Mastering Annexin V Flow Cytometry: A Comprehensive Guide to Troubleshooting Background Noise
This article provides researchers, scientists, and drug development professionals with a complete framework for understanding, identifying, and resolving background noise in Annexin V flow cytometry assays.
Mastering Annexin V Flow Cytometry: A Comprehensive Guide to Troubleshooting Background Noise
Abstract
This article provides researchers, scientists, and drug development professionals with a complete framework for understanding, identifying, and resolving background noise in Annexin V flow cytometry assays. Covering foundational principles from phosphatidylserine binding to advanced spectral overlap theory, the guide details optimized staining protocols and sample preparation techniques to prevent noise. It offers a systematic troubleshooting workflow for common issues like high autofluorescence and poor compensation, and concludes with robust validation strategies and comparative analyses against other apoptosis detection methods to ensure data accuracy and reliability in biomedical research.
Understanding the Sources and Impact of Background Noise in Apoptosis Detection
Annexin V staining is a cornerstone method for detecting apoptotic cells, providing critical insights into the mechanisms of programmed cell death for researchers and drug development professionals. The fundamental principle relies on the specific, high-affinity binding of the Annexin V protein to phosphatidylserine (PS), a phospholipid that undergoes translocation from the inner to the outer leaflet of the plasma membrane during the early stages of apoptosis. In normal, healthy cells, phosphatidylserine is maintained exclusively on the cytoplasmic surface of the plasma membrane. This asymmetrical distribution is actively regulated by flippase enzymes. During the execution phase of apoptosis, this regulation collapses, and a scramblase activity facilitates the rapid externalization of PS, making it a specific molecular flag for phagocytic cells—and for detection by Annexin V [1] [2].
The human vascular anticoagulant, Annexin V, is a 35–36 kDa, Ca2+-dependent phospholipid-binding protein [3] [1]. Once PS is exposed on the cell surface, fluorescent conjugates of Annexin V can bind to it, enabling the identification and quantification of apoptotic cells using flow cytometry or fluorescence microscopy. The difference in fluorescence intensity between apoptotic and non-apoptotic cells stained with fluorescent Annexin V conjugates is typically about 100-fold, making it a highly robust assay [1]. It is crucial to note that the binding is strictly dependent on calcium ions, and the presence of chelators like EDTA will abolish the interaction [4] [5].
Troubleshooting Guide: Common Problems and Solutions
A successful Annexin V experiment can be compromised by various factors, leading to unclear results, high background, or a lack of signal. The following table summarizes common issues and their targeted solutions to troubleshoot background noise and other artifacts in your flow cytometry data.
| Problem Phenomenon | Potential Causes | Recommended Solutions |
|---|---|---|
| High background / False positives in control group [6] [7] [5] | - Poor compensation causing fluorescence spillover [5].- Overly confluent or starved cells undergoing spontaneous apoptosis [6] [5].- Mechanical damage from harsh pipetting or over-trypsinization [6] [5].- Interference from fluorescent drugs or cell autofluorescence [6]. | - Re-adjust compensation using single-stain controls [7] [5].- Use healthy, log-phase cells and avoid over-confluency [6] [5].- Handle cells gently; use Accutase instead of trypsin/EDTA [5].- Choose a fluorophore with minimal spectral overlap (e.g., APC instead of FITC for GFP-expressing cells) [6] [5]. |
| No positive signal in treated group [6] [5] | - Insufficient drug concentration or treatment duration [5].- Failure to collect apoptotic cells in the supernatant [6] [5].- Operational error (e.g., forgot to add dye, washed after PI staining) [6] [4].- Reagent degradation due to improper storage [6]. | - Optimize treatment conditions with concentration and time gradients [5].- Always collect and combine cells from the culture supernatant with the trypsinized pellet [6].- Follow protocol strictly; do not wash after adding PI/7-AAD [4].- Use a positive control (e.g., camptothecin-treated cells) to verify kit functionality [1] [5]. |
| Unclear cell population clustering [6] | - High cellular autofluorescence [6] [7].- Excessive apoptosis leading to insufficient dye [6].- Poor overall cell health causing nonspecific PS exposure [6]. | - Switch to a brighter fluorophore or one with non-overlapping emission [6] [7].- Titrate and increase the amount of dye used [6].- Ensure proper cell culture conditions and gentle handling throughout the experiment [6]. |
| Only nuclear dye (PI) is positive [6] [5] | - The cells are primarily necrotic or in late-stage apoptosis [6].- The treatment was too intense, causing rapid cell death [6].- Cells were handled too roughly, damaging membranes [5]. | - Use gentler treatment conditions (e.g., lower drug concentration) [6].- Treat cells gently during harvesting and washing [6] [5].- Ensure cells are healthy and in good condition at the start of the experiment [5]. |
| Only Annexin V is positive [6] | - The nuclear dye (PI/7-AAD) was forgotten [6].- The cells are predominantly in early apoptosis [8].- The nuclear dye is inactive due to improper storage [6]. | - Repeat the experiment, confirming all dyes are added [6].- Verify apoptosis by other methods (e.g., morphology). This can be a normal result for early time points [8].- Check storage conditions of reagents; 7-AAD, for example, should be stored at -20°C [6]. |
| False PI staining from cytoplasmic RNA [9] | - PI binds to cytoplasmic RNA in large cells, misclassifying them as late apoptotic/necrotic [9]. | - Incorporate an RNase A treatment step (50 μg/mL) after fixation with 1% formaldehyde to digest RNA [9]. |
Detailed Experimental Protocols
Standard Annexin V Staining Protocol for Flow Cytometry
This protocol is adapted from industry standards and is designed for use with Annexin V conjugated to FITC and Propidium Iodide (PI) [4] [1].
Materials Required:
- Fluorochrome-conjugated Annexin V (e.g., Annexin V-FITC)
- Propidium Iodide (PI) Staining Solution or 7-AAD
- 10X Binding Buffer
- Phosphate Buffered Saline (PBS), calcium- and magnesium-free
- Flow cytometry tubes
Procedure:
- Prepare 1X Binding Buffer: Dilute the 10X binding buffer 1:9 with distilled water.
- Harvest Cells: Collect both the culture supernatant (containing detached dead cells) and the adherent cells. Use gentle dissociation methods like Accutase to avoid damaging the PS epitope. Critical: Avoid using trypsin containing EDTA, as it chelates the Ca²⁺ required for Annexin V binding [5].
- Wash Cells: Pellet the cells by centrifugation (300–500 × g for 5 minutes). Wash once with cold PBS and once with 1X Binding Buffer.
- Resuspend Cells: Resuspend the cell pellet in 1X Binding Buffer at a density of 1–5 × 10⁶ cells/mL. Transfer 100 µL of the cell suspension to a flow cytometry tube.
- Stain with Annexin V: Add 5 µL of fluorochrome-conjugated Annexin V to the 100 µL cell suspension. Mix gently and incubate for 10–15 minutes at room temperature in the dark.
- Add Viability Dye: After incubation, add 2 mL of 1X Binding Buffer and centrifuge. Discard the supernatant. Resuspend the cell pellet in 200 µL of 1X Binding Buffer. Add 5 µL of PI Staining Solution and incubate for 5–15 minutes on ice or at room temperature in the dark. Critical: Do not wash the cells after adding PI or 7-AAD, as this can remove the unbound dye and reduce the signal [4].
- Acquire Data: Analyze the samples by flow cytometry within 1 hour. Keep samples on ice and protected from light until acquisition.
Modified Annexin V/PI Protocol with RNase A Treatment
This modified protocol is essential for eliminating false-positive PI staining caused by binding to cytoplasmic RNA, which is prevalent in large cells like macrophages [9].
Procedure (follows standard protocol steps 1-6 above):
- Fix Cells: After staining with Annexin V and PI, resuspend the cells in 500 µL of 1X Binding Buffer and 500 µL of 2% formaldehyde to create a 1% final fixative solution. Fix on ice for 10 minutes.
- Wash: Add 1 mL of PBS to the sample, centrifuge (425 × g for 8 minutes), and decant the supernatant. Repeat this wash step.
- RNase Treatment: Resuspend the cell pellet by flicking the tube. Add 16 µL of a 1:100 diluted RNase A to achieve a final concentration of 50 µg/mL. Incubate for 15 minutes at 37°C.
- Wash and Analyze: Add 1 mL of PBS, centrifuge, and resuspend the pellet in an appropriate buffer for flow cytometry analysis [9].
The Scientist's Toolkit: Essential Research Reagents
The following table details key reagents and their critical functions in Annexin V-based apoptosis assays.
| Reagent | Function & Importance | Key Considerations |
|---|---|---|
| Annexin V Conjugate [1] | Binds with high affinity to externalized Phosphatidylserine (PS) in the presence of Ca²⁺, identifying apoptotic cells. | Available conjugated to various fluorophores (FITC, PE, APC, Alexa Fluor dyes). Choose one that fits your instrument's lasers and minimizes spectral overlap with other probes. |
| Viability Dye (PI, 7-AAD, FVDs) [8] [4] [1] | Distinguishes membrane integrity. PI/7-AAD are impermeant to intact membranes, staining only late apoptotic/necrotic cells. Fixable Viability Dyes (FVDs) allow subsequent fixation/permeabilization. | Do not wash after adding PI/7-AAD. FVD eFluor 450 is not recommended with some Annexin V kits due to potential spectral overlap [4]. |
| Binding Buffer [4] [1] | Provides the optimal calcium-containing environment for Annexin V-PS binding and maintains cell viability during staining. | Always use the recommended buffer. Avoid buffers containing EDTA or other calcium chelators, as they will inhibit binding [4]. |
| RNase A [9] | Digests cytoplasmic RNA to prevent false-positive staining by PI in large cells and primary cells with high RNA content. | Use after fixation in a modified protocol. A final concentration of 50 µg/mL is effective for most cell types [9]. |
| Camptothecin or Staurosporine [1] | Common apoptosis-inducing drugs used as reliable positive controls to verify the performance of your assay and reagents. | Include in every experiment to confirm that your system can detect a clear apoptotic signal. |
FAQs for Annexin V-Based Apoptosis Assays
Q1: Is the Annexin V assay species-specific? No. Since Annexin V binds to phosphatidylserine, a phospholipid that is highly conserved across species, the assay is not species-dependent and can be used with human, mouse, rat, and many other mammalian cells [5].
Q2: My cells express GFP. Which Annexin V conjugate should I use? You should avoid using Annexin V-FITC due to significant spectral overlap. Instead, select a conjugate with a distinct emission profile, such as PE, APC, or Alexa Fluor 647 [5].
Q3: Why is it critical to avoid EDTA during cell harvesting? Annexin V binding to PS is strictly calcium-dependent. Trypsin solutions that contain EDTA will chelate the required Ca²⁺ ions, thereby interfering with the binding and leading to false-negative results or weak signals. Use PBS without calcium and magnesium for washing, and consider gentler dissociation enzymes like Accutase [4] [5].
Q4: What are the essential controls for a flow cytometry experiment?
- Unstained Cells: For setting flow cytometry voltages and gating.
- Single-Stain Controls: Cells stained with Annexin V only and PI/7-AAD only. These are mandatory for accurate compensation to correct for fluorescence spillover between channels [7] [5].
- Untreated Control: To establish the baseline level of apoptosis.
- Treated & Stained Sample: Your experimental sample.
- Positive Control: Cells treated with a known apoptosis inducer (e.g., 10 µM camptothecin for 4 hours) to confirm the assay is working [1] [5].
Q5: How can I reduce high background fluorescence?
- Ensure thorough washing of cells before staining.
- Use Fc receptor blocking reagents to minimize non-specific antibody binding.
- Titrate your antibodies and Annexin V reagent to find the optimal concentration.
- Run an unstained control to assess the level of cellular autofluorescence, which can be higher in stressed or poorly healthy cells [7].
Data Analysis and Gating Strategy
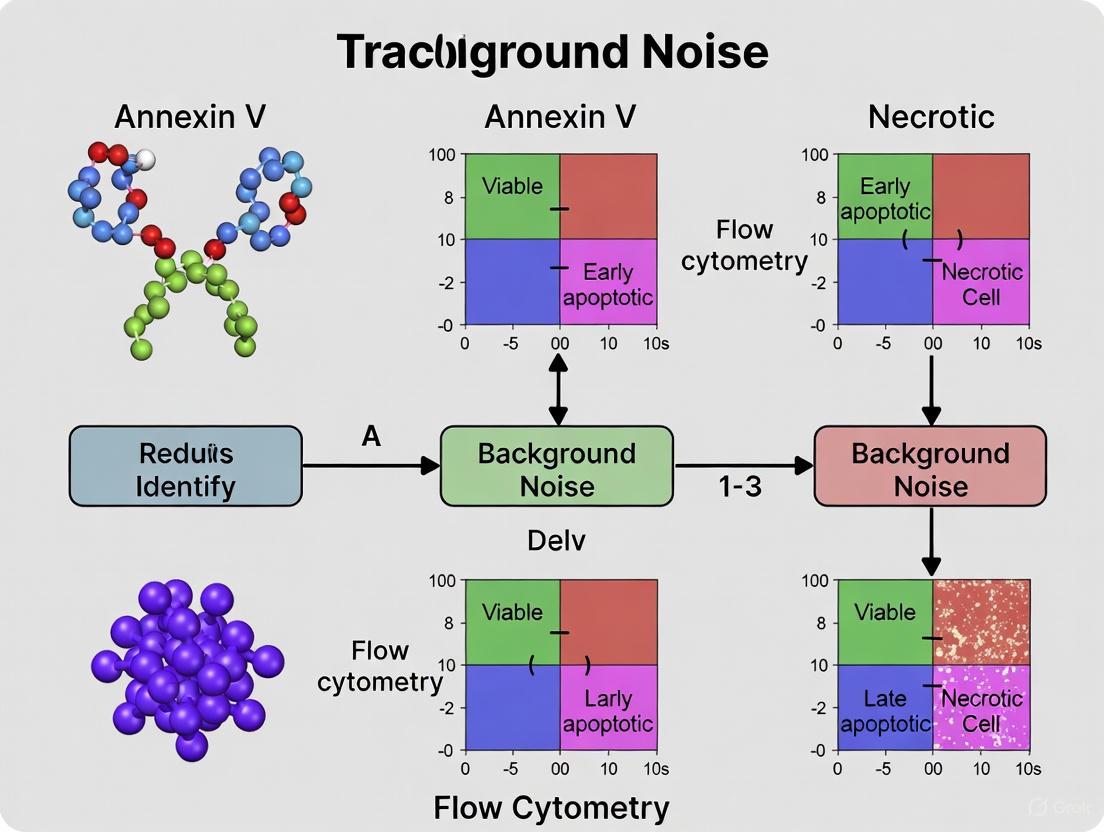
Proper gating is fundamental for accurate quantification of apoptotic populations. The standard approach involves creating a dot plot with Annexin V fluorescence on one axis and the viability dye (e.g., PI) on the other. This divides the cell population into four distinct quadrants [8] [1].
- Viable Cells (Annexin V⁻ / PI⁻): Located in the lower-left quadrant. These cells have an intact membrane and no externalized PS.
- Early Apoptotic Cells (Annexin V⁺ / PI⁻): Located in the lower-right quadrant. This population is the primary indicator of early apoptosis, showing PS externalization while maintaining membrane integrity that excludes PI.
- Late Apoptotic and Necrotic Cells (Annexin V⁺ / PI⁺): Located in the upper-right quadrant. These cells have both externalized PS and a compromised membrane, allowing PI to enter and stain the DNA. It is often impossible to distinguish late apoptotic from necrotic cells in this assay without additional kinetic or morphological data [8] [1].
- Necrotic/Damaged Cells (Annexin V⁻ / PI⁺): A small population in the upper-left quadrant may represent cells that have undergone primary necrosis or were mechanically damaged during processing, leading to membrane rupture before PS externalization [5].
In flow cytometry, background noise refers to any unwanted signal that interferes with the accurate detection and measurement of your specific fluorescent signal. This noise reduces the sensitivity and resolution of the instrument, making it difficult to distinguish between closely spaced cell populations and to detect weak signals [10]. For researchers using Annexin V-based apoptosis assays, understanding and mitigating background noise is critical for obtaining reliable, publication-quality data. This guide provides a comprehensive overview of noise sources and practical solutions tailored for scientists and drug development professionals.
Background noise in flow cytometry can be categorized into several distinct types, each with different origins and characteristics. The table below summarizes the primary noise types and their key features [10].
Table 1: Types of Background Noise in Flow Cytometry
| Noise Type | Origin | Key Characteristics | Impact on Data |
|---|---|---|---|
| Thermal Noise (Johnson Noise) | Random motion of electrons in conductors [10] | Temperature dependent; present in all electronics [10] | Reduces overall signal-to-noise ratio [10] |
| Shot Noise | Discrete nature of electric charge and random arrival of photons/electrons [10] | Signal-dependent; increases with average signal level [10] | Creates variance in measurements of identical samples [10] |
| Flicker Noise (1/f Noise) | Complex, not fully understood mechanisms in electronic components [10] | Dominant at lower frequencies [10] | Affects low-frequency signal stability [10] |
| Electronic Interference | External electromagnetic sources (power lines, radio transmitters, other devices) [10] | Often occurs at specific frequencies; time-dependent [10] | Causes erratic signals and high background [10] |
| Optical Noise | Stray light, autofluorescence, light scatter from particles/debris [10] | Light-based; wavelength and sample dependent [10] | Increases background fluorescence, obscures specific signal [10] |
| Reagent Noise | Non-specific antibody binding, aggregated antibodies, improperly labeled reagents, dye instability [10] | Reagent-specific; sample and time dependent [10] | Causes false positives and increased signal spread [10] |
| Biological Noise | Cellular autofluorescence, dead cells, non-specific antibody binding [11] [12] | Inherent to biological samples [11] | Masks specific staining, particularly for dim markers [11] |
Diagram 1: A taxonomy of background noise sources in flow cytometry, showing the three primary categories and their specific contributors.
Noise-Specific Troubleshooting FAQs
General Noise Reduction Strategies
Q: What are the most effective general strategies for reducing background noise in flow cytometry?
A: A multi-pronged approach is most effective for noise reduction [10]:
- Instrument Optimization: Adjust laser power, detector voltage, and amplifier gain to maximize signal-to-noise ratio [10].
- Appropriate Filtering: Use optical and electronic filters to block unwanted wavelengths and frequencies [10].
- Shielding and Grounding: Use shielded cables and proper grounding to reduce electronic interference [10].
- Sample Preparation: Filter samples and reagents to remove particles and debris that cause light scatter [10].
- Control Experiments: Always run proper controls (FMO, isotype, unstained) to identify and subtract background noise [10] [7].
Q: How can I determine if my noise is coming from the instrument versus my sample?
A: Follow this systematic diagnostic approach:
- Run calibration beads - If noise persists, the issue is likely instrumental [7].
- Analyze unstained cells - High background indicates autofluorescence or electronic noise [12].
- Check single-stained controls - Excessive spread in negative populations may indicate poor compensation [5] [7].
- Compare fresh vs. fixed cells - Increased noise in fixed cells may indicate fixation-induced autofluorescence [7].
Annexin V-Specific Noise Issues
Q: In my Annexin V/PI apoptosis assay, I'm seeing high background in the unstained control. What could be causing this?
A: High background in Annexin V assays can stem from several sources [5] [13]:
- Cell Health Issues: Overconfluent cultures, serum starvation, or rough handling during harvesting can cause spontaneous apoptosis and PS exposure [5].
- Trypsin/EDTA Use: Trypsin with EDTA chelates Ca²⁺, which is essential for Annexin V binding to phosphatidylserine, potentially causing artifactual results [5].
- Platelet Contamination: In blood samples, platelets contain PS and can bind Annexin V, producing misleading results [5].
- Instrument Contamination: Previous samples may not have been thoroughly cleaned from the flow cytometer [13].
- Delayed Analysis: Analyzing samples more than 1 hour after staining can increase background [5].
Q: My Annexin V/PI staining shows unclear cell population clustering. What should I investigate?
A: Poor population separation can result from [13]:
- Excessive Cellular Autofluorescence: Switch to a fluorochrome with less spectral overlap with autofluorescence [13].
- Insufficient Dye Concentration: The apoptosis signal may be weak relative to background; try increasing dye usage [13].
- Poor Cell State: If all cells show some PS exposure, the culture may be unhealthy [13].
- Improper Compensation: Re-adjust compensation using single-stain controls to prevent fluorescence spillover [5].
Diagram 2: Troubleshooting workflow for unclear population clustering in Annexin V/PI apoptosis assays, showing the primary categories of issues and their specific causes.
Experimental Protocols for Noise Identification and Reduction
Protocol: Establishing Proper Controls for Background Determination
Objective: To correctly identify and quantify background noise sources in Annexin V flow cytometry experiments.
Materials:
- Cells of interest (properly cultured and treated)
- Annexin V binding buffer (with Ca²⁺)
- Annexin V conjugated to fluorochrome (e.g., FITC)
- Propidium iodide (PI) or 7-AAD
- Flow cytometry staining tubes
- Centrifuge
- Flow cytometer with appropriate laser and filter configuration
- Prepare Control Tubes:
- Unstained Control: Cells + binding buffer only
- Single-Stain Controls: Cells + Annexin V only; Cells + PI only
- FMO (Fluorescence Minus One) Controls: For multicolor panels, include all antibodies except one
- Full-Stain Experimental Sample: Cells + all reagents
Staining Procedure:
- Harvest cells gently using EDTA-free dissociation enzymes to preserve membrane integrity [5].
- Wash cells twice with cold binding buffer.
- Resuspend cell pellet in 100 μL binding buffer.
- Add Annexin V conjugate to appropriate tubes (typically 5 μL).
- Incubate 15 minutes at room temperature in the dark.
- Add PI (1 μg/mL final concentration) to appropriate tubes.
- Add binding buffer to bring total volume to 500 μL.
- Analyze on flow cytometer within 1 hour.
Data Analysis:
- Use unstained cells to set photomultiplier tube (PMT) voltages.
- Use single-stain controls to set compensation.
- Use FMO controls to establish gating boundaries for dim populations.
Protocol: Titration of Antibodies and Reagents to Minimize Reagent Noise
Objective: To determine the optimal concentration of fluorescent reagents that maximizes signal-to-noise ratio.
Materials:
- Titration range of antibody or reagent
- Target cells with known expression of antigen
- Flow cytometry staining buffer (PBS + 1-5% BSA)
- Flow cytometry tubes
- Centrifuge
- Prepare a dilution series of your antibody (e.g., 0.125, 0.25, 0.5, 1, 2 times the manufacturer's recommended concentration).
- Aliquot equal numbers of cells (e.g., 1×10⁶) into each tube.
- Add the different antibody concentrations to respective tubes.
- Incubate according to standard protocol (time, temperature).
- Wash cells and resuspend in buffer for analysis.
- Analyze on flow cytometer, collecting sufficient events for statistical analysis.
- Calculate the stain index for each concentration: (Median Positive - Median Negative) / (2 × SD of Negative).
- Select the concentration that provides the highest stain index, indicating optimal signal-to-noise ratio.
The Scientist's Toolkit: Essential Reagents for Noise Reduction
Table 2: Key Research Reagent Solutions for Background Control
| Reagent/Category | Specific Examples | Primary Function in Noise Reduction |
|---|---|---|
| Annexin V Conjugates | Annexin V-FITC, Annexin V-PE, Annexin V-APC [5] | Detects phosphatidylserine exposure during apoptosis; different conjugates help avoid spectral overlap with autofluorescence or other probes [5]. |
| Viability Dyes | Propidium Iodide (PI), 7-AAD, DAPI [7] | Identifies dead cells which are notorious for non-specific antibody binding; enables gating out noisy dead cell signals [11] [7]. |
| Fc Receptor Blockers | Anti-CD16/32 (2.4G2), Human Fc Receptor Binding Inhibitor [11] | Blocks non-specific antibody binding via Fc receptors, reducing false positive signals [11]. |
| Blocking Reagents | BSA, Serum (FCS or species-matched), Glycine [8] [11] | Covers non-specific binding sites on cells and plastic surfaces, reducing background staining [8] [11]. |
| Cell Dissociation Reagents | EDTA-free enzymes, Accutase [5] | Gently dissociates adherent cells while preserving membrane integrity and preventing artifactual Annexin V binding [5]. |
| Compensation Beads | Antibody Capture Beads [7] | Provides consistent positive and negative populations for setting accurate compensation, reducing spread error [7]. |
| Fixable Viability Dyes | Fixable Viability Dyes eFluor series [11] | Allows fixation after staining without leakage of dye to other cells, maintaining clear live/dead discrimination [11]. |
Advanced Noise Mitigation Strategies
Spectral Flow Cytometry for Enhanced Noise Discrimination
Spectral flow cytometry represents an advanced approach to noise reduction by measuring the entire emission spectrum of fluorochromes rather than just peak emissions [12]. This enables:
- Better discrimination between autofluorescence and specific signals
- Improved unmixing of overlapping fluorochromes
- Direct measurement of cellular autofluorescence signatures
- Reduced compensation spread error
For Annexin V assays, spectral cytometry can particularly help when working with cells that have high intrinsic autofluorescence or when using drugs with fluorescent properties (e.g., doxorubicin) [12].
Fluorochrome Selection Strategy to Minimize Noise
The choice of fluorochrome significantly impacts background noise. Follow these guidelines for optimal selection [11] [12]:
- Avoid FITC for intracellular targets due to its charge characteristics that promote non-specific binding to cytoplasmic elements [11].
- Be cautious with cyanine dyes (Cy5, Cy7, and tandems) which can bind to某些 Fc receptors, particularly on monocytes [11].
- Consider PE and APC carefully, as small subsets of B and T cells specifically recognize these proteins as antigens [11].
- Match fluorochrome brightness to antigen density - use brightest fluorochromes for low-abundance targets [7].
- For cells with high autofluorescence, choose red-excited fluorochromes where autofluorescence is typically lower [12].
Diagram 3: Strategic approach to fluorochrome selection for minimizing background noise, showing the four key consideration categories and their specific selection criteria.
Effective management of background noise is fundamental to obtaining reliable flow cytometry data, particularly in sensitive applications like Annexin V-based apoptosis detection. By understanding the diverse sources of noise—from instrumental factors to biological variability—researchers can implement targeted strategies to minimize interference and enhance data quality. The protocols and guidelines provided here offer a systematic approach to noise identification, troubleshooting, and prevention that should enable scientists and drug development professionals to produce more accurate, reproducible results in their apoptosis research.
Diagnostic Guide: Identifying the Source of Background Noise
Accurately diagnosing the primary source of background noise is the critical first step in troubleshooting your Annexin V flow cytometry experiments. The table below outlines the characteristic signatures of autofluorescence and non-specific binding to aid in identification.
Table 1: Diagnostic Features of Major Noise Sources
| Feature | Autofluorescence | Non-Specific Binding |
|---|---|---|
| Primary Cause | Intrinsic properties of cells or experimental treatments [14] [15] | Antibody interactions with non-target structures or dead cells [15] [7] |
| Typical Signal Pattern | Broad emission spectrum across multiple detectors [15] | Signal confined to the specific fluorochrome's channel [15] |
| Affected Cell Populations | Often cell-type specific (e.g., neutrophils, epithelial cells) [15] [7] | Predominantly in dead cells or cells with high Fc receptor expression [16] [15] |
| Impact of Fixation | Can increase with over-fixation [16] [7] | Can be exacerbated by fixation, depending on the cause [15] |
| Best Control | Unstained cells from the same treatment group [15] [7] | Fluorescence Minus One (FMO) and isotype controls [7] |
The following workflow provides a systematic approach to diagnose and resolve background noise issues:
Diagram 1: Noise Source Diagnostic Workflow
Troubleshooting Autofluorescence
Autofluorescence occurs when cells or reagents intrinsically emit light, interfering with the detection of specific fluorescent signals. Below are frequently asked questions to guide your resolution of this issue.
FAQ 1: My cells have high intrinsic autofluorescence. How can I mitigate this? Some cell types, such as neutrophils and epithelial cells, are naturally more autofluorescent [15] [7]. To address this:
- Choose Optimal Fluorophores: Use fluorophores that emit in the red channel (e.g., APC, Alexa Fluor 647), where cellular autofluorescence is typically minimal [15].
- Employ Bright Fluorophores: In channels prone to autofluorescence (e.g., FITC), use very bright alternatives like Alexa Fluor 488 or Brilliant Blue 515 to improve the signal-to-noise ratio [15].
FAQ 2: My experimental treatment is causing autofluorescence. What can I do? Certain drugs (e.g., doxorubicin, tetracycline) or cells transfected with fluorescent proteins can contribute to background signal [14].
- Fluorophore Substitution: If your experimental system involves GFP, avoid using an Annexin V-FITC kit. Instead, select a kit labeled with PE, APC, or Alexa Fluor 647 to prevent spectral overlap [5].
- Control Strategy: Always include an unstained control that has undergone the same treatment (e.g., drug exposure, transfection) to accurately measure treatment-induced autofluorescence [7].
FAQ 3: Could my sample preparation be increasing autofluorescence? Yes, sample handling significantly impacts autofluorescence.
- Fixation: Avoid over-fixing cells, as this can increase autofluorescence. Do not exceed recommended fixation times (typically 30 minutes or less) [16] [7].
- Cell Health: Use healthy, log-phase cells and avoid conditions that induce stress or spontaneous apoptosis, which can alter cellular properties [5] [14].
Troubleshooting Non-Specific Binding
Non-specific binding results from antibody reagents interacting with cellular components in an undesired, off-target manner.
FAQ 4: I suspect non-specific binding from antibodies. How do I confirm and fix it?
- Fc Receptor Blocking: Many cell types express Fc receptors that bind the constant region (Fc) of antibodies. Incorporate an Fc receptor blocking step using normal serum, BSA, or commercial blocking reagents prior to antibody staining [15] [7].
- Antibody Titration: High antibody concentrations are a common cause of background. Titrate your antibodies to find the optimal concentration that provides a strong specific signal with minimal background [16] [15].
- Direct Staining Preference: Whenever possible, use directly conjugated antibodies rather than a primary-secondary system. This reduces the number of incubation and wash steps and eliminates background from cross-reactive secondary antibodies [15] [7].
FAQ 5: Dead cells are a major source of noise in my experiment. How can I manage this? Dead cells bind antibodies and dyes non-specifically, significantly increasing background [16] [15].
- Viability Dye Gating: Always include a viability dye (e.g., PI, 7-AAD, or a fixable viability dye) in your staining panel. During analysis, gate out the dead cells to remove their confounding signal [15] [7].
- Gentle Handling: Avoid procedures that increase cell death. Use gentle, non-enzymatic cell dissociation methods like Accutase where possible, as trypsin with EDTA can damage the cell membrane and affect Annexin V binding [5] [17]. Handle cells gently during pipetting and centrifugation to maintain membrane integrity [5] [14].
FAQ 6: My controls indicate non-specific binding, but I have already titrated my antibodies. What else can I check?
- Thorough Washing: Increase the volume, number, or duration of wash steps to ensure the removal of unbound antibody [16] [7].
- Check Compensation: Poor compensation can cause fluorescence spillover, making populations appear positive in multiple channels. Use single-stained controls to set compensation correctly on your flow cytometer [5] [7].
- Buffer Integrity: Ensure that buffers like Binding Buffer for Annexin V are correctly diluted, as abnormal osmotic pressure can induce apoptosis and secondary necrosis [14].
The Scientist's Toolkit: Essential Research Reagent Solutions
The following table lists key reagents and their roles in minimizing background noise for Annexin V apoptosis assays.
Table 2: Key Reagent Solutions for Noise Reduction
| Reagent / Tool | Primary Function | Role in Noise Reduction |
|---|---|---|
| Fc Receptor Blockers | Blocks Fc receptors on immune cells | Prevents non-specific antibody binding, a major source of high background [15] [7] |
| Viability Dyes (PI, 7-AAD) | Labels cells with compromised membranes | Enables gating and exclusion of dead cells, which bind antibodies non-specifically [5] [7] |
| Alternative Fluorophores (e.g., APC) | Fluorescent labels for detection | Red-shifted dyes minimize interference from cellular autofluorescence [5] [15] |
| Gentle Dissociation Reagents (Accutase) | Detaches adherent cells | Preserves membrane integrity, reducing false-positive apoptosis and necrosis signals [5] [17] |
| Calcium-Containing Binding Buffer | Provides optimal Annexin V binding conditions | Essential for specific Annexin V/PS interaction; its absence causes false negatives [5] [18] |
| Single-Stain Controls | Used for instrument compensation | Critical for correcting spectral overlap, which can be misinterpreted as positive signal [5] [7] |
FAQs: Addressing Common Instrumental Challenges
1. What is the single most important instrument setting for reducing background noise? The threshold (or discriminator) setting is critical for reducing background noise. It sets a minimum level that a signal must exceed to be counted as an event, effectively filtering out sub-threshold electronic noise and small debris [19]. Setting a threshold on a scatter parameter like Forward Scatter (FSC) prevents the system from processing and recording these insignificant signals, which drastically reduces file sizes and improves the clarity of your data by increasing the percentage of target cells in the acquired data [19].
2. How do PMT voltages directly affect my signal-to-noise ratio? Photomultiplier Tube (PMT) voltages directly control the amplification of the signal from your fluorochromes. If the voltage is too low, positive signals from your experiment will not be adequately amplified and may be lost or appear weak. If the voltage is too high, the amplification of background noise and autofluorescence can overwhelm your specific signal, leading to a poor signal-to-noise ratio and high background [7] [16]. Optimal PMT voltages, often determined using unstained cells and calibration beads, ensure that your positive signal is well-separated from the negative population.
3. Why is proper compensation crucial for a clean Annexin V assay? Spectral compensation is a mathematical correction for the unavoidable "spillover" fluorescence, where the emission of one fluorochrome is detected in the detector of another [11]. In an Annexin V assay using FITC and PI, improper compensation can make an Annexin V-FITC positive cell appear falsely positive for PI, or vice-versa [5]. This leads to misidentification of apoptotic stages and inaccurate data. Correct compensation ensures that fluorescence is assigned to the correct channel, which is fundamental for accurate quadrant placement in an Annexin V plot [5] [7].
4. My cell populations are not clearly separated in the Annexin V plot. Could this be an instrument issue? Yes, several instrument factors can cause unclear population separation. Poor compensation is a primary suspect, as spillover can smear populations diagonally [5] [7]. Additionally, if the lasers are misaligned, it can result in weak and variable signals [7]. Another common issue is using an inappropriate threshold that is too high, which can selectively remove genuine, smaller apoptotic cells from your analysis [19]. Finally, ensure your fluidics system is not clogged and is delivering a stable, single-cell stream, as irregularities here can cause inconsistent lighting and signal detection [20].
5. I see a high signal in my unstained control. Is this always a staining problem? Not always. While non-specific antibody binding can be a cause, high signal in an unstained control is frequently due to cellular autofluorescence, especially if cells are over-fixed, stressed, or from certain tissue types [7] [16]. Electronic noise from improperly set PMT voltages can also mimic a true signal [19]. To diagnose, compare your unstained cells to the system's background using only sheath fluid. Furthermore, including a viability dye is essential, as dead cells are notoriously "sticky" and bind antibodies non-specifically, contributing significantly to background [11] [7].
Troubleshooting Guide: Flow Cytometer Settings
| Problem | Potential Instrumental Cause | Recommended Solution |
|---|---|---|
| High Background Noise | Threshold set too low, allowing electronic noise and small debris to be recorded [19]. | Increase the FSC threshold to exclude sub-cellular debris [19]. |
| PMT voltage is set too high, over-amplifying background autofluorescence [7] [16]. | Titrate PMT voltages using unstained cells to place the negative population clearly on-scale [7]. | |
| Poor compensation, causing fluorescence spillover from bright channels into others [5] [7]. | Redo compensation calculations using single-stain controls that are brighter than your sample [7]. | |
| Weak or No Signal | PMT voltage is too low for the fluorochrome being detected [7]. | Increase the PMT voltage for the specific channel, ensuring the positive signal is not off-scale. |
| Laser misalignment or failure [7]. | Run calibration beads to check laser performance and alignment; service instrument if necessary [7]. | |
| Threshold set too high, excluding your cells of interest [19]. | Lower the threshold to ensure target cells are not being gated out during acquisition [19]. | |
| Unclear Population Separation | Incorrect spectral compensation causing spreading error [5] [7]. | Use properly titrated antibodies and create new single-stain compensation controls [5] [7]. |
| Unstable fluidics stream causing inconsistent cell lighting [20]. | Check for clogs, ensure proper sheath pressure, and de-clump the sample before running. | |
| Abnormal Event Rate | Flow cytometer clog or unstable fluidics [16]. | Stop, backflush, and clean the fluidics system according to the manufacturer's protocol. |
| Cell concentration is too high or too low [16]. | Adjust cell concentration to the manufacturer's recommended range (e.g., 1-5 x 10^6 cells/mL) [4]. |
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Annexin V Apoptosis Assays |
|---|---|
| Calibration Beads | Used to verify and optimize the performance of the flow cytometer, including laser alignment and PMT responsiveness, ensuring day-to-day consistency [7]. |
| Annexin V Binding Buffer | Provides the calcium-rich environment (Ca²⁺) essential for the specific binding of Annexin V to externalized phosphatidylserine (PS). Buffers containing EDTA must be avoided [5] [4]. |
| Viability Dye (PI, 7-AAD) | DNA-binding dyes that are excluded from live, intact cells. They identify late apoptotic and necrotic cells with compromised membranes, and are crucial for excluding "sticky" dead cells during analysis [5] [7]. |
| Fc Receptor Block | An antibody that blocks Fc receptors on cells (e.g., monocytes, macrophages) to prevent non-specific binding of your staining antibodies, thereby reducing background [11] [7]. |
| Single-Stain Controls | Cells or beads stained with a single fluorochrome used to calculate accurate spectral compensation, which is critical for cleanly separating Annexin V and viability dye signals [5] [7]. |
| EDTA-Free Dissociation Reagent | Enzymes like Accutase are preferred over trypsin-EDTA for detaching adherent cells, as EDTA chelates calcium and can inhibit the Annexin V binding step [5]. |
Workflow Diagram: Optimizing Signal-to-Noise Ratio
The following diagram outlines a logical workflow for diagnosing and resolving common signal-to-noise issues related to instrument settings.
In the context of a broader thesis on troubleshooting background noise in Annexin V flow cytometry research, understanding biological pitfalls is paramount. Background interference can compromise data interpretation, leading to inaccurate apoptosis quantification. This technical support guide addresses specific cell types and states inherently prone to high background, providing researchers, scientists, and drug development professionals with targeted troubleshooting strategies and methodological refinements to enhance experimental precision.
FAQ: Addressing Common Concerns
1. What are the primary biological sources of high background in Annexin V staining? The main biological sources include cellular autofluorescence, the presence of dead cells that non-specifically bind antibodies, interference from intrinsic cellular components like intracellular biotin, and undesirable interactions between fluorochromes and cellular receptors such as Fc receptors [11]. Certain cell types, including monocytes, dendritic cells, and some activated immune cells, exhibit higher autofluorescence and Fc receptor expression, exacerbating these issues [11] [7].
2. Which cell types are most problematic for non-specific antibody binding? Dead cells are notoriously problematic, as they become "sticky" and bind antibodies non-specifically [11]. This is partially due to exposed DNA and other intracellular contents [11]. Furthermore, monocytes and dendritic cells express high levels of Fc receptors, which can bind the Fc portion of antibodies, and in some cases, even the fluorochromes themselves, leading to significant background [11].
3. How does cellular autofluorescence interfere, and which cells have high autofluorescence? Autofluorescence is the inherent fluorescence of a cell, which can be confused with the specific signal from fluorochrome-conjugated Annexin V or antibodies [11]. This background signal is often more pronounced in cells containing granular components or with active metabolism, such as macrophages, hepatocytes, and some epithelial cells [7]. Using fresh cells and including an unstained control is critical for assessing the level of autofluorescence [7].
4. Can the choice of fluorochrome itself cause background issues with certain cells? Yes. Some fluorochromes can bind specifically to cellular receptors. For example:
- Phycoerythrin (PE) can be recognized as an antigen by a small subset of B cells and gamma-delta T cells [11].
- Cyanine dyes (e.g., Cy5, PE-Cy5, APC-Cy7) can bind to Fc-gamma receptors, particularly on cells like monocytes [11].
- FITC is a charged molecule and can bind non-specifically to cytoplasmic elements during intracellular staining [11].
- PE-Cy5.5 has been reported to bind with high specificity to mouse CD205 (DEC205) on dendritic cells [11].
5. Why might my untreated control cells show high background apoptosis? Poor cell health is a common culprit [21] [5]. This can be caused by over-confluent culture conditions, serum starvation, rough handling during experimental operations (like excessive pipetting or over-trypsinization), or prolonged incubation times that expose cells to stressful environments [21] [5]. Ensuring cells are healthy and handled gently throughout the protocol is essential for a clean baseline.
Troubleshooting Guide: Pitfalls and Solutions
The table below summarizes common biological pitfalls and their respective experimental solutions.
Table 1: Troubleshooting Cell Types and States Prone to High Background
| Problematic Cell Type/State | Underlying Cause of Interference | Recommended Solution |
|---|---|---|
| Dead Cells [11] | Non-specific antibody binding due to exposed cellular contents and damaged membranes. | Include a viability dye in every staining procedure [11]. Use fixable viability dyes to prevent leakage after fixation [11]. |
| Monocytes/Macrophages [11] | High expression of Fc Receptors (FcR) and autofluorescence. | Use Fc receptor blocking reagents (e.g., "Fc Block") or add unconjugated antibody to saturate FcR [11]. Consider avoiding cyanine dyes [11]. |
| Dendritic Cells [11] | High FcR expression and specific binding of PE-Cy5.5 to mouse CD205. | Use Fc receptor blocking. Avoid PE-Cy5.5 and related fluorochromes when studying mouse CD205+ cells [11]. |
| Cells with High Metabolic Activity (e.g., hepatocytes) [7] | Elevated autofluorescence. | Use fresh cells and run an unstained control to assess autofluorescence levels [7]. Choose bright fluorochromes for low-abundance targets [7]. |
| B Cells & Gamma-Delta T Cells (specific subsets) [11] | Antigen-receptor recognition of phycobiliproteins (PE, APC). | For studies of tiny cell subsets, avoid using PE or APC for the cells of interest [11]. |
| Cells in Poor Health [21] [5] | Spontaneous apoptosis and increased nonspecific staining. | Optimize culture conditions, avoid over-confluency, and use gentle detachment methods like Accutase instead of trypsin-EDTA [21] [5]. |
| Cells with High Intracellular Biotin [11] | Non-specific binding of streptavidin conjugates during intracellular staining. | For intracellular staining with biotinylated antibodies, pre-incubate fixed/permeabilized cells with unconjugated streptavidin to block endogenous biotin [11]. |
Experimental Protocols for Mitigating Background
Protocol 1: Fc Receptor Blocking for High-Autofluorescence Cells
Purpose: To minimize nonspecific antibody binding to Fc receptors, a major source of background in immune cells like monocytes and dendritic cells [11].
Materials:
- Fc receptor blocking antibody (e.g., anti-CD16/32 for mouse cells) OR unconjugated immunoglobulin of the same species and isotype as your experimental antibodies.
- Flow cytometry staining buffer (PBS with 1-5% BSA or serum).
Methodology:
- Harvest and Wash Cells: Prepare a single-cell suspension and wash with cold staining buffer.
- Resuspend in Blocking Solution: Resuspend the cell pellet in staining buffer containing the Fc block or unconjugated antibody. Use the manufacturer's recommended concentration, or titrate for optimal performance.
- Incubate: Incubate on ice for 10-15 minutes.
- Stain Without Wash: Proceed directly to the addition of your fluorochrome-conjugated antibody cocktail (e.g., Annexin V and surface antibodies) without an intervening wash step. This allows the blocking agent to remain present during the staining reaction [11].
Protocol 2: Dead Cell Exclusion and Viability Staining
Purpose: To accurately identify and exclude dead cells, which are a primary source of nonspecific binding and high background [11].
Materials:
- A fixable viability dye (e.g., Fixable Viability Stain 450/520/700).
- Phosphate Buffered Saline (PBS).
Methodology:
- Prepare Viability Dye: Reconstitute or dilute the fixable viability dye according to the manufacturer's instructions in PBS.
- Stain Cells Pre-Fixation: After surface staining (including Annexin V, which requires calcium and live cells), resuspend the cell pellet in the viability dye solution.
- Incubate: Incubate for 10-30 minutes on ice or at room temperature in the dark.
- Wash: Wash cells thoroughly with a large volume of flow cytometry staining buffer to remove unbound dye.
- Fix Cells (Optional): If required for downstream applications, fix the cells. Using a fixable dye ensures the viability signal is retained after fixation, preventing dye leakage and homogenously staining all cells, a problem associated with PI or 7-AAD post-fixation [11].
Workflow Visualization
The following diagram illustrates the logical decision process for troubleshooting high background based on cell type and state.
The Scientist's Toolkit: Research Reagent Solutions
Essential reagents for mitigating biological background in Annexin V assays are listed below.
Table 2: Key Reagents for Background Troubleshooting
| Reagent | Function/Purpose | Key Consideration |
|---|---|---|
| Fc Receptor Block [11] | Blocks nonspecific binding of antibodies to Fc receptors on immune cells. | Critical for staining monocytes, macrophages, and dendritic cells. |
| Fixable Viability Dye [11] [7] | Distinguishes live from dead cells; the "fixable" property prevents signal loss or transfer after fixation. | Superior to PI or 7-AAD for experiments involving cell fixation. |
| BSA or Serum [8] [11] | Added to wash and staining buffers to cover nonspecific protein binding sites on cells. | Reduces background from nonspecific antibody interactions. |
| EDTA-free Dissociation Reagent (e.g., Accutase) [5] | Gently detaches adherent cells without chelating calcium, which is essential for Annexin V binding. | Preserves membrane integrity and prevents loss of early apoptotic cells. |
| Annexin V Conjugate (non-FITC) (e.g., PE, APC) [5] | Detects phosphatidylserine exposure. Alternatives to FITC avoid issues with cellular autofluorescence or GFP-expressing cells. | Choose a fluorochrome compatible with your instrument and free of known interactions with your cell type. |
| Unconjugated Antibody [11] | Saturates Fc receptors and other nonspecific binding sites. A versatile alternative to specific Fc blocks. | Should match the species and isotype of the primary conjugated antibodies. |
| Compensation Beads [7] | Used with single-stained controls to accurately calculate spectral compensation on the flow cytometer. | Essential for clean separation of signals in multicolor experiments. |
Proactive Protocol Optimization for Minimizing Background Noise
In Annexin V flow cytometry research for apoptosis detection, the integrity of the plasma membrane is the cornerstone of reliable data. The fundamental principle of this assay hinges on the calcium-dependent binding of Annexin V to phosphatidylserine (PS), a phospholipid that translocates from the inner to the outer leaflet of the plasma membrane during early apoptosis [22] [5]. Any unintended damage to the plasma membrane during cell harvesting not only allows vital dyes like propidium iodide (PI) to enter the cell prematurely but can also cause non-specific exposure of PS, leading to false-positive staining and significant background noise [23] [5]. Consequently, gentle cell harvesting is not merely a recommendation but an essential practice to distinguish genuine apoptosis from procedure-induced artifacts, ensuring the accuracy and interpretability of your experimental results.
Key Principles for Preserving Membrane Integrity
The Impact of Cell Dissociation on Apoptosis Assays
The method chosen to harvest adherent cells is arguably the most critical variable in Annexin V staining. Traditional cell dissociation methods are a major source of membrane damage. Treating cells with trypsin or other reagents to detach adherent cells causes damage to the membrane, such that cells will be labeled with annexin V [23]. This is exacerbated when trypsin is used with EDTA, as EDTA chelates the calcium ions that are absolutely required for Annexin V to bind to PS, thereby directly interfering with the core assay principle [5]. The mechanical stress from scraping cells can cause similar physical damage to the plasma membrane.
Strategic Approach to Gentle Harvesting
A strategic approach to harvesting can mitigate these risks. The primary goal is to minimize both enzymatic and mechanical stress. Whenever possible, researchers should consider using gentle, EDTA-free dissociation enzymes like Accutase [5]. Furthermore, allowing cells a recovery period post-harvest is a highly effective strategy. After detachment, cells should be allowed to recover for 30–45 minutes in an incubator. During this period, swirl the tube or plate every few minutes to prevent re-attachment. This recovery phase permits the cell membrane to repair transient pores and restore phospholipid asymmetry, drastically reducing false-positive Annexin V binding [23].
Detailed Experimental Protocols
Standardized Protocol for Gentle Cell Harvesting and Staining
The following protocol is optimized for preserving membrane integrity during the preparation of adherent cells for Annexin V flow cytometry.
Materials Needed:
- Gentle Dissociation Reagent: EDTA-free enzyme solution (e.g., Accutase).
- Wash Buffer: Azide-free and serum/protein-free PBS, chilled.
- Annexin V Binding Buffer: 10X concentrate, diluted to 1X with distilled water before use.
- Staining Reagents: Fluorochrome-conjugated Annexin V and a viability dye (e.g., Propidium Iodide or 7-AAD).
- Centrifuge Tubes: 5 mL polystyrene round-bottom tubes.
Procedure:
- Gentle Cell Detachment:
- Remove culture media and gently wash the adherent cell layer with cold, azide-free PBS.
- Add the EDTA-free dissociation reagent (e.g., Accutase) and incubate at 37°C for the minimal time required to detach the cells (typically 5-10 minutes).
- Gently tap the flask to dislodge cells. Avoid pipetting or scraping to dissociate.
- Neutralize the enzyme by adding serum-containing media.
Cell Washing and Recovery:
- Transfer the cell suspension to a centrifuge tube and centrifuge at 300–400 x g for 5 minutes at room temperature.
- Discard the supernatant and gently resuspend the cell pellet in complete culture medium.
- Crucial Recovery Step: Incubate the cell suspension for 30–45 minutes in a 37°C incubator. Gently agitate the tube every 10–15 minutes to prevent clumping and re-attachment [23].
Post-Recovery Preparation and Staining:
- After recovery, centrifuge the cells and wash them once with cold PBS.
- Resuspend the cells in 1X Annexin V Binding Buffer at a concentration of 1–5 x 10^6 cells/mL.
- Transfer 100 µL of the cell suspension to a staining tube.
- Add 5 µL of fluorochrome-conjugated Annexin V, gently mix, and incubate for 10–15 minutes at room temperature in the dark [4] [24].
- Add 2–5 µL of a viability dye like Propidium Iodide (PI) or 7-AAD. Do not wash after adding the viability dye [4] [24].
- Add 400 µL of 1X Binding Buffer and analyze by flow cytometry within 1 hour.
Protocol for Complex Staining (Surface Markers & Viability)
For experiments requiring immunophenotyping alongside apoptosis detection, the staining order is critical to preserve antigen integrity and prevent false positives.
- Stain Cell Surface Antigens First: Perform staining for cell surface markers (e.g., CD3, CD4) using standard protocols in a staining buffer [4].
- Wash and Stain for Viability: Wash cells twice with azide-free PBS. Resuspend cells in PBS and add a fixable viability dye (FVD). Incubate for 30 minutes at 2–8°C in the dark. Wash cells twice with staining buffer [4].
- Annexin V Staining: Wash cells once with 1X Annexin V Binding Buffer. Resuspend in binding buffer and stain with Annexin V as described in the standard protocol (Steps 3–5). Wash once with binding buffer after incubation [4].
- Intracellular Staining (if required): If intracellular targets (e.g., transcription factors) need to be stained, proceed with fixation and permeabilization using a commercial buffer set after the Annexin V staining step [4].
The workflow for this multi-step assay is outlined in the diagram below.
Troubleshooting Guide: FAQs on Background Noise and Poor Results
Operation and Preparation
Q1: Why is there a high percentage of Annexin V-positive cells in my untreated control group? This is a classic sign of false positives. The most common causes are:
- Harsh Cell Harvesting: Over-trypsinization or mechanical scraping damaged the plasma membrane [5].
- Missing Recovery Step: Cells were not given time to recover post-detachment, leaving PS transiently exposed [23].
- Cell Health: Cells were over-confluent, starved, or otherwise stressed before the experiment, leading to spontaneous apoptosis [5].
- Calcium Chelation: Using trypsin with EDTA, which chelates Ca²⁺ and interferes with binding, or using buffers containing EDTA or other calcium chelators during the Annexin V experiment [4] [5].
Q2: My cell yield is low after gentle harvesting. What should I do? Ensure you are not discarding apoptotic cells that have detached during the culture period. When harvesting, always collect the supernatant (which may contain floating apoptotic cells) before detaching the adherent cells, and combine them for analysis [25] [5].
Q3: I am working with GFP-expressing cells. Which Annexin V conjugate should I use? Avoid FITC-labeled Annexin V due to significant spectral overlap with GFP. Choose conjugates with distinct emission spectra, such as PE, APC, or Alexa Fluor 647 [5].
Results and Analysis
Q4: After staining, I see a large population of cells that are both Annexin V and PI positive. What does this mean? While this population typically represents late-stage apoptotic or necrotic cells, a very high percentage in a sample that should be healthy suggests procedure-induced cell death. This can be caused by excessive centrifugation speed, overly vigorous pipetting, or delaying flow cytometry analysis for too long after staining [22] [5].
Q5: Why are there no positive signals in my drug-treated group? This indicates a potential failure to induce apoptosis or an error in the assay.
- Insufficient Treatment: The drug concentration or treatment duration may be too low [5].
- Lost Apoptotic Cells: You may have failed to collect and include the supernatant, which contains detached apoptotic cells [5].
- Reagent Failure: The Annexin V conjugate or viability dye may have degraded due to improper storage or is beyond its expiration date. Always include a positive control (e.g., cells treated with a known apoptosis inducer like staurosporine) to verify kit functionality [22] [5].
Q6: My cell populations on the flow cytometry plot are not clearly separated. How can I improve resolution?
- Check Compensation: Poorly compensated fluorescence spillover can blur population boundaries. Use single-stained controls (Annexin V only and PI only) to set compensation correctly on your flow cytometer [5] [24].
- Reduce Background: Cellular autofluorescence can interfere. If your cells have high autofluorescence, select an Annexin V conjugate with a fluorophore that emits in a different channel [5].
The table below summarizes these common issues and their solutions.
Table: Troubleshooting Common Annexin V Staining Problems
| Problem | Potential Cause | Recommended Solution |
|---|---|---|
| High background in control | Harsh trypsinization; No recovery step; EDTA use | Use gentle, EDTA-free detachment; Implement a 30–45 min recovery period [23] [5] |
| No positive signal in treated group | Apoptotic cells not collected; Insufficient drug | Always combine supernatant with trypsinized cells; Optimize treatment dose/duration [5] |
| Poor population separation | Autofluorescence; Poor compensation | Use a bright, non-overlapping fluorophore; Run single-stain controls for compensation [5] [24] |
| Unexpected double-positive cells | Mechanical damage; Delayed analysis | Use gentle pipetting; analyze samples within 1 hour of staining [5] |
Essential Reagents and Tools for the Researcher
The following toolkit is essential for performing reliable Annexin V apoptosis assays with minimal background noise.
Table: Essential Research Reagent Solutions for Annexin V Assays
| Item | Function | Key Consideration |
|---|---|---|
| EDTA-free Dissociation Reagent | Gently detaches adherent cells without damaging membrane integrity or chelating Ca²⁺. | Alternatives to trypsin-EDTA, like Accutase, are highly recommended [5]. |
| 10X Annexin V Binding Buffer | Provides the optimal calcium-containing environment for specific Annexin V-PS binding. | Critical to avoid buffers containing EDTA or other calcium chelators [4] [24]. |
| Fluorochrome-conjugated Annexin V | Binds to externalized phosphatidylserine (PS) to label apoptotic cells. | Choose a fluorophore compatible with your flow cytometer's lasers and filters, and that does not overlap with other labels (e.g., GFP) [4] [5]. |
| Membrane-Impermeant Viability Dye | Distinguishes between intact (viable) and compromised (necrotic/late apoptotic) membranes. | Propidium Iodide (PI) and 7-AAD are common choices. Do not wash cells after adding these dyes [22] [24]. |
| Fixable Viability Dyes (FVD) | Allows for dead cell exclusion in assays requiring subsequent fixation and permeabilization. | Must be used before fixation and Annexin V staining in multi-step protocols [4]. |
In Annexin V flow cytometry, the quality of your final data is determined at the very first step: cell harvesting. By adopting gentle, EDTA-free dissociation methods, implementing a crucial post-harvest recovery period, and meticulously controlling experimental conditions, researchers can effectively preserve plasma membrane integrity. This rigorous approach to sample preparation minimizes background noise and false positives, ensuring that the apoptotic signal you detect is a true biological phenomenon, not an artifact of your technique. Mastering these foundational practices is essential for generating robust, reliable, and publication-quality data in apoptosis research.
In Annexin V-based flow cytometry assays, the accuracy of your apoptosis data is fundamentally dependent on your buffer formulation. The binding of Annexin V to phosphatidylserine (PS) is a critical early apoptosis marker, and this interaction is exquisitely calcium-dependent. Using a buffer with incorrect calcium concentration, improper pH, or containing calcium-chelating agents can generate significant background noise, false positives, and unreliable quantification. This guide addresses the specific buffer-related challenges that researchers encounter and provides targeted troubleshooting solutions to ensure data integrity.
Troubleshooting Guide: Buffer-Related Background Noise
Table 1: Common Buffer-Related Problems and Solutions in Annexin V Staining
| Problem | Potential Cause | Recommended Solution | Key References |
|---|---|---|---|
| High background or false positive staining | Buffer contains EDTA or other calcium chelators [5] [4] | Use calcium-containing binding buffer and avoid trypsin with EDTA; use gentle, EDTA-free dissociation enzymes like Accutase [5]. | |
| Weak or no Annexin V signal | Insufficient calcium concentration in buffer [26] | Ensure binding buffer contains a final concentration of 2.5 mM CaCl₂ [24]; verify buffer preparation. | |
| Unstable staining over time | Incorrect buffer pH affecting Annexin V affinity [24] | Use a HEPES-buffered solution at pH 7.4 to maintain physiological conditions [24]. | |
| Poor cell viability affecting results | Mechanical or enzymatic damage during harvesting [5] | Harvest cells gently using non-enzymatic methods or EDTA-free enzymes; avoid over-trypsinization [5]. |
Frequently Asked Questions (FAQs)
1. Why is calcium absolutely essential for Annexin V binding? Annexin V binding to phosphatidylserine is a calcium-dependent process. The protein requires calcium ions to form a bridge between its binding sites and the negatively charged phosphatidylserine headgroups on the cell membrane. Research has shown that truncation of domain IV of Annexin A5 destroys its calcium-binding ability and severely impairs its affinity for PS, underscoring the critical role of calcium in this interaction [26]. Without adequate calcium, binding is minimal or non-existent.
2. What is a standard recipe for Annexin V binding buffer? A standard 1X binding buffer can be made from a 10X concentrate with the following formulation: 0.1 M HEPES (pH 7.4), 1.4 M NaCl, and 25 mM CaCl₂ [24]. When diluted to 1X, this provides the correct isotonic salt conditions and the critical 2.5 mM calcium chloride concentration needed for optimal Annexin V binding.
3. How do chelating agents like EDTA interfere with the assay? EDTA (Ethylenediaminetetraacetic acid) is a potent chelator of divalent cations like calcium. By binding to the free calcium in the buffer, EDTA effectively removes the essential co-factor required for Annexin V to attach to phosphatidylserine [5] [4]. This results in a weak or absent signal, compromising the experiment. Always check that any reagents used (e.g., cell harvesting solutions) are free of EDTA or other chelators.
4. Can I use PBS instead of a specialized binding buffer? No, standard phosphate-buffered saline (PBS) is not sufficient because it does not contain calcium. You must use a binding buffer that includes a defined concentration of calcium chloride (typically 2.5 mM) [4] [24]. Some protocols use PBS supplemented with 25 mM calcium chloride for this purpose [8].
Experimental Protocols for Optimal Buffer Use
Protocol 1: Standard Annexin V Staining with Commercial Kits
This protocol is adapted from major kit manufacturers and ensures proper buffer conditions [4] [24].
- Prepare Buffer: Dilute 10X binding buffer to 1X using distilled water. Ensure it is at room temperature.
- Harvest Cells: Gently harvest cells, avoiding the use of trypsin-EDTA. Use a gentle cell-dissociation agent like Accutase instead [5].
- Wash Cells: Wash cells once with cold PBS (without calcium or magnesium) and then once with the 1X binding buffer.
- Resuspend Cells: Resuspend the cell pellet in 1X binding buffer at a concentration of 1 x 10^6 cells/mL.
- Stain with Annexin V: Transfer 100 µL of cell suspension to a flow tube. Add 5 µL of fluorochrome-conjugated Annexin V. Mix gently and incubate for 15 minutes at room temperature in the dark.
- Add Viability Dye: Without washing, add 5 µL of Propidium Iodide (PI) or 7-AAD.
- Analyze: Add 400 µL of 1X binding buffer to the tube and analyze by flow cytometry within 1 hour.
Protocol 2: Annexin V Staining with Surface Marker Staining
This protocol is for researchers combining apoptosis detection with analysis of other cell surface proteins [8] [4].
- Stain Surface Antigens: First, stain the cell surface antigens of interest (e.g., CD44, CD24) using antibodies in a standard staining buffer. Follow the manufacturer's instructions.
- Wash: Wash cells twice with azide-free and serum/protein-free PBS.
- Wash with Binding Buffer: Wash cells once with 1X Annexin V binding buffer.
- Annexin V Staining: Resuspend the cell pellet in 1X binding buffer. Add Annexin V conjugate and incubate for 15 minutes at room temperature in the dark.
- Wash and Add Viability Dye: Wash cells once with 1X binding buffer, then resuspend in a small volume of buffer containing a viability dye like PI.
- Analyze: Proceed to flow cytometric analysis.
Table 2: Research Reagent Solutions for Annexin V Assays
| Reagent | Function | Key Considerations |
|---|---|---|
| Annexin V Binding Buffer | Provides calcium and proper ionic environment for specific Annexin V-PS binding. | Must contain 2.5 mM Ca²⁺; HEPES-buffered to pH 7.4; must be free of EDTA [4] [24]. |
| Propidium Iodide (PI) | Membrane-impermeant DNA dye to identify late apoptotic/necrotic cells. | Add after Annexin V staining without a wash step; must be present in buffer during acquisition [4] [27]. |
| 7-AAD | Alternative viability dye to PI; useful for multicolor panels. | Compatible with red laser-equipped cytometers; less prone to spillover into other channels than PI in some configurations [24]. |
| EDTA-free Dissociation Reagent | Gently detaches adherent cells for analysis without chelating calcium. | Preserves membrane integrity and prevents cleavage of Annexin V binding sites; e.g., Accutase [5]. |
| Fc Receptor Blocking Reagent | Reduces nonspecific antibody binding. | Crucial when combining with surface marker staining to minimize background noise [7]. |
Visualization of Calcium-Dependent Binding
The following diagram illustrates the critical role of calcium in the Annexin V binding mechanism and the consequences of buffer incompatibility.
Diagram 1: The critical role of calcium in Annexin V binding to phosphatidylserine (PS) exposed on apoptotic cells. The binding is entirely dependent on the presence of calcium ions in the buffer, which act as a essential bridge. Suboptimal buffer conditions, particularly the absence of calcium or the presence of chelators like EDTA, prevent this interaction and lead to failed experiments.
Core Concepts: Spectral Overlap and Compensation
What is spectral overlap and why is it a problem in flow cytometry?
Spectral overlap occurs when the emission spectrum of one fluorochrome spills over into the detection channel of another [28]. In a multicolor experiment, the light emitted by a fluorochrome is typically detected by a photomultiplier tube (PMT) dedicated to a specific wavelength range. However, because fluorochromes emit a range of wavelengths, the signal from a bright fluorochrome like PE can be detected in the PMT set for another fluorochrome, such as FITC [28]. If uncorrected, this spillover can lead to inaccurate data, false-positive results, and misinterpretation of cell populations [28].
Compensation is a mathematical correction applied during data analysis to subtract the contribution of spectral spillover from every other channel [28]. This process is essential for ensuring that the fluorescence signal in each detector accurately represents the specific fluorochrome it is intended to measure [28].
The diagram below illustrates the relationship between fluorochrome emission, spectral overlap, and the need for compensation.
How does spectral flow cytometry differ from conventional flow cytometry in handling overlap?
Spectral flow cytometry represents a significant advancement. Instead of measuring fluorescence intensity in discrete, predefined channels, it captures the full emission spectrum of every fluorochrome in the sample across all detectors for each laser [29]. Each fluorochrome has a unique spectral signature, much like a fingerprint. During analysis, a mathematical process called "unmixing" is used to deconvolute the composite signal from a cell and determine the contribution of each individual fluorochrome [29]. This allows for better distinction between fluorochromes with very similar emission maxima (e.g., APC and Alexa Fluor 647) and can simplify panel design by increasing the number of compatible fluorophores [29].
Fluorochrome Selection and Panel Design
What are the key considerations when selecting fluorochromes for a panel?
Designing a multicolor panel, especially one that includes Annexin V, requires strategic planning to minimize spectral spillover and background issues. The goal is to pair antigens and fluorochromes in a way that maximizes signal detection and minimizes compensation errors.
- Antigen Density and Fluorochrome Brightness: Pair bright fluorochromes (e.g., PE) with low-density antigens and dim fluorochromes (e.g., FITC) with high-density antigens [30].
- Instrument Configuration: Ensure your flow cytometer has the appropriate lasers and filters to excite and detect the fluorochromes you select.
- Spectral Overlap: Use a spectral viewer to predict potential spillover. Fluorochromes with minimal spillover into other channels are preferable.
- Experimental Context: For apoptosis detection, be aware that certain treatments can induce cellular autofluorescence. In such cases, using red-shifted fluorochromes (e.g., APC) is advantageous as autofluorescence is typically lower in these channels [30].
The table below summarizes common fluorochromes and their properties to aid in selection.
| Fluorochrome | Excitation Laser (Common) | Emission Max (nm) | Relative Brightness | Notes for Annexin V Assays |
|---|---|---|---|---|
| FITC | Blue (488 nm) | 520 | Dim | Common for Annexin V; high cellular autofluorescence can interfere [30]. |
| PE | Blue (488 nm) | 576 | Very Bright | Avoid if sample has high autofluorescence. |
| PE-Cy5 | Blue (488 nm) | 670 | Bright | Check for spillover into APC channel; common tandem dye, can be sensitive to light fixation [29]. |
| PE-Cy7 | Blue (488 nm) | 780 | Bright | Significant spillover into other red and near-IR channels. |
| APC | Red (640 nm) | 660 | Bright | Lower autofluorescence; good for high background samples [30]. |
| Alexa Fluor 647 | Red (640 nm) | 668 | Bright | Can often be distinguished from APC on spectral cytometers [29]. |
| 7-AAD | Blue (488 nm) | 675 | N/A | Viability dye; alternative to PI. |
| PerCP | Blue (488 nm) | 675 | Moderate | Photo-unstable; PerCP-eFluor 710 can be discriminated on spectral cytometers [29]. |
How should I choose an Annexin V conjugate for my specific experiment?
Your choice of Annexin V conjugate is critical and depends on your other reagents and instrument.
- Standard Apoptosis (Annexin V/PI): Annexin V-FITC with PI is the most common combination. Ensure your cytometer can detect FITC and PI without issue.
- GFP-Expressing Cells: Avoid Annexin V-FITC due to direct spectral overlap. Use Annexin V conjugated to PE, APC, or Alexa Fluor 647 instead [5].
- Multicolor Panels Beyond Viability: When incorporating Annexin V into a larger immunophenotyping panel, choose a conjugate that fits the brightness and spillover profile of your panel. Annexin V-APC or Annexin V-PE-Cy5 are often good choices to place in the red laser, freeing up the blue laser for other markers.
- Spectral Flow Cytometry: You have greater flexibility. Fluorochromes like APC and Alexa Fluor 647, which are difficult to use together on conventional cytometers, can be distinguished based on their unique spectral signatures [29].
Experimental Protocols for Optimal Results
Annexin V Staining Protocol for Minimal Background
The following detailed protocol is designed to preserve cell integrity and minimize factors that contribute to background noise [5] [31].
Workflow for Apoptosis Staining and Analysis
Materials Needed (The Scientist's Toolkit)
| Reagent/Material | Function | Key Considerations |
|---|---|---|
| Cells in log-phase growth | Experimental sample | Healthy, low spontaneous apoptosis. Avoid over-confluency [5]. |
| EDTA-free detachment reagent | Detaches adherent cells | EDTA chelates Ca²⁺, which is essential for Annexin V binding. Use Accutase or gentle cell scraping [5]. |
| Calcium-rich Binding Buffer | Staining buffer | Provides the necessary Ca²⁺ for Annexin V to bind to phosphatidylserine [31]. |
| Fluorochrome-conjugated Annexin V | Detects PS externalization | Protect from light. Choose conjugate (FITC, PE, APC) to fit your panel [5]. |
| Viability Dye (PI or 7-AAD) | Detects loss of membrane integrity | PI is common; 7-AAD is an alternative. Membrane-impermeable DNA-binding dyes [31]. |
| Flow Cytometer | Instrument for analysis | Ensure lasers and filters match your fluorochromes. |
Step-by-Step Methodology [5] [31]:
- Cell Preparation: Gently harvest cells using an EDTA-free enzyme solution like Accutase. For suspension cells, collect directly. Critical: Be gentle with pipetting and washing to avoid mechanical induction of phosphatidylserine (PS) exposure.
- Washing: Centrifuge cells at 300 x g for 5 minutes and resuspend the pellet in ice-cold PBS. Repeat once.
- Resuspension: Adjust cell concentration to 1 x 10⁶ cells/mL in the provided calcium-rich binding buffer.
- Staining: Aliquot 100 µL of cell suspension into a flow tube. Add the recommended volume of fluorochrome-conjugated Annexin V (e.g., 5 µL) and viability dye (e.g., 5 µL of PI).
- Incubation: Vortex tubes gently and incubate for 15 minutes at room temperature in the dark. Annexin V binding is light-sensitive.
- Analysis: After incubation, add 400 µL of additional binding buffer to each tube. Do not wash the cells, as this can remove the bound Annexin V. Keep samples on ice and analyze by flow cytometry within 1 hour.
How do I set up proper compensation controls?
Accurate compensation is non-negotiable for reliable multicolor data. The following controls are essential [28]:
- Unstained Cells: To set baseline fluorescence and PMT voltages.
- Single-Stain Controls: For every fluorochrome used in your panel (including Annexin V and PI), you need a sample stained with that fluorochrome alone.
- Preparation: Use compensation beads or cells stained with each individual antibody/ reagent. For Annexin V, it is best to use cells induced to undergo apoptosis (e.g., with staurosporine) to get a bright, positive population for the single-stain control.
- Fluorescence Minus One (FMO) Controls: These controls contain all fluorochromes except one. They are critical for setting correct gates and boundaries, especially when dealing with spread due to high compensation.
Troubleshooting Guide & FAQs
Frequently Asked Questions
Q: My unstained or negative control cells show high fluorescence in the Annexin V channel. What could be the cause? [5] [32] A: This high background can result from:
- Poor Cell Health: Over-confluent, starved, or contaminated cells undergo spontaneous apoptosis.
- Rough Handling: Over-trypsinization or excessive pipetting damages cells, causing PS exposure.
- Instrument Contamination: The flow cytometer fluidics system may not have been cleaned thoroughly from a previous experiment.
- Fluorescent Drug Interference: Compounds like doxorubicin are intrinsically fluorescent.
Q: I am using GFP-expressing cells and my Annexin V-FITC signal is compromised. What should I do? [5] A: This is a classic case of spectral overlap and interference. You must switch to an Annexin V conjugate that is excited by a different laser and does not emit in the same range as GFP. Annexin V-PE or Annexin V-APC are excellent alternatives.
Q: Why are there no Annexin V-positive cells in my treated sample, even though the cells look apoptotic under a microscope? [5] A:
- Lost Cells: Apoptotic cells become fragile and may be lost during washing steps. Always include the cell culture supernatant when harvesting.
- Insufficient Apoptosis: The drug concentration or treatment duration may be too low.
- Calcium Chelation: If you used trypsin-EDTA to harvest cells, the EDTA may have chelated the calcium in your binding buffer, preventing Annexin V binding. Switch to an EDTA-free detachment method.
Troubleshooting Common Problems
| Problem | Possible Causes | Solutions |
|---|---|---|
| High Background in Negative Control [5] [32] | Poor cell state; mechanical damage; fluorescent compound interference. | Use healthy, log-phase cells; handle gently; choose alternative fluorochrome (e.g., APC). |
| No Positive Signal in Treated Group [5] | Apoptotic cells in supernatant discarded; insufficient drug treatment; reagent degraded. | Collect all supernatant; optimize treatment conditions; use a positive control to validate kit. |
| Unclear Separation of Populations [5] [32] | High cellular autofluorescence; excessive apoptosis using up dye; poor instrument compensation. | Use bright, red-shifted dyes (e.g., PE, APC); increase dye concentration; check compensation with single-stain controls. |
| Only PI Positive, Annexin V Negative [5] | Cells are primarily necrotic due to harsh treatment or toxicity. | Reduce drug concentration or treatment intensity; ensure healthy cell starting population. |
| Only Annexin V Positive, PI Negative [32] | Cells are in early apoptosis; nuclear dye (PI) was forgotten or is degraded. | Confirm dye was added; check reagent storage conditions; this may be a valid result showing early apoptosis. |
Within the broader context of troubleshooting background noise in Annexin V flow cytometry research, mastering the practical execution of the staining protocol is paramount. Inconsistent incubation times, improper temperatures, or inadequate light protection are not minor oversights; they are direct contributors to high background fluorescence, increased false-positive rates, and compromised data integrity. This guide provides detailed, step-by-step protocols and directly addresses frequent pitfalls to ensure the generation of clean, reliable, and reproducible data for researchers and drug development professionals.
The Scientist's Toolkit: Essential Reagents and Materials
The following table details the core reagents required for a successful Annexin V apoptosis detection assay. Proper preparation and use of these components are critical for minimizing background noise.
Table 1: Key Research Reagent Solutions for Annexin V Staining
| Reagent/Material | Function & Importance | Key Considerations for Low Background |
|---|---|---|
| Fluorochrome-conjugated Annexin V | Binds to phosphatidylserine (PS) exposed on the outer leaflet of the cell membrane during early apoptosis [33] [34]. | Select a fluorochrome that does not overlap with cellular autofluorescence or other dyes used (e.g., avoid FITC if cells express GFP) [5]. |
| Viability Dye (PI, 7-AAD, or Fixable Viability Dyes) | Distinguishes viable from non-viable cells. Membrane-impermeable dyes like PI and 7-AAD only stain cells with compromised membranes (late apoptotic/necrotic) [33] [34]. | Do not wash after adding PI or 7-AAD, as this will remove the dye [4]. |
| 10X Binding Buffer | Provides the calcium-rich environment (2.5 mM CaCl₂) essential for the calcium-dependent binding of Annexin V to PS [24] [35]. | Always dilute to 1X and ensure it contains no EDTA or other calcium chelators, which will abolish Annexin V binding [4] [5]. |
| PBS (Calcium/Magnesium-Free) | Used for washing cells to remove residual media and serum proteins that can interfere with staining. | Must be azide- and serum/protein-free when used prior to fixable viability dye staining [4]. |
| Fixable Viability Dyes (FVD) | Covalently bind to amines in cells with compromised membranes, allowing for subsequent fixation and intracellular staining without loss of viability signal [4]. | FVD eFluor 450 is not recommended for use with Annexin V kits due to potential spectral overlap [4]. |
Core Staining Protocol and Workflow
The diagram below outlines the key decision points and steps in a standard Annexin V staining procedure, integrating viability assessment.
Annexin V Staining Experimental Workflow
Detailed Step-by-Step Instructions
Materials Required: 12 x 75 mm round-bottom tubes, 1X PBS, Annexin V conjugate (e.g., FITC, PE, APC), 10X Binding Buffer, viability dye (Propidium Iodide (PI), 7-AAD, or a Fixable Viability Dye) [4] [24].
- Preparation: Prepare a 1X solution of Binding Buffer by diluting the 10X concentrate with distilled water [4] [35].
- Cell Harvesting: Harvest cells (1-5 x 10⁵ per sample) by gentle centrifugation. For adherent cells, use gentle, non-enzymatic dissociation methods like EDTA to preserve membrane integrity. Harsh trypsinization can cause false-positive staining [5] [34]. Ensure you collect and include cells from the culture supernatant, as they may be apoptotic [36] [5].
- Washing: Wash cells once with cold 1X PBS and then once with 1X Binding Buffer. Centrifuge at 300-600 x g for 5 minutes at room temperature between washes [4] [35].
- Cell Suspension: Resuspend the cell pellet in 1X Binding Buffer at a concentration of 1-5 x 10⁶ cells/mL [24] [35].
- Annexin V Staining:
- Viability Dye Addition:
- Flow Cytometric Analysis:
Frequently Asked Questions (FAQs) & Troubleshooting
Q1: My unstained or negative control shows high background fluorescence. What could be the cause?
A: High background, or false positives, often stems from poor cell health or harsh handling.
- Poor Cellular Status: Over-confluent, starved, or contaminated cells can undergo spontaneous apoptosis or necrosis, leading to non-specific staining. Always use healthy, log-phase cells [36] [5].
- Rough Handling: Excessive pipetting, over-digestion of adherent cells (e.g., with trypsin-EDTA), or mechanical shear stress can disrupt the plasma membrane, allowing Annexin V to bind to internal PS or enabling PI uptake [36] [5].
- Improper Buffer: Using a binding buffer that was incorrectly diluted or contains calcium chelators like EDTA will prevent specific Annexin V binding and can promote cell death due to abnormal osmotic pressure [36] [4].
- Instrument Issues: A flow cytometer that was not cleaned thoroughly can contain fluorescent contaminants from previous runs [36].
Q2: I see no positive signal in my treated group. What should I check?
A: A lack of expected apoptosis signal can be due to biological or technical failures.
- Insufficient Apoptosis Induction: The drug concentration or treatment duration may be too low. Re-optimize treatment conditions and confirm apoptosis microscopically [36] [5].
- Missed Apoptotic Cells: Apoptotic cells often detach from the culture surface. Failing to collect and pellet the cell culture supernatant will result in the loss of this population [36] [5].
- Reagent Failure: Check that reagents have been stored correctly (e.g., some dyes require -20°C) and are not expired. Test with a positive control (e.g., cells treated with staurosporine) to verify kit functionality [36] [5].
- Operator Error: Confirm that all dyes were added. A common mistake is washing cells after adding PI, which removes the signal [5].
Q3: My cell populations are not clearly separated on the dot plot. How can I improve resolution?
A: Unclear clustering makes accurate gating difficult and is often linked to sample quality.
- Cell Autofluorescence: Some cell types have inherent fluorescence that can interfere with the detection signal. Consider switching to an Annexin V conjugate with a longer wavelength (e.g., APC instead of FITC) [36] [5].
- Poor Cell State: If all cells are in a poor state, widespread PS exposure can occur, making it hard to distinguish a clear negative population. Improve cell culture conditions and handle cells gently throughout the experiment [36].
- Excessive Apoptosis: If apoptosis is over-induced, the amount of externalized PS can be so high that it saturates the dye, leading to a continuum of staining rather than distinct populations. Titrate the apoptosis-inducing treatment to find milder conditions [36].
Q4: Why is it critical to protect samples from light during incubation?
A: Fluorochromes like FITC, PE, and PI are light-sensitive. Prolonged or intense light exposure causes photobleaching, which permanently diminishes the fluorescence intensity of the dye. This leads to weak signals and a loss of detection sensitivity. Furthermore, tandem dyes (e.g., PE-Cy7) are particularly susceptible to light-induced degradation, which can dissociate the acceptor and donor molecules, altering the emission spectrum and compromising compensation [4] [7]. Protecting samples from light throughout the staining procedure and during storage is essential for maintaining signal integrity.
Q5: What are the essential controls for setting up my flow cytometry experiment?
A: Proper controls are non-negotiable for accurate data interpretation and gating. Table 2: Required Flow Cytometry Controls for Annexin V Assays
| Control | Composition | Purpose |
|---|---|---|
| Unstained Cells | Cells in binding buffer only. | To adjust FSC/SSC and PMT voltages, and to measure cellular autofluorescence [24] [33]. |
| Single-Stain Controls | Cells stained with Annexin V only (no PI).Cells stained with PI only (no Annexin V). | Critical for compensation. Allows the instrument to calculate and subtract spectral overlap between the fluorescent channels [24] [33]. |
| Untreated/Negative Control | Healthy, untreated cells stained with both Annexin V and PI. | Defines the baseline for viable (Annexin V-/PI-) cells and identifies the level of spontaneous apoptosis in the population [24]. |
| Induced/Positive Control | Cells treated with a known apoptosis inducer (e.g., staurosporine) and stained. | Verifies that the assay is working correctly and helps identify the location of apoptotic populations on the dot plot [24] [33]. |
FAQs on Autofluorescence in Flow Cytometry
What causes intrinsic autofluorescence in cells and tissues, and which cell types are most problematic?
Intrinsic autofluorescence is caused by naturally occurring intracellular molecules such as flavin coenzymes (FAD, FMN) and nicotinamide adenine dinucleotide (phosphate) [NAD(P)H] [37]. This fluorescence is most pronounced in the green-to-yellow emission spectrum. Certain cell types are particularly prone to high autofluorescence, including neutrophils, hepatocytes, and epithelial cells, due to their metabolic activity and organelle content [37]. In tissues, this can be a significant challenge as these metabolically active cells contribute to a high background, masking specific fluorescent signals.
How can I confirm if my sample has high intrinsic autofluorescence?
The most straightforward method is to include an unstained control sample in every experiment [37] [38]. When you run this unstained control on your flow cytometer, you are observing the inherent fluorescent background of your cells. A population that shows a bright shift away from the axis in the channels where you plan to detect your fluorochromes indicates significant autofluorescence. This control is essential for setting your voltage and gating thresholds correctly to distinguish real positive signals from background noise.
My target antigen has low expression, and autofluorescence is drowning out the signal. What are my options?
For low-expression antigens, fluorochrome selection is critical. You should pair weak antigens with bright fluorochromes to amplify the specific signal above the autofluorescence level [37]. Bright fluorophores like PE (Phycoerythrin) or APC (Allophycocyanin) are highly recommended in this scenario [37] [38]. Furthermore, because autofluorescence is typically more intense in the green spectrum, using fluorochromes that emit in the red or far-red channels (e.g., APC, Alexa Fluor 700) can help, as cellular autofluorescence is minimal in these longer wavelengths [37].
I am working with fixed tissues. Does fixation affect autofluorescence?
Yes, fixation can increase autofluorescence. Prolonged storage in fixative solutions, especially paraformaldehyde (PFA), can elevate background fluorescence [37]. To minimize this, optimize your fixation protocol, typically using low concentrations of PFA (e.g., 1-2%) and limiting fixation time to less than 15-30 minutes when possible [37]. Always analyze your samples soon after staining and fixation, as prolonged storage can exacerbate the issue.
Besides fluorochrome choice, what other experimental steps can reduce background from autofluorescence?
Incorporating a viability dye is a crucial step. Dead cells and cellular debris often have very high autofluorescence. Using a viability dye like propidium iodide (PI) or 7-AAD allows you to gate out these non-viable cells during analysis, significantly cleaning up your data [37] [39]. Additionally, ensuring gentle sample handling to maintain high cell viability (>85%), using gentle dissociation enzymes like Accutase instead of trypsin, and thorough washing to remove debris all contribute to lower background [5] [39].
Troubleshooting Guide: High Background/Non-Specific Staining
| Problem Category | Specific Symptom | Possible Cause | Recommended Solution |
|---|---|---|---|
| Signal & Background | Weak or no signal from target antigen | Low-expression antigen paired with a dim fluorochrome [37]. | Use bright fluorochromes (e.g., PE, APC) for weak antigens [37]. |
| High background staining | High cellular autofluorescence [37]. | Use red-shifted fluorochromes (e.g., APC) and always include an unstained control [37]. | |
| Presence of dead cells and debris [37]. | Gate out dead cells using a viability dye (e.g., PI, 7-AAD) [37]. Filter cells through a strainer [39]. | ||
| Sample Preparation | High background scatter | Cells are lysed or damaged [37] [38]. | Optimize preparation; avoid vortexing/high-speed centrifugation; use fresh buffers [37] [38]. |
| Unclear cell population clustering | Poor cell health leading to nonspecific staining [40]. | Use healthy, log-phase cells; treat cells gently during harvesting [5] [40]. | |
| Cell clumping | DNA release from dead cells or calcium-dependent aggregation [39]. | Add DNase I to buffers; include EDTA in wash buffers [39]. | |
| Instrument Setup | High background in negative population | PMT voltage (gain) set too high [38]. | Use positive and negative controls to optimize voltage settings for each channel [37] [38]. |
| Inability to distinguish positive population | Fluorescence compensation is incorrect [5]. | Use single-stained controls or compensation beads to set accurate compensation [5] [39]. |
Experimental Protocols for Mitigating Autofluorescence
Protocol 1: Optimized Staining for High-Autofluorescence Samples
This protocol is designed to maximize signal-to-noise ratio when working with challenging samples like hepatocytes or neutrophils.
Materials Needed (Research Reagent Solutions):
- Annexin V Binding Buffer: A calcium-containing buffer (e.g., 10 mM HEPES, 140 mM NaCl, 2.5 mM CaCl₂, pH 7.4) essential for Annexin V binding to phosphatidylserine [41].
- Annexin V Conjugate: Fluorochrome-labeled Annexin V (e.g., Annexin V-APC, not FITC, for red-shifted detection) [37] [5].
- Viability Dye: Propidium Iodide (PI) or 7-AAD solution [41] [37].
- Cell Staining Buffer: PBS containing a protein carrier (e.g., 0.5-1% BSA) to reduce non-specific antibody binding.
- DNase I: To prevent cell clumping due to DNA release from dead cells [39].
- EDTA: Added to wash buffers to prevent calcium-dependent cell aggregation [39].
- Nylon Mesh Filter (70 μm): For removing clumps before running on the cytometer [39].
Step-by-Step Guide:
- Cell Harvesting: Harvest cells gently. For adherent cells, use a non-enzymatic dissociation buffer or a gentle enzyme like Accutase instead of trypsin-EDTA, as EDTA chelates calcium and trypsin can damage epitopes and increase autofluorescence [5] [39].
- Wash and Filter: Wash cells in cold cell staining buffer. To prevent clumping, add DNase I (1-10 µg/mL) and EDTA (1-5 mM) to your wash buffer. Pass the cell suspension through a 70 μm nylon mesh filter to obtain a single-cell suspension [39].
- Staining for Viability and Apoptosis:
- Resuspend cell pellet (1x10⁶ cells) in 100 µL of Annexin V Binding Buffer.
- Add Annexin V conjugate (e.g., Annexin V-APC) and viability dye (PI) as per manufacturer's instructions.
- Incubate for 15 minutes at room temperature in the dark.
- Add 400 µL of Annexin V Binding Buffer and keep on ice.
- Analyze by flow cytometry within 1 hour to prevent deterioration and increased background [41] [5].
- Controls: Always run an unstained control, a single-color control for Annexin V, and a single-color control for the viability dye to set up compensation accurately on the flow cytometer [41] [5].
Protocol 2: Data Acquisition and Analysis Setup
- Instrument Setup: Before running your experimental samples, use the unstained control to set the photomultiplier tube (PMT) voltages. Increase the voltage until the negative population is clearly visible on-scale, but avoid setting the gain too high, which will amplify background noise [37] [38].
- Compensation: Using your single-stained controls, apply compensation to correct for spectral overlap between the fluorescence channels. Proper compensation is vital to ensure that the signal in one channel does not "bleed through" into another, which is critical for accurate Annexin V/PI analysis [5] [42].
- Gating Strategy:
- Gate 1 (FSC-A vs. SSC-A): Select the main population of cells, excluding debris.
- Gate 2 (FSC-H vs. FSC-A): Apply a singlet gate to exclude cell doublets or aggregates.
- Gate 3 (Viability Dye vs. SSC-A): Gate on the viability dye-negative population to select only live cells for analysis. This is a crucial step to remove highly autofluorescent dead cells.
- Gate 4 (Annexin V vs. Viability Dye): Analyze the Annexin V binding within the live cell population to identify early apoptotic cells.
The following workflow visualizes the logical steps for troubleshooting and resolving high background from autofluorescence:
The Scientist's Toolkit: Key Reagent Solutions
| Item | Function/Benefit |
|---|---|
| Annexin V-APC Conjugate | Binds to externalized phosphatidylserine; the APC fluorophore emits in the red spectrum where cellular autofluorescence is minimal [37] [5]. |
| Propidium Iodide (PI) | Membrane-impermeant DNA dye used to label dead cells with compromised membranes; allows for gating out autofluorescent necrotic/late apoptotic cells [41] [37]. |
| 7-AAD | Alternative viability dye to PI; also used to exclude dead cells from analysis [5] [40]. |
| Annexin V Binding Buffer | Provides the calcium ions (Ca²⁺) necessary for Annexin V to bind to phosphatidylserine [41]. |
| Accutase | A gentle, enzyme-free cell dissociation solution; preferred over trypsin-EDTA as it better preserves cell surface epitopes and membrane integrity, reducing background [5] [39]. |
| DNase I | An enzyme that degrades DNA released from dead cells, preventing cell clumping and reducing background debris in samples [39]. |
| Bovine Serum Albumin (BSA) | A blocking agent added to staining buffers (typically 0.5-1%) to reduce non-specific antibody binding and lower background staining [37] [38]. |
| Compensation Beads | Uniform beads used with antibodies to create highly consistent single-color controls for setting fluorescence compensation on the flow cytometer, leading to more accurate data [39]. |
Systematic Troubleshooting Guide for High Background and Poor Resolution
FAQs: Troubleshooting Background Noise in Annexin V Flow Cytometry
FAQ 1: What are the most common sources of high background noise in Annexin V assays? High background noise, or false positives, often stems from several key areas related to sample handling and reagent use:
- Cell Membrane Integrity: Any damage to the plasma membrane allows Annexin V to bind to phosphatidylserine (PS) on the inner leaflet of the membrane, causing non-specific staining [34]. This damage can be caused by over-trypsinization of adherent cells, excessive centrifugation force, or using outdated or improperly stored reagents [34].
- Inappropriate Buffer Conditions: Annexin V binding to PS is calcium-dependent. Using buffers containing EDTA or other calcium chelators will inhibit binding and can cause unreliable results. Always use a calcium-rich 1X binding buffer [4].
- Viability Dye Interference: Using a fixable viability dye that is not compatible with your Annexin V conjugate can cause spectral overlap or unexpected interactions. For example, the FVD eFluor 450 is not recommended for use with standard Annexin V apoptosis detection kits [4].
- Delayed Analysis: Analyzing samples after prolonged periods (typically beyond 4 hours) can lead to a loss of cell viability and increased background staining from viability dyes like PI or 7-AAD [4].
FAQ 2: How can I distinguish between early apoptotic cells and necrotic cells using flow cytometry? The standard method is dual-staining with Annexin V and a membrane-impermeant dye like propidium iodide (PI) or 7-AAD [34]. This allows you to differentiate cell populations based on their staining profiles, as summarized in the table below.
Table: Interpreting Dual-Staining with Annexin V and Propidium Iodide (PI)
| Cell Population | Annexin V Staining | PI Staining | Biological Meaning |
|---|---|---|---|
| Viable/Live Cells | Negative | Negative | Healthy cells with intact membranes [34]. |
| Early Apoptotic Cells | Positive | Negative | Cells undergoing early apoptosis; PS is externalized, but the membrane is intact and excludes PI [34]. |
| Late Apoptotic/ Necrotic Cells | Positive | Positive | Cells in late-stage apoptosis (lost membrane integrity) or primary necrotic cells; both PS-exposed and permeable to PI [34]. |
FAQ 3: My Annexin V signal is weak. What could be the cause? A weak signal is frequently due to suboptimal reagent conditions.
- Calcium Chelators: Check all your buffers (including wash buffers) for the presence of EDTA, which will chelate the calcium required for Annexin V binding [4].
- Insufficient Reagent: Ensure you are using the recommended volume of Annexin V conjugate for your cell number [4] [34].
- Expired Reagents: Always use fresh reagents and buffers. Prepare the 1X binding buffer from the 10X stock close to the time of use [34].
FAQ 4: What are the essential controls for a reliable Annexin V experiment? Including the right controls is critical for accurate data interpretation and gating during flow cytometric analysis [43]. You should include:
- Unstained Cells: To assess autofluorescence.
- Annexin V Single Stain: To set the compensation and positive gate for Annexin V.
- Viability Dye Single Stain (e.g., PI): To set the compensation and positive gate for the viability dye.
- Untreated/Cells: To establish the baseline level of apoptosis in your population.
- Induced Cells: A sample treated with a known apoptosis-inducer (e.g., a drug) to serve as a positive control.
Experimental Protocols for Noise Reduction
Standard Annexin V Staining Protocol with PI
This protocol is optimized for use with specific detection kits to minimize background and ensure reproducibility [4] [34].
Materials:
- 1X PBS (without calcium or magnesium)
- 10X Binding Buffer (dilute to 1X with distilled water before use)
- Fluorochrome-conjugated Annexin V
- Propidium Iodide (PI) Staining Solution or 7-AAD
- Round-bottom tubes
Procedure:
- Prepare Cells: Harvest and wash cells once with 1X PBS, then once with 1X Binding Buffer [4].
- Resuspend Cells: Resuspend the cell pellet in 1X Binding Buffer at a density of 1-5 x 10⁶ cells/mL [4].
- Stain with Annexin V: Add 5 µL of Annexin V conjugate to 100 µL of the cell suspension. Mix gently [4] [34].
- Incubate: Incubate for 10-15 minutes at room temperature, protected from light [4].
- Wash: Add 2 mL of 1X Binding Buffer and centrifuge at 400-600 x g for 5 minutes. Discard the supernatant [4].
- Resuspend and Add PI: Resuspend cells in 200 µL of 1X Binding Buffer. Add 5 µL of PI Staining Solution. Do not wash after this step [4].
- Analyze: Analyze by flow cytometry within 4 hours while keeping samples on ice and protected from light [4].
Protocol for Use with Fixable Viability Dyes (FVD)
Using FVDs is superior for complex panels as they allow for fixation after staining. However, dye compatibility is critical [4].
Materials:
- All materials from the standard protocol.
- Azide-free and serum/protein-free PBS.
- Compatible Fixable Viability Dye (e.g., FVD eFluor 660, 506, or 780). Note: FVD eFluor 450 is not recommended [4].
Procedure (Key Differences):
- Wash Cells: Wash cells twice in azide-free and serum/protein-free PBS [4].
- Stain Viability: Resuspend cells in PBS and add 1 µL of FVD per 1 mL of cells. Vortex immediately and incubate for 30 minutes at 2-8°C in the dark [4].
- Wash FVD: Wash cells twice with Flow Cytometry Staining Buffer or an equivalent, then once with 1X Binding Buffer [4].
- Proceed with Annexin V Staining: Continue from Step 3 of the standard protocol [4].
The Scientist's Toolkit: Research Reagent Solutions
Table: Essential Reagents for Annexin V Flow Cytometry
| Reagent | Function/Description | Key Considerations |
|---|---|---|
| 1X Annexin V Binding Buffer | Provides the optimal calcium-rich environment for Annexin V to bind to phosphatidylserine (PS) [4] [34]. | Must be calcium-rich and free of EDTA or other calcium chelators [4]. |
| Fluorochrome-conjugated Annexin V | The primary probe that binds to externally exposed PS on apoptotic cells [4] [34]. | Available in various fluorochromes (FITC, PE, APC, etc.) for panel flexibility [4]. |
| Propidium Iodide (PI) | A membrane-impermeant DNA dye used to identify dead cells with compromised membranes [34]. | Do not wash out after adding; analyze immediately [4]. |
| Fixable Viability Dyes (FVD) | Covalently bind to intracellular amines in cells with compromised membranes; allow for cell fixation after staining [4]. | Critical: Choose a dye compatible with your Annexin V conjugate and laser/filter setup (e.g., avoid FVD eFluor 450) [4]. |
| Flow Cytometry Staining Buffer | A protein-based buffer used for washing and resuspending cells to reduce non-specific antibody binding [4]. | Used when performing complex panels with surface or intracellular markers [4]. |
Step-by-Step Diagnostic Workflow
The following diagnostic chart provides a systematic approach to identifying and resolving the most common sources of background noise in your Annexin V experiments.
Pro Tips for Data Interpretation
- Gating Strategy is Key: Always begin your analysis by gating on the target cell population using forward scatter (FSC) vs. side scatter (SSC) to exclude debris and dead cells [43]. FSC correlates with cell size, while SSC indicates internal complexity [44].
- Use Scatter Plots for Dual-Parameter Data: To visualize and quantify populations from your Annexin V/PI staining, use a two-parameter scatter plot (dot plot) with Annexin V on one axis and PI on the other. This allows you to clearly see and gate the four distinct quadrants representing viable, early apoptotic, and late apoptotic/necrotic cells [43].
Autofluorescence is a pervasive challenge in fluorescence-based techniques, manifesting as unwanted background signal that can obscure specific detection and complicate data analysis. This background noise arises from both endogenous tissue components and exogenous factors introduced during sample preparation. For researchers working with lipidic tissues and brain samples—key systems in neuroscience, cell biology, and drug development—managing autofluorescence is particularly crucial for obtaining accurate, interpretable results. This technical support resource provides comprehensive troubleshooting guidance and proven methodologies to identify, minimize, and resolve autofluorescence issues specifically within the context of Annexin V flow cytometry research and related applications.
Autofluorescence stems from multiple sources, which can be broadly categorized as endogenous (natural to the tissue) or exogenous (introduced during processing).
Endogenous sources include:
- Lipofuscin: An age-pigment composed of oxidatively modified protein and lipid degradation residues that accumulates in lysosomes of long-lived cells, especially in brain, cardiac, and skeletal muscle tissues [45] [46]. Its broad excitation and emission spectra interfere across multiple fluorescence channels, and it clusters around nuclei, often obscuring key cellular structures.
- Red Blood Cells (RBCs): Contain fluorescent hemoglobin that emits across multiple wavelengths, spanning the spectra of many common fluorescent reporters [47]. This is particularly problematic in embryonic tissues where perfusion isn't feasible.
- Structural Proteins: Collagen and elastin in extracellular matrix components naturally fluoresce [48] [47].
- Flavins and other cellular components [47].
Exogenous sources primarily arise from:
- Aldehyde Fixation: Formalin, formaldehyde, and glutaraldehyde form fluorescent Schiff bases with tissue amines [48] [47]. Glutaraldehyde induces stronger autofluorescence than paraformaldehyde, which in turn causes more than formaldehyde.
- Embedding Media: Certain paraffin and OCT compounds can chemically alter tissue molecules to induce fluorescence [47].
Table 1: Common Autofluorescence Sources and Their Characteristics
| Source | Affected Tissues | Spectral Properties | Notes |
|---|---|---|---|
| Lipofuscin | Brain, spinal cord, cardiac/skeletal muscle (aged tissues) | Broad emission spectrum (green to red) | Increases with age; clusters around nucleus [48] [45] [46] |
| Red Blood Cells | Highly vascularized tissues (spleen, liver, kidney), embryonic tissues | Multiple wavelengths (spanning green and red) | Particularly problematic in non-perfused embryonic tissue [48] [47] |
| Collagen/Elastin | Connective tissues, blood vessels | Broad spectrum | Common in many tissue types [48] [47] |
| Aldehyde Fixation | Any fixed tissue | Broad spectrum | Glutaraldehyde > paraformaldehyde > formaldehyde [47] |
Strategic Approaches for Different Tissue Types
Lipidic Tissues and Brain Samples
Lipidic tissues present unique challenges due to their high concentrations of autofluorescent components. The following strategies have proven effective:
Chemical Quenching Methods:
- TrueVIEW Autofluorescence Quenching Kit: This hydrophilic, non-fluorescent molecule binds electrostatically to collagen, red blood cells, and elastin, as well as aldehyde-fixed tissue. Treatment requires just 5 minutes at room temperature and is compatible with common fluorophores (GFP, AlexaFluor, FITC, DyLight, cyanines) [48].
- TrueBlack Lipofuscin Autofluorescence Quencher: Effectively quenches red blood cell autofluorescence across red and green wavelengths in fixed embryonic tissue without interfering with immunofluorescent signal intensity or introducing background staining [47]. Optimal concentration and incubation conditions should be determined empirically.
- Sudan Black B: A traditional lipid-soluble dye that binds to lipofuscin granules. While effective in some applications, it can increase background staining and fluorescence at high red wavelengths, and doesn't efficiently reduce red blood cell autofluorescence [47].
Photobleaching Technique: A highly effective alternative to chemical quenchers involves pre-treatment with white phosphor LED arrays. This method utilizes broad-spectrum emission to bleach fluorophores across multiple wavelength peaks prior to immunofluorescence staining [45].
Protocol: LED Photobleaching for Tissue Sections
- Construct a photobleaching apparatus using a white LED desk lamp with any diffusers removed.
- Prepare slide chambers using transparent petri dishes with 0.05% sodium azide in TBS to prevent microbial growth.
- Submerge tissue sections mounted on glass slides in the chamber.
- Cover with a reflective dome and illuminate for 48 hours at 4°C.
- Proceed with standard immunofluorescence protocols.
This method effectively reduces background and lipofuscin fluorescence without affecting probe fluorescence intensity [45].
Flow Cytometry Applications
In flow cytometry, autofluorescence manifests as high background that reduces sensitivity and resolution. Key mitigation strategies include:
- Fluorophore Selection: Use bright fluorophores that emit in the red channel (e.g., APC, Alexa Fluor 647) for highly autofluorescent cell types like neutrophils [16].
- Viability Staining: Include a viability dye to exclude dead cells during analysis, as they exhibit higher autofluorescence [16].
- Sample Handling: Keep samples on ice, avoid freeze-thaw cycles, and use gentle dissociation methods without EDTA when working with Annexin V, as it requires calcium for binding [5].
- Buffer Considerations: For microvesicle studies, use saline instead of phosphate-buffered saline (PBS) with Annexin V binding buffer, as PBS can produce insoluble calcium phosphate precipitates that generate false positive events [49].
Autofluorescence Resolution Strategy Selection
Troubleshooting FAQs for Annexin V Flow Cytometry
Q1: My control groups show false positive signals in Annexin V staining. What could be causing this?
- Poor compensation causing fluorescence overlap between channels
- Overconfluent or starved cells undergoing spontaneous apoptosis
- Mechanical damage from over-trypsinization or harsh pipetting
- Interfering fluorescence from drugs or cell autofluorescence
- Delayed analysis after staining or prolonged drug treatment [5]
Q2: I'm detecting only nuclear dye (PI) positivity without Annexin V binding. What does this indicate? This typically suggests poor cell health or excessive mechanical damage during processing. Use healthy, log-phase cells and gentle handling techniques. Avoid EDTA-containing trypsin as it chelates calcium needed for Annexin V binding [5].
Q3: Why do I see no positive signals in my treated groups despite expecting apoptosis?
- Insufficient drug concentration or treatment duration
- Apoptotic cells in the supernatant were discarded during processing
- Operational errors such as missing dye addition or washing after staining
- Kit degradation or improper storage [5]
Q4: How can I minimize autofluorescence specifically in flow cytometry experiments?
- Use bright fluorophores in red channels for highly autofluorescent cells
- Include viability dyes to exclude dead cells
- Keep samples on ice and avoid freeze-thaw cycles
- Filter samples to remove debris
- Use Fc receptor blocking reagents to reduce non-specific antibody binding [16]
Q5: What specific issues should I consider when working with human brain samples? Human brain tissue has significant autofluorescence compared to rodent models. Lipofuscin granules cluster around nuclei, interfering with detection of perinuclear structures. Consider spectral imaging with linear unmixing if available, or optimize chemical quenching protocols specifically for human tissue [46].
Detailed Experimental Protocols
TrueBlack Lipofuscin Autofluorescence Quenching Protocol
This protocol is optimized for fixed embryonic tissue but adaptable to various lipid-rich tissues [47]:
Tissue Preparation:
- Fix tissue in 4% formaldehyde (rather than glutaraldehyde)
- Cryoprotect in 30% sucrose solution
- Embed in OCT and section at 10-20μm thickness
TrueBlack Application:
- Dilute 20X TrueBlack stock in DMF to appropriate working concentration (typically 1X)
- Apply to tissue sections and incubate for determined optimal time (typically 30 seconds to 2 minutes)
- Rinse gently with buffer
Immunostaining:
- Proceed with standard immunofluorescence protocol
- Include appropriate controls without TrueBlack treatment
Flow Cytometry Annexin V Protocol with Autofluorescence Reduction
Adapted for high-background samples [5] [50]:
Cell Preparation:
- Use gentle, EDTA-free dissociation enzymes like Accutase
- Include viability dye (e.g., 7-AAD) for dead cell exclusion
- Keep cells on ice throughout processing
Staining:
- Resuspend cells in saline-based binding buffer rather than PBS
- Add Annexin V conjugate (avoid FITC if cells express GFP)
- Incubate 15 minutes in dark at room temperature
- Add viability dye shortly before analysis
- Analyze within 1 hour of staining
Controls:
- Unstained cells: for voltage settings and autofluorescence assessment
- Single-stain controls: for compensation
- Positive control: induced apoptosis for kit validation
Autofluorescence Resolution Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Autofluorescence Management
| Reagent/Kit | Primary Application | Key Features | Protocol Considerations |
|---|---|---|---|
| TrueVIEW Autofluorescence Quenching Kit (Vector Laboratories) | General immunofluorescence, aldehyde-fixed tissue | Hydrophilic molecule, 5-minute room temperature treatment, compatible with common fluorophores | Apply after antibody staining, mix 1:1:1 with mounting medium, includes DAPI counterstain option [48] |
| TrueBlack Lipofuscin Autofluorescence Quencher (Biotium) | Lipofuscin-rich tissues (brain, aged tissue), red blood cells | Effective across red and green wavelengths, doesn't mask antibody signal, reusable | Dilute 20X stock, optimize concentration and incubation time, compatible with various mounting media [47] |
| Sudan Black B | Traditional lipofuscin quenching | Lipid-soluble, binds lipofuscin granules | Can increase background in red channel, less effective on RBC autofluorescence [47] |
| White LED Photobleaching Setup | All tissue types, particularly post-mitotic tissues | Broad-spectrum emission, low cost, no chemical interference | 48-hour treatment at 4°C with 0.05% sodium azide, construct from off-the-shelf components [45] |
| Sodium Borohydride | Aldehyde-induced autofluorescence | Reduces Schiff bases formed during fixation | Can be caustic, may reduce antibody signal and damage tissue [47] |
| Annexin V Apoptosis Kits (PE/APC conjugated) | Flow cytometry with autofluorescent cells | Avoids FITC channel where autofluorescence is high | Use saline-based buffers, include viability dye, analyze immediately [5] [50] |
Advanced Techniques and Considerations
For persistent autofluorescence issues, consider these advanced approaches:
Spectral Imaging with Linear Unmixing: This optical method requires specialized laser scanning confocal microscopes and software capable of dividing emission light into ~10nm segments. By defining the emission profiles of autofluorescence, dyes, and fluorescent antibodies using single-color controls, signals can be computationally separated even when they overlap spatially and spectrally [46].
Buffer Optimization for Specific Applications: When studying microvesicles or performing Annexin V staining, avoid phosphate-buffered saline (PBS) in binding buffers as the phosphate can react with calcium to form insoluble Ca(H₂PO₄)₂ or Ca₃PO₄ precipitates that generate false nano-sized vesicles detectable by flow cytometry. Use saline instead for more accurate results [49].
Experimental Design Considerations: Always include appropriate controls:
- Unstained samples for autofluorescence assessment
- Single-color controls for compensation and spectral unmixing
- Positive controls with induced apoptosis for kit validation
- Fc receptor blocking controls to identify non-specific antibody binding [5] [46]
Successfully resolving autofluorescence in lipidic tissues and brain samples requires a systematic approach that begins with understanding the sources of background noise, implements appropriate quenching or avoidance strategies, and includes rigorous controls and optimization. The methods outlined here—from simple chemical quenching to advanced optical techniques—provide researchers with a comprehensive toolkit for overcoming these challenges. By applying these strategies specifically within the context of Annexin V flow cytometry and related applications, scientists can significantly enhance signal-to-noise ratio, resulting in more reliable, interpretable, and publication-quality data.
Fluorescence spillover, or spectral overlap, occurs when the emission signal of one fluorophore is detected in the channel of another. In Annexin V flow cytometry, this can cause inaccurate data interpretation, making it crucial to correct through compensation. This process uses single-stain controls to mathematically correct for the spillover, ensuring that fluorescence signals are accurately assigned to the correct detector.
Frequently Asked Questions (FAQs)
1. Why are single-stain controls necessary for Annexin V experiments? Single-stain controls are essential because they allow the flow cytometer to measure the exact amount of signal from one fluorophore that spills into another's detection channel. This information is used to calculate compensation values, ensuring that the fluorescence measured in each channel comes only from its intended dye. Without proper compensation, you cannot reliably distinguish early apoptotic (Annexin V positive) from late apoptotic/necrotic (PI or 7-AAD positive) cells [24] [7].
2. What can I use for single-stain controls? You can use either cells or compensation beads [7].
- Cells: Use the same cell type as your experimental samples. Ideally, they should be brightly and uniformly stained with a single fluorophore. For Annexin V and viability dyes, you will need:
- Cells stained with Annexin V conjugate only (e.g., Annexin V-FITC alone).
- Cells stained with the viability dye only (e.g., PI alone, 7-AAD alone) [24].
- Compensation Beads: These are synthetic beads that bind antibodies uniformly. They are highly recommended for their consistency and because they do not require a separate cell preparation for each control. Always use "unstained beads" as your negative control when using beads [7].
3. My unstained cells show high fluorescence. What could be the cause? High background in unstained cells can stem from several sources relevant to apoptosis assays:
- Cellular Autofluorescence: Cells like macrophages have inherent fluorescence [11] [7].
- Poor Cell Health: Dead cells are notoriously "sticky" and bind dyes non-specifically, dramatically increasing background [11]. Always include a viability dye in your panel to gate out dead cells during analysis.
- Contamination or Debris: Cellular debris or contaminated samples can increase background noise [51] [19].
- Instrument Background: Unclean fluidics or laser misalignment can cause background noise. Running a water blank can help identify this issue [51] [19].
4. After compensation, my negative population appears to have a negative value on the plot. Is this correct? Yes, this is a normal and expected result of the compensation algorithm. The compensation process subtracts the spilled-over signal from the negative channel, which can result in the median fluorescence intensity (MFI) of the negative population falling below zero. This confirms that the compensation has successfully corrected for the spillover [7].
Troubleshooting Guide
The table below outlines common problems, their potential causes, and solutions related to compensation and background in flow cytometry.
| Problem | Potential Cause | Recommended Solution |
|---|---|---|
| High background fluorescence | Too many dead cells in sample [11]. | Include a viability dye (e.g., PI, 7-AAD) and gate out dead cells during analysis [7]. |
| Antibody concentration is too high [11]. | Titrate antibodies to find the optimal concentration that maximizes signal and minimizes background. | |
| Non-specific binding via Fc receptors [11]. | Use an Fc receptor blocking reagent (e.g., "Fc Block") or add unconjugated antibody to saturate Fc receptors [11]. | |
| Poor separation of positive and negative populations | Inadequate compensation due to dim single-stain controls [7]. | Ensure single-stain controls are bright and have a clear, positive population. Collect at least 5,000 events for the positive population [7]. |
| Spectral overlap is too severe for the filter set [52]. | Use a panel design tool to select fluorophores with minimal overlap. Consider spectral flow cytometry for highly complex panels [53]. | |
| No signal in single-stain control | Reagent degradation or improper storage [51]. | Use fresh reagents and ensure they are stored correctly (e.g., 7-AAD should be stored at -20°C) [51]. |
| Operator error (e.g., forgot to add dye) [51] [5]. | Carefully follow a staining protocol and double-check all steps. | |
| Unexpected positivity in the untreated control | Spontaneous apoptosis from over-confluent cultures or harsh handling [5]. | Use healthy, log-phase cells and handle them gently during harvesting (use EDTA-free enzymes like Accutase) [51] [5]. |
| Fluorescence spillover from other sources (e.g., drugs like doxorubicin) [51]. | Be aware of fluorescent compounds in your experimental system and choose fluorophores that do not overlap with them. |
Experimental Protocol: Setting Up Single-Stain Controls
The following workflow details the critical steps for preparing and using single-stain controls for a standard Annexin V/FITC and PI apoptosis assay.
Single-Stain Control Setup Workflow
Materials Needed
| Item | Function |
|---|---|
| Healthy Cells | Provides the biological material for controls. Should be in good condition and match experimental cells. |
| Annexin V Conjugate | The fluorescently-labeled protein that binds to phosphatidylserine. For this protocol, Annexin V-FITC. |
| Viability Dye (PI/7-AAD) | Membrane-impermeant dye that stains nucleic acids in dead cells. |
| 1X Binding Buffer | Provides the calcium-rich environment required for Annexin V binding to phosphatidylserine. |
| Flow Cytometer | Instrument for analysis. Must be equipped with lasers and filters matching your fluorophores (e.g., 488 nm laser for FITC/PI). |
Step-by-Step Procedure
- Cell Preparation: Harvest and wash your cells (e.g., from culture) gently. Use cold PBS and avoid using trypsin containing EDTA, as it chelates calcium and inhibits Annexin V binding [5] [4]. Resuspend the cell pellet in 1X Binding Buffer at a concentration of 1 x 10^6 cells/mL [24] [4].
- Aliquot Cells: Transfer 100 µL of the cell suspension (containing ~1 x 10^5 cells) into each of three 5 mL flow cytometry tubes [24].
- Stain Controls:
- Tube 1 (Unstained Control): Add nothing. This tube is for setting detector voltages and identifying autofluorescence.
- Tube 2 (Annexin V-FITC Only): Add 5 µL of Annexin V-FITC. Gently vortex the tube to mix [24] [4].
- Tube 3 (PI Only): Add 2-5 µL of Propidium Iodide stock solution. Gently vortex to mix. Note: The optimal volume of PI may need to be titrated for your specific cell type [24].
- Incubate: Incubate the Annexin V-FITC tube for 15 minutes at room temperature in the dark. Incubate the PI tube for 5-15 minutes on ice or at room temperature in the dark [24] [4].
- Final Preparation:
- Acquisition: Analyze the samples on the flow cytometer as soon as possible, ideally within 1 hour. Use the unstained and single-stained controls to set up compensation on your flow cytometer software before running your experimental samples [24] [54].
The Scientist's Toolkit: Essential Research Reagents
The table below lists key materials required for successful Annexin V apoptosis detection with proper compensation.
| Item | Function | Critical Consideration |
|---|---|---|
| Annexin V Conjugate | Binds to exposed phosphatidylserine (PS) on apoptotic cells. | Choose a fluorophore compatible with your laser/filter setup and other markers (e.g., avoid FITC if cells express GFP) [5]. |
| Membrane-Impermeant Viability Dye (PI, 7-AAD) | Distinguishes late apoptotic/necrotic cells (permeant) from early apoptotic cells (impermeant). | Do not wash cells after adding PI/7-AAD. Analyze immediately [54] [4]. |
| Calcium-Enriched Binding Buffer | Provides the necessary Ca²⁺ for Annexin V-PS binding. | Avoid buffers containing EDTA or other calcium chelators, as they will prevent binding [5] [4]. |
| Compensation Beads | Provide a consistent and bright negative/positive signal for setting compensation. | Use instead of cells for more reproducible controls, especially for surface stains [7]. |
| Fc Receptor Blocking Reagent | Blocks non-specific antibody binding to Fc receptors on immune cells. | Reduces background staining, leading to a cleaner signal [11] [7]. |
FAQ: Understanding and Preventing False Positives
Q1: How does using trypsin with EDTA lead to false negatives in Annexin V assays? Annexin V binding to phosphatidylserine (PS) is calcium-dependent [5] [4]. EDTA, a calcium chelator, in trypsin solutions strips away the essential Ca²⁺ ions, thereby directly interfering with the Annexin V-PS interaction and compromising assay results [5] [55]. This can lead to a false negative because early apoptotic cells with exposed PS will not be stained.
Q2: Why can over-trypsinization or poor cell health cause false positives? Excessive trypsin digestion or mechanical stress (like vigorous pipetting) can physically damage the healthy cell membrane, causing non-apoptic phosphatidylserine (PS) exposure or letting viability dyes like PI enter the cell [5] [56]. Similarly, using over-confluent, starved, or otherwise unhealthy cells increases the rate of spontaneous apoptosis and general membrane fragility, leading to false positive signals [5] [7].
Q3: What are the best practices for harvesting adherent cells to minimize artifacts? To minimize harvesting-induced artifacts, consider these methods:
- Use Gentler Enzymes: Replace trypsin-EDTA with a gentler alternative like Accutase, which is less damaging to surface epitopes and the membrane [5] [57].
- Mechanical Scraping: While an option, note that scraping with a "rubber policeman" can also cause significant membrane damage and should be performed with caution [57].
- Optimize Protocol: Keep enzymatic digestion time as short as possible, handle cells gently, and always include the supernatant from the culture, as it contains dead and apoptotic cells that should be analyzed [5] [8].
Q4: How should controls be set up to identify false positives? Proper controls are essential for validating your results [56].
- Unstained Control: Cells without any dye to adjust FSC/SSC and detect autofluorescence.
- Single-Stain Controls: Apoptotic cells stained separately with Annexin V only and PI only. These are critical for accurate compensation on the flow cytometer [5] [7].
- Biological Control: Healthy, untreated cells stained with both dyes to establish the baseline for viable cells and identify background death caused by handling.
Troubleshooting Guide: Key Issues and Solutions
Table 1: Common Causes and Solutions for False Positives/Negatives
| Problem | Potential Cause | Recommended Solution |
|---|---|---|
| High False Positives | Cell harvesting with trypsin-EDTA [5] [57] | Use EDTA-free trypsin or gentler dissociation enzymes like Accutase; wash cells thoroughly after digestion [5] [56]. |
| Over-trypsinization or harsh mechanical handling [5] | Optimize digestion time; avoid vigorous pipetting; handle cells gently [5]. | |
| Poor overall cell health (over-confluent, starved) [5] | Use healthy, log-phase cells; ensure optimal culture conditions and appropriate seeding density [5]. | |
| Incorrect flow cytometry compensation [5] [7] | Use single-stain controls with apoptotic cells to properly adjust compensation and prevent signal spillover [5]. | |
| Unexpected False Negatives | EDTA in the trypsin solution chelating Ca²⁺ [5] [4] | Use calcium-containing binding buffers and avoid any buffers with EDTA or other calcium chelators during the staining steps [4]. |
| Loss of apoptotic cells during processing [5] | Always collect and include the culture supernatant when harvesting, as it contains detached apoptotic and dead cells [5] [8]. | |
| Inadequate apoptosis induction [5] | Verify drug treatment efficacy by using a positive control (e.g., drug-treated cells) and optimize treatment concentration/duration [5]. |
Experimental Protocol: A Recommended Workflow for Minimizing Artifacts
This protocol prioritizes gentle handling to preserve true apoptotic signals.
Cell Harvesting and Staining
- Harvesting: For adherent cells, use a gentle dissociation enzyme like Accutase instead of trypsin-EDTA. Quench the enzyme with complete medium. Critically, collect the culture supernatant containing any detached cells and combine it with the harvested cells [8] [57].
- Washing: Centrifuge the cell suspension and wash the pellet with calcium-rich PBS (with 25 mM CaCl₂) or 1X Annexin Binding Buffer. Avoid using standard PBS or buffers containing EDTA [8] [4].
- Staining: Resuspend the cell pellet (~1x10⁶ cells) in 100 µL of Annexin Binding Buffer.
- Annexin V Incubation: Add the fluorochrome-conjugated Annexin V (e.g., 5 µL), mix gently, and incubate for 10-15 minutes at room temperature in the dark [4].
- Viability Dye Addition: Add 2 mL of Binding Buffer, centrifuge, and discard the supernatant. Resuspend the cell pellet in 200 µL of fresh Binding Buffer. Add 5 µL of Propidium Iodide (PI) or 7-AAD. Do not wash after this step, as the viability dye must remain in the solution during acquisition [4].
- Analysis: Analyze the samples by flow cytometry within 1 hour to maintain cell viability and staining integrity [5] [4].
The following workflow diagram summarizes the key steps and decision points in this protocol:
Diagram: Optimized Workflow to Minimize False Positives
The Scientist's Toolkit: Essential Reagent Solutions
Table 2: Key Reagents for a Robust Annexin V Assay
| Reagent | Function & Importance | Key Consideration |
|---|---|---|
| Gentle Dissociation Reagent (e.g., Accutase) | Dissociates adherent cells with minimal proteolytic activity, preserving membrane integrity and surface epitopes better than trypsin-EDTA [5] [57]. | The optimal choice for maintaining cell health during harvesting. |
| Annexin V Binding Buffer | Provides the calcium-rich environment (Ca²⁺) essential for specific Annexin V binding to externalized phosphatidylserine (PS) [4] [55]. | Never substitute with standard PBS, as the lack of Ca²⁺ will cause false negatives. |
| Viability Dye (e.g., PI, 7-AAD) | Distinguishes late apoptotic/necrotic cells (membrane-compromised) from early apoptotic cells (membrane-intact) [5] [8]. | Do not wash cells after adding the dye. It must be present in the buffer during acquisition [4]. |
| Single-Stain Controls | Cells stained individually with Annexin V or the viability dye. These are non-negotiable for setting accurate fluorescence compensation on the flow cytometer [5] [7]. | Must be prepared from cells with confirmed apoptosis to ensure a strong positive signal for compensation. |
Why is proper PMT voltage optimization critical for Annexin V experiments?
Proper Photomultiplier Tube (PMT) voltage calibration is fundamental to flow cytometry. It ensures that the instrument detects fluorescence signals with high sensitivity and accurately separates the dim signals of negative populations from the positive ones. In Annexin V-based apoptosis detection, this is crucial for distinguishing early apoptotic cells (Annexin V positive, viability dye negative) from viable and late apoptotic populations. Incorrect voltages can lead to false positives or negatives, poor resolution of cell populations, and inaccurate quantification of apoptosis [5] [7].
FAQs on PMT Voltage and Background Noise
Q1: How do I set the initial PMT voltages for my Annexin V assay? Start by using unstained cells and cells stained with a single fluorochrome. Adjust the voltage for each detector so that the unstained cell population has a median fluorescence intensity (MFI) in the first log decade of the histogram. This provides adequate range to detect positive signals without compressing the negative population [7] [24].
Q2: My unstained control shows high fluorescence. Is this background noise or an instrument issue? High fluorescence in an unstained control is often due to cellular autofluorescence or poor cell health. Dying cells can exhibit nonspecific staining and increased background. Ensure you are using healthy, log-phase cells. You can also try using fluorochromes with longer emission wavelengths (e.g., APC, PE) to minimize interference from autofluorescence, which is often more pronounced in the FITC channel [5] [58] [7].
Q3: After setting voltages, my Annexin V and PI populations are not clearly separated. What should I check? This is frequently a compensation issue, not a voltage issue. Ensure you have created proper single-stained controls—cells stained with Annexin V-FITC only and PI only—to calculate the compensation matrix accurately. Fluorescence spillover from one channel into another can blur the distinction between populations if not properly compensated [5] [7] [24].
Q4: Can the chemicals I use affect background noise? Yes. Buffers containing EDTA or other calcium chelators will inhibit the calcium-dependent binding of Annexin V, leading to weak or false-negative signals [5] [4]. Always use the calcium-containing binding buffer provided with your Annexin V kit. Furthermore, harsh cell dissociation methods using trypsin-EDTA can damage the cell membrane and cause nonspecific Annexin V binding; use gentle, non-enzymatic dissociation agents like Accutase where possible [5] [58].
Troubleshooting Background Noise and Signal Issues
The table below summarizes common problems, their potential causes, and solutions related to instrument setup and sample preparation.
| Problem Phenomenon | Potential Cause | Recommended Solution |
|---|---|---|
| High background in unstained cells [58] [7] | Cell autofluorescence; Poor cell viability | Use healthy, log-phase cells; Switch to a red or far-red fluorescent Annexin V conjugate (e.g., PE, APC) to avoid autofluorescence. |
| Weak or no Annexin V signal [5] [4] | Incorrect PMT voltage; Calcium-free buffer | Re-titrate PMT voltages using a positive control; Ensure binding buffer contains calcium (use kit-provided buffer). |
| Poor separation between positive and negative populations [5] [7] | Suboptimal PMT voltage; Inadequate compensation | Adjust voltage to maximize the separation index (signal-to-noise); Use single-stain controls for proper compensation. |
| Only PI is positive, Annexin V is negative [5] [58] | Overly harsh cell handling; Cells are necrotic | Be gentle during cell harvesting and pipetting to preserve membrane integrity; Include a viability assessment. |
| Annexin V is positive, PI is negative in most cells [5] | Cells are in early apoptosis; PI dye was omitted | This is an expected pattern for early apoptosis; Verify that PI was added to the sample. |
Experimental Protocol: Calibration and Controls
A reliable Annexin V assay depends on a robust protocol with appropriate controls. The following workflow integrates critical calibration steps.
Workflow for Annexin V Staining with Integrated Controls
Detailed Step-by-Step Procedure
Cell Preparation and Staining
- Harvest cells gently. For adherent cells, collect the supernatant containing dead cells first, then use a gentle dissociation method to detach remaining cells. Combine all fractions [8] [59].
- Wash cells once with cold PBS and once with 1X Binding Buffer (ensure it contains calcium) [4] [24].
- Resuspend the cell pellet in 1X Binding Buffer at a concentration of 1 x 10^6 cells/mL [24].
- Transfer 100 µL of cell suspension (∼1 x 10^5 cells) to a flow cytometry tube.
- Add 5 µL of Annexin V conjugate (e.g., FITC) and the recommended volume of viability dye (e.g., 5 µL of PI or 7-AAD) [4] [24]. Note: For single-stain controls, add only one dye to separate aliquots of cells.
- Gently vortex the tubes and incubate for 15 minutes at room temperature in the dark [4] [60].
- Add 400 µL of 1X Binding Buffer to each tube. Do not wash after adding PI or 7-AAD, as this will remove the viability dye [4] [24].
- Analyze by flow cytometry within 1 hour [5] [24].
Mandatory Controls for Instrument Setup Proper controls are non-negotiable for setting PMT voltages and compensation correctly [5] [7] [24].
- Unstained Cells: Cells resuspended only in binding buffer. Used to set photodetector voltages and determine background autofluorescence.
- Single-Stain Controls:
- Annexin V Only: Apoptotic cells stained solely with the Annexin V conjugate (e.g., Annexin V-FITC). Critical for compensating spillover into the PI/7-AAD detector.
- Viability Dye Only: Cells stained solely with the viability dye (e.g., PI). Critical for compensating spillover into the Annexin V detector.
- Positive Control: Cells treated with an apoptosis-inducing agent (e.g., 1 µM staurosporine or camptothecin for 2-4 hours) [60] [59]. Validates the assay and helps set optimal PMT gains.
- Untreated Control: Healthy, untreated cells. Defines the baseline for spontaneous apoptosis.
The Scientist's Toolkit: Essential Research Reagents
| Item | Function in the Experiment |
|---|---|
| Calcium-Containing Binding Buffer | Provides the necessary Ca²⁺ for Annexin V to bind to externalized Phosphatidylserine (PS); using calcium-free buffers (e.g., PBS without Ca²⁺) is a common error [4] [24]. |
| Fluorochrome-conjugated Annexin V | Binds to PS on the outer membrane of apoptotic cells; available in FITC, PE, APC, and others to suit your instrument and panel [4] [50]. |
| Membrane-Impermeant Viability Dye | distinguishes cells with intact vs. compromised membranes; Propidium Iodide (PI) and 7-AAD are commonly used [5] [60] [24]. |
| Apoptosis Inducer (e.g., Staurosporine) | Used to generate a reliable positive control for assay validation and for setting up instrument parameters [60] [59]. |
| Single-Stain Controls | Cells individually stained with Annexin V or viability dye; essential for calculating fluorescence compensation on the flow cytometer [5] [7] [24]. |
Validating Your Assay and Comparing Apoptosis Detection Methods
The Role of Controls in Annexin V Assays
In Annexin V flow cytometry, proper controls are not merely optional; they are the foundation for valid data interpretation. They are essential for configuring your flow cytometer accurately and for distinguishing specific signal from background noise, which is the focus of this thesis. The primary controls can be categorized by their purpose:
- Instrument & Gating Controls: Unstained and single-stain controls allow for accurate compensation and setting of negative-positive boundaries.
- Specificity & Biological Controls: Biological controls, including untreated healthy cells and induced apoptotic cells, verify the assay's performance and the specificity of Annexin V binding.
Failing to include the correct controls is a common source of high background and uninterpretable results.
A Protocol for Control Preparation
The following procedure details the preparation of all necessary control samples for a robust Annexin V experiment. Always protect samples from light during incubation steps [4] [24].
Materials
- Cell Sample: Both untreated (healthy) and apoptosis-induced cell suspensions.
- Staining Reagents: Annexin V conjugate (e.g., FITC, PE), viability dye (e.g., PI, 7-AAD), and 1X Annexin Binding Buffer.
- Labware: Flow cytometry tubes, pipettes, centrifuge.
Step-by-Step Procedure
Prepare Cells: Harvest both untreated cells and cells in which you have induced apoptosis (e.g., using anti-Fas antibody for Jurkat cells or camptothecin) [61] [1]. For adherent cells, use gentle, non-enzymatic dissociation methods where possible and allow cells to recover for 30 minutes after detachment to prevent false positives from membrane damage [62]. Ensure you collect both supernatant and adherent cells to capture all apoptotic populations [25].
Aliquot Cells: For each control and experimental sample, transfer approximately 1-5 x 10^5 cells in 100 µL of 1X Binding Buffer to a labeled flow cytometry tube [24] [61].
Stain Control Tubes:
- Unstained Control: Cells + 400 µL Binding Buffer. No dyes added [59].
- Annexin V Single-Stain Control: Cells + Annexin V conjugate (e.g., 5 µL). Incubate 15 min at RT, then add 400 µL Binding Buffer [24] [61].
- Viability Dye Single-Stain Control: Cells + Viability Dye (e.g., 5 µL PI or 7-AAD). Add 400 µL Binding Buffer [24] [61]. Do not wash after adding viability dye.
- Biological Controls:
- Experimental Samples: Stain with both Annexin V and viability dye according to your specific protocol.
Analysis: Analyze all samples by flow cytometry as soon as possible, typically within 1 hour [24] [61]. Use the single-stain controls to set up compensation on your flow cytometer before running the experimental samples.
The relationships and purpose of each control in the experimental workflow are summarized in the diagram below.
The Scientist's Toolkit: Key Reagents for Control Experiments
The table below lists essential reagents and their specific functions in establishing rigorous controls.
| Item | Function in Control Experiments | Key Considerations |
|---|---|---|
| Annexin V Conjugate [1] | Used in single-stain and positive controls to detect phosphatidylserine exposure. | Choose a fluorophore that does not overlap with cellular autofluorescence or other dyes in the panel [5]. |
| Viability Dye (PI/7-AAD) [24] [61] | Used in single-stain controls to identify dead/necrotic cells and set compensation. | Do not wash out after staining; keep in buffer during acquisition [4] [24]. |
| 1X Annexin Binding Buffer [24] [61] | Provides the calcium-dependent environment required for specific Annexin V binding. | Avoid buffers containing EDTA or other calcium chelators, as they disrupt binding [4] [5]. |
| Apoptosis Inducer (e.g., anti-Fas, Camptothecin) [61] [59] | Used to generate the essential biological positive control (induced apoptotic cells). | Titrate concentration and treatment time to avoid excessive necrosis [63]. |
| Cell Dissociation Buffer (non-enzymatic) [62] | For gently detaching adherent cells for analysis, minimizing false-positive Annexin V staining. | Preferable to trypsin-EDTA; if using trypsin, allow a 30-minute recovery post-detachment [62]. |
FAQs and Troubleshooting Guide
Q1: My unstained control or untreated cells show a high percentage of Annexin V-positive cells. What is the cause?
This is a common sign of background noise or false positives. Potential causes and solutions include:
- Cause: Cell Handling and Health. Rough pipetting, over-trypsinization, or using cells in poor health (over-confluent, starved) can cause spontaneous apoptosis and membrane damage, allowing Annexin V to bind non-specifically [5] [63].
- Solution: Always handle cells gently. For adherent cells, use a gentle, non-enzymatic dissociation buffer and allow a 30-45 minute recovery in culture medium after trypsinization before staining [62]. Use healthy, log-phase cells.
- Cause: Contaminated or Improperly Prepared Buffer. If the binding buffer is incorrectly diluted, lacks calcium, or contains a calcium chelator like EDTA, it can lead to non-specific staining and high background [4] [63].
- Solution: Ensure the 1X binding buffer is prepared correctly according to the kit instructions and is free of contaminants.
Q2: I followed the protocol, but I see no positive signal in my induced apoptotic positive control. Why?
- Cause: Inadequate Apoptosis Induction or Lost Cells. The drug concentration or treatment time may be insufficient to induce detectable apoptosis. A very common mistake is discarding the floating cells in the culture supernatant, which often contain the apoptotic population [5].
- Solution: Re-optimize the concentration and duration of your apoptosis-inducing treatment. Always combine the culture supernatant with the trypsinized adherent cells when harvesting [25]. Use a known effective inducer like anti-Fas antibody for Jurkat cells as a positive control for your assay [61].
Q3: My single-stain controls look poor, making compensation difficult. What went wrong?
- Cause: Reagent Degradation or Omission. The fluorescent conjugates or viability dyes may have degraded due to improper storage (e.g., repeated freeze-thaw cycles, exposure to light). It is also possible the dye was accidentally omitted.
- Solution: Aliquot reagents to avoid repeated freeze-thaws and store them as recommended. Include a viability dye-only control to confirm the dye is functional [63]. Always use fresh controls for each experiment.
Q4: The cell populations in my dot plot are not clearly separated, making gating difficult.
- Cause: Cellular Autofluorescence or Poor Cell State. Some cell types have intrinsic autofluorescence that can overlap with your fluorophores, blurring the distinction between positive and negative populations. Poor overall cell health can also cause a continuum of staining [5] [63].
- Solution: If autofluorescence is an issue, switch to a brighter Annexin V conjugate (e.g., Alexa Fluor) or one with a longer wavelength (e.g., APC) to move away from the autofluorescence spectrum [1] [5]. Ensure cells are in an optimal state at the start of the experiment.
In Annexin V flow cytometry for apoptosis detection, consistent and accurate data interpretation is paramount. Defining quadrants and calculating apoptotic indices are foundational steps that transform raw fluorescence data into biologically meaningful results. A lack of standardization in these steps is a significant source of experimental variability and background noise, potentially compromising data reliability. This guide addresses the specific challenges researchers face when interpreting Annexin V data, providing clear standards for quadrant placement, control use, and index calculation to ensure reproducible and accurate quantification of apoptotic cell populations. Adhering to these standards minimizes interpretation artifacts and enhances the validity of your experimental conclusions, which is especially critical in drug development research where subtle changes in apoptosis can determine compound efficacy.
Core Principles of Annexin V Assay and Quadrant Analysis
The Annexin V assay detects the translocation of phosphatidylserine (PS) from the inner to the outer leaflet of the plasma membrane, an early event in apoptosis. This is combined with a viability dye, such as Propidium Iodide (PI) or 7-AAD, which penetrates cells with compromised membranes, typically characteristic of late apoptosis or necrosis [5].
The fundamental principle of the quadrant analysis is to distinguish four distinct cell populations based on their staining characteristics:
- Viable Cells (Annexin V-negative / Viability Dye-negative): Healthy cells with intact membranes and no exposed PS.
- Early Apoptotic Cells (Annexin V-positive / Viability Dye-negative): Cells undergoing apoptosis but maintaining membrane integrity.
- Late Apoptotic/Dead Cells (Annexin V-positive / Viability Dye-positive): Cells in late-stage apoptosis or post-apoptotic necrosis with exposed PS and compromised membranes.
- Necrotic/Damaged Cells (Annexin V-negative / Viability Dye-positive): This population is often minimal and may represent cellular debris, pre-existing necrosis, or a technical artifact.
The following workflow outlines the critical steps for proper data acquisition and analysis:
Essential Controls for Accurate Quadrant Placement
Proper quadrant placement is not arbitrary; it must be guided by experimental controls to avoid misinterpretation and background noise. The following controls are non-negotiable for defining the boundaries between negative and positive populations [24] [5]:
- Unstained Cells: This control is used to set the baseline autofluorescence and to adjust the flow cytometer's photomultiplier tube (PMT) voltages for the Forward Scatter (FSC) and Side Scatter (SSC), ensuring the cell population is properly positioned on the plot.
- Single-Stain Controls:
- Cells stained with Annexin V only (no PI): This sample defines the Annexin V-positive/PI-negative population. It is used to adjust for any fluorescence spillover from the Annexin V fluorochrome (e.g., FITC) into the PI detector.
- Cells stained with PI only (no Annexin V): This sample defines the Annexin V-negative/PI-positive population. It is used to adjust for any spillover from PI into the Annexin V detector.
- Induced Apoptosis Positive Control: A cell population treated with a known apoptosis inducer (e.g., UV irradiation, staurosporine) is critical for verifying that your assay is functioning correctly and provides a reference for what true positive staining looks like.
Step-by-Step Protocol for Control Setup and Acquisition
- Prepare Control Samples: For each experimental condition, prepare separate tubes for unstained, Annexin V single-stain, and PI single-stain controls [24] [25].
- Acquire Unstained Cells First: Run the unstained cells and adjust FSC and SSC voltages to place the cell population in the center of the dot plot. Then, adjust the fluorescence detectors for Annexin V and PI so that the unstained population is in the lower left quadrant (double-negative).
- Acquire Single-Stain Controls:
- Run the Annexin V-only control. The cell population should shift to the right (Annexin V-positive). Adjust the compensation between the Annexin V detector and the PI detector so that the population remains negative for PI.
- Run the PI-only control. The cell population should shift upward (PI-positive). Adjust the compensation between the PI detector and the Annexin V detector so that the population remains negative for Annexin V [5].
- Set Quadrant Gates: Once compensation is correctly set using the single-stain controls, place the quadrant lines such that the vast majority (>99%) of the unstained cells fall in the lower left quadrant. The Annexin V-only control should predominantly fall in the lower right quadrant, and the PI-only control should predominantly fall in the upper left quadrant.
- Acquire Experimental Samples: With compensation and quadrant gates fixed, acquire data for your experimental samples. Do not readjust gates or compensation between samples.
Troubleshooting Guide: Data Interpretation and Background Noise
Even with controls, data interpretation can be challenging. The table below summarizes common problems, their potential causes, and proven solutions.
Table 1: Troubleshooting Data Interpretation in Annexin V Assays
| Problem | Potential Causes | Recommended Solutions |
|---|---|---|
| High Background in Untreated Control [64] [5] | - Poor cell health before assay- Over-trypsinization (using EDTA)- Mechanical damage from harsh pipetting- Over-confluent or starved cells | - Use healthy, log-phase cells.- Use gentle, EDTA-free dissociation enzymes like Accutase.- Treat cells gently during all steps.- Ensure cells are at optimal density. |
| No Positive Signal in Treated Group [64] [5] | - Insufficient drug concentration/duration- Apoptotic cells in supernatant discarded- Reagent degradation or improper storage- Forgot to add dyes | - Include a positive control (e.g., staurosporine).- Always collect and combine supernatant with trypsinized cells.- Verify kit functionality with positive control.- Re-run experiment, confirming all reagents are added. |
| Only Late Apoptosis (No Early Apoptosis) [64] | - Overly intense treatment conditions (high drug conc., extreme conditions)- Rapid cell death (necrosis) | - Gentle treatment of cells; reduce drug concentration.- Limit use of organic solvents (e.g., keep DMSO < 0.5%). |
| Unclear Clustering/Poor Population Separation [64] [5] | - High cellular autofluorescence- Poor instrument compensation- Poor cell state leading to nonspecific staining | - Choose a fluorophore with minimal spectral overlap with autofluorescence (e.g., switch from FITC to PE or APC).- Re-optimize compensation using fresh single-stain controls.- Ensure good cell health and gentle handling. |
| Excessive "Necrotic" (PI-only) Population [7] [5] | - True necrosis due to toxic treatment- Excessive mechanical or enzymatic damage during processing- Cellular debris being counted as events | - Optimize treatment conditions.- Use gentler cell dissociation and pipetting techniques.- Gate out debris based on FSC/SSC characteristics. |
The Scientist's Toolkit: Essential Reagents and Materials
Successful apoptosis detection relies on the correct use of specific reagents. The following table details key materials and their critical functions.
Table 2: Key Research Reagent Solutions for Annexin V Flow Cytometry
| Reagent | Function | Critical Considerations |
|---|---|---|
| Annexin V Conjugate | Binds to exposed phosphatidylserine (PS) on the outer membrane. | Calcium-dependent; avoid buffers with EDTA or other calcium chelators [4]. Choose a fluorochrome (FITC, PE, APC) that avoids cellular autofluorescence and other fluorochromes in the panel [5]. |
| Viability Dye (PI, 7-AAD) | Distinguishes intact and compromised membranes. | PI and 7-AAD must remain in the buffer during acquisition; do not wash after adding them [4]. 7-AAD requires storage at -20°C to prevent degradation [64]. |
| 1X Binding Buffer | Provides the optimal calcium-containing environment for Annexin V binding. | Must be correctly diluted to maintain proper osmotic pressure; abnormal osmotic pressure can induce apoptosis [64]. |
| Fixable Viability Dye (FVD) | Allows for subsequent intracellular staining by providing a permanent viability marker. | FVD eFluor 450 is not recommended for use with Annexin V Apoptosis Detection Kits due to potential spectral overlap [4]. Staining is performed before Annexin V. |
| Annexin V (Unconjugated) | Used in a blocking control to demonstrate staining specificity. | Pre-incubation with unconjugated Annexin V should block subsequent binding of the conjugated Annexin V, confirming signal specificity [24]. |
Advanced Topics: Calculating Apoptotic Indices and Multiparameter Panels
Defining Apoptotic Indices
Once quadrants are correctly set, apoptotic indices can be quantified as follows:
- % Early Apoptosis: (Number of cells in Annexin V+ / Viability Dye- quadrant) / (Total cell count) × 100
- % Late Apoptosis: (Number of cells in Annexin V+ / Viability Dye+ quadrant) / (Total cell count) × 100
- % Total Apoptosis: % Early Apoptosis + % Late Apoptosis
For treatment studies, the basal level of apoptosis in the untreated control should be subtracted from the treated groups to determine the induced apoptosis [24].
Integrating Annexin V into Larger Multicolor Panels
When incorporating Annexin V into complex immunophenotyping panels, careful planning is required to manage fluorescence spillover. Spectral flow cytometry offers advantages here by using the full emission spectrum to better distinguish fluorophores with overlapping signals, such as PerCP and PerCP-eFluor 710 [29]. Key considerations include:
- Fluorophore Selection: Avoid using Annexin V-FITC if your cells express GFP. Use Annexin V conjugated to PE, APC, or Alexa Fluor 647 instead [5].
- Staining Order: For surface staining plus Annexin V and intracellular staining, the recommended order is: 1) Stain surface antigens, 2) Stain with Fixable Viability Dye, 3) Stain with Annexin V, 4) Fix and permeabilize, 5) Stain intracellular antigens [4].
- Viability Staining is Critical: In complex panels, dead cells are a major source of non-specific antibody binding. Using a viability dye is essential for clean data and accurate unmixing in spectral cytometry [65].
Frequently Asked Questions (FAQs)
Q1: My unstained control already shows a signal in the Annexin V channel. What should I do? A1: This is often due to cellular autofluorescence. First, ensure your flow cytometer was thoroughly cleaned. If the problem persists, it could be due to poor cell health, contamination, or interference from fluorescent substances (e.g., doxorubicin, transfected fluorescent plasmids). Replace the cells with a healthy culture or consider switching to an Annexin V conjugate with a longer wavelength (e.g., APC instead of FITC) to move away from the autofluorescence spectrum [64].
Q2: Are Annexin V kits species-specific? A2: No. Annexin V binds to phosphatidylserine (PS), which is a phospholipid conserved across mammalian species. Therefore, the kits are generally not species-dependent [5].
Q3: How does trypsinization affect Annexin V staining, and how can I mitigate this? A3: Trypsin that contains EDTA is problematic because it chelates calcium ions, which are essential for Annexin V binding to PS. This can lead to weak or false-negative results. To mitigate this, use gentle, EDTA-free dissociation enzymes like Accutase, and ensure cells are washed thoroughly with PBS or binding buffer after trypsinization to remove the EDTA [7] [5].
Q4: What is the purpose of an FMO (Fluorescence Minus One) control for Annexin V? A5: While single-stain controls are used for compensation, an FMO control (containing all antibodies in a multicolor panel except Annexin V) is invaluable for confirming the placement of the Annexin V-positive gate, especially when dealing with dim populations or high background. It helps distinguish true positive staining from spillover spreading or autofluorescence from other markers in the panel [7].
Q5: Why is it crucial to analyze cells immediately after staining? A6: Cell viability decreases over time when cells are left in staining buffer, especially in the presence of PI or 7-AAD. This can lead to a time-dependent increase in the late apoptotic/necrotic population. For consistent and reliable results, analyze the samples by flow cytometry within 1 hour of staining [4] [24].
FAQs: Core Principles and Method Selection
Q1: What are the fundamental biological processes detected by Annexin V/PI, TUNEL, and Caspase assays?
The three assays detect distinct biochemical events that occur at different stages of the apoptotic process:
- Annexin V/PI: Detects the loss of plasma membrane asymmetry. Annexin V binds to phosphatidylserine (PS), a phospholipid that translocates from the inner to the outer leaflet of the plasma membrane during early apoptosis. Propidium Iodide (PI) assesses membrane integrity, staining cells with compromised membranes (late apoptotic and necrotic cells) [66] [8].
- TUNEL (TdT-mediated dUTP Nick-End Labeling): Detects DNA fragmentation, a hallmark of late apoptosis. The assay labels the 3'-hydroxyl termini of DNA breaks with modified nucleotides using the enzyme Terminal Deoxynucleotidyl Transferase (TdT) [67] [68].
- Caspase Assays: Detect the activation of caspases, a family of cysteine proteases that are central effectors of apoptosis. These assays typically measure the cleavage of specific synthetic substrates by active caspase enzymes.
Q2: How do I choose the right assay for my apoptosis research question?
Your choice depends on the apoptotic stage you wish to investigate and your experimental model.
- Early Apoptosis: Use Annexin V/PI to identify cells that are still viable but have initiated apoptosis (Annexin V positive, PI negative) [66].
- Late Apoptosis: Both TUNEL and Annexin V/PI (Annexin V positive, PI positive) are suitable. TUNEL is particularly specific for the late-stage DNA cleavage event [67] [66].
- Apoptotic Pathway Activation: Use Caspase Assays to confirm the involvement of the core apoptotic machinery and to study signaling pathways [8].
- Multiparametric Analysis: Annexin V/PI is easily combined with antibody staining for other markers (e.g., cell surface proteins) in flow cytometry, allowing for complex immunophenotyping of apoptotic cells [8].
Troubleshooting Guide: Annexin V/PI Staining
Q1: I am observing a high background or false-positive signals in my Annexin V/PI flow cytometry data. What could be the cause? [69] [11] [7]
High background can stem from various sources related to sample preparation, instrument settings, and reagents.
- Poor Cell Status: Using unhealthy, stressed, or contaminated cells can lead to spontaneous apoptosis and necrosis, increasing background. Ensure cells are healthy and handled gently during harvesting (e.g., using non-enzymatic detachment for adherent cells) [69] [66].
- Rough Handling: Excessive digestion with trypsin or vigorous pipetting can damage the plasma membrane, causing false PI positivity and nonspecific Annexin V binding [69].
- Improper Reagent Use: Incorrect dilution of the Binding Buffer, leading to abnormal osmotic pressure, can induce apoptosis. Always follow the manufacturer's instructions for buffer preparation [69].
- Instrument and Reagent Background:
- Uncleaned Flow Cytometer: Residual fluorescent material from previous runs can contaminate your sample. Thoroughly clean the fluidics system [69].
- Cellular Autofluorescence: Some cell types or cells treated with certain compounds (e.g., doxorubicin) can autofluoresce. Using a fluorescent-minus-one (FMO) control helps define positive and negative populations accurately [11] [7].
- Dead Cells: Dead cells are "sticky" and bind antibodies and reagents nonspecifically. Include a viability dye in multicolor panels to identify and gate out these cells [11].
Q2: My Annexin V/PI experiment shows a lack of early apoptotic cells. What might be wrong? [69]
- Overly Harsh Treatment: If your apoptosis-inducing conditions are too intense (e.g., very high drug concentration, extreme physical conditions), cells may undergo rapid necrosis, bypassing the characteristic early apoptotic stage with PS externalization. Optimize treatment conditions to be gentler [69].
- Loss of Early Apoptotic Cells: For adherent cells, early apoptotic cells are often less adherent and may be lost in the culture supernatant. Ensure you collect and combine both the supernatant and the trypsinized cells when harvesting [69].
The following workflow outlines a systematic approach to diagnose and resolve common background issues in Annexin V/PI experiments:
Troubleshooting Guide: TUNEL Assay
Q1: I see weak or no fluorescent signal in my TUNEL staining, even though I expect apoptosis. How can I fix this? [67] [68] [70]
Weak signal often relates to problems with sample preparation or reagent activity.
- Inadequate Permeabilization: If the cell or nuclear membrane is not sufficiently permeabilized, the TdT enzyme and labeled dUTP cannot access the fragmented DNA. Optimize the concentration and incubation time of Proteinase K (e.g., 10-30 minutes at 20 μg/mL) [67] [68].
- Reagent Inactivation: The TdT enzyme is sensitive. Ensure it is stored properly and not expired. Prepare the TUNEL reaction mixture immediately before use and keep it on ice [68] [70].
- Improper Fixation: Using ethanol or methanol as a fixative can lead to inefficient labeling. Fix samples with 4% paraformaldehyde for an appropriate duration (e.g., 25 minutes at 4°C) [70].
- Fluorescence Quenching: Fluorochromes are light-sensitive. Perform all incubation and washing steps in the dark and observe samples promptly after staining [70].
Q2: My TUNEL assay has a high false-positive rate or strong background staining. What are the solutions? [67] [68] [70]
Nonspecific staining is frequently caused by excessive enzymatic reactions or endogenous activities.
- Over-digestion or Over-fixation: Excessive Proteinase K treatment or very long fixation times can physically damage DNA, creating breaks that are labeled nonspecifically. Follow recommended times for fixation and Proteinase K digestion precisely [68] [70].
- Prolonged TUNEL Reaction: Incubating with the TdT enzyme and labeled dUTP for too long increases background. A standard incubation is 60 minutes at 37°C [68] [70].
- Endogenous Enzyme Activity: Some tissues (e.g., smooth muscle) have high nuclease activity. Fix tissues immediately after collection to halt this activity [70].
- Insufficient Washing: Residual unbound reagent can cause high background. Increase the number of PBS washes after the TUNEL reaction (e.g., up to 5 times) [68].
The following diagram summarizes the key steps in a TUNEL assay protocol and highlights critical steps that commonly require optimization to avoid pitfalls:
Table 1: Quantitative Comparison of Apoptosis Detection Methods
| Feature | Annexin V/PI Staining | TUNEL Assay | Caspase Assays |
|---|---|---|---|
| Parameter Detected | Phosphatidylserine externalization & membrane integrity [66] [8] | DNA fragmentation (3'-OH ends) [67] [68] | Caspase enzyme activity [8] |
| Stage of Detection | Early (Annexin V+/PI-) to Late Apoptosis/Necrosis (Annexin V+/PI+) [66] | Late apoptosis [67] | Early execution phase |
| Typical Assay Time | ~1.5 hours (including staining and incubation) [66] | ~3 hours (excluding fixation and permeabilization) [68] | 1 - 4 hours (varies by kit) |
| Key Technical Considerations | Requires calcium; live cells needed for staining; gentle handling critical [69] [66] | Requires fixation/permeabilization; optimization of TdT enzyme concentration and reaction time is key [67] [68] | Can be performed on cell lysates (activity) or via staining (activation); specific to caspase family |
| Common Background Issues | Cell autofluorescence; dead cells; improper buffer; instrument debris [69] [11] | Incomplete washing; over-digestion with Proteinase K; over-fixation [68] [70] | Non-specific substrate cleavage; autofluorescence in plate readers |
Table 2: Research Reagent Solutions
| Reagent | Function in Apoptosis Detection | Key Considerations |
|---|---|---|
| Annexin V (FITC, PE conjugates) | Binds to externalized phosphatidylserine on apoptotic cells. Fluorescent conjugate allows detection by flow cytometry or microscopy [66]. | Calcium-dependent binding. Must be protected from light. Titration is recommended for optimal signal-to-noise ratio. |
| Propidium Iodide (PI) | DNA intercalating dye that stains cells with compromised plasma membranes (late apoptotic/necrotic). Impermeant to live cells [66] [8]. | Distinguishes late apoptotic/necrotic cells from early apoptotic. RNAse treatment may be needed for specific DNA staining. |
| 7-AAD / DAPI | Alternative viability dyes to PI. 7-AAD is used similarly in flow cytometry. DAPI stains DNA and is used for microscopy [69] [7]. | Check compatibility with instrument lasers and filters. Note storage conditions (e.g., 7-AAD at -20°C) [69]. |
| TdT Enzyme | Terminal deoxynucleotidyl transferase. Catalyzes the addition of labeled dUTP to the 3'-OH ends of fragmented DNA in the TUNEL assay [67] [68]. | Sensitive to improper storage and handling. Aliquot and avoid freeze-thaw cycles to prevent inactivation. |
| Proteinase K | Proteolytic enzyme used to permeabilize cells and nuclear membranes in TUNEL and other intracellular staining protocols [67] [68]. | Concentration and incubation time are critical. Too little leads to weak signal; too much causes false positives and morphology damage. |
| Binding Buffer | Provides the optimal calcium-containing environment for Annexin V to bind to phosphatidylserine [66]. | Must be correctly diluted to maintain proper osmotic pressure, or it can induce apoptosis [69]. |
In Annexin V-based flow cytometry assays, accurately determining sensitivity, specificity, and dynamic range is fundamental to obtaining reliable data on apoptotic cell death. These metrics define the assay's ability to correctly identify true apoptotic events (sensitivity), exclude non-apoptotic events (specificity), and detect changes across a meaningful quantitative range (dynamic range). However, numerous technical and biological factors can compromise these parameters, leading to increased background noise and reduced data quality. Understanding how to measure and optimize these metrics is therefore critical for researchers, scientists, and drug development professionals who rely on accurate apoptosis quantification for their work in cellular biology and therapeutic screening.
This guide addresses the most common challenges affecting assay performance in Annexin V flow cytometry, providing targeted troubleshooting FAQs and detailed protocols to help researchers achieve publication-quality results.
Troubleshooting FAQs: Addressing Common Performance Issues
Q1: My unstained or negative control shows high fluorescence. How does this impact sensitivity and specificity, and how can I resolve it?
- Impact: High background in negative controls directly reduces assay sensitivity (the ability to detect true positive apoptotic cells) by shrinking the separation between negative and positive populations. It can also lower specificity by increasing false positive rates [16] [71].
- Causes and Solutions:
- Cellular Autofluorescence: Some cell types are intrinsically fluorescent. Solution: Use fluorophores that emit in the red channel (e.g., PE, APC) instead of FITC, as autofluorescence is typically higher at shorter wavelengths [5] [16] [72].
- Poor Cell Health: Over-confluent cultures, serum starvation, or mechanical damage during handling can cause spontaneous apoptosis and PS exposure. Solution: Use healthy, log-phase cells and gentle handling; avoid over-trypsinization [5] [71].
- Fluorescent Compound Interference: Certain drugs (e.g., doxorubicin) or cells transfected with fluorescent proteins can contribute to background. Solution: Change to a fluorophore with non-overlapping emission spectra [5] [71].
- Inadequate Washing or Blocking: Unbound antibody or non-specific binding can increase background. Solution: Include an Fc receptor blocking step and ensure sufficient wash steps [16] [73].
Q2: I observe a low signal in my treated sample with no clear positive population. What could be affecting the sensitivity and dynamic range of my assay?
- Impact: This suggests insufficient sensitivity to detect the induced apoptotic response and a compressed effective dynamic range.
- Causes and Solutions:
- Insufficient Apoptosis Induction: Drug concentration or treatment duration may be too low. Solution: Perform a time-course and dose-response experiment to establish optimal treatment conditions [5].
- Loss of Apoptotic Cells: During sample preparation, early apoptotic cells, which are often fragile or detached, can be lost during washing steps. Solution: Always collect the culture supernatant when harvesting adherent cells and avoid washing cells after staining [5] [25].
- Reagent Degradation: Fluorophore-conjugated Annexin V is light-sensitive and can degrade if stored improperly. Solution: Protect all reagents from light during staining and storage, and use a positive control (e.g., cells treated with a known apoptosis inducer) to verify kit functionality [5] [73].
Q3: My compensation controls are poorly resolved, leading to spreading error. How does this affect specificity and how can I fix it?
- Impact: Improper compensation causes fluorescence spillover, which can misclassify cell populations (e.g., early apoptotic cells appearing as late apoptotic, or vice versa), severely compromising the specificity of the assay [5] [72].
- Solutions:
- Use Proper Controls: Generate single-stain controls using cells or compensation beads stained separately with Annexin V-FITC and PI (or other viability dyes). These are essential for the instrument to calculate accurate compensation matrices [5] [73] [8].
- Check Control Brightness: Ensure your single-stained controls are at least as bright as your experimental sample. Using apoptotic cells for these controls can improve compensation accuracy [5] [72].
- Verify with FMO Controls: Fluorescence Minus One (FMO) controls (staining with all fluorophores except one) help set accurate gates and confirm that your compensation is correct, especially for defining negative populations [72] [73].
Quantitative Data on Assay Performance
The table below summarizes key factors that influence the critical performance metrics of your Annexin V assay, based on established protocols and troubleshooting guides.
Table 1: Factors Influencing Key Performance Metrics in Annexin V Flow Cytometry
| Performance Metric | Definition | Key Influencing Factors | Optimal Range/Strategy |
|---|---|---|---|
| Sensitivity | Ability to detect true early apoptotic cells [74] | Cell health and handling [5] [71], Fluorophore brightness and antigen density [72], Apoptosis induction level [5] | Use healthy, log-phase cells; Pair Annexin V with bright fluorophores (e.g., PE, APC); Include supernatant when harvesting [5] |
| Specificity | Ability to distinguish between early apoptotic, late apoptotic, and necrotic cells [25] [50] | Accuracy of fluorescence compensation [5] [72], Purity of cell population (debris exclusion) [16], Specificity of PS binding (Ca²⁺ dependency) [5] | Use single-stain and FMO controls for gating [72] [73]; Use gentle, EDTA-free cell dissociation [5] |
| Dynamic Range | Quantitative range over which apoptosis can be reliably measured | Linearity of fluorescence signal with PS exposure, Instrument detector linearity [16], Resolution between cell populations | Use logarithmic (log) amplification on flow cytometer [16]; Ensure proper PMT voltage settings [16] [73] |
Advanced methodologies have demonstrated that real-time, live-cell imaging assays using Annexin V can achieve a 10-fold increase in sensitivity compared to traditional endpoint flow cytometry, primarily by eliminating the stress and artifacts introduced by sample handling and processing for flow analysis [74].
Essential Protocols for Metric Validation
Protocol 1: Standardized Annexin V/FITC & PI Apoptosis Assay
This protocol is critical for establishing a baseline performance for your assay's sensitivity, specificity, and dynamic range [25] [8].
- Cell Harvesting: For adherent cells, collect the culture supernatant (containing detached cells) and then trypsinize the remaining adherent cells using a gentle, EDTA-free enzyme like Accutase to preserve membrane integrity. Combine the cells from the supernatant and the trypsinized fraction. Centrifuge and wash cell pellet with PBS [5] [25] [8].
- Staining: Re-suspend ~2 x 10⁵ cells in 100 µL of Annexin V Binding Buffer (containing 1.8-2.0 mM CaCl₂). Add Annexin V-FITC and Propidium Iodide (PI) according to kit instructions. A typical concentration for Annexin V-FITC is 1 µg/mL, and for PI is 1 µg/mL [25] [8].
- Incubation: Incubate for 15 minutes at room temperature in the dark.
- Analysis: Without washing, analyze the cells by flow cytometry within 1 hour. Use the following gating strategy to define populations and calculate metrics:
- Viable Cells: Annexin V-FITC negative, PI negative.
- Early Apoptotic (Sensitivity Focus): Annexin V-FITC positive, PI negative.
- Late Apoptotic/Necrotic (Specificity Focus): Annexin V-FITC positive, PI positive.
Protocol 2: Multiparametric Analysis for Enhanced Specificity
This extended protocol allows for the simultaneous analysis of apoptosis and cell surface marker expression, providing a more specific assessment of apoptosis within defined subpopulations [50] [8].
- Induction and Harvest: Induce apoptosis in your cell model (e.g., treat MDA-MB-231 cells with 1 µM doxorubicin for 48 hours). Harvest cells as in Protocol 1, using PBS with 25 mM CaCl₂ for washing [8].
- Annexin V/PI Staining: Stain cells with Annexin V-FITC and PI as described in Protocol 1.
- Surface Marker Staining: After Annexin V/PI staining, wash cells once with PBS. Re-suspend cell pellet in a blocking solution (e.g., PBS with 5% BSA) for 10-15 minutes. Incubate with an APC-conjugated antibody against your surface marker of interest (e.g., anti-CD44-APC) for 30 minutes in the dark [8].
- Fixation: Fix cells with 80% methanol for 10 minutes on ice if subsequent intracellular staining is required. For surface staining only, proceed to the next step without fixation.
- Analysis: Analyze by flow cytometry. First, gate on your viable, early, and late apoptotic populations based on Annexin V and PI signals. Then, within each apoptotic gate, analyze the expression level of the APC-conjugated marker (e.g., CD44). This allows tracking of protein expression changes specifically in apoptotic cells [50] [8].
Visualizing the Troubleshooting Workflow
The following diagram outlines a logical, step-by-step process for diagnosing and resolving common issues that affect the performance metrics of your Annexin V assay.
The Scientist's Toolkit: Essential Research Reagents
The table below lists key reagents and materials required for a robust Annexin V apoptosis assay, along with their critical functions and technical notes.
Table 2: Essential Reagents for Annexin V-Based Apoptosis Detection
| Reagent/Material | Function | Technical Notes |
|---|---|---|
| Annexin V, Fluorophore-conjugated | Binds externally exposed phosphatidylserine (PS) on apoptotic cells [5] [25] | Not species-specific [5]. Light-sensitive; avoid exposure to light during use and storage [5]. |
| Viability Dye (PI, 7-AAD, DRAQ7, YOYO3) | Distinguishes late apoptotic/necrotic cells (membrane permeable) from early apoptotic cells (membrane impermeable) [5] [74] | PI is common but toxic for live-cell imaging. DRAQ7/YOYO3 are alternatives for kinetic assays [74]. |
| Calcium-Containing Binding Buffer | Provides Ca²⁺ ions essential for Annexin V-PS binding [5] [25] | EDTA in trypsin or other buffers chelates Ca²⁺ and must be washed away thoroughly [5] [73]. |
| Gentle Cell Dissociation Reagent | Detaches adherent cells without causing excessive membrane damage that leads to false positives [5] [71] | EDTA-free formulations (e.g., Accutase) are recommended over trypsin/EDTA [5]. |
| Compensation Controls | Allow for correction of spectral overlap between fluorophores [5] [72] [73] | Use single-stain controls (cells or beads) for each fluorophore. FMO controls aid in accurate gating [72] [73]. |
| Flow Cytometer with Appropriate Lasers/Filters | Instrument for detecting and quantifying fluorescence from stained cells. | Must be equipped with lasers and filters matching your chosen fluorophores (e.g., 488 nm laser for FITC and PI) [50] [8]. |
Visualizing Signal Specificity and Spillover
A fundamental understanding of how signals are meant to be specific and where interference occurs is key to optimizing specificity. The following diagram illustrates the principle of specific detection and a common source of noise.
Accurate detection of apoptosis is fundamental in biomedical research, particularly in drug development and cancer studies. The Annexin V flow cytometry assay is a powerful tool for this purpose, but its reliability can be compromised by background noise and artifacts. This guide details how to use microscopy as an orthogonal technique to validate your flow cytometry data, confirm the biological relevance of your findings, and troubleshoot common issues related to background signals.
Frequently Asked Questions (FAQs)
Q1: Why is my unstained control showing high background fluorescence in the Annexin V channel? High background in unstained samples can stem from several sources. Cell autofluorescence is a common cause, especially in metabolically active cells or those treated with certain drugs [5]. Cellular debris from poor sample preparation or excessive manipulation can also contribute to this noise [5]. To resolve this, ensure you are using healthy, log-phase cells and avoid over-trypsinization during harvesting. Using gentle, EDTA-free dissociation enzymes like Accutase can help preserve membrane integrity [5].
Q2: My flow data suggests apoptosis, but my cells look healthy under the microscope. What could be wrong? This discrepancy often points to a technical artifact in your flow setup rather than a true biological signal. The most common culprit is improper fluorescence compensation, where signal from one fluorochrome spills over into another detector, creating a false-positive population [5] [75]. First, re-check your compensation using single-stain controls. Then, use microscopy to visually confirm the presence of apoptotic hallmarks, such as cell shrinkage and membrane blebbing, in the suspected population [76].
Q3: How can I use microscopy to specifically validate Annexin V binding? Microscopy allows for direct visual correlation between Annexin V staining and cellular morphology. Using fluorescent microscopy, you can confirm that the green (or red) Annexin V signal is localized to the outer membrane of cells that also display classic features of early apoptosis, like membrane blebbing. The table below outlines key morphological features to confirm:
Table: Morphological Hallmarks of Cell Death for Microscopic Validation
| Cell Death Stage | Key Morphological Features | Annexin V Staining | Membrane Integrity |
|---|---|---|---|
| Viable Cell | Normal size, intact structure | Negative | Intact |
| Early Apoptosis | Cell shrinkage, membrane blebbing, chromatin condensation | Positive | Intact |
| Late Apoptosis | Further shrinkage, nuclear fragmentation, apoptotic bodies | Positive | Compromised |
| Necrosis | Cell swelling, loss of membrane integrity | May be negative or positive | Lost |
Q4: What are the best positive and negative controls for these experiments? Always include both controls to benchmark your assay.
- Positive Control: Treat a sample of your cells with a known apoptosis inducer, such as staurosporine (e.g., 0.5-1 µM for 2-6 hours) or hydrogen peroxide [77]. This validates that your reagents are working and shows what a true positive signal looks like in both flow and microscopy.
- Negative Control: Use healthy, untreated cells to establish the baseline for autofluorescence and non-specific binding [77].
Troubleshooting Guides
Problem: High Background Noise Obscuring Cell Populations
Potential Causes and Solutions:
Excessive Cellular Debris:
- Cause: Over-vigorous pipetting, using dead cells, or improper cell handling creates small particles that scatter light and fluoresce [5].
- Solution: Be gentle when handling cells. Use a pre-filter or strainer before loading the sample onto the flow cytometer. On the instrument, you can apply a threshold on the Forward Scatter (FSC) parameter to ignore signals from small particles [19]. Be cautious, as setting the threshold too high may exclude small apoptotic cells or debris that could contaminate sorted samples [19].
Sample Processing Errors:
- Cause: Inadequate washing of cells after staining can leave unbound Annexin V dye in solution, increasing background [5]. However, note that some commercial kits are "no-wash" and this step is not required.
- Solution: Follow the recommended protocol for your kit. If washing is specified, ensure it is performed thoroughly but gently. Analyze samples promptly (within 1 hour) after staining, as delays can lead to increased background and false positives [5] [77].
Problem: Discrepancy Between Flow Cytometry and Microscopy Data
Validation Protocol: Correlative Analysis using Flow Cytometry and Microscopy
This protocol helps you directly link flow cytometry data with morphological observation.
Experimental Workflow for Correlative Analysis
Materials Needed:
- Cells (e.g., MDA-MB-231 or your cell line of interest)
- Annexin V-FITC Apoptosis Detection Kit [50]
- Propidium Iodide (PI) stock solution (50 µg/mL) [77]
- Binding Buffer (with calcium) [77]
- Apoptosis inducer (e.g., 1 µM Doxorubicin or 1 µM Staurosporine) [50]
- Flow cytometer
- Fluorescence microscope
Step-by-Step Method:
- Cell Preparation and Treatment: Seed your cells and treat them with the apoptotic inducer for a predetermined time (e.g., 16-24 hours for doxorubicin). Include an untreated control.
- Cell Harvesting: For adherent cells, detach them gently using a non-enzymatic dissociation buffer (e.g., EDTA-free Accutase) to preserve phosphatidylserine (PS) on the membrane [5]. Avoid using trypsin-EDTA, as EDTA chelates calcium, which is essential for Annexin V binding [5].
- Staining: Wash the cells and resuspend them in binding buffer. Aliquot 1 x 10^5 cells per tube. Add Annexin V-FITC and PI according to your kit's instructions. Incubate for 15 minutes at room temperature in the dark [77].
- Parallel Analysis:
- Flow Cytometry: Analyze one aliquot of your stained sample on the flow cytometer. Use unstained and single-stained controls to set up compensation and gating [5] [75]. Identify the populations of viable, early apoptotic, and late apoptotic cells.
- Microscopy: Immediately after flow analysis, take another aliquot of the same stained sample and visualize it under a fluorescence microscope. Use FITC and TRITC/Rhodamine filter sets to observe Annexin V (green) and PI (red) signals, respectively. Capture images of multiple fields.
- Data Correlation: Compare the percentage of cells in each quadrant from your flow cytometry plot with the proportion of cells showing corresponding staining and morphology under the microscope. This confirms that the "Annexin V+/PI-" population from flow genuinely consists of cells with membrane blebbing and intact membranes, as seen visually.
The Scientist's Toolkit: Key Research Reagent Solutions
Table: Essential Reagents for Annexin V Assay Validation
| Reagent/Material | Function in Experiment | Key Considerations |
|---|---|---|
| Annexin V (FITC, PE, APC) | Binds externalized PS on apoptotic cells. | Avoid FITC if cells express GFP; choose PE or APC instead [5]. |
| Viability Dye (PI, 7-AAD) | Distinguishes late apoptotic/necrotic cells with compromised membranes. | PI is common; 7-AAD can be used with a violet laser [5]. |
| Calcium-Containing Binding Buffer | Provides necessary co-factor for Annexin V-PS binding. | Check buffer composition; absence of Ca²⁺ will cause assay failure [5] [77]. |
| Gentle Dissociation Agent (Accutase) | Detaches adherent cells without damaging membrane PS. | Prefer over trypsin-EDTA to prevent false negatives [5]. |
| Apoptosis Inducer (Staurosporine) | Provides a reliable positive control for the assay. | Use to validate kit performance and experimental conditions [77]. |
Advanced Techniques: Real-Time Assays and Pathway Mapping
For deeper investigation, consider real-time bioluminescent Annexin V assays, which use Annexin V fusion proteins with luciferase subunits for kinetic monitoring of PS exposure without wash steps [78]. Understanding the core apoptosis pathway helps in interpreting validation data.
Core Apoptosis Pathway and Key Markers
By systematically applying these correlative techniques, you can confidently distinguish true apoptosis from background noise and technical artifacts, ensuring the integrity of your data in critical research and drug development applications.
Conclusion
Effective troubleshooting of background noise in Annexin V flow cytometry requires a holistic approach that integrates foundational knowledge, optimized methodologies, systematic problem-solving, and rigorous validation. By understanding the core principles of phosphatidylserine binding and major noise sources like autofluorescence and spectral overlap, researchers can proactively design experiments to minimize interference. Adherence to optimized protocols for sample handling and staining is critical for preserving data integrity. When issues arise, a structured troubleshooting workflow enables precise identification and resolution of problems. Finally, comprehensive validation through proper controls and method comparison ensures the reliability and reproducibility of apoptosis data, which is fundamental for advancing drug discovery and clinical diagnostics. Future directions will likely involve the development of novel fluorochromes with reduced spectral overlap and automated analysis tools for enhanced noise discrimination in complex samples.
References
- 1. Annexin V Staining | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/cell-analysis/cell-viability-and-regulation/apoptosis/annexin-v-staining.html]
- 2. Phosphatidylserine exposure during apoptosis reflects ... [https://www.nature.com/articles/cdd201293]
- 3. Annexin V, the regulator of phosphatidylserine-catalyzed ... [https://pubmed.ncbi.nlm.nih.gov/9230931/]
- 4. Annexin V Staining Protocol for Flow Cytometry [https://www.thermofisher.com/us/en/home/references/protocols/cell-and-tissue-analysis/protocols/annexin-v-staining-flow-cytometry.html]
- 5. Common Problems and Solutions in Annexin V-Based Flow ... [https://www.yeasenbio.com/blogs/cell/common-problems-and-solutions-in-annexin-v-based-flow-cytometry-apoptosis-assays]
- 6. Analysis and Solution of Common Problems in Annexin V ... [https://www.elabscience.com/resources/cell-function-guidelines-and-q-a/981?srsltid=AfmBOoq6Ub6ovR1lWDLnI9c_8CuMRu0qfo-Lmcax9jiCVwisXF9U6-tW]
- 7. Flow Cytometry Troubleshooting Guide [https://www.bio-techne.com/applications/flow-cytometry/flow-cytometry-troubleshooting-guide]
- 8. Flow cytometry-based quantitative analysis of cellular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11260931/]
- 9. Modified Annexin V/Propidium Iodide Apoptosis Assay For ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3169266/]
- 10. Noise | Cytometry [https://cytometry.mlsascp.com/noise.html]
- 11. Flow Cytometry (FACS) Recommendations for Background ... [https://www.sinobiological.com/category/fcm-facs-background-control]
- 12. Principles of Advanced Flow Cytometry: A Practical Guide [https://pmc.ncbi.nlm.nih.gov/articles/PMC10802916/]
- 13. Analysis and Solution of Common Problems in Annexin V ... [https://www.elabscience.com/resources/cell-function-guidelines-and-q-a/981?srsltid=AfmBOopK0mainYAoOkV0DA2VHoYjSgdO4lD6bLE4J19z5XwUm_WlgZNV]
- 14. Analysis and Solution of Common Problems in Annexin V ... [https://www.elabscience.com/resources/cell-function-guidelines-and-q-a/981?srsltid=AfmBOopOg7vACM9RDESnLndOtHqia7JCmZbFvhrPxtmWJdBuuGpD-GKD]
- 15. Flow Cytometry Troubleshooting Guide [https://www.cellsignal.com/learn-and-support/troubleshooting/flow-cytometry-troubleshooting-guide?srsltid=AfmBOopOIEGQVY8GxZULWDV8uCt_5tPzQXLluyhUjX7nhAbcLXk4CP83]
- 16. Flow Cytometry Troubleshooting Guide [https://fluorofinder.com/flow-cytometry-troubleshooting/]
- 17. Cell Dissociation Enzymes Affect Annexin V/Flow- ... [https://ar.iiarjournals.org/content/42/6/2819]
- 18. Annexin V PI Staining Guide for Apoptosis Detection [https://www.bosterbio.com/blog/post/methods/flow-cytometry-with-annexin-v-pi-staining?srsltid=AfmBOoqJeuVgNUJsH0piwMT3Zk0x2zFBWLJjdjrnXqpaKuDiZ0Uflt9p]
- 19. How To Use A Threshold To Reduce Background Noise In ... [https://expertcytometry.com/use-threshold-to-reduce-background-noise-in-flow-cytometry/]
- 20. Overview of Flow Cytometry [https://www.cellsignal.com/applications/flow-cytometry/flow-cytometry-overview?srsltid=AfmBOooYtPenBjqhVUerBuodTnNi_xSZOFGKM9NPz08TIkobT_F7mVVZ]
- 21. Analysis and Solution of Common Problems in Annexin V ... [https://www.elabscience.com/resources/cell-function-guidelines-and-q-a/981?srsltid=AfmBOophA_K1HetQBHgklBA9v5qFGuFt8aJtZUgoRXQI611wLnNQQwgc]
- 22. Annexin V PI Staining Guide for Apoptosis Detection [https://www.bosterbio.com/blog/post/methods/flow-cytometry-with-annexin-v-pi-staining?srsltid=AfmBOoq7--f0DzU5e2Io-dB4Kqfi1sGXZ_79ZAQtp2W4D-SEzc8jAWJN]
- 23. Support— Flow Cytometry Troubleshooting [https://www.thermofisher.com/de/de/home/technical-resources/technical-reference-library/cell-analysis-support-center/flow-cytometry-support/flow-cytometry-support-troubleshooting.html]
- 24. Annexin V Staining Protocol [https://www.bdbiosciences.com/en-us/resources/protocols/annexin-v-staining-protocol]
- 25. Protocol for Apoptosis Assay by Flow Cytometry Using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4943750/]
- 26. Domain IV of Annexin A5 Is Critical for Binding Calcium and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6149819/]
- 27. PI Staining Guide for Apoptosis Detection | Boster Bio Annexin V [https://www.bosterbio.com/blog/post/methods/flow-cytometry-with-annexin-v-pi-staining]
- 28. Spectral Overlap | Cytometry [https://cytometry.mlsascp.com/spectral-overlap.html]
- 29. A Guide to Spectral Flow Cytometry Fluorophore Selection [https://www.thermofisher.com/us/en/home/life-science/cell-analysis/flow-cytometry/flow-cytometry-learning-center/flow-cytometry-resource-library/flow-cytometry-methods/spectral-flow-cytometry-fluorophore-selection.html]
- 30. Flow Cytometry Troubleshooting Guide [https://www.cellsignal.com/learn-and-support/troubleshooting/flow-cytometry-troubleshooting-guide?srsltid=AfmBOoq-sRBOMJcos3jaB1CDOgJ8pVJfW0YtkjD-1daS0ewXP5UyCwQ4]
- 31. Annexin V PI Staining Guide for Apoptosis Detection [https://www.bosterbio.com/blog/post/methods/flow-cytometry-with-annexin-v-pi-staining?srsltid=AfmBOoqFvZw-wn2gBQCDUBVDdhoqoh2IrWn9yyZ-j3oKkm-fqMm2naVw]
- 32. Analysis and Solution of Common Problems in Annexin V ... [https://www.elabscience.com/resources/cell-function-guidelines-and-q-a/981?srsltid=AfmBOopYy7uNx4cGXfCFNV7vv1TDtlYT_cAalIFtwddZEGRNS-D-11fC]
- 33. Annexin V PI Staining Guide for Apoptosis Detection [https://www.bosterbio.com/blog/post/methods/flow-cytometry-with-annexin-v-pi-staining?srsltid=AfmBOoruu8wDPXCeGl6QPtcH6zwMyXumDRUgK4yY8Vb0S3rskNNB3ZWe]
- 34. Annexin V staining assay protocol for apoptosis [https://www.abcam.com/en-us/technical-resources/protocols/annexin-v-for-apoptosis]
- 35. Annexin V and PI Staining for Apoptosis by Flow Cytometry [https://www.bio-techne.com/resources/protocols-troubleshooting/apoptosis-flow-annexin-v-protocol]
- 36. Analysis and Solution of Common Problems in Annexin V ... [https://www.elabscience.com/resources/cell-function-guidelines-and-q-a/981?srsltid=AfmBOorII_h0aSeFrMF3kEcSesz04oYk6l0xYjZPqyV9OG-nVlX2Mn3h]
- 37. Troubleshooting Flow Cytometry Issues [https://www.bosterbio.com/protocol-and-troubleshooting/flow-cytometry-troubleshooting?srsltid=AfmBOorYCi7ot2iU4VnM06jW5tv2RBca1VCDPVwNq84wXLex_wlC0ocH]
- 38. Flow cytometry troubleshooting [https://www.abcam.com/en-us/technical-resources/applications/flow-cytometry/troubleshooting]
- 39. Flow Cytometry Staining Protocols for High-Quality Data [https://www.technologynetworks.com/cell-science/infographics/flow-cytometry-staining-protocols-for-high-quality-data-404722]
- 40. Analysis and Solution of Common Problems in Annexin V ... [https://www.elabscience.com/resources/cell-function-guidelines-and-q-a/981?srsltid=AfmBOoqKqbt-N9jIJAjo47TqW1g0WbrTrtUmizB6-rR7zpV7AEOMiolv]
- 41. Annexin V PI Staining Guide for Apoptosis Detection [https://www.bosterbio.com/blog/post/methods/flow-cytometry-with-annexin-v-pi-staining?srsltid=AfmBOooJoWqZXwI5wEQJYf1N4BMCodjmfZDM2BDHAzpzyg9Bxm79czDF]
- 42. Publications Every Flow Cytometrist Should Know About [https://voices.uchicago.edu/ucflow/2022/04/12/best-flow-cytometry-publications/]
- 43. How to Interpret Flow Cytometry Data [https://www.fortislife.com/resources/antibody-resources/how-to-interpret-flow-cytometry-data]
- 44. Flow cytometry diagrams [http://www.assay-protocol.com/cell-biology/flow-cytometry/flowcytometry-diagrams.html]
- 45. Simple Elimination of Background Fluorescence in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5614400/]
- 46. Fixing Autofluorescence in Human Brain Samples [https://bitesizebio.com/26818/bright-minds-overcoming-autofluorescence-in-human-brain-samples/]
- 47. Suppression of Red Blood Cell Autofluorescence for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5657453/]
- 48. Reduce Tissue Autofluorescence And Dramatically ... [https://vectorlabs.com/blog/reduce-tissue-autofluorescence-and-dramatically-enhance-signal-to-noise-ratio/?srsltid=AfmBOopcYXxDmdZwCSz_L9NOlTa0KJKB-psE2DC-fTVBxYEHZP3u6iX7]
- 49. Saline is a more appropriate solution for microvesicles ... [https://www.oncotarget.com/article/15987/text/]
- 50. Flow cytometry-based quantitative analysis of cellular ... [https://www.sciencedirect.com/science/article/pii/S2405844024096968]
- 51. Analysis and Solution of Common Problems in Annexin V ... [https://www.elabscience.com/resources/cell-function-guidelines-and-q-a/981?srsltid=AfmBOoo5utCqPZAT3SM5y_ybxTaOEf4wuZ9Dr053Svtd93FVRF-Ndx92]
- 52. Flow Cytometry Part 3: Enhancing Signal-to-Noise Ratio [https://avantierinc.com/resources/application-note/improving-signal-to-noise-ratio-in-flow-cytometer-optics/]
- 53. Advances in Multiparametric Flow Cytometry and Data ... [https://www.labx.com/resources/advances-in-multiparametric-flow-cytometry-and-data-analysis-software/5522]
- 54. Annexin V PI Staining Guide for Apoptosis Detection [https://www.bosterbio.com/blog/post/methods/flow-cytometry-with-annexin-v-pi-staining?srsltid=AfmBOoo_wpBLiyi39OXd_VLrZgREC0KSLP1XSF1SCxxOhQsdVDeSoPvK]
- 55. Annexin V FAQ [https://www.linkedin.com/pulse/annexin-v-faq-elabscience]
- 56. Precautions for Annexin V Apoptosis Assay [https://www.elabscience.com/resources/cell-function-guidelines-and-q-a/982?srsltid=AfmBOooplZ8FQr-wZnsdWNyJlgJb8tq1jxZBSs9Vb8C7X5evRxBEgHW6]
- 57. Impact of cell harvesting methods on detection ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7798138/]
- 58. Analysis and Solution of Common Problems in Annexin V ... [https://www.elabscience.com/resources/cell-function-guidelines-and-q-a/981?srsltid=AfmBOoqOIzEIalV_hxW4S896IdMOhP3H3JIvTK8Wnb9a_q6Bznb4wkee]
- 59. Annexin V Stain Protocol | Flow Cytometry Core | ECU [https://medicine.ecu.edu/flow-core/annexin-v-stain-protocol/]
- 60. Annexin V PI Staining Guide for Apoptosis Detection [https://www.bosterbio.com/blog/post/methods/flow-cytometry-with-annexin-v-pi-staining?srsltid=AfmBOoppx-cXzPFcwJ-6UB0C_O4U4WBAPXvW-LtY7_Y4TMgAhb88Z7NV]
- 61. PE Annexin V [https://www.bdbiosciences.com/en-us/products/reagents/flow-cytometry-reagents/research-reagents/single-color-antibodies-ruo/pe-annexin-v.560930]
- 62. Annexin V, PE conjugate (R-PE Annexin V), 250 μL - FAQs [https://www.thermofisher.com/order/catalog/product/A35111/faqs]
- 63. Analysis and Solution of Common Problems in Annexin V ... [https://www.elabscience.com/resources/cell-function-guidelines-and-q-a/981?srsltid=AfmBOoquH-_Vx2UhOkg4UhH1adm0dI7fKF3LVQ1DJvgwvhCrmoV2B0N-]
- 64. Analysis and Solution of Common Problems in Annexin V ... [https://www.elabscience.com/resources/cell-function-guidelines-and-q-a/981?srsltid=AfmBOopYhWwNlf2E3wCM02dI8BI4V4zEYmIamsWRf07qyqEYhrbolCjP]
- 65. Spectral Panel Design [https://fluorofinder.com/spectral-panel-design-2/]
- 66. Annexin V PI Staining Guide for Apoptosis Detection [https://www.bosterbio.com/blog/post/methods/flow-cytometry-with-annexin-v-pi-staining?srsltid=AfmBOoqTYl1YfW_YesvtQKiX3JSiiJ2REy0EuBzmrezRGbh9TEDnO7RV]
- 67. How Much Do You Know About TUNEL Staining? A ... - Yeasen [https://www.yeasenbio.com/blogs/cell/how-much-do-you-know-about-tunel-staining-a-troubleshooting-guide-for-abnormal-staining-results]
- 68. Common Problems and Solutions in TUNEL Assay for ... [https://www.arcegen.com/blogs/cell-anti-contamination-assembly/common-problems-and-solutions-in-tunel-assay-for-detecting-cell-apoptosis]
- 69. Analysis and Solution of Common Problems in Annexin V ... [https://www.elabscience.com/resources/cell-function-guidelines-and-q-a/981?srsltid=AfmBOoo5ruvwrcnLgxhYjIr_QyJksMV9tYPfL9kXVaIWEHpsVOiT5OwL]
- 70. Analysis and Solution of Common Problems in TUNEL ... [https://www.elabscience.com/resource-cell_function-detail-988?srsltid=AfmBOooV0Vi2YtZkW-fCNzlLa2sp35i__suUpS7FD4knOUxkABEhHegJ]
- 71. Analysis and Solution of Common Problems in Annexin V ... [https://www.elabscience.com/resources/cell-function-guidelines-and-q-a/981?srsltid=AfmBOoqdcs-JHB8nDZF6M7Izne997mRr6CGH6yaDcGcdoeRtbP3Wmc-k]
- 72. Spectral Flow Cytometry Glossary of Terms [https://www.thermofisher.com/us/en/home/life-science/cell-analysis/flow-cytometry/flow-cytometry-learning-center/flow-cytometry-resource-library/flow-cytometry-methods/spectral-flow-cytometry-glossary.html]
- 73. Flow Cytometry Support—Troubleshooting [https://www.thermofisher.com/us/en/home/technical-resources/technical-reference-library/cell-analysis-support-center/flow-cytometry-support/flow-cytometry-support-troubleshooting.html]
- 74. Robust high-throughput kinetic analysis of apoptosis with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5261025/]
- 75. The 3 Most Common Flow Cytometry Fallacies [https://bitesizebio.com/35460/flow-cytometry-fallacies/]
- 76. Programmed cell death detection methods: a systematic ... [https://link.springer.com/article/10.1007/s10495-022-01735-y]
- 77. Annexin V PI Staining Guide for Apoptosis Detection [https://www.bosterbio.com/blog/post/methods/flow-cytometry-with-annexin-v-pi-staining?srsltid=AfmBOorC3PjNQOu7qtm-HhaJNYvv-8ty1FMQE0rTqnQEMc5_NPUpMGaK]
- 78. A real-time, bioluminescent annexin V assay for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6373262/]