DNA Fragmentation Assays vs. Apoptotic Body Formation: A Comparative Guide for Biomedical Research and Drug Development
This article provides a comprehensive comparison of two fundamental approaches in apoptosis detection: DNA fragmentation assays and apoptotic body formation analysis.
DNA Fragmentation Assays vs. Apoptotic Body Formation: A Comparative Guide for Biomedical Research and Drug Development
Abstract
This article provides a comprehensive comparison of two fundamental approaches in apoptosis detection: DNA fragmentation assays and apoptotic body formation analysis. Tailored for researchers, scientists, and drug development professionals, it explores the underlying biological mechanisms, details established and emerging methodologies, and offers practical troubleshooting guidance. By synthesizing foundational knowledge with application-focused insights, this resource aims to empower professionals in selecting the optimal techniques for their specific research context, from basic science to clinical diagnostics and therapeutic efficacy studies.
The Biology of Cellular Demise: Understanding Apoptotic DNA Fragmentation and Body Formation
Apoptosis, or programmed cell death, is a fundamental biological process essential for normal development and maintenance of tissue homeostasis in multicellular organisms. This highly regulated form of cell death occurs through a series of biochemical events leading to characteristic cellular changes including cell shrinkage, nuclear fragmentation, chromatin condensation, and formation of membrane-bound apoptotic bodies that are quickly phagocytosed by neighboring cells without inducing inflammation [1] [2]. The term "apoptosis" (pronounced ap-ə-TOH-sis) derives from the Ancient Greek word meaning "falling off," describing the process similar to petals or leaves falling from plants [2]. The average adult human loses approximately 50 to 70 billion cells daily through apoptosis, representing a crucial physiological process for eliminating damaged, infected, or unnecessary cells [2].
The significance of apoptosis extends far beyond developmental biology into pathology and therapeutic development. Proper regulation of apoptotic pathways is vital for health, with defective apoptosis contributing to numerous disease states. Excessive apoptosis is associated with neurodegenerative disorders, autoimmune diseases, and ischemic damage, while insufficient apoptosis can lead to cancer development and autoimmune disorders [1] [3]. The intricate balance between cell survival and death makes understanding apoptotic mechanisms particularly valuable for drug discovery and therapeutic interventions across multiple disease domains, especially in oncology where many treatments aim to reactivate apoptotic pathways in cancer cells [4] [5].
Molecular Mechanisms of Apoptosis
Core Signaling Pathways
Apoptosis proceeds through two principal signaling pathways that converge on a common execution phase, each characterized by distinct initiators and regulatory mechanisms.
The Extrinsic Pathway
The extrinsic pathway, also known as the death receptor pathway, is triggered by extracellular signals binding to cell surface death receptors belonging to the tumor necrosis factor (TNF) receptor superfamily. When ligands such as FasL or TNF-α bind to their respective receptors (Fas and TNFR1), the receptors trimerize and recruit adapter proteins including FADD (Fas-associated death domain) to form the Death-Inducing Signaling Complex (DISC) [2]. This complex then recruits and activates initiator caspases, primarily caspase-8, which subsequently activates downstream effector caspases that execute the apoptotic program. This pathway represents a critical mechanism for immune-mediated cell elimination and maintenance of cellular populations in tissues with high turnover rates [2].
The Intrinsic Pathway
The intrinsic pathway, or mitochondrial pathway, is initiated by intracellular stress signals including DNA damage, oxidative stress, growth factor deprivation, and endoplasmic reticulum stress. These stimuli cause mitochondrial outer membrane permeabilization (MOMP), leading to the release of pro-apoptotic proteins from the mitochondrial intermembrane space into the cytosol [2]. Key events include the release of cytochrome c, which binds to Apaf-1 (apoptotic protease activating factor 1) and procaspase-9 to form the apoptosome complex, activating caspase-9. Simultaneously, mitochondria release SMAC (second mitochondria-derived activator of caspases) and DIABLO proteins that neutralize inhibitor of apoptosis proteins (IAPs), thereby facilitating caspase activation [2]. The Bcl-2 family of proteins tightly regulates this pathway through the balance between pro-apoptotic (Bax, Bak, Bid) and anti-apoptotic (Bcl-2, Bcl-xL) members that control mitochondrial permeability [2].
Execution Phase
Both intrinsic and extrinsic pathways converge on the activation of executioner caspases, primarily caspase-3, -6, and -7, which orchestrate the systematic dismantling of the cell through cleavage of hundreds of cellular substrates [2]. These include structural proteins such as nuclear lamins and cytoskeletal components, DNA repair enzymes like poly(ADP-ribose) polymerase (PARP), and regulatory proteins. The coordinated proteolytic activity results in the characteristic morphological changes of apoptosis, including cell shrinkage, chromatin condensation, DNA fragmentation, and eventual formation of apoptotic bodies that display "eat-me" signals such as phosphatidylserine externalization for efficient phagocytic clearance [1] [2].
Detection Methodologies: DNA Fragmentation Versus Morphological Analysis
DNA Fragmentation Assays
DNA fragmentation represents a biochemical hallmark of apoptosis, occurring through the activation of calcium-dependent endonucleases that cleave nuclear DNA at internucleosomal regions, producing characteristic fragments in multiples of 180-200 base pairs [2] [5]. Several techniques have been developed to detect this specific DNA degradation pattern.
The TUNEL assay (Terminal deoxynucleotidyl transferase dUTP Nick End Labeling) identifies DNA strand breaks by incorporating labeled nucleotides at the 3'-ends of fragmented DNA, allowing visualization through fluorescence or colorimetric detection [5]. While highly sensitive for detecting late-stage apoptosis, TUNEL staining lacks absolute specificity for apoptosis, as some forms of necrosis and other cell death modalities may also generate positive signals under certain conditions [6].
The Sperm Chromatin Structure Assay (SCSA) represents a flow cytometry-based approach that assesses DNA fragmentation by measuring the susceptibility of nuclear DNA to acid-induced denaturation, providing a DNA Fragmentation Index (DFI) as a quantitative parameter [7]. This method has proven particularly valuable in andrology for assessing sperm quality, where elevated DFI correlates with reduced fertility potential and has been adapted for somatic cell apoptosis analysis [7].
Agarose gel electrophoresis of extracted DNA represents a classical biochemical approach for detecting the apoptotic "laddering" pattern, distinguishing it from the random DNA fragmentation observed in necrosis [6]. While this method provides definitive evidence of internucleosomal cleavage, it requires relatively large cell numbers and lacks the sensitivity and quantitative capabilities of more modern techniques.
Morphological Analysis of Apoptotic Bodies
Morphological assessment remains the "gold standard" for definitive identification of apoptosis, relying on characteristic structural changes that distinguish it from other forms of cell death [5] [8]. Key morphological features include cell shrinkage, chromatin condensation and margination, nuclear fragmentation, membrane blebbing, and formation of apoptotic bodies that contain intact organelles and nuclear fragments [2] [5].
Advanced imaging technologies have significantly enhanced our ability to detect and quantify these morphological changes. Quantitative Phase Imaging (QPI) enables label-free observation of subtle changes in cell mass distribution, density, and morphology in real-time, allowing discrimination between apoptosis and lytic forms of cell death based on dynamic morphological parameters [9]. Similarly, Multimodal Holographic Microscopy (MHM) combines holographic microscopy with fluorescence detection to monitor morphological changes preceding and during cell death while simultaneously verifying death mechanisms through specific molecular markers [8].
Flow cytometry approaches for morphological assessment typically employ light scattering measurements, where apoptotic cells demonstrate decreased forward scatter (indicating cell shrinkage) and increased side scatter (reflecting nuclear condensation and granularity) [5]. While providing high-throughput quantification, these methods often require correlation with additional apoptotic markers for definitive identification.
Table 1: Comparison of Apoptosis Detection Methodologies
| Method | Principle | Applications | Advantages | Limitations |
|---|---|---|---|---|
| TUNEL Assay | Labels 3'-OH ends of fragmented DNA | Histology, fluorescence microscopy | High sensitivity; works on fixed tissue | May label non-apoptotic DNA breaks; expensive reagents |
| SCSA/DNA Fragmentation Index | Flow cytometric analysis of DNA denaturation | Sperm quality assessment, clinical diagnostics | Quantitative; high throughput | Requires specialized equipment; limited to single cell suspensions |
| Agarose Gel Electrophoresis | Separates DNA fragments by size | Basic research; confirmation studies | Low cost; visually distinctive ladder pattern | Low sensitivity; semi-quantitative; requires many cells |
| Morphological Analysis (Microscopy) | Visual identification of structural changes | Gold standard for validation | Definitive identification; provides context | Subjective; time-consuming; requires expertise |
| Quantitative Phase Imaging | Label-free measurement of cellular mass distribution | Live cell imaging; drug screening | Non-invasive; real-time dynamics | Specialized equipment; complex data analysis |
| Annexin V/PI Staining | Detects PS externalization and membrane integrity | Early apoptosis detection; flow cytometry | Distinguishes early/late apoptosis & necrosis | Cannot distinguish apoptosis from other PS-exposing death |
Experimental Data Comparison
Quantitative Comparison of Detection Methods
Direct comparison of apoptosis detection methodologies reveals significant differences in sensitivity, specificity, and applicability across experimental contexts. Recent technological advances have enabled more precise discrimination between apoptosis and alternative cell death modalities, addressing long-standing challenges in accurate cell death classification.
Studies implementing real-time live cell imaging using FRET-based caspase sensors coupled with organelle-specific fluorescent markers have demonstrated exceptional accuracy in distinguishing apoptosis from primary necrosis. In one approach, researchers developed a neuroblastoma cell line stably expressing a FRET-based caspase sensor (ECFP-DEVD-EYFP) along with mitochondrial-targeted DsRed, enabling simultaneous monitoring of caspase activation and mitochondrial integrity [4]. This system achieved single-cell resolution for discriminating apoptosis (characterized by caspase activation with retained mitochondrial fluorescence) from necrosis (showing loss of FRET probe without caspase activation while maintaining mitochondrial fluorescence) [4].
Quantitative phase imaging studies have identified specific morphological dynamics that differentiate apoptotic and non-apoptotic cell death. Researchers established that cell density (pg/pixel) and Cell Dynamic Score (CDS) parameters could classify caspase-dependent and -independent cell death with 75.4% prediction accuracy, providing a label-free approach for cell death subroutine identification [9]. These morphological parameters detected by QPI demonstrated 76% accuracy in cell death detection compared to manual annotation, representing a robust non-fluorescent methodology [9].
Table 2: Quantitative Performance Metrics of Apoptosis Detection Methods
| Method | Sensitivity | Specificity for Apoptosis | Time Resolution | Multiplexing Capacity | Throughput |
|---|---|---|---|---|---|
| FRET-Based Caspase Sensing | High (single cell) | High (direct caspase activity) | Seconds to minutes | High (3+ parameters) | Medium |
| Annexin V/PI Assay | Medium | Medium (PS exposure not exclusive) | Minutes | Low to medium (2 parameters) | High |
| DNA Fragmentation Assays | Medium to high | Medium (later stage) | Hours | Low | Variable |
| Morphological Analysis (QPI) | High | High (pattern recognition) | Minutes | Medium | Medium |
| Holographic Microscopy | High | High (morphology + fluorescence) | Minutes | High | Low to medium |
| Flow Cytometry (light scatter) | Medium | Low (requires confirmation) | Minutes | High | High |
Experimental Protocols
FRET-Based Caspase Activation Assay
The following protocol adapts methodology from published studies for real-time discrimination of apoptosis and necrosis [4]:
Cell Line Preparation: Generate stable cell lines expressing FRET-based caspase sensor (ECFP-DEVD-EYFP) using lentiviral transduction followed by antibiotic selection. Isolate single-cell clones with homogeneous expression using fluorescence-activated cell sorting (FACS).
Optional Organelle Labeling: For enhanced discrimination, cotransfect with organelle-specific markers (e.g., Mito-DsRed for mitochondria) and select double-positive clones.
Live-Cell Imaging: Plate cells in glass-bottom dishes or flow chambers. Treat with experimental compounds and monitor using automated fluorescence microscopy systems capable of time-lapse imaging.
Image Acquisition: Collect donor (ECFP) and acceptor (EYFP) fluorescence channels simultaneously or sequentially with minimal delay. Include brightfield or phase contrast for morphological correlation.
Data Analysis: Calculate FRET ratio (ECFP/EYFP) over time. Identify apoptotic cells as those showing increased donor fluorescence and decreased acceptor fluorescence (FRET decrease) while maintaining organelle markers. Necrotic cells show simultaneous loss of both FRET components without ratio change while retaining organelle fluorescence.
DNA Fragmentation Analysis via SCSA
Adapted from sperm DNA fragmentation analysis, this protocol can be modified for somatic cell apoptosis assessment [7]:
Cell Preparation: Harvest cells and adjust concentration to 1-2 × 10^6 cells/mL in PBS.
Acid Denaturation: Mix 100 μL cell suspension with 200 μL acid detergent solution (pH 1.2) for 30 seconds.
Staining: Add 1.2 mL acridine orange staining solution (6 μg/mL in phosphate-citrate buffer, pH 6.0).
Flow Cytometry Analysis: Analyze samples within 3-5 minutes of staining using flow cytometry with 488 nm excitation. Measure green fluorescence (530/30 nm bandpass) for native DNA and red fluorescence (>630 nm longpass) for denatured DNA.
Data Interpretation: Calculate DNA Fragmentation Index (DFI) as the ratio of red to total (red + green) fluorescence intensity. Establish threshold values based on control samples, with DFI > 30% indicating significant fragmentation.
Research Reagent Solutions
Table 3: Essential Reagents for Apoptosis Detection
| Reagent/Category | Specific Examples | Function/Application | Detection Method |
|---|---|---|---|
| Caspase Substrates | DEVD-based FRET probes; Fluorogenic caspase-3/7 substrates (CellEvent) | Detection of caspase activation; apoptosis confirmation | Fluorescence microscopy; Flow cytometry |
| Membrane Integrity Markers | Propidium iodide; 7-AAD; TO-PRO family dyes | Distinguishes live, early apoptotic, and late apoptotic/necrotic cells | Flow cytometry; Fluorescence microscopy |
| Phosphatidylserine Detection | Annexin V conjugates (FITC, PE, APC) | Early apoptosis marker through PS externalization | Flow cytometry; Microscopy |
| DNA Fragmentation Assays | TUNEL assay kits; SCSA reagents | Late-stage apoptosis detection through DNA break labeling | Microscopy; Flow cytometry; Gel electrophoresis |
| Mitochondrial Dyes | JC-1; TMRM; MitoTracker Red | Assess mitochondrial membrane potential; intrinsic pathway activation | Fluorescence microscopy; Flow cytometry |
| Live Cell Imaging Reagents | SYTO dyes; Hoechst 33342; CellTracker probes | Viability assessment; nuclear morphology; cell tracking | Live cell imaging |
| Caspase Inhibitors | z-VAD-FMK; Q-VD-OPh; Specific caspase inhibitors | Mechanism studies; confirmation of caspase-dependent apoptosis | Functional assays |
Discussion and Pathological Significance
Apoptosis in Disease Pathogenesis
The precise regulation of apoptotic pathways is crucial for maintaining cellular homeostasis, with dysregulation contributing significantly to numerous pathological conditions. In cancer development, defective apoptosis represents a hallmark capability enabling tumorigenesis and resistance to therapy [1] [3]. The p53 tumor suppressor protein, frequently mutated in human cancers, serves as a critical initiator of apoptosis in response to DNA damage, highlighting the importance of apoptotic pathways in preventing malignant transformation [1]. Simultaneously, excessive apoptosis contributes to neurodegenerative disorders including Alzheimer's disease, Parkinson's disease, and Huntington's disease, where inappropriate neuronal loss leads to progressive neurological decline [1] [3].
The clinical significance of apoptosis extends to autoimmune disorders and infectious diseases, where dysregulated cell death can promote either excessive inflammation or pathogen persistence. In systemic lupus erythematosus, defective clearance of apoptotic cells results in exposure of intracellular antigens, triggering autoimmune responses against self-components [10]. Understanding these apoptotic dysfunctions provides valuable insights for developing targeted therapeutic interventions across diverse disease contexts.
Therapeutic Implications and Future Directions
The recognition of apoptosis as a therapeutic target has stimulated extensive drug development efforts, particularly in oncology. Many conventional chemotherapeutic agents induce apoptosis in cancer cells through DNA damage or disruption of metabolic pathways, while newer targeted therapies specifically engage apoptotic machinery [1] [5]. Current research focuses on developing small molecules that directly modulate key apoptotic regulators, including Bcl-2 family proteins and inhibitor of apoptosis proteins (IAPs) [1].
Emerging technologies continue to refine our ability to detect and characterize apoptosis in experimental and clinical contexts. Advanced imaging platforms such as multimodal holographic microscopy and high-content screening systems enable more precise discrimination between apoptosis and alternative cell death modalities, providing powerful tools for drug discovery and toxicology assessment [9] [8]. The integration of machine learning approaches with quantitative imaging data further enhances classification accuracy and predictive capabilities for cell death subroutines [9].
Future directions in apoptosis research include the development of more specific caspase inhibitors and activators, refined detection methodologies for clinical application, and exploration of non-apoptotic programmed cell death pathways that may offer alternative therapeutic targets for conditions where classical apoptosis is impaired [6]. As our understanding of cell death mechanisms continues to evolve, so too will our ability to harness these pathways for therapeutic benefit across the spectrum of human disease.
The controlled fragmentation of nuclear DNA is a defining biochemical hallmark of apoptosis, or programmed cell death [11] [12]. This process is not random but is meticulously executed by specific endonucleases that cleave genomic DNA at internucleosomal regions, producing characteristic fragments of approximately 180-200 base pairs [12]. This cleavage serves a vital biological purpose: it ensures the irreversible commitment of a cell to die and facilitates the clean packaging and disposal of the genetic material within apoptotic bodies, thereby maintaining genomic stability and preventing inflammatory responses [11] [13]. Two key enzymes, Caspase-Activated DNase (CAD/DFF40) and Deoxyribonuclease 1 Like 3 (DNAS1L3), have emerged as central players in this process. They operate within complementary biochemical pathways and cellular compartments to accomplish DNA degradation [14] [15] [16]. Understanding their distinct mechanisms, regulation, and interplay is fundamental for research in cell biology, oncology, and therapeutic development. This guide provides a comparative analysis of CAD and DNAS1L3, situating their functions within the broader context of DNA fragmentation assays and the morphological process of apoptotic body formation.
Comparative Mechanisms of CAD and DNAS1L3
CAD (also known as DNA Fragmentation Factor 40 or DFF40) and DNAS1L3 (also known as DNase γ) are the primary endonucleases responsible for DNA fragmentation during cell death, yet they are regulated through distinct pathways and exhibit different biochemical properties.
Table 1: Comparative Profile of CAD and DNAS1L3
| Feature | CAD / DFF40 | DNAS1L3 / DNase γ |
|---|---|---|
| Primary Activation Pathway | Caspase-dependent (Intrinsic & Extrinsic Apoptosis) | Caspase-independent; regulated by PARP-1 cleavage [16] |
| Primary Cellular Role | Oligonucleosomal DNA fragmentation in apoptosis [11] | DNA fragmentation in necrosis; collaborates with CAD in apoptosis [15] |
| Dependence | Dependent on Caspase-3 for activation [16] | Dependent on Ca²⁺ and Mg²⁺ for catalytic activity [16] |
| Inhibitor | DFF45/ICAD (Inhibitor of CAD) | Poly(ADP-ribosyl)ation by PARP-1 [16] |
| Subcellular Localization | Cytoplasmic (inactive complex); Nuclear (active) [16] | Endoplasmic Reticulum; translocates to nucleus during apoptosis [16] |
| Key Functional Readout | Characteristic DNA ladder on agarose gel [12] | Generation of cell-free DNA (cfDNA); longer fragments in its absence [14] |
The Caspase-Dependent Pathway of CAD (DFF40)
CAD is the canonical endonuclease responsible for the hallmark internucleosomal DNA cleavage observed in apoptosis [11]. It is synthesized and stored in the cytoplasm as an inactive complex bound to its specific inhibitor, DFF45 or ICAD [16]. When a cell receives an apoptotic signal—whether from the intrinsic (mitochondrial) or extrinsic (death receptor) pathway—caspase-3 is activated. Caspase-3 then cleaves DFF45/ICAD, leading to its dissociation and the subsequent activation of CAD [16] [12]. The active CAD enzyme translocates to the nucleus, where it cleaves DNA at the linker regions between nucleosomes, generating the classic "DNA ladder" pattern that is a gold standard for identifying apoptosis [12].
The Calcium/Magnesium-Dependent Pathway of DNAS1L3
DNAS1L3 operates through a caspase-independent mechanism. Its activity is regulated by calcium (Ca²⁺) and magnesium (Mg²⁺) and is critically inhibited by poly(ADP-ribosyl)ation, a post-translational modification mediated by the enzyme PARP-1 [16]. During apoptosis, PARP-1 is itself cleaved and inactivated by caspases. This cleavage event is thought to relieve the inhibition on DNAS1L3, allowing it to participate in nuclear DNA fragmentation [16]. DNAS1L3 is predominantly localized in the endoplasmic reticulum and translocates to the nucleus upon an apoptotic stimulus [16]. While CAD is sufficient for oligonucleosomal fragmentation, studies in nuclease-deficient mice have shown that DNAS1L3 is essential for generating the characteristic size profile of circulating cell-free DNA (cfDNA), digesting larger multinucleosomal fragments into the predominant mononucleosomal form [14] [15].
The following diagram illustrates the coordinated signaling pathways leading to the activation of CAD and DNAS1L3 during apoptosis.
Experimental Data and Research Findings
Empirical data, particularly from nuclease-deficient mouse models, have been instrumental in delineating the unique and cooperative functions of CAD and DNAS1L3.
Table 2: Summary of Key Experimental Findings from Nuclease-Deficient Models
| Experimental Model | Key Findings Related to DNA Fragmentation | Implication | Citation |
|---|---|---|---|
Dffb⁻/⁻ (CAD-deficient) Mice |
- Apoptosis occurs without oligonucleosomal fragmentation.- Cells show increased genomic instability and tumorigenic potential. | CAD is necessary and sufficient for internucleosomal DNA laddering but not for cell death. | [11] [14] |
Dnase1l3⁻/⁻ Mice |
- Longer cfDNA fragments observed in plasma.- Impaired generation of mononucleosomal cfDNA. | DNAS1L3 is critical for trimming large DNA fragments into the short cfDNA profile typical of circulation. | [14] [15] |
CAD⁻/⁻DNAS1L3⁻/⁻ (Double KO) Mice |
- Dramatic reduction in serum cfDNA following induced hepatocyte apoptosis and necrosis. | CAD and DNAS1L3 are the dominant endonucleases for cfDNA generation in vivo and can compensate for each other. | [15] |
| In vitro Biochemical Studies | - DNAS1L3 activity is inhibited by PARP-1 via poly(ADP-ribosyl)ation.- Activity is restored upon PARP-1 cleavage by caspases. | Establishes a direct molecular link between caspase activation and DNAS1L3 regulation. | [16] |
Essential Research Protocols
A fundamental technique for investigating CAD-mediated apoptosis is the DNA fragmentation assay, which visually detects the internucleosomal cleavage pattern.
Standard DNA Fragmentation Assay Protocol
This protocol is designed to isolate and visualize low-molecular-weight DNA from apoptotic cells [12].
Stage 1: Harvesting and Lysing Cells
- Pellet Cells: Collect cells by centrifugation.
- Lyse Cells: Resuspend the cell pellet in 0.5 mL of detergent buffer (10 mM Tris pH 7.4, 5 mM EDTA, 0.2% Triton X-100). Vortex the mixture.
- Incubate: Keep the lysate on ice for 30 minutes.
- Centrifuge: Spin at 27,000 x g for 30 minutes at 4°C. This pellets intact chromatin and high-molecular-weight DNA from healthy cells. The supernatant contains the fragmented DNA.
Stage 2: Precipitating DNA
- Salt and Alcohol Precipitation: To the supernatant, add 50 µL of 5 M NaCl, 600 µL of ethanol, and 150 µL of 3 M sodium acetate (pH 5.2). Mix thoroughly.
- Incubate: Place the tube at -80°C for 1 hour to precipitate the DNA.
- Centrifuge: Pellet the DNA at 20,000 x g for 20 minutes and carefully discard the supernatant.
- Dissolve and Digest: Redissolve the DNA pellet in 400 µL of extraction buffer (10 mM Tris, 5 mM EDTA).
- RNase Treatment: Add 2 µL of DNase-free RNase (10 mg/mL) and incubate at 37°C for 5 hours to remove RNA.
- Proteinase K Treatment: Add 25 µL of proteinase K (20 mg/mL) with 40 µL of buffer (100 mM Tris pH 8.0, 100 mM EDTA, 250 mM NaCl) and incubate overnight at 65°C to digest proteins.
- Purify DNA: Extract DNA with phenol/chloroform/isoamyl alcohol and precipitate again with ethanol.
Stage 3: Gel Electrophoresis and Visualization
- Resuspend DNA: Air-dry the final pellet and resuspend it in 20 µL of Tris-acetate EDTA buffer with loading dye.
- Run Gel: Separate the DNA fragments by electrophoresis on a 2% agarose gel containing 1 µg/mL ethidium bromide.
- Visualize: Examine the gel under UV light. A positive result for apoptosis is a "ladder" of bands at ~180 bp and its multiples.
The workflow for this protocol is summarized below.
The Scientist's Toolkit: Key Research Reagents
Successful investigation of DNA fragmentation mechanisms requires a set of specific reagents and tools.
Table 3: Essential Reagents for Studying DNA Fragmentation
| Reagent / Tool | Primary Function | Application Example | |
|---|---|---|---|
| Caspase-3 Inhibitors (e.g., Z-DEVD-FMK) | Selectively inhibits caspase-3 activity. | To block the CAD activation pathway and study caspase-independent DNA fragmentation. | [16] |
Nuclease-Deficient Mouse Models (Dffb⁻/⁻, Dnase1l3⁻/⁻) |
Genetically ablate specific endonucleases. | To dissect the unique in vivo roles of CAD and DNAS1L3 in apoptosis, necrosis, and cfDNA biology. | [14] [15] |
| Anti-Fas Antibody | Agonist that activates the extrinsic apoptosis pathway. | To induce robust, synchronized apoptosis in experimental models (e.g., mouse hepatocytes). | [15] |
| Acetaminophen Overdose Model | Induces specific hepatocyte necrosis in vivo. | To study the role of DNAS1L3 in DNA fragmentation during necrotic cell death. | [15] |
| DNase-Free RNase | Degrades RNA without damaging DNA. | A critical step in the DNA fragmentation protocol to prevent RNA contamination that obscures the DNA ladder. | [12] |
| Proteinase K | A broad-spectrum serine protease. | Digests nucleases and other proteins during DNA extraction to prevent DNA degradation after cell lysis. | [12] |
| Agonists/Antagonists of PARP-1 | Modulate the activity of PARP-1. | To probe the regulatory relationship between PARP-1 and DNAS1L3 activity. | [16] |
The mechanism of DNA fragmentation during cell death is a finely orchestrated process involving the complementary actions of CAD and DNAS1L3. CAD acts as the primary executioner for the characteristic apoptotic DNA ladder, a process tightly coupled to caspase activation. In parallel, DNAS1L3, regulated by calcium, magnesium, and PARP-1, plays a critical role in refining the fragment size of DNA, particularly in the generation of cfDNA, and serves as a key enzyme in necrotic cell death.
From a methodological perspective, the classic DNA fragmentation assay, which detects the CAD-generated ladder, remains a direct and valuable tool for confirming apoptosis. However, it is crucial to understand that this assay does not capture the full picture. The formation of apoptotic bodies—the morphological endpoint of apoptosis—and the involvement of other nucleases like DNAS1L3 represent related but distinct biological processes. A comprehensive analysis of cell death therefore requires a multi-faceted approach, correlating biochemical assays like the DNA ladder with other techniques such as TUNEL staining, caspase activity assays, and morphological analysis to fully appreciate the complex and cooperative mechanism of DNA fragmentation executed by CAD and DNAS1L3.
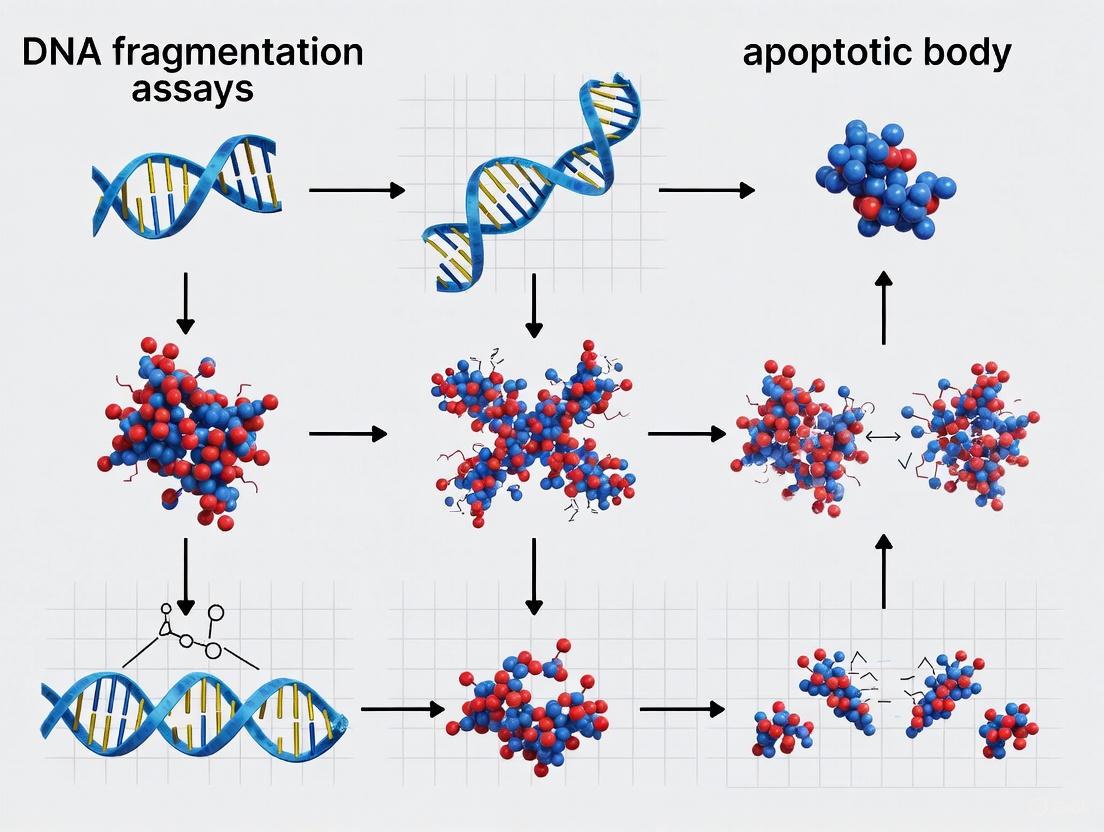
Apoptosis, or programmed cell death, is a fundamental biological process characterized by a series of distinctive and highly conserved morphological changes. Since its first description in 1972, these morphological features have remained the gold standard for identifying and defining apoptotic cell death [17]. The process is orchestrated by a genetic program that leads to an orderly cellular disassembly, contrasting sharply with the chaotic inflammatory death characteristic of necrosis [17]. The classical morphological hallmarks progress through several stages: initially, the cell shrinks and detaches from its neighbors; this is followed by chromatin condensation and nuclear fragmentation; finally, the cell membrane blebs and forms apoptotic bodies that are rapidly phagocytosed by neighboring cells [18] [17]. These features are conserved across diverse cell types and mammalian species, reflecting the fundamental nature of this cell death pathway in development, tissue homeostasis, and disease [19].
Understanding these morphological changes is particularly crucial in biomedical research and drug development, especially with the growing importance of the apoptosis market, which is projected to reach USD 6.08 billion by 2032, driven largely by oncology applications [20]. This guide provides a detailed comparison of two key apoptotic hallmarks—DNA fragmentation and apoptotic body formation—equipping researchers with the methodological and analytical frameworks needed to accurately assess these critical events in experimental systems.
The Morphological Spectrum of Apoptosis
Sequential Morphological Changes
The execution of apoptosis follows a characteristic morphological sequence that distinguishes it from other forms of cell death. The initial stage involves cell shrinkage, where the cell reduces its volume through controlled ion efflux and water loss, detaching from the extracellular matrix and neighboring cells [17]. This is followed by chromatin condensation, where nuclear chromatin aggregates at the nuclear periphery, forming crescent-shaped masses—a process regulated by factors such as Acinus and involving the degradation of nuclear matrix and lamina components [17].
The cell then enters the membrane blebbing phase, where the plasma membrane separates from the cytoskeleton, forming dynamic, non-retracting blebs at the cell surface. This process is regulated by ROCK-I-mediated phosphorylation of myosin light-chains and rearrangement of the actin cytoskeleton [17]. In the final stage, the cell undergoes formation of apoptotic bodies through extensive membrane blebbing and fragmentation. These sealed membrane vesicles, typically 1-5 μm in diameter, contain intact organelles and nuclear fragments and are impermeable to vital dyes, preventing the release of toxic cellular contents [21] [18] [17].
Table 1: Key Morphological Hallmarks of Apoptosis
| Morphological Feature | Key Characteristics | Regulatory Mechanisms |
|---|---|---|
| Cell Shrinkage | Reduction in cell volume, detachment from ECM | Ion efflux (K+, Na+, Cl−); inhibition of Na+/K+-ATPase [17] |
| Chromatin Condensation | Chromatin aggregation at nuclear periphery, nuclear pyknosis | Caspase-mediated degradation of nuclear lamina; Acinus activation [17] |
| Membrane Blebbing | Formation of dynamic surface protrusions | ROCK-I mediated myosin phosphorylation; actin cytoskeleton rearrangement [21] [17] |
| Apoptotic Body Formation | Membrane-bound vesicles (1-5 μm) with cellular contents | Actomyosin contraction; caspase-mediated protein cleavage [21] [18] |
Biochemical Executioners and Morphological Effectors
The morphological changes of apoptosis are driven by the activation of cysteine proteases called caspases, which cleave vital cellular substrates to produce the characteristic apoptotic phenotype [18]. Caspase activation leads to the cleavage of structural proteins such as nuclear lamins, resulting in nuclear fragmentation, and the activation of endonucleases that degrade DNA [18]. A key endonuclease, Caspase-Activated DNase (CAD), is responsible for cleaving chromatin at internucleosomal linker regions, generating the characteristic DNA ladder fragments of approximately 200 base pairs [17] [12].
The BCL-2 protein family serves as a critical regulator of these apoptotic events, controlling mitochondrial membrane permeability and the release of caspase-activating factors such as cytochrome c [22]. Mitochondrial events include depolarization of the mitochondrial membrane potential and opening of the mitochondrial permeability transition pore (MPTP), leading to the release of pro-apoptotic factors that amplify the death signal [12].
Diagram 1: Apoptotic Signaling and Morphological Execution Pathways. This diagram illustrates the key signaling pathways that initiate and execute the morphological changes in apoptosis, highlighting the central role of caspase activation and BCL-2 family regulation.
Comparative Analysis: DNA Fragmentation vs. Apoptotic Body Formation
DNA Fragmentation: Biochemical and Methodological Insights
DNA fragmentation represents a biochemical hallmark of apoptosis characterized by the activation of endonucleases that cleave nuclear DNA at internucleosomal regions. This process generates oligonucleosomal fragments of approximately 200 base pairs, which produce a characteristic "DNA ladder" when separated by agarose gel electrophoresis [12]. The DNA fragmentation protocol involves harvesting cells, lysing them with detergent buffers, precipitating DNA, and analyzing the fragmentation pattern via gel electrophoresis [12].
Recent research has revealed that DNA fragmentation occurs as a stepwise process. Initial cleavage generates fragments corresponding to mono-, di-, tri-, and tetra-nucleosomal units (~167 bp, ~360 bp, ~540 bp, and ~720 bp) inside apoptotic cells. Subsequently, these fragments undergo further digestion in the extracellular environment, producing the characteristic 10-bp periodic sub-nucleosomal fragments observed in cell-free DNA (cfDNA) [19]. This process is highly conserved across mammals and involves specific endonucleases including DFFB, DNASE1, and DNASE1L3 [19].
Table 2: DNA Fragmentation Analysis: Methodological Framework
| Parameter | DNA Fragmentation Assay | Alternative Methods |
|---|---|---|
| Primary Readout | DNA ladder pattern on agarose gel [12] | TUNEL fluorescence; Flow cytometry [17] [12] |
| Key Steps | Cell lysis, DNA precipitation, RNase/proteinase K treatment, gel electrophoresis [12] | Enzyme labeling, fluorescence detection [17] |
| Detection Capability | Late-stage apoptosis (post-DNA cleavage) [12] | Earlier detection possible with TUNEL [17] |
| Quantification Potential | Semi-quantitative [12] | Quantitative with flow cytometry [12] |
| Advantages | Direct visual confirmation; Cost-effective; No specialized equipment [12] | Higher sensitivity; Single-cell analysis; Multiplexing capability [17] [12] |
Apoptotic Body Formation: Mechanisms and Novel Insights
Apoptotic body formation represents the final morphological stage of apoptosis, where the cell fragments into membrane-bound vesicles that are rapidly cleared by phagocytes. Recent research has revealed a novel mechanism for apoptotic body formation called the "FOotprint Of Death" (FOOD) [21]. This process involves apoptotic cells leaving behind membrane-encased, F-actin-rich footprints tightly anchored to the substrate during cell retraction. These footprints subsequently vesicularize into FOOD-derived apoptotic extracellular vesicles (F-ApoEVs) approximately 2 μm in diameter [21].
The FOOD formation is regulated by the protein kinase ROCK1 and occurs across diverse cell types, apoptotic stimuli, and surface compositions. These F-ApoEVs expose phosphatidylserine—an "eat-me" signal—and function to flag the site of cell death to phagocytes for efferocytosis [21]. Interestingly, in viral infection settings, FOOD can harbor viral proteins and virions, potentially propagating infection to healthy cells [21]. This mechanism represents an alternative pathway for generating large apoptotic vesicles distinct from traditional apoptotic bodies formed through membrane blebbing.
Direct Comparative Analysis: Technical and Applications Perspective
Table 3: Comparative Analysis: DNA Fragmentation vs. Apoptotic Body Formation Assays
| Characteristic | DNA Fragmentation Assay | Apoptotic Body Detection |
|---|---|---|
| Morphological Stage Detected | Late stage (post-nuclear fragmentation) [12] | Intermediate to late stage (membrane remodeling) [21] [17] |
| Primary Methodology | Agarose gel electrophoresis [12] | Microscopy (light, electron, fluorescence) [17] |
| Key Identifying Features | DNA ladder (180-200 bp fragments) [12] | Membrane-bound vesicles (1-5 μm) [21] [17] |
| Temporal Resolution | End-point measurement [12] | Can be dynamic with time-lapse imaging [21] |
| Information Obtained | Biochemical confirmation of apoptosis [12] | Structural/morphological confirmation [17] |
| Ideal Application Context | Bulk population analysis; Drug screening [12] | Single-cell analysis; Mechanism studies [21] [17] |
| Limitations | Cannot detect early apoptosis; Semi-quantitative [12] | Requires specialized equipment; Subjectivity in identification [17] |
| Complementary Techniques | TUNEL assay; Caspase activation assays [17] [12] | Phosphatidylserine exposure; Membrane integrity assays [17] |
Advanced Research Applications and Protocols
Detailed Experimental Protocols
DNA Fragmentation Protocol
The standard DNA fragmentation assay provides a reliable method for detecting internucleosomal DNA cleavage. The procedure involves three main stages [12]:
Stage 1: Cell Harvesting and Lysis
- Pellet approximately 1-3 × 10^6 cells by centrifugation
- Lyse cells in 0.5 mL detergent buffer (10 mM Tris pH 7.4, 5 mM EDTA, 0.2% Triton X-100)
- Vortex the mixture thoroughly and incubate on ice for 30 minutes
- Centrifuge at 27,000 × g for 30 minutes to separate fragmented DNA (supernatant) from intact chromatin (pellet)
Stage 2: DNA Precipitation
- Divide supernatant into two 250 μL aliquots
- Add 50 μL ice-cold 5 M NaCl to each aliquot and vortex
- Add 600 μL ethanol and 150 μL 3 M sodium-acetate (pH 5.2)
- Incubate at -80°C for 1 hour to precipitate DNA
- Centrifuge at 20,000 × g for 20 minutes and carefully discard supernatant
- Pool DNA extracts by dissolving pellets in 400 μL extraction buffer (10 mM Tris, 5 mM EDTA)
- Add DNase-free RNase (2 μL of 10 mg/mL) and incubate for 5 hours at 37°C
- Add 25 μL proteinase K (20 mg/mL) and incubate overnight at 65°C
- Extract DNA with phenol/chloroform/isoamyl alcohol and precipitate with ethanol
Stage 3: Gel Electrophoresis and Visualization
- Air-dry DNA pellet and resuspend in 20 μL Tris-acetate EDTA buffer with 2 μL loading dye
- Separate DNA electrophoretically on a 2% agarose gel containing 1 μg/mL ethidium bromide
- Visualize DNA ladder pattern under ultraviolet transillumination
Diagram 2: DNA Fragmentation Assay Workflow. This diagram outlines the key steps in the DNA fragmentation protocol, from cell harvesting to the visualization of the characteristic apoptotic DNA ladder.
Apoptotic Body Analysis Protocol
Analysis of apoptotic bodies employs morphological assessment through various microscopy techniques [17]:
Light Microscopy Analysis
- Stain cells with Romanowski stains (e.g., Giemsa) or hematoxylin and eosin
- Identify apoptotic cells showing cell shrinkage, membrane blebbing, and formation of apoptotic bodies
- Apoptotic bodies appear as small, membrane-bound vesicles containing condensed chromatin
Fluorescence Microscopy
- Stain nuclei with Hoechst 33342 or DAPI (excitation ~350 nm, emission ~461 nm)
- Identify apoptotic cells with condensed, fragmented chromatin appearing as bright fluorescent masses
- Differentiate from normal nuclei based on chromatin condensation and nuclear fragmentation
Electron Microscopy
- Fix cells with glutaraldehyde and post-fix with osmium tetroxide
- Process samples through dehydration and embedding for ultrastructural analysis
- Identify characteristic features: intact membrane-bound vesicles containing organelles and nuclear fragments
Time-Lapse Imaging for Dynamic Assessment
- Utilize lattice light sheet microscopy or confocal microscopy for live-cell imaging
- Monitor the dynamic process of FOOD formation and F-ApoEV release [21]
- Track phosphatidylserine exposure using annexin A5 staining
Research Reagent Solutions
Table 4: Essential Research Reagents for Apoptosis Detection
| Reagent/Category | Specific Examples | Research Application |
|---|---|---|
| DNA Stains | Hoechst 33342, DAPI, Ethidium Bromide [17] [12] | Fluorescence microscopy; Gel visualization of DNA fragmentation [17] [12] |
| Cell Stains | Romanowski Stains, Hematoxylin & Eosin [17] | Light microscopy assessment of morphological changes [17] |
| Enzymes | DNase-free RNase, Proteinase K [12] | DNA purification for fragmentation assays [12] |
| Buffers & Solutions | Detergent Lysis Buffer, Tris-EDTA, Phenol/Chloroform [12] | Cell lysis and DNA extraction [12] |
| Apoptosis Inducers | BH3-mimetics (ABT-737, S63845), Staurosporine, Etoposide [21] [23] | Positive controls for apoptosis induction [21] |
| Specialized Kits | DNA Ladder Assay Kits, TUNEL Assay Kits [12] | Standardized apoptosis detection [12] |
Discussion and Research Implications
Technical Considerations and Limitations
When interpreting DNA fragmentation assays, researchers should note that this method is semi-quantitative and may not accurately reflect the exact proportion of apoptotic cells in heterogeneous populations [12]. The protocol requires careful handling to avoid DNA shearing or contamination, and the use of ethidium bromide poses safety concerns that require appropriate disposal procedures [12]. Additionally, certain cell types may not display the characteristic DNA ladder despite undergoing apoptosis, potentially leading to false negatives [17].
For apoptotic body detection, methodological challenges include the subjective nature of morphological identification and the potential confusion with other vesicular structures such as migrasomes or large extracellular vesicles from non-apoptotic processes [21]. The FOOD mechanism recently identified demonstrates that apoptotic bodies can be generated through distinct biogenesis pathways, adding complexity to their identification and interpretation [21]. Researchers can distinguish FOOD-derived F-ApoEVs from other structures using specific inhibitors—FOOD formation occurs in the presence of migrasome inhibitors (SMS2-IN-1, ISA-2011B), confirming its distinct mechanism [21].
Emerging Research Applications and Future Directions
The analysis of apoptotic morphology extends beyond basic research into substantial clinical applications, particularly in oncology. The global apoptosis market reflects this translational importance, valued at USD 4.04 billion in 2025 with significant growth driven by cancer drug development [20]. DNA fragmentation patterns in circulating cell-free DNA (cfDNA) have emerged as valuable non-invasive biomarkers for cancer detection and monitoring, with distinct fragmentation profiles observed in cancer patients including a higher proportion of short fragments and altered end preferences compared to healthy individuals [10] [19].
Recent discoveries of apoptotic body formation in unicellular organisms such as the cryptophyte alga Guillardia theta challenge evolutionary paradigms about programmed cell death and suggest deeper conservation of apoptotic mechanisms than previously recognized [23]. This finding indicates that the core apoptotic machinery, including apoptotic body formation, may have originated before the emergence of multicellularity.
The discovery of the FOOD mechanism opens new avenues for understanding how apoptotic cells communicate with their environment and influence tissue homeostasis, immune responses, and disease propagation [21]. These advances highlight the continuing importance of morphological analysis in apoptosis research, complementing molecular and biochemical approaches to provide a comprehensive understanding of cell death processes.
Researchers are encouraged to employ complementary techniques that assess both DNA fragmentation and apoptotic body formation to obtain comprehensive apoptosis data, as each method provides unique insights into different aspects of the apoptotic process. This multimodal approach is particularly valuable in drug discovery and development, where understanding the temporal sequence and morphological features of apoptosis can inform mechanism of action studies for novel therapeutics.
Programmed cell death, or apoptosis, is a highly regulated process essential for embryonic development, tissue homeostasis, and the elimination of damaged or infected cells [24] [2]. Apoptosis occurs through two principal signaling pathways—the extrinsic (death receptor) pathway and the intrinsic (mitochondrial) pathway—both culminating in the activation of caspases that execute cell death [25] [26]. Caspases (cysteine-dependent aspartate-directed proteases) are a family of endoproteases that serve as critical mediators and effectors of apoptotic cell death [27] [28]. They are synthesized as inactive zymogens (procaspases) and become activated through proteolytic cleavage and dimerization, initiating a cascade that leads to the controlled dismantling of cellular components [27]. Based on their functions in the apoptotic cascade, caspases are categorized as initiator caspases (including caspase-8, -9, and -10) or executioner caspases (caspase-3, -6, and -7) [27] [29]. This review provides a comparative analysis of the intrinsic, extrinsic, and executioner caspases, framing their roles within research methodologies that detect apoptosis, specifically DNA fragmentation assays and the analysis of apoptotic body formation.
Molecular Mechanisms of Caspase Activation
The Extrinsic Pathway and Initiator Caspase-8/10
The extrinsic apoptotic pathway initiates when extracellular death ligands, such as Fas Ligand (Fas-L) or TNF-Related Apoptosis-Inducing Ligand (TRAIL), bind to their corresponding death receptors (e.g., Fas, TNFR1, DR4, DR5) on the cell surface [26] [24]. This ligand-receptor interaction triggers receptor trimerization and the intracellular recruitment of the adaptor protein FADD (Fas-Associated Death Domain) via homophilic death domain interactions [27] [28]. FADD subsequently recruits procaspase-8 (and in humans, procaspase-10) through death effector domain (DED) interactions, forming a multi-protein complex known as the Death-Inducing Signaling Complex (DISC) [27] [24].
Within the DISC, procaspase-8 monomers are brought into close proximity, leading to their dimerization and auto-proteolytic activation—a process described by the "induced proximity model" [27]. Once activated, caspase-8 can directly cleave and activate downstream executioner caspases (caspase-3, -6, -7) in "Type I" cells [27]. In "Type II" cells, the apoptotic signal is amplified through the intrinsic pathway via caspase-8-mediated cleavage of the BH3-only protein Bid, generating truncated Bid (tBID), which translocates to mitochondria and promotes cytochrome c release [24] [28].
Table 1: Key Components of the Extrinsic Apoptotic Pathway
| Component | Type | Function in Pathway |
|---|---|---|
| Fas-L / TRAIL | Death Ligand | Binds and activates death receptors on the cell surface. |
| Fas / DR4 / DR5 | Death Receptor | Transduces the extracellular death signal into the cell. |
| FADD | Adaptor Protein | Recruits procaspase-8/10 to the activated receptor. |
| Caspase-8, -10 | Initiator Caspase | Initiates the caspase cascade; cleaves executioner caspases and Bid. |
| DISC | Protein Complex | Platform for initiator caspase activation. |
The Intrinsic Pathway and Initiator Caspase-9
The intrinsic apoptotic pathway is activated in response to intracellular stresses, including DNA damage, growth factor deprivation, oxidative stress, and irradiation [26] [27]. These stimuli cause an imbalance in the Bcl-2 protein family, shifting the equilibrium toward pro-apoptotic members [24]. The "executioner" proteins Bax and Bak are activated, often facilitated by "BH3-only" proteins like Bim, Puma, and Noxa, which neutralize anti-apoptotic proteins like Bcl-2 and Bcl-xL [29] [24]. Activated Bax and Bak oligomerize and integrate into the outer mitochondrial membrane, leading to Mitochondrial Outer Membrane Permeabilization (MOMP) [24].
MOMP results in the release of several mitochondrial intermembrane space proteins into the cytosol, including cytochrome c and SMAC (Second Mitochondria-derived Activator of Caspases) [24] [2]. Cytochrome c binds to the cytosolic protein APAF-1 (Apoptotic Protease-Activating Factor 1), which in the presence of ATP/dATP, oligomerizes to form a wheel-like signaling platform known as the apoptosome [27] [2]. The apoptosome recruits procaspase-9 via caspase recruitment domains (CARD), leading to its dimerization and activation [27]. SMAC promotes apoptosis by neutralizing Inhibitor of Apoptosis Proteins (IAPs), which would otherwise inhibit caspase activity [2].
Table 2: Key Components of the Intrinsic Apoptotic Pathway
| Component | Type | Function in Pathway |
|---|---|---|
| Bax / Bak | Pro-apoptotic Effectors | Mediate MOMP, leading to cytochrome c release. |
| BH3-only proteins | Pro-apoptotic Initiators | Sense stress and activate Bax/Bak or inhibit Bcl-2/Bcl-xL. |
| Cytochrome c | Mitochondrial Protein | Binds APAF-1 to nucleate apoptosome formation. |
| APAF-1 | Adaptor Protein | Oligomerizes to form the apoptosome platform. |
| Caspase-9 | Initiator Caspase | Activated by the apoptosome; cleaves executioner caspases. |
| SMAC/DIABLO | Mitochondrial Protein | Counteracts IAP-mediated caspase inhibition. |
The Execution Phase: Caspase-3, -6, and -7
The intrinsic and extrinsic pathways converge on the activation of the executioner caspases-3, -6, and -7 [27] [29]. Unlike initiator caspases, executioner caspases exist as inactive dimers and are activated through cleavage by initiator caspases (e.g., caspase-8 or -9) between their large and small subunits [27]. This cleavage induces a conformational change that creates a functional active site [27]. Once activated, a single executioner caspase can cleave and activate other executioner caspases, creating an accelerated feedback loop that ensures rapid and irreversible commitment to cell death [27].
Activated executioner caspases systematically dismantle the cell by cleaving hundreds of key structural and regulatory proteins [24]. Key cleavage events include:
- Cleavage of ICAD (Inhibitor of CAD): This releases the CAD nuclease, which is responsible for internucleosomal DNA fragmentation, a hallmark of apoptosis [24].
- Cleavage of structural proteins: Proteins such as lamin A/C and nuclear mitotic apparatus protein (NuMA) are cleaved, leading to the collapse of the nuclear envelope and nuclear fragmentation [29].
- Cleavage of ROCK1: This kinase is activated by caspase cleavage and induces actomyosin contraction, resulting in membrane blebbing and the formation of apoptotic bodies [24].
The following diagram illustrates the interconnected signaling pathways of intrinsic and extrinsic apoptosis leading to caspase activation:
Comparative Analysis of Caspase Functions and Experimental Detection
The distinct roles of initiator and executioner caspases, and the pathways that activate them, can be dissected using specific biochemical and morphological assays. The following table provides a comparative summary of these caspases, which is crucial for interpreting experimental data in apoptosis research.
Table 3: Comparative Analysis of Key Caspases in Apoptosis
| Caspase | Category | Activation Complex | Primary Activators | Key Substrates/Effectors | Main Functions |
|---|---|---|---|---|---|
| Caspase-8 | Initiator | DISC (Extrinsic) | Death Receptor Ligation | Caspase-3, Bid | Initiates extrinsic pathway; bridges to intrinsic pathway via tBID. |
| Caspase-9 | Initiator | Apoptosome (Intrinsic) | Cytochrome c/APAF-1 | Caspase-3 | Initiates intrinsic pathway in response to cellular stress. |
| Caspase-3 | Executioner | Cleavage by Casp-8/9 | Active Caspase-8/9 | PARP, ICAD, Lamin A/C | Principal executioner; mediates DNA fragmentation, nuclear disintegration. |
| Caspase-6 | Executioner | Cleavage by Casp-3/8/9 | Active Caspase-3/8/9 | Lamin A/C, Caspase-8 | Executioner; amplifies cascade; involved in nuclear membrane breakdown. |
| Caspase-7 | Executioner | Cleavage by Casp-3/8/9 | Active Caspase-3/8/9 | PARP | Executioner; cooperates with caspase-3 in substrate proteolysis. |
DNA Fragmentation Assays
DNA fragmentation is a biochemical hallmark of apoptosis, primarily executed by caspase-activated DNase (CAD) after caspase-3-mediated cleavage of its inhibitor, ICAD [24]. The TUNEL assay (Terminal deoxynucleotidyl transferase dUTP Nick End Labeling) is a widely used method to detect this DNA fragmentation in situ [29] [30]. The assay works by labeling the 3'-OH ends of fragmented DNA with modified dUTP (e.g., fluorescein-dUTP), which can be detected by fluorescence microscopy, flow cytometry, or immunohistochemistry [29]. While a hallmark of apoptosis, it is critical to note that DNA fragmentation can also occur during necrosis; therefore, TUNEL results must be corroborated with morphological analysis [29]. In apoptosis, TUNEL staining is associated with small, round apoptotic bodies, whereas in necrosis, the staining is more diffuse and associated with cell lysis [29]. Pulsed-field gel electrophoresis can further resolve the specific pattern of DNA fragmentation, distinguishing the large (~50 kbp) fragments and the internucleosomal (~180-200 bp) "DNA ladder" characteristic of apoptosis [30].
Analysis of Apoptotic Body Formation
The formation of apoptotic bodies is a key morphological endpoint of apoptosis, resulting from the executioner caspase-mediated cleavage of cellular structures [24]. This process includes cell shrinkage, chromatin condensation, nuclear fragmentation, and plasma membrane blebbing [29] [2]. A critical "eat-me" signal on apoptotic bodies is the externalization of phosphatidylserine (PtdSer), which is normally confined to the inner leaflet of the plasma membrane [24]. This exposure is mediated by caspase-dependent inactivation of the flippase ATP11A and activation of the scramblase Xkr8 [24].
The standard method for detecting this early/mid-stage event is Annexin V staining. Annexin V is a calcium-dependent phospholipid-binding protein with high affinity for PtdSer [29]. When conjugated to a fluorochrome, it can be used to label cells that have exposed PtdSer. Since the membrane remains intact in early apoptosis, Annexin V staining is typically performed in combination with a vital dye like propidium iodide (PI), which is excluded from live and early apoptotic cells but enters cells with compromised membranes (necrotic cells or late-stage apoptotic cells) [29]. Thus, early apoptotic cells are Annexin V positive and PI negative.
Recent research has identified a novel mechanism for generating large apoptotic extracellular vesicles (ApoEVs) that mark the site of cell death, termed the 'FOotprint Of Death' (FOOD) [31]. During apoptosis, adherent cells retract and leave behind actin-rich membrane tracks that subsequently vesicularise into large ApoEVs (~2 μm in diameter), exposing PtdSer and functioning to 'flag' the site of cell death for phagocytes [31]. The formation of FOOD and FOOD-derived ApoEVs (F-ApoEVs) is regulated by the kinase ROCK1 and represents a distinct biogenesis mechanism from classic apoptotic bodies and migrasomes [31].
The following workflow diagram integrates these key assays for detecting caspase-dependent apoptotic events:
The Scientist's Toolkit: Essential Reagents for Caspase and Apoptosis Research
Table 4: Key Research Reagents for Apoptosis and Caspase Analysis
| Reagent / Assay Kit | Primary Function | Experimental Application |
|---|---|---|
| Annexin V-FITC/PI Kit | Labels exposed phosphatidylserine and permeabilized cells. | Flow cytometry or microscopy to distinguish early apoptotic (Annexin V+/PI-), late apoptotic/necrotic (Annexin V+/PI+), and live cells (Annexin V-/PI-). |
| TUNEL Assay Kit | Labels 3'-OH ends of fragmented DNA. | In situ detection of apoptotic cells in tissue sections, cultured cells, or by flow cytometry. |
| Caspase Activity Assay Kits (e.g., Caspase-Glo) | Provides luminescent/fluorescent substrates to measure caspase enzymatic activity. | Quantitative measurement of initiator (caspase-8, -9) or executioner (caspase-3/7) caspase activity in cell lysates. |
| Caspase-Specific Antibodies (e.g., vs. Cleaved Caspase-3) | Detects the active, cleaved form of caspases. | Immunohistochemistry, immunofluorescence, and western blot to confirm caspase activation and localization. |
| BH3 Mimetics (e.g., ABT-737, Venetoclax) | Small molecules that inhibit anti-apoptotic Bcl-2 proteins. | Induce intrinsic apoptosis in cancer cells; used to study mitochondrial pathway regulation. |
| Pan-Caspase Inhibitor (e.g., z-VAD-FMK) | Irreversibly inhibits a broad range of caspases. | Determines the caspase-dependence of a cell death stimulus; used as a negative control. |
| Mitochondrial Membrane Potential Dyes (e.g., TMRE, JC-1) | Accumulates in polarized mitochondria; fluorescence decreases upon depolarization. | Detects early intrinsic apoptosis marked by loss of mitochondrial membrane potential (ΔΨm). |
The intricate biochemical pathways involving intrinsic, extrinsic, and executioner caspases form the core of the apoptotic cell death program. The extrinsic pathway, initiated by caspase-8, responds to external death signals, while the intrinsic pathway, mediated by caspase-9, integrates internal cellular damage. Both pathways converge on the activation of executioner caspases-3, -6, and -7, which orchestrate the biochemical and morphological hallmarks of apoptosis, including DNA fragmentation and the formation of apoptotic bodies. Within the context of apoptosis research, DNA fragmentation assays (like TUNEL) and the analysis of apoptotic body formation (via Annexin V staining and microscopy) serve as critical, complementary methodologies. The TUNEL assay detects a key biochemical consequence of executioner caspase activity, while morphological analysis confirms the non-inflammatory, organized nature of apoptotic death. A comprehensive understanding of these caspase pathways and their associated detection methods remains fundamental for advancing research in cancer biology, neurodegenerative diseases, and the development of novel therapeutics that modulate cell death.
The efficient clearance of apoptotic cells is a fundamental biological process essential for maintaining tissue homeostasis, shaping the immune response, and ensuring proper development. Within this field, two distinct analytical paradigms provide critical, yet different, insights into the machinery of programmed cell death: DNA fragmentation assays and the analysis of apoptotic body formation. DNA fragmentation assays are biochemical techniques that detect the hallmark internucleosomal cleavage of DNA, a key late-stage event in the apoptotic cascade. In contrast, the study of apoptotic bodies focuses on the morphological end-products of cell disassembly—the membrane-bound vesicles generated as the cell packages its contents for disposal and intercellular communication. This guide provides an objective comparison of these methodologies, detailing their principles, applications, and performance to aid researchers in selecting the optimal tools for their specific research context in cell biology, oncology, and drug development.
Analytical Framework: Principles and Technical Specifications
The following table provides a direct comparison of the core characteristics of DNA fragmentation assays and apoptotic body analysis.
Table 1: Core Characteristics of DNA Fragmentation Assays and Apoptotic Body Analysis
| Feature | DNA Fragmentation Assays | Apoptotic Body Analysis |
|---|---|---|
| Core Principle | Detection of biochemical DNA cleavage into oligonucleosomal fragments [12] [32] | Identification and characterization of membrane-bound vesicles formed during apoptotic cell disassembly [33] [34] |
| Primary Readout | DNA "ladder" on agarose gel; % DNA in tail (Comet); fluorescent signal (TUNEL) [12] [32] | Vesicle count, size, and content (e.g., nuclear fragments, mitochondria) via flow cytometry or microscopy [35] |
| Key Assay Formats | DNA Ladder, TUNEL, Comet Assay [32] | Flow Cytometry, Confocal Microscopy, Differential Centrifugation [33] [35] |
| Stage of Apoptosis Detected | Mid to late stage [12] | Late stage (following membrane blebbing) [34] |
| Tissue/Cell Type Flexibility | Suitable for most cell types (suspension and adherent) [12] [36] | Applicable to many, but not all, cell types (e.g., T cells, monocytes, fibroblasts) [35] |
| Quantitative Capability | Semi-quantitative (DNA Ladder) to Quantitative (TUNEL, Comet) [12] [32] | Quantitative (Flow Cytometry) to Semi-Quantitative (Microscopy) [35] |
| Throughput Potential | Low (DNA Ladder) to Medium (TUNEL, Comet) [12] | Medium (Flow Cytometry) to Low (Microscopy) [35] |
Experimental Protocols for Key Techniques
DNA Fragmentation Analysis via DNA Ladder Assay
The DNA ladder assay is a classic, semi-quantitative method for observing the characteristic internucleosomal DNA cleavage pattern of apoptosis [12] [32].
Protocol Steps [12]:
- Harvest and Lyse Cells: Pellet approximately 10⁶ cells and lyse in 0.5 mL of detergent buffer (10 mM Tris pH 7.4, 5 mM EDTA, 0.2% Triton X-100). Vortex and incubate on ice for 30 minutes.
- Separate Fragmented DNA: Centrifuge the lysate at 27,000 x g for 30 minutes. The fragmented DNA will be contained in the supernatant, while intact genomic DNA and nuclei are in the pellet.
- Precipitate DNA: Divide the supernatant into two aliquots. Add NaCl to a final concentration of ~0.7 M, followed by 2.5 volumes of ethanol and 0.5 volumes of 3 M sodium acetate (pH 5.2). Mix thoroughly and incubate at -80°C for 1 hour.
- Pellet and Purify DNA: Centrifuge at 20,000 x g for 20 minutes to pellet the DNA. Carefully discard the supernatant. Dissolve the pooled DNA pellets in Tris-EDTA buffer.
- Digest RNA and Protein: Add DNase-free RNase (e.g., 2 µL of a 10 mg/mL solution) and incubate at 37°C for several hours. Then add Proteinase K (e.g., 25 µL of 20 mg/mL) and incubate overnight at 65°C.
- Final Extraction and Precipitation: Extract DNA with phenol/chloroform/isoamyl alcohol and precipitate again with ethanol. Air-dry the final pellet.
- Visualize: Resuspend the DNA in loading buffer and separate on a 2% agarose gel containing a DNA stain like ethidium bromide. Visualize the characteristic DNA ladder under UV light.
Apoptotic Body Characterization via Flow Cytometry
Flow cytometry provides a robust, quantitative method to identify and characterize apoptotic bodies (ApoBDs) in a heterogeneous sample [35].
Protocol Steps [35]:
- Induce Apoptosis: Treat cells (e.g., Jurkat T cells, THP-1 monocytes) with an apoptotic inducer such as UV irradiation (150 mJ/cm²) or an anti-Fas antibody (62.5 ng/mL). Incubate for 2-6 hours.
- Stain for Surface and Internal Markers: Stain cells with a combination of fluorescent probes prior to or after induction of apoptosis:
- Annexin A5-FITC/PE/APC: Binds to phosphatidylserine (PS) exposed on the surface of ApoBDs and apoptotic cells.
- TO-PRO-3: A nucleic acid dye that enters cells with compromised membrane permeability, helping to differentiate late apoptotic and necrotic cells.
- Hoechst 33342: Stains DNA to identify ApoBDs containing nuclear material.
- MitoTracker Green: Stains mitochondria to identify ApoBDs containing this organelle.
- Cell Surface Marker Antibodies: Use antibodies against cell type-specific surface markers (e.g., CD3 for T cells, CD45 for leukocytes) to trace the origin of the ApoBDs.
- Sample Analysis: Resuspend the sample in annexin-binding buffer and analyze immediately on a flow cytometer.
- Gating Strategy: ApoBDs are typically identified based on their small size (low forward scatter, FSC) and low complexity (low side scatter, SSC), which distinguishes them from intact cells and debris. They are further characterized as Annexin A5-positive, TO-PRO-3-negative/positive, and positive for specific intracellular contents or surface markers.
Signaling Pathways and Experimental Workflows
DNA Fragmentation Signaling Pathway
The following diagram illustrates the key signaling events leading to DNA fragmentation during apoptosis, highlighting the points detected by major assays.
Diagram 1: DNA fragmentation signaling pathway.
Apoptotic Body Formation Workflow
This workflow outlines the key morphological steps in apoptotic body formation and the corresponding techniques for their analysis.
Diagram 2: Apoptotic body formation workflow.
The Scientist's Toolkit: Essential Research Reagents
Successful execution of these apoptosis detection methods relies on a suite of specific reagents and tools.
Table 2: Key Research Reagent Solutions for Apoptosis Analysis
| Reagent/Tool | Function in DNA Fragmentation Assays | Function in Apoptotic Body Analysis |
|---|---|---|
| Terminal deoxynucleotidyl transferase (TdT) | Enzyme used in TUNEL assay to label 3'-OH ends of fragmented DNA with modified nucleotides [32]. | Not typically used. |
| Formamidopyrimidine DNA glycosylase (Fpg) | Enzyme used in the Comet Assay to convert specific base damages into DNA breaks for measurement [37]. | Not typically used. |
| Annexin A5 (conjugated to fluorophores) | Can be used to correlate PS externalization (early apoptosis) with DNA fragmentation [36]. | Primary stain to detect phosphatidylserine on the surface of ApoBDs for flow cytometry and microscopy [35]. |
| DNase-free RNase | Critical for removing RNA that can contaminate the DNA sample and obscure the ladder pattern in gel electrophoresis [12]. | Not typically used. |
| Proteinase K | Digests proteins and nucleases to purify genomic DNA for the ladder assay [12]. | Not typically used. |
| TO-PRO-3 | Can be used as a DNA stain. | Nucleic acid dye used in flow cytometry to assess membrane permeability of particles, aiding in differentiating ApoBDs [35]. |
| Hoechst 33342 / DAPI | DNA stains for visualizing the nucleus; can be used in conjunction with other assays. | DNA stain used to identify the presence and distribution of nuclear material within ApoBDs [35]. |
| Cell Type-Specific Surface Markers (e.g., CD3, CD45) | Not typically used. | Antibodies against specific surface proteins (e.g., CD3 for T cells) used to trace the cellular origin of ApoBDs in a mixed population [35]. |
| ROCK I Inhibitor (e.g., GSK 269962) | Not typically used. | Chemical inhibitor used to study the role of the ROCK I kinase in mediating membrane blebbing during ApoBD formation [38] [34]. |
Performance Comparison and Functional Relevance
Data Output and Analytical Capabilities
The choice between these methodologies is often dictated by the specific research question, as they provide complementary data on the apoptotic process.
Table 3: Comparison of Assay Outputs and Functional Insights
| Aspect | DNA Fragmentation Assays | Apoptotic Body Analysis |
|---|---|---|
| Primary Data Generated | Qualitative ladder pattern; % DNA damage; fluorescence intensity [12] [32]. | Particle count, size distribution, surface markers, and internal cargo composition [35]. |
| Information on Clearance Mechanism | Indirect; confirms death but not how corpse is processed. | Direct; reveals the final packaged form of the cell for phagocytosis (efferocytosis) [33] [34]. |
| Role in Intercellular Communication | Not assessed. | Directly implicated; ApoBDs can shuttle proteins, nucleic acids (e.g., microRNAs), and other cargo between cells [34] [35]. |
| Link to Tissue Homeostasis | Disruption leads to inflammation and autoimmunity due to failed clearance [38]. | Essential for immune tolerance and tissue fitness; stem cells (e.g., HFSCs) directly engulf ApoBDs to maintain niche health [39]. |
| Best Suited For | Confirming and quantifying apoptotic death in cell populations; screening pro-apoptotic compounds [12]. | Studying the mechanisms of cell disassembly, efferocytosis, and the role of extracellular vesicles in disease and homeostasis [33] [35]. |
Limitations and Technical Challenges
A critical understanding of each method's limitations is required for experimental design and data interpretation.
- DNA Fragmentation Assays: The DNA ladder assay is semi-quantitative and lacks sensitivity for detecting low levels of apoptosis [12]. Both TUNEL and the Comet Assay can suffer from technical artifacts and require careful optimization to avoid false positives or overestimation of damage [12] [32]. These methods generally require destruction of the sample, precluding live-cell or temporal studies within the same sample.
- Apoptotic Body Analysis: ApoBDs are heterogeneous in size and content, making standardization challenging [35]. Their small size (1-5 μm) places them near the detection limit of some flow cytometers, requiring careful gating to distinguish them from debris [35]. Furthermore, not all apoptotic cells form ApoBDs; the process is cell-type and stimulus-dependent [33] [34].
DNA fragmentation assays and apoptotic body analysis are not competing techniques but rather complementary tools that illuminate different phases and facets of the apoptotic clearance process. DNA fragmentation assays serve as robust, well-established methods for confirming and quantifying the commitment to cell death via a key biochemical event. In contrast, apoptotic body analysis provides a dynamic window into the morphological execution and functional consequences of cell disassembly, including its critical role in efferocytosis and intercellular signaling. The selection between these approaches should be guided by the specific biological question—whether the focus is on the initiation and quantification of death itself, or on the fate of the dying cell and its impact on the surrounding tissue environment. As research continues to unveil the complex biological roles of apoptotic clearance, integrating both methodological paradigms will provide the most comprehensive understanding of cellular origins and fate in health and disease.
From Bench to Bedside: Methodologies for Detecting DNA Fragmentation and Apoptotic Bodies
The DNA ladder assay is a foundational technique in cell death research, serving as a historical hallmark for identifying apoptotic cells through the visualization of internucleosomal DNA fragmentation. This method remains widely used for its direct morphological evidence of programmed cell death, distinguishing apoptosis from necrosis by producing a characteristic ladder pattern on agarose gels, unlike the smeared pattern observed in necrotic cells [12] [40]. The assay detects a key biochemical event in apoptosis: the activation of endogenous endonucleases, such as CAD (Caspase-Activated DNase), which cleave genomic DNA at the linker regions between nucleosomes, generating fragments that are multiples of approximately 180-200 base pairs [12] [41]. While emerging electrochemical biosensors now offer higher sensitivity for early apoptosis markers like phosphatidylserine exposure and caspase-3 activation [42], the DNA ladder assay provides a straightforward, cost-effective, and visually intuitive method for confirming apoptosis in bulk cell populations, making it a staple in fields ranging from oncology to toxicology [12].
Principles and Biochemical Basis
The Molecular Mechanism of DNA Fragmentation
The DNA ladder observed in apoptotic cells results from a highly regulated biochemical process. During the execution phase of apoptosis, caspase-activated endonucleases are stimulated, primarily targeting DNA at internucleosomal regions. This cleavage produces discrete fragments consisting of single nucleosomes (∼180-200 bp), dinucleosomes (∼360-400 bp), and trinucleosomes (∼540-600 bp) [12] [40]. This specific fragmentation pattern stands in direct contrast to the random DNA degradation observed in necrotic cell death, which produces a continuous smear on agarose gels due to non-specific nuclease activity [40].
The following diagram illustrates the key biochemical pathway leading to DNA ladder formation:
Significance in Cell Death Classification
The DNA ladder assay provides critical discriminatory power in cell death classification. As a late-stage apoptotic marker, it typically appears after other biochemical events such as phosphatidylserine externalization (detectable by Annexin V staining) and caspase activation [12] [43]. Its key advantage lies in its ability to provide morphological evidence of the apoptotic process, complementing flow cytometry-based methods that quantify earlier events but don't visually demonstrate the internucleosomal cleavage pattern [41] [43]. However, researchers must recognize that this fragmentation represents an endpoint in apoptosis, making it less suitable for detecting early apoptotic events or reversible phases of programmed cell death [12] [40].
Experimental Protocol and Methodology
Standard DNA Ladder Assay Workflow
The following workflow outlines the core procedures for conducting a DNA ladder assay, integrating both conventional and updated protocols:
Detailed Procedural Steps
Cell Harvesting and Lysis
Begin by pelleting both adherent and floating cells, as apoptotic cells tend to detach from culture surfaces [41]. Resuspend the cell pellet in 0.5 mL of detergent-based lysis buffer (typically containing 10 mM Tris pH 7.4, 5 mM EDTA, and 0.2% Triton X-100) and incubate on ice for 30 minutes [12]. This step permeabilizes cell membranes while keeping nuclear membranes largely intact, allowing for the separation of cytoplasmic components from intact nuclei. Following lysis, centrifuge samples at 27,000 × g for 30 minutes to separate fragmented DNA (in supernatant) from high-molecular-weight genomic DNA and cellular debris (in pellet) [12].
DNA Precipitation and Purification
Transfer the supernatant containing fragmented DNA to a new tube and add ice-cold 5 M NaCl to precipitate proteins. For DNA precipitation, add 600 μL ethanol and 150 μL 3 M sodium-acetate (pH 5.2), then incubate at -80°C for 1 hour [12]. Centrifuge at 20,000 × g for 20 minutes to pellet DNA. For purified DNA, resuspend the pellet in Tris-EDTA buffer and treat with DNase-free RNase (2 μL of 10 mg/mL) for 5 hours at 37°C to remove RNA contamination [12]. Follow with proteinase K treatment (25 μL at 20 mg/mL) overnight at 65°C to digest nucleoproteins. Finally, extract DNA with phenol/chloroform/isoamyl alcohol (25:24:1) and precipitate with ethanol to obtain purified DNA fragments [12].
Gel Electrophoresis and Visualization
Air-dry the DNA pellet and resuspend in 20 μL Tris-acetate EDTA buffer supplemented with loading dye (0.25% bromophenol blue, 30% glycerol) [12]. Separate DNA fragments electrophoretically on a 1.5-2% agarose gel containing 1 μg/mL ethidium bromide or safer alternatives like SYBR Safe [41]. Run the gel at an appropriate voltage until sufficient separation of DNA markers is achieved, then visualize under UV transillumination [12]. The characteristic apoptotic ladder should appear as discrete bands at 180-200 bp intervals, with the smallest fragment at approximately 180-200 bp.
Research Reagent Solutions
The following table outlines essential reagents and their functions in the DNA ladder assay protocol:
| Reagent/Chemical | Function in Protocol | Specific Example/Concentration |
|---|---|---|
| Lysis Buffer | Cell membrane permeabilization and content release | 10 mM Tris pH 7.4, 5 mM EDTA, 0.2% Triton X-100 [12] |
| RNase A | Degradation of contaminating RNA | 2 μL of 10 mg/mL DNase-free RNase; 5h incubation at 37°C [12] |
| Proteinase K | Digestion of DNA-associated proteins | 25 μL at 20 mg/mL; overnight incubation at 65°C [12] |
| Phenol/Chloroform | Protein removal and DNA purification | Phenol/chloroform/isoamyl alcohol (25:24:1 ratio) [12] |
| Ethidium Bromide | DNA staining for visualization | 1 μg/mL in agarose gel; visualize by UV transillumination [12] |
| SYBR Safe | Alternative, safer DNA stain | Used according to manufacturer's instructions [41] |
Semi-Quantitative Analysis and Technical Considerations
Interpretation and Quantification Approaches
While primarily qualitative, the DNA ladder assay can provide semi-quantitative data when combined with densitometric analysis. The intensity of the DNA ladder bands correlates with the extent of apoptosis in the cell population [12]. Modern approaches use digital image processing software (e.g., ImageJ, GelGenie) to convert gel images into fluorescence intensity profiles, generating electropherogram-like data that allows for band intensity comparison [44]. This process involves background subtraction, lane identification, band detection, and intensity measurement to estimate relative DNA fragmentation levels between samples [44].
For improved quantification, some researchers employ spike-in controls like the synthetic CEREBIS construct, a 180 bp non-human DNA fragment designed to evaluate recovery efficiency during extraction processes [45]. However, studies show that normalization using such controls doesn't consistently reduce variability when using a single extraction method, though it may be beneficial when comparing different extraction techniques [45].
Technical Limitations and Sensitivity Considerations
The DNA ladder assay has specific technical constraints that researchers must acknowledge:
- Sensitivity Threshold: The method typically requires a minimum of 1 × 10⁶ cells undergoing apoptosis to generate a detectable ladder pattern, making it unsuitable for samples with low apoptotic rates [40].
- Semi-Quantitative Nature: While band intensity correlates with apoptosis extent, the assay doesn't provide precise quantification of apoptotic percentages [12].
- Stage Specificity: DNA fragmentation represents a relatively late event in apoptosis, limiting utility for detecting early apoptotic stages [12].
- Size Selectivity: Standard DNA extraction methods may preferentially recover certain fragment sizes, potentially biasing results [45].
Comparative Analysis with Other Apoptosis Detection Methods
Performance Comparison of Apoptosis Detection Methods
The following table compares the DNA ladder assay with other commonly used apoptosis detection techniques:
| Method | Principle | Detection Stage | Sensitivity | Throughput | Key Advantages | Main Limitations |
|---|---|---|---|---|---|---|
| DNA Ladder Assay | DNA fragmentation visualization | Late apoptosis | Moderate (requires ~10⁶ cells) [40] | Low | Direct morphological evidence; cost-effective; distinguishes apoptosis from necrosis [12] [41] | Semi-quantitative; labor-intensive; not for early apoptosis [12] |
| Annexin V/PI Staining | Phosphatidylserine exposure on cell surface | Early apoptosis (Annexin V+/PI-) to late (Annexin V+/PI+) [43] | High | Medium to high | Quantifies early apoptosis; distinguishes apoptotic stages [43] | Requires live cells; specialized equipment (flow cytometry) [12] |
| TUNEL Assay | Labeling of DNA strand breaks | Mid to late apoptosis | High | Medium | Detects DNA fragmentation in situ; higher sensitivity than DNA ladder [12] [40] | Potential false positives from necrotic cells or DNA repair [40] |
| Caspase Activity Assays | Caspase enzyme activity measurement | Early to mid apoptosis | High | Medium to high | Early detection; mechanistic insight [12] [42] | Does not confirm cell death completion [12] |
| Electrochemical Biosensors | Electrochemical detection of apoptosis biomarkers (e.g., caspase-3) [42] | Early to mid apoptosis | Very high (e.g., 0.04 pg mL⁻¹ for caspase-3) [42] | Potentially high | High sensitivity; potential for point-of-care use; real-time monitoring [42] | Emerging technology; not yet standardized for clinical use [42] |
Strategic Method Selection for Apoptosis Research
Choosing the appropriate apoptosis detection method depends on research goals, sample type, and available resources. The DNA ladder assay remains valuable for initial screening and confirmation of apoptosis, particularly when studying bulk cell populations and when resources are limited [12] [41]. Its strength lies in providing visual, morphological evidence of the characteristic internucleosomal cleavage pattern that distinguishes apoptosis from other forms of cell death [40].
For more sophisticated applications requiring early detection, quantification, or kinetic studies, flow cytometry-based methods (Annexin V/PI, caspase activity) offer superior sensitivity and statistical power [43]. The emerging electrochemical biosensors show promise for future clinical applications with their exceptional sensitivity for early apoptotic markers like caspase-3, detecting concentrations as low as 0.04 pg mL⁻¹ using peptide-based sensors [42].
The DNA ladder assay maintains its relevance in modern apoptosis research as a straightforward method for detecting the biochemical hallmark of programmed cell death. While newer technologies offer advantages in sensitivity, quantification, and early detection capabilities, the visual demonstration of internucleosomal DNA fragmentation provides compelling morphological evidence that complements more quantitative approaches. Understanding the principles, optimized protocols, and comparative strengths of this classic technique enables researchers to effectively incorporate it into a comprehensive cell death analysis strategy, particularly for initial screening and educational contexts where its cost-effectiveness and visual clarity remain advantageous.
The Terminal deoxynucleotidyl Transferase dUTP Nick End Labeling (TUNEL) assay stands as a foundational method in genetic toxicology and cell death research for directly detecting DNA fragmentation. This technique specifically identifies single and double-strand DNA breaks by enzymatically labeling their 3'-OH termini, serving as a crucial tool for researchers investigating apoptosis, genotoxicity, and sperm DNA integrity [46]. Within the broader context of DNA fragmentation assays, TUNEL provides distinct advantages over methods focused on detecting apoptotic body formation, as it directly targets the biochemical signature of DNA degradation rather than relying on morphological changes alone.
The fundamental principle underlying TUNEL involves the template-independent enzyme terminal deoxynucleotidyl transferase (TdT), which catalyzes the addition of modified deoxynucleotides (such as fluorescein-dUTP, BrdUTP, or EdUTP) to the 3'-hydroxyl ends of fragmented DNA [47] [46]. These incorporated nucleotides are then detected via fluorescence microscopy, flow cytometry, or colorimetric methods, allowing researchers to quantify the percentage of cells with significant DNA damage within a population.
Performance Comparison of DNA Fragmentation Assays
The selection of an appropriate DNA damage detection method depends on multiple factors including sensitivity, specificity, throughput, and technical requirements. The following table summarizes key characteristics of major assays used in research settings:
Table 1: Comparative Analysis of Major DNA Fragmentation Detection Assays
| Assay Method | Detection Principle | Key Advantages | Primary Limitations | Best Applications |
|---|---|---|---|---|
| TUNEL | Directly labels 3'-OH ends of DNA breaks with modified dUTP via TdT enzyme [46] | Detects both single- and double-strand breaks; Compatible with fluorescence microscopy, flow cytometry, and colorimetric detection [46] | May underestimate damage in highly compact chromatin [48]; Requires standardized protocol for reproducibility [49]; Cannot quantify damage magnitude within single cells [46] | Apoptosis detection, sperm DNA fragmentation studies, tissue section analysis |
| Comet Assay | Measures DNA migration under electrophoresis; damaged DNA forms "comet tail" [50] | Exceptionally sensitive to double-strand breaks [50]; Provides visual representation at single-cell level [50] | Labor-intensive quantification [51]; Subject to operator bias [52]; Limited to in vitro applications with cultured cells [51] | Genotoxicity testing, radiation biology, environmental toxicology |
| SCSA | Uses acridine orange fluorescence to detect DNA denaturation under acidic conditions [48] | High-throughput capability with flow cytometry; Reduced subjectivity compared to manual methods [48] | Requires expensive equipment; Complex workflow [48]; Indirect measurement of DNA damage [48] | Clinical sperm analysis, large cohort fertility studies |
| Novel TdT-Based Biosensors | Combines TdT labeling with signal amplification systems (e.g., CRISPR-Cas12a, strand displacement probes) [52] [48] | Exceptional sensitivity (detection as low as 0.001 nM) [48]; Molecular-level resolution; Quantitative breakpoint counting [52] | Emerging technology with limited standardization; Requires specialized reagents and equipment [52] | Precise DNA damage quantification, cryopreservation studies, pharmaceutical development |
Quantitative comparisons reveal significant performance differences. A comprehensive 2025 study analyzing 1,470 sperm samples found that while TUNEL and comet assay results correlated (R² = 0.34, P < 0.001), the comet assay identified 3,387 significantly differentially methylated regions compared to only 23 with TUNEL, suggesting superior sensitivity of the comet assay for detecting epigenetic impacts of DNA damage [50]. However, novel TdT-based methods demonstrate remarkable advancement, with one biosensor detecting DNA breakpoints at concentrations as low as 0.001 nM – significantly surpassing traditional methods in sensitivity [48].
Detailed Experimental Protocols
Standard TUNEL Assay Workflow for Cultured Cells
The following protocol represents a standardized approach for detecting apoptotic cells in culture, incorporating critical steps to ensure reproducibility:
Sample Preparation and Fixation:
- Culture cells on poly-lysine coated coverslips until desired confluence.
- Apply experimental treatments to induce apoptosis (e.g., 0.5 μM staurosporine for 4 hours for HeLa cells) [47].
- Aspirate medium and wash cells gently with phosphate-buffered saline (PBS).
- Fix cells with freshly prepared 4% paraformaldehyde in PBS for 30 minutes at room temperature.
- Permeabilize cells with 0.1% Triton X-100 in PBS for 15 minutes on ice [47] [46].
- Wash twice with PBS to remove residual fixative and permeabilization agents.
TUNEL Reaction:
- Prepare TUNEL reaction mixture according to manufacturer's instructions (typically containing TdT enzyme and modified nucleotide substrate).
- Apply sufficient reaction mixture to completely cover the fixed cells.
- Incubate in a humidified dark chamber at 37°C for 60-90 minutes.
- Terminate reaction by washing slides three times with PBS.
- For fluorescence detection, apply appropriate counterstain (e.g., DAPI for nuclear visualization) and mounting medium.
- Analyze using fluorescence microscopy or flow cytometry [47] [46].
Critical Considerations:
- Include positive controls (DNase I-treated samples) and negative controls (omitting TdT enzyme) with each experiment.
- For sperm cells, additional chromatin decondensation steps using dithiothreitol (DTT) may be necessary due to highly compact structure [46].
- Standardize cell counts and imaging settings across experimental conditions for quantitative comparisons.
Advanced TdT/Cas12a Biosensor Protocol
Emerging methodologies combine TUNEL principles with advanced detection systems. The following protocol for a TdT/Cas12a biosensor enables ultra-sensitive DNA break detection:
DNA Breakpoint Labeling:
- Extract genomic DNA using standardized kits (e.g., TIANGEN DP304) [48].
- Adjust sperm DNA concentration to 5 ng/μL for breakpoint detection.
- Prepare 15 μL reaction mixture containing:
- 1× TdT buffer
- 1× CoCl₂
- 0.1 mM dATP
- 0.5 U TdT enzyme
- 2 μL DNA sample (5 ng/μL)
- Incubate at 37°C for 30 minutes to allow TdT-mediated poly-A tailing at DNA breakpoints.
- Heat-inactivate TdT at 85°C for 10 minutes [48].
CRISPR-Cas12a Detection:
- Add CRISPR-Cas12a detection mixture to the reaction system containing:
- 20 nM LbCas12a enzyme
- 10 nM crRNA (designed to recognize poly-A tails)
- 200 nM FAM-BHQ1 fluorescent reporter probe
- 1× NEBuffer 2.1
- Conduct isothermal fluorescence measurement at 37°C for 60 cycles (30 seconds per cycle) using a real-time PCR instrument.
- Quantify DNA breakpoints based on fluorescence kinetics compared to standard curves [48].
Validation Steps:
- Establish standard curves using DNA samples with known breakpoint concentrations (0-0.3 nM).
- Include internal reference standards for quality control and reproducibility.
- Compare results with traditional TUNEL assays to validate sensitivity improvements.
Figure 1: Standard TUNEL Assay Workflow. This diagram illustrates the key steps in a conventional TUNEL assay protocol, from sample preparation through final analysis.
Figure 2: TdT/Cas12a Biosensor Workflow. This diagram shows the advanced biosensor approach that combines TdT labeling with CRISPR-Cas12a for enhanced sensitivity in DNA break detection.
Essential Research Reagent Solutions
Successful implementation of TUNEL assays requires specific reagent systems optimized for different sample types and detection platforms:
Table 2: Essential Reagents for TUNEL Assay Implementation
| Reagent/Category | Specific Examples | Function & Application Notes |
|---|---|---|
| TdT Enzyme Systems | Terminal deoxynucleotidyl transferase (TdT) with reaction buffer [52] [48] | Catalyzes template-independent addition of nucleotides to 3'-OH DNA ends; Requires CoCl₂ as cofactor for optimal activity [52] |
| Modified Nucleotides | Fluorescein-dUTP, BrdUTP, EdUTP (alkyne-modified dUTP) [47] [52] | Provides detectable labels for incorporated nucleotides; EdUTP enables flexible "click chemistry" detection strategies [47] |
| Detection Systems | Anti-BrdU antibodies, Alexa Fluor azides, streptavidin-peroxidase conjugates [47] | Enables visualization of incorporated nucleotides; Choice depends on platform (microscopy, flow cytometry, colorimetric detection) [47] |
| Sample Preparation Kits | Click-iT Plus TUNEL Assay, APO-BrdU TUNEL Assay [47] | Optimized reagent systems for specific sample types (tissues, cultured cells); Include fixation and permeabilization components [47] |
| Signal Amplification | CRISPR-Cas12a systems, strand displacement probes [52] [48] | Enhances detection sensitivity through enzymatic signal amplification; Enables detection of low-frequency DNA breaks [48] |
Emerging Methodologies and Future Directions
The field of DNA damage detection is rapidly evolving beyond traditional TUNEL methodologies. Recent advances focus on enhancing sensitivity, quantification capabilities, and clinical applicability through innovative molecular approaches.
Novel biosensor technologies that integrate TdT with CRISPR-Cas systems represent a significant leap forward in detection capabilities. The TdT/Cas12a platform demonstrates exceptional performance, detecting DNA breakpoints at concentrations as low as 0.001 nM – substantially more sensitive than conventional TUNEL assays [48]. This system leverages TdT to add poly-A tails exclusively at DNA breakpoints, followed by crRNA-guided Cas12a recognition and trans-cleavage activity that amplifies the fluorescent signal, enabling precise quantification of DNA damage at the molecular level.
Similarly, TdT-based strand displacement probes offer innovative approaches for comprehensive DNA damage assessment. One advanced system simultaneously detects both the mean number of DNA breakpoints (MDB) and apurinic/apyrimidinic (AP) sites, providing a more complete picture of DNA integrity [52]. This methodology incorporates standardized references that significantly improve detection accuracy and reproducibility, demonstrating superior clinical relevance in predicting recurrent pregnancy loss compared to traditional DNA fragmentation index (DFI) assessments [52].
Standardization efforts continue to address reproducibility challenges in TUNEL assays. Inter-laboratory studies demonstrate that through rigorous protocol standardization – including specific fixation conditions, additional washing steps, and consistent flow cytometer settings – TUNEL can achieve high correlation coefficients (r = 0.94) between different laboratories [49]. Such standardization is crucial for establishing TUNEL as a robust, clinically applicable assay for multicenter studies.
These technological advances, combined with improved standardization protocols, position DNA fragmentation analysis as an increasingly precise tool for diagnostic applications, toxicological assessment, and therapeutic development in both research and clinical settings.
Apoptotic bodies (ApoBDs) are subcellular, membrane-bound vesicles (generally 1–5 μm in diameter) generated during the late stages of apoptosis as cells disassemble [35]. Unlike other extracellular vesicles such as exosomes and microvesicles, ApoBDs formation is a tightly controlled process known as apoptotic cell disassembly, characterized by specific morphological steps: (1) plasma membrane blebbing, (2) generation of apoptotic membrane protrusions, and (3) fragmentation into individual ApoBDs [35]. For decades, the primary method for studying apoptosis in a research setting has been the DNA fragmentation assay, which detects internucleosomal DNA cleavage—a classic biochemical hallmark of apoptosis [53] [54]. However, a direct comparison reveals that analyzing ApoBDs via flow cytometry provides a more comprehensive biological readout. While DNA fragmentation assays report on a single, late apoptotic event, flow cytometric analysis of ApoBDs captures the complexity of the entire apoptotic disassembly process, including surface marker presentation and intracellular content packaging, offering deeper functional insights [35] [55].
The utility of flow cytometry in this context stems from its capacity for multiparameter measurements at a single-cell level, allowing researchers to distinguish ApoBDs from other particles (e.g., cells, debris, and other extracellular vesicles) and further characterize heterogeneous subpopulations based on size, granularity, and specific biomarkers [35] [5]. This guide provides a detailed comparison of the methodologies, markers, and applications of flow cytometry for ApoBD analysis, positioning it as a powerful alternative and complement to traditional DNA fragmentation assays.
Key Markers for Identification and Characterization
Accurate identification of ApoBDs relies on a combination of universal apoptotic markers and cell type-specific signatures. The following tables summarize the core markers used in flow cytometry analysis.
Table 1: Surface and Universal Markers for Apoptotic Body Detection
| Marker Type | Specific Marker | Detection Method | Function and Significance in ApoBDs |
|---|---|---|---|
| Lipid Exposure | Phosphatidylserine (PS) | Annexin V binding (e.g., A5-FITC, A5-APC) [35] [43] [56] | "Eat-me" signal; exposed on the outer leaflet of the plasma membrane during early apoptosis [57] [54]. |
| Membrane Integrity | Viability Dyes (TO-PRO-3, Propidium Iodide) | Nucleic acid dye uptake [35] [43] | Distinguishes early apoptotic (dye-negative) from late apoptotic/necrotic (dye-positive) cells and ApoBDs [43] [56]. |
| Apoptotic Enzyme | Activated Caspase-3 | FLICA assay; antibody detection [43] [53] | Key executioner caspase; its activation is a hallmark of apoptosis and can be detected in some ApoBDs [53] [55]. |
Table 2: Markers for Cell Origin and Intracellular Content Analysis
| Content Category | Specific Marker/Stain | Function and Distribution in ApoBDs |
|---|---|---|
| Cell Surface Markers (Origin) | CD45 (lymphocytes), CD146 (endothelial cells), CD3 (T cells) [35] | ApoBDs share the same surface markers as their cell of origin, allowing for tracking in mixed cultures [35]. |
| Nuclear Material | Hoechst 33342, DRAQ5 [35] [57] | Nuclear proteins and DNA are distributed to some, but not all, ApoBDs, a process influenced by the disassembly mechanism [35]. |
| RNA | SYTO RNASelect [35] | RNA can be packaged into specific ApoBD subpopulations [35]. |
| Mitochondria | MitoTracker Green [35] | Mitochondria are heterogeneously distributed into ApoBDs [35]. |
Experimental Protocols for Flow Cytometric Analysis
A Five-Step Workflow for Apoptotic Body Analysis
The following diagram illustrates a generalized protocol for the flow cytometric analysis of ApoBDs, integrating key steps from cell culture to data analysis.
Detailed Protocol: Surface Marker and Content Analysis
This protocol is adapted from a 2017 study that established a robust method for characterizing ApoBD subsets [35].
1. Induction of Apoptosis:
- Cell Lines: Use appropriate cell lines (e.g., THP-1 monocytic cells, Jurkat T cells, HUVEC).
- Inducers: Induce apoptosis via ultraviolet (UV) irradiation (e.g., 150 mJ/cm²) or anti-Fas treatment (e.g., 62.5 ng/ml).
- Incubation: Incubate cells at 37°C in a humidified atmosphere with 5% CO₂ for 2–6 hours in 1% BSA/RPMI media [35].
2. Pre-staining of Intracellular Contents (Prior to Induction):
- Staining Solution: Prior to apoptosis induction, stain cells with probes for intracellular contents.
- Nuclear Material: Use Hoechst 33342 at 5 μg/ml.
- Mitochondria: Use MitoTracker Green at 100 nM.
- RNA: Use SYTO RNASelect Green at 500 nM.
- Incubate according to manufacturer's instructions [35].
3. Staining for Flow Cytometry:
- Annexin V & Viability Dye: After induction, stain cells with a combination of fluorochrome-conjugated Annexin V (e.g., A5-FITC, A5-APC) and a viability dye like TO-PRO-3 in 1x Annexin V binding buffer for 10 minutes at room temperature, protected from light [35] [43].
- Surface Marker Staining: Concurrently or subsequently, stain with antibodies against cell type-specific surface markers (e.g., CD45-PeCy7 for hematopoietic cells, CD146-VioBlue for endothelial cells) to determine the cellular origin of ApoBDs [35].
4. Flow Cytometry Acquisition:
- Instrument Setup: Use a flow cytometer equipped with lasers and filters appropriate for the fluorochromes used.
- Controls: Always include unstained and single-stained controls for compensation.
- Gating Strategy:
- Gate on particles based on size (forward scatter, FSC) and granularity (side scatter, SSC) to exclude debris and larger cells.
- Within this gate, select the Annexin V-positive population.
- Further gate on the TO-PRO-3-negative (or low) population to distinguish ApoBDs from late apoptotic/necrotic cells and debris [35] [56].
- Acquire a sufficient number of events for statistical analysis.
5. Data Analysis:
- Analyze the distribution of intracellular contents (Hoechst, MitoTracker, SYTO RNAselect) and surface markers within the gated ApoBD population using flow cytometry analysis software (e.g., FlowJo) [35].
- The data will reveal heterogeneous subpopulations of ApoBDs containing different cargoes from the same parental cell.
Comparative Analysis with DNA Fragmentation Assays
While DNA fragmentation assays like TUNEL (Terminal deoxynucleotidyl transferase dUTP nick end labeling) have been a cornerstone in apoptosis research, flow cytometric ApoBD analysis offers distinct advantages and complementary data, as summarized below.
Table 3: Flow Cytometry vs. DNA Fragmentation for Apoptosis Analysis
| Feature | Flow Cytometry for ApoBDs | DNA Fragmentation Assays (e.g., TUNEL) |
|---|---|---|
| Analytical Focus | Holistic cellular disassembly process, vesicle formation, and content distribution. | Singular biochemical event of internucleosomal DNA cleavage [53] [54]. |
| Information Output | Multiparameter: size, granularity, PS exposure, membrane integrity, cell origin, cargo (DNA, RNA, organelles). | Single-parameter: presence of DNA strand breaks [57] [53]. |
| Functional Insight | High. Reveals "eat-me" signals (PS), potential for intercellular communication, and immunomodulatory capacity of ApoBDs [58] [55]. | Limited. Confirms apoptosis but offers little insight into downstream clearance or signaling functions. |
| Stage Detection | Can discriminate early (Annexin V+/TO-PRO-3-) and late (Annexin V+/TO-PRO-3-) stages [43] [56]. | Typically detects mid-to-late stages after endonuclease activation [57] [53]. |
| Throughput | High-throughput, rapid analysis of thousands of events per second [43] [5]. | Generally lower throughput, especially when combined with microscopy. |
| Key Application | Ideal for studying ApoBD heterogeneity, biogenesis, efferocytosis, and their role in disease and therapy [35] [55]. | Remains a gold standard for confirming apoptotic DNA degradation, often used in tissue sections [57]. |
The Scientist's Toolkit: Essential Research Reagents
Successful execution of the described protocols requires a set of core reagents. The following table lists essential solutions and their critical functions.
Table 4: Essential Reagents for Apoptotic Body Flow Cytometry
| Reagent Category | Specific Examples | Critical Function in the Protocol |
|---|---|---|
| Apoptosis Inducers | Staurosporine, UV Radiation, Anti-Fas Antibody | Triggers the controlled process of programmed cell death leading to ApoBD formation [35] [58]. |
| Viability & Membrane Probes | TO-PRO-3, Propidium Iodide (PI) | Assesses plasma membrane integrity; critical for distinguishing early apoptotic vesicles from late-stage debris [35] [43]. |
| PS Exposure Probes | Annexin V-FITC, Annexin V-APC | Binds to externalized phosphatidylserine, a primary marker for identifying apoptotic cells and ApoBDs [35] [56]. |
| Nuclear Stains | Hoechst 33342, DRAQ5 | Labels DNA, allowing for analysis of nuclear content distribution into ApoBDs [35] [57]. |
| Organelle & RNA Trackers | MitoTracker Green, SYTO RNASelect | Probes for tracking the packaging of specific intracellular components (mitochondria, RNA) into ApoBDs [35]. |
| Cell Surface Antibodies | CD45, CD3, CD146, CD44 | Fluorochrome-conjugated antibodies used to identify the cellular origin of ApoBDs in mixed populations [35] [58]. |
| Binding Buffer | 10x Annexin V Binding Buffer | Provides the optimal calcium-containing environment for efficient Annexin V binding to PS [43] [56]. |
Flow cytometry has evolved beyond mere apoptosis quantification into a powerful tool for dissecting the complex biology of apoptotic body formation and function. The ability to perform multiparameter analysis—simultaneously interrogating surface markers, intracellular cargo, and cellular origin—provides a level of detail that traditional DNA fragmentation assays cannot match. The presented protocols and comparative data underscore that ApoBDs are not homogeneous cellular waste but are instead heterogeneous vesicles with specific compositions and potential functions in immunomodulation and intercellular communication [35] [58] [55]. For researchers and drug development professionals, integrating this flow cytometry-based approach into their toolkit enables a more profound understanding of cell death mechanisms, which is critical for advancing therapeutic strategies in areas like cancer, autoimmune diseases, and regenerative medicine.
The study of apoptosis, or programmed cell death, relies heavily on the accurate identification of characteristic morphological hallmarks. Key events in apoptosis include cell shrinkage, chromatin condensation, nuclear fragmentation, and plasma membrane blebbing, culminating in the formation of apoptotic bodies [59]. Distinguishing apoptosis from other forms of cell death, such as necroptosis (which features cytoplasmic swelling and plasma membrane rupture) and pyroptosis (characterized by rapid plasma membrane rupture and the release of proinflammatory contents), is crucial and hinges on precise morphological analysis [59]. Confocal microscopy and electron microscopy are two powerful imaging techniques that provide the resolution and detail necessary for this task. This guide objectively compares their performance in visualizing apoptotic morphology within the context of research that also employs DNA fragmentation assays, providing supporting experimental data and protocols to inform researchers and drug development professionals.
Technical Comparison of Confocal and Electron Microscopy
The following table summarizes the core technical characteristics and applications of Confocal and Electron Microscopy for apoptosis research.
| Feature | Confocal Microscopy | Scanning Electron Microscopy (SEM) |
|---|---|---|
| Primary Principle | Point illumination and a pinhole aperture to block out-of-focus light, generating optical sections [60]. | A focused electron beam scans the surface, detecting secondary electrons to create topographical images [61]. |
| Key Strength | 3D imaging of live cells; specific fluorescence labeling of intracellular components [60]. | High-resolution surface topography visualization of membrane blebbing and apoptotic bodies [62]. |
| Resolution Limit | ~200 nm (lateral), limited by light diffraction [60]. | Sub-nanometer range (surface), offering superior detail for membrane structures [63]. |
| Sample Preparation | Can image live or fixed cells with fluorescent labeling [60]. | Requires fixation, dehydration, and conductive coating; not suitable for live cells [62]. |
| Morphological Hallmarks Detected | - Cell shrinkage (via 3D reconstruction)- Caspase activation (via fluorescent probes)- Phosphatidylserine externalization (via Annexin V) [62]. | - Membrane blebbing- Apoptotic body formation- Surface texture changes [62]. |
| Best for Apoptosis Stage | Early to late stages (dynamic processes and internal markers) [62]. | Mid to late stages (surface structure changes) [62]. |
| Quantitative Capability | High; enables photon-counting for absolute quantitation of fluorescence intensity and morphometric measurements [64] [65]. | Primarily qualitative for morphology; can be paired with other techniques for roughness parameters [66]. |
Experimental Data and Performance Comparison
Quantitative Imaging Performance
A comparative study of confocal reflectance microscopy and SEM for characterizing tissue surface roughness found that the measurements had a statistically significant difference for bovine tissue, though not for porcine and poultry tissue [66]. This highlights that the optimal tool can depend on the specific sample type. For fluorescence quantification, modern confocal systems like the FLUOVIEW FV5000 employ SilVIR photon-counting detector technology, which provides absolute quantitative measurements by counting individual photons. This, coupled with a Laser Power Monitor, eliminates key sources of variation and ensures reproducibility across experiments and laboratories, a critical feature for longitudinal studies of apoptotic progression [64].
Data Utilization in Imaging Studies
A systematic analysis of electron microscopy data utilization revealed a significant challenge: over a 10-year period at a core facility, more than 97% of the scientifically significant electron microscopy images generated remained unpublished [63]. This "lost data" phenomenon suggests that while SEM generates vast amounts of raw morphological data, only a select few images typically confirm a hypothesis. In contrast, quantitative confocal microscopy is inherently geared toward generating statistically analyzable data sets from entire samples, potentially leading to more comprehensive data utilization [65].
Detailed Experimental Protocols
Protocol: Imaging Apoptotic Morphology with Confocal Microscopy
This protocol is designed for detecting early and mid-stage apoptotic features in cultured cells using a laser scanning confocal microscope (LSCM).
- Sample Preparation: Culture cells on glass-bottom dishes or coverslips. Induce apoptosis using your chosen stimulus (e.g., chemotherapeutic agent). For fixed samples, wash with PBS and fix with 4% paraformaldehyde for 15 minutes. Permeabilize with 0.1% Triton X-100 if staining internal targets.
- Staining:
- Membrane Alterations (Early Apoptosis): Stain with FITC-conjugated Annexin V in a calcium-containing binding buffer for 15-20 minutes in the dark [62].
- Caspase Activation (Mid Apoptosis): Incubate with a primary antibody against active Caspase-3, followed by a fluorescently-labeled secondary antibody [62].
- Nuclear Morphology: Include a nuclear counterstain such as DAPI or Hoechst to visualize chromatin condensation and karyorrhexis.
- Image Acquisition:
- Use a high-NA immersion objective (e.g., 60x or 100x oil).
- Set the pinhole to 1 Airy Unit for optimal optical sectioning [65].
- Use sequential scanning mode to avoid cross-talk between fluorophores [65].
- For live-cell imaging of dynamics like membrane blebbing, use the resonant scanning mode for high-speed acquisition [64].
- Image Analysis: Use software to create 3D reconstructions from z-stacks. Quantify fluorescence intensity for Annexin V or active Caspase-3. Measure cell volume changes from 3D data.
Protocol: Visualizing Apoptotic Bodies with Scanning Electron Microscopy
This protocol details the preparation and imaging of cells for high-resolution surface topography analysis of late-stage apoptosis.
- Sample Preparation: Culture and treat cells on a suitable substrate (e.g., a glass coverslip). Wash cells gently with a physiological buffer (e.g., PBS).
- Fixation: Primary fixation with 2.5% glutaraldehyde in 0.1M cacodylate buffer for at least 1 hour at 4°C is critical for preserving morphology. Wash thoroughly with buffer.
- Dehydration: Gradually dehydrate the sample through a series of ethanol washes (e.g., 30%, 50%, 70%, 90%, 100%), with a final step in 100% ethanol.
- Critical Point Drying: Process the sample through a critical point dryer to avoid surface tension artifacts that occur during air drying.
- Sputter Coating: Mount the sample on a stub and coat with a thin layer (a few nanometers) of gold/palladium using a sputter coater to make the surface conductive.
- SEM Imaging: Place the sample in the microscope chamber. Image at an accelerating voltage of 5-15 kV. Use a secondary electron detector. Capture images at various magnifications to show overall cell population and high-resolution details of membrane blebs and apoptotic bodies.
Research Reagent Solutions
The table below lists key reagents and their functions for apoptosis imaging experiments.
| Reagent / Material | Function in Experiment |
|---|---|
| Annexin V (FITC conjugate) | Binds to phosphatidylserine exposed on the outer leaflet of the plasma membrane, a key early marker of apoptosis [62]. |
| Antibody to Active Caspase-3 | Detects the activated form of the key executioner caspase, confirming the engagement of the apoptotic pathway [62]. |
| DAPI or Hoechst | DNA-binding dyes that stain the nucleus, allowing visualization of nuclear condensation and fragmentation [60]. |
| Glutaraldehyde | A cross-linking fixative used for SEM that excellently preserves cellular ultrastructure and surface details [62]. |
| Gold/Palladium Target | Source for sputter coating, creating a conductive metal layer on biological samples to prevent charging in the SEM. |
| Subresolution Fluorescent Microspheres | Used to measure the point spread function (PSF) and ensure the confocal microscope is operating at optimal resolution [65]. |
Integrated Pathway and Workflow Analysis
The following diagram illustrates the core morphological events of apoptosis and how the different imaging techniques map onto this process.
The systematic dismantling of a cell during programmed cell death involves two key observable phenomena: DNA fragmentation, a biochemical hallmark of apoptosis, and the formation of apoptotic bodies, which are morphological endpoints. DNA fragmentation refers to the cleavage of nuclear DNA into internucleosomal fragments, typically in sizes of 180-200 base pairs, driven by the activation of specific endonucleases such as CAD (Caspase-Activated DNase). This process can be quantified through various assays to serve as a biomarker for the degree and mode of cell death. In parallel, apoptotic body formation describes the structural process where the dying cell condenses and fragments into membrane-bound vesicles containing cellular constituents, including the fragmented DNA. These bodies facilitate clean-up by phagocytes, preventing inflammatory responses.
Understanding the relationship between these two processes—one at the molecular level and the other at the morphological level—is crucial for researchers in cancer research, toxicology, and reproductive medicine. This guide provides a comparative analysis of the experimental methods used to detect and quantify these events, supported by current experimental data and protocols.
Comparative Analysis of Detection Methods
DNA Fragmentation Assays
DNA fragmentation assays measure the breakdown of a cell's nuclear DNA, a key indicator of apoptosis. The following table compares the major techniques used across different application contexts.
Table 1: Comparison of DNA Fragmentation Assays
| Assay Name | Primary Application Context | Principle | Key Performance Metrics | Advantages | Limitations |
|---|---|---|---|---|---|
| TUNEL(TdT dUTP Nick-End Labeling) [67] [68] | General Apoptosis Detection, Sperm DNA Fragmentation | Labels 3'-OH ends of DNA fragments with fluorescent nucleotides | Detects early-stage DNA breaks; High sensitivity to single-strand breaks [67] | Can be used on tissue sections, cells, and sperm; Adaptable for flow cytometry and microscopy [68] | Can overestimate damage; Poor concordance with other SDF tests (Lin's CCC <0.5) [67] |
| Comet Assay(Single Cell Gel Electrophoresis) [67] [69] [68] | Toxicology, Sperm DNA Fragmentation, Recurrent Pregnancy Loss Investigation | Electrophoresis of single cells to visualize DNA damage as a "comet tail" | Can differentiate single-strand (alkaline protocol) vs. double-strand breaks (neutral protocol, dsSDF) [69] | High sensitivity; Provides data at single-cell level [69] | Labor-intensive; Lower throughput than other methods [67] |
| SCSA(Sperm Chromatin Structure Assay) [67] [70] [71] | Sperm DNA Fragmentation, Male Fertility Assessment | Flow cytometry-based measurement of DNA denaturability in acidic conditions | Clinically validated with defined thresholds (e.g., DFI >30% associated with reduced embryo euploidy) [70] | High repeatability; Suitable for large sample sizes [71] | Requires flow cytometry; Primarily used for sperm analysis [70] |
| SCD Test(Sperm Chromatin Dispersion) [67] | Sperm DNA Fragmentation | Visualization of halos of dispersed DNA loops after protein denaturation | Shows moderate concordance with Comet Assay (Lin's CCC ~0.5) [67] | Simple and cost-effective; No need for complex equipment [67] | Less quantitative than SCSA or Comet [67] |
| cfDNA Fragmentomics(e.g., LIONHEART) [72] [73] | Cancer Detection & Therapy Monitoring | Sequencing of cell-free DNA (cfDNA) fragment patterns in blood | AUC 0.62-0.95 for pan-cancer detection; AUC 0.93 for predicting therapy failure [72] [73] | Non-invasive liquid biopsy; Can identify tissue of origin [72] | Requires sequencing and advanced bioinformatics [73] |
Apoptotic Body Formation Analysis
The formation of apoptotic bodies and other extracellular vesicles (EVs) represents a key morphological outcome of cell death. The detection methods and their performances are summarized below.
Table 2: Comparison of Apoptotic Body and Vesicle Analysis Methods
| Method Name | Application Context | Principle | Key Performance / Characteristics | Advantages | Limitations |
|---|---|---|---|---|---|
| Time-Lapse Microscopy(e.g., LLSM, CLSM) [31] | Basic Research, Viral Infection Studies | Direct visualization of vesicle formation and dynamics | Revealed ~40 F-ApoEVs generated per apoptotic cell within 4 hours [31] | Captures dynamic process in real-time; Reveals novel mechanisms (e.g., FOOD) [31] | Requires specialized, expensive equipment; Lower throughput |
| Flow Cytometry [74] [68] | Immunology, Cell Biology | Light scattering and fluorescence to identify vesicles in suspension | Can analyze up to 16 optical parameters at ~100,000 events/second [74] | High-throughput; Multi-parameter analysis [74] | Cannot analyze vesicles attached to substrate (e.g., FOOD) [31] |
| Scanning Electron Microscopy (SEM) [31] | Basic Research, Ultrastructural Analysis | High-resolution imaging of surface morphology | Reveals ultrastructure of FOOD as thin membrane surrounding the cell body [31] | Exceptional resolution for surface details [31] | No live-cell imaging; Complex sample preparation |
| Proteomic Analysis [31] | Biomarker Discovery, Functional Studies | Mass spectrometry of isolated vesicles | Identified enrichment of actin and adhesion proteins in F-ApoEVs [31] | Unbiased discovery of vesicle content and function [31] | Requires vesicle isolation; Destructive to samples |
Experimental Protocols for Key Methodologies
Protocol: Sperm DNA Fragmentation Assessment by SCSA
The Sperm Chromatin Structure Assay (SCSA) is a flow cytometry-based method for measuring sperm DNA Fragmentation Index (DFI), widely used in clinical andrology [70] [71].
Sample Collection and Staining:
- Collect semen sample and dilute with TNE buffer (0.01 M Tris-HCl, 0.15 M NaCl, 1 mM EDTA, pH 7.4) to a concentration of 1-2 x 10^6 sperm/mL.
- Mix a 200 µL aliquot of diluted sperm with 400 µL of acidic detergent solution (0.1% Triton X-100, 0.15 M NaCl, 0.08 N HCl, pH 1.2) for 30 seconds.
- Immediately add 1.2 mL of staining solution containing acridine orange (6 µg/mL in 0.1 M citric acid, 0.2 M Na2HPO4, 1 mM EDTA, 0.15 M NaCl, pH 6.0). Incubate for 3 minutes.
Flow Cytometric Analysis:
- Analyze samples using a flow cytometer equipped with a 488 nm argon laser.
- Measure green fluorescence (515-530 nm) from double-stranded DNA and red fluorescence (>630 nm) from denatured single-stranded DNA.
- Collect data from at least 5,000 events per sample.
Data Interpretation:
Protocol: Cancer Detection via cfDNA Fragmentomics (LIONHEART)
The LIONHEART method is a cross-dataset pan-cancer detection tool that correlates cfDNA fragment coverage with open chromatin sites [72].
Plasma Separation and cfDNA Extraction:
- Collect peripheral blood in Streck cell-free DNA BCT tubes.
- Centrifuge tubes at 1600× g for 10 minutes at 15°C to separate plasma.
- Transfer the plasma to a new tube and centrifuge again at 16,000× g for 10 minutes at room temperature to remove remaining cells.
- Extract cfDNA from 500 µL to 1 mL of plasma using the QIAamp Circulating Nucleic Acid Kit (Qiagen), following the manufacturer's protocol but omitting carrier RNA [73].
Whole-Genome Sequencing and Bias Correction:
- Perform low-coverage whole-genome sequencing (WGS) on the extracted cfDNA.
- Process the sequencing data to generate fragment coverage maps across the genome.
- Apply computational bias correction to the fragment coverage to account for technical artifacts and enable robust cross-dataset generalization [72].
Correlation with Chromatin Accessibility and Classification:
- Correlate the bias-corrected cfDNA fragment coverage profile with a reference panel of open chromatin sites from 898 cell and tissue types (from ENCODE, ATACdb, and TCGA).
- Use these correlation features to detect changes in the cell-free DNA cell type composition caused by the presence of cancer.
- Input the features into a machine learning model (trained on 1106 non-cancer controls and 1449 cancers) to distinguish cancer samples from non-cancer controls [72].
Protocol: Visualization and Quantification of FOOD-Derived ApoEVs
This protocol describes the visualization of a newly described mechanism for generating large apoptotic extracellular vesicles (F-ApoEVs) via the "FOotprint Of Death" (FOOD) [31].
Cell Culture and Apoptosis Induction:
- Plate adherent cells (e.g., A431, MEFs, or HeLa) on glass-bottom dishes or ECM-coated surfaces.
- Induce apoptosis using a relevant stimulus (e.g., BH3-mimetic cocktail ABT-737/S63845, UV irradiation, or etoposide).
Live-Cell Staining and Imaging:
- Stain cells with Annexin A5 (e.g., fluorescently conjugated) to label phosphatidylserine (PtdSer) exposure.
- Use 3D time-lapse confocal laser scanning microscopy (CLSM) or lattice light sheet microscopy (LLSM) to capture the dynamic process of cell retraction and FOOD formation. Image at a focal plane close to the cover glass (base).
Image Analysis and Quantification:
- Analyze time-lapse data to confirm the formation of sheet-like membranous FOOD structures that vesicularise into discrete F-ApoEVs.
- Quantify the number of F-ApoEVs generated per cell (median ~40 per cell over 4 hours), their diameter (~2 μm), and the area occupied by the FOOD structures [31].
Signaling Pathways and Experimental Workflows
Apoptotic Signaling and Vesicle Formation Pathways
The following diagram illustrates the core signaling pathways in apoptosis that lead to the key biochemical and morphological hallmarks discussed in this guide, including DNA fragmentation and the formation of different extracellular vesicles.
Diagram Title: Apoptosis Pathways and Detection Methods
Experimental Workflow for a Comprehensive Cell Death Analysis
This workflow outlines the steps for an integrated analysis of cell death, from sample preparation to data interpretation from multiple assays.
Diagram Title: Integrated Cell Death Analysis Workflow
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagent Solutions for DNA Fragmentation and Apoptotic Body Research
| Reagent / Kit | Primary Function | Application Context | Key Feature / Note |
|---|---|---|---|
| Streck Cell-Free DNA BCT Tubes | Stabilizes nucleated blood cells for plasma cfDNA analysis | Cancer Research (Liquid Biopsy) | Prevents dilution of cfDNA by genomic DNA from white blood cell lysis [73] |
| QIAamp Circulating Nucleic Acid Kit | Extraction of cell-free DNA from plasma/serum | Cancer Research (Liquid Biopsy) | High-sensitivity recovery of short cfDNA fragments; Carrier RNA can be omitted [73] |
| Annexin V (A5) Conjugates | Binds to phosphatidylserine (PtdSer) exposed on apoptotic membranes | General Apoptosis Detection, Vesicle Analysis | Key marker for early apoptosis and for identifying PtdSer-exposing FOOD/F-ApoEVs [31] |
| BH3 Mimetics (e.g., ABT-737, S63845) | Induces intrinsic apoptosis pathway by inhibiting BCL-2 family proteins | Basic Apoptosis Research, Cancer Biology | Highly specific triggers for studying canonical apoptotic morphology and FOOD formation [31] |
| Acridine Orange | Metachromatic dye for DNA/RNA staining; used in SCSA | Sperm DNA Fragmentation | Fluorescence shifts from green (dsDNA) to red (ssDNA) upon acid denaturation [71] |
| ROCK1 Inhibitor (e.g., Y-27632) | Inhibits Rho-associated kinase 1 (ROCK1) | Basic Apoptosis Research, Mechanism Studies | Tool to investigate ROCK1's role in FOOD formation and apoptotic membrane blebbing [31] |
Optimizing Apoptosis Assays: Overcoming Technical Challenges and Pitfalls
Accurately distinguishing between the major forms of programmed cell death—apoptosis, pyroptosis, and necroptosis—is fundamental to biomedical research, particularly in drug development and the study of inflammatory, infectious, and neurodegenerative diseases. A persistent challenge in the field is the accurate interpretation of biochemical and morphological hallmarks, such as DNA fragmentation and apoptotic body formation, which can be misinterpreted due to overlapping features and assay-specific artifacts. This guide objectively compares these cell death pathways within the context of DNA fragmentation assays and apoptotic body research, providing structured experimental data and protocols to aid researchers in making definitive distinctions.
Key Characteristics of Cell Death Pathways
The table below summarizes the core defining features of each cell death pathway, providing a foundation for their experimental differentiation.
Table 1: Key Characteristics of Apoptosis, Pyroptosis, and Necroptosis
| Feature | Apoptosis | Pyroptosis | Necroptosis |
|---|---|---|---|
| Physiological Role | Programmed cell deletion in development and homeostasis; immunologically silent [75] [76] | Innate immune defense against intracellular pathogens; highly inflammatory [75] [77] | Backup cell death pathway when apoptosis is blocked; promotes inflammation and tissue repair [75] |
| Morphology | Cell shrinkage, nuclear condensation, formation of membrane-bound apoptotic bodies [76] | Cell swelling, plasma membrane pore formation, cell lysis [77] | Organelle swelling, loss of plasma membrane integrity, cell lysis without condensation [75] [76] |
| Key Initiators | Caspase-8 (extrinsic), Caspase-9 (intrinsic) [76] [78] | Inflammatory Caspases (Caspase-1, human Caspase-4/5, mouse Caspase-11) [79] | RIPK1, RIPK3 (activated when Caspase-8 is inhibited) [75] [79] |
| Key Executors | Effector Caspases (Caspase-3/7), CAD/DFF40 [80] [76] [78] | Gasdermin D (GSDMD) N-terminal fragment [79] | Phosphorylated MLKL oligomers [75] |
| Membrane Integrity | Maintained until late stages (apoptotic body formation) [76] | Disrupted by GSDMD pores [79] | Disrupted by MLKL pores [75] |
| Inflammatory Response | Minimal ("silent"); promotes tolerance [75] | Robust; release of IL-1β and IL-18 [75] [77] | Robust; release of DAMPs and alarmins [75] |
| Classic Hallmarks | Phosphatidylserine (PS) externalization, apoptotic bodies, internucleosomal DNA fragmentation [80] [76] [81] | GSDMD pore formation, inflammasome activation, mature cytokine release [79] | Phosphorylation of RIPK3 and MLKL, necrosome formation [75] |
Signaling Pathways
The following diagrams illustrate the core molecular signaling pathways for apoptosis, pyroptosis, and necroptosis.
Apoptosis Signaling Pathways
Pyroptosis and Necroptosis Signaling
DNA Fragmentation and Apoptotic Body Formation
DNA fragmentation and the formation of apoptotic bodies are classic hallmarks of apoptosis, but their timing and context are critical for accurate interpretation.
Mechanism and Timing of DNA Fragmentation
The enzyme Caspase-Activated DNase (CAD) is the primary executor of apoptotic DNA fragmentation. CAD is normally inhibited by its chaperone, ICAD. During apoptosis, effector caspases (primarily caspase-3) cleave ICAD, releasing active CAD. CAD then cleaves nuclear DNA at the internucleosomal linker regions, generating fragments in multiples of ~180 base pairs [80]. This produces the characteristic "DNA ladder" pattern observed in gel electrophoresis.
A crucial consideration for researchers is that major DNA fragmentation is a late event in the apoptotic cascade. A time-lapse video microscopy study demonstrated that nuclear condensation occurs early in apoptosis, coinciding with initial surface blebbing. However, DNA strand breaks detectable by techniques like in situ nick-translation only appear much later, after the formation of separated apoptotic bodies or final cell lysis [82]. This temporal separation means that a cell can be committed to apoptosis and exhibit other morphological features before its DNA is extensively fragmented.
Comparative Analysis of Detection Assays
Different assays detect distinct aspects or stages of DNA fragmentation, leading to potential artifacts if not properly contextualized.
Table 2: Comparison of DNA Fragmentation and Apoptotic Body Detection Assays
| Assay | Target | Applications | Key Advantages | Common Artifacts & Limitations |
|---|---|---|---|---|
| DNA Laddering (Gel Electrophoresis) | Internucleosomal DNA cleavage (~180 bp fragments) [80] [78] | Bulk cell population analysis; confirming apoptotic commitment. | Simple, classic hallmark of apoptosis; distinguishes apoptosis (ladder) from necrosis (smear) [80]. | - Low sensitivity for heterogeneous samples.- Requires high apoptotic cell percentage.- Late-stage event; misses early apoptosis [82]. |
| TUNEL Assay | 3'-OH DNA ends in situ [80] [78] | Histology; flow cytometry; identification of apoptotic cells in tissue sections. | High sensitivity; single-cell resolution; can be combined with cell-specific markers [78]. | - Can label DNA breaks from non-apoptotic processes (e.g., necrosis, DNA repair, oxidative stress) [78].- Does not distinguish between different programmed cell death pathways. |
| Sub-G1 Peak Analysis (Flow Cytometry) | Fractional DNA content after extraction of low MW DNA [80] | High-throughput quantification of apoptotic cell populations. | Rapid, quantitative, suitable for large sample sizes. | - Late-S and G2 apoptotic cells may be misclassified [80].- False positives from mechanical cell damage or sample preparation. |
| Analysis of Apoptotic Bodies (Microscopy) | Membrane-bound cellular fragments containing condensed chromatin [76] | Morphological confirmation of apoptosis; time-lapse studies. | Direct visualization of a key apoptotic hallmark. | - Requires expert morphological identification.- Can be confused with other cellular debris or blebs from lytic cell death. |
Detailed Experimental Protocols
Annexin V/Propidium Iodide (PI) Staining for Flow Cytometry
The Annexin V binding assay is a standard method for detecting early- to mid-stage apoptosis by measuring the externalization of phosphatidylserine (PS) [83] [81].
Key Materials:
- Annexin V Conjugate: Fluorochrome-labeled (e.g., FITC, PE, APC) recombinant Annexin V protein.
- Propidium Iodide (PI) Staining Solution: DNA-binding dye for viability assessment.
- 10X Binding Buffer: Provides the calcium-dependent binding context for Annexin V.
- Cell Staining Buffer: Azide- and serum/protein-free PBS for wash steps.
Protocol:
- Cell Preparation: Harvest and wash cells twice in cold 1X PBS. Resuspend the cell pellet in 1X Binding Buffer at a concentration of 1-5 x 10⁶ cells/mL.
- Staining: Add 5 µL of fluorochrome-conjugated Annexin V to 100 µL of the cell suspension. Incubate for 10-15 minutes at room temperature, protected from light.
- Viability Staining: Add 2 mL of 1X Binding Buffer, centrifuge, and discard the supernatant. Resuspend the cells in 200 µL of 1X Binding Buffer. Add 5 µL of PI Staining Solution and incubate for 5-15 minutes on ice. Do not wash after adding PI.
- Analysis: Analyze the cells by flow cytometry within 4 hours. Use FITC (FL1) and PE (FL2) signal detectors for Annexin V and PI, respectively [83].
Interpretation of Results:
- Viable Cells: Annexin V⁻ / PI⁻
- Early Apoptotic Cells: Annexin V⁺ / PI⁻
- Late Apoptotic or Necrotic Cells: Annexin V⁺ / PI⁺
Critical Considerations:
- Calcium Dependence: Avoid buffers containing EDTA or other calcium chelators, as they inhibit Annexin V binding [83].
- Membrane Integrity: Annexin V can only reliably indicate apoptosis in cells with an intact plasma membrane. Destroying membrane integrity allows Annexin V to bind PS on the inner leaflet, leading to false positives [83].
- Specificity: Note that PS externalization is not exclusive to apoptosis and can occur in other forms of cell death, such as necroptosis [81].
TUNEL Assay for DNA Fragmentation
The TUNEL (TdT dUTP Nick-End Labeling) assay detects DNA strand breaks by labeling the 3'-OH termini with modified nucleotides.
Key Materials:
- Terminal Deoxynucleotidyl Transferase (TdT): The enzyme that catalyzes the addition of nucleotides to the 3'-OH ends of DNA.
- Labeled Nucleotides: Often fluorescein-dUTP or biotin-dUTP.
- Permeabilization Buffer: Typically containing Triton X-100 and sodium citrate.
- Blocking Solution: Such as bovine serum albumin (BSA) in PBS.
Protocol (for adherent cells on coverslips):
- Fixation: Fix cells in 4% paraformaldehyde in PBS for 15 minutes at room temperature.
- Permeabilization: Treat cells with 0.1% Triton X-100 in 0.1% sodium citrate buffer for 2 minutes on ice.
- Blocking: Incubate with 10% BSA in PBS for 1 hour at room temperature to block nonspecific binding.
- Labeling: Incubate coverslips with the TUNEL reaction mixture (containing TdT enzyme and labeled nucleotides) for 1 hour at 37°C.
- Visualization: Analyze by fluorescence microscopy. TUNEL-positive nuclei will exhibit fluorescent staining [78].
Critical Considerations:
- The TUNEL assay is highly sensitive but can produce false positives by labeling DNA breaks from non-apoptotic processes, including necrosis, DNA damage, and oxidative stress. It is crucial to correlate TUNEL results with other apoptotic markers, such as cell morphology [78].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Cell Death Research
| Reagent/Category | Specific Examples | Primary Function in Research |
|---|---|---|
| PS Binding Probes | Fluorochrome-conjugated Annexin V (FITC, PE, APC) [83] [81] | Detection of phosphatidylserine externalization as a marker of early apoptosis via flow cytometry or microscopy. |
| Viability Dyes | Propidium Iodide (PI), 7-AAD, Fixable Viability Dyes (e.g., FVD eFluor) [83] | Discrimination of cell membrane integrity; used to distinguish early apoptotic (dye-negative) from late apoptotic/necrotic (dye-positive) cells. |
| Caspase Inhibitors | z-VAD-FMK (pan-caspase inhibitor) [79] | Tool to inhibit apoptotic signaling; can shift cell fate towards necroptosis (e.g., in TNF-α induced models). |
| Necroptosis Inducers/Inhibitors | TNF-α + z-VAD (Inducer) [79], Necrostatin-1 (RIPK1 inhibitor) | Specific induction or inhibition of the necroptotic pathway for mechanistic studies. |
| Pyroptosis Inducers | LPS + ATP (for NLRP3 activation) [79] | Canonical activation of the inflammasome pathway to induce pyroptosis in macrophages. |
| Key Antibodies | Anti-cleaved Caspase-3, Anti-phospho-MLKL, Anti-cleaved GSDMD | Detection of activated executors of cell death via Western blot or immunofluorescence, allowing for specific pathway identification. |
The Emergence of PANoptosis
Recent research has revealed a more complex, interconnected landscape of cell death. PANoptosis is a novel, inflammatory programmed cell death pathway that is regulated by PANoptosomes—multiprotein complexes that integrate components from pyroptosis, apoptosis, and necroptosis. PANoptosis cannot be fully explained by any one of these three pathways alone [77] [84] [79].
For example, during viral infection, sensors like ZBP1 can nucleate a PANoptosome containing key molecules such as CASP8 (apoptosis), RIPK3/MLKL (necroptosis), and NLRP3/CASP1 (pyroptosis) [84]. This simultaneous activation leads to a catastrophic burst of inflammatory cell death, which has been implicated in pathologies including ischemic stroke, infectious diseases, and inflammatory bone disease [77] [84]. The existence of PANoptosis underscores the critical importance of using multi-parametric assays rather than relying on a single hallmark to conclusively identify a cell death modality.
In the study of programmed cell death, the DNA ladder assay remains a fundamental technique for detecting apoptosis, the process of programmed cell death essential for tissue homeostasis, development, and immune function [12] [68]. This method identifies the hallmark biochemical signature of apoptosis: internucleosomal DNA cleavage resulting in fragments that form a characteristic "ladder" pattern when separated by gel electrophoresis [41] [12]. Despite its widespread use in basic research, toxicology, and drug development, researchers frequently encounter technical challenges including weak signals and smearing that compromise data interpretation [85] [86]. This guide systematically addresses these issues by comparing detection methodologies, providing optimized protocols, and presenting quantitative data to support reliable apoptosis analysis, particularly in comparison with apoptotic body formation as a morphological endpoint.
Understanding Apoptosis: Biochemical vs. Morphological Hallmarks
Biochemical Signature: DNA Fragmentation
During apoptosis, the activation of caspase-activated DNase (CAD) cleaves chromosomal DNA at internucleosomal linker regions, generating DNA fragments in multiples of approximately 180-200 base pairs [12]. This specific cleavage pattern produces the characteristic DNA ladder distinguishable from the random DNA fragmentation observed in necrosis [12] [68].
Morphological Signature: Apoptotic Body Formation
Concurrent with DNA fragmentation, apoptotic cells undergo distinct morphological changes including cell shrinkage, chromatin condensation, and formation of membrane-bound apoptotic bodies containing nuclear fragments and organelles [12] [68]. These bodies are eventually phagocytosed by neighboring cells without inducing inflammation [68].
The following diagram illustrates the key events in apoptosis leading to DNA ladder formation and apoptotic body formation:
Experimental Protocols for DNA Ladder Detection
Standard DNA Fragmentation Protocol
Based on established methodologies [41] [12], the following protocol reliably detects apoptosis-induced DNA fragmentation:
Cell Harvesting and Lysis
- Pellet approximately 10⁶ cells by centrifugation at 5000 rpm for 5 minutes [41]
- Resuspend cell pellet in 500 μL lysis buffer (10 mM Tris pH 7.4, 5 mM EDTA, 0.2% Triton X-100) [12]
- Incubate on ice for 30 minutes to ensure complete cell lysis [12]
- Centrifuge at 27,000 × g for 30 minutes to separate fragmented DNA (supernatant) from intact chromatin (pellet) [12]
DNA Precipitation and Purification
- Transfer supernatant to a new tube and add 50 μL of 5 M NaCl [12]
- Add 600 μL cold ethanol and 150 μL 3 M sodium acetate (pH 5.2) [12]
- Mix thoroughly and incubate at -80°C for 1 hour [12]
- Centrifuge at 20,000 × g for 20 minutes to pellet DNA [12]
- Carefully discard supernatant without disturbing the pellet [12]
- Treat with DNase-free RNase (2 μL of 10 mg/mL) for 5 hours at 37°C to remove RNA contamination [12]
- Add 25 μL proteinase K (20 mg/mL) and incubate overnight at 65°C to digest proteins [12]
- Extract with phenol/chloroform/isoamyl alcohol (25:24:1) and precipitate with ethanol [12]
Gel Electrophoresis and Visualization
- Air-dry DNA pellet and resuspend in 20 μL Tris-acetate EDTA buffer with 2 μL loading dye (0.25% bromophenol blue, 30% glycerol) [12]
- Separate DNA on a 1.5-2% agarose gel containing 1 μg/mL ethidium bromide or SYBR-Safe DNA gel stain [41] [12]
- Run electrophoresis at 1-5 V/cm distance between electrodes until adequate separation is achieved [85]
- Visualize DNA fragments using an ultraviolet gel documentation system [41]
Modified Rapid DNA Extraction Protocol
For faster processing, researchers have developed an optimized protocol that reduces processing time while maintaining sensitivity [41]:
- Harvest both adherent and floating cells (including culture media) [41]
- Add 500 μL lysis buffer and incubate at 65°C for 5 minutes [41]
- Add 700 μL chloroform-isoamyl alcohol and centrifuge at 12,000 rpm for 5 minutes [41]
- Transfer aqueous phase and add equal volume of cold isopropanol [41]
- Centrifuge at 12,000 rpm for 5 minutes, discard supernatant, and air-dry pellet [41]
- Resuspend DNA in 50 μL distilled water [41]
The complete experimental workflow for DNA ladder detection is outlined below:
Quantitative Comparison of Apoptosis Detection Methods
Sensitivity and Specificity Analysis
Table 1: Comparison of Apoptosis Detection Methods
| Method | Detection Principle | Apoptosis Stage Detected | Sensitivity | Specificity | Time Required | Equipment Needs | Cost |
|---|---|---|---|---|---|---|---|
| DNA Ladder Assay | DNA fragmentation pattern | Late | Moderate | High | 24-48 hours | Gel electrophoresis system | Low |
| Apoptotic Body Formation (Microscopy) | Morphological changes | Mid to late | Moderate | Moderate | 2-6 hours | Fluorescence microscope | Moderate |
| Annexin V Staining | Phosphatidylserine externalization | Early | High | High | 1-2 hours | Flow cytometer | High |
| TUNEL Assay | DNA end-labeling | Mid to late | Very high | Moderate | 4-6 hours | Fluorescence microscope/Flow cytometer | High |
| Caspase Activity Assay | Caspase activation | Early to mid | High | High | 2-4 hours | Plate reader | Moderate |
Quantitative Detection Limits
Table 2: Detection Limits of DNA Ladder Assay vs. Alternative Methods
| Method | Minimum Apoptotic Cells Detectable | Quantitative Capability | Linearity Range | Interference from Necrosis |
|---|---|---|---|---|
| DNA Ladder Assay | 10-20% of population | Semi-quantitative | Limited | Low with proper technique |
| Apoptotic Body Counting (Microscopy) | 5-10% of population | Semi-quantitative | Limited | Moderate |
| Flow Cytometry (Annexin V/PI) | 1-5% of population | Quantitative | Broad | Can distinguish apoptosis/necrosis |
| Capillary Gel Electrophoresis | 5-10% of population | Fully quantitative | Broad | Low |
Troubleshooting Guide: Weak Signals and Smearing
Weak or Faint DNA Ladders
Weak signals represent one of the most common challenges in DNA ladder assays. The following table summarizes causes and solutions:
Table 3: Troubleshooting Weak or Faint DNA Ladders
| Cause | Detection Method | Solution | Effectiveness |
|---|---|---|---|
| Insufficient apoptotic cells | Microscopy: Few apoptotic bodies | Increase cell number (1×10⁷ recommended); Include floating cells in analysis [41] [12] | High |
| DNA degradation during preparation | Gel: Smearing across all lanes | Use DNase-free tips and tubes; Add nuclease inhibitors; Work on ice [85] | High |
| Incomplete DNA precipitation | Spectrophotometry: Low DNA yield | Ensure proper salt concentration; Extend -80°C incubation to 1+ hours [12] | High |
| Excessive DNA loss during purification | Spectrophotometry: Low DNA yield | Handle pellet carefully after ethanol precipitation; Avoid disturbing pellet [12] | Moderate |
| Inadequate staining | Gel: Visible ladder but faint | Use fresh ethidium bromide (1 μg/mL) or SYBR Safe; Extend staining time [41] | High |
| Low electrophoresis sensitivity | Gel: Faint ladder | Load 3-5 μL ready-to-use DNA ladder (0.5 μg) as control [85] | High |
Band Smearing and Resolution Issues
Band smearing compromises pattern interpretation and can be addressed as follows:
Table 4: Troubleshooting Band Smearing and Poor Resolution
| Cause | Detection Method | Solution | Effectiveness |
|---|---|---|---|
| Protein contamination | Gel: Wider, brighter bands with smeared tails | Extend proteinase K digestion (overnight at 65°C); Use fresh ladder [85] [12] | High |
| Excessive DNA loading | Gel: Overloaded wells with thick bands | Reduce DNA load to 0.5-1 μg per well; Dilute sample if necessary [85] | High |
| Gel concentration inappropriate | Gel: Poor separation of bands | Use 1.5-2% agarose for optimal resolution of 180-1000 bp fragments [85] [12] | High |
| Electrophoresis conditions | Gel: Band smiling or distortion | Run at 1-5 V/cm; Use constant current mode; Ensure adequate buffer volume [85] [86] | High |
| RNA contamination | Gel: Diffuse background staining | Extend RNase treatment (5 hours at 37°C); Use DNase-free RNase [12] | High |
| DNA degradation | Gel: Continuous smear without discrete bands | Check reagent quality; Use fresh cell culture; Avoid repeated freeze-thaw cycles [85] | High |
Advanced Technical Solutions and Methodological Comparisons
Alternative Electrophoresis Approaches
While conventional agarose gel electrophoresis remains widely used, capillary gel electrophoresis (CGE) offers enhanced quantification capabilities. CGE provides superior quantitative analysis of DNA forms with automated detection, reduced manual steps, and elimination of carcinogenic dye handling [87]. Studies demonstrate CGE can accurately quantify supercoiled (SC), open-circular (OC), and linear plasmid DNA isoforms with precision comparable to traditional methods [87].
Research Reagent Solutions
Table 5: Essential Reagents for DNA Ladder Assays
| Reagent | Function | Optimal Concentration | Technical Notes |
|---|---|---|---|
| Triton X-100/NP-40 | Cell membrane permeabilization | 0.2% in lysis buffer | Enables release of fragmented DNA [12] |
| Proteinase K | Protein digestion | 20 mg/mL, overnight at 65°C | Eliminates DNA-binding proteins that cause smearing [12] |
| DNase-free RNase | RNA removal | 10 mg/mL, 5 hours at 37°C | Reduces background staining [12] |
| Ethidium bromide/SYBR Safe | DNA visualization | 1 μg/mL in gel or staining solution | SYBR Safe reduces mutagenicity concerns [41] |
| Tris-EDTA buffer | DNA stabilization and resuspension | 10 mM Tris, 5 mM EDTA | Maintains DNA integrity during storage [12] |
| Agarose | Matrix for DNA separation | 1.5-2% for optimal resolution | Higher percentages improve small fragment separation [85] |
The DNA ladder assay remains a valuable, cost-effective method for detecting late-stage apoptosis, particularly when optimized to address common issues of weak signals and smearing. While this method provides definitive evidence of internucleosomal DNA cleavage—a biochemical hallmark of apoptosis—researchers should consider its limitations in quantification and stage-specific detection. For comprehensive apoptosis analysis, combining DNA ladder assays with complementary methods assessing apoptotic body formation (morphological analysis), phosphatidylserine externalization (Annexin V staining), or caspase activation (enzymatic assays) provides a more complete understanding of cell death mechanisms. Through systematic troubleshooting and method validation, researchers can overcome technical challenges and generate reliable, reproducible data for both basic research and drug development applications.
This guide provides an objective comparison of two critical methodologies in cell death research: DNA fragmentation assays and apoptotic body formation analysis. The focus is on how cryopreservation and fixation—essential steps for sample storage and preparation—impact the accuracy and reliability of these assays. Data synthesized from recent studies indicate that cryopreservation consistently increases DNA fragmentation, potentially confounding results, while the choice of fixation method is crucial for preserving morphological features like apoptotic bodies. The following sections present comparative quantitative data, detailed experimental protocols, and key reagent solutions to inform method selection for research and drug development.
Impact of Cryopreservation on Assay Parameters
Cryopreservation is a vital technique for preserving cells and gametes, but the freeze-thaw process induces significant cellular stress and damage. The table below summarizes its specific impact on parameters relevant to DNA fragmentation and apoptotic body formation assays.
Table 1: Quantifiable Impact of Cryopreservation on Sample Quality
| Assay Parameter | Pre-Cryopreservation State | Post-Cryopreservation State | Change | Context & Measurement Method |
|---|---|---|---|---|
| Sperm DNA Fragmentation Index (DFI) | 46.3% ± 18.3% | 60.0% ± 23.0% | ↑ 13.7% (p < 0.001) | Normozoospermic human samples (n=104); Sperm Chromatin Dispersion (SCD) test [88]. |
| Sperm Reactive Oxygen Species (ROS) | 3.2 × 10³ RLU/s | 14.7 × 10³ RLU/s | ↑ ~360% (p < 0.001) | Luminol-enhanced chemiluminescence [88]. |
| Caspase-3 Level (Apoptotic Marker) | Baseline | Post-thaw | Significant Increase | Human sperm samples; used as a marker for apoptosis [89]. |
| General Sperm Motility | High (Fertile group) | Post-thaw | Significant Decline | Compared to fresh samples; infertile samples more adversely affected [89]. |
Underlying Mechanisms of Cryopreservation Damage
The degradation in sample quality is primarily driven by two interconnected processes:
- Oxidative Stress: The freeze-thaw process disrupts cellular metabolism, leading to a surge in Reactive Oxygen Species (ROS) [90] [88]. Sperm are particularly vulnerable due to their limited cytoplasmic antioxidant defenses [90]. This oxidative stress causes lipid peroxidation of membranes and direct DNA damage, resulting in fragmentation [89] [88].
- Induction of Regulated Cell Death: Oxidative stress acts as a key trigger for Apoptosis, a regulated cell death pathway [90]. This is evidenced by the post-thaw increase in executioner enzymes like caspase-3 and the externalization of phosphatidylserine, both hallmarks of apoptosis [89] [90]. The formation of apoptotic bodies, a key metric for morphology-based assays, is a direct consequence of this activated apoptotic cascade [91] [12].
Diagram 1: How cryopreservation impacts key assay readouts. The process triggers oxidative stress, which activates apoptosis and causes direct DNA damage, leading to the primary outcomes measured in DNA fragmentation assays and apoptotic body analysis.
Comparative Analysis: DNA Fragmentation vs. Apoptotic Body Formation
The choice between analyzing DNA fragmentation or apoptotic body formation has significant implications for sensitivity, specificity, and susceptibility to pre-analytical artifacts.
Table 2: Method Comparison: DNA Fragmentation vs. Apoptotic Body Assays
| Feature | DNA Fragmentation Assays | Apoptotic Body Formation Analysis |
|---|---|---|
| What is Measured | Biochemical degradation of nuclear DNA into oligonucleosomal fragments [91] [12]. | Morphological changes: cell shrinkage, chromatin condensation, and membrane blebbing forming apoptotic bodies [92] [93]. |
| Key Detection Methods | Gel electrophoresis (DNA laddering) [12], TUNEL assay [94], Sperm Chromatin Dispersion (SCD) [88]. | Light/electron microscopy [94] [92], Annexin V staining (early marker) [92]. |
| Sensitivity to Cryopreservation | High. DNA is a direct target of cryo-induced oxidative stress, leading to significant artifactual increases in fragmentation [89] [88]. | Moderate. The process of apoptosis can be initiated by cryopreservation [94] [90], but morphology can be preserved by proper fixation. |
| Impact of Fixation | Low to Moderate. Must permeabilize cells for probe access (e.g., TUNEL). Over-fixation can mask DNA breaks [92]. | Critical. Fixation must perfectly preserve cellular architecture; aldehydes (e.g., formaldehyde) are standard to prevent degradation of apoptotic bodies [92]. |
| Quantification | Highly quantitative (e.g., DFI percentage from SCD) [88] or semi-quantitative (gel analysis) [12]. | Often qualitative or semi-quantitative (cell counting), though flow cytometry can be used with Annexin V [92]. |
| Primary Advantage | Objective, quantitative measure of a key apoptotic event. | Provides direct visual evidence of the apoptotic process in its cellular context. |
| Primary Limitation | Cannot distinguish between apoptotic fragmentation and non-specific DNA damage (e.g., from necrosis) [92]. | Requires expert interpretation; vulnerable to artifacts from improper handling or fixation [92]. |
Detailed Experimental Protocols
Protocol: DNA Fragmentation Analysis via Gel Electrophoresis
This protocol is a classic method for detecting the characteristic "ladder" pattern of apoptotic DNA [12].
Stage 1: Harvest and Lysate Preparation
- Pellet approximately 1-5 x 10^6 cells by centrifugation.
- Lyse the cell pellet in 0.5 mL of detergent buffer (10 mM Tris pH 7.4, 5 mM EDTA, 0.2% Triton X-100).
- Vortex the mixture and incubate on ice for 30 minutes.
- Centrifuge at 27,000 x g for 30 minutes to separate intact chromatin (pellet) from fragmented DNA (supernatant).
- Divide the supernatant into two aliquots and add 50 µL of ice-cold 5 M NaCl to each [12].
Stage 2: DNA Precipitation and Purification
- Add 600 µL of ethanol and 150 µL of 3 M sodium acetate (pH 5.2) to each aliquot to precipitate the DNA.
- Incubate at -80°C for 1 hour.
- Centrifuge at 20,000 x g for 20 minutes and carefully discard the supernatant.
- Pool the DNA pellets and dissolve them in 400 µL of extraction buffer (10 mM Tris, 5 mM EDTA).
- Add DNase-free RNase and incubate at 37°C for 5 hours.
- Add Proteinase K and incubate overnight at 65°C to digest proteins.
- Extract DNA with phenol/chloroform/isoamyl alcohol and precipitate with ethanol again [12].
Stage 3: Gel Electrophoresis and Visualization
- Air-dry the final DNA pellet and resuspend it in Tris-acetate-EDTA buffer with loading dye.
- Separate the DNA by electrophoresis on a 2% agarose gel containing ethidium bromide.
- Visualize the DNA fragments under UV light. A ladder of fragments in multiples of ~180-200 base pairs indicates apoptosis, whereas a smear suggests necrosis [12].
Workflow for Comparative Analysis
The following diagram outlines a standardized experimental workflow to compare the effects of sample preparation on both assay types.
Diagram 2: Experimental workflow for comparing preparation impacts. Cells are split and subjected to different preparation paths before analysis, allowing direct comparison of how cryopreservation affects assay results versus a freshly fixed control.
The Scientist's Toolkit: Key Research Reagent Solutions
Selecting appropriate reagents is fundamental to controlling variables in sample preparation and assay execution.
Table 3: Essential Reagents for Sample Preparation and Cell Death Assays
| Reagent / Solution | Function & Rationale |
|---|---|
| Permeable Cryoprotectants (e.g., Glycerol) | Penetrate cells, reduce intracellular ice crystal formation (a cause of Accidental Cell Death), and mitigate solute effects during slow freezing [90]. |
| Non-Permeable Cryoprotectants (e.g., Sucrose, Egg Yolk) | Increase extracellular osmolarity, promoting controlled dehydration and reducing osmotic shock. Egg yolk also protects membrane integrity [89]. |
| Formaldehyde / Paraformaldehyde | Cross-linking fixatives. Ideal for preserving cellular morphology and structures like apoptotic bodies for microscopic analysis [92]. |
| Triton X-100 / NP-40 | Non-ionic detergents used to permeabilize cell and nuclear membranes, allowing access of antibodies or enzymes (e.g., in TUNEL assay) to intracellular targets [12]. |
| Annexin V (FITC conjugate) | Binds to phosphatidylserine (PS). PS externalization is an early apoptotic event, detectable by flow cytometry or microscopy before loss of membrane integrity [92] [93]. |
| Proteinase K | A broad-spectrum serine protease. Critical for digesting and removing proteins that contaminate DNA preparations in fragmentation assays [12]. |
| DNase-free RNase | Essential for removing RNA from DNA samples prior to gel electrophoresis. Prevents RNA bands from obscuring the apoptotic DNA ladder pattern [12]. |
| Caspase-3 Antibodies | Enable detection of cleaved/activated caspase-3 via Western blot or immunofluorescence, providing specific biochemical evidence of ongoing apoptosis [89] [93]. |
In the field of cell death research, accurate detection and quantification are paramount for understanding disease mechanisms and therapeutic efficacy. DNA fragmentation assays and the analysis of apoptotic body formation represent two cornerstone methodologies for identifying apoptotic cells. DNA fragmentation assays, which detect internucleosomal DNA cleavage, and apoptotic body analysis, which identifies the membrane-bound vesicles formed during apoptosis, are widely used yet possess distinct technical profiles [12] [68]. This guide provides an objective, data-driven comparison of these methods, focusing on their inherent limitations in sensitivity, quantification capability, and stage specificity. Understanding these parameters is critical for researchers and drug development professionals to select the optimal assay for their specific experimental context and to correctly interpret the resulting data within the broader landscape of cell death research.
Comparative Analysis of Key Methodologies
DNA Fragmentation Assays
The DNA fragmentation assay, often visualized via agarose gel electrophoresis, is a classic technique for detecting apoptosis. Its fundamental principle relies on the activation of endonucleases during apoptosis, which cleave DNA at internucleosomal linker sites, generating a characteristic ladder pattern of fragments approximately 200 base pairs in length [12]. This ladder is distinct from the smeared pattern observed in necrotic cells, allowing for morphological distinction.
The experimental protocol typically involves several key stages [12]:
- Cell Harvesting and Lysis: Pelleted cells are lysed using a detergent-based buffer (e.g., containing Triton X-100, Tris, and EDTA) to release cellular contents.
- DNA Extraction and Precipitation: The lysate is centrifuged to separate fragmented DNA (in the supernatant) from intact genomic DNA (in the pellet). The fragmented DNA in the supernatant is then precipitated using ethanol and salts.
- Purification and Analysis: The DNA precipitate is treated with RNase to remove RNA and often with proteinase K to digest proteins. Further purification may involve phenol/chloroform extraction. The final DNA is resuspended and separated on a 2% agarose gel containing ethidium bromide for visualization under UV light.
Apoptotic Body Formation Analysis
Apoptotic bodies (ApoBDs) are membrane-bound extracellular vesicles formed during the final stages of apoptosis, considered a definitive hallmark of this cell death pathway [23]. They are characterized by their electron-dense composition, containing fragments of cytoplasm, shrunken organelles, and nuclear material, all encapsulated by the cell's plasma membrane [58] [23].
Analysis of ApoBDs can be performed using various technologies. Flow Cytometry and Fluorescence-Activated Cell Sorting (FACS) are powerful for quantifying ApoBDs based on their relative size and granularity, and can further classify them using DNA-binding dyes like DAPI to distinguish between DNA-containing and non-containing populations [23]. Transmission Electron Microscopy (TEM) provides high-resolution ultrastructural details, confirming hallmarks such as membrane encapsulation and dense cargo [58] [23]. Furthermore, imaging flow cytometry integrates the statistical power of flow cytometry with visual confirmation, allowing for the morphological verification of ApoBDs in a high-throughput manner [74].
Comparative Performance Data
The following tables summarize the direct, objective comparison of performance metrics and limitations between DNA fragmentation assays and apoptotic body analysis.
Table 1: Quantitative Performance Metrics of DNA Fragmentation vs. Apoptotic Body Assays
| Performance Parameter | DNA Fragmentation Assay | Apoptotic Body Analysis |
|---|---|---|
| Sensitivity | Low to Moderate; may not detect cells with low-level fragmentation [12] | High; capable of detecting individual vesicles [58] |
| Quantification Capability | Semi-quantitative; difficult to measure exact extent of cell death [12] | Quantitative via flow cytometry; enables precise population analysis [23] |
| Stage Specificity | Late-stage apoptosis marker [12] | Very late-stage apoptosis marker [23] |
| Typical Detection Limit | Requires ~5-10% of cell population to be apoptotic for clear ladder [12] | Capable of detecting rare events; sensitivity depends on instrumentation [74] |
| Throughput | Low; suitable for bulk cell population analysis [12] | High (flow cytometry); Low (TEM) [74] [23] |
Table 2: Key Limitations and Application Suitability
| Aspect | DNA Fragmentation Assay | Apoptotic Body Analysis |
|---|---|---|
| Key Limitations | Semi-quantitative, not suitable for high-throughput analysis, requires careful handling to avoid DNA loss, cannot distinguish apoptosis stages, subjective interpretation [12] | Requires specialized equipment (e.g., flow cytometer, TEM), complex sample preparation for TEM, not specific to early apoptosis [58] [74] |
| Best Suited For | Initial confirmation of apoptosis in bulk cell populations, low-budget projects, educational demonstrations [12] | Quantitative studies, high-throughput screening, functional studies of ApoBDs (e.g., immunomodulation), ultrastructural analysis [58] [23] |
| Cell Death Pathway Specificity | Specific for apoptosis vs. necrosis, but may not distinguish classical apoptosis from apoptosis-like PCD [74] | Considered a definitive hallmark of apoptosis; however, ApoBD-like vesicles have been reported in some non-metazoan PCD [23] |
Experimental Protocols
Detailed Protocol: DNA Fragmentation Assay
This protocol is adapted from Abcam's standard procedure for detecting apoptosis via DNA laddering [12].
Stage 1: Harvest and Lyse Cells
- Pellet approximately 1-5 x 10^6 cells by centrifugation.
- Resuspend the cell pellet thoroughly in 0.5 mL of lysis buffer (10 mM Tris pH 7.4, 5 mM EDTA, 0.2% Triton X-100).
- Vortex the mixture and incubate on ice for 30 minutes.
- Centrifuge at 27,000 x g for 30 minutes at 4°C to separate fragmented DNA (supernatant) from intact chromatin (pellet).
- Carefully transfer the supernatant to a new tube. Add 50 µL of ice-cold 5 M NaCl and vortex.
Stage 2: Precipitate DNA
- Add 600 µL of ethanol and 150 µL of 3 M sodium acetate (pH 5.2) to the supernatant. Mix by pipetting.
- Incubate at -80°C for 1 hour to precipitate the DNA.
- Centrifuge at 20,000 x g for 20 minutes at 4°C. Carefully discard the supernatant without disturbing the loose DNA pellet.
- Dissolve the DNA pellet in 400 µL of extraction buffer (10 mM Tris, 5 mM EDTA).
- Add 2 µL of DNase-free RNase (10 mg/mL) and incubate for 5 hours at 37°C.
- Add 25 µL of proteinase K (20 mg/mL) and 40 µL of digestion buffer (100 mM Tris pH 8.0, 100 mM EDTA, 250 mM NaCl). Incubate overnight at 65°C.
- Purify DNA by extraction with phenol/chloroform/isoamyl alcohol (25:24:1) and re-precipitate with ethanol.
Stage 3: Agarose Gel Electrophoresis
- Air-dry the DNA pellet and resuspend in 20 µL Tris-acetate EDTA (TAE) buffer with 2 µL of sample loading buffer (0.25% bromophenol blue, 30% glycerol).
- Separate the DNA fragments by electrophoresis on a 2% agarose gel containing 1 µg/mL ethidium bromide.
- Visualize the DNA ladder pattern using ultraviolet transillumination.
Workflow Visualization
The following diagram illustrates the key steps and decision points in the DNA fragmentation assay protocol.
Diagram 1: DNA Fragmentation Assay Workflow (13 words)
The Scientist's Toolkit: Essential Research Reagents
Successful execution of these assays requires specific reagents and tools. The following table details essential solutions and their functions.
Table 3: Key Research Reagent Solutions for DNA Fragmentation and Apoptotic Body Analysis
| Reagent / Solution | Function / Application | Key Characteristics |
|---|---|---|
| Cell Lysis Buffer (Tris, EDTA, Triton X-100) [12] | Disrupts cell membrane to release intracellular contents, including fragmented DNA. | Mild detergent-based buffer designed to lyse plasma membrane while keeping nuclear envelope largely intact for initial separation. |
| Acridine Orange [95] [7] | Metachromatic dye used in SCSA for flow cytometric quantification of sperm DNA Fragmentation Index (DFI). | Binds to dsDNA (green fluorescence) and ssDNA (red fluorescence), allowing calculation of DFI based on fluorescence emission shift. |
| Ethidium Bromide [12] | Intercalating nucleic acid stain for visualizing DNA fragments in agarose gels under UV light. | Standard dye for gel-based DNA detection; however, poses safety concerns and requires careful handling and disposal. |
| DNase-free RNase [12] | Enzyme treatment to degrade RNA in the DNA sample during purification. | Prevents RNA contamination that can obscure the DNA ladder pattern on the gel; must be free of DNase activity to preserve DNA integrity. |
| Proteinase K [12] | Broad-spectrum serine protease used to digest nucleases and other proteins in DNA samples. | Essential for degrading proteins that may interfere with DNA analysis or cause degradation; used after initial DNA precipitation. |
| Phenol/Chloroform/Isoamyl Alcohol [12] | Organic solvent mixture for purifying nucleic acids by removing protein contaminants. | Denatures and partitions proteins into the organic phase or interphase, leaving purified DNA in the aqueous phase. |
| Staurosporine (STS) [58] [23] | Pharmacological inducer of apoptosis used as a positive control in both DNA fragmentation and ApoBD studies. | Pan-kinase inhibitor that reliably triggers the intrinsic apoptotic pathway across various cell types. |
| SYTOX Green [23] | Cell-impermeant nucleic acid stain used to identify dead cells with compromised plasma membranes. | Useful for quantifying overall cell death in cultures where ApoBDs are being studied. |
Integrated Discussion
The comparative data reveals a clear trade-off between simplicity and quantitative rigor. The DNA fragmentation assay offers a low-cost, direct method for confirming apoptosis but is hampered by its semi-quantitative nature and low sensitivity [12]. Its limitation as a late-stage marker means it captures only the final executive phase of apoptosis, potentially missing critical earlier events and yielding a negative result in cases of caspase-independent apoptosis-like programmed cell death where extensive internucleosomal cleavage does not occur [74].
Conversely, apoptotic body analysis, particularly via flow cytometry, provides superior quantification and sensitivity [58] [23]. The discovery that ApoBDs themselves possess biological functions, such as immunomodulatory capacity, adds a layer of functional relevance to their detection [58]. However, ApoBD formation is an even later event in the apoptotic cascade than DNA fragmentation, and their analysis typically requires more sophisticated instrumentation [23].
A critical consideration for researchers is that no single assay can fully characterize the complex network of cell death pathways [74] [68]. For instance, a TUNEL assay, which also detects DNA breaks, may yield false positives if not carefully controlled, while Annexin V staining detects earlier membrane changes but cannot distinguish between apoptotic and necroptotic cells in later stages [74]. Therefore, a multiparametric approach, combining DNA fragmentation analysis or ApoBD detection with other assays like caspase activation markers or mitochondrial membrane potential assessment, is highly recommended for conclusive identification and quantification of apoptotic cells [74] [68].
Best Practices for Robust and Reproducible Apoptosis Detection
Apoptosis, or programmed cell death, is a fundamental biological process essential for maintaining tissue homeostasis, proper embryonic development, and immune system regulation. The precise detection and quantification of apoptosis are crucial in various research fields, particularly in oncology, neurobiology, and drug discovery. Dysregulation of apoptotic pathways is implicated in numerous diseases, including cancer, neurodegenerative disorders, and autoimmune conditions, making accurate apoptosis assessment vital for understanding disease mechanisms and developing novel therapeutics. According to recent market analysis, the North America apoptosis assay market was valued at USD 2.7 billion in 2024 and is projected to grow to USD 6.1 billion by 2034, reflecting the increasing importance of these assays in biomedical research and pharmaceutical development [96].
This guide provides a comprehensive comparison of two fundamental apoptosis detection methodologies: DNA fragmentation assays and analysis of apoptotic body formation. As research moves toward more sophisticated, high-throughput approaches, understanding the strengths, limitations, and appropriate application contexts of these methods becomes increasingly important for generating robust, reproducible data. We present experimental data, detailed protocols, and analytical frameworks to help researchers select the most appropriate detection method for their specific experimental needs.
Apoptosis Signaling Pathways: Molecular Foundations for Detection
Apoptosis occurs through two principal signaling pathways that converge on common execution elements. Understanding these pathways provides the foundation for selecting appropriate detection methods and interpreting results accurately.
The Intrinsic and Extrinsic Apoptosis Pathways
The intrinsic pathway (mitochondrial pathway) is triggered by internal cellular stressors such as DNA damage, oxidative stress, or growth factor deprivation. These stimuli cause mitochondrial outer membrane permeabilization (MOMP), leading to the release of cytochrome c into the cytosol. Cytochrome c then binds to Apaf-1, forming the apoptosome complex which activates caspase-9, subsequently initiating the caspase cascade [59] [97].
The extrinsic pathway (death receptor pathway) begins with the binding of extracellular death ligands (e.g., FasL, TRAIL) to their corresponding death receptors on the cell surface. This receptor-ligand interaction recruits adaptor proteins and initiator caspases (primarily caspase-8), forming the death-inducing signaling complex (DISC) which activates downstream effector caspases [59] [97].
Both pathways converge on the activation of executioner caspases (caspase-3, -6, and -7), which orchestrate the morphological and biochemical hallmarks of apoptosis, including DNA fragmentation, membrane blebbing, and formation of apoptotic bodies.
Key Biochemical Events in Apoptosis
Several characteristic biochemical events serve as detectable markers of apoptosis:
- Phosphatidylserine externalization: In viable cells, phosphatidylserine is restricted to the inner leaflet of the plasma membrane. During early apoptosis, it translocates to the outer leaflet, providing an "eat me" signal for phagocytes [98].
- Caspase activation: A cascade of caspase proteolysis represents a commitment to apoptotic cell death [59].
- DNA fragmentation: Activation of caspase-activated DNase leads to internucleosomal DNA cleavage, producing characteristic ~180 bp fragments [80].
- Membrane blebbing and apoptotic body formation: The cell undergoes dramatic structural changes, condensing and fragmenting into membrane-bound apoptotic bodies [9].
The following diagram illustrates the key events in apoptosis signaling pathways:
Comparative Analysis: DNA Fragmentation vs. Apoptotic Body Detection
This section provides a detailed comparison between DNA fragmentation assays and apoptotic body formation analysis, highlighting their respective technical characteristics, applications, and limitations.
Key Characteristics and Performance Metrics
Table 1: Comparison of DNA Fragmentation and Apoptotic Body Formation Detection Methods
| Parameter | DNA Fragmentation Assays | Apoptotic Body Detection |
|---|---|---|
| Detection Target | Internucleosomal DNA cleavage (~180 bp fragments) [80] | Membrane-bound cellular fragments with condensed chromatin [9] |
| Primary Detection Methods | DNA laddering, TUNEL assay, flow cytometry (sub-G1 peak) [80] [12] | Microscopy (phase contrast, QPI), flow cytometry, Annexin V staining [9] |
| Stage of Detection | Mid to late apoptosis [12] | Mid to late apoptosis [9] |
| Specificity for Apoptosis | High when combined with morphological confirmation [80] | Moderate (requires distinction from other fragmentation processes) [9] |
| Sensitivity | Varies by method: TUNEL > DNA laddering [80] | High with advanced imaging (QPI) [9] |
| Throughput Capability | Moderate to high (especially flow cytometry) [96] | Low to moderate (imaging-intensive) [9] |
| Quantitative Capability | Semi-quantitative (laddering) to quantitative (flow cytometry) [12] | Quantitative with advanced image analysis [9] |
| Key Advantages | Well-established, objective biochemical endpoint [80] | Provides morphological context, can be label-free [9] |
| Major Limitations | Cannot detect early apoptosis, may miss heterogeneous responses [12] | Subjectivity in identification, requires experience [9] |
| Resource Requirements | Low to moderate (laddering) to high (flow cytometry) [12] | Moderate (microscopy) to high (QPI systems) [9] |
Experimental Data and Performance Validation
Table 2: Experimental Performance Metrics from Comparative Studies
| Performance Metric | DNA Fragmentation (TUNEL) | DNA Fragmentation (Laddering) | Apoptotic Body (Phase Microscopy) | Apoptotic Body (QPI) |
|---|---|---|---|---|
| Detection Accuracy | 89-95% [80] | 75-85% [12] | 70-80% [9] | 76% [9] |
| Early Apoptosis Detection | Limited [80] | Not detectable [12] | Moderate [9] | High (with CDS parameter) [9] |
| Time to Result | 4-6 hours [12] | 24-48 hours [12] | Real-time to hours [9] | Real-time [9] |
| Sample Throughput | 40-60 samples/day [96] | 20-30 samples/day [12] | 10-20 samples/day [9] | 5-15 samples/day [9] |
| Inter-assay Variability | 10-15% CV [96] | 15-25% CV [12] | 20-30% CV [9] | 8-12% CV [9] |
| Cell Number Requirement | 1×10⁵ - 1×10⁶ [12] | 1×10⁶ - 5×10⁶ [12] | 1×10⁴ - 1×10⁵ [9] | 1×10³ - 1×10⁴ [9] |
| Cost per Sample | $25-50 [96] | $10-20 [12] | $5-15 [9] | $20-40 [9] |
Methodology Deep Dive: Experimental Protocols and Workflows
DNA Fragmentation Assay Protocol
The DNA laddering assay represents a classic biochemical method for apoptosis detection based on the characteristic internucleosomal DNA cleavage pattern. Below is a standardized protocol adapted from established methods [12]:
Stage 1: Cell Harvesting and Lysis
- Pellet approximately 1×10⁶ cells by centrifugation at 500 × g for 5 minutes.
- Resuspend cell pellet in 0.5 mL of detergent buffer (10 mM Tris pH 7.4, 5 mM EDTA, 0.2% Triton X-100).
- Vortex the mixture thoroughly and incubate on ice for 30 minutes.
- Centrifuge at 27,000 × g for 30 minutes at 4°C to separate fragmented DNA from intact chromatin.
- Transfer the supernatant containing fragmented DNA to a new tube and divide into two 250 μL aliquots.
- Add 50 μL of ice-cold 5 M NaCl to each aliquot and vortex.
Stage 2: DNA Precipitation
- Add 600 μL of absolute ethanol and 150 μL of 3 M sodium acetate (pH 5.2) to each aliquot.
- Mix thoroughly by pipetting and incubate at -80°C for 1 hour.
- Centrifuge at 20,000 × g for 20 minutes at 4°C and carefully discard supernatant.
- Pool DNA extracts by dissolving pellets in a total of 400 μL extraction buffer (10 mM Tris, 5 mM EDTA).
- Add 2 μL of 10 mg/mL DNase-free RNase and incubate for 5 hours at 37°C.
- Add 25 μL of 20 mg/mL proteinase K and 40 μL of digestion buffer (100 mM Tris pH 8.0, 100 mM EDTA, 250 mM NaCl), then incubate overnight at 65°C.
- Extract DNA with phenol/chloroform/isoamyl alcohol (25:24:1) and precipitate with ethanol.
Stage 3: Gel Electrophoresis and Visualization
- Air-dry the DNA pellet and resuspend in 20 μL Tris-acetate EDTA buffer with 2 μL loading buffer (0.25% bromophenol blue, 30% glycerol).
- Separate DNA fragments electrophoretically on a 2% agarose gel containing 1 μg/mL ethidium bromide.
- Visualize DNA ladder pattern under UV transillumination.
Troubleshooting Notes:
- Weak or absent DNA ladders may result from insufficient cell lysis, poor DNA recovery, or suboptimal apoptosis induction [12].
- Smearing on the gel may indicate overloading, incomplete protein digestion, or necrosis [12].
- Always include positive control (apoptosis-induced cells) and negative control (viable cells) for assay validation.
Apoptotic Body Detection via Quantitative Phase Imaging
Quantitative Phase Imaging offers a label-free approach for detecting apoptotic body formation based on morphological changes and cell mass distribution [9]:
Sample Preparation and Imaging:
- Culture cells on appropriate imaging chambers (e.g., μ-Slide I Lauer Family chambers).
- Maintain standard cultivation conditions (37°C, 5% CO₂) throughout imaging using environmental chambers.
- Acquire time-lapse images using a quantitative phase microscope (e.g., Q-PHASE system) with a 10× or 20× objective.
- Set image acquisition interval to 3-10 minutes depending on apoptosis kinetics.
- For correlative validation, include fluorescence markers for caspase activation (CellEvent Caspase-3/7 reagent) and membrane integrity (propidium iodide).
Image Analysis and Parameter Quantification:
- Apply segmentation algorithm to identify individual cells and track them through time-lapse sequences.
- Calculate cell density (pg/pixel) from phase measurements.
- Compute Cell Dynamic Score (CDS) based on average intensity change of cell pixels.
- Identify apoptotic cells using these parameters:
- CDS values typical for caspase-dependent apoptosis: 0.15-0.35
- CDS values typical for caspase-independent cell death: 0.35-0.65
- Characteristic morphological changes: cell shrinkage, membrane blebbing, apoptotic body formation
Validation and Quality Control:
- Compare QPI results with standard fluorescence markers for apoptosis.
- Train the detection algorithm with manually annotated datasets.
- Establish cell line-specific parameters as morphological changes can vary between cell types.
The following workflow diagram illustrates the key steps in apoptosis detection methodology:
The Scientist's Toolkit: Essential Reagents and Technologies
Selecting appropriate reagents and technologies is crucial for implementing robust apoptosis detection protocols. The following table summarizes key solutions available from leading vendors:
Table 3: Research Reagent Solutions for Apoptosis Detection
| Product Category | Key Vendors | Specific Products | Primary Applications | Technical Notes |
|---|---|---|---|---|
| Assay Kits & Reagents | Thermo Fisher Scientific [96] | Annexin V-FITC Apoptosis Detection Kit [96] | Flow cytometry, microscopy | High-quality reagents with consistent performance |
| Merck [96] | Validated antibodies, caspase substrates | Multiple platforms | Extensive validation for reproducibility | |
| Bio-Rad Laboratories [96] | Image Lab software with AI-assisted quantification | Western blot analysis | Automated analysis improves objectivity | |
| Instrumentation | Danaher (Beckman Coulter) [96] | Flow cytometers with apoptosis modules | High-throughput screening | Integration with automated systems |
| BD Biosciences [96] | High-content imaging systems | Morphological analysis | Multiplexing capabilities | |
| Emerging Technologies | Academic/Startup Innovations [98] | Microfluidic apoptosis chips | Point-of-care applications | Label-free, electronic detection |
| Telight [9] | Q-PHASE system | Label-free kinetic analysis | Real-time monitoring of apoptosis |
The comparative analysis of DNA fragmentation assays and apoptotic body detection methods reveals distinct advantages and limitations for each approach. DNA fragmentation methods, particularly TUNEL assays and flow cytometric detection of sub-G1 populations, provide objective, quantitative data with moderate to high throughput capability. These methods are well-suited for screening applications and quantitative assessment of apoptosis in population-based studies. Conversely, apoptotic body detection through advanced imaging techniques, especially QPI, offers unique insights into the morphological progression of apoptosis with single-cell resolution and label-free operation.
For robust and reproducible apoptosis detection, researchers should consider the following evidence-based recommendations:
- For drug screening and high-throughput applications: Implement TUNEL assays or flow cytometric methods focusing on DNA fragmentation or Annexin V staining [96] [80].
- For mechanistic studies with temporal resolution: Utilize live-cell imaging approaches, including QPI, to capture dynamic apoptotic processes [9].
- For maximum reliability: Employ orthogonal methods combining DNA fragmentation analysis with morphological assessment to confirm apoptosis specificity [80] [9].
- For resource-limited settings: Consider microchip-based detection systems that offer electronic readouts without extensive sample processing [98].
The future of apoptosis detection lies in integrated approaches that combine multiple detection modalities, leverage artificial intelligence for enhanced analysis, and utilize emerging technologies such as microfluidic platforms for single-cell analysis. As the field advances toward more personalized medicine applications, adaptability to different sample types and experimental contexts will remain paramount for accurate apoptosis assessment in both basic research and drug development pipelines.
Choosing the Right Tool: A Comparative Analysis of Apoptosis Detection Assays
The analysis of cell-free DNA (cfDNA) and its constituents has emerged as a transformative paradigm in molecular diagnostics, particularly in oncology. Two prominent analytical frameworks within this domain are DNA fragmentation assays, which interrogate the fragmentomic patterns of cfDNA, and assays centered on the detection of apoptotic bodies or apoptotic extracellular vesicles (ApoEVs). These approaches leverage distinct biological phenomena to achieve a common goal: the non-invasive detection and monitoring of diseases such as cancer. DNA fragmentation assays capitalize on the finding that cfDNA derived from tumor cells exhibits characteristic size distributions, end motifs, and genomic patterns that differ from cfDNA released by healthy cells [99]. In contrast, analyses of apoptotic bodies focus on membrane-bound vesicles shed during programmed cell death, which encapsulate a wealth of molecular information from their cell of origin, including proteins, nucleic acids, and organelles [21] [100]. This guide provides a systematic, side-by-side comparison of these two technological frameworks, evaluating their sensitivity, specificity, workflow, and clinical applicability to inform researchers and drug development professionals.
The following tables provide a direct comparison of the core characteristics and performance metrics of DNA fragmentation assays and apoptotic body-based detection.
Table 1: Core Technology and Analyte Comparison
| Feature | DNA Fragmentation Assays | Apoptotic Body (ApoEV) Detection |
|---|---|---|
| Primary Analyte | Cell-free DNA (cfDNA) in plasma/serum [99] | Apoptotic Extracellular Vesicles (ApoEVs) from biofluids [100] |
| Key Biomarkers | Fragment size, coverage, end motifs, copy number variation [99] | Vesicle-bound DNA, RNA, proteins (e.g., phosphatidylserine), organelle debris [100] |
| Underlying Biology | Aberrant fragmentation and packaging of nucleosomal DNA in malignant cells [99] | Organized cell disassembly during apoptosis, including membrane blebbing and "FOOD" formation [21] |
| Typical Specimen | Blood plasma | Blood plasma, other body fluids |
| Detection Platform | Next-generation sequencing (NGS) [99] | Flow cytometry, imaging, molecular analysis of cargo [100] |
Table 2: Analytical and Clinical Performance Metrics
| Performance Metric | DNA Fragmentation Assays | Apoptotic Body (ApoEV) Detection |
|---|---|---|
| Reported Sensitivity (Multi-cancer) | 95.5% (at 95% specificity) for early-stage PLC, CRC, LUAD [99] | Emerging biomarker; large-scale sensitivity data pending [100] |
| Limit of Detection | Can detect ctDNA at < 0.01% variant allele frequency [101] | Sensitivity enhanced by targeting large, DNA-rich ApoEVs [100] |
| Specificity | 95.0% (for multi-cancer detection) [99] | High potential specificity from unique ApoEV cargo; requires further validation [100] |
| Early-Stage (Stage I) Detection | >90% sensitivity for PLC, CRC, LUAD [99] | Potential for early detection as apoptosis is a key early event in tumorigenesis [100] |
| Tissue of Origin Accuracy | 93.1% overall accuracy for PLC, CRC, LUAD [99] | Theoretical potential via proteomic or nucleic acid profiling of ApoEV cargo [100] |
Detailed Experimental Protocols
Ultra-Sensitive Multi-Cancer Detection via cfDNA Fragmentomics
Objective: To detect multiple cancer types and identify the tissue of origin at high sensitivity and specificity using a machine learning model trained on plasma cfDNA fragmentomic features [99].
Workflow:
- Sample Collection & Processing: Collect peripheral blood from participants (e.g., patients with primary liver cancer, colorectal adenocarcinoma, lung adenocarcinoma, and healthy volunteers). Process blood to isolate plasma.
- DNA Extraction & Sequencing: Extract cell-free DNA from plasma. Prepare sequencing libraries and perform shallow whole-genome sequencing (WGS).
- Fragmentomic Feature Extraction: Process WGS data to extract five distinct cfDNA features:
- Fragment Size Coverage (FSC): Coverage patterns across specific fragment sizes.
- Fragment Size Distribution (FSD): Genome-wide distribution of cfDNA fragment lengths.
- End Motif (EDM): Frequency of 4-nucleotide sequences at the ends of cfDNA fragments.
- BreakPoint Motif (BPM): Motif frequencies at genomic breakpoints.
- Copy Number Variation (CNV): Somatic copy number alterations [99].
- Machine Learning Model Building:
- First-Level Model (Cancer Detection): Use the training cohort to build an ensemble stacked model. This integrates the five fragmentomic features using multiple algorithms (Generalized Linear Model, Gradient Boosting Machine, Random Forest, Deep Learning, XGBoost). The model is trained to differentiate cancer patients from healthy individuals.
- Second-Level Model (Cancer Origin): Using only the cancer samples from the training cohort, train a separate model to classify the tissue of origin (e.g., liver, colorectal, lung) [99].
- Model Validation: Validate the finalized model on a held-out test cohort that was not used during training. Performance metrics include Area Under the Curve (AUC), sensitivity, specificity, and accuracy for origin prediction.
Isolation and Analysis of Apoptotic Bodies (ApoEVs)
Objective: To isolate and characterize apoptotic bodies (ApoEVs) from cell culture supernatants or biofluids for downstream molecular analysis.
Workflow:
- Apoptosis Induction & ApoEV Generation:
- ApoEV Collection: Collect conditioned culture media containing ApoEVs after a specified incubation period.
- ApoEV Isolation & Purification: Isolate ApoEVs from the media using differential ultracentrifugation.
- Low-speed spins (e.g., 300 × g) to remove intact cells.
- Higher-speed spins (e.g., 2,000 × g) to pellet larger apoptotic bodies [100].
- Further purification via density gradient centrifugation can be performed to enhance purity.
- Characterization & Validation:
- Downstream Molecular Analysis:
Signaling Pathways and Molecular Mechanisms
Biogenesis of Apoptotic Bodies (ApoEVs)
The formation of apoptotic bodies is a tightly regulated process downstream of caspase activation. The following diagram illustrates the key molecular pathway.
The pathway is initiated by an apoptotic stimulus, which triggers the activation of caspase enzymes [102]. A critical downstream effector is Rho-associated kinase 1 (ROCK1), which, upon activation by caspases, phosphorylates myosin light chain. This drives actomyosin contraction, leading to membrane blebbing and cell contraction [21]. In adherent cells, this contractile force also causes the cell to retract and leave behind a membrane-encased, F-actin-rich structure anchored to the substrate, termed the "FOotprint Of Death" (FOOD). The FOOD subsequently undergoes vesicularisation into FOOD-derived ApoEVs (F-ApoEVs), which are ~2 μm in diameter and expose "eat-me" signals like phosphatidylserine [21]. These ApoEVs contain a rich molecular cargo (nuclear DNA, RNA, proteins) from the parent cell, which serves as the basis for biomarker development [100].
The Scientist's Toolkit: Key Research Reagents
The following table lists essential reagents and kits used in the featured experiments and this field of research.
Table 3: Essential Research Reagents and Materials
| Item Name/Type | Function/Application | Specific Example (if provided) |
|---|---|---|
| BH3-mimetic Cocktail | Chemically induces intrinsic apoptosis for ApoEV generation in vitro [21] | ABT-737 and S63845 [21] |
| Annexin A5 (Stained) | Binds externalized phosphatidylserine; used to detect and validate apoptotic cells and ApoEVs via microscopy/flow cytometry [21] | N/A |
| Differential Ultracentrifuge | Essential equipment for the isolation and purification of ApoEVs from biofluids or cell culture media based on size and density [100] | N/A |
| Next-Generation Sequencer | Platform for performing whole-genome sequencing to generate data for cfDNA fragmentomic analysis [101] [99] | N/A |
| cfDNA Extraction Kit | For the isolation of high-quality, uncontaminated cell-free DNA from blood plasma samples. | N/A |
| Cas12a RNP | CRISPR-associated protein used in novel, ultrasensitive ctDNA detection assays to cleave reporters upon target recognition [101] | Component of "one-pot" CRISPR assays [103] |
| Magnetic Nanoparticles | Used in electrochemical biosensors to capture and enrich target ctDNA fragments, enabling attomolar sensitivity [101] | Fe₃O₄–Au core–shell particles [101] |
In the field of cell death research, particularly in studies of apoptosis, scientists increasingly rely on multiple complementary techniques to validate their findings. The correlation between DNA fragmentation assays and the detection of apoptotic bodies provides a powerful two-pronged approach for confirming programmed cell death. DNA fragmentation represents a key biochemical event in apoptosis, characterized by the cleavage of nuclear DNA into specific oligonuclosomal fragments. Meanwhile, apoptotic body formation represents the terminal morphological manifestation of this process, where the cell shrinks and fragments into membrane-bound vesicles. This guide objectively compares the performance of these methodological approaches, providing experimental data and protocols to help researchers implement robust validation strategies for their apoptosis studies.
Morphological and Biochemical Hallmarks of Apoptosis
Apoptotic cell death is defined by a series of characteristic morphological and biochemical changes that distinguish it from other forms of cell death such as necrosis, necroptosis, or autophagy [13] [74]. The table below summarizes the key features of apoptosis compared to other cell death modalities:
Table 1: Comparative Characteristics of Major Cell Death Modalities
| Cell Death Mode | Distinctive Morphological Features | Distinctive Biochemical Features |
|---|---|---|
| Classical Apoptosis | Strong chromatin condensation, cell shrinkage, preservation of organelles, membrane blebbing, formation of apoptotic bodies [13] [74] | Absolute requirement of caspase activation, internucleosomal DNA fragmentation, phosphatidylserine exposure [13] [74] |
| Necrosis | Lack of geometric chromatin condensation, organelle and cell swelling, rapid rupture of plasma membrane [74] | Lack of protease cascade activation, random DNA degradation (no laddering), uncontrolled release of cell constituents [13] [74] |
| Autophagy | Partial chromatin condensation, formation of double/multilayered autophagosome vacuoles, cell membrane blebbing possible [13] [74] | Initially perceived as caspase-independent, possible cross-talk with apoptosis; lack of DNA fragmentation; increased lysosomal activity [13] [74] |
| Necroptosis | Necrosis-like morphology with cell swelling and membrane destruction, but regulated process [13] | Caspase-independent, regulated by specific signaling pathways (RIPK1/RIPK3/MLKL), can be inhibited by specific inhibitors [13] |
DNA Fragmentation Assays: Methodologies and Applications
Biochemical Principles and Significance
DNA fragmentation is a hallmark biochemical event in apoptosis, resulting from the activation of endonucleases that cleave DNA at internucleosomal linker regions. This process generates fragments of approximately 180-200 base pairs and multiples thereof, which produce a characteristic "ladder" pattern when separated by agarose gel electrophoresis [12]. This fragmentation occurs in a stepwise process involving initial intracellular cleavage producing fragments corresponding to nucleosomal units (mono-, di-, tri-, and tetra-nucleosomes), followed by further extracellular digestion that generates sub-nucleosomal fragments with 10-bp periodicity [19].
Standard DNA Fragmentation Protocol
The standard protocol for detecting DNA fragmentation via agarose gel electrophoresis involves three main stages [12]:
Table 2: DNA Fragmentation Analysis Protocol
| Stage | Key Steps | Technical Considerations |
|---|---|---|
| Cell Harvesting & Lysis | Pellet cells → Lyse in detergent buffer (10 mM Tris pH 7.4, 5 mM EDTA, 0.2% Triton) → Incubate on ice 30 min → Centrifuge at 27,000 × g for 30 min [12] | Maintain samples on ice throughout; ensure complete cell lysis; Triton X-100 can be substituted with 0.2% NP-40 [12] |
| DNA Precipitation | Add NaCl to supernatant → Precipitate with ethanol and sodium acetate → Incubate at -80°C for 1 h → Centrifuge → Treat with DNase-free RNase (37°C for 5 h) → Digest with proteinase K (65°C overnight) [12] | Use ice-cold reagents; pellets may be loose - handle with care; RNase treatment crucial to remove RNA contamination [12] |
| Gel Electrophoresis & Visualization | Air-dry pellet → Resuspend in Tris-acetate EDTA buffer → Separate on 2% agarose gel containing ethidium bromide → Visualize by UV transillumination [12] | Use 2% agarose for optimal resolution; include appropriate molecular weight markers; ethidium bromide alternative: safer DNA stains [12] |
Advanced Detection Methods
Beyond conventional gel electrophoresis, several advanced techniques offer enhanced sensitivity and quantification:
- TUNEL Assay (Terminal deoxynucleotidyl transferase dUTP Nick-End Labeling): Detects DNA strand breaks by incorporating labeled nucleotides at the 3'-ends of fragmented DNA. Can be combined with flow cytometry for quantification or microscopy for spatial localization [104] [105].
- γ-H2AX Detection: Phosphorylation of histone H2AX at serine 139 occurs concurrently with the initial appearance of high molecular weight DNA fragments during apoptosis, making it an early marker of DNA damage [106].
- Shallow Whole-Genome Sequencing (sWGS): Provides high-resolution analysis of fragment size distribution, revealing characteristic nucleosomal patterns (peaks at ~167 bp, ~360 bp, ~540 bp, and ~720 bp) [19].
Apoptotic Body Detection: Methodologies and Applications
Morphological Principles and Significance
Apoptotic bodies are membrane-bound vesicles formed during the final stages of apoptosis through a process of cellular fragmentation. They typically contain compact, electron-dense chromatin, intact organelles, and vary in size from approximately 0.5 to 5 μm in diameter [107]. The formation of apoptotic bodies represents the morphological culmination of the apoptotic process and serves to package cellular contents for efficient phagocytosis by neighboring cells, thereby preventing inflammatory responses [13] [107].
Novel Mechanisms: The "FOOTPRINT OF DEATH" (FOOD)
Recent research has identified a novel mechanism for generating large apoptotic extracellular vesicles called the "FOotprint Of Death" (FOOD). During apoptosis, adherent cells retract and leave behind actin-rich membrane tracks resembling a cellular footprint anchored to the substrate [31]. These structures subsequently vesicularize into large FOOD-derived Apoptotic Extracellular Vesicles (F-ApoEVs) approximately ~2 μm in diameter that expose phosphatidylserine ("eat-me" signal) and can function to 'flag' the site of cell death to phagocytes [31]. This process is regulated by the protein kinase ROCK1 and represents an alternative mechanism for generating large extracellular vesicles during apoptosis distinct from traditional apoptotic bodies or migrasomes [31].
Isolation and Quantification Protocols
Table 3: Apoptotic Body Isolation and Detection Methods
| Method | Procedure | Applications & Advantages |
|---|---|---|
| Differential Centrifugation | Series of sequential centrifugations (e.g., 2,500 × g for 15 min) to isolate apoptotic bodies from cell culture supernatants or plasma samples [107] | High recovery rates of intact apoptotic bodies; maintains structural integrity; suitable for various biological fluids [107] |
| Flow Cytometry Analysis | Size-based detection coupled with fluorescent labeling (e.g., annexin V for phosphatidylserine) [107] | High-throughput quantification; multi-parameter analysis; can distinguish apoptotic bodies from other extracellular vesicles [107] |
| Electron Microscopy | Visualization of ultrastructural features; typically shows round-shaped membrane structures with compact, electron-dense chromatin [107] | "Gold standard" for morphological confirmation; reveals characteristic apoptotic body structure [74] [107] |
| Dynamic Light Scattering | Size distribution analysis of vesicle preparations [107] | Rapid assessment of vesicle size distribution; minimal sample preparation required [107] |
Correlative Approach: Integrated Experimental Design
Strategic Method Combination
Implementing a correlative approach that combines DNA fragmentation assays with apoptotic body detection provides complementary data that strengthens experimental conclusions:
- DNA Fragmentation Assays provide biochemical evidence of apoptosis through detection of characteristic internucleosomal cleavage patterns [12] [19].
- Apoptotic Body Detection confirms the morphological execution of apoptosis and can be quantified to assess the extent of cell death [31] [107].
- Combined Approach overcomes limitations of individual methods, as some cells may undergo caspase-independent apoptosis with atypical DNA fragmentation but still produce apoptotic bodies [74].
Experimental Workflow Integration
The following diagram illustrates a logical workflow for integrating these methods in apoptosis validation:
Comparative Performance Data
Method Efficacy Across Cell Death Models
Table 4: Quantitative Comparison of Detection Methods Across Experimental Conditions
| Experimental Condition | DNA Fragmentation Detection | Apoptotic Body Formation | Correlative Concordance |
|---|---|---|---|
| Staurosporine Treatment (Intrinsic Pathway) | 85-95% detection by gel electrophoresis [105] | 70-80% cells showing apoptotic bodies [105] | High (90-95%) [105] |
| TRAIL Treatment (Extrinsic Pathway) | 75-85% detection by TUNEL [105] | 65-75% cells showing apoptotic bodies [105] | Moderate-High (85-90%) [105] |
| Serum Starvation | 60-70% detection by DNA laddering [19] | 50-60% cells showing apoptotic morphology [19] | Moderate (80-85%) [19] |
| Camptothecin Treatment | >90% detection by multiple methods [19] | >85% cells showing apoptotic bodies [19] | High (90-95%) [19] |
Research Reagent Solutions
Table 5: Essential Research Reagents for Apoptosis Detection Assays
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Apoptosis Inducers | Staurosporine, TRAIL, Camptothecin, BH3-mimetics (ABT-737, S63845) [31] [105] [19] | Positive controls for apoptosis induction via intrinsic or extrinsic pathways |
| DNA Staining Reagents | Ethidium bromide, Propidium iodide, DAPI, Hoechst stains [12] [92] | Visualization of nuclear chromatin condensation and DNA fragmentation patterns |
| Phosphatidylserine Detection | Fluorescently-labeled Annexin V conjugates [31] [92] | Flow cytometric detection of PS externalization on apoptotic cells and bodies |
| Caspase Activity Reagents Caspase substrates (DEVD-pNA), caspase inhibitors (Z-VAD-FMK) [13] [105] [92] | Confirmation of caspase-dependent apoptosis pathway activation | |
| Nuclease Inhibitors | DFFB inhibitors, DNASE1 inhibitors, DNASE1L3 inhibitors [19] | Mechanistic studies of DNA fragmentation pathways |
| Membrane Integrity Probes | Trypan blue, SYTOX Green, 7-AAD [92] | Distinction between apoptotic and necrotic cell death |
| Extracellular Vesicle Isolation Kits | Differential centrifugation kits, polymer-based precipitation kits [107] | Isolation of apoptotic bodies from cell culture media or biological fluids |
Technical Considerations and Limitations
Assay Limitations and Troubleshooting
Both DNA fragmentation and apoptotic body detection methods have specific limitations that researchers should consider:
- DNA Fragmentation Assays may yield false negatives in cells with caspase-independent apoptosis or in cell types with atypical endonuclease expression [74]. The gel electrophoresis approach is semi-quantitative and may not detect early stages of DNA cleavage [12].
- Apoptotic Body Detection can be complicated by the presence of other extracellular vesicles, such as microvesicles and exosomes, which have overlapping size ranges [107]. Additionally, some cell types (e.g., MEFs lacking Bax/Bak) may not efficiently form apoptotic bodies despite undergoing other apoptotic changes [31].
- Correlative Approaches mitigate these limitations by providing multiple lines of evidence, but require careful experimental timing as DNA fragmentation typically precedes apoptotic body formation [105] [19].
Emerging Technologies and Future Directions
Innovative cytometric technologies are enhancing our ability to correlate DNA fragmentation with apoptotic body formation:
- Imaging Flow Cytometry combines the statistical power of flow cytometry with visual confirmation of morphological features, allowing simultaneous detection of DNA fragmentation and apoptotic body formation in the same cells [74].
- Microfluidic Lab-on-a-Chip platforms enable high-throughput screening of apoptotic responses with minimal sample volumes [74].
- Advanced Fragmentomics approaches using whole-genome sequencing provide unprecedented resolution of DNA fragmentation patterns, revealing nucleosomal positioning and epigenetic features associated with apoptosis [19].
The correlative approach combining DNA fragmentation assays with apoptotic body detection provides a robust framework for validating apoptotic cell death in experimental systems. While DNA fragmentation analysis offers sensitive biochemical detection of specific apoptotic events, apoptotic body quantification provides confirmation of the characteristic morphological endpoint. The integration of these methods, along with emerging technologies such as imaging flow cytometry and advanced fragmentomics, creates a powerful validation strategy that minimizes false positives and provides comprehensive characterization of cell death mechanisms. Researchers should select specific method combinations based on their experimental models, timing considerations, and available technical resources to implement this correlative approach effectively.
In the fields of cell biology and reproductive medicine, accurately assessing cell death and genetic integrity is paramount. Two critical areas of analysis are the detection of apoptotic bodies, which signal the final stages of programmed cell death, and the measurement of DNA fragmentation, a key indicator of genomic damage, particularly in sperm cells. The choice between these analytical pathways is not trivial; it is fundamentally guided by the researcher's specific goals. Whether the aim is to understand cellular disintegration processes in disease models or to evaluate functional fertility potential, selecting the appropriate assay ensures that the data generated is both biologically relevant and actionable. This guide provides a structured comparison of these methodologies, empowering scientists to make informed, context-dependent decisions for their research.
Core Concepts and Biological Context
Apoptotic Body Formation and Detection
Apoptosis, or programmed cell death, is a tightly regulated process essential for development, immune function, and tissue homeostasis. Its final stages are characterized by cellular disassembly and the formation of subcellular, membrane-bound vesicles known as apoptotic bodies (ApoBDs) [35].
- Morphological Hallmarks: The process involves cell shrinkage, chromatin condensation, and dynamic membrane changes. The cell first forms membrane blebs, followed by more complex structures like apoptopodia (thin membrane protrusions) [31] [35]. A recently described structure, the "FOotprint Of Death" (FOOD), is a membrane-encased, F-actin-rich remnant that anchors to the substrate after an adherent cell retracts and subsequently vesicularises into large ApoEVs (FOOD-derived Apoptotic Extracellular Vesicles) [31].
- Key Regulatory Machinery: This morphological cascade is primarily executed by a family of proteases called caspases. The activation of initiator caspases (e.g., caspase-2, -8, -9, -10) triggers a cascade that activates effector caspases (e.g., caspase-3, -6, -7), leading to the cleavage of cellular substrates and the structural collapse of the cell [62]. The process is also regulated by the kinase ROCK1, which drives the actomyosin contraction necessary for membrane blebbing and cell retraction [31].
- Functional Significance: Apoptotic bodies are not merely cellular debris. They facilitate communication with phagocytes for efficient clearance (efferocytosis) and can carry DNA, RNA, and proteins to neighboring cells, influencing the tissue microenvironment [31] [35]. Their formation is therefore a definitive marker of apoptotic progression.
DNA Fragmentation: Causes and Consequences
DNA fragmentation refers to breaks in the DNA strands. In the context of apoptosis, it is a hallmark event caused by the caspase-activated endonucleases [62]. However, its significance extends far beyond, especially in male reproductive health.
- Sperm DNA Fragmentation (SDF): In sperm, DNA fragmentation is increasingly recognized as a critical biomarker of male fertility [108] [109]. It can manifest as single-strand breaks, double-strand breaks, base modifications, and telomere attrition [108].
- Primary Cause: Oxidative stress is the principal driver of SDF. During spermatogenesis, spermatozoa are particularly vulnerable to reactive oxygen species (ROS) due to their limited cytoplasmic antioxidant defenses [108]. Other contributing factors include abortive apoptosis, varicocele, advanced paternal age, smoking, and environmental pollutants [108].
- Functional Impact: Elevated SDF is strongly associated with impaired fertilization, poor embryo development, lower pregnancy rates in assisted reproductive technology (ART), and an increased risk of miscarriage [69] [108] [109]. It provides a functional assessment of sperm quality that goes beyond standard parameters like count and motility.
Comparative Analysis of Key Assays
The following tables summarize the primary techniques used for detecting apoptotic bodies and DNA fragmentation, highlighting their principles, applications, and limitations.
Table 1: Assays for Detecting Apoptotic Body Formation
| Assay Name | Principle of Detection | Key Readouts | Advantages | Limitations |
|---|---|---|---|---|
| Morphological Analysis (Microscopy) | Direct visualization of cellular morphological changes using electron, phase-contrast, or confocal microscopy [62]. | Cell shrinkage, chromatin condensation, membrane blebbing, apoptotic bodies [62]. | Reliable; provides a wealth of structural information [62]. | Time-consuming; subjective quantification; difficult for many samples [62]. |
| Annexin V Staining | Binds to phosphatidylserine (PS), a phospholipid that translocates to the outer leaflet of the plasma membrane in early apoptosis [62]. | PS exposure on the cell surface (including on ApoBDs), distinguishing apoptotic from necrotic cells when combined with a viability dye [62] [35]. | Easy, rapid quantitation of apoptosis in single cells [62]. | Requires intact tissues to be dissociated into single cells for analysis; PS exposure is not exclusive to apoptosis [62]. |
| Flow Cytometry of ApoBDs | Multiparameter analysis based on particle size, granularity, Annexin V binding, and uptake of nucleic acid dyes [35]. | Quantification and sub-classification of ApoBDs based on intracellular contents (DNA, mitochondria) and cell-origin surface markers [35]. | High-throughput, quantitative analysis of ApoBD subsets from a mixed cell population [35]. | Requires specialized flow cytometry setup and expertise to distinguish ApoBDs from other particles [35]. |
Table 2: Assays for Detecting DNA Fragmentation
| Assay Name | Principle of Detection | Key Readouts | Advantages | Disadvantages |
|---|---|---|---|---|
| TUNEL Assay (Terminal deoxynucleotidyl transferase dUTP Nick End Labeling) | Terminal transferase enzyme labels the 3'-OH ends of DNA breaks with tagged nucleotides [62]. | Direct labelling of DNA strand breaks from any form of cell death, detectable by microscopy or flow cytometry [62]. | Highly sensitive; can detect early apoptotic events; precise for cell death and DNA damage [62]. | Can label necrotic cells; multi-step procedure; results can be qualitative; specificity depends on fixation and pre-treatment [62]. |
| Sperm Chromatin Structure Assay (SCSA) | Flow cytometry-based assay using acridine orange staining; damaged DNA denatures and emits red fluorescence, while intact DNA emits green [7] [95]. | DNA Fragmentation Index (DFI) - the percentage of sperm with denatured DNA [7] [95]. | High throughput; objective and quantitative; considered a reliable standard for SDF [95]. | Requires flow cytometry equipment; does not distinguish between single- and double-strand breaks. |
| Comet Assay (Single Cell Gel Electrophoresis) | Individual cells embedded in agarose are lysed and subjected to electrophoresis; fragmented DNA migrates away from the nucleus, forming a "comet tail" [69]. | Tail length and intensity, which correlate with the level of DNA damage; can be run under alkaline (global damage) or neutral (double-strand breaks only) conditions [69]. | Highly sensitive; can detect low levels of damage; provides data at the single-cell level [69]. | Labor-intensive; low throughput; subjective analysis without specialized software. |
| Sperm Chromatin Dispersion (SCD) Test | Sperm with fragmented DNA fail to produce the characteristic halo of dispersed DNA loops when denatured and stained [109]. | The percentage of spermatozoa without a halo, indicating DNA fragmentation [109]. | Can be performed with a standard microscope; no need for flow cytometry [109]. | Halos are assessed manually, which can introduce subjectivity. |
Experimental Protocols
Protocol 1: Flow Cytometric Analysis of Apoptotic Bodies
This protocol enables the quantification and characterization of ApoBDs based on established methodologies [35].
- Induction of Apoptosis: Culture adherent or suspension cells and induce apoptosis using an appropriate stimulus (e.g., UV irradiation at 150 mJ/cm², BH3-mimetics, or etoposide) [31] [35].
- Sample Preparation: Collect cells and culture supernatant together to ensure all ApoBDs are harvested. Centrifuge at low speed (e.g., 300–500 x g) to pellet cells and large ApoBDs without damaging them.
- Staining:
- Resuspend the pellet in Annexin V Binding Buffer.
- Add fluorescently conjugated Annexin V to detect phosphatidylserine exposure.
- Add a cell-impermeable nucleic acid dye like TO-PRO-3 to assess membrane integrity and distinguish late apoptotic/necrotic cells.
- For intracellular content analysis, pre-stain cells with dyes like Hoechst 33342 (DNA) or MitoTracker Green (mitochondria) prior to apoptosis induction [35].
- Incubate for 10–15 minutes at room temperature in the dark.
- Flow Cytometry Acquisition: Analyze samples on a flow cytometer. ApoBDs are identified based on their small size (low forward scatter), low complexity (low side scatter), positive Annexin V staining, and low TO-PRO-3 staining (intact membrane) [35].
- Data Analysis: Gate on the ApoBD population and analyze the fluorescence intensity for different markers to determine subpopulations.
Protocol 2: Sperm Chromatin Structure Assay (SCSA) for DNA Fragmentation Index
This is a standardized protocol for assessing sperm DNA damage [7] [95].
- Semen Sample Preparation: Obtain semen samples after 2–7 days of sexual abstinence. Allow the sample to liquefy completely at 37°C.
- Sperm Concentration Adjustment: Dilute the semen sample with a buffer to a concentration of 1–2 x 10⁶ sperm/mL.
- Acid Denaturation: Treat 100 µL of the diluted sample with a low-pH acid solution to denature the DNA in sperm with fragmented chromatin.
- Staining: Add acridine orange stain. In sperm with intact DNA, the dye intercalates into double-stranded DNA and emits green fluorescence. In sperm with fragmented DNA, the dye associates with single-stranded DNA and emits red fluorescence.
- Flow Cytometry Analysis: Immediately analyze the stained sample using a flow cytometer. Measure the green (515–530 nm) and red (>630 nm) fluorescence for a minimum of 10,000 events per sample.
- DFI Calculation: The DNA Fragmentation Index (DFI) is calculated as the ratio of red fluorescence to total (red + green) fluorescence, expressed as a percentage: DFI (%) = [Red / (Red + Green)] x 100% [95].
Decision Workflows and Signaling Pathways
Diagram 1: Apoptotic Body Formation Pathway
Diagram 2: Assay Selection Decision Workflow
Research Reagent Solutions
The following table details essential reagents and kits for implementing the discussed assays.
Table 3: Key Research Reagents and Kits
| Item | Function/Application | Example Use Case |
|---|---|---|
| Recombinant Annexin V (conjugated to FITC, PE, APC, etc.) | Detects phosphatidylserine exposure on the outer membrane leaflet of apoptotic cells and ApoBDs [62] [35]. | Distinguishing apoptotic from necrotic cells in combination with a viability dye by flow cytometry or microscopy. |
| Caspase Activity Assays (Fluorometric or Colorimetric) | Measures the enzymatic activity of caspases (e.g., Caspase-3) using specific substrates that release a chromophore or fluorophore upon cleavage [62]. | Providing a specific biochemical confirmation of apoptosis initiation or execution. |
| Sperm Nuclear Integrity Staining Kit (for SCSA) | Contains buffers and acridine orange for the standardized denaturation and staining of sperm DNA for DFI calculation by flow cytometry [95]. | Objective and high-throughput assessment of sperm DNA fragmentation in fertility studies. |
| In Situ Cell Death Detection Kit (TUNEL) | Contains terminal deoxynucleotidyl transferase (TdT) and labeled nucleotides to label DNA strand breaks in situ [62]. | Detecting and quantifying apoptotic cells in tissue sections or cell smears via fluorescence microscopy. |
| Comet Assay Kit | Provides ready-to-use gels, lysis buffers, and electrophoresis solutions for single-cell DNA damage analysis under alkaline or neutral conditions [69]. | Sensitive detection of single- and double-strand DNA breaks in individual sperm or somatic cells. |
| ROCK1 Inhibitor (e.g., Y-27632) | Pharmacologically inhibits ROCK1 kinase activity. | Mechanistic studies to confirm the role of ROCK1 in apoptotic membrane blebbing and FOOD formation [31]. |
The strategic selection between apoptotic body assays and DNA fragmentation tests is a cornerstone of rigorous experimental design in cell death and reproductive research. Apoptotic body analysis remains the gold standard for confirming and quantifying the morphological execution of programmed cell death, providing insights into cell clearance and intercellular communication. In contrast, DNA fragmentation assays, especially in a fertility context, offer a powerful functional assessment of the genomic material's integrity, with a direct proven impact on embryonic development and clinical outcomes. By aligning the research question with the specific strengths and limitations of each assay—be it the high-throughput power of SCSA, the sensitivity of the Comet assay, the visual confirmation of microscopy, or the quantitative capacity of flow cytometry—researchers can ensure their findings are both robust and clinically or biologically meaningful.
Emerging Techniques and Technological Advancements in DNA Fragmentation and Apoptotic Body Analysis
The study of programmed cell death, or apoptosis, is a cornerstone of cell biology, cancer research, and therapeutic development. For decades, DNA fragmentation and the formation of apoptotic bodies have served as two fundamental hallmarks of this process. DNA fragmentation involves the enzymatic cleavage of nuclear DNA into characteristic oligonucleosomal fragments, while apoptotic body formation represents the morphological endpoint where the cell packages its contents into membrane-bound vesicles for disposal. Historically treated as sequential events in a single pathway, emerging research now reveals these as distinct, sometimes independent, processes regulated by separate molecular mechanisms. This guide provides a comparative analysis of the techniques used to study these phenomena, offering researchers a framework for selecting appropriate methodologies based on specific experimental needs and biological contexts.
Fundamental Biological Mechanisms
DNA Fragmentation: The Molecular Signature of Apoptosis
Apoptotic DNA fragmentation is a biochemical process characterized by the activation of specific endonucleases that cleave DNA at internucleosomal linker regions. The primary enzyme responsible is Caspase-Activated DNase (CAD), which is normally inhibited by its chaperone ICAD (Inhibitor of Caspase-Activated DNase). During apoptosis, executioner caspases (particularly caspase-3) cleave ICAD, releasing CAD to initiate DNA cleavage [80]. This systematic digestion produces DNA fragments of approximately 180-200 base pairs and their multiples, which form a characteristic "ladder" pattern when separated by agarose gel electrophoresis [80] [12].
The process occurs in a regulated manner, beginning with large-scale DNA fragmentation (≥50 kbp) that progresses to the internucleosomal cleavage that yields the classic ladder pattern [30]. This DNA degradation serves as both a marker of irreversible commitment to cell death and a means to prevent the transfer of genetic material to neighboring cells.
Apoptotic Body Formation: The Morphological Outcome
Apoptotic bodies are large extracellular vesicles (1-5 μm in diameter) formed during the final stages of apoptosis through a process termed apoptotic cell disassembly [31] [110]. This process involves three key morphological stages: (1) membrane blebbing, (2) formation of membrane protrusions (apoptopodia), and (3) fragmentation into discrete apoptotic bodies [110].
Recent research has identified a novel mechanism for apoptotic body formation called the "FOotprint Of Death" (FOOD), where retracting apoptotic cells leave behind actin-rich membrane tracks on substrates that subsequently vesicularize into large apoptotic bodies [31]. This process is regulated by the protein kinase ROCK1, which phosphorylates myosin light chain to drive actomyosin contraction [31].
The stability and fate of apoptotic bodies are regulated by NINJ1 (ninjurin-1), which oligomerizes on apoptotic bodies to mediate plasma membrane rupture and the release of inflammatory signals or viral particles in secondary necrosis [110]. This recent discovery provides new insights into how apoptotic bodies function in intercellular communication and disease propagation.
Comparative Analysis of Detection Methodologies
DNA Fragmentation Assays
Table 1: DNA Fragmentation Detection Methods
| Method | Principle | Sensitivity | Applications | Key Advantages | Limitations |
|---|---|---|---|---|---|
| DNA Laddering | Agarose gel separation of fragmented DNA | Semi-quantitative | Bulk cell analysis, apoptosis confirmation | Low cost, specific apoptotic pattern, simple protocol | Requires high apoptotic cell percentage, semi-quantitative [12] |
| TUNEL Assay | Terminal deoxynucleotidyl transferase labels DNA breaks | High (single-cell) | Tissue sections, flow cytometry, microscopy | Detects early fragmentation, quantitative potential | Possible false positives, requires specialized equipment [80] [111] |
| Comet Assay | Electrophoretic DNA migration from individual cells | Single-cell sensitivity | Genotoxicity studies, heterogeneous samples | Distinguishes single/double strand breaks, highly sensitive | Technically challenging, low throughput [69] [30] |
| Sub-G1 Analysis | Flow cytometric detection of hypodiploid DNA | Moderate | Cell cycle analysis, high-throughput screening | Quantitative, works with fixed cells | Cannot detect early apoptosis, may miss late-stage cells [80] |
Apoptotic Body Detection and Characterization
Table 2: Apoptotic Body Analysis Techniques
| Method | Principle | Resolution | Applications | Key Advantages | Limitations |
|---|---|---|---|---|---|
| Time-Lapse Microscopy | Direct visualization of morphological changes | Single-vesicle | Kinetic studies, mechanism discovery | Reveals dynamic process, high temporal resolution | Equipment intensive, low throughput [31] [112] |
| Flow Cytometry | Light scattering and fluorescence detection | Single-vesicle | Phenotyping, quantification | High-throughput, multi-parameter analysis | Limited morphological detail, size detection limits [31] [113] |
| Electron Microscopy | High-resolution ultrastructural imaging | Nanoscale | Detailed morphological analysis | Reveals ultrastructural details, definitive identification | Fixed samples only, technically demanding [31] [113] |
| Proteomic Analysis | Protein composition profiling | Molecular | Biomarker discovery, functional studies | Comprehensive molecular profiling, mechanistic insights | Requires vesicle isolation, complex data analysis [31] |
Experimental Protocols for Key Assays
DNA Fragmentation Analysis via Agarose Gel Electrophoresis
This protocol enables detection of the characteristic DNA ladder pattern in apoptotic cells [12]:
Cell Harvesting and Lysis
- Pellet approximately 10⁶ cells by centrifugation
- Resuspend in 0.5 mL lysis buffer (10 mM Tris pH 7.4, 5 mM EDTA, 0.2% Triton X-100)
- Vortex and incubate on ice for 30 minutes
- Centrifuge at 27,000 × g for 30 minutes to separate fragmented DNA (supernatant) from intact chromatin (pellet)
DNA Precipitation and Purification
- Divide supernatant into two 250 μL aliquots
- Add 50 μL of 5 M NaCl to each aliquot and vortex
- Add 600 μL ethanol and 150 μL 3 M sodium acetate (pH 5.2)
- Incubate at -80°C for 1 hour to precipitate DNA
- Centrifuge at 20,000 × g for 20 minutes and carefully discard supernatant
- Dissolve DNA pellets in 400 μL extraction buffer (10 mM Tris, 5 mM EDTA)
- Treat with DNase-free RNase (2 μL of 10 mg/mL) for 5 hours at 37°C
- Add 25 μL proteinase K (20 mg/mL) and incubate overnight at 65°C
- Extract with phenol/chloroform/isoamyl alcohol (25:24:1) and ethanol precipitate
Electrophoresis and Visualization
- Resuspend DNA in 20 μL Tris-acetate EDTA buffer with 2 μL sample buffer (0.25% bromophenol blue, 30% glycerol)
- Separate on 2% agarose gel containing 1 μg/mL ethidium bromide
- Visualize DNA fragments under UV transillumination
- Apoptotic samples display a characteristic ladder pattern with ~180 bp multiples
Apoptotic Body Isolation and Characterization
This differential centrifugation protocol enables isolation of apoptotic bodies for downstream analysis [110]:
Induction of Apoptosis and Sample Collection
- Treat cells with appropriate apoptotic inducer (e.g., BH3 mimetic cocktail: 2 μM ABT-737 + 10 μM S63845)
- Incubate for 4 hours at 37°C
- Collect culture supernatant containing apoptotic bodies
Differential Centrifugation
- Centrifuge at 300 × g for 10 minutes to remove intact cells and large debris
- Transfer supernatant to fresh tube
- Centrifuge at 2,000 × g for 20 minutes to pellet apoptotic bodies
- Wash pellet with PBS and repeat 2,000 × g centrifugation
- Resuspend apoptotic body pellet in appropriate buffer for downstream applications
Characterization and Validation
- Flow Cytometry: Analyze size distribution and phosphatidylserine exposure via annexin V binding
- Immunoblotting: Confirm presence of apoptotic markers (cleaved caspase-3, caspase-cleaved pannexin 1)
- Microscopy: Validate morphology and size (1-5 μm) using differential interference contrast or fluorescence microscopy
- Functional Assays: Assess phagocytosis by recipient cells or content release using LDH or FITC-dextran exclusion assays
Signaling Pathways and Molecular Regulation
The following diagrams illustrate the key signaling pathways regulating DNA fragmentation and apoptotic body formation:
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Apoptosis Research
| Reagent Category | Specific Examples | Function | Applications |
|---|---|---|---|
| Apoptosis Inducers | BH3 mimetics (ABT-737, S63845), Staurosporine, Etoposide | Trigger apoptotic signaling through specific pathways | Experimental apoptosis induction, mechanism studies [31] [112] |
| Caspase Substrates | Caspase-3/7 Glo Assay, Fluorogenic caspase substrates | Measure caspase activation | Early apoptosis detection, inhibitor studies [110] |
| DNA Staining Reagents | Propidium iodide, Acridine orange, Hoechst stains, SYTOX green | Label nucleic acids for detection and quantification | Cell viability, DNA content analysis, microscopy [111] [113] |
| Membrane Integrity Probes | Annexin V conjugates, FITC-dextran, LDH assay kits | Detect phosphatidylserine exposure and membrane permeability | Early/late apoptosis distinction, vesicle integrity [31] [110] |
| Kinase Inhibitors | ROCK1 inhibitors, Jasplakinolide | Modulate specific signaling pathways | Mechanism studies, apoptotic body formation inhibition [31] |
| EV Isolation Reagents | Differential centrifugation kits, Size-exclusion columns | Isolate extracellular vesicles including apoptotic bodies | Apoptotic body purification, downstream analysis [110] |
Applications and Research Implications
DNA Fragmentation in Clinical Assessment
DNA fragmentation assays have found particular utility in clinical diagnostics, especially in reproductive medicine. The sperm DNA fragmentation index (DFI) has been correlated with embryonic development outcomes, with DFI ≥30% associated with reduced blastocyst formation rates (53.72% vs 56.44% in DFI <15%) and increased risk of low birth weight (10.1% vs 3.9%) [114]. Furthermore, double-stranded DNA fragmentation assessed by comet assay shows strong association with recurrent pregnancy loss (AUC=0.909) [69], highlighting the clinical relevance of DNA integrity assessment.
Apoptotic Bodies in Disease Propagation and Intercellular Communication
Emerging research reveals that apoptotic bodies are not merely cellular debris but play active roles in intercellular communication and disease propagation. Apoptotic bodies generated during viral infection can harbor viral proteins and virions, propagating infection to healthy cells [31]. Their composition reflects their cell of origin, containing DNA, RNA, proteins, and organelles that can influence recipient cell behavior [110] [113].
Unexpectedly, apoptotic body formation is not limited to metazoans. Recent research has documented apoptotic body production in the cryptophyte alga Guillardia theta, with vesicle concentration increasing from 0.2×10⁶/mL in exponential phase to 0.75×10⁶/mL in death phase [113]. This challenges evolutionary paradigms about the origins of programmed cell death mechanisms.
The choice between DNA fragmentation assays and apoptotic body analysis depends fundamentally on research objectives. DNA fragmentation methods offer precise molecular detection of apoptosis, with TUNEL assays providing high sensitivity for early detection and comet assays revealing DNA damage spectra. Conversely, apoptotic body analysis illuminates the morphological consequences and intercellular communication aspects of apoptosis, with time-lapse microscopy capturing dynamic formation processes and proteomic approaches revealing cargo composition.
Emerging technologies are bridging these domains, enabling correlated assessment of both biochemical and morphological endpoints. The integration of single-cell analysis, advanced imaging, and multi-omics approaches will continue to refine our understanding of apoptosis, providing increasingly sophisticated tools for both basic research and therapeutic development.
Within the fields of oncology and drug development, the accurate detection of cell death is paramount for evaluating therapeutic efficacy and understanding disease mechanisms. Two fundamental biomarkers of apoptosis, or programmed cell death, are DNA fragmentation and the formation of apoptotic bodies. While both phenomena originate from the same physiological process, they offer distinct advantages and challenges in a diagnostic and screening context. DNA fragmentation assays provide a well-established, biochemical readout of apoptosis, characterized by the cleavage of DNA into oligonucleosomal fragments. In contrast, the analysis of apoptotic bodies, which are membrane-bound vesicles released during the late stages of apoptosis, represents an emerging frontier with potential for both diagnostic and therapeutic applications. This guide objectively compares the performance, experimental requirements, and applications of these two approaches, providing researchers with the data necessary to select the appropriate tool for their specific needs in drug screening and clinical diagnostics.
Fundamental Biological Principles and Signaling Pathways
The Apoptotic Process and Key Biomarkers
Apoptosis is a highly regulated form of programmed cell death essential for maintaining tissue homeostasis and eliminating damaged cells [115] [116]. It is characterized by a series of distinct morphological and biochemical changes, which include:
- Cell shrinkage and chromatin condensation: The cell reduces in volume, and its nuclear chromatin becomes densely packed [115].
- DNA fragmentation: Activation of endogenous endonucleases, such as CAD (caspase-activated DNase), cleaves nuclear DNA at internucleosomal linker regions, producing fragments of approximately 180-200 base pairs and their multiples [12] [115].
- Membrane blebbing and apoptotic body formation: The cell membrane undergoes controlled blebbing, eventually pinching off into small, membrane-bound vesicles known as apoptotic bodies (ABs). These vesicles contain condensed chromatin, organelles, and other cellular components [115] [55].
- Clearance: The apoptotic bodies are rapidly phagocytosed by neighboring cells or macrophages without triggering an inflammatory response [115].
Apoptosis Signaling Pathways
Two primary signaling pathways initiate apoptosis, both converging on the activation of executioner caspases.
DNA Fragmentation Assays: Protocols and Applications
Core Principle and Protocol
DNA fragmentation is a hallmark biochemical event in apoptosis, resulting from the activation of endonucleases that cleave chromosomal DNA into oligonucleosomal fragments [12] [115]. The classic DNA ladder assay detects these fragments through agarose gel electrophoresis. Below is a detailed protocol based on traditional and updated methodologies [12] [41].
Detailed Experimental Protocol: DNA Ladder Assay
Stage 1: Harvesting and Lysing Cells
- Pellet approximately 1-5 x 10^6 cells by centrifugation.
- Resuspend the cell pellet in 0.5 mL of detergent-based lysis buffer (e.g., 10 mM Tris pH 7.4, 5 mM EDTA, 0.2% Triton X-100).
- Vortex the mixture and incubate on ice for 30 minutes.
- Centrifuge at 27,000 x g for 30 minutes to separate fragmented DNA (in the supernatant) from intact chromatin (in the pellet) [12].
Stage 2: Precipitating and Purifying DNA
- Transfer the supernatant to a new tube and add ice-cold 5 M NaCl.
- Precipitate the DNA by adding ethanol and 3 M sodium-acetate (pH 5.2), then incubate at -80°C for 1 hour.
- Centrifuge at high speed (e.g., 20,000 x g) to pellet the DNA. Discard the supernatant carefully.
- Dissolve the DNA pellet and incubate with DNase-free RNase (e.g., 2 µL of 10 mg/mL) for several hours at 37°C to remove RNA.
- Add Proteinase K (e.g., 25 µL at 20 mg/mL) and incubate overnight at 65°C to digest proteins.
- Extract DNA with phenol/chloroform/isoamyl alcohol and perform a final ethanol precipitation [12].
Stage 3: Gel Electrophoresis and Visualization
- Air-dry the DNA pellet and resuspend it in Tris-acetate-EDTA (TAE) buffer.
- Load the DNA sample onto a 2% agarose gel containing a DNA stain like ethidium bromide or SYBR Safe.
- Separate the DNA by electrophoresis and visualize under UV light. A positive apoptotic result is indicated by a characteristic "ladder" pattern of DNA fragments at 180-200 bp intervals [12] [41].
Performance Data and Applications in Drug Screening
DNA fragmentation assays are widely used to evaluate the efficacy of chemotherapeutic agents and other pro-apoptotic compounds [12] [41].
Table 1: Quantitative Performance of DNA Fragmentation Assays in Drug Screening
| Cell Line/Model | Treatment (Apoptotic Inducer) | Key Assay | Result & Quantitative Data | Application Context |
|---|---|---|---|---|
| NIH-3T3 Cell Line [41] | 500 µM H2O2 for 48 hours | DNA Ladder Assay | Characteristic DNA ladder pattern observed on 1.5% agarose gel. | Validation of a modified, rapid DNA extraction protocol for drug screening. |
| General Tumor Cells [12] | Chemotherapeutic Agents | DNA Ladder Assay | Semi-quantitative assessment of apoptosis; used to evaluate treatment response. | Foundational research and oncology drug development to confirm apoptosis induction. |
| Sperm Cryopreservation [7] | Cryopreservation/Thawing | Sperm Chromatin Structure Assay (SCSA) | Increase in DNA Fragmentation Index (DFI) post-freezing; DFI >30% indicates significant fertility issues. | Assessment of cell damage in assisted reproductive technology; quality control in clinical settings. |
Apoptotic Body Analysis: Protocols and Applications
Core Principle and Protocol
Apoptotic bodies (ABs) are 50-5000 nm membrane-bound vesicles released during the final stage of apoptosis [55]. They are formed through caspase-3-mediated activation of kinases like ROCK1, leading to actomyosin contraction, membrane blebbing, and eventual vesicle budding [55]. Unlike DNA fragmentation, AB analysis focuses on the vesicular products of apoptosis, which carry diverse molecular cargo from the parent cell.
Detailed Experimental Protocol: Isolation and Characterization of Apoptotic Bodies The isolation of ABs is technically demanding due to their heterogeneous size and the lack of absolutely specific markers. The following multi-step protocol is standard in the field [55].
Induction and Collection
- Induce apoptosis in the cell population of interest (e.g., with chemotherapeutic drugs or UV irradiation).
- Collect the culture supernatant, which contains floating apoptotic cells and shed ABs.
- Centrifuge at low speed (e.g., 300 x g for 10 minutes) to pellet intact cells, leaving ABs in the supernatant.
Isolation and Purification
- Differential Centrifugation: Centrifuge the supernatant at higher speeds (e.g., 10,000 - 20,000 x g) to pellet ABs. This is the most common method but may co-pellet other vesicles and impurities [55].
- Filtration: Pass the supernatant through filters (e.g., 1.2 µm then 0.8 µm) to remove larger debris and microvesicles, enriching for ABs.
- Fluorescence-Activated Cell Sorting (FACS): For higher purity, ABs can be stained with fluorescently labeled Annexin V (which binds to phosphatidylserine exposed on the AB surface) and sorted [55].
Characterization and Analysis
- Flow Cytometry (FCM): Used for sizing, counting, and detecting surface markers (e.g., phosphatidylserine via Annexin V binding) [55].
- Nanoparticle Tracking Analysis (NTA): Determines the size distribution and concentration of ABs in a solution [55].
- Immunohistochemistry/Immunoblotting: Identifies specific protein markers present in ABs, such as caspases, histones, or tissue-specific antigens inherited from the parent cell [55].
- Cryo-Electron Microscopy (Cryo-TEM): Provides high-resolution morphological analysis of ABs [55].
Performance Data and Applications
AB analysis is emerging as a powerful tool in both diagnostics and therapeutics, particularly in the realm of liquid biopsy and targeted drug delivery.
Table 2: Quantitative Performance and Applications of Apoptotic Body Analysis
| Application Context | Source of Apoptotic Bodies | Key Analytical Method | Findings & Significance |
|---|---|---|---|
| Diagnostic: Liquid Biopsy [10] [55] | Patient blood plasma (e.g., from cancer patients) | AB isolation and cargo analysis (DNA, RNA, proteins) | ABs in biofluids reflect the physiological state of the originating tissue, carrying molecular information (e.g., tumor mutations) useful for non-invasive diagnosis and monitoring. |
| Therapeutic: Targeted Drug Delivery [55] | Engineered mesenchymal stem cells or other cell types | Tailoring of ABs via physical, chemical, or genetic methods | ABs demonstrate high biocompatibility and target accuracy due to inherited "find-me" and "eat-me" signals. Drug-loaded or surface-modified ABs show promise in alleviating inflammation and promoting tissue regeneration in disease models. |
| Functional Potency Assessment [55] | Osteoclast-derived EVs | Comparative bioactivity assays | Among all extracellular vesicles from osteoclasts, ABs showed the highest level of RANK, granting them the greatest osteogenic potency, highlighting their unique functional capacity. |
Direct Comparison: Assay Performance and Practical Considerations
For researchers selecting an appropriate method, a direct comparison of technical and performance characteristics is critical.
Table 3: DNA Fragmentation Assays vs. Apoptotic Body Analysis: A Categorical Comparison
| Parameter | DNA Fragmentation Assays | Apoptotic Body Analysis |
|---|---|---|
| What is Detected | Biochemical endpoint: Internucleosomal DNA cleavage. | Morphological endpoint: Membrane-bound vesicles released from dying cells. |
| Key Markers | DNA fragments (180-200 bp ladder). | Surface: Phosphatidylserine (Annexin V), caspases. Cargo: Proteins, nucleic acids, organelles [55]. |
| Sensitivity | Lower sensitivity; may not detect early apoptosis or small numbers of apoptotic cells [115]. | Potentially higher; sensitive techniques like FACS and NTA can detect rare events. |
| Specificity | Specific for late-stage apoptosis, but can produce false positives from necrotic DNA degradation [115]. | Highly specific for apoptosis, but isolation can be contaminated by other extracellular vesicles (e.g., microvesicles) [55]. |
| Throughput | Low to medium; gel-based methods are not high-throughput. | Medium to high; flow cytometry and NTA allow for higher throughput analysis. |
| Quantification | Semi-quantitative at best [12]. | Can be highly quantitative (e.g., particle concentration via NTA, cell counting via FCM). |
| Key Advantages | - Simple, cost-effective protocol [41]. - Directly visualizes a hallmark of apoptosis. - Requires minimal specialized equipment. | - Provides information on cellular origin via cargo. - Potential for functional studies and therapeutic application. - Suitable for liquid biopsy approaches [55]. |
| Key Limitations | - Cannot localize apoptosis to specific cells within a population. - Requires a large number of apoptotic cells. - Time-consuming and prone to DNA loss [12] [115]. | - Complex, multi-step isolation protocol. - Lack of universally specific markers makes pure isolation challenging [55]. - Requires sophisticated equipment (ultracentrifuges, FACS, NTA). |
| Best Suited For | - Initial confirmation of apoptosis induction in vitro. - Drug screening where a simple, binary (yes/no) apoptosis readout is sufficient. - Labs with limited budget or equipment. | - Advanced studies of intercellular communication. - Diagnostic biomarker discovery in liquid biopsies. - Developing novel, cell-free therapeutic delivery systems. |
The Scientist's Toolkit: Essential Reagent Solutions
Successful execution of these assays relies on a suite of specific reagents and tools.
Table 4: Key Research Reagent Solutions for Apoptosis Detection
| Reagent / Assay Kit | Function and Application | Experimental Context |
|---|---|---|
| Lysis Buffer (Tris, EDTA, Triton X-100) [12] | Disrupts cell and nuclear membranes to release fragmented DNA while leaving intact chromatin in the pellet. | Essential for the first step of the DNA ladder assay. |
| DNase-free RNase & Proteinase K [12] | Enzymes that digest RNA and proteins, respectively, to purify genomic DNA for a clean signal in gel electrophoresis. | Used in the DNA precipitation and purification stage of the DNA ladder assay. |
| Annexin V-FITC / PI Staining Kit [117] [116] | Annexin V binds to externalized phosphatidylserine (early apoptosis). Propidium iodide (PI) stains DNA in cells with compromised membranes (late apoptosis/necrosis). | Used in flow cytometry or image cytometry to distinguish between live, early apoptotic, and late apoptotic/necrotic cells. Can also be applied to AB characterization. |
| Caspase Activity Assay Kits [116] | Fluorogenic or colorimetric substrates that measure the enzymatic activity of key caspases (e.g., 3, 8, 9), an early event in apoptosis. | Used for early and mechanistic detection of apoptosis in response to drug treatments. |
| JC-1 Dye [117] [116] | A mitochondrial membrane potential dye. In healthy cells, it forms aggregates (red fluorescence); in apoptotic cells, it remains as monomers (green fluorescence). | Used to detect the loss of mitochondrial membrane potential, a key event in the intrinsic apoptotic pathway. |
| Antibodies to Apoptotic Markers (e.g., Caspase-3, Cytochrome c, Bcl-2 family) [116] | Enable detection and quantification of key apoptotic proteins via Western blot, immunohistochemistry, or flow cytometry. | Used for mechanistic studies to determine which apoptotic pathway is being activated. |
The choice between DNA fragmentation assays and apoptotic body analysis is not a matter of one being superior to the other, but rather which is most appropriate for the specific research question and context. DNA fragmentation assays remain a robust, accessible, and cost-effective method for definitively confirming apoptosis in bulk cell populations, making them a staple in initial drug screening and basic research. In contrast, the analysis of apoptotic bodies offers a more nuanced and powerful approach, unlocking opportunities in non-invasive diagnostics through liquid biopsy and pioneering new avenues in cell-free regenerative medicine and targeted drug delivery. As the field of regulated cell death continues to evolve, the integration of both methods will provide a more comprehensive understanding of therapeutic effects and disease mechanisms, ultimately accelerating the development of novel diagnostic and therapeutic strategies.
Conclusion
DNA fragmentation assays and apoptotic body formation analysis provide complementary yet distinct windows into the process of programmed cell death. While DNA laddering and TUNEL assays offer direct biochemical evidence of a key apoptotic hallmark, the study of apoptotic bodies reveals critical morphological and functional outcomes of cellular disassembly. The choice between these methods is not a matter of superiority but of purpose; DNA fragmentation assays are well-suited for bulk population analysis and confirmation of apoptosis, whereas apoptotic body characterization is indispensable for understanding cell-to-cell communication and immunogenic consequences. Future directions will likely involve greater integration of these techniques with single-cell 'omics' and high-content imaging, paving the way for more personalized applications in clinical diagnostics, monitoring therapeutic responses in oncology, and assessing male fertility, ultimately enhancing the precision of biomedical research and drug development.
References
- 1. Apoptosis (Programmed Cell Death) [https://my.clevelandclinic.org/health/articles/apoptosis]
- 2. Apoptosis [https://en.wikipedia.org/wiki/Apoptosis]
- 3. Apoptosis [https://www.genome.gov/genetics-glossary/apoptosis]
- 4. A quantitative real-time approach for discriminating ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5253725/]
- 5. Apoptosis and Beyond: Cytometry in Studies of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3263828/]
- 6. An alternative, nonapoptotic form of programmed cell death [https://pmc.ncbi.nlm.nih.gov/articles/PMC18926/]
- 7. Impact of cryopreservation agents on sperm quality, DNA ... [https://www.nature.com/articles/s41598-025-15456-0]
- 8. Multimodal Holographic Microscopy: Distinction between ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4372376/]
- 9. The Quantitative-Phase Dynamics of Apoptosis and Lytic ... [https://www.nature.com/articles/s41598-020-58474-w]
- 10. The comings and goings of cell-free DNA: Biological and ... [https://www.sciencedirect.com/science/article/pii/S2666634025003538]
- 11. The role of the DFF40/ CAD in genomic stability endonuclease [https://pubmed.ncbi.nlm.nih.gov/33387146/]
- 12. Apoptosis DNA fragmentation analysis protocol [https://www.abcam.com/en-us/technical-resources/protocols/apoptosis-dna-fragmentation]
- 13. Research progress on morphology and mechanism of ... [https://www.nature.com/articles/s41419-024-06712-8]
- 14. The Biology of Cell-free DNA Fragmentation and the Roles ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7010979/]
- 15. Cell-free DNA in blood circulation is generated ... [https://www.sciencedirect.com/science/article/abs/pii/S0006291X19312057]
- 16. A Key PARP-1-Regulated Event in Apoptosis - NCBI - NIH [https://www.ncbi.nlm.nih.gov/books/NBK6354/]
- 17. Morphological assessment of apoptosis [https://www.sciencedirect.com/science/article/abs/pii/S1046202307002150]
- 18. Morphologic and biochemical hallmarks of apoptosis [https://pubmed.ncbi.nlm.nih.gov/10728374/]
- 19. Circulating cell-free DNA fragmentation is a stepwise and ... [https://bmcbiol.biomedcentral.com/articles/10.1186/s12915-023-01752-6]
- 20. Apoptosis Market Size, Share and Opportunities, 2025-2032 [https://www.coherentmarketinsights.com/industry-reports/apoptosis-market]
- 21. The formation of the 'footprint of death' as a mechanism for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12528690/]
- 22. The BCL-2 protein family: from discovery to drug ... [https://www.nature.com/articles/s41418-025-01481-z]
- 23. Apoptotic bodies in phytoplankton suggest evolutionary ... [https://www.nature.com/articles/s41467-025-63956-4]
- 24. 50 years on and still very much alive: 'Apoptosis [https://www.nature.com/articles/s41416-022-02020-0]
- 25. Biochemistry, Extrinsic Pathway of Apoptosis - StatPearls - NCBI [https://www.ncbi.nlm.nih.gov/books/NBK560811/]
- 26. Apoptosis: A Comprehensive Overview of Signaling Pathways ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11592877/]
- 27. Caspase Functions in Cell Death and Disease - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3683896/]
- 28. Caspases as master regulators of programmed cell death [https://www.nature.com/articles/s12276-025-01470-9]
- 29. Mechanisms of Cell Death: Apoptosis - CST Blog [https://blog.cellsignal.com/mechanisms-of-cell-death-apoptosis]
- 30. DNA fragmentation and morphological changes in ... [https://www.sciencedirect.com/science/article/abs/pii/S0006291X02005880]
- 31. The formation of the 'footprint of death' as a mechanism for ... [https://www.nature.com/articles/s41467-025-64206-3]
- 32. An overview of apoptosis assays detecting DNA ... [https://pubmed.ncbi.nlm.nih.gov/30022463/]
- 33. Apoptotic Bodies: Mechanism of Formation, Isolation and ... [https://pubmed.ncbi.nlm.nih.gov/33779914/]
- 34. Apoptosis and apoptotic body: disease message and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6340950/]
- 35. Determining the contents and cell origins of apoptotic ... [https://www.nature.com/articles/s41598-017-14305-z]
- 36. Comparison of several techniques for the detection ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6495494/]
- 37. Comparison of comet-based approaches to assess ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10322757/]
- 38. Apoptotic cell clearance: basic biology and therapeutic potential [https://pmc.ncbi.nlm.nih.gov/articles/PMC4040260/]
- 39. Stem cells tightly regulate dead cell clearance to maintain ... [https://www.nature.com/articles/s41586-024-07855-6]
- 40. DNA Laddering - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/dna-laddering]
- 41. An update to DNA ladder assay for apoptosis detection [https://pmc.ncbi.nlm.nih.gov/articles/PMC4401164/]
- 42. Emerging Electrochemical Approaches for the Early Detection ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12355238/]
- 43. Flow cytometry-based apoptosis detection - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3863590/]
- 44. High-Resolution Analysis of DNA Electrophoretic ... [https://www.mdpi.com/2297-8739/12/11/296]
- 45. Evaluation of variability in cell-free DNA extraction ... [https://www.nature.com/articles/s41598-025-06563-z]
- 46. TUNEL Assay - an overview [https://www.sciencedirect.com/topics/medicine-and-dentistry/tunel-assay]
- 47. TUNEL Assays | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/cell-analysis/cell-viability-and-regulation/apoptosis/tunel-assays.html]
- 48. TdT/Cas12a-based biosensor for sensitive detection of ... [https://www.nature.com/articles/s41598-025-09768-4]
- 49. Inter- and intra-laboratory standardization of TUNEL assay ... [https://pubmed.ncbi.nlm.nih.gov/28245344/]
- 50. The correlation between sperm DNA methylation and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11882583/]
- 51. An alternative approach of TUNEL assay to specifically ... [https://link.springer.com/article/10.1007/s00418-024-02306-9]
- 52. Precise detection of sperm DNA breaks and AP sites based ... [https://www.sciencedirect.com/science/article/abs/pii/S000326702500902X]
- 53. CYTOMETRY OF APOPTOSIS. HISTORICAL ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3476471/]
- 54. Measuring apoptosis by microscopy and flow cytometry [https://www.sciencedirect.com/science/article/abs/pii/S1046202313000194]
- 55. Tailoring of apoptotic bodies for diagnostic and therapeutic ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-024-05451-w]
- 56. Flow cytometry-based quantitative analysis of cellular ... [https://www.sciencedirect.com/science/article/pii/S2405844024096968]
- 57. Comparison of Apoptosis Detection Markers Combined ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2716774/]
- 58. Comparison of in-vitro immunomodulatory capacity ... [https://www.sciencedirect.com/science/article/pii/S1567576925004709]
- 59. Programmed cell death detection methods - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC9308588/]
- 60. Confocal Microscopy Provides Powerful Imaging Insights [https://www.the-scientist.com/confocal-microscopy-provides-powerful-imaging-insights-73485]
- 61. A new combined approach using confocal and scanning ... [https://www.sciencedirect.com/science/article/pii/S2352409X19304262]
- 62. Comparison of Several Techniques for the Detection ... [https://www.creative-bioarray.com/support/comparison-of-several-techniques-for-the-detection-of-apoptotic-cells.htm]
- 63. Lost Data in Electron Microscopy [https://www.mdpi.com/2624-8549/7/5/160]
- 64. FLUOVIEW FV5000 Redefines the Boundaries of Confocal ... [https://evidentscientific.com/en/insights/innovations-in-microscopy-redefining-boundaries-of-confocal-and-multiphoton-imaging]
- 65. Quantitative confocal microscopy: Beyond a pretty picture [https://www.sciencedirect.com/science/article/abs/pii/B9780124201385000070]
- 66. "Comparison of Scanning Electron Microscopy and ... [https://digitalcommons.calpoly.edu/theses/2764/]
- 67. Comparison of TUNEL, SCSA, SCD Test and COMET Assay [https://pubmed.ncbi.nlm.nih.gov/41009547/]
- 68. Programmed cell death detection methods: a systematic ... [https://link.springer.com/article/10.1007/s10495-022-01735-y]
- 69. Double-stranded sperm DNA fragmentation assessed ... [https://www.sciencedirect.com/science/article/pii/S1472648325003918]
- 70. Effect of sperm DNA fragmentation on embryo euploidy rate in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12625451/]
- 71. Predictive model of sperm DNA fragmentation in infertile ... [https://www.frontiersin.org/journals/endocrinology/articles/10.3389/fendo.2025.1675168/full]
- 72. Cross-dataset pan-cancer detection by correlating cell-free ... [https://www.nature.com/articles/s41467-025-66503-3]
- 73. A Novel Cell-Free DNA Fragmentomic Assay and Its ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12606779/]
- 74. Cytometry in Cell Necrobiology Revisited. Recent Advances ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2975392/]
- 75. Necroptosis, pyroptosis and apoptosis: an intricate game ... [https://www.nature.com/articles/s41423-020-00630-3]
- 76. Apoptosis, Pyroptosis, and Necrosis: Mechanistic Description ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1087413/]
- 77. Identification of the pyroptosis, apoptosis, and necroptosis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12211273/]
- 78. Apoptotic DNA Fragmentation - an overview [https://www.sciencedirect.com/topics/neuroscience/apoptotic-dna-fragmentation]
- 79. A comparative study of apoptosis, pyroptosis, necroptosis, and ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0299577]
- 80. Apoptotic DNA fragmentation [https://en.wikipedia.org/wiki/Apoptotic_DNA_fragmentation]
- 81. Annexin V staining assay protocol for apoptosis [https://www.abcam.com/en-us/technical-resources/protocols/annexin-v-for-apoptosis]
- 82. Major DNA fragmentation is a late event in apoptosis [https://pubmed.ncbi.nlm.nih.gov/9212818/]
- 83. Annexin V Staining Protocol for Flow Cytometry [https://www.thermofisher.com/us/en/home/references/protocols/cell-and-tissue-analysis/protocols/annexin-v-staining-flow-cytometry.html]
- 84. Identification of PANoptosis-related genes as biomarkers in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12333936/]
- 85. Troubleshooting DNA Ladders [https://www.goldbio.com/blogs/articles/troubleshooting-dna-ladders?srsltid=AfmBOorLwSegEVyI5fyVwnu9ZDzmhwKyjmpM9w43GI9hhDioCtI3M4Lq]
- 86. Troubleshooting Common Electrophoresis Problems and ... [https://www.labx.com/resources/troubleshooting-common-electrophoresis-problems-and-artifacts/5539]
- 87. Rapid quantitative analysis of double-stranded plasmid ... [https://www.nature.com/articles/s41598-025-85132-w]
- 88. Cryopreservation increases sperm DNA fragmentation in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12523456/]
- 89. Impact of cryopreservation agents on sperm quality, DNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12358582/]
- 90. Sperm freezing damage: the role of regulated cell death [https://www.nature.com/articles/s41420-024-02013-3]
- 91. DNA fragmentation in apoptosis | Cell Research [https://www.nature.com/articles/7290049]
- 92. Guidelines for Regulated Cell Death Assays: A Systematic ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2021.634690/full]
- 93. Guidelines for Regulated Cell Death Assays - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC7970050/]
- 94. The role of apoptosis in cryopreserved animal oocytes and ... [https://www.sciencedirect.com/science/article/abs/pii/S0093691X21002429]
- 95. Correlation of the sperm DNA fragmentation index with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12101436/]
- 96. North America Apoptosis Assay Market Trends 2025–2034 [https://www.gminsights.com/industry-analysis/north-america-apoptosis-assay-market]
- 97. Apoptosis detection via automated algorithms to analyze ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7703573/]
- 98. Electronic detection of apoptotic cells on a microchip [https://www.sciencedirect.com/science/article/abs/pii/S0956566324007565]
- 99. An ultra-sensitive assay using cell-free DNA fragmentomics for ... [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-022-01594-w]
- 100. Apoptotic extracellular vesicles: emerging biomarkers for ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1619456/full]
- 101. Monitoring and Assessment of Circulating Tumor DNA in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12550271/]
- 102. Apoptotic cells promote circulating tumor cell survival and ... [https://www.nature.com/articles/s42003-025-08541-7]
- 103. Sensitive pathogen DNA detection by a multi-guide RNA ... [https://www.nature.com/articles/s41467-025-63094-x]
- 104. The Utility of Sperm DNA Fragmentation as a Diagnostic ... [https://www.mdpi.com/1422-0067/26/13/6314]
- 105. Cytoplasmic condensation is both necessary and sufficient ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2561226/]
- 106. Initiation of DNA Fragmentation during Apoptosis Induces ... [https://www.sciencedirect.com/science/article/pii/S0021925818300504]
- 107. Isolation and Quantification of Blood Apoptotic Bodies, a Non ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7395395/]
- 108. Sperm DNA Fragmentation and Yoga: A Narrative Review ... [https://www.sciencedirect.com/science/article/abs/pii/S0344033825004625]
- 109. Sperm DNA Fragmentation Impairs Early Embryo ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12386859/]
- 110. NINJ1 oligomerises on large apoptotic cell-derived ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1599809/full]
- 111. Assessment of apoptosis and necrosis by DNA fragmentation and morphological criteria - PubMed [https://pubmed.ncbi.nlm.nih.gov/18228342/]
- 112. Comparative Analysis of Different Methodological ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1867030/]
- 113. Apoptotic bodies in phytoplankton suggest evolutionary ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12462507/]
- 114. Impact of sperm DNA fragmentation index on assisted ... [https://pubmed.ncbi.nlm.nih.gov/39906032/]
- 115. Apoptosis detection: a purpose-dependent approach ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8208110/]
- 116. Apoptosis Assays | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/cell-analysis/cell-viability-and-regulation/apoptosis.html]
- 117. Apoptosis Detection: Methods, Assays & Analysis ... [https://www.revvity.com/ask/apoptosis]