Constructing Cytochrome C-GFP Reporter Cell Lines: A Comprehensive Guide from Design to High-Throughput Application
This article provides a complete roadmap for researchers and drug development professionals on the construction and application of cytochrome C-green fluorescent protein (GFP) reporter cell lines.
Constructing Cytochrome C-GFP Reporter Cell Lines: A Comprehensive Guide from Design to High-Throughput Application
Abstract
This article provides a complete roadmap for researchers and drug development professionals on the construction and application of cytochrome C-green fluorescent protein (GFP) reporter cell lines. We cover the foundational biology of cytochrome C in apoptotic pathways, detailed methodologies for stable cell line generation using lentiviral vectors, and solutions to common challenges like high background fluorescence and low transfection efficiency. Furthermore, we explore the validation of these tools through automated image analysis algorithms achieving over 90% precision and their application in high-throughput drug screening for cancer therapeutics. This guide synthesizes current best practices to empower scientists in implementing this powerful technology for dynamic, live-cell analysis of apoptosis.
The Biology of Apoptosis and Rationale for Cytochrome C-GFP Reporters
Understanding the Key Role of Cytochrome C in Intrinsic Apoptosis
Cytochrome c is a highly conserved, small, soluble heme-containing protein with a molecular weight of approximately 12-13 kDa, encoded by nuclear genes and synthesized as apocytochrome c before maturing in the mitochondrial intermembrane space [1] [2]. This essential protein serves two critical, yet opposing, functions within the cell. Its primary vital function is as an indispensable electron shuttle in the mitochondrial respiratory chain, where it transfers electrons between Complex III and Complex IV to support oxidative phosphorylation and maintain cellular ATP production [1] [3]. However, when cells receive apoptotic stimuli, cytochrome c undergoes a dramatic functional switch to become a central mediator of cell death [1] [4]. This dualism makes cytochrome c a crucial focal point for understanding cellular homeostasis and developing therapeutic strategies for diseases characterized by abnormal cell death, such as cancer and neurodegenerative disorders [5] [3] [2].
The transition between these opposing functions is regulated by cytochrome c's subcellular localization. Under normal conditions, cytochrome c is confined to the mitochondrial intermembrane and intercristae spaces, where it interacts with cardiolipin, an anionic phospholipid of the inner mitochondrial membrane [1] [4]. This interaction maintains cytochrome c in its respiratory function. However, during apoptosis, cytochrome c is released from mitochondria into the cytosol, where it acquires its pro-apoptotic function [4]. This release represents a critical commitment point in the intrinsic apoptotic pathway and is therefore a prime target for monitoring and manipulating apoptotic activity in research and therapeutic contexts.
The Molecular Mechanism of Cytochrome C in Intrinsic Apoptosis
Cytochrome C Release and Apoptosome Formation
The intrinsic apoptotic pathway, also known as the mitochondrial pathway, is initiated by diverse intracellular stressors including DNA damage, metabolic stress, unfolded protein accumulation, and radiation [4] [2]. These stimuli converge on mitochondria to trigger mitochondrial outer membrane permeabilization (MOMP), which allows proteins normally confined to the intermembrane space, including cytochrome c, to escape into the cytosol [4]. The release process involves at least two critical steps: first, the communication between intermembrane and intercristae spaces is facilitated, followed by the mobilization of cytochrome c from its binding to cardiolipin [4].
Two principal mechanisms mediate MOMP and cytochrome c release. The first involves Bcl-2 family proteins, where pro-apoptotic members like Bax and Bak oligomerize to form pores in the outer mitochondrial membrane [1] [4]. This oligomerization is triggered by activated BH3-only proteins (such as tBID) and can be inhibited by anti-apoptotic family members like Bcl-2 and Bcl-xL [1]. The second mechanism involves mitochondrial permeability transition (MPT), characterized by the opening of a non-specific pore across both mitochondrial membranes, leading to mitochondrial swelling and outer membrane rupture [1]. While cyclophilin D appears essential for MPT, components like VDAC and ANT may be dispensable for this process [1].
Once in the cytosol, cytochrome c initiates apoptosome formation by binding to Apaf-1 (apoptotic protease-activating factor-1) in a dATP/ATP-dependent manner [1] [2]. This binding triggers Apaf-1 oligomerization into a wheel-like heptameric complex that recruits and activates procaspase-9 [1] [2]. The resulting signaling platform, known as the apoptosome, facilitates the auto-activation of caspase-9, which then cleaves and activates downstream effector caspases, primarily caspase-3 and caspase-7 [5] [2].
Table 1: Key Molecular Components in Cytochrome C-Mediated Apoptosis
| Component | Function in Apoptosis | Localization |
|---|---|---|
| Cytochrome c | Apoptosome activation; caspase cascade initiation | Mitochondrial IMS → Cytosol |
| Apaf-1 | Apoptosome scaffold protein | Cytosol |
| Caspase-9 | Initiator caspase activated by apoptosome | Cytosol |
| Caspase-3/7 | Effector caspases executing cell death | Cytosol |
| Bcl-2/Bcl-xL | Anti-apoptotic; inhibits Bax/Bak | Mitochondrial membrane |
| Bax/Bak | Pro-apoptotic; mediates MOMP | Mitochondrial membrane (upon activation) |
| Cardiolipin | Tethers cytochrome c to mitochondrial membrane | Mitochondrial inner membrane |
Caspase Activation and Apoptotic Execution
The apoptosome serves as the molecular platform for activating the caspase cascade, a proteolytic pathway that systematically dismantles the cell [5]. Caspases are cysteine proteases that cleave their substrates at specific aspartic acid residues, and they are typically categorized as either initiator caspases (e.g., caspase-9) or executioner caspases (e.g., caspases-3, -6, -7) [5]. Once activated by the apoptosome, caspase-9 cleaves and activates the executioner caspases, which then orchestrate the morphological changes characteristic of apoptosis through limited proteolysis of specific cellular substrates [5] [2].
Key substrates of executioner caspases include PARP (poly-ADP ribose polymerase), whose cleavage disrupts DNA repair mechanisms; nuclear lamins, whose cleavage leads to nuclear envelope disintegration; and various cytoskeletal proteins [5]. This controlled proteolysis results in characteristic apoptotic features such as chromatin condensation, DNA fragmentation, membrane blebbing, and ultimately the formation of apoptotic bodies that are phagocytosed by neighboring cells without inducing inflammation [5]. The entire process represents a finely tuned mechanism for eliminating damaged or unnecessary cells while preserving tissue integrity.
Diagram 1: Cytochrome C-Mediated Intrinsic Apoptotic Pathway. This diagram illustrates the key molecular events from apoptotic stimuli to execution, highlighting cytochrome c's central role.
Cytochrome C in Disease and Therapy
Cytochrome C Dysregulation in Cancer
Dysregulation of cytochrome c-mediated apoptosis represents a hallmark of numerous pathological conditions, particularly cancer. In various malignancies, including breast cancer and glioma, reduced cytochrome c expression or impaired release has been observed, contributing to diminished apoptotic sensitivity and treatment resistance [3] [2]. Cancer cells often exhibit alterations in the cytochrome c/Apaf-1/caspase-9 axis that allow them to evade programmed cell death despite accumulating genetic damage [3]. For instance, in breast cancer tissues, cytochrome c demonstrates a redox imbalance, with increased levels of reduced cytochrome c that cannot effectively induce apoptosis [2]. This dysregulation is upregulated across all stages of cancer development and is associated with poorer patient survival outcomes [2].
The critical role of cytochrome c in cancer is further evidenced by the fact that many conventional cancer therapies, including chemotherapy, radiotherapy, and targeted therapies, ultimately depend on activating the mitochondrial apoptotic pathway to eliminate malignant cells [2]. These treatments frequently work by inducing cytochrome c release, thereby triggering caspase activation and apoptotic execution [2]. Consequently, tumors with defective cytochrome c function often demonstrate cross-resistance to diverse treatment modalities, highlighting this protein's central position in treatment response mechanisms.
Therapeutic Targeting of Cytochrome C Pathways
The molecular machinery of cytochrome c-mediated apoptosis presents multiple attractive targets for therapeutic intervention. Current strategies focus on either restoring cytochrome c function in apoptosis-resistant cancers or modulating its activity in conditions of excessive cell death. Numerous natural compounds and synthetic agents have demonstrated efficacy in promoting cytochrome c release and restoring apoptotic sensitivity in cancer cells [2]. For example, Moringa isothiocyanate from Moringa oleifera seeds induces pro-apoptotic proteins including cytochrome c, while apigenin (found in parsley and chamomile) activates intrinsic apoptotic pathways by inducing cytochrome c release [2].
Emerging therapeutic approaches include direct delivery of exogenous cytochrome c into the cytoplasm of cancer cells to bypass defects in cytochrome c release, effectively inducing apoptosis even in resistant malignancies [2]. Additionally, research is exploring the modulation of post-transl modifications that regulate cytochrome c's functions, particularly phosphorylation, which intricately controls its dual roles in respiration and apoptosis [3]. These innovative strategies highlight the therapeutic potential of targeting cytochrome c pathways, offering promising avenues for overcoming treatment resistance in challenging cancers like triple-negative breast cancer.
Table 2: Cytochrome C-Targeting Therapeutic Agents and Their Mechanisms
| Agent | Source | Mechanism of Action | Cancer Models |
|---|---|---|---|
| Moringa Isothiocyanate | Moringa oleifera seeds | Induces pro-apoptotic proteins (cyt c, p53, cleaved caspase-7) | MCF-7, MDA-MB-231 |
| Apigenin | Parsley, chamomile, citrus fruits | Activates intrinsic pathway (cyt c, Bax, caspase-3) | Breast cancer, lung cancer |
| Catalpol | Rehmannia glutinosa (Chinese medicine) | Loss of MMP, increased ROS, elevated cytoplasmic cyt c | MCF-7 (in vitro & in vivo) |
| CREE | Cimicifuga dahurica root | Upregulates Bax, caspase-9/3, cyt c | MCF-7, MDA-MB-231 |
| Diallyl Trisulfide (DATS) | Allium vegetables | Regulates cyt c release; induces apoptosis | Breast cancer |
Application Note: GFP Reporter Systems for Monitoring Apoptosis
Reporter System Design Principles
The development of fluorescent reporter systems represents a cutting-edge methodological approach for monitoring cytochrome c dynamics and apoptotic signaling in live cells. These systems typically utilize genetically encoded fluorescent proteins (particularly GFP variants) strategically targeted to specific subcellular compartments or linked to apoptotic signaling components [6] [7]. Two primary design strategies have emerged for apoptosis reporters: "bright-to-dark" systems, where fluorescence decreases upon apoptosis induction, and "dark-to-bright" systems, where fluorescence increases during apoptotic progression [7].
A particularly innovative approach involves inserting caspase cleavage motifs into the GFP protein itself. In one such design, researchers created a caspase-3 reporter by inserting DEVD-similar sequences (the cleavage motif for caspase-3) into specific structural positions of EGFP, considering factors like structural positioning and hydrophilicity [7]. When caspase-3 is activated during apoptosis, it cleaves the engineered GFP, resulting in decreased fluorescence in this "bright-to-dark" system, which has demonstrated superior sensitivity compared to alternative designs [7]. This design strategy offers the significant advantage of not requiring additional peptides, making it readily adaptable to various experimental systems and potentially suitable for high-throughput drug screening applications.
Mitochondrial-Targeted Reporters for Cytochrome C Release
Beyond caspase activation, reporter systems can also be designed to monitor earlier apoptotic events, such as cytochrome c release from mitochondria. This can be achieved by creating mitochondrially targeted fluorescent proteins that colocalize with cytochrome c under normal conditions but show altered distribution upon apoptosis induction [6]. For instance, researchers have developed stable cell lines expressing mitochondria-targeted GFP (mitoGFP) that demonstrates even distribution throughout the mitochondrial network, enabling quantitative analysis of mitochondrial morphology and biomass [6]. Using confocal microscopy and 3D image analysis, such systems can detect increases in mitochondrial volume, surface area, and number associated with mitochondrial biogenesis, as well as changes in mitochondrial morphology during early apoptosis [6].
The construction of such reporter lines has been greatly facilitated by CRISPR/Cas9-mediated homology-directed repair (HDR) in human pluripotent stem cells (hPSCs), which allows for precise integration of reporter genes into specific genomic loci [8]. This technique enables the generation of robust reporter cell lines that maintain pluripotency while expressing fluorescent markers under the control of endogenous regulatory elements, providing powerful tools for studying cytochrome c dynamics and apoptotic signaling during differentiation and disease modeling.
Protocol: Generating Cytochrome C-GFP Reporter Cell Lines
Reporter Construct Design and Assembly
This protocol provides a detailed methodology for generating cytochrome c-GFP reporter cell lines to monitor the intrinsic apoptotic pathway in live cells, adapted from established reporter generation strategies [6] [8].
Materials:
- High-quality hPSCs (hESC lines/hiPSCs) without spontaneous differentiation
- Gibson Assembly Master Mix (commercial or prepared as described below)
- KOD Xtreme Hot Start DNA Polymerase
- BbsI-HF and SmaI restriction enzymes
- QIAquick Gel Extraction and QIAprep Spin Miniprep Kits
- Primers designed using Primer3
- Matrigel hESC-Qualified Matrix
- StemFlex Medium supplemented with CloneR
- Accutase or 0.5 mM EDTA for passaging
- P3 Primary Cell 4D-Nucleofector X Kit L
Gibson Assembly Reagent Preparation:
- Prepare 5× Isothermal Buffer by combining:
- 0.75 g PEG-8000 (25% final)
- 1.5 mL 1 M Tris-HCl pH 7.5 (500 mM final)
- 75 μL 2 M MgCl₂ (50 mM final)
- 150 μL 1 M DTT (50 mM final)
- 30 μL each of 100 mM dATP, dTTP, dCTP, dGTP (1 mM each final)
- 150 μL 100 mM NAD (5 mM final)
- Add ddH₂O to 3 mL final volume
- Aliquot (80 μL) and store at -20°C for up to 6 months
- Prepare Gibson Assembly Master Mix by combining:
- 80 μL 5× Isothermal Buffer
- 0.16 μL T5 Exonuclease (10 U/μL)
- 5 μL Phusion DNA Polymerase (2 U/μL)
- 40 μL Taq DNA ligase (40 U/μL)
- 174.84 μL ddH₂O
- Aliquot (15 μL) and store at -20°C for up to 6 months
Reporter Construct Assembly:
- Design a donor plasmid containing:
- GFP sequence with mitochondrial targeting signal
- Caspase-3 cleavage site (DEVD) for apoptosis detection
- Homology arms (approximately 800 bp) targeting the safe harbor locus
- Selection marker (e.g., puromycin resistance)
- Amplify GFP cassette and homology arms using KOD Xtreme Hot Start DNA Polymerase
- Digest vector backbone with BbsI-HF and SmaI
- Purify all fragments using QIAquick Gel Extraction Kit
- Assemble using Gibson Assembly Master Mix (50°C for 60 minutes)
- Transform into competent E. coli and select on carbenicillin plates
- Verify correct assembly by colony PCR and Sanger sequencing
Cell Culture and Nucleofection
Cell Culture Preparation:
- Coat culture plates with Matrigel (1:100 dilution in DMEM/F12)
- Use 1 mL/well for 6-well plates, 0.5 mL/well for 12-well plates
- Incubate at 37°C for at least 1 hour before use
- Culture hPSCs in StemFlex Medium at 37°C, 5% CO₂
- Passage cells at 70-80% confluence using 0.5 mM EDTA or Accutase
- For single-cell cloning, use StemFlex Medium supplemented with 1× CloneR
CRISPR/Cas9 Nucleofection:
- Design sgRNA targeting safe harbor locus (e.g., AAVS1)
- Prepare ribonucleoprotein (RNP) complex by mixing:
- 10 μg Cas9 protein
- 5 μg sgRNA
- Incubate at room temperature for 10 minutes
- Harvest 1×10⁶ hPSCs using Accutase
- Resuspend cells in 100 μL P3 Primary Cell Nucleofector Solution
- Add RNP complex and 10 μg donor plasmid to cell suspension
- Nucleofect using 4D-Nucleofector with program CA-137
- Immediately transfer cells to Matrigel-coated plates with StemFlex Medium + CloneR
- After 48 hours, begin puromycin selection (0.5 μg/mL) for 7-10 days
Clone Screening and Validation
Single-Cell Clone Isolation:
- After puromycin selection, harvest cells as single cells using Accutase
- Plate at clonal density (500-1000 cells/10 cm dish) in StemFlex + CloneR
- Monitor colony formation for 10-14 days
- Pick individual colonies using pipette tip under microscope
- Expand clones in 96-well plates followed by 24-well and 12-well plates
Reporter Validation:
- Confirm genomic integration by PCR screening across both homology arms
- Verify correct insertion site by Southern blotting
- Assess GFP expression and mitochondrial localization via confocal microscopy
- Confirm colocalization with mitochondrial markers (e.g., MitoTracker Deep Red) [6]
- Test reporter functionality by inducing apoptosis with 1 μM staurosporine or 1 mM H₂O₂ [7]
- Monitor fluorescence changes over time using live-cell imaging
- Validate apoptosis induction by parallel Western blotting for caspase-3 cleavage and cytochrome c release
Functional Characterization:
- Differentiate validated reporter lines into relevant cell types
- Treat with apoptotic inducers (e.g., staurosporine, H₂O₂) in concentration- and time-dependent manner
- Monitor fluorescence intensity changes (time-lapse imaging for 24-48 hours)
- Quantify mitochondrial morphology parameters (volume, surface area, Feret ratio) [6]
- Assess cytochrome c release via subcellular fractionation and Western blotting
- Correlate fluorescence changes with biochemical apoptosis markers (caspase activation, PARP cleavage)
Diagram 2: Cytochrome C-GFP Reporter Generation Workflow. This diagram outlines the three-phase protocol for creating and validating reporter cell lines.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Cytochrome C Apoptosis Studies
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Apoptosis Inducers | Staurosporine, H₂O₂, AICAR | Activate intrinsic apoptotic pathway; AMPK activation [6] [7] |
| Caspase Substrates | DEVD-based fluorogenic substrates, Caspase-cleavable GFP | Detect and quantify caspase activity [7] |
| Mitochondrial Dyes | MitoTracker Deep Red, TMRE | Visualize mitochondria; measure membrane potential [6] |
| CRISPR/Cas9 Components | Cas9 protein, sgRNAs, Donor plasmids | Genome editing for reporter insertion [8] |
| Cell Culture Supplements | CloneR, StemFlex Medium, Matrigel | Enhance single-cell survival; maintain pluripotency [8] |
| Detection Antibodies | Anti-cytochrome c, Anti-cleaved caspase-3, Anti-PARP | Confirm apoptosis by Western blot, immunofluorescence |
| Fluorescent Proteins | mitoGFP, EGFP with caspase sites | Live-cell apoptosis reporting; localization studies [6] [7] |
| Selection Agents | Puromycin, G418 | Select for successfully transfected cells [8] |
GFP as a Quantitative Reporter of Gene Expression in Eukaryotic Cells
The Green Fluorescent Protein (GFP) has revolutionized molecular and cellular biology by enabling real-time visualization of gene expression and protein localization in living cells. Beyond its well-established role as a localization marker, GFP serves as a robust quantitative reporter of gene expression when properly validated and implemented. Research has demonstrated that fluorescence intensity from GFP correlates directly with underlying transcriptional activity, making it a powerful tool for investigating dynamic biological processes in eukaryotic systems [9].
When engineering reporter cell lines, the fundamental principle is that GFP fluorescence, measured by techniques such as flow cytometry or quantitative fluorescence microscopy, increases in direct proportion to both GFP gene copy number and mRNA abundance in individual eukaryotic cells [9]. This linear relationship enables researchers to monitor promoter activity, study gene regulation, and screen for pharmacological modulators of signaling pathways with high precision in real-time.
The application of GFP-based reporters has expanded to diverse research areas, including the study of cytochrome c dynamics during apoptosis, tracking cell-cell interactions in metastatic niches, and monitoring stress response pathways [7] [10] [11]. This protocol provides comprehensive methodologies for implementing GFP as a quantitative reporter, with particular emphasis on applications relevant to cytochrome c biology and drug discovery research.
Key Principles and Validation
Establishing Quantitative Relationships
For GFP to function as a reliable quantitative reporter, specific validation steps must be performed to establish the relationship between fluorescence intensity and gene expression parameters:
- Gene Copy Number Correlation: Deliver increasing copies of the GFP gene to cells using viral vectors and demonstrate that fluorescence intensity increases proportionally with gene copy number [9].
- mRNA Abundance Correlation: Perform parallel measurements of GFP fluorescence intensity and GFP mRNA levels (via qPCR) across different expression conditions to confirm direct correlation [9].
- Promoter Responsiveness: Validate that GFP expression accurately reports induction from inducible promoter systems (e.g., tetracycline-responsive or chemical-inducible promoters) [9].
Critical Experimental Considerations
Several technical factors must be controlled to ensure accurate quantification:
- Signal-to-Noise Optimization: High background fluorescence can compromise quantification accuracy. Implement strategies to improve signal-to-noise ratio during image acquisition [12].
- Proper Threshold Setting: Automated analysis requires careful threshold setting to distinguish true signal from background noise, particularly when GFP expression varies significantly between cells [12].
- Reference Markers: For cellular segmentation and masking, use complementary markers (e.g., membrane or cytoplasmic stains) that don't interfere with GFP detection rather than relying solely on the GFP signal itself [12].
Quantitative Applications and Reporter Systems
Apoptosis Reporting via Caspase-Responsive GFP
A mutagenesis-based approach has been developed to create GFP reporters for monitoring apoptosis through caspase-3 activation:
- Design Principle: Caspase-3 cleavage motifs (DEVD) are inserted into specific positions within the GFP protein structure based on structural positioning and hydrophilicity considerations [7].
- Mechanism: The inserted cleavage motif renders GFP fluorescently inactive upon caspase-3 activation, creating a "bright-to-dark" reporter system [7].
- Performance: This design demonstrates greater sensitivity compared to dark-to-bright apoptosis reporters and functions in a time- and concentration-dependent manner with apoptosis inducers like staurosporine and H₂O₂ [7].
- Applications: The system is adaptable to various cellular and organismal models without requiring additional peptides, making it valuable for drug screening applications [7].
Real-Time Monitoring of Signaling Pathways
Fluorescent reporters enable real-time tracking of pathway activation in living cells:
- CAFLUX HepG2 System: This reporter cell line expresses a histone H2B-GFP fusion protein under control of a dioxin-responsive cytochrome P450 1A1 (CYP1A1) promoter containing multiple dioxin-responsive elements (DREs) [13].
- Sensitivity: The system detects aryl hydrocarbon receptor (AhR) agonists with high sensitivity, achieving limits of detection of approximately 0.01 pM for TCDD and 0.1 pM for benzo[a]pyrene [13].
- Validation: Reporter activity correlates with endogenous CYP1A1 mRNA expression, confirming that GFP signals accurately reflect native transcriptional responses [13].
Simultaneous Monitoring of Multiple Pathways
Advanced reporter systems enable parallel tracking of interconnected cellular processes:
- Apoptosis-Autophagy Reporter: A single cell line can be engineered to express EGFP-LC3 (for autophagy monitoring) and RFP-tagged cytochrome c (for apoptosis monitoring), allowing simultaneous real-time imaging of both processes [14].
- Temporal Resolution: This approach reveals the sequence of cellular events, showing that rapid cell death often proceeds without autophagy induction, while cells with progressing autophagy survive longer without caspase-3 activation under treatment conditions [14].
Table 1: Quantitative Performance of GFP Reporter Systems
| Reporter Type | Detection Limit | Dynamic Range | Key Applications |
|---|---|---|---|
| Constitutive GFP | N/A | Proportional to gene copy number [9] | Promoter activity studies, transfection efficiency |
| Apoptosis Reporter (DEVD-GFP) | Caspase-3 activation | Time- and concentration-dependent [7] | Drug screening, mechanistic studies of cell death |
| CAFLUX HepG2 (H2B-GFP) | 0.01 pM TCDD [13] | Dose-dependent nuclear fluorescence [13] | AhR pathway activation, toxicological evaluation |
| sGRAPHIC | Single-cell interactions [11] | High-efficiency neighboring cell labeling [11] | Cell-cell interactions in metastatic niches |
Reporter System Construction and Implementation
Vector Design and Assembly
The construction of effective GFP reporter systems requires careful vector design:
- Promoter Selection: Choose promoters based on the biological process under investigation. Constitutive promoters (e.g., CMV) provide strong expression, while inducible or response element-containing promoters enable monitoring of specific pathways [13].
- Fluorophore Optimization: Enhanced GFP variants (e.g., S65T) provide improved brightness and photostability. For nuclear localization, create H2B-GFP fusion constructs [13] [15].
- Response Elements: Incorporate multiple copies of specific response elements (e.g., DREs for AhR signaling) upstream of the minimal promoter to enhance sensitivity and specificity [13].
Cell Line Engineering
Stable reporter cell lines are essential for consistent quantitative measurements:
- Lentiviral Transduction: Use lentiviral vectors (e.g., pFUGW backbone) for efficient gene delivery and stable integration [13].
- Selection and Cloning: Apply antibiotic selection followed by single-cell cloning to establish homogeneous reporter populations [14].
- Validation: Screen multiple clones for optimal response characteristics, including low basal expression and high inducibility [13] [14].
Advanced Labeling Strategies
Innovative approaches expand GFP applications in complex biological systems:
- sGRAPHIC System: This secretory split-GFP system uses glycosylphosphatidylinositol-anchored reconstitution-activated proteins to highlight intercellular connections. One cell population expresses a secretory C-terminal GFP fragment (sC-GR), while neighboring cells express a membrane-anchored N-terminal fragment (N-GR). GFP reconstitution occurs when cells are in proximity, efficiently labeling interacting cells [11].
- Advantages: sGRAPHIC demonstrates higher labeling efficiency compared to previous systems like Cherry-niche, enabling comprehensive analysis of cell-cell interactions in metastatic niches and other complex environments [11].
Experimental Protocols
Protocol 1: Flow Cytometric Quantification of GFP Expression
Purpose: To quantitatively measure GFP reporter expression at single-cell resolution.
Materials:
- GFP-expressing eukaryotic cells
- Appropriate culture medium
- Flow cytometer with 488-nm excitation laser and 530/30-nm emission filter
- Phosphate-buffered saline (PBS)
- Trypsin-EDTA solution (for adherent cells)
- Fixative (optional: 4% paraformaldehyde in PBS)
Procedure:
- Culture GFP-expressing cells under experimental conditions.
- Harvest cells at 70-80% confluence:
- For adherent cells: Wash with PBS, trypsinize, and resuspend in complete medium.
- For suspension cells: Collect by centrifugation and resuspend in PBS.
- Filter cells through a 35-40 μm mesh to obtain single-cell suspension.
- Keep samples on ice and protect from light.
- Acquire data on flow cytometer:
- Set threshold on forward scatter to exclude debris.
- Collect minimum of 10,000 events per sample.
- Use untransfected cells to set GFP negative population.
- Analyze data using flow cytometry software:
- Gate on viable cells based on forward and side scatter.
- Measure fluorescence intensity in GFP channel.
- Export geometric mean fluorescence intensity or median fluorescence intensity for statistical analysis.
Validation: Confirm linearity by demonstrating proportional increase in fluorescence with increasing gene copy number [9].
Protocol 2: Quantitative Image Analysis of GFP Expression
Purpose: To measure GFP fluorescence intensity in individual cells using ImageJ/Fiji.
Materials:
- Fluorescence microscopy images of GFP-expressing cells
- Fiji/ImageJ software
- Computer with sufficient RAM for image processing
Procedure:
- Open fluorescence image in Fiji/ImageJ.
- Set measurement parameters:
- Go to Analyze > Set Measurements
- Select "Area", "Integrated Density", and "Mean Gray Value"
- Select individual cells using appropriate selection tools:
- For regular shapes: Use rectangle or oval selections
- For irregular shapes: Use freehand selection tool
- Measure cell fluorescence:
- Select each cell and press "M" to record measurements
- Include background regions without cells
- Export data to spreadsheet software.
- Calculate Corrected Total Cell Fluorescence (CTCF):
- CTCF = Integrated Density - (Area of selected cell × Mean fluorescence of background readings) [16]
- Account for cell size variations, as rounded cells may show artificially high fluorescence due to concentrated signal in smaller area [16].
Troubleshooting:
- High background: Improve acquisition conditions or use background subtraction algorithms
- Variable expression: Ensure homogeneous reporter expression through clonal selection [14]
- Segmentation errors: Use complementary markers for cell masking rather than GFP signal alone [12]
Protocol 3: Cytochrome c-GFP Reporter Assay for Apoptosis Monitoring
Purpose: To detect sublethal cytochrome c release and persister cell formation using GFP-based reporters.
Background: Sublethal mitochondrial outer membrane permeabilization (MOMP) with limited cytochrome c release can generate drug-tolerant persister cells without triggering full apoptosis, mediated through HRI kinase activation and integrated stress response [10].
Materials:
- GFP reporter cells with cytochrome c localization signal
- Apoptosis inducers (e.g., BH3 mimetics: ABT-737, S63845)
- Control compounds (e.g., DMSO vehicle)
- Live-cell imaging setup with environmental control
- Image analysis software
Procedure:
- Seed cells in imaging-compatible plates at appropriate density.
- Treat with BH3 mimetics at predetermined sublethal concentrations:
- Include positive controls (full apoptosis inducers) and negative controls
- Use BAX/BAK/BOK knockout cells as specificity controls [10]
- Perform time-lapse imaging:
- Acquire images every 15-30 minutes for 24-48 hours
- Maintain physiological conditions (37°C, 5% CO₂)
- Analyze images for:
- Cytochrome c-GFP localization (mitochondrial vs. cytoplasmic)
- Morphological changes associated with apoptosis
- Persister cell identification (surviving cells with partial cytochrome c release)
- Correlate cytochrome c release patterns with:
- ATF4 synthesis (integrated stress response marker)
- Cell viability outcomes (persistence vs. death)
- Drug holiday responsiveness [10]
Validation: Confirm persister phenotype by demonstrating transient drug tolerance and increased metastatic potential in vivo [10].
Table 2: Research Reagent Solutions for GFP Reporter Studies
| Reagent/Cell Line | Function/Application | Key Features |
|---|---|---|
| pFUGW Lentiviral Vector [13] | Reporter construct delivery | Stable integration, high transduction efficiency |
| CAFLUX HepG2 Reporter [13] | AhR pathway activation studies | H2B-GFP nuclear localization, sensitive DRE detection |
| DEVD-Mutated GFP [7] | Apoptosis detection via caspase-3 | Bright-to-dark system, high sensitivity |
| sGRAPHIC Components [11] | Cell-cell interaction mapping | Split-GFP reconstitution, efficient neighboring cell labeling |
| EGFP-LC3/RFP-Smac [14] | Simultaneous autophagy-apoptosis monitoring | Real-time imaging of both processes in single cells |
Signaling Pathways and Experimental Workflows
Cytochrome c-Mediated Persister Formation Pathway
sGRAPHIC Cell-Cell Interaction Detection Workflow
AhR Signaling Reporter Pathway
Data Analysis and Interpretation
Quantitative Measurements and Normalization
Accurate quantification requires appropriate normalization strategies:
- Background Subtraction: Always subtract background fluorescence from control regions or untransfected cells.
- Cell Number Normalization: For population measurements, normalize to cell number or total protein content.
- Reference Standards: Include fluorescence standards or reference cells with known GFP expression levels for cross-experiment comparisons.
- Temporal Normalization: For time-course experiments, normalize to baseline measurements or control conditions.
Addressing Technical Variability
Several approaches minimize technical variability in GFP quantification:
- Clonal Selection: Use single-cell clones to ensure homogeneous reporter expression [14].
- Multiple Passages: Assess reporter stability across multiple cell passages to identify potential silencing or variegation.
- Control Elements: Include constitutive fluorescent reporters (e.g., with different emission spectra) as internal controls for transduction efficiency and cell number.
- Replicate Measurements: Perform sufficient biological and technical replicates to account for cell-to-cell variability.
Applications in Drug Discovery and Development
GFP reporter systems provide powerful platforms for pharmaceutical research:
- High-Content Screening: GFP-based assays enable automated screening of compound libraries for modulators of specific pathways [7] [13].
- Toxicology Assessment: Reporter cells like CAFLUX HepG2 allow evaluation of compound toxicity through pathway-specific activation [13].
- Mechanistic Studies: Real-time monitoring of biological processes reveals compound mechanism of action and kinetics [7] [14].
- Persister Cell Research: Cytochrome c-GFP reporters help identify strategies to overcome drug tolerance in cancer therapy [10].
The integration of GFP reporter systems with advanced imaging and computational analysis continues to expand our understanding of eukaryotic cell biology, providing unprecedented insights into dynamic cellular processes and accelerating therapeutic development.
The construction of a cytochrome c GFP reporter cell line is a powerful tool for visualizing mitochondrial dynamics and studying apoptosis in live cells. Cytochrome c plays a dual role in cellular processes: it is essential for the electron transport chain within mitochondria and is a key signaling molecule when released into the cytoplasm during apoptosis. Tagging cytochrome c with GFP allows for the real-time monitoring of its localization. However, the primary challenge lies in performing this tagging without disrupting its native structure and function. This application note details the principles and protocols for creating functional cytochrome c-GFP fusions, framed within the broader context of reporter cell line construction for drug discovery and basic research.
Core Design Principles for a Functional Cytochrome c-GFP Fusion
Creating a functional fusion construct requires careful consideration of several factors to ensure the tagged protein behaves like its wild-type counterpart. The following principles are critical for success.
Fusion Protein Topology and Linker Design
The placement of the GFP moiety and the linker sequence connecting it to cytochrome c are crucial for preserving function.
- Terminal Fusion: GFP is typically fused to the N- or C-terminus of cytochrome c. The terminal regions of cytochrome c are often more tolerant of modifications than the structured core, which contains the heme-binding site (CXXCH motif).
- Flexible Linker: A glycine-serine-rich flexible linker (e.g., GGGGS)³ should be used to connect cytochrome c and GFP. This linker provides spatial separation, reducing the risk of steric hindrance that could interfere with cytochrome c's interactions with its binding partners, such as cytochrome c₁ and Apaf-1.
Preservation of Critical Functional Domains
The integrity of specific domains and residues in cytochrome c is non-negotiable for its function.
- Heme-Binding Motif: The conserved CXXCH motif is essential for the covalent attachment of the heme group [17]. Any modification that alters this motif will abolish the protein's electron-carrying capability.
- Electron Transfer Surface: The surface of cytochrome c that interacts with Complex III and Complex IV must remain accessible. The GFP fusion should not occlude this interface.
Consideration of GFP Folding and Maturation
GFP must fold and form its chromophore to fluoresce. This process is temperature-dependent and can be relatively slow [18]. The fusion construct should be designed and expressed under conditions that permit proper GFP maturation without aggregating or interfering with the localization of cytochrome c to the mitochondrial intermembrane space.
Experimental Validation of Cytochrome c-GFP Function
Once a fusion construct is designed, its function must be rigorously validated through the following key experiments. The workflow for this validation is summarized in the diagram below.
Protocol 1: Validating Mitochondrial Localization
Objective: To confirm that the cytochrome c-GFP fusion protein correctly localizes to the mitochondrial intermembrane space.
Materials:
- Cells transfected with cytochrome c-GFP construct
- MitoTracker Red CMXRos (or similar dye)
- Confocal fluorescence microscope
Method:
- Culture transfected cells on glass-bottom culture dishes.
- Following the manufacturer's protocol, stain live cells with MitoTracker Red (50-100 nM) for 15-30 minutes at 37°C.
- Wash cells with pre-warmed culture medium.
- Image live cells using a confocal microscope. Excite GFP at ~488 nm and MitoTracker Red at ~579 nm.
- Analyze the images for colocalization of the green (cytochrome c-GFP) and red (mitochondria) signals, which indicates correct targeting.
Protocol 2: Assessing Electron Transport Chain Function
Objective: To verify that the cytochrome c-GFP fusion can functionally replace endogenous cytochrome c in the electron transport chain.
Materials:
- Yeast Strain: Saccharomyces cerevisiae null for endogenous cytochrome c (Δcyc1 Δcyc7) [19].
- Growth media: Rich fermentable medium (YPD) and non-fermentable medium (YPGlycerol or YPEthanol).
- Spectrophotometer or plate reader for measuring cell growth (OD600).
Method:
- Transform the cytochrome c-GFP construct into the yeast null strain. Include controls: empty vector (negative control) and wild-type cytochrome c (positive control).
- Plate serial dilutions of transformed yeast cells onto solid YPD and YPGlycerol media.
- Incubate plates at 30°C for 2-5 days and observe growth.
- Interpretation: Functional cytochrome c is required for respiration. Growth on the non-fermentable carbon source (glycerol) indicates that the cytochrome c-GFP fusion can support respiratory growth, proving its role in electron transport [19]. Quantitative growth data can be obtained by monitoring OD600 in liquid culture.
Table 1: Quantitative Validation of Electron Transport Function
| Construct Expressed | Growth on YPD (Fermentation) | Growth on YPGlycerol (Respiration) | Respiratory Competence |
|---|---|---|---|
| Empty Vector | Positive | No Growth | Not Competent |
| Wild-type Cytochrome c | Positive | Positive | Fully Competent |
| Cytochrome c-GFP | Positive | Positive [19] | Fully Competent |
Protocol 3: Testing Apoptotic Release
Objective: To determine if the cytochrome c-GFP fusion is released from mitochondria upon induction of apoptosis.
Materials:
- Stable reporter cell line expressing cytochrome c-GFP
- Apoptosis inducers (e.g., Staurosporine, UV irradiation)
- Fluorescence microscope with time-lapse capability
Method:
- Seed reporter cells and allow them to adhere.
- Induce apoptosis by adding a chemical inducer (e.g., 1 µM Staurosporine) or by applying UV irradiation.
- Immediately begin time-lapse imaging using a fluorescence microscope to monitor the localization of the GFP signal over time.
- Interpretation: In healthy cells, the signal will be punctate, reflecting its mitochondrial localization. Upon apoptosis induction, a diffuse, cytoplasmic GFP signal will appear as cytochrome c is released from the mitochondria. Note that the large size of the GFP moiety (~27 kDa) may sterically hinder its release through certain pores, as observed in yeast models with Bax expression [19] [20]. This makes the reporter ideal for visualizing initial release events but may not perfectly mimic the kinetics of untagged cytochrome c.
The Scientist's Toolkit: Key Research Reagents
Table 2: Essential Reagents for Cytochrome c Reporter Line Development
| Reagent / Tool | Function / Role in Experiment | Key Consideration |
|---|---|---|
| Cytochrome c-Null Yeast | In vivo model for testing electron transport function of fusion constructs [19]. | Provides a clean background without endogenous cytochrome c interference. |
| Bax-Expressing Plasmid | Induces mitochondrial outer membrane permeabilization to test cytochrome c release [19]. | A standard tool for probing apoptotic function. |
| MitoTracker Dyes | Live-cell staining of mitochondria to confirm fusion protein localization. | Choose a dye with a fluorescence spectrum distinct from GFP (e.g., red fluorescent). |
| Kozak Sequence | Regulatory element added to the 5' end of the gene to enhance translation initiation in mammalian cells [21]. | Optimizes expression levels in the final reporter cell line. |
| Leader Sequence | A signal peptide that can be fused to direct protein trafficking and improve folding [21]. | May enhance mitochondrial targeting efficiency. |
Application in a Broader Research Context
A functional cytochrome c-GFP reporter cell line is a cornerstone for advanced research, enabling:
- Drug Discovery: Screening for compounds that modulate the mitochondrial apoptosis pathway, either as pro-apoptotic anti-cancer agents or as cytoprotective agents for neurodegenerative diseases. The release of cytochrome c-GFP serves as a direct, quantifiable readout.
- Toxicology Studies: Assessing if new chemical entities induce mitochondrial toxicity by triggering premature cytochrome c release.
- Basic Biology: Investigating the dynamics of mitochondrial membrane permeabilization in real-time and the cross-talk between metabolic state and cell death signaling.
The relationships between cytochrome c's functions and its applications as a reporter are illustrated below.
The successful design of a cytochrome c-GFP reporter hinges on a principled approach that respects the protein's structural and functional constraints. By employing a terminal fusion strategy with a flexible linker and rigorously validating mitochondrial localization, electron transport capability, and apoptotic release, researchers can generate a robust and powerful tool. This protocol provides a roadmap for creating a reliable reporter system that will yield physiologically relevant insights into cell death and metabolism, directly serving the needs of scientists in basic research and drug development.
The study of cellular dynamics, particularly in the context of critical processes like apoptosis, has been revolutionized by advanced live-cell monitoring technologies. Traditional endpoint assays, while valuable, provide only static snapshots of dynamic biological processes, potentially missing transient events and introducing artifacts from fixation and staining procedures. The development of cytochrome C GFP reporter cell lines represents a significant innovation, enabling researchers to monitor mitochondrial outer membrane permeabilization (MOMP)—a key commitment step in apoptosis—in real-time, within living cells, and without the need for disruptive staining protocols. This application note details the substantial advantages these advanced methodologies offer over traditional assays, supported by quantitative data and detailed protocols for implementation.
Modern live-cell imaging systems now provide the ability to regulate environmental conditions precisely, maintaining health and viability of cells while monitoring molecular and cellular dynamics from single cell to organismal level [22]. This technological advancement, coupled with novel reporter systems and dyes, has created unprecedented opportunities for studying complex biological mechanisms in their native physiological state.
Key Advantages Over Traditional Methods
True Physiological Relevance with Minimal Perturbation
Live-cell monitoring maintains cells in their native state, avoiding the artifacts introduced by fixation and staining protocols that inherently disrupt cellular architecture and function.
- Preservation of Native Biology: Fixation and staining protocols inherently stress cells and disrupt their native state and architecture [23]. In contrast, live-cell approaches maintain physiological conditions, allowing observation of processes as they naturally occur.
- Reduced Cellular Perturbation: Technologies like ChromaLIVE, a non-toxic live cell painting dye, demonstrate biological inertness, with cells cultured in its presence for weeks remaining healthy and unperturbed [23]. This ensures that profiling data reflect the true biological response rather than reactions to assay conditions.
Enhanced Kinetic Profiling for Comprehensive Insight
The ability to capture dynamic, time-resolved data enables detection of transient biological events that would be missed in fixed-timepoint assays.
- Detection of Transient Phenotypes: Live cell painting with ChromaLIVE can reveal time-to-onset of drug effects and discriminate between fast-acting and slow-acting compounds [23]. This dynamic view provides biological information simply not accessible through conventional fixed-point methods.
- Identification of Critical Windows: By enabling multiple timepoint acquisitions, live-cell monitoring detects broader windows of bioactivity—early, late, and transient—significantly improving bioactivity detection compared to single fixed timepoints [23].
Superior Sensitivity and Specificity
Advanced live-cell methods demonstrate remarkable performance in distinguishing true biological signals from background noise.
Table 1: Performance Comparison of Live-Cell vs. Traditional Methods
| Method | Sensitivity Advantage | Key Performance Metrics | Application Benefits |
|---|---|---|---|
| Live Cell Painting with Acridine Orange | Detects cellular responses at concentrations up to 40× lower than conventional MTT assays [24] | Clusters drugs based on induced phenotypes; captures texture, intensity, and granularity features [24] | Enhanced early-stage drug discovery and toxicity assessment |
| ChromaLIVE Live Cell Painting | Improved bioactivity detection through multiple timepoints [23] | Similar or higher mean Average Precision (mAP) scores compared to cell painting [23] | Optimal balance between false positives and false negatives in screening |
| CAFLUX Reporter Systems | Dose-dependent nuclear GFP fluorescence with detection limits of ~0.01 pM for TCDD [13] | Correlation with endogenous mRNA expression [13] | Sensitive monitoring of pathway activation in real-time |
Streamlined Workflows and Enhanced Compatibility
Live-cell methods often feature simplified protocols while supporting more complex biological models.
- Effortless Workflows: Live cell painting with ChromaLIVE requires just one mix-and-read dye step with zero wash steps, unlike traditional cell painting's multi-step, labor-intensive process involving fixation and multiple washes [23]. This streamlined approach saves significant time and resources while reducing experimental variability.
- Compatibility with Sensitive Models: The gentle, non-toxic nature of modern live-cell dyes makes them ideal for delicate but physiologically relevant models including patient-derived cells, iPSCs, neurons, and 3D organoids [23]. These systems are often incompatible with traditional fixation and staining protocols.
Cytochrome C GFP Reporter Construction and Applications
Reporter Design Principles for Apoptosis Monitoring
The construction of cytochrome C GFP reporters leverages molecular principles similar to other successfully implemented reporter systems, adapting them to specifically monitor mitochondrial apoptosis.
- Fluorescent Protein Selection: For optimal performance, selection of appropriate fluorescent proteins is critical. Recent comparative studies indicate that mStayGold variants stand out as far superior to EGFP or mEmerald with a functional lifetime at least 8-10-fold longer [25]. This enhanced photostability is particularly valuable for long-term time-lapse imaging of apoptotic processes.
- Subcellular Targeting: Effective cytochrome C reporters require precise subcellular targeting to mitochondria. This is typically achieved by incorporating mitochondrial targeting sequences (MTS) derived from proteins such as cytochrome C oxidase subunit VIII to ensure proper localization of the fluorescent protein to the mitochondrial intermembrane space.
- Cleavage-Sensitive Design: Similar to the apoptosis reporter described in [7], which uses caspase-3 cleavage motifs, cytochrome C reporters can be engineered with cleavage sites that respond to apoptotic activation. Upon apoptosis induction, cytochrome C release from mitochondria can be tracked through changes in fluorescence localization or intensity.
Diagram Title: Cytochrome C GFP Reporter Mechanism During Apoptosis
Experimental Protocol: Implementing Cytochrome C GFP Reporters
Protocol: Live-Cell Monitoring of Apoptosis Using Cytochrome C GFP Reporter Cell Lines
Background This protocol describes the implementation of cytochrome C GFP reporter cell lines for monitoring mitochondrial apoptosis in real-time. The approach enables kinetic assessment of compound-induced toxicity and mechanistic studies of cell death pathways without fixation or staining artifacts.
Materials and Reagents
- Cytochrome C GFP reporter cell line (constructed as detailed in Section 3.1)
- Appropriate cell culture medium and supplements
- 96-well black polystyrene microplates with μClear flat bottom (e.g., Greiner Bio-One, #655090) [26]
- Test compounds for apoptosis induction (e.g., Staurosporine, H2O2) [7]
- Positive control apoptosis inducers
- Live-cell imaging compatible medium (e.g., FluoroBrite DMEM) [26]
- Automated live-cell imaging system with environmental control (e.g., ImageXpress systems) [22]
Procedure
- Cell Culture and Plating
- Culture cytochrome C GFP reporter cells under standard conditions until approximately 80% confluency is reached [26].
- Detach cells using appropriate dissociation reagent and count viable cells using trypan blue exclusion.
- Seed 8 × 10² to 1 × 10³ viable cells per well in 96-well black μClear plates. To prevent edge effects, do not plate cells in peripheral wells; instead, fill these with sterile PBS [26].
- Allow plates to rest in a laminar flow hood for 20 minutes to ensure even cell adherence.
- Incubate plates for 24 hours in a humidified incubator (37°C, 5% CO₂) to allow complete cell attachment and recovery.
Compound Treatment and Experimental Setup
- Prepare serial dilutions of test compounds in live-cell imaging compatible medium.
- Carefully aspirate culture medium from wells and add compound solutions, including appropriate vehicle controls and positive controls for apoptosis induction.
- For kinetic studies, include multiple replicate plates for fixed-timepoint validation assays if required.
Live-Cell Image Acquisition
- Place plate into automated imaging system with integrated environmental control (maintaining 37°C, 5% CO₂, and humidity).
- Configure acquisition settings for time-lapse imaging:
- Acquisition intervals: Every 15-30 minutes for short-term assays (0-8h); every 1-2 hours for longer-term assays (8-72h)
- Total acquisition time: 24-72 hours depending on experimental objectives
- Multiple imaging positions per well to ensure adequate cell numbers for statistical analysis
- GFP channel for reporter signal (EX 469/35 nm, EM 525/39 nm) [26]
- Brightfield or phase contrast for morphological assessment
- Initiate automated time-lapse acquisition.
Image Analysis and Data Processing
- Use image analysis software (e.g., CellProfiler, CellReporterXpress) for automated cell segmentation and tracking [26] [22].
- Quantify cytochrome C release events by measuring changes in GFP fluorescence distribution:
- Mitochondrial-to-cytosolic fluorescence ratio
- Timing of fluorescence redistribution
- Percentage of cells exhibiting cytochrome C release over time
- Extract kinetic parameters:
- Time to initial cytochrome C release
- Rate of apoptosis propagation through cell population
- EC₅₀ values for compound-induced apoptosis
Troubleshooting Notes
- Optimize expression levels of cytochrome C GFP reporter to avoid artifacts from overexpression.
- Validate system response with known apoptosis inducers at multiple concentrations.
- Include control for phototoxicity by comparing results with minimal light exposure conditions.
Advanced Research Applications
Integration with High-Content Screening Platforms
The cytochrome C GFP reporter system is particularly valuable in high-content screening (HCS) environments where it enables multiparametric analysis of apoptotic responses across diverse compound libraries.
- Multiparametric Phenotypic Profiling: Live cell painting with acridine orange has demonstrated capability to cluster drugs based on induced phenotypes in hepatocytes, capturing features related to texture, intensity, and granularity that are crucial for determining compound profiles [24]. Similar approaches can be integrated with cytochrome C GFP reporters for enhanced mechanistic insights.
- High-Throughput Compatible Workflows: The streamlined, one-step protocols developed for live cell painting align well with high-throughput screening requirements [23]. These approaches can be adapted for cytochrome C GFP reporter assays to enable large-scale compound profiling.
Three-Dimensional Model Systems
Advanced live-cell monitoring techniques show particular utility in complex 3D model systems where traditional endpoint assays often fail.
- Enhanced Compatibility with 3D Cultures: The non-toxic nature of modern live-cell dyes makes them ideal for delicate 3D organoids and spheroids, preserving their intricate architecture while enabling effective profiling of cellular responses [23]. This compatibility extends to cytochrome C GFP reporter systems, allowing apoptosis monitoring in more physiologically relevant 3D contexts.
- Deep Tissue Imaging Applications: Technologies like sGRAPHIC have demonstrated efficient labeling of cell-cell interactions in deep tissues [11], suggesting potential for adapting similar approaches to monitor cytochrome C release in complex tissue models and in vivo settings.
Essential Research Reagent Solutions
Table 2: Key Research Reagents for Live-Cell Apoptosis Monitoring
| Reagent/Category | Specific Examples | Function and Application | Performance Notes |
|---|---|---|---|
| Live-Cell Dyes | ChromaLIVE [23], Acridine Orange [24] | Multiparametric cellular staining in live cells | Non-toxic, compatible with long-term imaging; AO provides two-channel readout of nucleic acids and acidic compartments |
| Advanced FPs | mStayGold variants [25], mCherry [11] | Genetically encoded reporters for long-term imaging | mStayGold offers 8-10× longer functional lifetime vs EGFP; optimal for kinetic studies |
| Reporter Systems | FUCCI cell cycle indicators [27], sGRAPHIC [11] | Cell cycle monitoring and cell-cell interaction mapping | FUCCI distinguishes G1 (red) vs S/G2/M (green) phases; sGRAPHIC labels neighboring cells via split-GFP reconstitution |
| Detection Tools | CAFLUX HepG2 reporters [13], Apoptosis reporters [7] | Pathway-specific activation monitoring | CAFLUX enables real-time nuclear monitoring of AhR activation; caspase reporters detect apoptosis via cleavage |
| Instrumentation | ImageXpress systems [22], Cytation platforms [26] | Automated live-cell imaging and analysis | Integrated environmental control, high acquisition speeds, robust focusing for long-term assays |
Live-cell, dynamic, and dye-free monitoring methodologies represent a paradigm shift in cellular analysis, offering substantial advantages over traditional fixed-cell and endpoint assays. The development and implementation of cytochrome C GFP reporter cell lines exemplifies this progress, enabling researchers to capture the dynamic process of mitochondrial apoptosis in real-time without introducing artifacts from fixation or staining. The quantitative data presented demonstrates enhanced sensitivity, kinetic resolution, and physiological relevance achievable through these advanced approaches.
As live-cell technologies continue to evolve, integrating these methodologies into drug discovery and basic research pipelines will accelerate the identification of novel therapeutic candidates and deepen our understanding of fundamental biological processes. The protocols and reagents detailed in this application note provide researchers with practical guidance for implementing these powerful technologies in their own investigative workflows.
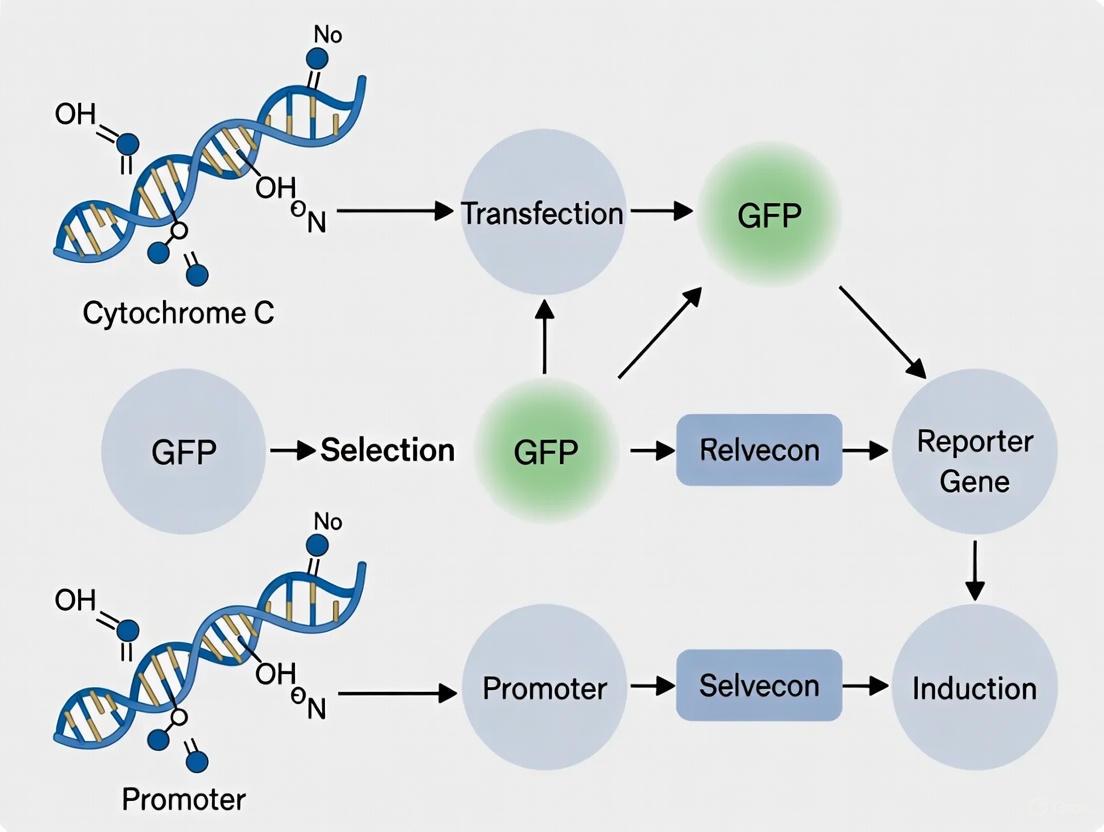
A Step-by-Step Protocol for Reporter Cell Line Construction and Use
Within the broader scope of constructing cytochrome C reporter cell lines, the design of the expression vector is a critical determinant of experimental success. The primary application of such a cell line is to act as a sensitive biosensor for apoptosis detection in research and drug development. This process is fundamentally linked to the release of cytochrome c from the mitochondrial intermembrane space into the cytosol, a committed step in the intrinsic apoptotic pathway [28]. A well-engineered cytochrome C-GFP fusion construct allows for the real-time, visual monitoring of this translocation event via fluorescence microscopy or flow cytometry. This Application Note provides a detailed protocol for the construction and validation of these essential research tools, leveraging the latest insights in mitochondrial biology and reporter design.
Scientific Background and Principle
Cytochrome c in Apoptosis
Cytochrome c is normally sequestered within the cristae of the mitochondrial intermembrane space (IMS), where it is electrostatically bound to the inner mitochondrial membrane (IMM) [28]. During apoptosis, mitochondrial outer membrane permeabilization (MOMP) occurs, facilitated by pores formed by proteins like BAX/BAK. Subsequently, the inner mitochondrial membrane undergoes remodeling, a process recently shown to be regulated by the tumor suppressor LACTB. This remodeling is crucial for the efficient release of cytochrome c into the cytosol [28]. Once in the cytosol, cytochrome c initiates the formation of the apoptosome, leading to caspase activation and execution of the cell death program.
Reporter Design Principle
The core principle of the reporter is to fuse the gene encoding cytochrome c to a Green Fluorescent Protein (GFP) variant. The localization of the fusion protein can then be tracked in live cells. In healthy cells, the mitochondrial pattern of GFP fluorescence will be observed, co-localizing with specific mitochondrial markers. Upon induction of apoptosis, the release of the cytochrome C-GFP fusion protein from the mitochondria results in a diffuse, cytosolic fluorescence pattern. This bright-to-dark transition for mitochondrial fluorescence, as opposed to dark-to-bright systems for cytosolic reporters, has been demonstrated to offer high sensitivity in detecting apoptotic events [7].
The following diagram illustrates this core principle and the downstream experimental workflow.
Vector Engineering and Component Selection
The functionality of the reporter is contingent upon the careful selection of each genetic element in the expression vector.
Core Vector Components
A standard vector backbone must include essential elements for replication and selection in the desired host (e.g., mammalian cells). Key components include an Origin of Replication (ORI) suitable for the production system (e.g., SV40 for mammalian), a resistance marker (e.g., puromycin, blasticidin, or G418) for stable cell line selection, and a Multiple Cloning Site (MCS) for inserting the expression cassette [29].
Promoter and Enhancer Selection
The choice of promoter dictates the strength and specificity of expression. For constitutive expression in a wide range of mammalian cells, the CMV (Cytomegalovirus) promoter is a robust choice [21]. The addition of an enhancer element can further boost transcription levels without directional constraints [29].
Regulatory Elements for Translation Efficiency
To ensure high-level protein synthesis, specific regulatory sequences should be incorporated upstream of the start codon:
- Kozak Sequence: A strong Kozak sequence (e.g., GCCACC) is crucial for efficient translation initiation in eukaryotic cells. Research in CHO cells has demonstrated that optimizing the Kozak sequence can increase recombinant protein expression by over 1.2-fold [21].
- Leader Sequence: Adding a leader peptide sequence can improve protein folding and trafficking. Studies have shown that a combination of Kozak and Leader sequences can synergistically enhance target protein yield by more than 2-fold [21].
Cytochrome C-GFP Fusion Design
The cytochrome c gene should be fused in-frame to the N-terminus of a bright GFP variant (e.g., EGFP). It is critical to ensure that the fusion does not disrupt the native structure and function of cytochrome c, particularly its heme-binding domain. A flexible peptide linker (e.g., (GGGGS)₂) between the two proteins can help maintain independent folding and functionality.
Selection of Expression Vector Type
The choice between a transient and a stable expression vector is fundamental and depends on the application's requirements.
Table 1: Comparison of Transient vs. Stable Expression Vectors
| Feature | Transient Expression Vector | Stable Expression Vector |
|---|---|---|
| Working Principle | Vector exists episomally; rapid, high-level transient expression. | Vector integrates into host genome via random integration or targeted systems (e.g., lentivirus). |
| Expression Profile | High-level expression peaks around 24-72 hours post-transfection, then declines. | Continuous, stable expression over many cell generations. |
| Key Advantages | Fast results; simple operation; low cost; suitable for rapid screening. | Long-term, consistent expression; genetically defined clones; ideal for long-term studies and bioproduction. |
| Primary Limitations | Expression is transient and heterogeneous; potential for cellular toxicity. | Low transformation efficiency; lengthy and costly cell line development process. |
| Ideal Applications | Rapid validation of vector function and apoptosis induction experiments. | Generation of clonal reporter cell lines for high-throughput drug screening and mechanistic studies. [29] |
For a stably integrating reporter cell line, lentiviral vectors are highly effective. They offer a broad host range (including non-dividing cells), stable integration into the host genome, and relatively large cargo capacity [29]. The use of a lentiviral system, as demonstrated in the construction of the CAFLUX HepG2 reporter line, allows for the efficient generation of clonal cell populations with consistent reporter expression [13].
Recommended Experimental Protocol
Vector Construction and Preparation
- Synthesize or Clone the coding sequence for human cytochrome c.
- Assemble the Expression Cassette in a suitable plasmid backbone (e.g., a lentiviral transfer plasmid for stable lines). The cassette should be: Promoter (CMV) - Kozak - Leader - Cytochrome C - (Linker) - EGFP - Transcription Terminator.
- Sequence Verify the entire final construct to ensure the fusion is in-frame and that no mutations have been introduced.
Generation of Stable Reporter Cell Line
- Package Lentivirus by co-transfecting the transfer plasmid (from 4.1) with packaging plasmids (e.g., psPAX2, pMD2.G) into a producer cell line like HEK293FT [13].
- Harvest Viral Supernatant 48-72 hours post-transfection, concentrate if necessary, and determine the viral titer.
- Transduce Target Cells (e.g., HeLa, U2-OS, or specialized cell lines relevant to your research) with the virus at a low Multiplicity of Infection (MOI ~3-5) to encourage single-copy integration.
- Select Transduced Cells by adding the appropriate antibiotic (e.g., 1-2 µg/mL Puromycin) 48 hours post-transduction. Maintain selection for at least 5-7 days.
- Isolate Single Clones by limiting dilution or fluorescence-activated cell sorting (FACS) based on GFP brightness.
- Expand Clonal Lines and validate them for correct mitochondrial localization of the cytochrome C-GFP signal.
Validation and Functional Assay
- Confirm Mitochondrial Localization: Treat reporter cells with a mitochondrial dye (e.g., MitoTracker Red) and confirm co-localization via confocal microscopy.
- Induce Apoptosis: Treat validated reporter cells with established apoptosis inducers:
- Staurosporine: 0.5-1 µM for 2-6 hours.
- ABT-737 (Bcl-2 inhibitor) + S63845 (MCL-1 inhibitor): Use at optimized concentrations (e.g., 1 µM each) for 4-16 hours [28].
- Monitor Cytochrome c Release: Image cells over time using live-cell fluorescence microscopy. Quantify the transition from a punctate (mitochondrial) to a diffuse (cytosolic) fluorescence pattern. Flow cytometry can also be used to measure a decrease in cellular fluorescence intensity as GFP disperses from mitochondria, a hallmark of bright-to-dark reporters [7].
- Correlate with Apoptotic Markers: Perform parallel assays to confirm apoptosis, such as Western blotting for caspase-3 cleavage or PARP cleavage [28].
The following workflow summarizes the key steps in generating and validating the reporter cell line.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Research Reagents for Reporter Construction and Assay
| Reagent / Solution | Function / Explanation |
|---|---|
| pFUGW Lentiviral Vector | A common lentiviral backbone used for constructing stable, GFP-expressing cell lines. Allows for efficient integration and long-term expression [13]. |
| CMV Promoter | A strong, constitutive viral promoter that drives high-level expression of the cytochrome C-GFP fusion protein in a wide range of mammalian cell types [21]. |
| Kozak & Leader Sequences | Regulatory elements placed upstream of the start codon to significantly enhance the translation initiation efficiency and proper folding of the recombinant fusion protein [21]. |
| Staurosporine | A broad-spectrum protein kinase inhibitor commonly used as a potent and reliable chemical inducer of the intrinsic apoptotic pathway for assay validation [28]. |
| ABT-737 & S63845 | Small-molecule inhibitors targeting Bcl-2 and MCL-1, respectively. Used in combination to specifically activate the mitochondrial apoptosis pathway by inducing BAX/BAK pore formation [28]. |
| MitoTracker Red | A cell-permeant dye that selectively labels active mitochondria. Used in validation experiments to confirm the correct mitochondrial localization of the cytochrome C-GFP fusion protein in untreated cells. |
Troubleshooting and Data Interpretation
- No Fluorescence: Verify plasmid construction and sequencing. Check promoter and cell line compatibility. Ensure antibiotic selection was effective.
- Improper Localization (Cytosolic in Untreated Cells): The fusion may be disrupting cytochrome c folding or the mitochondrial targeting sequence (if the native sequence is not sufficient for the fusion protein). Re-evaluate the fusion design and linker.
- No Release Upon Apoptosis Induction: Confirm the activity of apoptosis inducers using a positive control (e.g., Western blot for caspase-3). Titrate inducer concentration and duration. Consider that LACTB-mediated IMM remodeling may be a prerequisite for efficient cytochrome c release in some cell types [28].
- High Background Cytosolic Signal: This could indicate leaky expression or partial mislocalization. Select a clonal cell line with tight mitochondrial localization and low cytosolic background. Using a destabilized GFP variant (e.g., GFP-ASV) can reduce background from accumulated protein [30].
The strategic design of a cytochrome C-GFP fusion construct, paying close attention to promoter strength, regulatory elements like Kozak and Leader sequences, and the choice of a stable expression system, is paramount for developing a robust and sensitive apoptosis reporter cell line. The protocols outlined herein provide a reliable roadmap for constructing and validating this powerful tool, which can significantly advance research in cell biology, toxicology, and drug discovery.
Within the context of constructing a cytochrome C GFP reporter cell line for apoptosis research, the generation of a stable cell line is a critical foundational step. Unlike transient transfection, which offers only short-term gene expression, stable cell lines permanently integrate the reporter construct into the host genome, ensuring consistent, long-term expression over many cell generations [31] [32]. This stability is indispensable for extended functional studies, sustained expression in gene therapy models, and large-scale protein production, providing the reproducible and reliable data required for rigorous scientific inquiry and drug development [31] [32]. The process of developing such a cell line, specifically one designed to report on cytochrome C release—a pivotal event in the mitochondrial apoptosis pathway—demands a meticulous approach to transfection, selection, and validation to ensure the reporter system accurately reflects underlying biological processes.
Technical Foundation and Workflow
The general workflow for generating a stable cell line is methodical, involving sequential stages from transfection to final validation. A stable cell line is defined as a population of cells that have genetically incorporated a gene of interest, allowing it to be passed on to daughter cells during division [32]. This is in stark contrast to transient transfection, where introduced DNA is expressed for only a short period without genomic integration [32] [33]. For a cytochrome C GFP reporter, the goal is to create a cell line where the GFP gene is stably integrated and strategically designed to indicate apoptotic activity.
The overarching workflow can be visualized as follows, illustrating the key stages from initial preparation to a fully characterized clone:
Figure 1: Stable Cell Line Development Workflow. This diagram outlines the sequential stages from project initiation to the creation of a validated master cell bank, as commonly applied in recombinant protein production and reporter cell line generation [34].
Core Principle for Apoptosis Reporting
A cytochrome C GFP reporter cell line typically functions on a relocalization principle. In healthy cells, cytochrome C is confined to the mitochondrial intermembrane space, and a GFP fused to it will show a punctate, mitochondrial pattern. Upon induction of apoptosis, cytochrome C is released into the cytosol, leading to a diffuse GFP signal throughout the cell. This visual shift from punctate to diffuse fluorescence serves as a real-time, live-cell indicator of apoptosis activation [7]. This is distinct from caspase-activated reporters that rely on fluorescence unmasking or induction [7] [13].
Experimental Protocols
Protocol 1: Determining Antibiotic Selection Conditions
Before transfection, a kill curve must be established to determine the minimum antibiotic concentration that kills all non-transfected (parental) cells within 10-14 days. This is crucial for effective selection [32].
Procedure:
- Plate Preparation: Split a confluent culture of the host cells (e.g., HEK 293, CHO, or HepG2) and seed them at a low density (e.g., 1:10 to 1:20 split) into multiple culture dishes or wells.
- Antibiotic Dilution: Prepare a series of antibiotic concentrations in complete growth medium. A common starting range for common antibiotics is provided in Table 1.
- Application: Replace the cell culture medium with the medium containing the different antibiotic concentrations. Include a control well with no antibiotic.
- Incubation and Monitoring: Incubate the cells for 10 days, replacing the selective medium every 3-4 days.
- Assessment: Examine the dishes daily for cell death. After 10 days, use a cell viability method (e.g., trypan blue staining with an automated cell counter or hemocytometer) to count the viable cells in each concentration.
- Analysis: Plot the number of viable cells versus antibiotic concentration. The optimal selective concentration is the lowest concentration that results in 100% cell death in the control, non-transfected population within 10-14 days [32].
Table 1: Common Antibiotics for Stable Selection
| Antibiotic | Common Working Concentration Range | Mechanism of Action |
|---|---|---|
| Geneticin (G418) | 100 - 1000 µg/mL | Interferes with protein synthesis in eukaryotic cells by binding to the 80S ribosome. |
| Puromycin | 0.5 - 10 µg/mL | An aminonucleoside antibiotic that inhibits protein synthesis by causing chain termination. |
| Hygromycin B | 50 - 500 µg/mL | An aminocyclitol antibiotic that inhibits protein synthesis by disrupting translocation. |
| Blasticidin | 1 - 50 µg/mL | Inhibits protein synthesis by preventing peptide bond formation. |
| Zeocin | 50 - 1000 µg/mL | A glycopeptide antibiotic that cleaves DNA, causing cell death. |
Source: Adapted from Thermo Fisher Scientific stable transfection guide [32].
Protocol 2: Stable Transfection and Clonal Isolation
This protocol outlines the process following the establishment of a kill curve.
Materials:
- Plasmid DNA containing:
- Cytochrome C-GFP fusion gene.
- A selectable marker (e.g., puromycin resistance gene).
- Appropriate transfection reagent (e.g., lipofection) or electroporation system.
- Host cells (e.g., HEK 293, CHO, HepG2).
- Pre-tested selection antibiotic.
Procedure:
- Transfection: Transfect the cells using your method of choice (e.g., lipofection, electroporation) following the manufacturer's protocol. A 5:1 to 10:1 molar ratio of the gene-of-interest plasmid to the selection marker plasmid is recommended if they are on separate vectors [32].
- Control Transfection: Perform a parallel control transfection with a plasmid containing only the selectable marker and a "mock" transfection with no DNA.
- Recovery: Incubate the cells for 48-72 hours to allow for transient expression of the antibiotic resistance gene.
- Initiation of Selection: After the recovery period, passage the cells and re-seed them into fresh medium containing the pre-determined optimal concentration of the selection antibiotic. Cells should be sub-confluent for effective selection [32].
- Maintenance: Culture the cells under selection pressure for 2-5 weeks, replacing the drug-containing medium every 3-4 days. Cell death in the control transfections should be evident after 3-9 days.
- Clonal Isolation: Once distinct, healthy colonies (or "islands") of resistant cells have formed (typically containing 500-1000 cells), they can be isolated.
- For Adherent Cells: Use cloning cylinders or sterile toothpicks to physically isolate individual colonies. Trypsinize the cells within the cylinder and transfer them to a well of a 24- or 48-well plate.
- For Suspension Cells: Use limiting dilution in 96-well plates to statistically ensure single cell per well [32].
- Expansion: Continue to maintain the isolated clones in selective medium as they are expanded for further analysis.
Protocol 3: Clone Validation and Characterization
Validation is critical to confirm that the selected clones not only survive antibiotic selection but also express the cytochrome C-GFP reporter correctly and functionally.
1. Molecular Validation:
- Genomic Integration PCR: Isolate genomic DNA from expanded clones. Use PCR with primers specific to the GFP sequence or the cytochrome C fusion to confirm the presence of the integrated transgene.
- mRNA Expression (RT-qPCR): Isolate total RNA and perform reverse transcription quantitative PCR (RT-qPCR) to verify that the integrated transgene is being actively transcribed [33].
2. Protein Expression and Function Validation:
- Fluorescence Microscopy: Visually inspect clones for GFP fluorescence. In a valid cytochrome C-GFP reporter line, the fluorescence should display a punctate, mitochondrial pattern under normal conditions [7] [13].
- Functional Apoptosis Assay: Treat the clonal cells with a known apoptosis inducer (e.g., Staurosporine (0.1-1 µM) or H2O2 (100-500 µM)) for several hours. Monitor the cells under a fluorescence microscope for the characteristic relocalization of GFP signal from punctate to diffuse, indicating cytochrome C release. An example of a robust functional validation is demonstrated in the CAFLUX HepG2 reporter system, which showed dose-dependent fluorescence changes upon exposure to toxicants [13].
- Western Blot: Confirm the expression of the full-length cytochrome C-GFP fusion protein using GFP-specific or cytochrome C-specific antibodies.
3. Clonality Assurance:
- Single-Cell Confirmation: Ensure that the expanded clone was derived from a single cell by transferring a single cell into a 96-well plate and confirming it can yield an antibiotic-resistant colony [32].
Table 2: Key Validation Assays for a Cytochrome C GFP Reporter Cell Line
| Validation Tier | Assay | Expected Outcome for a Validated Clone |
|---|---|---|
| Genomic | PCR for Transgene | Clear amplification of a DNA fragment of the expected size. |
| Transcriptional | RT-qPCR for GFP mRNA | Detectable and stable levels of GFP transcript. |
| Protein & Localization | Fluorescence Microscopy (Basal) | Punctate, mitochondrial pattern of GFP fluorescence. |
| Functional | Fluorescence Microscopy (Post-Apoptosis Induction) | Shift from punctate to diffuse GFP signal upon treatment. |
| Specificity | Western Blot | A single band of the expected molecular weight for the fusion protein. |
The Scientist's Toolkit: Essential Research Reagents
Successful development of a reporter cell line relies on a suite of key reagents and materials. The following table details essential components and their functions.
Table 3: Essential Reagents for Stable Reporter Cell Line Generation
| Reagent / Material | Function / Explanation |
|---|---|
| Expression Vector | A plasmid backbone containing the cytochrome C-GFP fusion gene, a strong promoter (e.g., CMV), and a prokaryotic origin of replication for amplification. |
| Selectable Marker | A gene (e.g., for puromycin or blasticidin resistance) that allows for the elimination of non-transfected cells during antibiotic selection [32] [33]. |
| Transfection Reagent | A chemical-based reagent (e.g., lipofection) or a non-chemical method (e.g., electroporation) to introduce plasmid DNA into host cells [33]. |
| Selection Antibiotics | Purified antibiotics (e.g., Puromycin, G418) used at a predetermined concentration to select for successfully transfected cells [32]. |
| Appropriate Host Cell Line | Mammalian cells (e.g., HEK293, CHO, HepG2) are preferred for their ability to perform complex post-translational modifications and support stable gene expression [31] [35]. |
| Fluorescence Microscope | Essential for visualizing GFP expression, confirming correct sub-cellular localization (punctate mitochondrial pattern), and monitoring the functional response to apoptosis inducers. |
| Apoptosis Inducers | Chemical agents such as Staurosporine or H2O2 used to functionally validate the reporter system by triggering cytochrome C release and the subsequent change in GFP localization [7] [13]. |
Underlying Signaling Pathway
The cytochrome C GFP reporter is designed to detect activation of the intrinsic (mitochondrial) apoptosis pathway. Understanding this pathway is key to designing proper validation experiments. The pathway and the reporter's role within it can be summarized as follows:
Figure 2: The Intrinsic Apoptosis Pathway and Reporter Detection Point. This diagram illustrates the key events in the mitochondrial apoptosis pathway, culminating in cell death. The cytochrome C-GFP reporter provides a visual readout at the critical step of cytochrome C release, allowing for real-time detection of apoptosis initiation. The formation of the apoptosome complex involving Apaf-1 is a critical step in this cascade [21] [7].
The generation of a stable cytochrome C GFP reporter cell line is a multi-stage process that demands careful planning and rigorous validation. By following the detailed protocols for transfection, antibiotic selection, and clonal isolation—and by employing a comprehensive validation strategy that includes molecular, protein-based, and crucial functional assays—researchers can create a powerful tool for apoptosis research. This robust cellular model enables the real-time, live-cell detection of a key apoptotic event, making it invaluable for mechanistic studies of cell death, high-throughput drug screening for pro-apoptotic or anti-apoptotic compounds, and toxicological evaluations. The resulting stable cell line provides a consistent and reproducible system that can significantly enhance the reliability and depth of research into the mitochondrial apoptosis pathway.
Cell lines are indispensable tools in oncology research, providing reproducible and accessible models for studying cancer biology and therapeutic strategies. The lung cancer cell line PC9 and the breast cancer cell line T47D are two prominent models, each offering unique experimental applications and characteristics. PC9 cells, which harbor an EGFR mutation, are particularly valuable for studying targeted therapies and drug resistance mechanisms in lung adenocarcinoma [36]. T47D cells, derived from a breast ductal carcinoma, are characterized by their expression of estrogen and progesterone receptors, making them a critical model for investigating hormone-responsive breast cancer and the effects of endocrine disruptors [37] [38]. This document details the application of these cell lines within the broader context of constructing cytochrome C GFP reporter systems for apoptosis research, providing detailed protocols and resources for the scientific community.
Cell Line Characteristics and Research Applications
The PC9 and T47D cell lines are defined by their distinct origins, genetic profiles, and primary research uses, making them suitable for different experimental questions in oncology.
Table 1: Characteristics of PC9 and T47D Cancer Cell Lines
| Characteristic | PC9 Cell Line | T47D Cell Line |
|---|---|---|
| Cancer Type | Lung Adenocarcinoma [36] | Breast Ductal Carcinoma [37] |
| Origin | Not specified in search results | Pleural Effusion [37] |
| Key Genetic/Molecular Features | EGFR mutation [36] | Expression of estrogen receptors, progesterone receptors, and the WNT7B oncogene [37] |
| Primary Research Applications | Drug sensitivity studies, CTC detection models, research on EGFR-targeted therapies [36] | Study of endocrine disruptors, hormone response, anti-estrogenic therapies, and xenograft models [37] [38] |
Application Notes and Protocols
Protocol 1: Predicting Chemotherapy Response Using Machine Learning with Lung Cancer Cell Lines
Background: Machine learning (ML) models trained on multi-omics data from cell lines can predict patient responses to chemotherapy, enabling personalized treatment strategies [39].
Detailed Methodology:
- Data Acquisition: Obtain RNA-seq data and drug sensitivity data (e.g., IC₅₀ values) for lung cancer cell lines, including PC9, from public databases such as the Genomics of Drug Sensitivity in Cancer (GDSC) [39].
- Model Training and Validation:
- Clinical Correlation: Correlate predicted "sensitive" versus "resistant" classifications with patient overall survival data to validate the clinical relevance of the model [39].
- Functional Validation: Perform siRNA-mediated knockdown of model-identified key genes (e.g., TMED4, DYNLRB1) in PC9 cells to assess the impact on chemosensitivity to agents like cisplatin and gemcitabine [39].
Protocol 2: Establishing an AI-Based Circulating Tumor Cell (CTC) Recognition System
Background: Circulating tumor cells are rare in patient blood, making it difficult to acquire sufficient images to train accurate AI recognition models. Using cancer cell lines like PC9 can overcome this limitation [36].
Detailed Methodology:
- Cell Line Preparation: Culture PC9 cells in RPMI-1640 medium supplemented with 10% fetal bovine serum at 37°C in 5% CO₂ [36].
- CTC-Chip Capture and Staining:
- Prepare a microfluidic CTC-Chip coated with an anti-EpCAM antibody to capture cells [36].
- Fix captured cells with 4% formalin and permeabilize with 0.25% Triton [36].
- Perform immunofluorescence staining: incubate with primary antibodies (anti-cytokeratin and anti-CD45), followed by secondary antibodies (e.g., Alexa Fluor 594 anti-rabbit IgG and Alexa Fluor 488 anti-rat IgG) and a nuclear stain (Hoechst 33342) [36].
- Image Acquisition and Processing: Acquire fluorescent images using a microscope. Use Otsu's method for image segmentation to extract individual cell images based on the nuclear stain [36].
- AI Model Training via Transfer Learning:
- Pre-training: Train a Convolutional Neural Network (CNN) on a large dataset of images from PC9 and other lung cancer cell lines [36].
- Fine-tuning: Apply transfer learning by further training the pre-trained model on a small set of clinical CTC images (as few as 17) to significantly boost recognition accuracy [36].
Table 2: Key Steps for AI-Based CTC Recognition
| Step | Process | Key Details | Purpose |
|---|---|---|---|
| 1. Cell Capture | Antigen-antibody interaction | Use CTC-Chip functionalized with anti-EpCAM antibody [36] | Isolate rare CTCs from blood samples |
| 2. Staining | Immunofluorescence | Stain for Cytokeratin (CK+, CD45-, and Hoechst+ [36] | Identify CTCs and distinguish from leukocytes |
| 3. Imaging | Fluorescence microscopy | Acquire multi-channel images for each cell [36] | Generate data for AI model |
| 4. AI Training | Transfer Learning | Pre-train CNN on cell line images, then fine-tune with clinical CTC images [36] | Achieve high accuracy (>99.5%) with limited patient data |
Protocol 3: Evaluating Estrogenic and Anti-Estrogenic Activity in 2D and 3D Models
Background: The T47D cell line is an ideal model for studying the effects of endocrine disruptors and therapeutic compounds due to its high expression of estrogen receptors (ERα and ERβ) [38].
Detailed Methodology:
- Cell Culture:
- 2D Culture: Maintain T47D cells in DMEM medium without phenol red, supplemented with 5% charcoal-stripped fetal bovine serum (CS-FBS) for experiments [38].
- 3D Spheroid Culture: Use low-attachment plates and specialized media (e.g., DMEM-F12 without phenol red with CS-FBS) to generate tumor spheroids that better emulate in vivo conditions [38].
- Compound Treatment:
- Viability and Gene Expression Analysis:
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents and Resources
| Reagent / Resource | Function / Application | Examples / Details |
|---|---|---|
| CTC-Chip | Microfluidic device for highly sensitive capture of circulating tumor cells from blood samples. | Coated with anti-EpCAM antibody; used for CTC isolation prior to AI-based imaging [36]. |
| Machine Learning Algorithms | Computational prediction of drug sensitivity and treatment response from multi-omics data. | Random Forest and Support Vector Machine algorithms have shown superior performance in predicting lung cancer chemotherapy response [39]. |
| sGRAPHIC System | Genetically encoded optical-labeling system for interrogating cell-cell interactions in metastatic niches. | Utilizes split-GFP reconstitution to label neighboring cells; useful for studying cancer-tissue resident cell interactions [11]. |
| CRISPR/Cas9 Technology | Genome editing tool for functional validation of genes involved in drug resistance or cancer pathways. | Applied to modulate EGFR signaling in lung cancer and correct pathogenic mutations in cell lines [40] [41]. |
| Anti-EpCAM Antibody | Capture antibody for isolating epithelial-derived cells, including CTCs and cancer cell lines. | Key component for functionalizing the CTC-Chip to capture cells like PC9 [36]. |
| Charcoal-Stripped FBS | Fetal bovine serum processed to remove small molecules like hormones. | Essential for hormone-related studies in T47D cells to eliminate confounding effects of serum hormones [38]. |
The PC9 and T47D cell lines are powerful and versatile models that address distinct challenges in cancer research. PC9 cells are instrumental in advancing personalized medicine for lung cancer through drug prediction models and innovative CTC detection technologies. Meanwhile, T47D cells provide critical insights into the mechanisms of hormone-responsive breast cancer and the impact of environmental disruptors. The protocols and resources detailed herein provide a framework for employing these cell lines within advanced research paradigms, including the development of reporter systems for dynamic biological processes.
Programmed cell death, or apoptosis, is a fundamental biological process essential for tissue homeostasis, development, and immune system regulation. Dysregulation of apoptotic controls can lead to pathological conditions including cancer, autoimmune diseases, and developmental defects [42]. Apoptosis occurs through two main pathways: the intrinsic pathway, triggered by internal cellular stress such as radiation or chemotherapeutic drugs, and the extrinsic pathway, activated by external signals binding to death receptors on the cell membrane [42]. Both pathways converge on the activation of executioner caspases that orchestrate cellular destruction.
A key event in the intrinsic apoptotic pathway is the release of cytochrome-C (Cyt-C) from mitochondria into the cytosol. This release triggers the formation of an apoptosome complex, leading to the activation of caspase-9, which subsequently cleaves and activates downstream executioner caspases including caspase-3 and caspase-7 [42]. To visualize these dynamic apoptotic events in live cells, researchers have developed innovative reporter systems using fluorescent protein tags that enable real-time monitoring of biomarker translocation without the need for additional dyes or fixatives [42] [7].
Table 1: Key Apoptotic Events and Corresponding Reporter Strategies
| Apoptotic Event | Biological Significance | Reporter Strategy |
|---|---|---|
| Cytochrome-C Release | Initiation of intrinsic apoptosis | Cyt-C-GFP translocation from mitochondria to cytosol |
| Caspase-3 Activation | Execution phase of apoptosis | Cleavage of DEVD sequence and nuclear translocation of EYFP |
| Caspase-8 Activation | Initiation of extrinsic apoptosis | Cleavage of IETD sequence and nuclear translocation of EYFP |
| Mitochondrial Membrane Permeabilization | Commitment to cell death | Loss of fluorescent marker retention |
Reporter Cell Line Construction
Cytochrome-C-GFP Reporter Construction
The construction of cytochrome-C-GFP reporter cell lines involves molecular engineering to fuse the green fluorescent protein (GFP) sequence with the gene encoding cytochrome-C. Prior studies have established that tagging GFP onto Cyt-C does not affect the biological kinetics of Cyt-C, and the localization of the conjugated Cyt-C-GFP into the mitochondrial membrane can be verified with mitochondria-specific dyes [42]. This reporter construction allows live monitoring of Cyt-C release from mitochondria during apoptosis initiation, serving as a spatial fluorescent signal translocation pattern that reports on apoptotic activation [42].
The reporter cell lines are typically generated in relevant cancer cell models, such as PC9 non-small-cell lung cancer cells and T47D ductal carcinoma cells, enabling cell type-specific investigation of apoptotic responses [42]. These engineered cells are characterized by their response to apoptotic stimuli, with the spatial fluorescent signal translocation patterns serving as reporters of activations of apoptotic events, specifically Cyt-C release [42].
Caspase Reporter Systems
Beyond cytochrome-C monitoring, researchers have developed complementary caspase reporter systems. The caspase-3 reporter is commercially available in plasmid form and contains caspase-specific cleavage sites (DEVD for caspase-3) bridging a nuclear export signal (NES) to a nuclear localization sequence (NLS) tagged to EYFP [42]. Similarly, the caspase-8 reporter plasmid can be generated as a modification to the caspase-3 plasmid, utilizing the IETD cleavage site specific to caspase-8 [42].
These caspase reporter molecules permit the EYFP to remain in the cytosol until caspase activation occurs. Once the respective caspase is activated, it cleaves the EYFP-NLS sequence, allowing its transport to the nucleus [42]. This translocation from cytosol to nucleus provides a visually detectable signal of caspase activation.
An alternative approach involves designing apoptosis reporters through mutagenesis-based insertion of caspase-3 cleavage motifs directly into GFP protein. This creates a bright-to-dark system where fluorescence of DEVD-inserted GFP is inactivated by caspase-3 activation, demonstrating greater sensitivity for apoptosis detection compared to dark-to-bright systems [7].
Figure 1: Apoptotic Signaling Pathways Visualized for Reporter System Development
CRISPR/Cas9-Mediated Reporter Generation
Modern reporter cell line construction often utilizes CRISPR/Cas9-mediated homology-directed repair (HDR) for precise genetic engineering. This approach is particularly valuable for generating reporter lines in human pluripotent stem cells (hPSCs), which are highly desirable for studying differentiation, lineage tracing, and target cell selection [8]. The process involves:
- Guide RNA Design: Targeting the specific genomic locus of interest
- Donor Plasmid Construction: Containing the reporter cassette with homologous arms
- Delivery System: Electroporation or transfection to introduce CRISPR components
- Selection and Cloning: Antibiotic selection and single-cell cloning to isolate pure reporter lines [8]
This method provides a robust and cost-effective approach to perform knock-in in human pluripotent stem cells, overcoming technical bottlenecks such as DNA transduction, low homology recombination rate, and single-cell cloning that have traditionally made this effort challenging in hPSCs [8].
Automated Algorithm Implementation
Algorithm Development and Rationale
The development of a vision-based, tunable automated algorithm in MATLAB addresses critical limitations in current image analysis methods for apoptotic event detection [42]. Traditional approaches have suffered from several bottlenecks: (1) reliance on proprietary software requiring heavy manual intervention and biased threshold adjustments; (2) lack of mathematical accuracy, where visually distinct images pass undetected by algorithms; and (3) utilization of multiple fluorophores that limit downstream assay options [42].
The automated algorithm forgoes simple image statistics in favor of more robust analytics that strongly coincide with the human perspective of identifying biomarker translocation [42]. This implementation can be scaled to single-cell analysis, high-throughput analysis of single cells, or high-throughput batch analysis, making it adaptable to various experimental needs and throughput requirements. The method specifically addresses the need for rapid, efficient, and robust quantitative analyses of dynamic apoptotic events in high-throughput screening workflows [42].
Image Processing Workflow
The algorithmic implementation follows a structured workflow for robust signal translocation analysis:
- Image Acquisition: Capture high-quality fluorescence images of reporter cells
- Cell Segmentation: Identify and delineate individual cells within images
- Subcellular Localization: Determine the spatial distribution of fluorescent signals
- Feature Extraction: Quantify relevant parameters indicating translocation events
- Classification: Categorize cells based on established translocation criteria [42]
This workflow enables the algorithm to achieve a precision greater than 90% and a sensitivity higher than 85% in detecting apoptotic events, representing a significant improvement over conventional methods [42].
Figure 2: Automated Image Analysis Workflow for Translocation Quantification
Feature Extraction and Analysis
The algorithm identifies extractable features and criteria that provide valuable, robust information for detecting signal translocation. Key analytical parameters include:
- Spatial signal distribution patterns
- Intensity profiles across cellular compartments
- Temporal dynamics of signal movement
- Morphological changes associated with apoptosis
These features enable the algorithm to distinguish between distinct stages of apoptosis and provide quantitative metrics for comparative analysis across experimental conditions [42]. The tunable nature of the algorithm allows researchers to adjust sensitivity parameters based on specific experimental requirements and cell types.
Experimental Protocols
Reporter Cell Line Culture and Maintenance
Materials:
- Reporter cell lines (PC9 lung cancer or T47D breast cancer cells with Cyt-C-GFP, caspase-3, or caspase-8 reporters)
- Roswell Park Memorial Institute (RPMI) culture medium
- Matrigel hESC-Qualified Matrix (for stem cell reporters)
- StemFlex Medium or mTESR1/E8 medium (for hPSC reporters)
- 0.5 mM EDTA solution for passaging [42] [8]
Procedure:
- Culture reporter cells in appropriate medium at 37°C with 5% CO₂
- For hPSC reporters, coat culture vessels with Matrigel (1:100 dilution in DMEM/F12)
- Passage cells at 70-80% confluence using 0.5 mM EDTA or Accutase
- For single-cell cloning of hPSC reporters, use StemFlex Medium supplemented with 1× CloneR
- Maintain mycoplasma-free cultures with regular testing
- Validate reporter functionality periodically with positive controls [42] [8]
Apoptosis Induction and Live-Cell Imaging
Materials:
- Apoptosis inducers (staurosporine, H₂O₂, TRAIL, or chemotherapeutic agents)
- Live-cell imaging chamber with environmental control
- Conventional epifluorescence microscope or confocal system
- Image acquisition software [42] [7]
Procedure:
- Plate reporter cells at appropriate density in imaging-compatible vessels
- Allow cells to adhere and recover for 24 hours
- Treat with apoptotic stimuli at optimized concentrations (e.g., 0.5-1μM staurosporine)
- Place cells in environmental chamber maintaining 37°C and 5% CO₂
- Acquire time-lapse images at regular intervals (e.g., every 15-30 minutes)
- Capture multiple fields of view for statistical robustness
- Maintain consistent imaging parameters throughout experiment [42] [7]
Image Analysis Using Automated Algorithm
Materials:
- MATLAB software with image processing toolbox
- Custom algorithm for translocation analysis
- High-performance computing workstation
- Data storage and management system [42]
Procedure:
- Import image sequences into MATLAB environment
- Run cell segmentation module to identify individual cells
- Execute subcellular localization to determine fluorescent signal distribution
- Apply feature extraction to quantify translocation parameters
- Implement classification algorithm to identify apoptotic events
- Export quantitative data for statistical analysis
- Generate visualization outputs for result verification [42]
Table 2: Algorithm Performance Metrics for Apoptosis Detection
| Performance Parameter | Reported Value | Measurement Basis |
|---|---|---|
| Precision | >90% | Accurate identification of true translocation events |
| Sensitivity | >85% | Detection of genuine apoptotic cells |
| Throughput Capacity | Several 100s to 1000s of samples | Scalable analysis framework |
| Single-Cell Analysis | Supported | Individual cell tracking and classification |
| Multiplexing Capability | Single fluorophore | Compatible with downstream assays |
Research Reagent Solutions
Table 3: Essential Research Reagents for Reporter Cell Line Generation and Analysis
| Reagent/Category | Specific Examples | Function in Experiment |
|---|---|---|
| Reporter Plasmids | Cyt-C-GFP, Caspase-3 Reporter (DEVD), Caspase-8 Reporter (IETD) | Engineered to visualize specific apoptotic events via fluorescence translocation |
| Cell Lines | PC9 lung cancer, T47D breast cancer, H1/H9 hESCs, iPSCs | Cellular models for reporter construction and apoptosis studies |
| Culture Matrices | Matrigel hESC-Qualified Matrix | Provides substrate for cell attachment and growth, particularly for hPSCs |
| Culture Media | StemFlex Medium, mTESR1, E8 medium, RPMI | Optimized nutrient support for different cell types |
| CRISPR Components | Cas9 enzyme, guide RNAs, HDR donor templates | Precision genetic engineering for reporter insertion |
| Apoptosis Inducers | Staurosporine, H₂O₂, TRAIL, Doxorubicin | Activate intrinsic or extrinsic apoptotic pathways |
| Detection Enzymes | Trypsin, Bowman-Birk trypsin inhibitor | Used in vesicle-based translocation assays |
| Image Analysis Software | MATLAB with custom algorithm | Automated quantification of signal translocation |
Data Analysis and Interpretation
Quantitative Metrics for Apoptosis Detection
The automated algorithm generates multiple quantitative metrics to assess apoptotic progression:
- Translocation Efficiency: Percentage of cells exhibiting cytochrome-C release or caspase activation
- Temporal Kinetics: Time from stimulus to initial translocation event
- Spatial Dynamics: Pattern and completeness of biomarker redistribution
- Population Heterogeneity: Variation in apoptotic response within cell populations [42]
These metrics provide comprehensive insights into apoptotic dynamics that extend beyond simple binary classification of live versus dead cells, enabling researchers to capture the continuum of apoptotic responses.
Validation and Quality Control
Rigorous validation ensures accurate interpretation of translocation data:
- Positive Controls: Treat cells with established apoptosis inducers (e.g., staurosporine)
- Negative Controls: Include untreated cells and caspase inhibitor treatments
- Algorithm Calibration: Compare automated classification with manual scoring
- Specificity Testing: Verify expected subcellular localization patterns [42] [7]
Quality control measures should be implemented throughout the experimental workflow, from cell culture to image analysis, to ensure reproducible and reliable results.
Application in Drug Screening
The combination of reporter cell lines with automated analysis algorithms creates a powerful platform for high-throughput drug screening. This approach enables:
- Temporal Resolution: Monitoring dynamic drug responses over time rather than single endpoints
- Spatial Information: Capturing intracellular events with subcellular resolution
- Mechanistic Insights: Distinguishing between different apoptotic pathways
- Toxicity Assessment: Identifying compound-specific cytotoxic profiles [42]
This integrated system addresses critical bottlenecks in pharmaceutical development by providing rapid, efficient, and robust quantitative analyses of dynamic apoptotic events essential in high-throughput screening workflows [42]. The single-fluorophore design leaves room for using other fluorophores of similar color as part of secondary assays, enhancing multiplexing capabilities in drug discovery applications.
The integration of genetically encoded fluorescent reporters into high-throughput screening (HTS) platforms has revolutionized early-stage drug discovery and toxicological assessment. These tools enable researchers to monitor specific cellular pathways, stress responses, and phenotypic changes in real-time within living systems. The construction of reporter cell lines, particularly those leveraging indicators of fundamental cellular processes like cytochrome c release, provides a critical window into mechanistic toxicology and therapeutic efficacy. This application note details the implementation of such reporters, with protocols designed for researchers and drug development professionals operating within industrial and academic screening environments.
The Scientist's Toolkit: Essential Research Reagent Solutions
The successful implementation of a high-throughput screening campaign relies on a carefully selected suite of reagents and tools. The table below catalogs essential solutions for reporter-based assays.
Table 1: Key Research Reagent Solutions for Reporter-Based Screening
| Reagent Solution | Function & Application in Screening |
|---|---|
| Reporter Plasmid Vectors | Engineered constructs (e.g., lentiviral pFUGW-based) containing promoter elements (DRE, ERE) driving fluorescent protein expression (GFP, H2B-GFP) for monitoring specific pathway activation [43] [13]. |
| Stable Reporter Cell Lines | Genetically engineered cells (e.g., mES, HepG2, HeLa) with stably integrated reporters for DNA damage, oxidative stress, UPR, or pathway-specific activity (e.g., AhR, NRF-1), ensuring assay reproducibility [6] [44] [13]. |
| HTS-Compatible Viability Assays | Homogeneous assays measuring metabolic activity (e.g., resazurin reduction, ATP levels) as orthogonal endpoints to fluorescent reporter signals, often multiplexed with imaging readouts [45]. |
| 3D Culture Matrices & Scaffolds | Commercialized plates and scaffolds (e.g., for spheroid culture) that support the establishment of robust 3D cellular models which better recapitulate in vivo drug responses [45]. |
| Pathway-Specific Agonists/Antagonists | Reference compounds (e.g., AICAR for AMPK/NRF-1, TCDD for AhR, staurosporine for apoptosis) used for reporter system validation and as experimental controls [7] [6] [13]. |
Reporter Cell Line Construction: Core Methodologies and Quantitative Outcomes
The cornerstone of a successful screening campaign is a robust and well-characterized reporter cell line. The following section outlines the construction and validation of diverse reporter systems, with quantitative data summarized for comparison.
Reporter Design and Engineering Strategies
Reporter constructs are designed to translate a biological event into a quantifiable fluorescent signal. A primary strategy involves cloning a specific promoter sequence or response element upstream of a gene encoding a fluorescent protein.
- Pathway-Specific Reporters: The CAFLUX HepG2 reporter was generated by placing a histone H2B-Green Fluorescent Protein (H2B-GFP) fusion gene under the control of a synthetic promoter containing multiple Dioxin-Responsive Elements (DREs). This system allows nuclear-localized GFP accumulation upon activation of the Aryl Hydrocarbon Receptor (AhR) pathway by compounds like TCDD [13].
- Protein Localization Reporters: For studying subcellular events like cytochrome c release during apoptosis, a "split-GFP" system can be employed. In one advanced configuration, a larger GFP fragment (GFP1-10) is targeted to the mitochondrial matrix, while a smaller tag (GFP11) is fused to a protein of interest (e.g., cytochrome c). Fluorescence only reconstitutes if the tagged protein enters the matrix, providing a direct readout of mitochondrial membrane permeabilization [46].
- Caspase-Activity Reporters: An alternative, highly sensitive apoptosis reporter was developed by mutagenically inserting a caspase-3 cleavage motif (DEVD) directly into the GFP protein itself. Upon caspase-3 activation, the fluorescent protein is inactivated, creating a "bright-to-dark" signal transition that is highly amenable to high-throughput imaging [7].
Stable Cell Line Generation and Validation
Following construct design, stable cell lines are generated to ensure consistent, long-term reporter expression.
- Lentiviral Transduction: The CAFLUX HepG2 reporter line was created using a lentiviral construct packaged in HEK 293FT cells, which was then used to transduce HepG2 hepatocellular carcinoma cells. Stable clones were isolated via limiting dilution and screened for their GFP response to a reference agonist (TCDD) [13].
- Validation via Orthogonal Assays: Reporter functionality must be rigorously confirmed. This includes:
- Dose-Response Analysis: Treating cells with known agonists across a concentration range to establish a Limit of Detection (LOD), as demonstrated by the CAFLUX system's LOD of 0.01 pM for TCDD [13].
- Correlation with Endogenous Markers: Confirming that the reporter signal (e.g., GFP fluorescence) correlates with the expression of the corresponding endogenous gene (e.g., CYP1A1 mRNA levels measured by qPCR) [13].
- Pathway Inhibition: Demonstrating that the reporter signal is suppressed by specific antagonists (e.g., curcumin for AhR), confirming pathway specificity [13].
Table 2: Quantitative Performance of Exemplar Fluorescent Reporter Systems
| Reporter System / Cell Line | Inducing Stimulus / Target | Key Performance Metrics & Outcomes | Application in Screening |
|---|---|---|---|
| Caspase-3 Sensitive GFP Mutant [7] | Apoptosis (Caspase-3 activation) | - "Bright-to-dark" signal.- Greater sensitivity than "dark-to-bright" systems.- Response to Staurosporine & H₂O₂ is time/dose-dependent. | Real-time apoptosis detection in mechanistic research and drug development. |
| ToxTracker mES Reporters [44] | DNA damage, Oxidative stress, UPR | - Panel of 6 GFP reporters.- Outstanding sensitivity/specificity in validation.- Discriminates between primary chemical reactivity modes. | Mechanistic in vitro human hazard assessment of chemicals. |
| CAFLUX HepG2 (H2B-GFP) [13] | AhR activation (CYP1A1) | - LOD: ~0.01 pM for TCDD; ~0.1 pM for B[a]P.- Nuclear-localized signal for easy quantification.- Responsive to agonists and antagonists. | Toxicological evaluation, drug discovery, and modulator screening. |
| NRF-1 mitoGFP HeLa Reporter [6] | Mitochondrial biogenesis (NRF-1) | - Inducible mitoGFP expression with AICAR (0.5 mM max).- Increased mitochondrial biomass, volume, and network filaments.- Reversible upon AICAR removal. | Live-cell analysis of mitochondrial regulation and dynamics. |
Experimental Protocols
Protocol 1: High-Throughput Imaging-Based Drug Screen
This protocol is adapted from a screen that identified inducers of ERα signaling in triple-negative breast cancer, demonstrating the application of a GFP reporter in a high-throughput setting [43].
Workflow:
Diagram Title: HTS Screening Workflow
Materials:
- Reporter cell line (e.g., SUM149PT ERE-GFP [43])
- 384-well microtiter plates, tissue culture treated
- Automated liquid handling system
- Small molecule compound library (e.g., 9,501 compounds [43])
- High-content fluorescence microscope with automated stage
- Image analysis software (e.g., CellProfiler, custom pipelines)
Procedure:
- Cell Seeding: Seed reporter cells into 384-well plates at a density optimized for confluency after the assay duration (e.g., 1,000-2,000 cells/well in 50 µL medium) using an automated liquid handler. Incubate for 24 hours.
- Compound Addition: Pin-transfer or acoustically dispense compounds from the library into assay plates. Include DMSO-only wells as negative controls and known pathway agonists as positive controls.
- Incubation: Incubate cells with compounds for a predetermined period (e.g., 48 hours [43]) at 37°C/5% CO₂ in a humidified incubator.
- Image Acquisition: Using a high-content imager, automatically acquire fluorescence (reporter signal, e.g., GFP) and bright-field images from multiple sites per well to ensure adequate cell sampling.
- Image Analysis: Process images with an automated pipeline to quantify the GFP signal per well, normalized to cell count (from nuclear stain or bright-field segmentation). Apply a threshold (e.g., Z-score > 3) to identify "hits" – compounds that significantly induce the reporter signal.
- Hit Validation: Re-test primary hits in a dose-response format (e.g., 8-point concentration series) to confirm activity and determine potency (EC₅₀). Classify compounds based on their effect on cell proliferation (proliferative, cytostatic, toxic) [43].
Protocol 2: Validation of a Cytochrome c Release Reporter Using a Split-GFP System
This protocol outlines the strategy for employing a bi-genomic split-GFP system to visualize cytochrome c release, a key apoptotic event [46].
Workflow:
Diagram Title: Split-GFP Cytochrome c Assay
Materials:
- SWAT-tagged yeast collection (or mammalian cells) with cytochrome c gene fused to 3xGFP11 tag at C-terminus [46]
- Mating partner strain with mitochondrial DNA (mtDNA) encoding the GFP1-10 fragment [46]
- Apoptosis-inducing agents (e.g., Staurosporine, H₂O₂ [7])
- Confocal or high-resolution fluorescence microscope
- Mitochondrial dye (e.g., MitoTracker Deep Red) for colocalization
Procedure:
- Reporter Strain Generation:
- For yeast: Use the SWAp-Tag (SWAT) method to create a haploid strain where the endogenous cytochrome c gene is C-terminally fused to a 3xGFP11 tag [46].
- Mate this strain with a haploid strain of the opposite mating type that carries the mtDNA-encoded GFP1-10 fragment.
- Select for diploid cells that contain both the nuclear-encoded GFP11-cytochrome c and the mtDNA-encoded GFP1-10.
- Treatment and Induction:
- Grow the resulting reporter strain to mid-log phase in appropriate medium.
- Treat cells with a pro-apoptotic stimulus (e.g., 1 µM Staurosporine) for a time-course (e.g., 0-6 hours). Include untreated controls.
- Image Acquisition and Analysis:
- For fixed time points or live-cell imaging, collect z-stacks using a confocal microscope.
- Co-stain with MitoTracker Deep Red to visualize the entire mitochondrial network and confirm the mitochondrial localization of the initial signal.
- Pre-apoptosis: Fluorescence should be absent or very low, as cytochrome c is confined to the intermembrane space and cannot interact with matrix-localized GFP1-10.
- Post-apoptotic induction: The release of cytochrome c-GFP11 into the matrix allows reconstitution of GFP fluorescence. Quantify the increase in GFP signal intensity within mitochondrial regions over time as a direct metric of cytochrome c release.
Data Analysis and Interpretation in High-Throughput Studies
The transition from 2D to 3D cell culture models in HTS, while providing more physiologically relevant data, introduces significant complexity in image acquisition and analysis [45].
- 3D Imaging Challenges: Light scattering and absorption in thick samples, reagent penetration, and volumetric collapse during fixation can confound analysis. Macro-level readouts (e.g., spheroid growth, overall viability) are often more feasible than subcellular resolution in these models [45].
- Multiplexing and Orthogonal Readouts: Reporter signals should be complemented with other metrics. For example, in a screen for ERα inducers, the primary GFP reporter readout was supplemented with flow cytometry, qPCR for downstream targets, and global proteomics to confirm pathway activation [43].
- Pathway Cross-Talk: Chemicals often induce multiple types of cellular damage. Assays like the expanded ToxTracker, which uses six reporters monitoring distinct pathways (DNA damage, oxidative stress, UPR, p53 stress), allow for the integration of multiple signals to accurately interpret a compound's primary biological reactivity [44].
Solving Common Challenges in Reporter Cell Line Development and Assays
Mitigating High Background Fluorescence from Culture Medium Components
High background fluorescence from culture medium components presents a significant challenge in fluorescence microscopy and high-content screening (HCS), particularly when working with sensitive reporter systems such as cytochrome C GFP reporter cell lines. This interference can obscure specific signals, reduce the signal-to-noise ratio, and compromise data interpretation in drug discovery research [47]. Autofluorescence from media components elevates fluorescent backgrounds, making it difficult to distinguish target fluorescence from background, especially within spectral ranges used by common fluorescent proteins like GFP [47]. This application note provides detailed protocols and data-driven solutions for identifying and mitigating these interference issues, with specific application to cytochrome C GFP reporter research.
Culture media components can contribute significantly to background fluorescence, particularly in live-cell imaging applications. Certain components possess intrinsic fluorescent properties that interfere with detection systems:
- Riboflavins and flavins: Fluoresce in the ultraviolet through green fluorescent protein (GFP) variant spectral ranges (excitation 375-500 nm, emission 500-650 nm) [47]
- Phenol red: Exhibits fluorescence that can interfere with detection
- Undefined supplements: Serum-containing supplements like fetal bovine serum (FBS) introduce complex mixtures of fluorescent compounds
- Human platelet lysates (hPL): May contain fluorescent biomarkers including myeloperoxidase, glycocalicin, and fibrinogen [48]
The problematic spectral overlap is particularly evident for GFP-based reporters, where media autofluorescence directly competes with the target signal.
Table 1: Common Fluorescent Culture Media Components and Their Spectral Properties
| Component | Excitation Range (nm) | Emission Range (nm) | Primary Impacted Reporters |
|---|---|---|---|
| Riboflavins | 375-500 | 500-650 | GFP, YFP, and variants |
| Phenol red | 450-550 | 550-600 | GFP, YFP |
| FBS components | 350-700 | 400-700 | Broad-spectrum interference |
| hPL components | 400-650 | 450-700 | RFP, GFP, and others |
Impact on Cytochrome C GFP Reporter Systems
For cytochrome C GFP reporter cell lines, background fluorescence presents particular challenges in monitoring dynamic processes such as:
- Apoptosis detection: Tracking cytochrome C translocation from mitochondria to cytosol [42]
- Drug screening assays: Identifying compounds inducing cytochrome C release
- Kinetic studies: Monitoring real-time changes in cytochrome C localization
Media-induced background can obscure the subtle cytoplasmic GFP signal following cytochrome C release, potentially leading to false negatives or underestimated responses in drug screening applications [42] [47].
Quantitative Comparison of Culture Supplements
Performance Metrics Across Supplement Types
Comprehensive analysis of various culture supplements reveals significant differences in composition and performance characteristics relevant to fluorescence-based assays:
Table 2: Quantitative Analysis of Culture Media Supplements and Fluorescence Impact
| Supplement Type | Growth Factor Content | Batch Variability | Autofluorescence Potential | Cell Growth Support | Cost Consideration |
|---|---|---|---|---|---|
| FBS | High, undefined | High | High | Excellent | Low |
| hPL | High, defined | Moderate to high | Moderate to high | Excellent | Moderate |
| SFM-1 | Defined | Low | Low | Variable | High |
| SFM-2 | Defined | Low | Low | Good | High |
| CDM | Fully defined | Very low | Very low | Good to excellent | High |
Analysis of seven commercially available serum-free media (SFM) revealed that two contained significant levels of human serum components, including myeloperoxidase, glycocalicin, and fibrinogen, effectively reclassifying them as human platelet lysate equivalents despite "serum-free" marketing [48]. This underscores the importance of rigorous supplement characterization for fluorescence-sensitive applications.
Growth Factor Content Analysis
Quantitative assessment of growth factors across supplement types using ELISA-based methods:
Table 3: Growth Factor Concentrations in Culture Supplements (Mean Values)
| Supplement | IGF-1 (ng/mL) | PDGF-AB (ng/mL) | TGF-ß1 (ng/mL) | VEGF (ng/mL) |
|---|---|---|---|---|
| FBS | 110-150 | 25-40 | 40-60 | 0.5-2 |
| hPL | 80-120 | 60-90 | 80-120 | 5-15 |
| SFM-1 | 20-40 | <5 | <5 | <1 |
| SFM-2 | 30-50 | 10-20 | 10-20 | 1-3 |
| CDM | Recombinant form | Recombinant form | Recombinant form | Recombinant form |
While growth factor content varied significantly between categories, this did not directly correlate with mesenchymal stem cell growth kinetics or maximal cell yield [48], suggesting that defined formulations can support robust cell growth without fluorescence-inducing complex supplements.
Experimental Protocols for Media Evaluation and Optimization
Protocol 1: Assessment of Media Autofluorescence
Purpose: Quantify background fluorescence of culture media components to identify interference with cytochrome C GFP reporter systems.
Materials:
- Test media/supplements: FBS, hPL, SFM, CDM
- Black-walled, clear-bottom 96-well plates
- Fluorescence plate reader with appropriate filters (GFP: ex 488 nm, em 510 nm)
- Spectrophotometer for absorbance measurements (optional)
Procedure:
- Dispense 100 µL of each test medium into replicate wells (n=6)
- Include negative controls (PBS or balanced salt solution)
- Measure fluorescence using settings matching experimental parameters
- For GFP reporters: Ex 488±10 nm, Em 510±10 nm, gain adjusted to mid-range
- Record values and calculate signal-to-background ratio
- For additional characterization, measure absorbance from 350-700 nm
Data Analysis:
- Calculate mean fluorescence intensity (MFI) for each medium
- Determine signal-to-background ratio: S/B = MFIsample / MFInegative control
- Values <2.0 indicate acceptable background for most applications
- Values >5.0 suggest potential interference with sensitive reporters
Protocol 2: Adaptation of Cytochrome C GFP Reporter Cells to Low-Fluorescence Media
Purpose: Transition reporter cell lines from serum-containing to serum-free or chemically defined media while maintaining reporter functionality and cell viability.
Materials:
- Cytochrome C GFP reporter cells (e.g., PC9 lung cancer or T47D breast cancer derivatives) [42]
- Base medium (DMEM/F12, RPMI, or other appropriate formulation)
- Current serum-containing medium (e.g., with 10% FBS)
- Target low-fluorescence medium (SFM or CDM)
- Recombinant TrypLE or other animal-free dissociation reagent [49]
- 6-well culture plates, T-25 flasks
Procedure:
- Begin with cells in exponential growth phase in serum-containing medium
- Day 1: Plate cells at 30-40% confluence in 70:30 ratio (serum-containing:target medium)
- Day 3: Passage cells using recombinant TrypLE into 50:50 ratio
- Day 5: Passage into 30:70 ratio
- Day 7: Transition to 100% target medium
- Monitor cell morphology, viability, and growth rate daily
- After 2 passages in 100% target medium, validate cytochrome C GFP reporter function using known induces (e.g., apoptosis inducers)
Validation:
- Assess GFP expression patterns in response to apoptotic stimuli
- Compare translocation dynamics between original and adapted cultures
- Verify maintenance of growth characteristics and viability
- Confirm expected cytochrome C release patterns using control compounds [42]
Protocol 3: Media Component Fluorescence Profiling
Purpose: Systematically evaluate individual media components for contribution to background fluorescence.
Materials:
- Individual media components (amino acids, vitamins, salts, growth factors)
- Basal medium without supplements
- Fluorescence spectrometer or plate reader with scanning capability
Procedure:
- Prepare individual component solutions at 10X working concentration
- Dilute in PBS to 1X concentration for testing
- Scan fluorescence across excitation 350-600 nm, emission 400-700 nm
- Identify components with significant fluorescence in reporter spectrum
- Test potential substitutes for problematic components
Troubleshooting:
- If specific components show high background, investigate purified alternatives
- Consider concentration reduction for non-essential fluorescent components
- Evaluate synthetic replacements for biological derivatives
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Reagents for Low-Background Fluorescence Imaging
| Reagent Category | Specific Products/Examples | Function | Advantages for Fluorescence Assays |
|---|---|---|---|
| Chemically Defined Media | Custom formulation, Commercial SFM | Cell growth medium | Eliminates serum-derived autofluorescence; reduced variability |
| Animal-Free Dissociation Reagents | Recombinant TrypLE | Cell passaging | Avoids fluorescent contaminants in trypsin preparations |
| Low-Fluorescence Supplements | Recombinant insulin, transferrin, selenium | Growth support | Defined composition with minimal background |
| Validated Antibodies | Phage-derived recombinant antibodies [49] | Target detection | High specificity; animal-free production |
| Fluorescence Validated Plates | Black-walled, clear-bottom microplates | Assay vessel | Minimize cross-talk and background |
| Reference Control Compounds | Bedaquiline, Clofazimine, Q203 [50] | Assay validation | Known inducers of cytochrome C release in reporter systems |
Workflow Diagrams for Media Optimization
Media Selection and Validation Workflow
Media Selection Workflow: Systematic approach for identifying and implementing low-fluorescence media for cytochrome C GFP reporter systems.
Cytochrome C GFP Reporter Response Pathway
Reporter Response Pathway: Key events in cytochrome C GFP reporter activation following apoptotic stimuli.
Mitigating background fluorescence from culture medium components is essential for optimizing performance of cytochrome C GFP reporter systems in drug discovery research. Implementation of chemically defined media, rigorous component screening, and systematic cell adaptation protocols significantly enhance signal-to-noise ratios while maintaining physiological relevance. The protocols and data presented herein provide a framework for researchers to address fluorescence interference challenges, thereby improving data quality and reliability in high-content screening applications. As the field moves toward more defined culture systems, these approaches will become increasingly integral to reproducible cytochrome C translocation studies and apoptosis research.
Optimizing Transfection Efficiency for Robust Reporter Expression
The construction of a cytochrome C GFP reporter cell line is a powerful approach for investigating mitochondrial function and apoptosis in live cells. The efficacy of such research is, however, fundamentally dependent on achieving high transfection efficiency to ensure robust and reliable reporter expression. Efficient delivery of the reporter construct into the host cell is crucial, as it directly impacts the signal-to-noise ratio, the accuracy of quantitative measurements, and the overall biological relevance of the data obtained. This application note provides detailed protocols and key optimization strategies for maximizing transfection efficiency, specifically within the context of developing and utilizing cytochrome C GFP reporter cell lines for advanced research and drug discovery.
Critical Factors Influencing Transfection Efficiency
Successful transfection is a multivariate process. Optimizing the following factors is essential for achieving high levels of reporter expression while maintaining cell health.
Cell Health and Culture Conditions
The starting point for any successful transfection is a healthy, actively dividing cell culture.
- Cell Viability and Passage Number: Cells should be at least 90% viable prior to transfection and should be used in their logarithmic growth phase. It is strongly recommended to use cells that have undergone fewer than 30 passages after thawing a stock culture, as excessive passaging can detrimentally affect transfection efficiency [51]. For optimal reproducibility, aliquots of low-passage-number cells should be stored frozen and thawed as needed [51].
- Cell Confluency: For adherent cells, a confluency of 70–90% at the time of transfection is generally optimal [51] [52] [53]. Overly confluent cells can become contact-inhibited, leading to poor nucleic acid uptake, while too few cells may not grow robustly due to a lack of cell-to-cell contact [51].
- Culture Medium and Serum: The use of fresh, appropriate medium is critical. While serum can enhance cell health, its presence can interfere with the formation of cationic lipid-DNA complexes. For lipid-based transfections, complexes should be formed in serum-free medium, but the transfection can often be performed in serum-containing medium [51]. The quality and consistency of the serum lot can significantly impact results [51].
Nucleic Acid Quality and Vector Design
The integrity and design of the nucleic acid to be delivered are paramount.
- DNA Quality: Plasmid DNA must be of high purity, with an A260/A280 ratio of 1.7–1.9 and low endotoxin levels. Endotoxins can cause cytotoxicity and provoke unintended cellular responses, compromising experimental outcomes [53].
- Vector Optimization: The design of the reporter vector itself can dramatically influence expression levels. Incorporating regulatory elements such as a Kozak sequence (GCCRCC) upstream of the start codon enhances translation initiation efficiency [21]. Furthermore, the addition of a Leader sequence can improve protein folding and trafficking. Research in CHO cells has demonstrated that combining Kozak and Leader sequences can increase the expression of reporter proteins like eGFP by more than twofold compared to a baseline vector [21].
Transfection Method and Reagent Optimization
The choice of transfection method and its precise optimization are perhaps the most critical steps.
- Transfection Methods: Common methods include chemical methods (e.g., cationic lipids, polymers), physical methods (e.g., electroporation, microinjection), and biological methods (viral vectors) [52] [54] [55]. Cationic lipid-based reagents are widely popular due to their relative ease of use, broad applicability, and low toxicity [55]. They work by coating the nucleic acid, neutralizing its negative charge, and facilitating cellular uptake via endocytosis [55].
- Reagent:DNA Ratio: The ratio of transfection reagent to DNA mass (µl/µg) must be determined empirically for each cell line and reagent. This ratio determines the charge and size of the complexes formed, directly impacting both transfection efficiency and cytotoxicity [53]. A typical optimization experiment tests a range of ratios (e.g., 1:1, 2:1, 3:1) [52].
- Incubation Time: The incubation period of the transfection complexes with the cells requires optimization. Insufficient time may limit uptake, while prolonged exposure can increase cytotoxic effects. For sensitive cells, reducing the incubation time (e.g., to 6-8 hours) before replacing the medium can help maintain cell viability [52].
Table 1: Key Optimization Parameters for Transfection
| Parameter | Optimal Condition / Recommendation | Rationale / Impact |
|---|---|---|
| Cell Health | >90% viability, <30 passages post-thaw | Ensures robust, metabolically active cells for high DNA uptake [51] [53]. |
| Cell Confluency | 70-90% for adherent cells | Balances active division with available space, avoiding contact inhibition [51] [53]. |
| DNA Quality | A260/A280 = 1.7-1.9, low endotoxin | Prevents cytotoxicity and unintended cellular stress responses [53]. |
| Reagent:DNA Ratio | Empirically determined (e.g., 1:1 to 3:1) | Critical for forming optimal complexes; affects efficiency & toxicity [52] [53]. |
| Complex Incubation | 0-15 min formation; 6-24h with cells | Allows complex formation & cellular uptake while limiting cytotoxicity [52] [53]. |
Experimental Protocol: Optimizing Transfection for a Cytochrome C GFP Reporter
The following protocol provides a step-by-step guide for optimizing transfection conditions in a 96-well plate format, enabling the systematic testing of multiple variables with minimal reagent use.
Materials and Reagents
- Cell Line: Your target cell line for cytochrome C GFP reporter construction (e.g., HEK293, HeLa, or a relevant primary cell).
- Reporter Plasmid: Plasmid encoding cytochrome C fused to GFP. For example, a construct where GFP is fused to a subunit of cytochrome c oxidase (e.g., CoxVIIIa) has been successfully used for mitochondrial studies [56].
- Transfection Reagent: A cationic lipid-based reagent such as FuGENE HD [53] or Lipofectamine 3000 [51].
- Cell Culture Plates: 96-well tissue culture-treated plates.
- Assay Kits:
- Fluorescence-based cell viability assay (e.g., CellTiter-Fluor [53]).
- Reporter assay (e.g., if using a luciferase secondary reporter).
Table 2: Research Reagent Solutions for Reporter Cell Line Development
| Reagent / Material | Function / Application | Example Product / Note |
|---|---|---|
| Cationic Lipid Reagent | Forms complexes with nucleic acids for cell delivery. | FuGENE HD, Lipofectamine 3000 [53] [51]. |
| High-Quality Plasmid Prep Kit | Ensures pure, endotoxin-free DNA for transfection. | PureYield Plasmid Purification Systems [53]. |
| Cell Viability Assay | Multiplexes with reporter assay to monitor cytotoxicity. | CellTiter-Fluor Cell Viability Assay [53]. |
| Reporter Gene Assay (RGA) | Quantifies biological activity of the expressed reporter. | Luciferase Assay Systems [57] [53]. |
| CRISPR/Cas9 System | For genomic integration or knockout of apoptotic genes (e.g., Apaf1) to enhance protein yield and cell survival [57] [21]. |
Step-by-Step Optimization Procedure
Day 1: Cell Seeding
- Harvest cells in the logarithmic growth phase to ensure a single-cell suspension.
- Count the cells and assess viability using trypan blue exclusion; viability should be >95% [53].
- Seed the cells in the 96-well plate at a density that will achieve 70-90% confluency at the time of transfection (typically 24 hours later). For many common lines, this is 1–2 x 10⁴ cells per well. To account for plate edge effects, it is advisable to fill the perimeter wells with sterile PBS or culture medium only.
Day 2: Transfection
- Prepare DNA:Reagent Complexes: In sterile tubes, prepare transfection complexes according to the reagent manufacturer's guidelines, but test a range of reagent-to-DNA ratios (e.g., 1:1, 2:1, 3:1 µl/µg). A constant mass of DNA (e.g., 0.2 µg per well) should be used. Incubate the complexes at room temperature for 15 minutes [53].
- Apply Complexes to Cells: Add the complexes drop-wise to the respective wells of the 96-well plate. Gently swirl the plate to ensure even distribution.
- Incubate: Return the plate to the 37°C CO₂ incubator. The incubation time with the complexes should also be optimized (e.g., 6-24 hours), after which the medium may be replaced with fresh, pre-warmed complete medium to minimize toxicity.
Day 3 or 4: Analysis (24-48 hours post-transfection)
- Assay Reporter Expression and Viability: Use a multiplexed assay to measure both reporter activity (e.g., GFP fluorescence intensity via microscopy or flow cytometry) and cell viability in the same well [53]. For example, the CellTiter-Fluor assay can be used followed by a luciferase assay if applicable [53].
- Data Analysis: Plot the reporter activity and cell viability for each tested condition. The optimal condition is the one that delivers the highest reporter activity with minimal impact on cell health.
Diagram 1: Transfection Optimization Workflow
Advanced Strategy: Enhancing Expression via Vector and Host Cell Engineering
Beyond standard optimization, robust reporter expression can be engineered at the molecular and cellular level.
Vector Engineering with Regulatory Elements: As demonstrated in CHO cells, the strategic insertion of a Kozak sequence (GCCACC) upstream of the GFP start codon in your cytochrome C reporter plasmid can enhance translation initiation. Combining this with an appropriate Leader sequence can further improve protein expression levels, potentially doubling the output of the fluorescent reporter protein [21].
Host Cell Engineering via CRISPR/Cas9: To improve the health and productivity of the host cell during and after transfection, consider knocking out genes involved in apoptosis. For instance, knocking out the Apaf1 (Apoptotic protease-activating factor 1) gene can inhibit the mitochondrial apoptosis pathway. Apaf1 is a key component of the apoptosome, which forms in response to cytochrome c release and activates caspase-9 [21]. Using CRISPR/Cas9 to generate an Apaf1 knockout cell line can create a more resilient host that is less prone to transfection-induced apoptosis, thereby increasing the yield of recombinant protein and the stability of the resulting reporter cell line [57] [21].
Diagram 2: Engineering for Robust Expression
Optimizing transfection efficiency is a critical, multi-faceted process for the successful development and application of cytochrome C GFP reporter cell lines. By systematically addressing cell culture conditions, nucleic acid quality, and transfection reagent parameters, researchers can achieve high levels of reporter expression with minimal cytotoxicity. Furthermore, leveraging modern molecular techniques such as vector optimization with regulatory elements and host cell engineering via CRISPR/Cas9-mediated gene knockout provides powerful strategies to push beyond standard efficiency plateaus. The protocols and strategies outlined herein provide a robust framework for researchers to generate high-quality data in their investigations of mitochondrial dynamics and apoptotic signaling.
The development of cytochrome-C GFP (Cyt-C-GFP) reporter cell lines represents a significant advancement in high-throughput screening workflows for apoptosis detection. These engineered cell lines allow for the live monitoring of apoptotic events, such as the release of cytochrome-C from mitochondria, without the need for additional dyes or fixatives [42]. The core mechanism relies on the spatial translocation of a fluorescent signal—when apoptosis is initiated, Cyt-C-GFP moves from the mitochondria into the cytosol, creating a measurable change in fluorescence distribution [42]. The accurate detection of this critical biomarker translocation is heavily dependent on precise optical filter selection to maximize signal-to-noise ratio, minimize background autofluorescence, and ensure robust, quantitative analysis. This application note provides detailed methodologies for selecting and validating filter combinations to optimize GFP detection within the specific context of apoptosis research using Cyt-C GFP reporter systems.
The Role of GFP Reporter Cell Lines in Apoptosis Research
Reporter Cell Line Construction and Apoptosis Signaling
Apoptosis, or programmed cell death, occurs through two primary pathways: the intrinsic (mitochondrial) pathway and the extrinsic (death receptor) pathway [42]. The intrinsic pathway, often initiated by cellular stress or chemotherapeutic drugs, is characterized by mitochondrial outer membrane permeabilization (MOMP) and the release of cytochrome-C (Cyt-C) into the cytosol [42]. Once in the cytosol, Cyt-C triggers the formation of the apoptosome, leading to the activation of caspase-9 and downstream executioner caspases like caspase-3 and caspase-7, ultimately resulting in cell death.
To visualize this process in real-time, researchers have constructed reporter cell lines by fusing the gene for Green Fluorescent Protein (GFP) to the gene for cytochrome-C [42]. Prior studies have confirmed that tagging GFP onto Cyt-C does not adversely affect the biological kinetics of Cyt-C, and the fusion protein correctly localizes to the mitochondrial intermembrane space [42]. Upon apoptosis induction, the release of the Cyt-C-GFP fusion protein from mitochondria into the cytosol serves as a direct, spatially resolved reporter of one of the earliest commitment steps in the intrinsic apoptotic pathway.
The following diagram illustrates the key stages in the use of a Cyt-C-GFP reporter for detecting the intrinsic apoptosis pathway.
Figure 1: Apoptosis Detection Workflow with a Cyt-C-GFP Reporter. The diagram outlines the key biological and technical steps from the initiation of apoptosis to its automated quantification. The apoptotic stimulus triggers mitochondrial pore formation and the release of the Cyt-C-GFP fusion protein. This spatial translocation of fluorescence from a punctate mitochondrial pattern to a diffuse cytosolic pattern is detected via fluorescence microscopy and quantified using automated algorithms.
Key Advantages of Single-Color Reporter Systems
The use of a single-color fluorophore like GFP in these reporter systems offers several distinct benefits for high-throughput screening (HTS) [42]:
- Downstream Compatibility: It leaves other fluorescence channels available for subsequent assays, enabling multiplexed experimental designs.
- Live-Cell Dynamics: It facilitates the real-time monitoring of dynamic apoptotic events in living cells, providing temporal resolution that fixed-cell, end-point assays cannot offer.
- Simplified Optics: It reduces the complexity of filter requirements compared to multi-color experiments, though it makes the precise selection of the GFP filter set even more critical.
Fundamentals of Fluorescence Filter Sets
Components of a Fluorescence Filter Cube
A standard epifluorescence microscope uses a filter cube positioned between the light source, the sample, and the detector. This cube contains three critical optical components that work in concert [58] [59]:
- Excitation Filter (Ex): This is a bandpass filter placed in the light path from the source. Its function is to selectively transmit only the specific wavelength range that optimally excites the fluorophore, while blocking other wavelengths.
- Dichroic Beamsplitter (DB): This is a specialized mirror, positioned at a 45-degree angle, that reflects the selected excitation light down onto the sample. It then transmits the longer-wavelength emitted fluorescence from the sample toward the detector, while blocking reflected excitation light.
- Emission Filter (Em): Also a bandpass filter, this component is placed in the light path to the detector. It functions to transmit the desired emission wavelengths from the fluorophore while effectively blocking any residual scattered excitation light, which is typically orders of magnitude brighter than the emission.
Table 1: Core Functions of Fluorescence Filter Set Components [58] [59]
| Component | Primary Function | Common Filter Types |
|---|---|---|
| Excitation Filter | Selects optimal wavelengths to excite the fluorophore. | Bandpass, Short-Wave Pass Edge, Multi-Bandpass. |
| Dichroic Beamsplitter | Reflects excitation light; transmits emission light. | Long-Wave Pass edge filter optimized for 45° incidence. |
| Emission Filter | Isolates fluorescence emission; blocks excitation light. | Bandpass, Long-Wave Pass Edge, Multi-Bandpass. |
Filter Selection Trade-Offs: Signal vs. Isolation
The design of a filter set always involves a balance between maximizing the detected signal and minimizing optical noise [58].
- Maximizing Signal: Using excitation and emission filters with wider bandwidths captures more light, leading to a brighter image. This is beneficial for dim samples or when minimizing light exposure to live cells is critical.
- Minimizing Noise (Isolation): Using narrower bandwidths that are well-separated spectrally reduces bleed-through from autofluorescence or other fluorophores, improving contrast and specificity. However, this can unacceptably reduce the signal strength [58].
For a single GFP reporter, the goal is to find a balance that provides a strong, specific signal suitable for the detection method (e.g., camera vs. photomultiplier tube) and the required downstream analysis, such as the automated algorithms described in the research [42].
Quantitative Filter Set Selection for GFP
Recommended Filter Parameters for GFP and Common Variants
The optimal filter set is determined by the specific spectral profile of the fluorescent protein used. For Cyt-C-GFP reporters, the starting point is typically enhanced GFP (EGFP). The following table provides standard filter parameters for EGFP and other common GFP variants, which can serve as a baseline for configuration.
Table 2: Exemplary Filter Set Parameters for Common Green Fluorescent Proteins [60]
| Fluorescent Protein | Excitation Filter CWL/BW (nm) | Dichroic Mirror Cut-On (nm) | Emission Filter CWL/BW (nm) | Relative Brightness (% of EGFP) |
|---|---|---|---|---|
| GFP (wt) | 450/50 | 480LP | 510/50 | 48% |
| EGFP | 470/40 | 495LP | 515/30 | 100% |
| Emerald | 470/40 | 495LP | 515/30 | 116% |
| Azami Green | 470/40 | 495LP | 520/30 | 121% |
| CopGFP | 470/40 | 490LP | 510/30 | 125% |
| AcGFP | 470/40 | 490LP | 510/30 | 82% |
Abbreviations: CWL: Center Wavelength, BW: Bandwidth, LP: Longpass.
For a standard EGFP-based reporter, a typical and effective filter combination is a 470/40 nm excitation filter, a 495 nm longpass dichroic mirror, and a 515/30 nm emission filter [60]. This set is designed to closely match the peak absorption (~488 nm) and emission (~509 nm) spectra of EGFP.
Advanced Considerations for Cyt-C-GFP Apoptosis Detection
The quantitative analysis of signal translocation in apoptosis reporter cell lines, as developed by researchers using automated algorithms in MATLAB, requires a high-quality, high-contrast initial image [42]. Filter selection directly impacts the performance of such algorithms.
- Precision and Sensitivity: The cited research achieved algorithm precision greater than 90% and sensitivity higher than 85% for detecting apoptotic events [42]. Proper filter selection is a prerequisite for achieving this level of analytical performance, as it ensures that the raw image data accurately represents the underlying biological phenomenon.
- Signal-to-Noise Ratio (SNR): For detecting the diffuse cytosolic signal of released Cyt-C-GFP against the brighter punctate mitochondrial background, a high SNR is critical. Bandpass emission filters (e.g., 515/30 nm) are generally preferred over longpass filters for this application, as they provide better isolation of the GFP signal from cellular autofluorescence [58] [59].
Experimental Protocol: Validation of Filter Performance for Cyt-C-GFP Reporters
Workflow for Filter Set Validation
The following protocol ensures that the selected filter set is optimally configured for detecting cytochrome-C release in your specific experimental system.
Figure 2: Experimental Workflow for Filter Set Validation. This protocol outlines the key steps for confirming that a fluorescence filter set is correctly configured for a Cyt-C-GFP reporter assay, from initial setup to final optimization and documentation.
Detailed Methodology
Step 1: Confirm Spectral Profiles
- Before selecting filters, verify the exact excitation and emission spectra of the GFP variant used in your Cyt-C-GFP reporter cell line. Use online tools like the Fluorescence SpectraViewer (available from Thermo Fisher and other vendors) to model the spectra and simulate performance with different filter sets [58]. Remember that spectral profiles can be influenced by the local environment (e.g., pH, polarity) [58] [60].
Step 2: Select and Install Filter Set
- Based on the spectral data, install the recommended filter set on your epifluorescence microscope. For EGFP-based reporters, start with a standard set (e.g., Excitation: 470/40 nm, Dichroic: 495 LP, Emission: 515/30 nm) [60].
Step 3: Image Untreated Control Cells
- Plate and image your Cyt-C-GFP reporter cells (e.g., PC9 or T47D lines as used in the cited research [42]) under normal conditions.
- Expected Result: A punctate, mitochondrial pattern of fluorescence.
- Protocol: Culture cells in appropriate medium (e.g., RPMI-1640 for PC9 cells with 10% FBS [42]) on glass-bottom dishes. Acquire images using a high-numerical-aperture (NA) objective (e.g., 60x or 100x oil immersion) to resolve individual mitochondria. Set exposure times and laser power to avoid pixel saturation.
Step 4: Image Apoptosis-Induced Cells
- Treat reporter cells with an apoptotic inducer relevant to your system.
- Protocol: Treat cells with a known intrinsic apoptosis inducer, such as 1 µM Staurosporine or 1 µM Doxorubicin [42]. Perform live-cell imaging over a time course (e.g., 0-24 hours) to capture the dynamics of Cyt-C release. Maintain cells at 37°C and 5% CO₂ during imaging.
- Expected Result: A progressive shift from a punctate to a diffuse, homogeneous cytosolic fluorescence pattern indicates successful cytochrome-C release.
Step 5: Quantify Signal Translocation
- Use automated image analysis software to quantify the fluorescence translocation.
- Protocol: Implement a version of the automated algorithm described in the research [42]. The methodology should:
- Segment individual cells.
- Calculate a cytoplasmic-to-mitochondrial fluorescence ratio or a similar metric for spatial distribution.
- Classify cells as apoptotic or non-apoptotic based on a validated threshold.
- A successful filter set will enable the algorithm to achieve high precision (>90%) and sensitivity (>85%) in this classification [42].
Step 6: Optimize and Document Settings
- If the signal is weak or the background is high, iteratively adjust the filter set or image acquisition settings (e.g., exposure time, gain). Once optimized, document all filter specifications and acquisition parameters meticulously to ensure experimental reproducibility.
The Scientist's Toolkit: Key Reagents and Materials
Table 3: Essential Research Reagents for Cyt-C-GFP Reporter Assays
| Item | Specification / Example | Function in the Experiment |
|---|---|---|
| Reporter Cell Line | PC9 or T47D cells with stably integrated Cyt-C-GFP [42]. | Reports intrinsic apoptosis via translocation of fluorescence from mitochondria to cytosol. |
| Apoptosis Inducer | Staurosporine, Doxorubicin, TRAIL [42]. | Triggers the intrinsic or extrinsic apoptotic pathway to activate the reporter. |
| Microscope Filter Set | Excitation: 470/40 nm, Dichroic: 495 LP, Emission: 515/30 nm [60]. | Isolates GFP fluorescence; critical for achieving high signal-to-noise in detection. |
| Cell Culture Medium | RPMI-1640 or DMEM, supplemented with 10% FBS and antibiotics [42]. | Supports the growth and maintenance of the reporter cell line. |
| Live-Cell Imaging Chamber | Temperature and CO₂-controlled chamber. | Maintains cell viability and normal physiology during time-lapse imaging. |
| Image Analysis Software | MATLAB, ImageJ (Fiji) with custom algorithms [42]. | Automates the quantification of fluorescence translocation for robust, high-throughput analysis. |
Ensuring Consistent GFP Expression through Culture Stability and Passage Control
For researchers constructing cytochrome C GFP reporter cell lines, maintaining consistent fluorescent protein expression is not merely a technical convenience but a fundamental prerequisite for generating reliable and reproducible data. The integrity of this data, particularly in high-stakes fields like drug development, hinges on the stability of the reporter gene throughout extended culture periods. A primary, yet often underestimated, factor influencing this stability is culture passage number. Evidence compellingly demonstrates that as cell lines undergo repeated subculturing, they experience alterations in morphology, growth rates, and critically, transfection efficiency and protein expression levels [61]. These passage-dependent phenotypic drifts can significantly compromise experimental outcomes, leading to variable reporter signal intensity and confounding the interpretation of results related to cellular events, such as apoptosis studied via cytochrome C-GFP fusions [19]. This application note provides a structured framework for monitoring and controlling culture stability, ensuring that GFP expression in reporter cell lines remains a dependable tool for scientific discovery.
The Critical Impact of Passage Number on Cell Lines
The passage number of a cell line refers to the number of times a culture has been subcultured, or passaged, from one vessel to another. As this number increases, cumulative genetic and phenotypic changes can alter the baseline characteristics of the cell population.
Documented Evidence of Passage Effects
The scientific literature contains robust evidence of passage-dependent effects across various cell lines. The table below summarizes key findings from peer-reviewed studies and technical reports.
Table 1: Documented Effects of High Passage Number on Cell Lines
| Cell Line | Observed Passage-Dependent Change | Implication for Research |
|---|---|---|
| MIN-6 (Mouse insulinoma) | Significant differential expression of nearly 1,000 genes between low (P18) and high (P40) passage cells [61]. | Alters the differentiation state and functional profile of the cell model. |
| LNCaP (Human prostate cancer) | Regulation of androgen receptor activity by the PI3K/Akt pathway changes with passage [61]. | May lead to inconsistent responses to stimuli and irreproducible signaling data. |
| Caco-2 & MCF7 | High-passage Caco-2 showed increased GFP reporter expression after transfection, while high-passage MCF7 showed a decrease [61]. | Effect is cell line-specific and unpredictable; highlights the need for individual validation. |
| General Continuous Cell Lines | Genomic instability, dedifferentiation, and loss of tissue-specific function [61]. | The cell population no longer accurately represents the original biological source. |
The underlying mechanisms driving these changes often involve the evolutionary processes of natural selection within a heterogeneous cell population. Cells with a faster growth rate can overgrow others, leading to a population that may no longer represent the original starting material [61]. For transformed cell lines used in reporter assays, pre-existing mutations in checkpoint genes like p53 can exacerbate this genomic instability over time.
Quantitative Monitoring of GFP Reporter Expression
Ensuring consistent performance from a cytochrome C GFP reporter cell line requires establishing baseline data and routinely monitoring key parameters against these benchmarks.
Establishing a Growth Curve and Monitoring Schedule
A fundamental tool for maintaining culture consistency is the growth curve. Performing a growth curve analysis provides critical information for determining the optimal time for subculturing, estimating plating efficiency, and calculating population doubling times [61]. Researchers should establish a growth curve for their specific cytochrome C GFP reporter line at a low, reference passage number and routinely compare the growth properties of working cultures against this standard.
Table 2: Key Parameters for Routine Monitoring of Reporter Cell Lines
| Parameter | Monitoring Method | Acceptance Criteria |
|---|---|---|
| Cellular Morphology | Frequent visual observation under a microscope; maintain a digital image library for comparison [61]. | No abrupt changes in cell size, shape, or granulation. |
| Growth Rate & Doubling Time | Growth curve analysis performed at regular intervals (e.g., every 5-10 passages) [61]. | Consistent with the established baseline growth curve. |
| GFP Expression Intensity | Flow cytometry (Mean Fluorescence Intensity) or fluorescence microscopy [13] [62]. | Stable MFI and proportion of GFP-positive cells within an acceptable range (e.g., ±15% of baseline). |
| Passage Number | Meticulous record-keeping of subculturing events. | Maintain cultures within a pre-defined, validated passage range. |
Protocols for Maintaining Stable GFP Expression
Protocol 1: Routine Passage and Monitoring of Cytochrome C GFP Reporter Cells
This protocol outlines the steps for the routine maintenance and quality control of stably transduced cytochrome C GFP reporter cell lines.
Materials & Reagents:
- Cytochrome C GFP reporter cell line (e.g., HepG2, HEK293T)
- Complete cell culture medium
- Dulbecco's Phosphate-Buffered Saline (PBS), without Mg²⁺ and Ca²⁺ [62]
- Trypsin-EDTA solution [62]
- Culture vessels (T-flasks, plates)
- Centrifuge
- Inverted fluorescence microscope
- Flow cytometer (optional, for quantitative analysis)
Procedure:
- Observation: Prior to passaging, observe cells under an inverted fluorescence microscope. Check for overall health, confluence, and a qualitative assessment of GFP fluorescence. Document morphology with images.
- Harvesting:
- Aspirate the culture medium from the flask.
- Wash the cell monolayer gently with sterile PBS to remove residual serum.
- Add a pre-warmed Trypsin-EDTA solution (e.g., 0.5 mL for a T25 flask) and incubate at 37°C for 2-5 minutes [62].
- Confirm cell detachment under the microscope and inhibit trypsin by adding complete culture medium.
- Subculturing:
- Gently resuspend the cells to achieve a single-cell suspension.
- Take a small aliquot for counting and viability assessment (e.g., using a TC20 cell counter [62]).
- Centrifuge the remaining cell suspension (5 minutes at 300 x g) and resuspend the pellet in fresh, pre-warmed complete medium [62].
- Seed new culture vessels at the recommended density. Record the split ratio and the new passage number.
- Quantitative Monitoring (Every 3-5 passages):
- Use a sample of the cell suspension for flow cytometry analysis.
- Analyze the percentage of GFP-positive cells and the Mean Fluorescence Intensity (MFI). Compare these values to the established baseline for your cell line.
Protocol 2: Validating Reporter Response via Apoptosis Induction
This protocol describes a method to confirm the functional integrity of the cytochrome C GFP reporter system by inducing apoptosis and monitoring GFP localization.
Materials & Reagents:
- Stably transduced cytochrome C GFP reporter cells
- Apoptosis-inducing agent (e.g., Staurosporine)
- Paraformaldehyde (4%) for fixation [62]
- Hoechst or DAPI nuclear stain
- Confocal or high-resolution fluorescence microscope
Procedure:
- Seed cells on glass-bottom culture dishes or coverslips and allow them to adhere overnight.
- Treat cells with an appropriate concentration of the apoptosis-inducing agent. Include an untreated control.
- Incubate for the required time (e.g., 2-6 hours, to be optimized).
- Fix cells with 4% paraformaldehyde for 15 minutes at room temperature, then wash with PBS [62].
- Stain nuclei with Hoechst or DAPI.
- Image cells using a confocal microscope. In healthy cells, the cytochrome C-GFP fusion should show a punctate, mitochondrial pattern. Upon apoptosis induction, the release of cytochrome C into the cytoplasm should be visible as a diffuse, pan-cellular GFP signal. A failure to see this expected pattern may indicate issues with the reporter construct or cellular health.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Cytochrome C GFP Reporter Line Development and Maintenance
| Reagent / Tool | Function / Application | Example |
|---|---|---|
| Lentiviral Vector System | Stable integration of the cytochrome C-GFP reporter construct into the host cell genome for consistent long-term expression [13] [62]. | pFUGW-based vector [13]; pHAGE2-Ef1a-eGFP-IRES-PuroR [62]. |
| Selection Antibiotic | Selection and maintenance of stably transduced cell pools, eliminating non-expressing cells. | Puromycin [62], Blasticidin [21]. |
| Fluorescence-Activated Cell Sorter (FACS) | Isolation of a pure population of high-GFP-expressing cells (single-cell cloning) and quantitative monitoring of expression stability [63]. | BD FACS Canto II [62]. |
| CRISPR-Cas9 System | Validating gene editing outcomes or creating isogenic control lines; can be used to engineer the host genome to improve protein production [21] [63]. | SpCas9-NLS, gene-specific sgRNA [62]. |
| Linear Polyethylenimine (PEI) | Transfection reagent for delivering plasmid DNA, including reporter constructs and gene-editing machinery [62]. | Linear PEI, MW 25,000 [62]. |
Visualizing Workflow and Signaling Pathways
Workflow for Establishing a Stable Reporter Cell Line
The diagram below outlines the key steps in developing and validating a cytochrome C GFP reporter cell line, integrating strategies for maintaining consistency through passage control.
Mitochondrial Apoptosis Pathway & Cytochrome C Release
This diagram illustrates the core signaling pathway under investigation with a cytochrome C GFP reporter, highlighting the key event the reporter is designed to visualize.
Assessing Performance: Validation, Specificity, and Comparative Analysis
Within the framework of cytochrome C GFP reporter cell line construction research, the accurate quantification of apoptotic events is paramount. The transition of cytochrome C from the mitochondria to the cytosol is a definitive early marker of the intrinsic apoptotic pathway [42]. The development of reporter cell lines, where cytochrome C is tagged with Green Fluorescent Protein (GFP), allows for the live-cell, spatial-temporal monitoring of this critical event. However, the full potential of this technology is only realized with robust, automated image analysis algorithms capable of translating fluorescent signal translocation into reliable, quantitative data. This application note details a validated methodology that synergizes a cytochrome C-GFP reporter cell line with a tunable automated algorithm to achieve exceptional performance metrics of >90% precision and >85% sensitivity in apoptosis detection, providing a powerful tool for high-throughput drug screening and mechanistic studies [42].
Core Validation Metrics and Biological Context
The performance of an apoptosis detection system is foundational to its utility in research and drug discovery. The cited study, which forms the basis of this protocol, established a rigorous validation framework for its automated algorithm using cytochrome C-GFP (Cyt-C) and caspase reporter cell lines in both lung (PC9) and breast (T47D) cancer models [42]. The achieved metrics are summarized in the table below.
Table 1: Key Validation Metrics for Automated Apoptosis Detection Algorithm
| Performance Metric | Reported Performance | Experimental Context |
|---|---|---|
| Precision | >90% | Quantification of fluorescent signal translocation in single or multiple cells. |
| Sensitivity | >85% | Detection of Cyt-C release and caspase activation upon apoptotic stimuli. |
| Cell Lines Used | PC9 (Lung Cancer), T47D (Breast Cancer) | Validation across different cellular backgrounds. |
| Key Apoptotic Stimuli | Staurosporine, H2O2 | Induction of intrinsic apoptotic pathway. |
The biological relevance of this approach is underscored by the central role of TP53-mediated apoptosis in cancer therapeutics. In breast cancer, for instance, TP53 mutations occur in approximately 30% of cases, a frequency that rises to 60-80% in triple-negative breast cancer (TNBC) [64]. These mutations disrupt normal apoptotic machinery, leading to therapy resistance. Therefore, assays that precisely measure the functional output of apoptotic pathways, such as cytochrome C release, provide critical insights into a tumor's therapeutic vulnerability and potential resistance mechanisms [64].
Detailed Experimental Protocol
Cytochrome C-GFP Reporter Cell Line Construction and Culture
This protocol enables the creation of a stable cell line for live-cell imaging of cytochrome C translocation.
- Objective: To generate a stable cell line expressing cytochrome C fused to GFP for monitoring mitochondrial release during apoptosis.
- Materials:
- Human cancer cells (e.g., PC9 lung cancer or T47D breast cancer cells).
- Cytochrome C-GFP fusion construct.
- Retroviral or lentiviral packaging system.
- Appropriate cell culture media (e.g., RPMI for PC9 cells).
- Selection antibiotic (e.g., Puromycin).
- Fluorescence-activated cell sorter (FACS).
- Procedure:
- Construct Transduction: Transduce the target cells with the viral particles containing the cytochrome C-GFP construct.
- Selection and Clonal Expansion: Apply the appropriate selection antibiotic to the culture medium for 1-2 weeks to select for successfully transduced cells. Subsequently, use FACS to isolate single cells or cell populations exhibiting high GFP fluorescence intensity and expand them clonally [42] [6].
- Functional Validation: Treat the stable reporter cells with a known apoptotic inducer (e.g., 1 µM Staurosporine for 2-6 hours). Confirm the translocation of the GFP signal from a punctate mitochondrial pattern to a diffuse cytosolic pattern using fluorescence microscopy [42].
Automated Image Acquisition and Analysis for Apoptosis Quantification
This protocol uses a custom algorithm to achieve high-precision, high-sensitivity detection of apoptosis.
- Objective: To automatically and quantitatively analyze the translocation of cytochrome C-GFP signal as a measure of apoptosis.
- Materials:
- Stable cytochrome C-GFP reporter cell line.
- Apoptotic inducing agent (e.g., Staurosporine, H2O2).
- Live-cell imaging chamber with controlled CO₂ and temperature.
- Epifluorescence or confocal microscope.
- MATLAB software with the custom-developed algorithm [42].
- Procedure:
- Image Acquisition:
- Plate reporter cells in a multi-well imaging plate.
- Induce apoptosis by adding the chosen stimulus. Include negative control wells (vehicle only).
- Acquire time-lapse fluorescence images at regular intervals (e.g., every 15-30 minutes) post-induction using a microscope equipped with a live-cell imaging chamber.
- Algorithmic Analysis:
- Input: Load the acquired time-lapse image series into the MATLAB environment.
- Feature Extraction: The algorithm identifies and tracks individual cells over time, extracting features related to the spatial distribution of the fluorescent signal. It moves beyond simple intensity statistics to capture the texture and pattern changes associated with cytochrome C release [42].
- Signal Translocation Classification: For each cell and time point, the algorithm classifies the state of cytochrome C based on the extracted features, determining whether it is "mitochondrial" (punctate) or "cytosolic" (diffuse).
- Output: The algorithm generates quantitative data, including the percentage of cells undergoing apoptosis at each time point and the timing of the event for individual cells.
- Image Acquisition:
Diagram: Automated Analysis Workflow
Pathway Logic and Experimental Rationale
Understanding the signaling pathway being monitored is crucial for experimental design and data interpretation. The intrinsic apoptotic pathway, triggered by cellular stress, is the primary process detected by the cytochrome C-GFP reporter.
Diagram: Cytochrome C in Intrinsic Apoptosis
The Scientist's Toolkit: Essential Research Reagents and Materials
The successful implementation of this high-performance apoptosis detection assay relies on a set of core reagents and tools. The following table lists the essential components.
Table 2: Key Research Reagent Solutions for Apoptosis Detection
| Item Name | Function / Description | Example Use-Case in Protocol |
|---|---|---|
| Cytochrome C-GFP Reporter Construct | Plasmid for creating stable cell lines; encodes cytochrome C fused to GFP. | Stably integrated into PC9 or T47D cells to visualize mitochondrial cytochrome C release. |
| Apoptotic Inducers (Staurosporine, H₂O₂) | Chemical stimuli that trigger the intrinsic apoptotic pathway. | Used to validate reporter function and induce apoptosis in experimental assays. |
| Annexin V-FITC Apoptosis Detection Kit | Commercial kit using Annexin V binding to phosphatidylserine for early apoptosis detection. | Can be used as a secondary, orthogonal method to validate cytochrome C release data [65] [66]. |
| MATLAB with Custom Algorithm | Software environment and code for automated image analysis of signal translocation. | Processes time-lapse images to quantify apoptosis with >90% precision and >85% sensitivity [42]. |
| Flow Cytometry Instrument | Instrument for high-throughput, single-cell analysis of fluorescence. | Used for initial sorting of high-expressing reporter cells and can be used for end-point apoptosis analysis. |
Technical Notes and Integration in Broader Research
- Advantages over Traditional Methods: This single-color, live-cell reporter system eliminates the need for fixatives or additional dyes, allowing for dynamic, longitudinal studies of the same cell population. The automated algorithm overcomes the bias and limitations of manual thresholding and simple image statistics, providing a robust and scalable solution for high-throughput screening [42].
- Integration with Broader Biomarker Discovery: The precision of this assay makes it highly valuable for validating therapeutic strategies that target apoptotic pathways. For example, it can be used to assess the efficacy of PARP inhibitors in BRCA-mutated cells or to study the synthetic lethal interactions that are a major focus of modern targeted cancer therapy [67]. Furthermore, the quantitative data generated is amenable to integration with multi-biomarker machine learning models, enhancing predictive capabilities for complex disease outcomes [68].
- Troubleshooting: A key validation step is confirming that the GFP tag does not interfere with the native function of cytochrome C in the electron transport chain or its release during apoptosis. Prior studies have established that this is not the case, but this should be contextually verified [42]. High background fluorescence in the cytosolic compartment post-induction may indicate high levels of secondary necrosis; optimizing the timing and concentration of the apoptotic stimulus is critical.
Correlating GFP Fluorescence with mRNA Abundance for Quantitative Accuracy
Within the context of cytochrome C GFP reporter cell line construction, establishing a robust correlation between green fluorescent protein (GFP) fluorescence and underlying mRNA abundance is critical for quantitative accuracy in reporting transcriptional activity. This relationship is foundational for interpreting data from reporter systems, where GFP serves as a proxy for gene expression driven by specific promoters, such as that of the cytochrome C gene (Cycs) [69]. However, this correlation is not automatic; it is influenced by a complex interplay of post-transcriptional regulation, translation efficiency, and protein stability [70]. This Application Note details protocols and analytical strategies to empirically validate and ensure that measured fluorescence accurately reflects mRNA levels in your reporter system.
The Critical Link Between Reporter mRNA and Fluorescence
The pathway from gene activation to detectable GFP fluorescence is multi-staged. A transcriptional stimulus, such as neuronal activity activating the Cycs promoter, leads to the production of GFP mRNA [69]. This mRNA is then translated into GFP protein, which must properly fold to become fluorescent [71].
The 3' untranslated region (3'UTR) of the mRNA plays a particularly significant role in this process. It is a major hub for post-transcriptional regulation, housing elements that control RNA stability, localization, and translational efficiency [70]. Consequently, the choice of 3'UTR in your reporter construct can profoundly impact the relationship between mRNA abundance and protein output.
Furthermore, it cannot be assumed that fluorescence intensity linearly correlates with mRNA concentration across all experimental conditions. Factors such as the health of the cells, availability of cellular resources for translation, and the maturation time of the GFP chromophore can all decouple fluorescence from mRNA levels. Therefore, direct measurement of both parameters is necessary for confident quantification.
Application to Cytochrome C Reporter Systems
The cytochrome C gene (Cycs) proximal enhancer is a well-established model for studying activity-dependent gene expression in neurons. This enhancer contains binding sites for transcription factors like CREB and NRF1, linking neuronal activity to mitochondrial function [69].
Reporter Construct Design
In foundational research, a 235 bp fragment of the rat Cycs proximal enhancer (from -73 to -308 relative to the transcription start site) has been successfully used to drive reporter expression [69]. The experimental workflow for implementing and validating such a cytochrome C GFP reporter is systematic.
Key Considerations for Cycs Systems
- Promoter Specificity: The Cycs enhancer used is responsive to calcium influx through NMDA receptors and L-type channels, and its activity involves ERK1/2 signaling [69]. These specific pathway dependencies should be considered when designing stimulation experiments.
- Validation: In the original study, reporter gene expression driven by this element correlated with increased amplitude of miniature postsynaptic currents (mEPSCs), demonstrating its functional relevance to synaptic strength [69].
Experimental Protocols
Below are detailed protocols for the key experiments required to correlate GFP fluorescence with mRNA abundance.
Protocol 1: Quantifying GFP Fluorescence in Live Cells
This protocol is adapted from plate reader-based assays for quantifying GFP in cell cultures [72] [73].
Materials:
- Reporter cell line (e.g., hippocampal neurons with Cycs-GFP reporter) [69]
- Black-walled, clear-bottom 96-well plate (e.g., ThermoFisher #M33089) [72]
- Multi-mode microplate reader capable of fluorescence and OD600 measurements (e.g., Agilent BioTek Cytation 1) [72]
- Culture medium (e.g., Dulbecco’s Modified Eagle Medium supplemented with 10% FBS) [69]
- Phosphate Buffered Saline (PBS)
Method:
- Seed Cells: Plate cells into the 96-well plate at a density optimized for your cell line. Include control wells with non-transfected/untreated cells for background autofluorescence measurement.
- Apply Stimulus: Treat cells with the experimental stimulus (e.g., 20 µM bicuculline for neuronal activation) [69].
- Plate Reader Setup:
- Data Acquisition: Run the protocol. Ensure the gain is set to a level that avoids signal saturation.
- Data Normalization:
- Subtract the average fluorescence and OD600 of the control (media-only) wells from all sample wells.
- Further normalize the GFP signal from experimental wells by subtracting the signal from non-transfected control wells grown and treated under identical conditions to correct for cellular autofluorescence [72].
- Finally, divide the background- and autofluorescence-corrected GFP signal by the corrected OD600 to obtain a density-normalized fluorescence value.
Protocol 2: Measuring GFP mRNA Abundance by RT-qPCR
This protocol describes mRNA quantification from cell samples, a critical step for correlation.
Materials:
- RNA extraction kit (e.g., kits from Zymo Research, Qiagen, or Thermo Fisher)
- DNase I, RNase-free
- Reverse transcription kit (e.g., High-Capacity cDNA Reverse Transcription Kit)
- qPCR reagents (e.g., SYBR Green or TaqMan Master Mix)
- Primers specific for GFP and a housekeeping gene (e.g., GAPDH, β-actin)
Method:
- RNA Extraction:
- Lyse cells directly in the culture dish or well using an appropriate lysis buffer.
- Extract total RNA according to your kit's instructions.
- Treat the extracted RNA with DNase I to remove any contaminating genomic DNA.
- Quantify RNA concentration and purity using a spectrophotometer.
- Reverse Transcription:
- Use equal amounts of total RNA (e.g., 500 ng - 1 µg) from each sample for cDNA synthesis in a 20 µL reaction.
- Perform the reverse transcription as per the kit protocol.
- Quantitative PCR:
- Dilute the synthesized cDNA 1:5 to 1:10 with nuclease-free water.
- Prepare qPCR reactions in triplicate for each sample. A typical 20 µL reaction contains:
- 10 µL of 2X Master Mix
- Forward and reverse primers (final concentration 200-500 nM each)
- 2-5 µL of diluted cDNA template
- Nuclease-free water to 20 µL.
- Run the qPCR with the following cycling conditions:
- Step 1: 95°C for 10 minutes (polymerase activation)
- Step 2: 40 cycles of:
- 95°C for 15 seconds (denaturation)
- 60°C for 1 minute (annealing/extension)
- Include a melt curve stage to confirm primer specificity.
- Data Analysis:
- Calculate the average Cq value for the GFP and housekeeping gene triplicates.
- Use the ΔΔCq method to determine the relative fold change in GFP mRNA expression in experimental groups compared to the control group.
Quantitative Data and Correlation Analysis
The table below summarizes key parameters and expected outcomes from a well-correlated experiment, drawing from published methodologies.
Table 1: Expected Quantitative Relationships in a Validated Reporter System
| Experimental Parameter | Measurement Method | Expected Outcome with Good Correlation | Notes |
|---|---|---|---|
| GFP Fluorescence Intensity | Microplate reader (Ex/Em ~488/509 nm) [73] | Linear increase with mRNA levels over dynamic range | Signal must be normalized to cell density and background |
| GFP mRNA Abundance | RT-qPCR (Cq value for GFP) | Linear increase with transcriptional stimulus | Normalize to housekeeping gene (e.g., GAPDH) |
| Correlation Coefficient (R²) | Linear regression (Fluorescence vs. mRNA) | R² > 0.9 indicates a strong linear relationship | Lower values suggest post-transcriptional decoupling |
| Dynamic Range | Dose-response of stimulus | Fluorescence and mRNA show parallel log-linear increases | System should be sensitive to relevant stimuli [69] |
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for Reporter Assay Development and Validation
| Reagent / Tool | Function / Description | Example Use Case |
|---|---|---|
| pFUGW Lentiviral Vector | Lentiviral backbone for stable integration of reporter construct [13] | Creating stable Cycs-GFP HepG2 or neuronal reporter cell lines [13] |
| Enhanced GFP (EGFP/EmGFP) | Optimized GFP variants for brighter fluorescence in mammalian cells [71] | Primary reporter protein for high-sensitivity detection |
| Kozak & Leader Sequences | Regulatory elements placed upstream of the GFP start codon to enhance translation initiation [21] | Boosting GFP protein output without increasing mRNA levels [21] |
| Specific 3'UTR Library | A collection of different 3'UTR sequences to study their impact on mRNA stability and translation [70] | Identifying a 3'UTR that optimizes the correlation between Cycs-GFP mRNA and fluorescence |
| DeNovix DS-11/QFX | Spectrophotometer/Fluorometer for precise GFP quantification in cell lysates or purified samples [73] | Generating standard curves and accurately measuring total GFP concentration |
Troubleshooting Common Discrepancies
A weak correlation between mRNA and fluorescence can arise from several sources. The following flowchart guides the systematic troubleshooting of this issue, focusing on the mitochondrial apoptosis pathway relevant to cytochrome C biology.
Problem: High mRNA, Low Fluorescence. This often indicates a problem at the protein level.
- Solution 1: Inhibit Apoptosis. Apoptosis can lead to the degradation of GFP. Consider inhibiting the mitochondrial apoptosis pathway. Knocking out Apaf1, a key component of the apoptosome that is activated by cytochrome c, has been shown to reduce apoptosis and increase recombinant protein yields in CHO cells [21].
- Solution 2: Optimize Translation. Ensure your construct contains a strong Kozak sequence (e.g., GCCACC) upstream of the GFP start codon. The addition of a Kozak sequence alone has been shown to increase eGFP expression by 1.26-fold, while a combination of Kozak and Leader sequences increased it by 2.2-fold [21].
Problem: Low Correlation After Stimulus.
- Solution: Evaluate the 3'UTR. The 3'UTR is a major determinant of mRNA stability and translational efficiency [70]. If the endogenous Cycs 3'UTR contains regulatory elements that respond to your stimulus, it could decouple mRNA from protein. Test your promoter with a synthetic, inert 3'UTR to isolate the promoter's activity.
Rigorous correlation of GFP fluorescence with mRNA abundance is not merely a validation step but a fundamental requirement for generating quantitatively accurate data from cytochrome C GFP reporter cell lines. By employing the protocols and analytical frameworks outlined here—including careful construct design, parallel measurement of fluorescence and mRNA, and systematic troubleshooting—researchers can ensure their reporter system faithfully reflects transcriptional dynamics. This precision is essential for advancing research in neuronal metabolism, mitochondrial function, and high-throughput drug screening.
Comparative Analysis with Other Reporter Systems: Caspase-3/-8 Reporters
Within the broader research on constructing cytochrome C GFP reporter cell lines for monitoring the intrinsic apoptosis pathway, the development of parallel reporters for caspase-3 and caspase-8 provides a critical toolkit for dissecting the complex interplay of apoptotic signaling. The cytochrome C reporter serves as a early indicator of mitochondrial outer membrane permeabilization, a key commitment point in the intrinsic pathway [42]. In contrast, caspase-8 reporters illuminate initiation of the extrinsic death receptor pathway, while caspase-3 reporters mark the final convergence point of both pathways, serving as a definitive indicator of execution-phase apoptosis [42] [74] [75]. This application note provides a comparative analysis of these reporter systems, offering detailed protocols and performance data to guide researchers in selecting and implementing the optimal tools for their investigative or screening workflows.
Reporter System Technologies and Performance
Caspase reporters are engineered using diverse fluorescent strategies, each with distinct operational principles and performance characteristics suitable for different experimental needs.
Table 1: Performance Comparison of Apoptosis Reporter Systems
| Reporter Type | Key Technology | Detection Method | Key Advantage | Reported Sensitivity/Performance |
|---|---|---|---|---|
| Caspase-3/-7 (ZipGFP) | Split-GFP reassembly after DEVD cleavage | Fluorescence intensity increase (Green) | Very low background, irreversible signal, marks historical events [75] | Single-cell resolution in 3D spheroids and organoids [75] |
| Caspase-3 (FLIM-FRET) | FRET pair linked by DEVD sequence | Fluorescence Lifetime Imaging (FLIM) | Intensity- and depth-independent; ideal for 3D/in vivo models [76] | Effective in 2D, 3D spheroids, and in vivo tumor xenografts [76] |
| Caspase-8 (Translocation) | NES-DEVD-NLS-EYFP | Signal translocation from cytosol to nucleus | Live-cell monitoring without dyes or fixatives [42] | >90% precision, >85% sensitivity in automated analysis [42] |
| Caspase-3 (SFCAI/VC3AI) | Cyclized Venus activated by DEVD cleavage | Fluorescence intensity increase (Yellow) | Minimal background; "switch-on" functionality [77] | Specific activation in MCF-7 cells (caspase-3 deficient) [77] |
| Cytochrome C-GFP | GFP fused to Cytochrome C | Signal translocation from mitochondria to cytosol | Direct monitoring of intrinsic pathway initiation [42] | Early apoptosis detection prior to caspase activation [42] |
Beyond the technologies summarized in Table 1, alternative designs include bright-to-dark reporters, where an inserted DEVD motif inactivates EGFP fluorescence, which is restored upon caspase-3 cleavage [7]. Furthermore, genetic systems like CasExpress in Drosophila can permanently mark cells that have survived caspase-3 activation, revealing that survival after caspase activation is a widespread phenomenon during development [78].
Detailed Experimental Protocols
Protocol: Caspase-3/-7 ZipGFP Reporter Assay in 3D Cultures
This protocol enables real-time, high-content analysis of apoptosis in physiologically relevant models [75].
A. Generation of Stable Reporter Cell Lines:
- Vector Transduction: Transduce target cells (e.g., MiaPaCa-2, HUVECs, or patient-derived organoids) with a lentiviral vector constitutively expressing the ZipGFP-based caspase-3/-7 reporter and a separate mCherry fluorescent marker.
- Selection and Expansion: Select and expand successfully transduced cells using appropriate antibiotics or fluorescence-activated cell sorting (FACS) based on mCherry expression to establish a homogenous population.
B. 3D Spheroid/Organoid Culture and Treatment:
- Spheroid Formation: Seed reporter cells in low-attachment 96-well plates to promote self-assembly into 3D spheroids, or embed cells in Cultrex or Matrigel to support organoid growth.
- Apoptosis Induction: Treat 3D cultures with the apoptotic stimulus of choice (e.g., 10-100 nM carfilzomib or 50-200 µM oxaliplatin). Include control groups treated with vehicle (e.g., DMSO) and, for specificity validation, a group co-treated with a pan-caspase inhibitor (e.g., 20 µM zVAD-FMK).
- Real-Time Imaging: Transfer cultures to a live-cell imaging system (e.g., IncuCyte). Acquire GFP (caspase activity) and mCherry (cell presence/viability) images every 2-4 hours for 72-120 hours.
- Data Analysis: Quantify total GFP and mCherry fluorescence intensity per well or spheroid. Normalize the GFP signal to the mCherry signal to account for changes in cell number. Caspase activation is indicated by a time-dependent increase in the GFP/mCherry ratio.
Protocol: Caspase-8 Translocation Assay with Automated Analysis
This protocol uses a vision-based algorithm for robust, high-throughput quantification of caspase-8 activation via subcellular translocation [42].
A. Reporter Cell Line Generation and Characterization:
- Construct Design: Create a caspase-8 reporter by fusing EYFP to a nuclear localization sequence (NLS), linked via a peptide containing the caspase-8 cleavage site IETD to a nuclear export signal (NES).
- Stable Expression: Generate stable reporter cell lines (e.g., in PC9 lung cancer or T47D breast cancer cells) using lentiviral transduction or the PiggyBac transposon system.
- Validation: Treat cells with a known caspase-8 activator (e.g., TRAIL) and confirm EYFP translocation from the cytosol to the nucleus via confocal microscopy.
B. Image Acquisition and Automated Analysis:
- Live-Cell Imaging: Plate reporter cells in a 96- or 384-well plate. After experimental treatment, image live cells at regular intervals on a high-throughput microscope, exciting EYFP with a standard GFP filter set.
- Algorithmic Tuning: Implement a custom, tunable algorithm in MATLAB or similar environment. The algorithm should:
- Identify individual cell nuclei.
- Define a cytoplasmic region around each nucleus.
- Calculate the ratio of nuclear to cytoplasmic EYFP fluorescence intensity (N/C ratio).
- Thresholding and Quantification: Establish a threshold N/C ratio for positive caspase-8 activation. A significant increase in the N/C ratio indicates cleavage of the linker and nuclear import of EYFP. The algorithm can achieve >90% precision and >85% sensitivity in classifying apoptotic cells [42].
Diagram 1: Signaling and detection workflow for caspase-8 and caspase-3 reporters.
The Scientist's Toolkit: Key Research Reagents
Table 2: Essential Reagents for Caspase Reporter Assays
| Reagent / Resource | Function / Application | Example Use-Case / Note |
|---|---|---|
| Lentiviral Vectors (pLVX) | Stable integration of reporter constructs into host cell genome [76] | Constitutive expression of ZipGFP or translocation reporters. |
| PiggyBac Transposon System | Non-viral method for stable genomic integration of large constructs [42] | Used in constructing cytochrome C-GFP and caspase reporter cell lines. |
| TRAIL (TNF-related apoptosis-inducing ligand) | Activates the extrinsic apoptosis pathway via death receptors DR4/DR5 [42] [79] | Specific activator for studying caspase-8-initiated apoptosis. |
| Carfilzomib / Oxaliplatin | Inducers of intrinsic apoptosis (proteasome inhibitor / chemotherapeutic) [75] | Used to validate caspase-3/-7 reporter activation in 2D and 3D models. |
| zVAD-FMK (pan-caspase inhibitor) | Irreversible caspase inhibitor; validates caspase-dependent reporter activation [77] [75] | Essential control to confirm signal specificity. |
| z-DEVD-FMK (caspase-3/7 inhibitor) | Specific, irreversible inhibitor of executioner caspases [77] | Confirms that reporter activation is due to caspase-3/-7 activity. |
| FuGENE 6 Transfection Reagent | Non-liposomal transfection of plasmid DNA into mammalian cells [76] | For transient expression or during stable cell line generation. |
| Cultrex / Matrigel | Basement membrane extract for 3D cell culture [75] | Provides a physiological scaffold for spheroid and organoid growth. |
Integrating caspase-3/-8 reporters with a cytochrome C GFP reporter creates a powerful multiplexed platform for comprehensive apoptosis analysis. One can track the temporal sequence from cytochrome C release (intrinsic commitment) to caspase activation and eventual cell demise. Furthermore, the subcellular spatial localization of active caspase-8—diffuse cytosolic in full apoptosis versus concentrated in membrane lipid rafts during T-cell activation—can be a critical functional determinant [74]. Advanced applications include coupling these reporters with proliferation dyes to study apoptosis-induced proliferation or with endpoint immunostaining for surface calreticulin to investigate immunogenic cell death [75].
In conclusion, the selection of a caspase reporter system should be guided by the specific research question. Translocation and ZipGFP reporters offer high specificity and are ideal for high-content screening, while FLIM-FRET reporters provide superior quantification in complex 3D and in vivo environments. When used within a strategic framework that includes the cytochrome C reporter, these tools enable a deep and mechanistic dissection of cell death pathways in basic research and drug discovery.
Within the broader scope of developing a cytochrome c (Cyt c) GFP reporter cell line, functional validation of its release from mitochondria is a critical step. This process confirms the integrity of the apoptotic signaling pathway and the reporter's ability to accurately detect this key event. Cyt c release is a hallmark of the intrinsic apoptotic pathway, and its translocation to the cytosol triggers the formation of the apoptosome and the subsequent activation of executioner caspases [80]. This application note details complementary protocols for confirming Cyt c release using live-cell immunofluorescence and a cell-free SEIRA assay, providing researchers with robust methods for validating their reporter systems.
Experimental Protocols
Live-Cell Imaging and Immunofluorescence for Cyt c Release
This protocol is designed to monitor and quantify Cyt c release in real-time using a GFP reporter, or to confirm release via fixed-cell immunofluorescence.
Materials & Reagents
- Cell line expressing Cyt c-GFP reporter (e.g., generated via CRISPR/Cas9 HDR as in [8])
- Apoptosis inducers: Staurosporine (0.1-1 µM) or other relevant compounds (e.g., MYLS22/Opitor-0 [81])
- Culture medium appropriate for the cell line
- Phosphate-Buffered Saline (PBS)
- Fixative: 4% paraformaldehyde (PFA) in PBS
- Permeabilization/Blocking Buffer: PBS containing 0.1% Triton X-100 and 5% normal goat serum
- Primary Antibody: Anti-cytochrome c antibody (e.g., from Novus [82])
- Secondary Antibody: Fluorophore-conjugated (e.g., Cy3) highly cross-adsorbed antibody
- Nuclear Counterstain: DAPI
- Mounting medium
Procedure
- Seed cells onto Matrigel-coated [8] glass-bottom dishes or 384-well plates [82] at an appropriate density for 24-48 hours of growth.
- Induce Apoptosis: Treat cells with the chosen apoptosis inducer. For a positive control, use 1 µM Staurosporine for 2-6 hours. Include a vehicle control (e.g., DMSO).
- For Live-Cell Imaging (Cyt c-GFP Reporter):
- Place the dish on a pre-warmed stage of a confocal fluorescence microscope.
- Acquire time-lapse images every 5-10 minutes for up to 6 hours post-induction to capture the kinetics of Cyt c-GFP release, which can occur within approximately 5 minutes [83].
- For Fixed-Cell Immunofluorescence (Endogenous Cyt c):
- At designated time points post-induction, wash cells twice with warm PBS.
- Fix cells with 4% PFA for 15 minutes at room temperature.
- Wash three times with PBS.
- Permeabilize and Block with Permeabilization/Blocking Buffer for 1 hour.
- Incubate with primary antibody diluted in blocking buffer overnight at 4°C.
- Wash three times with PBS.
- Incubate with secondary antibody and DAPI for 1 hour at room temperature in the dark.
- Wash three times with PBS and mount with mounting medium.
- Image Acquisition: Use a high-resolution confocal microscope. For each field of view, capture images in the GFP (reporter), Cy3 (immunostained Cyt c), and DAPI (nucleus) channels.
Data Analysis
- In validated Cyt c-GFP reporter cells, a relocalization of fluorescence from a punctate mitochondrial pattern to a diffuse cytoplasmic pattern indicates release [82].
- In immunofluorescence, compare the staining pattern in induced cells versus controls. A diffuse cytoplasmic pattern confirms release, while a punctate pattern indicates mitochondrial retention.
- Quantify the percentage of cells with released Cyt c by counting cells with diffuse fluorescence across multiple fields of view (typically >200 cells per condition).
Cell-Free SEIRA Assay for Cytochrome c
This cell-free assay utilizes surface-enhanced infrared absorption (SEIRA) spectroscopy to detect and characterize Cyt c, providing a biochemical complement to cellular imaging [84].
Materials & Reagents
- Gold Nanocluster Probe: Glutathione-coated Au25(SG)18 nanoclusters [84]
- Purified cytochrome c protein
- Apoptotic cell lysate cytosolic fractions (prepared by differential centrifugation)
- IR-transparent prism (e.g., diamond)
Procedure
- Sample Preparation:
- Prepare the Au25(SG)18/cyt c assembly by mixing the gold nanocluster probe with the sample containing cytochrome c (purified protein or cytosolic fraction).
- For a standard assay, boost the stoichiometry of the assembly to enhance the SEIRA signal [84].
- Incubate the mixture for a few minutes at room temperature.
- Infrared Sensing:
- Place a single drop of the assembly on a diamond prism.
- Acquire infrared spectra immediately.
- Data Acquisition:
- Measure the infrared vibrational signatures of the sample. The entire process from sample preparation to data acquisition can be completed within minutes [84].
Data Analysis
- The presence of cytochrome c is indicated by its characteristic infrared absorption bands.
- The signal evolution is enhanced by more than two orders of magnitude due to the Au25 sensor/cyt c assembly [84].
- Use curve-fitting analysis to determine structural modifications of the assembled cytochrome c, which can be verified with complementary techniques like circular dichroism [84].
Research Reagent Solutions
Table 1: Essential Research Reagents for Cytochrome c Release Studies
| Reagent/Category | Specific Examples | Function & Application |
|---|---|---|
| Chemical Inducers | MYLS22, Opitor-0 [81] | OPA1 GTPase inhibitors; induce cristae remodeling and sensitize cells to cytochrome c release. |
| Apoptosis Inducers | Staurosporine, H₂O₂ [7] | General inducers of intrinsic apoptosis; used as positive controls in validation experiments. |
| Reporter Tags | Green Fluorescent Protein (GFP) [7] [8] | Tag for cytochrome c to create a live-cell reporter for real-time tracking of localization. |
| Detection Nanosensor | Au25(SG)18 Gold Nanoclusters [84] | Cell-free SEIRA substrate for enhanced infrared detection and structural analysis of cytochrome c. |
| Key Antibodies | Anti-cytochrome c [82] | Immunodetection of endogenous cytochrome c in fixed cells via immunofluorescence. |
| CRISPR/Cas9 Tools | HDR Donor Template, Cas9/gRNA RNP [8] | For precise knock-in of reporter constructs (e.g., Cyt c-GFP) into specific genomic loci in host cells. |
Expected Results & Data Interpretation
Quantitative Data from Cyt c Release Studies
Table 2: Benchmarking Cytochrome c Release in Experimental Models
| Experimental System / Treatment | Key Readout | Reported Effect / Kinetics | Reference |
|---|---|---|---|
| Cyt c-GFP Release (General) | Release Kinetics | ~5 minutes duration for complete release during apoptosis. | [83] |
| Bright-to-Dark Caspase Reporter | Apoptosis Sensitivity | Showed greater sensitivity for apoptosis detection compared to dark-to-bright systems. | [7] |
| OPA1 Inhibition (MYLS22) | Therapy Sensitization | Enhanced cytochrome c release and restored sensitivity to anti-Bcl-2 therapy in cancer cells. | [81] |
| Au25(SG)18 SEIRA Assay | Detection Signal | SEIRA signal increased by >2 orders of magnitude with optimized assembly stoichiometry. | [84] |
| Apaf1 Knockout in CHO cells | Recombinant Protein Yield | Increased production of recombinant proteins by inhibiting cytochrome c-mediated apoptosis. | [21] |
When performing validation, researchers should expect to observe a clear transition from a punctate to a diffuse fluorescence pattern in the Cyt c-GFP reporter cell line upon induction of apoptosis. The kinetics of this release should be rapid. In the cell-free SEIRA assay, a successful detection will show the characteristic infrared signature of cytochrome c, with a significant signal enhancement confirming the assay's sensitivity.
Workflow Visualization
Diagram 1: Integrated workflow for functional validation of cytochrome c release, combining cellular imaging and cell-free analytical techniques.
Diagram 2: Cytochrome c signaling pathway and the Apaf-1-mediated cell fate switch between apoptosis and inflammation, a key functional context for reporter validation [80].
Conclusion
The construction of cytochrome C-GFP reporter cell lines represents a significant advancement in cell biology and drug discovery, enabling the real-time, quantitative analysis of apoptotic events in living cells. By integrating a robust biological understanding with refined methodological protocols, optimized troubleshooting approaches, and rigorous validation standards, researchers can create powerful tools for high-throughput screening. The future of this technology points towards more sophisticated multi-parametric reporter systems, increased application in personalized medicine for patient-specific drug response profiling, and broader use in evaluating cardiomyotoxicity and other off-target effects of chemotherapeutic agents. This solid foundation will continue to drive innovations in biomedical research and clinical therapeutic development.
References
- 1. Cytochrome c: the Achilles' heel in apoptosis - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11114681/]
- 2. The role and application potential of cytochrome c in breast ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12589747/]
- 3. Cytochrome c and cancer cell metabolism: A new perspective [https://www.sciencedirect.com/science/article/pii/S1319016424002457]
- 4. Mechanisms of cytochrome c release from mitochondria [https://www.nature.com/articles/4401950]
- 5. Caspases as master regulators of programmed cell death [https://www.nature.com/articles/s12276-025-01470-9]
- 6. A new live-cell reporter strategy to simultaneously monitor ... [https://www.nature.com/articles/srep17217]
- 7. Designing an apoptosis reporter by mutagenesis-based ... [https://www.sciencedirect.com/science/article/pii/S2090123225004795]
- 8. Protocol for the Generation of Human Pluripotent Reporter ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7566848/]
- 9. Green fluorescent protein is a quantitative reporter of gene ... [https://pubmed.ncbi.nlm.nih.gov/15640280/]
- 10. Sublethal Cytochrome-c release generates drug-tolerant ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9450215/]
- 11. Secretory GFP reconstitution labeling of neighboring cells ... [https://www.nature.com/articles/s41467-023-43855-2]
- 12. How to use Fiji to automatically analyze GFP expression ... [https://forum.image.sc/t/how-to-use-fiji-to-automatically-analyze-gfp-expression-levels-for-each-cell/3990]
- 13. Development and Application of a CAFLUX HepG2 Reporter ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12564245/]
- 14. A reporter cell line for real-time imaging of autophagy and ... [https://www.sciencedirect.com/science/article/abs/pii/S0378427420300606]
- 15. Green fluorescent protein (GFP) as gene expression ... [https://www.sciencedirect.com/science/article/abs/pii/S0378109799002189]
- 16. Measuring cell fluorescence using ImageJ - The Open Lab Book [https://theolb.readthedocs.io/en/latest/imaging/measuring-cell-fluorescence-using-imagej.html]
- 17. Biogenesis of cytochromes c and c1 in the electron transport ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11991887/]
- 18. GFP tagging sheds light on protein translocation [https://pmc.ncbi.nlm.nih.gov/articles/PMC11115126/]
- 19. A cytochrome c-GFP fusion is not released from ... [https://pubmed.ncbi.nlm.nih.gov/10767430/]
- 20. A cytochrome c–GFP fusion is not released from ... [https://www.sciencedirect.com/science/article/pii/S0014579300014046]
- 21. Optimization of a novel expression system for recombinant ... [https://www.nature.com/articles/s41598-024-76995-6]
- 22. Live Cell Imaging, Live Cell Applications, Time-Lapse ... [https://www.moleculardevices.com/applications/live-cell-imaging]
- 23. 5 Reasons Why Drug Discovery Leaders Choose Live ... [https://www.saguarobio.com/fr/blogs/resources/5-reasons-why-drug-discovery-leaders-choose-live-cells-for-their-cell-painting-assay]
- 24. Live Cell Painting: A New Frontier in Cellular Profiling [https://www.springscience.com/case-study/live-cell-painting-a-new-frontier-in-cellular-profiling]
- 25. Quantitative comparison of fluorescent proteins ... - PubMed [https://pubmed.ncbi.nlm.nih.gov/40326462/]
- 26. Image-Based Profiling in Live Cells Using Live Cell Painting [https://pmc.ncbi.nlm.nih.gov/articles/PMC12514141/]
- 27. A concise guide to fluorescent cell cycle reporters for live- ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2025.1702230/full]
- 28. The tumor suppressor LACTB remodels mitochondria to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12609066/]
- 29. Vector Selection Guide [https://synbio-tech.com/vector-guide]
- 30. A dual-reporter system for investigating and optimizing ... [https://www.nature.com/articles/s41467-021-26337-1]
- 31. Stable Cell Line Generation for Reliable Research [https://www.betalifesci.com/blogs/news/stable-cell-line-generation-for-reliable-research?srsltid=AfmBOooJEaOLEB891x0z96LEvaPsiU7IkX-t1bzsEYJ3-OO-Zn69WpX_]
- 32. Stable Cell Line Generation | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/references/gibco-cell-culture-basics/transfection-basics/applications/stable-transfection.html]
- 33. Cell Line Generation: Methods, Uses & Key Considerations [https://www.bosterbio.com/blog/post/learn-about-cell-line-generation?srsltid=AfmBOopWmY_l5Xpc5HDkzDumT8QT4JNMt8Qr1efVN8taEvyTBIoTGvyP]
- 34. Cell Line Development Workflow, Recombinant Proteins [https://www.moleculardevices.com/applications/cell-line-development-workflow]
- 35. Stable Cell Line Development Market [https://www.futuremarketinsights.com/reports/stable-cell-line-development-market]
- 36. Establishing a Highly Accurate Circulating Tumor Cell ... [https://www.mdpi.com/2072-6694/17/14/2289]
- 37. T47D Cells [https://www.cytion.com/us/T47D-Cells/300353]
- 38. a comparative study of T47D and MCF7 cell lines in 2D ... [https://www.frontiersin.org/journals/toxicology/articles/10.3389/ftox.2025.1547640/full]
- 39. Integrative machine learning approach for forecasting lung ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12329548/]
- 40. CRISPR-mediated modulation of EGFR signaling in lung ... [https://www.sciencedirect.com/science/article/pii/S2468294225001285]
- 41. Cancer Cell Lines and How CRISPR is Transforming ... [https://www.synthego.com/blog/crispr-cancer-cell-lines/]
- 42. Apoptosis detection via automated algorithms to analyze ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7703573/]
- 43. A high-throughput drug screen reveals means to ... [https://www.nature.com/articles/s41388-022-02429-0]
- 44. The Extended ToxTracker Assay Discriminates Between ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5009621/]
- 45. High throughput fluorescence imaging approaches for drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4863443/]
- 46. A systematic bi-genomic split-GFP assay illuminates the ... [https://elifesciences.org/reviewed-preprints/98889]
- 47. Interference and Artifacts in High-content Screening - NCBI - NIH [https://www.ncbi.nlm.nih.gov/books/NBK615088/]
- 48. Comparison of culture media supplements identifies serum ... [https://stemcellres.biomedcentral.com/articles/10.1186/s13287-025-04561-6]
- 49. Promoting ethical and reproducible cell culture [https://www.frontiersin.org/journals/toxicology/articles/10.3389/ftox.2025.1670513/full]
- 50. A fluorescence-based reporter for monitoring expression of ... [https://www.nature.com/articles/s41598-017-10944-4]
- 51. Factors Influencing Transfection Efficiency [https://www.thermofisher.com/us/en/home/references/gibco-cell-culture-basics/transfection-basics/factors-influencing-transfection-efficiency.html]
- 52. Optimizing Cell Transfection Conditions in "Four Steps" [https://www.linkedin.com/pulse/optimizing-cell-transfection-conditions-four-steps-say-goodbye-07mqc]
- 53. Optimize Transfection of Cultured Cells [https://worldwide.promega.com/resources/pubhub/optimize-transfection-of-cultured-cells/]
- 54. Transfection Efficiency Assays [https://www.aracelibio.com/articles/transfection-efficiency-assays/]
- 55. Overview of Transfection Methods [https://www.promega.com/resources/guides/cell-biology/transfection/]
- 56. Lifetime imaging of GFP at CoxVIIIa reports respiratory ... [https://www.nature.com/articles/srep46055]
- 57. Advancements in the application of reporter gene cell lines ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12460214/]
- 58. Selecting Optical Filters for Fluorescence Microscopy— ... [https://www.thermofisher.com/us/en/home/references/molecular-probes-the-handbook/technical-notes-and-product-highlights/selecting-optical-filters-for-fluorescence-microscopy.html]
- 59. Best Fluorescence Filters for Microscopy and Spectroscop [https://deltaopticalthinfilm.com/knowledge/tech-notes-and-papers/fluorescence-filters/]
- 60. Choosing Filter Combinations for Fluorescent Proteins [https://www.microscopyu.com/tutorials/choosing-filter-combinations-for-fluorescent-proteins]
- 61. Passage Number Effects in Cell Lines [https://www.atcc.org/resources/technical-documents/passage-number-effects-in-cell-lines]
- 62. Protocol to rapidly screen CRISPR-Cas9 gene editing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12282252/]
- 63. GFP-on mouse model for interrogation of in vivo gene editing [https://www.nature.com/articles/s41467-025-61449-y]
- 64. TP53 apoptosis biomarkers in breast cancer [https://www.sciencedirect.com/science/article/abs/pii/S0009898125005571]
- 65. North America Apoptosis Assay Market Trends 2025–2034 [https://www.gminsights.com/industry-analysis/north-america-apoptosis-assay-market]
- 66. Apoptosis Assay Market Size Report, 2025 – 2034 [https://www.gminsights.com/industry-analysis/apoptosis-assay-market]
- 67. Perspectives on cancer therapy—synthetic lethal precision ... [https://www.nature.com/articles/s41420-025-02418-8]
- 68. A multi-biomarker machine learning approach for early ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12355783/]
- 69. The cytochrome c gene proximal enhancer drives activity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3296065/]
- 70. The quantitative impact of 3′UTRs on gene expression [https://pmc.ncbi.nlm.nih.gov/articles/PMC12207410/]
- 71. Reporter & Specialty Vectors for Mammalian Expression ... [https://www.thermofisher.com/us/en/home/technical-resources/technical-reference-library/protein-expression-support-center/reporter-specialty-vectors-support/reporter-specialty-vectors-support-getting-started.html]
- 72. GFP fluorescence assay [https://bio-protocol.org/exchange/minidetail?id=16724981&type=30]
- 73. GFP Quantification | Technical Note 170 [https://www.denovix.com/tn-170-gfp-quantification-with-denovix-ds-11-series/]
- 74. Spatial differences in active caspase-8 defines its role in T cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2665918/]
- 75. Integrated real-time imaging of executioner caspase ... [https://www.nature.com/articles/s41420-025-02662-y]
- 76. A Caspase-3 Reporter for Fluorescence Lifetime Imaging ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6025355/]
- 77. Visualization of caspase-3-like activity in cells using a ... [https://www.nature.com/articles/ncomms3157]
- 78. CasExpress reveals widespread and diverse patterns of cell survival of... [https://elifesciences.org/articles/10936]
- 79. A novel caspase 8 selective small molecule potentiates ... [https://www.nature.com/articles/srep09893]
- 80. Apaf-1 is an evolutionarily conserved DNA sensor that ... [https://www.nature.com/articles/s41421-024-00750-4]
- 81. Small molecule OPA1 inhibitors amplify cytochrome c ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12227059/]
- 82. A reporter cell line for the automated quantification of ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2022.1031204/full]
- 83. Cytochrome c is released in a single step during apoptosis [https://pubmed.ncbi.nlm.nih.gov/15933725/]
- 84. based probe for enhanced infrared sensing of cytochrome c [https://www.sciencedirect.com/science/article/abs/pii/S0026265X25022295]