Intrinsic vs. Extrinsic Pathway Biomarkers: From Mechanisms to Clinical Applications
This article provides a comprehensive comparison of biomarkers for intrinsic and extrinsic pathway activation, crucial for researchers and drug development professionals.
Intrinsic vs. Extrinsic Pathway Biomarkers: From Mechanisms to Clinical Applications
Abstract
This article provides a comprehensive comparison of biomarkers for intrinsic and extrinsic pathway activation, crucial for researchers and drug development professionals. It covers the fundamental definitions and biological mechanisms, explores advanced methodological approaches for their assessment, addresses common challenges in interpretation and optimization, and delivers a critical validation of their predictive power across therapeutic areas. By integrating insights from oncology, autoimmune diseases, and toxicology, this review serves as a definitive guide for the precise application of these biomarkers in personalized medicine and drug development.
Decoding the Blueprint: Core Concepts and Biological Mechanisms of Intrinsic and Extrinsic Pathways
In biological systems, the concepts of intrinsic and extrinsic pathways represent fundamental paradigms for understanding how cells process information and execute programmed responses. These pathways form the cornerstone of cellular communication, governing processes as diverse as programmed cell death (apoptosis), neuronal regeneration, and immune system regulation. The intrinsic pathway refers to signaling cascades that originate from within the cell, typically in response to internal stressors or damage signals. In contrast, the extrinsic pathway describes signaling initiated by external stimuli or ligands originating from other cells, representing intercellular communication mechanisms [1] [2].
This conceptual framework transcends specific biological contexts, providing researchers with a unified model for understanding cellular decision-making. From a therapeutic perspective, distinguishing between these pathways is crucial for drug development, as it enables targeted interventions that either modulate a cell's internal environment or manipulate extracellular signaling networks. This comparative guide examines the defining characteristics, molecular mechanisms, and experimental approaches for studying these pathways across biological systems, with particular emphasis on their implications for biomedical research and therapeutic development.
Defining the Pathways: Core Principles and Molecular Mechanisms
The Extrinsic Pathway: Externally Triggered Signaling
The extrinsic pathway functions as a communication system between cells, where signaling is initiated by extracellular ligands binding to specific cell surface receptors. This pathway is particularly crucial in immune regulation and tissue homeostasis, allowing one cell to directly influence the fate of another [2].
The canonical extrinsic apoptosis pathway begins when death receptors of the tumor necrosis factor receptor superfamily (TNFRSF) bind to their cognate trimeric ligands. These receptors contain a specialized protein interaction module known as a "death domain" that is approximately 80 amino acids long. Upon ligand binding, receptor oligomerization occurs, leading to the formation of a membrane-bound death-inducing signaling complex (DISC). This complex serves as a platform for recruiting and activating initiator caspases, particularly caspase-8, which then proteolytically activates downstream effector caspases such as caspase-3, committing the cell to apoptosis [1] [2].
The Intrinsic Pathway: Internally Generated Signals
The intrinsic pathway operates as a cell's internal monitoring system, activating in response to intracellular stress signals including DNA damage, oxidative stress, hypoxia, and survival factor deprivation. This pathway integrates various stress signals through sensors like the p53 protein, which acts as a critical activator of the intrinsic pathway by transcriptionally regulating pro-apoptotic and anti-apoptotic Bcl-2 family members [1].
A pivotal event in the intrinsic apoptosis pathway is mitochondrial outer membrane permeabilization (MOMP), which is regulated by the balance between pro-apoptotic and anti-apoptotic Bcl-2 family proteins. Pro-apoptotic proteins such as BAX and BAK form pores in the mitochondrial membrane, leading to the release of cytochrome c and other pro-apoptotic factors into the cytosol. Cytochrome c then binds to Apaf-1, forming a complex called the apoptosome that activates caspase-9, which in turn activates downstream effector caspases [1] [3].
Table 1: Fundamental Characteristics of Intrinsic and Extrinsic Pathways
| Characteristic | Extrinsic Pathway | Intrinsic Pathway |
|---|---|---|
| Origin of Signal | Extracellular; from other cells | Intracellular; from within the cell |
| Key Initiators | Death ligands (FasL, TNF-α, TRAIL) | Cellular stress (DNA damage, oxidative stress) |
| Molecular Hubs | Death receptors (Fas, TNFR1), DISC | Mitochondria, p53, Bcl-2 family proteins |
| Key Adaptors | FADD, TRADD | Apaf-1, cytochrome c |
| Initiator Caspases | Caspase-8, Caspase-10 | Caspase-9 |
| Regulatory Proteins | FLIP, cIAPs | Bcl-2 family, IAPs, SMAC/Diablo |
| Primary Functions | Immune regulation, tissue homeostasis | Response to cellular damage, development |
Comparative Analysis: Key Biological Contexts
Apoptosis: The Paradigm System
The distinction between intrinsic and extrinsic pathways is most clearly defined in apoptosis, where both pathways converge on caspase activation but differ fundamentally in their initiation mechanisms. The extrinsic apoptotic pathway begins when death ligands such as Fas ligand (FasL) or TNF-related apoptosis-inducing ligand (TRAIL) bind to their cognate receptors, rapidly triggering caspase activation within seconds of ligand binding [1].
In contrast, the intrinsic apoptotic pathway (also called the mitochondrial pathway) responds to internal damage cues through mitochondrial outer membrane permeabilization, leading to cytochrome c release and apoptosome formation. This pathway involves a more delayed response as it requires transcriptional and post-translational regulation of Bcl-2 family proteins [1] [3].
Notably, cross-talk exists between these pathways. For example, activated caspase-8 in the extrinsic pathway can cleave the Bcl-2 family protein BID, generating truncated BID (tBID) that translocates to mitochondria and amplifies the apoptotic signal through the intrinsic pathway [1] [4].
Neuronal Development and Injury Responses
In neuronal systems, intrinsic and extrinsic pathways exhibit distinct roles in development, plasticity, and response to injury. Research using mutant mice has revealed that in sympathetic neurons, apoptosis induced by trophic factor deprivation primarily follows the intrinsic pathway, depending on BAX translocation and cytochrome c release despite the expression of both intrinsic and extrinsic pathway components [4].
Recent studies on spinal cord injury recovery demonstrate how modulating both pathways simultaneously can enhance therapeutic outcomes. Deletion of RhoA (an extrinsic pathway modulator responsive to extracellular inhibitory signals) combined with deletion of Pten (an intrinsic pathway inhibitor of neuronal growth) in corticospinal neurons, coupled with neuronal activation, promoted significantly greater axonal growth and functional recovery than either intervention alone [5]. This highlights the therapeutic potential of coordinated targeting of both pathways.
Immune Regulation and Disease Pathogenesis
In immunology, the extrinsic pathway plays a crucial role in immune surveillance, with natural killer (NK) cells and cytotoxic T lymphocytes (CTLs) using death receptor ligands to eliminate virally infected or transformed cells [2]. Meanwhile, the intrinsic pathway regulates immune cell homeostasis, controlling the contraction phase of immune responses after pathogen clearance.
In autoimmune disorders like systemic sclerosis, recent single-cell RNA-sequencing analyses have revealed that disease severity involves complex rewiring of both cell-intrinsic and cell-extrinsic signaling networks across multiple cell types, including fibroblasts, myeloid cells, and keratinocytes. This suggests that both autonomous cellular signaling and intercellular communication contribute to disease progression [6].
Table 2: Pathway Involvement Across Biological Processes
| Biological Process | Dominant Pathway | Key Molecular Players | Cellular Outcomes |
|---|---|---|---|
| Developmental Apoptosis | Primarily Intrinsic | Bcl-2 family, Caspase-9 | Sculpting tissues, eliminating excess cells |
| Immune-Mediated Cell Killing | Primarily Extrinsic | Death receptors, Caspase-8 | Elimination of infected or transformed cells |
| Neuronal Plasticity/Regeneration | Both | RhoA (extrinsic), Pten (intrinsic) | Axonal growth, synaptic rewiring |
| Stress-Induced Apoptosis | Primarily Intrinsic | p53, BAX, Cytochrome c | Elimination of damaged cells |
| Autoimmune Pathogenesis | Both | Cell-type specific biomarkers | Inflammation, fibrosis, tissue damage |
Experimental Approaches: Methodologies and Reagents
Pathway Analysis Techniques
Investigating intrinsic and extrinsic pathways requires specialized methodologies that can distinguish between these overlapping but distinct signaling cascades. Key experimental approaches include:
Genetic Manipulation: Studies using knockout mice, such as Bax⁻/⁻, Bak⁻/⁻, Bim⁻/⁻, Bid⁻/⁻, and Bad⁻/⁻ neurons, have been instrumental in defining functional redundancies and specific roles of Bcl-2 family members in intrinsic pathway regulation [4]. Similarly, analysis of lpr and gld mice, which have mutations in Fas and FasL respectively, has helped elucidate extrinsic pathway components.
Single-Cell RNA Sequencing: Advanced computational approaches like LASSO-based predictive machine learning applied to single-cell RNA-sequencing data can identify predictive biomarkers of disease severity across cell types, revealing both cell-intrinsic and cell-extrinsic signaling networks [6].
Neuronal Stimulation and Circuit Mapping: Combining genetic modifications with chemogenetic neuronal stimulation (e.g., DREADD technology) allows researchers to test the functional outcomes of pathway manipulations. This approach demonstrated that pairing RhoA/Pten deletion with chemogenetic stimulation enhanced presynaptic bouton formation and motor recovery after spinal cord injury [5].
Research Reagent Solutions
Table 3: Essential Research Reagents for Pathway Analysis
| Reagent Category | Specific Examples | Research Applications |
|---|---|---|
| Genetic Modulators | AAV8-fDIO-Cre, AAVretro-DIO-hM3Dq-mCherry | Cell-type specific gene deletion, chemogenetic neuronal stimulation |
| Pathway Activators/Inhibitors | Death ligands (FasL, TRAIL), Bcl-2 inhibitors, Caspase inhibitors | Selective pathway activation or inhibition |
| Detection Antibodies | Phosphospecific antibodies, Bcl-2 family antibodies, Death receptor antibodies | Protein localization, post-translational modification analysis |
| Animal Models | Bax⁻/⁻, Bak⁻/⁻, Bim⁻/⁻, Bid⁻/⁻, lpr (Fas mutant), gld (FasL mutant) mice | Defining pathway components in physiological contexts |
| Computational Tools | LASSO-based predictive modeling, correlation network analyses | Identification of predictive biomarkers and signaling networks |
Signaling Pathway Visualizations
Diagram 1: Intrinsic and Extrinsic Apoptosis Signaling Pathways. The extrinsic pathway (top) initiates from extracellular death ligands binding to cell surface receptors, while the intrinsic pathway (bottom) begins with intracellular stress signals. Both pathways converge on caspase-3/7 activation, with cross-talk occurring through BID cleavage.
Diagram 2: Experimental Workflow for Pathway Analysis. This workflow illustrates the integrated approach combining genetic manipulation, stimulation paradigms, single-cell sequencing, computational analysis, and functional validation to delineate intrinsic and extrinsic pathway contributions.
The conceptual framework distinguishing intrinsic and extrinsic pathways provides a powerful paradigm for understanding cellular communication and fate decisions across biological systems. Rather than existing as isolated entities, these pathways exhibit complex interactions and cross-talk that integrate internal cellular states with external environmental cues. For researchers and drug development professionals, this distinction offers strategic opportunities for therapeutic intervention.
Future research directions will likely focus on developing more sophisticated tools for selectively modulating these pathways, particularly in a cell-type-specific manner. The integration of single-cell technologies with computational approaches will continue to reveal novel biomarkers and signaling networks that operate across intrinsic and extrinsic dimensions. Furthermore, combinatorial approaches that simultaneously target both pathways—as demonstrated in spinal cord injury models—hold significant promise for enhancing therapeutic efficacy in complex diseases.
As our understanding of these fundamental biological pathways deepens, so too does our ability to precisely manipulate cellular behavior for therapeutic benefit, ushering in a new era of targeted interventions for cancer, neurological disorders, autoimmune conditions, and regenerative medicine applications.
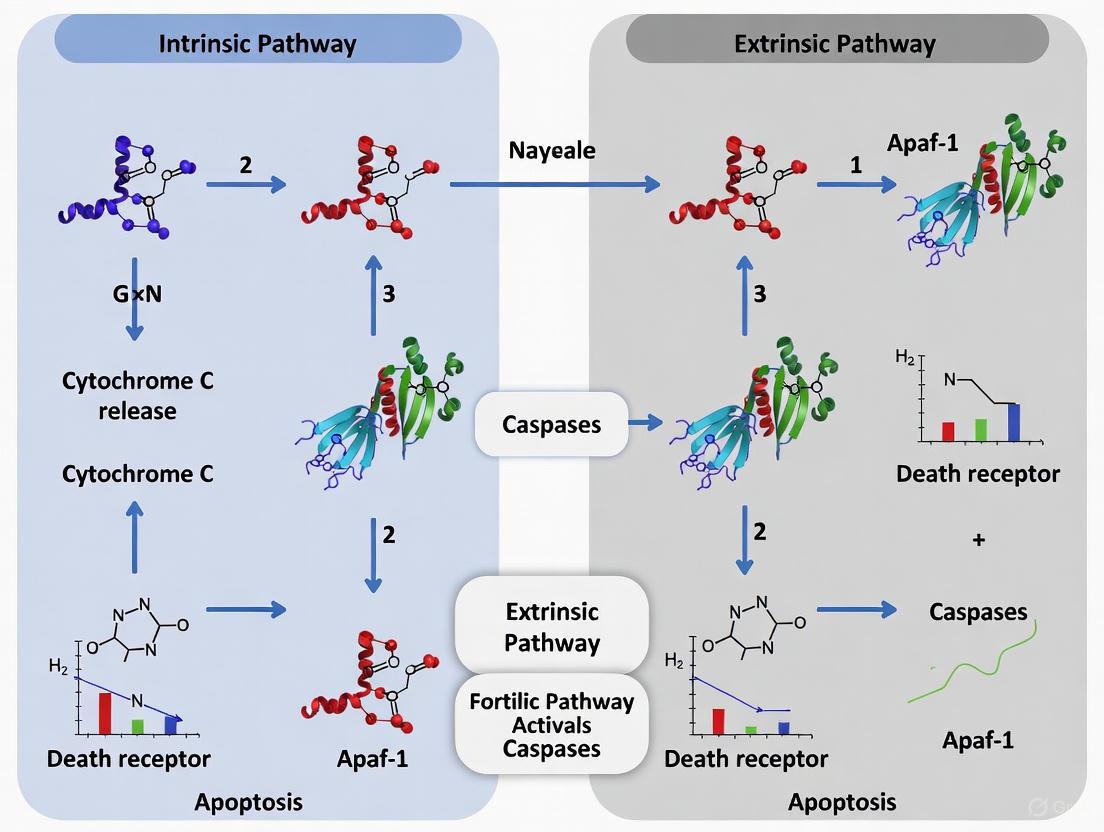
Programmed cell death, or apoptosis, is a fundamental process critical for tissue homeostasis, embryonic development, and the removal of damaged or unwanted cells. In mammalian systems, apoptosis proceeds primarily through two evolutionarily conserved signaling pathways: the mitochondrial (intrinsic) pathway and the death receptor (extrinsic) pathway [7] [8]. While these pathways are initiated by distinct stimuli and involve different upstream components, they ultimately converge to activate a cascade of proteases that systematically dismantle the cell. Disruption of either pathway can lead to pathological conditions; insufficient apoptosis contributes to cancer progression and autoimmune diseases, while excessive apoptosis is implicated in neurodegenerative disorders and stroke [7] [9]. This guide provides a detailed comparative analysis of the intrinsic and extrinsic apoptotic pathways, focusing on key biomarkers, experimental methodologies, and the crucial cross-talk that integrates these systems. Aimed at researchers and drug development professionals, this resource synthesizes current understanding to support targeted investigations into apoptotic signaling cascades.
Pathway Comparison: Core Components and Mechanisms
The intrinsic and extrinsic apoptosis pathways, while functionally convergent, are characterized by unique initiators, regulators, and execution mechanisms. The table below provides a systematic comparison of their core components.
Table 1: Comparative Analysis of the Intrinsic and Extrinsic Apoptosis Pathways
| Feature | Mitochondrial (Intrinsic) Pathway | Death Receptor (Extrinsic) Pathway |
|---|---|---|
| Initiating Stimuli | Internal stress: DNA damage, hypoxia, oxidative stress, cytokine deprivation, cytotoxic agents [10] [1] | External ligands: FasL, TNF-α, TRAIL binding to cell surface death receptors [11] [1] |
| Key Initiators | p53, Bcl-2 family proteins (BH3-only sensors, Bax/Bak) [9] [1] | Death Receptors (Fas, TNFR1, DR4/5), FADD, caspase-8 [11] [1] |
| Activation Complex | Apoptosome (Cytochrome c + Apaf-1 + caspase-9) [7] [1] | DISC (Death-Inducing Signaling Complex) [11] [1] |
| Key Initiator Caspase | Caspase-9 [7] [9] | Caspase-8 [11] [9] |
| Regulatory Proteins | Bcl-2, Bcl-xL (anti-apoptotic); Bax, Bak, Bid, Bad, Bim, Puma (pro-apoptotic) [10] [9] [1] | FLIP (inhibits caspase-8), cIAPs (Inhibitor of Apoptosis Proteins) [11] [1] |
| Mitochondrial Involvement | Central event: MOMP, release of cytochrome c, Smac/Diablo [7] [1] | Optional amplification: via caspase-8-mediated cleavage of Bid to tBid [11] [8] |
| Primary Function | Elimination of damaged or stressed cells [1] | Immune regulation, deletion of infected or abnormal cells [7] [1] |
The intrinsic pathway functions as a sensor of internal cellular well-being. Stress signals such as DNA damage or growth factor withdrawal are monitored by sensors like p53, which transcriptionally activates pro-apoptotic Bcl-2 family members like Bax, Puma, and Noxa [1]. The critical execution point is Mitochondrial Outer Membrane Permeabilization (MOMP), which is controlled by the balanced interactions of the Bcl-2 protein family. Anti-apoptotic members (e.g., Bcl-2, Bcl-xL) preserve membrane integrity, while activated pro-apoptotic effectors Bax and Bak form pores, leading to the release of cytochrome c and other apoptogenic factors [7] [9]. Cytochrome c in the cytosol binds to Apaf-1, triggering the formation of the apoptosome and the activation of caspase-9, which then initiates the downstream caspase cascade [7] [1].
In contrast, the extrinsic pathway is activated from outside the cell by ligand-mediated engagement of death receptors. Upon binding their cognate ligands (e.g., FasL to Fas), the receptors oligomerize and recruit the adaptor protein FADD and the initiator procaspase-8 to form the Death-Inducing Signaling Complex (DISC) [11] [1]. Within the DISC, caspase-8 is activated through proximity-induced autocatalysis. In some cell types (classified as Type I), active caspase-8 is sufficient to directly cleave and activate effector caspases like caspase-3. In others (Type II cells), the signal is amplified through the intrinsic pathway via caspase-8-mediated cleavage of the BH3-only protein Bid. Truncated Bid (tBid) translocates to mitochondria, promoting MOMP and engaging the intrinsic amplification loop [7] [11] [8].
Experimental Data and Biomarker Analysis
Empirical data from various models highlight the distinct yet interconnected nature of these pathways. The following table summarizes key biomarkers and representative experimental findings.
Table 2: Key Apoptotic Biomarkers and Experimental Evidence
| Biomarker / Assay | Role/Function | Experimental Context & Findings |
|---|---|---|
| BID | BH3-only protein; critical linker (crosstalk) cleaved by caspase-8 to form tBid, which activates mitochondrial pathway [11] [8] | ICH (Intracerebral Hemorrhage) Model: Identified as a key biomarker in apoptosis post-ICH. Validation in a rat model showed significant involvement, regulated by miR-1225-3p [12]. |
| Caspase-8 | Initiator caspase in extrinsic pathway; activated at the DISC [11] [1] | Daunorubicin Treatment (Leukemia): Activation confirmed in CCRF-CEM and MOLT-4 T-lymphoblastic leukemia cells, indicating engagement of the extrinsic pathway [10]. |
| Caspase-9 | Initiator caspase in intrinsic pathway; activated by the apoptosome [7] [1] | Daunorubicin Treatment (Leukemia): Activation observed in CCRF-CEM and MOLT-4 cells, indicating concurrent intrinsic pathway activation [10]. |
| Cytochrome c Release | Hallmark of MOMP; triggers apoptosome formation [7] [1] | Direct Irradiation: Upstream of caspase-9 activation, a definitive marker of intrinsic pathway engagement following direct cellular damage [13]. |
| Bax/Bak | Pro-apoptotic executioner proteins; mediate MOMP [9] [1] | Ionizing Radiation: Pro-apoptotic Bax was up-regulated in response to direct irradiation, promoting mitochondrial dysfunction [13]. |
| Phosphatidylserine Externalization | "Eat-me" signal on the outer leaflet of the plasma membrane [9] | Annexin V Assay: Used to detect early apoptosis in daunorubicin-treated leukemia cells and camptothecin-treated Jurkat cells [10] [9]. |
| DNA Fragmentation | Late-stage apoptotic event; result of CAD nuclease activation [1] | TUNEL Assay: Effectively detected apoptosis in a developing mouse embryo (E14.5) and in a rat ICH model [12] [9]. |
Case Study: Daunorubicin-Induced Apoptosis in Leukemia Cells
Research on daunorubicin-treated leukemia cell lines provides a clear example of how both pathways can be engaged with cell-type-specific variations. In T-lymphoblastic leukemia cells (CCRF-CEM and MOLT-4), treatment with 10 μM daunorubicin for 4 hours followed by a recovery period induced apoptosis through both intrinsic and extrinsic pathways. Evidence included the activation of both caspase-8 and caspase-9, along with changes in mitochondrial membrane potential (Δψm) [10]. In contrast, under the same conditions, SUP-B15 (B-lymphoblastic leukaemia) cells exhibited signs of apoptosis but without a loss of Δψm, suggesting cell death was primarily driven by the extrinsic pathway, independent of mitochondrial amplification [10]. This underscores that the requirement for mitochondrial involvement in death receptor signaling is cell-type-dependent.
Methodologies for Apoptosis Detection
Accurate assessment of apoptosis requires a multi-parametric approach to distinguish it from other forms of cell death and to identify the initiating pathway.
- Annexin V/Propidium Iodide (PI) Staining: This assay detects the externalization of phosphatidylserine, an early event in apoptosis. Annexin V binds to phosphatidylserine, while PI is a vital dye that stains cells with compromised membranes (necrotic cells or late-stage apoptotic cells). Live cells are Annexin V-/PI-; early apoptotic cells are Annexin V+/PI-; and late apoptotic/necrotic cells are Annexin V+/PI+ [10] [9].
- Caspase Activity Measurement: Activation of specific initiator caspases can help delineate the pathway involved. This can be done using fluorogenic substrate assays or flow cytometry with pancaspase inhibitors like Apostat [10]. Selective caspase-8 or caspase-9 inhibitors can further define the contribution of each pathway.
- Mitochondrial Membrane Potential (Δψm) Assay: The loss of Δψm is an early event in the intrinsic pathway. Fluorescent dyes like DiOC₆ or TMRE accumulate in active mitochondria based on membrane potential. A decrease in fluorescence signal indicates mitochondrial depolarization, a key step in MOMP [10] [9].
- TUNEL Assay: The TUNEL (Terminal deoxynucleotidyl transferase dUTP Nick End Labeling) assay detects DNA fragmentation, a hallmark of late-stage apoptosis. It labels the 3'-OH ends of fragmented DNA, which can be visualized by fluorescence microscopy or flow cytometry [12] [9].
- Western Blot Analysis: This technique is crucial for detecting the cleavage and activation of key apoptotic proteins, such as caspases (e.g., cleavage of procaspase-3 to active caspase-3), and Bid (cleavage to tBid), as well as changes in the expression levels of Bcl-2 family proteins [12] [10].
- Immunofluorescence (IF): IF allows for the subcellular localization of proteins. It can be used to visualize cytochrome c release from mitochondria, the translocation of Bax to mitochondria, or the cleavage and activation of caspases, providing spatial information about pathway activation [12] [9].
Pathway Crosstalk and Integration
The intrinsic and extrinsic pathways are not isolated; significant cross-talk exists, primarily mediated by the BH3-only protein Bid [7] [8]. As detailed in Figure 1, in Type II cells, activation of the death receptor pathway leads to caspase-8 activation. Caspase-8 then cleaves Bid into its active truncated form, tBid. tBid translocates to the mitochondria, where it promotes MOMP by activating Bax and Bak or antagonizing anti-apoptotic Bcl-2 proteins, thereby engaging the intrinsic pathway to amplify the apoptotic signal [11] [8]. This cross-talk ensures a robust and irreversible commitment to cell death when signals from either pathway are insufficient on their own.
The following DOT code defines a diagram illustrating the crosstalk between the extrinsic and intrinsic apoptotic pathways.
Figure 1: Crosstalk between Apoptotic Pathways. The extrinsic pathway can amplify the death signal via caspase-8-mediated cleavage of Bid to tBid, which engages the intrinsic mitochondrial pathway. MOMP: Mitochondrial Outer Membrane Permeabilization.
The Scientist's Toolkit: Key Research Reagents
The following table lists essential reagents and kits for studying apoptosis, drawing from methodologies cited in the research.
Table 3: Essential Research Reagents for Apoptosis Investigation
| Reagent / Kit | Primary Function / Target | Application & Notes |
|---|---|---|
| Annexin V-Based Kits | Binds to externalized Phosphatidylserine | Flow cytometry or IF detection of early apoptosis. Must be used with a viability dye (e.g., PI) to distinguish from late apoptosis/necrosis [10] [9]. |
| TUNEL Assay Kit | Labels 3'-OH ends of fragmented DNA | Detection of late-stage apoptosis via IF, IHC, or flow cytometry. Not specific to apoptosis; requires morphological confirmation [12] [9]. |
| Caspase Activity Assays | Measure activation of specific caspases (e.g., 3, 8, 9) | Use fluorogenic substrates or inhibitors (e.g., Apostat). Differentiates pathway involvement (caspase-8 for extrinsic, caspase-9 for intrinsic) [10]. |
| Mitochondrial Membrane Potential Dyes (e.g., DiOC₆, TMRE, JC-1) | Accumulate in polarized mitochondria | Flow cytometry or microscopy to detect loss of Δψm, an early event in the intrinsic pathway [10] [9]. |
| Antibodies for Western Blot/IF | Target cleaved/activated proteins (e.g., Cleaved Caspase-3, Cleaved PARP, tBid) | Confirms protein cleavage as a marker of activation. IF allows subcellular localization (e.g., cytochrome c release) [12] [9]. |
| Bcl-2 Family Antibody Sampler Kits | Detect pro- and anti-apoptotic Bcl-2 family members | Monitor expression changes and post-translational modifications of key regulators of the intrinsic pathway [9]. |
| Chemical Inducers/Inhibitors | Activate or inhibit specific pathway components | e.g., Caspase-8 inhibitor Z-IETD-FMK; Bax/Bak activator ABT-737. Useful for dissecting pathway contributions. |
The following DOT code defines a diagram outlining a typical experimental workflow for differentiating apoptotic pathways.
Figure 2: Experimental Workflow for Pathway Differentiation. A decision-tree approach to delineate the primary apoptotic pathway engaged, utilizing sequential assays from early to late markers.
Cell-intrinsic and cell-extrinsic factors represent two fundamental categories of biological signals that coordinate gene regulation in development and disease. Cell-intrinsic factors originate from within the cell, including genetic programs, transcription factors, and metabolic states, while cell-extrinsic factors comprise external signals such as circulating hormones, cytokines, and cell-cell contact mediators. Understanding the interplay between these factors provides critical insights into disease mechanisms and therapeutic development. This guide compares experimental approaches for studying these regulatory systems, with particular focus on apoptosis and blood coagulation as paradigmatic examples of intrinsic and extrinsic pathway activation.
Defining Intrinsic and Extrinsic Pathways: Core Concepts and Biological Systems
Apoptosis: A Paradigm of Programmed Cell Death
The apoptotic pathway demonstrates the fundamental distinction between intrinsic and extrinsic activation mechanisms. Both pathways culminate in caspase activation and controlled cellular dismantling but initiate through distinct mechanisms and biomarkers [9].
Intrinsic Pathway (Mitochondrial Pathway): Triggered by internal cellular damage or stress, this pathway involves mitochondrial outer membrane permeabilization (MOMP), cytochrome c release, and caspase-9 activation. Key regulators include BCL-2 family proteins, which contain conserved BCL-2 homology (BH) domains that determine pro- or anti-apoptotic function [14].
Extrinsic Pathway (Death Receptor Pathway): Initiated by extracellular death ligands (TNF-α, FasL) binding to cell surface receptors, leading to formation of the death-inducing signaling complex (DISC) and caspase-8 activation [9].
Coagulation: Proteolytic Cascade in Blood Clotting
The coagulation system similarly operates through distinct intrinsic and extrinsic pathways that converge on common effectors [15].
Intrinsic Coagulation Pathway: Activated by exposed endothelial collagen and involving factors XII, XI, IX, and VIII. Assessed clinically via partial thromboplastin time (PTT) [15] [16].
Extrinsic Coagulation Pathway: Triggered by external trauma and tissue factor release, involving factor VII. Measured clinically via prothrombin time (PT) [15] [17].
Comparative Analysis of Pathway Biomarkers
Table 1: Key Biomarkers in Intrinsic and Extrinsic Apoptotic Pathways
| Parameter | Intrinsic Apoptosis | Extrinsic Apoptosis |
|---|---|---|
| Initiating Stimuli | Cellular stress, DNA damage, oxidative stress | Death ligands (FasL, TNF-α, TRAIL) |
| Membrane Receptors | Not receptor-mediated | Death receptors (Fas, TNFR, TRAIL-R) |
| Adaptor Molecules | Apaf-1 | FADD, TRADD |
| Key Initiator Caspases | Caspase-9 | Caspase-8, Caspase-10 |
| Regulatory Proteins | BCL-2 family (Bax, Bak, Bcl-2, Bcl-xL) | FADD, c-FLIP |
| Mitochondrial Involvement | Central (MOMP, cytochrome c release) | Secondary (via Bid cleavage) |
| Biomarker Detection Methods | Cytochrome c release, Bax/Bak activation, caspase-9 cleavage | DISC formation, caspase-8 activation, death ligand levels |
Table 2: Key Biomarkers in Intrinsic and Extrinsic Coagulation Pathways
| Parameter | Intrinsic Coagulation | Extrinsic Coagulation |
|---|---|---|
| Initiating Stimuli | Exposed endothelial collagen, negatively charged surfaces | Tissue factor release from external injury |
| Key Factors | XII, XI, IX, VIII | VII, III (tissue factor) |
| Activation Time | Longer cascade | Shorter, more rapid activation |
| Clinical Tests | Partial Thromboplastin Time (PTT) | Prothrombin Time (PT) |
| Diagnostic Applications | Hemophilia A/B diagnosis, heparin monitoring | Liver disease, warfarin monitoring, vitamin K deficiency |
| Convergence Point | Factor X activation | Factor X activation |
Experimental Protocols for Pathway Analysis
Protocol 1: Thrombin Generation Measurement in Whole Blood
This protocol assesses coagulation capacity in mouse whole blood, adaptable for human samples [18].
Sample Collection:
- Collect blood into 3.2% trisodium citrate (9:1 blood:citrate ratio)
- Discard first drop to avoid contamination
- Process samples within 4 hours of collection
Reaction Setup:
- Use 96-well round-bottom plates
- Final reaction mixture: 65μL per well
- Add 10μL citrated whole blood to wells
- Mix with 23μL stimulator (kaolin, ellagic acid, or polyphosphate for intrinsic pathway activation)
- Add 20μL fluorogenic substrate mixture (Z-Gly-Gly-Arg-AMC)
- Include 12μL calcium chloride to reverse citrate anticoagulation
Measurement Parameters:
- Lag time: Duration until thrombin generation initiates
- Time to peak: Time until maximum thrombin generation
- Peak thrombin: Maximum thrombin concentration achieved
- Endogenous thrombin potential (ETP): Total thrombin generated over time
Validation Controls:
- Include FXII-deficient blood (F12-/-) for specificity confirmation
- Use direct oral anticoagulants (DOACs) as inhibition controls
- Employ specific inhibitors (rHA-Infestin-4 for FXIIa) to verify mechanism
Protocol 2: Apoptosis Pathway Analysis via Caspase Activation
This methodology distinguishes intrinsic versus extrinsic apoptosis activation [9].
Cell Stimulation:
- For extrinsic pathway: Treat cells with death ligands (FasL, TNF-α, TRAIL) at appropriate concentrations (typically 10-100 ng/mL) for 2-24 hours
- For intrinsic pathway: Indce with cellular stressors (staurosporine 0.1-1 μM, UV irradiation, or chemotherapeutic agents)
Membrane Changes Detection (Early Apoptosis):
- Use Annexin V-FITC staining (1:100 dilution in binding buffer)
- Counterstain with propidium iodide (1 μg/mL) to distinguish early vs. late apoptosis
- Analyze by flow cytometry within 1 hour of staining
- Critical Note: Annexin V alone cannot distinguish apoptosis from other cell death forms; always include viability markers
Mitochondrial Membrane Potential Assessment:
- Stain cells with TMRE (tetramethylrhodamine ethyl ester; 100-500 nM)
- Incubate for 15-30 minutes at 37°C
- Analyze fluorescence decrease indicating membrane potential loss
- Include CCCP (10-50 μM) as positive control for depolarization
Caspase Activity Measurement:
- Use fluorogenic caspase substrates (DEVD-AFC for caspase-3, IETD-AFC for caspase-8, LEHD-AFC for caspase-9)
- Measure cleavage product fluorescence hourly for 4-8 hours
- Include caspase inhibitors (Z-VAD-FMK, 20 μM) as specificity controls
Western Blot Analysis:
- Probe for cleaved caspases (anti-cleaved caspase-3, -8, -9 antibodies)
- Detect BCL-2 family members (Bax, Bak, Bid cleavage)
- Assess cytochrome c release from mitochondrial fractions
Pathway Signaling Diagrams
Figure 1: Intrinsic and Extrinsic Apoptosis Pathway Signaling. The intrinsic pathway (red) initiates from internal cellular stress, while the extrinsic pathway (blue) begins with extracellular death ligands. Cross-talk occurs via Bid cleavage (yellow).
Figure 2: Intrinsic and Extrinsic Coagulation Pathway Signaling. Both pathways converge at Factor X activation in the common pathway (green), culminating in fibrin clot formation.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Intrinsic and Extrinsic Pathway Studies
| Reagent Category | Specific Examples | Research Applications | Function |
|---|---|---|---|
| Pathway Activators | Kaolin, ellagic acid, polyphosphate [18] | Intrinsic coagulation studies | Contact activation of Factor XII |
| Tissue factor, recombinant Factor VIIa [15] | Extrinsic coagulation studies | Initiate tissue factor pathway | |
| Staurosporine, UV irradiation, chemotherapeutics [9] | Intrinsic apoptosis induction | Cellular stress induction | |
| Recombinant death ligands (FasL, TNF-α, TRAIL) [9] | Extrinsic apoptosis studies | Death receptor activation | |
| Inhibitors & Modulators | rHA-Infestin-4 [18] | FXIIa-specific inhibition | Contact pathway blockade |
| Direct oral anticoagulants (DOACs) [18] | Coagulation pathway inhibition | Thrombin/FXa inhibition | |
| Z-VAD-FMK (pan-caspase inhibitor) [9] | Apoptosis inhibition | Caspase activity blockade | |
| Venetoclax (BCL-2 inhibitor) [14] | Intrinsic apoptosis promotion | BH3 mimetic, disrupts BCL-2 interactions | |
| Detection Reagents | Fluorogenic substrates (Z-G-G-R-AMC) [18] | Thrombin generation assays | Thrombin activity measurement |
| Annexin V conjugates [9] | Early apoptosis detection | Phosphatidylserine exposure | |
| TMRE, JC-1 dyes [9] | Mitochondrial membrane potential | MOMP detection in intrinsic apoptosis | |
| Anti-cleaved caspase antibodies [9] | Apoptosis pathway mapping | Specific caspase activation | |
| Cell Models | FXII-deficient (F12-/-) blood [18] | Coagulation pathway studies | Intrinsic pathway specificity |
| Primary cells vs. cell lines | Pathway mechanism validation | Physiological relevance assessment |
Discussion: Research Applications and Therapeutic Implications
Disease-Specific Pathway Alterations
The differential activation of intrinsic and extrinsic pathways has significant disease implications. In depression research, the extrinsic coagulation pathway demonstrates altered activity, with studies identifying significantly upregulated fibrinogen chains (FGA, FGB, FGG) and downregulated Factor VII in patient plasma [19]. These findings suggest cross-talk between coagulation and inflammation in neuropsychiatric disorders.
Suicidal behavior in major depressive disorder shows distinct extrinsic coagulation pathway activation, with altered inflammatory and coagulatory protein profiles in attempters versus non-attempters [20]. This pattern reveals a proinflammatory and prothrombotic phenotype associated with specific behavioral manifestations.
Network Biomarkers in Pathway Analysis
Emerging approaches integrate multiple biomarkers into network-based analyses. Network biomarkers consider protein-protein or gene-gene interactions rather than individual molecules, while dynamic network biomarkers (DNBs) monitor these interactions across disease progression stages [21]. This approach is particularly valuable for:
- Identifying pre-disease states through correlation and fluctuation patterns
- Understanding therapeutic mechanisms via network perturbation analysis
- Developing pathway-specific diagnostics with improved sensitivity/specificity
Therapeutic Targeting Strategies
Pathway-specific targeting requires distinct approaches:
Intrinsic Pathway Targeting: BCL-2 inhibition with venetoclax in hematologic malignancies demonstrates selective killing through intrinsic apoptosis activation [14]. Similarly, FXII-targeting agents show antithrombotic potential without bleeding risk in coagulation [18].
Extrinsic Pathway Targeting: Death receptor agonists (e.g., TRAIL receptor agonists) and extrinsic coagulation inhibitors (e.g., anti-tissue factor antibodies) provide extrinsic pathway modulation.
The integration of intrinsic and extrinsic pathway biomarkers enables comprehensive disease mapping and therapeutic development across multiple pathological conditions.
Apoptosis, or programmed cell death, is a fundamental process essential for maintaining tissue homeostasis and eliminating damaged or unnecessary cells in multicellular organisms [14]. The intrinsic apoptotic pathway, also known as the mitochondrial pathway, represents one of the two main routes of apoptosis initiation and is centrally controlled by the B-cell lymphoma-2 (BCL-2) protein family [22] [23]. This pathway is characterized by mitochondrial outer membrane permeabilization (MOMP), which serves as a critical commitment point to cellular destruction [23]. Upon MOMP, several pro-apoptotic proteins are released from the mitochondrial intermembrane space into the cytosol, including cytochrome c, second mitochondria-derived activator of caspases (SMAC, also known as DIABLO), apoptosis-inducing factor (AIF), and endonuclease G [24] [25]. These proteins orchestrate the systematic dismantling of the cell through caspase-dependent and independent mechanisms.
The intrinsic pathway is primarily activated in response to intracellular stressors, including DNA damage, growth factor deprivation, oxidative stress, and oncogene activation [23] [26]. Given its crucial role in maintaining cellular integrity and eliminating compromised cells, dysregulation of this pathway is a hallmark of cancer and other diseases [22] [14]. Cancer cells often develop mechanisms to evade apoptosis, frequently through overexpression of anti-apoptotic BCL-2 family members or mutations in key components of the pathway [22]. Consequently, biomarkers of the intrinsic apoptotic pathway have emerged as critical tools for cancer diagnosis, prognosis, and therapeutic targeting. This guide provides a comprehensive comparison of three central biomarker categories: cytochrome c, SMAC/Diablo, and BCL-2 family proteins, focusing on their molecular functions, experimental detection, and clinical relevance.
Molecular Functions and Mechanisms
BCL-2 Family Proteins: The Regulators of MOMP
The BCL-2 protein family serves as the central regulatory unit of the intrinsic apoptotic pathway, governing the critical decision point of MOMP [22] [23]. This family consists of three structurally and functionally distinct subgroups classified based on their BCL-2 homology (BH) domains and their effects on apoptosis:
- Anti-apoptotic proteins (BCL-2, BCL-XL, BCL-W, MCL-1, A1/BFL-1) contain four BH domains (BH1-BH4) and promote cell survival by inhibiting the activation of pro-apoptotic effectors [22] [14]. These proteins function by sequestering pro-apoptotic family members, thereby maintaining mitochondrial membrane integrity.
- Pro-apoptotic pore-formers (BAX, BAK, BOK) multi-domain proteins that, when activated, oligomerize to form pores in the mitochondrial outer membrane, leading to MOMP [22] [23].
- Pro-apoptotic BH3-only proteins (BIM, BID, PUMA, NOXA, BAD, BMF, BIK, HRK) function as sensitizers or activators that initiate the apoptotic cascade by neutralizing anti-apoptotic proteins or directly activating BAX and BAK [22] [14].
The balance between these competing factions determines cellular fate. In healthy cells, anti-apoptotic members maintain mitochondrial integrity by binding and neutralizing activated pro-apoptotic proteins. During apoptosis induction, BH3-only proteins are activated transcriptionally and/or post-translationally, tipping the balance toward MOMP [22] [23]. The "primed for death" state describes cancer cells that overexpress anti-apoptotic proteins but remain highly susceptible to BH3 mimetics because they are poised to undergo apoptosis once these anti-apoptotic proteins are inhibited [22].
Cytochrome c: The Caspase Activator
Cytochrome c is a well-studied mitochondrial protein with a dual role in cellular metabolism and apoptosis [24] [25]. In healthy cells, it functions as an essential component of the electron transport chain, localized between the inner and outer mitochondrial membranes where it transfers electrons from complex III to complex IV [24]. Following MOMP, cytochrome c is released into the cytosol, where it undergoes a profound functional switch from metabolic regulator to apoptotic activator [24] [26].
In the cytosol, cytochrome c binds to apoptotic protease-activating factor-1 (APAF-1), triggering a conformational change that enables APAF-1 to oligomerize into a wheel-like signaling complex known as the apoptosome [24]. The apoptosome then recruits and activates procaspase-9 through caspase recruitment domains (CARD), initiating the caspase cascade that leads to cellular dismantling [25]. Activated caspase-9 cleaves and activates downstream effector caspases (caspase-3 and -7), which execute apoptosis by proteolytically degrading hundreds of cellular substrates [24] [25].
SMAC/Diablo: The IAP Antagonist
SMAC (second mitochondria-derived activator of caspases), also known as DIABLO (direct IAP binding protein with low pI), is a crucial mitochondrial protein that promotes apoptosis by counteracting inhibitor of apoptosis proteins (IAPs) [27] [25]. This 239-amino acid protein is synthesized as a precursor with an N-terminal mitochondrial targeting sequence that directs it to the mitochondrial intermembrane space [27]. Upon mitochondrial import, the N-terminal 55 residues are cleaved, generating the mature, functional protein [27] [25].
The mature form of SMAC/Diablo exists as an arch-shaped homodimer that exposes an N-terminal tetrapeptide motif (Ala-Val-Pro-Ile, or AVPI) essential for its function [27]. Following MOMP, SMAC/Diablo is released into the cytosol where its AVPI motif binds to baculoviral IAP repeat (BIR) domains in IAP family proteins, particularly XIAP, cIAP1, and cIAP2 [27] [25]. This interaction neutralizes IAP-mediated caspase inhibition, thereby permitting apoptosis progression. Specifically, SMAC/Diablo binding displaces caspases from XIAP's BIR2 and BIR3 domains, relieving the suppression of caspase-3, -7, and -9 activity [27] [25]. Beyond this primary function, SMAC/Diablo can also promote the ubiquitin-mediated degradation of IAPs through its RING domain and may have IAP-independent roles in apoptosis [27].
Table 1: Comparative Functions of Core Intrinsic Pathway Biomarkers
| Biomarker | Subcellular Location (Inactive) | Activation Mechanism | Primary Molecular Function | Key Interacting Partners |
|---|---|---|---|---|
| BCL-2 Family | Mitochondrial membrane, cytosol | BH3-only protein activation | Regulation of MOMP | Other BCL-2 members, mitochondrial membranes |
| Cytochrome c | Mitochondrial intermembrane space | Release after MOMP | Caspase activation via apoptosome formation | APAF-1, caspase-9, cardiolipin |
| SMAC/Diablo | Mitochondrial intermembrane space | Release after MOMP, N-terminal processing | IAP neutralization | XIAP, cIAP1, cIAP2 |
Experimental Detection and Methodologies
Immunohistochemistry and Tissue Microarray Analysis
Immunohistochemistry (IHC) represents a widely employed technique for detecting intrinsic pathway biomarkers in tissue specimens, allowing for simultaneous protein localization and morphological assessment. A comprehensive study analyzing gastric adenocarcinoma tissues demonstrated the utility of IHC combined with tissue microarray (TMA) technology for evaluating DIABLO, AIF, cytochrome c, and cleaved caspase-3 expression [28]. The experimental protocol involved:
TMA Construction and Staining Protocol:
- Tissue sections (4 μm) were cut from paraffin-embedded gastric adenocarcinoma and normal adjacent mucosa blocks from 87 patients [28].
- Cylindrical cores (1 mm diameter) were extracted from donor blocks and transferred to a recipient TMA block using specialized equipment [28].
- Sections (3 μm) were mounted on coated slides, deparaffinized in xylene, and rehydrated through graded ethanol [28].
- Antigen retrieval was performed by heating slides in citrate buffer (pH 6.0) for 30 minutes [28].
- Endogenous peroxidase activity was blocked with 3% hydrogen peroxide for 20 minutes [28].
- Primary antibodies were applied and incubated overnight at 4°C: DIABLO (1:200), AIF (1:400), cytochrome c (1:500), cleaved caspase-3 (1:100) [28].
- Detection was performed using biotinylated link universal and streptavidin-HRP with DAB as the chromogen, followed by counterstaining with Harris hematoxylin [28].
Scoring and Interpretation: The researchers employed a semi-quantitative scoring system evaluating both the percentage of immunoreactive cells (0: <10%; 1: 10-<25%; 2: 25-<50%; 3: >50%) and staining intensity (0: negative; 1: weak; 2: moderate; 3: strong) [28]. The final immunoreactive score (0-9) was calculated by multiplying these two values, with scores of 0-3 considered negative and 4-9 positive [28]. This approach revealed distinct expression patterns, with cytochrome c detected in 68.9% of tumors compared to 54.4% of normal tissues, while cleaved caspase-3 was present in 24.1% of tumors versus only 3.4% of normal tissues [28].
Biomarker Quantification and Pathway Activation Scoring
Advanced computational approaches have been developed to quantify pathway activation levels based on biomarker expression patterns. The Pathway Activation Level (PAL) algorithm represents a sophisticated method that translates expression data into quantitative measures of pathway deregulation while considering pathway architecture [29]. This approach offers significant advantages over single-molecule biomarker analysis:
PAL Calculation Methodology:
- PAL is calculated as a weighted sum of logarithms of case-to-normal ratios for expression levels of all pathway genes [29].
- Weight coefficients range from -1 to 1 based on the activator or repressor role of each gene product within the pathway [29].
- An algorithmic approach assigns activator-repressor role coefficients to each pathway component based on pathway molecular architecture and interaction types [29].
- Gene-centric pathways can be automatically constructed as interacting networks around central genes of interest using whole-interactome models [29].
This methodology has demonstrated superior performance in cancer classification and prognostic assessment compared to individual gene expression biomarkers, with one study identifying 7,441 potential RNA biomarker associations for gene-centric pathways versus 24,349 for individual genes across 21 cancer types [29]. PAL values also exhibit better stability against experimental noise and reduced batch effects in both transcriptomic and proteomic data [29].
Comparative Biomarker Analysis in Cancer
Expression Patterns Across Cancer Types
The expression and clinical significance of intrinsic pathway biomarkers vary substantially across different cancer types, reflecting tissue-specific mechanisms of apoptosis regulation. BCL-2 family proteins demonstrate particularly diverse expression patterns that correlate with disease progression and treatment response:
Hematological Malignancies:
- BCL-2 is overexpressed in the majority of chronic lymphocytic leukemia (CLL) cases due to BCL-2 gene hypomethylation and loss of miR-15/16 at 13q14 [22].
- Approximately 80-90% of follicular lymphomas and one-third of diffuse large B-cell lymphomas (DLBCL) exhibit constitutive BCL-2 expression caused by t(14;18) chromosomal translocation [22].
- Double-hit lymphomas, characterized by MYC and BCL-2 rearrangements, represent an aggressive DLBCL subtype with particularly poor prognosis [22].
Solid Tumors:
- SMAC/Diablo overexpression has been associated with increased sensitivity to apoptosis in various solid tumors, including head and neck squamous cell carcinoma, hepatocellular carcinoma, breast cancer, and glioblastoma [27].
- In gastric adenocarcinoma, cytochrome c expression was detected in 68.9% of tumor specimens compared to 54.4% of normal tissues, while DIABLO positivity was observed in 45.6% of tumors and 46.8% of normal tissues [28].
- Reduced SMAC/Diablo expression in cancer cells may inhibit apoptosis, thereby promoting survival and treatment resistance [28].
Table 2: Biomarker Alterations in Selected Cancer Types
| Cancer Type | BCL-2 Family Alterations | Cytochrome c Status | SMAC/Diablo Status | Clinical Implications |
|---|---|---|---|---|
| CLL | BCL-2 overexpression (hypomethylation, miR-15/16 loss) | Not well characterized | Controversial, mimetics show cytotoxic effects | Venetoclax sensitivity, resistance development |
| Follicular Lymphoma | BCL-2 overexpression (t(14;18) translocation) | Not well characterized | Not well characterized | Venetoclax monotherapy poor response |
| Gastric Adenocarcinoma | Not well characterized | 68.9% tumor positivity | 45.6% tumor positivity | Potential diagnostic utility |
| Solid Tumors (Various) | MCL-1 upregulation in resistance | Not well characterized | Overexpression in multiple types | Sensitivity to apoptosis |
Predictive and Prognostic Value
Biomarkers of the intrinsic apoptotic pathway provide valuable information for predicting treatment response and patient outcomes across diverse malignancies:
BCL-2 Family Biomarkers:
- BCL-2 expression levels significantly influence tumor susceptibility to BCL-2 inhibitors like venetoclax, with highest efficacy observed in CLL and AML [22].
- Overexpression of alternative anti-apoptotic proteins (MCL-1, BCL-XL) represents a common resistance mechanism to venetoclax, sequestering BIM displaced from BCL-2 [22].
- NOTCH1 mutations in CLL with trisomy 12 lead to decreased IRF4 and increased MCL-1 expression, contributing to venetoclax resistance [22].
- BH3 profiling serves as a functional biomarker for predicting chemotherapy sensitivity by measuring mitochondrial priming [23].
SMAC/Diablo Biomarkers:
- SMAC/Diablo expression levels correlate with increased sensitivity to apoptosis in multiple tumor types [27].
- Small-molecule SMAC mimetics demonstrate enhanced cytotoxic effects when combined with conventional chemotherapy, death receptor agonists, and targeted therapies [27].
- SMAC mimetics can increase tumor cell sensitivity to treatment, potentially allowing for dose reduction and minimized side effects [27].
Cytochrome c and Caspase Activation:
- Cleaved caspase-3 expression, a downstream consequence of cytochrome c release and apoptosome formation, was significantly elevated in gastric tumors (24.1%) compared to normal tissue (3.4%), indicating attempted apoptosis execution [28].
- Ki-67 proliferation index correlated with caspase-3 activation in gastric cancer, suggesting coordinated regulation of proliferation and cell death pathways [28].
Therapeutic Applications and Clinical Translation
Targeted Therapies and Resistance Mechanisms
Components of the intrinsic apoptotic pathway represent promising therapeutic targets, particularly in hematological malignancies:
BCL-2 Inhibition:
- Venetoclax (ABT-199), a highly selective oral BCL-2 antagonist, is FDA-approved for relapsed/refractory CLL with 17p deletion and for older adults with newly diagnosed AML who are unsuitable for intensive chemotherapy [22].
- Venetoclax functions as a BH3 mimetic that displaces pro-apoptotic proteins like BIM from BCL-2, activating BAX/BAK and triggering cytochrome c release [22].
- Resistance to venetoclax develops through multiple mechanisms, including upregulation of alternative anti-apoptotic proteins (MCL-1, BCL-XL), mutations in BCL-2 that reduce drug binding, and failure to execute apoptosis despite successful BCL-2 inhibition [22].
SMAC Mimetics:
- Small-molecule SMAC mimetics designed to mimic the AVPI N-terminal motif of SMAC/Diablo have been developed to promote apoptosis in cancer cells [27].
- These compounds bind to BIR domains of IAPs, particularly XIAP, cIAP1, and cIAP2, preventing caspase inhibition and promoting cell death [27].
- Several SMAC mimetics (including SM-406 and Genentech compounds) are undergoing clinical trials, with preclinical studies demonstrating enhanced efficacy when combined with conventional therapies [27].
- Potential side effects include elevated cytokine and chemokine levels in normal tissues, reflecting the role of IAPs in inflammatory signaling [27].
Biomarker-Driven Treatment Strategies
The development of robust biomarkers has enabled more personalized therapeutic approaches targeting the intrinsic apoptotic pathway:
BH3 Profiling:
- This functional assay measures mitochondrial priming by exposing isolated mitochondria to BH3 peptides and quantifying membrane depolarization or cytochrome c release [23].
- BH3 profiling can identify tumors dependent on specific anti-apoptotic proteins, guiding selection of appropriate targeted therapies [23].
- The technique has been proposed as a biomarker strategy for predicting chemotherapy sensitivity in diverse cancer types [23].
Pathway Activation Scoring:
- Algorithmic pathway analysis using PAL provides a comprehensive assessment of intrinsic pathway status beyond individual biomarker expression [29].
- Gene-centric pathways have demonstrated superior biomarker performance compared to individual genes for tumor classification and outcome prediction [29].
- Computer-built pathways represent a credible alternative to manually curated pathways, reducing investigator bias and enabling rapid biomarker development [29].
Research Reagent Solutions
Table 3: Essential Research Reagents for Intrinsic Pathway Biomarker Analysis
| Reagent Category | Specific Examples | Research Applications | Key Features |
|---|---|---|---|
| Recombinant Proteins | Recombinant Human BCL2 (His-tagged) [24] | Protein-protein interaction studies, antibody production | Various protein lengths (1-239 aa, 101-239 aa, 50-150 aa) |
| Antibodies for IHC | DIABLO mouse monoclonal (Cell Signaling) [28] | Tissue localization and expression analysis | 1:200 dilution for gastric cancer TMA |
| AIF rabbit polyclonal (H-300) [28] | Mitochondrial and cytoplasmic staining | 1:400 dilution, cytoplasmic expression pattern | |
| Cytochrome c goat polyclonal (C-20) [28] | Detection of mitochondrial release | 1:500 dilution, cytoplasmic expression | |
| Cleaved caspase-3 rabbit polyclonal [28] | Apoptosis execution marker | 1:100 dilution, cytoplasmic pattern | |
| Cell Lysates | Human BCL2 Knockdown Cell Lysate [24] | Western blot controls, functional studies | HeLa cell source |
| Human CASP9 Knockdown Cell Lysate [24] | Apoptosome formation studies | HeLa cell source | |
| Research Kits | LSAB+ System-HRP [28] | IHC detection | Biotin-streptavidin amplification |
| Liquid DAB+ Substrate Chromogen [28] | IHC visualization | Permanent brown staining |
Visualizing the Intrinsic Apoptotic Pathway
The following diagram illustrates the key molecular events and biomarkers of the intrinsic apoptotic pathway:
Diagram 1: Intrinsic Apoptotic Pathway Signaling. This diagram illustrates the sequence of events from initial cellular stress to apoptosis execution, highlighting the central regulatory role of BCL-2 family proteins, mitochondrial outer membrane permeabilization (MOMP), and the coordinated actions of cytochrome c and SMAC/Diablo.
The intrinsic apoptotic pathway biomarkers—BCL-2 family proteins, cytochrome c, and SMAC/Diablo—provide critical insights into cellular life-and-death decisions with profound implications for cancer biology and therapy. These biomarkers function as an integrated system rather than isolated entities, with BCL-2 proteins regulating the initial commitment step of MOMP, cytochrome c activating the caspase cascade through apoptosome formation, and SMAC/Diablo removing inhibitory blocks on apoptosis execution. Their coordinated analysis offers superior diagnostic and prognostic information compared to individual biomarker assessment, as demonstrated by advanced computational approaches like pathway activation level scoring.
The clinical translation of these biomarkers is particularly evident in hematological malignancies, where BCL-2 inhibition with venetoclax has revolutionized treatment for specific patient subsets. However, resistance mechanisms highlight the complexity of apoptotic regulation and the need for comprehensive biomarker assessment that includes alternative anti-apoptotic family members and functional mitochondrial priming. Ongoing development of SMAC mimetics and combination strategies offers promising avenues for enhancing therapeutic efficacy across diverse cancer types. As biomarker technologies continue to evolve, particularly with algorithmic pathway analysis and functional assays, personalized targeting of the intrinsic apoptotic pathway will likely expand, providing new opportunities for cancer therapy and overcoming treatment resistance.
The extrinsic apoptosis pathway, a fundamental form of programmed cell death, is initiated at the cell surface through specific death receptors and propagates intracellular signals via defined molecular interactions [30]. This pathway is critical for maintaining cellular homeostasis, eliminating damaged or dangerous cells, and proper immune system function [31]. The core molecular machinery of this pathway consists of death receptors, adaptor proteins, and initiator caspases that together determine cellular fate [32]. Research into these components provides crucial biomarkers for understanding disease mechanisms, particularly in cancer, neurodegenerative disorders, and inflammatory conditions [31] [32]. This guide objectively compares the key biomarkers—death receptors, caspase-8, and FADD—that researchers utilize to monitor and quantify extrinsic pathway activation, providing experimental data and methodologies relevant for drug development professionals.
Core Biomarkers of the Extrinsic Pathway
Death Receptors: The Initiation Complex
Death receptors are transmembrane proteins belonging to the tumor necrosis factor (TNF) receptor superfamily that contain a conserved cytoplasmic death domain (DD) essential for apoptosis signaling [30]. These receptors function as the entry point for extrinsic death signals by binding specific extracellular ligands such as FasL (for CD95/Fas), TRAIL (for TRAIL-R1/DR4 and TRAIL-R2/DR5), and TNF (for TNFR1) [30] [33].
Key Comparative Features:
- Structural Organization: All death receptors share characteristic cysteine-rich extracellular domains and a conserved intracellular death domain (∼80 amino acids) that mediates homotypic protein interactions [30].
- Signaling Mechanism: Upon ligand-induced trimerization, death receptors undergo conformational changes that facilitate the recruitment of intracellular adaptor proteins [30] [33].
- Research Significance: Different death receptors activate overlapping but distinct downstream pathways. For example, TNFR1 recruitment of FADD requires the intermediate adapter TRADD, while CD95 and TRAIL receptors recruit FADD directly [30] [33].
Table 1: Comparative Characteristics of Major Death Receptors
| Death Receptor | Alternative Names | Ligand | Adapter Recruitment | Primary Research Applications |
|---|---|---|---|---|
| CD95/Fas | TNFRSF6, Apo-1 | FasL | Direct FADD binding | Autoimmunity, liver disease, cancer immunotherapy |
| TRAIL-R1 | DR4 | TRAIL | Direct FADD binding | Cancer research (selective tumor cell apoptosis) |
| TRAIL-R2 | DR5 | TRAIL | Direct FADD binding [33] | Cancer therapeutics, metastasis inhibition |
| TNFR1 | TNFRSF1a | TNF | FADD via TRADD intermediate [30] | Inflammation, sepsis, neurodegenerative diseases |
FADD: The Critical Adaptor Protein
Fas-associated death domain (FADD) serves as the essential adaptor protein that bridges activated death receptors to downstream effector molecules [34]. FADD contains two primary domains: a C-terminal death domain (DD) that interacts with death receptors and an N-terminal death effector domain (DED) that engages initiator caspases [34] [32].
Research Applications:
- Molecular Recruitment: FADD's DD binds to the trimerized death receptors through homotypic interactions with a 3:1 (receptor:FADD) stoichiometry [30].
- Complex Formation: The DED of FADD undergoes a conformational change upon receptor binding, exposing its binding surface to recruit procaspase-8 through DED-DED interactions [34].
- Experimental Evidence: Structural studies using mutagenesis (e.g., F122A/I128A mutations in caspase-8 DED) demonstrate that specific residues are critical for stable FADD-caspase-8 complex formation [34].
Caspase-8: The Initiator Protease
Caspase-8 is the primary initiator caspase in the extrinsic pathway, functioning as a cysteine-aspartic protease that initiates the apoptotic cascade [30] [32]. It exists as an inactive zymogen (procaspase-8) in living cells, becoming activated through proximity-induced dimerization and autocleavage upon recruitment to the death-inducing signaling complex (DISC) [30].
Key Research Characteristics:
- Structural Domains: Caspase-8 contains two N-terminal death effector domains (DEDs), a large protease subunit (p18/p20), and a small protease subunit (p10) [32].
- Activation Mechanism: Within the DED filaments of the DISC, procaspase-8 molecules form homodimers that undergo autoproteolytic cleavage at D384 (human) and D387 (mouse) for partial activation, followed by further processing to release the fully mature enzyme [30].
- Regulatory Checkpoints: Phosphorylation at specific residues (e.g., Y380) can regulate caspase-8 activity without affecting DISC recruitment, providing a regulatory mechanism for apoptosis control [30].
Table 2: Quantitative Biomarker Measurements in Experimental Models
| Biomarker | Detection Method | Sample Type | Key Measurable Parameters | Experimental Notes |
|---|---|---|---|---|
| Death Receptors | Flow cytometry, IHC | Cell surface, tissue sections | Receptor density, ligand binding affinity | TRAIL-R2 activation recruits FADD/caspase-8 within 1 minute [33] |
| FADD | Western blot, GST pull-down | Cell lysates, purified complexes | Protein-protein interaction kinetics, phosphorylation status | DED-DED interaction KD = 250-1000 nM by Biacore [34] |
| Caspase-8 | ELISA, IHC, activity assays | Plasma, serum, tissue extracts | Procaspase-8, cleaved fragments, enzymatic activity | Cleavage at D384/D387 indicates activation; phosphorylation at Y380 inhibits apoptosis [30] |
| FADD-Caspase-8 Complex | Co-immunoprecipitation, BIO-TEM | Immunoprecipitated DISC | Complex formation, stoichiometry, spatial organization | FADD:caspase-8 ratio averages 1:6 in stoichiometric studies [30] |
Experimental Protocols for Biomarker Analysis
DISC Immunoprecipitation and Analysis
This protocol isolates and characterizes the Death-Inducing Signaling Complex (DISC) to study early events in extrinsic apoptosis activation [33].
Methodology:
- Cell Stimulation: Treat cells (e.g., BJAB, Jurkat, K562, or MCF-7) with death receptor ligands (e.g., TRAIL at 100-500 ng/mL) for 1-60 minutes at 37°C [33].
- Cell Lysis: Use mild lysis buffers (1% Triton X-100, 20 mM Tris-HCl pH 7.4, 150 mM NaCl) with protease inhibitors to preserve protein complexes.
- Immunoprecipitation: Incubate lysates with specific death receptor antibodies (e.g., anti-TRAIL-R2) coupled to protein A/G beads for 2-4 hours at 4°C [33].
- Complex Analysis: Wash beads thoroughly and elute bound proteins for Western blotting to detect co-precipitated FADD, caspase-8, and other DISC components.
Key Applications: Determining composition and kinetics of DISC assembly; comparing receptor signaling specificity; testing drug effects on complex formation [33].
FADD-Caspase-8 Interaction Mapping
This biophysical approach characterizes the direct molecular interactions between FADD and caspase-8 death effector domains [34].
Methodology:
- Protein Purification: Express and purify recombinant FADD DED (residues 1-83) and caspase-8 DEDs (residues 1-188) using His-tagged or GST-tagged systems in E. coli [34].
- Site-Directed Mutagenesis: Generate specific point mutations in caspase-8 DED (e.g., Y8D/I128A, E12A/I128A, F122A/I128A) to identify critical interaction residues [34].
- Binding Assays:
- GST Pull-down: Incubate GST-tagged FADD DED with His-tagged caspase-8 DED mutants, capture on glutathione sepharose, and detect bound proteins by Western blot [34].
- Surface Plasmon Resonance: Immobilize FADD DED on CM5 chip and measure binding kinetics with caspase-8 DED concentrations (250-5000 nM) using Biacore systems [34].
- Structural Analysis: Determine crystal structures of caspase-8 DED mutants (e.g., F122A/I128A) at 1.7 Å resolution to visualize interaction interfaces [34].
Key Applications: Identifying critical interaction residues; quantifying binding affinities; guiding drug discovery targeting DED interactions.
Caspase-8 Activity Monitoring
This functional assay measures caspase-8 enzymatic activity as a direct indicator of extrinsic pathway activation [31].
Methodology:
- Sample Preparation: Prepare cell lysates or fractionated cellular compartments from treated and control cells.
- Activity Assay: Use fluorogenic or colorimetric substrates (e.g., IETD-pNA) that release detectable signals upon cleavage by active caspase-8.
- Quantification: Measure signal generation over time and compare to standard curves of recombinant active caspase-8.
- Specificity Controls: Include caspase-8 specific inhibitors to confirm signal specificity.
Key Applications: Pharmacodynamic monitoring of drug efficacy; determining apoptotic thresholds; correlating caspase activation with cell death outcomes [31].
Signaling Pathway Visualization
Figure 1: Extrinsic Apoptosis Pathway Molecular Interactions. This diagram illustrates the core signaling cascade from death receptor activation to apoptosis execution, highlighting the critical role of FADD and caspase-8 as central biomarkers. Regulatory mechanisms involving cFLIP are shown in green/red to indicate their dual role in promoting or inhibiting apoptosis depending on cellular context [30] [32].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Extrinsic Pathway Studies
| Reagent Category | Specific Examples | Research Application | Functional Notes |
|---|---|---|---|
| Recombinant Proteins | His-tagged FADD DED (1-83), Caspase-8 DEDs (1-188) [34] | Biophysical interaction studies, structural biology | Enables precise measurement of DED-DED interactions without full-length protein complexity |
| Cell Line Models | FADD-deficient Jurkat cells, Caspase-8-deficient cells [33] | Genetic validation of pathway components | Essential for establishing necessity of specific biomarkers in death receptor signaling |
| Activation Ligands | Recombinant TRAIL, Anti-Fas agonistic antibodies [33] | Specific pathway activation | TRAIL particularly valuable for cancer studies due to selective tumor cell apoptosis |
| Detection Antibodies | Anti-FADD, Anti-Caspase-8 (cleaved and total), Anti-death receptors [34] [33] | Western blot, immunoprecipitation, flow cytometry | Cleavage-specific antibodies critical for distinguishing active vs. inactive caspase-8 |
| Inhibitors/Mutants | cFLIP overexpression constructs, Caspase-8 phosphorylation mutants (Y380F) [30] [32] | Pathway modulation studies | Phosphorylation mutants help elucidate regulatory mechanisms controlling caspase-8 activity |
Comparative Analysis and Research Applications
The core biomarkers of the extrinsic pathway function as an integrated system with distinct but interconnected roles. Death receptors serve as the specific recognition components, FADD as the essential adaptor, and caspase-8 as the enzymatic activator. Each biomarker provides unique research insights:
Death Receptors offer target specificity, with different receptors activating context-dependent signaling outcomes. TRAIL receptors are particularly valuable in cancer research due to their ability to selectively induce apoptosis in transformed cells while sparing normal cells [33].
FADD represents a convergence point in extrinsic signaling, with its essential role demonstrated across multiple death receptor pathways. Structural studies reveal that specific residues (F122, I128) in caspase-8 DED are critical for FADD binding, providing targets for potential therapeutic intervention [34].
Caspase-8 serves as the critical decision point between apoptosis and alternative cell fates. Its activation status provides a definitive biomarker for extrinsic pathway engagement, while its regulatory modifications (phosphorylation, cFLIP binding) offer insights into pathway modulation in different disease contexts [30] [32].
The integrated analysis of these biomarkers provides researchers with a comprehensive toolkit for investigating cell death mechanisms, screening potential therapeutics, and understanding disease pathologies involving dysregulated apoptosis.
Cellular behavior is governed by a complex interplay between intrinsic and extrinsic signaling pathways. The integration point of these signals determines critical cellular outcomes, including proliferation, differentiation, and programmed cell death (apoptosis). The extrinsic pathway of apoptosis initiates outside the cell when extracellular conditions dictate cell death, while the intrinsic pathway activates internally in response to cellular stress or damage [35]. Understanding the molecular convergence of these pathways is essential for advancing biomarker research and developing targeted therapies, particularly in oncology and immunology. This guide provides a comparative analysis of key biomarkers and experimental methodologies used to dissect these integrated signaling networks.
Key Signaling Pathways and Their Molecular Regulators
Core Apoptosis Pathways
The apoptotic signaling network represents a paradigm of intrinsic-extrinsic pathway integration. The extrinsic apoptosis pathway begins with ligand binding to death receptors (e.g., Fas, TNFR1) at the cell surface, leading to formation of the Death-Inducing Signaling Complex (DISC) and activation of caspase-8 [35]. This initiates a proteolytic cascade that executes the apoptotic program. Meanwhile, the intrinsic apoptosis pathway activates in response to internal stressors like DNA damage, oxidative stress, or oncogene activation, culminating in mitochondrial outer membrane permeabilization (MOMP) and release of cytochrome c [35]. The convergence point between these pathways occurs at several levels, particularly through the caspase activation cascade and BID cleavage, which amplifies the death signal.
Mechanotransduction Pathways
Beyond biochemical signaling, mechanical cues from the cellular microenvironment serve as critical extrinsic signals that influence intrinsic pathway activation. Cells sense physical properties like matrix stiffness, fluid shear stress, and compressive stress through mechanosensors including integrins, Piezo channels, and YAP/TAZ signaling [36]. These mechanical signals undergo mechanotransduction into biochemical responses that regulate fundamental cellular processes including morphogenesis, immunity, and tumor progression [36]. The tumor microenvironment exhibits distinct biomechanical properties that influence therapeutic responses, particularly in desmoplastic cancers like pancreatic ductal adenocarcinoma [36].
Wnt Signaling in Cellular Homeostasis
The Wnt signaling pathway exemplifies how intrinsic and extrinsic signals integrate to regulate tissue homeostasis and development. This pathway activates through extrinsic Wnt ligands binding to Frizzled receptors, which transduce signals to intrinsic effectors including β-catenin [37]. In the canonical pathway, signal transduction inhibits the β-catenin destruction complex (APC, Axin, GSK3β), allowing β-catenin accumulation and nuclear translocation to activate target genes [37]. Dysregulated Wnt signaling occurs in many cancers through mutations in pathway components like APC or CTNNB1 (encoding β-catenin), leading to constitutive pathway activation independent of extrinsic signals [37].
Table 1: Key Signaling Pathways and Their Components
| Pathway | Extrinsic Signals | Membrane Receptors | Intrinsic Signal Transducers | Cellular Outcomes |
|---|---|---|---|---|
| Extrinsic Apoptosis | FasL, TRAIL, TNF-α | Fas, DR4/5, TNFR1 | FADD, caspase-8, caspase-3 | Membrane blebbing, DNA fragmentation, phagocytosis |
| Intrinsic Apoptosis | Cellular stress (DNA damage, hypoxia) | N/A | p53, Bcl-2 family, cytochrome c, caspase-9 | Mitochondrial permeability, caspase activation |
| Wnt Signaling | Wnt proteins | Frizzled, LRP5/6 | β-catenin, APC, GSK-3β, TCF/LEF | Proliferation, differentiation, stem cell maintenance |
| Mechanotransduction | Matrix stiffness, fluid shear stress | Integrins, Piezo channels | YAP/TAZ, Rho GTPases | Gene expression, cytoskeletal reorganization, migration |
Comparative Analysis of Pathway Biomarkers
Apoptosis Pathway Biomarkers
Biomarker profiling enables researchers to distinguish pathway activation and identify convergence points. The extrinsic pathway features characteristic biomarkers including death receptors (Fas, TNFR1), adapter proteins (FADD, TRADD), and initiator caspases (caspase-8, -10) [35]. The intrinsic pathway biomarkers include stress sensors (p53), Bcl-2 family proteins (BAX, BAK, Bid), mitochondrial components (cytochrome c, SMAC/Diablo), and caspase-9 [35]. Integrated biomarkers that reflect pathway convergence include executioner caspases (caspase-3, -7), DNA fragmentation factors (CAD/ICAD), and cleaved substrates like PARP.
Table 2: Biomarkers of Intrinsic and Extrinsic Apoptosis Pathways
| Biomarker Category | Extrinsic Pathway Markers | Intrinsic Pathway Markers | Convergence Point Markers |
|---|---|---|---|
| Initiation Proteins | Fas, TNFR1, TRAIL-R1/2 | p53, Bim, Bax, Bak | tBid, caspase-2 |
| Adapter/Mediator Proteins | FADD, TRADD, Daxx | Apaf-1, cytochrome c | PIDDosome |
| Protease Activators | Caspase-8, caspase-10 | Caspase-9 | Caspase-3, caspase-7 |
| Inhibitory Proteins | FLIP, cIAP1/2 | Bcl-2, Bcl-xL, Mcl-1 | IAP family, survivin |
| Mitochondrial Factors | N/A | SMAC/Diablo, Omi/HtrA2, AIF | N/A |
| Effector Proteins | N/A | N/A | ICAD/CAD, cleaved PARP |
Wnt Pathway Biomarkers
Wnt pathway biomarkers provide critical insights into developmental signaling and carcinogenesis. Key ligands and receptors include various Wnt proteins and Frizzled receptors, particularly FZD7 in gastric cancer [37]. Intracellular transducers include β-catenin, APC, Axin, and GSK-3β, with β-catenin stabilization and nuclear translocation serving as a primary activation marker [37]. Negative regulators encompass extracellular antagonists like Dickkopf (DKK) and secreted Frizzled-related proteins (sFRPs), while target genes include c-Myc, cyclin D1, and survivin [37]. Mutational analysis of APC and CTNNB1 genes provides predictive biomarkers for Wnt pathway activation across multiple cancer types.
Biomarkers in Targeted Protein Degradation
The emerging field of targeted protein degradation reveals new dimensions of intrinsic-extrinsic signaling integration. PROTAC molecules function as extrinsic诱导剂 that bridge target proteins to E3 ubiquitin ligases, exploiting intrinsic degradation machinery [38]. Key regulatory biomarkers in this process include ubiquitin chain types (K29/K48-branched chains), E2 conjugating enzymes (UBE2G, UBE2R), and regulatory factors like TRIP12 that promote branched ubiquitination [38]. Signaling pathways that modulate degradation efficiency include PARP ribosylation, HSP90-mediated protein stabilization, and PERK-regulated unfolded protein response, identified through chemical enhancer screens [38].
Experimental Methodologies for Pathway Analysis
Mechanotransduction Assays
Biomechanical profiling employs specialized technologies to quantify cellular responses to physical forces. Atomic force microscopy (AFM) enables precise measurement of mechanical properties including tensile stress, compressive stress, and matrix stiffness at nanoscale resolution [36]. Optical tweezers provide manipulation of individual molecules and cellular components with piconewton force sensitivity, while micropipette aspiration assesses cellular mechanical properties through controlled deformation [36]. Advanced methods for residual stress quantification include fluorescent oil microdroplet injection and computational modeling of stress distributions within tissues and tumors [36].
Molecular Profiling Techniques
High-throughput sequencing enables comprehensive analysis of pathway activation states through transcriptomic profiling. Single-cell RNA sequencing reveals cell-type-specific biomarkers of disease severity, as demonstrated in systemic sclerosis research identifying keratinocyte, myeloid, and fibroblast subpopulations with distinct signaling networks [6]. Machine learning approaches including LASSO regression, random forest, and support vector machines analyze complex genomic data to identify predictive biomarkers of pathway activation and therapeutic response [39] [6]. Protein-protein interaction mapping through STRING database analysis and co-expression network construction via Weighted Gene Co-expression Network Analysis (WGCNA) elucidate signaling modules and cross-talk mechanisms [39].
Functional Degradation Assays
Targeted protein degradation studies employ specialized reporter systems to quantify pathway modulation. The HiBiT tagging system enables sensitive monitoring of protein stability through knock-in of a small luciferase fragment into endogenous loci, allowing real-time tracking of PROTAC-induced degradation [38]. Chemical screening approaches identify modulators of degradation efficiency, exemplified by discovery of PARG, HSP90, and PERK inhibitors as enhancers of BRD4 degradation [38]. Mechanistic validation employs complementary techniques including ubiquitination assays, ternary complex formation analysis, and chromatin dissociation studies to pinpoint regulatory steps [38].
Figure 1: Integration of Intrinsic and Extrinsic Signaling Pathways. This diagram illustrates how diverse extrinsic signals converge with intrinsic pathway components to regulate fundamental cellular processes including apoptosis, gene expression, and protein degradation.
Research Reagent Solutions
Pathway Modulation Tools
Small molecule inhibitors enable precise dissection of signaling nodes and convergence mechanisms. Apoptosis modulators include caspase inhibitors (Z-VAD-FMK), Bcl-2 family inhibitors (venetoclax, ABT-737), and IAP antagonists [35]. Wnt pathway modulators comprise inhibitors of tankyrase (XAV939), porcupine (LGK974), and β-catenin-responsive transcription [37]. Targeted degradation tools include PROTAC molecules for BRD4 (MZ1, ARV-771, dBET6) and estrogen receptor (ARV-471), plus degradation enhancers like PARG inhibitor PDD00017273 and HSP90 inhibitor luminespib [38].
Detection and Analysis Reagents
Biomarker detection reagents facilitate quantitative assessment of pathway activation states. Antibody-based tools include phospho-specific antibodies for activated signaling intermediates, cleavage-specific antibodies for caspase substrates, and conformation-specific antibodies for Bcl-2 family proteins [35]. Reporter systems encompass HiBiT tagging for protein stability monitoring, fluorescent biosensors for second messengers, and luciferase reporter constructs for pathway activity readouts [38]. Multiplex assays enable simultaneous measurement of multiple biomarkers through technologies like Luminex bead-based arrays and proximity extension assays [39].
Table 3: Essential Research Reagents for Signaling Pathway Analysis
| Reagent Category | Specific Examples | Research Applications | Key Suppliers |
|---|---|---|---|
| Pathway Activators | Recombinant death ligands (FasL, TRAIL), Wnt proteins, Bioactive matrices | Induce specific pathway activation, Study signal initiation | R&D Systems, PeproTech |
| Small Molecule Inhibitors | Caspase inhibitors (Z-VAD-FMK), Bcl-2 inhibitors (Venetoclax), PROTACs (MZ1, ARV-771) | Pathway dissection, Therapeutic mechanism studies | Selleckchem, MedChemExpress |
| Detection Antibodies | Phospho-specific antibodies, Cleavage-specific caspase substrates, Conformation-specific Bcl-2 antibodies | Assess pathway activation states, Monitor protein modifications | Cell Signaling Technology, Abcam |
| Reporter Systems | HiBiT tagging systems, Luciferase pathway reporters, FRET biosensors | Real-time pathway monitoring, High-throughput screening | Promega, Addgene |
| Mechanical Analysis Tools | AFM cantilevers, Tunable stiffness substrates, Microfluidic shear devices | Quantify mechanical properties, Study mechanotransduction | Bruker, CellScale |
| Multiplex Assays | Luminex bead arrays, Proximity extension assays, High-throughput sequencers | Simultaneous biomarker measurement, Comprehensive profiling | Qiagen, Olink |
The integration point of intrinsic and extrinsic signals represents a crucial regulatory nexus in health and disease. Biomarker-driven approaches that capture pathway activation states and convergence mechanisms enable more precise therapeutic targeting across diverse conditions including cancer, autoimmune disorders, and fibrotic diseases. Advanced technologies in single-cell analysis, mechanical profiling, and targeted degradation continue to reveal new dimensions of signaling cross-talk, presenting opportunities for innovative therapeutic strategies. The continuing methodological refinement in detecting and quantifying these integrated signals will accelerate translation of basic pathway insights into clinically impactful interventions.
From Bench to Bedside: Analytical Techniques and Translational Applications
The precise identification of intrinsic and extrinsic pathway activation biomarkers represents a frontier in molecular pathology and therapeutic development. Traditional bulk analysis methods, which average signals across heterogeneous cell populations, often obscure critical cellular dynamics and rare but biologically significant events. The integration of single-cell RNA sequencing (scRNA-seq), multiplex immunohistochemistry (mIHC), and sophisticated machine learning (ML) algorithms is redefining this landscape. These technologies enable researchers to dissect complex biological pathways with unprecedented resolution, capturing cell-specific responses within their native spatial context. This guide provides a comparative analysis of these advanced tools, detailing their experimental protocols, performance metrics, and practical applications in pathway-oriented biomarker research.
Technology Comparison and Performance Data
The following table summarizes the core characteristics, strengths, and limitations of each technology for detecting pathway activation biomarkers.
Table 1: Technology Comparison for Pathway Biomarker Detection
| Technology | Primary Application in Pathway Research | Key Performance Metrics (from cited studies) | Key Advantages | Major Limitations |
|---|---|---|---|---|
| Single-Cell RNA-Seq | Unbiased discovery of cell-type-specific transcriptional signatures and novel pathway components. | - AUC of 0.89 for predicting Alzheimer's patients using a 34-gene panel from integrated cfRNA and scRNA-seq data [40].- Identification of neutrophil-specific biomarkers (MCEMP1, CLEC4D) for ischemic stroke via integrated bulk and single-cell analysis [41]. | - Reveals cellular heterogeneity and rare cell populations.- Discovers novel genes and pathways without prior hypothesis. | - Loss of native spatial context (unless combined with spatial transcriptomics).- Complex and costly data generation and analysis. |
| Multiplex IHC/IF | Spatial quantification of protein-level pathway activation within the tissue microenvironment. | - Predictive AUCs ~0.8 for response to anti-PD-(L1) therapy, outperforming PD-L1 IHC alone [42].- Identification of conserved receptor-ligand pairs (e.g., CCL5-SDC1/4) in pancreatic cancer [43]. | - Preserves spatial architecture and cell-cell interactions.- Directly measures protein expression and post-translational modifications. | - Limited multiplexing capacity without specialized platforms.- Requires high-quality, specific antibodies. |
| Machine Learning | Integration of multi-omic data to build predictive models of pathway activity and patient outcomes. | - Random Forest model achieved AUC of 0.915 for pan-cancer detection using a 12-exosomal-RNA signature [44].- SMAGS method improved sensitivity from 0.31 to 0.57 at 98.5% specificity for colorectal cancer detection [45] [46]. | - Integrates diverse data types (e.g., transcriptomic, proteomic, clinical).- Identifies complex, non-linear biomarker interactions. | - Requires large, high-quality datasets for training.- "Black box" nature can complicate biological interpretation. |
Experimental Protocols for Pathway Biomarker Discovery
Single-Cell RNA-Seq Workflow for Transcriptional Pathway Analysis
The following protocol, derived from studies on ischemic stroke and Alzheimer's disease, outlines a standard pipeline for scRNA-seq analysis focused on pathway activity [41] [47].
- Single-Cell Suspension Preparation: Tissues are dissociated into single-cell suspensions using enzymatic or mechanical methods, with viability and cell integrity rigorously checked.
- Library Preparation & Sequencing: Single cells are captured, and cDNA libraries are constructed using platforms such as the 10x Genomics Chromium system. The libraries are then sequenced to an appropriate depth.
- Computational Data Analysis:
- Quality Control (QC): Filter out low-quality cells based on metrics like the number of detected genes, total counts per cell, and mitochondrial gene percentage [47].
- Pre-processing & Normalization: Normalize data to account for technical variability (e.g., using SCTransform) and perform data integration if multiple samples are analyzed [47].
- Dimensionality Reduction & Clustering: Principal Component Analysis (PCA) is performed, followed by graph-based clustering on the top principal components. Cells are visualized in 2D using UMAP or t-SNE to identify distinct cell populations [41] [47].
- Cell Type Annotation & Differential Expression: Cluster identity is determined by referencing known marker genes. Differentially expressed genes (DEGs) between conditions (e.g., disease vs. control) are identified within each cell type to pinpoint cell-specific pathway alterations [41].
- Pathway & Interaction Analysis: DEG lists are subjected to Gene Ontology (GO) and pathway enrichment analyses (e.g., KEGG). Cell-cell communication networks (e.g., via ligand-receptor interactions) can be inferred to understand pathway crosstalk [41] [43].
Diagram 1: scRNA-seq analysis workflow for biomarker discovery.
Multiplex IHC/IF Protocol for Spatial Protein Biomarker Quantification
This protocol, based on Society for Immunotherapy of Cancer (SITC) best practices and a PDAC study, details the process for quantifying protein-based pathway biomarkers in situ [42] [43].
- Tissue Preparation and Staining:
- Sectioning: Formalin-fixed, paraffin-embedded (FFPE) or frozen tissues are sectioned onto slides.
- Staining Cycle: The process involves iterative cycles of staining. For each cycle: apply primary antibody, apply secondary antibody (if required), apply tyramide signal amplification (TSA) opal fluorophores, and perform microwave-based antibody stripping to remove the antibodies before the next cycle. This repeats for 5-8 markers [42] [43].
- Counterstaining and Mounting: Nuclei are counterstained with DAPI, and slides are mounted.
- Image Acquisition:
- Whole slides or selected regions of interest (ROIs) are scanned using a fluorescence slide scanner (e.g., VS200) with calibrated exposure times for each channel to prevent signal bleed-through [42].
- Quantitative Digital Pathology Analysis:
- Spectral Unmixing: Separate the overlapping fluorescence signals into distinct channels for each marker [42].
- Tissue and Cell Segmentation: Classify tissue regions (e.g., tumor vs. stroma) based on a marker like Pan-CK. Detect individual cell nuclei using DAPI staining [43].
- Cell Phenotyping: Classify cells based on the expression of markers (e.g., CD8+ T cell, CD68+ macrophage) and detect cells positive for multiple markers (e.g., CD8+PD1+ T cells) [43].
- Spatial Analysis: Quantify distances between different cell types (e.g., PD1+ cells within 30μm of PD-L1+ regions) or densities of specific cells within defined tissue compartments [43].
Diagram 2: Multiplex IHC/IF staining and analysis workflow.
Essential Research Reagent Solutions
Successful execution of these advanced assays requires a suite of reliable reagents and platforms. The following table details key solutions used in the cited studies.
Table 2: Key Research Reagent Solutions for Advanced Biomarker Detection
| Category | Specific Product/Platform | Function in Experiment |
|---|---|---|
| Single-Cell Genomics | 10x Genomics Chromium System [47] | Captures single cells and prepares barcoded sequencing libraries for transcriptome analysis. |
| Multiplex Staining | PANO Multiplex IHC Kit (Panovue) [43] | Provides reagents for sequential staining, amplification, and stripping in mIHC/IF protocols. |
| Image Analysis Software | QuPath (Open Source) [43] | Performs whole-slide image analysis, including cell detection, phenotyping, and spatial analysis. |
| Antibody Validation | Validated Primary Antibodies (e.g., from Abcam, CST) [43] | Ensures specific and reproducible detection of protein targets in multiplexed panels. |
| Data Integration & ML | R packages (e.g., limma, DESeq2, Seurat) [41] [44] |
Provides computational environment for differential expression, data integration, and model building. |
Integrated Data Analysis and Machine Learning Applications
Machine learning serves as the powerful engine that transforms high-dimensional data from scRNA-seq and mIHC into predictive models and robust biomarkers.
- Feature Selection and Model Training: In a multi-cancer study, researchers used RNA-seq data from blood-derived exosomes to identify differentially expressed genes. Machine learning techniques, including LASSO regression and Random Forest, were applied to refine 33 initial candidates down to a 12-exosomal-RNA signature (ETR.sig) for pan-cancer detection [44]. The final Random Forest model achieved an AUC of 0.915, demonstrating high diagnostic accuracy [44].
- Optimizing for Clinical Utility: Standard logistic regression may not yield optimal performance for specific clinical needs. The Sensitivity Maximization At a Given Specificity (SMAGS) method is a novel ML framework that modifies the logistic regression loss function to find a linear decision rule that maximizes sensitivity at a clinically required high specificity (or vice versa) [45] [46]. Applied to a colorectal cancer dataset, SMAGS improved sensitivity from 0.31 to 0.57 at a fixed specificity of 98.5%, a significant 14% improvement crucial for early detection where false positives must be minimized [46].
- Multi-Omic Data Integration: In Alzheimer's disease research, a non-invasive diagnostic approach was developed by integrating plasma-derived cell-free RNA (cfRNA) sequencing data with brain-derived scRNA-seq data [40]. This cross-validation identified 34 dysregulated genes consistent in both blood and brain. Machine learning models (SVM, Random Forest, Logistic Regression) trained on the cfRNA expression of these 34 genes accurately predicted AD patients with an AUC of up to 0.89, showcasing the power of integrating liquid biopsy with single-cell atlases [40].
The concerted application of single-cell RNA sequencing, multiplex IHC, and machine learning represents a paradigm shift in biomarker discovery for intrinsic and extrinsic pathway activation. scRNA-seq offers an unbiased, high-resolution view of transcriptional pathways across diverse cell types. Multiplex IHC provides the essential spatial protein context, revealing how pathway activity is organized within tissues. Finally, machine learning integrates these complex datasets to generate predictive, clinically actionable biomarkers. While each technology has distinct strengths and limitations, their synergistic use, as demonstrated across numerous studies in neurology and oncology, provides a more complete and mechanistically informed understanding of disease pathways, accelerating the development of targeted diagnostics and therapies.
LASSO-based Predictive Models for Uncovering Cell-Type-Specific Biomarkers of Disease Severity
In the evolving landscape of precision medicine, the identification of robust biomarkers that accurately reflect disease severity remains a critical challenge. Traditional bulk transcriptomic analyses often obscure cell-specific contributions to disease pathogenesis, limiting their clinical utility. The integration of machine learning approaches, particularly the least absolute shrinkage and selection operator (LASSO), with high-resolution single-cell technologies has emerged as a powerful methodology for deciphering cell-type-specific biomarkers across various disease contexts. This approach enables researchers to address the intricate balance between model complexity and interpretability while managing high-dimensional biological data.
LASSO's implementation in biomarker discovery is particularly valuable due to its intrinsic feature selection capability, which helps prevent overfitting by applying L1 regularization to shrink less important coefficients to zero. This review comprehensively examines the application of LASSO-based predictive models across diverse pathological conditions—from autoimmune disorders to infectious diseases—comparing their efficacy in identifying intrinsic and extrinsic pathway activation biomarkers. By synthesizing experimental protocols, key findings, and practical resources, this analysis aims to equip researchers with the knowledge needed to advance biomarker discovery in their respective fields.
LASSO Algorithm Fundamentals and Implementation
Mathematical Principles and Feature Selection Mechanism
The LASSO algorithm operates on a fundamental mathematical principle of linear regression with L1 regularization. Specifically, it modifies the typical least squares optimization by adding a penalty proportional to the absolute value of the regression coefficients. The objective function minimizes the sum: RSS + λΣ|βj|, where RSS represents the residual sum of squares, βj denotes the coefficients, and λ is the tuning parameter that controls the strength of the penalty. This L1 penalty has the crucial effect of forcing the coefficients of less relevant variables to become exactly zero, thereby performing automatic variable selection while maintaining model predictability.
The implementation of LASSO in biomarker discovery typically involves k-fold cross-validation (commonly 5-fold or 10-fold) to determine the optimal λ value that minimizes prediction error. This process, as demonstrated in multiple studies, identifies the lambda value where the mean cross-validated error is minimized [6] [48]. The resulting model retains only the most predictive features, effectively reducing dimensionality and enhancing biological interpretability. This characteristic makes LASSO particularly suitable for analyzing high-dimensional genomic and proteomic data where the number of features (p) vastly exceeds the number of observations (n).
Comparative Advantages in Biological Research
LASSO offers several distinct advantages over other feature selection methods in biological research contexts. Unlike univariate filtering approaches that assess variables independently, LASSO considers multivariate relationships, capturing complex interactions among biomarkers. Compared to ridge regression (L2 regularization), which only shrinks coefficients but never sets them to zero, LASSO produces more sparse solutions that are easier to interpret biologically. Furthermore, unlike random forest or other tree-based methods that may identify complex non-linear relationships but offer less transparency, LASSO generates linear models with clearly quantified contributions for each selected feature.
The practical implementation of LASSO has been facilitated by its integration into standard statistical packages. For example, researchers commonly utilize the glmnet package in R, which provides efficient algorithms for fitting LASSO models even with large-scale datasets [48]. The cv.glmnet function specifically enables automated cross-validation to determine the optimal penalty parameter, making the methodology accessible to researchers without deep computational backgrounds. This accessibility has contributed to its widespread adoption across diverse disease contexts from autoimmune conditions to infectious diseases.
Application Across Disease Models
Autoimmune and Inflammatory Diseases
In systemic sclerosis (SSc), researchers applied LASSO regression to single-cell RNA-sequencing data from 24 patients across the disease severity spectrum, quantified by the modified Rodnan skin score (MRSS) [6]. The analysis revealed cell-type-specific predictive biomarkers of MRSS across multiple cell types, including previously unidentified roles for keratinocytes in addition to established contributions from fibroblast and myeloid cells. The study identified novel gene biomarkers in keratinocytes, with subsequent validation confirming the association of KRT6A and S100A8 protein expression with SSc skin disease severity [6]. This application demonstrated LASSO's capability to uncover previously uncharacterized cell-intrinsic and cell-extrinsic signaling networks underlying disease severity.
In COVID-19 research, LASSO was employed to analyze blood protein profiles from 420 individuals to differentiate between test-negative healthy, asymptomatic, and symptomatic individuals [48]. The algorithm identified two key protein factors—Serpin A10 and Complement C9—that effectively discriminated between asymptomatic and symptomatic patients. This analysis further revealed distinct immunopathological profiles, with symptomatic patients showing lower levels of CD4+ T naïve, CD4+ T effector & memory, and CD8+ T naïve cells, along with higher levels of CD14+ classical monocytes compared to asymptomatic patients [48]. The integration of these findings with single-cell RNA sequencing data demonstrated that CD16+ non-classical monocytes, major producers of C1QA/B/C, appeared to contribute to the observed Complement C9 levels, providing mechanistic insights into disease pathogenesis.
Table 1: LASSO-Based Biomarker Discovery Across Disease Contexts
| Disease Context | Data Type | Key Biomarkers Identified | Cell Types Involved | Reference |
|---|---|---|---|---|
| Systemic Sclerosis | Single-cell RNA-seq | KRT6A, S100A8, SFRP2 | Keratinocytes, fibroblasts, myeloid cells | [6] |
| COVID-19 Severity | Blood protein factors, scRNA-seq | Serpin A10, Complement C9 | CD14+ classical monocytes, CD16+ non-classical monocytes, T cell subsets | [48] |
| Intracerebral Hemorrhage | Bulk RNA-seq, miRNA sequencing | BID, miR-1225-3p | Neurons, glial cells | [12] |
Cancer and Cerebrovascular Applications
In cancer research, while the specific application of LASSO was not detailed in the available sources, the broader context of molecular pathway activation biomarkers highlights the relevance of such approaches for personalized selection of target drugs [49]. The emergence of pathway activation biomarkers represents a new generation of diagnostic tools that leverage high-throughput gene expression profiling and bioinformatic algorithms to quantitatively measure the degree of pathway activation, potentially informing therapeutic decisions.
For intracerebral hemorrhage (ICH), researchers employed a complementary bioinformatics approach to identify differentially expressed genes associated with apoptosis following hemorrhagic stroke [12]. Though not explicitly using LASSO, this methodology shared similar analytical goals in feature selection from high-dimensional genomic data. The study identified BID as a critical regulator of apoptosis following ICH, with validation in a rat model confirming its significance as a key biomarker in the apoptotic process post-hemorrhage [12]. The construction of an mRNA-miRNA interaction network further suggested that miR-1225-3p may be an important regulator of BID expression, demonstrating the multi-omics integration potential in such analyses.
Experimental Design and Methodologies
Single-Cell RNA Sequencing Workflows
The application of LASSO-based models to single-cell RNA-sequencing data requires meticulous experimental design and execution. In the systemic sclerosis study, researchers performed scRNA-seq on skin biopsies from 24 SSc patients across the severity spectrum and implemented strict quality control measures [6]. The standard workflow encompasses several critical phases beginning with tissue dissociation and single-cell isolation, followed by library preparation using platforms such as 10x Genomics. Sequencing is typically performed to sufficient depth (50,000-100,000 reads per cell) to capture the transcriptomic diversity.
Following data generation, the bioinformatic pipeline involves quality control to remove low-quality cells and potential multiplets, normalization to account for technical variability, and dimensionality reduction using techniques such as principal component analysis (PCA). Cell types are then annotated based on the expression of canonical marker genes, creating the foundation for cell-type-specific analysis [6] [48]. The normalized gene expression matrices for each cell type serve as input for the LASSO regression model, with disease severity scores (e.g., MRSS) as the response variable. This approach enables the identification of genes whose expression within specific cell types most strongly predicts clinical severity.
Proteomic Profiling and Integration Approaches
For COVID-19 severity biomarker discovery, researchers employed a multi-omics integration strategy combining blood protein factor profiling with single-cell transcriptomics [48]. The experimental protocol began with blood collection from 420 individuals, including test-negative healthy controls, asymptomatic SARS-CoV-2-positive individuals, and symptomatic COVID-19 patients. Protein factor levels were quantified using immunoassays or proteomic profiling techniques, with rigorous normalization to account for technical variation.
The LASSO implementation involved several methodical steps. First, researchers performed statistical pre-screening of protein factors using t-tests to identify potentially significant analytes. The dataset was then divided into training and test sets (typically 70:30 ratio) to enable model validation [48]. The LASSO algorithm was applied to the training set with 5-fold cross-validation to determine the optimal lambda value that minimized prediction error. The resulting model was evaluated on the held-out test set using area under the curve (AUC) metrics to assess classification performance between asymptomatic and symptomatic individuals. Finally, the identified protein biomarkers were contextualized through integration with scRNA-seq data to identify potential cellular sources.
Table 2: Key Experimental Parameters in LASSO-Based Biomarker Studies
| Experimental Component | Systemic Sclerosis Study | COVID-19 Severity Study |
|---|---|---|
| Sample Size | 24 patients | 420 individuals |
| Data Type | Single-cell RNA-sequencing | Blood protein factors, scRNA-seq |
| Severity Metric | Modified Rodnan Skin Score (MRSS) | Symptomatic vs. Asymptomatic classification |
| LASSO Validation | Cross-validation, functional validation | Train-test split (70:30), cross-validation |
| Key Outcomes | Cell-type-specific gene biomarkers (KRT6A, S100A8) | Protein biomarkers (Serpin A10, Complement C9) with cellular sources |
Signaling Pathways and Biomarker Networks
Cell-Intrinsic and Cell-Extrinsic Circuitry
LASSO-based analyses have revealed complex interactions between cell-intrinsic and cell-extrinsic pathways in disease severity. In systemic sclerosis, the application of correlation network analyses coupled with LASSO models identified novel cross-talk between immune pathways that implicated keratinocytes alongside fibroblasts and myeloid cells as key players in pathogenesis [6]. These networks encompass both autocrine signaling within specific cell types and paracrine interactions between different cellular compartments, creating a complex signaling ecosystem that drives disease progression.
The extrinsic coagulation pathway has emerged as a significant component in tumor microenvironments, particularly in colorectal cancer [50]. Although not identified through LASSO, these findings highlight how extrinsic pathway activation—initiated by tissue factor (TF) release—contributes to disease pathophysiology beyond its traditional role in coagulation. Similarly, in COVID-19, the complement system—an extrinsic pathway of immune activation—was identified through LASSO analysis as a key discriminator of symptom status [48], with Complement C9 emerging as a significant biomarker and CD16+ non-classical monocytes implicated as cellular sources.
Visualization of Analytical Workflows
The following diagram illustrates the integrated computational and experimental workflow for LASSO-based biomarker discovery:
Diagram 1: Integrated Workflow for LASSO-Based Biomarker Discovery. This schematic outlines the key stages from sample collection through experimental validation, highlighting the integration of multi-omics data with computational feature selection.
Research Reagent Solutions Toolkit
Table 3: Essential Research Reagents and Platforms for LASSO-Based Biomarker Studies
| Reagent/Platform | Specific Examples | Research Application | Function in Workflow |
|---|---|---|---|
| Single-cell RNA-seq Platform | 10x Genomics Chromium | Single-cell partitioning & barcoding | High-throughput transcriptome profiling at single-cell resolution |
| Protein Quantification Assay | ELISA, Olink, Luminex | Blood protein factor measurement | Quantification of protein biomarkers in biofluids |
| Cell Type Marker Antibodies | CD3, CD4, CD8, CD14, CD16 | Immunophenotyping by flow cytometry/FACS | Cell identification, sorting, and validation |
| Bioinformatics Tools | Seurat, glmnet, DoubletFinder | scRNA-seq analysis, LASSO modeling | Data preprocessing, clustering, and predictive modeling |
| Validation Reagents | Primary antibodies for WB/IF, TUNEL assay kits | Biomarker confirmation | Protein detection, localization, and functional assessment |
Comparative Analysis and Future Directions
The application of LASSO-based models across different disease contexts reveals both consistent strengths and context-specific adaptations. In autoimmune diseases like systemic sclerosis, the integration with single-cell transcriptomics has enabled the decomposition of complex tissue environments into constituent cell types, permitting the identification of cell-specific biomarkers that would be obscured in bulk analyses [6]. In infectious diseases like COVID-19, the combination with proteomic profiling has facilitated the identification of circulating biomarkers with diagnostic and prognostic potential [48]. These complementary approaches highlight how LASSO methodology can be adapted to different data types while maintaining its core feature selection advantages.
Future developments in this field will likely focus on multi-omics integration, combining transcriptomic, proteomic, epigenomic, and clinical data within unified modeling frameworks. Additionally, methodological advances may incorporate interaction terms to capture non-linear relationships and time-series analyses to model dynamic biomarker changes throughout disease progression. As single-cell technologies continue to evolve, providing increasingly comprehensive molecular measurements, LASSO and related regularized regression approaches will remain essential tools for distilling this complexity into clinically actionable biomarkers.
The consistent demonstration that cell-type-specific biomarkers outperform bulk tissue signatures across multiple disease contexts underscores the importance of cellular resolution in biomarker discovery. Furthermore, the identification of previously underappreciated cell types—such as keratinocytes in systemic sclerosis and non-classical monocytes in COVID-19—highlights how unbiased computational approaches can reveal novel biological insights with potential therapeutic implications.
The evaluation of transporter-mediated drug-drug interactions (DDIs) has become an integral component of drug development, contributing critical data for benefit-risk assessment and clinical management strategies. Membrane transporters, proteins that control the movement of endogenous and exogenous substances across cellular barriers, significantly influence drug disposition, efficacy, and safety. Among the numerous transporters identified, Organic Anion Transporting Polypeptides (OATP) 1B1 and 1B3 have emerged as clinically relevant due to their role in hepatic drug uptake. Traditional DDI assessment approaches have relied on static in vitro-in vivo extrapolation methods followed by clinical studies with probe substrate drugs. However, these methods sometimes yield false positive or false negative predictions, creating a need for more reliable approaches [51].
The emergence of endogenous biomarkers as tools for assessing transporter activity represents a significant advancement in the field. These biomarkers are physiological compounds that serve as substrates for clinically relevant uptake and efflux transporters. Their quantification can provide a direct, non-invasive means to evaluate the transporter inhibitory potential of investigational drugs without always requiring administration of exogenous probe drugs. Among the most promising endogenous biomarkers are coproporphyrin I and III (CP-I and CP-III), which have demonstrated considerable value in assessing OATP1B activity. The strategic incorporation of endogenous biomarker assessment in clinical development programs enables more informed DDI risk assessment and helps formulate prudent clinical management strategies [51] [52].
The utilization of endogenous biomarkers has expanded beyond facilitating DDI assessment to include understanding alterations in transporter activity in organ dysfunction and various disease states. This review comprehensively examines the current status of endogenous biomarkers, with a particular focus on coproporphyrins for OATP1B transporter assessment, providing comparative analysis with alternative approaches, detailed experimental methodologies, and their growing role in precision medicine.
Coproporphyrins as Endogenous Biomarkers for OATP1B Transporters
Biological Basis and Mechanistic Role
Coproporphyrins I and III are organic anions and porphyrin metabolites derived from the heme biosynthesis pathway. These endogenous compounds undergo a specific transport pathway wherein they are taken up into hepatocytes primarily by OATP1B1 and OATP1B3 transporters, then eliminated via biliary efflux mediated by Multidrug Resistance-Associated Protein 2 and 3 (MRP2/MRP3) or through urinary excretion [51]. This specific transport pathway makes them ideal biomarker candidates for OATP1B function, as any inhibition of these uptake transporters directly impacts their plasma concentrations.
The transporter specificity of CP-I and CP-III differs somewhat, providing complementary information. Genetic studies have revealed that unlike CP-III, plasma concentrations of CP-I demonstrate sensitivity to SLCO1B1 genotypes, particularly the c.521T>C polymorphism, suggesting a closer association with OATP1B1 transport activity. CP-III appears to have a broader transporter interaction profile, potentially involving both OATP1B1 and OATP1B3. This differential specificity has been leveraged in more refined analyses to delineate inhibition patterns for specific OATP1B transporters [51] [53].
Comparative Performance of CP-I and CP-III as OATP1B Biomarkers
Extensive research has established the validity of both CP-I and CP-III as biomarkers for OATP1B function, though with some distinguishing characteristics:
Table 1: Comparative Characteristics of Coproporphyrin Biomarkers
| Characteristic | Coproporphyrin I (CP-I) | Coproporphyrin III (CP-III) |
|---|---|---|
| Primary Transporters | OATP1B1, OATP1B3 | OATP1B1, OATP1B3 |
| Genetic Sensitivity | Sensitive to SLCO1B1 c.521T>C | Less sensitive to SLCO1B1 genotypes |
| Baseline Levels | 0.02-100 ng/mL in human plasma | 0.02-100 ng/mL in human plasma |
| Response to Rifampin | 2.8-3.7-fold AUC increase | 2.4-3.1-fold AUC increase |
| Specificity | Higher specificity for OATP1B1 | Broader transporter involvement |
| Analytical Measurement | LC-MS/MS (m/z 655.3→596.3) | LC-MS/MS (m/z 655.3→596.3) |
Recent evidence suggests that CP-I may offer superior selectivity as an OATP1B biomarker compared to CP-III. A 2024 case study on cedirogant demonstrated that CP-I could effectively delineate OATP1B inhibition from inhibition of other transporters like Breast Cancer Resistance Protein (BCRP) in complex DDIs. The study established that an OATP1B1 R-value of >1.5 and [Cmax,u]/[OATP1B1 IC50] of >0.1 are associated with a >1.25-fold increase in CP-I Cmax ratio, providing quantitative thresholds for OATP1B inhibition assessment [53].
The response profile of these biomarkers has been characterized through numerous clinical studies with known OATP1B inhibitors. Administration of rifampin, a potent OATP1B inhibitor, consistently increases plasma exposure of both CP-I and CP-III by approximately 2.5 to 4-fold across different ethnic populations, demonstrating their sensitivity to transporter inhibition [54]. Furthermore, studies in subjects with SLCO1B1 polymorphisms have shown that those with increased function variants (c.388AG and c.388GG genotypes) have lower basal CPI concentrations, consistent with enhanced transporter activity [54].
Comparative Analysis with Alternative Assessment Methods
Endogenous Biomarkers vs. Probe Drugs
The traditional approach to clinical DDI assessment for transporters has involved administration of probe drugs such as rosuvastatin for OATP1B1/BCRP/OAT3 or metformin for OCT2/MATE transporters. While this approach remains valuable, endogenous biomarkers offer several distinct advantages:
Table 2: Endogenous Biomarkers versus Probe Drugs for Transporter DDI Assessment
| Assessment Factor | Endogenous Biomarkers | Probe Drugs |
|---|---|---|
| Need for Dosing | No additional administration required | Requires administration of exogenous compound |
| Study Design | Can be incorporated into early-phase studies | Often requires dedicated DDI studies |
| Multi-transporter Scenarios | Can help delineate complex mechanisms | May involve overlapping substrate specificities |
| Population Applicability | Can assess transporter activity in patient populations | Typically limited to healthy volunteers |
| Temporal Assessment | Allows continuous monitoring of transporter activity | Provides snapshot assessment |
| Regulatory Acceptance | Emerging acceptance for decision-making | Well-established regulatory framework |
A key advantage of endogenous biomarkers is their ability to deconvolute complex DDIs involving multiple transporters. For instance, when the investigational drug ritlecitinib was co-administered with rosuvastatin (a substrate of BCRP, OATP1B1, and OAT3), the resulting 13% decrease in rosuvastatin exposure alone provided limited mechanistic insight. However, the concurrent assessment of CP-I for OATP1B1 and pyridoxic acid (PDA) for OAT3 showed no changes in these biomarkers, allowing researchers to exclude OATP1B1 and OAT3 inhibition as mechanisms and attribute the modest change to factors other than transporter inhibition [55].
Comparison with Other Endogenous Biomarkers
Beyond coproporphyrins for OATP1B transporters, several other endogenous biomarkers have been identified for various transporters:
Table 3: Endogenous Biomarkers for Various Transporters
| Transporter | Endogenous Biomarker | Performance and Utility |
|---|---|---|
| OATP1B1/1B3 | Coproporphyrin I and III | Well-validated, sensitive to strong and moderate inhibitors |
| OAT1/3 | Pyridoxic Acid (PDA) | Useful for renal OAT assessment |
| OCT2/MATE1/2-K | N1-methylnicotinamide (1-NMN), N1-methyladenosine (m1A) | Superior to creatinine for specific MATE inhibition |
| OCT1 | Isobutyryl-L-carnitine (IBC) | Emerging biomarker for hepatic OCT1 |
The performance of these biomarkers varies based on their specificity, dynamic range, and sensitivity to inhibition. For renal transporter assessment, clinical studies have demonstrated that 1-NMN and m1A serve as superior biomarkers for MATE1/2-K inhibition compared to creatinine, as changes in their renal clearance closely correlate with metformin clearance changes when co-administered with the MATE inhibitor pyrimethamine [56]. Similarly, pyridoxic acid (PDA) has emerged as a valuable biomarker for organic anion transporters (OAT1/3), providing a non-invasive means to assess renal uptake transporter inhibition [55].
Experimental Protocols and Methodologies
Analytical Method for Coproporphyrin Quantification
Robust analytical methods are crucial for reliable quantification of endogenous biomarkers. The following protocol describes a validated LC-MS/MS method for coproporphyrin I and III quantification in human plasma [57]:
Sample Preparation:
- Utilize mixed-mode anion exchange solid-phase extraction (Oasis Max 96-well plates, 30 mg)
- Employ stable isotope-labeled internal standards (CP-I-d4 and CP-III-d4) for quantification
- Critical attention to light protection during processing due to photosensitivity of porphyrins
Chromatographic Separation:
- Column: Ace Excel 2 C18 PFP (3μm, 2.1×150mm) maintained at 60°C
- Mobile Phase: A) 10mM ammonium formate with 0.1% formic acid; B) 100% acetonitrile
- Gradient elution with optimized separation of CP-I and CP-III isomers
- Flow rate: 0.3-0.5 mL/min with total run time of 8-10 minutes
Mass Spectrometric Detection:
- Ionization: Electrospray ionization (ESI) in positive mode
- Mass transitions: m/z 655.3→596.3 for both CP-I and CP-III; m/z 659.3→600.3 for internal standard
- Lower Limit of Quantification (LLOQ): 20 pg/mL for both analytes
- Calibration range: 0.02-100 ng/mL with correlation coefficients >0.988
Validation Parameters:
- Extraction recovery: ~70% for both CP-I and CP-III
- Inter-day precision: CV <9%
- Accuracy: 84.3-103.9%
- Stability assessment under various processing and storage conditions
Clinical Study Design for Biomarker Application
The strategic incorporation of endogenous biomarker assessment in clinical studies follows these key principles [51] [55]:
Early Phase Integration:
- Biomarker assessment can be incorporated into single and multiple ascending dose studies
- Dose-dependent changes in biomarker levels provide early readout of transporter inhibition
- Enables in vivo inhibition constant (Ki) determination for more accurate DDI prediction
DDI Mechanism Delineation:
- When complex DDIs are observed with probe drugs, endogenous biomarkers help identify specific transporter involvement
- Example: Rosuvastatin DDI with cedirogant showed 141% increase in Cmax – CP-I assessment confirmed no OATP1B involvement, pointing to BCRP inhibition [53]
Cross-Study Applications:
- Biomarker data can be pooled across studies to strengthen statistical power
- Population approaches can quantify the impact of intrinsic factors (genetics, organ impairment) on transporter activity
Diagram 1: Experimental workflow for biomarker analysis
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of endogenous biomarker strategies requires specific reagents and methodological tools:
Table 4: Essential Research Reagents for Coproporphyrin Research
| Reagent/Material | Specification | Application and Function |
|---|---|---|
| Reference Standards | CP-I and CP-III (≥95% purity) | Method development, calibration standards |
| Internal Standards | Stable isotope-labeled (CP-I-d4, CP-III-d4) | Normalization of extraction efficiency and matrix effects |
| SPE Cartridges | Mixed-mode anion exchange (Oasis MAX) | Selective extraction and clean-up from biological matrices |
| LC Column | C18 with pentafluorophenyl functionality (e.g., Ace Excel 2 C18 PFP) | Isomeric separation of CP-I and CP-III |
| Mobile Phase | Ammonium formate with formic acid in water/acetonitrile | Optimal ionization and chromatographic separation |
| Quality Controls | Pooled human plasma with validated CP levels | Method validation and batch acceptance |
| Inhibition Controls | Rifampin or other known OATP1B inhibitors | Positive control for biomarker response |
Additional methodological considerations include light-protected containers and processing conditions to prevent photodegradation of porphyrins, matrix-matched calibration standards to account for plasma effects, and appropriate storage conditions (-70°C or below) to maintain analyte stability [57].
Endogenous biomarkers, particularly coproporphyrins for OATP1B transporters, have fundamentally transformed the approach to transporter-mediated DDI assessment in drug development. The robust analytical methods, established clinical validation, and growing regulatory acceptance position these biomarkers as powerful tools for mechanistic understanding of complex DDIs. The comparative advantage of endogenous biomarkers lies in their ability to provide continuous assessment of transporter activity without the need for dedicated probe drug studies, enabling more efficient drug development programs.
Future directions in the field include the identification and validation of novel biomarkers for additional transporters, the standardization of analytical approaches across laboratories, and the integration of biomarker data into physiologically-based pharmacokinetic models for improved DDI prediction. Furthermore, the application of endogenous biomarkers extends beyond DDI assessment to understanding the impact of organ impairment, genetic polymorphisms, and disease states on transporter function, offering exciting possibilities for personalized medicine approaches [52] [58].
As the field evolves, the strategic incorporation of endogenous biomarker assessment in clinical pharmacology programs will continue to enhance our understanding of transporter-mediated drug disposition, ultimately contributing to the development of safer and more effective therapeutics.
Diagram 2: Mechanistic pathway of coproporphyrin disposition and biomarker response to inhibition
The clinical landscape of oncology has been fundamentally transformed by the development of immune checkpoint inhibitors (ICIs), which block regulatory pathways such as PD-1, PD-L1, and CTLA-4 to reactivate T-cell-mediated anti-tumor immunity [59]. Despite demonstrating remarkable success across various malignancies, a significant clinical challenge persists: only a minority of patients experience durable benefits from these therapies [59] [60]. This variability in treatment response has catalyzed extensive research into predictive biomarkers that can identify patients most likely to respond to ICI therapy, thereby optimizing treatment selection and improving clinical outcomes [61].
The current biomarker landscape encompasses both established biomarkers with regulatory approval and emerging next-generation approaches. Three biomarkers—programmed death-ligand 1 (PD-L1) expression, microsatellite instability (MSI), and tumor mutational burden (TMB)—have received FDA approval for clinical use in patient selection for ICI therapy [61]. However, each presents limitations in predictive accuracy and practical implementation, driving investigation into novel biomarkers and integrated models [61] [60]. These biomarkers can be broadly conceptualized within a framework of intrinsic tumor characteristics (such as genetic alterations and antigen presentation machinery) and extrinsic factors (including tumor microenvironment composition and systemic immune mediators) that collectively determine therapeutic response [59] [62].
This review provides a comprehensive comparison of established and emerging biomarker technologies for predicting response to immune checkpoint inhibitors, with particular emphasis on their underlying biological pathways, methodological approaches for assessment, and performance characteristics across diverse cancer types. We systematically evaluate experimental protocols, analytical validation requirements, and clinical implementation considerations to inform biomarker-guided immunotherapy development.
Classification and Definitions of Immuno-oncology Biomarkers
Biomarkers in immuno-oncology serve distinct clinical purposes, with accurate classification being fundamental to their appropriate application in both research and clinical settings. According to established definitions from the National Institutes of Health (NIH), a biomarker represents "a characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention" [63]. Within cancer immunotherapy, biomarkers are typically categorized based on their specific clinical utility, with predictive and prognostic biomarkers representing two fundamentally distinct categories [63].
Predictive biomarkers identify patients who are more likely to experience favorable outcomes from a specific treatment modality. For ICIs, predictive biomarkers indicate which patients will derive clinical benefit from immune checkpoint blockade [63] [61]. These biomarkers function by detecting biological characteristics that correlate with enhanced anti-tumor immune activation upon checkpoint inhibition. A truly predictive biomarker demonstrates differential treatment effect, where biomarker-positive patients respond better to the investigational therapy (e.g., ICIs) compared to standard treatments, while biomarker-negative patients show limited or no benefit from the same therapy [63]. Examples include PD-L1 expression, which predicts response to anti-PD-1/PD-L1 antibodies, and MSI status, which predicts efficacy across multiple tumor types [61].
Prognostic biomarkers, in contrast, provide information about the natural history of the disease and overall clinical outcome regardless of specific therapeutic interventions [63]. These biomarkers stratify patients based on their inherent disease aggressiveness or baseline outcome probability, independently of treatment selection. A prognostic biomarker identifies patients with differing risks of specific outcomes such as disease progression or mortality, irrespective of the treatments administered [63]. While some biomarkers possess both predictive and prognostic properties, this distinction is crucial for appropriate clinical interpretation and therapeutic decision-making.
Table 1: Classification of Biomarkers in Immuno-oncology
| Biomarker Type | Definition | Clinical Utility | Examples in Immuno-oncology |
|---|---|---|---|
| Predictive | Identifies patients more likely to respond to a specific treatment | Guides therapy selection; predicts differential treatment effect | PD-L1 expression, MSI status, TMB [63] [61] |
| Prognostic | Provides information about disease course regardless of treatment | Stratifies patients by disease aggressiveness; informs prognosis | Gene signature chips (Oncotype DX, MammaPrint) [63] |
| Pharmacodynamic | Demonstrates biological response to therapeutic intervention | Confirms target engagement; establishes proof of mechanism | CRP reduction in inflammatory diseases, FDG-PET changes [63] |
| Surrogate Endpoint | Substitutes for clinical endpoints in therapeutic evaluation | Accelerates drug development; supports regulatory approval | LDL cholesterol, HbA1c [63] |
The clinical application of predictive biomarkers for ICIs must account for the complex quantitative nature of treatment response, which is determined by multiple intrinsic and extrinsic factors [61]. This complexity is reflected in the observation that predicted responders identified by the three FDA-approved biomarkers (PD-L1, MSI, and TMB) show only minimal overlap, suggesting that each biomarker captures different biological contributors to ICI response [61]. This understanding has driven the development of integrated biomarker models that combine multiple features to improve predictive accuracy.
Established FDA-Approved Predictive Biomarkers
PD-L1 Expression
Programmed death-ligand 1 (PD-L1) expression represents the most extensively validated predictive biomarker for ICIs targeting the PD-1/PD-L1 axis. PD-L1 is expressed on tumor cells and various immune cells within the tumor microenvironment, where it interacts with PD-1 receptors on T cells to transmit inhibitory signals that suppress anti-tumor immunity [59]. Mechanistically, PD-1 functions through its ligands, PD-L1 and PD-L2, by inhibiting T-cell receptor (TCR) signaling through the phosphorylation of immunoreceptor tyrosine-based switch motifs (ITSM) and recruitment of Src homology region 2 domain-containing phosphatase-1 (SHP-1) and -2 (SHP-2), which dephosphorylate ZAP70 and CD3ζ, thereby impairing TCR activation [59].
The predictive utility of PD-L1 is typically assessed through immunohistochemistry (IHC) staining of tumor tissue specimens, with various companion diagnostic assays utilizing different antibodies, scoring systems, and expression cutoffs across cancer types [61] [60]. For advanced non-small-cell lung cancer (NSCLC), urothelial cancer, and triple-negative breast cancer, specific PD-L1 expression thresholds have been established to guide treatment decisions [60]. Despite its widespread clinical implementation, PD-L1 testing faces several limitations, including tumor heterogeneity, dynamic expression patterns, sampling bias due to tissue requirements, and variable performance across different cancer types [62] [61].
Microsatellite Instability (MSI)
Microsatellite instability (MSI) has emerged as a tissue-agnostic predictive biomarker for ICIs, leading to the first FDA-approved histology-independent indication for cancer therapy. MSI results from deficiencies in the DNA mismatch repair (MMR) system, which normally corrects errors occurring during DNA replication [61]. Deficient MMR (dMMR) significantly promotes the expression of several immune checkpoint ligands, including PD-1, PD-L1, CTLA-4, LAG-3, and IDO, creating a hypermutated tumor phenotype with increased neoantigen burden that enhances immunogenicity and T-cell recognition [60].
MSI status is typically determined through polymerase chain reaction (PCR)-based analysis of microsatellite regions or through IHC assessment of MMR protein expression (MLH1, MSH2, MSH6, and PMS2) [61]. Tumors are classified as MSI-high (MSI-H) or MSI-stable (MSS) based on the number of unstable loci. The robust predictive value of MSI-H/dMMR status across multiple tumor types has established it as a cornerstone biomarker for immunotherapy selection, though its prevalence varies significantly across cancer types [61].
Tumor Mutational Burden (TMB)
Tumor mutational burden (TMB) represents a quantitative measure of the total number of mutations per megabase (mut/Mb) of DNA sequenced in a tumor specimen [61]. High TMB is hypothesized to generate increased neoantigen formation, enhancing tumor immunogenicity and facilitating T-cell recognition and attack, particularly when immune checkpoints are inhibited [61]. The predictive value of TMB was initially demonstrated in melanoma and lung cancer, which typically exhibit higher mutation rates, and has since been validated across additional tumor types.
TMB assessment requires comprehensive genomic profiling, typically through next-generation sequencing (NGS) panels that capture coding regions across a substantial portion of the genome [61]. The transition of TMB from research tool to clinically validated biomarker has faced challenges related to standardization of measurement approaches, definition of optimal thresholds across cancer types, and integration with other biomarker data [61]. Despite these challenges, TMB has received regulatory approval as a predictive biomarker for ICIs in specific clinical contexts.
Table 2: FDA-Approved Predictive Biomarkers for Immune Checkpoint Inhibitors
| Biomarker | Detection Method | Key Cancers with Demonstrated Utility | Limitations |
|---|---|---|---|
| PD-L1 Expression | Immunohistochemistry (IHC) with various companion diagnostics | NSCLC, melanoma, urothelial cancer, TNBC [61] [60] | Tumor heterogeneity, dynamic expression, sampling bias, variable cutoffs [62] [61] |
| Microsatellite Instability (MSI) | PCR-based analysis or IHC for MMR proteins | Colorectal, endometrial, gastrointestinal cancers (tissue-agnostic) [61] | Low prevalence in some tumor types, technical standardization needs |
| Tumor Mutational Burden (TMB) | Next-generation sequencing (NGS) | Melanoma, lung cancer, bladder cancer (specific contexts) [61] | Lack of standardized cutoff, cost, variability across platforms |
Emerging Next-Generation Predictive Biomarkers
Exosomal PD-L1 (exo-PD-L1)
Exosomal PD-L1 (exo-PD-L1) has emerged as a promising liquid biopsy-based biomarker that addresses several limitations of tissue-based PD-L1 assessment [62]. Tumor-derived exosomes are small extracellular vesicles (30-150 nm) that facilitate intercellular communication by transporting bioactive molecules, including PD-L1, to immune cells [62]. Unlike membrane-bound PD-L1, exosomal PD-L1 is systemically distributed through bodily fluids such as blood and capable of suppressing T-cell activity at distant sites, potentially contributing to systemic immune suppression and resistance to ICIs [62].
The biogenesis of exo-PD-L1 begins with inward invagination of the parent cell's plasma membrane, forming early endosomes that mature into multivesicular bodies (MVBs) containing intraluminal vesicles (ILVs) [62]. During MVB formation, PD-L1 is incorporated into ILVs with preserved topological orientation, exposing the extracellular domain on the exosome surface upon release. This conservation ensures that exo-PD-L1 retains its ability to engage PD-1 receptors on T cells, triggering PD-1-mediated intracellular signaling that inhibits PI3K-AKT and MAPK pathways, thereby restricting T-cell proliferation, activation, and survival [62].
Functionally, exo-PD-L1 demonstrates dynamic responsiveness to inflammatory cues. Interferon-gamma (IFN-γ) significantly stimulates exo-PD-L1 release as an immune evasion mechanism in response to cytokine secretion by CD8+ T cells, macrophages, and natural killer cells [62]. Elevated circulating exo-PD-L1 levels have been associated with poor prognosis, resistance to ICB, and increased tumor burden across various cancers [62]. Notably, removing exosomal PD-L1 enhances sensitivity to anti-PD-L1 therapy in mouse tumor models, suggesting both prognostic and potential therapeutic implications [62].
Table 3: Emerging Next-Generation Predictive Biomarkers for ICI Response
| Biomarker | Type | Detection Method | Advantages |
|---|---|---|---|
| Exosomal PD-L1 | Liquid biopsy | Immunoaffinity capture, ultracentrifugation, microfluidic devices [62] | Dynamic monitoring, systemic immune snapshot, non-invasive |
| T-cell Inflamed GEP | Gene expression profile | RNA sequencing, nanostring technology | Captures tumor microenvironment state, quantitative measurement |
| TIDE | Computational model | RNA sequencing data analysis | Models T-cell dysfunction and exclusion mechanisms |
| PathNetDRP | Network-based biomarker | Integration of PPI networks, pathway data, machine learning [60] | Incorporates biological context, identifies novel biomarkers |
Gene Expression Profiles and Signatures
Gene expression profiling has advanced beyond single-gene biomarkers to encompass multi-gene signatures that capture complex biological processes within the tumor microenvironment. The T-cell-inflamed gene expression profile (GEP) represents one such signature that quantifies the presence of an activated immune microenvironment, including antigen presentation, chemokine expression, T-cell inflammation, and interferon-γ signaling [61]. This signature has demonstrated predictive value for ICI response across multiple cancer types and has shown superior performance compared to PD-L1 IHC in some analyses [61].
The T-cell dysfunction and exclusion gene signature (TIDE) represents another computational approach that models two primary mechanisms of tumor immune evasion: T-cell dysfunction in tumors with high infiltration of cytotoxic T cells, and T-cell exclusion in tumors with low T-cell abundance [61] [60]. TIDE has demonstrated predictive accuracy surpassing that of PD-L1 expression or TMB alone and has identified novel candidate regulators of ICI resistance [60]. Similarly, the IMPRES predictor utilizes pairwise transcriptomic relations between immune checkpoint genes to generate a predictive signature that has achieved high accuracy in melanoma across multiple datasets [60].
Integrated and Network-Based Biomarker Approaches
The recognition that ICI response represents a complex quantitative trait determined by multiple factors has driven the development of integrated biomarker models that combine diverse data types. The PathNetDRP framework represents a novel approach that integrates protein-protein interaction (PPI) networks, biological pathway information, and machine learning to identify functionally relevant biomarkers for ICI response prediction [60]. Unlike conventional methods that focus solely on gene expression differences, PathNetDRP applies the PageRank algorithm to individual pathways to quantify gene contributions within their biological context, enabling more precise biomarker identification [60].
Validation across multiple independent cancer cohorts demonstrated that PathNetDRP achieved strong predictive performance, with area under the receiver operating characteristic curves (AUCs) increasing from 0.780 to 0.940 compared to conventional methods [60]. This framework not only improved predictive accuracy but also provided biological insights into key immune-related pathways, reinforcing its potential for identifying clinically relevant biomarkers [60]. Similarly, combined biomarkers such as TMB+GEP and MPS+TIDE have shown improved predictive outcomes compared to single predictors, highlighting the value of integrated approaches [61].
Experimental Protocols and Methodologies
Detection Methods for Established Biomarkers
PD-L1 Immunohistochemistry Protocol: PD-L1 expression analysis typically follows standardized IHC protocols using validated companion diagnostic assays. The general workflow involves: (1) collection of formalin-fixed paraffin-embedded (FFPE) tumor tissue sections (4-5 μm thickness); (2) deparaffinization and rehydration through xylene and graded alcohol series; (3) antigen retrieval using citrate or EDTA-based buffers at high temperature; (4) peroxidase blocking to quench endogenous peroxidase activity; (5) incubation with primary anti-PD-L1 antibodies (clone-specific for each diagnostic assay); (6) detection using horseradish peroxidase-conjugated secondary antibodies and chromogenic substrates; (7) counterstaining with hematoxylin; and (8) scoring by qualified pathologists using assay-specific criteria (e.g., tumor proportion score or combined positive score) [61] [60].
MSI Testing Protocol: MSI status can be determined through either PCR-based analysis or IHC for MMR proteins. The PCR-based approach involves: (1) DNA extraction from matched tumor and normal tissues; (2) amplification of standardized microsatellite markers (typically including BAT-25, BAT-26, NR-21, NR-24, and MONO-27); (3) fragment analysis by capillary electrophoresis; and (4) interpretation based on the number of unstable markers, with instability at ≥2 markers (≥30% for the 5-marker panel) defining MSI-H status [61]. Alternatively, MMR IHC involves staining for MLH1, MSH2, MSH6, and PMS2 proteins, with loss of nuclear expression in tumor cells indicating dMMR status [61].
TMB Measurement Protocol: Tumor mutational burden assessment requires: (1) DNA extraction from FFPE tumor tissue and matched normal specimens; (2) library preparation using comprehensive genomic panels covering at least 1 Mb of coding genome; (3) next-generation sequencing at appropriate depth (typically >500x coverage); (4) bioinformatic processing including alignment, variant calling, and filtering to remove germline polymorphisms and driver mutations; (5) calculation of TMB as the total number of nonsynonymous somatic mutations per megabase of genome sequenced; and (6) classification using validated cutoffs (typically ~10 mut/Mb, though this varies by cancer type and assay) [61].
Emerging Biomarker Detection Workflows
Exosomal PD-L1 Isolation and Detection: The protocol for exo-PD-L1 analysis includes: (1) collection of plasma or serum samples (typically 1-10 mL); (2) preprocessing by centrifugation at 2,000-3,000 × g to remove cells and debris; (3) exosome isolation using ultracentrifugation (100,000-120,000 × g for 70-120 minutes), size-exclusion chromatography, or immunoaffinity capture; (4) characterization of exosomes by nanoparticle tracking analysis, transmission electron microscopy, or Western blotting for exosomal markers (CD63, CD81, TSG101); (5) exo-PD-L1 quantification using enzyme-linked immunosorbent assay (ELISA), flow cytometry with bead-based capture, or microfluidic devices; and (6) data normalization to total exosome protein content or particle number [62].
Network-Based Biomarker Analysis (PathNetDRP): The PathNetDRP framework implements a multi-step computational protocol: (1) ICI-related gene selection via PageRank algorithm applied to protein-protein interaction networks; (2) identification of ICI-related biological pathways through hypergeometric testing of candidate genes; (3) calculation of PathNetGene scores using network analysis on identified pathways; (4) biomarker selection based on statistical significance and biological relevance; and (5) validation through cross-validation and independent cohort analysis [60]. This approach integrates multiple data types, including ICI target information, PPI networks, pathway databases, and gene expression data from ICI-treated patients to identify robust predictive biomarkers [60].
Figure 1: PathNetDRP Biomarker Discovery Workflow. This diagram illustrates the computational framework for identifying predictive biomarkers through integration of network biology and machine learning approaches [60].
The Scientist's Toolkit: Essential Research Reagents and Technologies
Table 4: Essential Research Reagents and Technologies for Biomarker Discovery
| Reagent/Technology | Function | Application Examples |
|---|---|---|
| FFPE Tissue Sections | Preserves tissue architecture and biomolecules for histological analysis | PD-L1 IHC, DNA extraction for TMB and MSI analysis [61] |
| Companion Diagnostic Anti-PD-L1 Antibodies | Specific detection of PD-L1 protein in tissue sections | PD-L1 IHC using validated assays (e.g., 22C3, 28-8, SP142 clones) [61] [60] |
| Next-Generation Sequencing Panels | Comprehensive genomic profiling for mutation detection | TMB calculation, MSI status determination through genomic analysis [61] |
| Exosome Isolation Kits | Enrichment of extracellular vesicles from biofluids | Exosomal PD-L1 isolation from plasma/serum samples [62] |
| Protein-Protein Interaction Databases | Curated network data for computational biology | PathNetDRP framework for biomarker discovery [60] |
| Gene Expression Profiling Platforms | Genome-wide transcriptome analysis | T-cell inflamed GEP, TIDE signature calculation [61] [60] |
Figure 2: PD-1/PD-L1 Signaling Pathway. This diagram illustrates the molecular mechanisms of T-cell inhibition through PD-1/PD-L1 interaction, highlighting key signaling pathways affected by immune checkpoint activation [59].
Comparative Performance of Biomarker Technologies
The predictive accuracy of biomarker technologies varies considerably based on cancer type, analytical platform, and patient population. Direct comparison studies have revealed that each biomarker class demonstrates distinctive strengths and limitations in clinical application.
Established Biomarkers: PD-L1 IHC shows variable performance across cancer types, with generally higher positive predictive value in NSCLC compared to other malignancies [61]. MSI status demonstrates exceptional predictive value with response rates exceeding 50% in MSI-H tumors across multiple histologies, but its low prevalence in many common cancers limits its overall utility [61]. TMB shows more continuous correlation with response outcomes, with optimal thresholds varying across cancer types and technical platforms [61]. Importantly, these three FDA-approved biomarkers identify predominantly non-overlapping patient populations, suggesting they capture distinct biological aspects of ICI response [61].
Emerging Biomarkers: Comparative analyses indicate that gene expression signatures such as T-cell inflamed GEP and TIDE demonstrate superior predictive performance compared to PD-L1 IHC alone [61]. The TIDE model has shown better accuracy than either PD-L1 expression or TMB alone in multiple cancer types [60]. Similarly, the melanocytic plasticity signature (MPS) has demonstrated superior predictive performance compared to PD-L1, TMB, and TIDE in melanoma [61]. Integrated approaches that combine multiple biomarkers, such as TMB+GEP and MPS+TIDE, have consistently shown improved predictive outcomes compared to individual biomarkers, highlighting the value of multi-analyte assessment [61].
Network-Based Approaches: The PathNetDRP framework has demonstrated robust predictive performance across multiple independent cancer cohorts, with cross-validation AUCs increasing from 0.780 to 0.940 compared to conventional differential expression analysis [60]. This improvement highlights the value of incorporating biological network information and pathway context into biomarker discovery. Additionally, network-based approaches have identified novel biomarker candidates with strong predictive performance, expanding the repertoire of potential biomarkers beyond those identified through conventional methods [60].
The rapidly evolving landscape of predictive biomarkers for immune checkpoint inhibitors reflects both the complexity of anti-tumor immunity and the innovative approaches being developed to decipher it. While established biomarkers such as PD-L1, MSI, and TMB provide foundational tools for patient selection, their limitations have catalyzed the development of next-generation approaches that capture broader biological dimensions of treatment response.
The integration of multi-analyte biomarkers represents a promising direction for enhancing predictive accuracy. Combined biomarkers such as TMB+GEP and MPS+TIDE have demonstrated improved performance compared to individual predictors, suggesting that comprehensive assessment of tumor-immune system interactions requires multidimensional measurement [61]. Similarly, network-based approaches like PathNetDRP that incorporate biological pathway information and protein-protein interactions have shown enhanced predictive capability while providing mechanistic insights into ICI response mechanisms [60].
The emergence of liquid biopsy-based biomarkers, particularly exosomal PD-L1, addresses critical limitations of tissue-based assessment, including spatial heterogeneity, dynamic changes in expression, and practical constraints of serial tissue sampling [62]. The ability to monitor biomarker status throughout treatment courses provides opportunities for dynamic adaptation of therapy and early detection of resistance mechanisms.
Future biomarker development will likely focus on increasingly sophisticated integration of multi-omics data, refined computational modeling of tumor-immune interactions, and standardization of analytical and reporting frameworks across platforms. Additionally, the application of artificial intelligence and machine learning to extract latent patterns from complex biomarker data holds promise for further enhancing predictive accuracy. As these technologies mature, the vision of truly personalized immunotherapy selection based on comprehensive biomarker assessment moves closer to clinical reality.
The study of pathway-specific biomarkers has revolutionized our understanding of complex disease mechanisms, particularly in autoimmune and inflammatory conditions. The concepts of intrinsic and extrinsic pathway activation provide a powerful framework for dissecting disease pathogenesis, stratification, and therapeutic targeting. In autoimmune contexts, these pathways reveal distinct molecular signatures that drive phenotypic diversity, while in sepsis-induced coagulopathy, they delineate the complex interplay between inflammation and hemostasis. This review employs a comparative approach to analyze pathway-specific biomarkers in systemic sclerosis (SSc) and sepsis-induced coagulopathy (SIC), two conditions characterized by dysregulated intrinsic and extrinsic activation patterns. By examining these disparate conditions through the unifying lens of pathway biomarkers, we identify common principles of disease classification, prognostic assessment, and targeted intervention that advance the field of precision medicine.
Biomarker Classification and Pathophysiological Significance
Table 1: Comparative Analysis of Intrinsic and Extrinsic Pathway Biomarkers
| Feature | Systemic Sclerosis (Autoimmunity) | Sepsis-Induced Coagulopathy (Coagulation) |
|---|---|---|
| Extrinsic Pathway Initiators | Anti-topoisomerase I antibodies, Anti-RNA polymerase III antibodies, Environmental triggers [64] | Tissue factor exposure, Bacterial endotoxins, Monocyte activation [65] [66] |
| Intrinsic Pathway Components | HLA-DR/DQ alleles, IRF5, STAT4, IRAK1 genetic factors [67] [64] | Neutrophil extracellular traps (NETs), Platelet activation, Contact pathway [65] [66] |
| Key Amplification Mechanisms | TGF-β signaling, IL-6, Endothelin-1, PDGFR activation [64] [68] | Thrombin generation, Cytokine release (TNF-α, IL-6), Phosphatidylserine exposure [65] [66] |
| Regulatory Checkpoints | PPAR-γ, Adiponectin, Maresin 1 [64] | Antithrombin, Protein C, TFPI [65] |
| End-Organ Damage Markers | KL-6, Surfactant protein-D (lung), NT-proBNP (cardiac) [64] | Lactate (perfusion), Organ-specific enzymes (e.g., creatinine) [66] |
The conceptual framework of intrinsic and extrinsic pathways, while originating in apoptosis research [9], provides valuable models for understanding disease mechanisms across pathophysiology. In systemic sclerosis, the dichotomy manifests through distinct autoimmune activation patterns, where extrinsic factors include environmental triggers and intrinsic factors encompass genetic susceptibility elements. Similarly, in sepsis-induced coagulopathy, the extrinsic pathway represents the primary tissue factor-mediated initiation of coagulation, while intrinsic components contribute to amplification and propagation of thrombotic responses through neutrophil and platelet activation [65] [66]. This classification system enables researchers to categorize biomarkers based on their position within pathogenic cascades, facilitating more targeted diagnostic and therapeutic approaches.
Systemic Sclerosis: Pathway Biomarkers and Clinical Correlations
Autoantibodies as Extrinsic Pathway Biomarkers
In systemic sclerosis, autoantibodies represent the most well-validated extrinsic pathway biomarkers with direct clinical utility. Anti-nuclear antibodies (ANA) are present in more than 90% of SSc patients, with specific patterns conferring distinct phenotypic associations [64]. Anti-centromere antibodies define a patient subset characterized by limited cutaneous involvement, increased risk of pulmonary arterial hypertension, and generally better prognosis. In contrast, anti-topoisomerase I antibodies (anti-Scl-70) predict diffuse cutaneous involvement, rapid skin thickness progression, and significant interstitial lung disease [64]. Anti-RNA polymerase III antibodies represent another critical extrinsic biomarker associated with rapid skin progression, gastric antral vascular ectasia, scleroderma renal crisis, and an increased risk of synchronous cancer [64]. These autoantibodies are mutually exclusive in most cases, suggesting distinct pathogenic pathways with different genetic and environmental triggers.
Genetic and Molecular Biomarkers of Intrinsic Activation
The intrinsic pathway in SSc is governed by genetic susceptibility elements and downstream molecular effectors. HLA class II alleles (HLA-DRB1, HLA-DQB1) constitute primary genetic biomarkers, with specific polymorphisms determining autoantibody specificity and disease susceptibility [64]. Beyond the MHC complex, non-HLA genetic factors including IRF5, STAT4, and CD226 polymorphisms influence innate and adaptive immune signaling, tilting the balance toward pro-fibrotic responses [67] [64]. At the molecular level, TGF-β stands as a master regulator of fibrosis, activating downstream Smad signaling and connective tissue growth factor (CTGF) that collectively drive fibroblast activation and extracellular matrix deposition [64] [68]. Interleukin signaling networks (particularly IL-6, IL-17, IL-22) further amplify the intrinsic pro-fibrotic response, while deficient PPAR-γ signaling and reduced adiponectin levels impair natural resolution mechanisms [64].
Table 2: Experimental Methodologies for SSc Biomarker Detection
| Methodology | Biomarkers Detected | Protocol Overview | Applications in SSc |
|---|---|---|---|
| Immunofluorescence/ANA Testing | Anti-centromere, Anti-topoisomerase I, Anti-RNA polymerase III | HEp-2 cell substrate, patient serum incubation, fluorescent conjugate detection [64] | Diagnosis, classification, prognosis |
| ELISA/Multiplex Assays | TGF-β, IL-6, IL-17, Adiponectin, KL-6 | Solid-phase antibody capture, enzymatic or fluorescent detection, standard curve quantification [64] [68] | Disease activity monitoring, treatment response |
| Genetic Sequencing | HLA alleles, IRF5, STAT4 polymorphisms | DNA extraction, PCR amplification, Sanger or next-generation sequencing [64] | Risk stratification, personalized medicine |
| Nailfold Capillaroscopy | Microvascular abnormalities | Microscopic examination of nailfold capillaries, pattern classification [64] | Early diagnosis, vascular assessment |
| RNA Expression Profiling | Transcriptional signatures, miRNA patterns | RNA extraction, microarray or RNA-seq, bioinformatic analysis [68] | Molecular phenotyping, novel biomarker discovery |
Experimental Models and Methodological Approaches
Research into SSc biomarkers employs sophisticated methodological approaches ranging from traditional immunoassays to cutting-edge multi-omics technologies. Autoantibody detection remains foundational, typically using indirect immunofluorescence on HEp-2 cells with pattern recognition and specific confirmatory assays [64]. For cytokine and chemokine profiling, multiplex ELISA platforms enable simultaneous quantification of numerous inflammatory mediators in limited sample volumes, revealing distinct signatures associated with disease subsets [64]. Genetic studies utilize genome-wide association approaches followed by targeted sequencing to identify susceptibility loci and potential therapeutic targets. Functional validation of biomarkers often employs in vitro systems of fibroblast activation or endothelial dysfunction, coupled with animal models that recapitulate specific aspects of scleroderma pathology. Emerging technologies focus on extracellular vesicle characterization, microRNA profiling, and metabolomic approaches that offer novel insights into disease mechanisms and potential diagnostic applications [68].
Sepsis-Induced Coagulopathy: Pathway-Specific Diagnostic and Prognostic Markers
Extrinsic Pathway Initiation in SIC
Sepsis-induced coagulopathy initiates primarily through extrinsic pathway activation, with tissue factor (TF) serving as the cornerstone biomarker and mechanistic driver [65] [66]. Under physiological conditions, TF is sequestered from circulation, but during sepsis, microbial pathogens and inflammatory cytokines induce its expression on monocytes, endothelial cells, and neutrophil-derived extracellular vesicles. TF exposure triggers the extrinsic coagulation cascade through factor VII activation, generating thrombin and fibrin deposition [66]. Bacterial endotoxins, particularly lipopolysaccharide from Gram-negative organisms, further amplify this process through direct monocyte activation and cytokine release [65]. The central role of TF makes it a theoretically attractive biomarker; however, technical challenges in measurement have limited its routine clinical application. Instead, downstream markers of TF activity, including soluble fibrin and fibrin monomers, provide practical alternatives for assessing extrinsic pathway activation in septic patients [66].
Intrinsic and Amplification Mechanisms in Coagulopathy
While the extrinsic pathway initiates coagulation in sepsis, intrinsic and amplification mechanisms significantly contribute to disease progression. Neutrophil extracellular traps (NETs) represent a crucial intrinsic component, releasing prothrombotic DNA-histone complexes that activate platelets and provide scaffolding for fibrin deposition [65] [66]. The contact activation system (intrinsic pathway) may further augment coagulation through factor XII activation by polyphosphates and NET components [66]. Simultaneously, natural anticoagulant pathways become suppressed, with marked reductions in antithrombin, protein C, and tissue factor pathway inhibitor (TFPI) creating a prothrombotic milieu [65]. Fibrinolytic shutdown represents another critical amplification mechanism, driven by increased plasminogen activator inhibitor-1 (PAI-1) that tips the balance toward fibrin accumulation and microvascular thrombosis [66]. These interconnected processes create a self-amplifying cycle of coagulation and inflammation that characterizes progressive SIC.
Table 3: Laboratory Biomarkers in Sepsis-Induced Coagulopathy
| Biomarker Category | Specific Markers | Pathophysiological Significance | Diagnostic Utility |
|---|---|---|---|
| Traditional Coagulation Tests | Platelet count, Prothrombin time (PT), Fibrinogen | Consumption coagulopathy, factor deficiency | ISTH DIC scoring, SIC identification [65] [66] |
| Fibrinolysis Markers | D-dimer, Fibrin degradation products (FDP), PAI-1 | Fibrin formation and degradation, fibrinolysis shutdown | Diagnosis, prognosis, mortality risk [66] |
| Cell-Specific Markers | Neutrophil extracellular traps (NETs), Monocyte tissue factor, Platelet microparticles | Cellular activation, immunothrombosis | Early detection, mechanistic insight [65] [66] |
| Anticoagulant Factors | Antithrombin, Protein C, Thrombomodulin | Natural anticoagulant depletion, endothelial damage | Severity assessment, therapeutic monitoring [65] |
| Emerging Biomarkers | Soluble fibrin, Thrombin-antithrombin complexes, Viscoelastic parameters | Real-time coagulation status, hypercoagulability | Early diagnosis, treatment guidance [66] |
Diagnostic Approaches and Scoring Systems
The diagnosis of sepsis-induced coagulopathy integrates multiple biomarkers through validated scoring systems that enhance prognostic accuracy. The International Society on Thrombosis and Haemostasis (ISTH) recommends a two-step approach beginning with the sepsis-induced coagulopathy (SIC) score, followed by the overt DIC score for positive cases [66]. The SIC score incorporates platelet count, prothrombin time (PT), and Sequential Organ Failure Assessment (SOFA) score, enabling earlier identification of coagulopathy before progression to irreversible DIC [66]. Traditional biomarkers including platelet count, PT, and fibrinogen provide fundamental assessment but lack specificity individually. D-dimer has emerged as a more specific fibrin degradation marker, with elevated levels reflecting both coagulation activation and fibrinolysis [66]. Viscoelastic testing such as thromboelastography offers a holistic assessment of clot formation, strength, and dissolution, providing real-time functional evaluation that complements conventional tests [65]. The integration of these multimodal approaches facilitates timely diagnosis and risk stratification.
Comparative Analysis of Experimental Approaches
Methodological Commonalities and Distinctions
Despite studying different disease processes, SSc and SIC biomarker research share important methodological principles while employing distinct technical approaches. Both fields utilize multi-parameter scoring systems that integrate clinical and laboratory parameters for classification and prognostication. However, SSc research emphasizes autoantibody profiling and genetic markers for long-term risk stratification, while SIC investigations prioritize rapid-turnaround coagulation tests for immediate therapeutic decisions. Technologically, ELISA and related immunoassays represent workhorse methodologies in both fields, though SSc research increasingly incorporates genomic, transcriptomic, and proteomic platforms for biomarker discovery [68], while SIC studies focus on functional coagulation assays and viscoelastic testing [66]. Validation approaches differ accordingly, with SSc biomarkers requiring long-term observational studies to establish prognostic value, while SIC biomarkers necessitate rapid assessment of predictive value for thrombotic events and mortality.
Research Reagent Solutions for Pathway Biomarker Investigation
Table 4: Essential Research Reagents for Pathway Biomarker Studies
| Reagent Category | Specific Examples | Research Applications | Technical Considerations |
|---|---|---|---|
| Antibody Reagents | Anti-human cytokine antibodies, Phospho-specific antibodies, Flow cytometry panels | Protein detection, signaling pathway analysis, cell phenotyping | Validation required, species cross-reactivity [64] [9] |
| ELISA/Multiplex Kits | Cytokine panels, Autoantibody detection, Coagulation factor assays | Biomarker quantification, patient stratification | Standard curve accuracy, dynamic range [64] [66] |
| Genetic Analysis Tools | PCR primers, Sequencing panels, SNP detection assays | Genetic association studies, mutation screening | Quality control, contamination prevention [64] |
| Functional Assay Kits | Caspase activity assays, Thrombin generation tests, Mitochondrial membrane potential dyes | Pathway activation assessment, drug screening | Appropriate controls, timing optimization [66] [9] |
| Cell Culture Models | Primary fibroblasts, Endothelial cells, Monocyte cell lines | Mechanistic studies, therapeutic testing | Passage number effects, activation state [64] [68] |
The comparative analysis of pathway biomarkers in systemic sclerosis and sepsis-induced coagulopathy reveals convergent principles despite distinct pathophysiology. Both conditions demonstrate the critical importance of differentiating intrinsic and extrinsic pathway activation for precise disease classification, prognostication, and therapeutic targeting. In SSc, this paradigm enables stratification of patients based on autoantibody profiles and genetic susceptibility markers, while in SIC, it facilitates timed interventions based on specific coagulation pathway involvement. Emerging technological approaches including multi-omics profiling, extracellular vesicle analysis, and digital biomarker development show promise across both fields [69] [68]. The future of pathway biomarker research lies in integrated multimodal assessment that combines traditional assays with novel platforms to create comprehensive molecular signatures. These advances will enable truly personalized medicine approaches, targeting specific pathway components based on individual patient biomarker profiles to improve outcomes in these complex conditions.
Systemic Sclerosis Biomarker Pathways
Sepsis-Induced Coagulopathy Pathways
The study of complex biological systems requires a move beyond single-layer analysis. Multi-omics integration represents a transformative approach in biomedical research, simultaneously analyzing data from multiple molecular layers—including genomics, transcriptomics, and proteomics—to construct comprehensive models of biological activity. Within apoptosis research, this approach is particularly valuable for elucidating the complex interplay between intrinsic and extrinsic pathway activation, revealing how genetic predispositions, gene expression regulation, and protein-level execution collectively govern programmed cell death. This holistic view is crucial for identifying robust biomarkers and therapeutic targets in cancer and neurodegenerative diseases where apoptotic dysregulation is a key pathological feature.
Two primary computational strategies have emerged for integrating these diverse datasets: multi-staged integration and meta-dimensional integration. Multi-staged approaches analyze data sequentially, where findings from one omics layer (e.g., genomic variants) inform the analysis of another (e.g., transcript expression), making them particularly suited for identifying driver genes and establishing causal relationships in apoptotic pathways. In contrast, meta-dimensional methods combine all data types simultaneously to create new composite biomarkers or sample classifications, potentially revealing novel apoptotic subtypes based on coordinated molecular profiles across layers [70]. The choice between these strategies depends heavily on the research objective, whether identifying key regulatory genes like BID in intracerebral hemorrhage apoptosis [12] or establishing clinically relevant tumour classifications.
Comparative Analysis of Multi-Omics Integration Tools
Selecting appropriate computational tools is fundamental to successful multi-omics integration. A comprehensive comparative study of nine integration tools using real and simulated cancer datasets provides critical performance insights [70]. The evaluation assessed multi-staged integration tools at the gene, function, and pathway levels, while meta-dimensional tools were judged on sample classification accuracy. Performance varies significantly based on data representation, sample size, and signal-to-noise ratio, necessitating careful tool selection based on specific research goals and data characteristics.
Table 1: Comparison of Multi-Staged Integration Tools for Driver Gene Identification
| Tool Name | Primary Analysis Objective | Omics Layers Integrated | Performance Assessment | Key Strengths |
|---|---|---|---|---|
| Similarity Network Fusion (SNF) | Sample classification based on molecular patterns | Genomic, transcriptomic, proteomic, epigenomic | High accuracy in tumour subtyping | Robust to noise, identifies patient subgroups |
| iClusterPlus | Latent variable identification for subtyping | Multiple omics data types | Effective sample clustering | Handles different data types effectively |
| Multi-omics Factor Analysis (MOFA) | Latent factor representation | Multiple omics data types | Captures major sources of variation | Interpretable factors, handles missing data |
| Painter | Gene-level multi-omics data visualization | Genomic, transcriptomic | Effective gene-level integration | Intuitive visual output for candidate genes |
Table 2: Meta-Dimensional Integration Tools for Sample Classification
| Tool Name | Classification Approach | Data Input Requirements | Performance Metrics | Optimal Use Cases |
|---|---|---|---|---|
| Regularized Generalized Canonical Correlation Analysis (RGCCA) | Multiblock component analysis | Multiple omics datasets | High classification accuracy | Small sample sizes, strong signals |
| Integrative Bayesian Analysis | Probabilistic modeling | Multiple omics datasets | Robust to noise | Uncertainty quantification |
| Multi-Kernel Learning | Kernel-based integration | Multiple similarity matrices | Excellent with large samples | Complex non-linear relationships |
Beyond these established tools, emerging visualization packages like MultiModalGraphics enhance interpretability by creating annotated scatterplots and heatmaps with embedded statistical summaries such as fold-changes, p-values, and q-values [71]. This R/Bioconductor package interoperates seamlessly with analytical frameworks like limma and voom, streamlining workflows from raw data processing to publication-ready visualization of multi-omics data.
Experimental Protocols for Multi-Omics Validation in Apoptosis Research
Integrated Spatial Multi-Omics Workflow
A groundbreaking 2025 study demonstrated a novel wet-lab and computational framework for performing spatial transcriptomics and spatial proteomics on the same tissue section, overcoming limitations of adjacent section analysis [72]. This protocol ensures perfect spatial registration between molecular layers:
- Sample Preparation: Human lung carcinoma FFPE tissue sections (5µm) are mounted on appropriate slides.
- Sequential Multi-Omics Processing:
- Spatial Transcriptomics: Process with Xenium In Situ gene expression platform using a targeted gene panel (e.g., 289-gene human lung cancer panel) following manufacturer protocols [72].
- Spatial Proteomics: Following Xenium, perform hyperplex immunohistochemistry (hIHC) using the COMET platform with off-the-shelf primary antibodies for 40 protein markers [72].
- Histological Staining: Conduct manual H&E staining on the post-processed section.
- Computational Integration:
- Co-register DAPI images from Xenium and COMET to the H&E image using a non-rigid spline-based algorithm in Weave software.
- Apply cell segmentation masks (CellSAM for proteomics, DAPI expansion for transcriptomics) to associate transcript spots and protein intensities with individual cells.
- Generate unified dataset containing both transcript counts and protein marker intensities per cell.
- Downstream Analysis:
- Calculate Spearman correlation between transcript and protein levels for markers present in both modalities.
- Perform dimension reduction (UMAP) and Louvain clustering on integrated data.
- Conduct region-specific analysis of immune and tumour markers using pathology annotations.
This integrated approach revealed systematically low transcript-protein correlations at cellular resolution, highlighting the importance of multi-layer validation in biomarker studies [72].
Apoptosis-Focused Multi-Omics Analysis Protocol
Research identifying BID as a key apoptosis biomarker following intracerebral hemorrhage demonstrates a bioinformatics-focused approach [12]:
- Data Acquisition:
- Obtain human ICH and normal brain datasets from GEO database (accessions: GSE24265, GSE171144, GSE43618).
- Identify differentially expressed genes (DEGs) using GEO2R with parameters |log2(fold change)| > 1 and p-value < 0.05.
- Functional Enrichment:
- Perform GO and KEGG pathway enrichment on DEGs using clusterProfiler.
- Conduct Gene Set Enrichment Analysis (GSEA) to identify apoptosis-related pathways.
- Apoptosis-Specific Analysis:
- Cross-reference DEGs with apoptosis gene sets from MSigDB to identify apoptosis-related DEGs.
- Construct Protein-Protein Interaction (PPI) networks and apply MECDE algorithm to identify hub genes (e.g., BID).
- Experimental Validation:
- Establish rat ICH model through autologous blood injection into basal ganglia.
- Validate BID protein expression changes via Western blot and immunofluorescence.
- Confirm apoptotic activity through TUNEL staining in ICH versus sham groups.
- Regulatory Network Analysis:
- Construct mRNA-miRNA interaction networks to identify potential regulators (e.g., miR-1225-3p) of key apoptosis genes.
Visualization of Multi-Omics Data and Apoptosis Pathways
Effective visualization is crucial for interpreting complex multi-omics data and apoptotic signaling networks. The following diagrams adhere to specified color contrast requirements using the approved palette and Graphviz DOT language.
Multi-Omics Integration Strategy Workflow
Intrinsic and Extrinsic Apoptosis Pathways Integration
Essential Research Reagent Solutions for Multi-Omics Apoptosis Studies
Successful multi-omics integration requires carefully selected reagents and platforms optimized for preserving biomolecular integrity across analytes. The following table details essential research reagents and their functions in apoptosis-focused multi-omics studies.
Table 3: Research Reagent Solutions for Multi-Omics Apoptosis Studies
| Reagent/Platform | Specific Function | Application in Apoptosis Research |
|---|---|---|
| Xenium In Situ | Targeted spatial transcriptomics with 289-gene panels | Mapping expression of apoptosis genes (BID, BAX, BCL-2) in tissue context [72] |
| COMET Hyperplex IHC | Spatial proteomics with 40-plex protein detection | Quantifying protein levels of caspase cleavage, death receptors in same section [72] |
| Lunaphore COMET | Automated sequential immunofluorescence staining | High-throughput spatial proteomics for apoptotic marker validation [72] |
| CellSAM | Deep learning-based cell segmentation | Integrating nuclear (DAPI) and membrane (PanCK) markers for precise cellular analysis [72] |
| Weave Software | Multi-omics data registration and visualization | Co-registering ST, SP, and H&E data for single-cell correlation analysis [72] |
| ArchR | scRNA-seq integration with epigenomic data | Transferring cell type labels between transcriptomic and epigenomic datasets [73] |
| MultiModalGraphics R Package | Statistical annotation embedding in visualizations | Creating publication-ready heatmaps and scatterplots with p-values and fold-changes [71] |
| TUNEL Assay Kits | Fluorescent detection of DNA fragmentation | Gold-standard validation of apoptotic cells in tissue sections [12] |
| MSigDB Apoptosis Gene Sets | Curated collections of apoptosis-related genes | Reference standards for pathway enrichment analysis in omics data [12] |
| Phospho-Specific Antibodies | Detection of activated signaling molecules | Assessing phosphorylation status of key apoptotic regulators (e.g., BID) [12] |
Multi-omics integration represents a paradigm shift in apoptosis research, moving beyond single-layer analysis to reveal the complex interplay between genetic predisposition, transcriptional regulation, and protein-level execution of cell death programs. The comparative analysis presented here demonstrates that tool selection must align with specific research objectives, with multi-staged approaches excelling at driver gene identification like BID in ICH models [12], while meta-dimensional methods provide superior sample classification power [70]. Emerging technologies enabling true simultaneous spatial multi-omics on single tissue sections [72] address critical limitations of adjacent section analysis, while sophisticated visualization packages enhance interpretability of complex datasets [71]. As these methodologies continue to mature, multi-omics integration will increasingly enable the identification of master apoptotic regulators and context-specific biomarkers, ultimately advancing targeted therapeutic development for cancer, neurodegenerative disorders, and other conditions characterized by apoptotic dysregulation.
Navigating Complexity: Challenges, Pitfalls, and Strategy Optimization
Addressing Specificity and Sensitivity Limitations in Traditional Biomarker Assays
In the pursuit of precision medicine, biomarkers serve as essential tools for disease detection, diagnosis, prognosis, and predicting response to therapeutic interventions. A biomarker is formally defined as "a defined characteristic that is measured as an indicator of normal biological processes, pathogenic processes, or biological responses to an exposure or intervention" [74]. However, traditional biomarker assays often face significant limitations in their sensitivity (the ability to correctly identify true positives) and specificity (the ability to correctly identify true negatives). These limitations can lead to false diagnoses, missed treatment opportunities, and ineffective monitoring of disease progression.
The context of intrinsic and extrinsic pathway activation research provides a compelling framework for exploring these challenges. Whether examining coagulation cascades, apoptotic pathways, or immune response mechanisms, the fundamental principle remains that pathway activation biomarkers represent a more robust approach compared to single-molecule measurements. Unlike conventional biomarkers that often rely on individual molecules, pathway activation levels quantitatively capture the collective behavior of multiple pathway components, offering enhanced specificity and sensitivity for assessing biological processes [75]. This article examines the limitations of traditional biomarker assays and explores emerging solutions through the lens of pathway activation biomarkers, providing experimental data and methodological details to guide researchers in this evolving field.
Comparative Analysis of Biomarker Assay Performance
Performance Metrics for Biomarker Evaluation
The evaluation of biomarker assays requires multiple statistical metrics to comprehensively assess their clinical utility. Sensitivity measures the proportion of true positives correctly identified by the test, while specificity measures the proportion of true negatives correctly identified [74]. Positive predictive value (PPV) indicates the proportion of test-positive patients who actually have the disease, and negative predictive value (NPV) represents the proportion of test-negative patients who truly do not have the disease. Both PPV and NPV are influenced by disease prevalence. The diagnostic odds ratio (DOR) combines sensitivity and specificity into a single metric, with higher values indicating better discriminatory power [76]. Discrimination, often measured by the area under the receiver operating characteristic (ROC) curve (AUC), quantifies how well a marker distinguishes cases from controls, with values ranging from 0.5 (no better than chance) to 1.0 (perfect discrimination) [74].
Limitations of Single-Marker Approaches
Traditional biomarker assays often focus on single molecules, which inherently limits their sensitivity and specificity due to biological complexity and heterogeneity. For instance, in predicting response to PD-1/PD-L1 immunotherapy, standalone PD-L1 immunohistochemistry (IHC) demonstrates variable performance across different tumor types and is influenced by pathologist experience and scoring methods [76]. Similarly, in diagnosing periprosthetic joint infection (PJI), conventional tests like erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) show sensitivity and specificity ranging from 68% to 95% and 66% to 88%, respectively, leaving considerable room for diagnostic error [77].
Table 1: Performance of Traditional Biomarker Assays in Various Clinical Contexts
| Biomarker Assay | Clinical Context | Sensitivity | Specificity | Limitations |
|---|---|---|---|---|
| ESR and CRP [77] | Periprosthetic Joint Infection Diagnosis | 68-95% | 66-88% | Limited accuracy for definitive diagnosis |
| PD-L1 IHC [76] | Predicting Immunotherapy Response | Variable by tumor type | Variable by tumor type | Influenced by pathologist experience and scoring methods |
| Synovial Fluid Cell Count [77] | Periprosthetic Joint Infection Diagnosis | 68% | 95% | Requires additional confirmatory tests |
| TMB (Tumor Mutational Burden) [76] | Predicting Immunotherapy Response | Varies by platform and thresholds | Varies by platform and thresholds | Lacks standardized thresholds across platforms |
Enhanced Performance of Pathway-Based and Multi-Marker Approaches
Pathway-based biomarkers and multi-marker panels demonstrate superior performance by capturing the integrated activity of multiple pathway components rather than relying on single molecules. The alpha defensin-1 (AD-1) biomarker assay for periprosthetic joint infection diagnosis exemplifies this advancement, showing 100% sensitivity and 95% specificity in a comparative study, outperforming traditional tests like ESR, CRP, and synovial fluid cell count [77]. Similarly, in immuno-oncology, multiplex immunohistochemistry/immunofluorescence (mIHC/IF) demonstrates enhanced sensitivity (0.76) for predicting response to PD-1/PD-L1 inhibitors, while microsatellite instability (MSI) testing shows high specificity (0.90) [76].
Table 2: Performance of Advanced and Multi-Marker Assays
| Biomarker Assay | Clinical Context | Sensitivity | Specificity | Advantages |
|---|---|---|---|---|
| Alpha Defensin-1 (AD-1) [77] | Periprosthetic Joint Infection Diagnosis | 100% | 95% | Superior to traditional tests |
| Multiplex IHC/IF (mIHC/IF) [76] | Predicting Immunotherapy Response | 0.76 | Moderate | High sensitivity for tumor microenvironment analysis |
| Microsatellite Instability (MSI) [76] | Predicting Immunotherapy Response | Moderate | 0.90 | High specificity, especially in gastrointestinal tumors |
| Combined PD-L1 IHC + TMB [76] | Predicting Immunotherapy Response | 0.89 | Improved over single markers | Enhanced predictive power through complementary mechanisms |
The integration of multiple biomarkers into panels further enhances diagnostic performance. Combined approaches, such as PD-L1 IHC with tumor mutational burden (TMB), achieve sensitivity of 0.89, significantly improving upon individual marker performance [76]. This synergy occurs because different biomarkers often capture complementary aspects of disease biology, providing a more comprehensive assessment than any single marker could achieve alone.
Experimental Assessment of Pathway Activation Biomarkers
Methodologies for Pathway Activation Quantification
The quantitative assessment of pathway activation represents a paradigm shift from traditional biomarker approaches. Pathway activation levels (PALs) are calculated by integrating data from multiple measurable pathway components, typically using high-throughput proteomic or transcriptomic profiles [75]. The fundamental principle involves measuring concentration changes across all measurable pathway components and integrating these measurements into a single quantitative score that reflects the overall pathway activation state. This approach leverages enclosed bioinformatic algorithms that transform expression data into pathway activation scores, which take both positive and negative values corresponding to the extent of pathway upregulation or downregulation [75].
The experimental workflow typically begins with sample preparation from relevant tissues or biofluids, followed by RNA or protein extraction. For transcriptomic analysis, platforms such as RNA sequencing (RNA-Seq) or DNA microarrays are employed to generate gene expression profiles. For proteomic analysis, mass spectrometry or immunoassays are used to quantify protein levels. The resulting data undergoes quality control and normalization before being processed through specialized bioinformatic algorithms that calculate pathway activation scores based on predefined molecular signatures [75]. These algorithms typically employ mathematical models that weight individual component contributions according to their importance within the pathway architecture.
Alpha Defensin-1 Assay Protocol
The alpha defensin-1 assay for periprosthetic joint infection (PJI) diagnosis provides a compelling case study of a successful pathway-informed biomarker assay. The experimental protocol involves collecting synovial fluid from the potentially infected joint through aspiration under aseptic conditions [77]. The sample is processed using a proprietary immunoassay that detects human alpha defensin-1 peptides. The test can be performed qualitatively using a lateral flow technology with results available within 10 minutes, or quantitatively using liquid chromatography-mass spectrometry (LC-MS) for higher precision [77].
In the validation study, the assay was performed on 61 synovial fluid samples from 57 patients undergoing evaluation for PJI. The diagnosis was established using the Musculoskeletal Infection Society (MSIS) criteria as the reference standard. Among 19 confirmed infections, the AD-1 assay demonstrated 100% sensitivity (95% CI: 79%-100%) and 95% specificity (95% CI: 83%-99%), with no false negatives and only two false positives [77]. This performance surpassed traditional tests including synovial fluid cell count, culture, erythrocyte sedimentation rate, and C-reactive protein, though the improvement did not reach statistical significance in this sample size, except when comparing sensitivity to erythrocyte sedimentation rate.
Multiplex Immunofluorescence Protocol
Multiplex immunohistochemistry/immunofluorescence (mIHC/IF) represents a technological advancement for assessing multiple biomarkers simultaneously within the tissue context. The experimental protocol begins with formalin-fixed paraffin-embedded (FFPE) tissue sections mounted on slides. The process involves sequential rounds of staining with primary antibodies against multiple targets (e.g., immune cell markers, checkpoint proteins), followed by appropriate secondary antibodies conjugated with fluorescent dyes [76].
After each staining round, image acquisition is performed using a multispectral microscope capable of capturing the fluorescence signals. The process typically includes an antibody stripping step between rounds to remove antibodies before the next staining cycle. Computational algorithms then deconvolve the multispectral images to quantify marker expression and determine cellular phenotypes and spatial relationships within the tumor microenvironment [76].
In the network meta-analysis comparing predictive biomarkers for immunotherapy response, mIHC/IF exhibited the highest sensitivity (0.76, 95% CI: 0.57-0.89) and the second-highest diagnostic odds ratio (5.09, 95% CI: 1.35-13.90), supporting its superior performance for predicting response to anti-PD-1/PD-L1 therapy [76].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Research Reagent Solutions for Biomarker Assay Development
| Reagent/Technology | Function | Application Examples |
|---|---|---|
| Alpha Defensin-1 Immunoassay [77] | Detection of antimicrobial peptides in synovial fluid | Diagnosis of periprosthetic joint infections |
| Multiplex IHC/IF Platforms [76] | Simultaneous detection of multiple biomarkers in tissue sections | Tumor microenvironment analysis for immunotherapy prediction |
| Next-Generation Sequencing (NGS) [76] | Comprehensive assessment of tumor mutational burden and genetic alterations | Genomic profiling for therapy selection |
| RNA Sequencing Platforms [75] | Genome-wide transcriptome analysis | Pathway activation scoring from gene expression data |
| Mass Spectrometry Systems [75] | High-throughput protein identification and quantification | Proteomic profiling for pathway activation analysis |
| Pathway Analysis Algorithms [75] | Bioinformatics tools for calculating pathway activation levels | Quantitative assessment of intracellular pathway activity |
Signaling Pathways and Experimental Workflows
Conceptual Framework of Pathway Activation Biomarkers
Experimental Workflow for Multi-Marker Validation
The evolution from single-marker assays to pathway-based approaches represents a significant advancement in addressing the sensitivity and specificity limitations of traditional biomarkers. Pathway activation biomarkers offer enhanced robustness by capturing the integrated activity of multiple pathway components, reflecting the biological complexity of disease processes more accurately than individual molecule measurements [75]. The experimental evidence demonstrates that approaches such as the alpha defensin-1 assay for joint infection diagnosis and multiplexed tissue analysis for immunotherapy prediction consistently outperform conventional single-analyte tests.
Future directions in biomarker development will likely involve increased integration of multi-omics data, refinement of bioinformatic algorithms for pathway analysis, and standardization of analytical frameworks across platforms. As the field progresses, the validation of these advanced biomarkers will require rigorous analytical method validation followed by clinical qualification to establish their utility as surrogate endpoints in drug development and clinical practice [78]. The continued collaboration between experimental researchers, clinical investigators, bioinformaticians, and statisticians will be essential to translate these promising approaches into clinically validated tools that enhance patient care and therapeutic outcomes.
Overcoming False Positives/Negatives in Transporter Inhibition Assessment with Endogenous Biomarkers
In the field of drug development, accurately predicting drug-drug interactions (DDIs) mediated by renal membrane transporters is critical for patient safety. Current pharmaceutical industry approaches rely on in vitro inhibition potency parameters (e.g., IC₅₀ or Kᵢ values) and the ratio of unbound maximum plasma concentration to IC₅₀ (Cmax,u/IC₅₀) to assess DDI risk [79]. Regulatory-guided "static" decision trees then determine whether a formal clinical DDI study is required [79].
However, this approach suffers from significant limitations with high false-positive prediction rates. Recent analyses reveal that using European Medicines Agency (EMA) criteria, the current approach yields positive predictive values of only 64% for organic cation transporter 2 (OCT2), 47% for multidrug and toxin extrusion proteins (MATE1/2-K), and 52% for organic anion transporters (OAT1 and OAT3) [79]. This means that nearly half of the compounds flagged as potential DDI risks may not actually cause clinically significant interactions in humans, leading to unnecessary, costly, and time-consuming clinical DDI studies.
Endogenous Biomarkers: A Paradigm Shift in DDI Assessment
Endogenous biomarkers represent a transformative approach to assessing transporter-mediated DDIs. These are naturally occurring compounds in the body that serve as substrates for specific drug transporters. By monitoring changes in the plasma and urine levels of these biomarkers when an investigational drug is administered, researchers can directly assess the drug's inhibitory potential toward specific transporters in early phase I studies, potentially avoiding dedicated DDI studies [80] [79].
This approach offers several advantages over current methods:
- Real-time assessment: Endogenous biomarkers provide a direct readout of transporter inhibition in clinical settings
- Cost efficiency: They can be integrated into early clinical trials without requiring separate DDI studies
- Physiological relevance: They reflect actual transporter function in humans rather than extrapolations from in vitro models
Key Endogenous Biomarkers for Renal Transporters
Table 1: Key Endogenous Biomarkers for Renal Transporters
| Transporter | Endogenous Biomarker | Matrix | Clinical Utility |
|---|---|---|---|
| OCT2/MATE1/2-K | Creatinine | Plasma, Urine | Monitoring renal secretion of cationic drugs [79] |
| OAT1/OAT3 | Dimethylarginine (DMGV) | Plasma, Urine | Assessing interactions with anionic substances [79] |
| OAT1/OAT3 | Glycochenodeoxycholate sulfate (GCDCA-S) | Plasma, Urine | Predicting methotrexate-type interactions [79] |
| OAT1/OAT3 | Taurine | Plasma, Urine | Evaluating NSAID-diuretic interactions [79] |
Experimental Protocols for Biomarker Validation
Clinical Validation Approach
The utility of endogenous biomarkers for assessing transporter inhibition is established through rigorous clinical validation protocols:
- Study Design: Controlled clinical trials with known transporter inhibitors (e.g., cimetidine for MATEs, probenecid for OATs) [79]
- Sample Collection: Serial blood and urine samples collected before, during, and after inhibitor administration
- Biomarker Quantification: Liquid chromatography-mass spectrometry (LC-MS/MS) methods to precisely measure biomarker concentrations
- Data Analysis: Comparison of biomarker exposure (AUC) and renal clearance (CLr) in the presence and absence of inhibitors
Assessment Framework
Researchers have proposed a conceptual framework for integrating endogenous biomarkers into early clinical development [79]:
- Biomarker Qualification: Establish baseline levels and variability in healthy volunteers
- Inhibitor Response: Characterize biomarker changes with known inhibitors
- NME Assessment: Evaluate investigational drugs using established biomarker thresholds
- Decision Making: Use biomarker data to inform the need for dedicated DDI studies
Comparative Performance: Current Methods vs. Endogenous Biomarkers
Table 2: Performance Comparison of DDI Assessment Methods
| Assessment Method | False Positive Rate | Time Requirements | Cost Implications | Clinical Relevance |
|---|---|---|---|---|
| Current In Vitro + Static Model | 36-53% [79] | Medium (requires follow-up clinical studies) | High (dedicated DDI studies often needed) | Indirect extrapolation |
| Endogenous Biomarker Approach | Significantly reduced (emerging data) | Short (integrated into early trials) | Lower (avoids dedicated studies) | Direct clinical measurement |
| Combined Approach | Optimal balance | Flexible | Cost-effective | Comprehensive assessment |
Signaling Pathways and Experimental Workflows
Renal Transporter Pathways
Diagram 1: Renal transporter pathways in proximal tubule cells
Endogenous Biomarker Workflow
Diagram 2: Endogenous biomarker assessment workflow
The Scientist's Toolkit: Essential Research Reagents
Table 3: Research Reagent Solutions for Transporter Biomarker Studies
| Reagent/Resource | Function | Application Notes |
|---|---|---|
| LC-MS/MS Systems | Quantitative biomarker analysis | Essential for precise measurement of endogenous biomarkers in biological matrices [79] |
| Transporter-Expressing Cell Lines | In vitro inhibition screening | HEK293 or MDCK cells overexpressing specific human transporters (OCT2, MATE1/2-K, OAT1/3) [79] |
| Validated Inhibitors | Positive controls for transporter inhibition | Cimetidine (MATE), Probenecid (OAT), Pyrimethamine (MATE) for assay qualification [79] |
| Stable Isotope-Labeled Standards | Internal standards for MS quantification | Deuterated or ¹³C-labeled versions of endogenous biomarkers for accurate quantification [79] |
| Clinical Biospecimen Collection Kits | Standardized sample processing | Maintain biomarker stability during collection, processing, and storage [79] |
The integration of endogenous biomarkers into transporter inhibition assessment represents a significant advancement in overcoming the limitations of current approaches. By providing direct clinical evidence of transporter inhibition, these biomarkers can substantially reduce the high false-positive rates associated with traditional in vitro to in vivo extrapolation methods [79].
While endogenous biomarkers show tremendous promise, further work is needed to fully standardize their implementation, establish definitive cutoff values for clinical decision-making, and expand their application to a broader range of transporters. As the field evolves, the combination of endogenous biomarker assessment with refined in vitro approaches promises to create a more accurate, efficient, and cost-effective paradigm for evaluating transporter-mediated DDIs in drug development.
The conceptual framework proposed by industry experts [79] provides a roadmap for implementing endogenous biomarkers in early clinical development, potentially streamlining the assessment of renal transporter-mediated DDIs and reducing unnecessary dedicated DDI studies. This approach represents the future of transporter inhibition assessment, moving from conservative predictive models to direct clinical measurement of transporter function.
Disentangling Cell-Autonomous Signals from Microenvironmental Influences in Data Interpretation
In modern biomedicine, a fundamental challenge lies in accurately determining whether an observed cellular behavior—such as gene expression patterns, apoptosis resistance, or treatment response—stems from factors intrinsic to the cell itself or from signals originating in its extracellular microenvironment. This distinction between cell-autonomous (intrinsic) and non-cell-autonomous (extrinsic) mechanisms is crucial for developing targeted therapies and accurate diagnostic biomarkers [81] [82]. The complexity of cellular ecosystems, particularly in pathological states like cancer, means that observed phenotypes often emerge from dynamic interactions between a cell's genetic programming and external cues from its surroundings [83] [84].
Failure to properly distinguish these influences can lead to misinterpretation of experimental data and inefficient therapeutic strategies. For instance, a cell might exhibit apoptosis resistance due to its own mutated genes (intrinsic) or because of survival signals from neighboring cells (extrinsic)—scenarios requiring fundamentally different intervention approaches [84]. This comparative guide examines experimental frameworks and biomarkers that enable researchers to dissect these complex relationships across different biological contexts, from cancer immunology to leukemia research.
Fundamental Concepts: Defining Intrinsic and Extrinsic Regulation
Cell-autonomous (intrinsic) regulation refers to cellular behaviors determined by the cell's own genetic programming and internal machinery, independent of external signals. These mechanisms include genetic mutations, epigenetic modifications, and internally regulated signaling pathways that function regardless of the cellular environment [81] [82].
Non-cell-autonomous (extrinsic) regulation encompasses cellular behaviors directed by signals originating outside the cell, including soluble factors (cytokines, growth factors), physical interactions with neighboring cells, extracellular matrix components, and metabolic conditions within the tissue microenvironment [83] [84].
In practice, many cellular phenotypes arise through interactive mechanisms where intrinsic predispositions and extrinsic signals combine to determine ultimate outcomes. For example, oncogenic mutations (intrinsic) may alter how a cell responds to microenvironmental growth factors (extrinsic), creating unique vulnerability profiles [84].
Experimental Frameworks for Disentanglement
Chimera-Based Decomposition Framework
Interspecies chimeras provide a powerful experimental system for quantitatively decomposing evolutionary and developmental traits into intrinsic, extrinsic, and interactive components. This approach involves grafting donor cells from one species into a host embryo of another species, then comparing how these cells behave in species-matched versus species-mismatched environments [81] [82].
Table 1: Quantitative Framework for Decomposing Trait Divergence in Chimeric Models
| Component | Definition | Interpretation | Experimental Measurement |
|---|---|---|---|
| Intrinsic Divergence | Trait differences determined by the cell of origin | Cell-autonomous regulation | Compare donor cells of different genotypes in identical host environments |
| Extrinsic Divergence | Trait differences determined by the extracellular environment | Non-cell-autonomous regulation | Compare identical genotypes in different host environments |
| Intrinsic-Extrinsic Interaction | Trait differences where cell genotype modifies response to environment | Context-dependent regulation | Difference between observed values and expected additive effects |
The mathematical framework for this decomposition uses four key measurements: (1) mouse cells in mouse environment, (2) rat cells in rat environment, (3) mouse cells in rat environment, and (4) rat cells in mouse environment. By comparing these configurations, researchers can calculate the proportion of divergence attributable to intrinsic factors, extrinsic factors, and their interactions [82].
Application of this framework to gene expression data from rat-mouse chimeras revealed that most expression divergence is cell-intrinsic, though extrinsic factors play significant roles in specific contexts. For example, a rat-like extracellular environment extrinsically up-regulated expression of key transcriptional regulators of the endoplasmic reticulum stress response in some cell types, which subsequently affected downstream target genes [82].
Causal Disentanglement in Single-Cell Omics
Recent advances in single-cell technologies enable disentanglement of intrinsic and extrinsic factors at unprecedented resolution. The CausCell framework combines structural causal modeling with diffusion models to perform causal disentanglement and counterfactual generation on single-cell omics data [85].
This approach assumes each cell's data is generated by two concept types: observed concepts (e.g., cell type, spatial location) and unexplained concepts (unknown biological factors). The framework incorporates factual information about causal structures between biological concepts to generate reliable disentangled cellular representations with three key advantages [85]:
- Explainability: Uses structural causal models to recover latent concepts with semantic meanings and their causal relationships
- Generalisability: Employs diffusion models as backbone, providing strong generative capabilities
- Controllability: Enables controllable generation by manipulating disentangled representations while preserving causal consistency
The experimental workflow involves encoding input gene expression profiles into exogenous embeddings, processing through a structural causal model layer to produce endogenous embeddings that capture biological concepts in a causally structured latent space, then mapping these to predict concept labels using separate predictors for each concept [85].
Comparative Analysis of Pathway Activation Biomarkers
Coagulation Pathway Biomarkers
The coagulation system provides a well-characterized model for studying distinct intrinsic and extrinsic pathways that converge on common effectors. These pathways can be differentiated using specific biomarkers and functional assays [15] [17] [86].
Table 2: Comparative Analysis of Intrinsic vs. Extrinsic Coagulation Pathways
| Characteristic | Intrinsic Pathway | Extrinsic Pathway | Common Pathway |
|---|---|---|---|
| Activation Trigger | Spontaneous internal vascular damage, exposed collagen [15] | External trauma, tissue factor release [15] | Convergence point of both pathways |
| Key Factors | XII, XI, IX, VIII [17] | VII, III (tissue factor) [17] | X, V, II, I, XIII [17] |
| Diagnostic Tests | Partial Thromboplastin Time (PTT): 25-40 seconds (normal) [17] | Prothrombin Time (PT): 11-15 seconds (normal) [17] | Both PT and PTT |
| Cellular Sites | Platelets, monocytes [86] | Tissue factor-expressing cells (endothelial cells, monocytes, macrophages) [86] | Platelets, monocytes |
| Activation Kinetics | Slower activation cascade [15] | Rapid response [15] | Final common pathway |
| Factor X Activation Efficiency | High efficiency on monocyte/macrophage membranes (Km = 12.1-14.6 nM) [86] | Lower efficiency on monocyte/macrophage membranes (Km = 90.6-117.0 nM) [86] | N/A |
Notably, monocytes and macrophages uniquely possess membrane sites supporting both intrinsic and extrinsic pathway assembly, making them crucial for comparative studies of these activation mechanisms. Kinetic studies demonstrate that factor X activation via the intrinsic pathway is more efficient than through the extrinsic pathway on these cells [86].
Cancer Microenvironment Biomarkers
In oncology, distinguishing cell-autonomous oncogenic signals from microenvironmental influences is critical for understanding therapy resistance and developing effective treatments. The tumor microenvironment creates complex extrinsic signals that interact with cancer cells' intrinsic programming [83] [84].
Table 3: Biomarkers of Intrinsic vs. Extrinsic Regulation in Cancer
| Biomarker Category | Intrinsic (Cell-Autonomous) Examples | Extrinsic (Microenvironmental) Examples | Experimental Applications |
|---|---|---|---|
| Genomic Biomarkers | EGFR, ALK, BRAF mutations [83] | N/A (Extrinsic factors don't alter DNA sequence) | Targeted therapy selection |
| Proteomic Biomarkers | BCL-2 family protein expression in JMML [84] | Cytokine-induced proteins (ICAM-1) [83] | Apoptosis resistance profiling |
| Immunological Biomarkers | PD-L1 expression on tumor cells [83] | Cytokine levels (IL-6, IL-8, TNF-α) [83] | Immunotherapy response prediction |
| Metabolic Biomarkers | Oncogene-driven metabolic reprogramming | Nutrient availability, hypoxia markers | Metabolic vulnerability assessment |
| Apoptosis Regulation | BCL-XL and MCL-1 directly regulated by oncogenic RAS signaling [84] | Microenvironmental signals influencing BCL-2 family members [84] | BH3-mimetic therapy selection |
In juvenile myelomonocytic leukemia (JMML), research demonstrates how apoptosis resistance mechanisms differ by cell type and are influenced by both intrinsic and extrinsic factors. Monocytes and granulocytes show overexpression of anti-apoptotic BCL-2 family members mediated by both oncogenic RAS signaling (intrinsic) and microenvironmental signals (extrinsic) [84]. Interestingly, stem and progenitor cells expressing the same oncogenic PTPN11 mutant showed no increased apoptosis resistance, highlighting the cell-type-specific nature of these regulatory mechanisms [84].
The Scientist's Toolkit: Essential Research Reagents and Methodologies
Key Research Reagent Solutions
Table 4: Essential Research Reagents for Disentanglement Studies
| Reagent Category | Specific Examples | Research Application | Experimental Function |
|---|---|---|---|
| Chimera Models | Rat-mouse blastocyst chimeras [81] [82] | Developmental and evolutionary studies | Decompose trait divergence into intrinsic/extrinsic components |
| Single-Cell Omics Platforms | scRNA-seq, spatial transcriptomics [83] [85] | Tumor microenvironment analysis | Resolve cellular heterogeneity and spatial relationships |
| BH3 Mimetics | ABT737 (BCL-2/BCL-XL/BCL-w inhibitor), A1155463 (BCL-XL specific), S63845 (MCL-1 inhibitor) [84] | Apoptosis resistance profiling | Identify anti-apoptotic protein dependencies |
| Cytokine Analysis Tools | Cytokine bead arrays, neutralizing antibodies [84] | Microenvironmental signaling analysis | Identify and block extrinsic survival signals |
| Pathway Reporters | Phospho-flow cytometry (pMAPK, pmTOR) [84] | Signaling pathway activation | Measure intrinsic pathway activity in different environments |
| Coagulation Assays | PT/INR, PTT reagents [15] [17] | Hemostasis research | Differentiate intrinsic vs. extrinsic pathway defects |
Experimental Protocols for Key Methodologies
Chimera-Based Trait Decomposition Protocol [82]:
- Generate reciprocal interspecies chimeras via blastocyst injection (e.g., ratmouse)
- Ensure donor cell contribution remains below 10% to maintain species-mismatched environment
- Collect target tissues from stage-matched embryos
- Perform single-cell RNA sequencing with species-specific barcoding
- Assign cell identities using reference transcriptomes
- Apply quantitative framework to decompose expression divergence:
- Intrinsic = (Mousedonorinrat - Rathost) OR (Ratdonorinmouse - Mousehost)
- Extrinsic = (Mousedonorinrat - Mousehost) OR (Ratdonorinmouse - Rathost)
- Calculate interaction effects as differences between observed and expected additive values
Apoptosis Dependence Profiling Protocol [84]:
- Isolate primary cells of interest (e.g., CD11b+ myeloid cells, LSK stem/progenitor cells)
- Culture in defined conditions with or without microenvironmental factors
- Treat with panel of BH3 mimetics at optimized concentrations:
- ABT737: 0.1-1μM for BCL-2/BCL-XL/BCL-w inhibition
- A1155463: 0.1-0.5μM for selective BCL-XL inhibition
- S63845: 0.1-1μM for MCL-1 inhibition
- Incubate for 6-24 hours depending on cell type
- Perform Annexin V/7AAD staining and flow cytometry analysis
- Calculate specific apoptosis: 100 × (% live cellsuntreated - % live cellstreated) / % live cells_untreated
- Compare patterns across cell types and culture conditions to identify intrinsic vs. extrinsic regulation
Causal Disentanglement in Single-Cell Data Protocol [85]:
- Preprocess single-cell omics data (quality control, normalization, batch correction)
- Define observed concepts (cell type, spatial location, treatment condition)
- Specify known causal structure between concepts using directed acyclic graphs
- Train CausCell framework with novel loss function integrating:
- Evidence lower bound (ELBO) for reconstruction accuracy
- Independence constraint for unexplained concepts
- Supervised constraint for biological interpretability
- Generate counterfactual cells by intervening on specific concepts
- Validate disentanglement quality using quantitative benchmarks:
- Disentanglement metrics: concept predictability, separability
- Reconstruction metrics: mean squared error, structural similarity
Disentangling cell-autonomous signals from microenvironmental influences remains a fundamental challenge with direct therapeutic implications. The experimental frameworks and biomarkers compared in this guide demonstrate that most biological outcomes emerge from complex interactions between intrinsic cellular programming and extrinsic environmental signals rather than either mechanism alone [81] [84] [82].
The most promising approaches integrate multiple methodologies—chimera models for decomposition, single-cell omics for resolution, and causal modeling for interpretation—to address this complexity. These integrated strategies are particularly valuable for developing biomarkers that can predict therapeutic responses in variable microenvironments, such as determining which cancer patients will benefit from BH3 mimetics based on both tumor-intrinsic BCL-2 family expression and extrinsic survival signals [84].
As these technologies advance, particularly in single-cell spatial analysis and causal machine learning models, researchers will gain increasingly precise tools for determining the relative contributions of intrinsic and extrinsic factors across diverse biological contexts and disease states. This precision will ultimately enable more targeted interventions that account for both the cellular actors and their environmental contexts in complex biological systems.
In the evolving landscape of biomedical research, biomarkers serve as critical tools for diagnosis, prognosis, and therapeutic guidance. While single biomarkers have historically been the cornerstone of clinical decision-making, a growing body of evidence reveals significant limitations in their accuracy and generalizability. This guide objectively compares the performance of single biomarkers against multi-marker classifiers, framing the analysis within research on intrinsic and extrinsic apoptosis pathway activation. Supported by experimental data and detailed methodologies, we demonstrate how combined classifiers, leveraging advanced computational approaches, provide a more robust framework for understanding complex disease mechanisms and personalizing treatment. This paradigm shift is particularly relevant for researchers, scientists, and drug development professionals navigating the challenges of biomarker validation and clinical translation.
A biomarker is formally defined as "a defined characteristic that is measured as an indicator of normal biological processes, pathogenic processes, or responses to an exposure or intervention" [74] [87]. These biological molecules, found in blood, other body fluids, or tissues, provide invaluable insights into disease states and therapeutic responses. In clinical practice and drug development, biomarkers have been traditionally deployed as solitary indicators—single proteins, genetic mutations, or metabolic products intended to detect diseases, predict outcomes, or guide treatments [63].
The reliance on single biomarkers stems from practical considerations: simplified assay development, straightforward clinical interpretation, and regulatory approval pathways. Established examples include prostate-specific antigen (PSA) for prostate cancer monitoring, B-type natriuretic peptide (BNP) in heart failure, and single gene mutations like BRCA1/2 for hereditary cancer risk assessment [74] [63]. However, this reductionist approach often fails to capture the complexity of pathological processes, particularly in multifactorial diseases where multiple biological pathways interact to determine clinical outcomes.
Within apoptosis research—a fundamental process in development, homeostasis, and disease—the distinction between intrinsic (mitochondrial) and extrinsic (death receptor) pathway activation exemplifies this limitation. While individual biomarkers like BID (a key mediator cross-talking between pathways) or specific caspase activations provide valuable snapshots, they cannot fully characterize the dynamic interplay between these pathways in conditions such as intracerebral hemorrhage, cancer, or neurodegenerative disorders [12]. The scientific community is increasingly recognizing that complex biological systems require multidimensional assessment, pushing the field toward integrated classifier approaches that combine multiple biomarkers to achieve superior diagnostic and prognostic performance.
Theoretical Foundations: Limitations of Single Biomarkers
Incomplete Pathway Representation
Single biomarkers offer a fragmented view of complex biological systems. Diseases often involve multiple interconnected pathways, and focusing on a single marker inevitably misses critical aspects of the pathological process. In apoptosis, for instance, the extrinsic pathway initiates through death receptors (e.g., Fas, TNF receptors) activating caspase-8, while the intrinsic pathway involves mitochondrial outer membrane permeabilization and cytochrome c release activating caspase-9 [12]. While a biomarker like BID expression might indicate apoptotic activity, it cannot definitively distinguish pathway predominance or quantify crosstalk between mechanisms. This limitation is particularly problematic for therapeutic development, where targeted interventions often aim at specific pathway components.
Statistical and Methodological Limitations
From a statistical perspective, single biomarkers frequently demonstrate inadequate performance characteristics for clinical application. The Youden index (J = sensitivity + specificity - 1) is a commonly used metric for diagnostic accuracy, ranging from 0 (uninformative) to 1 (perfect separation) [88]. Single biomarkers often achieve suboptimal Youden indices due to biological heterogeneity and overlapping distributions between diseased and healthy populations. One study noted that "diagnosis based on one single biomarker may not provide sufficient accuracy," particularly for diseases with multifactorial etiology or population-specific variations [88]. This statistical limitation becomes clinically significant when biomarkers are used for critical decisions regarding treatment selection or patient stratification.
Biological and Analytical Vulnerabilities
Single biomarkers are susceptible to numerous confounding factors that can compromise their reliability. Pre-analytical variables (sample collection, processing), analytical performance (assay precision, accuracy), and biological heterogeneity (diurnal variation, comorbid conditions) can all influence measurements [74] [63]. Additionally, regulatory perspectives highlight that "scientific justification behind biomarkers and interpretation of biomarker measurements are not always reliable and appropriate" [87]. Without complementary markers to provide context or verification, single biomarkers may lead to misinterpretation of biological states or therapeutic responses.
Combined Classifiers: A Multidimensional Solution
Conceptual Framework and Advantages
Combined classifiers represent a paradigm shift from reductionist to systems-level approaches in biomarker research. Rather than relying on a single measurement, these classifiers integrate multiple biomarkers—often from different biological domains—using statistical or machine learning algorithms to generate a composite score with superior performance characteristics [88] [89]. The theoretical foundation rests on the principle that multidimensional assessment can more completely capture complex pathological processes.
The advantages of combined classifiers are substantial. First, they can integrate markers representing different aspects of a disease process—for instance, combining biomarkers of intrinsic apoptosis (e.g., cytochrome c), extrinsic apoptosis (e.g., caspase-8 activation), and downstream execution phases (e.g., caspase-3 cleavage) to fully characterize cell death dynamics [12]. Second, they improve statistical power for distinguishing disease states; one study on Alzheimer's trials found that "using all biomarkers jointly" allowed selection of subjects most likely to decline, substantially boosting power for detecting treatment effects [90]. Third, they offer redundancy that mitigates the impact of analytical variability in any single measurement.
Methodological Approaches for Classifier Development
Multiple methodological frameworks exist for developing combined classifiers, each with distinct strengths and applications:
Linear Combination Methods: Parametric and nonparametric approaches linearly combine biomarkers to optimize accuracy metrics like the Youden index or area under the ROC curve [88]. While computationally efficient, they may miss complex interactions between variables.
Machine Learning Approaches: Flexible algorithms like Support Vector Machines (SVM), random forests, and neural networks can model both linear and nonlinear relationships [89] [90]. These are particularly valuable when combining high-dimensional data from omics technologies [91].
Large Margin Classification Frameworks: Recent approaches formulate combination functions in reproducing kernel Hilbert spaces (RKHS), allowing both linear and nonlinear combinations via specified kernel functions for optimized classification [88].
The choice of methodology depends on the number of available biomarkers, sample size, and the anticipated complexity of relationships between markers and clinical outcomes.
Comparative Analysis: Single versus Combined Biomarker Performance
Quantitative Performance Metrics
Table 1: Comparative Performance of Single versus Combined Biomarkers in Diagnostic Accuracy
| Biomarker Type | Application Area | Sensitivity (%) | Specificity (%) | Youden Index | AUC | Reference |
|---|---|---|---|---|---|---|
| Single MRI measure | Alzheimer's classification | 71.2 | 73.5 | 0.447 | 0.79 | [90] |
| PET-FDG alone | MCI classification | 68.9 | 70.1 | 0.390 | 0.75 | [90] |
| CSF Aβ42 alone | MCI classification | 72.5 | 69.8 | 0.423 | 0.76 | [90] |
| SVM combined biomarkers | Alzheimer's classification | 89.4 | 88.7 | 0.781 | 0.94 | [90] |
| Flexible combination | Liver disorder diagnosis | 85.2 | 87.6 | 0.728 | 0.91 | [88] |
The performance advantage of combined classifiers is evident across multiple diagnostic domains. In Alzheimer's disease, a Support Vector Machine algorithm combining MRI measures, PET-FDG, CSF biomarkers, ApoE genotype, and clinical variables achieved substantially better classification accuracy than any single biomarker alone [90]. Similarly, in general diagnostic applications, flexible approaches allowing both linear and nonlinear combinations demonstrated significant improvements in the Youden index compared to traditional single-marker approaches [88].
Application in Apoptosis Pathway Research
Table 2: Biomarker Performance in Intrinsic/Extrinsic Apoptosis Pathway Detection
| Biomarker | Pathway Association | Detection Method | Strength | Limitation |
|---|---|---|---|---|
| BID | Cross-talk (intrinsic/extrinsic) | Western blot, IHC | Indicates pathway integration | Cannot quantify individual pathway contribution |
| Caspase-8 activation | Extrinsic initiation | Activity assay, cleavage | Specific to death receptor signaling | Misses intrinsic pathway activation |
| Cytochrome c release | Intrinsic initiation | ELISA, IHC | Specific to mitochondrial pathway | Requires cell fractionation |
| Bax/Bak oligomerization | Intrinsic execution | FRET, cross-linking | Proximal marker of MOMP | Technically challenging |
| Combined classifier I/E ratio | Pathway predominance | Computational integration | Quantifies pathway contribution | Requires multiple assays |
Research on intracerebral hemorrhage demonstrates the particular value of combined approaches in apoptosis. While BID was identified as a key gene involved in apoptosis following ICH, its full explanatory power emerged only when considered within a network of interacting biomarkers [12]. A classifier incorporating multiple apoptosis-related genes provided more comprehensive insights into cell death mechanisms than any single marker could achieve.
Experimental Protocols for Biomarker Combination Studies
Protocol 1: Developing a Flexible Biomarker Combination
This protocol follows methodology from studies aiming to maximize the Youden index through flexible biomarker combinations [88]:
Sample Preparation: Collect biospecimens (serum, tissue, etc.) from well-characterized cohorts representing disease and control states. For apoptosis studies, this may include tissue samples from disease models (e.g., ICH rat model) and appropriate controls [12].
Biomarker Measurement: Quantify candidate biomarkers using appropriate analytical platforms (ELISA, Western blot, PCR, etc.). For apoptosis pathway differentiation, include markers specific to intrinsic (e.g., cytochrome c, Bax), extrinsic (e.g., caspase-8, Fas), and executioner (caspase-3) phases.
Data Preprocessing: Normalize data to account for technical variability. Address missing values through appropriate imputation methods.
Classifier Construction: Formulate the combination function in a reproducing kernel Hilbert space (RKHS) framework. The optimization problem is: [ \max{g,c} \frac{1}{2n} \sum{i=1}^{n} \hat{w}(yi)(1 + yi \text{sign}(g(\mathbf{x}i)-c)) - 1 ] where (g(\mathbf{x})) is the combination function, (c) is the cut-point, (\mathbf{x}i) is the biomarker vector for subject (i), and (y_i \in {1,-1}) indicates disease status [88].
Validation: Evaluate performance using cross-validation or external validation cohorts. Compare the combined classifier against individual biomarkers using ROC analysis and compute the improvement in Youden index.
Protocol 2: Machine Learning Approach for Pathway Classification
This protocol adapts methodology from studies using machine learning to combine biomarkers for classification [89] [90]:
Feature Selection: Identify informative biomarkers using filter methods (correlation with outcome), wrapper methods (objective function-based), or embedded methods (incorporated in classifiers) [89]. For apoptosis pathway classification, prioritize markers with known pathway specificity.
Classifier Training: Implement a Support Vector Machine (SVM) algorithm with radial basis function kernel. The objective is to find the hyperplane that maximizes the margin between classes in the transformed feature space.
Hyperparameter Optimization: Use grid search with cross-validation to optimize parameters such as regularization (C) and kernel parameters.
Pathway Activation Scoring: Derive a continuous score representing the probability of intrinsic versus extrinsic pathway predominance based on the classifier output.
Performance Assessment: Evaluate using metrics such as precision, recall, and F1-score in addition to traditional ROC analysis. Assess stability through resampling methods.
Apoptosis Pathway Research Workflow
The following diagram illustrates a generalized experimental workflow for developing combined classifiers in intrinsic/extrinsic apoptosis pathway research:
Visualization of Apoptosis Pathways and Biomarker Integration
Intrinsic and Extrinsic Apoptosis Signaling Pathways
The following diagram illustrates key biomarkers in intrinsic and extrinsic apoptosis pathways and their points of integration, highlighting potential targets for combined classifier development:
Essential Research Reagents and Solutions
Table 3: Research Reagent Solutions for Apoptosis Biomarker Studies
| Reagent Category | Specific Examples | Research Function | Application Notes |
|---|---|---|---|
| Primary Antibodies | Anti-BID, Anti-Caspase-8 (cleaved), Anti-Cytochrome c, Anti-Bax | Detection of specific apoptosis biomarkers via Western blot, IHC | Validate specificity for intended targets; optimize dilution |
| Activity Assays | Caspase-3/7 Glo, Caspase-8 Luminescent, Caspase-9 Colorimetric | Quantify enzymatic activity of key apoptosis mediators | Prefer luminescent for sensitivity in multiplex setups |
| ELISA Kits | Human Cytochrome c ELISA, M30 Apoptosis Assay | Quantify specific biomarkers in solution | Check cross-reactivity with related proteins |
| PCR Assays | BID TaqMan, Caspase-8 SYBR Green, Apoptosis PCR Arrays | Measure gene expression of apoptosis regulators | Use validated probe-primer sets for quantification |
| Cell Death Detection | Annexin V FITC, TUNEL Assay, Propidium Iodide | Detect and quantify apoptotic cells | Combine markers for early/late apoptosis distinction |
The evidence for transitioning from single biomarkers to combined classifiers is compelling and multifaceted. While single biomarkers will continue to have value in specific, well-defined contexts, their limitations in sensitivity, specificity, and biological comprehensiveness render them inadequate for understanding complex pathological processes like the interplay between intrinsic and extrinsic apoptosis pathways. Combined classifiers, leveraging advances in computational biology and machine learning, offer superior accuracy, robustness, and clinical utility. For researchers and drug development professionals, embracing this multidimensional approach is not merely an optimization but a necessary evolution to address the complexity of human disease and realize the promise of personalized medicine. Future efforts should focus on standardizing analytical frameworks, improving computational accessibility, and developing regulatory pathways for multivariate biomarker approval.
The advancement of precision medicine is fundamentally reliant on robust, reproducible biomarkers. For researchers investigating intrinsic and extrinsic pathway activation, the lack of standardized assays and universally accepted cut-off values presents a major translational hurdle. Biomarkers for pathway activity, which measure the integrated output of signaling cascades rather than single molecules, are particularly vulnerable to inter-platform variability due to their computational derivation from complex multi-analyte data [49]. This variability undermines data comparability across research sites and clinical trials, ultimately slowing the development of pathway-targeted therapies. This guide objectively compares the performance of different harmonization approaches and the technologies that support them, providing a framework for selecting and validating robust assays for pathway activation studies.
The Landscape of Biomarker Assay Platforms
Multiple technology platforms are employed to measure biomarkers, each with distinct performance characteristics, costs, and throughput capacities. The choice of platform directly impacts the success of any harmonization effort.
Comparative Analysis of Key Assay Technologies
The table below summarizes the core technologies used for biomarker analysis, highlighting their applicability to pathway-level biomarker research.
Table 1: Comparison of Core Biomarker Assay Technologies
| Technology | Principle | Throughput | Key Strengths | Key Limitations | Suitability for Pathway Biomarkers |
|---|---|---|---|---|---|
| ELISA [92] | Antibody-based colorimetric detection | Low to Medium | Gold standard; high specificity; widely accessible | Narrow dynamic range; low-plex; antibody-dependent | Low. Suitable for single proteins, not complex pathway signatures. |
| Meso Scale Discovery (MSD) [92] | Electrochemiluminescence detection | Medium to High | High sensitivity (100x ELISA); broad dynamic range; multiplexing | Requires specialized instrumentation | Medium. Useful for measuring panels of pathway-related proteins. |
| Liquid Chromatography Tandem Mass Spectrometry (LC-MS/MS) [92] | Separation by mass/charge ratio | Medium | High specificity and precision; can detect hundreds of proteins | High cost; complex operation; requires expertise | High. Ideal for large-scale, discovery-phase proteomics for pathway analysis. |
| Next-Generation Sequencing (NGS) [93] | DNA/RNA sequencing | High | Comprehensive; enables genomic and transcriptomic pathway analysis | Data complexity; bioinformatic burden | High. Essential for gene expression-based pathway activity scores. |
The Economic and Operational Case for Advanced Platforms
While traditional ELISA is the historical gold standard, advanced platforms like MSD and LC-MS/MS offer superior performance, particularly for complex biomarker work. MSD provides up to 100 times greater sensitivity than ELISA and supports multiplexing, which allows for the simultaneous measurement of multiple analytes from a single small sample volume [92]. This capability is critical for constructing pathway activity scores that require input from several proteins or genes.
From a cost perspective, multiplexing presents a significant advantage. For example, measuring a panel of four inflammatory biomarkers (IL-1β, IL-6, TNF-α, and IFN-γ) costs approximately $61.53 per sample using individual ELISAs, compared to only $19.20 per sample using an MSD multiplex assay—a saving of $42.33 per sample [92]. For large-scale studies, this cost efficiency, combined with richer data output, makes advanced platforms highly compelling despite higher initial instrumentation costs.
Experimental Protocols for Harmonization and Validation
Demonstrating the equivalence of biomarker measurements across different laboratories and platforms requires rigorous experimental designs. The following protocols are considered best practices.
Protocol 1: Inter-Laboratory Concordance Testing
This protocol, modeled after the NCI's Designated Laboratory (DL) Network for the ComboMATCH trial, is designed to ensure that different labs produce comparable results from the same sample [93].
- Objective: To achieve an inter-laboratory concordance rate of ≥80% for biomarker classification (e.g., "pathway active" vs. "pathway inactive").
- Materials:
- Reference Standards: Well-characterized, pooled patient-derived samples (e.g., cell line lysates, pooled serum) with a predefined range of biomarker expression.
- Participating Laboratories: Multiple CLIA-certified/accredited labs using their own validated platforms (e.g., different NGS panels, MSD instruments) [93].
- Methodology:
- Sample Distribution: Distribute identical aliquots of reference standards to all participating laboratories.
- Blinded Analysis: Each lab processes and analyzes the samples using their standard operating procedures.
- Data Collection: Labs report raw data, normalized values, and final biomarker calls (e.g., positive/negative or continuous activity score).
- Concordance Calculation: Compare all results against a reference method or a consensus result from all participants. Calculate the percent agreement for categorical calls or the correlation coefficient for continuous scores.
- Expected Outcome: Labs demonstrating ≥80% concordance are deemed to have a harmonized assay. Those below the threshold must investigate and optimize their analytical or computational workflows [93].
Protocol 2: Cross-Platform Pathway Activity Score Validation
This protocol validates a computational method for deriving a pathway activity score from gene expression data, ensuring it is robust across different measurement platforms (e.g., microarray vs. RNA-seq).
- Objective: To demonstrate that a pathway activity score is consistent and clinically predictive regardless of the underlying gene expression platform.
- Materials:
- Biological Samples: A set of patient tumors with paired gene expression data from both microarray and RNA-seq platforms.
- Pathway Analysis Tool: Software like PathOlogist, which calculates pathway activity by considering interaction types and directionality, not just gene set averages [94].
- Clinical Outcome Data: Patient response data to a relevant targeted therapy.
- Methodology:
- Data Acquisition: Obtain gene expression profiles for the same sample set generated on two different platforms (e.g., Microarray and RNA-seq).
- Pathway Score Calculation: Input both gene expression datasets into the PathOlogist tool to calculate activity scores for the pathway of interest (e.g., HIF-1α pathway, BRCA pathway).
- Correlation Analysis: Assess the correlation (e.g., Pearson's r) between the pathway activity scores derived from the two platforms. A high correlation (e.g., r > 0.9) indicates platform independence.
- Clinical Validation: Test whether the pathway activity score from each platform significantly predicts therapy response (e.g., via ROC analysis or Cox proportional hazards model) [94].
- Expected Outcome: A validated, platform-agnostic pathway activity score that can be used as a predictive biomarker in clinical trials aggregating data from multiple sources.
Case Studies in Real-World Harmonization
Success Story: The NCI CIMAC-CIDC Network
The Cancer Immunomonitoring and Analysis Centers–Cancer Immunologic Data Commons (CIMAC-CIDC) network, a Cancer Moonshot initiative, provides a successful model for assay harmonization in immunotherapy clinical trials. The network established four academic laboratories to profile patient specimens using standardized, validated assays. The core of its success lies in the harmonization of a core set of assays, which drastically reduced data variability and now facilitates powerful cross-trial analyses [93]. This approach directly addresses the challenge of comparing pathway activation biomarkers generated in different research environments.
The Challenge of Traditional Biomarkers: AKI Diagnosis
The field of acute kidney injury (AKI) highlights the consequences of poor harmonization. Despite the discovery of numerous promising early biomarkers like Neutrophil Gelatinase-Associated Lipocalin (NGAL) and Kidney Injury Molecule-1 (KIM-1), their clinical translation is hampered by a lack of standardization. For example, different studies report varying optimal cut-off values for plasma NGAL (e.g., >546 ng/mL for predicting severe AKI), and its performance is confounded by systemic inflammation [95]. This lack of uniform standards prevents the establishment of universal clinical decision points, limiting their utility in practice.
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful harmonization requires not just a good protocol but also high-quality, consistent reagents and tools.
Table 2: Key Reagents and Resources for Biomarker Harmonization
| Item / Resource | Function & Utility in Harmonization | Example Use Case |
|---|---|---|
| Reference Standard Samples | Provides a universal benchmark for comparing assay performance across labs and over time. | Used in the NCI's SPOT/Dx Working Group to achieve inter-laboratory standardization in NGS assays [93]. |
| Multiplex Immunoassay Panels (e.g., MSD U-PLEX) | Allows simultaneous measurement of multiple pathway components from a single, small-volume sample, improving data consistency. | Profiling a panel of inflammatory cytokines to generate a composite immune activation score [92]. |
| Pathway Analysis Software (e.g., PathOlogist) | Computes pathway activity scores using the structure and directionality of molecular interactions, creating more robust biomarkers than single genes. | Identifying high BRCA-pathway activity as a biomarker for MAD2L1 essentiality in breast cancer [94]. |
| CLIA-Certified Laboratory Networks | Provides a pre-qualified ecosystem of labs that have demonstrated proficiency and concordance in specific assays. | The NCI DL Network ensures that patients are accurately matched to trials like ComboMATCH based on their molecular profile [93]. |
Harmonizing assays and biomarker cut-offs is not merely a technical exercise but a fundamental requirement for the future of pathway-based precision medicine. As the research demonstrates, pathway-level biomarkers offer superior robustness against technical noise compared to single-gene markers [94]. Success depends on a multi-faceted strategy: adopting advanced, multiplexed technologies like MSD and LC-MS/MS for richer, more cost-effective data; implementing rigorous harmonization protocols like inter-lab concordance testing; and leveraging universal reference materials and computational tools.
The growing trend of outsourcing to specialized Contract Research Organizations (CROs) provides researchers with access to these cutting-edge technologies and expertise without massive capital investment, thereby accelerating the path to reliable, clinically actionable biomarkers [92]. For researchers focused on intrinsic and extrinsic pathway activation, committing to these harmonization principles is essential for generating data that can truly translate into effective, personalized cancer therapies.
In molecular diagnostics and therapeutic development, the concepts of intrinsic and extrinsic biomarkers provide a powerful framework for understanding complex biological systems. Intrinsic biomarkers originate from within an organism's innate biological processes, such as genetic signatures, internally produced proteins, or metabolic byproducts. In contrast, extrinsic biomarkers often involve responses to external stimuli, environmental exposures, or administered substances that reveal system functionality. The strategic integration of these complementary biomarker classes enables researchers to develop more comprehensive diagnostic and prognostic models that account for both inherent biological predispositions and external influences.
The distinction between intrinsic and extrinsic factors extends across multiple biological domains. In apoptosis, the intrinsic pathway initiates from internal cellular damage and stress signals, while the extrinsic pathway begins with external death ligands activating cell surface receptors [35]. Similarly, in coagulation, the intrinsic pathway activates through surface contact exposure, whereas the extrinsic pathway triggers via tissue factor released from vascular damage [96]. Beyond these classic examples, contemporary drug development now recognizes the critical importance of evaluating how intrinsic factors (e.g., genetics, age, organ function) and extrinsic factors (e.g., diet, concomitant medications, environmental exposures) influence therapeutic responses and disease outcomes [97].
Harnessing the combined predictive power of intrinsic and extrinsic biomarkers represents a frontier in precision medicine. This comparative guide examines current methodologies, experimental protocols, and analytical frameworks for integrating these biomarker classes, with a focus on practical applications for researchers and drug development professionals seeking to optimize diagnostic and prognostic accuracy.
Comparative Analysis of Biomarker Integration Approaches
Table 1: Comparison of biomarker integration strategies across biological domains
| Biological Domain | Intrinsic Biomarkers/Signatures | Extrinsic Biomarkers/Signatures | Integrated Approach Benefits |
|---|---|---|---|
| Apoptosis Signaling | Cytochrome C, SMAC/Diablo, AIF from mitochondria [35] | Fas Ligand, TNF-α, TRAIL death receptors [35] | Comprehensive cell death assessment; therapeutic targeting |
| Coagulation Cascade | Factor XII, XI, IX, VIII (surface contact activation) [96] | Tissue Factor, Factor VII (vascular damage) [96] | Complete coagulation profiling; bleeding disorder diagnosis |
| Cancer Progression | Gene expression signatures from TCGA [98] | Essential survival genes from DepMap RNAi screens [98] | Improved prediction of tumor progression and treatment response |
| Renal Cell Carcinoma | Genomic and transcriptomic alterations [99] | SAA2, CFB from experimental metastasis models [99] | Enhanced prediction of metastasis and relapse |
| Drug Transport | Endogenous biomarkers (creatinine, metabolites) [79] | Exogenous probe drugs (metformin, methotrexate) [79] | Streamlined assessment of drug-drug interactions |
| Neuropsychiatry | Genetic predispositions for MDD | Extrinsic coagulation factors in suicidal behavior [20] | Biological suicide risk assessment in depression |
Table 2: Performance metrics of integrated biomarker signatures in disease prediction
| Disease Context | Single Biomarker Performance | Combined Signature Performance | Validation Cohort |
|---|---|---|---|
| Lung Adenocarcinoma (LUAD) | Variable predictive power [98] | PGS accurately stratified high-risk patients (AUC improved) [98] | TCGA + 4 independent GEO datasets |
| Glioblastoma (GBM) | MGMT promoter methylation inconsistent [98] | PGS predicted survival more accurately than single biomarkers [98] | TCGA + Rembrandt + 6 fresh tumors |
| Renal Cell Carcinoma | Limited predictive value for progression [99] | Computational model with CFB and SAA2 predicted metastasis [99] | Multiple patient cohorts |
| Major Depressive Disorder | Clinical assessment alone | Extrinsic coagulation pathway predicted suicidal behavior [20] | Two independent patient cohorts |
Experimental Workflows for Signature Discovery and Validation
Integrated Genomic and Functional Screening Pipeline
The discovery of robust biomarker signatures requires the integration of complementary data types. A proven workflow for cancer progression signature identification combines genomic data from patient samples with functional genomics data from experimental models:
Step 1: Data Acquisition - Retrieve gene expression profiles from The Cancer Genome Atlas (TCGA) and essential survival gene datasets from The Cancer Dependency Map (DepMap), which catalogs genes driving cancer progression through genome-wide RNAi screens [98].
Step 2: Signature Identification - Apply bioinformatics pipelines to identify genes highly associated with cancer progression, designated as progression gene signatures (PGSs). For lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), and glioblastoma (GBM), this approach revealed distinct PGSs for each cancer type [98].
Step 3: Analytical Validation - Evaluate signature performance using receiver operating characteristic (ROC) curve analysis. Research demonstrates that PGSs predict patient survival more accurately than previously identified single biomarkers [98].
Step 4: Independent Verification - Validate signatures in independent microarray datasets from Gene Expression Omnibus and freshly dissected tumor samples. For GBM, this included verification in six freshly dissected tumors from patients who underwent surgical resection [98].
Experimental and Computational Modeling for Cancer Progression
For renal cell carcinoma (RCC), an innovative approach combining experimental models with computational analysis has proven effective:
Step 1: Progressive Model Generation - Generate cell lines of increasing aggressiveness by serial passaging of mouse renal cancer RENCA cells in mice, creating multiple cell lines depicting major steps of tumor progression [99].
Step 2: Multi-Omics Profiling - Conduct large-scale transcriptome, genome, and methylome analyses of the progressively aggressive cell lines. These analyses reveal distinct clustering of cell lines without genomic variation, indicating epigenetic drivers of progression [99].
Step 3: Clinical Correlation - Perform clinical correlations of the experimental datasets with patient data, identifying SAA2 and CFB as soluble prognostic and predictive biomarkers of therapeutic response [99].
Step 4: Computational Modeling - Develop and validate a computational model predicting tumor progression and relapse based on the experimental data. Machine learning confirmed the importance of CFB and SAA2 together, which had the highest impact on distant metastasis-free survival [99].
Endogenous Biomarker Assessment for Drug Transport
The assessment of drug-drug interactions (DDIs) mediated by renal transporters has evolved to incorporate endogenous biomarkers:
Step 1: In Vitro Screening - Generate transporter inhibition potency parameters (IC₅₀ or Kᵢ values) for new molecular entities (NMEs) regarding uptake of known substrates for key renal transporters (OCT2, MATE1/2-K, OAT1, OAT3) [79].
Step 2: Static Risk Assessment - Apply regulatory-guided decision trees using the ratio of unbound maximum plasma concentration (Cmax,u) to IC₅₀ to assess DDI risk and determine need for clinical DDI studies [79].
Step 3: Clinical Biomarker Evaluation - Monitor plasma-based and urine-based endogenous biomarkers in early phase I studies to predict transporter-mediated DDIs. This approach can complement or potentially avoid conventional clinical DDI studies [79].
Step 4: Decision Framework Application - Implement a conceptual framework for assessment that integrates endogenous biomarker data to support more informed decisions about dedicated DDI studies [79].
Visualization of Integrated Workflows
Integrated Biomarker Discovery Workflow
Intrinsic and Extrinsic Apoptosis Pathways
Analytical Tools for Signature Optimization
CombiROC for Multi-Marker Analysis
The combinatorial analysis of multiple biomarkers requires specialized computational tools. CombiROC is an interactive web tool designed to help researchers accurately determine optimal marker combinations from diverse omics data [100]. This application addresses the challenge of selecting multimarker signatures, which traditionally requires integration of data signatures with sophisticated statistical methods.
Key features of CombiROC include:
- Sensitivity/Specificity Filtering: Data from different domains (proteomics, transcriptomics) can be analyzed using adjustable sensitivity/specificity filters
- Combinatorial Analysis: Computes sensitivity and specificity for all marker combinations, bypassing limitations imposed by different experimental approaches
- Interactive Visualization: Displays performance metrics through bubble plots where bubble size is proportional to the number of markers in each combination
- ROC Curve Generation: Automatically generates ROC curves with AUC calculations for selected marker panels
- Validation Support: Facilitates confirmation analyses to evaluate candidate biomarker panel performance and avoid overfitting
The application was validated with published data, confirming previously described marker combinations and identifying new, potentially superior combinations [100]. CombiROC is freely available at http://CombiROC.eu with a full online tutorial for users to practice with pre-loaded real datasets.
Machine Learning for Signature-Based Diagnostics
Advances in microfluidic biomarker screening technologies coupled with machine learning have enabled sophisticated signature-based diagnostics [101]. This approach utilizes unique combinations of multiple biomarkers or diagnostic "fingerprints" rather than discrete analyte measurements, improving both diagnostic accuracy and specificity.
Machine learning applications in signature-based diagnostics:
- Pattern Recognition: Identify complex relationships between biomarkers within a signature that may not be apparent through traditional statistical methods
- Dimensionality Reduction: Process high-dimensional data from multiplexed microfluidic devices that simultaneously screen multiple biomarkers
- Classification Models: Accurately classify disease states from healthy patients using biomarker signature profiles
- Image Processing: Analyze complex data outputs from advanced microfluidic detection mechanisms
These advanced data processing techniques are particularly valuable for analyzing enveloped biomarkers such as circulating tumor cells (CTCs) and exosomes, which provide both extra- and intra-cellular markers for analysis [101].
Research Reagent Solutions
Table 3: Essential research reagents and resources for intrinsic/extrinsic biomarker studies
| Reagent/Resource | Category | Specific Function | Example Applications |
|---|---|---|---|
| TCGA Datasets | Data Resource | Provides genomic, transcriptomic, and clinical data from patient samples | Identification of intrinsic cancer signatures [98] |
| DepMap/Project Achilles | Functional Resource | Genome-wide RNAi screens across cancer cell lines | Identification of essential survival genes [98] |
| CombiROC | Analytical Tool | Web-based combinatorial analysis of marker performance | Optimizing sensitivity/specificity of multi-marker panels [100] |
| Primary GBM Cells | Biological Model | Freshly dissected tumor cells from patients | Validation of biomarker signatures in relevant biological context [98] |
| RENCA Cell Line | Biological Model | Mouse renal cancer cells for serial passaging | Modeling progressive tumor aggressiveness [99] |
| Microfluidic Platforms | Technological Tool | Highly parallelized biomarker screening and isolation | Signature-based diagnostic development [101] |
| Antibody Panels | Detection Reagent | Target surface proteins on enveloped biomarkers | Isolation of CTCs and exosomes via immunoaffinity [101] |
Regulatory Pathways for Biomarker Integration
The integration of novel biomarkers into drug development follows structured regulatory pathways within the FDA's Center for Drug Evaluation and Research (CDER) [102]. Understanding these pathways is essential for researchers and drug development professionals seeking to translate biomarker signatures into clinically useful tools.
The two primary pathways for biomarker integration are:
Drug Approval Process - Biomarkers, whether established or novel, can be used within the context of a specific drug development program. Drug developers may use biomarkers as part of clinical trials to answer questions pertinent to a particular drug. For novel biomarkers, the drug developer is responsible for all aspects of biomarker development [102].
Biomarker Qualification Program (BQP) - This pathway is for biomarkers that may be used in multiple drug development programs. Once qualified, a biomarker can be used under its qualified context of use (COU) during the development of any candidate drug [102].
Additional regulatory mechanisms include:
- Critical Path Innovation Meetings (CPIMs) - Opportunities to discuss methodologies or technologies that might enhance drug development, including biomarkers in early development phases [102]
- Letters of Support (LOS) - Briefly describe CDER's thoughts on a biomarker's potential value and encourage further development, enhancing visibility and stimulating additional studies [102]
The strategic integration of intrinsic and extrinsic biomarkers represents a paradigm shift in diagnostic and prognostic modeling. The comparative analyses presented in this guide demonstrate that multi-marker signatures consistently outperform single biomarkers across diverse biological contexts and disease states. The experimental workflows, analytical tools, and reagent solutions outlined provide researchers with practical frameworks for implementing these integrated approaches.
Future developments in this field will likely focus on several key areas:
- Advanced Multi-Omics Integration: Combining genomic, proteomic, metabolomic, and epigenomic data to create more comprehensive biological signatures
- Dynamic Monitoring: Developing technologies for longitudinal tracking of biomarker signatures to capture disease progression and treatment response
- Standardization and Validation: Establishing rigorous standards for analytical and clinical validation of multi-marker signatures across diverse patient populations
- Regulatory Science: Evolving regulatory pathways to accommodate the unique characteristics of complex biomarker signatures
As these developments unfold, the integration of intrinsic and extrinsic biomarkers will continue to enhance the predictive power in both clinical diagnostics and therapeutic development, ultimately advancing the goals of precision medicine.
Evidence and Evaluation: Validating Biomarker Utility and Comparative Performance
The transition from preclinical discovery to clinical application represents one of the most challenging hurdles in drug development, with only approximately 15% of drugs advancing from Phase II to final approval [103]. A significant factor in these failures is incorrect drug target identification, accounting for over 50% of efficacy failures in late-stage clinical trials [103]. Validation frameworks serve as critical tools to bridge this translational divide by establishing standardized evidence-building processes that ensure research findings from model systems reliably predict human biological responses. These frameworks provide structured approaches for verifying that experimental tools generate accurate data (verification), confirming that analytical methods measure what they intend to measure (analytical validation), and demonstrating that measurements reflect relevant biological or clinical states (clinical validation) [104].
The growing complexity of biomedical research, particularly with the emergence of digital measures and complex computational models, has heightened the need for robust validation methodologies. Global regulatory bodies including the FDA and EMA have responded with increased scrutiny, issuing guidance documents that emphasize transparency, reliability, and context-specific validation for tools used in medicinal product development [105]. The EU AI Act further classifies medical AI systems as "high-risk," mandating rigorous validation and comprehensive documentation [105]. Within this evolving landscape, validation frameworks have become indispensable for establishing credibility and facilitating the translation of scientific discoveries into clinical applications, particularly in the comparison of intrinsic and extrinsic pathway activation biomarkers across experimental systems.
Comparative Analysis of Validation Frameworks
The V3 Framework: From Clinical to Preclinical Application
The Digital Medicine Society (DiMe) established the V3 framework as a comprehensive approach for validating digital health technologies in clinical research. This framework structures the validation process into three distinct evidence-building pillars: Verification, Analytical Validation, and Clinical Validation [106]. Verification ensures that digital technologies correctly capture and store raw data according to specifications. Analytical Validation assesses the precision and accuracy of algorithms that transform raw data into meaningful biological metrics. Clinical Validation confirms that these digital measures accurately reflect relevant clinical, biological, or functional states within a specified context of use [106] [104].
The 3Rs Collaborative's Translational Digital Biomarkers (TDB) initiative subsequently adapted this clinical framework for preclinical applications, creating an "in vivo V3 Framework" that tailors the validation process to the unique requirements of animal research [106] [104]. This adaptation maintains the three core pillars while adjusting for challenges specific to preclinical environments, such as sensor verification in variable laboratory conditions and biological validation that establishes relevance to animal models rather than human patients [104]. The framework emphasizes that responsibility for applying these validation standards is shared by technology developers, vendors, and end-users, with specific obligations depending on technology maturity and intended study use [104].
Framework Comparison Table
Table 1: Comparison of Validation Frameworks for Preclinical and Clinical Application
| Framework Component | DiMe V3 Framework (Clinical) | 3RsC In Vivo V3 Framework (Preclinical) | AI Validation (Clinical Trials) |
|---|---|---|---|
| Primary Scope | Digital measures in human clinical trials and healthcare | Digital measures in animal model research | AI-assisted workflows in clinical research |
| Verification Focus | Proper data capture by consumer-grade sensors (wearables, mobile devices) | Proper data capture in variable lab environments (cages, implants) | Data input integrity and preprocessing |
| Analytical Validation Focus | Algorithm performance on human-derived data | Algorithm performance accounting for species-specific behaviors and physiology | AI model performance, bias assessment, and reliability |
| Clinical/Biological Validation Focus | Correlation with human clinical outcomes and states | Reflection of relevant biological states in animal models | Clinical relevance and accurate data interpretation |
| Regulatory Context | FDA bioanalytical method validation guidance | Adapted from clinical frameworks; evolving regulatory acceptance | FDA draft guidance on credibility assessment; EU AI Act |
| Key Challenges | Patient compliance, real-world variability | Environmental interference, species translation | Bias mitigation, explainability, rapid technology evolution |
| Typical Outputs | Clinical-grade digital biomarkers | Translationally relevant digital measures | Validated, compliant AI tools for patient selection, data analysis |
Cross-Framework Validation Principles
Despite their different applications, common principles unite various validation approaches. First, context of use is paramount across all frameworks, defining the specific manner and purpose of application for any technology or methodology [104]. Second, documentation and traceability requirements appear consistently, emphasizing maintained audit trails for decisions and outputs [105]. Third, risk-based approaches govern the extent of validation needed, with higher-stakes applications warranting more rigorous validation processes [105]. These shared principles enable a continuous validation thread from early discovery through clinical development, particularly important for complex biological measurements like pathway activation biomarkers.
Pathway Activation Biomarkers in Translational Research
Intrinsic and Extrinsic Apoptosis Pathways
The intrinsic and extrinsic apoptosis pathways represent well-characterized biological processes frequently measured as indicators of therapeutic efficacy in oncology and other disease areas. The extrinsic pathway begins outside the cell when extracellular death ligands (such as FasL) bind to cell surface death receptors (such as Fas), initiating receptor oligomerization and formation of the Death-Inducing Signaling Complex (DISC) [35]. This complex activates caspase-8, which then cleaves and activates executioner caspases-3, -6, and -7, committing the cell to apoptotic death [35] [9].
The intrinsic pathway initiates from within the cell in response to internal stressors including DNA damage, oxidative stress, or cytotoxic compounds. These signals cause pro-apoptotic Bcl-2 family proteins (such as Bax and Bak) to permeabilize the mitochondrial outer membrane, releasing cytochrome c into the cytoplasm [35] [107]. Cytochrome c then binds to Apaf-1, forming the apoptosome complex that activates caspase-9, which in turn activates the same executioner caspases triggered by the extrinsic pathway [35] [107] [9].
Table 2: Key Biomarkers for Apoptosis Pathway Analysis
| Pathway | Initiation Biomarkers | Amplification/Integration Biomarkers | Execution Biomarkers | Functional Assays |
|---|---|---|---|---|
| Extrinsic Apoptosis | Death receptor oligomerization (Fas, TNFR1), FADD recruitment, Caspase-8 activation | BID cleavage, Mitochondrial involvement | Caspase-3/7 activation, PARP cleavage, DNA fragmentation | Death ligand sensitivity, DISC immunoprecipitation, Caspase-8 activity |
| Intrinsic Apoptosis | Cytochrome c release, Bax/Bak activation, Bcl-2 phosphorylation, p53 activation | Apoptosome formation, Caspase-9 activation | Caspase-3/7 activation, PS externalization, Nuclear condensation | Mitochondrial membrane potential (ΔΨm), BH3 profiling, Cytochrome c release |
| Common/Execution Phase | Caspase-3/7 activation, PARP cleavage, DNA fragmentation, Membrane blebbing | TUNEL assay, Annexin V staining, Caspase activity assays |
Coagulation Pathway Biomarkers
Similar to apoptosis, coagulation occurs through complementary intrinsic and extrinsic pathways that converge on a common terminal process. The extrinsic coagulation pathway initiates when vascular injury exposes tissue factor (TF), which binds and activates factor VII, ultimately leading to factor X activation [15]. The intrinsic pathway activates through exposed endothelial collagen, sequentially activating factors XII, XI, and IX before also converging on factor X [15] [107]. Both pathways then proceed through the common pathway where factor Xa converts prothrombin to thrombin, which subsequently converts fibrinogen to fibrin to form stable clots [15].
Research examining differential pathway contributions to thrombin generation has demonstrated the value of quantitative pathway assessment. One study used an anti-FIXa aptamer (RB006) to specifically inhibit the intrinsic pathway and BAX499 to downregulate tissue factor pathway inhibitor (TFPI) and enhance extrinsic pathway activity [108]. This approach revealed that intrinsic pathway inhibition significantly reduced thrombin generation in adult and maternal plasma but had minimal effect in cord plasma, indicating developmental differences in pathway utilization [108].
Pathway Visualization Diagrams
Diagram 1: Intrinsic and Extrinsic Apoptosis Pathway Convergence. The intrinsic pathway (yellow) initiates from intracellular stress, while the extrinsic pathway (green) begins with extracellular death ligands. Both converge on common executioner caspases (blue).
Diagram 2: Coagulation Cascade Pathway Convergence. The extrinsic pathway (green) initiates with tissue factor exposure, while the intrinsic pathway (yellow) begins with collagen exposure. Both converge at Factor Xa activation in the common pathway (blue).
Experimental Protocols for Pathway Validation
Thrombin Generation Assay for Coagulation Pathway Analysis
The calibrated automated thrombogram (CAT) assay provides a robust method for quantitatively assessing the functional contribution of intrinsic and extrinsic coagulation pathways to overall thrombin generation [108]. This protocol enables comparative evaluation of pathway utilization across different physiological states (adult, maternal, cord plasma) and assessment of pharmacological interventions on specific pathways.
Materials and Reagents:
- Platelet-poor plasma samples (citrated)
- Thrombinoscope reagent (2 pM tissue factor)
- Fluorogenic substrate (Z-Gly-Gly-Arg-AMC)
- Thrombin calibrator
- Low-affinity fluorogenic compound for calibration
- Anti-FIXa aptamer (RB006, 24 μg/ml final concentration) for intrinsic pathway inhibition
- BAX499 (200 nM final concentration) for TFPI inhibition (extrinsic pathway enhancement)
- 96-well microtiter plates
- Plate fluorometer capable of continuous measurement (390/460 nm filter)
Procedure:
- Prepare platelet-poor plasma by double centrifugation at 2000 × g for 20 minutes and aliquot for storage at -80°C until batch analysis.
- Thaw plasma samples rapidly at 37°C and maintain on ice until assay initiation.
- Add 80 μl of plasma to each well of the microtiter plate. Include calibrator wells with 20 μl thrombin calibrator added to 80 μl plasma.
- For pathway inhibition studies, pre-incubate plasma with RB006 (24 μg/ml) for intrinsic pathway inhibition or BAX499 (200 nM) for extrinsic pathway enhancement for 5 minutes at room temperature.
- Initiate thrombin generation by adding 20 μl of 2 pM tissue factor-based PPP reagent.
- Start the reaction by automated addition of 20 μl/well of CaCl₂-substrate buffer.
- Continuously monitor fluorescence for 90 minutes with readings every 20 seconds.
- Process acquired data using Thrombinoscope software with calibration against the thrombin calibrator well.
- Export lag time (initiation phase), peak thrombin (Cmax), and rate (velocity index) parameters for statistical analysis.
Data Interpretation: Comparative analysis of RB006 effects reveals the relative contribution of the intrinsic pathway to thrombin generation, while BAX499 effects indicate the degree of TFPI-mediated restraint on the extrinsic pathway [108]. This protocol demonstrated that intrinsic pathway inhibition significantly reduces thrombin generation in adult and maternal plasma but has minimal effect in cord plasma, indicating developmental differences in coagulation pathway utilization [108].
Apoptosis Pathway Activation Assessment
Comprehensive apoptosis assessment requires multiplexed approaches to distinguish intrinsic and extrinsic pathway activation and quantify downstream execution events.
Materials and Reagents:
- Cell culture samples (treated and untreated controls)
- Annexin V binding buffer
- Fluorochrome-conjugated Annexin V
- Propidium iodide (PI) or other viability dyes
- Caspase substrates and inhibitors
- Lysis buffer for protein extraction
- Antibodies for: active caspase-8 (extrinsic initiator), active caspase-9 (intrinsic initiator), cleaved caspase-3 (executioner), PARP cleavage
- TUNEL assay reagents
- Mitochondrial membrane potential dyes (TMRE, JC-1)
Procedure: Early Apoptosis Assessment (Annexin V/PI Assay):
- Harvest cells by gentle trypsinization or non-enzymatic methods.
- Wash cells twice with cold PBS and resuspend in Annexin V binding buffer.
- Add fluorochrome-conjugated Annexin V and incubate for 15 minutes in the dark.
- Add PI immediately before analysis by flow cytometry.
- Analyze samples within 1 hour using flow cytometry with appropriate fluorescence channels.
Caspase Activity Assessment:
- Prepare cell lysates using non-denaturing lysis buffer.
- Measure caspase activity using fluorogenic substrates (e.g., DEVD-AFC for caspase-3, IETD-AFC for caspase-8, LEHD-AFC for caspase-9).
- Monitor fluorescence release over time using a plate reader.
- Include caspase-specific inhibitors as controls for assay specificity.
Mitochondrial Membrane Potential Assessment:
- Load cells with TMRE (tetramethylrhodamine ethyl ester) at 50-200 nM for 30 minutes.
- Wash cells and analyze by flow cytometry or fluorescence microscopy.
- Include CCCP (carbonyl cyanide m-chlorophenyl hydrazone) as a positive control for membrane depolarization.
Western Blot Analysis of Apoptotic Proteins:
- Separate proteins by SDS-PAGE and transfer to membranes.
- Probe with antibodies against pathway-specific markers: active caspase-8 (extrinsic), active caspase-9 (intrinsic), cleaved caspase-3 (common executioner), PARP cleavage (downstream effect).
- Use chemiluminescence detection with appropriate secondary antibodies.
Data Interpretation: Predominant activation of caspase-8 with minimal caspase-9 activation indicates extrinsic pathway initiation, while the reverse pattern suggests intrinsic pathway activation. Combined caspase-8 and -9 activation indicates cross-talk between pathways. TMRE fluorescence loss confirms mitochondrial involvement characteristic of intrinsic apoptosis.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Reagents for Pathway Validation Studies
| Reagent Category | Specific Examples | Research Application | Validation Role |
|---|---|---|---|
| Pathway-Specific Inhibitors | Anti-FIXa aptamer (RB006), BAX499 (TFPI inhibitor) | Coagulation pathway contribution analysis [108] | Functional assessment of specific pathway necessity |
| Caspase Activity Assays | Fluorogenic substrates (DEVD-AFC, IETD-AFC, LEHD-AFC), Active caspase antibodies | Apoptosis pathway initiation and execution measurement [9] | Quantification of pathway-specific activation events |
| Mitochondrial Function Probes | TMRE, JC-1, MitoTracker Red, Cytochrome c antibodies | Intrinsic apoptosis pathway activation [35] [9] | Verification of mitochondrial membrane permeabilization |
| Death Receptor Ligands | Recombinant FasL, TRAIL, TNF-α | Extrinsic apoptosis pathway activation [35] [9] | Controlled stimulation of receptor-mediated apoptosis |
| Flow Cytometry Reagents | Annexin V conjugates, Propidium iodide, Cell viability dyes | Early vs. late apoptosis discrimination [9] | Multiparameter cell death stage assessment |
| DNA Fragmentation Assays | TUNEL assay kits, DNA laddering detection | Late-stage apoptosis confirmation [9] | Detection of irreversible apoptotic commitment |
| Computational Tools | Rosalind tensor factorization, Knowledge graph integration | Therapeutic target prediction and validation [103] | Prioritization of pathway targets for experimental testing |
The journey from preclinical models to clinical trial corroboration demands rigorous validation frameworks that span biological scales and experimental systems. The adaptation of the V3 framework from clinical to preclinical applications represents a significant advancement in creating a common language and standardized evidence requirements for translational research [106] [104]. Similarly, computational approaches like Rosalind's tensor factorization on heterogeneous graphs demonstrate how modern bioinformatics can prospectively identify therapeutic targets with an impressive 1 in 4 prediction accuracy for eventually proven therapeutic relationships [103].
For intrinsic and extrinsic pathway research specifically, robust validation requires both specific biochemical measurements and functional pathway interrogation through selective inhibition or activation. The thrombin generation assay with pathway-specific inhibitors [108] and multiplexed apoptosis assessment [9] provide exemplars of how quantitative pathway contribution can be measured and translated across model systems. As validation frameworks continue to evolve alongside technological advancements, their consistent application will be essential for strengthening the predictive power of preclinical research and ultimately improving the success rate of therapeutic development.
The pursuit of precise, reliable, and early disease detection and prognosis is a cornerstone of modern biomedical research. Within this endeavor, biomarkers—measurable indicators of biological processes or pharmacological responses to therapeutic intervention—play a pivotal role. Historically, research and clinical practice have often relied on single biomarkers for diagnostic or prognostic tasks. However, the inherent complexity and multifactorial nature of most diseases mean that a single molecule is frequently insufficient to capture the full pathological spectrum, leading to limitations in sensitivity and specificity [109]. Consequently, the strategy of combining multiple biomarkers into a single integrated panel has gained significant traction. This approach aims to provide a more holistic view of the disease state by aggregating complementary information from different biological pathways. This guide provides a systematic, evidence-based comparison of the performance of individual biomarkers versus multi-marker panels across various disease contexts, including sepsis, Alzheimer's disease, and several cancer types. The analysis is framed within the specific context of research on intrinsic and extrinsic pathway activation, which are fundamental mechanisms in cellular processes like apoptosis and immune response.
Comparative Performance Data Across Disease Models
Quantitative data from recent studies consistently demonstrate the superior performance of combined biomarker panels over individual markers. The following tables summarize key head-to-head comparisons across different diseases.
Table 1: Diagnostic Performance of Individual vs. Combined Biomarkers
| Disease | Biomarker Type | Individual Biomarker Performance (AUC) | Combined Panel Performance (AUC) | Study Details |
|---|---|---|---|---|
| Sepsis Diagnosis [109] | Presepsin | 0.821 | PHO Panel: 0.892 | ICU patients (n=411); Panel: Presepsin, HE4, OI |
| HE4 | 0.803 | |||
| Oxygenation Index (OI) | 0.752 | |||
| Alzheimer's Disease (Amyloid Positivity) [110] | pTau217 | > 0.90 | Combined Panel: > 0.92 | Cohort (n=371); Panel: pTau217, pTau181, GFAP, NFL, Aβ42/40, total tau |
| Hepatocellular Carcinoma (HCC) Detection [111] | RANSE2, TNF-α, or MAP3K7 (Individual performance not specified) | 3-Marker Panel: 0.98 (Accuracy: 98.4%) | Patients & controls (n=47); Panel: RANSE2, TNF-α, MAP3K7 | |
| Sepsis Mortality Prediction [112] | Individual tsRNAs | 0.827 - 0.837 | tsRNA Signature: 0.967 | ICU sepsis patients (n=26); Serum small RNA profiling |
| Individual miRNAs | 0.797 - 0.850 | miRNA Panel: 0.902 |
Table 2: Prognostic and Advanced Lesion Detection Performance
| Disease / Context | Application | Individual Marker / Standard Test | Combined Panel Performance | Study Details |
|---|---|---|---|---|
| Sepsis [109] | 30-Day Mortality Prediction | Presepsin, HE4, OI (Individual AUCs not specified) | PHO Panel AUC: 0.706 | Superior to individual biomarkers |
| Colorectal Cancer (CRC) [113] | Advanced Precancerous Lesion (APL) Detection | FIT (Fecal Immunochemical Test) | Sensitivity: 23.8% | Meta-analysis of 44 studies |
| Cologuard (mt-sDNA panel) | Sensitivity: 42.4% | Panel: KRAS mutation, methylated BMP3 & NDRG4, FIT | ||
| SDC2 + SFRP1/2 (Methylated) | Sensitivity: 89.2% | Stool-based ctDNA panel | ||
| SDC2 + TFPI2 (Methylated) | Sensitivity: 100% | Stool-based ctDNA panel | ||
| Cardiovascular Disease & Chronic Kidney Disease (CKD) [114] | 2-Year Cardiovascular Event Prediction | Not specified | C-statistic: 0.77 | Patients undergoing catheterization (n=446); Panel: KIM-1, NT-proBNP, Osteopontin, TIMP-1 |
Detailed Experimental Protocols from Key Studies
Protocol 1: Sepsis Diagnosis and Prognosis with the PHO Panel
This protocol outlines the methodology for validating the Presepsin, HE4, and Oxygenation Index (OI) panel for sepsis in an ICU setting [109].
- Study Population and Design: A prospective, single-center study was conducted with 411 ICU patients, stratified into a non-sepsis group (n=165) and a sepsis/septic shock group (n=246), based on Sepsis-3 criteria.
- Sample Collection and Clinical Parameters: Residual serum and plasma samples collected within 24 hours of ICU admission were used. Clinical and laboratory data extracted within the same timeframe included:
- Clinical Scores: SOFA score, Glasgow Coma Scale (GCS) score.
- Oxygenation Index (OI): Calculated as the ratio of arterial oxygen partial pressure (PaO₂) to fractional inspired oxygen (FiO₂).
- Conventional Biomarkers: White blood cell count, procalcitonin (PCT), C-reactive protein (CRP).
- Organ Function Markers: Creatinine, bilirubin, platelet count.
- Biomarker Quantification:
- Presepsin and HE4: Levels were measured from stored serum samples using electrochemiluminescence assays.
- Machine Learning and Statistical Analysis:
- Model Training: Four machine learning algorithms (Decision Tree, Linear Support Vector Classifier, Logistic Regression, and Gradient Boosting) were evaluated using 5-fold cross-validation.
- Performance Evaluation: The Gradient Boosting model identified Presepsin, HE4, and OI as top predictors. The performance of individual biomarkers and the combined PHO panel was assessed using Receiver Operating Characteristic (ROC) curve analysis, reporting the Area Under the Curve (AUC), sensitivity, and specificity.
Protocol 2: Early Pancreatic Cancer Detection via a Glycan and Protein Panel
This protocol describes a multi-laboratory effort to develop a blood test for early pancreatic ductal adenocarcinoma (PDAC) [115].
- Study Design and Specimen Validation: To ensure robust and reproducible results, the study employed a common set of specimens with consistent collection methods, uniform sample handling, blinded distribution, and independent biostatistical analysis across participating labs.
- Biomarker Selection and Measurement: The panel combined biomarkers previously identified as relevant to pancreatic cancer:
- CA19-9: The current gold-standard carbohydrate antigen for PDAC.
- CA199.STRA (STRA): A sugar identified in 2016, structurally associated with CA19-9.
- LRG1: A protein biomarker (Leucine-Rich Alpha-2-Glycoprotein 1).
- Performance Analysis: The diagnostic accuracy of the individual biomarkers and the multi-marker panel was evaluated and compared. The primary metrics were the improvement in accurate detection rate and the reduction in false-positive rate compared to CA19-9 alone.
Signaling Pathways and Biomarker Logic
The rationale for combining biomarkers often lies in their connection to distinct but interconnected biological pathways. The following diagrams illustrate key intrinsic and extrinsic pathways relevant to biomarker research.
Apoptosis Signaling Pathways
Diagram 1: Apoptosis Pathway Crosstalk. This diagram illustrates the core intrinsic (mitochondrial) and extrinsic (death receptor) apoptosis pathways. The extrinsic pathway can amplify the intrinsic pathway via caspase-8-mediated cleavage of BID (a key biomarker identified in ICH research [12]), demonstrating a critical point of crosstalk. Combined biomarker panels can simultaneously measure activators from both pathways, providing a more comprehensive assessment of cell death status.
Host Response and Organ Dysfunction in Sepsis
Diagram 2: Sepsis Biomarker Logic. The superior performance of the PHO panel in sepsis [109] is logical because it captures complementary information from different aspects of the disease: Presepsin reflects the intensity of the innate immune response, HE4 is linked to tissue injury and fibrotic processes, and the Oxygenation Index directly measures consequent organ (lung) dysfunction.
The Scientist's Toolkit: Essential Research Reagents
Successful development and validation of biomarker panels require a specific set of reagents and tools. The following table details key solutions used in the featured studies.
Table 3: Key Research Reagent Solutions for Biomarker Panel Studies
| Reagent / Solution | Function / Application | Example from Featured Research |
|---|---|---|
| Electrochemiluminescence Assays | Quantitative measurement of protein biomarkers (e.g., Presepsin, HE4) in serum/plasma. Known for high sensitivity and broad dynamic range. | Used for quantifying Presepsin and HE4 levels in the sepsis PHO panel study [109]. |
| High-Throughput Sequencing Platforms | Comprehensive profiling of RNA populations, including small non-coding RNAs (miRNAs, tsRNAs). | Illumina NovaSeq 6000 used for serum small RNA profiling in sepsis mortality study [112]. |
| Multiplex Immunoassays / Targeted Proteomics | Simultaneous measurement of multiple protein biomarkers from a single sample, conserving valuable specimen volume. | Used in the cardiovascular risk study to measure a panel of 4 protein biomarkers [114]. |
| RNA Extraction Kits (Serum/Plasma) | Specialized kits for isolation of high-quality, cell-free RNA from biofluids, which is crucial for liquid biopsy applications. | miRNeasy Serum/Plasma Kit (Qiagen) used for extracting total RNA from serum [112]. |
| Demethylation & RNA Modification Kits | Enzymatic treatment (e.g., with AlkB demethylase) to remove common RNA modifications, improving adapter ligation efficiency in small RNA sequencing. | An optimized library prep protocol included this step to enhance detection of tRNA-derived small RNAs (tsRNAs) [112]. |
| Machine Learning Algorithms | Computational tools for identifying the most predictive biomarker combinations and building classification models from high-dimensional data. | Gradient Boosting, Decision Tree, and Logistic Regression used to develop and validate the PHO panel [109]. |
Systemic sclerosis (SSc) is a complex autoimmune fibrotic disorder characterized by vascular dysfunction, immune system activation, and progressive fibrosis of the skin and internal organs. Disease heterogeneity presents significant challenges for prognosis and treatment personalization. The modified Rodnan skin score (MRSS) serves as a primary clinical measure for assessing skin disease severity in SSc, particularly in the diffuse cutaneous variant (dcSSc). While fibroblasts and immune cells have traditionally been the focus of SSc research, recent evidence has identified keratinocytes as active contributors to disease pathology through cell-intrinsic and cell-extrinsic signaling circuits [6].
This case study examines two emerging keratinocyte-derived biomarkers—KRT6A and S100A8—evaluating their performance against established and alternative biomarkers for assessing skin disease severity in SSc. We focus specifically on their roles within intrinsic and extrinsic pathway activation networks, providing objective comparison data and detailed experimental protocols to guide researchers and drug development professionals in biomarker validation and application.
Biomarker Performance Comparison
The following tables provide a comprehensive comparison of established and emerging biomarkers for SSc, with emphasis on their association with skin disease severity.
Table 1: Biomarker Performance Characteristics for SSc Skin Severity
| Biomarker | Cellular Source | Association with MRSS | Mechanistic Insight | Key Supporting Evidence |
|---|---|---|---|---|
| KRT6A | Keratinocytes | Positive correlation [6] | Barrier alarmin; promotes inflammation via JAK1-STAT3 activation [116] | Machine learning identification; IHC validation in human SSc skin [6] |
| S100A8/A9 | Keratinocytes, Myeloid cells | Positive correlation [6] | DAMP; TLR4 activation; MMP stimulation [117] | Protein validation in SSc patient keratinocytes; correlation with severity [6] |
| Anti-Topo I | Serum (Autoantibody) | Associated with dcSSc, severe skin disease [118] [119] | Targets DNA topoisomerase I; poor prognosis marker | Clinical cohort studies; prognostic stratification [118] [119] |
| Anti-RNA Pol III | Serum (Autoantibody) | Associated with rapid skin thickening [118] [119] | Targets RNA polymerase III; rapid disease progression | Clinical cohort studies; renal crisis association [118] [119] |
| CXCL4 | Platelets, Immune cells | Positive correlation [119] | Chemokine; fibroblast activation; endothelial dysfunction | Multicenter prospective studies [119] |
| IL-6 | Immune cells, Fibroblasts | Positive correlation [119] | Pro-fibrotic cytokine; promotes myofibroblast differentiation | Serum levels correlate with early progressive skin sclerosis [119] |
Table 2: Technical Validation Data for Key SSc Skin Severity Biomarkers
| Biomarker | Detection Method | Sample Type | Sensitivity/Specificity for Severe Skin Disease | Longitudinal Monitoring Utility |
|---|---|---|---|---|
| KRT6A | IHC, scRNA-seq, Western Blot | Skin biopsy, Keratinocyte cultures | High specificity for active progressive disease [6] [116] | Experimental evidence supports monitoring potential [116] |
| S100A8/A9 | ELISA, IHC, Proteomics | Serum, Skin biopsy, Plasma | Correlates with inflammatory activity [6] [117] | Established for monitoring in other inflammatory diseases [117] [120] |
| Anti-Topo I | ELISA, Immunoprecipitation | Serum | ~40% sensitivity for dcSSc; >95% specificity for SSc [118] | Prognostic value at diagnosis; limited for short-term monitoring |
| Anti-RNA Pol III | ELISA, Line immunoassay | Serum | ~20% sensitivity for dcSSc; >98% specificity for SSc [118] | Predicts rapid progression; associated with cancer development |
| miR-150 | qRT-PCR, Sequencing | Serum | Reduced levels correlate with severe manifestations [118] | Potential for monitoring disease progression |
Experimental Protocols for Biomarker Validation
Keratinocyte-Specific Biomarker Identification Using scRNA-seq and Machine Learning
Objective: Identify keratinocyte-specific biomarkers predictive of SSc skin severity using single-cell RNA sequencing and machine learning approaches [6].
Methodology Details:
- Patient Recruitment and Sampling: Skin biopsies collected from 24 SSc patients across MRSS severity spectrum and matched healthy controls
- Single-Cell RNA Sequencing:
- Tissue dissociation and single-cell suspension preparation
- 10X Genomics platform for scRNA-seq library preparation
- Sequencing on Illumina platforms (minimum 50,000 reads per cell)
- Machine Learning Analysis:
- LASSO (Least Absolute Shrinkage and Selection Operator) regression with L1 regularization
- Input features: cell type-specific gene expression patterns
- Prediction target: MRSS values
- Cross-validation to prevent overfitting
- Correlation Network Analysis:
- Construction of cell-intrinsic and cell-extrinsic co-expression networks
- Identification of key signaling pathways and intercellular communication
Validation Approaches:
- Immunohistochemistry for protein-level confirmation on independent patient cohorts
- Western blot analysis of keratinocyte cell cultures
- Correlation of biomarker expression with clinical disease progression data
KRT6A Functional Validation in Skin Inflammation Models
Objective: Investigate the functional role of KRT6A in skin inflammation and validate its mechanistic contribution to SSc pathogenesis [116].
Methodology Details:
- In Vivo Models:
- LL37-induced rosacea-like inflammation in BALB/c mice
- Imiquimod-induced psoriasis-like skin inflammation
- Intradermal injection of KRT6A-targeting adeno-associated virus (AAV-shKRT6A) or lentivirus for overexpression
- Clinical assessment of skin inflammation (redness area and score measurement)
- Molecular Mechanism Studies:
- Co-immunoprecipitation assays in HaCaT keratinocytes to identify KRT6A interaction partners
- JAK1-STAT3 pathway activation analysis by Western blot
- Assessment of RNF41-mediated JAK1 ubiquitination
- Proinflammatory cytokine profiling (IL-6, IL-1β, TNF-α) by ELISA
- Human Tissue Validation:
- Immunohistochemistry on skin biopsies from SSc patients and healthy controls
- Quantitative analysis of KRT6A expression in epidermal layers
Key Reagents:
- siKRT6A and control sequences (mouse-siKRT6A-1: CTCAGCTCTTCTACCATCA; human-siKRT6A-1: CAACAAGTTTGCCTCCTTCAT)
- Anti-KRT6A antibody (Abcam) for IHC
- JAK1 and STAT3 inhibitors for mechanistic studies
Signaling Pathways and Molecular Mechanisms
KRT6A-Mediated JAK1-STAT3 Inflammatory Signaling
Pathway Title: KRT6A-JAK1-STAT3 Inflammatory Signaling
This diagram illustrates the mechanistic pathway through which keratinocyte-derived KRT6A promotes skin inflammation in SSc. Following epidermal barrier disruption, KRT6A expression increases significantly. Rather than functioning solely as a structural protein, KRT6A acts as a barrier alarmin that impairs the interaction between the E3 ubiquitin ligase RNF41 and JAK1 [116]. This disruption of normal protein degradation leads to JAK1 stabilization and subsequent STAT3 phosphorylation. Activated STAT3 translocates to the nucleus and drives the expression of pro-inflammatory cytokines including IL-6 and TNF-α, creating a sustained inflammatory microenvironment that promotes fibrosis and correlates with clinical skin severity in SSc [6] [116].
S100A8/A9 as a Damage-Associated Molecular Pattern in SSc
Pathway Title: S100A8/A9-Mediated Inflammation and Tissue Damage
S100A8/A9 (calprotectin) functions as a damage-associated molecular pattern (DAMP) released by activated keratinocytes and myeloid cells in SSc [6] [117]. This heterodimer activates Toll-like receptor 4 (TLR4) signaling, leading to NF-κB pathway activation and subsequent production of pro-inflammatory cytokines including IL-1β, IL-6, and TNF-α [117] [120]. Additionally, S100A8/A9 directly stimulates matrix metalloproteinase (MMP) expression in target cells, contributing to tissue remodeling and joint destruction in inflammatory arthritis models [117] [120]. The resulting inflammatory microenvironment promotes immune cell recruitment, tissue damage, and fibrosis, with serum and tissue levels correlating with both inflammatory activity and structural damage in SSc [6].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Keratinocyte Biomarker Studies
| Reagent/Category | Specific Examples | Research Application | Technical Considerations |
|---|---|---|---|
| Keratinocyte Cell Models | Primary human keratinocytes, HaCaT immortalized line | In vitro mechanistic studies, pathway analysis | Primary cells better reflect physiology; immortalized lines for reproducibility |
| SSc Animal Models | LL37-induced inflammation, Imiquimod-induced psoriasis | In vivo functional validation of biomarker role | Model selection depends on specific research question (inflammatory vs. fibrotic) |
| KRT6A Modulators | siKRT6A (CAACAAGTTTGCCTCCTTCAT), AAV-shKRT6A | Functional studies of KRT6A in inflammation | Multiple sequences recommended to control for off-target effects |
| S100A8/A9 Detection | In-house mouse S100A8/A9 ELISA, Commercial ELISAs | Quantification in serum, tissue extracts, conditioned media | Species-specific assays critical for accurate measurement |
| Pathway Inhibitors | JAK inhibitors (Tofacitinib), STAT3 inhibitors | Mechanistic studies of KRT6A signaling pathway | Use multiple inhibitors targeting different pathway components for validation |
| IHC Antibodies | Anti-KRT6A (Abcam), Anti-S100A8/A9 | Spatial protein localization in patient tissues | Optimize antigen retrieval for formalin-fixed paraffin-embedded tissues |
| scRNA-seq Platform | 10X Genomics, Illumina sequencing | Cell type-specific biomarker discovery | Single-cell resolution essential for identifying keratinocyte-specific signals |
Discussion: Comparative Biomarker Utility in Research and Development
The integration of keratinocyte-derived biomarkers KRT6A and S100A8 into SSc research represents a significant advancement in understanding disease pathogenesis. While autoantibodies like anti-topoisomerase I and anti-RNA polymerase III remain valuable for patient classification and prognosis [118] [119], keratinocyte-specific biomarkers offer distinct advantages for understanding underlying molecular mechanisms and assessing treatment responses.
KRT6A demonstrates particular promise as it bridges epidermal barrier dysfunction with innate immune activation through the newly identified JAK1-STAT3 mechanism [116]. This pathway not only provides insight into disease pathogenesis but also suggests potential therapeutic targets. The machine learning approach used to identify KRT6A highlights how computational methods can extract novel insights from complex single-cell datasets [6].
S100A8/A9 offers complementary value as a measurable inflammatory DAMP that correlates with disease activity [6] [117]. Its established role in other inflammatory arthritides [117] [120] and association with joint destruction in seronegative arthritis models suggests potential utility for monitoring both cutaneous and articular manifestations in SSc.
From a drug development perspective, these keratinocyte-derived biomarkers enable more precise patient stratification for clinical trials and provide pharmacodynamic markers for targeted therapies. The JAK1-STAT3 pathway associated with KRT6A is particularly amenable to therapeutic intervention with existing JAK inhibitors, potentially enabling rapid translation of these findings into clinical applications.
This case study demonstrates that KRT6A and S100A8 represent validated biomarkers that capture critical aspects of SSc skin severity through distinct but complementary mechanisms. Their identification underscores the importance of keratinocytes in SSc pathogenesis and provides new tools for mechanistic studies and therapeutic development. When selected based on specific research objectives and used with appropriate methodologies, these biomarkers significantly enhance our ability to investigate disease mechanisms, stratify patient populations, and evaluate novel therapeutic approaches in systemic sclerosis.
Future directions should include standardized assay development, validation in larger prospective cohorts, and integration with other biomarker classes to create comprehensive prognostic models that reflect the multifaceted nature of SSc pathogenesis.
Immune checkpoint inhibitors (ICIs) targeting the PD-1/PD-L1 axis have revolutionized cancer treatment, yet they benefit only a subset of patients—approximately 20-30% across various cancer types [59] [121]. This clinical challenge has intensified the search for reliable predictive biomarkers to guide treatment decisions. Currently, programmed death-ligand 1 (PD-L1) expression and tumor mutational burden (TMB) serve as the most established biomarkers in clinical practice [122] [123]. However, both have significant limitations: PD-L1 expression exhibits spatial and temporal heterogeneity and is susceptible to subjective interpretation in immunohistochemical staining [121], while TMB represents a quantitative measure of mutations that fails to account for their qualitative immunogenic potential [121] [124].
The emerging understanding of tumor-intrinsic factors influencing immune responses has revealed that specific genomic alterations can dramatically reshape the tumor immune microenvironment (TIME) and modulate ICI efficacy [125] [126]. Against this backdrop, a novel Gene mutation-based Predictive Signature (GPS) classifier, incorporating mutations in three genes—STK11, FAT1, and ERBB4—has been developed as a potential solution to the limitations of conventional biomarkers [121]. This case study provides a comprehensive comparison between the GPS classifier and traditional biomarkers (PD-L1 and TMB), evaluating their respective performances in predicting immunotherapy outcomes within the context of intrinsic and extrinsic pathway activation biomarkers.
Biomarker Fundamentals: Mechanisms and Limitations
Conventional Biomarkers: PD-L1 and TMB
PD-L1 Expression is currently the most widely used biomarker for ICI therapy. The biological rationale stems from the PD-1/PD-L1 axis mechanism where PD-L1 on tumor cells binds to PD-1 on T cells, transmitting an inhibitory signal that suppresses T-cell activation and facilitates immune evasion [127] [123]. ICIs blocking this interaction can restore anti-tumor immunity. However, PD-L1 expression has demonstrated variable predictive value across cancer types, with some PD-L1-negative patients responding to therapy and some PD-L1-positive patients showing resistance [121] [123]. Technical challenges include lack of standardization in assay platforms, scoring systems, and cutoff values across different cancer types [122].
Tumor Mutational Burden (TMB) measures the total number of somatic mutations per megabase of DNA. The underlying hypothesis posits that higher mutation loads increase the probability of generating immunogenic neoantigens that can be recognized by T cells, thereby enhancing response to checkpoint blockade [123] [124]. While TMB-high status (often defined as ≥10 mutations/Mb) has received FDA approval as a pan-tumor biomarker for pembrolizumab, limitations persist. TMB calculation includes all nonsynonymous mutations regardless of their functional impact on immunogenicity, and the optimal cutoff varies across cancer types [121] [124]. Furthermore, TMB assessment suffers from technical variability across sequencing platforms and panel sizes, complicating clinical implementation [121].
The GPS Classifier: A Novel Gene Signature
The GPS classifier represents a paradigm shift from quantitative mutation counting to focused assessment of specific functional mutations. This signature was developed through machine learning analysis of non-synonymous mutations in lung adenocarcinoma patients treated with ICIs [121]. The classifier incorporates three genes with distinct biological impacts on tumor-immune interactions:
STK11 (Serine/Threonine Kinase 11): A tumor suppressor gene whose loss is associated with an immunosuppressive tumor microenvironment, characterized by reduced T-cell infiltration and primary resistance to PD-1 axis inhibitors [125] [128]. STK11 mutations are frequently co-occur with KRAS mutations and drive a "cold" tumor phenotype resistant to immunotherapy [125].
FAT1: A tumor suppressor gene encoding a protocadherin involved in planar cell polarity and Wnt signaling. FAT1 mutations exert a positive influence on ICB response, though their precise mechanism requires further elucidation [121].
ERBB4: A member of the epidermal growth factor receptor family, with mutations that similarly correlate positively with immunotherapy benefit [121].
The GPS algorithm classifies patients into three distinct categories based on their mutation profile: GPS-pos (G1) (presence of FAT1 or ERBB4 mutations), GPS-zero (G2) (no mutations in any of the three genes), and GPS-neg (G3) (presence of STK11 mutation) [121].
Figure 1: GPS Classifier Components and Their Biological Implications. STK11 mutations (red) drive immunosuppression and ICI resistance, while FAT1 and ERBB4 mutations (green) are associated with favorable immunotherapy responses.
Experimental Comparison: Methodologies and Performance
Development and Validation of the GPS Classifier
The GPS classifier was developed through a rigorous analytical process utilizing multiple machine learning approaches. The initial training cohort comprised 179 lung adenocarcinoma (LUAD) patients treated with ICIs from the Rizvi cohort, with sequencing data obtained via MSK-IMPACT panels [121]. To ensure balanced comparison, researchers implemented propensity score matching (PSM) to control for potential confounders including sex, age, smoking history, treatment lines, and treatment type between responders (durable clinical benefit, DCB) and non-responders (no durable benefit, NDB) [121].
Machine Learning Framework: After removing synonymous and low-frequency mutations, a nonsynonymous mutation matrix of 165 samples across 47 genes was generated. Five distinct machine learning algorithms—Naive Bayes (NB), Random Forest (RF), Support Vector Machine (SVM), Linear Discriminant Analysis (LDA), and XGBoost—were applied to identify mutations most predictive of treatment response [121]. Feature selection was performed using recursive feature elimination (RFE) within a leave-one-out cross-validation (LOOCV) framework. The optimal gene ensemble was determined by identifying overlapping candidates across multiple models, resulting in the final three-gene signature (STK11, FAT1, ERBB4) [121].
Validation Studies: The predictive capacity of GPS was subsequently validated in an independent cohort of 75 NSCLC patients (the Hellmann cohort) treated with combined PD-1 and CTLA-4 blockade [121]. This independent validation confirmed the classifier's robust performance across different patient populations and immunotherapy regimens.
Figure 2: GPS Classifier Development Workflow. The classifier was developed through propensity score matching, multiple machine learning approaches, and validated in an independent cohort.
Performance Comparison Across Biomarkers
Direct comparison of the GPS classifier with conventional biomarkers reveals significant differences in predictive power across multiple validation studies:
Table 1: Performance Comparison of Predictive Biomarkers for Immunotherapy Response
| Biomarker | Methodology | Predictive Value | Limitations |
|---|---|---|---|
| GPS Classifier | NGS-based detection of STK11, FAT1, and ERBB4 mutations | ORR: 100% in GPS-pos vs. 0% in GPS-neg patients [121]; Superior predictive accuracy vs. PD-L1 and TMB in multivariate analysis [121] | Limited validation across diverse cancer types; Biological mechanisms of FAT1 and ERBB4 not fully elucidated |
| PD-L1 Expression | IHC (22C3 PharmDx assay); TPS scoring | Borderline significance in predicting response (p=0.0532) in validation cohort [121]; Variable predictive value across tumor types | Spatial and temporal heterogeneity; Subjective interpretation; Multiple scoring systems |
| TMB | NGS sequencing; mutations per megabase | Associated with improved outcomes but inferior to GPS in multivariate analysis [121]; Fails to predict response in some cancer types [121] | Includes non-immunogenic mutations; Platform-dependent; No universal cutoff |
In the validation cohort, the GPS classifier demonstrated remarkable stratification power, with 100% objective response rate (ORR) in GPS-pos patients compared to 0% in GPS-neg patients [121]. Furthermore, GPS-positive patients showed significantly prolonged progression-free survival (PFS) compared to other groups (p=0.0009) [121].
Multivariate logistic regression analysis confirmed that GPS served as the only significant independent predictor for ICB outcome (OR=2.627, 95% CI=1.172-5.887, p=0.019) after adjusting for various clinicopathological variables including age, sex, smoking, EGFR status, TP53/KRAS co-mutation, KEAP1 status, and TMB [121].
Biological Mechanisms Underpinning GPS Components
The biological plausibility of the GPS classifier strengthens its predictive value, particularly for STK11, which has been extensively characterized as a key modulator of the tumor immune microenvironment:
STK11/LKB1 Mechanisms: STK11 mutations occur in approximately 16.3% of lung adenocarcinomas and are strongly associated with primary resistance to PD-1 pathway inhibitors [128]. The inactivation of this tumor suppressor gene drives profound immunosuppression through multiple mechanisms:
- Development of an immunologically "cold" tumor microenvironment with significantly reduced CD8+ T-cell infiltration [128]
- Downregulation of PD-L1 expression on tumor cells despite high TMB [125]
- Alteration of metabolic pathways that contribute to an immunosuppressive environment [125]
- Frequent co-occurrence with KRAS mutations, defining a particularly aggressive NSCLC subtype with poor outcomes following ICI therapy [125]
The strength of the STK11 mutation as a negative predictor is highlighted by SHAP (SHapley Additive exPlanations) value analysis, where it ranked first among all mutations in terms of negative impact on ICB response [121].
Table 2: Clinical Impact of Specific Mutations on Immunotherapy Response
| Genetic Alteration | Frequency in LUAD | Impact on TIME | Effect on ICI Response | HR for Poor Outcome |
|---|---|---|---|---|
| STK11 mutation | 16.3% [128] | "Cold" TIME; Reduced T-cell infiltration [128] | Primary resistance [125] | KRAS/STK11 co-mutation: HR 1.63 for OS [125] |
| KEAP1 mutation | Often co-occurs with KRAS [125] | Immunosuppressive metabolism [125] | Reduced benefit from CIT [125] | KRAS/KEAP1 co-mutation: HR 1.57 for OS [125] |
| STK11+KEAP1 co-mutation | ~10% of KRAS-mutant [125] | Highly immunosuppressive [125] | Marked resistance [125] | HR 1.93 for OS vs wild-type [125] |
| FAT1 mutation | Component of GPS [121] | Not fully characterized | Positive predictor [121] | Favorable DCB rate [121] |
| ERBB4 mutation | Component of GPS [121] | Not fully characterized | Positive predictor [121] | Favorable DCB rate [121] |
Technical Implementation and Research Applications
Experimental Protocols for Biomarker Assessment
GPS Classifier Detection Protocol:
- Sample Preparation: DNA extraction from FFPE tumor samples using validated kits (e.g., QIAamp DNA FFPE Kit) [122]
- Sequencing Library Preparation: Utilize targeted NGS panels (e.g., MSK-IMPACT) with hybridization capture-based target enrichment [121] [124]
- Sequencing: Perform high-depth sequencing (>500x) on Illumina platforms (e.g., HiSeq 3000) [122]
- Bioinformatic Analysis:
- Align sequencing reads to reference genome (hg19) using BWA
- Call variants using established pipelines (MuTect2 for SNVs, GATK for InDels)
- Annotate variants and filter for STK11, FAT1, and ERBB4 mutations
- Classification: Categorize patients as GPS-pos (FAT1 or ERBB4 mutant), GPS-neg (STK11 mutant), or GPS-zero (no mutations) [121]
Comparative Biomarker Assessment Methodologies:
Table 3: Methodological Comparison of Biomarker Detection Platforms
| Parameter | GPS Classifier | TMB Assessment | PD-L1 IHC |
|---|---|---|---|
| Required Sample | DNA from tumor tissue | DNA from tumor tissue | FFPE tissue section |
| Platform | Targeted NGS (≥1 Mb) | Large NGS panel or WES | Immunohistochemistry |
| Analysis | Automated mutation calling + classification | Mutation count normalized to panel size | Pathologist interpretation of TPS |
| Standardization | Universal classification system | Varies by panel size/gene content | Multiple assays (22C3, SP142, etc.) |
| Turnaround Time | ~7-10 days | ~7-14 days | ~2-3 days |
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagents for Biomarker Investigation
| Reagent/Category | Specific Examples | Research Application |
|---|---|---|
| Targeted NGS Panels | MSK-IMPACT, FoundationOne CDx | Simultaneous assessment of GPS genes, TMB, and other genomic alterations [121] [124] |
| DNA Extraction Kits | QIAamp DNA FFPE Kit | High-quality DNA extraction from archival tumor samples [122] |
| PD-L1 IHC Assays | 22C3 PharmDx, SP142, SP263 | Standardized PD-L1 protein expression analysis [122] |
| Immunophenotyping Panels | Multiplex IHC (mIHC), Flow cytometry | Characterization of tumor immune microenvironment (CD8+ T cells, Tregs, macrophages) [121] |
| Cell Culture Models | Tumor organoid-PBMC co-culture systems | Functional validation of biomarker-predicted responses ex vivo [121] |
Discussion: Implications for Biomarker Development
Clinical Applicability and Limitations
The GPS classifier represents a significant advancement in the field of predictive biomarkers for immunotherapy by addressing fundamental limitations of current standards. Where TMB quantifies mutation quantity without regard to functional impact, the GPS classifier incorporates specific mutations with demonstrated biological significance in modulating tumor-immune interactions [121] [124]. This approach aligns with growing understanding that specific mutations, such as those in STK11, can serve as powerful determinants of immunotherapy response regardless of overall mutation burden [125] [128].
From a practical standpoint, the GPS classifier offers technical advantages for clinical implementation. Its binary mutation-based classification system eliminates the need for arbitrary cutoffs that plague TMB interpretation [124]. Furthermore, it can be readily incorporated into existing NGS panels already widely used in oncology practice, potentially increasing accessibility without requiring additional specialized testing [121].
However, several limitations warrant consideration. The GPS classifier requires validation across more diverse patient populations and cancer types beyond NSCLC. Additionally, the biological mechanisms through which FAT1 and ERBB4 mutations enhance immunotherapy response remain incompletely characterized, meriting further investigation [121]. Finally, as our understanding of tumor-immune interactions evolves, additional genetic determinants may need incorporation into refined versions of the classifier.
Future Research Directions
Several promising research directions emerge from this comparison:
Integration with Tumor Microenvironment Analysis: Combining GPS classification with immune contexture assessment (e.g., T-cell infiltration density, myeloid cell populations) may provide enhanced predictive power [128] [126]. Research indicates that STK11-mutant tumors not only resist immunotherapy but exhibit fundamentally different immune microenvironments characterized by neutrophil infiltration and T-cell exclusion [125] [128].
Expansion to Additional Cancer Types: While extensively validated in NSCLC, the performance of the GPS classifier across other malignancies common in clinical trials (e.g., bladder cancer, melanoma, colorectal cancer) requires systematic evaluation [124].
Therapeutic Strategies for Resistant Profiles: For GPS-negative patients with STK11 mutations, alternative therapeutic approaches are urgently needed. Preclinical models suggest potential susceptibility to metabolic inhibitors or combination therapies that reverse immunosuppression, representing a critical area for translational investigation [125].
The GPS classifier represents a paradigm shift in biomarker development for cancer immunotherapy, moving beyond quantitative approaches to incorporate functionally significant genomic determinants of treatment response. While PD-L1 expression and TMB remain established biomarkers with clinical utility, the GPS classifier demonstrates superior predictive performance in direct comparisons, particularly for identifying both exceptional responders and those with primary resistance to immune checkpoint blockade [121].
The integration of this three-gene signature into clinical trial designs and biomarker-validation studies offers the potential to significantly improve patient selection for immunotherapy. Furthermore, its biological foundation in specific immune-modulatory pathways provides insights for developing novel combination strategies to overcome resistance, particularly for STK11-mutant tumors. As the field advances toward personalized immuno-oncology, the GPS classifier exemplifies the evolution of biomarkers from general correlates of inflammation to specific mediators of tumor-immune interactions.
The evaluation of biomarker performance is a critical step in translational research, particularly in studies comparing intrinsic and extrinsic pathway activation. Biomarkers, defined as measurable characteristics indicating normal biological processes, pathogenic processes, or responses to an exposure or intervention, serve various functions including disease detection, diagnosis, prognosis, and prediction of therapeutic response [74]. In the context of pathway research, accurately quantifying a biomarker's ability to distinguish between different biological states is fundamental to determining its clinical and research utility.
The process of biomarker validation follows a rigorous pathway from discovery to clinical application, requiring careful statistical consideration at each stage. Proper evaluation ensures that biomarkers selected for further development provide genuine biological insight and potential clinical value. For researchers comparing biomarkers across different pathways, selecting appropriate statistical measures is not merely a methodological formality but a fundamental aspect of deriving meaningful conclusions from experimental data [74].
This guide focuses on the most established and emerging metrics for biomarker evaluation, with particular emphasis on the Area Under the Receiver Operating Characteristic (ROC) Curve (AUC) and its alternatives. We provide objective comparisons of these measures to assist researchers in selecting the most appropriate quantification methods for their specific experimental contexts.
Core Accuracy Measures: ROC Curve and AUC
The ROC Curve and its Interpretation
The Receiver Operating Characteristic (ROC) curve is a fundamental graphical tool for evaluating the discriminatory power of diagnostic tests or biomarkers. It visually represents the relationship between a biomarker's sensitivity (true positive rate) and 1-specificity (false positive rate) across all possible threshold values [129]. The curve is created by plotting sensitivity against 1-specificity as the threshold value (c) varies from -∞ to +∞ [130].
For a binary disease outcome D (where D=1 indicates diseased and D=0 indicates non-diseased), and a continuous biomarker X, the sensitivity at threshold c is defined as SEN(c) = P(X > c|D=1) = 1 - FD(c), where FD is the cumulative distribution function of the biomarker among cases. The specificity is SPE(c) = P(X ≤ c|D=0) = FD̄(c), where FD̄ is the cumulative distribution function among controls [130]. The ROC curve thus provides an exhaustive visualization of the trade-off between sensitivity and specificity at all possible classification thresholds.
The Area Under the ROC Curve (AUC) serves as the most popular overall discrimination accuracy index, providing a single numerical summary of a biomarker's diagnostic performance [131]. The AUC value corresponds to the probability that a randomly selected individual with the target disease has a marker value greater than a randomly selected individual free of the disease [130]. Mathematically, this can be expressed as AUC = P(XD > XD̄), where XD and XD̄ represent biomarker measurements from diseased and non-diseased populations, respectively.
The AUC ranges from 0 to 1, where 0.5 indicates performance equivalent to random chance, and 1 represents perfect discrimination between diseased and non-diseased individuals [74]. In practical terms, biomarkers with AUC values below 0.7 are generally considered to have weak discriminatory power, while those between 0.7-0.8, 0.8-0.9, and above 0.9 are considered fair, good, and excellent, respectively [132].
Partial AUC (pAUC) for Focused Evaluation
In many research scenarios, particularly when high sensitivity or specificity is clinically required, the partial Area Under the Curve (pAUC) provides a more targeted evaluation metric. The pAUC characterizes classification performance over a clinically relevant region of the ROC curve, rather than the entire curve [130] [133]. This is particularly valuable when a biomarker serves specific purposes such as population screening, where high specificity is essential to minimize false positives [133].
The pAUC is defined mathematically as the integral of the ROC curve over a specified interval of false positive rates. For a given specificity region (1-t, 1) for some predetermined t ∈ (0,1), the pAUC of a linear combination of biomarkers a^TX can be expressed as:
[ pAUC(a) = \int_0^t F(a,u)du ]
where F(a,u) represents the sensitivity at specificity 1-u [133]. This focused assessment prevents potentially misleading conclusions that might arise from evaluating performance across the entire ROC curve when only a specific operating region is clinically relevant.
Table 1: Core Accuracy Measures for Biomarker Evaluation
| Measure | Definition | Interpretation | Best Use Cases |
|---|---|---|---|
| Sensitivity | Proportion of true cases correctly identified | High sensitivity reduces missed cases | When false negatives are particularly concerning |
| Specificity | Proportion of true controls correctly identified | High specificity reduces false alarms | When false positives are particularly concerning |
| ROC Curve | Plot of sensitivity vs. 1-specificity across all thresholds | Visualizes overall discrimination performance | Initial biomarker assessment and threshold selection |
| AUC | Area under the entire ROC curve | Probability a case has higher value than control | Overall performance summary; biomarker comparison |
| Partial AUC | Area under a specific region of the ROC curve | Performance in clinically relevant range | When only certain operating ranges are meaningful |
Limitations of AUC and the Need for Alternative Metrics
Statistical and Conceptual Limitations
Despite its widespread use, the AUC statistic has several well-documented limitations that researchers must consider when evaluating biomarkers. The AUC summarizes performance across all possible thresholds, which may include regions that are clinically irrelevant [129]. This can mask important performance characteristics in specific ranges of interest, potentially leading to overstated claims about a biomarker's utility in practical applications.
Another significant limitation is the AUC's focus on the ranking of predictions rather than the absolute differences in risk estimates [129]. It is possible for a new biomarker to produce substantial changes in absolute risk estimates for individuals at clinically important risk levels without significantly improving the AUC. Conversely, small changes in risk among many low-risk individuals might improve the AUC without meaningful clinical impact. This property makes the AUC relatively insensitive to improvements that specifically benefit high-risk groups, which are often the primary target for clinical interventions.
From a statistical perspective, comparison of AUCs between different biomarkers or models often suffers from low statistical power, making it difficult to detect genuine improvements in performance [134]. This insensitivity can lead researchers to prematurely discard promising biomarkers that provide meaningful but subtle improvements in classification accuracy.
Challenges in Clinical Interpretation
The AUC lacks direct clinical interpretability in terms of how a biomarker might affect patient management decisions. While it provides an overall measure of discrimination, it does not directly inform clinicians about how the biomarker should be implemented in practice or what clinical actions should follow from its measurements [129]. This limitation becomes particularly problematic when attempting to translate biomarker research findings into clinical practice.
Furthermore, the AUC is not affected by calibration, or how well the predicted risks match observed outcomes [129]. A biomarker could have excellent discrimination (high AUC) but poor calibration, leading to systematically overestimated or underestimated risks that would impair clinical decision-making. This separation between discrimination and calibration means that AUC alone provides an incomplete picture of a biomarker's potential utility.
Beyond AUC: Alternative Performance Measures
Net Reclassification Improvement (NRI)
The Net Reclassification Improvement (NRI) addresses specific limitations of the AUC by focusing on clinically relevant risk categories. The NRI quantifies how well a new biomarker reclassifies individuals into more appropriate risk categories compared to an existing model [129]. It computes the net proportion of individuals moving to more appropriate risk strata separately for cases and controls.
The NRI is calculated as:
[ NRI = P(\text{up-classification}|\text{case}) - P(\text{down-classification}|\text{case}) + P(\text{down-classification}|\text{control}) - P(\text{up-classification}|\text{control}) ]
This measure directly assesses whether a new biomarker improves classification accuracy in ways that could influence clinical management, particularly when risk strata correspond to established treatment thresholds. However, the NRI's value can be affected by the number and size of pre-specified risk categories, and these should be clearly justified based on clinical considerations [129].
Decision Curve Analysis and Net Benefit
Decision curve analysis provides a framework for evaluating biomarker performance based on clinical consequences rather than statistical discrimination alone. The net benefit metric incorporates the relative clinical consequences of false positives and false negatives by specifying a threshold probability at which a patient would opt for treatment [135]. This approach explicitly acknowledges that the clinical value of a biomarker depends not only on its accuracy but also on the trade-offs between different types of classification errors.
Net benefit is calculated as:
[ \text{Net Benefit} = \frac{\text{True Positives}}{n} - \frac{\text{False Positives}}{n} \times \frac{pt}{1-pt} ]
where (p_t) is the threshold probability and n is the total sample size. By evaluating net benefit across a range of clinically reasonable threshold probabilities, researchers can determine whether a biomarker improves clinical decision-making across various clinical scenarios and patient preferences [135].
Likelihood-Based Measures and Information Criteria
Likelihood-based measures provide a fundamental approach to assessing whether new biomarkers improve model fit. The likelihood ratio test directly evaluates whether adding a biomarker to a model significantly increases the likelihood of observing the data [129]. This method tests whether the improvement in model fit justifies the additional complexity introduced by the new biomarker.
Information criteria such as the Akaike Information Criterion (AIC) and Bayesian Information Criterion (BIC) extend this approach by applying penalties for model complexity [129]. The AIC applies a penalty of 2 degrees of freedom per parameter, while the BIC applies a typically larger penalty of ln(N), where N is the sample size. These criteria are particularly valuable when comparing non-nested models or when conducting biomarker selection from a larger set of candidates.
Table 2: Comparison of Biomarker Performance Metrics
| Metric | Strengths | Limitations | Appropriate Contexts |
|---|---|---|---|
| AUC | Threshold-independent; intuitive probability interpretation; widely accepted | Insensitive to clinically important risk changes; summarizes all thresholds including irrelevant ones | Initial biomarker screening; overall performance comparison |
| Partial AUC | Focuses on clinically relevant operating range | Requires pre-specified range of interest; not a comprehensive summary | When only specific sensitivity/specificity ranges matter |
| NRI | Direct clinical interpretability; focuses on meaningful risk categories | Dependent on category definitions; can be biased if categories poorly chosen | When clear clinical decision thresholds exist |
| Net Benefit | Incorporates clinical consequences of decisions; directly addresses utility | Requires specification of threshold probabilities; less familiar to researchers | When clinical utility and decision consequences are paramount |
| AIC/BIC | Fundamental measure of model improvement; penalizes complexity | No direct clinical interpretation; requires nested models for tests | Model selection; assessing statistical value of new biomarkers |
Experimental Design and Methodological Considerations
Two-Phase Case-Control Studies for Biomarker Evaluation
Two-phase case-control sampling designs are widely used for the evaluation of candidate biomarkers, particularly when biomarker measurement is costly or invasive [130]. In this design, a large cohort is first randomly selected from the target population, with disease status and easily measured covariates collected for all subjects. In the second phase, subsamples of cases and controls are drawn from the phase-one cohort, often with sampling probabilities that depend on covariates or sampling strata.
A critical methodological consideration in these studies is that biased sampling schemes can lead to invalid inference if not properly accounted for. When controls are frequency-matched to cases on risk factors, or when sampling probabilities vary across strata, traditional empirical estimators of classification accuracy can be severely biased [130]. Inverse probability weighting (IPW) methods have been developed to address this issue, providing unbiased estimation of ROC curves, AUC, and partial AUC by weighting observations inversely to their probability of selection into the phase-two sample [130].
Handling Multiple Biomarkers and High-Dimensional Settings
In many contemporary research settings, particularly with advances in omics technologies, studies involve evaluating large numbers of biomarkers rather than single candidates. The challenge of combining multiple biomarkers to improve diagnostic accuracy has received significant methodological attention [131]. Both parametric and nonparametric methods have been developed for finding optimal linear combinations of biomarkers that maximize the AUC.
When dealing with large numbers of relatively weak biomarkers (typically with AUCs between 0.5-0.7), traditional combination methods may perform poorly, particularly when the number of biomarkers approaches the sample size [131]. Regularization methods such as ridge regression or LASSO (Least Absolute Shrinkage and Selection Operator) can help prevent overfitting in these settings by imposing penalties on the size of combination coefficients [131]. These approaches are particularly valuable in high-dimensional biomarker discovery research, such as genomic or proteomic studies.
Statistical Testing and Biomarker Selection
Formal statistical testing procedures play a crucial role in biomarker evaluation, particularly when selecting promising candidates from a larger set. When assessing multiple biomarkers simultaneously, adjustment for multiple testing is essential to control the risk of false discoveries [132]. Simultaneous confidence intervals for multiple AUCs provide a principled approach to this problem, maintaining the overall coverage probability across all comparisons [132].
For small sample sizes, which are common in early biomarker development studies, Wild Bootstrap approaches have been developed that provide robust inference for AUC comparisons [132]. These resampling methods yield better control of type I error rates compared to asymptotic methods, particularly when dealing with high accuracy biomarkers or imbalanced case-control ratios.
Experimental Protocols for Biomarker Evaluation
Standard Protocol for ROC Analysis
Sample Collection and Preparation: Collect representative samples from clearly defined case and control populations. Ensure appropriate storage conditions to preserve biomarker integrity.
Blinded Measurement: Perform biomarker measurements blinded to case-control status to prevent assessment bias. Implement randomization procedures to control for batch effects and technical variability [74].
Data Quality Control: Assess measurement precision, accuracy, and detect potential outliers using appropriate statistical methods.
ROC Curve Construction: Calculate sensitivity and specificity at all observed biomarker values. Plot the ROC curve with sensitivity on the y-axis and 1-specificity on the x-axis.
AUC Estimation: Calculate the AUC using the trapezoidal rule or maximum likelihood estimation under binormal assumptions. Compute confidence intervals using appropriate methods (e.g., DeLong's method or bootstrap).
Comparison with Existing Biomarkers: If applicable, statistically compare AUCs using paired tests that account for the correlation between measurements from the same subjects.
Protocol for Evaluating Biomarker Combinations
Pre-specification of Biomarker Set: Define the set of candidate biomarkers to be combined based on biological rationale or preliminary data.
Selection of Combination Method: Choose an appropriate combination method based on the number of biomarkers, sample size, and distributional assumptions. Options include logistic regression, linear discriminant analysis, or regularized methods for high-dimensional settings [131].
Optimization Procedure: Estimate combination coefficients to maximize the desired performance metric (AUC, partial AUC, or other criteria). For linear combinations under normality assumptions, the optimal coefficients are given by ( \lambda = (\Sigma1 + \Sigma0)^{-1} \mu ), where ( \mu = \mu1 - \mu0 ) is the mean difference between cases and controls, and ( \Sigma0 ), ( \Sigma1 ) are covariance matrices [131].
Validation: Assess the performance of the combined biomarker using cross-validation or external validation datasets to obtain unbiased performance estimates.
Clinical Utility Assessment: Evaluate whether the combined biomarker provides meaningful improvement over existing methods using metrics such as NRI or decision curve analysis.
Signaling Pathways and Analytical Workflows
The following diagram illustrates the key statistical concepts and their relationships in biomarker evaluation, highlighting the progression from fundamental measures to more advanced applications:
Diagram 1: Biomarker Evaluation Metrics Framework
Research Reagent Solutions for Biomarker Validation
Table 3: Essential Research Reagents and Materials for Biomarker Studies
| Reagent/Material | Function/Purpose | Application Notes |
|---|---|---|
| Clinical Specimens | Source of biomarkers for measurement | Proper collection, processing, and storage critical for biomarker stability |
| Reference Standard Materials | Calibration and quality control | Enables standardization across experiments and laboratories |
| Assay Kits | Quantitative measurement of specific biomarkers | Selection should consider precision, sensitivity, and dynamic range |
| Statistical Software | Data analysis and visualization | R, SAS, or Python with specialized packages for ROC analysis |
| Laboratory Information | Sample tracking and data management | Maintains chain of custody and integrates clinical with biomarker data |
The evaluation of biomarker performance requires careful selection of appropriate statistical measures that align with the research question and potential clinical application. While the AUC remains a valuable overall measure of discrimination, researchers should supplement it with additional metrics that address its limitations, particularly when assessing biomarkers for specific pathway research or clinical implementation contexts.
The optimal approach to biomarker evaluation involves a comprehensive assessment using multiple complementary metrics, with selection based on the specific goals of the research. For intrinsic and extrinsic pathway activation studies, this might include AUC for initial screening, partial AUC for focused performance assessment in relevant operating ranges, and net reclassification improvement or decision curve analysis when clinical utility is a primary consideration.
By applying these principles and methodologies, researchers can conduct more rigorous and informative biomarker evaluations, leading to more reliable conclusions about the comparative performance of biomarkers across different biological pathways and ultimately accelerating the translation of biomarker research into clinical practice.
In modern biomedical research and drug development, biomarkers serve as indispensable tools for objectively measuring and evaluating biological processes, pathogenic states, or pharmacological responses to therapeutic interventions. These biological molecules—which can include proteins, nucleic acids, lipids, and carbohydrates—provide critical insights that bridge basic scientific discovery and clinical application [136]. The precise classification and application of biomarkers have become increasingly important for understanding disease heterogeneity, predicting treatment outcomes, and developing personalized therapeutic strategies.
The complexity of biomarker science necessitates clear differentiation between various biomarker types based on their specific applications. Within clinical and research contexts, three categories have proven particularly valuable: prognostic biomarkers, which provide information about a patient's overall disease outcome regardless of therapy; predictive biomarkers, which identify individuals more likely to respond to specific treatments; and pharmacodynamic biomarkers, which determine whether a drug is hitting its intended target and producing the desired biological effect [137] [136]. Understanding these distinctions is fundamental to their appropriate application in both research and clinical settings.
This comparative analysis examines the distinct roles, applications, and methodological considerations for prognostic, predictive, and pharmacodynamic biomarkers, with particular emphasis on their utility in investigating intrinsic and extrinsic pathway activation. By synthesizing current research and experimental approaches, this guide provides researchers, scientists, and drug development professionals with a framework for selecting, implementing, and interpreting these powerful biological indicators across various research contexts.
Fundamental Definitions and Comparative Framework
Conceptual Definitions and Key Distinctions
Biomarkers represent measurable indicators of biological states or conditions. Their classification depends primarily on their specific application rather than their molecular nature [136]. A prognostic biomarker offers information about the likely course of a disease in an untreated individual, essentially foreseeing the natural history of the condition. For example, PIK3CA mutation status in HER-2 positive metastatic breast cancer serves as a prognostic biomarker, where individuals with mutated PIK3CA exhibit lower rates of disease-free survival regardless of treatment [136].
In contrast, a predictive biomarker identifies patients who are likely or unlikely to benefit from a specific therapeutic intervention. The classic example is EGFR mutation status in non-small-cell lung cancer, which predicts response to erlotinib treatment [136]. Meanwhile, a pharmacodynamic biomarker demonstrates that a drug has engaged its intended target and is having a biological effect. Phosphorylated AKT (pAKT) levels fulfill this role for PI3K inhibitor treatments in cancer, confirming pathway inhibition [136].
Table 1: Core Definitions and Applications of Biomarker Types
| Biomarker Type | Primary Function | Clinical/Research Utility | Representative Examples |
|---|---|---|---|
| Prognostic | Provides information about overall disease outcome regardless of therapy | Patient stratification, natural history studies, trial design | PIK3CA mutations in breast cancer; Portal vein diameter in cirrhosis [138] [136] |
| Predictive | Identifies likelihood of response to a specific therapeutic intervention | Treatment selection, personalized medicine, companion diagnostics | EGFR mutations for erlotinib in NSCLC; BCL-2 overexpression for venetoclax [14] [136] |
| Pharmacodynamic | Indicates biological response to therapeutic intervention | Target engagement confirmation, dose optimization, mechanism validation | pAKT reduction with PI3K inhibitors; caspase activation with pro-apoptotic drugs [136] [139] |
Methodological Considerations for Biomarker Validation
The validation of each biomarker type requires distinct methodological approaches and statistical considerations. Prognostic biomarker validation typically involves observational studies tracking untreated patients over time to establish correlations between the biomarker and disease outcomes [138]. Predictive biomarker validation necessitates randomized controlled trials where biomarker-positive and biomarker-negative patients are assigned to different treatment groups to demonstrate differential treatment effects [137]. Pharmacodynamic biomarker validation often involves pre-clinical and early-phase clinical studies with repeated biomarker measurements before, during, and after treatment to establish target engagement [136].
The context of use critically determines the validation requirements for each biomarker. Regulatory agencies like the FDA have established frameworks for evaluating biomarker validity based on their intended application, with different levels of evidence required for biomarkers used in diagnostic, prognostic, predictive, or pharmacodynamic contexts [136]. For all biomarker types, analytical validation (establishing accuracy, precision, and reproducibility of the measurement) must precede clinical validation (establishing relationships to clinical endpoints).
Biomarkers in Intrinsic and Extrinsic Apoptosis Pathway Research
The intrinsic and extrinsic apoptosis pathways represent fundamental biological processes that can be leveraged for biomarker development. The extrinsic pathway (death receptor pathway) initiates apoptosis through extracellular signals binding to death receptors such as FAS (CD95), leading to caspase-8 activation and subsequent execution of apoptosis [14] [139]. The intrinsic pathway (mitochondrial pathway) responds to intracellular stress signals, involving BCL-2 family proteins, mitochondrial membrane permeabilization, cytochrome c release, and caspase-9 activation [14].
These pathways offer rich opportunities for biomarker development because their components are frequently dysregulated in diseases, particularly cancer, and in response to therapeutic interventions. For example, in hepatitis C virus infection, researchers have documented alterations in both extrinsic (FAS, caspase-8) and intrinsic (caspase-9, cytochrome c) pathway components in peripheral blood mononuclear cells, with expression patterns varying according to disease stage [139]. This highlights how pathway-specific biomarkers can reflect both disease progression and treatment response.
Experimental Data and Biomarker Performance
Research across various disease contexts has yielded quantitative data on apoptosis pathway biomarkers. In cirrhosis, prognostic biomarkers like portal vein diameter (HR = 7.39 [4.90, 11.15]) and spleen size (HR = 5.79 [2.00, 16.80]) show significant predictive value for decompensation risk [138]. In alcoholic cirrhosis, inflammatory biomarkers including extracellular vesicles (HR = 5.09 [2.01, 12.86]) and keratin-18 (HR = 1.77 [1.14, 2.75]) demonstrate prognostic utility [138].
In hepatitis C infection, studies of peripheral blood mononuclear cells revealed significant alterations in apoptosis biomarkers, with group 1 (early disease) showing a 3-fold increase in caspase-8 mRNA and 4-fold increase in protein, while group 4 (advanced disease) demonstrated 2-fold increases in caspase-9 mRNA and 1.5-fold increases in protein [139]. These findings illustrate how different biomarker profiles may emerge at various disease stages.
Table 2: Biomarker Performance in Intrinsic and Extrinsic Apoptosis Pathways
| Biomarker | Pathway | Change/Effect Size | Context/Disease | Biomarker Type |
|---|---|---|---|---|
| Caspase-8 | Extrinsic | 3-fold mRNA, 4-fold protein increase | HCV infection (early stage) [139] | Prognostic/Pharmacodynamic |
| Caspase-9 | Intrinsic | 2-fold mRNA, 1.5-fold protein increase | HCV infection (advanced) [139] | Prognostic/Pharmacodynamic |
| Caspase-3 | Execution | 4-6-fold mRNA, 5-7-fold protein increase | HCV infection [139] | Pharmacodynamic |
| BCL-2 | Intrinsic | Overexpression delays cell death | Various cancers [14] | Prognostic/Predictive |
| FAS Receptor | Extrinsic | 7.5-fold increase (early), 5-fold decrease (late) | HCV infection [139] | Prognostic |
| Cytochrome c | Intrinsic | 5-fold increase | HCV infection (advanced) [139] | Prognostic/Pharmacodynamic |
Experimental Protocols for Biomarker Assessment
Standardized Methodologies for Pathway Analysis
Robust experimental protocols are essential for reliable biomarker assessment in intrinsic and extrinsic apoptosis pathways. For gene expression analysis of apoptosis-related markers, the reverse transcription quantitative polymerase chain reaction (RT-qPCR) method provides sensitive quantification. The standard protocol involves: RNA extraction from target cells or tissues, cDNA synthesis through reverse transcription, qPCR amplification with gene-specific primers, and normalization to housekeeping genes [139]. This approach enabled researchers to detect significant increases in caspase-8, caspase-9, and caspase-3 expression in PBMCs from HCV patients compared to controls [139].
For protein-level analysis, enzyme-linked immunosorbent assay (ELISA) offers specific quantification of apoptosis proteins. The standard protocol includes: sample preparation and protein extraction, plate coating with capture antibodies, sample incubation and target protein binding, detection with specific secondary antibodies, and colorimetric measurement with appropriate substrates [139]. This method confirmed corresponding protein-level increases for caspases whose mRNA was elevated in HCV infection [139].
Advanced techniques for apoptosis biomarker detection include flow cytometry for surface death receptor expression, immunohistochemistry for tissue localization, Western blot for protein quantification, and functional assays like mitochondrial membrane potential measurements for intrinsic pathway activity. Each method offers distinct advantages for specific biomarker applications and requires appropriate validation controls.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for Apoptosis Biomarker Studies
| Reagent/Category | Specific Examples | Research Function | Application Context |
|---|---|---|---|
| qPCR Reagents | Custom primers for caspases, BCL-2 family, FAS [139] | mRNA expression quantification | Prognostic biomarker discovery; Pathway activation |
| ELISA Kits | Caspase-8, caspase-9, cytochrome c kits [139] | Protein quantification and detection | Pharmacodynamic studies; Treatment response |
| Antibodies | Anti-FAS, anti-BCL-2, anti-caspase antibodies [139] | Protein detection and localization | IHC, Western blot, flow cytometry |
| Cell Viability Assays | MTT, Annexin V/PI staining | Apoptosis quantification and differentiation | Functional validation of biomarker findings |
| Sample Prep Tools | Omni LH 96 homogenizer [140] | Standardized sample processing | Biomarker reproducibility and precision |
| Pathway Modulators | BCL-2 inhibitors (venetoclax), caspase inhibitors | Experimental pathway manipulation | Predictive biomarker validation; Mechanism studies |
Comparative Analysis and Clinical Applications
Strategic Implementation in Research and Development
The strategic application of prognostic, predictive, and pharmacodynamic biomarkers significantly enhances research efficiency and clinical development success. In drug discovery, pharmacodynamic biomarkers provide early proof-of-concept by confirming target engagement before expensive late-stage trials [136]. For example, monitoring phosphorylated AKT (pAKT) reduction provides immediate feedback on PI3K inhibitor efficacy during early development [136].
Predictive biomarkers enable patient stratification for targeted therapies, increasing trial efficiency and likelihood of success. This approach is particularly valuable in oncology, where biomarkers like EGFR mutations identify patients most likely to respond to specific therapies [136]. The BCL-2 inhibitor venetoclax exemplifies this approach, with response highly dependent on BCL-2 expression levels as a predictive biomarker [14].
Prognostic biomarkers aid in trial design by identifying patients with different expected outcomes, enabling enrichment strategies or stratification. In cirrhosis research, biomarkers like portal vein diameter and spleen size help identify high-risk populations for targeted intervention studies [138]. The integration of multiple biomarker types creates a comprehensive framework for understanding therapeutic interventions from biological mechanism to clinical outcome.
Emerging Trends and Future Directions
Biomarker science continues to evolve with technological advancements and conceptual innovations. Emerging approaches include multi-omics integration, combining genomic, proteomic, and metabolomic data for comprehensive biomarker panels [140]. Liquid biopsy technologies enable non-invasive biomarker assessment through circulating tumor DNA, extracellular vesicles, and other blood-based markers, particularly valuable for serial pharmacodynamic monitoring [140].
Artificial intelligence and machine learning are transforming biomarker discovery and validation, identifying complex patterns in high-dimensional data beyond human analytical capability [140]. These technologies accelerate the identification of novel biomarker signatures for intrinsic and extrinsic pathway activation across diverse disease contexts.
The biomarker field is moving toward increasingly dynamic assessments, monitoring pathway activation in real-time rather than relying on static measurements. This approach provides richer insights into biological processes and therapeutic responses, particularly for dynamic pathways like apoptosis that evolve over time and in response to interventions. As these advancements mature, they will further refine the distinct applications of prognostic, predictive, and pharmacodynamic biomarkers in both research and clinical practice.
Conclusion
The comparative analysis of intrinsic and extrinsic pathway biomarkers underscores their indispensable yet distinct roles in deciphering disease mechanisms and guiding therapeutic interventions. While intrinsic biomarkers often reflect core cellular stress and genetic events, extrinsic biomarkers capture critical microenvironmental and systemic signals. The future lies not in choosing one over the other, but in strategically integrating them into multi-parametric classifiers. Advancements in machine learning on high-dimensional data, the refinement of endogenous biomarkers for real-time activity assessment, and the rigorous validation of these combined signatures in diverse clinical populations will be pivotal. This synergistic approach will ultimately accelerate the development of precise diagnostic tools and personalized treatment strategies across a spectrum of human diseases, from cancer to autoimmune disorders.
References
- 1. Intrinsic and Extrinsic Pathways of Apoptosis | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/antibodies/antibodies-learning-center/antibodies-resource-library/cell-signaling-pathways/cellular-apoptosis-pathway.html]
- 2. Biochemistry, Extrinsic Pathway of Apoptosis - StatPearls - NCBI Bookshelf [https://www.ncbi.nlm.nih.gov/books/NBK560811/]
- 3. Apoptosis - Wikipedia [https://en.wikipedia.org/wiki/Apoptosis]
- 4. Intrinsic and extrinsic pathway signaling during neuronal apoptosis: lessons from the analysis of mutant mice - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2173286/]
- 5. Modulation of Extrinsic and Intrinsic Signaling Together with Neuronal Activation Enhances Forelimb Motor Recovery after Cervical Spinal Cord Injury | eNeuro [https://www.eneuro.org/content/12/3/ENEURO.0359-24.2025]
- 6. Cell Type-Specific Biomarkers of Systemic Sclerosis Disease Severity... [https://pubmed.ncbi.nlm.nih.gov/37096444/]
- 7. Death Receptor Signals to Mitochondria - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2941887/]
- 8. Death receptor signals to mitochondria - PubMed [https://pubmed.ncbi.nlm.nih.gov/15640619/]
- 9. Apoptosis | Intrinsic & Extrinsic Pathways | Mechanisms of Cell Death [https://blog.cellsignal.com/mechanisms-of-cell-death-apoptosis]
- 10. Intrinsic and extrinsic apoptosis responses in leukaemia cells following daunorubicin treatment | BMC Cancer | Full Text [https://bmccancer.biomedcentral.com/articles/10.1186/s12885-021-08167-y]
- 11. Death Receptor Signaling | Cell Signaling Technology [https://www.cellsignal.com/pathways/death-receptor-signaling]
- 12. Frontiers | Unveiling BID: a key biomarker in apoptosis ... [https://www.frontiersin.org/journals/neurology/articles/10.3389/fneur.2025.1533558/full]
- 13. is Apoptosis early by low doses of ionising radiation in... signalled [https://pubmed.ncbi.nlm.nih.gov/23454491/]
- 14. Emerging biomarkers and potential therapeutics of the BCL-2 protein... [https://jmhg.springeropen.com/articles/10.1186/s43042-024-00485-7]
- 15. Physiology, Coagulation Pathways - StatPearls - NCBI Bookshelf [https://www.ncbi.nlm.nih.gov/books/NBK482253/]
- 16. Bleeding and blood clotting - Intrinsic ... | Britannica Pathway [https://www.britannica.com/science/bleeding/Intrinsic-pathway-of-blood-coagulation]
- 17. Biochemistry, Clotting Factors - StatPearls - NCBI Bookshelf [https://www.ncbi.nlm.nih.gov/books/NBK507850/]
- 18. Frontiers | Intrinsic coagulation pathway-mediated thrombin generation in mouse whole blood [https://www.frontiersin.org/journals/cardiovascular-medicine/articles/10.3389/fcvm.2022.1008410/full]
- 19. Potential biomarkers: differentially expressed proteins of the extrinsic coagulation pathway in plasma samples from patients with depression - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8806736/]
- 20. The Extrinsic Coagulation Pathway : a Biomarker for Suicidal... [https://pubmed.ncbi.nlm.nih.gov/27605454/]
- 21. mapping and Pathway of development -specific disease ... biomarkers [https://pmc.ncbi.nlm.nih.gov/articles/PMC4407592/]
- 22. - BCL inhibitors in hematological malignancies: 2 that... biomarkers [https://pmc.ncbi.nlm.nih.gov/articles/PMC11790632/]
- 23. MOMP, cell suicide as a BCL-2 family business | Cell Death & Differentiation [https://www.nature.com/articles/cdd2017179]
- 24. Mitochondrial Control of Apoptosis - Creative BioMart [https://www.creativebiomart.net/resource/signal-pathway-mitochondrial-control-of-apoptosis-356.htm?page=41]
- 25. Apoptosis II: Beyond Cytochrome c; the Other Mitochondrial Proteins: R&D Systems [https://www.rndsystems.com/resources/articles/apoptosis-ii-beyond-cytochrome-c-other-mitochondrial-proteins]
- 26. The Intrinsic of Apoptosis | SpringerLink Pathway [https://link.springer.com/chapter/10.1007/978-1-59745-221-2_1]
- 27. Diablo homolog - Wikipedia [https://en.wikipedia.org/wiki/Diablo_homolog]
- 28. Immunoexpression of DIABLO , AIF and Cytochrome in Gastric... c [https://ar.iiarjournals.org/content/33/2/647]
- 29. Algorithmically Reconstructed Molecular Pathways as the New Generation of Prognostic Molecular Biomarkers in Human Solid Cancers [https://www.mdpi.com/2227-7382/11/3/26]
- 30. Caspase-8; regulating life and death - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5417704/]
- 31. Biomarkers of apoptosis - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2538762/]
- 32. Caspase-8 in inflammatory diseases: a potential therapeutic ... [https://cmbl.biomedcentral.com/articles/10.1186/s11658-024-00646-x]
- 33. TRAIL receptor-2 signals apoptosis through FADD and ... [https://www.nature.com/articles/ncb0400_241]
- 34. DED Interaction of FADD and Caspase-8 in the Induction of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9628938/]
- 35. and Intrinsic of... | Thermo Fisher Scientific - RU Extrinsic Pathways [https://www.thermofisher.com/ru/ru/home/life-science/antibodies/antibodies-learning-center/antibodies-resource-library/cell-signaling-pathways/cellular-apoptosis-pathway.html]
- 36. Deciphering mechanical cues in the microenvironment: from... [https://biomarkerres.biomedcentral.com/articles/10.1186/s40364-025-00727-9]
- 37. Wnt signaling in cancer: from biomarkers to targeted therapies and clinical translation | Molecular Cancer | Full Text [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-025-02306-w]
- 38. modulate... | Nature Communications Intrinsic signaling pathways [https://www.nature.com/articles/s41467-024-49519-z?error=cookies_not_supported&code=a4a8faec-b9ed-4362-a567-54158ad718ec]
- 39. Frontiers | Comprehensive integration of diagnostic biomarker analysis and immune cell infiltration features in sepsis via machine learning and bioinformatics techniques [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1526174/full]
- 40. Integrative single - cell and cell-free plasma RNA transcriptomics... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12162594/]
- 41. Frontiers | Comprehensive transcriptomic analysis integrating bulk and... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2025.1582252/full]
- 42. Society for Immunotherapy of Cancer: updates and best practices for multiplex immunohistochemistry (IHC) and immunofluorescence (IF) image analysis and data sharing - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11749220/]
- 43. Immune profiling and prognostic model of pancreatic cancer using... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-023-04051-4]
- 44. Identification of blood-derived exosomal tumor RNA signatures as... [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-025-02271-4]
- 45. A Novel Sensitivity Maximization at a Given Specificity Method for Binary Classifications - PubMed [https://pubmed.ncbi.nlm.nih.gov/39618306/]
- 46. A Novel Sensitivity Maximization at a Given Specificity Method for Binary Classifications - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11875929/]
- 47. Current best practices in single - cell - RNA analysis: a tutorial seq [https://pubmed.ncbi.nlm.nih.gov/31217225/]
- 48. Immunopathological markers and cell linked to COVID-19... types [https://bmcinfectdis.biomedcentral.com/articles/10.1186/s12879-024-10139-z]
- 49. Molecular pathway - New type of activation for tumor... biomarkers [https://pubmed.ncbi.nlm.nih.gov/29935311/]
- 50. The role of extrinsic clotting pathway activation in the colorectal cancer microenvironment - Research Explorer The University of Manchester [https://research.manchester.ac.uk/en/studentTheses/the-role-of-extrinsic-clotting-pathway-activation-in-the-colorect]
- 51. Expanding Role of Endogenous Biomarkers for Assessment of Transporter Activity in Drug Development: Current Applications and Future Horizon - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11280074/]
- 52. Membrane Transporters in Drug Development and as Determinants of Precision Medicine - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11464068/]
- 53. ‐I as a Selective Coproporphyrin Can Be... | CoLab OATP 1 B Biomarker [https://colab.ws/articles/10.1002%2Fcpt.3399]
- 54. Further Studies to Support the Use of Coproporphyrin I and III as... [https://pubmed.ncbi.nlm.nih.gov/29777022/]
- 55. Utilization of Rosuvastatin and Endogenous in Evaluating... Biomarkers [https://link.springer.com/article/10.1007/s11095-023-03564-3]
- 56. Identification of Appropriate Endogenous Biomarker for Risk Assessment of Multidrug and Toxin Extrusion Protein‐Mediated Drug‐Drug Interactions in Healthy Volunteers - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7891601/]
- 57. of an LC-MS method to quantify Development I and... coproporphyrin [https://pubmed.ncbi.nlm.nih.gov/29241088/]
- 58. Membrane transporters in drug and as determinants of... development [https://www.nature.com/articles/s41573-023-00877-1?error=cookies_not_supported]
- 59. Current trends in sensitizing immune for cancer... checkpoint inhibitors [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-024-02179-5]
- 60. PathNetDRP: a novel biomarker discovery framework using pathway... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-025-06125-0]
- 61. FDA-Approved and Emerging Next Generation Predictive Biomarkers ... [https://pubmed.ncbi.nlm.nih.gov/34164344/]
- 62. Frontiers | Exosomal PD-L1 detection in cancer predictive ... biomarker [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1603855/full]
- 63. Study Designs and Statistical Analyses for Biomarker Research - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3444086/]
- 64. in Pathogenesis, Diagnosis, and Treatment of SSc | JIR Biomarkers [https://www.dovepress.com/biomarkers-in-the-pathogenesis-diagnosis-and-treatment-of-systemic-scl-peer-reviewed-fulltext-article-JIR]
- 65. Sepsis-Induced Coagulopathy: An Update on Pathophysiology, Biomarkers, and Current Guidelines - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9961497/]
- 66. Biomarkers of sepsis-induced coagulopathy: diagnostic insights and potential therapeutic implications | Annals of Intensive Care | Full Text [https://annalsofintensivecare.springeropen.com/articles/10.1186/s13613-025-01434-2]
- 67. in Biomarkers : An Overview Systemic Sclerosis [https://pubmed.ncbi.nlm.nih.gov/37886934/]
- 68. Existing and novel biomarkers for precision medicine in systemic ... [https://www.nature.com/articles/s41584-018-0021-9?error=cookies_not_supported&code=85155b64-c437-4b26-a822-8682dd72e489]
- 69. Digital Biomarker Summit USA 2025 | May 13, 2025 [https://digitalbiomarkersusa.panagorapharma.com/]
- 70. A comparative study of multi - omics tools for cancer... integration [https://pubmed.ncbi.nlm.nih.gov/31774481/]
- 71. MultiModalGraphics: an R package for graphical integration of... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-025-06265-3]
- 72. Frontiers | An integrated approach for analyzing spatially resolved... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2025.1614288/full]
- 73. Chapter 25 Annotate by Integration with scRNA-seq | Single Cell... [https://bookdown.org/ytliu13207/SingleCellMultiOmicsDataAnalysis/annotate-by-integration-with-scrna-seq.html]
- 74. Biomarker Discovery and Validation: Statistical Considerations - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8012218/]
- 75. Using proteomic and transcriptomic data to assess activation of... [https://pubmed.ncbi.nlm.nih.gov/34340765/]
- 76. Frontiers | Comparison of different predictive biomarker testing... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2023.1265202/full]
- 77. The alpha defensin-1 biomarker can be used to evaluate the... assay [https://pubmed.ncbi.nlm.nih.gov/25256621/]
- 78. Validation of Analytical Methods for Biomarkers Employed in Drug Development - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2744124/]
- 79. Utilising Endogenous in Drug Development to Streamline... Biomarkers [https://link.springer.com/article/10.1007/s40262-024-01385-0]
- 80. to Assess Drug-Drug Interactions by Drug... Endogenous Biomarkers [https://pubmed.ncbi.nlm.nih.gov/28738769/]
- 81. Disentangling cell-intrinsic and cell-extrinsic factors underlying evolution - ScienceDirect [https://www.sciencedirect.com/science/article/pii/S2666979X25001478]
- 82. Disentangling cell-intrinsic and extrinsic factors underlying evolution - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11118348/]
- 83. Frontiers | Immunotherapy biomarkers in brain metastases: insights... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1600261/full]
- 84. Oncogenic and microenvironmental drive signals type specific... cell [https://www.nature.com/articles/s41419-025-07479-2?error=cookies_not_supported]
- 85. Causal disentanglement for single-cell representations and controllable counterfactual generation | Nature Communications [https://www.nature.com/articles/s41467-025-62008-1]
- 86. Functional difference between intrinsic and extrinsic coagulation pathways. Kinetics of factor X activation on human monocytes and alveolar macrophages - ScienceDirect [https://www.sciencedirect.com/science/article/pii/S0021925818929431]
- 87. Biomarkers: Opportunities and Challenges for Drug Development in the Current Regulatory Landscape - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7727038/]
- 88. Flexible combination of multiple diagnostic biomarkers to improve... [https://bmcmedresmethodol.biomedcentral.com/articles/10.1186/s12874-015-0085-z]
- 89. Machine Learning Approaches for Biomarker Discovery Using Gene Expression Data - Bioinformatics - NCBI Bookshelf [https://www.ncbi.nlm.nih.gov/books/NBK569564/]
- 90. Boosting power for clinical trials using classifiers based on multiple biomarkers - PubMed [https://pubmed.ncbi.nlm.nih.gov/20541286/]
- 91. Progress in multi-omics studies of osteoarthritis | Biomarker Research [https://biomarkerres.biomedcentral.com/articles/10.1186/s40364-025-00732-y]
- 92. Beyond ELISA: the future of biomarker validation - Drug Target Review [https://www.drugtargetreview.com/article/154316/beyond-elisa-the-future-of-biomarker-validation/]
- 93. Activities and Resources - NCI Assay Harmonization [https://dctd.cancer.gov/drug-discovery-development/assays/harmonization]
- 94. Predicting and affecting response to cancer therapy based on pathway-level biomarkers | Nature Communications [https://www.nature.com/articles/s41467-020-17090-y]
- 95. Advances in the diagnosis of early biomarkers for acute kidney injury... [https://bmcnephrol.biomedcentral.com/articles/10.1186/s12882-025-04040-3]
- 96. Coagulation Cascade | Intrinsic + Extrinsic | Geeky Medics [https://geekymedics.com/the-coagulation-cascade/]
- 97. Diversity and Inclusion in Drug : Rethinking Development and... Intrinsic [https://pmc.ncbi.nlm.nih.gov/articles/PMC9540180/]
- 98. An integrated approach to biomarker discovery reveals gene signatures highly predictive of cancer progression | Scientific Reports [https://www.nature.com/articles/s41598-020-78126-3]
- 99. Experimental and computational modeling for signature and biomarker discovery of renal cell carcinoma progression - PubMed [https://pubmed.ncbi.nlm.nih.gov/34670568/]
- 100. CombiROC: an interactive web tool for selecting accurate marker combinations of omics data | Scientific Reports [https://www.nature.com/articles/srep45477]
- 101. Scalable signature-based molecular diagnostics through on-chip biomarker profiling coupled with machine learning - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7785517/]
- 102. Pathways for Biomarker in Integration | FDA Drug Development [https://www.fda.gov/drugs/biomarker-qualification-program/pathways-biomarker-integration-drug-development]
- 103. of therapeutic targets predicted by tensor... Preclinical validation [https://www.nature.com/articles/s41598-020-74922-z?error=cookies_not_supported&code=e899f100-bb45-4e41-986c-e5bf7ace22ee]
- 104. for in vivo digital measures - PMC Validation framework [https://pmc.ncbi.nlm.nih.gov/articles/PMC11751004/]
- 105. From Promise to Proof: Validating AI-Assisted Workflows in Clinical ... [https://www.linical.com/articles-research/from-promise-to-proof-validating-ai-assisted-workflows-in-clinical-trials]
- 106. The Translational Divide and Digital Biomarker Validation Part 1: Why... [https://www.linkedin.com/pulse/translational-divide-digital-biomarker-validation-why-szczepan-baran-bmg0e]
- 107. Intrinsic Pathway - an overview | ScienceDirect Topics [https://www.sciencedirect.com/topics/engineering/intrinsic-pathway]
- 108. Differential Contributions of Intrinsic and Extrinsic Pathways to Thrombin Generation in Adult, Maternal and Cord Plasma Samples | PLOS One [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0154127]
- 109. of Presepsin, HE4, and Oxygenation... Combined Biomarker Panel [https://www.dovepress.com/combined-biomarker-panel-of-presepsin-he4-and-oxygenation-index-for-se-peer-reviewed-fulltext-article-JIR]
- 110. Optimizing timing and cost-effective use of plasma biomarkers in... [https://alzres.biomedcentral.com/articles/10.1186/s13195-025-01851-2]
- 111. Predictive three-biomarker panel in peripheral blood mononuclear cells for detecting hepatocellular carcinoma - PubMed [https://pubmed.ncbi.nlm.nih.gov/38553531/]
- 112. Frontiers | Serum small RNA profiling identifies prognostic biomarkers ... [https://www.frontiersin.org/journals/medicine/articles/10.3389/fmed.2025.1665726/full]
- 113. Diagnostic value of genetic and epigenetic biomarker for... panels [https://pmc.ncbi.nlm.nih.gov/articles/PMC12098509/]
- 114. Performance of a multi-biomarker panel for prediction of cardiovascular event in patients with chronic kidney disease - PubMed [https://pubmed.ncbi.nlm.nih.gov/36202172/]
- 115. Innovative biomarker panel boosts early pancreatic cancer detection - Bioanalysis Zone [https://www.bioanalysis-zone.com/innovative-biomarker-panel-boosts-early-pancreatic-cancer-detection/]
- 116. Keratin 6 promotes A inflammation through JAK1-STAT3 activation... skin [https://jbiomedsci.biomedcentral.com/articles/10.1186/s12929-025-01143-9]
- 117. S100A8/A9, a potent serum and molecular imaging biomarker for synovial inflammation and joint destruction in seronegative experimental arthritis - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5078998/]
- 118. Update on biomarkers in systemic sclerosis: tools for diagnosis and treatment - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4554742/]
- 119. Potential Biomarkers in Systemic Sclerosis: A Literature Review and Update - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7690387/]
- 120. S100A8/A9, a potent serum and molecular imaging biomarker for synovial inflammation and joint destruction in seronegative experimental arthritis - PubMed [https://pubmed.ncbi.nlm.nih.gov/27776554/]
- 121. Integration of multiple machine learning approaches develops a gene... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11865301/]
- 122. Comprehensive characterization of PD - L and... 1 expression [https://bmcpulmmed.biomedcentral.com/articles/10.1186/s12890-025-03687-w]
- 123. Biomarkers of response to PD-1 pathway blockade | British Journal of Cancer [https://www.nature.com/articles/s41416-022-01743-4]
- 124. Novel tumor mutation score versus tumor mutation burden in predicting... [https://atm.amegroups.org/article/view/39912/html]
- 125. Clinical perspectives on the value of testing for STK and... 11 [https://pmc.ncbi.nlm.nih.gov/articles/PMC11655323/]
- 126. Editorial: New advancement in tumor microenvironment remodeling and cancer therapy - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10955374/]
- 127. Frontiers | Resistance to PD-L1/PD-1 Blockade Immunotherapy. A Tumor-Intrinsic or Tumor-Extrinsic Phenomenon? [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2020.00441/full]
- 128. Less immune cell infiltration and worse prognosis after immunotherapy for patients with lung adenocarcinoma who harbored STK11 mutation - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S1567576920309255]
- 129. Quantifying the added value of new biomarkers : how and how not [https://diagnprognres.biomedcentral.com/articles/10.1186/s41512-018-0037-2]
- 130. Evaluating classification performance of biomarkers in two-phase... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6317859/]
- 131. Combining large number of weak biomarkers based on AUC - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5823017/]
- 132. A Wild Bootstrap approach for the selection of biomarkers in early diagnostic trials | BMC Medical Research Methodology | Full Text [https://bmcmedresmethodol.biomedcentral.com/articles/10.1186/s12874-015-0025-y]
- 133. Biomarker selection for medical diagnosis using the partial area under the ROC curve | BMC Research Notes | Full Text [https://bmcresnotes.biomedcentral.com/articles/10.1186/1756-0500-7-25]
- 134. evaluation - c- Biomarker ( statistic ) and alternatives AUC [https://discourse.datamethods.org/t/biomarker-evaluation-c-statistic-auc-and-alternatives/6956]
- 135. Statistical inference for net benefit measures in biomarker validation studies - PubMed [https://pubmed.ncbi.nlm.nih.gov/31732971/]
- 136. What Is A Biomarker ? Definition, Types, Applications [https://bitesizebio.com/26559/biomarkers-explained/]
- 137. versus Prognostic value of predictive in oncology biomarkers [https://pubmed.ncbi.nlm.nih.gov/18396036/]
- 138. Frontiers | Prognostic for biomarkers decompensation in... predicting [https://www.frontiersin.org/journals/medicine/articles/10.3389/fmed.2025.1650124/full]
- 139. Altered of apoptotic markers of both extrinsic and intrinsic ... pathways [https://virologyj.biomedcentral.com/articles/10.1186/1743-422x-9-314]
- 140. What Are Biomarkers? Definition, Applications and Updates for 2025 [https://blog.omni-inc.com/blog/what-are-biomarkers-and-whats-new-in-2025]