Intrinsic vs. Extrinsic Apoptosis: Molecular Mechanisms, Caspase Activation Pathways, and Therapeutic Implications
This article provides a comprehensive analysis of the molecular mechanisms governing caspase activation in the intrinsic (mitochondrial) and extrinsic (death receptor) apoptosis pathways.
Intrinsic vs. Extrinsic Apoptosis: Molecular Mechanisms, Caspase Activation Pathways, and Therapeutic Implications
Abstract
This article provides a comprehensive analysis of the molecular mechanisms governing caspase activation in the intrinsic (mitochondrial) and extrinsic (death receptor) apoptosis pathways. Tailored for researchers and drug development professionals, it details the distinct initiator caspases, activating complexes, and regulatory controls for each pathway. The scope extends from foundational concepts and key experimental methods to common challenges in pathway interrogation, validation techniques, and the growing understanding of pathway crosstalk. With a focus on clinical relevance, the content also explores the therapeutic potential of targeting these caspase activation pathways in diseases such as cancer and neurodegeneration.
Core Mechanisms: Decoding the Initiator Caspases and Activating Complexes in Apoptotic Pathways
Apoptosis, a form of programmed cell death (PCD), is an energy-dependent, biochemically-mediated process essential for development, tissue homeostasis, and the elimination of damaged or infected cells [1] [2]. The controlled dismantling of a cell via apoptosis is primarily executed by a family of cysteine proteases known as caspases, which cleave their substrates at specific aspartic acid residues [3]. The activation of these caspases represents a critical commitment point in the cell's demise and occurs through two principal, well-defined initiation routes: the intrinsic and extrinsic pathways [4] [2]. While these pathways are distinct in their initial triggers and early signaling events, they ultimately converge on the activation of effector caspases that orchestrate the characteristic morphological changes of apoptosis, including cell shrinkage, chromatin condensation, DNA fragmentation, and formation of apoptotic bodies [5] [6].
This review delineates the precise triggers and molecular mechanisms of the intrinsic and extrinsic pathways of apoptosis, with a specific focus on the hierarchical activation of caspases. Understanding the nuances of these pathways is of paramount importance for researchers and drug development professionals aiming to modulate cell death in pathological contexts such as cancer, autoimmune disorders, and neurodegenerative diseases [5] [7].
The Intrinsic Pathway: Activation by Intracellular Stress Signals
The intrinsic pathway, also known as the mitochondrial pathway, is activated in response to a diverse array of internal cellular insults [4] [6]. These intracellular stressors signal that the cell is damaged beyond repair or is no longer necessary for the organism's survival.
Key Triggers of the Intrinsic Pathway
The intrinsic pathway is initiated by the following core intracellular stresses:
- DNA Damage: Caused by agents such as ionizing radiation, ultraviolet light, or chemotherapeutic drugs. This damage activates the tumor suppressor protein p53, which acts as a critical sensor and mediator [4] [8].
- Oxidative Stress: Resulting from an overabundance of reactive oxygen species (ROS) that cause oxidative damage to cellular components [4] [7].
- Endoplasmic Reticulum (ER) Stress: Occurs due to the accumulation of misfolded proteins within the ER [3].
- Growth Factor Withdrawal: The absence of essential survival signals that normally inhibit apoptotic pathways [6].
- Hypoxia: Oxygen deprivation within the cellular microenvironment [4].
- Cellular Stress: Biochemical stress that disrupts normal cellular metabolism [6].
These triggers converge on a central regulatory checkpoint: the mitochondria.
The Bcl-2 Family: Gatekeepers of Mitochondrial Integrity
The initiation and regulation of the intrinsic pathway are governed by the Bcl-2 protein family, which functions as a critical molecular switch determining cellular fate [8] [6]. The balance between pro-apoptotic and anti-apoptotic members of this family dictates whether a cell will survive or undergo apoptosis.
Table 1: Key Members of the Bcl-2 Protein Family
| Function | Protein Members | Mechanism of Action |
|---|---|---|
| Anti-apoptotic | Bcl-2, Bcl-XL, Bcl-W, Mcl-1 [8] [7] | Preserve mitochondrial outer membrane integrity by preventing pore formation and cytochrome c release. |
| Pro-apoptotic Effectors | Bax, Bak [8] [2] | Form pores in the mitochondrial outer membrane, leading to Mitochondrial Outer Membrane Permeabilization (MOMP). |
| Pro-apoptotic Initiators (BH3-only) | Bid, Bim, Puma, Noxa, Bad [8] [7] | Sense cellular stress and directly or indirectly activate Bax/Bak by neutralizing anti-apoptotic Bcl-2 proteins. |
Upon activation by intracellular stress, BH3-only proteins like Puma and Noxa are transcriptionally upregulated by p53 [8]. These proteins then inhibit the anti-apoptotic members Bcl-2 and Bcl-XL. This inhibition frees the pro-apoptotic effectors Bax and Bak, which oligomerize and integrate into the outer mitochondrial membrane, leading to MOMP [8] [2]. MOMP is widely considered the "point of no return" for the intrinsic apoptotic pathway [8].
Caspase Activation via the Apoptosome
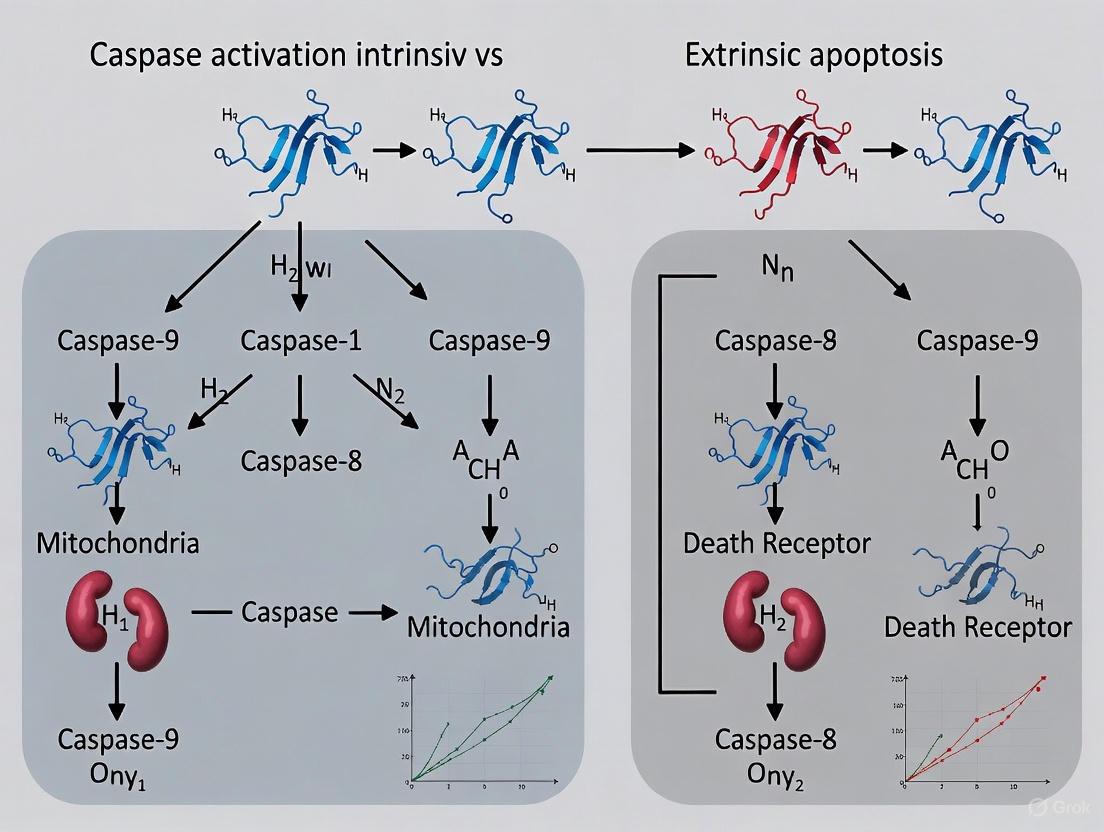
MOMP triggers the release of several pro-apoptotic proteins from the mitochondrial intermembrane space into the cytosol, most critically cytochrome c [4] [2]. In the cytosol, cytochrome c binds to the protein Apoptotic Protease-Activating Factor 1 (Apaf-1) in the presence of dATP/ATP. This binding induces a conformational change in Apaf-1, prompting it to oligomerize and form a wheel-like signaling platform known as the apoptosome [4] [9]. The apoptosome recruits and activates the initiator caspase-9 through caspase recruitment domain (CARD) interactions [3] [9]. Once activated within the apoptosome, caspase-9 cleaves and activates the downstream effector caspases, primarily caspase-3 and caspase-7, which then proceed to dismantle the cell by cleaving a multitude of structural and functional proteins [10] [9].
The following diagram illustrates the sequence of events in the intrinsic apoptosis pathway:
Figure 1: The Intrinsic Apoptosis Pathway. Intracellular stresses activate p53 and/or BH3-only proteins, leading to Bax/Bak-mediated MOMP, cytochrome c release, apoptosome formation, and caspase-9-dependent activation of effector caspases.
The Extrinsic Pathway: Activation by Extracellular Death Ligands
In contrast to the intrinsic pathway, the extrinsic pathway is initiated by external signals that originate from outside the target cell [4] [1]. This pathway is most often triggered by immune cells, such as Cytotoxic T Lymphocytes (CTLs) and Natural Killer (NK) cells, to eliminate virally infected, damaged, or cancerous cells [1].
Key Triggers and Receptor-Ligand Interactions
The canonical extrinsic pathway begins with the binding of specific extracellular death ligands to their corresponding cell-surface death receptors (DRs) [1]. These receptors belong to the Tumor Necrosis Factor Receptor (TNFR) superfamily and are characterized by a conserved intracellular protein-protein interaction motif known as the "death domain" (DD) [8] [1].
Table 2: Principal Death Receptors and Their Ligands
| Death Receptor | Ligand | Primary Source of Ligand |
|---|---|---|
| Fas (CD95) | FasL (Fas Ligand) | Activated T lymphocytes, NK cells [1] |
| TNFR1 (Tumor Necrosis Factor Receptor 1) | TNF-α (Tumor Necrosis Factor-alpha) | Activated macrophages [2] |
| DR4/TRAIL-R1, DR5/TRAIL-R2 | TRAIL (TNF-Related Apoptosis-Inducing Ligand) | Immune cells; used therapeutically [9] [2] |
Caspase Activation via the Death-Inducing Signaling Complex (DISC)
The binding of a trimeric death ligand to its receptor induces receptor trimerization and clustering of the intracellular death domains [4]. This cluster serves as a platform to recruit the adapter protein FADD (Fas-Associated protein with Death Domain) [9] [1]. FADD, in turn, recruits the initiator caspase-8 (and in humans, caspase-10) via interactions between death effector domains (DEDs) [9] [1]. This assembly of the death receptor, FADD, and procaspase-8 forms the Death-Inducing Signaling Complex (DISC) [8] [1].
Within the DISC, the high local concentration of procaspase-8 molecules leads to their autocatalytic activation through "induced proximity" [10] [9]. Active caspase-8 then propagates the death signal by directly cleaving and activating the effector caspases, caspase-3 and caspase-7, committing the cell to apoptosis [10] [1].
The following diagram illustrates the sequence of events in the extrinsic apoptosis pathway:
Figure 2: The Extrinsic Apoptosis Pathway. Extracellular death ligands bind to cell-surface death receptors, leading to DISC formation and caspase-8 activation. Caspase-8 can directly activate effector caspases or, in some cells, amplify the signal via Bid cleavage and the intrinsic mitochondrial pathway.
Pathway Amplification and Cross-Talk
The efficiency of the extrinsic pathway varies between cell types. In so-called Type I cells, the amount of active caspase-8 generated at the DISC is sufficient to directly activate effector caspases [8]. However, in Type II cells, the DISC formation is weaker, and the apoptotic signal requires amplification. This is achieved through molecular cross-talk with the intrinsic pathway [8] [1]. In these cells, active caspase-8 cleaves the BH3-only protein Bid, converting it into its active truncated form, tBid [8] [2]. tBid then translocates to the mitochondria, where it activates Bax and Bak, inducing MOMP, cytochrome c release, and apoptosome-mediated activation of caspase-9. This engages the intrinsic pathway to amplify the initial death signal and ensure robust activation of effector caspases [1] [2].
Comparative Analysis: Intrinsic vs. Extrinsic Pathways
Table 3: Comparative Overview of Intrinsic and Extrinsic Apoptosis Pathways
| Feature | Intrinsic Pathway | Extrinsic Pathway |
|---|---|---|
| Primary Trigger | Intracellular stress (DNA damage, oxidative stress, ER stress) [4] [6] | Extracellular death ligands (FasL, TRAIL, TNF-α) [4] [1] |
| Initial Caspase | Caspase-9 [10] [9] | Caspase-8, Caspase-10 [10] [1] |
| Key Signaling Complex | Apoptosome (Apaf-1 + Cytochrome c) [4] [9] | DISC (Death Receptor + FADD + Caspase-8) [8] [1] |
| Central Regulatory Proteins | Bcl-2 Family (Bax/Bak, BH3-only) [8] [6] | FADD, FLIP (inhibitor) [4] [1] |
| Key Organelle | Mitochondria [4] [2] | Plasma Membrane [1] |
| Cross-talk Mechanism | Not Applicable | Caspase-8-mediated cleavage of Bid [8] [2] |
Experimental Toolkit for Apoptosis Research
Studying these pathways requires a suite of well-established reagents and methodologies to detect activation markers, inhibit specific components, and measure functional outcomes.
Key Research Reagents and Assays
Table 4: Essential Reagents and Methodologies for Apoptosis Research
| Reagent / Assay | Target / Function | Experimental Application |
|---|---|---|
| Annexin V Staining | Phosphatidylserine (PS) externalization [5] | Flow cytometry or microscopy to detect early apoptotic cells. |
| TUNEL Assay | DNA fragmentation [4] | Labeling of cleaved DNA ends to detect late-stage apoptosis in tissue sections or cells. |
| Caspase Activity Assays | Cleavage of specific synthetic substrates (e.g., DEVD for caspase-3) [10] | Fluorometric or colorimetric measurement of caspase activation in cell lysates. |
| Western Blotting | Cleaved caspases, PARP cleavage, Bcl-2 family proteins, cytochrome c release [10] | Detection of protein expression, cleavage, and subcellular localization. |
| Mitochondrial Membrane Potential Probes (JC-1, TMRE) | Mitochondrial membrane integrity (ΔΨm) [4] | Flow cytometry or fluorescence microscopy to detect MOMP. |
| zVAD-fmk | Pan-caspase inhibitor [5] [10] | To confirm caspase-dependent apoptosis. |
| Small Molecule BH3 Mimetics (e.g., ABT-199/Venetoclax) | Inhibit anti-apoptotic Bcl-2 proteins [5] [8] | To sensitize cells to intrinsic apoptosis, used therapeutically in cancer. |
| Recombinant Death Ligands (e.g., TRAIL, FasL) | Activate death receptors [8] [1] | To induce the extrinsic pathway in vitro. |
Detailed Experimental Protocol: Discriminating Pathway Activation
To determine whether apoptosis is occurring via the intrinsic or extrinsic pathway, researchers can employ a combination of the following methodological approaches:
1. Assessing Initiator Caspase Activation:
- Methodology: Perform Western blot analysis on cell lysates treated with an apoptotic stimulus.
- Key Targets:
- Procaspase-8 Cleavage: The appearance of cleaved, active caspase-8 fragments (p43/p41 and p18) is a hallmark of DISC formation and extrinsic pathway activation [10] [1].
- Procaspase-9 Cleavage: The cleavage of procaspase-9 to its active form indicates apoptosome formation and intrinsic pathway activation [10] [9].
- Interpretation: Preferential cleavage of caspase-8 suggests extrinsic activation, while cleavage of caspase-9 points to intrinsic activation. In Type II cells, both may be observed due to cross-talk.
2. Measuring Mitochondrial Involvement:
- Methodology:
- Subcellular Fractionation & Western Blot: Fractionate cells into cytosolic and mitochondrial fractions and probe for cytochrome c. Its presence in the cytosol is a definitive marker of MOMP and intrinsic pathway engagement [4] [2].
- Mitochondrial Membrane Potential Assay: Use fluorescent dyes like JC-1. A collapse in ΔΨm (shift from red to green fluorescence) indicates MOMP and is characteristic of the intrinsic pathway [4].
- Interpretation: Loss of ΔΨm or cytosolic cytochrome c release confirms the involvement of the mitochondrial pathway, which is central to intrinsic apoptosis and the Type II extrinsic amplification loop.
3. Genetic and Pharmacological Inhibition:
- Methodology: Use specific inhibitors or siRNA/gene knockout models prior to applying an apoptotic stimulus.
- Extrinsic Pathway Inhibition: Utilize caspase-8-specific inhibitors (e.g., Z-IETD-FMK) or FADD-deficient cells [10] [1]. If apoptosis is blocked, the pathway is likely extrinsic-dependent.
- Intrinsic Pathway Inhibition: Use the caspase-9 inhibitor Z-LEHD-FMK or cells deficient in Apaf-1 or Bax/Bak [10] [9]. If apoptosis is suppressed, the intrinsic pathway is critical.
- Interpretation: This functional approach identifies which initiator caspase and signaling complex are necessary for cell death in a given context.
The intrinsic and extrinsic pathways of apoptosis represent two sophisticated, evolutionarily conserved molecular machines for initiating programmed cell death. They are distinguished by their fundamental triggers: the intrinsic pathway responds to a panorama of intracellular stresses that signal internal damage, while the extrinsic pathway is activated by extracellular ligands that communicate a death sentence from the immune system. Both pathways culminate in the activation of a cascade of caspases, yet they achieve this through distinct initiator caspases (-9 and -8, respectively) and unique multi-protein activation platforms (the apoptosome and the DISC). The intricate cross-talk between these pathways, particularly through the cleavage of Bid, ensures a robust and fail-safe cellular response. For the research and drug development community, a precise understanding of these triggers and mechanisms is not merely academic; it provides the essential foundation for developing targeted therapies that can selectively induce apoptosis in cancer cells or protect healthy cells from inappropriate death in degenerative diseases.
The onset of intrinsic apoptosis is governed by the formation of a critical signaling platform known as the apoptosome, which activates initiator caspases to execute programmed cell death. This complex represents a fundamental control point where cellular damage is converted into a proteolytic cascade, culminating in the dismantling of the cell. Within the broader context of caspase activation research, the apoptosome exemplifies the "induced proximity" model of initiator caspase activation, which stands in contrast to the death-induced signaling complex (DISC)-mediated activation in the extrinsic pathway [11] [12]. The apoptosome specifically activates caspase-9, which then propagates the death signal by cleaving and activating effector caspases-3 and -7 [11]. Dysregulation of this complex contributes to numerous human diseases; insufficient apoptosome formation can drive carcinogenesis and therapy resistance, while excessive activity may underlie degenerative conditions [13] [14]. This technical guide examines the molecular architecture, assembly mechanisms, and regulatory controls of the apoptosome, providing researchers with a comprehensive resource for understanding this essential component of the intrinsic apoptosis pathway.
Molecular Composition and Assembly
Structural Components of the Apoptosome
The apoptosome is composed of apoptotic protease-activating factor 1 (Apaf-1), cytochrome c, and procaspase-9, which assemble into a heptameric complex in response to intrinsic death signals [15] [16]. Apaf-1 contains multiple structured domains that mediate apoptosome assembly and function, as detailed in Table 1.
Table 1: Domain Architecture of Human Apaf-1
| Domain | Structure | Function | Regulatory Significance |
|---|---|---|---|
| CARD | N-terminal caspase recruitment domain | Homotypic interaction with procaspase-9 CARD | Forms disk-like spiral for caspase recruitment and activation [17] [16] |
| NBD | Nucleotide-binding domain (AAA+ family) | Binds dATP/ATP | Nucleotide exchange triggers conformational change for oligomerization [15] [18] |
| HD1 | Helical domain 1 | Associates with NBD | Part of the central hub that forms oligomerization interface [15] [17] |
| WHD | Winged helix domain | Connects NBD/HD1 to HD2 | Participates in ring formation within the central hub [15] [18] |
| HD2 | Helical domain 2 | Extended arm domain | Connects central hub to WD40 repeats [15] [17] |
| WD40 Repeats | 15 repeats forming two β-propellers | Binds cytochrome c; maintains autoinhibition | Sensor domain; cytochrome c binding relieves autoinhibition [15] [17] |
In healthy cells, Apaf-1 exists as an autoinhibited monomer, with its WD40 domains folded back onto the CARD and NBD domains, preventing spontaneous oligomerization [15] [18]. This inactive state is stabilized by bound ADP/dADP [15]. The transition to an active apoptosome requires specific molecular triggers that relieve this autoinhibition.
Stepwise Assembly Mechanism
The assembly of the functional apoptosome follows a precisely regulated sequence of molecular events, illustrated in Figure 1 below.
Figure 1: Stepwise Assembly of the Human Apoptosome
Cytochrome c Release and Binding: Intrinsic apoptotic stimuli (e.g., DNA damage, oxidative stress) trigger mitochondrial outer membrane permeabilization (MOMP), leading to cytochrome c release into the cytosol [11] [7]. Cytochrome c then binds to the WD40 repeats of Apaf-1, initiating a conformational change that partially relieves autoinhibition [15].
Nucleotide Exchange: Cytochrome c binding promotes the exchange of ADP/dADP for dATP/ATP in the NBD of Apaf-1 [15] [16]. Recent structural studies using cryo-EM have revealed that dATP binding triggers extensive conformational changes that enable Apaf-1 oligomerization [15].
Oligomerization: Following nucleotide exchange, Apaf-1 undergoes a dramatic conformational shift from a compact, autoinhibited monomer to an extended conformation that oligomerizes into a heptameric, wheel-like complex [15] [16]. This complex, known as the apoptosome, measures approximately 270 Å in diameter and 75 Å in height [16].
Procaspase-9 Recruitment: The central hub of the apoptosome recruits procaspase-9 through homotypic CARD-CARD interactions between Apaf-1 and procaspase-9 [17] [16]. This recruitment leads to the formation of a CARD disk that sits atop the central platform, facilitating caspase-9 activation.
Table 2: Key Molecular Triggers in Apoptosome Assembly
| Trigger | Source | Binding Site | Functional Outcome |
|---|---|---|---|
| Cytochrome c | Mitochondrial intermembrane space | WD40 repeats of Apaf-1 | Relieves autoinhibition; primes Apaf-1 for oligomerization [15] [11] |
| dATP/ATP | Cellular nucleotide pool | NBD of Apaf-1 | Drives conformational changes required for oligomerization [15] [18] |
| Procaspase-9 | Cytoplasmic zymogen | Apaf-1 CARD domain | Completes holo-apoptosome formation; becomes activated [16] [18] |
Structural Organization and Caspase-9 Activation
High-Resolution Architecture
Recent advances in cryo-electron microscopy have yielded near-atomic resolution structures of the human apoptosome, providing unprecedented insights into its organization [15] [16]. The overall architecture features a central hub comprised of seven Apaf-1 NBD/HD1/WHD domains arranged in a symmetric ring, with seven spokes radiating outward, each consisting of HD2 and the WD40 repeats with bound cytochrome c [15]. The CARD domains form an acentric disk-like structure above the central hub, arranged in a shallow spiral that accommodates three to four procaspase-9 CARDs [17] [16]. This creates a stoichiometric mismatch between the CARD spiral (asymmetric) and the platform (7-fold symmetric), suggesting a dynamic activation mechanism.
Mechanisms of Caspase-9 Activation
The precise mechanism by which the apoptosome activates caspase-9 remains an area of active investigation, with several models proposed based on structural and biochemical evidence, as illustrated in Figure 2 below.
Figure 2: Models of Caspase-9 Activation on the Apoptosome
The induced proximity model posits that the apoptosome serves primarily as a platform to concentrate procaspase-9 monomers, facilitating their autoactivation [13] [12]. An evolved version, the proximity-induced dimerization model, suggests that the platform promotes caspase-9 dimerization, which is essential for activation [13] [11]. In contrast, the induced conformation model proposes that direct interaction with the apoptosome induces conformational changes in caspase-9 that create a catalytically competent active site [13] [12]. Recent evidence also supports a molecular timer model, where procaspase-9 autoprocessing following activation regulates the duration of apoptosome signaling by reducing its affinity for the complex [13] [11].
Notably, caspase-9 activation occurs without proteolytic processing in the initial stages, as cleavage is not required for enzymatic activity [11] [12]. The uncleaved caspase-9 homodimer represents the active form on the apoptosome, with proteolytic processing serving to regulate the caspase-9-apoptosome interaction rather than directly enabling activation [13] [11].
Regulatory Mechanisms
Endogenous Regulators
Apoptosome function is tightly controlled through multiple regulatory mechanisms to ensure appropriate apoptotic responses. As summarized in Table 3, these include phosphorylation, endogenous inhibitors, and alternative splicing.
Table 3: Endogenous Regulators of Caspase-9 and Apoptosome Activity
| Regulator | Type | Mechanism of Action | Functional Outcome |
|---|---|---|---|
| ERK1/2 | Kinase | Phosphorylates caspase-9 at Thr125 | Inhibits caspase-9 processing and activity [13] |
| DYRK1A | Kinase | Phosphorylates caspase-9 at Thr125 | Suppresses caspase-9 activation [13] |
| CDK1-cyclinB1 | Kinase complex | Phosphorylates caspase-9 at Thr125 | Cell cycle-dependent regulation of apoptosis [13] |
| p38α | Kinase | Phosphorylates caspase-9 at Thr125 | Stress-responsive regulation [13] |
| XIAP | Inhibitor protein | BIR3 domain binds processed caspase-9 | Direct inhibition of caspase-9 activity [14] [11] |
| Alternative Splicing | mRNA processing | Generates caspase-9 variants (e.g., caspase-9b) | Dominant-negative regulation of apoptosis [13] |
Phosphorylation at Thr125 represents a key inhibitory mechanism, located in the hinge region near the N-terminus of the caspase-9 large subunit [13]. While this phosphorylation does not prevent caspase-9 recruitment to Apaf-1, it suppresses caspase-9 processing and activity, potentially by serving as a dominant-negative regulator that modulates recruitment of non-phosphorylated caspase-9 to the apoptosome [13].
Evolutionary Variations
Comparative analysis of apoptosomes across model organisms reveals significant evolutionary divergence in composition and regulation (Table 4), providing insights into conserved and species-specific features.
Table 4: Comparative Analysis of Apoptosomes Across Species
| Organism | Apaf-1 Homolog | Oligomeric State | Caspase Activated | Cytochrome c Requirement | Unique Features |
|---|---|---|---|---|---|
| H. sapiens | Apaf-1 | Heptamer | Caspase-9 | Required | WD40 domains bind cytochrome c; CARD disk spiral [17] [16] |
| D. melanogaster | Dark/Dapaf-1/Hac-1 | Octamer | Dronc | Not required | Stable double-ring structure; different regulation [17] [18] |
| C. elegans | CED-4 | Tetramer of dimers | CED-3 | Not applicable | No WD40 domains; regulated by CED-9 binding [17] [18] |
The C. elegans CED-4 apoptosome represents the simplest architecture, functioning as a tetramer of dimers without WD40 domains and directly regulated through interaction with CED-9 [17] [18]. In Drosophila, Dark forms an octameric apoptosome that activates Dronc, generally without requiring cytochrome c, though tissue-specific requirements exist [17] [18]. These evolutionary differences highlight the adaptability of the core apoptosis machinery while maintaining the fundamental function of caspase activation.
Experimental Analysis and Research Tools
Key Methodologies for Apoptosome Study
The investigation of apoptosome structure and function relies on specialized biochemical and structural approaches, with cryo-electron microscopy revolutionizing our understanding in recent years.
Table 5: Key Experimental Protocols for Apoptosome Research
| Method | Application | Key Steps | Technical Considerations |
|---|---|---|---|
| Cryo-EM Structure Determination | High-resolution apoptosome architecture [15] [16] | 1. Apoptosome assembly with Apaf-1, cytochrome c, dATP2. Grid preparation and vitrification3. Data collection on cryo-EM4. Single-particle analysis and 3D reconstruction | Resolution not isotropic (3-4Å hub vs. 4.5-10Å arms); enables atomic modeling of key interfaces [15] [16] |
| Apoptosome Assembly & Activity Assay | Functional analysis of apoptosome formation and caspase activation [16] | 1. Express and purify full-length Apaf-12. Assemble with cytochrome c, dATP, procaspase-93. Fractionate by size exclusion chromatography4. Measure caspase-9 activity (LEHDase assay) | Requires proper nucleotide conditions; activity measurements confirm functional assembly [16] |
| Stoichiometry Analysis | Determining component ratios in active complex [16] [18] | 1. Gradient centrifugation of apoptosome2. SDS-PAGE and quantitative staining3. Mass spectrometry analysis4. Biochemical quantification | Reveals 3-5 procaspase-9 molecules per apoptosome on average; suggests dynamic recruitment [16] |
The protocol for cryo-EM structure determination has been particularly transformative, enabling researchers to visualize the apoptosome at near-atomic resolution (3.8-4.1 Å) [15] [16]. This approach involves assembling the apoptosome from purified Apaf-1, cytochrome c, and dATP, followed by vitrification and imaging under cryogenic conditions. Single-particle analysis of tens to hundreds of thousands of particles allows high-resolution reconstruction, revealing molecular details of cytochrome c binding, nucleotide coordination, and CARD disk formation [15] [16].
Essential Research Reagents
Table 6: Key Research Reagents for Apoptosome Studies
| Reagent | Function/Application | Key Features & Utility |
|---|---|---|
| Recombinant Apaf-1 | Structural and functional studies | Full-length protein required for proper assembly; often expressed in baculovirus system [15] |
| Cytochrome c | Apoptosome assembly trigger | Horse cytochrome c commonly used; binds WD40 repeats to relieve autoinhibition [15] [16] |
| dATP/ATP | Nucleotide cofactor | dATP preferred for efficient assembly; drives conformational changes [15] [18] |
| Procaspase-9 | Apoptosome substrate | Two-chain form used in structural studies; CARD domain essential for recruitment [16] |
| LEHD-AFC | Caspase-9 activity substrate | Fluorogenic peptide for measuring enzymatic activity (LEHDase assay) [16] |
| Venetoclax (ABT-199) | BCL-2 inhibitor research tool | Induces intrinsic apoptosis; demonstrates therapeutic targeting of pathway [14] |
Therapeutic Implications and Research Applications
Disease Associations and Therapeutic Targeting
Dysregulation of the apoptosome contributes to various human diseases. In cancer, reduced caspase-9 activity and Apaf-1 expression are associated with resistance to chemotherapeutic agents in head and neck squamous cell carcinoma and testicular cancer [13]. Polymorphisms in the CASPASE-9 gene have been linked to increased susceptibility to lung, bladder, pancreatic, colorectal, and gastric cancers [13]. Conversely, excessive apoptosome activity may contribute to degenerative conditions, as evidenced by elevated caspase-9 expression in degenerated intervertebral discs and correlation of CASPASE-9 polymorphisms with discogenic low back pain [13]. Activated caspase-9 has also been observed in end-stage Huntington's disease, suggesting apoptotic contribution to neuronal death [13].
Therapeutic strategies targeting the apoptosome pathway include BH3 mimetics such as venetoclax, which inhibits BCL-2 to promote cytochrome c release and apoptosome formation [14]. Additionally, DR5 agonist antibodies and TRAIL analogues aim to bypass defective intrinsic apoptosis signaling in cancer cells, though clinical success has been limited by resistance mechanisms [14]. Emerging approaches seek to overcome resistance by combining TRAIL pathway activators with IAP inhibitors to enhance caspase activation [14].
Non-Apoptotic Functions
Beyond its canonical role in cell death, emerging evidence suggests that caspase-9 participates in non-apoptotic processes, including myoblast differentiation [13]. Caspase-9 and caspase-3 activities have been implicated in determining myoblast differentiation fate, while caspase-9 knockdown shows potential therapeutic application in bovine skeletal muscle atrophy [13]. These findings expand the functional repertoire of apoptosome components beyond traditional cell death paradigms and highlight the importance of contextual regulation.
The apoptosome represents a critical signaling node in the intrinsic apoptosis pathway, integrating damage signals into a proteolytic cascade through its sophisticated macromolecular architecture. Recent structural insights have illuminated the mechanisms of cytochrome c-mediated activation, nucleotide-driven conformational changes, and the unique asymmetric organization of caspase-9 activation domains. The regulatory complexity of this system, encompassing phosphorylation, endogenous inhibitors, and evolutionary adaptations, underscores its importance in maintaining cellular homeostasis. Ongoing research continues to refine our understanding of apoptosome function while identifying novel therapeutic opportunities for manipulating this essential cell death machinery in human disease.
The Death-Inducing Signaling Complex (DISC) represents the crucial molecular platform that initiates the extrinsic pathway of apoptosis, a form of programmed cell death essential for development, tissue homeostasis, and immune function. Formed upon activation of death receptors by their cognate ligands, the DISC serves as the assembly point for a cascade of protein interactions that ultimately determine cellular fate. Within the broader context of caspase activation research, the extrinsic pathway—orchestrated by the DISC—stands in contrast to the intrinsic (mitochondrial) pathway, differing in both initiation mechanisms and regulatory dynamics [5] [19]. While the intrinsic pathway responds to internal cellular damage through mitochondrial outer membrane permeabilization and caspase-9 activation, the extrinsic pathway transduces external death signals via direct caspase-8 and caspase-10 activation at the DISC [3] [20]. This whitepaper provides an in-depth technical analysis of the DISC architecture, focusing on the core components FADD, caspase-8, and caspase-10, their complex interactions, and the experimental approaches used to elucidate their functions.
Molecular Composition and Stoichiometry of the DISC
The DISC is a multimolecular complex that assembles in a highly ordered sequence following death receptor activation. The core components include oligomerized death receptors (such as CD95/Fas or TRAIL receptors), the adaptor protein FADD (Fas-Associated protein with Death Domain), and initiator caspases—primarily caspase-8 and its homolog caspase-10 [21] [22]. Quantitative mass spectrometry analyses have revealed a critical insight into DISC organization: contrary to early 1:1 stoichiometry models, FADD is substoichiometric relative to both death receptors and DED-containing proteins, with up to a 9-fold excess of caspase-8 compared to FADD within the native TRAIL DISC [23]. This finding prompted the proposed DED chain model, where a single FADD molecule nucleates a helical filament of multiple caspase-8 molecules through sequential death effector domain interactions [23] [24].
Table 1: Core Components of the Death-Inducing Signaling Complex (DISC)
| Component | Domain Structure | Key Function in DISC | Regulatory Proteins |
|---|---|---|---|
| Death Receptors (e.g., CD95, TRAIL-R) | Death Domain (DD), transmembrane domain | Receptor for extracellular death ligands; initiates DISC assembly | - |
| FADD | Death Domain (DD), Death Effector Domain (DED) | Central adaptor; bridges death receptors and initiator caspases | - |
| Caspase-8 | Two DEDs, large (p18) and small (p10) catalytic subunits | Primary initiator caspase; undergoes activation through DED chain-induced dimerization | cFLIP isoforms |
| Caspase-10 | Two DEDs, large and small catalytic subunits | Regulatory homolog of caspase-8; negatively regulates caspase-8 and promotes NF-κB signaling | cFLIP isoforms |
| cFLIP | DEDs, caspase-like domain (proteolytically inactive) | Key regulator of caspase-8 activation; multiple isoforms with opposing functions | - |
The recruitment of procaspase-8 and procaspase-10 to the DISC occurs through homotypic death effector domain (DED) interactions, where the N-terminal tandem DEDs of these initiator caspases bind to the single DED of FADD [24]. This assembly facilitates the activation of caspase-8 through a unique mechanism involving anti-parallel dimerization of its catalytic domains, driven by the structural constraints imposed by the DED filament architecture [24]. Caspase-10, while structurally similar to caspase-8 and sharing overlapping substrate specificities, has emerged as a crucial regulatory component rather than a redundant executor of cell death [21] [22].
Structural Mechanisms of Caspase Activation Within the DISC
DED Filament Architecture and Caspase-8 Activation
Recent structural insights from cryo-electron microscopy (cryo-EM) have revolutionized our understanding of caspase activation within the DISC. Full-length procaspase-8 forms filamentous complexes nucleated by FADD, with increasing numbers of caspase-8 molecules extending the filament length [24]. These complexes exhibit a triple helix structure in which the tandem DEDs (tDEDs) of caspase-8 form a helical core that orients the catalytic domains for activation. The FADD-nucleated tDED filament is essential for properly orienting the procaspase-8 catalytic domains, enabling their activation through anti-parallel dimerization [24]. This dimerization event triggers a series of autoproteolytic cleavage events at specific aspartic acid residues, resulting in the maturation of caspase-8 into its active heterotetrameric form (p182-p102) capable of initiating the apoptotic cascade.
Diagram 1: DED Chain Model of Caspase-8 Activation. The death receptor recruits FADD, which nucleates a caspase-8 DED filament through Type I interactions. This filament orients the catalytic domains for dimerization and activation.
The Regulatory Role of Caspase-10 in DISC Signaling
Contrary to earlier assumptions of functional redundancy with caspase-8, caspase-10 has been identified as a negative regulator of caspase-8-mediated cell death. Experimental evidence from siRNA knockdown studies demonstrates that caspase-10 depletion enhances CD95L-induced cell death in multiple cell lines, including HeLa and SK-Mel melanoma cells [21]. This inhibitory function occurs through caspase-10's ability to reduce DISC association and activation of caspase-8 independently of competition for FADD binding. Rather than directly competing with caspase-8, caspase-10 appears to modulate the overall architecture and signaling output of the DISC. Significantly, caspase-10 recruitment to the DISC depends on the scaffold function of caspase-8, as demonstrated in caspase-8-knockout cells where DISC formation is critically impaired [21] [22].
Beyond its role in apoptosis regulation, caspase-10 contributes to rewiring DISC signaling toward NF-κB activation and cell survival. Both caspase-10 and caspase-8 exhibit redundant catalytic activity in gene induction, suggesting a coordinated regulatory mechanism where these caspases balance death and survival signaling [21] [22]. This functional relationship extends to cooperative regulation with cFLIP, where caspase-10 and cFLIP isoforms coordinately determine CD95L-mediated signaling outcomes toward either apoptosis or survival.
Diagram 2: Caspase-10-Mediated Switching of DISC Signaling. Caspase-10 negatively regulates caspase-8-mediated apoptosis and promotes NF-κB activation and cell survival, dependent on the scaffold function of caspase-8.
Experimental Approaches for DISC Analysis
Key Methodologies and Protocols
Research elucidating DISC composition and function has employed sophisticated biochemical, genetic, and structural approaches. Key experimental protocols include:
DISC Immunoprecipitation and Quantitative Mass Spectrometry: The native DISC can be isolated from cells stimulated with death receptor ligands (e.g., CD95L or TRAIL) using immunoprecipitation with receptor-specific antibodies [23] [22]. Quantitative label-free LC-MS/MS analysis of immunoprecipitated complexes enables precise determination of component stoichiometry. This approach revealed the substoichiometric relationship between FADD and caspase-8, fundamentally changing DISC assembly models [23]. The protocol typically involves: (1) cell stimulation with cross-linked ligand for specific time periods (5-30 minutes); (2) lysis with mild non-denaturing detergents; (3) immunoprecipitation with death receptor-specific antibodies; (4) complex purification and tryptic digestion; and (5) LC-MS/MS analysis with label-free quantification.
Gene Knockdown and Knockout Approaches: siRNA-mediated knockdown and CRISPR-Cas9 knockout cell lines have been instrumental in defining individual component functions [21]. Caspase-10 knockdown experiments utilizing multiple specific siRNAs demonstrated enhanced CD95L-induced cell death, revealing its inhibitory role. Similarly, caspase-8-knockout cells have proven essential for establishing the scaffold function of caspase-8 in DISC formation, as these cells show defective recruitment of both cFLIP and caspase-10 [21] [22].
Structural Analysis by Cryo-Electron Microscopy: Cryo-EM has provided unprecedented insights into the architecture of FADD:caspase-8 complexes [24]. The protocol involves: (1) co-expression of FLAG-tagged FADD and catalytically inactive full-length caspase-8 in mammalian cells; (2) affinity purification of complexes using FLAG resin; (3) glycerol gradient centrifugation to separate complexes of different lengths; (4) negative-stain EM for initial characterization; and (5) high-resolution cryo-EM data collection and single-particle analysis for 3D reconstruction.
Research Reagent Solutions
Table 2: Essential Research Reagents for DISC Investigation
| Reagent/Category | Specific Examples | Application/Function | Experimental Notes |
|---|---|---|---|
| Cell Lines | HeLa, SK-Mel melanoma, BJAB, Jurkat, HaCaT, Caspase-8-knockout 293F | Model systems for DISC study | Cell line selection critical due to variable caspase-10 expression and function [21] |
| Gene Modulation Tools | siRNA targeting caspase-10/caspase-8, CRISPR-Cas9 knockout systems, Doxycycline-inducible shRNA | Functional analysis of specific components | Multiple siRNAs recommended to confirm phenotype specificity [21] |
| Death Receptor Ligands | Recombinant CD95L/FasL, TRAIL | DISC activation and assembly | Cross-linked ligands often required for efficient receptor activation |
| Affinity Purification Reagents | FLAG-tagged FADD, death receptor-specific antibodies, protein A/G beads | DISC isolation and composition analysis | Mild lysis conditions (1% NP-40 or CHAPS) preserve complex integrity [23] |
| Structural Biology Tools | Catalytically inactive caspase-8 (C360A), Cryo-EM grids, Negative stains | Structural analysis of complexes | Catalytically inactive mutants enable study of activation mechanisms [24] |
| Detection Antibodies | Anti-caspase-8, anti-caspase-10, anti-FADD, anti-cFLIP | Western blot, immunoprecipitation validation | Critical for assessing recruitment and processing in DISC |
Discussion: Therapeutic Implications and Future Directions
The intricate regulation of DISC signaling through the coordinated actions of FADD, caspase-8, and caspase-10 presents compelling therapeutic opportunities. In cancer, where apoptosis evasion is a hallmark, modulating DISC components could restore cell death sensitivity. Specifically, strategies to inhibit caspase-10's anti-apoptotic function or disrupt its recruitment to the DISC might sensitize tumor cells to death receptor-targeted therapies [21] [22]. Conversely, in autoimmune or degenerative diseases characterized by excessive cell death, enhancing caspase-10's protective signaling could provide therapeutic benefit. The scaffold function of caspase-8 represents another potential intervention point, as modulating its structural role could influence the entire DISC assembly process.
Future research directions should focus on elucidating the precise structural determinants of caspase-10's regulatory function and its interplay with cFLIP isoforms. The development of selective caspase-10 inhibitors or activators would provide valuable tools for both basic research and therapeutic applications. Additionally, exploring the DISC's role in non-apoptotic signaling, particularly in NF-κB activation and inflammatory responses, may reveal novel functions for these core components in immune regulation and disease pathogenesis. As our structural understanding of the DISC continues to evolve through techniques like cryo-EM, new opportunities for targeted therapeutic intervention will undoubtedly emerge, offering potential for more precise modulation of cell death pathways in human disease.
The DISC represents a sophisticated molecular machine that integrates extracellular death signals into precise cellular responses through the coordinated actions of FADD, caspase-8, and caspase-10. Moving beyond simplistic models of linear activation, contemporary research reveals a complex regulatory network where component stoichiometry, structural organization, and competitive interactions determine signaling outcomes. The DED chain model, with its helical caspase-8 filaments nucleated by FADD, provides a structural framework for understanding initiator caspase activation. Meanwhile, the emerging role of caspase-10 as a negative regulator of caspase-8 and promoter of survival signaling adds crucial nuance to our understanding of DISC function. These insights not only advance fundamental knowledge of apoptosis regulation but also open new avenues for therapeutic intervention in cancer, autoimmune disorders, and degenerative diseases where programmed cell death is dysregulated.
Apoptosis, or programmed cell death, is a fundamental process essential for development and tissue homeostasis in multicellular organisms. Its deregulation can lead to autoimmune diseases, cancer, or debilitating degenerative diseases [25]. A core event in apoptosis is the activation of caspases, a family of cysteine proteases that cleave key cellular proteins, leading to the characteristic morphological changes of cell death [25] [26]. The two principal pathways initiating apoptosis are the intrinsic (mitochondrial) pathway and the extrinsic (death receptor) pathway, which converge on the execution-phase caspases [27] [28]. The intrinsic pathway is regulated by the BCL-2 protein family at the level of the mitochondria, while the extrinsic pathway is initiated at the plasma membrane and critically modulated by c-FLIP (cellular FLICE-inhibitory protein). Understanding the mechanisms of these key regulators is not only crucial for basic cell biology but also for developing novel therapies, particularly for cancer, where these pathways are often disrupted [29] [30].
Table 1: Core Characteristics of Intrinsic and Extrinsic Apoptosis
| Feature | Intrinsic Apoptosis | Extrinsic Apoptosis |
|---|---|---|
| Primary Initiator | Internal stresses (e.g., DNA damage, growth-factor withdrawal) | External signals (e.g., FasL, TRAIL) via death receptors (DRs) |
| Key Regulatory Point | Mitochondrial Outer Membrane Permeabilization (MOMP) | Death-Inducing Signaling Complex (DISC) formation |
| Central Regulators | BCL-2 Family Proteins (Pro- and Anti-apoptotic) | Caspase-8, c-FLIP, FADD |
| Key Initiator Caspase | Caspase-9 | Caspase-8 |
| Amplification Loop | - | Caspase-8-mediated cleavage of Bid to tBid, engaging mitochondria |
The BCL-2 Family and Regulation of the Intrinsic Pathway
The BCL-2 Family Network
The BCL-2 family of proteins constitutes a critical intracellular checkpoint in the intrinsic apoptotic pathway. These proteins integrate diverse death and survival signals to decide whether a cell will live or die by controlling the release of cytochrome c from the mitochondrial intermembrane space [30] [31]. The family is defined by the presence of BCL-2 homology (BH) domains and is structurally and functionally divided into three main subgroups [25] [28]:
- Anti-apoptotic proteins (e.g., BCL-2, BCL-XL, MCL-1, BCL-w, Bfl-1): These proteins contain four BH domains (BH1-BH4) and are characterized by their globular α-helical bundle structure, which forms a hydrophobic surface groove. This groove serves as the binding site for the BH3 domains of pro-apoptotic family members. Their canonical function is to preserve mitochondrial outer membrane integrity and prevent the release of apoptogenic factors [25] [30].
- Multi-domain pro-apoptotic effector proteins (BAX, BAK, and to some extent BOK): These proteins contain BH1-BH3 domains. In healthy cells, BAX is largely cytosolic or loosely associated with mitochondria, while BAK is integrated into the outer mitochondrial membrane. Upon activation, they undergo a conformational change, homo-oligomerize, and form pores in the mitochondrial outer membrane [25] [27].
- BH3-only proteins (e.g., Bid, Bim, Bad, Puma, Noxa): These are sentinels for cellular damage and stress. They are activated by various mechanisms such as transcriptional upregulation (e.g., Puma, Noxa by p53), post-translational modification (e.g., dephosphorylation of Bad), or proteolytic cleavage (e.g., Bid by caspase-8) [25] [32]. They either directly activate BAX/BAK or function as "sensitizers" by neutralizing anti-apoptotic proteins.
Mitochondrial Outer Membrane Permeabilization (MOMP)
MOMP is the pivotal, "point-of-no-return" event in the intrinsic pathway [27] [32]. It is orchestrated by the BCL-2 family and leads to the release of several pro-apoptotic proteins from the mitochondrial intermembrane space, including cytochrome c and SMAC/DIABLO [25] [27].
The current model of MOMP involves the following steps:
- Activation: In response to cellular stress, activated BH3-only proteins engage the anti-apoptotic proteins or directly activate BAX/BAK.
- BAX/BAK Activation: BAX and BAK undergo a conformational change, exposing their N-terminal domains. For BAX, this involves translocation from the cytosol and insertion into the mitochondrial outer membrane [25].
- Oligomerization and Pore Formation: Activated BAX and BAK form homo-oligomers that create pores in the outer mitochondrial membrane. Cells doubly deficient for Bax and Bak are resistant to a wide array of apoptotic stimuli, underscoring their essential role [25] [27].
- Release of IMS Proteins: These pores allow for the efflux of cytochrome c, which in the cytosol binds to Apaf-1 to form the apoptosome, leading to the activation of caspase-9 and the downstream caspase cascade. SMAC/DIABLO promotes apoptosis by neutralizing inhibitor of apoptosis proteins (IAPs) [27] [32].
Anti-apoptotic proteins like BCL-2 and MCL-1 block MOMP by sequestering the activator BH3-only proteins and/or activated, monomeric forms of BAX and BAK, preventing their oligomerization [25].
Diagram 1: BCL-2 Family Regulation of the Intrinsic Apoptotic Pathway. Cellular stress activates BH3-only proteins, which either directly activate BAX/BAK or neutralize anti-apoptotic proteins. Anti-apoptotic proteins sequester both BH3-only proteins and activated BAX/BAK. When activation signals prevail, BAX/BAK oligomerize to cause MOMP, leading to the release of cytochrome c and SMAC.
c-FLIP and Regulation of the Extrinsic Pathway
Death Receptor Signaling and the DISC
The extrinsic apoptotic pathway is initiated by the ligation of death receptors (e.g., Fas/CD95, TRAIL receptors) at the cell surface [26] [33]. This triggers the assembly of a multi-protein signaling complex known as the Death-Inducing Signaling Complex (DISC). The core components of the DISC are the death receptor, the adaptor protein FADD (Fas-Associated protein with Death Domain), and the initiator procaspase-8 (and/or -10) [26] [29]. Within the DISC, procaspase-8 molecules are brought into close proximity, leading to their dimerization, auto-proteolytic processing, and activation [26].
The Dual-Role of c-FLIP
c-FLIP is a key regulatory protein of the extrinsic pathway. It is structurally homologous to procaspase-8 and -10, containing two N-terminal death effector domains (DEDs) that enable its recruitment to the DISC. However, it lacks (or has an inactive) caspase protease domain [26] [33]. Three main splice variants exist in humans: c-FLIPL (long, 55 kDa), c-FLIPS (short, 26 kDa), and c-FLIPR (24 kDa) [33].
The role of c-FLIP is complex and concentration-dependent:
- Anti-apoptotic Role (High Expression): At high expression levels, c-FLIP variants, particularly c-FLIPS and c-FLIPR, act as dominant-negative inhibitors by competing with procaspase-8 for binding to FADD at the DISC. This prevents the dimerization and activation of procaspase-8, thereby blocking apoptosis initiation [26] [33].
- Pro-apoptotic Role (Low/Physiological Expression): At low, physiological levels (approximately 1% of procaspase-8 concentration), c-FLIPL has a surprising pro-apoptotic function. It is enriched in the DISC and forms a heterodimer with procaspase-8. This heterodimerization potently enhances the catalytic activity of procaspase-8, leading to its processing and the efficient initiation of apoptosis [26]. This pro-apoptotic function is essential during embryonic development, as demonstrated by the lethal phenotype of c-flip −/− mice, which resembles that of fadd −/− and caspase-8 −/− mice [26] [34].
The processing of procaspase-8 at the DISC leads to the activation of downstream executioner caspases (e.g., caspase-3, -7). In some cells (designated Type I cells), this direct activation is sufficient to induce apoptosis. In other cells (Type II cells), the signal requires amplification through the mitochondrial pathway. In Type II cells, active caspase-8 cleaves the BH3-only protein Bid to its truncated form, tBid, which then translocates to mitochondria to activate BAX/BAK, leading to MOMP and amplification of the death signal [29] [27] [33].
Diagram 2: c-FLIP's Dual Role in Regulating the Extrinsic Apoptotic Pathway. Death receptor ligation leads to DISC formation and recruitment of c-FLIP. At low levels, c-FLIPL promotes caspase-8 activation. Active caspase-8 can directly activate executioner caspases (Type I cells) or cleave Bid to tBid, engaging the mitochondrial amplification loop (Type II cells). At high levels, c-FLIP inhibits caspase-8 activation.
Experimental Methods and Research Tools
Key Experimental Protocols
BH3 Profiling
BH3 profiling is a functional assay that measures the mitochondrial "priming" of cells for apoptosis, providing a window into the interactions between BCL-2 family members within a cell [25].
Detailed Methodology:
- Mitochondrial Isolation: Cells (from a cell line or a patient sample) are permeabilized with digitonin, or mitochondria are isolated via differential centrifugation.
- Peptide Incubation: The isolated mitochondria are incubated with synthetic peptides corresponding to the BH3 domains of various BH3-only proteins (e.g., Bim, Bad, Noxa, HRK). A positive control peptide (e.g., from Bim or the small molecule agonist ABT-737) is used to induce maximal MOMP, while a negative control (DMSO) is used to measure baseline.
- MOMP Measurement: The release of cytochrome c from the mitochondria is quantified as a readout for MOMP. This can be measured by several methods:
- Western Blot: Analyzing the cytochrome c content in the mitochondrial pellet versus the supernatant.
- ELISA: Quantifying cytochrome c in the supernatant.
- Flow Cytometry: Using fluorescent dyes that detect the loss of mitochondrial membrane potential (ΔΨm) in permeabilized cells.
- Data Interpretation: The pattern of which BH3 peptides cause MOMP reveals the apoptotic block and dependency on specific anti-apoptotic proteins. For example:
- Sensitivity to Bad-BH3 indicates BCL-2/BCL-XL/BCL-w dependence.
- Sensitivity to Noxa-BH3 indicates MCL-1 dependence.
- If only activator peptides (Bid, Bim) cause MOMP, it suggests a lack of "primed" anti-apoptotic proteins (Class A block).
- If no peptides cause MOMP, it suggests a defect in Bax/Bak function (Class B block) [25].
Diagram 3: BH3 Profiling Experimental Workflow.
Analyzing Caspase-8 Activation via c-FLIP
The role of c-FLIP in caspase-8 activation can be dissected using hetero-dimerization systems and DISC immunoprecipitation [26].
Detailed Methodology (Induced Dimerization System):
- Construct Design: Create fusion proteins where the protease domain of procaspase-8 (Fv-CASP-8) and the protease-like domain of c-FLIPL (FRB-FLIP) are fused to inducible dimerization domains (e.g., FKBP-Fv and FRB, which bind upon addition of a divalent ligand like AP20187 or rapamycin).
- In Vitro Reconstitution: Produce the fusion proteins via in vitro transcription/translation in the presence of [³⁵S]-methionine.
- Induced Dimerization and Processing: Incubate the proteins with the dimerizing drug for set time periods (e.g., 0, 2, 4 hours).
- Analysis: Resolve the proteins by SDS-PAGE and visualize processing (cleavage into signature fragments like p10) via autoradiography. The enhancement of procaspase-8 processing upon hetero-dimerization with c-FLIPL, compared to its auto-processing alone, demonstrates the catalytic role of c-FLIPL [26].
Detailed Methodology (DISC Immunoprecipitation):
- Stimulation: Treat cells expressing the death receptor of interest (e.g., CD95) with the cognate ligand (e.g., CD95L) for a short duration.
- Cell Lysis and Immunoprecipitation: Lyse cells with mild, non-denaturing detergents. Immunoprecipitate the DISC using an antibody against the death receptor or an epitope tag.
- Western Blot Analysis: Analyze the immunoprecipitated complex by western blotting for key components: procaspase-8, c-FLIP, and FADD. The presence and processing of procaspase-8 and c-FLIPL in the DISC can be assessed, revealing the molecular composition under different experimental conditions [26] [29].
The Scientist's Toolkit: Key Research Reagents
Table 2: Essential Reagents for Apoptosis Research
| Reagent / Tool | Primary Target/Function | Key Applications |
|---|---|---|
| ABT-263 (Navitoclax) | BH3-mimetic; inhibits BCL-2, BCL-XL, BCL-w [25] [30] | Inducing intrinsic apoptosis; studying anti-apoptotic dependencies; combination therapy [29]. |
| ABT-199 (Venetoclax) | BH3-mimetic; selective BCL-2 inhibitor [30] [28] | Targeting BCL-2-dependent malignancies (e.g., CLL, AML); studying BCL-2-specific biology [30] [28]. |
| S63845 | BH3-mimetic; MCL-1 inhibitor [29] [30] | Inducing apoptosis in MCL-1-dependent cells; combination therapy to overcome resistance [29]. |
| FLIPinB/FLIPinBγ | Small molecule targeting c-FLIPL in the caspase-8/c-FLIPL heterodimer [29] | Enhancing caspase-8 activation in the DISC; sensitizing cells to TRAIL/CD95L-induced apoptosis [29]. |
| Recombinant LZ-CD95L | Aggregated, highly active form of Fas Ligand [29] | Robust activation of the Fas death receptor pathway; studying DISC formation and extrinsic apoptosis. |
| BH3 Peptides | Synthetic peptides corresponding to BH3 domains of BH3-only proteins [25] | BH3 profiling; determining mitochondrial priming and anti-apoptotic protein dependency. |
| Caspase-Glo 3/7 Assay | Luminescent substrate for caspases-3 and -7 [29] | Quantifying the activity of executioner caspases as a direct measure of apoptosis induction. |
| Annexin-V / Sytox Orange | Annexin-V binds phosphatidylserine (early apoptosis); Sytox stains DNA in late apoptotic/necrotic cells [29] | Flow cytometry-based quantification of apoptotic cell populations. |
The BCL-2 family and c-FLIP are master regulators of the intrinsic and extrinsic apoptotic pathways, respectively. Their precise interplay determines cellular fate in response to a vast array of stresses and signals. The translational impact of understanding these regulators is profound, as evidenced by the clinical success of the BCL-2-selective BH3-mimetic venetoclax in treating hematological malignancies [30] [28]. Current research is focused on overcoming resistance mechanisms, such as upregulation of MCL-1 or BCL-XL, through combination therapies [29] [30]. Similarly, pharmacological targeting of c-FLIP with molecules like FLIPinB represents a promising strategy to overcome resistance to death receptor agonists and conventional chemotherapy [29]. The continued elucidation of the complex mechanisms governing caspase activation through these pathways will undoubtedly yield novel and more effective targeted therapies for cancer and other diseases characterized by dysregulated apoptosis.
Within the complex signaling networks of programmed cell death, the intrinsic and extrinsic apoptotic pathways converge decisively upon the activation of a critical set of effector proteins. This review delineates the central and distinct roles of the executioner caspases-3 and -7 in this terminal demolition phase of apoptosis. While historically viewed as functionally redundant, emerging evidence reveals that these proteases perform non-overlapping roles, with caspase-3 acting as the primary executioner responsible for the cleavage of a vast majority of cellular substrates. This whitepaper synthesizes current molecular understanding of their activation mechanisms, substrate specificity, and functional hierarchy, providing researchers and drug development professionals with a detailed technical guide, complete with experimental data and methodologies, to inform therapeutic strategies targeting apoptotic dysregulation.
Apoptosis, a form of programmed cell death essential for development and cellular homeostasis, is orchestrated through two primary signaling cascades: the intrinsic (mitochondrial) and extrinsic (death receptor) pathways [35]. Despite differing in their initiation mechanisms, these pathways converge to activate a conserved set of effector molecules that execute the final, irreversible stages of cell death. Central to this process are the executioner caspases, cysteine-aspartate-specific proteases that mediate the systematic dismantling of the cell [35] [36].
Caspases are broadly categorized as either initiators (e.g., caspase-8, -9) or effectors (e.g., caspase-3, -6, -7). Initiator caspases are activated in response to pro-apoptotic signals and propagate the death signal by processing and activating the downstream effector caspases [37]. The effector caspases then undertake the demolition phase of apoptosis by cleaving over 400 cellular protein substrates, leading to the characteristic biochemical and morphological changes associated with apoptotic cell death [38]. Among the effector caspases, caspase-3 and caspase-7 are the most prominent and are universally activated during apoptosis. For years, their high sequence similarity and nearly indistinguishable activity toward certain synthetic peptides fostered a perception of functional redundancy [38]. However, as this review will elaborate, evidence from gene knockout studies and biochemical profiling reveals that they perform distinct, non-redundant roles in the apoptotic cascade [38] [39].
Caspase-3 and -7: From Activation to Substrate Cleavage
Activation in the Converged Apoptotic Pathway
The activation of caspase-3 and -7 represents the critical point of convergence for the intrinsic and extrinsic pathways.
- Extrinsic Pathway Activation: The extrinsic pathway is triggered by the ligation of plasma membrane death receptors (e.g., Fas, TNFR1). This leads to the formation of the Death-Inducing Signaling Complex (DISC), which activates initiator caspase-8. Active caspase-8 can directly cleave and activate procaspase-3 and -7 [35].
- Intrinsic Pathway Activation: The intrinsic pathway is initiated by intracellular stressors such as DNA damage, leading to mitochondrial outer membrane permeabilization (MOMP) and the release of cytochrome c into the cytosol. Cytochrome c, together with Apaf-1 and dATP/ATP, forms the "apoptosome," a multi-protein complex that activates caspase-9. This initiator caspase then proteolytically processes caspase-3 and -7 [35] [40].
Once activated, caspase-3 can further propagate the caspase cascade through feedback loops, such as the processing of caspase-2, -6, and even further activation of caspase-9, ensuring an irreversible commitment to cell death [38] [37].
Hierarchical and Non-Redundant Substrate Specificity
Although caspase-3 and -7 share the preferred tetra-peptide cleavage motif DEVD, they exhibit significant differences in their efficiency toward natural protein substrates. A comparative analysis using purified recombinant enzymes and cell-free extracts has delineated their distinct substrate profiles [38].
Table 1: Substrate Cleavage Efficiency of Caspase-3 versus Caspase-7
| Substrate Protein | Caspase-3 Efficiency | Caspase-7 Efficiency | Functional Consequence of Cleavage |
|---|---|---|---|
| PARP | High | High | Disables DNA repair; facilitates cellular dismantling |
| RhoGDI | High | High | Cytoskeletal reorganization |
| Bid | High | Low/None | Amplifies apoptotic signal via mitochondria |
| XIAP | High | Low | Relieves caspase inhibition |
| Gelsolin | High | Low | Mediates cytoskeletal breakdown |
| Caspase-6 | High | Low | Propagates caspase cascade |
| Caspase-9 | High | Low | Amplifies caspase activation via feedback |
| Cochaperone p23 | Low | High | Disrupts protein folding; function in apoptosis unclear |
As illustrated, caspase-3 is the more promiscuous and potent executioner, necessary for the cleavage of a wide array of structurally and functionally diverse proteins, including those involved in propagating the caspase cascade itself [38] [39]. In contrast, caspase-7 demonstrates a more restricted substrate profile, with a few unique targets like cochaperone p23 [38].
Experimental Evidence and Methodologies
The distinct roles of caspase-3 and -7 have been elucidated through a series of definitive experimental approaches, including immuno-depletion studies and the use of specific inhibitors.
Immuno-depletion in Cell-Free Systems
A foundational methodology for dissecting the individual contributions of executioner caspases involves the use of cell-free extracts, typically from Jurkat cells, where specific caspases are selectively removed via immuno-depletion.
- Protocol Outline: Cytosolic extracts are prepared and incubated with antibodies specific to caspase-3, -6, or -7, followed by precipitation with protein A/G beads to remove the target caspase. The depleted extracts are then stimulated with cytochrome c and dATP to initiate intrinsic apoptosis. The resulting proteolysis of candidate substrates is analyzed by Western blotting, while nuclear morphology (condensation, DNA fragmentation) is assessed microscopically [39].
- Key Findings: Extracts depleted of caspase-3 showed a near-complete abolition of cleavage for numerous substrates, including DFF45/ICAD, XIAP, gelsolin, and lamin B. Furthermore, caspase-3 depletion prevented key morphological events like chromatin margination and DNA fragmentation. In stark contrast, depletion of caspase-7 (or caspase-6) had minimal impact on these parameters, establishing caspase-3 as the primary executioner protease in this system [39].
Caspase-3 Inhibition in Postmortem Studies
The critical role of caspase-3 in a functional pathological process has been demonstrated in studies of postmortem muscle softening in Northern pike (Esox lucius), which shares biochemical features with apoptotic proteolysis [40].
- Experimental Workflow: Fresh fish muscle was treated with the specific caspase-3 inhibitor Ac-DEVD-CHO or vehicle control. Samples were analyzed over a 15-day storage period.
- Measurements and Results:
- Mitochondrial Function: Caspase-3 inhibition reduced mitochondrial membrane permeability transition pore (MPTP) opening, stabilized mitochondrial membrane potential (ΔΨm), and reduced mitochondrial swelling.
- Apoptotic Cascade: The inhibitor significantly reduced the release of cytochrome c from mitochondria and subsequent activation of caspase-9 and caspase-3.
- Structural Protein Degradation: The degradation of key myofibrillar proteins (desmin, troponin-T, titin, nebulin) was markedly suppressed in the inhibitor-treated group, directly linking caspase-3 activity to the structural collapse of tissue [40].
Table 2: Key Research Reagents for Studying Executioner Caspases
| Reagent Name | Type | Primary Function in Research | Example Application |
|---|---|---|---|
| Ac-DEVD-CHO | Caspase-3 Inhibitor | Potent, cell-permeable inhibitor of caspase-3 activity | Inhibiting caspase-3-dependent protein degradation in postmortem studies [40] |
| zVAD-fmk | Pancaspase Inhibitor | Broad-spectrum, irreversible caspase inhibitor | Determining caspase-dependence of cell death; used in active-site titration [38] [41] |
| DEVD-AFC | Fluorogenic Substrate | Synthetic substrate cleaved by caspase-3 and -7; releases fluorescent AFC | Quantifying caspase-3/7 enzyme activity in extracts or live cells [38] |
| Anti-Caspase-3/-7 Antibodies | Immunological Reagent | Selective immuno-depletion or detection of specific caspases | Depleting caspases from cell extracts for functional studies [39] |
| Recombinant Caspase-3/-7 | Enzyme | Highly purified active enzymes for in vitro assays | Direct comparison of substrate specificity and cleavage kinetics [38] |
Cross-Talk with Other Cell Death Mechanisms
Emerging evidence indicates that the functional roles of caspase-3 and -7 extend beyond classical apoptosis into other regulated cell death pathways, particularly pyroptosis.
In models of multiple sclerosis (MS) and its animal model, experimental autoimmune encephalomyelitis (EAE), microglial cells undergoing pyroptosis—a pro-inflammatory, lytic cell death traditionally executed by gasdermin D (GSDMD)—show clear evidence of caspase-3 and -7 activation [42]. siRNA-mediated knockdown of CASP3 or CASP7 in human microglia suppressed nigericin-induced plasma membrane rupture, a hallmark of pyroptosis. Furthermore, canonical caspase-3/7 substrates (PARP, DFF45, ROCK1) were cleaved during this process, and caspase-3 was found to colocalize with GSDMD in microglia within active MS and EAE lesions [42]. This demonstrates a convergence of apoptotic and pyroptotic machinery in neuroinflammation, where executioner caspases contribute to GSDMD-mediated pyroptosis, blurring the traditional boundaries between cell death pathways.
Visualizing Pathway Convergence and Caspase Activation
The following diagram synthesizes the signaling pathways that lead to the activation of caspase-3 and -7, integrating the intrinsic and extrinsic routes and their points of cross-talk.
Diagram 1: Convergence of Intrinsic and Extrinsic Apoptotic Pathways on Executioner Caspases-3 and -7. The diagram illustrates how distinct initiation signals converge to activate caspase-3 and -7, highlighting the central role of caspase-3 and its feedback amplification of the cascade. The cross-talk mediated by tBid connects the two pathways.
The convergence of the intrinsic and extrinsic apoptotic pathways upon the activation of caspase-3 and -7 represents a critical commitment point in programmed cell death. Far from being redundant, these executioner caspases perform specialized functions, with caspase-3 acting as the primary and most promiscuous executioner, essential for the cleavage of a majority of cellular substrates and the manifestation of classic apoptotic morphology. Caspase-7, while activated simultaneously, appears to have a more restricted, complementary role. This refined understanding, supported by robust immuno-depletion and inhibitor studies, is further complicated by emerging evidence of their involvement in non-apoptotic processes like pyroptosis. For researchers and drug developers, these insights underscore the necessity of targeting specific executioner caspases based on their distinct substrate profiles and pathological contexts to effectively modulate cell death in disease.
From Bench to Bedside: Techniques for Detecting Caspase Activity and Developing Caspase-Targeted Therapeutics
Caspases, a family of cysteine-dependent aspartate-specific proteases, are the central executioners of apoptosis, a programmed cell death process crucial for development, homeostasis, and disease pathogenesis [20] [43]. These enzymes are synthesized as inactive zymogens (pro-caspases) and undergo proteolytic cleavage to form active enzymes that dismantle the cell through targeted cleavage of structural and regulatory proteins [44] [45]. Detection of caspase activation provides a critical window into cellular decision-making processes, enabling researchers to quantify and visualize cell death events in diverse experimental contexts.
The two principal branches of apoptosis—intrinsic and extrinsic—converge on caspase activation through distinct mechanisms. The extrinsic pathway initiates from extracellular signals mediated by death receptors on the cell surface, leading to the formation of the Death-Inducing Signaling Complex (DISC) and activation of initiator caspase-8 [46] [4]. In contrast, the intrinsic pathway emerges from intracellular stress signals such as DNA damage or oxidative stress, triggering mitochondrial outer membrane permeabilization, cytochrome c release, and formation of the apoptosome complex, which activates initiator caspase-9 [46] [4]. Both pathways ultimately converge on the activation of executioner caspases-3, -6, and -7, which orchestrate the systematic dismantling of cellular structures [44] [4].
This technical guide provides an in-depth comparison of two fundamental methodological approaches for detecting caspase activation: antibody-based detection and fluorescent reporter systems. By examining their principles, applications, and limitations within the context of intrinsic versus extrinsic apoptosis research, we aim to equip researchers with the knowledge to select optimal detection strategies for their specific experimental needs.
Antibody-Based Detection Methods
Principles and Workflow
Antibody-based methods leverage the specific binding between antibodies and caspase antigens to detect presence, activation, and cellular localization. These techniques primarily include immunofluorescence, western blotting, and flow cytometry, each offering distinct advantages for different research scenarios [47] [44].
The core principle underlying antibody-based detection involves recognizing specific caspase epitopes, often targeting either the inactive pro-caspase form or the cleaved active fragments. For example, antibodies can distinguish between full-length caspase-3 and its cleaved subunits, providing direct evidence of protease activation [44]. In immunofluorescence protocols, fixed and permeabilized cells are incubated with primary antibodies against specific caspases, followed by fluorescently-labeled secondary antibodies that enable visualization under microscopy [47]. This approach preserves spatial information, allowing researchers to correlate caspase activation with subcellular localization and morphological changes characteristic of apoptosis.
Western blotting represents another widely-used antibody-based approach that detects caspase cleavage events through size separation on SDS-PAGE gels [44]. This method enables simultaneous assessment of multiple caspases from the same cellular population, providing a comprehensive characterization of the cell death processes [44]. A significant advantage of western blotting is its ability to distinguish between the inactive zymogen and active cleaved forms based on molecular weight shifts, offering unambiguous evidence of caspase activation.
Detailed Experimental Protocol: Caspase Immunofluorescence
The following protocol provides a standardized workflow for detecting caspase activation via immunofluorescence in fixed cell samples [47]:
- Sample Preparation and Fixation: Culture cells on glass coverslips and treat with apoptosis-inducing agents. Rinse with PBS and fix with 4% paraformaldehyde for 15 minutes at room temperature.
- Permeabilization: Incubate fixed samples in PBS containing 0.1% Triton X-100 or 0.1% NP-40 for 5 minutes at room temperature to allow antibody access to intracellular epitopes.
- Blocking: Apply 200μL of blocking buffer (PBS/0.1% Tween 20 supplemented with 5% serum from the host species of the secondary antibody) for 1-2 hours at room temperature in a humidified chamber to minimize non-specific antibody binding.
- Primary Antibody Incubation: Incubate with 100μL of primary antibody (e.g., anti-Caspase-3 rabbit monoclonal antibody diluted 1:200 in blocking buffer) overnight at 4°C in a humidified chamber. Include a no-primary-antibody control to assess background staining.
- Secondary Antibody Incubation and Mounting: Wash slides three times for 10 minutes each in PBS/0.1% Tween 20. Apply 100μL of appropriate fluorescently-conjugated secondary antibody (e.g., goat anti-rabbit Alexa Fluor 488 conjugate diluted 1:500 in PBS) for 1-2 hours at room temperature, protected from light. After final washes, mount slides with anti-fade mounting medium and image using fluorescence microscopy.
Applications and Advantages
Antibody-based methods offer several distinct advantages for caspase detection. Immunofluorescence provides exceptional spatial resolution at the single-cell level, enabling researchers to visualize caspase activation within specific subcellular compartments and correlate it with morphological hallmarks of apoptosis [47]. This approach is particularly valuable when investigating heterogeneous cellular responses to death stimuli or when co-localization with other markers is required.
The ability to conduct multiplexed experiments represents another significant advantage, as researchers can simultaneously detect multiple caspases or combine caspase detection with other apoptotic markers using antibodies with different fluorophores [47]. This capability facilitates the dissection of complex caspase activation hierarchies and their relationships with other cellular events. Furthermore, antibody-based methods do not require genetic manipulation of cells, making them accessible for primary cell cultures and clinical samples where establishing stable reporter lines may be impractical.
Table 1: Key Antibody-Based Detection Techniques for Caspase Activation
| Method | Detection Principle | Key Applications | Spatial Resolution | Throughput |
|---|---|---|---|---|
| Immunofluorescence | Antibody binding with fluorescent detection | Subcellular localization, co-localization studies | High (single-cell) | Low to moderate |
| Western Blotting | Size-based separation and antibody detection | Confirm cleavage events, parallel caspase assessment | None (population average) | Moderate |
| Flow Cytometry | Antibody binding in suspension with fluorescence quantification | Population analysis, quantification of heterogeneous responses | Low (population-level) | High |
| Immunohistochemistry | Antibody binding with chromogenic/fluorescent detection in tissues | Caspase detection in tissue architecture, clinical pathology | High (tissue and cellular) | Low |
Fluorescent Reporter Systems
Principles and Design Strategies
Fluorescent reporter systems represent a powerful alternative for detecting caspase activation in live cells, offering real-time kinetic data and enabling longitudinal studies of cell death dynamics. These genetically-encoded biosensors typically exploit the proteolytic activity of caspases to trigger measurable changes in fluorescence properties [48] [49].
The most common design strategy involves FRET-based reporters, which consist of two fluorescent proteins with compatible spectral properties connected by a short linker containing the caspase cleavage sequence DEVD (for caspases-3 and -7) [49]. In the intact reporter, excitation of the donor fluorophore results in FRET to the acceptor fluorophore, producing a characteristic emission ratio. Upon caspase activation and cleavage of the DEVD sequence, the physical separation of the fluorophores eliminates FRET, resulting in decreased acceptor emission and increased donor emission [49]. This ratiometric change provides a robust indicator of caspase activity that is largely independent of reporter concentration.
An innovative advancement in caspase reporter technology is the ZipGFP-based system, which utilizes a split-GFP architecture where the eleventh β-strand is tethered to the rest of the GFP molecule via a flexible linker containing the caspase cleavage motif [48]. Under basal conditions, the forced proximity of the β-strands prevents proper folding and chromophore maturation, resulting in minimal background fluorescence. Upon caspase-mediated cleavage, the β-strands separate and spontaneously refold into the native GFP structure, generating a fluorescent signal that irreversibly marks cells that have experienced caspase activation [48]. This design minimizes background noise and enhances signal stability, particularly advantageous for long-term imaging studies.
Detailed Experimental Protocol: FLIM-FRET Caspase-3 Reporter
Fluorescence Lifetime Imaging Microscopy combined with FRET provides a sophisticated approach for quantifying caspase-3 activation independent of reporter concentration or imaging depth, making it ideal for complex 3D models and in vivo applications [49]:
- Stable Cell Line Generation: Create lentiviral vectors expressing the LSS-mOrange-DEVD-mKate2 FRET reporter. Transduce target cells (e.g., MDA-MB-231 metastatic breast cancer cells) and select for stable expression using blasticidin drug selection or fluorescence-activated cell sorting to establish a homogeneous expressing population.
- Sample Preparation and Imaging: For 2D cultures, plate reporter cells on glass-bottom dishes and treat with apoptotic inducers. For 3D spheroids, embed reporter cells in Cultrex or Matrigel to form structures. For in vivo imaging, implant reporter cells into appropriate mouse models (e.g., mammary fat pad for tumor studies).
- FLIM Data Acquisition: Image samples using a multiphoton microscope equipped with FLIM capability. Excite LSS-mOrange at 440-460nm and measure fluorescence lifetime decays using time-correlated single-photon counting. Collect sufficient photons per pixel (typically 500-1000) to ensure accurate lifetime calculations.
- Data Analysis and Interpretation: Calculate fluorescence lifetimes on a pixel-by-pixel basis using specialized software. Cells with inactive caspase-3 display shorter LSS-mOrange lifetimes due to FRET, while caspase-3 activation increases the fluorescence lifetime resulting from cleavage of the DEVD linker and separation of the FRET pair.
Applications and Advantages
Fluorescent reporter systems excel in applications requiring temporal resolution and kinetic analysis of caspase activation. The ability to monitor caspase activity in real-time provides invaluable insights into the dynamics of cell death, including the timing, synchrony, and propagation of apoptotic signals within cell populations [48]. This temporal dimension is particularly important for distinguishing primary apoptotic events from secondary necrosis and for understanding the sequence of molecular events in different cell death pathways.
The compatibility of reporter systems with live-cell imaging enables longitudinal tracking of individual cells throughout the apoptosis process, revealing cell-to-cell heterogeneity in caspase activation kinetics that would be masked in population-averaged assays [48]. Furthermore, stable expression of caspase reporters in cells facilitates their application in complex physiological models, including 3D organoids and in vivo imaging, where traditional endpoint assays would be impractical or insufficient [48].
Table 2: Comparison of Fluorescent Reporter Strategies for Caspase Detection
| Reporter Type | Mechanism of Action | Caspase Targets | Key Advantages | Optimal Applications |
|---|---|---|---|---|
| FRET-Based | Cleavage eliminates energy transfer between fluorophores | Caspase-3/7 (DEVD), Caspase-8 (IETD) | Ratiometric quantification, reversible in some designs | Kinetic studies, high-throughput screening |
| Split GFP/ZipGFP | Cleavage enables fluorescent protein reconstitution | Caspase-3/7 (DEVD) | Low background, irreversible signal accumulation | Long-term fate mapping, 3D models |
| Localization-Based | Cleavage releases localization sequences | Various executioner caspases | Subcellular resolution, pathway-specific activation | Compartment-specific activation studies |
| Transcription-Based | Cleavage activates transcription factors | Specific initiator caspases | Signal amplification, persistent marking | Low-abundance caspase detection |
Comparative Analysis: Antibodies vs. Reporters
Method Selection Guide
Choosing between antibody-based methods and fluorescent reporters requires careful consideration of experimental goals, model systems, and practical constraints. The following comparative analysis highlights key performance characteristics across critical methodological dimensions:
Table 3: Direct Comparison of Antibody-Based and Reporter Methods for Caspase Detection
| Parameter | Antibody-Based Methods | Fluorescent Reporters |
|---|---|---|
| Temporal Resolution | Endpoint measurements only | Real-time, continuous monitoring |
| Spatial Resolution | Excellent (subcellular) | Good to excellent |
| Sample Throughput | Moderate to high | Low to moderate |
| Quantification Ease | Moderate (requires controls for quantification) | Excellent (inherently quantifiable) |
| Cellular Context | Fixed/dead cells | Live cells, dynamic processes |
| Multiplexing Capacity | High (multiple antigens) | Moderate (spectral limits) |
| Technical Complexity | Low to moderate | High (requires genetic manipulation) |
| Experimental Duration | Hours to days | Days to weeks (including cell generation) |
| Cost Considerations | Moderate (antibody expenses) | High (imaging equipment, vector generation) |
For studies focused on intrinsic apoptosis, where mitochondrial events precede caspase activation, fluorescent reporters offer advantages in capturing the temporal relationship between caspase activation and other hallmarks such as mitochondrial membrane potential loss. Conversely, for extrinsic apoptosis initiated by death receptor engagement, antibody-based methods can effectively document the rapid activation of caspase-8 and its downstream substrates in fixed samples collected at precise timepoints [46] [4].
Pathway-Specific Applications
The distinct initiation mechanisms of intrinsic and extrinsic apoptosis present unique considerations for caspase detection method selection:
In extrinsic pathway studies, where death receptor ligation triggers rapid caspase-8 activation at the DISC complex, antibody-based detection of cleaved caspase-8 provides unambiguous evidence of pathway engagement [46] [4]. The ability to simultaneously detect receptor components, caspase-8 cleavage, and downstream substrates in fixed samples makes immunofluorescence particularly valuable for dissecting DISC assembly and function. However, FRET-based reporters specific for caspase-8 (containing IETD sequences) enable real-time assessment of initiator caspase activation kinetics following death receptor engagement.
For intrinsic pathway investigations, where caspase activation follows mitochondrial outer membrane permeabilization and apoptosome formation, the delayed but amplified nature of caspase-9 and executioner caspase activation makes fluorescent reporters ideal for capturing the commitment phase to apoptosis [4]. The ZipGFP caspase-3/7 reporter has been successfully deployed in 3D spheroid and patient-derived organoid models to visualize apoptosis heterogeneity in response to chemotherapeutic agents that trigger the intrinsic pathway [48].
The Scientist's Toolkit: Essential Research Reagents
Successful detection of caspase activation requires appropriate research tools and reagents tailored to the selected methodology. The following table summarizes key reagents for both antibody-based and reporter-based approaches:
Table 4: Essential Research Reagents for Caspase Detection
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| Primary Antibodies | Anti-caspase-3 (cleaved), Anti-caspase-8, Anti-caspase-9 | Target-specific detection in immunoassays |
| Secondary Antibodies | Alexa Fluor conjugates, HRP conjugates | Signal amplification and detection |
| Fluorescent Reporters | FRET-DEVD probes, ZipGFP caspase reporters | Live-cell caspase activity sensing |
| Activity-Based Probes | AB50 (caspase-3/7), LE22 (caspase-3/6/7) | Direct labeling of active caspases for biochemical analysis |
| Caspase Inhibitors | z-VAD-fmk (pan-caspase), DEVD-CHO (caspase-3/7) | Specificity controls, mechanistic studies |
| Apoptosis Inducers | Anti-DR5 antibody, carfilzomib, staurosporine | Positive controls for caspase activation |
| Detection Substrates | Chemiluminescent substrates, fluorogenic caspase substrates | Signal generation in biochemical assays |
Signaling Pathway Visualizations
Apoptosis Signaling Pathways
Diagram 1: Intrinsic and Extrinsic Apoptosis Pathways Converge on Caspase Activation. The extrinsic pathway initiates from extracellular death ligands, while the intrinsic pathway responds to intracellular stress signals. Both pathways converge on executioner caspase activation [46] [4].
Caspase Detection Workflows
Diagram 2: Comparative Workflows for Caspase Detection Methods. Antibody-based methods require cell fixation and multiple incubation steps, while reporter systems enable live-cell imaging but necessitate genetic modification [47] [48] [49].
The selection between antibody-based detection and fluorescent reporter systems for monitoring caspase activation represents a critical methodological decision that directly influences experimental outcomes and interpretations. Antibody-based approaches offer superior spatial resolution and accessibility for fixed samples, making them ideal for endpoint analyses, clinical specimens, and multiplexing applications. In contrast, fluorescent reporters provide unparalleled temporal resolution and enable longitudinal tracking of caspase dynamics in live cells, particularly valuable for kinetic studies and complex physiological models.
Within the framework of intrinsic versus extrinsic apoptosis research, both techniques contribute unique insights. Antibody methods effectively capture the rapid, receptor-proximal events of extrinsic apoptosis initiation, while fluorescent reporters excel at documenting the delayed but amplified caspase activation characteristic of the intrinsic pathway. The emerging integration of these complementary approaches, coupled with advanced detection technologies such as FLIM and high-content imaging, continues to expand our understanding of caspase biology in health and disease. As research progresses toward increasingly complex model systems and therapeutic applications, the strategic selection and implementation of these detection methodologies will remain fundamental to advancing apoptosis research.
Apoptosis, or programmed cell death, is a fundamental process regulated through two primary signaling pathways: the intrinsic and extrinsic pathways. Both pathways converge on the activation of caspases, a family of cysteine proteases that act as the main executioners of cell death [11]. The intrinsic pathway is initiated from within the cell by internal stresses such as DNA damage, oxidative stress, or growth factor deprivation, leading to mitochondrial outer membrane permeabilization (MOMP) and the release of pro-apoptotic proteins like cytochrome c into the cytosol [4]. The extrinsic pathway, in contrast, is triggered by extracellular signals binding to death receptors on the cell surface, leading to the formation of the Death-Inducing Signaling Complex (DISC) [11]. This technical guide provides detailed methodologies for assaying key events in each pathway, enabling researchers to dissect the complex mechanisms of caspase activation.
The Intrinsic Apoptosis Pathway: Key Events and Assays
The intrinsic apoptosis pathway is characterized by mitochondrial dysfunction, a pivotal event in cellular stress response. Two key assay targets for monitoring this pathway are changes in mitochondrial membrane potential (MMP) and the release of cytochrome c.
Mitochondrial Membrane Potential (MMP) Assays
MMP is an essential component for maintaining the electrochemical gradient needed for ATP synthesis. Its dissipation is an early indicator of mitochondrial dysfunction and intrinsic apoptosis activation [50] [51].
Fluorescent Dyes for MMP Detection
Table 1: Fluorescent Dyes for Mitochondrial Membrane Potential Assays
| Dye Name | Detection Method | Readout | Healthy vs. Apoptotic Mitochondria | Compatible Platforms | Fixable? |
|---|---|---|---|---|---|
| JC-1 | Ratiometric (dual-emission) | Emission shift: 529 nm (monomer, green) to 590 nm (J-aggregates, red) | Healthy: High red/green ratio; Apoptotic: Low red/green ratio | Fluorescence microscopy, Flow cytometry | No [52] |
| TMRM / TMRE | Single-emission, intensity-based | Fluorescence intensity at ~574 nm | Healthy: Bright fluorescence; Apoptotic: Dim fluorescence | Live-cell imaging, Flow cytometry, Fluorescence microscopy | No [52] [51] |
| DiOC₂(3) | Ratiometric | Emission shift: ~497 nm (monomer) to >650 nm (aggregate) | Healthy: Red-shifted emission; Apoptotic: Green emission | Flow cytometry | No [52] |
| m-MPI | Ratiometric (dual-emission) | Emission at 535 nm (monomer, green) and 590 nm (aggregate, red) | Healthy: High red/green ratio; Apoptotic: Low red/green ratio | High-throughput screening (1536-well plates) | No [50] |
Quantitative High-Throughput Screening (qHTS) Protocol for MMP
This protocol adapts the MMP assay for high-throughput screening in 1536-well plates using the water-soluble m-MPI indicator [50].
- Cell Culture and Plating: Harvest HepG2 cells from 80-90% confluent T225 flasks using Trypsin-EDTA. Centrifuge at 900 rpm for 4 minutes and resuspend in culture medium. Plate cells at a density of 2,000 cells per well in 5 μL of culture medium into black-walled, clear-bottom 1536-well plates using a multidispensor [50].
- Incubation and Compound Treatment: Incubate assay plates overnight at 37°C, 5% CO2 for cell adhesion. Treat adhered cells with 23 nL of test compounds or controls (e.g., FCCP as a positive control for depolarization) using a pintool workstation. Incubate plates at 37°C for 1 h or 5 h depending on the experimental design [50].
- Dye Loading and Detection: After compound incubation, add 5 μL of 2x m-MPI dye-loading solution to each well. Incubate plates at 37°C for 30 minutes. Measure fluorescence intensity using a plate reader with appropriate filter sets: 485 nm excitation/535 nm emission for green fluorescent monomers and 540 nm excitation/590 nm emission for red fluorescent aggregates [50].
- Data Analysis: Express data as the ratio of 590 nm/535 nm emissions, which serves as an indicator of MMP. The positive control FCCP should concentration-dependently decrease MMP with characteristic IC50 values (e.g., 44 nM and 116 nM for 1 h and 5 h treatments, respectively) [50].
Real-Time Live-Cell MMP Monitoring with the Incucyte System
The Incucyte MMP Orange Reagent Kit enables kinetic measurements of MMP in live cells within a cell culture incubator, compatible with multiplexing with other cell health reagents [51].
- Protocol: Seed cells in a multiwell plate and allow to adhere. Add the Incucyte MMP Orange Reagent according to the manufacturer's recommended concentration. Add experimental compounds (e.g., FCCP for depolarization, Oligomycin A for hyperpolarization). Place the plate in the Incucyte live-cell analysis system for continuous monitoring. Fluorescence intensity is quantified automatically over time, with depolarization indicated by decreased fluorescence and hyperpolarization by increased fluorescence [51].
- Multiplexing Capability: The MMP Orange Reagent can be multiplexed with Incucyte Caspase-3/7 reagents, Annexin V for phosphatidylserine exposure, or Cytotox dyes for simultaneous monitoring of multiple cell death parameters [51].
Cytochrome c Release Assays
Cytochrome c release from mitochondria is a hallmark of intrinsic apoptosis activation. Upon release into the cytosol, cytochrome c binds to Apaf-1, forming the apoptosome complex that activates caspase-9, which in turn activates effector caspases [11] [4].
Flow Cytometry-Based Cytochrome c Release Assay
This method enables quantitative analysis of cytochrome c release in a large number of cells, overcoming limitations of Western blotting and immunocytochemistry [53].
- Cell Treatment and Permeabilization: Induce apoptosis in cells (e.g., HL-60 cells with staurosporine or thymocytes with dexamethasone). Collect cells at appropriate time points. Wash cells with PBS and selectively permeabilize the plasma membrane using a low concentration of digitonin (optimized for each cell type) for a brief period (e.g., 5 minutes) on ice. This step allows the release of cytosolic cytochrome c while retaining mitochondrial cytochrome c [53].
- Fixation and Immunostaining: After selective permeabilization, fix cells with paraformaldehyde to preserve cellular structures. Permeabilize the mitochondrial membranes with a stronger detergent (e.g., saponin or Triton X-100) to allow antibody access. Incubate with anti-cytochrome c primary antibody, followed by appropriate fluorescently-labeled secondary antibody [53].
- Flow Cytometry Analysis: Analyze labeled cells by flow cytometry. Cells that have retained cytochrome c in their mitochondria will show high fluorescence intensity, while cells that have released cytochrome c will display low fluorescence. The percentage of cells in each population can be quantified over time to track apoptosis progression [53] [54].
Diagram Title: Intrinsic Apoptosis Pathway and Assay Targets
The Extrinsic Apoptosis Pathway: DISC Analysis
The extrinsic apoptosis pathway initiates when extracellular death ligands (e.g., FasL, TRAIL) bind to their cognate death receptors, leading to the formation of the Death-Inducing Signaling Complex (DISC) and activation of initiator caspases-8 and -10 [10] [11].
DISC Immunoprecipitation Protocol
DISC immunoprecipitation allows for the isolation and analysis of the native protein complex that forms upon death receptor activation.
- Cell Stimulation and Lysis: Treat cells (typically 1-5 × 10⁷ cells per condition) with the death ligand of interest (e.g., FasL, TRAIL) for varying time points (typically 0-30 minutes) to activate the death receptors. Prepare a control sample without stimulation. Rapidly lyse cells using a mild, non-denaturing lysis buffer (e.g., 1% Triton X-100 or CHAPS in PBS) containing protease and phosphatase inhibitors to preserve protein interactions. Keep samples at 4°C throughout the process [10] [11].
- Immunoprecipitation: Pre-clear the cell lysates by incubation with protein A/G beads for 30 minutes at 4°C. Incubate the pre-cleared lysates with antibody against the death receptor cytoplasmic domain (e.g., anti-Fas, anti-DR5) or against associated proteins (e.g., anti-FADD) overnight at 4°C with gentle rotation. Add protein A/G beads and incubate for an additional 2-4 hours to capture the immune complexes. Pellet beads by gentle centrifugation and wash extensively with lysis buffer to remove non-specifically bound proteins [10] [11].
- Analysis of Immunoprecipitated Complex: Elute bound proteins by boiling in SDS-PAGE sample buffer. Analyze by Western blotting for DISC components: caspase-8 (both pro-form and processed forms), FADD, FLIP, and the death receptor. Quantify the recruitment and processing of caspase-8 to assess DISC activity. Alternatively, silver staining or mass spectrometry can be used to identify novel DISC components [10] [11].
Caspase Activation Hierarchy in Apoptosis Pathways
Understanding the sequence of caspase activation events is crucial for interpreting apoptosis assay results.
Table 2: Caspase Activation Hierarchy and Key Regulatory Points
| Pathway | Initiator Caspases | Activator Complex | Effector Caspases | Key Regulatory Factors | Experimental Notes |
|---|---|---|---|---|---|
| Extrinsic | Caspase-8, Caspase-10 | DISC (Death-Inducing Signaling Complex) | Caspase-3, Caspase-7 | FLIP (inhibits caspase-8 activation), FADD | Caspase-8 can directly activate caspase-3 or engage mitochondrial amplification via BID cleavage [10] [11] |
| Intrinsic | Caspase-9 | Apoptosome (Apaf-1 + cytochrome c) | Caspase-3, Caspase-7 | Bcl-2 family proteins, IAPs, SMAC/Diablo | Caspase-9 activation leads to caspase-3/7 activation, which then processes caspases-2 and -6 [10] [11] |
| Regulatory Cross-Talk | Partial redundancy exists between caspases-3 and -7; they show distinct substrate preferences despite overlapping functions [10] |
Diagram Title: Extrinsic Apoptosis Pathway and DISC Assay
Research Reagent Solutions
Table 3: Essential Reagents and Kits for Apoptosis Pathway Assays
| Assay Type | Product Name | Key Components | Primary Applications | Vendor Examples |
|---|---|---|---|---|
| MMP Assays | MitoProbe JC-1 Assay Kit | JC-1 dye, DMSO, CCCP (control) | Flow cytometry-based MMP detection | Thermo Fisher Scientific [52] |
| Incucyte MMP Orange Reagent Kit | MMP Orange Dye, FCCP, Oligomycin A | Live-cell, kinetic MMP imaging | Sartorius [51] | |
| m-MPI Mitochondrial Membrane Potential Indicator | m-MPI dye, assay buffer | High-throughput screening (1536-well plates) | Codex BioSolutions [50] | |
| Cytochrome c Release | Anti-cytochrome c antibodies | Primary antibodies against cytochrome c | Immunodetection in flow cytometry, microscopy | Multiple vendors |
| DISC Analysis | Death receptor antibodies (anti-Fas, anti-DR5) | Antibodies for immunoprecipitation | DISC immunoprecipitation studies | Multiple vendors |
| FADD antibodies | Antibodies against DISC components | Western blot analysis of DISC | Multiple vendors | |
| Caspase Activity | Caspase-8 substrates & antibodies | Fluorogenic substrates, specific antibodies | DISC activity measurements | Multiple vendors |
| General Apoptosis | Annexin V conjugates | Annexin V with various fluorophores | Phosphatidylserine exposure detection | Bio-Rad, Thermo Fisher [55] [56] |
| CellTiter-Glo Luminescent Assay | Luciferase-based reagents | Cell viability assessment | Promega [50] |
The assays described in this guide—MMP measurement, cytochrome c release detection, and DISC immunoprecipitation—provide robust methods for specifically investigating the intrinsic and extrinsic apoptosis pathways. The quantitative data generated from these techniques enable researchers to dissect the complex regulatory mechanisms controlling caspase activation and cell fate decisions. With the growing importance of apoptosis research in drug discovery and development, particularly in oncology and neurodegenerative diseases, these pathway-specific assays continue to be refined through technological advancements in high-throughput screening, live-cell imaging, and multiplexed assay formats [55] [56]. The integration of these methods with emerging technologies such as AI-powered analysis and automated platforms will further enhance our understanding of apoptotic mechanisms and accelerate the development of novel therapeutics targeting these critical cell death pathways.
Caspases, an evolutionarily conserved family of cysteine-dependent aspartate-directed proteases, serve as master regulators of programmed cell death (PCD) and inflammation [57] [20]. These enzymes are synthesized as inactive zymogens (procaspases) and undergo proteolytic activation at specific aspartic acid residues in response to apoptotic signals [57]. Caspases are strategically positioned at the convergence point of the two principal apoptotic pathways: the intrinsic (mitochondrial) and extrinsic (death receptor) pathways [58] [59]. The intrinsic pathway is initiated by internal cellular stressors such as DNA damage, oxidative stress, or growth factor deprivation, leading to mitochondrial outer membrane permeabilization (MOMP) and release of cytochrome c into the cytosol [20] [60]. Cytochrome c then facilitates the formation of the apoptosome complex, comprising Apaf-1 and procaspase-9, resulting in caspase-9 activation [61] [20]. In contrast, the extrinsic pathway is triggered by extracellular death ligands such as FasL, TNF-α, and TRAIL binding to their corresponding death receptors on the cell surface [58] [14]. This ligand-receptor interaction promotes assembly of the death-inducing signaling complex (DISC), which recruits and activates initiator caspases-8 and -10 [20] [14].
Once activated, these initiator caspases (caspase-8, -9, -10) propagate the death signal by cleaving and activating executioner caspases (caspase-3, -6, -7), which in turn mediate the systematic dismantling of the cell through cleavage of hundreds of cellular substrates [20] [58]. Given their pivotal role in controlling cell survival and death, caspases have emerged as attractive therapeutic targets for a wide spectrum of diseases, including neurodegenerative disorders, ischemic injuries, autoimmune conditions, and cancer [57] [60] [14]. This whitepaper provides a comprehensive technical overview of pharmacological caspase inhibitors, with particular emphasis on their mechanisms of action, specificity profiles, and experimental applications within the context of intrinsic versus extrinsic apoptosis research.
Caspase Inhibitor Classes: Mechanisms and Specificity Profiles
Peptide-Based Caspase Inhibitors
Peptide-based inhibitors represent the first generation of synthetic caspase inhibitors designed to mimic the natural substrate recognition motifs of caspases [57]. These compounds typically consist of a short peptide sequence (tetrapeptide or pentapeptide) corresponding to the cleavage site of known caspase substrates, conjugated to an electrophilic functional group that covalently binds the catalytic cysteine residue in the caspase active site [57].
Table 1: Characterization of Common Peptide-Based Caspase Inhibitors
| Inhibitor Name | Peptide Motif | Warhead Group | Primary Caspase Targets | Reversibility | Research Applications |
|---|---|---|---|---|---|
| Z-VAD-FMK | Val-Ala-Asp | Fluoromethyl ketone (FMK) | Pan-caspase inhibitor | Irreversible | Broad-spectrum apoptosis inhibition |
| Q-VD-OPh | Val-Asp | Phenoxy | Pan-caspase inhibitor | Irreversible | In vivo studies (reduced toxicity) |
| Ac-DEVD-CHO | Asp-Glu-Val-Asp | Aldehyde | Caspase-3/-7 | Reversible | In vitro enzyme assays |
| Ac-YVAD-CHO | Tyr-Val-Ala-Asp | Aldehyde | Caspase-1 | Reversible | Inflammation research |
| Z-VDVAD-FMK | Val-Asp-Val-Ala-Asp | Fluoromethyl ketone (FMK) | Caspase-2 | Irreversible | DNA damage response studies |
The peptide moiety determines caspase specificity, with DEVD sequences selectively targeting caspase-3/7 (mimicking the PARP cleavage site), while YVAD sequences preferentially inhibit caspase-1 (mimicking the pro-IL-1β cleavage site) [57]. The electrophilic "warhead" group covalently modifies the thiol group of the catalytic cysteine residue, with different warheads conferring distinct pharmacological properties [57]. Aldehyde-based inhibitors (e.g., Ac-DEVD-CHO) form reversible thiohemiacetal adducts and are primarily used for in vitro applications due to poor metabolic stability and cell permeability [57]. In contrast, fluoromethyl ketone (FMK) derivatives (e.g., Z-VAD-FMK) form irreversible thioether bonds with the catalytic cysteine and exhibit improved cell permeability, making them suitable for cellular applications [57]. Q-VD-OPh, a broad-spectrum caspase inhibitor incorporating a phenoxy group, demonstrates enhanced efficacy and significantly reduced toxicity in vivo compared to earlier generation inhibitors, even at high concentrations (up to 500-1000 μM) [57].
Allosteric Caspase Inhibitors
Allosteric caspase inhibitors represent a more recent class of compounds that modulate enzyme activity by binding to regulatory sites distinct from the catalytic cleft, thereby inducing conformational changes that suppress catalytic function [61] [62]. High-throughput screening efforts identified the first class of non-peptide allosteric inhibitors (Comp-A, B, C, D), which share a common pyridinyl, copper-containing multi-ring structure [61]. These compounds inhibit multiple caspases with sub-micromolar IC₅₀ values but exhibit significantly reduced potency against other cysteine proteases such as cathepsin C and papain (IC₅₀ > 5 μM) [61].
Table 2: Profile of Characterized Allosteric Caspase Inhibitors
| Inhibitor Name/Class | Chemical Structure | Mechanism of Action | Caspase Targets | IC₅₀ Values | Specificity Notes |
|---|---|---|---|---|---|
| Compound A | Pyridinyl copper complex | Binds dimerization interface | Caspase-3, -7, -8, -9, -1 | Sub-micromolar | >10-fold selectivity over cathepsin C |
| Compound B | Pyridinyl copper complex | Binds dimerization interface | Caspase-3, -7, -8, -9, -1 | Sub-micromolar | >10-fold selectivity over cathepsin C |
| Compound C | Pyridinyl copper complex | Binds dimerization interface | Caspase-3, -7, -8, -9, -1 | Sub-micromolar | >10-fold selectivity over papain |
| Compound D | Covalent dimer of pyridinyl complexes | Binds dimerization interface | Caspase-3, -7, -8, -9, -1 | Sub-micromolar | Similar potency for caspase-7 and papain |
| Pifithrin-μ (PFTμ) | Small molecule | Promiscuous caspase inhibitor | Multiple caspases | Not specified | Also inhibits TP53 |
Structural analyses via X-ray crystallography have revealed that these allosteric inhibitors bind specifically to the dimerization interface of caspases, a common structural element shared by all active caspases [61]. This binding mode disrupts the catalytic activity by altering the conformation of the active site without directly occupying the substrate-binding cleft [61] [62]. Biochemical studies demonstrate that these compounds effectively abrogate caspase-8 dimerization, providing mechanistic insight into their inhibitory action [61]. The allosteric mechanism offers several potential advantages over active-site-directed inhibitors, including improved selectivity and the ability to target inactive zymogen forms, which may exhibit distinct structural features compared to their activated counterparts [62].
Experimental Protocols for Caspase Inhibition Studies
High-Throughput Screening for Allosteric Inhibitors
The discovery of novel allosteric caspase inhibitors has been facilitated by reconstituted biochemical assays adapted for high-throughput screening (HTS) platforms. The following protocol outlines the methodology employed to identify the pyridinyl copper-based allosteric inhibitors:
Protocol 1: HTS for Inhibitors of Cytochrome c-Mediated Caspase Activation
Assay Reconstitution: Prepare the intrinsic pathway components by combining purified recombinant proteins at near-physiological concentrations: Apaf-1 (50 nM), cytochrome c (10 μM), caspase-9 (50 nM), procaspase-3 (100 nM), and dATP (1 mM) in assay buffer [61].
Automated Screening: Adapt the reconstituted system to a 384-well plate format using an automated screening platform. Incubate test compounds (10 μM final concentration) with the pathway components for 30 minutes at 37°C [61].
Activity Measurement: Initiate the reaction by adding the fluorogenic caspase-3 substrate Ac-DEVD-AFC (10 μM final concentration). Monitor fluorescence emission (λex = 400 nm, λem = 505 nm) continuously over 60 minutes to quantify caspase-3 activation kinetics [61].
Hit Identification: Apply a threshold of ≥30% inhibition relative to DMSO-treated controls for initial hit selection. For confirmed hits, determine IC₅₀ values through dose-response titration (typically 0.01-100 μM range) [61].
Target Deconvolution: Identify the direct caspase target through follow-up assays using individually recombinant active caspases (caspase-3, -7, -8, -9, -1) to assess direct inhibition independent of the activation complex [61].
This HTS approach achieved a high signal-to-noise ratio of 19:1 with Z′-factors ranging from 0.59 to 0.75, indicating a robust screening assay suitable for large compound library evaluation [61].
Engineering Activation-Based Screening Platforms
Recent innovations in caspase inhibitor screening involve engineering activatable caspase constructs that enable identification of zymogen-selective inhibitors. The following protocol describes the development of a TEV-activatable caspase-10 system:
Protocol 2: TEV-Activatable Caspase-10 Screening Platform
Protein Engineering: Generate engineered procaspase-10 (proCASP10TEV Linker) by replacing the endogenous caspase cleavage sites (D415 and D435) with tobacco etch virus (TEV) protease recognition sequences (ENLYFQG) using site-directed mutagenesis [63].
Protein Purification: Express the engineered procaspase-10 in E. coli and purify using nickel-affinity chromatography followed by size-exclusion chromatography. Confirm the absence of pre-activated caspase in preparations through Rho-DEVD-AOMK labeling and SDS-PAGE analysis [63].
Assay Optimization: Characterize TEV-dependent activation by incubating proCASP10TEV Linker (333 nM) with TEV protease (667 nM) in the presence of fluorogenic substrate Ac-VDVAD-AFC (10 μM). Validate minimal background activity in the absence of TEV protease and comparable kinetics to recombinant active caspase-10 [63].
High-Throughput Screening: Implement a ∼100,000 compound screen in 384-well format, incubating test compounds with proCASP10TEV Linker prior to TEV protease addition. Define hit criteria as compounds exhibiting Z-score less than -3 relative to DMSO controls [63].
Counter-Screening: Eliminate non-specific hits and TEV protease inhibitors through counter-screening against directly activated caspase-10 and TEV protease alone [63].
This innovative platform enables discovery of inhibitors that preferentially target the zymogen conformation, potentially offering enhanced selectivity by exploiting structural differences between inactive procaspases and their activated forms [63].
Signaling Pathways and Molecular Mechanisms
The following diagrams illustrate the molecular mechanisms of caspase inhibitors in the context of intrinsic and extrinsic apoptotic pathways.
Diagram 1: Caspase Inhibition in Intrinsic and Extrinsic Apoptotic Pathways. Allosteric inhibitors (green) bind the dimerization interface of multiple caspases, while peptide-based inhibitors (green) target the catalytic site. Both inhibitor classes suppress caspase activation in intrinsic (left) and extrinsic (right) pathways. The dotted lines represent inhibitory actions.
Research Reagent Solutions Toolkit
Table 3: Essential Research Reagents for Caspase Inhibition Studies
| Reagent Category | Specific Examples | Research Applications | Key Features & Considerations |
|---|---|---|---|
| Broad-Spectrum Caspase Inhibitors | Z-VAD-FMK, Q-VD-OPh | Initial apoptosis confirmation, pan-caspase inhibition studies | Q-VD-OPh offers reduced cellular toxicity; Z-VAD-FMK widely validated but more toxic |
| Specific Peptide Inhibitors | Ac-DEVD-CHO (caspase-3/7), Ac-YVAD-CHO (caspase-1), Z-VDVAD-FMK (caspase-2) | Mechanistic studies of specific caspase functions | Aldehyde derivatives reversible, suitable for enzymatic assays; FMK derivatives irreversible, cell-permeable |
| Allosteric Inhibitors | Compounds A-D (pyridinyl copper complexes) | Studies requiring non-active site inhibition, structural biology | Sub-micromolar potency, bind dimerization interface, preferential inhibition of caspases over other proteases |
| Activity Assay Systems | Fluorogenic substrates (Ac-DEVD-AFC, Ac-VDVAD-AFC), Rho-DEVD-AOMK | Quantitative caspase activity measurement, high-throughput screening | AFC-based substrates for kinetic measurements; AOMK probes for active-site labeling |
| Engineered Caspase Proteins | proCASP10TEV Linker, Caspase-9-LZ | Screening for zymogen-selective inhibitors, mechanistic studies | TEV-cleavable constructs enable controlled activation; leucine zipper fusions force dimerization |
| Cell-Based Assay Systems | UV-treated HeLa cells, TNF-α + CHX treated U937 cells | Validation of inhibitor efficacy in cellular context | Multiple cell lines recommended to assess cell-type specific effects |
This curated toolkit provides researchers with essential reagents for comprehensive caspase inhibition studies, from initial target identification to mechanistic characterization and cellular validation.
Pharmacological caspase inhibitors represent invaluable tools for dissecting apoptotic signaling pathways and developing novel therapeutic strategies. Peptide-based inhibitors, particularly those incorporating irreversible warheads like FMK, continue to serve as workhorse reagents for basic apoptosis research due to their well-characterized specificity and commercial availability [57]. However, their therapeutic application has been hampered by poor pharmacokinetic properties and off-target effects [57]. Allosteric inhibitors offer a promising alternative mechanism for modulating caspase activity, with potential advantages in selectivity and safety profiles [61] [62]. The emerging strategy of targeting caspase zymogens rather than active enzymes represents a particularly innovative approach to enhance inhibitor selectivity, leveraging structural differences between inactive procaspases and their activated forms [63].
Despite these advances, significant challenges remain in translating caspase inhibitors into clinical applications. Only a limited number of synthetic caspase inhibitors have advanced to clinical trials, with none achieving approved clinical use to date, primarily due to inadequate efficacy, poor target specificity, or adverse side effects [57]. Future research directions should focus on elucidating the non-apoptotic functions of caspases, understanding compensatory pathways activated upon caspase inhibition, and developing more sophisticated targeting strategies that account for the complex interplay between different cell death modalities [57] [20] [60]. As our understanding of caspase biology continues to evolve, particularly their roles in non-apoptotic processes and interactions with other cell death pathways, next-generation caspase inhibitors with enhanced specificity and therapeutic potential will undoubtedly emerge.
Caspases are an evolutionarily conserved family of cysteine-dependent proteases that cleave their substrates at specific aspartic acid residues, playing a central role in programmed cell death (PCD) and inflammation [3]. These enzymes are crucial for maintaining cellular homeostasis, with dysregulated caspase functions being linked to a wide array of diseases, including liver disorders and neurodegenerative conditions [57] [3]. Caspases are broadly categorized based on their function in apoptosis (caspases-2, -3, -6, -7, -8, -9, -10) and inflammation (caspases-1, -4, -5, -12) [64]. The historic belief of caspases as mediators of apoptosis and inflammation has rendered them attractive therapeutic targets for several diseases [57].
Apoptosis occurs through two main signaling pathways: the intrinsic pathway (mitochondrial), activated by internal cellular stress, and the extrinsic pathway (death receptor), initiated by extracellular signals [4] [65]. Both pathways converge on the activation of effector caspases that execute the final stages of cell death [66]. Inhibiting caspases offers a novel therapeutic strategy for treating apoptosis-related and inflammatory diseases, though developing effective and safe caspase therapeutics has faced consistent challenges [57] [64].
Caspase Activation in Intrinsic versus Extrinsic Apoptosis
Molecular Pathways of Apoptosis
The following diagram illustrates the key components and sequence of events in the intrinsic and extrinsic apoptotic pathways.
Key Differences Between Intrinsic and Extrinsic Apoptosis
Table 1: Characteristics of Intrinsic and Extrinsic Apoptotic Pathways
| Feature | Intrinsic Pathway | Extrinsic Pathway |
|---|---|---|
| Initiation | Internal cellular stress (DNA damage, oxidative stress) [4] | External death ligands (FasL, TNF-α) binding death receptors [4] |
| Key Regulators | Bcl-2 family proteins, p53 [4] [65] | Death receptors, FADD, FLIP [4] |
| Activation Complex | Apoptosome (Apaf-1, cytochrome c, caspase-9) [64] | DISC (Death-Inducing Signaling Complex) [64] |
| Initiator Caspase | Caspase-9 [4] [64] | Caspase-8, -10 [64] |
| Mitochondrial Involvement | Central (MOMP, cytochrome c release) [4] | Optional (via Bid cleavage) [4] |
| Physiological Context | Development, cellular damage, oncogene activation [65] | Immune regulation, removal of infected cells [65] |
Caspase Inhibitors in Liver Disease
Clinical Evidence and Therapeutic Potential
The role of caspases in liver disease presents a complex paradox. While excessive apoptosis is implicated in various forms of liver failure, evidence suggests that caspase activation may also be associated with spontaneous recovery in acute liver failure (ALF) [67]. A pivotal study investigating 70 ALF patients using novel biomarkers for caspase activation revealed that patients with spontaneous recovery from ALF showed significantly higher activation of caspases than patients who required transplantation or died, despite the latter patients exhibiting extensive DNA fragmentation and signs of non-apoptotic cell death [67]. In the spontaneous survivors, increased caspase activation was accompanied by elevated levels of TNF-α and IL-6, important cytokines involved in liver regeneration [67].
This finding suggests that caspase activation and apoptosis are involved in ALF of patients with spontaneous recovery, whereas caspase-independent cell death might be more relevant in irreversible forms of liver failure [67]. These findings have significant implications for therapeutic interventions, suggesting that measurement of caspase activation might be of prognostic value to predict the outcome of acute liver failure [67].
Caspase Inhibitors in Clinical Trials for Liver Disease
Several caspase inhibitors have entered clinical trials for liver diseases. Emricasan (IDN-6556), an irreversible pan-caspase inhibitor, was developed for the treatment of liver diseases [57]. Preclinical and clinical studies showed its efficacy; however, extended treatment with IDN-6556 revealed side effects, and its clinical development was terminated for undisclosed reasons [57]. Current clinical trials listed by the Advanced Liver Therapies Research Center include:
Table 2: Caspase Inhibitors in Clinical Trials for Liver Disease
| Drug/Candidate | Target | Clinical Condition | Development Status |
|---|---|---|---|
| Emricasan (IDN-6556) | Pan-caspase inhibitor | Liver diseases | Clinical development terminated due to side effects [57] |
| IDN-6554 | Caspase inhibitor | NASH with Cirrhosis | Currently enrolling [68] |
| IDN-6556 | Caspase inhibitor | NASH with Cirrhosis | Currently enrolling [68] |
The challenges faced in developing caspase inhibitors for liver diseases include inadequate efficacy, poor target specificity, or adverse side effects [57]. A significant proportion of these failures lies in the incomplete understanding of various caspase functions beyond apoptosis [57].
Caspase Inhibitors in Neurodegenerative Disorders
Preclinical Evidence in Alzheimer's Disease Models
Caspase-1 inhibition has emerged as a promising therapeutic strategy for neurodegenerative disorders, particularly Alzheimer's disease (AD). The association of several inflammation-related genetic markers with AD and the early activation of pro-inflammatory pathways in AD suggest inflammation as a plausible therapeutic target [69]. Inflammatory Caspase-1 has a significant impact on AD-like pathophysiology, and Caspase-1 inhibitor VX-765 reverses cognitive deficits in AD mouse models [69].
A groundbreaking study demonstrated that a one-month pre-symptomatic treatment of Swedish/Indiana mutant amyloid precursor protein (APPSw/Ind) J20 and wild-type mice with VX-765 delayed both APPSw/Ind- and age-induced episodic and spatial memory deficits [69]. VX-765 treatment delayed inflammation without considerably affecting soluble and aggregated amyloid beta peptide (Aβ) levels, and episodic memory scores correlated negatively with microglial activation [69]. These results suggest that Caspase-1-mediated inflammation occurs early in the disease and indicates that VX-765 may be a useful drug to prevent the onset of cognitive deficits and brain inflammation in AD [69].
Pharmacokinetics and Experimental Protocols
The experimental workflow and key findings from the VX-765 preclinical study in Alzheimer's models are summarized below:
The pharmacokinetic profile of VX-765 revealed that both VX-765 and its active metabolite VRT-043198 reached plasma concentrations 100- and 20,000-fold, brain concentrations 50- and 170-fold, and CSF concentrations 100- and 4,000-fold their respective in vitro Caspase-1 IC₅₀ values within 0.25 hours post-injection [69]. VRT-043198 levels surpassed VX-765 concentrations, indicating rapid conversion of the prodrug [69]. Although brain VX-765 and VRT-043198 levels were short-lived, the data confirmed that both compounds reach the brain at concentrations sufficient to inhibit Caspase-1 after a single intraperitoneal injection [69].
Clinical Trial Status for Neurodegenerative Indications
While VX-765 (belnacasan) showed promise for inflammatory diseases and entered clinical trials, development for rheumatoid arthritis and osteoarthritis was terminated due to liver toxicity induced by high doses in animal models [57]. Similarly, clinical trials for VX-765 were terminated because of liver toxicity concerns, despite its potential efficacy [57]. The drug has been previously evaluated in human CNS clinical trials, raising hope for its potential application in preventing cognitive deficits in AD [69].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Caspase and Apoptosis Studies
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Caspase Inhibitors | Z-VAD-FMK (pan-caspase), Q-VD-OPh, VX-765 (Caspase-1 specific) [57] | Investigate caspase functions; potential therapeutic candidates [57] |
| Activity Assays | Caspase-Glo Assays, fluorometric substrates (e.g., DEVD-AFC) [57] | Measure caspase activation and enzymatic activity in cells/tissues |
| Apoptosis Detection | TUNEL Assay, Annexin V staining, mitochondrial membrane potential dyes (TMRE) [65] | Detect and quantify apoptotic cells; assess mitochondrial health [65] |
| Cleavage-Specific Antibodies | Anti-cleaved caspase-3, anti-cleaved PARP, anti-cleaved cytokeratin-18 [70] | Detect caspase activity and substrate cleavage in situ (IHC, WB) [70] |
| Pathway-Specific Reagents | BH3 mimetics (Venetoclax), death receptor agonists/antagonists [65] | Modulate specific apoptotic pathways; therapeutic development [65] |
Challenges and Future Perspectives
The development of caspase inhibitors as therapeutics has faced consistent challenges, with only a few candidates progressing to clinical trials and none yet achieving successful clinical use [57]. The primary obstacles include inadequate efficacy, poor target specificity, or adverse side effects [57]. A significant limitation is the incomplete understanding of the non-apoptotic and non-inflammatory roles of caspases and how they contribute to cell function and disease [57].
Emerging evidence has shown that upon targeted caspase inhibition, alternative signaling pathways and caspase-independent cell death processes can be activated [57]. Furthermore, caspases and their targeted proteins mediate multiple cellular processes far beyond their apoptotic and inflammatory functions [57]. This complexity necessitates further studies on understanding caspase function in disease models to effectively develop their inhibitors as treatments for different pathologies [57].
Future directions should focus on developing more specific caspase inhibitors with improved safety profiles, identifying biomarkers for patient stratification, and determining optimal treatment timing in disease progression. The paradoxical finding that caspase activation is associated with spontaneous recovery in acute liver failure [67] underscores the need for a more nuanced understanding of caspase biology in different disease contexts. Similarly, the promising results with Caspase-1 inhibition in Alzheimer's models [69] highlight the potential of targeting inflammatory caspases in neurodegenerative disorders, despite previous setbacks in clinical development.
The relentless proliferation of cancer cells is not merely a consequence of uncontrolled division but is equally attributable to a failure of programmed cell death, or apoptosis. This meticulously regulated process is governed by two primary signaling pathways: the extrinsic pathway, initiated by extracellular death ligands binding to cell surface receptors, and the intrinsic pathway, activated by internal cellular stresses such as DNA damage [5] [66]. Both pathways converge on the activation of a cascade of cysteine-aspartic proteases, or caspases, which execute the final stages of cellular dismantling [71]. A critical regulatory node within the intrinsic pathway is the BCL-2 family of proteins, which arbitrates cell fate by controlling mitochondrial outer membrane permeabilization (MOMP) and the subsequent release of cytochrome c, a pivotal step in caspase activation [30] [72]. Parallel to this, the Inhibitor of Apoptosis (IAP) proteins function as a secondary regulatory layer, directly suppressing caspase activity [73]. The strategic disruption of these upstream regulators, BCL-2 and IAP proteins, represents a paradigm shift in cancer therapy, moving from genotoxic agents to targeted drugs that directly engage the apoptotic machinery to eliminate malignant cells.
Molecular Mechanisms of Apoptotic Regulation
The BCL-2 Family: Guardians of the Mitochondrial Pathway
The BCL-2 protein family constitutes a tripartite apoptotic switch, comprising both pro-survival and pro-apoptotic members that interact through a complex network to determine cellular commitment to death [30] [71].
- Anti-apoptotic Proteins: This group includes BCL-2, BCL-XL, MCL-1, BCL-w, and A1/BFL-1. These proteins share four BCL-2 Homology (BH) domains (BH1-BH4). Their canonical function is to preserve mitochondrial integrity by sequestering pro-apoptotic activators and directly restraining the executioner proteins BAX and BAK, thereby preventing MOMP [30] [72] [74]. Their activity is often heightened in cancer cells, conferring a survival advantage.
- Pro-apoptotic Effectors: The multi-domain proteins BAX and BAK serve as the essential gatekeepers of MOMP. Upon activation, they oligomerize within the mitochondrial outer membrane, forming pores that lead to the release of cytochrome c and other apoptogenic factors into the cytosol [72]. Once released, cytochrome c facilitates the formation of the apoptosome, a complex that activates caspase-9, initiating the caspase cascade [5].
- BH3-only Proteins: This sensor group includes BIM, BID, PUMA, BAD, NOXA, and others. They share only the BH3 domain and are activated in response to diverse cellular stresses. They promote apoptosis by neutralizing specific anti-apoptotic proteins. For instance, PUMA and BIM are "promiscuous" binders that can engage all major anti-apoptotic members, while NOXA selectively targets MCL-1 and A1, and BAD selectively targets BCL-2, BCL-XL, and BCL-w [30] [75]. In the prevailing "indirect activation" model, their primary role is to displace anti-apoptotic proteins from BAX/BAK, thereby unleashing these executioners to induce MOMP [71].
The following diagram illustrates the complex interactions and balance of power within the BCL-2 family that regulates the intrinsic apoptotic pathway.
Inhibitor of Apoptosis (IAP) Proteins: Suppressors of Caspase Activity
IAP proteins, including XIAP, cIAP1, and cIAP2, constitute a second tier of apoptotic regulation. Their primary mechanism of action is the direct binding and inhibition of active caspases, particularly caspase-3, -7, and -9, thus functioning as a brake on the apoptotic execution phase [73]. A key endogenous regulator of IAPs is the mitochondrial protein Smac/DIABLO, which is released alongside cytochrome c during MOMP. Smac binds to IAP proteins, displacing them from caspases and thereby promoting apoptosis [5]. The overexpression of IAP proteins is a common mechanism by which cancer cells evade cell death, making them a attractive therapeutic target.
Therapeutic Targeting Strategies and Clinical Agents
BH3-mimetics: Targeting the BCL-2 Family
BH3-mimetics are small molecule drugs designed to mimic the function of native BH3-only proteins. By occupying the hydrophobic groove on anti-apoptotic BCL-2 proteins, they disrupt protein-protein interactions, leading to BAX/BAK activation, MOMP, and the initiation of apoptosis [30] [76]. The clinical development of these agents has been a landmark in targeted cancer therapy.
Table 1: Evolution and Clinical Status of Key BCL-2 Family Inhibitors
| Compound | Primary Targets | Development Status | Key Indications & Notes | References |
|---|---|---|---|---|
| Venetoclax (ABT-199) | BCL-2 | FDA-Approved | CLL, AML; High selectivity avoids thrombocytopenia. | [76] [74] |
| Navitoclax (ABT-263) | BCL-2, BCL-XL, BCL-w | Phase I/II | CLL, SCLC; Dose-limiting thrombocytopenia from BCL-XL inhibition. | [30] [76] |
| Lisaftoclax (APG-2575) | BCL-2 | Phase I/II | Hematologic cancers; Newer selective BCL-2 inhibitor. | [76] |
| Obatoclax (GX15-070) | BCL-2, BCL-XL, MCL-1 | Phase I/II | Hematologic cancers, solid tumors; Broad-targeting, limited by toxicity. | [76] |
| AZD0466 | BCL-2, BCL-XL | Phase I/II | Hematologic cancers, solid tumors; PROTAC-based degrader. | [76] |
| APG-1252 (Pelcitoclax) | BCL-2, BCL-XL | Phase I/II | NSCLC, NHL; Dual inhibitor. | [76] |
The success of venetoclax has spurred the development of next-generation strategies to overcome the limitations of earlier compounds. A major challenge has been the on-target toxicity associated with inhibiting BCL-XL (thrombocytopenia) and MCL-1 (cardiac toxicity) [30]. Innovative approaches such as PROTACs (Proteolysis Targeting Chimeras) and antibody-drug conjugates (ADCs) are being explored to achieve tumor-specific targeting of these proteins, which could be transformational for many cancer subtypes [30] [72].
IAP Antagonists (Smac-mimetics): Releasing the Caspase Brake
Smac-mimetics are designed to replicate the function of the endogenous Smac protein. By binding to IAP proteins, they antagonize their anti-apoptotic activity, leading to the release and activation of caspases and the potentiation of apoptosis [73]. These agents can promote cell death not only by relieving caspase inhibition but also by triggering the auto-ubiquitination and degradation of cIAP1 and cIAP2, which can alter pro-survival NF-κB signaling pathways [73]. Agents like LCL161 and AT-406 have been investigated in clinical trials, often in combination with other chemotherapeutic agents [73].
Experimental Assessment of Apoptotic Regulators
Core Methodologies for Evaluating Apoptosis Induction
Rigorous in vitro assessment is critical for validating the efficacy and mechanism of action of novel apoptotic regulators. The following protocol outlines a standard workflow for evaluating BH3-mimetics and IAP antagonists.
Detailed Experimental Protocol:
Cell Treatment: Seed appropriate cancer cell lines (e.g., hematologic lines for BCL-2 inhibitors) in 96- or 384-well plates. The following day, treat cells with a dose range of the investigational compound (e.g., BH3-mimetic or IAP antagonist). A typical 10-point, 1:3 serial dilution starting from 10 µM is suitable for an initial dose-response curve. Include controls for baseline viability (DMSO vehicle) and maximum killing (e.g., 1 µM Staurosporine). Incubate for 24-72 hours [73].
Viability and Apoptosis Measurement:
- Cell Viability: Quantify overall cell health using colorimetric (MTT) or luminescent (CellTiter-Glo) assays according to manufacturer protocols. CellTiter-Glo measures ATP levels, providing a direct correlation with metabolically active cells.
- Apoptosis Detection: Use Annexin V/propidium iodide (PI) staining followed by flow cytometry. Annexin V binds to phosphatidylserine externalized on the outer leaflet of the plasma membrane in early apoptosis, while PI stains DNA in late apoptotic and necrotic cells with compromised membranes. Caspase activation can be measured concurrently using fluorescently-labeled caspase inhibitors (FLICA) kits [73].
Mechanism of Action Studies:
- Western Blotting: Analyze changes in protein levels of key BCL-2 family members (BCL-2, MCL-1, BIM, BAX) and IAPs (XIAP, cIAP1). Monitor cleavage of caspase-3 and its substrate PARP as definitive markers of apoptosis execution. Cytochrome c release can be assessed by comparing mitochondrial and cytosolic fractions.
- Co-Immunoprecipitation (Co-IP): Validate direct target engagement by immunoprecipitating the anti-apoptotic protein (e.g., BCL-2) from treated cells and probing for disrupted interaction with its pro-apoptotic partners (e.g., BIM) [30] [71].
- BH3 Profiling: This functional assay measures mitochondrial priming. Isolate mitochondria from treated cells and expose them to synthetic BH3 peptides (e.g., from BIM, BAD, NOXA). The pattern of cytochrome c release indicates which anti-apoptotic proteins are dominant and whether the treatment has successfully primed the mitochondria for death [30].
Genotoxicity Assessment of Direct Apoptosis Inducers
A proposed advantage of direct apoptosis inducers over conventional chemotherapy is their potential for lower genotoxicity, as they do not directly damage DNA to kill cells. This can be evaluated using the following assays [73]:
- HPRT Clonogenic Assay: Measures the frequency of mutations at the hypoxanthine-guanine phosphoribosyltransferase (HPRT) locus in clonogenically competent cells. Cells are exposed to the drug, allowed to recover, and then grown in 6-thioguanine (6-TG)-containing media. Only HPRT-mutant cells survive, allowing for quantification of mutation frequency.
- γH2AX Staining: Phosphorylated histone H2AX (γH2AX) is a sensitive marker for DNA double-strand breaks. Detection by flow cytometry or immunoblotting provides a quantitative measure of DNA damage.
- COMET Assay (Single Cell Gel Electrophoresis): Detects single and double-strand DNA breaks at the level of individual cells. Cells embedded in agarose are lysed, subjected to electrophoresis, and stained. DNA damage is visualized as a "comet tail," with the tail length and intensity correlating with the extent of damage.
A key study demonstrated that IAP antagonists (LCL161, AT-406) and physiologically achievable doses of the BH3-mimetic ABT-737 showed negligible genotoxicity, unlike traditional DNA-damaging agents like doxorubicin and etoposide [73].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for Investigating Apoptosis and Targeted Therapy
| Research Reagent / Assay | Primary Function | Experimental Utility | References |
|---|---|---|---|
| ABT-737 | BH3-mimetic (BCL-2, BCL-XL, BCL-w inhibitor) | Lab tool compound for proof-of-concept studies; not orally bioavailable. | [76] [73] |
| Recombinant Human TRAIL | Death receptor agonist (extrinsic pathway) | Positive control for inducing apoptosis; used in combination studies. | [5] [73] |
| Annexin V / PI Staining Kit | Flow cytometry-based apoptosis detection | Distinguishes early apoptotic (Annexin V+/PI-), late apoptotic (Annexin V+/PI+), and necrotic (Annexin V-/PI+) cells. | [5] [73] |
| Caspase-Glo Assay | Luminescent measurement of caspase activity | Quantifies activation of initiator (caspase-8, -9) and effector (caspase-3/7) caspases. | [5] |
| γH2AX Antibody | Immunodetection of DNA double-strand breaks | Key reagent for assessing genotoxic stress via flow cytometry or immunofluorescence. | [73] |
| BH3 Peptides (e.g., BIM, BAD, NOXA) | Synthetic peptides for functional mitochondrial assays | Used in BH3 profiling to determine dependence on specific anti-apoptotic proteins. | [30] |
The targeted inhibition of upstream apoptotic regulators like BCL-2 and IAP proteins has irrevocably altered the therapeutic landscape, particularly in hematologic malignancies. Venetoclax stands as a testament to the success of translating fundamental knowledge of protein-protein interactions into a life-extending clinical therapy [30] [74]. The future of this field lies in overcoming the challenges of on-target toxicities and resistance. Resistance to venetoclax can arise through upregulation of alternative anti-apoptotic proteins like MCL-1 or BCL-XL, driving the need for combination therapies and novel agents [76]. The clinical trajectory is now moving towards rational combination regimens (e.g., venetoclax with azacitidine in AML) and highly innovative tumor-specific targeting strategies [30] [74]. Modalities such as PROTACs, which catalytically degrade target proteins, and ADCs, which deliver cytotoxic payloads directly to cancer cells, hold immense promise for targeting previously undruggable proteins like MCL-1 or for selectively inhibiting BCL-XL in tumors while sparing platelets [30] [72]. As these sophisticated strategies mature, they will undoubtedly expand the applicability of apoptotic targeted therapy, ultimately improving outcomes for patients across a broader spectrum of cancers.
Navigating Experimental Complexities and Overcoming Therapeutic Challenges in Apoptosis Research
Addressing Caspase-3 and -7 Functional Redundancy in Data Interpretation
For decades, caspase-3 and caspase-7 have been widely perceived as functionally redundant executioner caspases due to nearly indistinguishable activity profiles against synthetic peptide substrates. However, emerging evidence from genetic, biochemical, and proteomic studies reveals profound functional specialization between these proteases. This technical guide examines the distinct roles of caspase-3 and caspase-7 within apoptotic signaling networks, providing researchers with frameworks for accurate data interpretation in cell death research. We synthesize current understanding of their differential substrate specificity, structural determinants, and unique contributions to intrinsic and extrinsic apoptosis, offering methodological considerations for investigating their non-redundant functions in both cell death and non-apoptotic processes.
The historical view of caspase-3 and caspase-7 as functionally interchangeable executioners stems from their similar activation mechanisms and nearly identical cleavage preferences toward synthetic tetrapeptide substrates, particularly DEVD-based sequences [38]. Both enzymes are activated downstream of initiator caspases (caspase-8/-10 in extrinsic pathways; caspase-9 in intrinsic pathways) and share structural similarities as members of the caspase family of cysteine proteases [20] [77]. However, this redundancy paradigm fails to explain the distinct phenotypes observed in knockout mouse models, where caspase-3 deficiency causes severe brain developmental defects while caspase-7 deficiency results in viable mice with more subtle phenotypes [38].
Critical analysis of caspase function reveals that synthetic substrate preferences do not accurately predict natural protein substrate specificity. While caspase-3 and caspase-7 both efficiently cleave canonical substrates like PARP, they exhibit marked differences in processing many other cellular proteins [38]. This guide examines the experimental evidence distinguishing caspase-3 and caspase-7 functionality, providing researchers with frameworks for proper data interpretation when investigating apoptotic pathways in both physiological and therapeutic contexts.
Functional Distinctions Between Caspase-3 and Caspase-7
Differential Substrate Specificity and Cleavage Efficiency
Comparative studies using purified recombinant enzymes reveal striking differences in substrate preference between caspase-3 and caspase-7. While both caspases cleave PARP, RhoGDI, and ROCK I with similar efficiency, caspase-3 demonstrates broader substrate promiscuity and more efficient cleavage of many key apoptotic substrates [38].
Table 1: Differential Substrate Cleavage by Caspase-3 and Caspase-7
| Substrate Protein | Caspase-3 Activity | Caspase-7 Activity | Functional Consequences |
|---|---|---|---|
| Bid | Efficient cleavage | Minimal cleavage | Reduces mitochondrial amplification |
| XIAP | Efficient cleavage | Less efficient | Affects caspase inhibition |
| Gelsolin | Efficient cleavage | Less efficient | Alters cytoskeletal reorganization |
| Caspase-6 | Efficient processing | Minimal processing | Impacts caspase cascade propagation |
| Caspase-9 | Efficient feedback processing | Minimal processing | Alters apoptosome function |
| Cochaperone p23 | Less efficient | Efficient cleavage | Affects protein folding complexes |
This differential specificity extends to gasdermin proteins, where caspase-3 cleaves GSDME to induce pyroptosis, while caspase-7 performs non-canonical cleavage of GSDMB and GSDMD that suppresses pyroptotic cell death [20]. These distinctions highlight the unique roles each caspase plays in coordinating different cell death modalities.
Structural Determinants of Functional Differences
The functional divergence between caspase-3 and caspase-7 originates from structural differences despite their 56% sequence identity [38] [78]. Research identifying the functional regions defining different activity reveals that seven specific amino acid regions contribute to their distinct functionalities within cells [78].
Table 2: Structural and Functional Comparisons Between Caspase-3 and Caspase-7
| Characteristic | Caspase-3 | Caspase-7 |
|---|---|---|
| Sequence identity | Reference | 56% compared to caspase-3 |
| Synthetic substrate preference | DEVD-AFC, LEHD-AFC | DEVD-AFC primarily |
| Homodimer formation | Distinct regions critical for specific homodimer-forming activity | Different regions mediating dimerization |
| Intracellular protease activity | Stronger against cellular substrates | Weaker against most cellular substrates |
| Key structural regions | 7 specific regions defining stronger activity | Corresponding regions confer different properties |
Chimeric constructs swapping these regions between caspase-3 and caspase-7 demonstrate that three-dimensional structural features at the homodimer interface determine their functional differences [78]. These structural insights explain why the proteolytic landscapes in stressed viable cells fully depend on executioner caspase activity, with caspase-3 and caspase-7 contributing distinct cleavage patterns [79].
Non-Redundant Biological Roles in Apoptosis
Genetic studies reveal that caspase-3 and caspase-7 fulfill specialized roles during apoptotic execution. Caspase-3 deficiency renders mouse embryonic fibroblasts less sensitive to intrinsic cell death stimuli, while caspase-7 deficiency has minimal impact on cell death sensitivity but significantly impairs apoptotic cell detachment [80].
During intrinsic apoptosis, caspase-9 cleaves Bid at aspartic acid 59 to generate tBid, which drives mitochondrial remodeling and ROS production. Caspase-3 subsequently limits ROS production and is essential for efficient cell killing, while caspase-7 regulates cell detachment without significantly affecting death kinetics [80]. This functional specialization explains why double knockout of both caspases is required to completely ablate executioner caspase function in many systems.
Methodological Considerations for Caspase Research
Experimental Approaches for Distinguishing Caspase Functions
Researchers should employ multiple complementary methods to accurately assess the specific contributions of caspase-3 versus caspase-7 in biological systems:
Genetic Knockout Models: Utilize caspase-3-deficient, caspase-7-deficient, and double knockout cell lines to dissect individual functions. Studies show that caspase-3-/- MEFs are partially resistant to intrinsic apoptosis, while caspase-7-/- MEFs maintain normal death kinetics but exhibit impaired detachment [80].
Immunodepletion Studies: Remove specific caspases from cell-free extracts to assess substrate cleavage dependencies. Immunodepletion of caspase-3 abolishes cytochrome c/dATP-induced proteolysis of most substrates, while caspase-7 depletion has minimal effects [38].
Activity-Based Probes: Employ specific inhibitors and fluorescent substrates that can distinguish caspase-3 and caspase-7 activities in complex mixtures. While both enzymes cleave DEVD-based substrates, caspase-3 shows superior activity toward LEHD-based substrates [38].
Proteomic Profiling: Conduct quantitative mass spectrometry to identify cleavage events dependent on each caspase. Recent studies reveal that approximately 90% of discrete cleavage events in apoptotic cells depend on caspase-3/caspase-7, with distinct substrates preferences for each protease [79].
Research Reagent Solutions
Table 3: Essential Research Reagents for Caspase Functional Studies
| Reagent Category | Specific Examples | Research Applications | Considerations |
|---|---|---|---|
| Caspase inhibitors | z-VAD-fmk (pan-caspase), BocD-fmk (effector caspase) | Distinguishing caspase-dependent processes | BocD-fmk inhibits both caspase-3 and caspase-7 |
| Specific substrates | DEVD-AFC, LEHD-AFC, VEID-AFC | Enzyme activity measurements | Caspase-3 cleaves LEHD-AFC more efficiently than caspase-7 |
| Antibodies for detection | Anti-cleaved caspase-3, anti-caspase-7 | Immunoblotting, immunohistochemistry | Confirm specificity for intended targets |
| Recombinant enzymes | Active caspase-3, caspase-7 | In vitro cleavage assays | Active-site titrate for quantitative comparisons |
| Cell lines | Caspase-3-/- MEFs, caspase-7-/- MEFs, DKO cells | Genetic functional analysis | Validate knockout status and potential compensations |
| Activity-based probes | FLICA reagents, biotinylated inhibitors | Cellular localization and activity profiling | Consider membrane permeability and specificity |
Common Pitfalls in Data Interpretation
Researchers should avoid these common misinterpretations when studying caspase-3 and caspase-7:
Assuming DEVDase activity reflects only one caspase: DEVD-based substrates and inhibitors detect both caspase-3 and caspase-7 activity, requiring additional methods to distinguish their individual contributions.
Overgeneralizing from synthetic substrates: Cleavage efficiency toward synthetic peptides does not predict natural protein substrate processing, as exosite interactions significantly influence natural substrate cleavage [38].
Ignoring cell-type specific expression: Relative expression levels of caspase-3 and caspase-7 vary significantly between cell types and tissues, potentially creating functional compensation in some contexts [38] [79].
Overlooking non-apoptotic functions: Both caspases participate in non-lethal cellular processes, including differentiation and stress adaptation, where their activities may be regulated differently than in apoptosis [79].
Caspase Functions in Therapeutic Contexts
The functional distinctions between caspase-3 and caspase-7 have significant implications for therapeutic development, particularly in oncology where apoptotic pathways are frequently dysregulated.
Venetoclax (BCL-2 inhibitor) treatment for leukemia depends on intact apoptotic signaling through both initiator and executioner caspases [14]. Resistance mechanisms can involve alterations in caspase expression or function, highlighting the need to understand specific caspase requirements in different cancer types.
TRAIL receptor agonists and DR5 antibodies designed to activate extrinsic apoptosis pathways show variable efficacy across cancer models, partly due to differences in how efficiently caspase-3 and caspase-7 are activated downstream of death receptor engagement [14]. Pancreatic cancers frequently resist TRAIL-induced apoptosis through overexpression of IAP family proteins that inhibit both caspase-3 and caspase-7 [14].
The distinct substrate profiles of caspase-3 and caspase-7 suggest that targeted activation of specific executioner caspases might achieve more precise therapeutic outcomes with reduced off-target effects.
Caspase-3 and caspase-7 represent functionally distinct executioner caspases with specialized roles in apoptotic and non-apoptotic processes. Caspase-3 serves as the primary executioner protease with broader substrate specificity and greater efficiency toward key apoptotic substrates, while caspase-7 processes a more limited substrate set and regulates specific aspects of cell death including detachment. Researchers must employ appropriate experimental designs that account for these functional differences rather than assuming redundancy based on similar activation mechanisms or synthetic substrate preferences. Accurate interpretation of caspase-related data requires consideration of their structural differences, substrate specificities, and the cellular context in which they function. Future research should further elucidate the non-apoptotic functions of these caspases and explore their therapeutic potential through targeted modulation in disease states.
Challenges in Differentiating Apoptosis from Other Regulated Cell Death Forms like Pyroptosis and Necroptosis
Regulated cell death (RCD) is essential for maintaining cellular homeostasis, and its dysregulation underpins numerous diseases, including cancer, neurodegenerative disorders, and inflammatory conditions [7] [81] [58]. Among the various RCD modalities, apoptosis, pyroptosis, and necroptosis represent the most extensively characterized pathways. A paramount challenge in modern cell biology lies in the accurate differentiation between these pathways, given their overlapping components, contextual crosstalk, and convergent morphological features [82] [83]. This challenge is acutely relevant in caspase-centric research, where the same initiator caspases can participate in multiple death pathways, blurring the lines between them [20]. For instance, caspase-8 is a critical initiator of extrinsic apoptosis but can also suppress necroptosis and, under certain conditions, initiate pyroptosis [20] [82]. This intricate interplay complicates the interpretation of experimental results and the development of targeted therapeutics. This whitepaper delineates the core challenges in distinguishing these RCD forms, with a specific focus on caspase activation paradigms, and provides a structured framework of molecular, biochemical, and morphological criteria to guide researchers and drug development professionals.
Molecular Mechanisms and Key Differentiators
Core Signaling Pathways and Effector Molecules
The intrinsic and extrinsic pathways of apoptosis, alongside pyroptosis and necroptosis, are defined by unique initiators, executors, and morphological outcomes.
- Apoptosis: This non-lytic, immunologically silent form of RCD is executed by caspases. The extrinsic pathway is initiated by the binding of extracellular death ligands (e.g., FasL, TRAIL) to cell surface death receptors, leading to the formation of the Death-Inducing Signaling Complex (DISC) and activation of initiator caspase-8 or -10 [7] [14] [4]. The intrinsic pathway is triggered by internal cellular stresses (e.g., DNA damage, oxidative stress), which cause mitochondrial outer membrane permeabilization (MOMP) and the release of cytochrome c. Cytochrome c, together with Apaf-1, forms the apoptosome, activating initiator caspase-9 [7] [9] [58]. Both pathways converge on the activation of executioner caspases-3, -6, and -7, which orchestrate the controlled dismantling of the cell [20] [58].
- Pyroptosis: This lytic, pro-inflammatory form of RCD is a defense mechanism against intracellular pathogens and damage. It is primarily mediated by inflammatory caspases (caspase-1, or human caspases-4/5 and mouse caspase-11) [20] [84]. These caspases cleave members of the gasdermin (GSDM) family, most notably GSDMD. The N-terminal fragment of GSDMD oligomerizes to form pores in the plasma membrane, leading to ion dysregulation, cell swelling, and eventual rupture, with the release of pro-inflammatory cytokines like IL-1β and IL-18 [20] [83].
- Necroptosis: This lytic, pro-inflammatory RCD pathway often serves as a backup mechanism when caspase-8 is inhibited. It is regulated by receptor-interacting serine/threonine-protein kinase 1 (RIPK1) and RIPK3, which form the necrosome. This complex phosphorylates the effector protein mixed lineage kinase domain-like protein (MLKL) [82] [84]. Phosphorylated MLKL oligomerizes and translocates to the plasma membrane, where it executes membrane disruption, leading to uncontrolled release of cellular contents and potent inflammation [82] [83].
Table 1: Comparative Overview of Key RCD Modalities
| Feature | Apoptosis | Pyroptosis | Necroptosis |
|---|---|---|---|
| Primary Stimuli | Death receptors (Fas, TNFR), DNA damage, growth factor withdrawal [4] [58] | Intracellular PAMPs/DAMPs, pathogenic infection [20] [83] | Death receptors, TLRs; often when caspases are inhibited [82] [84] |
| Key Initiators | Caspase-8/-9/-10 [20] [9] | Caspase-1/-4/-5/-11 [20] [84] | RIPK1, RIPK3 (Caspase-8 inhibition) [82] |
| Key Effectors | Caspase-3/-6/-7 [20] [58] | Gasdermin D (GSDMD) [20] [83] | Phospho-MLKL [82] [84] |
| Morphology | Cell shrinkage, membrane blebbing, chromatin condensation, apoptotic bodies [58] [83] | Cell swelling, plasma membrane pore formation, eventual lysis [20] [83] | Organelle swelling, plasma membrane rupture without condensation [82] [83] |
| Immunogenicity | Non-inflammatory / tolerogenic [82] [83] | Highly inflammatory [20] [84] | Highly inflammatory [82] [83] |
| Key Inflammatory Outputs | None ("silent" clearance) [82] | Release of IL-1β, IL-18, HMGB1, LDH [20] [84] | Release of DAMPs, HMGB1, LDH [82] [83] |
The Central Role and Complexity of Caspases
Caspases are cysteine proteases that cleave substrates at specific aspartic acid residues and are the central regulators of RCD, presenting a major node of crosstalk and challenge for differentiation [20]. Their function extends across the death pathways in complex ways:
- Caspase-8: Acts as a master molecular switch. It is the key initiator of extrinsic apoptosis but also cleaves and inactivates RIPK1 and RIPK3, thereby acting as a potent inhibitor of necroptosis. Furthermore, it can cleave gasdermins (GSDMC, GSDMD) under specific contexts to induce pyroptosis [20] [82].
- Caspase-3: The primary executioner of apoptosis can also cleave GSDME (and under certain conditions, GSDMB and GSDMD), converting an apoptotic signal into a pyroptotic-like, lytic outcome [20]. This demonstrates that the final cell death morphology is not determined solely by the initiator caspase but also by the available downstream substrates.
- Inflammatory vs. Apoptotic Caspases: While caspase-1 is dedicated to pyroptosis, human caspases-4/5 and mouse caspase-11 can initiate pyroptosis independently of caspase-1 by directly cleaving GSDMD in response to cytosolic LPS [20] [84].
The following diagram illustrates the complex interplay and crosstalk between the key components of apoptosis, pyroptosis, and necroptosis, highlighting the central role of caspases.
Diagram Title: Crosstalk Among Apoptosis, Pyroptosis, and Necroptosis Pathways
Experimental Differentiation of RCD Modalities
Accurately determining the specific mode of cell death in an experimental system requires a multi-parametric approach. Reliance on a single assay is insufficient due to the significant crosstalk between pathways.
Morphological and Biochemical Hallmarks
The most fundamental differentiation lies in the lytic versus non-lytic nature of the death.
- Apoptosis Assays: Key features include cell shrinkage, chromatin condensation (visualized by Hoechst or DAPI staining), and phosphatidylserine (PS) externalization, detected by Annexin V staining before loss of membrane integrity. DNA fragmentation can be specifically confirmed by a TUNEL assay. Crucially, early apoptotic cells are Annexin V-positive and propidium iodide (PI)-negative, indicating an intact plasma membrane [58] [83].
- Pyroptosis & Necroptosis Assays: As lytic pathways, both result in a loss of plasma membrane integrity. This can be measured by the release of lactate dehydrogenase (LDH) or uptake of propidium iodide (PI). To distinguish between them, specific markers are required: cleaved GSDMD (by Western blot or immunofluorescence) is a definitive marker for pyroptosis, while phosphorylated MLKL is a key marker for necroptosis [82] [84] [83]. The release of specific cytokines like IL-1β is also highly indicative of pyroptosis [20].
Table 2: Key Assays for Differentiating RCD Modalities
| Assay / Reagent | Target/Principle | Application in RCD Differentiation | Key Interpretations |
|---|---|---|---|
| Annexin V / PI Staining | Binds externalized PS (Annexin V) and DNA in cells with permeable membrane (PI) [58] | Distinguishes early apoptosis from late apoptosis/necrosis/lytic death [58] | Annexin V+/PI-: Early apoptosis.\nAnnexin V+/PI+: Late apoptosis or secondary necrosis.\nAnnexin V-/PI+: Primary necrosis/necroptosis/pyroptosis. |
| LDH Release Assay | Measures lactate dehydrogenase enzyme released from cytosol upon membrane rupture [84] | Confirms lytic cell death (pyroptosis, necroptosis) [84] [83] | High LDH release indicates plasma membrane rupture, inconsistent with early apoptosis. |
| Caspase Activity Assays | Fluorogenic substrates or antibodies specific for active caspase forms [20] [58] | Determines if death is caspase-dependent. | Activation of caspase-8/-9/-3 suggests apoptosis. Activation of caspase-1/-4/-5/-11 suggests pyroptosis. |
| Western Blot (Key Markers) | Protein-level detection of cleaved/phosphorylated effectors [84] | Definitive pathway identification. | Cleaved Caspase-3 & PARP: Apoptosis.\nCleaved GSDMD: Pyroptosis.\nPhospho-MLKL: Necroptosis. |
| TUNEL Assay | Labels fragmented DNA (a hallmark of apoptosis) [58] | Supports apoptosis identification. | Positive signal is characteristic of, but not exclusive to, apoptosis. |
| Inhibitor Studies | Pharmacological blockade of specific pathway nodes. | Functional confirmation of pathway involvement. | z-VAD-fmk (pan-caspase inhibitor): Inhibits apoptosis, may enhance necroptosis.\nNecrostatin-1 (RIPK1 inhibitor): Inhibits necroptosis.\nDisulfiram or NSA: Inhibits GSDMD pore formation, blocks pyroptosis. |
A Practical Experimental Workflow
A robust strategy for identifying RCD involves a sequential workflow that combines pharmacological inhibition, morphological assessment, and biochemical confirmation. The following diagram outlines a logical, step-by-step protocol for differentiating between these pathways in a research setting.
Diagram Title: A Decision Workflow for Differentiating RCD
The Scientist's Toolkit: Essential Reagents for RCD Research
The following table compiles key reagents and their applications, as featured in contemporary research, for the study of apoptosis, pyroptosis, and necroptosis.
Table 3: Research Reagent Solutions for Cell Death Studies
| Reagent / Tool | Function / Target | Specific Application in RCD Research |
|---|---|---|
| z-VAD(OMe)-FMK (z-VAD) | Irreversible pan-caspase inhibitor [84] | Used to determine caspase-dependence of cell death. Inhibition of death suggests apoptotic involvement. Can unmask or potentiate necroptosis by inhibiting caspase-8 [82] [84]. |
| Necrostatin-1 (Nec-1) | Allosteric inhibitor of RIPK1 kinase activity [82] [83] | A key tool for specifically inhibiting the necroptosis pathway. Used to functionally confirm RIPK1-dependent necroptosis [84]. |
| Recombinant TRAIL / Agonistic DR5 Antibodies | Activate the extrinsic apoptosis pathway by binding Death Receptors [14] | Used to selectively induce apoptosis via the extrinsic pathway. Useful for studying resistance mechanisms and DISC formation [14]. |
| Venetoclax (ABT-199) | BH3-mimetic; inhibits anti-apoptotic BCL-2 [14] | Used to selectively trigger the intrinsic (mitochondrial) apoptosis pathway in cells dependent on BCL-2 for survival. A prime example of targeted apoptosis induction [14]. |
| Disulfiram / Necrosulfonamide (NSA) | Inhibitor of GSDMD pore formation [83] | Used to specifically block the execution phase of pyroptosis, allowing dissection of upstream signaling from downstream lytic events. |
| Smac Mimetics (e.g., LCL161) | Mimic Smac/DIABLO to antagonize IAP proteins [82] [84] | Sensitize cells to apoptosis (by relieving caspase inhibition) and can promote necroptosis by depleting cIAP1/2. Context-dependent tool [82]. |
| Lipopolysaccharide (LPS) + ATP | Canonical NLRP3 inflammasome activator [84] | A standard experimental protocol to induce pyroptosis in macrophages. LPS primes the cells (signal 1), and ATP triggers NLRP3 inflammasome assembly and caspase-1 activation (signal 2) [84]. |
| TNF-α + z-VAD (TSZ) | Combined cytokine and caspase inhibitor [84] | A standard experimental protocol to induce necroptosis in many cell types. TNF-α provides the death signal, and z-VAD blocks caspase-8-mediated apoptosis, diverting the signal to necroptosis [84]. |
Therapeutic Implications and Concluding Perspectives
The precise differentiation of RCD is not merely an academic exercise; it has profound implications for therapeutic development. The dysregulation of these pathways is a hallmark of cancer, inflammatory diseases, and neurodegeneration [7] [14] [58]. For example, cancer cells often evade apoptosis by overexpressing anti-apoptotic proteins like BCL-2. The development of BH3 mimetics like venetoclax represents a successful therapeutic strategy to directly target this resistance and re-activate intrinsic apoptosis [14]. Conversely, in chronic inflammatory diseases, excessive pyroptosis or necroptosis can drive pathology. In these contexts, inhibitors of GSDMD, RIPK1, or NLRP3 inflammasomes are being actively pursued as novel anti-inflammatory therapeutics [82] [83].
The primary challenge remains the context-dependent crosstalk between these pathways, where inhibition of one can lead to the engagement of another, potentially compromising therapeutic efficacy or causing adverse effects [7] [82]. Future research must leverage the integrated experimental framework outlined herein to dissect these complex interactions in disease-specific contexts. A deep understanding of the molecular switches, like caspase-8, that govern the balance between apoptosis, pyroptosis, and necroptosis will be crucial for developing the next generation of smart, targeted therapies that can precisely modulate cell death for therapeutic benefit.
Caspase inhibitors, despite their significant therapeutic potential across a spectrum of diseases including inflammatory, neurodegenerative, and metabolic disorders, have consistently faced substantial barriers to clinical success. This whitepaper delineates the core limitations—inadequate efficacy, poor target specificity, and adverse effects—that have hindered the transition of these compounds from bench to bedside. Framed within the context of intrinsic and extrinsic apoptosis research, we detail the molecular underpinnings of these challenges, summarize critical experimental data, and provide standardized protocols for evaluating novel inhibitors. The complex and multifaceted roles of caspases in cellular homeostasis, which extend beyond canonical apoptosis into lytic cell death pathways like pyroptosis and PANoptosis, render their selective inhibition particularly challenging. A deeper understanding of caspase functions in specific disease models and the development of innovative, state-specific inhibitors are paramount for future breakthroughs in therapeutic caspase modulation.
Caspases are an evolutionarily conserved family of cysteine-dependent aspartate-specific proteases that serve as critical regulators of programmed cell death (PCD) and inflammation [57] [3]. Their activity is orchestrated through two primary apoptotic pathways:
- The Extrinsic Pathway: Initiated by the binding of extracellular death ligands (e.g., FasL, TRAIL) to cell surface death receptors, leading to the formation of the Death-Inducing Signaling Complex (DISC) and the activation of initiator caspases-8 and -10 [3] [85].
- The Intrinsic Pathway: Activated by intracellular stressors such as DNA damage or oxidative stress, this pathway involves mitochondrial outer membrane permeabilization (MOMP) and the release of cytochrome c. Cytochrome c then facilitates the formation of the apoptosome, which activates initiator caspase-9, subsequently triggering the executioner caspases-3 and -7 [3] [58].
The following diagram illustrates the interplay between these pathways and the points of caspase inhibitor intervention:
Core Limitations of Caspase Inhibitors
Inadequate Therapeutic Efficacy
The failure of caspase inhibitors to demonstrate robust efficacy in clinical settings often stems from the complexity of cell death pathways and compensatory mechanisms.
- Pathway Redundancy and Crosstalk: Inhibition of one caspase or one cell death pathway can lead to the activation of alternative pathways. For instance, pharmacological inhibition or genetic deletion of caspase-8, a key initiator of extrinsic apoptosis, can result in a switch to receptor-interacting protein kinase (RIPK)-dependent necroptosis, another form of lytic cell death [85] [43]. This demonstrates that cells possess backup mechanisms to ensure death occurs even when one pathway is blocked.
- Non-Apoptotic Caspase Functions: Emerging research reveals that caspases play key roles in diverse cellular processes beyond apoptosis, such as differentiation, proliferation, and migration [57] [63]. Inhibiting them for a disease-related function may inadvertently disrupt these vital homeostatic processes, limiting the therapeutic window and overall efficacy.
- Disease Model Discrepancies: Promising results in animal models have frequently failed to translate to human trials. For example, the caspase-1 inhibitor VX-740 showed efficacy in preclinical models of rheumatoid arthritis and osteoarthritis but was terminated in clinical trials due to liver toxicity, revealing a gap between animal and human physiology [57] [86].
Poor Target Specificity and Selectivity
Achieving selectivity for a specific caspase is a monumental challenge due to the high degree of structural and sequence homology within the caspase family.
- Structural Homology: The active sites of caspases are highly conserved, making it difficult to design small molecules that can distinguish between closely related family members [57] [63]. A compound designed to inhibit one caspase often exhibits significant off-target inhibition of others, leading to unintended biological consequences.
- Peptide-Based Inhibitor Limitations: Many early-generation inhibitors are peptide-based and mimic the caspase cleavage sequence. While this provides a basis for binding, their peptidic nature often results in poor pharmacokinetic properties, including low metabolic stability and inadequate membrane permeability [57] [87].
- Zymogen State vs. Active State: Most conventional inhibitors target the active conformation of caspases. Recent strategies focus on developing inhibitors that bind the inactive zymogen (pro-caspase) state, which exhibits greater structural diversity among caspase members and offers a potential avenue for improved selectivity [63].
Adverse Effects and Toxicity Profiles
The physiological roles of caspases in normal development and tissue homeostasis make their systemic inhibition prone to mechanism-based toxicities.
- Mechanism-Based Toxicity: Caspases are indispensable for normal development and immune function. For example, caspase-8 deficiency in mice results in embryonic lethality due to cardiac defects, highlighting its critical non-redundant role [87]. Systemic inhibition risks disrupting these essential processes.
- Liver Toxicity: Hepatotoxicity has been a recurring issue for several caspase inhibitors. VX-740 (pralnacasan) and VX-765 (belnacasan) both faced clinical trial termination or suspension due to liver toxicity observed in animal models or human participants [57] [86].
- Impact on Innate Immunity: Inflammatory caspases (caspase-1, -4, -5, -11) are crucial for host defense against pathogens by processing pro-inflammatory cytokines like IL-1β and IL-18. Their broad inhibition can compromise the innate immune response, increasing susceptibility to infections [57] [85].
Table 1: Clinical Failures of Select Caspase Inhibitors
| Inhibitor Name | Primary Target | Intended Indication | Stage of Failure | Reasons for Failure |
|---|---|---|---|---|
| VX-740 (Pralnacasan) | Caspase-1 | Rheumatoid Arthritis, Osteoarthritis | Phase II/III | Liver toxicity in animal models [57] |
| VX-765 (Belnacasan) | Caspase-1 | Epilepsy, Inflammatory Diseases | Phase II | Liver toxicity [57] [86] |
| IDN-6556 (Emricasan) | Pan-Caspase | Liver Diseases (e.g., NASH, fibrosis) | Phase II/III | Undisclosed reasons; inadequate efficacy in some trials [57] [87] |
Experimental Analysis of Inhibitor Limitations
Standardized Protocol for Evaluating Inhibitor Specificity
To systematically assess the limitations of caspase inhibitors, researchers employ a combination of biochemical and cellular assays.
Protocol: Profiling Inhibitor Specificity Across the Caspase Family
Recombinant Caspase Enzyme Assays:
- Purpose: To determine the potency (IC50) and selectivity of an inhibitor against a panel of purified recombinant caspase enzymes.
- Method: Incubate serial dilutions of the test inhibitor with individual active recombinant caspases (e.g., caspase-1, -2, -3, -6, -7, -8, -9, -10) in an appropriate reaction buffer. Initiate the reaction by adding a fluorogenic substrate (e.g., Ac-DEVD-AFC for caspase-3). Monitor the release of the fluorescent group in real-time using a plate reader.
- Data Analysis: Calculate the percentage inhibition at each concentration and determine the IC50 value for each caspase. A selectivity index can be calculated by comparing IC50 values for the primary target versus off-target caspases.
Cellular Apoptosis/Necroptosis Switching Assay:
- Purpose: To identify if caspase inhibition triggers a switch to alternative cell death pathways like necroptosis.
- Method:
- Use human cancer cell lines (e.g., HT-29, FADD-deficient Jurkat cells) known to be susceptible to necroptosis.
- Pre-treat cells with a broad-spectrum caspase inhibitor (e.g., Z-VAD-FMK, 20-50 µM) or a vehicle control for 1 hour.
- Induce death by adding a death receptor agonist (e.g., TNF-α, 50 ng/mL) in combination with a SMAC mimetic (e.g., birinapant, 100 nM) and a caspase inhibitor (if not already present).
- After 18-24 hours, quantify cell death by measuring plasma membrane integrity using propidium iodide (PI) staining and flow cytometry. Necroptosis is confirmed by rescue with the RIPK1 inhibitor Necrostatin-1 (10-30 µM).
Target Engagement and Cellular Pathway Analysis:
- Purpose: To verify that the inhibitor engages its intended target in cells and to map the consequent molecular events.
- Method: Use cell lines or primary cells relevant to the disease context. Treat cells with the inhibitor and the apoptotic stimulus. Analyze downstream effects by:
- Western Blotting: Probe for cleavage of specific caspase substrates (e.g., PARP-1 for caspase-3, IL-1β for caspase-1) and markers of other death pathways (e.g., phosphorylation of MLKL for necroptosis).
- Activity-Based Protein Profiling (ABPP): Use fluorescently-labeled active-site probes (e.g., biotin-DEVD-AOMK) to monitor the activity state of multiple caspases in cell lysates, providing a direct readout of target engagement and potential off-target effects [63].
Quantitative Data from Experimental Studies
The following table compiles key experimental findings that highlight the challenges associated with caspase inhibitors.
Table 2: Experimental Profile of Characterized Caspase Inhibitors
| Inhibitor | Reported Specificity | Key Experimental Findings | Identified Limitations |
|---|---|---|---|
| Z-VAD-FMK | Pan-caspase | Inhibits apoptosis in GH3 cells treated with MPP+ [87]. | High toxicity in vivo; promotes a switch to necroptosis in L929 cells upon TNF-α stimulation [58]. |
| Q-VD-OPh | Pan-caspase | Effective at maintaining T cell ratios in SIV-infected macaques; less toxic than Z-VAD-FMK in vitro, working at up to 500 µM [57]. | As a broad-spectrum inhibitor, it still lacks specificity for individual caspases, potentially disrupting non-apoptotic functions. |
| VX-765 (Belnacasan) | Caspase-1 | Reduced Caspase-1 activity, chondrocyte senescence, and MMP13 secretion in an in vitro model of osteoarthritis [86]. | Clinical development halted due to liver toxicity; inhibits processing of IL-1β, potentially suppressing innate immunity [57] [86]. |
| IDN-6556 (Emricasan) | Pan-caspase | Showed efficacy in preclinical models of liver injury; progressed to Phase III trials for liver fibrosis [87]. | Terminated due to undisclosed reasons; long-term treatment associated with side effects, potentially related to broad caspase inhibition [57]. |
The experimental workflow for a comprehensive caspase inhibitor study is outlined below:
The Scientist's Toolkit: Research Reagent Solutions
This section details essential reagents and their functions for researching caspase biology and inhibitor properties.
Table 3: Essential Reagents for Caspase and Inhibitor Research
| Reagent / Assay | Function and Application | Key Considerations |
|---|---|---|
| Fluorogenic Substrates(e.g., Ac-DEVD-AFC) | Synthetic peptides linked to a fluorophore (e.g., AFC). Caspase cleavage releases the fluorophore, allowing real-time kinetic measurement of enzyme activity. Used in recombinant and cellular assays. | Substrate sequence determines specificity (e.g., DEVD for caspase-3/7, VDVAD for caspase-2). Always confirm substrate specificity for the caspase of interest. |
| Active-Site Directed Probes(e.g., biotin-/AFC-DEVD-AOMK) | Irreversibly bind active caspase sites, enabling labeling, pull-down, or visualization. Critical for Activity-Based Protein Profiling (ABPP) to monitor inhibitor target engagement and off-targets in complex proteomes [63]. | AOMK-based probes are irreversible. Newer, more selective probes are continuously being developed for specific caspases. |
| Broad-Spectrum Inhibitors(Z-VAD-FMK, Q-VD-OPh) | Tool compounds to establish the general involvement of caspases in a cell death process. Q-VD-OPh is generally preferred for in vitro work due to its lower cellular toxicity [57]. | Results must be interpreted with caution due to lack of specificity. Rescue of cell death suggests, but does not prove, apoptotic involvement, due to potential pathway switching. |
| Selective Chemical Inhibitors(VX-765, specific zymogen inhibitors) | Used to dissect the role of specific caspases (e.g., caspase-1 with VX-765) [86]. Zymogen-state inhibitors represent a newer strategy for achieving selectivity [63]. | Always confirm selectivity in the specific cellular model being used, as potency can vary. Counter-screen for effects on related proteases. |
| Genetic Tools(siRNA, shRNA, CRISPR-Cas9) | To knock down or knock out specific caspase genes, providing genetic validation for pharmacological studies. Essential for controlling for off-target effects of small molecules. | Consider compensation by other caspases or pathways in stable knockout lines. Transient knockdown may be preferable for acute studies. |
The journey to develop clinically successful caspase inhibitors has been fraught with challenges, primarily rooted in the biological complexity of the caspase family itself. The limitations of inadequate efficacy, poor specificity, and adverse effects are interconnected, stemming from pathway redundancy, high structural homology, and critical physiological roles of the targets. Future efforts must pivot towards several innovative strategies: First, the development of state-specific inhibitors, particularly those targeting the more variable zymogen states, holds promise for achieving unprecedented selectivity [63]. Second, a deeper understanding of the non-apoptotic functions of caspases in specific tissues and disease contexts is essential to predict and mitigate mechanism-based toxicities [57] [85]. Finally, exploring localized delivery systems or conditional activation of inhibitors could minimize systemic exposure and off-target effects. Overcoming these hurdles will require a concerted effort combining structural biology, chemical proteomics, and sophisticated disease modeling to finally realize the long-held therapeutic potential of caspase modulation.
For decades, the activation of executioner caspases (caspase-3 and -7) has been considered the definitive "point of no return" in the apoptotic pathway, an irreversible commitment to cell death [88]. This paradigm is fundamentally shifting. Research now reveals that caspase activation is not exclusively a death sentence; it can occur in sub-lethal contexts and even be reversed. Two key concepts challenging the traditional view are non-apoptotic caspase functions and anastasis (Greek for "rising to life")—the process by which cells recover after initiating apoptosis, including after executioner caspase activation [88] [89] [90].
This guide synthesizes current knowledge for researchers and drug development professionals, framing these concepts within the broader context of intrinsic versus extrinsic apoptosis research. Understanding when caspase activation leads to death, non-apoptotic signaling, or recovery is critical for accurately interpreting experimental data and developing therapeutic strategies, especially in cancer and neurodegenerative diseases.
The Conventional Role of Caspases in Apoptosis
Caspases (cysteine-aspartic proteases) are evolutionarily conserved enzymes that cleave cellular substrates after specific aspartic acid residues. They are central regulators of programmed cell death (PCD), maintaining cellular homeostasis [20] [91].
Core Apoptotic Pathways
The two main apoptotic pathways converge on caspase activation:
- Extrinsic Pathway: Initiated by external death ligands (e.g., TNFα, FasL) binding to cell surface death receptors. This leads to the formation of a FADDosome complex, which activates initiator caspase-8 [20].
- Intrinsic Pathway: Triggered by internal cellular stresses (e.g., DNA damage, oxidative stress). It involves mitochondrial outer membrane permeabilization (MOMP), cytochrome c release, and formation of the apoptosome complex, which activates initiator caspase-9 [20] [91].
Both pathways ultimately activate the executioner caspases-3, -6, and -7. These enzymes then cleave hundreds of cellular substrates, such as PARP and nuclear lamins, leading to the characteristic morphological changes of apoptosis, including cell shrinkage, membrane blebbing, and DNA fragmentation [20] [91].
Table 1: Key Caspases in Programmed Cell Death Pathways
| Caspase | Primary Role/Pathway | Key Functions & Interactions |
|---|---|---|
| Caspase-8 | Initiator; Extrinsic Apoptosis | Activates executioner caspases; cleaves BID; molecular switch between apoptosis, necroptosis, and pyroptosis [20]. |
| Caspase-9 | Initiator; Intrinsic Apoptosis | Activates executioner caspases-3/-7; can inhibit necroptosis by cleaving RIPK1 [20]. |
| Caspase-3/7 | Executioner; Apoptosis | Key effectors cleaving structural & regulatory proteins (e.g., PARP); also cleave gasdermins to induce pyroptosis [20] [48]. |
| Caspase-1/4/5/11 | Inflammatory; Pyroptosis | Cleave GSDMD to trigger pore formation, leading to inflammatory lytic cell death [20] [91]. |
Anastasis: Cellular Recovery from the Brink of Death
Anastasis is defined as cellular recovery from the brink of apoptotic death, even after executioner caspase activation [88]. This process is an active, transcriptionally driven recovery, not merely a stalling of the death program.
Evidence and Experimental Induction
- Initial Discovery: First named in 2012, experiments showed that mammalian cells exposed to transient apoptotic stimuli (e.g., ethanol, staurosporine) could activate caspase-3, shrink, and membrane-bleb, yet recover a normal morphology upon stimulus removal [88].
- In Vivo Evidence: Using a CaspaseTracker biosensor in Drosophila, researchers demonstrated that egg chambers subjected to environmental stresses (cold shock, protein starvation) activated caspases and exhibited apoptotic morphology. Upon return to normal conditions, these egg chambers recovered, produced functional progeny, and were permanently labeled by the biosensor, confirming anastasis had occurred [89].
Anastasis can be induced by various stimuli, including chemical inducers (ethanol, DMSO, death receptor ligands), physical stress (cold shock), and physiological stress (protein starvation) [88]. It has been observed in cancer cell lines (HeLa, H4), immortalized lines (NIH3T3), and primary cells (hepatocytes, cardiomyocytes) [88].
Molecular Mechanism and Cellular Consequences
Transcriptome profiling reveals anastasis is an active, multi-stage process [88]:
- Early Stage: Characterized by a burst of transcription for transcription factors, stress response genes, and re-entry into the cell cycle.
- Late Stage: Involves cytoskeletal rearrangement and increased cell migration.
This demonstrates that recovering cells are not dysfunctional "zombies" but can proliferate and migrate, which has critical implications. While anastasis could promote tissue repair (e.g., rescuing neurons or cardiomyocytes from transient injury), it may also pose a significant oncogenic risk. Surviving cells can harbor genomic instability and chromosome rearrangements, potentially leading to tumorigenesis and cancer recurrence [88] [89].
Figure 1: The Process of Anastasis. This pathway illustrates the critical decision point where removal of an apoptotic stimulus allows a cell to undergo anastasis and recover, rather than die.
Non-Apoptotic Functions of Caspases
Beyond apoptosis and anastasis, caspases function in a wide range of normal, non-lethal physiological processes. This activity is often localized or restricted in level to prevent cell death [88] [89].
Table 2: Documented Non-Apoptotic Roles of Caspases
| Caspase | Non-Apoptotic Role | Biological Context |
|---|---|---|
| Caspase-3/7 | Synaptic plasticity, learning & memory [89]. | Neuronal function. |
| Caspase-3 | Dendrite pruning & remodeling [88]. | Neural development. |
| Caspase(-like) activity | Sperm individualization [88] [89]. | Drosophila development. |
| Caspase-8 | Inhibition of necroptosis; regulatory role in innate immunity [20]. | Cell survival & inflammation. |
| Caspase-1 | Cell differentiation processes [88]. | Development. |
Distinguishing Non-Apoptotic Activation from Anastasis:
- Non-apoptotic caspase activation is typically regulated, occurs in specific developmental stages or cell types, and the cell is never at risk of dying [88].
- Anastasis follows genuine, stress-induced apoptotic signaling where the cell is on the verge of death but recovers [88]. In vivo, the CaspaseTracker showed that developmental anastasis occurs sporadically, while non-apoptotic activation is more patterned [89].
The Scientist's Toolkit: Methods for Detecting Caspase Activity
Interpreting caspase context requires methods to detect activity, not just protein presence. The field has evolved from endpoint assays to dynamic, real-time biosensors.
Table 3: Key Research Reagents and Methods for Caspase Detection
| Reagent/Method | Principle | Key Applications & Advantages | Example Reagents/Assays |
|---|---|---|---|
| Antibody-Based Methods | Detect caspase protein or cleavage products (e.g., cleaved caspase-3, cleaved PARP). | Western Blot, Immunofluorescence. Standard but often endpoint analysis [91]. | Anti-cleaved caspase-3 antibodies. |
| Fluorogenic Substrates | Cell-permeable peptides (e.g., DEVD) linked to a fluorophore. Caspase cleavage releases fluorescent signal. | Live-cell imaging, High-throughput screening (HTS). Measures activity in real-time [91] [92]. | DEVD-NucView488, Caspase-Glo. |
| FRET-Based Biosensors | Two fluorophores linked by a caspase-cleavable sequence. FRET signal decreases upon cleavage. | Real-time kinetics in single living cells. High spatiotemporal resolution [48] [93]. | SCAT3, CFP-DEVD-Venus. |
| Split-FP Biosensors | Fragments of a fluorescent protein linked by a cleavable sequence. Fluorescence reconstitutes upon cleavage. | Stable cell lines, robust signal-to-noise, marks cells permanently. Ideal for long-term tracking [48]. | ZipGFP-based caspase-3/7 reporter. |
| Genetic Fate-Mapping Biosensors | Caspase-cleavable sequence controls a transcriptional activator. Permanent marker expression upon activity. | In vivo tracking of historical caspase activity (anastasis). Unmasks non-apoptotic roles [89]. | CaspaseTracker (mCD8-DQVD-Gal4). |
Experimental Protocol: Real-Time Caspase Activation and Anastasis Assay
This protocol outlines how to use a stable fluorescent reporter to monitor caspase dynamics and potential recovery in cell culture.
Objective: To dynamically track caspase-3/7 activation and assess cell fate (death vs. anastasis) in response to a transient apoptotic stimulus.
Materials:
- Stable reporter cell line expressing a caspase-3/7 biosensor (e.g., ZipGFP-DEVD-mCherry) [48].
- Apoptosis inducer: e.g., 1µM Staurosporine (STS) or clinical therapeutic (e.g., Carfilzomib) [48] [93].
- Pan-caspase inhibitor: e.g., Z-VAD-FMK (20-50 µM) for control [92] [48].
- Live-cell imaging system (e.g., IncuCyte or confocal microscope with environmental chamber).
- Culture vessels suitable for microscopy.
Procedure:
- Seed & Culture: Seed reporter cells into an appropriate culture vessel and allow to adhere overnight.
- Pre-image: Acquire baseline images of GFP (caspase signal) and mCherry (cell presence) channels.
- Treat & Image:
- Transient Treatment Group: Replace medium with media containing the apoptotic inducer (e.g., 1µM STS). Incubate for a defined, sub-lethal period (e.g., 2-6 hours, requires optimization) [88] [93].
- Wash & Recovery: Remove the inducer-containing media, wash cells gently with PBS, and add fresh complete media.
- Continuous Treatment Control: Maintain cells in inducer-containing media.
- Inhibitor Control: Pre-treat cells with Z-VAD-FMK for 1 hour before adding the inducer to confirm caspase-specific signal.
- Initiate time-lapse imaging, acquiring images every 30-60 minutes for 24-48 hours.
- Data Analysis:
- Quantify the GFP fluorescence intensity (Caspase activation) and mCherry fluorescence (Cell presence/viability) over time.
- In the continuous treatment group, expect a strong GFP signal followed by a loss of mCherry signal as cells die.
- Evidence of anastasis in the transient treatment group is indicated by a strong GFP signal that plateaus or eventually decreases, while the mCherry signal is maintained or recovers, and cells resume normal morphology and proliferation [88] [48].
Figure 2: Experimental Workflow for Live-Cell Anastasis Assay. This diagram outlines the key steps for setting up an experiment to detect caspase activation and subsequent cell recovery.
Implications for Therapeutic Drug Development
The revised understanding of caspase activation has profound implications for cancer therapy and drug development.
- Therapy Resistance: The ability of cancer cells to undergo anastasis presents a novel mechanism for therapy resistance and tumor repopulation. Cells that recover after exposure to chemo- or radiotherapy may contribute to relapse [88] [48].
- Apoptosis-Induced Proliferation (AIP): Apoptotic cells can release mitogenic signals that stimulate the proliferation of neighboring surviving cells. Real-time imaging platforms now allow the study of this pro-survival mechanism, which is a significant driver of tumor repopulation after treatment [48].
- Caspase-Independent Agents: Due to the potential for anastasis and common mutations in caspase-dependent pathways in cancers, there is a growing interest in developing agents that induce caspase-independent cell death. For example, chimeric proteins like GnRH-AIF are designed to trigger apoptosis-inducing factor (AIF)-mediated, caspase-independent apoptosis, offering a potential workaround for resistant cancers [94].
The biological context of caspase activation is paramount. The same executioner caspases can orchestrate irreversible death, mediate subtle physiological functions, or be actively silenced to allow cellular recovery via anastasis. For researchers, this demands careful experimental design employing real-time biosensors and fate-mapping tools to distinguish between these outcomes. For the drug development industry, these concepts unveil new mechanisms of treatment resistance and highlight the need for therapeutic strategies that either exploit or inhibit anastasis and non-apoptotic caspase signaling. Moving beyond the simplistic "death marker" paradigm is essential for advancing both fundamental cell biology and the next generation of therapeutics.
The Impact of Pathway Crosstalk and PANoptosis on Experimental Outcomes and Therapeutic Efficacy
The historical classification of programmed cell death (PCD) into distinct pathways such as apoptosis, necroptosis, and pyroptosis has been fundamentally challenged by the emerging paradigm of PANoptosis. This novel, inflammatory PCD pathway demonstrates extensive crosstalk and coordination among classical death mechanisms, driven by multi-protein complexes known as PANoptosomes. For researchers investigating caspase activation in intrinsic versus extrinsic apoptosis, this crosstalk presents both a significant experimental challenge and a transformative therapeutic opportunity. This whitepaper provides a comprehensive technical analysis of PANoptosis molecular mechanisms, detailed experimental methodologies for detecting pathway crosstalk, and critical implications for therapeutic development. Understanding these interconnected networks is essential for designing robust experiments and developing effective treatments for cancer, neurodegenerative disorders, and other diseases characterized by dysregulated cell death.
Programmed cell death (PCD) is a genetically regulated process essential for development, homeostasis, and eliminating damaged or infected cells [5]. The classical view identified several distinct PCD pathways: apoptosis (characterized by cell shrinkage, membrane blebbing, and caspase activation without significant inflammation), necroptosis (a regulated inflammatory cell death with necrotic morphology), and pyroptosis (an inflammatory lytic cell death mediated by gasdermin family proteins) [7] [65]. Apoptosis itself occurs through two well-established routes: the extrinsic pathway initiated by extracellular death ligands binding to cell surface receptors, and the intrinsic pathway initiated by intracellular stress signals leading to mitochondrial outer membrane permeabilization (MOMP) [14] [5] [95].
However, accumulating evidence reveals these pathways do not operate in isolation. Extensive crosstalk occurs at multiple molecular levels, challenging the traditional boundaries between distinct PCD forms [7]. This understanding led to the identification of PANoptosis, first conceptualized by Kanneganti et al. in 2019 [96] [97]. PANoptosis is defined as a unique, innate immune, inflammatory cell death pathway that is triggered by specific stimuli and regulated by PANoptosome complexes, which simultaneously activate key molecules from pyroptosis, apoptosis, and/or necroptosis [96] [97] [98]. This integrated death pathway cannot be fully accounted for by any single PCD mechanism alone, representing a significant shift in our understanding of cell fate decisions and their impact on health and disease.
Molecular Mechanisms of Apoptosis and PANoptosis
Core Apoptotic Pathways: Intrinsic and Extrinsic Signaling
The precise molecular machinery of apoptosis provides the foundational framework for understanding more complex PCD crosstalk.
Extrinsic Apoptosis: This pathway initiates outside the cell when death ligands (e.g., FasL, TRAIL, TNF-α) bind to cognate death receptors (e.g., Fas, DR4/5, TNFR1) on the plasma membrane [14] [7]. This binding induces receptor trimerization and recruitment of adapter proteins like FADD (Fas-associated death domain) and pro-caspase-8 to form the Death-Inducing Signaling Complex (DISC). Within the DISC, caspase-8 undergoes auto-activation, subsequently activating downstream effector caspases-3, -6, and -7, which execute the apoptotic program through cleavage of hundreds of cellular substrates [14] [65].
Intrinsic Apoptosis: This mitochondrial pathway activates in response to internal cellular stressors, including DNA damage, oxidative stress, and endoplasmic reticulum (ER) stress [5] [95]. These stressors shift the balance of Bcl-2 family proteins in favor of pro-apoptotic members (e.g., Bax, Bak, Bid, Bim, Puma), which induce MOMP [14] [65]. MOMP permits the release of mitochondrial intermembrane space proteins, most critically cytochrome c, into the cytosol. Cytochrome c binds Apaf-1, triggering apoptosome formation and activation of caspase-9, which then cleaves and activates effector caspases [5] [95].
Table 1: Core Components of Apoptotic Pathways
| Component Category | Extrinsic Pathway Elements | Intrinsic Pathway Elements |
|---|---|---|
| Initiators | Death Receptors (Fas, DR4/5), Death Ligands (FasL, TRAIL), FADD, Caspase-8 | Cellular stress (DNA damage, ER stress), Bcl-2 family proteins |
| Key Mediators | Active Caspase-8, Caspase-10 | Bax/Bak oligomers, Cytochrome c, Apaf-1, Caspase-9 |
| Executioners | Caspase-3, -6, -7 | Caspase-3, -6, -7 |
| Regulators | c-FLIP, Decoy Receptors | Bcl-2, Bcl-xL, Mcl-1, IAPs, SMAC/DIABLO |
PANoptosis: An Integrated Cell Death Framework
PANoptosis represents a convergence point for PCD pathways, characterized by the assembly of a PANoptosome—a complex molecular scaffold that integrates sensors, adapters, and catalytic effectors from multiple death pathways [96] [97]. The composition of the PANoptosome varies depending on the triggering stimulus but typically includes components from pyroptosis (e.g., ASC, caspase-1), apoptosis (e.g., FADD, caspase-8), and necroptosis (e.g., RIPK1, RIPK3) [96] [98].
Several distinct PANoptosomes have been identified, each named for its core sensor:
- ZBP1-PANoptosome: Activated by sensing viral nucleic acids, comprising ZBP1, RIPK3, RIPK1, FADD, caspase-8, NLRP3, ASC, and caspase-1 [96] [97].
- AIM2-PANoptosome: Triggered by cytoplasmic double-stranded DNA, containing AIM2, ZBP1, ASC, caspase-1, FADD, caspase-8, RIPK3, and RIPK1 [97].
- RIPK1-PANoptosome and NLRP12-PANoptosome: Additional complexes responding to specific inflammatory triggers [97] [98].
The following diagram illustrates the molecular architecture of a canonical PANoptosome and its downstream death pathways:
Critical Nodes of Pathway Crosstalk
The crosstalk between apoptosis, pyroptosis, and necroptosis occurs at several key molecular nodes within the PANoptosome, creating a robust, interconnected network that defies simplistic linear modeling.
Caspase-8 as a Master Switch: Caspase-8 exemplifies pathway integration, functioning as a critical molecular switch. It can initiate apoptosis by activating caspase-3, trigger pyroptosis by directly cleaving GSDMD, and regulate necroptosis by cleaving RIPK1 and RIPK3 [96]. When caspase-8 is inhibited, the cell can default to RIPK3/MLKL-mediated necroptosis [7] [98].
RIPK1 and RIPK3 as Nexus Points: RIPK1 possesses kinase-dependent and scaffolding functions that can promote either apoptosis or necroptosis depending on cellular context and caspase-8 activity [96]. RIPK3, a central necroptosis mediator, also contributes to NLRP3 inflammasome activation, thereby bridging necroptosis and pyroptosis [98].
Inflammasome Platforms as Integration Hubs: Inflammasomes, particularly NLRP3, serve as core platforms for PANoptosome assembly in neurodegenerative diseases. In Alzheimer's disease models, Aβ/Tau activates the NLRP3 inflammasome, promoting ASC oligomerization and recruiting pro-caspase-1 and caspase-8 to form the "NLRP3-PANoptosome," leading to IL-1β release and mitochondrial apoptosis simultaneously [96].
Experimental Considerations for Detecting Pathway Crosstalk
Technical Challenges and Validation Requirements
Research into PANoptosis presents unique methodological challenges. A key principle is that the activation of multiple death pathways must be confirmed through several parallel detection methods, as relying on a single marker is insufficient to distinguish PANoptosis from isolated PCD [97]. For example, DNA fragmentation detected by TUNEL assay occurs in both late apoptosis and necrosis, while loss of mitochondrial membrane potential can indicate either apoptosis or necroptosis [65]. Furthermore, the dynamic and stimulus-specific nature of PANoptosome assembly requires careful experimental design to account for cell-type-specific responses and temporal progression of cell death.
Comprehensive Detection Strategies
Robust detection of PANoptosis requires a multi-modal approach targeting hallmark features of all three constituent death pathways. The table below summarizes key detection methodologies and their specific applications in identifying PANoptosis components.
Table 2: Experimental Methods for Detecting PANoptosis Components
| Target Pathway | Detection Method | Specific Target/Readout | Technical Considerations |
|---|---|---|---|
| Apoptosis | Western Blot / IHC | Cleaved Caspase-3, -8, -9; Cleaved PARP | Distinguish initiator vs. executioner caspase activation [65]. |
| TUNEL Assay | DNA fragmentation | Not apoptosis-specific; also positive in necrosis [65]. | |
| Annexin V/PI Staining | Phosphatidylserine externalization (early apoptosis) vs. membrane permeability (late apoptosis/necrosis) | Requires flow cytometry and careful interpretation of quadrant analysis [65]. | |
| Pyroptosis | Western Blot / IHC | Cleaved GSDMD (GSDMD-NT), Cleaved Caspase-1 (p20), mature IL-1β | GSDMD-NT is a definitive pyroptosis marker [96] [99]. |
| ELISA / LDH Release | Extracellular IL-1β/IL-18, LDH | Measures inflammatory outcome and lytic cell death [99]. | |
| Necroptosis | Western Blot / IHC | Phospho-MLKL (pMLKL), RIPK3 activation | pMLKL is a key necroptosis executioner [96] [97]. |
| PANoptosis Integration | Co-immunoprecipitation / Proximity Ligation | PANoptosome assembly (e.g., ZBP1-RIPK3-CASP8 interaction) | Confirms physical interaction of components from different pathways [96] [97]. |
| Multiplex Inhibitor Assay | Cell death in presence of Z-VAD (pan-caspase inh.), Nec-1 (RIPK1 inh.), GSDMD inhibitor | Death persisting despite combined inhibition indicates compensatory activation [7]. |
The following workflow diagram outlines a logical, multi-step experimental approach for confirming PANoptosis:
Essential Research Reagents and Tools
A sophisticated toolkit of chemical inhibitors, antibodies, and genetic tools is mandatory for dissecting PANoptosis. The table below catalogs essential research reagents.
Table 3: Research Reagent Solutions for PANoptosis Studies
| Reagent Category | Specific Examples | Primary Function/Target | Experimental Application |
|---|---|---|---|
| Small Molecule Inhibitors | Z-VAD-FMK (or Q-VD-OPh) | Pan-caspase inhibitor (Caspase-1, -3, -8, -9) | Blocks apoptotic and pyroptotic signaling; can enhance necroptosis [96] [5]. |
| Necrostatin-1 (Nec-1) | RIPK1 kinase inhibitor | Specifically inhibits necroptosis [7]. | |
| Emricasan | Irreversible pan-caspase inhibitor | Blocks Caspase-3, -8, and -9 activation [96]. | |
| MCC950 | NLRP3 inflammasome inhibitor | Inhibits NLRP3 assembly and activation, reducing pyroptosis [96]. | |
| Activating Antibodies | Anti-DR4/DR5 Agonist Antibodies (e.g., Conatumumab, Mapatumumab) | Activate extrinsic apoptosis pathway | Induce DISC formation and caspase-8 activation [14]. |
| Critical Antibodies for Detection | Anti-Cleaved Caspase-3, -8 | Activated caspases (Apoptosis) | Confirm apoptosis execution and initiation [65]. |
| Anti-GSDMD (NT-terminal) | Active GSDMD fragment (Pyroptosis) | Definitive marker for pyroptosis execution [96] [99]. | |
| Anti-phospho-MLKL (pMLKL) | Activated MLKL (Necroptosis) | Key marker for necroptosis execution [96] [97]. | |
| Anti-RIPK1, Anti-RIPK3 | Necroptosis regulators | Assess necroptosis pathway involvement [97]. | |
| Genetic Tools | siRNA/shRNA/CRISPR-Cas9 | ZBP1, AIM2, NLRP3, RIPK1, RIPK3, Caspase-8 | Knockdown/out key PANoptosome components to validate function [96] [97]. |
Therapeutic Implications and Clinical Translation
The interconnected nature of PANoptosis has profound implications for therapeutic interventions, particularly in cancer and neurodegenerative diseases, where modulating cell death is a primary treatment goal.
Impact on Cancer Therapy
In oncology, the crosstalk between cell death pathways significantly influences treatment efficacy and resistance. Cancer cells often exploit this crosstalk to evade therapy. For instance, pancreatic cancer cells resist TRAIL-induced apoptosis by overexpressing IAP family proteins (e.g., cIAP-1, XIAP) that block caspase activation [14]. However, combining TRAIL pathway activators like TLY012 with agents that target other pathways (e.g., ONC201) can overcome this resistance by synergistically activating multiple death mechanisms [14]. Similarly, the BCL-2 inhibitor venetoclax, approved for leukemia, promotes intrinsic apoptosis by mimicking BH3-only proteins, but its efficacy can be modulated by the status of other PANoptosis components [14] [95]. Therapeutically, simultaneously targeting multiple nodes within the PANoptosis network may provide a more effective strategy to overcome the inherent redundancy and compensatory mechanisms that lead to drug resistance.
Implications for Neurological and Ischemic Diseases
In neurological contexts, PANoptosis often plays a detrimental role by exacerbating neuroinflammation and neuronal loss. In Alzheimer's disease, β-amyloid oligomers can activate the ZBP1-PANoptosome in microglia, leading to mitochondrial ROS bursts and tau hyperphosphorylation [96]. In Parkinson's disease, α-synuclein fibrils activate the NLRP3/ASC axis to trigger PANoptosis in dopaminergic neurons [96]. In cerebral ischemia, damage signals activate sensors like ZBP1 and AIM2, initiating PANoptosis that worsens brain injury [99] [97]. Consequently, therapeutic inhibition of PANoptosis emerges as a promising neuroprotective strategy. Preclinical studies suggest that targeting upstream initiators (e.g., NLRP3 with MCC950) or key effectors (e.g., caspases with Q-VD-OPh) can mitigate tissue damage [96] [99].
Emerging Therapeutic Strategies
Future therapeutic development must account for the complexity of PANoptosis. Potential strategies include:
- Multi-target Inhibitors: Developing single agents or combination therapies that concurrently target multiple PANoptosis components (e.g., simultaneously inhibiting NLRP3 and RIPK1) [96] [98].
- Context-Specific Agonists: In cancer, designing agents that selectively induce PANoptosis in malignant cells while sparing healthy tissues, potentially by leveraging differential expression of death receptors or metabolic vulnerabilities [14] [100].
- Biomarker-Driven Therapy: Identifying biomarkers that predict a patient's likelihood of responding to specific death pathway modulators, enabling personalized treatment approaches [95] [98].
The paradigm of PANoptosis represents a fundamental shift in our understanding of cellular suicide programs. For researchers focused on caspase activation in intrinsic versus extrinsic apoptosis, this new framework necessitates a broader experimental perspective that considers the extensive crosstalk and compensatory mechanisms between all programmed cell death pathways. The experimental and therapeutic implications are significant: robust research requires multi-parameter validation, and effective treatments may need to target the integrated PANoptosis network rather than individual pathways. As our understanding of these complex interactions deepens, so too will our ability to precisely modulate cell death for therapeutic benefit across a spectrum of human diseases.
Direct Comparison and Integration: Crosstalk, Non-Apoptotic Functions, and Pathway Validation
Programmed cell death, or apoptosis, is a fundamental process essential for development, tissue homeostasis, and immune defense. At the molecular heart of apoptosis lie caspases, a family of cysteine-aspartic proteases that cleave cellular substrates to orchestrate controlled cellular dismantling [101]. These enzymes are synthesized as inactive zymogens (pro-caspases) and require precise activation to initiate the apoptotic cascade [11]. Apoptosis proceeds primarily via two distinct yet sometimes interconnected signaling pathways: the intrinsic (mitochondrial) pathway and the extrinsic (death receptor) pathway [102] [103]. The intrinsic pathway is activated by internal cellular stressors, such as DNA damage or oxidative stress, while the extrinsic pathway is triggered by extracellular death ligands binding to cell surface receptors [102]. This whitepaper provides a detailed technical comparison of these pathways, focusing on their molecular initiators, key adaptor proteins, and the caspase cascades they activate, providing a resource for researchers and drug development professionals working in this critical field.
Pathway Architecture: Core Components and Cascades
The Extrinsic Apoptosis Pathway
The extrinsic pathway is initiated outside the cell through death receptors belonging to the tumor necrosis factor (TNF) receptor superfamily, such as Fas (CD95), TNFR1, and TRAIL-R [103]. The core mechanism involves:
- Molecular Initiators: Death ligands (e.g., FasL, TNF-α, TRAIL) binding to their cognate transmembrane death receptors [102] [103].
- Key Adaptors and Complexes: Ligand binding induces receptor trimerization and recruitment of the adaptor protein FADD (Fas-Associated protein with Death Domain) via homophilic death domain interactions. FADD then recruits procaspase-8 (and in humans, procaspase-10) via death effector domain (DED) interactions, forming a multi-protein complex known as the Death-Inducing Signaling Complex (DISC) [11] [101].
- Caspase Activation: Within the DISC, procaspase-8 molecules are brought into close proximity, leading to their dimerization, autoproteolytic cleavage, and full activation [11] [101]. This "induced proximity" model is the hallmark of initiator caspase activation.
The Intrinsic Apoptosis Pathway
The intrinsic pathway is activated by internal cellular insults, including DNA damage, radiation, oxidative stress, or cytokine deprivation [102] [103]. The core mechanism involves:
- Molecular Initiators: Intracellular stress signals, often mediated by the tumor suppressor p53, which upregulates pro-apoptotic genes [102].
- Key Adaptors and Complexes: The critical event is mitochondrial outer membrane permeabilization (MOMP), controlled by the Bcl-2 family of proteins [103]. Anti-apoptotic members (e.g., Bcl-2, Bcl-xL) preserve membrane integrity, while pro-apoptotic members (e.g., BAX, BAK) oligomerize to form pores upon activation [103]. This leads to the release of cytochrome c from the mitochondrial intermembrane space into the cytosol.
- Caspase Activation: Cytochrome c binds to the adaptor protein Apaf-1 (Apoptotic Protease-Activating Factor 1), which in the presence of dATP/ATP, oligomerizes to form a wheel-like signaling platform called the apoptosome [11] [101]. The apoptosome recruits and activates procaspase-9 via CARD-CARD interactions [11].
The Execution Phase
Both pathways converge to activate the executioner caspases. Active initiator caspase-8 (extrinsic) or caspase-9 (intrinsic) cleaves and activates the effector caspases-3, -6, and -7 [101]. These executioners then systematically cleave over 600 cellular substrates, including structural proteins like lamins and cytoskeletal components, and proteins involved in DNA repair, such as PARP, leading to the characteristic morphological hallmarks of apoptosis: cell shrinkage, chromatin condensation, DNA fragmentation, and formation of apoptotic bodies [104] [103] [101].
There is crosstalk between the two pathways. In some cell types (Type II cells), the extrinsic pathway amplifies the death signal through caspase-8-mediated cleavage of the BH3-only protein BID into truncated tBID. tBID translocates to the mitochondria, activating BAX and BAK to trigger MOMP and engage the intrinsic pathway, thereby amplifying the caspase cascade [102] [11] [103].
Figure 1: Apoptosis Signaling Pathways Overview. This diagram illustrates the key components and flow of signals in the extrinsic (yellow) and intrinsic (green) apoptosis pathways, culminating in the execution phase (red).
Comparative Analysis: Molecular Components
Table 1: Comparative Overview of Intrinsic and Extrinsic Apoptosis Pathways
| Feature | Extrinsic Pathway | Intrinsic Pathway |
|---|---|---|
| Initiating Stimulus | Extracellular death ligands (e.g., FasL, TRAIL) [102] [103] | Intracellular stress (e.g., DNA damage, radiation, oxidative stress) [102] [103] |
| Molecular Initiator | Death Receptors (e.g., Fas, TNFR1) [103] | p53, Bcl-2 family proteins (BAX, BAK) [102] [103] |
| Key Adaptor Protein | FADD (Fas-Associated Death Domain) [11] [101] | Apaf-1 (Apoptotic Protease-Activating Factor 1) [11] [101] |
| Signaling Complex | DISC (Death-Inducing Signaling Complex) [11] [101] | Apoptosome [11] [101] |
| Initiator Caspase | Caspase-8, Caspase-10 (human) [3] [101] | Caspase-9 [3] [101] |
| Regulatory Complex | FADDosome [20] | PIDDosome (for caspase-2 activation) [11] |
| Primary Regulation | c-FLIP inhibits caspase-8 activation at the DISC [102] | Bcl-2 family proteins control MOMP; IAPs inhibit caspases [103] |
| Crosstalk Mechanism | Caspase-8 cleaves BID to form tBID, activating the mitochondrial pathway [102] [103] | N/A |
Table 2: Caspase Classification and Functions in Apoptosis
| Caspase | Role/Type | Activation Complex | Key Functions | Domain |
|---|---|---|---|---|
| Caspase-8 | Initiator (Extrinsic) [101] | DISC / FADDosome [11] [20] | Activates executioner caspases; cleaves BID [3] [103] | DED [3] |
| Caspase-10 | Initiator (Extrinsic) [101] | DISC [101] | Involved in extrinsic apoptosis; may regulate caspase-8 [3] | DED [3] |
| Caspase-9 | Initiator (Intrinsic) [101] | Apoptosome [11] [101] | Activates executioner caspases-3 and -7 [3] [103] | CARD [3] |
| Caspase-2 | Initiator (Intrinsic) [101] | PIDDosome [11] | DNA damage response; cleaves BID [3] [11] | CARD [3] |
| Caspase-3 | Executioner [101] | Cleaved by initiator caspases | Primary executioner; cleaves PARP, ICAD, etc. [103] [101] | Short Prodomain [43] |
| Caspase-6 | Executioner [101] | Cleaved by initiator caspases | Cleaves lamin proteins [101] | Short Prodomain [43] |
| Caspase-7 | Executioner [101] | Cleaved by initiator caspases | Cleaves PARP; overlaps with caspase-3 [3] [101] | Short Prodomain [43] |
Experimental Analysis: Methodologies and Techniques
Key Experimental Protocols for Pathway Analysis
1. DISC Immunoprecipitation for Extrinsic Pathway Analysis:
- Objective: To isolate and identify components of the Death-Inducing Signaling Complex (DISC) formed upon death receptor activation.
- Methodology: Cells are stimulated with a death ligand (e.g., FasL or TRAIL) for varying time points. Cells are then lysed under mild, non-denaturing conditions to preserve protein complexes. The DISC is immunoprecipitated using antibodies specific to the death receptor (e.g., anti-Fas antibody) or an epitope-tagged receptor. Co-precipitated proteins are eluted and analyzed by Western blotting to detect key components like FADD, procaspase-8, and its cleaved products [11].
- Key Controls: Use of unstimulated cells and isotype control antibodies for immunoprecipitation is critical.
2. Cytochrome c Release Assay for Intrinsic Pathway Analysis:
- Objective: To monitor mitochondrial outer membrane permeabilization (MOMP), a defining event in the intrinsic pathway.
- Methodology: Cells are treated with intrinsic apoptotic stimuli (e.g., UV radiation, chemotherapeutic agents). At designated times, cells are gently fractionated via differential centrifugation to separate the heavy membrane fraction (containing mitochondria) from the cytosolic fraction (S-100). The presence of cytochrome c in the cytosolic fraction is then detected by Western blotting. Its appearance in the cytosol indicates successful MOMP [11] [103].
- Key Controls: Blotting for a mitochondrial marker (e.g., COX IV) in the cytosolic fraction ensures purity and lack of contamination.
3. Caspase Activity Assays:
- Objective: To measure the enzymatic activity of specific caspases during apoptosis.
- Methodology:
- Fluorometric/Spectrophotometric Assays: Cell lysates are incubated with caspase-specific peptide substrates conjugated to a fluorophore (e.g., AFC) or chromophore (e.g., pNA). Cleavage of the substrate releases the reporter, generating a measurable signal. Substrate specificity (e.g., DEVD for caspases-3/7, IETD for caspase-8, LEHD for caspase-9) allows discrimination between initiator and executioner activities [104].
- Western Blot Analysis: Detection of caspase cleavage (e.g., procaspase-3 to cleaved caspase-3) provides evidence of activation [11].
- Key Controls: Use of specific caspase inhibitors (e.g., Z-VAD-FMK, pan-caspase inhibitor) confirms the specificity of the signal.
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents for Apoptosis Research
| Reagent / Assay | Function / Application | Specific Example |
|---|---|---|
| Recombinant Death Ligands | Activate the extrinsic pathway by binding death receptors. | Recombinant Human FasL/TRAIL [102] |
| Chemotherapeutic Agents | Induce DNA damage and activate the intrinsic pathway. | Etoposide, Actinomycin D [102] |
| Caspase-Specific Inhibitors | Pharmacologically inhibit caspase activity to determine functional roles. | Z-VAD-FMK (pan-caspase), Z-DEVD-FMK (caspase-3/7) [43] |
| Mitochondrial Fractionation Kits | Isolate mitochondrial and cytosolic fractions to assay MOMP. | Assays for cytochrome c release [103] |
| Phospho-Specific Antibodies | Detect activation of signaling proteins by Western blot. | Anti-phospho-BID, Anti-cleaved Caspase-3, Anti-cleaved PARP [11] |
| qPCR Arrays | Profile expression of apoptosis-related genes. | RT² Profiler PCR Array Human Apoptosis [103] |
| Flow Cytometry Assays | Quantify apoptosis in cell populations using fluorescent probes. | Annexin V/PI staining, FLICA caspase activity probes [103] |
Figure 2: Key Experimental Workflows. This diagram outlines three fundamental methodologies for analyzing extrinsic (DISC), intrinsic (MOMP), and executioner (caspase activity) phases of apoptosis.
Discussion: Therapeutic Implications and Future Directions
The precise understanding of caspase activation pathways holds immense therapeutic potential. In cancer, where apoptosis is often evaded, strategies include developing agents to trigger the extrinsic pathway (e.g., recombinant TRAIL) or small molecules that inhibit anti-apoptotic Bcl-2 proteins (e.g., Venetoclax) to promote the intrinsic pathway [3] [103]. Conversely, in neurodegenerative diseases where excessive apoptosis occurs, caspase inhibitors are being explored as neuroprotective agents [3] [101]. The complexity is heightened by the contextual roles of caspases; for instance, caspase-8 can serve as a molecular switch between apoptosis, necroptosis, and pyroptosis, suggesting its modulation must be highly specific [3] [20]. Future research will continue to elucidate the intricate regulatory networks and post-translational modifications controlling these pathways, paving the way for more targeted and effective therapies in oncology, neurology, and immunology.
Apoptosis, a programmed cell death mechanism, is essential for development, homeostasis, and disease defense in multicellular organisms [3] [105]. The two principal apoptosis pathways—extrinsic (death receptor-mediated) and intrinsic (mitochondrial)—converge on a common execution phase mediated by effector caspases [106] [107]. A critical molecular bridge connecting these pathways is formed by the caspase-8-mediated cleavage of the BH3-interacting domain death agonist (BID) protein [105]. This cleavage event allows death receptor signals to amplify through the mitochondrial pathway, ensuring efficient apoptosis in cell types where a direct caspase cascade is insufficient [106] [108].
The significance of this crosstalk is particularly evident in Type II cells, where low levels of active caspase-8 generated at the death-inducing signaling complex (DISC) cannot directly activate effector caspases sufficiently due to inhibitors like XIAP [106] [107]. Instead, caspase-8 cleaves BID to produce truncated BID (tBid), which translocates to mitochondria and triggers mitochondrial outer membrane permeabilization (MOMP), leading to cytochrome c release and apoptosome formation [106] [3] [108]. This review synthesizes current knowledge on the molecular mechanisms, experimental evidence, and regulatory aspects of this critical apoptotic crosstalk node, providing a comprehensive resource for researchers investigating caspase signaling and therapeutic targeting.
Molecular Mechanisms: From Caspase-8 Activation to BID Cleavage
Caspase-8 Activation at the DISC
The extrinsic apoptosis pathway initiates when death ligands (e.g., FasL, TRAIL) bind to their cognate receptors, triggering receptor trimerization and recruitment of the adaptor protein FADD through death domain interactions [107]. FADD then recruits procaspase-8 via death effector domain (DED) interactions, forming the core of the DISC [107] [109]. Within this complex, caspase-8 zymogens undergo dimerization—a process fundamental to their activation [109].
Unlike effector caspases that exist as pre-formed dimers, initiator caspases like caspase-8 are monomers that require induced proximity-driven dimerization on oligomeric platforms to gain catalytic activity [109]. This dimerization enables autoproteolytic cleavage at specific aspartic acid residues (D216, D374, D384 in human caspase-8), generating the fully active heterotetramer composed of two p18 and two p10 subunits [108] [110]. The initial cleavage at D374 yields p43 and p12 subunits, with subsequent cleavages producing the mature enzyme [108]. Recent optogenetic studies have validated that controlled oligomerization alone is sufficient to activate caspase-8, confirming the induced proximity model [110].
BID Structure and Cleavage by Caspase-8
BID is a 22 kDa pro-apoptotic member of the Bcl-2 family characterized by eight α-helices, with two central hydrophobic helices (αH6 and αH7) surrounded by six amphipathic ones [105] [111]. This structural organization resembles pore-forming bacterial toxins and provides insights into its membrane-targeting function [111]. Between αH2 and αH3, BID contains an unstructured loop (amino acids 42-79) that houses the caspase-8 cleavage site [106] [105].
Caspase-8 cleaves BID primarily at Asp-60 (D60) in humans, although some studies suggest alternative cleavage sites (e.g., Leu56 or Gly60) depending on cellular context [106] [105]. This proteolytic event separates the N-terminal fragment (p7) from the C-terminal fragment (p15), known as truncated BID (tBid) [105]. Despite this cleavage, the overall structure of BID remains largely unchanged, suggesting that proteolysis alone does not fully activate the protein [106] [111]. Following cleavage, tBid undergoes myristoylation at Gly60, a post-translational modification that facilitates its association with mitochondrial membranes [105].
Table 1: Key Molecular Components in Caspase-8/BID Apoptotic Signaling
| Component | Type/Structure | Function in Apoptotic Signaling |
|---|---|---|
| Caspase-8 | Initiator caspase (DED domains, p55/p53 zymogen) | Extrinsic pathway initiator; cleaves and activates effector caspases and BID |
| BID | BH3-only Bcl-2 protein (8 α-helices, unstructured loop 42-79) | Critical crosstalk node; connects death receptor signaling to mitochondrial pathway |
| tBid | C-terminal BID fragment (p15) after cleavage | Mitochondrial-targeted activator; induces MOMP through Bax/Bak activation |
| FADD | Adaptor protein (Death Domain, DED domains) | DISC scaffold; bridges death receptors and caspase-8 recruitment |
| Bax/Bak | Multi-BH domain pro-apoptotic proteins | MOMP effectors; form pores in mitochondrial outer membrane |
Mitochondrial Targeting and MOMP Induction
Following cleavage and myristoylation, tBid translocates to the mitochondrial outer membrane (MOM), where its hydrophobic αH6 and αH7 helices embed into the lipid bilayer [105]. This mitochondrial association is crucial for tBid function, as demonstrated by studies showing that tBid mutants lacking these helices exhibit diminished apoptotic activity despite possessing an intact BH3 domain [106].
At the MOM, tBid activates the pro-apoptotic effector proteins Bax and Bak through both direct and indirect mechanisms [105] [112]. tBid binding induces conformational changes in Bax/Bak that promote their oligomerization and integration into the MOM, forming pores that facilitate cytochrome c release [112]. This permeabilization allows cytochrome c to escape into the cytosol, where it promotes apoptosome formation with Apaf-1 and procaspase-9, leading to caspase-9 activation and subsequent amplification of the caspase cascade [106] [3].
Figure 1: Caspase-8/BID Signaling Pathway. This diagram illustrates the molecular events from death receptor activation to apoptosis execution via caspase-8-mediated BID cleavage and mitochondrial amplification.
Experimental Evidence and Key Findings
Genetic Studies Establishing Essential Roles
Gene editing approaches have been instrumental in elucidating the non-redundant functions of caspase-8 and BID in extrinsic apoptosis. A pivotal study using Bid-deficient (Bid KO) colon cancer cells demonstrated complete resistance to TRAIL-induced apoptosis, which was rescued by wild-type Bid reconstitution but not by caspase-resistant BidD60E or BH3-defective BidG94E mutants [106]. This genetic evidence established that both caspase-8 cleavage and an intact BH3 domain are essential for BID's pro-apoptotic function.
Further insights came from the generation of Bid/Bax/Bak triple knockout (TKO) cells, which revealed that BID is primarily cleaved by caspase-8 rather than effector caspases during TRAIL-induced apoptosis [106]. This finding resolved longstanding questions about which caspase is responsible for BID cleavage in death receptor signaling and highlighted caspase-8's specific role in this process. Complementary in vivo studies showed that Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis, underscoring the physiological relevance of this pathway [112].
Compartmentalization of Caspase-8 and BID Activation
Recent research has revealed that caspase-8 activation and BID cleavage are spatially regulated processes. Contrary to the traditional view that caspase-8 functions exclusively at the DISC, studies have identified stable caspase-8/BID complexes on mitochondrial membranes [108]. During type II apoptosis, caspase-8 translocates to mitochondria, where it associates with the mitochondrial-specific lipid cardiolipin—an anionic phospholipid predominantly located at contact sites between mitochondrial membranes [113].
This compartmentalization creates a mitochondrial platform for BID cleavage, ensuring that tBid is generated precisely where it is needed for MOMP induction [108] [113]. Inhibition of caspase-8 mitochondrial enrichment impairs both caspase-8 activation and BID cleavage, establishing this localization as functionally significant [108]. The dependence on cardiolipin was further demonstrated in cells from Barth syndrome patients, which exhibit reduced mature cardiolipin and consequent resistance to Fas-induced apoptosis [113].
Table 2: Key Experimental Evidence for Caspase-8/BID Crosstalk
| Experimental Approach | Key Finding | Citation/Model |
|---|---|---|
| Bid KO and reconstitution | BidD60E (caspase-resistant) fails to restore TRAIL-induced apoptosis | HCT116 colon cancer cells [106] |
| Bid/Bax/Bak TKO cells | BID is primarily cleaved by caspase-8, not effector caspases, in TRAIL signaling | HCT116-derived TKO cells [106] |
| Mitochondrial caspase-8 inhibition | Blocks BID cleavage and apoptosis despite DISC formation | HeLa cells [108] |
| Cardiolipin manipulation | Mature cardiolipin essential for caspase-8 mitochondrial translocation and BID cleavage | Barth syndrome models [113] |
| Optogenetic caspase-8 activation | Caspase-8 oligomerization sufficient to induce BID cleavage and apoptosis | HEK293T and HeLa cells [110] |
Structural Insights into BID Activation
Structural studies have provided mechanistic explanations for how caspase-8 cleavage activates BID. Nuclear magnetic resonance (NMR) analysis revealed that BID maintains its overall globular structure after caspase-8 cleavage, with the two fragments remaining non-covalently associated [111]. This finding initially posed a puzzle regarding how cleavage activates BID.
The solution emerged from studies demonstrating that cleavage enables subsequent myristoylation at the newly exposed N-terminal glycine of tBid (Gly60) [105]. This lipid modification, coupled with exposure of the hydrophobic αH6 and αH7 helices, promotes tBid targeting and insertion into mitochondrial membranes [105] [111]. Once membrane-integrated, tBid's BH3 domain becomes accessible to interact with Bax and Bak, facilitating their activation through both direct binding and relief of inhibition by anti-apoptotic Bcl-2 family members [112].
Research Methods and Experimental Approaches
Genetic Manipulation Strategies
Contemporary research on caspase-8/BID signaling employs sophisticated gene editing techniques to establish functional relationships. The following protocol outlines key methodological approaches:
Generation of Bid-Deficient Cells Using TALEN or CRISPR/Cas9
- Design TALEN pairs targeting early exons of the human BID gene or CRISPR sgRNAs with high on-target efficiency [106]
- Transfect HCT116 or other suitable cell lines with gene editing constructs
- Isolate single-cell clones by limiting dilution and validate knockout via immunoblotting and sequencing
- For reconstitution studies, clone wild-type and mutant BID (e.g., D60E, G94E) into retroviral vectors (e.g., pMSCV-PIG) and transduce Bid KO cells [106]
Triple Knockout (TKO) Cell Generation
- Sequential or simultaneous targeting of BID, BAX, and BAK genes in HCT116 cells
- Validate complete knockout of all three proteins and confirm resistance to various apoptotic stimuli
- Use TKO cells for definitive assessment of BID requirement in death receptor-mediated apoptosis [106]
Biochemical and Cellular Assays
Monitoring BID Cleavage and Caspase-8 Activation
- Treat cells with TRAIL (100 ng/mL) or other death ligands for 0-8 hours
- Prepare whole-cell lysates in EBC buffer (50 mM Tris, 120 mM NaCl, 0.5% NP-40) with protease inhibitors
- Perform immunoblotting with anti-BID, anti-caspase-8, anti-PARP, and anti-β-actin antibodies [106]
- For mitochondrial fractionation, use differential centrifugation to isolate heavy membrane fractions and probe for caspase-8 localization [108]
Optogenetic Caspase-8 Activation
- Construct optogenetic tools by fusing caspase-8 with photolyase homology region (PHR) domain of Arabidopsis cryptochrome 2 (Opto-Casp8-V1) or separately fusing caspase-8 with PHR and CIB1 domains (Opto-Casp8-V2) [110]
- Transfect constructs into HEK293T or HeLa cells and expose to blue light (30-50 μmol m⁻² s⁻¹) for specified durations
- Assess caspase-8 oligomerization by confocal microscopy and cleavage by immunoblotting
- Quantify apoptosis using Annexin V/PI staining and flow cytometry [110]
Figure 2: Experimental Workflow for Studying Caspase-8/BID Signaling. This diagram outlines the complementary methodological approaches used to investigate the caspase-8/BID crosstalk mechanism.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Studying Caspase-8/BID Signaling
| Reagent/Tool | Specific Example | Application/Function |
|---|---|---|
| Gene Editing Systems | TALEN targeting human BID; CRISPR/Cas9 with sgRNAs for BAX/BAK | Generation of isogenic knockout cell lines to establish protein requirements |
| Apoptosis Inducers | Recombinant TRAIL (100 ng/mL); Anti-Fas agonistic antibodies | Specific activation of death receptor pathways to study extrinsic apoptosis |
| Caspase Inhibitors | Z-VAD-FMK (pan-caspase); Z-IETD-FMK (caspase-8 specific) | Pharmacological inhibition to determine caspase-dependent processes |
| Antibodies for Detection | Anti-BID (cell signaling); Anti-caspase-8; Anti-PARP; Anti-cytochrome c | Immunoblotting and immunofluorescence to monitor cleavage and localization |
| Optogenetic Tools | Opto-Casp8-V1/V2 (CRY2/CIB1-caspase-8 fusions) | Precise spatiotemporal control of caspase-8 activation using blue light |
| Mitochondrial Dyes | MitoTracker Red CMXRos; JC-1 | Assessment of mitochondrial membrane potential and integrity |
Regulatory Context and Therapeutic Implications
Integration with Other Cell Death Pathways
Caspase-8 serves as a critical molecular switch between apoptosis, necroptosis, and pyroptosis [3] [109]. When caspase-8 activity is compromised (e.g., by pharmacological inhibition or viral proteins), death receptor signaling often shifts to RIPK1/RIPK3-mediated necroptosis—a lytic, inflammatory form of cell death [107] [109]. This switch occurs because caspase-8 normally cleaves and inactivates components of the necroptotic machinery, particularly RIPK1 and RIPK3 [3] [107].
The balance between apoptotic and necroptotic signaling is further modulated by cFLIP isoforms, which heterodimerize with caspase-8 at the DISC [109]. These heterodimers exhibit altered substrate specificity, preferentially cleaving necroptosis components while showing reduced activity toward apoptotic substrates like BID and caspase-3 [109]. This regulatory mechanism ensures that apoptosis proceeds efficiently when caspase-8 activity is robust, while directing signaling toward necroptosis when caspase-8 is limited.
Pathophysiological Relevance and Therapeutic Targeting
Dysregulation of the caspase-8/BID pathway contributes to various human diseases. In cancer, impaired caspase-8 activation or BID mutation can confer resistance to death receptor-mediated apoptosis, facilitating tumor survival and progression [106] [105]. Conversely, excessive BID activation has been implicated in pathological cell death observed in neurodegenerative disorders, ischemia-reperfusion injury, and viral infections [105].
Several viruses have evolved strategies to subvert this apoptotic pathway. For instance, hepatitis B virus (HBV), herpes simplex virus (HSV), and SARS-CoV-2 encode proteins that interfere with BID activation or function, thereby prolonging host cell survival to support viral replication [105]. Understanding these viral evasion mechanisms provides insights for developing novel antiviral strategies that restore apoptotic sensitivity in infected cells.
Therapeutic approaches targeting the caspase-8/BID axis include:
- TRAIL receptor agonists that exploit the caspase-8/BID pathway to kill cancer cells while sparing normal cells [106]
- SMAC mimetics that antagonize XIAP, reducing the threshold for mitochondrial amplification in Type II cells [107]
- Small molecules that directly activate BAX/BAK or inhibit anti-apoptotic Bcl-2 proteins, bypassing upstream defects in death receptor signaling [112]
The caspase-8-mediated cleavage of BID represents a critical signaling node that amplifies extrinsic apoptotic signals through the intrinsic pathway. This crosstalk mechanism ensures robust cell death execution in Type II cells, where direct caspase activation is insufficient. Key features of this pathway include its spatial regulation through mitochondrial compartmentalization, its modulation by competing cell death programs, and its pathophysiological relevance in cancer, infection, and degenerative disorders.
Ongoing research continues to elucidate the structural basis of BID activation, the regulatory networks that balance apoptosis with alternative cell death pathways, and the therapeutic potential of targeting this pathway in human disease. As our understanding of these molecular mechanisms deepens, so too will opportunities for developing more precise interventions that modulate apoptotic signaling for therapeutic benefit.
Caspases, a family of cysteine-aspartate proteases, are universally recognized as master regulators of apoptotic cell death. However, emerging research has illuminated critical roles for these enzymes in diverse physiological processes that extend far beyond cell death, including cellular differentiation and the control of inflammation. This whitepaper synthesizes current evidence validating these non-apoptotic caspase functions, framing them within the established context of intrinsic and extrinsic apoptotic pathways. We provide a technical guide detailing experimental methodologies for discerning sublethal caspase activities, complete with summarized quantitative data, visualized signaling pathways, and essential research reagent solutions. For researchers and drug development professionals, understanding these non-canonical roles is paramount, as they present novel therapeutic avenues for treating developmental, neurodegenerative, and inflammatory diseases without triggering unintended cell death.
The historical classification of caspases as either apoptotic initiators (caspase-2, -8, -9, -10), apoptotic executioners (caspase-3, -6, -7), or inflammatory mediators (caspase-1, -4, -5, -11) is insufficient to describe their full functional repertoire [85] [114]. It is now evident that apoptotic caspases can drive inflammatory lytic cell death, and that low-level, spatially restricted caspase activation can direct fundamental cellular processes without inducing apoptosis [115] [3]. This paradigm shift necessitates robust experimental frameworks to validate these non-apoptotic functions. The core principle is that localized activation, tight regulation by cellular inhibitors like XIAP, and limited substrate cleavage distinguish these roles from full-blown apoptotic execution [115]. This guide details the mechanisms and validation strategies for these roles, anchoring them in the molecular language of caspase-8-mediated extrinsic and caspase-9-mediated intrinsic pathways familiar to all cell death researchers.
Molecular Mechanisms of Non-Apoptotic Caspase Functions
Caspases in Cellular Differentiation
Caspase activity is a critical regulator of differentiation in multiple cell lineages, particularly in the hematopoietic and nervous systems. The key differentiator from apoptosis is the tight control over the extent and localization of caspase activation.
Erythroid and Myeloid Differentiation: During erythropoiesis, a transient and subtle activation of caspase-3 occurs in basophilic erythroblasts. This is spatially and temporally controlled by the chaperone HSP70, which migrates to the nucleus to protect the master regulator GATA-1 from caspase-mediated cleavage [116]. This limited activity is thought to prepare the cell for nucleus and organelle expulsion during terminal differentiation. Similarly, in megakaryocytes, spatially restricted caspase-3 activation promotes proplatelet maturation and platelet shedding [116]. Furthermore, caspase-8 activation downstream of colony-stimulating factor-1 (CSF-1) signaling in monocytes contributes to their differentiation into resting macrophages by modulating NF-κB activity [116].
Neuronal Development: In the developing nervous system, caspases are indispensable for axonal and dendritic pruning, a process that refines neuronal connectivity without killing the parent neuron [115]. In Drosophila, the initiator caspase DRONC and effector caspases are required for dendritic pruning of sensory neurons during metamorphosis [115]. In vertebrates, caspases-3, -6, and -9 are involved in pruning retinal ganglion cell axons and in axon degeneration induced by neurotrophic factor deprivation in sensory neurons [115]. Crucially, caspase activation in these contexts is confined to the axonal or dendritic compartment being eliminated.
Caspases in Inflammation
The role of caspases in inflammation extends far beyond the traditional maturation of IL-1β and IL-18 by caspase-1.
Alternative Inflammasome Activation: Human monocytes can secrete mature IL-1β in response to lipopolysaccharide (LPS) via a caspase-8-dependent pathway, which operates independently of the canonical caspase-1 inflammasome and does not lead to cell death [116]. This reveals a non-apoptotic, pro-inflammatory role for an initiator caspase of the extrinsic pathway.
Crosstalk in PANoptosis: Recent research has identified extensive molecular crosstalk between cell death pathways, leading to the concept of PANoptosis—a lytic, innate immune cell death pathway driven by multi-protein complexes called PANoptosomes [85]. These complexes can simultaneously recruit and activate multiple caspases, including caspase-1, -3, -7, and -8, demonstrating that apoptotic caspases are integral players in inflammatory cell death [85] [3].
The table below summarizes key non-apoptotic functions of specific caspases.
Table 1: Validated Non-Apoptotic Functions of Caspases
| Caspase | Non-Apoptotic Function | Biological Context | Key Mechanisms |
|---|---|---|---|
| Caspase-3 | Erythroid Differentiation [116] | Hematopoiesis | Transient activation; HSP70 protects GATA-1 from cleavage. |
| Caspase-3 | Axon/Dendrite Pruning [115] | Neuronal Development | Localized activation in distal axons/dendrites. |
| Caspase-8 | Monocyte to Macrophage Differentiation [116] | Immune Cell Function | Forms molecular platform with CSF-1R; modulates NF-κB. |
| Caspase-8 | Alternative IL-1β Secretion [116] | Inflammation | Activated by non-canonical inflammasome in response to LPS. |
| Caspase-9 | Axon Pruning [115] | Neuronal Development | Localized activation in axons deprived of NGF. |
| DRONC (Caspase-9 homologue) | Dendritic Pruning [115] | Drosophila Development | Requires DARK (APAF-1 homologue) for activation. |
Experimental Protocols for Validating Sublethal Caspase Activity
Distinguishing non-apoptotic caspase functions from apoptosis requires specific methodologies designed to detect low-level, localized activity and functional outcomes unrelated to cell death.
Monitoring Caspase Activity in Live Cells
Protocol: Using Genetically Encoded Caspase Reporters
This protocol utilizes FRET-based or switch-on fluorescent biosensors to visualize caspase activity in real-time within live cells and complex 3D environments [117] [118].
Reporter Design:
- FRET-based Reporter: A construct is designed with donor and acceptor fluorescent proteins (e.g., LSSmOrange and mKate2) linked by a caspase-cleavage sequence (e.g., DEVD for caspase-3-like activity). When the reporter is intact, FRET occurs. Upon caspase cleavage, the separation of the fluorophores leads to a loss of FRET, which can be measured by fluorescence lifetime imaging microscopy (FLIM) [118]. FLIM is preferred for 3D models as it is independent of probe concentration and light scattering.
- Switch-on Indicator (e.g., VC3AI): A circularly permuted fluorescent protein is cyclized using intein technology, with a caspase-cleavage site embedded in the linker. Cleavage by caspases restores fluorescence [117].
Cell Line Generation:
- Stably transduce the cell type of interest (e.g., neuronal lines, hematopoietic progenitors) with the reporter construct using lentiviral or PiggyBac transposon systems [117] [118].
- Select a homogeneous population using drug selection (e.g., blasticidin) or fluorescence-activated cell sorting (FACS).
Imaging and Analysis:
- Culture reporter cells under conditions that induce differentiation (e.g., with CSF-1 for monocytes) or pruning (e.g., NGF deprivation for sensory neurons).
- For FRET-FLIM, image cells over time and measure the fluorescence lifetime of the donor. A significant increase in donor lifetime indicates caspase cleavage [118].
- For switch-on indicators, directly monitor the emergence of fluorescence [117].
- Critical Control: Co-incubate with pan-caspase inhibitors (e.g., Z-VAD-fmk) or specific inhibitors (e.g., Z-DEVD-fmk for caspase-3/7) to confirm the signal is caspase-dependent [117].
Functional Validation in Neuronal Pruning
Protocol: Assessing Caspase Role in Axon Degeneration [115]
This method uses compartmentalized chambers to spatially separate apoptotic signals in the cell body from pruning signals in the axon.
Establishment of Compartmentalized Cultures:
- Plate primary sensory neurons (e.g., from Dorsal Root Ganglia) in compartmentalized chambers (e.g., Campenot chambers or microfluidic devices). The cell bodies reside in one compartment, while the axons grow through a physical barrier into a separate compartment.
Localized Treatment:
- Apply a pruning stimulus, such as NGF deprivation, exclusively to the axonal compartment. This induces axonal degeneration without activating apoptosis in the cell body.
- Maintain the cell body compartment in NGF-rich medium to ensure neuronal survival.
Inhibition and Analysis:
- Treat the axonal compartment with caspase inhibitors (Z-VAD-fmk, Z-DEVD-fmk) or introduce dominant-negative caspase mutants via viral transduction to assess the requirement for caspase activity.
- Readouts:
- Morphological: Quantify axonal fragmentation over time using immunofluorescence for axonal markers (e.g., β-III-tubulin).
- Biochemical: Perform western blot analysis of axonal lysates for cleaved, active caspase-3 or -6.
- Genetic: Utilize neurons from caspase-3, -6, or -9 knockout mice, which show significant resistance to axonal degeneration upon NGF deprivation [115].
The logical workflow for investigating non-apoptotic caspase functions is summarized below.
The Scientist's Toolkit: Essential Research Reagents
Successfully investigating non-apoptotic caspase functions relies on a specific toolkit of reagents and model systems. The table below details key resources.
Table 2: Research Reagent Solutions for Non-Apoptotic Caspase Studies
| Reagent / Model | Function and Utility | Example Application |
|---|---|---|
| Pharmacological Inhibitors | ||
| Z-VAD-fmk (Pan-caspase) [117] | Irreversibly binds catalytic site of most caspases. | Confirm caspase-dependency of a process; control for off-target apoptotic effects. |
| Z-DEVD-fmk (Caspase-3/7) [117] | Specific inhibitor of effector caspase activity. | Determine contribution of caspase-3/7 to differentiation or pruning. |
| Genetically Encoded Reporters | ||
| FRET-based Caspase Sensor (e.g., LSSmOrange-DEVD-mKate2) [118] | Detects caspase-3/7 activity via loss of FRET, quantified by FLIM. | Real-time, high-resolution imaging of caspase activation in live cells and in vivo. |
| Switch-on Indicator (e.g., VC3AI) [117] | Becomes fluorescent upon caspase-3/7 cleavage. | Simple, sensitive detection of caspase activity in single cells and populations. |
| Genetic Models | ||
| Caspase Knockout Mice (e.g., Casp3⁻/⁻, Casp6⁻/⁻) [115] | Provides cells devoid of specific caspase activity. | Study requirement of specific caspases in neuronal pruning or differentiation. |
| Compartmentalized Cell Cultures (e.g., Microfluidic Devices) [115] | Enables spatial separation of cell bodies and axons. | Study localized caspase activation in axon pruning without somatic apoptosis. |
| Differentiation-Inducing Cytokines | ||
| Colony-Stimulating Factor-1 (CSF-1) [116] | Promotes monocyte to macrophage differentiation. | Model for studying caspase-8 activation in cell differentiation. |
| Erythropoietin (EPO) [116] | Drives erythroid progenitor differentiation. | Model for studying transient caspase-3 activation in erythropoiesis. |
Integrated Signaling Pathways in Non-Apoptotic Contexts
Non-apoptotic caspase signaling often engages modules of the classic apoptotic pathways but regulates them to avoid a full death response. The diagram below integrates these pathways in the context of differentiation and inflammation.
The evidence is compelling: caspases are multifunctional enzymes whose roles encompass vital physiological processes like differentiation and inflammation, operating independently of their well-characterized death-inducing activities. The experimental frameworks outlined in this guide—emphasizing the detection of localized activity, the use of specific inhibitors and genetic models, and the functional assessment in validated cellular systems—provide a roadmap for researchers to rigorously investigate these non-apoptotic functions. As the field moves forward, a deeper understanding of how cells harness the proteolytic power of caspases for life-sustaining functions will undoubtedly reveal new biology and unlock innovative therapeutic strategies that modulate caspase activity with precision, aiming to treat disease without the cost of cell loss.
PANoptosis represents a transformative concept in cell death research, bridging the historical divide between the studies of intrinsic and extrinsic apoptosis. This novel, inflammatory programmed cell death (PCD) pathway is characterized by the simultaneous activation of pyroptosis, apoptosis, and necroptosis, and is coordinated by multifaceted protein complexes known as PANoptosomes [119] [120]. Within these complexes, caspase-6 and caspase-8 function as critical molecular switches, integrating signals from various PCD pathways and dictating cellular fate in response to pathogenic infections, cellular damage, and other stressors [121] [122]. This whitepaper provides an in-depth analysis of the molecular architecture of PANoptosis, with a specialized focus on the mechanistic roles of caspase-6 and caspase-8, and provides the scientific community with structured data, experimental protocols, and visualization tools to advance research in this emerging field.
The conventional understanding of programmed cell death has long been segmented into distinct pathways: the non-inflammatory, immunologically silent process of apoptosis, and the lytic, inflammatory pathways of pyroptosis and necroptosis [119]. Apoptosis itself is bifurcated into the extrinsic pathway, initiated by cell surface death receptors and reliant on caspase-8, and the intrinsic pathway, triggered by internal cellular stresses and mediated by caspase-9 [20] [123]. However, recent biochemical, genetic, and molecular evidence has revealed extensive crosstalk and redundancy between these pathways, challenging their historical classification as independent linear routes [121] [120].
This crosstalk led to the discovery and characterization of PANoptosis, an unique inflammatory PCD pathway activated by specific triggers (e.g., viral infection, cytokine storms) and regulated by a macromolecular complex dubbed the PANoptosome [119] [122]. The totality of the biological effects observed in PANoptosis cannot be fully explained by pyroptosis, apoptosis, or necroptosis alone, and critically, inhibiting any one of these three pathways fails to prevent cell death [120] [123]. This paradigm shift positions caspase-6 and caspase-8 as central integrators and switchers within the PANoptosome, effectively bridging signaling between the intrinsic and extrinsic death machinery [121] [124] [125].
The Molecular Architecture of PANoptosis
Core Components of the PANoptosome
The PANoptosome is a cytosolic multiprotein complex that serves as the execution platform for PANoptosis. Its composition, while adaptable to specific stimuli, generally incorporates three classes of proteins [120] [125]:
- Sensors: Proteins such as Z-DNA binding protein 1 (ZBP1), NLRP3, and AIM2 that detect pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs).
- Adapters: Proteins like ASC and FADD that bridge sensor proteins to effector molecules via homotypic domain interactions.
- Catalytic Effectors: Enzymes including caspase-1, caspase-6, caspase-8, RIPK1, and RIPK3 that execute the core cell death programs.
To date, several stimulus-specific PANoptosomes have been identified, including the ZBP1-PANoptosome, AIM2-PANoptosome, RIPK1-PANoptosome, and NLRP12-PANoptosome [120] [123] [125].
PANoptosis in Disease and Homeostasis
PANoptosis functions as a double-edged sword in human physiology and disease. It provides a robust host defense mechanism against pathogens like influenza A virus (IAV) and Francisella novicida by eliminating infected cells and triggering protective inflammation [119] [120]. Conversely, dysregulated PANoptosis is implicated in the pathology of numerous conditions, including acute lung injury/acute respiratory distress syndrome (ALI/ARDS), cancer, ischemic reperfusion injury, and autoimmune diseases [120] [123]. In cancer, for instance, PANoptosis can suppress tumor growth by killing cancer cells, but it can also shape a tumor-promoting microenvironment [123] [125].
Caspase-6 and Caspase-8 as Molecular Switches
Caspase-8: The Master Regulator at the Crossroads
Caspase-8, an initiator caspase in the extrinsic apoptotic pathway, serves as a quintessential molecular switch within PANoptosis, balancing the activation of apoptosis, necroptosis, and pyroptosis [20] [122].
- Apoptosis Activation: Upon activation by death receptors, caspase-8 directly cleaves and activates executioner caspases-3 and -7, precipitating apoptotic cell death [119] [20].
- Necroptosis Suppression: Caspase-8 exerts a tonic inhibitory effect on necroptosis by proteolytically cleaving key necroptotic mediators, including RIPK1 and RIPK3 [119] [20]. When caspase-8 is inhibited genetically or pharmacologically, this brake is released, allowing the RIPK1-RIPK3-MLKL axis to drive necroptosis.
- Pyroptosis and PANoptosis Integration: Within the PANoptosome, caspase-8 contributes to the activation of the NLRP3 inflammasome and caspase-1, thereby promoting pyroptosis [119] [120]. It also cleaves gasdermin family members, such as GSDMC, to induce pyroptosis under certain conditions [20].
Table 1: Multifunctional Roles of Caspase-8 in Cell Death
| Role | Molecular Mechanism | Pathway | Context |
|---|---|---|---|
| Apoptosis Initiator | Direct activation of caspases-3/7; cleavage of BID to trigger intrinsic amplification | Apoptosis | Death Receptor signaling [119] [20] |
| Necroptosis Suppressor | Proteolytic cleavage and inactivation of RIPK1 and RIPK3 | Necroptosis | Homeostatic maintenance; Caspase inhibition [119] [20] |
| Pyroptosis Contributor | Activation of NLRP3 inflammasome; cleavage of GSDMC | Pyroptosis/PANoptosis | PANoptosome assembly [120] [20] |
| PANoptosis Integrator | Core component of multiple PANoptosomes (e.g., ZBP1, AIM2) | PANoptosis | Viral infection, cellular stress [121] [120] |
Caspase-6: The Bridging Activator
Historically classified as an executioner caspase in apoptosis, caspase-6 has been re-evaluated as a critical activator and bridging molecule within the PANoptotic cascade [121] [122].
- Activator of Caspase-8: Biochemical studies have established that caspase-6 is a direct activator of caspase-8 in the intrinsic apoptosis pathway initiated by cytochrome c release, forming a positive feedback loop that amplifies cell death signals [124].
- PANoptosome Assembly: During IAV infection, caspase-6 promotes the physical interaction between the sensor ZBP1 and the kinase RIPK3, thereby facilitating the assembly of the ZBP1-PANoptosome. Intriguingly, this function appears to be independent of caspase-6's proteolytic activity [120] [125].
- Neuronal Death and Regeneration: Beyond PANoptosis, a caspase-6/caspase-8 axis has been identified as a key mediator of neuronal apoptosis and axonal regeneration failure following central nervous system injury [126].
Table 2: Diverse Functions of Caspase-6 in Programmed Cell Death
| Function | Molecular Mechanism | Pathway | Significance |
|---|---|---|---|
| Caspase-8 Activation | Direct proteolytic cleavage of procaspase-8 | Intrinsic Apoptosis | Amplifies mitochondrial death signal [124] |
| PANoptosome Scaffold | Facilitating ZBP1-RIPK3 interaction (protease-independent) | PANoptosis | ZBP1-PANoptosome assembly during IAV infection [120] [125] |
| Neuronal Death Mediator | Activation of caspase-8 downstream of injury | Apoptosis | Contributes to neuronal loss and inhibits axon regeneration [126] |
| Gasdermin Processing | Cleavage of GSDMB at D91 | Pyroptosis/Apoptosis | Can inhibit pyroptotic function under specific conditions [20] |
Experimental Analysis of PANoptosis
Key Experimental Protocols
Objective: To investigate the role of caspase-6 and caspase-8 in neuronal survival and axonal regeneration in vitro and in vivo [126].
Methodology:
- Retinal Whole-Mount Cultures: Retinas from postnatal day 14 rats are flat-mounted and cultured. Treatment involves the application of specific caspase inhibitors:
Z-VEID-FMK: A selective caspase-6 inhibitor.Z-IETD-FMK: A selective caspase-8 inhibitor.Z-VAD-FMK: A pan-caspase inhibitor.DMSO: Vehicle control.- Cell survival is quantified over 4 days using propidium iodide staining and fluorescence intensity analysis [126].
- Retinal Explant Cultures: Explants are placed on inhibitory substrates (e.g., myelin or CSPG) and treated with caspase inhibitors or a ROCK inhibitor (
Y-27632). Neurite outgrowth is assessed after 18 hours by phalloidin staining and quantified for fiber number and length [126]. - In Vivo Optic Nerve Injury and Intraocular Injection: Adult rats undergo optic nerve transection (for survival studies) or crush (for regeneration studies). Caspase inhibitors or vehicle are administered via intraocular injection at post-injury time points preceding the onset of apoptosis. RGC survival is assessed by retrograde labeling with FluoroGold, and axonal regeneration is evaluated histologically [126].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Studying Caspase-6 and Caspase-8 in PANoptosis
| Reagent / Tool | Function / Specificity | Example Application |
|---|---|---|
| Z-VEID-FMK | Cell-permeable, irreversible caspase-6 inhibitor | Inhibiting caspase-6 activity in retinal cultures to probe its role in neuronal death [126]. |
| Z-IETD-FMK | Cell-permeable, irreversible caspase-8 inhibitor | Assessing the contribution of caspase-8 to cell death and regeneration in neuronal injury models [126]. |
| SIMA 13a | Sulfonamide isatin Michael acceptor; non-peptide caspase-6 inhibitor | In vivo inhibition of caspase-6 with potentially improved pharmacokinetics [126]. |
| Caspase-6 KO Mice | Genetic ablation of caspase-6 | Studying host response to IAV infection and ZBP1-PANoptosome assembly [120] [125]. |
| Caspase-8 KO Mice | Genetic ablation of caspase-8 | Investigating the switch from apoptosis to necroptosis and PANoptosis [119] [20]. |
| Phospho-MLKL Antibodies | Detect activated, phosphorylated MLKL | Confirming the occurrence of necroptosis in the PANoptotic cascade [119] [123]. |
| Anti-GSDMD Antibodies | Detect full-length and cleaved GSDMD | Validating the induction of pyroptosis in experimental models [119] [120]. |
Visualizing the PANoptotic Network
The following diagrams, generated using Graphviz DOT language, illustrate the core signaling pathways and molecular relationships in PANoptosis.
The discovery of PANoptosis represents a fundamental shift in our understanding of cellular self-destruction. The model of functionally isolated death pathways has been superseded by one of an integrated and highly redundant network, with caspase-6 and caspase-8 acting as pivotal molecular switches at its core. Their roles extend far beyond their historical classifications: caspase-8 serves as a master regulator balancing death modalities, while caspase-6 acts as a crucial bridge and activator, particularly within the PANoptosome.
Targeting these caspases and the PANoptotic pathway holds immense therapeutic potential. In cancer, inducing PANoptosis could overcome the resistance to single-pathway pro-apoptotic drugs [123] [125]. Conversely, in hyperinflammatory diseases, inhibiting specific nodes of the PANoptosome may prevent excessive tissue damage. Future research must focus on elucidating the precise structural interactions within different PANoptosomes, mapping the complete regulatory networks, and developing highly specific agonists and antagonists for translational application. The tools, data, and visualizations provided herein offer a foundation for these endeavors, empowering researchers to further decode the complexities of integrated cell death.
Utilizing Genetic Models (Knockout Mice) and Biochemical Assays for Pathway Validation
Caspases, cysteine-dependent aspartate-specific proteases, are the central executioners of programmed cell death, playing distinct yet interconnected roles in the intrinsic (mitochondrial) and extrinsic (death receptor) apoptotic pathways [5] [85]. The extrinsic pathway is initiated by the binding of death ligands (e.g., FasL, TNF-α) to cell surface receptors, leading to the formation of the Death-Inducing Signaling Complex (DISC) and activation of initiator caspases-8 and -10 [127] [5]. Conversely, the intrinsic pathway is triggered by internal cellular stress signals (e.g., DNA damage, oxidative stress), causing mitochondrial outer membrane permeabilization (MOMP) and release of cytochrome c, which forms the apoptosome with Apaf-1 to activate initiator caspase-9 [127] [85]. Both pathways converge on the activation of executioner caspases-3, -6, and -7, which dismantle the cell through cleavage of key structural and functional proteins [3] [128]. Validating the complex interplay within and between these pathways requires an integrated approach, combining the physiological context provided by genetic models like knockout mice with the mechanistic precision of biochemical assays.
Genetic Models: Deciphering Caspase Function In Vivo
Knockout mouse models have been indispensable for delineating the non-redundant, physiological functions of individual caspases within apoptotic pathways, revealing phenotypes that range from embryonic lethality to subtle cellular defects.
Key Knockout Mouse Models and Phenotypes
The table below summarizes the phenotypic consequences of caspase deletion in mice, highlighting their critical roles in development and tissue homeostasis.
Table 1: Phenotypes of Caspase Knockout Mouse Models
| Caspase | Primary Role | Knockout Phenotype | Implication for Apoptosis Pathways |
|---|---|---|---|
| Caspase-8 | Extrinsic Initiator [85] | Embryonic lethality due to impaired heart development and vascular defects [128]. | Absolute requirement for extrinsic apoptosis in development [128]. |
| Caspase-9 | Intrinsic Initiator [85] | Perinatal lethality; severe brain malformations (exencephaly) due to reduced neuronal apoptosis [127] [128]. | Central role in intrinsic, mitochondrial pathway [127]. |
| Caspase-3 | Executioner [85] | Perinatal lethality; brain overgrowth and disorganization [128]. | Key executioner downstream of both pathways [128]. |
| Caspase-7 | Executioner [85] | Viable; subtle phenotypes, but perinatal lethality when co-deleted with Caspase-3 [128]. | Functional overlap with Caspase-3, but not complete redundancy [128]. |
| Caspase-2 | Initiator/Orphan [128] | Viable; female mice exhibit excessive oocyte reserves; increased susceptibility to metabolic syndrome and steatohepatitis [127] [128]. | Context-dependent pro- and anti-apoptotic roles; links apoptosis to metabolism and genomic integrity [128]. |
Species-Specific Considerations: A critical factor in experimental design is the recognition that the caspase repertoire differs between humans and mice. Humans have caspase-10, which is involved in extrinsic apoptosis and can negatively regulate caspase-8, but this gene is absent in mice [128]. Conversely, mouse caspase-11 is considered an orthologue of human caspases-4 and -5, which are involved in non-canonical pyroptosis [85] [128]. These differences must be accounted for when translating findings from mouse models to human physiology and disease.
Biochemical Assays for Pathway Dissection
Biochemical assays provide a reductionist approach to define molecular mechanisms, measure catalytic activity, and validate substrate specificity, complementing the in vivo findings from genetic models.
Orthogonal Protease Activation Systems
A powerful method for probing the specific functions of individual caspases involves engineered activation systems. One such approach uses a small-molecule-activated Tobacco Etch Virus (TEV) protease, known as the SNIPer, in conjunction with caspase alleles where the native cleavage site has been replaced with a TEV recognition sequence [129] [63]. This system allows for the selective, orthogonal activation of a specific caspase (e.g., caspase-3, -6, or -7) in human cells without triggering the entire endogenous network. Studies using this method revealed that activation of caspase-3 or -7 is sufficient to induce apoptosis, whereas caspase-6 activation is not, demonstrating a key functional distinction between executioner caspases [129].
High-Throughput Screening (HTS) for Inhibitor Discovery
HTS assays are crucial for developing selective chemical tools. Recent work has focused on targeting the inactive zymogen (pro-form) of caspases to achieve greater selectivity, given the high structural homology of active caspases. For example, researchers have engineered a TEV-activatable caspase-10 construct (proCASP10TEV Linker) with low background activity for HTS. This assay successfully identified a class of thiadiazine-containing compounds that act as procaspase-10 inhibitors [63]. Such state-specific inhibitors are invaluable for dissecting the unique roles of closely related caspases like caspase-8 and -10 in extrinsic apoptosis.
Immunofluorescence for Caspase Activation
Immunofluorescence provides spatial resolution of caspase activation within fixed cells or tissues. The typical protocol involves:
- Permeabilization: Treating fixed samples with PBS/0.1% Triton X-100 to allow antibody access.
- Blocking: Incubating with a buffer containing serum (e.g., 5% goat serum) to reduce non-specific binding.
- Primary Antibody Incubation: Applying a caspase-specific primary antibody (e.g., anti-active caspase-3) overnight at 4°C.
- Secondary Antibody Incubation: Using a fluorophore-conjugated secondary antibody for detection.
- Imaging: Visualizing with a fluorescence microscope to identify cells with active caspases, often correlating with classic apoptotic morphology [47].
This method is ideal for validating apoptosis in specific cell types within a heterogeneous sample or for co-localization studies with other markers.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for Caspase Pathway Validation
| Research Reagent / Tool | Function and Application |
|---|---|
| Caspase Knockout Mice | In vivo validation of caspase-specific functions in development, tissue homeostasis, and disease models [127] [128]. |
| TEV-Activatable Caspase Alleles | Orthogonal, precise activation of specific caspases in cells to delineate non-redundant functions and sufficient triggers for apoptosis [129] [63]. |
| Fluorogenic Substrates (e.g., Ac-DEVD-AFC) | Biochemical measurement of caspase enzyme activity. Cleavage releases a fluorescent group (AFC), allowing kinetic quantification [63]. |
| Selective Caspase Inhibitors | Chemical tool compounds to probe the functional contribution of a specific caspase to a death pathway (e.g., procaspase-10 inhibitors) [63]. |
| Caspase-Specific Antibodies | Detection and localization of caspase expression and activation states via immunofluorescence, western blot, or flow cytometry [47]. |
Integrated Workflow for Pathway Validation
The following diagram illustrates a synergistic experimental strategy that combines genetic and biochemical tools to validate the role of a specific caspase in apoptosis.
A multi-faceted approach is paramount for validating the complex roles of caspases in intrinsic and extrinsic apoptosis. Knockout mouse models provide an irreplaceable physiological context, revealing the consequences of caspase loss at the organismal level. These in vivo findings must be mechanistically dissected using precise biochemical tools, including orthogonal activation systems, selective inhibitors, and activity-based assays. The integration of genetic and biochemical data, as outlined in this guide, provides the most robust framework for defining caspase functions, with critical implications for developing novel therapeutics targeting apoptosis in cancer, neurodegenerative disorders, and beyond.
Conclusion
The intrinsic and extrinsic apoptosis pathways, while initiated by distinct stimuli and molecular complexes, are intricately connected through critical crosstalk mechanisms and converge on the activation of effector caspases-3 and -7. A deep understanding of their unique caspase activation logic, regulatory networks, and the emerging complexities of non-apoptotic functions and PANoptosis is paramount. Future directions in biomedical research must focus on developing more specific caspase modulators, advancing our knowledge of cell death pathway integration in disease models, and translating these insights into effective therapeutic strategies that can selectively induce or inhibit apoptosis in cancer, neurodegenerative disorders, and beyond.
References
- 1. Biochemistry, Extrinsic Pathway of Apoptosis - StatPearls - NCBI [https://www.ncbi.nlm.nih.gov/sites/books/NBK560811/]
- 2. Apoptosis [https://en.wikipedia.org/wiki/Apoptosis]
- 3. Caspases as master regulators of programmed cell death [https://pmc.ncbi.nlm.nih.gov/articles/PMC12229594/]
- 4. Intrinsic and Extrinsic Pathways of Apoptosis [https://www.thermofisher.com/us/en/home/life-science/antibodies/antibodies-learning-center/antibodies-resource-library/cell-signaling-pathways/cellular-apoptosis-pathway.html]
- 5. Apoptosis: A Comprehensive Overview of Signaling Pathways ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11592877/]
- 6. Apoptosis - Intrisinic Pathway - External [https://teachmeanatomy.info/physiology/biochemistry/apoptosis/]
- 7. Insights on the crosstalk among different cell death ... [https://www.nature.com/articles/s41420-025-02328-9]
- 8. Your Complete Guide to Intrinsic & Extrinsic Pathways [https://www.assaygenie.com/blog/apoptosis-unveiled-your-complete-guide-to-intrinsic-extrinsic-pathways/?srsltid=AfmBOoqqGx-0uMcMcS5uqAcq3BXstyyJhCwGx8IKTZGmG_DC1lU0JjaA]
- 9. Caspase Activation Pathways: an Overview - NCBI - NIH [https://www.ncbi.nlm.nih.gov/books/NBK14011/]
- 10. Caspase activation pathways: some recent progress [https://www.nature.com/articles/cdd200959]
- 11. Cellular Mechanisms Controlling Caspase Activation and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3660825/]
- 12. Apoptosome: a platform for the activation of initiator caspases [https://www.nature.com/articles/4402028]
- 13. Caspase-9: structure, mechanisms and clinical application [https://pmc.ncbi.nlm.nih.gov/articles/PMC5410359/]
- 14. Targeting apoptotic pathways for cancer therapy - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11245162/]
- 15. Atomic structure of the apoptosome: mechanism of cytochrome c [https://pmc.ncbi.nlm.nih.gov/articles/PMC4691890/]
- 16. A near atomic structure of the active human apoptosome [https://elifesciences.org/articles/17755]
- 17. New insights into apoptosome structure and function - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6030056/]
- 18. Apoptosome structure, assembly and procaspase activation [https://pmc.ncbi.nlm.nih.gov/articles/PMC3644875/]
- 19. Intrinsic and Extrinsic Pathways of Apoptosis [https://www.thermofisher.com/sa/en/home/life-science/antibodies/antibodies-learning-center/antibodies-resource-library/cell-signaling-pathways/cellular-apoptosis-pathway.html]
- 20. Caspases as master regulators of programmed cell death [https://www.nature.com/articles/s12276-025-01470-9]
- 21. Caspase-10 Negatively Regulates Caspase-8-Mediated Cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5413585/]
- 22. Caspase-10 Negatively Regulates Caspase-8-Mediated ... [https://www.sciencedirect.com/science/article/pii/S2211124717304850]
- 23. A Death Effector Domain Chain DISC Model Reveals ... [https://www.sciencedirect.com/science/article/pii/S1097276512003875]
- 24. Cryo-EM structural analysis of FADD:Caspase-8 ... [https://www.nature.com/articles/s41467-020-20806-9]
- 25. Control of mitochondrial apoptosis by the Bcl-2 family - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2714431/]
- 26. c-FLIPL is a dual function regulator for caspase-8 activation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC125398/]
- 27. MOMP, cell suicide as a BCL-2 family business [https://www.nature.com/articles/cdd2017179]
- 28. The role of BCL-2 family proteins in regulating apoptosis ... [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2022.985363/full]
- 29. Pharmacological targeting of c-FLIP L and Bcl-2 family ... [https://www.nature.com/articles/s41598-020-76079-1]
- 30. The BCL2 family: from apoptosis mechanisms to new ... [https://www.nature.com/articles/s41392-025-02176-0]
- 31. BCL-2 family members and the mitochondria in apoptosis [https://genesdev.cshlp.org/content/13/15/1899]
- 32. Regulation of mitochondrial membrane permeabilization ... [https://www.sciencedirect.com/science/article/abs/pii/S0955067404001450]
- 33. Roles of c-FLIP in Apoptosis, Necroptosis, and Autophagy [https://pmc.ncbi.nlm.nih.gov/articles/PMC4219646/]
- 34. Requirement for Casper (c-FLIP) in Regulation of Death ... [https://www.sciencedirect.com/science/article/pii/S1074761300802149]
- 35. Apoptotic signaling cascades [https://www.sciencedirect.com/science/article/abs/pii/S0278584603000162]
- 36. Caspases, apoptosis and aging [https://www.sciencedirect.com/science/article/abs/pii/S1568163703000266]
- 37. Serial killers: ordering caspase activation events in apoptosis [https://www.nature.com/articles/4400601]
- 38. Executioner caspase-3 and caspase-7 are functionally distinct ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2529079/]
- 39. Executioner Caspase-3, -6, and -7 Perform Distinct, Non- ... [https://www.sciencedirect.com/science/article/pii/S0021925819671826]
- 40. The effect of caspase-3 in mitochondrial apoptosis ... [https://www.sciencedirect.com/science/article/abs/pii/S0308814621017738]
- 41. Caspases, the enemy within, and their role in oxidative ... [https://pubmed.ncbi.nlm.nih.gov/15241246/]
- 42. 7 promotes microglial pyroptosis in models of multiple sclerosis [https://jneuroinflammation.biomedcentral.com/articles/10.1186/s12974-020-01902-5]
- 43. Caspases: structural and molecular mechanisms and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12052993/]
- 44. Evaluation of Caspase Activation to Assess Innate Immune ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10202388/]
- 45. Detection of Active Caspases During Apoptosis Using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6091214/]
- 46. Biochemistry, Extrinsic Pathway of Apoptosis - StatPearls - NCBI [https://www.ncbi.nlm.nih.gov/books/NBK560811/]
- 47. Protocol for detection of caspases using immunofluorescence [https://www.abcam.com/en-us/technical-resources/protocols/immunofluorescence-protocol-to-detect-caspases]
- 48. Integrated real-time imaging of executioner caspase ... [https://www.nature.com/articles/s41420-025-02662-y]
- 49. A Caspase-3 Reporter for Fluorescence Lifetime Imaging ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6025355/]
- 50. Mitochondrial Membrane Potential Assay - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5375165/]
- 51. Mitochondrial Membrane Potential Assay [https://www.sartorius.com/en/applications/life-science-research/live-cell-assays/mitochondrial-membrane-potential]
- 52. Assays for Mitochondria Function [https://www.thermofisher.com/us/en/home/life-science/cell-analysis/cell-viability-and-regulation/apoptosis/mitochondria-function.html]
- 53. Immunodetection of cytochrome c by flow cytometry after ... [https://pubmed.ncbi.nlm.nih.gov/16680678/]
- 54. A new quantitative assay for cytochrome c release in ... [https://www.nature.com/articles/4401263]
- 55. North America Apoptosis Assay Market Trends 2025–2034 [https://www.gminsights.com/industry-analysis/north-america-apoptosis-assay-market]
- 56. Apoptosis Assays Market Size, Share, Trends and Industry ... [https://www.consegicbusinessintelligence.com/apoptosis-assays-market]
- 57. A long way to go: caspase inhibitors in clinical use [https://www.nature.com/articles/s41419-021-04240-3]
- 58. Apoptosis: A Comprehensive Overview of Signaling ... [https://www.mdpi.com/2073-4409/13/22/1838]
- 59. Extrinsic and Intrinsic Apoptosis Signal Pathway Review [https://www.intechopen.com/chapters/38236]
- 60. Reactivating apoptotic pathways in cancer: A review of ... [https://www.sciencedirect.com/science/article/abs/pii/S0014299925007198]
- 61. A Class of Allosteric, Caspase Inhibitors Identified by High ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3428514/]
- 62. Allosteric modulation of caspases [https://www.sciencedirect.com/science/article/abs/pii/S0163725811001604]
- 63. An activation-based high throughput screen identifies caspase ... [https://pubs.rsc.org/en/content/articlehtml/2025/cb/d5cb00017c]
- 64. Caspases as therapeutic targets - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3918066/]
- 65. Mechanisms of Cell Death: Apoptosis - CST Blog [https://blog.cellsignal.com/mechanisms-of-cell-death-apoptosis]
- 66. The concept of intrinsic versus extrinsic apoptosis [https://pubmed.ncbi.nlm.nih.gov/35147165/]
- 67. Caspase activation is associated with spontaneous ... [https://pubmed.ncbi.nlm.nih.gov/18393389/]
- 68. Advanced Liver Therapies Research Clinical Trials [https://www.bcm.edu/research/research-centers/advanced-liver-therapies-research/clinical-trials]
- 69. Pre-symptomatic Caspase-1 inhibitor delays cognitive ... [https://www.nature.com/articles/s41467-020-18405-9]
- 70. Detection of elevated caspase activation and early ... [https://www.sciencedirect.com/science/article/abs/pii/S0171933504701387]
- 71. The Bcl-2-regulated apoptosis switch: mechanism and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2754308/]
- 72. A new era in cancer therapy: targeting the Proteasome-Bcl-2 ... [https://jeccr.biomedcentral.com/articles/10.1186/s13046-025-03505-5]
- 73. Inhibition of Bcl-2 or IAP proteins does not provoke ... [https://www.sciencedirect.com/science/article/abs/pii/S0027510715000871]
- 74. Bcl-2 inhibition in the treatment of hematologic malignancies [https://www.frontiersin.org/journals/hematology/articles/10.3389/frhem.2023.1307661/full]
- 75. Harnessing PUMA's lethal potential: BCL-2 family ... [https://www.sciencedirect.com/science/article/pii/S246829422500111X]
- 76. Current Advances and Future Strategies for BCL-2 Inhibitors [https://pmc.ncbi.nlm.nih.gov/articles/PMC10605442/]
- 77. Apoptosis Dependent and Independent Functions of Caspases [https://www.ncbi.nlm.nih.gov/books/NBK6198/]
- 78. Cell Biology Identification of Functional Regions Defining ... [https://www.sciencedirect.com/science/article/pii/S0021925820599262]
- 79. The proteolytic landscape of cells exposed to non-lethal ... [https://www.nature.com/articles/s41420-021-00539-4]
- 80. Caspase-9, caspase-3 and caspase-7 have distinct roles ... [https://bmcmolcellbiol.biomedcentral.com/articles/10.1186/1471-2121-14-32]
- 81. Comprehensive landscape of cell death mechanisms [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2025.1611055/full]
- 82. Necroptosis, pyroptosis and apoptosis: an intricate game of ... [https://www.nature.com/articles/s41423-020-00630-3]
- 83. Apoptosis, Pyroptosis, and Necroptosis—Oh My! The Many ... [https://www.sciencedirect.com/science/article/abs/pii/S002228362100615X]
- 84. A comparative study of apoptosis, pyroptosis, necroptosis, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10898734/]
- 85. structural and molecular mechanisms and functions in cell ... [https://www.nature.com/articles/s41421-025-00791-3]
- 86. Targeting Caspase-1 in osteoarthritis: multi-omics insights ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1677801/full]
- 87. Caspase Inhibitor - an overview [https://www.sciencedirect.com/topics/pharmacology-toxicology-and-pharmaceutical-science/caspase-inhibitor]
- 88. Q&A: Cellular near death experiences—what is anastasis ? [https://bmcbiol.biomedcentral.com/articles/10.1186/s12915-017-0441-z]
- 89. In vivo CaspaseTracker biosensor system for detecting anastasis and... [https://www.nature.com/articles/srep09015?error=cookies_not_supported&code=b01a081e-c53c-4f4d-9a5b-1cbd065cd456]
- 90. In vivo CaspaseTracker biosensor system for detecting anastasis and... [https://pubmed.ncbi.nlm.nih.gov/25757939/]
- 91. A Comprehensive Exploration of Caspase Detection Methods [https://pmc.ncbi.nlm.nih.gov/articles/PMC11121653/]
- 92. Live Cell Imaging of Caspase Activation for High Content ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3613133/]
- 93. Quantitative analysis of caspase-3 activation by fitting ... [https://www.sciencedirect.com/science/article/abs/pii/S0968432809001103]
- 94. Inducing Targeted, Caspase-Independent Apoptosis with ... [https://www.mdpi.com/2072-6694/17/7/1179]
- 95. Impact of Complex Apoptotic Signaling Pathways on ... [https://www.mdpi.com/2072-6694/16/5/984]
- 96. PANoptosis: Cross-Talk Among Apoptosis, Necroptosis, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12184785/]
- 97. PANoptosis in neurological disorders: mechanisms ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1579360/full]
- 98. Targeting panoptosis: a narrative review of its therapeutic ... [https://bmcnephrol.biomedcentral.com/articles/10.1186/s12882-025-04339-1]
- 99. The molecular mechanisms and therapeutic implications of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12474704/]
- 100. Betulinic Acid and Apoptosis-Involved Pathways [https://www.sciencedirect.com/science/article/pii/S001429992501115X]
- 101. Caspase [https://en.wikipedia.org/wiki/Caspase]
- 102. Intrinsic and Extrinsic Pathways of Apoptosis [https://www.thermofisher.com/mx/en/home/life-science/antibodies/antibodies-learning-center/antibodies-resource-library/cell-signaling-pathways/cellular-apoptosis-pathway.html]
- 103. Apoptosis Signaling Pathway: Stages, Types and Key ... [https://geneglobe.qiagen.com/us/knowledge/pathways/apoptosis-signaling]
- 104. Caspase structure, proteolytic substrates, and function ... [https://www.nature.com/articles/4400598.pdf]
- 105. Bid Protein: A Participant in the Apoptotic Network with Roles ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11942203/]
- 106. Cleavage by Caspase 8 and Mitochondrial Membrane ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4882451/]
- 107. Caspase-8; regulating life and death - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5417704/]
- 108. BID is cleaved by caspase-8 within a native complex on the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3132005/]
- 109. Functions of Caspase 8: the Identified and the Mysterious [https://pmc.ncbi.nlm.nih.gov/articles/PMC4099255/]
- 110. Optogenetic induction of caspase-8 mediated apoptosis by ... [https://www.nature.com/articles/s41598-023-50561-y]
- 111. Solution Structure of BID, an Intracellular Amplifier ... [https://www.sciencedirect.com/science/article/pii/S0092867400805723]
- 112. Pro-apoptotic cascade activates BID, which oligomerizes ... [https://www.nature.com/articles/4400783]
- 113. Caspase-8 goes cardiolipin: a new platform to provide ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2582901/]
- 114. Caspases in cell death, inflammation and disease - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6611727/]
- 115. Physiological functions of non-apoptotic caspase activity in the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5988915/]
- 116. Non-apoptotic functions of caspases in myeloid cell ... [https://www.nature.com/articles/cdd201719]
- 117. Visualization of caspase-3-like activity in cells using a ... [https://www.nature.com/articles/ncomms3157]
- 118. Fluorescence Lifetime Imaging of a Caspase-3 Apoptosis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5729923/]
- 119. PANoptosis: a unique inflammatory cell death modality - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9586465/]
- 120. Mechanisms of PANoptosis and relevant small-molecule ... [https://www.nature.com/articles/s41419-023-06370-2]
- 121. Caspases in PANoptosis [https://pubmed.ncbi.nlm.nih.gov/39985853/]
- 122. Caspases in PANoptosis [https://www.sciencedirect.com/science/article/abs/pii/S245231862500011X]
- 123. PANoptosis: bridging apoptosis, pyroptosis, and ... [https://www.nature.com/articles/s41417-024-00765-9]
- 124. absolute requirement for removal of caspase-6 prodomain [https://www.nature.com/articles/4401065]
- 125. The molecular mechanisms, roles, and potential applications ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12069359/]
- 126. Involvement of Caspase-6 and Caspase-8 in Neuronal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6622648/]
- 127. Regulation of Oocyte Apoptosis: A View from Gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9861590/]
- 128. Exploring caspase functions in mouse models | Apoptosis [https://link.springer.com/article/10.1007/s10495-024-01976-z]
- 129. Activation of Specific Apoptotic Caspases with an ... [https://www.sciencedirect.com/science/article/pii/S009286741000783X]