulated form of cell death resulting from extreme physicochemical stress, characterized by cellular swelling, organelle breakdown, and plasma membrane rupture that elicits inflammatory responses [80]. However, the discovery of necroptosis has established that certain forms of necrosis follow defined molecular pathways [78] [79].
Necroptosis represents a regulated form of necrotic cell death that can be activated when caspase activity is inhibited, serving as a backup cell death pathway [78] [79]. This pathway is initiated by death receptors (e.g., TNFR1), Toll-like receptors (TLR3, TLR4), or other mediators which lead to the phosphorylation of receptor-interacting protein kinase 1 (RIPK1) and RIPK3, forming the necrosome complex [78]. This complex subsequently phosphorylates mixed lineage kinase domain-like protein (MLKL), which oligomerizes and translocates to the plasma membrane, executing membrane disruption and lytic cell death [78].
Ferroptosis: Iron-Dependent Lipid Peroxidation
Ferroptosis is an iron-dependent form of regulated cell death characterized by overwhelming lipid peroxidation, distinct from other death modalities in both biochemistry and morphology [44] [78] [81]. The execution of ferroptosis hinges on the failure of the glutathione-dependent antioxidant defense system, specifically the inactivation of glutathione peroxidase 4 (GPX4), which normally functions to reduce lipid hydroperoxides to lipid alcohols, thereby preventing toxic lipid peroxide accumulation [44] [81]. Key molecular features include:
System xc- Inhibition: Disruption of the cystine/glutamate antiporter system xc- depletes cellular cysteine, impairing glutathione synthesis and antioxidant capacity [81].
Iron Dependency: Intracellular ferrous iron (Fe²⁺) participates in Fenton chemistry, generating reactive oxygen species that propagate lipid peroxidation chain reactions [81].
Mitochondrial Alterations: Ferroptotic cells display characteristic mitochondrial abnormalities, including reduced cristae and outer membrane rupture [81].
Notably, inducers like RSL3 directly target GPX4, while erastin inhibits system xc-, both converging on lipid peroxidation and eventual membrane failure [44] [81].
PARP-1 Truncation: A Molecular Signature in Cell Death
PARP-1 Structure and Normal Function
PARP-1 is a multifunctional nuclear enzyme comprising three primary domains: an N-terminal DNA-binding domain (DBD) containing two zinc finger motifs that recognize DNA strand breaks; a central automodification domain (AMD); and a C-terminal catalytic domain (CAT) responsible for poly(ADP-ribose) (PAR) synthesis [3] [1] [82]. Under physiological conditions, PARP-1 serves as a primary DNA damage sensor, detecting DNA strand breaks and catalyzing the transfer of ADP-ribose units from NAD+ to target proteins, including itself, facilitating DNA repair through base excision repair and other pathways [3] [51] [82]. This automodification generates extensive PAR chains that serve as signaling platforms for recruiting additional DNA repair factors [3] [1].
PARP-1 Cleavage in Apoptosis
During apoptosis, PARP-1 serves as a primary substrate for executioner caspases-3 and -7, which cleave the enzyme at a specific aspartate-glutamate-valine-aspartate (DEVD) motif located within the DBD, generating signature fragments of approximately 24 kDa and 89 kDa [1] [82]. This proteolytic event represents a biochemical hallmark of apoptosis and serves critical functional consequences:
Inhibition of DNA Repair: The 24-kDa fragment, containing the DBD with intact zinc fingers, translocates to the nucleus and binds irreversibly to DNA strand breaks, functioning as a trans-dominant inhibitor of DNA repair by blocking access of intact PARP-1 and other repair factors to damage sites [1].
Energy Conservation: Cleavage prevents PARP-1 overactivation and consequent NAD+/ATP depletion, thereby conserving cellular energy for the ordered execution of the apoptotic program [3] [1].
Execution Promotion: The 89-kDa fragment, containing the AMD and CAT domains, exhibits altered subcellular localization and may actively promote apoptotic chromatin fragmentation [44] [1].
The detection of these characteristic PARP-1 fragments via Western blot analysis serves as a definitive biomarker for caspase-dependent apoptosis in experimental systems [1] [82].
PARP-1 in Necrosis and Ferroptosis
In contrast to apoptosis, PARP-1 plays a distinct role in necrotic cell death. Under conditions of severe genotoxic stress, PARP-1 becomes hyperactivated, leading to excessive consumption of NAD+ and subsequent ATP depletion, ultimately causing cellular energy crisis and necrotic cell demise [3] [51]. This PARP-1-dependent necrosis involves the translocation of apoptosis-inducing factor (AIF) from mitochondria to the nucleus, triggering large-scale DNA fragmentation [3].
Emerging evidence indicates complex crosstalk between PARP-1 and ferroptosis. Recent studies demonstrate that the ferroptosis inducer RSL3 triggers PARP-1 cleavage through dual mechanisms: direct caspase-3 activation and modulation of METTL3-mediated m6A RNA modification of PARP-1, reducing its translational efficiency [44]. This intersection between ferroptotic and apoptotic signaling highlights PARP-1's role as a molecular integrator of cell death pathways.
Table 1: Comparative Analysis of Cell Death Modalities
| Feature | Apoptosis | Necrosis/Necroptosis | Ferroptosis |
|---|---|---|---|
| Morphology | Cell shrinkage, chromatin condensation, apoptotic bodies | Cellular swelling, organelle breakdown, membrane rupture | Mitochondrial shrinkage, reduced cristae, membrane rupture |
| Key Mediators | Caspases, BCL-2 family, cytochrome c | RIPK1/RIPK3, MLKL (necroptosis) | GPX4, lipid ROS, iron |
| PARP-1 Cleavage | Caspase-3/7 mediated (89-kDa/24-kDa fragments) | PARP-1 hyperactivation (energy depletion) | Caspase-dependent cleavage via RSL3 |
| Inflammation | Non-inflammatory | Highly inflammatory | Inflammatory |
| Energy Dependency | ATP-dependent | ATP-depletion | ATP-independent |
| Primary Physiological Role | Development, homeostasis | Pathological response, backup cell death | Anti-cancer defense, neurodegeneration |
Experimental Methodologies for Cell Death Detection
PARP-1 Cleavage Analysis
Western blot analysis remains the gold standard for detecting PARP-1 cleavage fragments, utilizing antibodies targeting specific epitopes within the N-terminal (for 24-kDa fragment) or C-terminal (for 89-kDa fragment) regions [1] [82]. Experimental protocols typically involve:
- Cell Lysis: Preparation of whole cell or nuclear extracts using RIPA buffer supplemented with protease inhibitors to prevent post-lysis proteolysis.
- Electrophoresis: Separation of proteins (20-50 μg per lane) on 8-12% SDS-polyacrylamide gels to resolve full-length (116-kDa) and cleaved (89-kDa) PARP-1 fragments.
- Transfer and Immunoblotting: Protein transfer to PVDF membranes, blocking with 5% non-fat milk, and incubation with primary anti-PARP-1 antibodies (e.g., 1:1000 dilution) overnight at 4°C.
- Detection: Incubation with HRP-conjugated secondary antibodies and visualization using enhanced chemiluminescence substrates.
Quantitative analysis of cleavage fragments can be performed via densitometry, with the ratio of cleaved to full-length PARP-1 serving as an indicator of apoptotic activation [1].
Multiparameter Cell Death Assessment
Comprehensive characterization of cell death modalities requires integrated assessment of multiple biochemical markers:
- Apoptosis Detection: Annexin V/propidium iodide (PI) staining by flow cytometry to detect phosphatidylserine externalization; caspase-3/7 activity assays using fluorogenic substrates; cytochrome c release assays; and DNA fragmentation analysis (TUNEL) [81].
- Necroptosis Markers: Phospho-MLKL immunoblotting; LDH release assays to measure plasma membrane integrity; and inhibition studies using necroptosis-specific inhibitors (necrostatin-1) [78] [81].
- Ferroptosis Assays: Intracellular Fe²⁺ detection using FerroOrange; lipid peroxidation measurement with BODIPY 581/591 C11 or Liperfluo; GPX4 activity assays; and rescue experiments with ferroptosis inhibitors (ferrostatin-1, liproxstatin-1) [44] [81].
Table 2: Essential Research Reagents for Cell Death Detection
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Viability/Cytotoxicity | Cell Counting Kit-8 (WST-8), MTT, Calcein-AM | Measures metabolic activity and cell viability |
| Apoptosis Detection | Annexin V Apoptosis Kit, caspase-3/7 substrates, Z-VAD-FMK (inhibitor) | Detects phosphatidylserine exposure and caspase activity |
| Necrosis Markers | LDH Cytotoxicity Assay, DAPI/PI staining, anti-RIP1/RIP3 antibodies | Assesses plasma membrane integrity and necrotic signaling |
| Ferroptosis Tools | FerroOrange (Fe²⁺), Liperfluo (lipid peroxides), Erastin/RSL3 (inducers) | Measures iron accumulation and lipid peroxidation |
| PARP-1 Analysis | Anti-PARP-1 antibodies (full-length and cleaved), PARP inhibitors (olaparib) | Detects PARP-1 cleavage and functional inhibition |
| Pathway Modulators | Necrostatin-1 (necroptosis inhibitor), Ferrostatin-1 (ferroptosis inhibitor) | Specific pathway inhibition for mechanistic studies |
PARP-1 Signaling Pathways in Cell Death
The following diagrams illustrate the central role of PARP-1 in mediating different cell death pathways, with particular emphasis on the generation and function of its truncated fragments.
PARP-1 Cleavage in Cell Death Pathways
Research Applications and Therapeutic Implications
The differential cleavage patterns of PARP-1 provide valuable insights for basic research and therapeutic development. In cancer biology, PARP-1 cleavage serves as a biomarker for assessing chemotherapeutic efficacy, with resistant tumors often demonstrating impaired cleavage despite treatment [44] [51]. PARP inhibitors have emerged as promising therapeutic agents, particularly in BRCA-deficient cancers, where they induce synthetic lethality by exploiting DNA repair deficiencies [44] [51]. Recent evidence suggests that combining PARP inhibitors with ferroptosis inducers like RSL3 may overcome therapeutic resistance in aggressive malignancies, highlighting the clinical relevance of understanding cross-talk between cell death pathways [44].
In neurodegenerative diseases, excessive PARP-1 activation contributes to neuronal injury through energy depletion and parthanatos, a PARP-1-dependent cell death pathway [1]. Inhibiting PARP-1 activity or promoting its cleavage may represent neuroprotective strategies. Furthermore, the detection of specific PARP-1 fragments in biological samples holds diagnostic potential for distinguishing between apoptotic, necrotic, and ferroptotic cell death in pathological contexts [1] [82].
The comparative analysis of apoptosis, necrosis, and ferroptosis reveals both distinct and overlapping features in their molecular mechanisms and physiological consequences. PARP-1 and its proteolytic fragments serve as critical determinants and biomarkers of cellular fate, with specific cleavage patterns characterizing different death modalities. The 89-kDa and 24-kDa fragments generated by caspase-mediated cleavage represent definitive signatures of apoptosis, while PARP-1 hyperactivation contributes to necrotic cell death through bioenergetic catastrophe. Emerging evidence of PARP-1 cleavage in ferroptosis underscores the complex interplay between distinct cell death pathways. Understanding these differential effects and the role of PARP-1 fragments provides a sophisticated framework for investigating disease mechanisms and developing targeted therapeutic interventions that modulate cell death signaling for clinical benefit.
Within the broader thesis exploring the biological functions of truncated PARP-1 (tPARP1) fragments in cell death research, in vivo validation represents a critical step in translating mechanistic findings into therapeutic potential. The cleavage of full-length PARP1, a 116-kDa nuclear enzyme, by executioner caspases during apoptosis generates a characteristic 89-kD C-terminal fragment (tPARP1) and a 24-kD DNA-binding fragment [19]. This proteolytic event is considered a hallmark of apoptosis and was initially viewed merely as an inert marker of cell death execution. However, emerging evidence indicates that tPARP1 is not a passive bystander but possesses distinct biological activities that influence cell death pathways, DNA damage signaling, and innate immune responses [5] [19]. This technical guide synthesizes current research on the validation of tPARP1 functions using mouse xenograft and radiation response models, providing detailed methodologies and datasets for researchers investigating PARP1 biology in cancer and cell death.
Biological Context of tPARP1
Formation and Traditional Understanding
PARP1 is cleaved by caspase-3 at D214 during apoptosis, separating its N-terminal DNA-binding domain (containing two zinc finger motifs) from its C-terminal catalytic domain [5] [19]. The traditional interpretation suggested that this cleavage inactivated PARP1 to prevent excessive NAD+ and ATP consumption, thereby facilitating the apoptotic process [19]. The 24-kD fragment was thought to act as a trans-dominant inhibitor by irreversibly binding to DNA strand breaks and blocking DNA repair processes [19].
Emerging Non-Canonical Functions
Recent studies have revealed non-canonical functions of tPARP1 that extend beyond its role as an apoptosis marker. The tPARP1 fragment, which retains the BRCT domain, WGR domain, and catalytic domain, translocates from the nucleus to the cytoplasm during apoptosis [5]. Interestingly, evolutionary analysis reveals that PARP1 orthologs in several lower organisms naturally lack the first two zinc finger motifs, suggesting that tPARP1 may represent a functional isoform with specific biological activities conserved through evolution [5].
A seminal study demonstrated that tPARP1 recognizes and mono-ADP-ribosylates the RNA polymerase III (Pol III) complex in the cytosol during poly(dA-dT)-stimulated apoptosis [5]. This tPARP1-mediated modification of Pol III facilitates IFN-β production and enhances apoptosis, revealing a novel role for tPARP1 in connecting apoptotic signaling with innate immune activation [5]. This finding positions tPARP1 as an active participant in cell death pathways rather than merely a consequence of proteolytic inactivation.
In Vivo Validation Models and Therapeutic Applications
Mouse Xenograft Models for PARP1-Targeted Therapies
Mouse xenograft models have been instrumental in validating PARP1-targeted therapies and their combinations with radiation. The following table summarizes key in vivo studies investigating PARP inhibition in combination with radiotherapy:
Table 1: In Vivo Studies of PARP Inhibition with Radiotherapy
| Cancer Type | Model Details | Treatment Protocol | Key Findings | Source |
|---|---|---|---|---|
| Rectal Cancer | Patient-derived organoid xenografts | MEK inhibitor + PARP inhibitor + radiation | Combined MEK-PARP inhibition enhanced radiosensitivity; efficacy confirmed in xenografts | [83] |
| Small Cell Lung Cancer (SCLC) | SCRX-Lu149 PDX models (chemonaive & chemoresistant) | Olaparib (concurrent + maintenance) with RT | Maintenance olaparib post-RT significantly improved time to volumetric endpoint (51.5 days vs 32 days in chemonaive) | [84] |
| Neuroblastoma | IMR-05 xenografts in NSG mice | [211At]PTT (PARP1-targeted alpha therapy) | Transient PARP1 upregulation post-therapy; enhanced DNA damage (γH2AX) | [85] |
| Ewing Sarcoma | SK-N-MC flank tumor xenografts | Olaparib + low-dose RT (4 Gy) | PARP inhibition combined with RT stopped tumor growth | [86] |
Radiation Response and DNA Damage Modulation
The combination of PARP inhibition with radiation has demonstrated significant efficacy across multiple xenograft models. In SCLC models, the maintenance of olaparib following concurrent PARP inhibitor and radiation treatment (COM approach) resulted in substantially greater DNA damage compared to pulsed treatment (PUL approach), as measured by alkaline comet assay (Olive tail moment: COM mean 7.2 vs. PUL mean 2.8 at 48 hours, p < 0.0001) and γH2AX foci formation [84]. This enhanced DNA damage correlated with improved tumor control, supporting the importance of sustained PARP inhibition in radiotherapy protocols.
In neuroblastoma xenografts, PARP1-targeted alpha-particle therapy using [211At]parthanatine ([211At]PTT) induced transient upregulation of PARP1 expression, as visualized by [18F]fluorthanatrace ([18F]FTT) PET/CT imaging [85]. Tumor uptake of [18F]FTT increased by 34.1% from baseline to day 2 post-treatment (mean difference: 3.0%ID/g, p = 0.03), returning to baseline by day 6, indicating dynamic changes in PARP1 expression following targeted radiation [85]. This transient PARP1 upregulation in sublethally damaged cells potentially creates a therapeutic window for fractionated radiation approaches.
Experimental Protocols for Key Methodologies
Xenograft Therapeutic Efficacy Study
Objective: To evaluate the efficacy of combined PARP inhibition and radiotherapy in patient-derived xenograft models.
Materials:
- NOD/SCID IL2Rγ−/− (NSG) mice, 6-8 weeks old
- SCLC PDX tumors (e.g., SCRX-Lu149 chemonaive and chemoresistant lines)
- Olaparib (PARP inhibitor)
- Irradiation equipment (e.g., 137Cs γ-ray source)
- Calipers for tumor measurement
Methodology:
- Tumor Engraftment: Implant 1 million tumor cells mixed with 50% Matrigel subcutaneously into mouse flanks.
- Treatment Groups: Randomize mice into 5 arms (n=6-9/group):
- Vehicle control
- Olaparib alone (50 mg/kg daily, oral gavage)
- Radiotherapy alone (2 Gy × 5 fractions)
- Pulsed olaparib (concurrent with RT only)
- Combination: concurrent + maintenance olaparib
- Radiation Protocol: Administer focal RT to tumors (2 Gy/fraction, 5 fractions) using appropriate shielding.
- Olaparib Administration: For pulsed group: administer 3 hours before RT, withdraw 1 hour post-RT. For combination group: maintain olaparib daily post-RT completion.
- Endpoint Monitoring: Measure tumor volumes biweekly using formula: V = 0.5 × L × W². Record time to reach volumetric endpoint (e.g., 1000 mm³).
- Statistical Analysis: Compare time-to-endpoint using log-rank test; analyze tumor growth curves by repeated measures ANOVA [84].
PARP1 Expression and DNA Damage Assessment
Objective: To quantify PARP1 expression and DNA damage following PARP1-targeted radiotherapy.
Materials:
- [18F]FTT (PARP1-specific PET tracer)
- [211At]PTT (PARP1-targeted alpha therapy agent)
- Anti-γH2AX antibody (DNA damage marker)
- Anti-PARP1 antibody
- Zeiss Widefield microscope for immunofluorescence
Methodology:
- Baseline Imaging: Perform [18F]FTT PET/CT imaging at baseline (60 min post-IV injection of 7.4 MBq [18F]FTT).
- Therapy Administration: Administer 370 kBq [211At]PTT IV 24 hours after baseline imaging.
- Longitudinal Imaging: Repeat [18F]FTT PET/CT at days 2 and 6 post-therapy.
- Image Analysis: Quantify tumor uptake as %ID/g using MIM Software; calculate percent change from baseline.
- Immunofluorescence Validation:
- Resect tumors at baseline, day 2, and day 6 post-treatment.
- Snap-freeze in OCT, section at 10 µm thickness.
- Fix in 4% PFA, permeabilize with 0.1% Triton X-100.
- Block with 10% goat serum, incubate with primary antibodies (PARP1 1:1000; γH2AX 1:5000).
- Incubate with Alexa Fluor-conjugated secondary antibodies.
- Mount with ProLong Glass containing NucBlue (Hoechst 33344).
- Image using Zeiss Widefield microscope; analyze fluorescence intensity normalized to Hoechst-positive nuclei using Fiji/ImageJ [85].
Mechanistic Analysis of tPARP1 Functions
Objective: To characterize tPARP1 interactions with RNA Polymerase III and innate immune signaling.
Materials:
- PARP1-deficient 293T cells
- SFB-tagged mutant tPARP1 (E988A)
- Poly(dA-dT) for apoptosis induction
- Antibodies against POLR3A, POLR3B, POLR3F
Methodology:
- Cell Modeling:
- Express SFB-tagged mtPARP1 in PARP1-deficient 293T cells.
- Induce apoptosis by transfecting poly(dA-dT) to mimic pathogenic DNA.
- Interaction Analysis:
- Perform co-immunoprecipitation with anti-POLR3A, anti-POLR3B, anti-POLR3F antibodies.
- Validate interactions by Western blot.
- Domain Mapping:
- Generate truncation mutants of tPARP1 lacking specific domains.
- Identify interaction domains via co-IP (BRCT domain critical for Pol III binding).
- Functional Assays:
- Measure IFN-β production via ELISA or qPCR.
- Assess apoptosis by Annexin V-FITC/PI staining and flow cytometry [5].
Signaling Pathways and Molecular Mechanisms
The molecular mechanisms underlying tPARP1 functions and PARP inhibitor-mediated radiosensitization involve multiple interconnected pathways:
Diagram 1: Molecular Mechanisms of tPARP1 and PARP Inhibition in DNA Damage Response
The diagram above illustrates the complex interplay between tPARP1 formation and PARP inhibitor mechanisms following DNA damage. Radiation and genotoxic stress induce DNA damage, triggering both caspase activation and full-length PARP1 recruitment. Caspase-mediated cleavage generates tPARP1, which translocates to the cytosol, interacts with RNA Polymerase III, and catalyzes ADP-ribosylation, leading to innate immune activation and enhanced apoptosis [5]. Concurrently, PARP inhibitors trap PARP1 on DNA and inhibit single-strand break repair, resulting in persistent double-strand breaks that activate cGAS-STING signaling and chemokine production while inducing synthetic lethality in HR-deficient cells [55] [84].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagents for tPARP1 and PARP Inhibition Studies
| Reagent/Cell Line | Specific Example | Function/Application | Source/Reference |
|---|---|---|---|
| PARP Inhibitors | Olaparib, Talazoparib, Veliparib | Catalytic inhibition and PARP trapping; radiosensitization | [83] [84] |
| PARP1-Targeted Radiotherapeutics | [211At]Parthanatine ([211At]PTT) | Alpha-particle emitter targeting PARP1 for precise DNA damage induction | [85] |
| PARP1 Imaging Agents | [18F]Fluorthanatrace ([18F]FTT) | PARP1-specific PET tracer for target engagement monitoring | [85] |
| Cell Lines | IMR-05 neuroblastoma, SBC5 SCLC, RD-ES Ewing sarcoma | Disease-specific models for PARP inhibition and radiation studies | [85] [86] [84] |
| Patient-Derived Xenografts | SCRX-Lu149 (SCLC), Rectal cancer organoids | Clinically relevant models preserving tumor heterogeneity | [83] [84] |
| DNA Damage Antibodies | Anti-γH2AX, Anti-pATM, Anti-pDNA-PKcs | Detection and quantification of DNA damage response | [85] [86] |
| Apoptosis Detection Kits | Annexin V-FITC/PI, Cleaved Caspase-3 IHC | Assessment of apoptotic cell death | [5] [86] |
| PARP1 Cleavage-Specific Antibodies | Anti-tPARP1 (89 kD fragment) | Specific detection of caspase-cleaved PARP1 | [5] [19] |
Data Analysis and Interpretation Guidelines
Quantitative Metrics for Therapeutic Efficacy
When evaluating tPARP1-related therapies in xenograft models, several quantitative metrics provide robust assessment of treatment efficacy:
Tumor Growth Inhibition: Calculate tumor volume (0.5 × L × W²) and compare growth curves across treatment groups. The combination of MEK and PARP inhibition with radiation in rectal cancer models demonstrated significant tumor growth inhibition compared to single modalities [83].
Time to Volumetric Endpoint: Defined as time to reach predetermined tumor volume (e.g., 1000 mm³). In SCLC PDX models, maintenance olaparib following RT significantly extended time to endpoint compared to pulsed treatment (85 vs. 43 days in SBC5 models, p = 0.0002) [84].
DNA Damage Quantification: Olive tail moment in comet assay and γH2AX foci counts provide quantitative measures of DNA damage persistence. Combination PARP inhibitor and radiation treatment demonstrated significantly higher Olive tail moments (7.2 vs. 2.8, p < 0.0001) compared to pulsed treatment [84].
Assessment of PARP1 Expression Dynamics
Longitudinal monitoring of PARP1 expression provides insights into target engagement and adaptive responses:
PET Imaging Quantification: [18F]FTT uptake measured as %ID/g. In neuroblastoma models, uptake increased from 8.6 ± 3.5%ID/g at baseline to 11.6 ± 4.8%ID/g at day 2 post-therapy (34.1% increase, p = 0.03) [85].
Immunofluorescence Analysis: Normalize PARP1 fluorescence intensity to Hoechst-positive nuclei. Correlation between [18F]FTT uptake and PARP1 immunofluorescence demonstrated strong agreement (Pearson's r = 0.73, p = 0.02) [85].
The validation of tPARP1 functions in mouse xenograft and radiation response models has revealed complex roles for PARP1 fragments beyond their traditional association with apoptosis execution. The emerging paradigm recognizes tPARP1 as an active signaling molecule that modulates immune responses through RNA polymerase III interaction and ADP-ribosylation [5]. Furthermore, the combination of PARP inhibitors with radiation leverages multiple mechanisms including PARP trapping, DNA damage persistence, and immune activation through cGAS-STING signaling [84].
Future research directions should focus on optimizing therapeutic sequencing and identifying biomarkers for patient selection. The differential efficacy of maintenance versus pulsed PARP inhibitor administration following radiation highlights the importance of treatment scheduling [84]. Additionally, the development of PARP1-specific imaging agents like [18F]FTT enables non-invasive monitoring of target engagement and adaptive responses, potentially guiding personalized therapy approaches [85].
The integration of these findings into the broader thesis on tPARP1 biological functions underscores the multifaceted roles of PARP1 fragments in cell death pathways and positions tPARP1 as both a biomarker and potential therapeutic target in DNA damage response pathways.
Conclusion
Truncated PARP-1 fragments are not merely inert cleavage products but active mediators of cell death with distinct biological functions. The 89-kD fragment executes novel cytosolic roles in innate immune activation, while the 24-kD fragment dominantly inhibits DNA repair. The cleavage of PARP1 serves as a critical molecular switch between apoptotic and necrotic cell fates. Future research should focus on developing specific biomarkers based on tPARP1 signatures, creating therapeutics that selectively enhance tPARP1 formation in resistant cancers, and exploring the intersection of tPARP1 with other cell death modalities like ferroptosis. These approaches hold significant promise for overcoming current limitations in PARP inhibitor therapy and developing novel treatments for cancer and other diseases characterized by dysregulated cell death.
References
- 1. PARP-1 cleavage fragments: signatures of cell-death ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3022541/]
- 2. Cleaved PARP (Asp214) Antibody #9541 [https://www.cellsignal.com/products/primary-antibodies/cleaved-parp-asp214-antibody/9541?srsltid=AfmBOopKQy9X3w2xiLmd4tG9dJULFBKNuYeAfWZYxJ1dYMWlF4giZyd1]
- 3. Biological function, mediate cell death pathway and their ... [https://www.frontiersin.org/journals/nutrition/articles/10.3389/fnut.2022.1093939/full]
- 4. The 89-kDa PARP1 cleavage fragment serves as a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7948984/]
- 5. Truncated PARP1 mediates ADP-ribosylation of RNA ... [https://www.nature.com/articles/s41421-021-00355-1]
- 6. Cleaved PARP (Asp214) (D64E10) Rabbit Monoclonal ... [https://www.cellsignal.com/products/primary-antibodies/cleaved-parp-asp214-d64e10-rabbit-monoclonal-antibody/5625?srsltid=AfmBOooOKmtrxuf10HIodzNjsGT3jwIzn4lpsEz6V-3BCHCqjPdgq3s9]
- 7. Activation and Cleavage of Caspase-3 in Apoptosis Induced ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6793169/]
- 8. Cellular Mechanisms Controlling Caspase Activation and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3660825/]
- 9. Activation and Caspase-mediated Inhibition of PARP - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC99613/]
- 10. Structural Basis for the Inhibition of Caspase-3 by XIAP [https://www.sciencedirect.com/science/article/pii/S0092867401002744]
- 11. Cooperative dynamics of PARP-1 zinc-finger domains in ... [https://www.nature.com/articles/s41598-024-73707-y]
- 12. The BRCT Domain of PARP1 Binds Intact DNA and Mediates ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8769213/]
- 13. The deubiquitination-PARylation positive feedback loop of the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12256264/]
- 14. Evolutionary history of the poly(ADP-ribose) polymerase gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2964712/]
- 15. Regulatory apoptotic fragment of PARP1 complements ... [https://www.sciencedirect.com/science/article/abs/pii/S1568786423001477]
- 16. The zinc-finger domains of PARP1 cooperate to recognise ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4826610/]
- 17. Mechanism and evolution of the Zn-fingernail required for ... [https://www.nature.com/articles/s41467-020-18773-2]
- 18. The 89-kDa PARP1 cleavage fragment serves as a ... - PubMed [https://pubmed.ncbi.nlm.nih.gov/33168626/]
- 19. PARP-1 cleavage fragments: signatures of cell-death ... [https://biosignaling.biomedcentral.com/articles/10.1186/1478-811X-8-31]
- 20. The 89-kDa PARP1 cleavage fragment serves as a ... [https://www.sciencedirect.com/science/article/pii/S0021925820000320]
- 21. Vesicular translocation of PARP-1 to cytoplasm causes ... [https://www.frontiersin.org/journals/cellular-neuroscience/articles/10.3389/fncel.2024.1363154/full]
- 22. Alternate therapeutic pathways for PARP inhibitors and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8080675/]
- 23. Is PARP1 a Hero or Villain? [https://bellbrooklabs.com/parp1-hero-or-villain/?srsltid=AfmBOooE4bfTGmpS6gTJgcRa3np5McgmfjIPDIOcXsgJoymGH7uoog1a]
- 24. The DNA-Binding Domain of Human PARP-1 Interacts with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3094755/]
- 25. Roles and therapeutic potential of PARP-1 in ... [https://www.sciencedirect.com/science/article/abs/pii/S0006295225006380]
- 26. Tumor-targeted top1 inhibitor delivery with optimized parp ... [https://www.nature.com/articles/s41467-025-64509-5]
- 27. Poly(ADP-ribose) polymerase-1 and its cleavage products ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4013233/]
- 28. Truncated PARP1 mediates ADP-ribosylation of RNA ... [https://pure.johnshopkins.edu/en/publications/truncated-parp1-mediates-adp-ribosylation-of-rna-polymerase-iii-f/]
- 29. Anti-PARP1 antibody [EPR18461] (ab191217) [https://www.abcam.com/en-us/products/primary-antibodies/parp1-antibody-epr18461-ab191217]
- 30. PARP1 antibody (66520-1-PBS) [https://www.ptglab.com/products/PARP1-Antibody-66520-1-PBS.htm?srsltid=AfmBOopJtfNlL3UFFwg6xNOhaOc3vGsWryqK1IAVZOIXCo0oPCSdLvQ2]
- 31. PARP1 Monoclonal Antibody (123) (436400) [https://www.thermofisher.com/antibody/product/PARP1-Antibody-clone-123-Monoclonal/436400]
- 32. PARP1 genomics: chromatin immunoprecipitation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3164790/]
- 33. Poly(ADP-ribose) polymerase-1 and its ambiguous role in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9877585/]
- 34. Truncated PARP1 mediates ADP-ribosylation of RNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8764063/]
- 35. Integrated proteomics identifies PARP inhibitor-induced ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9636579/]
- 36. Review The quest to identify ADP-ribosylation readers [https://www.sciencedirect.com/science/article/abs/pii/S0968000424002056]
- 37. Characterization of the necrotic cleavage of poly(ADP- ... [https://www.nature.com/articles/4400851]
- 38. TOPBP1 as a potential predictive biomarker for enhanced ... [https://cellandbioscience.biomedcentral.com/articles/10.1186/s13578-025-01350-9]
- 39. RSL3 promotes PARP1 apoptotic functions by distinct ... [https://cmbl.biomedcentral.com/articles/10.1186/s11658-025-00785-9]
- 40. PARP-1 in liver diseases: Molecular mechanisms ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12486748/]
- 41. Roles and therapeutic potential of PARP-1 in ... [https://pubmed.ncbi.nlm.nih.gov/41022359/]
- 42. PARP inhibitor resistance: the underlying mechanisms and ... [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-020-01227-0]
- 43. Mechanisms of PARP1 inhibitor resistance and their ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9773381/]
- 44. RSL3 promotes PARP1 apoptotic functions by distinct ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12492750/]
- 45. RSL3 promotes PARP1 apoptotic functions by distinct ... [https://pubmed.ncbi.nlm.nih.gov/41039223/]
- 46. Organelle-Specific Mechanisms in Crosstalk between ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9836800/]
- 47. Interplay of ferroptotic and apoptotic cell death and its ... [https://www.nature.com/articles/s41418-025-01514-7]
- 48. The Enigmatic Function of PARP1: From PARylation Activity to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6953017/]
- 49. PARP-1 cleavage fragments: Signatures of cell-death ... [https://augusta.elsevierpure.com/en/publications/parp-1-cleavage-fragments-signatures-of-cell-death-proteases-in-n]
- 50. Characterization of the necrotic cleavage of poly(ADP- ... [https://pubmed.ncbi.nlm.nih.gov/11536009/]
- 51. PARP-1, a determinant of cell survival in response to DNA ... [https://www.sciencedirect.com/science/article/pii/S0301472X03000833]
- 52. Significance of Caspase-Mediated Cleavage of PARP-1 in ... [https://grantome.com/grant/NIH/R36-AG031572-01]
- 53. Computational Chemistry Advances in the Development of ... [https://www.mdpi.com/1424-8247/18/11/1679]
- 54. Mapping the Subtype-Specific PARP1 ADP-ribosylated ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12621782/]
- 55. Targeting PARP1: A Promising Approach for Next- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12162764/]
- 56. A Two-tiered compensatory response to loss of DNA repair ... [https://www.aging-us.com/article/100127/text]
- 57. Poly(ADP-ribose) polymerase-1 mediated caspase ... [https://www.sciencedirect.com/science/article/abs/pii/S0891584905001632]
- 58. Clinical approaches to overcome PARP inhibitor resistance [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-025-02355-1]
- 59. The CYLD–PARP1 feedback loop regulates DNA damage ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11725943/]
- 60. Research progress on ferroptosis and PARP inhibitors in ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1598279/full]
- 61. Combined DNA-PK and PARP Inhibition as a Therapeutic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12618835/]
- 62. Glucose Deprivation Converts Poly(ADP-ribose) Polymerase ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3868765/]
- 63. Poly(ADP-ribose) polymerase-dependent energy depletion ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4104885/]
- 64. NAD+ consumption by PARP1 in response to DNA damage ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6740200/]
- 65. Calpain-generated natural protein fragments as short-lived ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3948289/]
- 66. Caspases and their substrates | Cell Death & Differentiation [https://www.nature.com/articles/cdd201744]
- 67. An atlas of caspase cleavage events in differentiating ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11344277/]
- 68. Granzyme B [https://en.wikipedia.org/wiki/Granzyme_B]
- 69. Granzyme B - an overview [https://www.sciencedirect.com/topics/medicine-and-dentistry/granzyme-b]
- 70. The three-dimensional structure of human granzyme to... B compared [https://pubmed.ncbi.nlm.nih.gov/11325591/]
- 71. Structural Biochemistry/SitePrediction - Wikibooks, open books for an... [https://en.wikibooks.org/wiki/Structural_Biochemistry/SitePrediction]
- 72. Poly(ADP-ribosyl)ation by PARP1: reaction mechanism and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6486540/]
- 73. Molecular mechanisms of cell by parthanatos: More questions... death [https://pmc.ncbi.nlm.nih.gov/articles/PMC11445734/]
- 74. PARP1 regulates the protein stability and proapoptotic ... [https://www.nature.com/articles/cddis2016345]
- 75. Therapeutic Strategies and Biomarkers to Modulate PARP ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7226473/]
- 76. Potent EGFR/ PARP - 1 inhibition by spirooxindole-triazole hybrids for... [https://pubs.rsc.org/en/content/articlelanding/2025/ra/d4ra05966b]
- 77. Truncated PARP1 mediates ADP-ribosylation of RNA ... [https://pubmed.ncbi.nlm.nih.gov/35039483/]
- 78. Targeting regulated cell death: Apoptosis, necroptosis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11921819/]
- 79. Regulated cell death pathways and their roles in ... [https://www.nature.com/articles/s12276-023-01069-y]
- 80. Cell death triad: Integrating apoptotic, autophagic, and ... [https://www.sciencedirect.com/science/article/pii/S1877117325001048]
- 81. Detect Cell Death: Apoptosis, Necrosis & Ferroptosis [https://www.dojindo.com/contents/detect_celldeath_apoptosis_necrosis_ferroptosis.html]
- 82. Potential biological role of poly (ADP-ribose) polymerase ... [https://rbej.biomedcentral.com/articles/10.1186/1477-7827-7-143]
- 83. Combined MEK and PARP inhibition enhances radiation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12432356/]
- 84. PARP inhibitor radiosensitization enhances anti-PD-L1 ... [https://www.nature.com/articles/s41467-025-57257-z]
- 85. PARP1-targeted alpha therapy enhances target expression [https://ejnmmires.springeropen.com/articles/10.1186/s13550-025-01256-0]
- 86. Combining poly(ADP-ribose) polymerase 1 (PARP ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3823674/]