Mitochondrial Membrane Potential: A Central Regulator in Mitophagy and Quality Control Mechanisms
This article comprehensively explores the critical role of mitochondrial membrane potential (ΔΨm) as a primary sensor and regulator in mitochondrial quality control and mitophagy.
Mitochondrial Membrane Potential: A Central Regulator in Mitophagy and Quality Control Mechanisms
Abstract
This article comprehensively explores the critical role of mitochondrial membrane potential (ΔΨm) as a primary sensor and regulator in mitochondrial quality control and mitophagy. Tailored for researchers and drug development professionals, it synthesizes foundational knowledge on how ΔΨm governs the PINK1-Parkin pathway and integrates with mitochondrial dynamics. It further delves into methodological approaches for monitoring ΔΨm, discusses common challenges in interpreting its fluctuations, and validates its significance through comparative analysis in disease models, particularly in neurodegenerative and cardiovascular disorders. The review aims to bridge molecular mechanisms with therapeutic applications, highlighting mitochondrial membrane potential as a promising target for pharmacological intervention.
The Gatekeeper of Health: How Mitochondrial Membrane Potential Governs Quality Control
Mitochondrial Membrane Potential (ΔΨm) as the Energetic Foundation for Cellular Life
The mitochondrial inner membrane potential (ΔΨm) is a fundamental biophysical parameter, generated by the electron transport chain's proton-pumping activity, that forms the cornerstone of cellular energy transduction [1]. This electrical gradient is a critical component of the proton motive force used by ATP synthase to phosphorylate ADP, thereby converting electrochemical energy into chemical energy stored in ATP [1]. However, the functional significance of ΔΨm extends far beyond its classical role in oxidative phosphorylation. Even under hypoxic conditions that preclude ATP synthesis, mitochondria maintain ΔΨm through ATP hydrolysis, underscoring its essential non-energetic functions [1]. These include the import of nuclear-encoded proteins and metal cations, export of anions, regulation of reactive oxygen species (ROS) generation, and—most critically—serving as a key regulator in mitochondrial quality control mechanisms [1]. The membrane potential thus represents a vital physiological index that integrates mitochondrial functional state with cellular homeostasis, making it a focal point for understanding pathogenesis and developing therapeutic interventions for numerous diseases.
Quantitative Foundations: Measuring and Interpreting ΔΨm
Table 1: Key Quantitative Parameters of Mitochondrial Membrane Potential (ΔΨm)
| Parameter | Typical Value/Range | Measurement Context | Biological Significance |
|---|---|---|---|
| Resting ΔΨm Magnitude | ~140-180 mV (negative inside) | Mammalian cells under physiological conditions [1] | Maintains proton motive force for ATP synthesis; drives protein import |
| TMRE Fluorescence Intensity Ratio (KO/WT) | ~1.3-1.5 fold increase | IF1-KO vs. WT HEK293 cells [2] | Indicates chronic hyperpolarization in genetic models |
| ΔΨm Contribution from Glycolytic ATP Hydrolysis | Significant decrease in galactose medium | IF1-KO cells in glucose vs. galactose [2] | Reveals alternative mechanisms of ΔΨm maintenance beyond ETC |
| Ca2+ Clearance Rate | Faster kinetics in hyperpolarized mitochondria | IF1-KO vs. WT permeabilized cells [2] | Demonstrates functional consequence on mitochondrial calcium buffering |
| ATP Hydrolytic Activity | Significantly increased | IF1-KO isolated mitochondria [2] | Confirms loss of inhibitory regulation on ATP synthase reverse activity |
Table 2: Experimental Models of ΔΨm Perturbation and Pathophysiological Correlates
| Model/Context | ΔΨm Alteration | Key Molecular Players | Downstream Consequences |
|---|---|---|---|
| IF1-Knockout Cells | Chronic hyperpolarization [2] | ATP5IF1 (IF1), ATP synthase [2] | Nuclear DNA hypermethylation; phospholipid remodeling; transcriptional reprogramming [2] |
| Parkinson's Disease Models | Loss/depolarization triggers mitophagy [3] [4] | PINK1, Parkin, p62 [3] [4] | Selective autophagic clearance of damaged mitochondria [3] [5] |
| Myocardial Ischemia | Fluctuations in ΔΨm during I/R injury [4] | BNIP3, FUNDC1, Parkin [4] | Mitophagic activation; determines cardiomyocyte fate (survival vs. death) [4] |
| Chemical Exposure | Induced hyperpolarization [2] | Environmental chemicals | Epigenetic modifications mimicking genetic hyperpolarization models [2] |
| Cancer Cells (Glioblastoma, Ovarian) | Elevated resting ΔΨm [2] | Unclear; potentially IF1 depletion [2] | Enhanced proliferation; metabolic adaptation [2] |
Molecular Mechanisms: ΔΨm as Master Regulator of Mitochondrial Quality Control
ΔΨm in Mitophagy: The PINK1-Parkin Signaling Axis
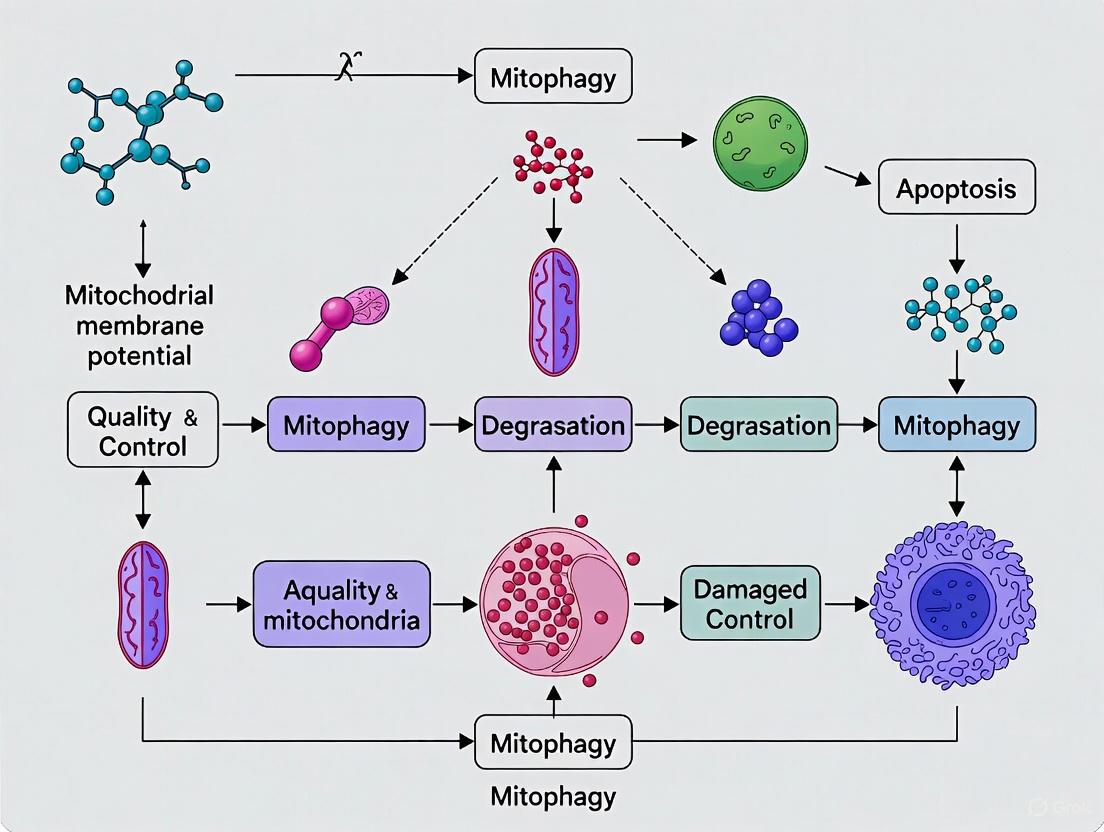
The most characterized pathway linking ΔΨm to mitochondrial quality control is the PINK1-Parkin mediated mitophagy pathway. Under normal conditions with preserved ΔΨm, PTEN-induced putative kinase 1 (PINK1) is continuously imported into mitochondria through the TIM/TOM complex, where it undergoes cleavage by matrix-processing peptidase and PRESENILIN-associated rhomboid-like protein (PARL), followed by proteasomal degradation [4]. However, upon mitochondrial damage and consequent ΔΨm dissipation, PINK1 import is impaired, leading to its accumulation on the outer mitochondrial membrane (OMM) [4] [5]. Here, PINK1 dimerizes and auto-phosphorylates at Ser228 and Ser402, activating its kinase activity [4].
The activated PINK1 then phosphorylates ubiquitin at Ser65 and recruits the E3 ubiquitin ligase Parkin from the cytosol [4]. PINK1-mediated phosphorylation of Parkin's ubiquitin-like domain at Ser65 activates its E3 ligase activity [4]. Activated Parkin then ubiquitinates numerous OMM proteins, including mitofusins (Mfn1/2) and VDAC1, creating phospho-ubiquitin chains that serve as "eat-me" signals for autophagic machinery [3] [4]. These ubiquitin decorations are recognized by autophagy receptors such as p62/SQSTM1, which simultaneously bind to lipidated LC3 (LC3-II) on forming autophagosomal membranes, thereby targeting damaged mitochondria for selective autophagic degradation [3] [4].
Diagram 1: PINK1-Parkin mitophagy pathway triggered by ΔΨm dissipation.
ΔΨm in Mitochondrial Dynamics: Fission-Fusion Balance
Mitochondrial membrane potential is intricately linked to mitochondrial dynamics—the coordinated processes of fission and fusion that determine mitochondrial morphology and network architecture. The dynamic nature of mitochondria allows adjustment of morphology to specific cellular processes, with mitochondrial architecture determined by the opposing actions of fission proteins (Drp1, Mff) and fusion proteins (Mfn1/2, Opa1) [3]. Notably, mitochondrial fusion is strictly dependent on ΔΨm, as the activity of the inner membrane fusion protein Opa1 is voltage-dependent [3].
Healthy mitochondria undergo continuous fission and fusion cycles, but depolarized mitochondria are prevented from re-fusing into the mitochondrial network, effectively segregating them for quality control [3]. This coupling between membrane potential and dynamics provides a powerful mechanism for identifying and eliminating damaged mitochondria. The inability of depolarized mitochondria to fuse retains them in a post-fission state, making them preferential targets for autophagic degradation [3] [5].
Alternative Mitophagy Pathways: Receptor-Mediated Mechanisms
Beyond the PINK1-Parkin axis, ΔΨm-sensitive alternative mitophagy pathways exist, particularly relevant in specialized cellular contexts. During erythrocyte maturation, the protein NIX/BNIP3L mediates selective elimination of mitochondria independently of Parkin [3] [5]. Similarly, FUNDC1 serves as a mitophagy receptor on the OMM that responds to hypoxic conditions [4]. While these pathways may not directly sense ΔΨm through the same mechanism as PINK1, they nonetheless contribute to the overall quality control network that maintains mitochondrial health and is influenced by mitochondrial energetic status.
Experimental Approaches: Methodologies for ΔΨm Investigation
Protocol: TMRE/TMRM-Based ΔΨm Measurement in Live Cells
Principle: Cationic fluorescent dyes like tetramethylrhodamine ethyl ester (TMRE) and methyl ester (TMRM) accumulate in the mitochondrial matrix in a ΔΨm-dependent manner through the Nernst equation relationship [2].
Reagents:
- TMRE or TMRM (100-500 nM working concentration)
- MitoTracker Green (50-100 nM) for normalization
- Carbonyl cyanide m-chlorophenyl hydrazone (CCCP; 10-20 μM) as uncoupler control
- Appropriate cell culture medium without serum
Procedure:
- Culture cells on glass-bottom dishes or plates suitable for fluorescence microscopy.
- Prepare loading solution containing TMRE/TMRM in pre-warmed culture medium.
- Incubate cells with dye solution for 15-30 minutes at 37°C in the dark.
- Replace with fresh dye-free medium or maintain dye concentration during imaging (equilibrium mode).
- For normalized measurements, co-stain with MitoTracker Green (non-voltage-sensitive) for 15 minutes [2].
- Acquire fluorescence images using appropriate filter sets (excitation/emission ~549/575 nm for TMRE/TMRM; ~490/516 nm for MitoTracker Green).
- For quantitative analysis, measure fluorescence intensity in regions of interest corresponding to mitochondria and normalize to MitoTracker Green signal to account for mitochondrial mass [2].
- Include controls with CCCP (protonophore that collapses ΔΨm) to confirm ΔΨm-dependence of signal.
Data Interpretation: Increased TMRE/TMRM fluorescence intensity (normalized to MitoTracker Green) indicates higher ΔΨm, while decreased signal suggests depolarization. The TMRE/MTG ratio in IF1-KO cells typically shows 1.3-1.5-fold increase compared to wild-type controls [2].
Protocol: Assessment of ΔΨm in Permeabilized Cells for ETC-Specific Analysis
Principle: Permeabilizing the plasma membrane allows direct control over substrates provided to mitochondria, isolating ETC-specific effects on ΔΨm.
Reagents:
- Plasma membrane permeabilizing agent (e.g., digitonin, 10-50 μg/mL)
- Intracellular buffer (e.g., 120 mM KCl, 10 mM NaCl, 1 mM KH2PO4, 20 mM HEPES-Tris, pH 7.2)
- Substrates: Succinate (complex II; 10 mM), glutamate/malate (complex I; 5 mM each)
- TMRM or TMRE for ΔΨm measurement
- Fura-FF AM for simultaneous Ca2+ uptake monitoring [2]
Procedure:
- Culture cells on appropriate imaging dishes.
- Load with TMRM/TMRE and optionally Fura-FF AM according to manufacturer protocols.
- Permeabilize cells with digitonin-containing intracellular buffer for 1-2 minutes.
- Replace with fresh intracellular buffer containing respiratory substrates.
- Monitor TMRM fluorescence (ΔΨm) and Fura-FF fluorescence (Ca2+) simultaneously over time [2].
- Add Ca2+ pulses to assess mitochondrial calcium uptake capacity, which is ΔΨm-dependent.
Data Interpretation: Faster Ca2+ clearance rates in hyperpolarized mitochondria (e.g., IF1-KO) reflect the influence of ΔΨm on mitochondrial calcium buffering capacity [2].
Table 3: Research Reagent Solutions for ΔΨm and Quality Control Studies
| Reagent/Category | Specific Examples | Function/Application | Key Considerations |
|---|---|---|---|
| ΔΨm-Sensitive Dyes | TMRE, TMRM, JC-1, Rhodamine 123 | Quantitative measurement of membrane potential | Concentration-dependent artifacts; use quench/dequench modes appropriately |
| Mitochondrial Mass Indicators | MitoTracker Green, MitoTracker Deep Red | Normalization for mitochondrial content | MitoTracker Green is voltage-insensitive; Deep Red has some voltage sensitivity |
| ΔΨm Disruptors (Controls) | CCCP, FCCP, Valinomycin | Positive controls for depolarization | Complete vs. partial uncoupling; concentration optimization required |
| Genetic Models | IF1-KO cells, PINK1-KO, Parkin-KO [2] | Study specific pathway components | Isogenic controls essential; potential compensatory mechanisms |
| Mitophagy Reporters | mt-Keima, Rosella, Mito-QC | Direct monitoring of mitophagic flux | pH-sensitive fluorescent proteins; lysosomal delivery confirmation |
| Antibodies for Key Proteins | Anti-PINK1, Anti-Parkin, Anti-TOM20 | Protein localization and abundance assessment | Validate specificity; phosphorylation-specific antibodies available |
The Scientist's Toolkit: Key Research Reagents and Solutions
Table 4: Advanced Research Tools for Mitochondrial Quality Control Studies
| Technology Platform | Specific Application | Research Utility |
|---|---|---|
| CRISPR/Cas9 Gene Editing | Generation of IF1-KO, PINK1-KO, Parkin-KO cell lines [2] | Define specific gene functions in ΔΨm regulation and quality control |
| AAV-Mediated Gene Delivery | Tissue-specific expression of mitophagy reporters (mt-Keima) | In vivo monitoring of mitochondrial quality control processes |
| Nanoparticle-Based Targeting | Mitochondria-targeted drug delivery systems [6] | Therapeutic modulation of ΔΨm and mitophagy in disease contexts |
| Super-Resolution Microscopy | Visualization of mitochondrial ultrastructure and protein localization [7] | Nanoscale analysis of mitochondrial morphology and contact sites |
| Multi-Omics Approaches | Integration of transcriptomics, proteomics, metabolomics [8] | Systems-level understanding of ΔΨm-dependent signaling networks |
| AI/ML Predictive Modeling | Analysis of genotype-phenotype correlations in mitochondrial disease [8] | Identification of novel biomarkers and therapeutic targets |
Integrated Regulation: Crosstalk Between ΔΨm, Mitophagy, and Mitochondrial Biogenesis
The coordination between ΔΨm-mediated mitophagy and mitochondrial biogenesis ensures maintenance of a healthy mitochondrial population. This crosstalk represents a critical homeostatic circuit where the removal of damaged mitochondria is balanced by the generation of new organelles [9].
Transcriptional Regulation: Mitochondrial biogenesis is primarily regulated by the PGC-1α/NRF1/TFAM axis. Peroxisome proliferator-activated receptor γ coactivator 1-α (PGC-1α) serves as a master regulator that coordinates the activity of nuclear respiratory factors (NRF1 and NRF2), which in turn activate the expression of mitochondrial transcription factor A (TFAM) and other nuclear-encoded mitochondrial genes [9]. TFAM is essential for mitochondrial DNA replication, transcription, and maintenance [9].
Retrograde Signaling: ΔΨm influences these biogenesis pathways through multiple retrograde signaling mechanisms. The membrane potential affects mitochondrial calcium uptake, which activates Ca2+-sensitive kinases and regulates transcriptional coactivators [9]. Additionally, ΔΨm-dependent ROS production can modulate redox-sensitive transcription factors such as NFE2L2/NRF2, which itself can stimulate mitobiogenesis through activation of NRF1 [9].
Diagram 2: Quality control cycle linking ΔΨm to mitophagy and biogenesis.
This integrated system ensures that mitochondrial quality control is not merely a destructive process but part of a regenerative cycle that maintains optimal mitochondrial fitness. Disruption of either arm of this balance—excessive mitophagy or inadequate biogenesis—can lead to pathological mitochondrial depletion, while insufficient mitophagy with continued biogenesis results in accumulation of damaged organelles [9].
The central role of ΔΨm in mitochondrial quality control presents promising therapeutic avenues for numerous human diseases. In neurodegenerative disorders like Parkinson's disease, where PINK1-Parkin mediated mitophagy is impaired, strategies to enhance ΔΨm stability or facilitate mitophagic clearance of damaged mitochondria hold therapeutic potential [3] [5]. In cardiovascular contexts, modulating ΔΨm and mitophagy during ischemia/reperfusion injury could protect cardiomyocytes and prevent cell death [4]. Emerging technologies including mitochondria-targeted nanoparticles for drug delivery, AI-driven predictive models of mitochondrial function, and precision medicine approaches based on metabolic profiling offer innovative strategies to target ΔΨm therapeutically [6] [8]. As our understanding of the intricate relationships between mitochondrial membrane potential, quality control mechanisms, and cellular homeostasis continues to deepen, so too will our ability to develop targeted interventions for the myriad diseases characterized by mitochondrial dysfunction.
Mitochondria are traditionally recognized as the power plants of the cell, generating adenosine triphosphate (ATP) through oxidative phosphorylation. However, their functions extend far beyond mere energy production to include regulation of apoptosis, calcium storage, oxidative stress balance, and signal transduction [10] [11]. The functional integrity of mitochondria is paramount for cellular homeostasis, particularly in high-energy-demand tissues such as neurons, cardiac muscle, and skeletal muscle. To maintain this integrity, cells employ a sophisticated network of mechanisms collectively known as the mitochondrial quality control (MQC) system [10] [12]. This system regulates mitochondrial homeostasis through coordinated processes including dynamics, biogenesis, repair, and selective degradation [12].
The MQC system is especially crucial in the context of the mitochondrial membrane potential (ΔΨm), which serves as a key indicator of mitochondrial health and a central regulator of quality control decisions [13]. Disruption of MQC mechanisms contributes significantly to the pathogenesis of various diseases, including neurodegenerative disorders, cardiovascular pathologies, metabolic syndromes, and diabetic complications [11] [13] [12]. This review provides a comprehensive technical overview of the MQC system, with particular emphasis on the role of mitochondrial membrane potential in regulating mitophagy and overall quality control, while presenting key experimental methodologies and reagents essential for research in this field.
Core Mechanisms of the Mitochondrial Quality Control System
Mitochondrial Dynamics: The Foundation of MQC
Mitochondrial dynamics, comprising fission and fusion events, form the foundational layer of MQC by regulating mitochondrial morphology, distribution, and functional complementarity [10]. These processes are mediated by highly conserved dynamin-family GTPases.
Mitochondrial fusion promotes the mixing of mitochondrial contents, allowing functional complementation between partially damaged mitochondria [10]. Outer mitochondrial membrane (OMM) fusion is mediated by mitofusins (MFN1 and MFN2), while inner mitochondrial membrane (IMM) fusion is driven by optic atrophy 1 (OPA1) [10]. Fusion enhances ATP production capacity and facilitates the exchange of mitochondrial DNA (mtDNA), promoting genetic complementation [10].
Mitochondrial fission enables the division of mitochondrial networks, facilitating the segregation of damaged components for removal and the distribution of mitochondria to daughter cells during division [10]. Fission is primarily mediated by dynamin-related protein 1 (DRP1), which is recruited from the cytoplasm to the OMM by adaptor proteins including mitochondrial fission factor (Mff), mitochondrial dynamics proteins (MiD49 and MiD51), and fission 1 (Fis1) [10].
Table 1: Core Machinery Regulating Mitochondrial Dynamics
| Process | Core Regulators | Accessory Proteins | Post-Translational Modifications |
|---|---|---|---|
| Fusion | MFN1, MFN2, OPA1 | MSTO1, SLC25A46, MTCH2 | Ubiquitination (MFNs), Proteolytic cleavage (OPA1) |
| Fission | DRP1 | Mff, MiD49, MiD51, Fis1, INF2, Spire1C | Phosphorylation, Ubiquitination, SUMOylation |
The balance between fission and fusion is critically influenced by mitochondrial membrane potential. A localized loss of ΔΨm can lead to uncoupled fusion where OMM fusion occurs independently of IMM fusion [10]. Furthermore, the proteolytic cleavage of the long isoform of OPA1 (L-OPA1) to generate S-OPA1, which promotes a shift toward fission, is regulated by ΔΨm dissipation [10].
Mitophagy: Selective Clearance of Damaged Mitochondria
Mitophagy, the selective autophagic degradation of mitochondria, represents a crucial pathway for removing damaged organelles and is centrally regulated by changes in mitochondrial membrane potential [13] [14]. Two primary mechanistic pathways govern mitophagy: ubiquitin-dependent and ubiquitin-independent pathways.
The ubiquitin-dependent pathway is primarily mediated by the PINK1-Parkin signaling axis [14]. Under normal conditions with preserved ΔΨm, PINK1 is imported into mitochondria and rapidly degraded. However, upon mitochondrial depolarization (loss of ΔΨm), PINK1 stabilizes on the OMM where it activates Parkin, an E3 ubiquitin ligase [14]. Parkin then ubiquitinates numerous OMM proteins, generating "eat-me" signals that are recognized by autophagy adaptor proteins such as p62, OPTN, and NDP52, leading to autophagosome engulfment [14].
Ubiquitin-independent pathways are mediated by mitophagy receptors on the OMM that directly interact with LC3 on developing autophagosomes [14]. Key receptors include:
- FUNDC1: Regulates hypoxic mitophagy [14].
- BNIP3 & NIX: Mediate mitophagy during hypoxia and erythrocyte differentiation [14].
- BCL2L13 & FKBP8: Function as mammalian homologs of yeast mitophagy receptors [14].
- PHB2: An IMM receptor exposed upon OMM rupture [14].
Diagram 1: Mitophagy Pathways Regulated by Membrane Potential
In diabetic retinopathy, research using mitophagy-reporter mice (mitoQC-Ins2Akita) and pMitoTimer has demonstrated that mitochondrial loss occurs due to an inability of biogenesis to compensate for diabetes-exacerbated mitophagy [13]. Interestingly, with prolonged diabetes, PINK1-dependent mitophagy deteriorates, leading to the accumulation of mitochondria primed for degradation and the development of retinal senescence [13].
Mitochondrial Biogenesis and Protein Homeostasis
Mitochondrial biogenesis involves the synthesis of new mitochondrial components and is regulated by a transcriptional network centered on peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α) [12]. PGC-1α activates nuclear respiratory factors (NRF1/NRF2), which in turn promote the expression of mitochondrial transcription factor A (TFAM), essential for mtDNA replication and transcription [12]. This process is modulated by energy status, redox changes, and environmental cues.
Mitochondrial protein homeostasis is maintained through sophisticated import machinery for nuclear-encoded proteins and internal quality control systems. The mitochondrial unfolded protein response (UPRmt) and intramitochondrial proteases, such as OMA1 and AFG3L2, detect and degrade misfolded proteins, preventing proteotoxic stress [12]. Defects in protein import or folding can activate OMA1, leading to IMM remodeling and influencing dynamics [12].
Experimental Approaches for Investigating MQC
Quantitative Analysis of Mitochondrial Morphology
Advanced imaging and computational analysis enable high-throughput, quantitative assessment of mitochondrial morphology. Automated high-content fluorescence microscopy can classify mitochondrial objects into distinct morphological categories based on interconnectedness and shape [15].
Table 2: Mitochondrial Morphological Categories for Quantitative Analysis
| Morphological Category | Description | Functional Association |
|---|---|---|
| Puncta | Small, fragmented organelles | Often associated with fission or fragmentation |
| Rod | Intermediate, tubular shapes | Transitional state or healthy smaller mitochondria |
| Network | Elongated, interconnected tubules | Indicates active fusion and health |
| Large & Round | Swollen, circular organelles | Suggests pathological swelling or dysfunction |
A protocol for evaluating CCCP-induced mitochondrial stress involves staining cells with MitoTracker dyes, acquiring z-stack images via wide-field or confocal microscopy, and using open-source software like Fiji for segmentation and classification [15] [16]. Mitochondrial objects are identified through segmentation algorithms, and morphometric features (area, perimeter, aspect ratio, form factor) are calculated to train a classifier for automatic categorization [15].
Diagram 2: Workflow for Mitochondrial Morphology Analysis
This method has been validated using various mitochondrial toxicants, such as t-butyl hydroperoxide (TBHP), rotenone, and oligomycin, which consistently reduce mitochondrial networked areas while increasing the proportion of large & round mitochondria, indicating swelling [15].
Assessing Mitochondrial Membrane Potential and Function
Mitochondrial membrane potential (ΔΨm) is a key parameter of mitochondrial health and a central regulator of MQC. It can be quantified using potentiometric fluorescent dyes such as tetramethylrhodamine methyl ester (TMRM) [15]. The protocol involves:
- Cell staining: Loading cells with TMRM in culture medium.
- Image acquisition: Using fluorescence microscopy with appropriate filter sets.
- Intensity quantification: Measuring fluorescence intensity within mitochondrial regions identified by segmentation.
Depolarizing agents like FCCP decrease TMRM fluorescence (to 0.33-fold of vehicle control), while ATP synthase inhibitors like oligomycin cause hyperpolarization (5.25-fold increase) [15]. This method can be combined with respirometry to correlate morphological and potential changes with respiratory function [15].
The Scientist's Toolkit: Key Research Reagents and Models
Table 3: Essential Research Reagents for MQC Investigation
| Reagent/Model | Category | Primary Function/Application |
|---|---|---|
| MitoTracker Deep Red FM | Fluorescent dye | Labels mitochondria for morphology and mass analysis [15] |
| TMRM | Potentiometric dye | Quantifies mitochondrial membrane potential (ΔΨm) [15] |
| MitoQC mice | Animal model | Reports on mitophagy activity in vivo [13] |
| pMitoTimer | Molecular tool | Visualizes mitochondrial age and turnover dynamics [13] |
| CCCP/FCCP | Chemical uncoupler | Induces mitochondrial depolarization to stress MQC [13] [16] |
| Mdivi-1 | Small molecule inhibitor | Inhibits DRP1 to block mitochondrial fission [10] |
| Oligomycin | ATP synthase inhibitor | Induces hyperpolarization; tests MQC response [15] |
| Rotenone/Antimycin A | ETC inhibitors | Induce ROS production and MQC stress [15] |
The Mitochondrial Quality Control system represents an integrated network of processes that maintain functional mitochondrial populations, with mitochondrial membrane potential serving as a central regulator. The experimental methodologies and reagents outlined provide powerful approaches for investigating MQC in health and disease. Continued research into the intricate interplay between MQC mechanisms, particularly how mitochondrial membrane potential integrates these processes, will be crucial for developing novel therapeutic strategies for the multitude of diseases associated with mitochondrial dysfunction.
Mitochondrial membrane potential (ΔΨm) serves as a central regulator of cellular fate, functioning as a critical nexus between the dynamic processes of mitochondrial fission and fusion and the ultimate pathways of quality control and cell death. This whitepaper explores the sophisticated interplay between mitochondrial dynamics and ΔΨm, examining how these processes collectively govern mitochondrial integrity, function, and cellular survival. We present comprehensive quantitative data on ΔΨm regulation, detailed experimental protocols for assessing mitochondrial dynamics and membrane potential, and visualization of key signaling pathways. Within the context of mitochondrial quality control research, we further discuss how dysregulation of this interplay contributes to pathological states and present emerging therapeutic strategies targeting these interconnected processes for drug development applications.
Mitochondrial membrane potential (ΔΨm), representing the electrical gradient across the inner mitochondrial membrane, serves as a primary indicator of mitochondrial functional status and a key driver of ATP synthesis. Simultaneously, mitochondrial networks undergo continuous remodeling through opposing processes of fission (division) and fusion (joining), which are essential for maintaining mitochondrial health, distribution, and function. The interdependence between ΔΨm and mitochondrial dynamics creates a sophisticated regulatory circuit that determines mitochondrial destiny: dysfunctional organelles are targeted for degradation via mitophagy, while functional mitochondria are preserved through quality control mechanisms.
Research has established that mitochondrial dynamics (fusion and fission) and mitophagy play crucial roles in cellular stress response and are increasingly recognized as contributors to disease mechanisms, including cancer drug resistance [17]. The dynamic balance between fission and fusion allows mitochondria to adapt to metabolic demands and mitigate stress, with ΔΨm serving as both a regulator and readout of these processes. Understanding this interplay is paramount for developing targeted therapies for conditions ranging from neurodegenerative diseases to cancer, where mitochondrial dysfunction is a central feature.
Molecular Mechanisms of Mitochondrial Fission and Fusion
Mitochondrial Fusion Machinery
Mitochondrial fusion is a two-step process involving sequential fusion of the outer and inner mitochondrial membranes, mediated by conserved GTPase proteins.
Outer Membrane Fusion: Governed by mitofusins (MFN1 and MFN2), which form homo- and hetero-oligomeric complexes between adjacent mitochondria. The HR2 domain at the C-terminal of MFN1/MFN2 folds into an antiparallel coiled-coil dimer, initiating membrane tethering, with GTP hydrolysis providing energy for fusion completion [17]. MFN2 exhibits additional extra-mitochondrial functions, including endoplasmic reticulum tethering and participation in PINK1/Parkin-mediated mitophagy signaling.
Inner Membrane Fusion: Mediated by optic atrophy 1 (OPA1) protein, which exists in long (L-OPA1) and short (S-OPA1) isoforms generated through proteolytic processing. L-OPA1 is anchored to the inner membrane, while S-OPA1 is soluble in the intermembrane space. These isoforms form higher-order helical assemblies that work cooperatively to remodel membrane curvature and drive fusion [17]. The appropriate balance between L-OPA1 and S-OPA1 is critical for efficient fusion, with excess S-OPA1 inhibiting the process.
Mitochondrial Fission Machinery
Mitochondrial fission enables the division of damaged or overly elongated mitochondria and is crucial for mitochondrial distribution and quality control.
Primary Fission Mechanism: Dynamin-related protein 1 (DRP1) serves as the central fission GTPase, recruited from the cytosol to mitochondrial fission sites where it oligomerizes into spirals around the mitochondrial tubule. Recent research reveals that fission occurs through a two-stage process: first, DRP1 spirals constrict the mitochondrial membrane to form a narrow neck, followed by disassembly of the DRP1 scaffold which drives membrane bending through a snap-through instability mechanism, ultimately leading to division [18].
Regulation of Fission: DRP1 recruitment and activity are regulated by phosphorylation, with phosphorylation at Ser616 promoting mitochondrial translocation and fission activity. The ROS-Drp1-mediated mitochondrial fission pathway represents a key mechanism where reactive oxygen species can activate DRP1 to drive excessive fission, contributing to apoptotic pathways [19]. Mitochondrial fission process 1 (MTFP1) protein also plays a regulatory role in fission dynamics, with its dysfunction leading to mitochondrial fragmentation and associated pathologies [20].
Table 1: Core Proteins Regulating Mitochondrial Dynamics
| Protein | Location | Function | Regulatory Mechanisms |
|---|---|---|---|
| MFN1/MFN2 | Outer Mitochondrial Membrane | Mediates outer membrane fusion, ER-mitochondria tethering (MFN2) | GTP hydrolysis, ubiquitination by Parkin, transcriptional regulation |
| OPA1 | Inner Mitochondrial Membrane | Mediates inner membrane fusion, cristae organization | Proteolytic processing by OMA1/YME1L, membrane potential sensitivity |
| DRP1 | Cytosol (recruited to mitochondria) | Mediates mitochondrial fission | Phosphorylation (Ser616 activates), SUMOylation, ubiquitination, interaction with MFF/Fis1 |
| MTFP1 | Inner Mitochondrial Membrane | Regulates fission process, mitochondrial permeability | Expression levels, interactions with mPTP components |
Quantitative Assessment of ΔΨm: Methodologies and Measurements
ΔΨm serves as a crucial indicator of mitochondrial health, with its quantitative assessment being essential for understanding mitochondrial function in both physiological and pathological contexts.
Fluorescence-Based Measurement Techniques
The most widely employed approach for measuring ΔΨm in live cells utilizes lipophilic, cationic fluorescent dyes that distribute across membranes according to the Nernst equation.
TMRM (Tetramethylrhodamine Methyl Ester) Protocol: Cells are loaded with 200 nM TMRM for 30 minutes in modified Hank's Balanced Salt Solution (HBSS) or complete growth media, followed by washing and maintenance in 50 nM TMRM to preserve equilibrium distribution [21]. Imaging is performed using laser scanning confocal microscopy with 561 nm excitation and 590-610 nm emission detection. The non-quenching mode is employed for quantitative measurements, where fluorescence intensity is proportional to dye accumulation.
Absolute Quantification Method: A biophysical model accounting for probe compartmentation and dynamics enables conversion of fluorescence readings to absolute ΔΨm values in millivolts. This approach incorporates factors including ΔΨm, plasma membrane potential (ΔΨp), matrix:cell volume ratio, binding coefficients, and optical dilution. The calibration involves measuring fluorescence intensities under different conditions to deconvolute the contributions of various parameters to the final signal [22].
Quantitative ΔΨm Values in Physiological Contexts
Research utilizing these quantitative approaches has revealed important insights into ΔΨm regulation across different cell types and conditions.
Table 2: Quantitative ΔΨm Measurements in Different Cellular Contexts
| Cell Type/Condition | ΔΨm Value (mV) | Measurement Technique | Biological Significance |
|---|---|---|---|
| Cultured Rat Cortical Neurons (resting) | -139 ± 5 | Absolute TMRM calibration [22] | Baseline for neuronal energy metabolism |
| Cortical Neurons (metabolic activation) | -158 ± 7 | Absolute TMRM calibration [22] | Ca²⁺-dependent substrate activation |
| Cortical Neurons (increased ATP demand) | -108 ± 4 | Absolute TMRM calibration [22] | Response to sustained depolarization |
| Cancer Cells (HepG2) | Heterogeneous | Semi-quantitative TMRM [21] | Metabolic adaptation in proliferation |
| Fibroblasts | More homogeneous | Semi-quantitative TMRM [21] | Stable metabolic requirements |
Studies have demonstrated that ΔΨm heterogeneity is more pronounced in cancer cells compared to fibroblasts, reflecting the metabolic adaptability of tumor cells [21]. This heterogeneity is modulated primarily by intramitochondrial factors rather than plasma membrane potential or cell cycle phase, and can be reduced by pharmacological inhibition of electron transport chain complexes or ATP synthase.
The Interplay Between Dynamics and ΔΨm in Quality Control
ΔΨm Regulation of Mitochondrial Dynamics
ΔΨm exerts significant influence over mitochondrial dynamics through multiple mechanisms:
OPA1 Processing: ΔΨm loss stimulates OPA1 cleavage by the protease OMA1, converting fusion-competent L-OPA1 to S-OPA1 fragments, thereby inhibiting inner membrane fusion and promoting mitochondrial fragmentation [17].
DRP1 Recruitment: Depolarization can enhance DRP1 recruitment to mitochondria through calcium-dependent signaling pathways and phosphorylation events, promoting fission of damaged organelles.
Dynamics Regulation of ΔΨm
Conversely, mitochondrial dynamics proteins directly modulate ΔΨm:
Fusion and ΔΨm Stability: MFN2 deficiency reduces ΔΨm and respiratory capacity, while OPA1 overexpression enhances mitochondrial polarization and cristae organization, supporting more efficient ATP production [17].
Fission and ΔΨm Depolarization: Excessive DRP1-mediated fission promotes ΔΨm loss, particularly under pathological conditions such as silver nanoparticle-induced neurotoxicity, where ROS-Drp1-mitochondrial fission axis activation leads to decreased mitochondrial membrane potential and ATP synthesis [19].
The following diagram illustrates the core signaling pathway governing the interplay between mitochondrial dynamics and membrane potential:
Integration with Mitophagy and Quality Control
The interplay between dynamics and ΔΨm forms the foundation of mitochondrial quality control:
Depolarization-Induced Mitophagy: PINK1/Parkin pathway activation occurs specifically on depolarized mitochondria, where PINK1 stabilization leads to Parkin recruitment and ubiquitination of mitochondrial proteins, including mitofusins, marking them for autophagic clearance [23].
Fission in Quality Control: DRP1-mediated fission facilitates the separation of damaged, depolarized mitochondrial segments from the healthy network, enabling selective targeting of compromised organelles for mitophagy while preserving functional mitochondria [19].
This quality control mechanism is particularly crucial in post-mitotic cells such as cardiomyocytes and neurons, where mitochondrial dysfunction can have severe consequences. In myocardial ischemic stress, for example, mitophagy serves as a critical quality control mechanism to eliminate damaged mitochondria and preserve cardiac function [24].
Experimental Approaches and Research Toolkit
Key Research Reagents and Methodologies
Investigating the interplay between mitochondrial dynamics and ΔΨm requires a comprehensive toolkit of reagents and methodologies.
Table 3: Essential Research Reagents for Investigating Mitochondrial Dynamics and ΔΨm
| Reagent/Category | Specific Examples | Function/Application | Key Considerations |
|---|---|---|---|
| ΔΨm Indicators | TMRM, Rhodamine 123, JC-1 | Quantitative/semi-quantitative ΔΨm measurement | Mode (quench/non-quench), loading concentration, calibration requirements |
| Plasma Membrane Potential Indicators | DiBAC₄(3), FLIPR Membrane Potential Assay | Control for ΔΨp contribution to cationic dye distribution | Compatibility with ΔΨm indicators, loading conditions |
| Fission Inhibitors | Mdivi-1 | DRP1 inhibition to study fission consequences | Specificity, concentration-dependent effects |
| Fusion Manipulators | MFN/OPA1 expression constructs, siRNA | Genetic manipulation of fusion machinery | Efficiency of transfection, compensatory mechanisms |
| OXPHOS Modulators | Antimycin A, Oligomycin, CCCP | Induce controlled mitochondrial dysfunction | Concentration, time of exposure, specificity |
| ROS Modulators | N-acetylcysteine (NAC), H₂O₂ | Manipulate redox state to study ROS-dynamics interplay | Direct vs. indirect effects, concentration ranges |
| Mitophagy Reporters | mt-Keima, LC3-GFP | Monitor mitophagic flux | Validation with lysosomal inhibitors, specificity controls |
Integrated Experimental Workflow
A comprehensive assessment of mitochondrial dynamics and ΔΨm involves multiple experimental steps, as visualized in the following workflow:
Pathophysiological Implications and Therapeutic Targeting
Dysregulation of the mitochondrial dynamics-ΔΨm interplay contributes significantly to disease pathogenesis and represents a promising therapeutic target.
Disease Associations
Cancer: Cancer cells exhibit heterogeneous ΔΨm and altered dynamics that contribute to drug resistance. Mitochondrial fusion can enhance chemoresistance by stabilizing mitochondrial networks and maintaining energy production under stress, while excessive fission can sensitize to apoptosis in some contexts [17].
Neurodegenerative Disorders: Impaired dynamics and ΔΨm regulation are hallmarks of neurodegenerative diseases. In hippocampal neurons, ROS-Drp1-mediated mitochondrial fission contributes to apoptosis induced by neurotoxic agents like silver nanoparticles [19].
Cardiovascular Diseases: In myocardial ischemic stress, mitophagy serves as a quality control mechanism, with therapeutic strategies designed to augment protective mitophagy showing promise in preclinical models [24].
COPD: Mitochondrial quality control mechanisms become dysregulated in chronic obstructive pulmonary disease, leading to mitochondrial dysfunction characterized by excessive ROS production and disrupted dynamics [25].
Therapeutic Targeting Strategies
Emerging therapeutic approaches focus on modulating the dynamics-ΔΨm interplay:
DRP1 Inhibition: Compounds like Mdivi-1 that inhibit excessive DRP1-mediated fission can reduce pathological mitochondrial fragmentation and apoptosis, as demonstrated in neurotoxicity models [19].
Fusion Enhancement: Strategies to promote mitochondrial fusion through OPA1 stabilization or mitofusin activation may improve mitochondrial function in conditions characterized by mitochondrial fragmentation.
ΔΨm Modulation: Compounds that mildly depolarize mitochondria could reduce ROS production while maintaining sufficient energy output, representing a potential approach for conditions involving oxidative stress.
Recent advances in understanding the fundamental mechanisms of mitochondrial fission, including the two-stage process involving DRP1 assembly and disassembly, open new possibilities for targeted interventions with greater specificity and fewer off-target effects [18].
The intricate interplay between mitochondrial fission, fusion, and ΔΨm represents a critical regulatory nexus determining cellular destiny through its integration with quality control mechanisms. Quantitative assessment of these processes reveals their tight coordination in health and their dysregulation in disease. Continued refinement of research methodologies, including absolute ΔΨm quantification and targeted manipulation of dynamics proteins, will enhance our understanding of these relationships. Therapeutic strategies that precisely modulate these interconnected processes hold significant promise for addressing numerous pathological conditions characterized by mitochondrial dysfunction, from neurodegenerative diseases to cancer, though careful consideration of context-dependent effects remains essential for successful clinical translation.
The PINK1-Parkin pathway represents a crucial mitochondrial quality control system that senses damage and initiates the selective removal of dysfunctional mitochondria via autophagy. This in-depth technical guide examines the molecular mechanisms by which loss of mitochondrial membrane potential (ΔΨm) triggers a coordinated ubiquitin-dependent signaling cascade leading to mitophagy. We explore how PINK1 functions as a sophisticated sensor of mitochondrial health, how Parkin is transformed from an autoinhibited cytosolic enzyme into an active ubiquitin ligase, and the downstream events that culminate in mitochondrial degradation. This review also provides detailed experimental methodologies and key research tools essential for investigating this pathway, offering researchers a comprehensive resource for studying mitochondrial quality control mechanisms relevant to neurodegenerative diseases, metabolic disorders, and therapeutic development.
Mitophagy, the selective autophagic clearance of damaged or superfluous mitochondria, serves as a fundamental cellular quality control mechanism essential for maintaining mitochondrial homeostasis and cellular health [26]. This process represents a crucial component of the mitochondrial quality control network, which also includes mitochondrial dynamics (fusion and fission) and proteostatic mechanisms [27] [28]. The proper regulation of mitophagy is particularly critical in high-energy demand tissues, and its dysfunction has been implicated in numerous pathological conditions including neurodegenerative diseases like Parkinson's disease, metabolic disorders, and aging-related conditions [26] [29].
The PINK1-Parkin axis constitutes the most extensively characterized pathway for ubiquitin-dependent mitophagy in mammalian cells [26] [30]. This pathway centers on two key proteins: PTEN-induced putative kinase 1 (PINK1), a mitochondrial serine/threonine kinase, and Parkin, a cytosolic E3 ubiquitin ligase. Mutations in both proteins are associated with autosomal recessive early-onset Parkinson's disease, highlighting their neuroprotective functions and the critical importance of mitochondrial quality control in neuronal survival [31] [32]. The current model positions PINK1 as the primary sensor of mitochondrial damage that recruits and activates Parkin to selectively tag damaged mitochondria for autophagic degradation [32] [33].
Table 1: Key Proteins in the PINK1-Parkin Mitophagy Pathway
| Protein | Gene | Function | Domain Architecture |
|---|---|---|---|
| PINK1 | PARK6 | Mitochondrial damage sensor kinase | MTS, TM, Kinase domain |
| Parkin | PARK2 | E3 ubiquitin ligase | Ubl, RING0, RING1, IBR, RING2 |
| Ubiquitin | UBB | Signaling molecule | Conserved ubiquitin fold |
| MFN1/2 | MFN1/2 | Mitochondrial fusion | GTPase, TM domains |
| TOM Complex | Multiple subunits | Mitochondrial protein import | TOM20, TOM22, TOM40, TOM7 |
Molecular Mechanisms of PINK1-Parkin Pathway Activation
PINK1 as a Mitochondrial Damage Sensor
PINK1 functions as the primary sensor of mitochondrial damage through a sophisticated import-proteolysis coupling mechanism that directly monitors mitochondrial health status [31] [26]. In healthy, polarized mitochondria, PINK1 is constitutively synthesized in the cytosol and imported into mitochondria through the TOM/TIM23 complex translocation system. The N-terminal mitochondrial targeting sequence (MTS) directs PINK1 to the inner mitochondrial membrane, where it undergoes sequential proteolytic processing by the mitochondrial processing peptidase (MPP) and presenilin-associated rhomboid-like protein (PARL) [31]. This processing generates a cleaved, unstable ~52 kDa fragment that is retro-translocated to the cytosol and rapidly degraded by the proteasome via the N-end rule pathway, maintaining minimal PINK1 levels under basal conditions [31] [32].
Upon mitochondrial depolarization or damage, the import of PINK1 is halted, leading to its accumulation on the outer mitochondrial membrane (OMM) [31] [26]. The current model indicates that PINK1 stabilization occurs through interactions with the TOM complex, particularly with the TOM7 subunit, which prevents PINK1 retro-translocation and degradation [31]. At the OMM, PINK1 forms a stable 720 kDa complex with TOM and undergoes trans-autophosphorylation at Ser228, which triggers conformational changes that activate its kinase activity [26]. This OMM-stabilized PINK1 then phosphorylates ubiquitin molecules attached to OMM proteins at Ser65, creating phospho-ubiquitin (pUb) moieties that serve as the initial "eat-me" signal for Parkin recruitment [31] [33].
Parkin Recruitment and Activation
Parkin exists in an autoinhibited conformation in the cytosol under basal conditions, with multiple intramolecular interactions preventing its enzymatic activity [26]. The protein contains several functional domains: an N-terminal ubiquitin-like (Ubl) domain, a repressor element of Parkin (REP), and really interesting new gene (RING) domains including RING0, RING1, an in-between-RING (IBR) domain, and RING2 [26]. In this inactive state, the Ubl domain interacts with RING1, while a catalytic Cys431 in RING2 is blocked by REP, preventing ubiquitin transfer [26].
The activation of Parkin occurs through a multi-step process initiated by phospho-ubiquitin binding [33] [26]. Phospho-ubiquitin generated by PINK1 binds to Parkin at a specific site between RING0 and RING1, triggering conformational changes that release the Ubl domain and expose the E2 interaction surface in RING1 [26]. This initial activation step is followed by PINK1-mediated phosphorylation of Parkin at Ser65 within its Ubl domain, which further stabilizes the active conformation [33]. The structural remodeling ultimately releases RING2, allowing it to receive ubiquitin from E2 ubiquitin-conjugating enzymes and transfer it to substrate proteins on the mitochondrial surface [26].
Table 2: Key Phosphorylation Events in PINK1-Parkin Pathway Activation
| Phosphorylation Target | Phosphorylation Site | Kinase | Functional Consequence |
|---|---|---|---|
| PINK1 | Ser228 | PINK1 (autophosphorylation) | Kinase activation, N-lobe destabilization |
| Ubiquitin | Ser65 | PINK1 | Creates Parkin recruitment signal |
| Parkin | Ser65 | PINK1 | Releases autoinhibition, enhances E3 ligase activity |
| MFN1/2 | Multiple sites | PINK1 | Enhances Parkin binding and ubiquitination |
Ubiquitin Chain Assembly and Amplification
Once activated, Parkin initiates a robust ubiquitination cascade on numerous outer mitochondrial membrane proteins [31] [32]. Primary substrates include mitofusins (MFN1 and MFN2), mitochondrial Rho GTPases (MIRO1/2), voltage-dependent anion channels (VDAC1/2/3), and components of the TOM complex [31] [32]. Parkin predominantly generates Lys6, Lys11, and Lys63-linked ubiquitin chains on these substrates, with the specific chain topology determining downstream signaling outcomes [32].
This ubiquitination creates a positive feedback loop that amplifies the mitophagy signal [26]. Newly deposited ubiquitin chains serve as additional substrates for PINK1-mediated phosphorylation, generating more phospho-ubiquitin molecules that recruit additional Parkin molecules to the damaged mitochondrion [26]. This cooperative mechanism ensures rapid and comprehensive labeling of damaged mitochondria while minimizing false activation signals. The extensive ubiquitination also inhibits mitochondrial fusion by targeting mitofusins for degradation, effectively isolating damaged mitochondria from the healthy network and facilitating their selective removal [32].
Experimental Approaches for Studying PINK1-Parkin Mitophagy
Inducing and Monitoring Mitophagy
Researchers have established standardized protocols for activating and quantifying PINK1-Parkin-mediated mitophagy in cellular models. The most common method for inducing mitochondrial depolarization involves treatment with protonophores such as carbonyl cyanide m-chlorophenyl hydrazone (CCCP) or carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP), typically at concentrations ranging from 10-20 μM for 1-24 hours depending on the cell type and experimental requirements [31] [33]. These compounds collapse the mitochondrial membrane potential by equilibrating protons across the inner mitochondrial membrane, thereby mimicking the physiological signal for PINK1 stabilization.
Multiple approaches exist for monitoring mitophagy progression:
Parkin Translocation Assay: Cells expressing GFP- or RFP-tagged Parkin are treated with CCCP and monitored by live-cell imaging or fixed at various time points for immunofluorescence analysis. Parkin translocation typically occurs within 5-30 minutes post-treatment [33].
Western Blot Analysis of PINK1 Stabilization: Mitochondrial fractions or whole cell lysates are prepared at different time points after mitochondrial depolarization. Full-length PINK1 (∼63 kDa) accumulation is detected using specific antibodies, with processing intermediates (∼55 kDa) appearing in healthy mitochondria [31].
Phos-tag SDS-PAGE for Phosphorylation Detection: This specialized electrophoretic technique detects phosphorylated Parkin and ubiquitin by incorporating Phos-tag acrylamide into SDS-PAGE gels, which retards the migration of phosphorylated species [33].
Mitophagy Reporter Assays: Fluorescent reporters such as mt-Keima or Rosella allow quantitative assessment of mitophagy flux through pH-sensitive fluorescence changes or colocalization with lysosomal markers [32].
Key Research Reagents and Tools
Table 3: Essential Research Reagents for Studying PINK1-Parkin Mitophagy
| Reagent Category | Specific Examples | Application/Function | Experimental Notes |
|---|---|---|---|
| Mitochondrial Depolarizers | CCCP, FCCP, Valinomycin, Oligomycin/Antimycin A | Induce ΔΨm loss and PINK1 stabilization | CCCP (10-20 μM) most widely used; treatment duration varies by cell type |
| Proteasome Inhibitors | MG132, Epoxomicin, Bortezomib | Block degradation of PINK1 fragments and ubiquitinated proteins | Used to study ubiquitination events (10 μM MG132 for 4-6h) |
| Kinase Inhibitors | Kinetin, Cmpd-17 (PINK1 inhibitors) | Validate PINK1-specific phosphorylation events | Limited cell permeability for some inhibitors |
| Parkin Mutants | S65A (phosphodead), C431F (catalytic dead), pathogenic mutants (T240R, etc.) | Study Parkin activation mechanism and pathobiology | S65A blocks phosphorylation; pathogenic mutants show impaired translocation |
| Cell Lines | HeLa, SH-SY5Y, MEFs (PINK1-/-/Parkin-/-) | Model systems for mitophagy studies | HeLa cells have low endogenous Parkin; knockout MEFs for rescue experiments |
| Antibodies | Anti-PINK1, anti-Parkin, anti-phospho-ubiquitin (Ser65), anti-TOM20, anti-COX IV | Detection and localization of pathway components | Phospho-specific antibodies require validation |
Downstream Events and Mitochondrial Fate
Following extensive ubiquitination by Parkin, damaged mitochondria are targeted for degradation through a series of well-orchestrated events. Autophagy adaptor proteins including p62/SQSTM1, optineurin (OPTN), NDP52, NBR1, and TAX1BP1 are recruited to the mitochondrial surface through their ubiquitin-binding domains [32] [26]. These adaptors simultaneously interact with LC3 (and other ATG8 family proteins) on forming autophagosomal membranes, effectively tethering the damaged mitochondrion to the autophagy machinery [26]. The autophagy machinery then engulfs the mitochondria in a double-membraned autophagosome, which subsequently fuses with lysosomes to form mitolysosomes where mitochondrial components are degraded by acidic hydrolases [26] [30].
The role of the proteasome in this process appears complex and context-dependent. Several studies demonstrate that proteasomal inhibition impairs Parkin-mediated mitophagy, suggesting that proteasomal degradation of certain OMM proteins facilitates mitophagy progression [32]. The AAA+ ATPase p97/VCP is recruited to ubiquitinated mitochondria and may extract proteins from the OMM to promote autophagic engulfment [32]. Additionally, histone deacetylase 6 (HDAC6) recognizes ubiquitinated mitochondria and promotes autophagosome-lysosome fusion through actin remodeling, while also facilitating mitochondrial aggregation in the perinuclear region [32].
The PINK1-Parkin pathway represents an elegant cellular mechanism for maintaining mitochondrial quality through the selective elimination of damaged organelles. The core mechanism involves PINK1 accumulation on depolarized mitochondria, phosphorylation of ubiquitin, Parkin recruitment and activation, extensive ubiquitination of OMM proteins, and finally, recognition by autophagy adaptors leading to lysosomal degradation. This pathway not only provides fundamental insights into mitochondrial quality control but also offers therapeutic targets for numerous human diseases linked to mitochondrial dysfunction.
Future research directions include elucidating the structural basis of PINK1 activation on the TOM complex, understanding the spatial and temporal regulation of ubiquitin chain types in mitophagy signaling, identifying novel physiological and pathological stimuli beyond ΔΨm loss that activate this pathway, and developing specific small molecule modulators for therapeutic applications. The continued investigation of the PINK1-Parkin axis will undoubtedly yield important insights into cellular quality control mechanisms and their relevance to human health and disease.
Mitochondrial membrane potential (ΔΨm) serves as a fundamental regulator of cellular energy status and a critical sensor for mitochondrial quality control. This technical review examines the molecular mechanisms by which the receptor-mediated mitophagy pathways—specifically those orchestrated by FUNDC1, BNIP3, and NIX—detect and respond to ΔΨm dynamics to maintain mitochondrial homeostasis. We synthesize recent structural and functional evidence revealing how these receptors initiate autophagosome biogenesis through distinct molecular machinery, with particular emphasis on their roles in pathological contexts such as neurodegenerative diseases, ischemic injury, and cancer. The data presented herein underscore the therapeutic potential of targeting these pathways for conditions characterized by mitochondrial dysfunction.
The maintenance of mitochondrial membrane potential (ΔΨm) is critical for mitochondrial function, governing processes from ATP production to protein import and calcium homeostasis [2]. Loss of ΔΨm represents a well-established trigger for the ubiquitin-dependent PINK1/Parkin mitophagy pathway, which identifies and tags depolarized mitochondria for degradation. In parallel, receptor-mediated mitophagy pathways employ a distinct mechanism: mitochondria-anchored receptors that directly tether autophagy machinery to mitochondrial substrates without requiring ubiquitination [34] [35].
The outer mitochondrial membrane (OMM) proteins FUNDC1, BNIP3, and NIX (also known as BNIP3L) have emerged as central regulators of ΔΨm-sensitive mitophagy, functioning as molecular sensors that translate alterations in mitochondrial physiology into selective autophagic degradation. These receptors respond to diverse cellular stresses—including hypoxia, oxidative stress, and metabolic perturbations—that ultimately converge on ΔΨm regulation. Recent advances have elucidated their unique structural features, activation mechanisms, and downstream effectors, positioning them as key nodes in the mitochondrial quality control network with profound implications for health and disease [36] [37] [38].
Molecular Mechanisms of Mitophagy Receptors
FUNDC1 Signaling Pathway
FUNDC1 is a mitochondrial outer membrane protein characterized by a conserved LC3-interacting region (LIR) that enables direct binding to autophagy proteins LC3 and GABARAP. Its activity is primarily regulated through post-translational modifications that alter its affinity for autophagy machinery:
- Phosphorylation Regulation: Under basal conditions, FUNDC1 is phosphorylated at Ser13 by CK2 and Tyr18 by Src kinase, which inhibits its interaction with LC3. During hypoxia or mitochondrial stress, phosphatases (including PGAM5) dephosphorylate these sites while ULK1 phosphorylates Ser17, enhancing LC3 binding and mitophagy initiation [34].
- Pathway Initiation: Unlike BNIP3/NIX, FUNDC1 directly recruits the upstream FIP200/ULK1 complex through a FIP200-interacting (FIR) motif, facilitating autophagosome biogenesis at mitochondrial surfaces [37].
- Functional Significance: FUNDC1-mediated mitophagy protects against ischemic injury in multiple tissue contexts. In spinal cord injury models, FUNDC1 overexpression enhanced mitophagy, inhibited mitochondria-dependent apoptosis, improved mitochondrial membrane potential, and promoted functional recovery [39].
BNIP3 and NIX Signaling Pathways
BNIP3 and NIX are atypical BH3-only proteins that function as homodimers in the OMM. They employ a distinct molecular mechanism from FUNDC1 for mitophagy initiation:
- Activation Cycle: In their inactive state, BNIP3 and NIX exist as phosphorylated monomers. Upon mitophagy induction, dephosphorylation triggers dimerization, creating the active receptor form with enhanced LC3/GABARAP binding capacity [35].
- Alternative Pathway: Recent groundbreaking research reveals that BNIP3 and NIX cannot directly bind FIP200. Instead, they initiate mitophagy by recruiting WIPI2 and WIPI3 proteins, core components of the autophagy machinery that typically function downstream of ULK1 complex activation. This represents a non-canonical initiation pathway that bypasses traditional upstream regulators [37].
- Mitochondrial Dynamics Coordination: BNIP3 induces mitochondrial translocation of Drp1, promoting mitochondrial fission that facilitates mitophagy. Inhibition of Drp1 reduces BNIP3-mediated mitophagy and increases cell death, indicating functional integration between fission and mitophagy pathways [40].
Table 1: Comparative Molecular Mechanisms of Mitophagy Receptors
| Feature | FUNDC1 | BNIP3/NIX |
|---|---|---|
| Primary Activation Signals | Hypoxia, ROS, ischemia | Hypoxia, erythroid differentiation, energy stress |
| Structural Organization | Monomer with LIR domain | Phosphorylation-regulated dimer |
| Upstream Machinery Recruitment | Direct FIP200/ULK1 binding via FIR motif | WIPI2/WIPI3 recruitment; FIP200-independent |
| Key Regulatory Modifications | Ser13 (CK2), Tyr18 (Src), Ser17 (ULK1) phosphorylation | Ser212 phosphorylation regulates dimerization |
| LC3/GABARAP Preference | Binds both LC3 and GABARAP | Preferential GABARAP binding |
| Interaction with Mitochondrial Dynamics | Associates with OPA1 and DRP1 | Recruits DRP1 to mitochondria |
Integration with Mitochondrial Dynamics and ΔΨm Sensing
Mitophagy receptors operate within a broader mitochondrial quality control system that continuously monitors ΔΨm through several integrated mechanisms:
- Fission-Fusion Balance: Mitochondrial fission, mediated by Drp1 recruitment to mitochondrial membranes, enables the isolation of depolarized mitochondrial segments for selective removal. BNIP3 directly promotes Drp1 translocation, creating a molecular link between mitophagy initiation and fission machinery [40] [10].
- Membrane Potential Sensing: While these receptors do not directly sense ΔΨm like PINK1, they respond to physiological changes associated with ΔΨm loss, including increased ROS production and metabolic alterations. BNIP3 and NIX mitigate mitochondrial ROS accumulation, providing protection against ferroptosis—a ROS-dependent cell death pathway [36].
- Cross-talk with Ubiquitin Pathway: Under oxidative stress conditions, BNIP3 can promote Parkin translocation to mitochondria, suggesting potential integration points between receptor-mediated and ubiquitin-dependent mitophagy pathways [40].
Diagram 1: Molecular initiation pathways of receptor-mediated mitophagy. Note the distinct upstream machinery recruitment between FUNDC1 and BNIP3/NIX pathways.
Quantitative Data and Functional Comparisons
Table 2: Functional Roles and Disease Associations of Mitophagy Receptors
| Parameter | FUNDC1 | BNIP3 | NIX |
|---|---|---|---|
| Primary Physiological Functions | Hypoxic mitophagy, mitochondrial quality control | Hypoxic mitophagy, mitochondrial fragmentation | Erythroid maturation, developmental mitophagy |
| Key Binding Partners | LC3, FIP200, OPA1, DRP1 | LC3, WIPI3, DRP1, Parkin | LC3, WIPI2, DRP1 |
| Protective Roles | Reduces Aβ1-42 by 35% in AD models, improves functional recovery after SCI | Protects against ferroptosis, limits mtROS | Essential for mitochondrial clearance in reticulocytes |
| Disease Associations | Alzheimer's disease, spinal cord injury, cardiac ischemia | Cancer, neurodegeneration, heart failure | Anemia, neurodegeneration, Parkinson's disease |
| Experimental Models | APP/PS1 mice, rat SCI models, OGD cell models | Bnip3-/- mice, ferroptosis models, cardiac myocytes | Nix-/- mice, erythroid differentiation models |
The functional significance of these receptors is evidenced by quantitative data from disease models:
- Neuroprotection: In Alzheimer's disease models, restoration of FUNDC1 activity reduces soluble Aβ1-42 by 35% and suppresses GSK-3β-mediated Tau phosphorylation [34]. Postmortem AD brains show reduced FUNDC1 activity with hyperphosphorylated inhibitory sites correlating with pathological burden.
- Ferroptosis Protection: BNIP3/NIX double knockout cells exhibit enhanced sensitivity to ferroptosis, which is completely rescued by wild-type BNIP3 and NIX, but not by mitophagy-deficient mutants, demonstrating the essential role of their mitophagic function in preventing this iron-dependent cell death [36].
- Developmental Clearance: During erythroid maturation, NIX-mediated mitophagy enables complete mitochondrial clearance, with Nix-/- mice exhibiting anemia due to defective erythrocyte maturation [35] [38].
Experimental Methodologies
Key Research Reagent Solutions
Table 3: Essential Research Reagents for Studying Receptor-Mediated Mitophagy
| Reagent Category | Specific Examples | Research Application | Key Findings Enabled |
|---|---|---|---|
| Genetic Models | BNIP3/NIX DKO HeLa cells [36], FUNDC1 knockout/overexpression models [39], Nix-/- mice [35] | Loss-of-function and gain-of-function studies | Established essential roles in ferroptosis protection, neuronal survival, and erythroid development |
| Biochemical Assays | Purified receptor domains (soluble GFP/GST-tagged) [37], in vitro binding assays, phosphorylation analysis | Molecular mechanism studies | Revealed distinct initiation pathways (FIP200 vs WIPI recruitment) and phosphorylation regulation |
| Chemical Tools | Deferiprone (DFP) [37], Menadione (MN) [41], 3-methyladenine (3-MA) [39] | Inducing or inhibiting specific pathway components | Demonstrated pathway-specific mitophagy induction and functional outcomes |
| Visualization Reagents | GFP-LC3, Mito-DsRed, TMRE/TMRM (ΔΨm sensors), JC-1 [39] | Live-cell imaging and mitophagy quantification | Visualized mitochondrial recruitment of autophagy proteins and ΔΨm changes |
Critical Experimental Protocols
Reconstitution of Mitophagy Initiation
For analyzing molecular interactions between mitophagy receptors and autophagy machinery:
- Protein Purification: Express and purify soluble domains of mitophagy receptors (e.g., residues 1-134 for NIX, 1-138 for BNIP3) with GFP or GST tags to mimic monomeric or dimeric states [37].
- Interaction Mapping: Employ microscopy-based bead assays with purified autophagy components (FIP200 Claw domain, WIPI proteins, LC3/GABARAP family members).
- Functional Validation: Test binding specificity by introducing LIR/FIR motif mutations (e.g., FUNDC1 LIR mutant, NIX L27A) and confirm loss of interaction.
- Kinase/Phosphatase Studies: Incubate receptors with purified kinases (TBK1, ULK1, Src, CK2) or lambda protein phosphatase to assess phosphorylation-dependent regulation.
This approach revealed that FUNDC1 and BCL2L13 directly bind FIP200, while BNIP3 and NIX utilize WIPI proteins for autophagy initiation [37].
In Vivo Functional Assessment
For evaluating receptor function in disease models:
- Gene Manipulation: Employ AAV-mediated overexpression (e.g., AAV-DJ-FUNDC1 in spinal cord injury models) or lentiviral shRNA knockdown in target tissues [39].
- Functional Outcomes: Assess recovery using standardized behavioral scales (e.g., Basso-Beatie-Bresnahan scoring for locomotor function in SCI).
- Histopathological Analysis: Quantify neuronal survival (Nissl staining), tissue organization (H&E), and mitochondrial morphology (electron microscopy).
- Biochemical Markers: Evaluate mitophagy flux (LC3-II/I ratio, p62 degradation), apoptosis (caspase cleavage, Bcl-2/Bax ratio), and mitochondrial function (JC-1 for ΔΨm, ROS assays).
Pathophysiological Implications and Therapeutic Targeting
Dysregulation of receptor-mediated mitophagy contributes significantly to human disease, offering promising therapeutic targets:
- Neurodegenerative Disorders: In Alzheimer's disease, impaired FUNDC1 function exacerbates Aβ and Tau pathology. Postmortem AD brains show approximately 30-50% reduction in basal mitophagy levels compared to normal populations [34]. Restoration of mitophagy flux reduces both pathological hallmarks, suggesting enhancement of receptor activity as a potential therapeutic strategy.
- Ischemic Injuries: FUNDC1-mediated mitophagy protects against neuronal loss in spinal cord injury by inhibiting mitochondria-dependent apoptosis and improving mitochondrial function. Overexpression enhances autophagosome formation and functional recovery, with significant improvements in locomotor scores observed within 72 hours to 7 days post-injury [39].
- Metabolic Diseases: BNIP3 and NIX regulate mitochondrial quality in high-metabolic-demand tissues. In cardiac progenitor cells, simultaneous knockdown of both BNIP3L/NIX and FUNDC1 leads to dysfunctional mitochondrial networks with increased susceptibility to oxidative stress-mediated death [35].
- Cancer Implications: The balance of receptor-mediated mitophagy can influence tumor progression, with BNIP3 acting as a context-dependent oncogene or tumor suppressor. Cancer cells often exhibit altered ΔΨm regulation, with hyperpolarized mitochondria observed in glioblastoma and ovarian cancer [2].
Diagram 2: Pathophysiological consequences of receptor-mediated mitophagy dysfunction. Multiple pathological triggers converge on receptor dysregulation, leading to mitochondrial dysfunction and disease-specific manifestations.
The receptor-mediated mitophagy pathways governed by FUNDC1, BNIP3, and NIX represent sophisticated molecular systems for maintaining mitochondrial quality control in response to ΔΨm alterations. While significant progress has been made in elucidating their distinct activation mechanisms and downstream effectors, several frontiers demand further investigation:
- Structural Basis: High-resolution structures of full-length receptors in complex with their binding partners would illuminate precise molecular interactions and inform targeted therapeutic development.
- Hierarchical Relationships: The contextual dominance and cross-regulation between different receptor pathways under varying physiological conditions remains incompletely mapped.
- Therapeutic Translation: Small molecule modulators targeting receptor activity or phosphorylation states hold promise for conditions characterized by mitochondrial dysfunction but require refined development and validation.
- Organellar Networks: Emerging evidence suggests coordination between mitochondrial quality control and other organellar systems, including ER-mitochondria contact sites and lysosomal function, representing an expanding research frontier.
The molecular dissection of these pathways continues to reveal unexpected complexity and flexibility in the autophagy machinery, providing both challenges and opportunities for therapeutic intervention in the expanding spectrum of diseases linked to mitochondrial dysfunction.
Mitochondrial membrane potential (ΔΨm), generated by the proton gradient across the inner mitochondrial membrane during oxidative phosphorylation, serves as the paramount indicator of mitochondrial health and the primary trigger for mitophagy. The collapse of ΔΨm acts as an initiating signal for dedicated quality control systems that identify, isolate, and target damaged mitochondria for degradation via lysosomal pathways. This whitepaper delineates the central role of ΔΨm dissipation in activating core mitophagy pathways, including the canonical PINK1-Parkin axis and receptor-mediated alternative routes. We synthesize the molecular mechanisms, present quantitative data on regulatory dynamics, and detail experimental methodologies for interrogating ΔΨm-dependent mitophagy. Furthermore, we explore the therapeutic implications of targeting this fundamental process in neurodegenerative diseases and cancer, providing a comprehensive resource for researchers and drug development professionals advancing mitochondrial quality control research.
The mitochondrial membrane potential (ΔΨm), typically ranging from -150 to -180 mV, represents a critical electrochemical gradient essential for ATP production, protein import, and metabolite transport. This potential is not merely a prerequisite for energy transduction but functions as a sophisticated cellular biosensor that continuously monitors mitochondrial functional integrity. The collapse of ΔΨm serves as the earliest detectable molecular event signaling mitochondrial distress, preceding overt organelle dysfunction. In the context of quality control, ΔΨm dissipation transitions from a damage sensor to a definitive "eat-me" signal, initiating a cascade of molecular events that ultimately designate compromised mitochondria for autophagic degradation. This primacy of ΔΨm in mitophagy initiation establishes it as a critical regulatory node whose manipulation offers promising therapeutic potential across numerous disease contexts, particularly neurodegenerative conditions like Parkinson's and Alzheimer's disease where mitochondrial quality control is compromised [34] [42].
Molecular Mechanisms: How ΔΨm Dissipation Triggers Mitophagy
The Canonical PINK1/Parkin Pathway
In healthy, polarized mitochondria, PTEN-induced putative kinase 1 (PINK1) is continuously imported through the translocase of the outer membrane (TOM) and inner membrane (TIM23) complexes. Upon entry into the inner membrane, PINK1 undergoes cleavage by the mitochondrial protease presenilin-associated rhomboid-like protein (PARL) and is subsequently degraded by the proteasome, maintaining low basal levels [43] [44]. However, when ΔΨm collapses, this import pathway fails, leading to PINK1 accumulation on the outer mitochondrial membrane (OMM). Here, PINK1 undergoes autophosphorylation and forms stable dimers that recruit and activate the cytosolic E3 ubiquitin ligase Parkin [43] [45].
Activated Parkin then ubiquitinates numerous OMM proteins, including mitofusins (MFN1/2), VDAC1, and TOM20, generating phosphorylated ubiquitin chains. These chains serve as recognition sites for autophagy adaptor proteins—optineurin (OPTN), nuclear dot protein 52 (NDP52), and sequestosome-1 (p62/SQSTM1)—which simultaneously bind ubiquitin chains via their ubiquitin-binding domains and LC3 on developing phagophores through LC3-interacting regions (LIRs) [34] [44]. This dual engagement effectively tethers damaged mitochondria to the growing autophagosomal membrane, ensuring their selective encapsulation.
Table 1: Key Proteins in PINK1/Parkin-Mediated Mitophagy and Their Functions
| Protein | Function | Regulation by ΔΨm |
|---|---|---|
| PINK1 | Serine/threonine kinase; Damage sensor | Stabilized on OMM upon ΔΨm loss |
| Parkin | E3 ubiquitin ligase; Amplifies "eat-me" signal | Recruited to mitochondria by PINK1; Activated via phosphorylation |
| OPTN/NDP52 | Autophagy adaptors; Bridge ubiquitinated mitochondria to LC3 | Recruited to phospho-ubiquitin chains on OMM |
| TBK1 | Kinase; Enhances adaptor affinity for ubiquitin and LC3 | Activated by PINK1/Parkin signaling; Phosphorylates OPTN |
| LC3/GABARAP | Phagophore membrane proteins; Receptor for LIR motifs | Processed and lipidated during autophagy initiation |
This PINK1-Parkin amplification system creates a sensitive response mechanism where minimal ΔΨm dissipation can trigger complete mitochondrial removal, preventing the propagation of dysfunctional organelles [45] [44].
ΔΨm-Sensitive Receptor-Mediated Pathways
Beyond the ubiquitin-dependent PINK1/Parkin axis, multiple ubiquitin-independent pathways同样 respond to ΔΨm collapse through dedicated mitophagy receptors on the OMM. The FUN14 domain-containing 1 (FUNDC1) pathway represents a particularly sophisticated ΔΨm-responsive mechanism. Under normal conditions, FUNDC1 is phosphorylated at Ser13 (by CK2) and Tyr18 (by Src kinase), inhibiting its interaction with LC3. When ΔΨm dissipates, phosphatases such as PGAM5 dephosphorylate these sites while ULK1 phosphorylates Ser17, markedly enhancing FUNDC1's affinity for LC3 and promoting autophagosome engagement [34].
Similar ΔΨm-sensitive mechanisms govern other receptors, including BNIP3 and NIX/BNIP3L, which are transcriptionally upregulated during hypoxia but also respond to depolarization through conformational changes that expose their LIR domains [34] [42]. These receptor-mediated pathways provide complementary, tissue-specific backup systems that ensure robust mitochondrial quality control even when PINK1/Parkin signaling is compromised, as occurs in certain forms of Parkinson's disease [42].
Diagram 1: ΔΨm Collapse Triggers Multiple Mitophagy Pathways. The loss of mitochondrial membrane potential stabilizes PINK1 on the OMM, initiating the PINK1-Parkin ubiquitin-dependent pathway while simultaneously activating receptor-mediated pathways through FUNDC1 dephosphorylation. These convergent mechanisms ultimately recruit LC3 to engulp damaged mitochondria.
Quantitative Dynamics of ΔΨm in Mitophagy Regulation
The relationship between ΔΨm dissipation and mitophagy initiation follows precise quantitative parameters that define activation thresholds, kinetics, and coordination with other mitochondrial quality control processes. Research indicates that a ΔΨm reduction of approximately 50% or more is required to stabilize sufficient PINK1 on the OMM to activate Parkin-mediated mitophagy [44]. This threshold mechanism prevents unnecessary degradation of mildly compromised mitochondria that may recover function.
Time-course analyses reveal that PINK1 stabilizes on the OMM within 5-30 minutes of ΔΨm collapse, with Parkin recruitment occurring within 30-60 minutes in cultured cells. Complete ubiquitination of OMM proteins follows over 1-3 hours, with LC3 recruitment and autophagosome formation typically observed within 3-6 hours post-depolarization [44]. The entire process from ΔΨm dissipation to lysosomal degradation can be completed within 12-24 hours under experimental conditions.
Table 2: Quantitative Parameters of ΔΨm-Dependent Mitophagy
| Parameter | Value/Range | Experimental Context |
|---|---|---|
| ΔΨm Threshold for Initiation | ~50% reduction | CCCP-treated HeLa cells [44] |
| PINK1 Stabilization | 5-30 minutes | Antimycin A/Oligomycin treatment [43] |
| Parkin Recruitment | 30-60 minutes | Live imaging of YFP-Parkin [44] |
| Ubiquitination Completion | 1-3 hours | Western blot of OMM proteins [44] |
| Autophagosome Formation | 3-6 hours | Immunofluorescence of LC3 puncta [44] |
| Complete Degradation | 12-24 hours | Mito-Keima assay [46] |
Notably, ΔΨm dissipation coordinates mitophagy with mitochondrial dynamics through shared regulatory components. Depolarization-induced PINK1 activation not only triggers mitophagy but also inhibits mitochondrial fusion by promoting proteasomal degradation of mitofusins, thereby isolating damaged organelles from the healthy network [34]. This coordination ensures that compromised mitochondria are efficiently segregated before degradation.
Experimental Methodologies for Monitoring ΔΨm-Dependent Mitophagy
ΔΨm Measurement Techniques
Accurate quantification of ΔΨm is essential for establishing its causal relationship with mitophagy initiation. Tetramethylrhodamine ethyl ester (TMRE) and tetramethylrhodamine methyl ester (TMRM) remain the gold-standard fluorescent indicators for ΔΨm measurement due to their potential-dependent accumulation in the mitochondrial matrix. These cationic dyes exhibit increased fluorescence intensity with higher ΔΨm, allowing quantitative assessment through flow cytometry or live-cell imaging. The JC-1 dye offers an alternative ratiometric approach, forming red fluorescent J-aggregates in polarized mitochondria while remaining green and monomeric in depolarized organelles, providing an internal calibration reference [46].
Critical experimental considerations include using low dye concentrations (typically 20-100 nM) to avoid artifacts, establishing baseline measurements before perturbations, and validating results with ΔΨm dissipators (e.g., CCCP, FCCP) and stabilizers (e.g., cyclosporin A). Concurrent measurement of mitochondrial mass markers (e.g., TOM20, COX IV) helps distinguish true ΔΨm loss from mitochondrial volume changes.
Mitophagy Detection Assays
The Mito-Keima assay represents one of the most specific methods for quantifying mitophagy flux. Keima is a pH-sensitive fluorescent protein that exhibits excitation maxima at 440 nm in neutral environments (mitochondria) and 586 nm in acidic environments (lysosomes). When targeted to mitochondria (Mito-Keima), the ratio of 586/440 nm excitation fluorescence provides a direct measure of mitochondrial delivery to lysosomes, independent of autophagosome formation [46]. This assay enables tracking of mitophagy dynamics over time in live cells and can be combined with ΔΨm measurements for correlative analysis.
LC3-based assays monitor the recruitment of this essential autophagy protein to mitochondria following ΔΨm collapse. Immunofluorescence detection of LC3 puncta colocalizing with mitochondrial markers (e.g., TOM20, COX IV) provides spatial information about mitophagy initiation, while Western blot analysis of LC3-II conversion offers quantitative assessment of autophagosome formation. These approaches are often complemented with lysosomal inhibitors (e.g., bafilomycin A1) to differentiate between increased autophagosome formation versus impaired lysosomal degradation [46].
Diagram 2: Experimental Workflow for Monitoring ΔΨm-Dependent Mitophagy. A comprehensive approach combining ΔΨm measurements with mitophagy detection assays enables robust quantification of the relationship between membrane potential dissipation and mitochondrial degradation.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for ΔΨm and Mitophagy Studies
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| ΔΨm Indicators | TMRE, TMRM, JC-1, MT-1 MitoMP Detection Kit | Quantitative measurement of mitochondrial membrane potential |
| Mitophagy Reporters | Mito-Keima, Mito-QC, GFP-LC3/RFP-LC3 | Specific detection of mitochondrial delivery to lysosomes |
| Chemical Inducers | CCCP, FCCP, Oligomycin/A, Antimycin A | Experimental ΔΨm dissipation to induce mitophagy |
| Small Molecule Modulators | SP11 (Fis1 inhibitor), Fundc1-targeted agents | Targeted manipulation of specific mitophagy pathways |
| Pathway Inhibitors | Bafilomycin A1 (lysosomal inhibitor), 3-MA (autophagy inhibitor) | Blockade of specific mitophagy stages to measure flux |
| Antibodies | Anti-PINK1, Anti-Parkin, Anti-LC3, Anti-TOM20 | Immunodetection of key mitophagy proteins and colocalization |
Therapeutic Implications and Future Directions
The primacy of ΔΨm in mitophagy initiation presents compelling therapeutic opportunities for modulating mitochondrial quality control in human disease. In neurodegenerative disorders like Parkinson's disease, where PINK1-Parkin signaling is often compromised, targeting alternative ΔΨm-sensitive pathways offers promising alternative strategies. Small molecule activators of PINK1 or Parkin have demonstrated potential in preclinical models, enhancing clearance of damaged mitochondria and ameliorating disease phenotypes [45].
The recent discovery of SP11, a small molecule that binds to Fis1's Cys41 site and prevents abnormal mitochondrial fragmentation under oxidative stress, exemplifies this therapeutic approach. By specifically inhibiting stress-induced mitochondrial fragmentation without disrupting physiological division, SP11 and similar compounds represent a targeted strategy for maintaining mitochondrial integrity in conditions like Parkinson's disease and amyotrophic lateral sclerosis [47].
In Alzheimer's disease, where FUNDC1 dysregulation contributes to pathogenesis, restoring its phosphorylation dynamics presents another ΔΨm-centered therapeutic avenue. Postmortem AD brains show reduced FUNDC1 activity with hyperphosphorylated Ser13/Tyr18, correlating with Aβ plaque burden and Tau neurofibrillary tangles. Notably, restoring FUNDC1 dephosphorylation in APP/PS1 mice reduces soluble Aβ1-42 by 35% and suppresses GSK-3β-mediated Tau phosphorylation, highlighting the therapeutic potential of modulating this ΔΨm-sensitive pathway [34].
Emerging nanotechnology approaches further expand these opportunities through mitochondrial-targeted drug delivery systems. Liposomes, polymer nanoparticles, and inorganic nanoparticles functionalized with mitochondrial-targeting ligands (e.g., triphenylphosphonium, mitochondrial-penetrating peptides) can enhance drug delivery to mitochondria, potentially enabling more precise modulation of ΔΨm and mitophagy with reduced off-target effects [48].
Non-pharmacological interventions like exercise also demonstrate therapeutically relevant effects on ΔΨm-sensitive mitophagy pathways. Regular physical activity activates mitophagy through AMPK/ULK1 and PINK1/Parkin signaling, enhancing mitochondrial function and antioxidant capacity in Parkinson's disease models [42]. This suggests that lifestyle interventions may complement pharmacological approaches for maintaining mitochondrial quality control.
The expanding toolkit for investigating ΔΨm and mitophagy, including advanced fluorescent probes, genetic reporters, and small molecule modulators, continues to accelerate both basic research and therapeutic development. As our understanding of the quantitative relationships between ΔΨm dissipation and mitophagy initiation deepens, so too will opportunities for targeted interventions across the spectrum of mitochondrial diseases.
Tools and Techniques: Probing Membrane Potential for Research and Therapy
Fluorescent Dyes and Live-Cell Imaging for Dynamic ΔΨm Quantification
Mitochondrial membrane potential (ΔΨm) is the electrical potential difference across the inner mitochondrial membrane, representing the central intermediate in oxidative energy metabolism. This potential drives ATP synthesis through oxidative phosphorylation and serves as a key indicator of mitochondrial health and functional status. Quantitative measurement of dynamic changes in ΔΨm provides crucial insights into cellular energy regulation, particularly within the context of mitochondrial quality control mechanisms. Disruption of ΔΨm is intimately linked to the initiation of mitophagy, the selective autophagic degradation of damaged mitochondria, making accurate ΔΨm quantification essential for understanding cellular homeostasis in health and disease.
This technical guide examines the principles and methodologies for quantitative ΔΨm assessment using fluorescent dyes and live-cell imaging, with emphasis on applications in mitochondrial quality control research. We provide researchers with detailed protocols, quantitative comparisons of available tools, and integration strategies for connecting ΔΨm measurements to broader mitochondrial quality control mechanisms, including their relevance for drug development in neurodegenerative diseases, cardiovascular conditions, and metabolic disorders.
Fluorescent Probes for ΔΨm Quantification
Probe Selection Criteria
Choosing appropriate fluorescent probes represents the foundational step in obtaining reliable ΔΨm measurements. Several critical factors must be considered during probe selection, including binding affinity, photostability, toxicity, quantization capability, and compatibility with other fluorescent markers in multiplexed assays. Additionally, researchers must balance the need for high signal-to-noise ratio against the requirement to maintain physiological expression levels of fluorescent proteins to avoid perturbing the native molecular networks under investigation [49].
Commercially Available ΔΨm Probes
Table 1: Characteristics of Common ΔΨm Fluorescent Probes
| Probe Name | Excitation/Emission Maxima | Detection Mode | Key Advantages | Quantitative Capability | References |
|---|---|---|---|---|---|
| TMRM | 548/573 nm | Quench/Non-quench | Reversible binding, suitable for kinetics | Yes (absolute values in mV) | [22] [50] |
| LDS 698 | 648/668 nm | Multiple imaging platforms | High sensitivity to subtle changes, photostable | Semi-quantitative | [51] |
| JC-1 | 514/529 nm (monomer); 585/590 nm (J-aggregate) | Ratiometric | Built-in ratio metric capability | Semi-quantitative | [22] |
| Rhod-2 AM | 552/581 nm | Intensity-based | Can be combined with Ca²⁺ measurements | Semi-quantitative (relative changes) | [50] |
Advanced Probe Properties and Applications
Tetramethylrhodamine methyl ester (TMRM) remains the gold standard for quantitative ΔΨm measurements due to its Nernstian behavior and reversible binding properties, allowing calculation of absolute membrane potential values in millivolts [22]. The recently characterized LDS 698 dye offers exceptional advantages for detecting subtle ΔΨm changes in live cells, with demonstrated utility across fluorescence microscopy, flow cytometry, and plate reader assays. Its robustness, photostability, and non-toxicity enable prolonged live-cell imaging sessions essential for capturing mitochondrial dynamics [51].
For ratiometric measurements, JC-1 provides an internal calibration mechanism through its concentration-dependent formation of J-aggregates, which exhibit distinct spectral shifts at higher membrane potentials. However, its non-equilibrium accumulation presents interpretation challenges for quantitative applications [22]. Newer generation dyes continue to emerge with improved photophysical properties and reduced cellular toxicity profiles.
Quantitative Measurement Methodologies
Absolute ΔΨm Quantification Using TMRM
The most rigorous approach for absolute ΔΨm quantification involves modeling fluorescent potentiometric probe compartmentation and dynamics using a biophysical framework. This method accounts for multiple factors influencing probe distribution, including plasma membrane potential (ΔΨp), matrix-to-cell volume ratio, binding coefficients, activity coefficients, background fluorescence, and optical dilution effects [22].
The fundamental principle underlying this approach is the Nernstian equilibrium distribution of lipophilic cations between compartments:
[ \Delta\Psi = -\frac{RT}{F} \ln\frac{[C{in}]}{[C{out}]} ]
Where R is the gas constant, T is temperature, F is Faraday's constant, and [Cin] and [Cout] represent the intra- and extramitochondrial dye concentrations, respectively.
Table 2: Key Parameters for Absolute ΔΨm Calibration
| Parameter | Description | Measurement Method | Impact on Calculation |
|---|---|---|---|
| Matrix:Cell Volume Ratio | Fractional volume occupied by mitochondria | Confocal microscopy with mitochondrial markers | Directly affects concentration calculations |
| Binding Coefficients | High- and low-affinity binding sites | Fluorescence titration experiments | Influences apparent dye concentration |
| Activity Coefficients | Effective ionic activities | Theoretical estimation based on matrix composition | Affects Nernst equation application |
| ΔΨp Contribution | Plasma membrane potential | Parallel measurement with bis-oxonol dyes | Critical for deconvoluting mitochondrial signal |
Experimental Protocol for Absolute ΔΨm Assay
Step 1: Cell Culture and Preparation
- Culture primary cortical neurons or other cell types of interest on poly-ornithine-coated coverglasses or chambered coverglasses [22].
- Maintain optimal cell density (typically 50-70% confluence) for individual cell resolution while providing sufficient signal intensity.
Step 2: Dye Loading and Incubation
- Prepare TMRM working solution in imaging buffer (typically 20-50 nM for non-quench mode).
- Incubate cells with TMRM for 30-40 minutes at physiological temperature to ensure equilibrium distribution.
- Include PMPI (2-5 nM) for simultaneous ΔΨp measurement if required by experimental design [22].
Step 3: Live-Cell Imaging Setup
- Maintain uncompromised incubation conditions (37°C, 5% CO₂) throughout imaging [49].
- Implement reliable autofocus mechanism to maintain focus during prolonged acquisitions.
- Balance spatial resolution requirements against photo-toxicity concerns [49].
- Set appropriate temporal resolution based on biological process timescales (typically 30-60 second intervals) [49].
Step 4: Image Acquisition and Calibration
- Acquire time-lapse fluorescence images using optimized microscope settings.
- Perform parallel volume ratio measurements using confocal microscopy with mitochondrial markers.
- Determine binding coefficients through fluorescence titration experiments.
- Apply mathematical model to deconvolute ΔΨm from raw fluorescence intensities [22].
Step 5: Validation and Quality Control
- Compare imaged cell viability with non-imaged controls to assess photo-toxicity [49].
- Validate measurements using pharmacological agents (e.g., FCCP for depolarization).
- Perform sensitivity analysis to estimate measurement errors (typically <11 mV for absolute values) [22].
Data Analysis and Interpretation
Using this calibrated approach, resting ΔΨm in cultured rat cortical neurons measures approximately -139 mV, with physiological regulation observed between -108 mV and -158 mV during metabolic activation [22]. The maximal rate of mitochondrial ATP production approximately doubles with each 10 mV increase in ΔΨm, while reactive oxygen species emission rises exponentially at strongly polarized potentials, highlighting the critical relationship between ΔΨm magnitude and functional outputs [22].
Integration with Mitochondrial Quality Control Assessment
ΔΨm in Mitophagy Regulation
Mitochondrial membrane potential serves as a key regulator in mitochondrial quality control systems, particularly in initiating mitophagy. The PINK1-Parkin pathway, the best characterized mitophagy mechanism, directly responds to ΔΨm loss. In healthy mitochondria with normal ΔΨm, PINK1 is imported through the TIM/TOM complex and subsequently degraded. When ΔΨm collapses, PINK1 import is impaired, leading to its accumulation on the outer mitochondrial membrane where it recruits Parkin and initiates ubiquitin-dependent mitophagy [52] [53].
Simultaneous monitoring of ΔΨm together with mitophagy markers provides critical insights into the temporal relationship between mitochondrial dysfunction and quality control activation. This integrated approach is particularly valuable for evaluating potential therapeutic compounds that modulate mitophagy in neurodegenerative diseases where impaired mitochondrial clearance contributes to pathology [54] [53].
Multi-Parameter Assessment of Mitochondrial Health
Comprehensive mitochondrial assessment requires moving beyond single-parameter measurements. Combining ΔΨm quantification with additional parameters provides a systems-level view of mitochondrial function:
- Reactive Oxygen Species (ROS): Use MitoSOX Red for mitochondrial superoxide detection [50]
- Mitochondrial Calcium: Employ Rhod-2 AM for matrix calcium measurements [50]
- Mitophagy Flux: Monitor using LC3-II puncta formation or mitophagy reporters [55] [53]
- Morphological Dynamics: Assess fission/fusion balance through mitochondrial network analysis [27] [52]
Advanced high-content analysis platforms enable simultaneous multiparametric data collection from the same cell population, revealing functional interactions between different aspects of mitochondrial biology [56].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for ΔΨm and Quality Control Studies
| Reagent Category | Specific Examples | Primary Function | Application Notes |
|---|---|---|---|
| ΔΨm Indicators | TMRM, LDS 698, JC-1 | Quantitative and semi-quantitative ΔΨm measurement | TMRM for absolute values; LDS 698 for subtle changes [51] [22] |
| ROS Detection | MitoSOX Red, CellROX | Mitochondrial superoxide and oxidative stress measurement | Use with antioxidant controls for specificity [56] [50] |
| Calcium Indicators | Rhod-2 AM, X-Rhod-1 | Mitochondrial calcium monitoring | AM esters for cell permeability [50] |
| Mitophagy Reporters | mt-Keima, LC3-GFP/RFP | Mitophagy flux quantification | mt-Keima pH-sensitive for lysosomal delivery [55] |
| Morphology Stains | MitoTracker dyes, HCS Mitochondrial Health Kit | Mitochondrial network visualization | Fixed vs. live-cell compatible variants [56] |
| Viability Indicators | HCS LIVE/DEAD kits, Hoechst 33342 | Cell health and cytotoxicity assessment | Essential for data normalization [56] |
| High-Content Analysis | HCS NuclearMask stains, CellMask | Automated image analysis and segmentation | Enable high-throughput screening [56] |
Technical Considerations and Pitfalls
Live-Cell Imaging Optimization
Successful quantitative live-cell imaging requires careful attention to technical细节 that are often under-appreciated in conventional cell biology applications. Maintaining cell health throughout imaging sessions is paramount, as photo-damaged cells may not display immediate morphological phenotypes yet exhibit compromised physiology that invalidates experimental results [49].
Critical optimization parameters include:
Expression Level Control: Fluorescent proteins should be expressed at levels comparable to their natural counterparts to avoid perturbing molecular networks. Strong constitutive promoters (e.g., CMV) often produce non-physiological expression levels that can alter system behavior [49].
Spatial Resolution Balance: Higher spatial resolution requires more intense illumination, increasing photo-toxicity. Optimal settings should provide sufficient resolution for quantitative analysis while minimizing cellular stress [49].
Temporal Resolution Strategy: The imaging interval must adequately sample the biological process while avoiding excessive photo-exposure. For many mitochondrial processes, 30-60 second intervals provide reasonable balance [49].
Microenvironmental Stability: Maintaining uncompromised incubation conditions (temperature, CO₂, humidity) throughout imaging is essential for physiological relevance [49].
Methodological Validation in Mitophagy Studies
With increasing interest in chemical mitophagy modulators for therapeutic development, methodological rigor is essential. Recent analyses reveal frequent methodological flaws in mitophagy studies that lead to unreliable conclusions [55]. The Mitophagy Modulator Characterization System (MMCS) provides a framework for standardized assessment, emphasizing:
- Multiple orthogonal assays to confirm mitophagy induction
- Dose-response relationships and time course analyses
- Specificity controls using genetic and pharmacological approaches
- Functional validation in disease-relevant models [55]
Particularly for CNS applications, adequate representation of neuronal complexity is essential, as mitophagy regulation differs significantly between cell types [54].
Quantitative dynamic assessment of ΔΨm using fluorescent dyes and live-cell imaging provides powerful insights into mitochondrial function within the broader context of quality control mechanisms. The integration of absolute ΔΨm measurements with multiparametric analyses of related mitochondrial features enables comprehensive profiling of mitochondrial health in physiologically relevant environments.
As research continues to elucidate the complex relationships between ΔΨm dysregulation and human disease, particularly in neurodegeneration, cardiovascular pathology, and metabolic disorders, refined methodologies for ΔΨm quantification will play increasingly important roles in both basic research and drug development. The technical guidelines presented here provide a foundation for implementing robust, quantitative approaches to ΔΨm measurement that yield physiologically meaningful data while avoiding common methodological pitfalls.
Future methodological developments will likely focus on enhancing spatial and temporal resolution, improving multiplexing capabilities for parallel monitoring of additional parameters, and increasing throughput for drug screening applications. Additionally, better integration of ΔΨm measurements with other aspects of mitochondrial quality control will provide more holistic understanding of how mitochondrial functional status dictates cellular fate decisions.
Mitochondrial membrane potential (ΔΨm), the electrochemical gradient across the inner mitochondrial membrane, serves as a fundamental regulator of cellular bioenergetics and viability. Generated by proton pumps of the electron transport chain (ETC), this potential of 100-150 mV not only drives ATP synthesis but also facilitates metabolite transport, protein import, and reactive oxygen species (ROS) production [57] [58] [59]. Perhaps most critically within the context of quality control, ΔΨm serves as a key signal enabling the disposal of dysfunctional mitochondria through mitophagy, a selective autophagy pathway that eliminates damaged organelles [5] [59]. The precise manipulation of ΔΨm using pharmacological and genetic probes thus provides researchers with a powerful experimental approach to investigate mitochondrial quality control mechanisms and their relevance to human disease.
The investigation of ΔΨm in live cells has been complicated by a historical lack of tools for its direct manipulation without significant off-target effects [57] [60] [59]. This technical guide comprehensively details current methodological approaches for modulating ΔΨm using pharmacological uncouplers, ETC inhibitors, and emerging genetically encoded tools, with particular emphasis on their application in studying mitochondrial quality control pathways. We provide structured quantitative comparisons, detailed experimental protocols, and visualization of key signaling pathways to equip researchers with practical resources for implementing these techniques in their investigative workflows.
Theoretical Foundation: OXPHOS and ΔΨm Dynamics
Fundamental Principles of Oxidative Phosphorylation
Oxidative phosphorylation (OXPHOS) constitutes two autonomous modules: the electron transport chain (ETC), which reduces oxygen to water and generates ΔΨm, and ATP synthase, which consumes ΔΨm to produce ATP [58]. The ETC drives protons from the mitochondrial matrix to the intermembrane space, creating a proton motive force (Δp) consisting primarily (approximately 80%) of the electrical component (ΔΨm) with a smaller contribution from the pH gradient (ΔpH) [58]. ATP synthase consumes this potential energy by importing protons back to the matrix to phosphorylate ADP. Mitochondria where ΔΨm generation by the ETC is coupled to its consumption by ATP synthase are termed "coupled mitochondria," whereas those with reduced ATP synthase contribution to ΔΨm consumption are "leaky," and those with no ATP synthase-mediated consumption are "uncoupled" [58].
Table 1: Mitochondrial Bioenergetics States and ΔΨm Characteristics
| State | ΔΨm Level | Oxygen Consumption | ATP Production | Primary Characteristics |
|---|---|---|---|---|
| Coupled (State 4) | High | Low | Minimal | Resting state with limited ADP availability |
| Active Phosphorylation (State 3) | Moderate | High | Maximal | Active ATP synthesis with ample substrates |
| Uncoupled | Low | High | None | Proton leak dominates, energy dissipated as heat |
ΔΨm as a Signal for Mitochondrial Quality Control
Mitochondrial membrane potential serves as a critical determinant in mitochondrial quality control decisions, particularly in regulating PINK1-Parkin mediated mitophagy [4] [5]. Under physiological conditions, PTEN-induced kinase 1 (PINK1) is continuously imported into mitochondria and degraded. However, when ΔΨm is dissipated, PINK1 import is impaired, leading to its accumulation on the outer mitochondrial membrane where it recruits and activates the E3 ubiquitin ligase Parkin [4]. This pathway represents a fundamental mechanism whereby cells can identify and eliminate damaged mitochondria based on their inability to maintain membrane potential.
Figure 1: PINK1-Parkin Mitophagy Pathway Activation by ΔΨm Collapse. Under normal ΔΨm, PINK1 is imported and degraded. Mitochondrial depolarization prevents PINK1 import, leading to OMM accumulation, Parkin recruitment, ubiquitination of mitochondrial proteins, and ultimately mitophagic clearance. [4] [5]
Pharmacological and Genetic Probes for ΔΨm Manipulation
Mitochondrial Uncouplers
Uncouplers function as protonophores, dissipating the proton gradient across the inner mitochondrial membrane and thereby reducing ΔΨm without affecting ETC function. This results in increased oxygen consumption as the ETC attempts to restore the gradient, with energy released as heat rather than captured as ATP [60] [59].
Table 2: Characteristics of Common Mitochondrial Uncouplers
| Uncoupler | Mechanism | Effective Concentration | Key Applications | Limitations |
|---|---|---|---|---|
| FCCP | Proton ionophore | 1-5 μM (titration required) | Maximal respiration assessment | Inhibits cell proliferation at >2.5 μM; affects plasma membrane potential [60] [61] |
| Bam15 | Protonophore | 5-10 μM | Obesity, fatty liver disease research | Cell proliferation inhibition at >5 μM [60] [62] |
| DNP | Protonophore | 10-100 μM | Historical weight loss agent; metabolic studies | Narrow therapeutic window; safety concerns [62] [59] |
| UCP1 (Genetic) | Fatty acid-activated proton conductance | Doxycycline-inducible expression | Specific ΔΨm manipulation without proliferation effects | Requires fatty acids for activity (e.g., 300 μM oleate) [57] [60] [59] |
Electron Transport Chain Inhibitors
ETC inhibitors target specific complexes within the respiratory chain, ultimately affecting ΔΨm by reducing proton pumping activity. Their effects are complex and cell-type dependent, as demonstrated in vascular smooth muscle cells where ETC inhibitors induced both depolarization and mitochondrial fragmentation [61].
Table 3: Electron Transport Chain Inhibitors and Their Effects on ΔΨm
| Inhibitor | Target | Effect on ΔΨm | Effect on Respiration | Key Experimental Findings |
|---|---|---|---|---|
| Oligomycin | ATP synthase (Complex V) | Increases ΔΨm (hyperpolarization) | Decreases | Hyperpolarization results from reduced proton influx; inhibits cell proliferation rescueable by UCP1 [60] [58] [61] |
| Rotenone | Complex I | Decreases by 10-50% | Decreases | Induces mitochondrial fragmentation; alters NAD+/NADH ratio [63] [61] |
| Antimycin A | Complex III | Decreases by 10-50% | Decreases | Induces fragmentation; increases superoxide production from upstream sites [61] |
| Piericidin | Complex I | Decreases | Decreases | Inhibits cell proliferation not rescueable by UCP1 [60] |
Genetically Encoded Tools
Recent advances have introduced UCP1 as a genetically encoded tool for specific ΔΨm manipulation without the off-target effects associated with chemical uncouplers [57] [60] [59]. When exogenously expressed in mammalian cells, UCP1 localizes to the mitochondrial inner membrane and, in the presence of physiological concentrations of fatty acids (e.g., 300 μM oleate), induces uncoupled respiration and lowers ΔΨm to a similar extent as chemical uncouplers [60]. Critically, UCP1 expression does not inhibit cell proliferation—a significant advantage over chemical uncouplers [60] [59]. This tool has been validated in demonstrating that elevated ΔΨm drives the Integrated Stress Response induced by ATP synthase inhibition [57] [59].
Experimental Approaches and Methodologies
Measurement of ΔΨm Using Fluorescent Indicators
Accurate determination of ΔΨm requires understanding the limitations and proper application of fluorescent dyes. The cationic dyes TMRM and TMRE accumulate in mitochondria driven by the negative charge, with fluorescence intensity reflecting ΔΨm [60] [58] [64]. However, researchers must consider that these probes report on both plasma membrane potential and ΔΨm, requiring careful experimental controls [58]. Recent advances include genetically encoded voltage indicators (GEVIs) that offer improved specificity for in vivo applications [64].
Protocol: TMRM Assay for ΔΨm Measurement
- Cell Preparation: Seed cells at appropriate density on culture-treated plates or glass-bottom dishes for microscopy.
- Dye Loading: Incubate cells with 20-100 nM TMRM in culture medium for 20-30 minutes at 37°C.
- Washing: Replace dye-containing medium with pre-warmed dye-free medium or Hanks' Balanced Salt Solution (HBSS).
- Baseline Measurement: Acquire fluorescence using flow cytometry (excitation/emission: 548/573 nm) or fluorescence microscopy.
- Inhibitor Treatment: Apply pharmacological probes (e.g., 5 μM FCCP for maximal depolarization, 1-10 μM oligomycin for hyperpolarization).
- Data Analysis: Normalize fluorescence to baseline or FCCP-treated controls. Note that FCCP treatment completely dissipates ΔΨm, providing a minimum fluorescence value [60] [58].
Validating Uncoupler Activity Through Bioenergetic Assessment
Confirmation of uncoupler activity requires demonstrating increased oxygen consumption that is resistant to ATP synthase inhibition, coupled with increased fermentative metabolism.
Protocol: Validation of Uncoupling Activity
- Oxygen Consumption Rate (OCR): Using a Seahorse XF Analyzer or similar system, measure baseline OCR, then sequential treatment with:
- 1-2 μM oligomycin to inhibit ATP synthase
- Titrated uncoupler (e.g., 1-5 μM FCCP or UCP1 expression with oleate)
- 0.5 μM rotenone + 0.5 μM antimycin A to inhibit ETC
- Extracellular Acidification Rate (ECAR): Measure concurrently with OCR as a proxy for lactic acid production.
- Interpretation: Active uncouplers increase OCR in the presence of oligomycin and increase ECAR, indicating a shift to fermentative metabolism when respiratory ATP production is inefficient [60].
Experimental Workflow for Investigating ΔΨm in Quality Control
Figure 2: Comprehensive Experimental Workflow for ΔΨm Studies. Integrated approach for investigating ΔΨm manipulation encompassing intervention, validation, and assessment of functional consequences. [57] [4] [60]
The Scientist's Toolkit: Key Research Reagents
Table 4: Essential Research Reagents for ΔΨm Manipulation Studies
| Reagent Category | Specific Examples | Function/Application | Key Considerations |
|---|---|---|---|
| Chemical Uncouplers | FCCP, Bam15, DNP | Dissipate ΔΨm to study uncoupled respiration | Titrate carefully; monitor off-target effects on proliferation [60] [62] |
| ETC Inhibitors | Oligomycin, Rotenone, Antimycin A, Piericidin | Inhibit specific ETC complexes to probe respiratory function | Induce mitochondrial fragmentation; cell-type specific effects [60] [61] |
| ΔΨm Indicators | TMRM, TMRE, Rhodamine 800 | Fluorescent detection of ΔΨm | Affected by both plasma and mitochondrial membrane potentials [58] [64] |
| Genetically Encoded Tools | UCP1, mt-GEVIs | Specific ΔΨm manipulation or measurement | UCP1 requires fatty acids; GEVIs enable in vivo application [60] [64] |
| Mitophagy Reporters | mt-Keima, LC3-GFP | Monitor mitophagic flux | Provide quantitative assessment of quality control activation [4] [5] |
Applications in Disease Research and Therapeutic Development
The strategic manipulation of ΔΨm has revealed important pathophysiological mechanisms and emerging therapeutic opportunities. Mitochondrial uncouplers demonstrate substantial anti-cancer effects in preclinical models, with some showing selective toxicity toward cancer cells [62]. These effects likely stem from disruption of metabolic signaling pathways essential for cancer cell proliferation, including AMPK/mTOR regulation, and altering cellular ATP levels [62]. In pulmonary hypertension, vascular smooth muscle cells exhibit mitochondrial hyperpolarization and resistance to ETC inhibitor-induced depolarization, suggesting fundamental differences in bioenergetic regulation [61].
Metabolic remodeling in response to ETC inhibition extends beyond ATP generation to include profound effects on nucleotide metabolism. Cells with ETC defects suppress de novo purine synthesis while enhancing purine salvage pathways, with tumors exhibiting low oxidative metabolism showing enhanced expression of the salvage enzyme HPRT1 [63]. This remodeling creates metabolic vulnerabilities that could be therapeutically exploited, as blocking HPRT1 sensitizes cancer cells to ETC inhibition [63].
The development of UCP1 as a specific tool for ΔΨm manipulation has enabled researchers to dissect stress response pathways with improved precision, demonstrating that elevated ΔΨm specifically drives the Integrated Stress Response induced by ATP synthase dysfunction [57] [59]. This approach provides a template for investigating ΔΨm in diverse pathological contexts without the confounding effects of chemical uncouplers.
Pharmacological and genetic probes for ΔΨm manipulation represent indispensable tools for investigating mitochondrial biology and quality control mechanisms. While chemical uncouplers and ETC inhibitors continue to provide valuable insights, their off-target effects necessitate careful experimental design and interpretation. The emerging generation of genetically encoded tools offers unprecedented specificity for ΔΨm manipulation, enabling researchers to establish causal relationships between mitochondrial depolarization/hyperpolarization and downstream cellular responses. As our understanding of ΔΨm's role in health and disease continues to evolve, these methodological approaches will remain fundamental to advancing both basic mitochondrial biology and the development of novel therapeutic strategies targeting mitochondrial dysfunction.
High-Content Screening and Mathematical Modeling of ΔΨm Dynamics in Toxicity Assessment
Mitochondrial membrane potential (ΔΨm) is the electrochemical gradient across the inner mitochondrial membrane, serving as a fundamental indicator of mitochondrial health and functional state. Generated primarily by the electron transport chain (ETC), this inside-negative potential is not only essential for ATP production but also actively regulates critical quality control processes, including mitochondrial fusion and mitophagy [65] [66]. Within the framework of Mitochondrial Quality Control (MQC), ΔΨm acts as a key signaling node: its dissipation can trigger the PINK1/Parkin pathway to initiate the removal of damaged mitochondria, while its maintenance is a prerequisite for mitochondrial fusion, allowing functional complementation between organelles [67] [10]. The bi-directional relationship between ΔΨm and mitochondrial dynamics means that dysfunction in one often precipitates failure in the other, creating a vicious cycle of impairment that underpins various pathological states and drug-induced toxicities [65] [66].
The assessment of ΔΨm has therefore emerged as a critical parameter in toxicological studies, enabling researchers to identify compounds that disrupt mitochondrial function either intentionally or as off-target effects. This technical guide explores the integration of high-content screening (HCS) methodologies with mathematical modeling approaches to quantitatively analyze ΔΨm dynamics, providing a robust framework for predicting compound toxicity and elucidating mechanisms of action within the broader context of MQC.
High-Content Screening Methodologies for ΔΨm Quantification
Fluorescent Probes and Live-Cell Imaging Strategies
High-content screening of ΔΨm employs fluorescent dyes that accumulate in the mitochondrial matrix in a potential-dependent manner. Tetramethylrhodamine methyl ester (TMRM) and ethyl ester (TMRE) represent the gold standard for these measurements due to their reliability and reduced artifacts compared to other probes [68]. These cationic dyes can be used in two distinct modes:
- Quenching Mode: Using high dye concentrations where fluorescence self-quenches upon accumulation in the matrix. Depolarization causes dye release and increased fluorescence.
- Non-Quenching Mode: Using low dye concentrations (5-20 nM) where fluorescence intensity directly correlates with ΔΨm. Depolarization reduces signal intensity [68].
The selection of imaging mode depends on the experimental requirements, with non-quenching mode being more suitable for detecting subtle, real-time changes in ΔΨm.
Table 1: Key Fluorescent Probes for ΔΨm Measurement in High-Content Screening
| Probe Name | Excitation/Emission (nm) | Working Mode | Key Characteristics | Common Applications |
|---|---|---|---|---|
| TMRM/TMRE | ~549/573 | Quenching & Non-quenching | Minimal ETC inhibition; reliable kinetics | Live-cell kinetics; high-content analysis |
| JC-1 | 514/529 (monomer); 585/590 (aggregate) | Ratio-metric | Exists as monomer (green) at low ΔΨm and forms aggregates (red) at high ΔΨm | Distinguishing populations with different ΔΨm |
| Rhodamine 123 | ~507/529 | Non-quenching | Reversible binding; can be used for long-term imaging | General viability and ΔΨm screening |
| DASPMI | ~475/605 | Non-quenching | High photostability; used in super-resolution studies | Specialized microscopy applications |
Experimental Workflow for Multiparametric HCS
A comprehensive HCS approach for simultaneous analysis of ΔΨm and mitochondrial morphology involves several critical stages [65] [66] [68]:
Cell Preparation and Staining: Plate cells in appropriate vessels (e.g., 96-well or 384-well plates). For live-cell imaging, culture cells in phenol-red free medium. Load with TMRM (typically 20-50 nM for non-quenching mode, 100-500 nM for quenching mode) for 20-30 minutes at 37°C. Include a mitochondrial marker (e.g., Mitotracker Green) at 50-100 nM to visualize morphology.
Pharmacological Challenges: To validate the system and probe mitochondrial function, include controls with:
- Oligomycin (1-5 µM): ATP synthase inhibitor that induces hyperpolarization in functional mitochondria.
- FCCP (0.5-2 µM): Protonophore that completely collapses ΔΨm, serving as a positive control for depolarization.
Image Acquisition: Utilize automated microscopy systems capable of time-lapse imaging for kinetic assessments. Maintain environmental control (37°C, 5% CO₂) throughout acquisition. Acquire multiple fields per well to ensure statistical robustness.
Image Analysis and Data Extraction: Process images through a segmentation pipeline to identify mitochondrial objects. Extract multiple parameters including:
This workflow has been successfully adapted for various models including 2D monolayers, 3D spheroids, co-cultures, and primary cells such as isolated muscle fibers [68].
Diagram 1: High-content screening workflow for ΔΨm and morphology analysis. The process integrates experimental and computational stages for multiparametric assessment of mitochondrial function.
Data Analysis and Machine Learning Approaches
Advanced computational methods transform raw image data into biologically meaningful insights. Machine learning algorithms can classify mitochondrial phenotypes and identify subtle patterns not discernible through manual analysis [65]. For instance, principal component analysis (PCA) can reduce dimensionality while preserving essential information, enabling the identification of compound-specific fingerprints based on their effects on ΔΨm and morphology [65] [66].
Multiparametric analysis is particularly powerful as it captures the interconnected nature of mitochondrial parameters – for example, demonstrating how ETC dysfunction often coincides with both ΔΨm loss and morphological aberrations such as excessive fragmentation [65].
Mathematical Modeling of ΔΨm in Mitochondrial Quality Control
Conceptual Framework for Mitochondrial Dynamics
Mathematical models provide a quantitative framework to understand how ΔΨm regulates and is regulated by MQC processes. A systems biology approach conceptualizes the mitochondrial population as existing in different functional states:
- Healthy (H): High ΔΨm, capable of fusion and ATP production
- Moderately Damaged (M): Reduced ΔΨm, limited fusion capability
- Severely Damaged (S): Depolarized, targeted for mitophagy
- Fused (F): Networks formed by fusion of H and M mitochondria [69]
These compartments interconvert through processes including damage accumulation (H→M→S), fusion (H+M→F), fission (F→H+M+S), mitophagy (S→degradation), and biogenesis (increasing H and M) [69].
Ordinary Differential Equation Model
A recently published ODE model captures these dynamics using the following equations, where variables represent volume densities of each mitochondrial state normalized to carrying capacity (K) [69]:
Where:
- h, m, s, f: normalized volume densities of Healthy, Moderately damaged, Severely damaged, and Fused mitochondria
- n: total normalized mitochondrial density (h+m+s+f)
- β: damage rate (normalized to mitogenesis rate)
- γ: fusion rate
- δ: fission rate
- ε: mitophagy rate [69]
This normalized model reveals several critical insights into how ΔΨm-dependent processes maintain mitochondrial fitness, particularly under accelerated damage conditions similar to toxicant exposure.
Table 2: Key Parameters in Mitochondrial Quality Control Mathematical Models
| Parameter | Biological Process | Relationship to ΔΨm | Impact on System |
|---|---|---|---|
| β (Damage Rate) | Accumulation of mitochondrial defects | Often triggered by ΔΨm collapse | Higher β increases burden on quality control systems |
| γ (Fusion Rate) | Mitochondrial fusion | Requires adequate ΔΨm [66] | Increased γ promotes functional complementation |
| δ (Fission Rate) | Mitochondrial division | Precedes segregation of depolarized units | Higher δ facilitates isolation of damaged mitochondria |
| ε (Mitophagy Rate) | Clearance of damaged mitochondria | Triggered by persistent ΔΨm loss | Increased ε enhances removal of dysfunctional organelles |
Model Insights for Toxicity Assessment
Analysis of the steady-state behavior of this ODE system reveals that both fission and mitophagy are essential for maintaining a healthy mitochondrial population under conditions of accelerated damage [69]. The model predicts that:
- In the absence of fission (δ=0), damaged mitochondria cannot be effectively segregated for removal, leading to their accumulation.
- Without mitophagy (ε=0), severely damaged mitochondria persist, creating a source of oxidative stress and propagating dysfunction.
- Increasing fission and/or mitophagy rates can partially compensate for elevated damage rates (high β), suggesting potential therapeutic strategies for mitochondrial toxicity [69].
These computational insights align with experimental observations that ΔΨm heterogeneity—maintained through balanced fusion and fission—correlates with improved mitochondrial fitness and resistance to stress-induced depolarization [10].
Diagram 2: Mitochondrial quality control state transitions. The model describes how mitochondria move between functional states through damage, repair, and quality control processes influenced by ΔΨm.
Integration for Predictive Toxicology
Correlating HCS Data with Model Predictions
The power of this integrated approach emerges when HCS experimental data informs mathematical model parameters, creating a predictive framework for toxicity assessment. For example:
- Parameter Estimation: HCS measurements of ΔΨm loss kinetics following compound exposure can quantify damage rates (β).
- Morphological Analysis: Quantifying fragmentation dynamics provides estimates of fission/fusion balance (γ and δ).
- Outcome Prediction: The parameterized model can then simulate long-term outcomes that might be impractical to test experimentally, such as chronic low-dose exposure effects.
This integration is particularly valuable for classifying compounds based on their mechanisms of toxicity. Some toxicants directly depolarize mitochondria (high β), while others might impair quality control by disrupting fission (low δ) or mitophagy (low ε) without immediate ΔΨm effects [69] [70].
Application to Drug Development
The MitoTox database, which contains over 1,400 compounds with documented mitochondrial toxicity, provides a valuable resource for validating HCS-modeling approaches [70]. This database catalogs compounds with known mitochondrial off-target effects, including:
- Troglitazone: Withdrawn antidiabetic that inhibits complex I
- Cerivastatin: Lipid-lowering drug associated with rhabdomyolysis via complex III effects
- Various environmental toxicants: Including pesticides (paraquat, rotenone) and heavy metals (mercury, cadmium) [70]
By establishing characteristic "fingerprints" of ΔΨm dynamics and morphological changes for known toxicants, HCS-modeling platforms can predict potential mitochondrial toxicity early in drug development, guiding structural optimization to mitigate these risks.
Table 3: Key Research Reagent Solutions for ΔΨm and Quality Control Studies
| Reagent Category | Specific Examples | Function/Application | Technical Notes |
|---|---|---|---|
| ΔΨm Indicators | TMRM, TMRE, JC-1, Rhodamine 123 | Quantitative measurement of mitochondrial membrane potential | TMRM preferred for kinetics; JC-1 for ratio-metric analysis |
| Morphology Markers | Mitotracker Green, GFP-targeted to mitochondria | Visualization of mitochondrial structure and network | Use with non-overlapping emission for multiplexing with ΔΨm probes |
| Pharmacological Modulators | FCCP (depolarizing agent), Oligomycin (hyperpolarizing agent) | Experimental controls and pathway interrogation | Validate concentrations for specific cell types |
| Mitophagy Inducers | PMI (P62-mediated mitophagy inducer) [67] [71] | ΔΨm-independent activation of mitophagy | Useful for probing alternative quality control pathways |
| Mitochondrial Toxins | Rotenone (CI inhibitor), Antimycin A (CIII inhibitor) | Positive controls for mitochondrial dysfunction | Include in screening assays as reference compounds |
| Computational Resources | MitoTox database [70] | Reference for known mitochondrial toxicants | Contains >1,400 compounds with mechanistic information |
The integration of high-content screening and mathematical modeling establishes a powerful paradigm for evaluating ΔΨm dynamics in toxicological assessment. This approach moves beyond static snapshots to capture the dynamic interplay between membrane potential and quality control processes, providing mechanistic insights that inform safety assessment and drug development.
Future developments in this field will likely include:
- More sophisticated models incorporating spatial heterogeneity of ΔΨm within single mitochondria
- Automated platforms combining HCS with functional respirometry
- Expanded databases linking specific ΔΨm signatures to clinical toxicity outcomes
As these methodologies mature, they will enhance our ability to predict compound effects on mitochondrial health, ultimately contributing to the development of safer therapeutics with reduced off-target effects on this critical organelle.
Mitochondria are essential organelles that serve as the primary energy producers in eukaryotic cells, generating adenosine triphosphate (ATP) through oxidative phosphorylation. Beyond their role as cellular powerhouses, mitochondria regulate critical processes including apoptosis, calcium homeostasis, and reactive oxygen species (ROS) generation [25]. The inner mitochondrial membrane (IMM) maintains an electrochemical gradient known as the mitochondrial membrane potential (ΔΨm), typically ranging from -150 to -180 mV, which is fundamental for energy transduction and mitochondrial function [4] [72]. This membrane potential serves as both a critical marker of mitochondrial health and a strategic gateway for therapeutic intervention.
In many pathological conditions, particularly in cancer cells, this membrane potential becomes significantly elevated compared to normal cells [72] [73]. Cancer cells frequently exhibit a more hyperpolarized mitochondrial membrane, a characteristic that provides an opportunity for selective targeting. This differential in membrane potential forms the fundamental basis for employing delocalized lipophilic cations (DLCs) as mitochondrial-targeting vehicles. These compounds preferentially accumulate in mitochondria of diseased cells, offering a promising strategy for developing precision therapeutics that can selectively intervene in mitochondrial dysfunction while sparing healthy tissues [73] [74].
Mitochondrial Membrane Potential in Quality Control and Mitophagy
Mitochondrial Quality Control Systems
Mitochondrial quality control encompasses a multi-tiered system of mechanisms that preserve mitochondrial integrity and function. These include:
- Mitophagy: Selective autophagy of damaged mitochondria
- Mitochondrial dynamics: Balanced processes of fusion and fission
- Mitochondrial biogenesis: Generation of new mitochondria
- Protein quality control: Unfolded protein response and proteasome-mediated degradation [75]
These systems collectively maintain a functional mitochondrial network by monitoring and responding to mitochondrial damage. When mitochondria become impaired, they undergo specific changes that signal their need for removal or repair [5].
Membrane Potential in Mitophagy Activation
The mitochondrial membrane potential plays a decisive role in initiating quality control responses, particularly the PINK1-Parkin mitophagy pathway. Under normal conditions with preserved membrane potential, PTEN-induced kinase 1 (PINK1) is continuously imported into mitochondria and degraded. However, when mitochondrial damage causes depolarization, PINK1 stabilizes on the outer mitochondrial membrane (OMM) where it auto-phosphorylates and recruits the E3 ubiquitin ligase Parkin from the cytoplasm [4].
This PINK1-Parkin interaction triggers a signaling cascade that marks damaged mitochondria for autophagic clearance. Activated Parkin ubiquitinates numerous OMM proteins, and PINK1 further phosphorylates these ubiquitin tags, creating a positive feedback loop that amplifies mitophagy signals [4] [5]. The system ensures selective removal of dysfunctional mitochondria that have lost membrane potential, thereby preventing the accumulation of toxic mitochondria and preserving cellular viability [4] [75].
Table 1: Key Proteins in Mitophagy Pathways
| Protein | Function | Role in Mitophagy |
|---|---|---|
| PINK1 | Serine/threonine-protein kinase | Sensor of mitochondrial damage; accumulates on depolarized mitochondria |
| Parkin | E3 ubiquitin ligase | Ubiquitinates OMM proteins to标记 damaged mitochondria |
| Mfn2 | Mitochondrial fusion protein | Receptor for Parkin recruitment; phosphorylation weakens fusion activity |
| p62/SQSTM1 | Autophagy receptor | Links ubiquitinated mitochondria to LC3 on autophagosomes |
Delocalized Lipophilic Cations: Fundamental Mechanisms
Structural and Chemical Properties
Delocalized lipophilic cations (DLCs) are characterized by a positive charge spread over a large hydrophobic molecular area through resonance stabilization. This unique electronic structure requires lower enthalpy for desolvation, enabling them to traverse lipid bilayers with far lower activation energy than hydrophilic cations [76]. Common DLC classes include:
- Triphenylphosphonium (TPP⁺)-based compounds
- Rhodamine derivatives (including Rhodamine 123 and Rhodamine B)
- Cyanine (Cy)-based derivatives [76]
These structural features allow DLCs to passively diffuse across biological membranes and accumulate in the mitochondrial matrix, driven by the highly negative membrane potential [76] [73].
Accumulation Mechanisms and Selectivity
The preferential accumulation of DLCs in mitochondria occurs through a membrane potential-driven process. The substantial transmembrane potential (negative inside) creates an electrophoretic force that attracts and concentrates lipophilic cations within the mitochondrial matrix. This accumulation can reach concentrations 100-1000 times higher in mitochondria compared to the extracellular medium [73].
In cancer cells, which frequently exhibit elevated mitochondrial membrane potentials, this accumulation is further enhanced, providing a therapeutic window for selective targeting. Research demonstrates that DLC-conjugated compounds can exploit this differential to achieve targeted effects in malignant versus normal cells [72] [73] [74].
Experimental Evidence and Case Studies
DLC-Conjugated Anionic Polymers
A significant advancement in mitochondrial targeting emerged from the unexpected discovery that DLC-conjugated anionic polymers exhibit superior mitochondrial targeting compared to cationic or neutral polymers. This finding was counterintuitive given that cell membranes generally bear a net negative charge that would theoretically repel anionic compounds [76].
In one comprehensive study, researchers synthesized a library of methacrylate polymers with different surface charges and conjugated them with Cyanine 3 (Cy3), a delocalized lipophilic cation. Through systematic evaluation across multiple cell types (HeLa, HUVECs, hTERT-MSC, and C2C12), they observed that Cy3-conjugated anionic polymers (NEG or SO₃) exhibited significantly higher uptake efficiency than cationic (POS) or charge-neutral (PEG and MPC) analogs [76].
Table 2: Cellular Uptake Efficiency of Cy3-Conjugated Polymers Across Cell Types
| Polymer Type | Surface Charge | HeLa Cells | HUVECs | hTERT-MSC | C2C12 |
|---|---|---|---|---|---|
| NEG | Anionic | ++++ | ++++ | ++++ | ++++ |
| SO₃ | Anionic | ++++ | ++++ | ++++ | ++++ |
| POS | Cationic | + | + | + | + |
| PEG | Neutral | + | + | + | + |
| MPC | Neutral | + | + | + | + |
Colocalization studies confirmed that these anionic polymers specifically localized to mitochondria rather than other organelles such as lysosomes or endoplasmic reticulum. Importantly, these DLC-conjugated anionic polymers circumvented endosomal entrapment—a significant limitation for many delivery systems—and rapidly accumulated in mitochondria within one hour of administration [76].
Mechanistic investigations revealed that this uptake was membrane potential-dependent. Treatment with FCCP (a mitochondrial uncoupler that dissipates membrane potential) decreased polymer uptake, while oligomycin (which increases membrane potential) enhanced uptake. Furthermore, inhibition of mitochondrial pyruvate carriers with UK5099 reduced accumulation, suggesting the involvement of these transporters in the uptake process [76].
Therapeutic Applications: Camptothecin Derivatives
The therapeutic potential of DLC-mediated mitochondrial targeting has been demonstrated in cancer drug development. Researchers have designed and synthesized mitochondria-targeted camptothecin (CPT) derivatives by conjugating the anticancer drug with various DLCs, including triphenylphosphonium, F16, and rhodamine B [74].
These derivatives exhibited significantly enhanced antiproliferative activity against HCT116 colorectal cancer cells compared to the parent camptothecin compound. Particularly, compounds 8a and 8c, connected to rhodamine B, showed IC₅₀ values of 0.21 μM and 0.18 μM respectively, representing a substantial improvement over conventional camptothecin. Importantly, these mitochondria-targeted derivatives demonstrated lower toxicity toward normal liver cells, highlighting their selective therapeutic potential [74].
Cellular imaging experiments confirmed the excellent mitochondria-targeting capability of these compounds, while flow cytometry analysis revealed that they induce apoptosis in a concentration-dependent manner. This approach exemplifies how DLC-mediated mitochondrial targeting can enhance drug efficacy while reducing off-target effects [74].
Photodynamic Enhancement of DLC Efficacy
Research on the DLC compound D112 has revealed another strategic approach to enhance the selective toxicity of DLC-based therapies. D112 accumulates in transformed cells where it interacts with mitochondrial DNA, inhibits Complex I respiration, and induces reactive oxygen species (ROS) production [73].
Notably, photo-activation of D112 potentiated selective ROS production and increased the window of toxicity toward cancer cells over non-transformed cells. This combination approach leverages both the inherent mitochondrial accumulation of DLCs and external activation to achieve spatial and temporal control over therapeutic activity, representing a promising strategy for precision cancer therapy [73].
Experimental Protocols and Methodologies
Assessing DLC-Mediated Mitochondrial Targeting
Protocol 1: Colocalization Studies for Mitochondrial Localization
- Cell Preparation: Seed cells (e.g., HeLa, SK-MEL-2) on glass coverslips in appropriate culture medium and grow to 60-70% confluence.
- Staining:
- Incubate cells with DLC-conjugated compound of interest at optimal concentration (e.g., 1-10 μM) for desired time (typically 1-4 hours).
- Add MitoTracker Green (100-200 nM) or other mitochondrial dyes during the final 30 minutes of incubation.
- For counterstaining other organelles, include LysoTracker for lysosomes or ER-Tracker for endoplasmic reticulum.
- Fixation and Imaging:
- Wash cells with PBS and fix with 4% paraformaldehyde for 15 minutes at room temperature.
- Mount coverslips and image using confocal microscopy with appropriate laser lines and emission filters.
- Analysis:
- Calculate Pearson's correlation coefficient or Mander's overlap coefficient using image analysis software (e.g., ImageJ with Coloc2 plugin).
- Values >0.7 indicate strong colocalization with mitochondria [76].
Protocol 2: Membrane Potential Dependence Assay
- Cell Treatment:
- Pre-treat cells with membrane potential modulators:
- FCCP (1-10 μM, 30 minutes) to dissipate ΔΨm
- Oligomycin (1-5 μM, 30 minutes) to hyperpolarize mitochondria
- Include untreated controls and vehicle controls.
- Pre-treat cells with membrane potential modulators:
- Compound Exposure:
- Incubate cells with DLC-conjugated compound for standardized time (e.g., 1-2 hours).
- Analysis:
Functional Assessment of Mitochondrial Targeting
Protocol 3: Oxygen Consumption Measurements
- Cell Preparation: Harvest cells and resuspend in appropriate assay medium (e.g., MiR05) at 1-2×10⁶ cells/mL.
- Respirometry:
- Use high-resolution respirometry systems (e.g., O2k-FluoRespirometer).
- Perform substrate-uncoupler-inhibitor titration (SUIT) protocol:
- Measure routine respiration (endogenous substrates)
- Add ADP for OXPHOS capacity
- Titrate Complex I inhibitors (e.g., rotenone) and Complex II substrates (succinate)
- Add uncoupler (FCCP) for ETS capacity
- Add inhibitors to determine residual oxygen consumption
- DLC Treatment:
- Incubate cells with DLC compounds before or during measurements.
- Monitor changes in respiratory parameters, particularly Complex I activity [73].
Protocol 4: ROS Production Assay
- Cell Staining:
- Incubate cells with ROS-sensitive fluorescent probes (CellROX Green, CM-H₂DCFDA, or MitoSOX Red for mitochondrial ROS) according to manufacturer's protocols.
- Treatment and Analysis:
- Expose stained cells to DLC compounds with or without ROS scavengers (e.g., N-acetylcysteine).
- Measure fluorescence intensity via flow cytometry or fluorescence microscopy.
- Correlate ROS production with apoptotic markers (annexin V, caspase activation) [73].
Visualization of Key Mechanisms
DLC Accumulation and Mitophagy Pathway
Diagram 1: DLC Mechanism and Mitophagy Pathway (Width: 760px)
Experimental Workflow for DLC Evaluation
Diagram 2: DLC Conjugate Evaluation Workflow (Width: 760px)
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Mitochondrial Targeting Research
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| DLC Compounds | TPP⁺ (Triphenylphosphonium), Rhodamine derivatives (123, B), Cyanine dyes (Cy3), F16 | Mitochondrial targeting moieties for conjugation |
| Membrane Potential Probes | JC-1, TMRM, TMRE, MitoTracker Red CMXRos | Measure ΔΨm; validate targeting dependence |
| ROS Detection Probes | MitoSOX Red, CellROX Green, CM-H₂DCFDA | Detect reactive oxygen species production |
| Mitochondrial Inhibitors | FCCP (uncoupler), Oligomycin (ATP synthase inhibitor), Rotenone (Complex I inhibitor) | Modulate membrane potential; investigate mechanisms |
| Mitophagy Reporters | Mito-QC, LC3-GFP, Keima reporters | Monitor mitophagy flux and mitochondrial turnover |
| Polymer Scaffolds | Anionic methacrylate polymers, PLGA nanoparticles, Dendrimers | Delivery vehicles for DLC conjugation |
| Cell Lines | HeLa, HCT116, MCF-7, primary cells, ρ⁰ cells | Model systems for evaluating targeting and efficacy |
The strategic application of delocalized lipophilic cations represents a transformative approach in mitochondrial medicine, leveraging fundamental biophysical principles—particularly the mitochondrial membrane potential—to achieve precise subcellular targeting. The convergence of DLC technology with advanced delivery platforms, especially anionic polymers, has overcome historical barriers in mitochondrial therapeutics, including endosomal entrapment and insufficient accumulation.
Future developments in this field will likely focus on enhancing the specificity and controllability of mitochondrial targeting systems. The integration of DLC approaches with external activation mechanisms (such as photoactivation) and disease-specific biomarkers promises to further refine the therapeutic window. Additionally, combining mitochondrial-targeted strategies with emerging modalities in gene editing and immunometabolism may unlock new therapeutic paradigms for addressing the fundamental mitochondrial dysfunction underlying diverse human pathologies.
As our understanding of mitochondrial biology deepens and nanomedicine continues to advance, DLC-based targeting platforms stand poised to make significant contributions to precision medicine, offering hope for more effective treatments for cancer, neurodegenerative disorders, and metabolic diseases rooted in mitochondrial dysfunction.
Marine and Terrestrial Natural Products as a Source for ΔΨm-Modulating Compounds
Mitochondrial membrane potential (ΔΨm) is a critical parameter of mitochondrial health, serving as a fundamental driver for ATP synthesis and a key regulator in quality control mechanisms such as mitophagy. Its dysregulation is a hallmark of aging and numerous neurological disorders. This whitepaper examines the capacity of marine and terrestrial natural products to modulate ΔΨm, thereby influencing mitochondrial quality control. We synthesize current research on bioactive compounds, detailing their mechanisms of action, experimental evidence, and potential as neurotherapeutics. Structured tables compare the effects of diverse compounds, standardized protocols for assessing ΔΨm are provided, and signaling pathways are visualized to guide future drug discovery efforts for age-associated neurological diseases.
Mitochondrial membrane potential (ΔΨm), generated by the proton gradient across the inner mitochondrial membrane during oxidative phosphorylation, is indispensable for energy production and cellular survival [77] [52]. It acts as a central indicator of mitochondrial functional status and is a critical regulatory signal within the mitochondrial quality control network. This network encompasses mitochondrial biogenesis, dynamics (fusion and fission), and mitophagy—a selective autophagic process that eliminates dysfunctional mitochondria [53] [52].
The preservation of a healthy ΔΨm is particularly vital in the brain, where neurons, due to their high energy demands and post-mitotic nature, are exceptionally vulnerable to bioenergetic deficits [77]. In age-associated neurological disorders such as Alzheimer's disease (AD) and Parkinson's disease (PD), mitochondrial dysfunction is a common feature, often characterized by a loss of ΔΨm [77] [53]. This depolarization can initiate quality control pathways; most notably, it triggers the PINK1-Parkin pathway of mitophagy [4] [5]. When ΔΨm is dissipated, PTEN-induced putative kinase 1 (PINK1) stabilizes on the outer mitochondrial membrane, recruiting the E3 ubiquitin ligase Parkin. This recruitment marks the damaged organelle for autophagic degradation, preventing the accumulation of toxic mitochondria [4] [5]. Consequently, compounds capable of modulating ΔΨm represent a promising therapeutic strategy to bolster mitochondrial quality control, mitigate neuronal damage, and slow disease progression.
Terrestrial Natural Products and Their Effects on ΔΨm
Terrestrial plants and microorganisms have yielded numerous bioactive compounds that exert significant effects on ΔΨm, offering potential pathways to modulate mitochondrial quality control.
Table 1: Terrestrial Natural Products Modulating ΔΨm and Mitochondrial Function
| Compound | Source | Effects on ΔΨm & Bioenergetics | Impact on Mitochondrial Quality Control | Experimental Models |
|---|---|---|---|---|
| Quercetin [78] | Fruits, vegetables (e.g., onions, apples) | ≥50 µM decreases ΔΨm, uncouples OXPHOS, stimulates O₂ consumption [78] | May induce removal of depolarized mitochondria via mitophagy; pro-apoptotic in cancer cells [78] | Isolated rat heart mitochondria, various cell lines (e.g., U937, THP-1) [78] |
| Resveratrol [78] | Grapes, berries | Modulates ΔΨm indirectly by improving OXPHOS and inducing biogenesis; inhibits F0F1-ATPase at high conc. (0.7-70 µM) [78] | Enhances mitochondrial biogenesis via PGC-1α deacetylation; promotes renewal of healthy pool [78] | Rat models, isolated rat brain mitochondria, human coronary endothelial cells [78] |
| Curcumin [78] | Turmeric (Curcuma longa) | Acts as protonophoric uncoupler, decreasing ATP biosynthesis; effect varies by tissue [78] | Reported to upregulate antioxidant defenses, mitigating oxidative stress on mitochondria [78] | Isolated rat liver/brain mitochondria, E. coli models [78] |
Key Mechanistic Insights
- Quercetin demonstrates a concentration-dependent dual role. At lower concentrations, it may exhibit antioxidant properties, but at higher concentrations (≥50 µM), it acts as an uncoupler and depolarizing agent, potentially stimulating the removal of dysfunctional mitochondria [78].
- Resveratrol primarily promotes a healthy mitochondrial pool by activating biogenesis through the PGC-1α pathway, thereby indirectly supporting a robust ΔΨm. Its inhibitory effect on ATP synthase at high concentrations suggests a complex, context-dependent interaction with bioenergetic machinery [78].
- Curcumin's function as a protonophore directly dissipates the proton gradient, reducing ΔΨm. This action can be detrimental in healthy cells but may selectively target compromised cells, such as in cancer [78].
Marine Natural Products and Their Effects on ΔΨm
The extreme and competitive marine environment drives the evolution of unique biochemical pathways, making marine organisms a prolific source of bioactive compounds with distinct mechanisms for modulating ΔΨm.
Table 2: Marine Natural Products Modulating ΔΨm and Mitochondrial Function
| Compound | Source | Effects on ΔΨm & Bioenergetics | Impact on Mitochondrial Quality Control | Experimental Models |
|---|---|---|---|---|
| Piscidin-1 [79] | Hybrid striped bass | Reduces mitochondrial function, OXPHOS complex levels, and ΔΨm [79] | Induces apoptosis via mitochondrial pathway; depolarization precedes cell death [79] | Oral squamous cell carcinoma (OSCC) cells [79] |
| Aurilides [80] | Marine mollusks and cyanobacteria | Induces mitochondrial fragmentation and cristae disorganization [80] | Potent pro-apoptotic activity; disrupts mitochondrial integrity and dynamics [80] | Various cancer cell lines [80] |
| Microwave-extracted Clam Polysaccharide (MCP) [79] | Clam (Ruditapes philippinarum) | Lowers intrinsic ΔΨm, facilitates cytochrome c release [79] | Shifts tumor-associated macrophages to M1-type; induces mitochondrial apoptosis in cancer cells [79] | HT-29 colorectal cancer cells, RAW 264.7 macrophages [79] |
Key Mechanistic Insights
- Piscidin-1 and Aurilides exemplify marine compounds that directly target mitochondrial integrity, inducing depolarization and disrupting dynamics to trigger apoptosis. This makes them promising candidates for anticancer therapeutics [80] [79].
- Microwave-extracted Clam Polysaccharide (MCP) demonstrates a multi-faceted mechanism, not only directly inducing mitochondrial apoptosis in cancer cells but also modulating the tumor immune microenvironment, showcasing the diverse therapeutic potential of marine bioactives [79].
Experimental Protocols for Assessing ΔΨm
Standardized methodologies are crucial for reliably evaluating the impact of natural products on ΔΨm. The following protocols are widely employed in the field.
Protocol 1: Assessment Using Fluorescent Dyes (e.g., JC-1, TMRM)
This is a common method for quantifying ΔΨm in live cells.
- Principle: Cationic, fluorescent dyes accumulate in the mitochondrial matrix in a ΔΨm-dependent manner. JC-1 forms red fluorescent aggregates in polarized mitochondria and green monomers in depolarized mitochondria. Tetramethylrhodamine methyl ester (TMRM) exhibits a fluorescence intensity shift.
- Procedure:
- Cell Preparation: Seed cells onto glass-bottom dishes or multi-well plates and culture until ~70% confluent.
- Treatment & Staining: Treat cells with the natural product or vehicle control for a predetermined time. Load cells with JC-1 (e.g., 2 µM) or TMRM (e.g., 20 nM) in culture medium for 30 minutes at 37°C, 5% CO₂.
- Washing & Imaging: Gently wash cells with warm PBS to remove excess dye. Add fresh pre-warmed medium.
- Image Acquisition: Capture fluorescence images using a confocal or epifluorescence microscope.
- For JC-1: Use 488 nm excitation; measure emission at 530 nm (green monomers) and 590 nm (red aggregates). Calculate the red/green fluorescence intensity ratio.
- For TMRM: Use 543 nm excitation and 565-625 nm emission. A decrease in intensity indicates depolarization.
- Data Analysis: Quantify fluorescence intensities from multiple cells/fields. A decrease in the JC-1 red/green ratio or TMRM intensity signifies a loss of ΔΨm [77] [79].
Protocol 2: Functional Assessment in Isolated Mitochondria
This protocol evaluates direct effects on mitochondrial bioenergetics.
- Principle: Isolating mitochondria from tissues like rat liver or brain allows direct measurement of oxygen consumption rates (OCR) and membrane potential changes in response to compounds.
- Procedure:
- Mitochondrial Isolation: Homogenize fresh tissue in ice-cold isolation buffer and purify mitochondria via differential centrifugation.
- Respiratory Measurements: Resuspend mitochondria in assay buffer. Using a Clark-type oxygen electrode or Seahorse Analyzer, measure basal OCR.
- ΔΨm Measurement: In parallel, incubate mitochondria with a ΔΨm-sensitive dye (e.g., JC-1 or Rhodamine 123) and substrates (e.g., succinate). Add the natural product and monitor fluorescence change over time.
- Data Interpretation: An uncoupler like Quercetin stimulates State 2 respiration and decreases dye fluorescence, indicating depolarization. An ATP synthase inhibitor may hyperpolarize membranes initially [78].
Diagram 1: Natural Product Modulation of ΔΨm and Mitophagy. This figure illustrates how marine and terrestrial natural products can directly induce mitochondrial depolarization to trigger PINK1-Parkin mediated mitophagy or indirectly promote a healthy mitochondrial pool via enhanced biogenesis.
The Scientist's Toolkit: Key Research Reagents
This section details essential reagents and their functions for investigating ΔΨm and mitophagy.
Table 3: Essential Research Reagents for ΔΨm and Mitophagy Studies
| Reagent / Assay | Function / Utility | Key Considerations |
|---|---|---|
| JC-1 Dye [79] | Ratiometric fluorescent probe for ΔΨm; distinguishes polarized (red) from depolarized (green) mitochondria. | The red/green ratio is more reliable than either channel alone; sensitive to loading conditions. |
| TMRM / TMRE | Cationic dyes that distribute according to ΔΨm; fluorescence intensity indicates polarization level. | Used in quench or non-quench modes; requires careful calibration for quantitative analysis. |
| Oxygenph System (e.g., Clark Electrode) | Measures mitochondrial oxygen consumption rate (OCR) in isolated mitochondria. | Direct functional readout of electron transport chain activity; requires fresh mitochondrial preparations. |
| Carbonyl Cyanide m-chlorophenyl hydrazone (CCCP) [5] | Potent mitochondrial uncoupler; used as a positive control for maximal ΔΨm dissipation and mitophagy induction. | Highly effective; use at calibrated concentrations to avoid non-specific toxicity. |
| Antibodies for Mitophagy Markers (PINK1, Parkin, LC3-II) [4] | Western blot detection of key proteins in the PINK1-Parkin mitophagy pathway. | LC3-II lipidation and PINK1 stabilization on OMM are key indicators. |
| Seahorse XF Analyzer | Measures real-time OCR and extracellular acidification rate (ECAR) in live cells. | Provides a multi-parametric bioenergetic profile; ideal for screening compound effects. |
Marine and terrestrial natural products represent an immense and structurally diverse resource for discovering novel ΔΨm-modulating compounds. As detailed in this whitepaper, bioactives like Quercetin, Resveratrol, Piscidin-1, and Aurilides interact directly with mitochondrial components to influence ΔΨm, thereby impacting downstream quality control mechanisms, most notably mitophagy. The experimental frameworks and research tools outlined provide a foundation for systematic investigation.
Future research should prioritize overcoming the challenges associated with natural product development, particularly poor bioavailability and tissue-specific delivery, potentially through nanoformulations and synthetic biology approaches [80] [81]. Furthermore, exploring the synergistic effects of natural product combinations and validating their efficacy in complex physiological models, such as 3D organoids, will be crucial for translating these findings into viable neurotherapeutics. Harnessing the power of these natural compounds offers a promising path toward developing disease-modifying treatments for neurodegenerative disorders by targeting the fundamental mechanisms of mitochondrial quality control.
Mitochondrial membrane potential (ΔΨm), generated by the electrochemical gradient across the inner mitochondrial membrane, serves as a fundamental regulator of cellular energy capacity and a critical sensor of mitochondrial health. Within the mitochondrial quality control (MQC) network, ΔΨm depolarization acts as a primary signal for initiating mitophagy, while its maintenance is essential for ATP production and calcium homeostasis. This whitepaper synthesizes current research demonstrating how ΔΨm dysregulation contributes to the pathogenesis of neurodegenerative, cardiovascular, and metabolic diseases. Furthermore, we explore emerging therapeutic strategies that target ΔΨm to restore MQC, detailing specific experimental methodologies and reagent tools essential for translational research and drug development in this evolving field.
The mitochondrial quality control system represents a sophisticated network of processes that maintain mitochondrial integrity, including biogenesis, dynamics (fusion and fission), and mitophagy [82] [10]. At the core of this regulatory system lies ΔΨm, typically ranging from -150 to -180 mV, which drives ATP synthesis through the proton motive force across the inner mitochondrial membrane [52]. Beyond its bioenergetic function, ΔΨm serves as a key indicator of mitochondrial fitness within MQC pathways, where its collapse often triggers selective autophagic removal of damaged organelles [10] [52].
The "spatiotemporal-threshold" model of the mitochondrial quality control–cell death axis establishes ΔΨm as a critical determinant of cellular fate [52]. According to this model, sustained ΔΨm loss represents a "point-of-no-return" that initiates irreversible commitment to cell death pathways, including apoptosis, pyroptosis, and ferroptosis. Consequently, therapeutic maintenance of ΔΨm presents a promising strategy for preserving mitochondrial function across multiple disease contexts with disrupted energy metabolism.
ΔΨm as a Diagnostic and Therapeutic Biomarker
Quantitative Assessment of ΔΨm
Researchers employ multiple methodological approaches to quantify ΔΨm in experimental models, each with specific applications and limitations. The following table summarizes key techniques and their research applications.
Table 1: Core Methodologies for ΔΨm Quantification in Research Settings
| Method | Principle | Research Applications | Key Considerations |
|---|---|---|---|
| Tetramethylrhodamine Ester (TMRE) | Lipophilic cationic dye accumulates in mitochondria proportional to ΔΨm [83] | Flow cytometry, fluorescence microscopy [83] | Concentration-dependent; requires proper controls for quantification |
| Tetramethylrhodamine Methyl Ester (TMRM) | Potential-sensitive distribution between mitochondria and cytoplasm [84] | Identification of metabolically robust T cells and stem cells [84] | Can be used in quenching or non-quenching modes |
| JC-1 Assay | Forms red fluorescent J-aggregates at high ΔΨm; green monomers at low ΔΨm [25] | Distinguishing high and low ΔΨm populations; apoptosis studies | Ratio metric measurement (red/green) provides internal control |
| SCENITH Assay | Measures protein translation changes in response to metabolic inhibitors [83] | Quantifying metabolic dependencies in primary cells | Functional assessment of oxidative phosphorylation reliance |
| Seahorse Extracellular Flux Analysis | Measures oxygen consumption rate (OCR) as indicator of mitochondrial respiration [83] | Evaluating maximal respiration and spare respiratory capacity | Indirect assessment of ΔΨm via functional capacity |
Experimental Protocols for ΔΨm Assessment
Protocol 1: TMRE Staining for Flow Cytometry
- Cell Preparation: Harvest and wash cells in PBS, counting to achieve 0.5-1×10^6 cells/sample [83]
- Staining: Resuspend cells in pre-warmed culture media containing 20-100 nM TMRE
- Incubation: Incubate for 15-30 minutes at 37°C in the dark
- Control Preparation: Prepare control samples with 10 μM carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) for 15 minutes to dissipate ΔΨm
- Analysis: Wash cells twice with PBS, resuspend in cold PBS, and analyze immediately via flow cytometry using FL2 channel (or appropriate filter for TMRE fluorescence)
- Data Interpretation: Higher TMRE fluorescence indicates elevated ΔΨm; normalize to FCCP-treated controls [83]
Protocol 2: Seahorse XF Analyzer for Mitochondrial Function
- Cell Preparation: Seed cells in XF assay plates at optimized density (typically 20,000-80,000 cells/well for adherent cells)
- Sensor Cartridge Hydration: Hydrate XF sensor cartridge in XF calibrant at 37°C in non-CO₂ incubator overnight
- Media Exchange: Replace growth media with XF assay media (pH 7.4) supplemented with 1 mM pyruvate, 2 mM glutamine, and 10 mM glucose
- Incubation: Incubate cells for 45-60 minutes at 37°C in non-CO₂ incubator
- Mitochondrial Stress Test Protocol:
- Baseline measurements (3 cycles)
- Oligomycin injection (1-2 μM) to inhibit ATP synthase
- FCCP injection (0.5-1.5 μM) to uncouple mitochondria and measure maximum respiration
- Rotenone/Antimycin A injection (0.5 μM each) to inhibit complexes I and III
- Data Analysis: Calculate basal respiration, ATP production, proton leak, maximal respiration, and spare respiratory capacity [83]
Diagram 1: Experimental Workflow for ΔΨm Assessment (Title: ΔΨm Assessment Workflow)
Disease-Specific Therapeutic Applications
Neurodegenerative Diseases
Pathophysiological Basis: In neurodegenerative diseases including Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD), and amyotrophic lateral sclerosis (ALS), mitochondrial dysfunction represents a hallmark feature [82] [85]. Progressive ΔΨm dissipation occurs in response to pathological protein accumulation including amyloid-β, hyperphosphorylated Tau, and α-synuclein, disrupting electron transport chain efficiency and increasing reactive oxygen species (ROS) production [82]. This ΔΨm collapse precedes neuronal apoptosis and contributes to synaptic dysfunction.
Therapeutic Strategies:
- Natural Product Interventions: Several natural compounds demonstrate efficacy in maintaining ΔΨm in neurodegenerative models. Specific mechanisms include enhancing mitochondrial biogenesis via PGC-1α activation, reducing oxidative damage, and improving electron transport chain function [82].
- Nanoparticle-Mediated Delivery: Advanced nanocarriers functionalized with mitochondrial-targeting ligands (e.g., triphenylphosphonium, TPP+) facilitate blood-brain barrier penetration and specific mitochondrial accumulation of therapeutic agents [85]. This approach enhances drug efficacy while minimizing systemic exposure.
- MQC Enhancement: Therapeutic approaches targeting mitochondrial dynamics regulators including Drp1, OPA1, and mitofusins show promise in stabilizing ΔΨm and preventing neuronal apoptosis [10] [52].
Cardiovascular Diseases
Pathophysiological Basis: Cardiovascular diseases including heart failure, ischemic heart disease, and cardiomyopathies involve characteristic ΔΨm dysregulation through multiple mechanisms [86]. Calcium overload in hypertension drives mitochondrial permeability transition pore (mPTP) opening and ΔΨm collapse, while VDAC dysfunction impairs metabolite flux and energetics. Additionally, MCU complex overactivation exacerbates calcium-mediated ΔΨm dissipation in ischemia-reperfusion injury [86].
Therapeutic Strategies:
- VDAC-Targeted Approaches: VDAC1 inhibition (e.g., ABT-737) prevents apoptosis induction, while VDAC2 enhancement improves mitochondrial-calcium coupling and contractile function in failing hearts [86].
- MCU Modulation: Pharmacological MCU inhibition using compounds including Ru360 prevents calcium overload and mPTP opening, preserving ΔΨm during ischemic injury [86].
- Metabolic Reprogramming: Therapeutic strategies targeting mitochondrial transport proteins (VDACs, ANT, MCU) reestablish metabolic flexibility and prevent ΔΨm loss through improved substrate utilization [86].
Table 2: Mitochondrial Transport Proteins as Therapeutic Targets in Cardiovascular Diseases
| Target Protein | Localization | Role in ΔΨm Regulation | Therapeutic Approach | Experimental Evidence |
|---|---|---|---|---|
| VDAC1 | Outer Mitochondrial Membrane | Regulates metabolite flux; oligomerization promotes ΔΨm loss and apoptosis [86] | VDAC1 inhibitors (ABT-737) | Reduces apoptosis in heart failure models [86] |
| VDAC2 | Outer Mitochondrial Membrane | Facilitates calcium signaling; maintains ΔΨm via calcium coupling [86] | VDAC2 enhancement | Restores contractile function in failing hearts [86] |
| MCU Complex | Inner Mitochondrial Membrane | Calcium overload triggers ΔΨm collapse and mPTP opening [86] | MCU inhibitors (Ru360) | Protects against ischemia-reperfusion injury [86] |
| ANT | Inner Mitochondrial Membrane | ATP/ADP exchange affects ΔΨm maintenance [86] | ANT activators | Improves ATP synthesis efficiency in heart failure [86] |
| UCP2/3 | Inner Mitochondrial Membrane | Regulates proton leak and ROS production; affects ΔΨm [86] | UCP2/3 activators | Reduces oxidative stress in atherosclerosis models [86] |
Metabolic and Other Diseases
Clonal Hematopoiesis: In Dnmt3a-mutant hematopoietic stem and progenitor cells (HSPCs), elevated ΔΨm represents a therapeutic vulnerability rather than a deficit [83]. Mutant HSPCs demonstrate increased maximal respiration, spare respiratory capacity, and elevated ΔΨm associated with DNA hypomethylation and enhanced expression of electron transport chain components [83]. This elevated ΔΨm enables selective targeting using ΔΨm-exploiting compounds.
Chronic Obstructive Pulmonary Disease (COPD): In COPD pathogenesis, oxidative stress from cigarette smoke exposure reduces ΔΨm, impairing ATP production and promoting inflammatory responses [25]. Therapeutic approaches targeting mitochondrial quality control mechanisms aim to restore ΔΨm and mitigate disease progression.
Therapeutic Strategy for Elevated ΔΨm Conditions: Long-chain alkyl-TPP molecules including MitoQ and d-TPP selectively accumulate in high-ΔΨm mitochondria, causing reduced respiration and mitochondrial-driven apoptosis in target cells [83]. This approach effectively ablates the competitive advantage of Dnmt3a-mutant HSPCs in aged recipient mice while sparing wild-type cells with normal ΔΨm [83].
Diagram 2: ΔΨm Dysregulation in Disease Pathogenesis (Title: ΔΨm in Disease Pathogenesis)
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for ΔΨm-Targeted Investigations
| Reagent/Category | Specific Examples | Research Application | Key Considerations |
|---|---|---|---|
| ΔΨm-Sensitive Dyes | TMRE, TMRM, JC-1, Rhodamine 123 | Quantitative ΔΨm measurement via flow cytometry, microscopy | Concentration optimization critical; include proper controls (FCCP) [83] [84] |
| TPP+ Conjugates | MitoQ, d-TPP, MitoTEMPO | Selective targeting of high-ΔΨm mitochondria; drug delivery | Leverage ΔΨm for organelle-specific accumulation [83] |
| Ion Channel Modulators | Ru360 (MCU inhibitor), VBIT-4 (VDAC inhibitor) | Investigating calcium-mediated ΔΨm regulation | Specificity varies; use multiple concentrations [86] |
| Metabolic Inhibitors | Oligomycin, FCCP, Rotenone, Antimycin A | Seahorse assays; metabolic dependency studies | Sequential injection required for mitochondrial stress test [83] |
| Genetic Tools | CRISPR/Cas9 for MQC genes (PINK1, Parkin, DRP1, OPA1) | Mechanistic studies of ΔΨm regulation in MQC | Confirm efficiency with multiple sgRNAs |
| Natural Compounds | Resveratrol, Berberine, Curcumin | ΔΨm stabilization in neurodegenerative models | Multiple mechanisms of action; dose optimization required [82] |
Therapeutic targeting of ΔΨm represents a promising approach for addressing mitochondrial dysfunction across multiple disease contexts. The expanding toolkit of ΔΨm-sensitive compounds, particularly those exploiting the negative membrane potential for mitochondrial accumulation, offers unprecedented specificity in modulating MQC pathways. Future research directions should focus on developing tissue-specific delivery systems, particularly for neurodegenerative applications where blood-brain barrier penetration remains challenging [85]. Additionally, personalized approaches considering individual variations in mitochondrial function and ΔΨm baseline characteristics may enhance therapeutic efficacy while minimizing off-target effects.
As our understanding of the spatiotemporal dynamics of ΔΨm regulation deepens, particularly through advanced imaging techniques and real-time monitoring technologies, the precision of ΔΨm-targeted therapeutics will continue to improve. Integration of ΔΨm assessment into standard diagnostic and drug development pipelines holds significant promise for addressing the growing burden of diseases characterized by mitochondrial dysfunction.
Navigating Complex Dynamics: Challenges in Interpreting Membrane Potential Signals
Distinguishing Adaptive Hyperpolarization from Pathological Depolarization
Mitochondrial membrane potential (ΔΨm), an electrical gradient across the inner mitochondrial membrane, serves as a fundamental regulator of cellular energy status and mitochondrial quality control. While often simplified as a binary indicator of mitochondrial health, ΔΨm operates as a dynamic signaling platform that mediates critical cellular decisions. This technical review examines the nuanced interpretation of ΔΨm fluctuations, distinguishing adaptive hyperpolarization and depolarization from pathological manifestations. We explore how specific ΔΨm signatures integrate with mitochondrial quality control mechanisms, particularly mitophagy, and provide methodologies for accurate experimental assessment. The precise discrimination of these states holds significant implications for drug development targeting neurodegenerative diseases, stroke, and other conditions characterized by mitochondrial dysfunction.
The mitochondrial membrane potential (ΔΨm), typically ranging from -150 to -180 mV (negative inside), constitutes approximately 80% of the proton motive force (PMF) that drives ATP synthesis [58]. Beyond its canonical role in energy transduction, ΔΨm functions as a dynamic signaling hub that regulates reactive oxygen species (ROS) production, calcium handling, and critically, mitochondrial quality control decisions [87]. The maintenance of ΔΨm is essential for neuronal health, given the brain's substantial energy demands, consuming over 20% of total body oxygen metabolism despite representing only 2% of body weight [88].
Mitochondrial quality control (MQC) encompasses a multi-tiered system preserving mitochondrial network integrity through mechanisms including mitochondrial dynamics (fission and fusion), the ubiquitin-proteasome system, and mitophagy—the selective autophagic degradation of damaged mitochondria [11]. ΔΨm serves as a primary signal in determining mitochondrial fate, where sustained depolarization often marks dysfunctional organelles for removal via mitophagy [87] [89]. However, the relationship between ΔΨm changes and mitochondrial fate is complex, as both hyperpolarization and depolarization can represent either adaptive physiological responses or pathological states depending on context, magnitude, and duration.
Molecular Mechanisms of ΔΨm Regulation
Generation and Maintenance of ΔΨm
ΔΨm is generated through the electron transport chain (ETC), where complexes I, III, and IV pump protons from the mitochondrial matrix to the intermembrane space, creating an electrochemical gradient [87]. This potential is primarily consumed by ATP synthase to phosphorylate ADP, with the balance between proton pumping and reflux determining the steady-state ΔΨm value [58]. The finite range of physiologically maintainable ΔΨm creates a system where modest fluctuations regulate function, while extreme or sustained deviations trigger quality control pathways.
Table 1: Key Proteins Regulating Mitochondrial Membrane Potential
| Protein | Function | Impact on ΔΨm |
|---|---|---|
| ETC Complexes I-V | Electron transfer, proton pumping, ATP synthesis | Generate and consume ΔΨm |
| Uncoupling Proteins (UCPs) | Controlled proton leak | Dissipate ΔΨm as heat |
| Adenine Nucleotide Translocase (ANT) | ATP/ADP exchange across IMM | Consumes ΔΨm (1 H+ per ATP exported) |
| PINK1 | Kinase sensing ΔΨm loss | Accumulates upon depolarization, initiates mitophagy |
| Parkin | E3 ubiquitin ligase | Amplifies PINK1 signal, tags mitochondria for degradation |
ΔΨm as a Signaling Hub in Quality Control Decisions
ΔΨm regulates mitochondrial quality control through several mechanisms. The PINK1-Parkin pathway represents the best-characterized mechanism linking ΔΨm to mitophagy. In healthy, polarized mitochondria, PINK1 is imported through the TIM23 complex and constitutively degraded. When ΔΨm dissipates, PINK1 import is impaired, leading to its accumulation on the outer mitochondrial membrane where it recruits and activates Parkin, an E3 ubiquitin ligase [26] [87]. Parkin then ubiquitinates numerous outer membrane proteins, recruiting autophagy adapters like p62/SQSTM1, NDP52, and optineurin that link ubiquitinated mitochondria to the LC3-containing autophagosomal membrane [26] [89].
Beyond mitophagy initiation, ΔΨm influences mitochondrial dynamics that precede quality control decisions. Mitochondrial fission, mediated by Drp1, facilitates the isolation of damaged mitochondrial segments, while fusion, mediated by Mfn1, Mfn2, and OPA1, allows content mixing and complementation [90] [88]. Following fission, daughter mitochondria with preserved ΔΨm typically re-fuse with the network, while those with sustained depolarization are targeted for mitophagy [87]. This quality control mechanism is particularly crucial in neurons, where dysfunctional mitochondria contribute to neurodegenerative pathologies [11].
Discriminating Adaptive and Pathological ΔΨm States
Adaptive Hyperpolarization
Adaptive hyperpolarization represents a physiological response to increased energy demand or signaling requirements. In pancreatic β-cells, glucose-induced hyperpolarization enhances ATP production and calcium signaling to promote insulin secretion [58]. This hyperpolarization occurs within a controlled range and is transient, resolving when the stimulus diminishes. Similarly, neuronal activation can trigger localized mitochondrial hyperpolarization to support synaptic plasticity and dendritic remodeling [87]. Such adaptive responses typically exhibit moderate magnitude, reversibility, and correlation with increased ATP production or specific signaling functions.
Adaptive Depolarization
Controlled depolarization can serve adaptive functions in specific contexts. In hepatocytes, acute ethanol metabolism induces widespread mitochondrial depolarization that facilitates NAD+ regeneration, supporting alcohol dehydrogenase (ADH) and aldehyde dehydrogenase (ALDH2) activity in what is termed "swift increase in alcohol metabolism" (SIAM) [91]. This depolarization is transient and reverses as ethanol is cleared. Similarly, mild uncoupling through UCP activation can prevent excessive ROS generation by dissipating the proton gradient, serving as a protective mechanism against oxidative damage [87]. These adaptive depolarizations are typically self-limiting and resolve upon removal of the initiating stimulus.
Pathological Depolarization
Sustained, irreversible depolarization typically indicates mitochondrial dysfunction and often precedes cell death pathways. In stroke models, excessive mitochondrial fission mediated by Drp1 activation leads to pathological fragmentation and depolarization, exacerbating neurovascular unit dysfunction [90]. Similarly, in neurodegenerative diseases, persistent depolarization is associated with defective mitophagy, allowing damaged mitochondria to accumulate and propagate oxidative damage [11]. Pathological depolarization is characterized by magnitude, duration, association with cytochrome c release, and failure to recover following stimulus removal.
Table 2: Characteristics of Adaptive versus Pathological ΔΨm States
| Parameter | Adaptive Hyperpolarization | Adaptive Depolarization | Pathological Depolarization |
|---|---|---|---|
| Magnitude | Moderate (10-30 mV increase) | Moderate (20-40 mV decrease) | Severe (>50 mV decrease) |
| Duration | Transient (minutes to hours) | Transient (hours) | Sustained (irreversible) |
| Context | Increased energy demand, signaling | Metabolic adaptation, uncoupling | Toxicity, ischemia, neurodegeneration |
| ATP Synthesis | Maintained or increased | Transiently decreased | Severely compromised |
| ROS Production | Modest increase | Decreased via uncoupling | Markedly increased |
| Outcome | Signal resolution, return to baseline | Restoration of ΔΨm | Mitophagy initiation or cell death |
| Examples | β-cell glucose response, synaptic plasticity | Ethanol metabolism (SIAM), mild uncoupling | Stroke, Parkinson's disease models |
Experimental Approaches for ΔΨm Assessment
Methodological Considerations
Accurate ΔΨm measurement requires understanding the limitations of fluorescent potentiometric dyes. Widely used probes like tetramethylrhodamine methyl ester (TMRM), JC-1, and others exhibit ΔΨm-dependent accumulation in the mitochondrial matrix, but their fluorescence is influenced by factors beyond ΔΨm, including mitochondrial mass, membrane permeability, and non-specific binding [58]. Rigorous ΔΨm assessment should incorporate complementary approaches rather than relying on a single methodology.
Critical methodological principles include:
- Validation with controls: Use established uncouplers (FCCP) and ATP synthase inhibitors (oligomycin) to confirm ΔΨm dependence.
- Quantitative calibration: Employ potassium gradients with valinomycin to establish quantitative ΔΨm values.
- Multi-parameter assessment: Combine ΔΨm measurements with simultaneous assessment of ATP production, oxygen consumption rate (OCR), and mitochondrial calcium.
- Context interpretation: Recognize that ΔΨm changes must be interpreted within the specific bioenergetic context, as hyperpolarization may indicate either enhanced coupling or impaired ATP synthesis demand [58].
Integrated Quality Control Assessment
Discriminating adaptive from pathological ΔΨm states requires correlation with mitochondrial quality control markers. Experimental workflows should simultaneously assess:
- PINK1/Parkin recruitment via immunofluorescence or biosensors
- Mitophagic flux using mt-Keima or LC3-II colocalization assays
- Mitochondrial dynamics via time-lapse imaging of fission/fusion events
- Functional outputs including OCR, ATP production, and ROS generation
This multi-parameter approach enables researchers to determine whether ΔΨm changes represent functional adaptations or pathological states requiring quality control intervention.
Diagram 1: Decision matrix for interpreting ΔΨm fluctuations in quality control contexts
Research Reagent Solutions
Table 3: Essential Research Tools for ΔΨm and Quality Control Studies
| Reagent Category | Specific Examples | Function/Application | Key Considerations |
|---|---|---|---|
| Potentiometric Dyes | TMRM, JC-1, TMRE | ΔΨm-dependent accumulation | Concentration optimization crucial; ratiometric JC-1 preferred for quantitative work |
| Genetically-encoded Probes | mito-GEVI, CEPIA | Ratiometric ΔΨm measurement | Enable long-term imaging; target specific mitochondrial subpopulations |
| PINK1/Parkin Reporters | pH-sensitive GFP-Parkin | Mitophagy initiation tracking | Correlate ΔΨm loss with quality control activation |
| Chemical Modulators | FCCP (uncoupler), Oligomycin (ATP synthase inhibitor) | Experimental ΔΨm manipulation | Titration required; assess multiple concentrations |
| Oxygen Consumption Systems | Seahorse XF Analyzer | Coupling efficiency assessment | Combine with ΔΨm for comprehensive bioenergetic profile |
Discriminating between adaptive and pathological changes in mitochondrial membrane potential requires a multi-parametric approach that considers context, magnitude, duration, and functional consequences. Adaptive ΔΨm fluctuations serve physiological roles in energy management, signaling, and metabolic adaptation, while pathological deviations trigger quality control mechanisms or initiate cell death pathways. The integration of ΔΨm measurements with assessments of mitochondrial dynamics, mitophagic flux, and overall cellular health provides the most meaningful interpretation for research and drug development applications. As our understanding of ΔΨm as a dynamic signaling platform evolves, so too will our ability to target specific ΔΨm states for therapeutic benefit in neurological disorders, metabolic diseases, and age-related conditions characterized by mitochondrial dysfunction.
The long-standing paradigm in mitochondrial quality control has posited that mitochondrial fission is an absolute prerequisite for the efficient removal of damaged mitochondria via mitophagy. This review synthesizes recent conflicting evidence that challenges this conventional wisdom, demonstrating that the relationship between fission and mitophagy is context-dependent and influenced by stress type, cellular environment, and the specific mitophagy pathways activated. We examine the mechanistic role of mitochondrial membrane potential (ΔΨm) as a critical determinant in this process, where loss of ΔΨm not only marks mitochondria for degradation but also renders them fusion-incompetent, creating isolated targets for autophagic machinery. Through comprehensive analysis of experimental data across different cell types and disease models, we provide a nuanced framework for understanding the complex interplay between mitochondrial dynamics and quality control, with significant implications for therapeutic targeting in neurodegenerative diseases, metabolic disorders, and ischemia-reperfusion injury.
Mitochondrial quality control maintains a healthy mitochondrial network through coordinated processes including mitochondrial dynamics (fusion and fission) and mitophagy—the selective autophagic degradation of damaged mitochondria [92]. The traditional view holds that mitochondrial fission is essential for mitophagy because it facilitates the isolation of damaged mitochondrial segments from the interconnected network, creating smaller, discrete units that can be efficiently engulfed by autophagosomes [93]. This perspective is supported by substantial evidence demonstrating that mitochondrial fission often precedes mitophagy and that inhibition of fission machinery can impair mitochondrial degradation under certain conditions [94] [93].
The central regulator of mitochondrial fission, dynamin-related protein 1 (DRP1), mediates membrane constriction through GTP hydrolysis, while fission protein 1 (FIS1) and mitochondrial fission factor (MFF) serve as mitochondrial recruitment factors [95] [92]. Conversely, mitochondrial fusion is regulated by mitofusins 1 and 2 (MFN1/2) on the outer membrane and optic atrophy 1 (OPA1) on the inner membrane [95]. The prevailing model suggests that mitochondrial damage triggers DRP1-mediated fission, generating a depolarized daughter mitochondrion that is targeted for mitophagy while the healthy portion reconnects to the network [93].
However, emerging research reveals a more complex relationship, demonstrating that under specific conditions, mitophagy can proceed independently of fission, suggesting cell type-specific and stress-specific variations in this fundamental quality control mechanism [96]. This review examines the evidence both supporting and challenging the fission-prerequisite hypothesis, with particular focus on the role of mitochondrial membrane potential as a critical switch governing mitochondrial fate.
The Evidence: Establishing the Fission-Mitophagy Connection
Mechanistic Basis for Fission as a Prerequisite
The theoretical foundation for fission as a prerequisite for mitophagy rests on both physical and biochemical considerations. From a physical standpoint, the mitochondrial network in many cell types presents as an interconnected tubular system that would be challenging to engulf as a single unit. Mitochondrial fission creates discrete, manageable-sized organelles that can be efficiently surrounded by autophagosomal membranes [93]. From a biochemical perspective, fission enables the asymmetric distribution of damaged components, allowing the cell to isolate and selectively target compromised mitochondrial segments while preserving functional portions [92].
Table 1: Key Evidence Supporting Fission as a Mitophagy Prerequisite
| Experimental System | Key Findings | Molecular Mechanisms | Citation |
|---|---|---|---|
| HeLa cells with DRP1 overexpression | 70% decrease in mitochondrial mass | Enhanced fission facilitates mitophagic clearance | [93] |
| HeLa cells with FIS1 overexpression | ~50% reduction in mitochondrial volume | Increased fission promotes mitochondrial autophagy | [93] |
| INS1 β-cells with FIS1 RNAi | 70% reduction in autophagosomes containing mitochondria | Fission inhibition impairs mitophagy | [93] |
| INS1 β-cells with DRP1 dominant-negative (K38A) | 75% reduction in mitochondrial-containing autophagosomes | Impaired fission prevents mitophagic engulfment | [93] |
| C2C12 myoblasts with DNM1L knockdown | Reduced mitophagic flux and impaired differentiation | Links fission deficiency to defective mitophagy in myogenesis | [94] |
Biochemical studies have revealed that mitochondrial fission generates bioenergetic heterogeneity between daughter mitochondria. Live-cell imaging in INS1 and COS7 cells demonstrates that approximately 5% of fission events produce a persistently depolarized daughter mitochondrion, while most generate transient depolarization [93]. This sustained loss of ΔΨm in the impaired daughter unit serves as a key "eat-me" signal for mitophagy pathways, particularly the PINK1-Parkin system [4] [93].
Molecular Mechanisms Linking Fission to Mitophagy
The molecular machinery connecting fission to mitophagy involves several coordinated events. First, mitochondrial damage or depolarization leads to PINK1 stabilization on the outer mitochondrial membrane, where it phosphorylates ubiquitin and recruits the E3 ubiquitin ligase Parkin [4]. Parkin then ubiquitinates numerous outer mitochondrial membrane proteins, including mitofusins, which are subsequently degraded, preventing fusion and isolating damaged mitochondria [4].
Simultaneously, DRP1 is recruited to mitochondria through interaction with MFF and FIS1, leading to mitochondrial constriction and fission [92]. The resulting mitochondrial fragments are then recognized by autophagic adaptor proteins such as p62/SQSTM1, which bind both ubiquitin on mitochondrial surfaces and LC3 on forming autophagosomes, completing the targeting process [4].
The following diagram illustrates this coordinated process:
Diagram Title: Fission-Dependent Mitophagy Pathway
The Counterevidence: Fission-Independent Mitophagy Pathways
Experimental Challenges to the Paradigm
Despite the substantial evidence supporting fission as a mitophagy prerequisite, rigorous experimental studies have demonstrated scenarios where mitophagy proceeds effectively without fission. A pivotal investigation using primary cortical neurons with conditional DRP1 knockout subjected to oxygen-glucose deprivation and reoxygenation (OGD/R)—an in vitro model of cerebral ischemia-reperfusion injury—revealed that DRP1 deletion prevented mitochondrial fragmentation but did not alter mitophagic flux [96]. This finding directly challenges the necessity of fission for mitophagy, at least in neuronal systems under ischemic stress.
Table 2: Evidence Supporting Fission-Independent Mitophagy
| Experimental System | Key Findings | Molecular Mechanisms | Citation |
|---|---|---|---|
| Primary cortical neurons with DRP1 KO | Normal mitophagic flux despite blocked fragmentation | PINK1-Parkin activation independent of fission | [96] |
| Renal ischemia-reperfusion model | mtROS-ATM-CHK2 pathway activates mitophagy without fission requirement | DNA damage response bypasses fission machinery | [97] |
| C. elegans models | Alternative mitophagy pathways (e.g., FUNDC1-mediated) | Receptor-mediated mitophagy less dependent on fission | [30] |
| Pancreatic β-cells | Stable ΔΨm during solitary phase suggests alternative degradation pathways | Spontaneous depolarization without fission | [93] |
Further supporting fission-independent pathways, recent research has identified a mitochondrial ROS (mtROS)-triggered mechanism involving the DNA damage response pathway. This pathway activates mitophagy through ATM-CHK2 signaling, which phosphorylates multiple mitophagy components including ATAD3A (regulating PINK1 stability), OPTN (enhancing autophagosomal targeting), and Beclin-1 (promoting autophagosome formation) [97]. This cascade operates effectively without requiring mitochondrial fragmentation as an initial step.
Alternative Mechanisms for Mitochondrial Selection
In the absence of fission, cells employ alternative strategies to designate individual mitochondria for degradation. The mitochondrial membrane potential (ΔΨm) appears central to these mechanisms, as depolarization not only activates PINK1-Parkin signaling but also renders mitochondria fusion-incompetent [93]. This fusion incompetence effectively creates "virtual fission" by isolating damaged mitochondria from the network without physical scission.
Receptor-mediated mitophagy pathways, such as those involving FUNDC1 and BNIP3, may also operate with reduced dependence on fission [30] [4]. These resident outer mitochondrial membrane proteins contain LC3-interacting regions that directly recruit autophagosomal membranes to mitochondria, potentially bypassing the need for fragmentation, particularly under hypoxic conditions [30].
The following diagram illustrates key fission-independent pathways:
Diagram Title: Fission-Independent Mitophagy Pathways
The Central Regulator: Mitochondrial Membrane Potential
ΔΨm as a Fate Determinant
The mitochondrial membrane potential (ΔΨm) serves as a critical switch governing both mitochondrial dynamics and mitophagy, potentially explaining the context-dependent relationship between fission and mitophagy. Depolarization below a specific threshold renders mitochondria fusion-incompetent by inhibiting MFN2 function, effectively isolating them from the mitochondrial network regardless of fission status [93]. This isolation creates manageable targets for autophagic engulfment without requiring physical scission.
The PINK1-Parkin pathway demonstrates exquisite sensitivity to ΔΨm. Under normal conditions with maintained ΔΨm, PINK1 is imported into mitochondria and rapidly degraded. However, upon depolarization, PINK1 import is blocked, leading to its accumulation on the outer mitochondrial membrane where it recruits and activates Parkin [4]. This elegant sensing mechanism directly couples the degradation signal to mitochondrial energetic status.
Temporal Dynamics of ΔΨm in Mitophagy
Live-cell imaging studies reveal complex temporal dynamics between ΔΨm and mitophagy. While sustained depolarization reliably triggers mitophagy, transient depolarization episodes often occur after fission events without progressing to degradation [93]. This suggests that ΔΨm loss must persist for a critical duration to commit a mitochondrion to degradation, potentially through sustained PINK1 stabilization and Parkin activation.
The following experimental workflow illustrates methods for investigating these relationships:
Diagram Title: Experimental Workflow for ΔΨm-Mitophagy Studies
Experimental Approaches and Methodologies
Key Research Models and Reagents
Investigating the fission-mitophagy relationship requires sophisticated experimental tools, including specialized cell lines, genetic models, and biochemical reagents. The following table outlines essential research solutions for this field:
Table 3: Research Reagent Solutions for Fission-Mitophagy Studies
| Reagent/Cell Model | Application | Key Features | Citation |
|---|---|---|---|
| MitoQC reporter mice (C57BL/6-Gt(ROSA)26Sortm1(CAG-mCherry/GFP)Ganl/J) | Quantifying mitophagic flux | pH-sensitive GFP-mCherry mitochondrial tag; mCherry signal persists in lysosomes | [96] |
| DRP1 conditional knockout models | Testing fission requirement | Tissue-specific or inducible DRP1 deletion | [96] |
| C2C12 myoblasts with adenoviral transduction | Studying differentiation-linked mitophagy | Compatible with DNM1L, BNIP3, PPARGC1A manipulation | [94] |
| PINK1/Parkin translocation assays | Monitoring pathway activation | Immunofluorescence with antibodies against PINK1, Parkin, ATPB | [96] |
| OGD/R (oxygen-glucose deprivation/reoxygenation) | Modeling cerebral I/R injury | In vitro ischemia-reperfusion simulation | [96] |
| CCCP (carbonyl cyanide m-chlorophenyl hydrazone) | Inducing mitochondrial depolarization | Chemical uncoupler for maximal ΔΨm dissipation | [5] |
| Machine learning mitochondrial classification | Objective morphology quantification | Unbiased classification into network, unbranched, swollen, punctate | [96] |
Protocol for Assessing Fission-Mitophagy Relationship
Based on methodologies from cited studies, the following integrated protocol can assess the fission-mitophagy relationship:
Cell Model Preparation: Utilize primary cortical neurons from MitoQC reporter mice or C2C12 myoblasts. For genetic manipulation, transduce with adenoviruses expressing DRP1 variants (wild-type, dominant-negative K38A, or shRNA) at MOI 30-100 24 hours pre-differentiation or pre-stress [96] [94].
Stress Induction: For neuronal models, apply oxygen-glucose deprivation (1-4 hours) followed by reoxygenation (1-6 hours). For chemical induction, treat with 10-20μM CCCP for 1-4 hours [96].
Mitophagic Flux Quantification:
- Image live cells using confocal microscopy with 63× oil objective
- Count mCherry-positive/GFP-negative puncta (MitoQC signal) per cell
- Co-stain with LAMP1 antibodies to confirm lysosomal localization
- Calculate Mander's correlation coefficient for colocalization analysis [96]
Mitochondrial Morphology Analysis:
- Acquire high-resolution mitochondrial images (TOM20, ATPB, or COX8 labeling)
- Apply machine learning classification system categorizing mitochondria as network, unbranched, swollen, or punctate
- Quantify distribution across morphological categories [96]
Biochemical Validation:
ΔΨm Monitoring: Use TMRE or JC-1 staining with live-cell imaging to correlate depolarization events with fission and mitophagy initiation [93].
Discussion and Future Perspectives
The relationship between mitochondrial fission and mitophagy emerges as context-dependent rather than universally deterministic. While fission undoubtedly facilitates mitophagy in many scenarios—particularly by generating bioenergetically heterogeneous daughters and creating physically manageable substrates for autophagy—compelling evidence demonstrates that alternative pathways can bypass this requirement under specific conditions.
Several factors likely influence the fission dependence of mitophagy:
Cell Type: Neurons, with their complex architecture and elongated mitochondrial distributions, may employ different quality control strategies compared to more compact cells like HeLa or C2C12 myoblasts [95] [96].
Mitophagy Pathway: PINK1-Parkin-mediated mitophagy may have different fission requirements compared to receptor-mediated pathways (FUNDC1, BNIP3) or those activated by alternative ubiquitin ligases [30] [4].
Nature and Intensity of Stress: Acute, profound stress (e.g., complete OGD) may trigger different mechanisms compared to chronic, low-level stress (e.g., hyperglycemia) [4].
Metabolic Status: Cellular energy status and AMPK activation influence both mitochondrial dynamics and autophagic capacity, potentially modulating the fission-mitophagy relationship [20].
Future research should focus on developing more sophisticated tools to precisely manipulate mitochondrial dynamics in spatial and temporal contexts, enabling clearer dissection of cause-effect relationships. Additionally, investigating how different mitochondrial subpopulations (e.g., synaptic vs. somatic mitochondria in neurons) vary in their quality control mechanisms may reveal further complexity in the fission-mitophagy relationship.
From a therapeutic perspective, understanding when fission is required for mitophagy has significant implications for diseases involving mitochondrial dysfunction. In conditions where excessive fission contributes to pathology but mitophagy remains protective, strategies that selectively inhibit pathological fission while preserving mitophagic capacity would be advantageous.
The question "Is mitochondrial fission always a prerequisite for mitophagy?" does not yield a simple binary answer. The evidence reveals a nuanced relationship in which fission serves as a facilitating mechanism rather than an absolute requirement across all contexts. Mitochondrial membrane potential emerges as a central regulator that may determine mitophagy commitment independently of fission status in specific scenarios. As research methodologies advance, particularly in live-cell imaging and genetic manipulation, our understanding of this fundamental biological process will continue to evolve, potentially revealing additional layers of complexity in mitochondrial quality control and offering new therapeutic avenues for mitochondrial diseases.
Mitophagy, the selective autophagic clearance of mitochondria, is a cornerstone of mitochondrial quality control (MQC) essential for cellular homeostasis. While its protective role in eliminating damaged mitochondria is well-established, emerging evidence reveals that mitophagy can undergo pathogenic activation, contributing to disease progression under specific conditions. This whitepaper explores the dual nature of mitophagy outcomes, framed within the critical context of mitochondrial membrane potential (ΔΨm), a key regulator in MQC. We delineate the molecular mechanisms and cellular contexts that transform this cytoprotective process into a pathogenic driver in neurological disorders, metabolic diseases, and muscular atrophy. The analysis incorporates structured quantitative data, detailed experimental methodologies, and visual signaling pathways to provide researchers and drug development professionals with a comprehensive technical resource for navigating this complex biological phenomenon.
Mitophagy serves as a critical quality control mechanism, selectively targeting damaged or superfluous mitochondria for lysosomal degradation to maintain a healthy mitochondrial network [26] [92]. This process is essential for cellular homeostasis, particularly in high-energy-demanding tissues such as neurons, cardiac muscle, and skeletal muscle. Under physiological conditions, mitophagy acts as a protective mechanism, preventing the accumulation of dysfunctional mitochondria that produce excessive reactive oxygen species (ROS) and release pro-apoptotic factors [98] [93].
However, the relationship between mitophagy and cellular health is not straightforward. Recent advances reveal that mitophagy outcomes are fundamentally context-dependent, with the same process that normally maintains homeostasis potentially becoming pathogenic when dysregulated [98]. Both excessive and insufficient mitophagy can disrupt mitochondrial homeostasis and contribute to disease pathogenesis. In spinal cord injury (SCI), for example, abnormal mitophagy significantly contributes to secondary injury processes, leading to impaired adenosine triphosphate (ATP) production, ion imbalance, excessive ROS production, neuroinflammation, and neuronal cell death [98]. Similarly, in Alzheimer's disease (AD), mitophagy deficits establish a vicious cycle with amyloid-β (Aβ) and Tau pathology, ultimately resulting in neuronal damage and death [99].
The mitochondrial membrane potential (ΔΨm) serves as a crucial biological switch that dictates mitophagic activity. As the driving force for mitochondrial ATP synthesis, ΔΨm is a sensitive indicator of mitochondrial health, with depolarization below a certain threshold triggering mitophagic removal [93]. This whitepaper examines the precise molecular mechanisms through which ΔΨm-regulated mitophagy transitions from preserving cellular integrity to driving pathogenesis across various disease contexts.
Molecular Mechanisms and Regulatory Pathways
Core Mitophagy Pathways and Their Regulation
Mitophagy activation occurs through several molecular pathways that converge on the lysosomal degradation of mitochondria. The major pathways include:
PINK1-Parkin-Mediated Pathway
The PINK1-Parkin pathway represents the most extensively characterized mechanism of mitophagy in mammalian cells [26] [30]. This ubiquitin-dependent pathway operates through a finely tuned sequence:
- Damage Sensing: Under normal conditions, PINK1 (PTEN-induced putative kinase 1) is continuously imported into healthy mitochondria through the TOM/TIM complexes, where it undergoes proteolytic cleavage by mitochondrial processing peptidase (MPP) and presenilin-associated rhomboid-like (PARL) protein, followed by degradation [26]. When mitochondria are damaged and ΔΨm is lost, PINK1 import is impaired, leading to its accumulation on the outer mitochondrial membrane (OMM).
- Signal Amplification: Stabilized PINK1 undergoes autophosphorylation and phosphorylates ubiquitin at Ser65. This phosphorylated ubiquitin recruits Parkin, an E3 ubiquitin ligase, from the cytosol to the damaged mitochondria [26]. Parkin then undergoes structural remodeling that activates its E3 ligase activity, initiating extensive ubiquitination of OMM proteins including mitofusins (MFN1/2), mitochondrial Rho-GTPase 1 (Miro1), and voltage-dependent anion channel 1 (VDAC1) [26] [100].
- Effector Recruitment: Ubiquitinated proteins are recognized by autophagy adapters including sequestosome 1 (P62/SQSTM1), optineurin (OPTN), and nuclear dot protein 52 (NDP52/CALCOCO2), which possess both ubiquitin-binding domains and LC3-interacting regions (LIR) that tether the damaged mitochondria to the expanding phagophore [26].
The following diagram illustrates the PINK1-Parkin mediated mitophagy pathway:
Receptor-Mediated Pathways
Beyond the PINK1-Parkin pathway, cells employ several receptor-mediated mechanisms for mitophagy that operate independently of ubiquitination [26] [30]. These pathways utilize OMM proteins that function as mitophagy receptors by directly interacting with LC3 through LIR motifs:
- FUNDC1: Regulates hypoxia-induced mitophagy, with its activity controlled by phosphorylation-dephosphorylation events [30].
- BNIP3 and NIX/BNIP3L: Participate in hypoxia-induced mitophagy and mitochondrial elimination during erythroid differentiation [26].
- PHB2: An inner mitochondrial membrane protein exposed upon mitochondrial damage that interacts with LC3 [26].
- FIS1: While traditionally associated with mitochondrial fission, FIS1 downregulation can trigger alternative mitophagy pathways through abnormal accumulation of STX17 in mitochondria [101].
Interplay with Mitochondrial Dynamics
Mitophagy is intricately linked with mitochondrial dynamics—the continuous cycles of fission and fusion that govern mitochondrial morphology and distribution [93] [92]. Mitochondrial fission, mediated by Drp1 and its adapters (FIS1, MFF, MiD49/51), enables the fragmentation of damaged mitochondrial segments from the healthy network, facilitating their mitophagic removal [92] [101]. Conversely, mitochondrial fusion, orchestrated by MFN1/2 (OMM) and OPA1 (IMM), promotes functional complementation between mitochondria, potentially rescuing mildly damaged units from degradation [92].
The relationship between mitochondrial dynamics and mitophagy represents a critical decision point in mitochondrial fate. As illustrated below, mitochondrial fission often generates heterogeneous daughter units, with one maintaining ΔΨm and remaining in the network, while the other depolarizes and becomes targeted for mitophagy [93].
Mitochondrial Membrane Potential as a Central Regulator
ΔΨm serves as the fundamental parameter that integrates mitochondrial functional state with mitophagic activity. As the electrochemical gradient across the inner mitochondrial membrane, ΔΨm is not only essential for ATP production but also acts as a key damage sensor that triggers quality control responses [93].
Several mechanisms link ΔΨm to mitophagy initiation:
- PINK1 Stability Regulation: ΔΨm directly controls PINK1 import and stability. Under normal polarization, PINK1 is efficiently imported and degraded. Upon depolarization, PINK1 accumulation on the OMM initiates the PINK1-Parkin pathway [26].
- Fusion Competence: ΔΨm determines mitochondrial fusion capability, with depolarized mitochondria becoming fusion-incompetent and segregated from the network [93] [92].
- Phosphorylation Cascades: ΔΨm loss triggers kinase-mediated phosphorylation events that activate mitophagy receptors and adapters [26] [100].
The precise threshold of ΔΨm that triggers mitophagy varies by cell type and context, but typically a reduction of >15 mV from baseline can initiate the process [93]. This threshold behavior ensures that only severely damaged mitochondria are targeted for degradation while allowing for functional recovery of moderately stressed organelles.
Context-Dependent Pathogenic Transitions
Neurological Disorders
In the central nervous system, the balance of mitophagy is crucial for neuronal health, with both deficiency and excess contributing to neurodegeneration.
Alzheimer's Disease (AD): Mitophagy deficits establish a vicious cycle with hallmark AD pathologies. Impaired mitochondrial clearance leads to accumulation of damaged organelles that produce excessive ROS, promoting amyloid-β production and Tau hyperphosphorylation [99]. These pathological proteins further disrupt mitochondrial function, creating a self-reinforcing cycle of degeneration. Additionally, impaired mitophagy activates the NLRP3 inflammasome, driving neuroinflammation that characterizes AD progression [99].
Spinal Cord Injury (SCI): The temporal regulation of mitophagy following SCI dramatically illustrates its context-dependent nature. Initially, mitophagy activation may serve protective functions by removing damaged mitochondria. However, persistent or excessive mitophagy contributes to secondary injury through depletion of functional mitochondria, leading to ATP deficiency, ionic imbalance, and neuronal cell death [98]. Both excessive and insufficient mitophagy can impede recovery, highlighting the critical importance of balanced regulation [98].
Metabolic and Muscular Disorders
Skeletal Muscle Atrophy: Mitochondrial dynamics and mitophagy are coordinately regulated in skeletal muscle, with imbalances driving atrophy pathogenesis. Denervation, aging, or disease states can disrupt this balance, leading to either insufficient mitophagy (accumulating damaged mitochondria) or excessive mitophagy (depleting healthy mitochondria) [101]. Drp1-mediated mitochondrial fission is typically a prerequisite for mitophagy, while impaired fusion due to MFN2 downregulation inhibits mitophagy flux, promoting muscle aging phenotypes [101].
Diabetes Mellitus: Dysfunctional mitophagy contributes to the pathogenesis of diabetes and its complications by disrupting metabolic homeostasis. Natural compounds like ginsenosides and resveratrol demonstrate therapeutic potential by enhancing mitophagy and restoring mitochondrial function via PINK1/Parkin, BNIP3/NIX, and FUNDC1 pathways [30].
Table 1: Disease Contexts of Dysregulated Mitophagy
| Disease Context | Primary Mitophagy Defect | Consequences | Therapeutic Implications |
|---|---|---|---|
| Alzheimer's Disease | Deficient mitophagy | Accumulation of damaged mitochondria, enhanced Aβ & Tau pathology, neuroinflammation | Mitophagy enhancers may break vicious cycle of degeneration |
| Spinal Cord Injury | Biphasic dysregulation (early excess/late deficiency) | Secondary injury, ATP depletion, neuronal death | Timely modulation required; both inhibition and enhancement considered |
| Skeletal Muscle Atrophy | Imbalance with mitochondrial dynamics | Fragmented network, ROS accumulation, protein degradation | Targeting mitochondrial dynamics to restore mitophagy balance |
| Diabetes Mellitus | Impaired mitophagic flux | Insulin resistance, oxidative stress, chronic inflammation | Natural compounds (e.g., resveratrol, berberine) as mitophagy modulators |
Experimental Approaches and Methodologies
Assessing Mitophagy Activity and Flux
Researchers employ multiple complementary approaches to monitor mitophagy in experimental systems:
Mitochondrial Membrane Potential Monitoring
ΔΨm is typically measured using potentiometric fluorescent dyes that accumulate in polarized mitochondria:
- TMRE (Tetramethylrhodamine, ethyl ester): A cell-permeant dye that accumulates in active mitochondria in a ΔΨm-dependent manner. Depolarized mitochondria show reduced TMRE fluorescence [93] [100].
- JC-1: Exhibits potential-dependent accumulation in mitochondria, indicated by a fluorescence emission shift from green (~529 nm) to red (~590 nm). The red/green fluorescence ratio provides a quantitative measure of ΔΨm.
Protocol: Cells are loaded with TMRE (20-50 nM) or JC-1 (2-5 μM) in culture medium for 15-30 minutes at 37°C, followed by washing and immediate imaging or flow cytometry analysis. Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP; 5-10 μM), a mitochondrial uncoupler, serves as a positive control for depolarization [100].
Mitophagy Flux Assays
Several engineered systems enable specific tracking of mitophagic flux:
- mt-Keima: A pH-sensitive fluorescent protein targeted to mitochondria that exhibits a excitation shift from 440 nm (neutral pH) to 586 nm (acidic pH) upon delivery to lysosomes, allowing quantification of mitochondria in acidic compartments [100].
- Mito-QC: A tandem mCherry-GFP tag targeted to mitochondria. The GFP signal is quenched in acidic lysosomes while mCherry remains stable, differentiating mitochondria in autophagosomes (GFP+/mCherry+) from those in lysosomes (GFP-/mCherry+) [100].
Protocol: Cells expressing mt-Keima or Mito-QC are treated with experimental conditions, then analyzed by confocal microscopy or flow cytometry. For flux measurements, parallel samples can be treated with lysosomal inhibitors (e.g., bafilomycin A1, 100 nM) to block degradation and quantify accumulated mitophagic intermediates.
Molecular Pathway Analysis
Immunofluorescence and biochemical approaches assess specific pathway components:
- PINK1 Stabilization: Western blotting of mitochondrial fractions detects PINK1 accumulation (∼63 kDa) following mitochondrial depolarization [26].
- Parkin Translocation: Live-cell imaging of GFP-Parkin or immunostaining of endogenous Parkin reveals its recruitment to depolarized mitochondria [26] [100].
- Ubiquitination and Phosphorylation: Phos-tag gels and ubiquitin linkage-specific antibodies detect phosphorylation events (e.g., ubiquitin Ser65, Parkin Ser65) and ubiquitin chain formation on mitochondrial proteins [26] [100].
Manipulating Mitophagy Pathways
Genetic and pharmacological tools enable specific perturbation of mitophagy pathways:
Genetic Manipulations:
- PINK1 Knockout: B6.129S4-Pink1tm1Shn/J mice (Jackson Laboratory #017946) provide PINK1-deficient cells [100].
- Parkin Knockout: B6.129S4-Prkntm1Shn/J mice (Jackson Laboratory #006582) generate Parkin-null systems [100].
- MFN2 Mutants: Phosphomimetic (T111E, S378E, S442E) and non-phosphorylatable (T111A, S378A, S442A) MFN2 mutants dissect phosphorylation-specific regulation [100].
Pharmacological Modulators:
- FCCP: Mitochondrial uncoupler (5 μM, 1-8 hours) induces depolarization and PINK1-Parkin-mediated mitophagy [100].
- Mdivi-1: Drp1 inhibitor (50 μM) blocks mitochondrial fission and subsequent mitophagy [92].
- Valinomycin: K+ ionophore that specifically depolarizes mitochondria to induce mitophagy.
Table 2: Essential Research Reagents for Mitophagy Studies
| Reagent/Category | Specific Examples | Function/Application | Experimental Notes |
|---|---|---|---|
| ΔΨm Indicators | TMRE, JC-1, TMRM | Quantitative measurement of mitochondrial membrane potential | Use at low concentrations (20-50 nM) to avoid artifacts; confirm with FCCP control |
| Genetic Models | PINK1 KO mice, Parkin KO mice, MFN2 phosphomutants | Dissecting specific pathway components | MFN2 mutants reveal phosphorylation barcoding of mitochondrial fate |
| Mitophagy Reporters | mt-Keima, Mito-QC (mCherry-GFP), Rosella | Quantifying mitophagic flux | mt-Keima allows ratiometric measurement; Mito-QC differentiates autophagosomes vs. lysosomes |
| Pathway Activators | FCCP, CCCP, Valinomycin, Oligomycin/Antimycin A | Inducing mitochondrial depolarization and mitophagy | FCCP (5 μM) most widely used; treatment duration determines mitophagy extent |
| Pathway Inhibitors | Mdivi-1 (Drp1 inhibitor), Bafilomycin A1 (lysosomal inhibitor) | Blocking specific mitophagy steps | Mdivi-1 (50 μM) inhibits fission; BafA1 (100 nM) blocks degradation for flux measurements |
| Antibodies | PINK1, Parkin, Phospho-ubiquitin (Ser65), LC3, TOMM20 | Detecting pathway components and markers | Phospho-specific antibodies critical for monitoring activation states |
The Scientist's Toolkit: Research Reagent Solutions
The following table provides a comprehensive overview of essential research tools for investigating context-dependent mitophagy outcomes:
Table 3: Advanced Research Reagents for Mitophagy Studies
| Reagent Category | Specific Examples | Function/Application | Experimental Considerations |
|---|---|---|---|
| Pathway-Specific Reporters | PINK1FRET, Ubiquitin phosphorylation sensors | Real-time monitoring of specific pathway activation | Genetically encoded biosensors for live-cell imaging of kinase activity |
| Inducible Systems | Tetracycline-inducible Parkin expression, AAV-mito-QC | Temporal control over mitophagy components | Allows separation of expression effects from experimental treatments |
| Organelle-Specific Dyes | MitoTracker Red CMXRos (potential-sensitive), MitoSOX Red (ROS-specific) | Multiparameter mitochondrial assessment | CMXRos retention depends on ΔΨm; MitoSOX detects mitochondrial superoxide |
| Proteostasis Tools | OMA1/OPA1 cleavage reporters, LON protease inhibitors | Monitoring mitochondrial protein quality control | Connects proteostatic stress to mitophagy activation |
| Lipid Biosensors | Cardiolipin externalization probes (NAO) | Detecting early mitophagy signals | Cardiolipin externalization to OMM serves as "eat-me" signal in some pathways |
| Super-Resolution Imaging | STED, STORM of mitochondrial networks | Ultrastructural analysis of mitochondrial dynamics | Reveals sub-diffraction details of fission/fusion and autophagosome formation |
The dual nature of mitophagy—as both guardian and executioner of cellular homeostasis—underscores the critical importance of context in determining functional outcomes. Mitochondrial membrane potential serves as the central regulator that integrates mitochondrial functional state with mitophagic activity, creating decision points that determine cellular fate. The transition from protective to pathogenic mitophagy involves complex interactions between the PINK1-Parkin pathway, receptor-mediated mechanisms, and mitochondrial dynamics.
For researchers and drug development professionals, several key considerations emerge:
Temporal Dynamics: Mitophagy modulation requires precise timing, as exemplified by the biphasic nature of mitophagy in spinal cord injury where both excessive and insufficient activity can be detrimental [98].
Tissue Specificity: Mitophagy regulation differs across tissues, necessitating cell-type-specific approaches. Neurons, muscle cells, and pancreatic β-cells each demonstrate unique mitophagy characteristics and vulnerabilities [93] [30] [101].
System Integration: Mitophagy does not operate in isolation but functions within a broader MQC network that includes mitochondrial biogenesis, dynamics, and proteostasis [92]. Effective therapeutic strategies must consider these interconnected systems.
Technical Advancements: Future progress depends on developing more sophisticated tools for monitoring mitophagy flux in vivo, assessing specific pathway activation, and achieving temporal and spatial control over mitophagy modulation.
The emerging understanding of context-dependent mitophagy outcomes opens new therapeutic opportunities for neurological disorders, metabolic diseases, and muscular atrophy. By targeting specific regulatory nodes within mitophagy pathways—particularly those linked to ΔΨm sensing and signaling—researchers can develop precision interventions that maintain the protective functions of mitophagy while preventing its pathogenic activation.
In the study of complex cellular processes like mitophagy—the selective autophagic degradation of mitochondria—researchers increasingly rely on compound-based assays to probe biological mechanisms. The initiation of mitophagy is critically dependent on mitochondrial membrane potential (Δψm), as depolarization serves as a fundamental signal triggering the removal of dysfunctional mitochondria [93] [5]. Experimental compounds designed to modulate mitophagy often target this membrane potential, but their effectiveness is governed by pharmacokinetic (PK) properties that determine compound concentration at the target site over time. Pharmacokinetic decay refers to the time-dependent decrease in compound concentration due to absorption, distribution, metabolism, and excretion (ADME) processes. In mitophagy research, where mitochondrial quality control mechanisms operate on dynamic timescales, failing to account for PK decay can lead to significant misinterpretations of experimental outcomes. This technical guide examines the critical pitfalls in neglecting pharmacokinetic considerations and provides methodologies to enhance assay reliability in mitochondrial membrane potential and quality control research.
Fundamental Concepts: Pharmacokinetic Principles in Mitochondrial Research
The Role of Mitochondrial Membrane Potential in Mitophagy
Mitochondrial membrane potential (Δψm) serves as a central regulator in mitochondrial quality control, particularly in initiating mitophagy. Depolarization below a critical threshold indicates impaired mitochondrial function and acts as a prerequisite for mitophagy activation [93]. The PINK1-Parkin pathway, a primary mitophagy mechanism, is exquisitely sensitive to Δψm changes. Under normal conditions, PINK1 is imported into mitochondria and rapidly degraded. Upon depolarization, PINK1 stabilizes on the outer mitochondrial membrane where it recruits and activates the E3 ubiquitin ligase Parkin, initiating a signaling cascade that targets damaged mitochondria for autophagic degradation [26]. This membrane potential-dependent mechanism means that compounds affecting Δψm must maintain appropriate concentrations within a specific window to yield biologically relevant results.
Pharmacokinetic Decay and Its Experimental Implications
Pharmacokinetic decay follows recognizable patterns typically characterized by parameters such as half-life (t½), clearance (CL), and volume of distribution (Vd). In experimental settings, the effective concentration of a compound diminishes non-linearly over time, potentially creating a disconnect between the initial administered dose and the concentration present when measuring outcomes. For mitophagy assays, this is particularly problematic because:
- Mitophagy is a time-dependent process requiring sustained modulation of membrane potential
- Brief compound exposure may insufficiently engage the PINK1-Parkin amplification loop
- Suboptimal concentrations may produce partial depolarization without committing to mitophagy
The metabolism of experimental compounds occurs through various pathways, including oxidation, demethylation, and sulfidation, as demonstrated in PK studies of triazole-based FKBP12 ligands where rapid metabolism led to short half-lives (12.6 ± 3.3 minutes) [102]. Without accounting for these metabolic processes, researchers may misinterpret negative results as biological phenomena rather than pharmacokinetic limitations.
Technical Pitfalls: Consequences of Neglecting Pharmacokinetic Decay
False Negatives in Mitophagy Induction
A primary pitfall involves false negative results arising from insufficient compound exposure duration. If a mitophagy-inducing compound has a short half-life due to rapid hepatic metabolism or efflux transport, it may not maintain sufficient intracellular concentration to trigger the complete mitophagy cascade. For instance, since mitochondrial fission events occur approximately every 22 minutes and depolarization after fission is often transient [93], a compound with rapid clearance might miss the critical window for engagement with the fission machinery.
Inaccurate EC₅₀ Determinations
Incorrect potency calculations frequently occur when PK decay is unaccounted for. The reported half-maximal effective concentration (EC₅₀) values for mitophagy inducers may appear higher than their true potency if metabolic degradation significantly reduces bioactive compound levels during the assay period. This inaccuracy propagates through subsequent research, affecting structure-activity relationships and lead optimization efforts.
Mischaracterization of Compound Mechanisms
Compounds with similar effects on mitochondrial membrane potential but different pharmacokinetic profiles may be erroneously categorized together. A compound with moderate potency but excellent exposure might outperform a highly potent but rapidly-cleared compound, leading to incorrect conclusions about structure-function relationships. Furthermore, intermittent compound exposure due to PK decay may produce conflicting effects on mitochondrial dynamics, as brief depolarization might not commit to mitophagy but could still impair mitochondrial function through other mechanisms.
Table 1: Common Experimental Artifacts Resulting from Unaccounted Pharmacokinetic Decay
| Artifact Type | Underlying Mechanism | Impact on Data Interpretation |
|---|---|---|
| Apparent Time-Dependent Reversal | Compound concentration falls below effective threshold due to metabolism/clearance | Misinterpreted as biological adaptation or feedback mechanism |
| Inconsistent Dose-Response | Variable compound exposure across assay durations | Incorrect potency and efficacy estimations |
| Cell-Type Specific Effects | Differential expression of metabolizing enzymes or transporters | Wrongly attributed to unique biological pathways rather than PK differences |
| Poor Translational Correlation | Disparate metabolic rates between in vitro systems and in vivo models | Failed translation from cell culture to animal models or clinical applications |
Methodological Approaches: Accounting for PK Decay in Experimental Design
Direct Measurement of Compound Stability
Incorporating compound stability assessment into experimental protocols provides critical data for interpreting results:
Microsomal Stability Assays: Incubate test compounds with liver microsomes (from human or relevant species) to quantify metabolic turnover [103]. This approach identifies rapidly metabolized compounds early in the testing cascade.
Protocol:
- Prepare incubation mixture containing liver microsomes (0.5-1 mg/mL), test compound (1-5 µM), and NADPH-regenerating system in phosphate buffer
- Incubate at 37°C with shaking
- Remove aliquots at predetermined time points (0, 5, 15, 30, 60 minutes)
- Terminate reactions with ice-cold acetonitrile
- Analyze compound concentration via LC-MS/MS
- Calculate half-life using the formula: t½ = 0.693 / k, where k is the elimination rate constant
Hepatocyte Stability Assays: Utilize intact primary hepatocytes to provide a more comprehensive metabolic profile including both Phase I and Phase II metabolism [103].
Continuous Concentration Monitoring
Advanced techniques enable real-time monitoring of compound concentration:
Radiolabeling Approaches: Incorporate radioactive isotopes (³H, ¹⁴C) into test compounds to track their fate during experiments [102]. This method provides unparalleled sensitivity for quantifying parent compound and metabolites.
PET Imaging Applications: Positron Emission Tomography (PET) using carbon-11 (¹¹C) or fluorine-18 (¹⁸F) labeled compounds enables non-invasive monitoring of compound distribution and retention [102]. Although more common in vivo, the principles can be adapted for sophisticated in vitro systems.
Temporal Mapping of Assay Endpoints
Instead of single endpoint measurements, implement time-course analyses to correlate compound exposure with biological effects:
Protocol for Mitophagy Time-Course:
- Treat cells with test compound across a range of concentrations
- At multiple time points (e.g., 1, 2, 4, 8, 16, 24 hours), assess:
- Mitochondrial membrane potential (using TMRE or JC-1 dyes)
- PINK1 stabilization and Parkin recruitment (via immunofluorescence)
- Mitophagic flux (using mt-Keima or LC3-II accumulation assays)
- Parallel samples should be collected for LC-MS/MS analysis of compound concentration
- Correlate compound concentration with magnitude of mitophagic response
Table 2: Research Reagent Solutions for Pharmacokinetic Assessment
| Reagent/Assay | Function | Application in Mitophagy Research |
|---|---|---|
| Liver Microsomes | Provide cytochrome P450 enzymes for metabolic stability assessment | Predict hepatic clearance of mitophagy modulators |
| Caco-2 Cell Model | Assess intestinal permeability and efflux transporter susceptibility | Determine oral bioavailability of compounds |
| MDCK-MDR1 Cells | Evaluate P-glycoprotein-mediated efflux | Identify compounds with potential CNS penetration issues for neurological mitophagy studies |
| CYP Isoform Inhibitors | Chemical inhibition of specific metabolic pathways | Identify enzymes responsible for compound degradation |
| Stable Isotope Labels (deuterium, ¹³C) | Modify metabolic soft spots to improve stability | Extend half-life of mitophagy probes without altering target engagement |
| CYP Recombinant Enzymes | Identify specific cytochrome P450 isoforms involved in metabolism | Guide chemical optimization to reduce metabolic clearance |
Computational and AI-Based Solutions
In Silico Prediction of PK Properties
Artificial intelligence and machine learning approaches now enable reasonably accurate prediction of ADME properties early in compound development [104]. These tools can prioritize compounds with favorable PK profiles before synthesis:
QSAR Modeling: Quantitative Structure-Activity Relationship models trained on large chemical datasets (e.g., ChEMBL) can predict metabolic stability, plasma protein binding, and membrane permeability [103] [105].
Deep Learning Approaches: Convolutional Neural Networks (CNNs) and Recurrent Neural Networks (RNNs) can extract complex patterns from molecular structures to predict human liver microsomal stability and other PK parameters [104].
PK-PD Modeling Integration
Pharmacokinetic-Pharmacodynamic (PK-PD) modeling mathematically links compound exposure to biological effect, providing a powerful framework for designing mitophagy assays:
Protocol for Implementing PK-PD Modeling:
- Determine in vitro metabolic stability parameters
- Measure target engagement potency (e.g., EC₅₀ for depolarization)
- Establish relationship between target engagement and functional response (mitophagy induction)
- Develop integrated PK-PD model using software such as Phoenix WinNonlin or R-based packages
- Use the model to simulate optimal dosing regimens for experimental assays
- Validate model predictions with targeted in vitro experiments
Special Considerations for Mitochondrial Membrane Potential Assays
Addressing Compound Interference with Detection Methods
Many membrane potential-sensitive dyes (e.g., TMRE, JC-1) may compete with test compounds for mitochondrial uptake or exhibit spectral overlaps:
Protocol for Control Experiments:
- Measure fluorescence of potential test compounds across relevant emission spectra
- Assess potential quenching or enhancement of dye signals by test compounds
- Perform dye loading both before and after compound treatment to identify timing artifacts
- Validate key findings with multiple detection methods (e.g., combine fluorescent dyes with TMRM quantification via flow cytometry)
Temporal Considerations in Membrane Potential Measurements
Since mitochondrial membrane potential exhibits dynamic fluctuations and fission events can generate heterogeneous depolarization [93], sampling time critically influences results:
Protocol for Longitudinal Assessment:
- Implement continuous monitoring using genetically encoded biosensors (e.g., mito-GFP)
- For endpoint assays, standardize timing relative to compound addition and cellular processes
- Account for cellular circadian rhythms by conducting experiments at consistent times of day
- Consider mitochondrial network dynamics—individual mitochondria undergo fission every 16-22 minutes [93]
Quality Control and Validation Strategies
Analytical Method Validation
Ensure robust quantification of test compounds in biological matrices:
Protocol for LC-MS/MS Method Validation:
- Establish linearity over expected concentration range (typically 1-1000 nM)
- Determine precision (intra-day and inter-day) with CV < 15%
- Assess accuracy (85-115% of nominal values)
- Evaluate matrix effects and extraction efficiency
- Verify stability under experimental conditions (37°C in culture medium)
Benchmarking Against Pharmacological Standards
Include reference compounds with well-characterized PK/PD profiles in mitophagy assays:
Recommended Reference Compounds:
- CCCP: Potent uncoupler with rapid membrane depolarization but potential cytotoxicity
- Oligomycin: ATP synthase inhibitor that indirectly affects membrane potential
- Valinomycin: K⁺ ionophore that specifically depolarizes mitochondria
- Nigericin: K⁺/H⁺ exchanger affecting both membrane potential and pH
Visualization of Key Concepts
Diagram 1: Interplay Between Pharmacokinetics and Mitophagy Signaling. This pathway illustrates how pharmacokinetic processes influence compound availability for engaging mitochondrial targets, with critical pitfalls highlighted.
Diagram 2: Experimental Workflow for PK-Informed Mitophagy Assays. This workflow integrates pharmacokinetic assessment throughout the experimental process to enhance data reliability.
Pharmacokinetic decay represents a critical yet frequently overlooked variable in compound-based assays of mitochondrial membrane potential and mitophagy. The dynamic nature of both mitochondrial quality control processes and compound disposition necessitates integrated experimental approaches that account for temporal changes in bioactive compound concentrations. By implementing the methodological frameworks outlined in this technical guide—including direct stability assessment, temporal mapping, computational prediction, and PK-PD modeling—researchers can significantly enhance the reliability and translational relevance of their findings in mitochondrial research. As the field advances toward more sophisticated modulation of mitophagy for therapeutic applications, rigorous pharmacokinetic characterization will be indispensable for distinguishing true biological effects from experimental artifacts.
Optimizing Assay Conditions to Avoid False Positives in ΔΨm Measurement
Mitochondrial membrane potential (ΔΨm), generated by the electron transport chain, is a key parameter necessary for healthy mitochondrial functioning. It serves as an essential component of the driving force behind mitochondrial ATP synthesis and plays a fundamental role in mitochondrial homeostasis through selective elimination of dysfunctional mitochondria [106]. Within the context of mitochondrial quality control (MQC), ΔΨm is particularly crucial as it serves as a key signal determining mitochondrial fate. A distinctive feature of the early stages of apoptosis and other pathological processes is the disruption of normal mitochondrial function, often marked by a collapse in ΔΨm [106] [107]. Furthermore, ΔΨm is a critical regulator of mitophagy, the selective autophagic clearance of damaged mitochondria. The current paradigm indicates that loss of ΔΨm is a primary trigger for the PINK1/Parkin pathway, where dissipated ΔΨm leads to PINK1 accumulation on the outer mitochondrial membrane, subsequently activating the ubiquitin ligase PARKIN to recruit autophagy machinery [107] [92].
However, research advancements have revealed that mitophagy can also occur through ΔΨm-independent pathways. Compounds like the P62-mediated mitophagy inducer (PMI) can force mitochondria into autophagy without collapsing ΔΨm or recruiting Parkin, acting downstream of the traditional PINK1/Parkin pathway [108]. This complexity underscores why accurate ΔΨm measurement is paramount for correctly interpreting mitochondrial health and function in research. False positives in ΔΨm measurement can lead to significant misinterpretation of a compound's mechanism of action, particularly in drug discovery screens where mitochondrial toxicity is a concern. This technical guide provides detailed methodologies and considerations for optimizing assay conditions to ensure accurate, reliable ΔΨm measurements within the framework of MQC research.
Multiple technical factors can compromise ΔΨm assay integrity, leading to false positive or negative results that misrepresent the true state of mitochondrial polarization.
Dye-Related Artifacts and Limitations
Fluorescent dyes are powerful tools for assessing ΔΨm, but their limitations must be acknowledged. Probes such as JC-1, JC-10, TMRM, and TMRE accumulate in mitochondria in a potential-dependent manner, but they vary significantly in their sensitivity and susceptibility to artifacts. JC-1, for instance, can form precipitates when diluted into aqueous buffers, creating artifacts that interfere with accurate measurement [106]. JC-10, a derivative of JC-1, was developed to address this specific issue, as it does not precipitate in aqueous buffers, thereby eliminating associated artifacts and providing a higher signal-to-background ratio [106]. It is also critical to recognize that different dyes have distinct mechanisms of action; some exhibit concentration-dependent quenching, while others, like TMRM and TMRE, show minimal self-quenching and low cytotoxicity, making them favorable for dynamic measurements [106] [109].
The proper selection of controls is equally vital for data interpretation. Uncoupling agents like FCCP (carbonyl cyanide-p-trifluoromethoxyphenylhydrazone) that dissipate the proton gradient cause complete mitochondrial depolarization, serving as a robust negative control. Conversely, Oligomycin A, an ATP synthase inhibitor, typically leads to hyperpolarization due to the inhibition of proton flow through Complex V, providing a positive control for increased ΔΨm [109]. The table below summarizes key dyes and their properties for informed selection.
Table 1: Characteristics of Common ΔΨm-Sensitive Fluorescent Dyes
| Dye Name | Detection Method | Key Advantages | Known Limitations/Artifacts |
|---|---|---|---|
| JC-1 | Fluorescence shift (monomer 525 nm → aggregate 590 nm) | Qualitative (color change) and quantitative (ratio) data [106] | Can precipitate in aqueous buffers, causing artifacts; lower signal-to-background than JC-10 [106] |
| JC-10 | Fluorescence shift (green 525 nm → orange 590 nm) | Enhanced solubility, robust, higher signal-to-background, detects subtle changes [106] | Similar to JC-1 but with improved performance characteristics [106] |
| TMRM / TMRE | Intensity-based quantification | Minimal self-quenching, low cytotoxicity, reasonable photostability [106] [109] | Signal is also dependent on mitochondrial mass; requires careful concentration optimization [110] |
| m-MPI | Fluorescence shift (green 535 nm → red 590 nm) | Water-soluble, homogenous assay format, suitable for high-throughput screening [111] | Similar to JC-1/JC-10 in principle, but optimized for microplate readers [111] |
Cell Culture and Biological Considerations
The metabolic state of cells is a major biological factor influencing ΔΨm and must be carefully controlled. Cells primarily relying on glycolysis due to high glucose conditions will have a different basal ΔΨm and response to stressors compared to cells forced to rely on oxidative phosphorylation. Research has demonstrated that adapting cells to low glucose medium supplemented with fatty acids (e.g., oleic acid) induces a metabolic shift from glycolysis to oxidative phosphorylation, which increases oxygen consumption rate (OCR) and ATP levels while decreasing extracellular acidification rate (ECAR), a marker of glycolysis [112]. This optimization ensures that cells solicit mitochondria for energy production, making ΔΨm measurements more physiologically relevant, especially for studies on mitochondrial toxicity.
Cell confluence is another critical variable. Studies show that as cell confluence increases, cells can shift from glycolysis to oxidative phosphorylation to produce ATP, becoming more sensitive to mitochondrial inhibitors like oligomycin [112]. Performing assays at a standardized, high confluence (e.g., 95%) helps ensure metabolic consistency. Furthermore, genetic and pharmacological models can intrinsically alter ΔΨm. For instance, cells genetically depleted of the ATP synthase inhibitory factor 1 (IF1) display chronic mitochondrial hyperpolarization, while environmental chemicals can also induce hyperpolarization [110]. Researchers must be aware of their model's inherent properties to avoid misinterpreting baseline states as experimental effects.
Optimized Experimental Protocols and Workflows
Quantitative High-Throughput Screening (qHTS) Protocol for MMP
This protocol, adapted for a 1536-well plate format, is designed to minimize artifacts and enable multiplexing with viability assays [111].
- Cell Seeding: Plate cells at an optimized density (e.g., 2,000 cells/well for HepG2) in 5 μL of culture medium into 1536-well plates. Using a reagent dispenser ensures uniformity.
- Incubation: Incubate assay plates overnight at 37°C under a humidified atmosphere with 5% CO2 to allow for complete cell adhesion.
- Compound Treatment: Transfer test compounds and controls (e.g., FCCP, Oligomycin A) to assay plates using a pintool workstation. Including a range of concentrations is critical for identifying toxic effects.
- Compound Exposure: Incubate assay plates at 37°C for a defined period (e.g., 1 h or 5 h). Kinetic measurements over time provide more information than single endpoints.
- Dye Loading: Add 5 μL of a 2X dye-loading solution (e.g., m-MPI) to each well using a flying reagent dispenser (FRD). Homogenous dispensing is key to well-to-well consistency.
- Dye Incubation: Incubate assay plates at 37°C for 30 minutes to allow for dye accumulation.
- Fluorescence Measurement: Measure fluorescence intensity using a plate reader with appropriate filter sets. For ratiometric dyes like m-MPI or JC-10, measure both emission wavelengths (e.g., 535 nm and 590 nm).
- Data Expression: Calculate the ratio of the emissions (590 nm/535 nm) as the primary indicator of ΔΨm. A decrease in this ratio indicates depolarization.
- Viability Multiplexing (Optional but Recommended): Immediately after the MMP assay, add a cell viability reagent (e.g., CellTiter-Glo) to the same wells. Incubate and measure luminescence to quantify ATP as a viability marker. This step is crucial for distinguishing true ΔΨm loss from general cytotoxicity.
Real-Time Kinetic MMP Assay
For monitoring transient changes, real-time assays are superior.
- Dye Addition: Add the MMP reagent (e.g., Incucyte MMP Orange Dye, TMRM) directly to the cell culture medium.
- Baseline Measurement: Place the plate in a live-cell imaging system (e.g., Incucyte) inside a cell culture incubator and acquire baseline fluorescence and phase-contrast images.
- Compound Treatment: Add compounds of interest and controls without removing the plate from the incubator.
- Kinetic Data Acquisition: Automatically collect fluorescence and morphological data at regular intervals (e.g., every 1-2 hours) over the desired timeframe (e.g., 24-48 hours).
- Analysis: Quantify mean fluorescence intensity per well or per cell over time. Normalize data to the baseline measurement. A decrease in fluorescence indicates depolarization, while an increase suggests hyperpolarization [109].
Figure 1: A workflow for optimizing ΔΨm assays to prevent false positives, covering key steps from cell culture to data validation.
Data Interpretation and Validation Strategies
Statistical Rigor and Hit Validation
In screening environments, applying robust statistical methods is essential for distinguishing true hits from false positives. The Z-factor is a key metric for assessing assay quality, with values above 0.5 indicating an excellent assay. It is important to note that different assay parameters can have varying reliabilities; for example, while ATP and redox potential assays might achieve Z-factors of 0.58 and 0.85 respectively, a ΔΨm assay might have a lower Z-factor of 0.01, indicating a higher potential for false positives and negatives [112]. This underscores the necessity of multiparametric assessment.
Compounds should be classified as hits only when they induce a signal change significantly beyond the vehicle control (e.g., DMSO ± 2σ) and deviate from normal distribution [112]. Hierarchical clustering of hits based on their profiles across multiple parameters (e.g., ΔΨm, ATP levels, redox potential) can help identify clusters of compounds with shared mechanisms and distinguish general toxins from specific modulators.
Multiplexing with Viability and Orthogonal Assays
A primary strategy to avoid false positives is to multiplex the ΔΨm assay with a cell viability readout. A decrease in ΔΨm coupled with a simultaneous, sharp decline in cellular ATP (as measured by a luminescent viability assay) strongly suggests that the depolarization is a secondary consequence of general cell death rather than a primary mitochondrial effect [111]. True mitochondrial toxicants may cause ΔΨm loss before or without immediate cell death. Furthermore, correlating ΔΨm data with other mitochondrial parameters provides a more comprehensive picture. Key complementary assays include:
- Oxygen Consumption Rate (OCR): Measured using a Seahorse Analyzer, it directly reports on mitochondrial electron transport chain function [112].
- Extracellular Acidification Rate (ECAR): An indicator of glycolysis, which can increase when oxidative phosphorylation is impaired [112].
- Imaging of Mitochondrial Morphology: Using fluorescent markers to visualize mitochondrial fragmentation, a common feature of dysfunction [109] [113].
Table 2: Strategies for Validating ΔΨm Data and Avoiding False Conclusions
| Challenge | Validation Strategy | Interpretation of Validated Result |
|---|---|---|
| Distinguishing specific ΔΨm loss from general cytotoxicity | Multiplex with cell viability assay (e.g., ATP content) [111]. | ΔΨm loss WITHOUT viability loss suggests specific mitochondrial toxicity. Concurrent loss suggests cytotoxicity. |
| Confirming bioenergetic impact | Measure Oxygen Consumption Rate (OCR) and Extracellular Acidification Rate (ECAR) [112]. | Reduced OCR supports electron transport chain dysfunction. Increased ECAR suggests a shift to glycolysis. |
| Verifying mitophagy induction | Use orthogonal assays: monitor PINK1/Parkin recruitment or LC3-II colocalization [107] [108]. | Confirms if ΔΨm loss is functionally linked to the mitophagy pathway. |
| Identifying assay-specific artifacts | Use a second, structurally distinct ΔΨm dye (e.g., confirm TMRM results with JC-10) [106] [111]. | Consistent results across dye chemistries confirm the ΔΨm phenotype. |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for ΔΨm and Mitophagy Research
| Reagent / Tool | Function / Mechanism | Application in Assay Validation |
|---|---|---|
| JC-10 Dye | Ratiometric, potential-dependent probe that forms orange J-aggregates in healthy mitochondria and green monomers upon depolarization [106]. | Primary ΔΨm measurement; preferred over JC-1 due to enhanced solubility and reduced artifacts [106]. |
| TMRM / TMRE | Cell-permeable cationic dyes that accumulate in active mitochondria; intensity-based measurement [106] [109]. | Kinetic, real-time measurement of ΔΨm in live cells; low cytotoxicity [109]. |
| FCCP | Protonophore uncoupler that dissipates the proton gradient, collapsing ΔΨm [111] [109]. | Standard positive control for complete depolarization. |
| Oligomycin A | ATP synthase inhibitor that can cause hyperpolarization by blocking proton flow through Complex V [109]. | Positive control for increased ΔΨm (hyperpolarization). |
| PMI (P62-mediated mitophagy inducer) | Activates mitophagy by increasing P62 expression without collapsing ΔΨm or recruiting Parkin [108]. | Tool to study ΔΨm-independent mitophagy pathways and confirm specificity of compounds. |
| Liproxstatin-1 | Potent ferroptosis inhibitor that suppresses lipid peroxidation [113]. | Control for distinguishing ΔΨm changes specific to ferroptosis, a cell death pathway with distinct mitochondrial features [113]. |
Accurate measurement of mitochondrial membrane potential is non-negotiable for rigorous research in mitochondrial quality control and drug discovery. False positives arising from technical artifacts, inappropriate cell culture conditions, or confounding cytotoxic effects can severely compromise data interpretation and lead to incorrect conclusions about compound mechanisms. By implementing the optimized protocols outlined in this guide—including careful dye selection, metabolic conditioning of cells, multiplexing with viability assays, and validating hits with orthogonal approaches—researchers can significantly enhance the reliability of their ΔΨm data. This disciplined experimental framework ensures that observed changes in ΔΨm accurately reflect biological reality, thereby strengthening the validity of subsequent conclusions regarding mitophagy, mitochondrial health, and chemical modulator effects.
The mitochondrial membrane potential (ΔΨm) is a fundamental biophysical parameter that functions as a central regulator of cellular fate. It governs mitochondrial energy production, reactive oxygen species (ROS) generation, and calcium (Ca²⁺) homeostasis, creating an intricate signaling network that determines cellular health. This technical guide explores the core principles and quantitative relationships within this triad, framing them within the context of mitochondrial quality control and mitophagy research. Dysregulation of this interplay is a hallmark of pathological conditions, making its accurate interpretation critical for understanding disease mechanisms and identifying novel therapeutic targets in drug development.
The mitochondrial membrane potential (ΔΨm), a electrical gradient across the inner mitochondrial membrane (IMM), is the primary component of the proton-motive force driving adenosine triphosphate (ATP) synthesis. Beyond its bioenergetic role, ΔΨm serves as a key signaling hub, integrating and amplifying cellular stress signals. The loss of ΔΨm is a definitive marker of mitochondrial dysfunction and a pivotal trigger for mitophagy, the selective autophagic clearance of damaged mitochondria [4] [28]. The core interdependencies are bidirectional: ΔΨm drives Ca²⁺ uptake into the mitochondrial matrix, while Ca²⁺ overload can induce depolarization; similarly, physiological ROS signaling can regulate ΔΨm, but excessive ROS production can lead to its irreversible collapse [114] [115] [116]. This guide provides a framework for modeling these dynamic interactions, with a focus on experimental data interpretation for research scientists.
Core Quantitative Relationships and Data Interpretation
The interactions between ΔΨm, ROS, and Ca²⁺ are governed by quantifiable parameters. The tables below summarize key quantitative data and their functional consequences for researchers to utilize in experimental modeling.
Table 1: Quantitative Parameters of Mitochondrial Membrane Potential (ΔΨm)
| Parameter | Typical/Measured Value | Experimental Context | Functional Consequence |
|---|---|---|---|
| Magnitude of ΔΨm | ~180 mV (negative inside) | Isolated mitochondria & in vivo measurements [115] | Drives ATP synthesis & mitochondrial Ca²⁺ uptake |
| ΔΨm Threshold for mPTP opening | Depolarization to low, specific level | Ca²⁺ overload & oxidative stress models [114] | Permeability transition, cytochrome c release, apoptosis |
| FCCP/CCCP Uncoupling Concentration | 1-5 µM (stimulates respiration); Higher doses inhibit [115] | Experimental dissipation of ΔΨm | Increased O₂ consumption; decreased mitochondrial ROS at low doses |
| Impact of ΔΨm on ROS Production | Inverse correlation at high potential; Complex with metabolic state [114] [117] | Measurements with potentiometric dyes & ROS probes | High ΔΨm can increase superoxide leak from ETC; Ca²⁺-induced depolarization can decrease or increase ROS depending on context |
Table 2: Interplay Between Calcium and Reactive Oxygen Species (ROS)
| Parameter / Mechanism | Measured Effect / Relationship | Experimental Context & Citation |
|---|---|---|
| Ca²⁺ Regulation of ROS | ||
| - Mitochondrial Metabolic Rate | Ca²⁺ stimulates metabolism, correlating with increased mitochondrial ROS generation [114] | Modulation of Krebs cycle & OxPhos enzymes |
| - Mitochondrial Ca²⁺ Overload | Can induce mPTP opening, leading to massive ROS burst [114] | In vitro Ca²⁺ challenge assays |
| - NADPH Oxidase (Nox5, Duox) Activation | Direct activation via Ca²⁺ binding to EF hands or Ca²⁺/Calmodulin [118] | Receptor-mediated Ca²⁺ signaling pathways |
| ROS Regulation of Ca²⁺ | ||
| - Redox Modification of Ca²⁺ Channels/Pumps | Altered activity of RyR, IP3R, SERCA pumps [114] [118] | Thiol-oxidation studies on isolated proteins/cells |
| - Mode of ROS Signaling | Physiological (nM H₂O₂) vs. Pathological (µM H₂O₂) have divergent effects on Ca²⁺ flux [119] [118] | Use of antioxidants, Nox inhibitors, and H₂O₂ scavengers |
Key Signaling Pathways and Experimental Workflows
The Mitophagy Trigger Pathway
The PINK1/Parkin pathway is a primary mechanism of mitophagy, directly activated by a loss of ΔΨm. The following diagram and protocol detail its key steps and measurement.
Experimental Protocol: Inducing and Quantifying PINK1/Parkin Mitophagy
- Objective: To model and measure the initiation of mitophagy in response to ΔΨm dissipation.
- Cell Line: HeLa or HEK293T cells are commonly used due to their well-characterized Parkin recruitment response.
- Reagents:
- ΔΨm Depolarization Agent: Carbonyl cyanide m-chlorophenyl hydrazone (CCCP), 10-20 µM.
- Fluorescent Dyes:
- Tetramethylrhodamine, methyl ester (TMRM) or Tetramethylrhodamine, ethyl ester (TMRE) for ΔΨm.
- MitoTracker Green (or similar) for total mitochondrial mass.
- Antibodies for PINK1, Parkin, and ubiquitin for immunofluorescence.
- LC3B antibody to mark autophagosomes.
- Procedure:
- Culture and Transfection: Culture cells on glass-bottom dishes. If using Parkin-overexpressing models, transfect with a plasmid encoding fluorescently tagged Parkin (e.g., Parkin-mCherry).
- Treatment: Treat cells with CCCP for 1-4 hours. Include a DMSO-only vehicle control.
- Staining: Load cells with TMRM/TMRE (e.g., 20-100 nM) and MitoTracker Green (e.g., 50-200 nM) in culture medium for 20-30 minutes at 37°C.
- Live-Cell Imaging: Image cells over time using a confocal microscope.
- ΔΨm Loss: Monitor the fluorescence intensity of TMRM/TMRE (quenches with depolarization).
- Parkin Translocation: Quantify the recruitment of fluorescent Parkin to mitochondria (appears as punctate pattern).
- Immunofluorescence (Fixed Cells): Fix cells at specific time points post-CCCP. Co-stain with Parkin and a mitochondrial marker (e.g., TOMM20). Score the percentage of cells with mitochondrial Parkin puncta.
- Western Blot: Analyze whole-cell lysates for PINK1 accumulation, Parkin translocation (OMM fraction), and LC3 lipidation (LC3-II).
- Data Interpretation:
- A successful induction of mitophagy is confirmed by the temporal sequence of: (1) ΔΨm loss (TMRM signal loss), (2) PINK1 stabilization on OMM, (3) Parkin translocation to mitochondria, and (4) subsequent colocalization of LC3 with ubiquitinated mitochondria.
- The kinetics of this process provide a quantitative measure of mitophagic efficiency.
The Calcium-ROS Cross-Talk Amplification Loop
This pathway describes the vicious cycle where Ca²⁺ and ROS potentiate each other's production, often leading to mitochondrial dysfunction and permeability transition.
Experimental Protocol: Measuring the ROS-Ca²⁺ Feedforward Loop
- Objective: To simultaneously monitor cytosolic/mitochondrial Ca²⁺ and ROS levels in real-time to model their interplay.
- Cell Line: Primary cardiomyocytes or neuronal cells are highly relevant, but HEK293 or HeLa can be used.
- Reagents:
- Ca²⁺ Indicators:
- Fluo-4 AM (cytosolic Ca²⁺)
- Rhod-2 AM (mitochondrial Ca²⁺)
- ROS Indicators:
- H₂DCFDA (general cellular ROS)
- MitoSOX Red (mitochondrial superoxide)
- Inducers: Histamine or ATP (to induce ER Ca²⁺ release), H₂O₂ (exogenous ROS).
- Inhibitors: NAC (antioxidant), BAPTA-AM (Ca²⁺ chelator), Ru360 (mitochondrial Ca²⁺ uniporter inhibitor).
- Ca²⁺ Indicators:
- Procedure:
- Dye Loading: Load cells with the appropriate combination of fluorescent dyes according to manufacturer protocols (e.g., Fluo-4 AM with MitoSOX Red).
- Live-Cell Imaging: Use a fluorescence microscope or plate reader capable of rapid kinetic measurements and multiple fluorescence channels.
- Stimulation:
- Protocol A (Ca²⁺-induced ROS): Stimulate cells with a Ca²⁺-mobilizing agonist (e.g., ATP) and monitor cytosolic Ca²⁺ (Fluo-4) followed by mitochondrial ROS (MitoSOX) in the same cell.
- Protocol B (ROS-induced Ca²⁺ release): Treat cells with a bolus of H₂O₂ (e.g., 100-500 µM) and monitor ROS (H₂DCFDA) followed by cytosolic Ca²⁺ (Fluo-4) release.
- Pharmacological Intervention: Pre-treat cells with inhibitors like NAC or BAPTA-AM before stimulation to dissect the dependency of one signal on the other.
- Data Interpretation:
- A positive feedforward loop is indicated when an increase in Ca²⁺ is swiftly followed by a rise in ROS, and conversely, when exogenous ROS triggers Ca²⁺ release.
- The lag time between the peaks of each signal and the effect of specific inhibitors can help identify the primary trigger and major sites of interaction (e.g., ER vs. mitochondria) in a given model.
The Scientist's Toolkit: Essential Research Reagents
This table catalogs critical reagents for investigating ΔΨm, ROS, and Ca²⁺ signaling.
Table 3: Research Reagent Solutions for Mitochondrial Signaling Studies
| Reagent / Tool | Category | Primary Function & Mechanism | Example Use-Case |
|---|---|---|---|
| CCCP / FCCP | ΔΨm Modulator | Protonophore uncoupler; dissipates ΔΨm by shuttling protons across IMM [115]. | Induce mitophagy; study bioenergetics. |
| TMRM / TMRE | ΔΨm Sensor | Potentiometric, cationic dye accumulated in mitochondria proportional to ΔΨm; fluorescence quenches with depolarization [28]. | Live-cell imaging of mitochondrial health. |
| MitoSOX Red | ROS Sensor | Cell-permeable dye targeted to mitochondria; fluorescent upon oxidation by superoxide [117]. | Quantify mitochondrial superoxide production. |
| H₂DCFDA | ROS Sensor | Measures general cellular ROS (particularly H₂O₂); becomes fluorescent upon oxidation [118]. | Detect broad oxidative stress. |
| Rhod-2 AM | Ca²⁺ Sensor | Ratiometric dye with net positive charge, preferentially loading into mitochondria; fluorescence increases with Ca²⁺ binding [118]. | Monitor mitochondrial Ca²⁺ transients. |
| Ru360 | Ca²⁺ Inhibitor | Potent, specific inhibitor of the mitochondrial calcium uniporter (MCU) [114]. | Block mitochondrial Ca²⁺ uptake to dissect its role. |
| N-Acetylcysteine (NAC) | Antioxidant | Precursor to glutathione; boosts cellular antioxidant capacity, scavenges ROS [119]. | Attenuate ROS signaling to test its necessity. |
| Cyclosporin A | mPTP Inhibitor | Binds cyclophilin D to inhibit mitochondrial permeability transition pore (mPTP) opening [114]. | Test for mPTP involvement in cell death. |
| siRNA/shRNA vs. PINK1 | Genetic Tool | Knocks down PINK1 expression, a key initiator of the Parkin-mediated mitophagy pathway [4] [28]. | Validate specificity of mitophagy pathway. |
Modeling the interplay between ΔΨm, ROS, and Ca²⁺ is essential for advancing our understanding of mitochondrial quality control. The quantitative relationships, signaling pathways, and experimental tools outlined in this guide provide a foundation for rigorous data interpretation. Accurately dissecting these interactions is paramount for identifying the molecular switches that determine cell fate, from adaptive mitophagy to pathological cell death, thereby opening new avenues for therapeutic intervention in a wide range of diseases, including neurodegenerative disorders, cardiovascular diseases, and cancer.
From Bench to Bedside: Validating Membrane Potential's Role in Disease Pathogenesis
The PINK1/Parkin pathway represents a critical mechanism for mitochondrial quality control, serving as a cornerstone in the pathogenesis of Parkinson's disease (PD). This pathway functions as a sophisticated surveillance system that detects and eliminates damaged mitochondria through a process known as mitophagy, which is exclusively triggered by the loss of mitochondrial membrane potential (ΔΨm). Mutations in PINK1 (PTEN-induced kinase 1) and Parkin proteins account for a significant proportion of autosomal recessive early-onset Parkinson's disease, establishing this pathway as fundamental to neuronal health. This technical review examines the molecular orchestration of ΔΨm-dependent mitophagy, the pathological consequences of its failure, and the experimental frameworks utilized to investigate this quality control system. We further explore emerging therapeutic strategies and the current challenges in translating these findings into clinical applications, providing researchers with comprehensive methodological guidance and critical analysis of the field's trajectory.
Parkinson's disease is the second most common neurodegenerative disorder worldwide, characterized pathologically by the progressive loss of dopaminergic neurons in the substantia nigra pars compacta and the presence of Lewy bodies containing aggregated α-synuclein [45]. The socioeconomic burden of PD is substantial, with the total economic burden in the U.S. alone recorded at $51.9 billion in 2017, and global prevalence projected to increase 1.5-fold by 2035 due to demographic aging trends [45]. While the majority of PD cases are classified as sporadic, the study of rare familial forms has provided invaluable insights into disease mechanisms, particularly regarding mitochondrial dysfunction [120].
The landmark discovery by Schapira's team in 1989 first reported significantly reduced activity of mitochondrial respiratory chain Complex I in postmortem brain tissues of PD patients, establishing a fundamental molecular link between mitochondrial impairment and PD pathogenesis [45]. Subsequent genetic investigations have further corroborated this framework by demonstrating direct associations between pathogenic mutations in mitochondrial quality control genes, notably PINK1 and Parkin, and familial PD cases [45]. The proteins encoded by these genes coordinate a specialized form of autophagy termed mitophagy, which selectively targets damaged mitochondria for degradation [120] [23].
Table 1: Key Genetic Factors in Familial Parkinson's Disease Linked to Mitochondrial Dysfunction
| Gene | Protein Function | Inheritance Pattern | Primary Mitochondrial Role |
|---|---|---|---|
| PINK1 (PARK6) | Serine/threonine kinase | Autosomal recessive | Mitochondrial damage sensor |
| Parkin (PARK2) | E3 ubiquitin ligase | Autosomal recessive | Signal amplifier in mitophagy |
| DJ-1 (PARK7) | Redox-sensitive chaperone | Autosomal recessive | Oxidative stress protection |
| LRRK2 (PARK8) | Kinase | Autosomal dominant | Regulation of vesicular trafficking |
| α-synuclein (PARK1/4) | Presynaptic protein | Autosomal dominant | Mitochondrial membrane interaction |
Mitochondria are essential organelles that not only generate ATP through oxidative phosphorylation but also regulate calcium homeostasis, redox balance, and apoptotic signaling [121]. In neurons, which have exceptionally high energy demands and are post-mitotic, the maintenance of a healthy mitochondrial network is particularly crucial. The PINK1/Parkin pathway has emerged as a central regulator of mitochondrial quality control, with its dysfunction representing a key paradigm for understanding PD pathogenesis at the molecular level [122] [121].
Molecular Mechanisms of ΔΨm-Dependent Mitophagy
The PINK1/Parkin Signaling Cascade
The PINK1/Parkin-mediated mitophagy pathway represents the most extensively characterized mechanism for selective mitochondrial clearance. This process is stringently regulated by mitochondrial membrane potential (ΔΨm), making it a exquisite sensor of mitochondrial health [26] [121]. Under physiological conditions with intact ΔΨm, PINK1 is continuously imported into mitochondria through the translocase of the outer membrane (TOM) and inner membrane (TIM23) complexes [26]. Following import, PINK1 undergoes proteolytic processing by the mitochondrial processing peptidase (MPP) and the presenilin-associated rhomboid-like (PARL) protease, resulting in its rapid degradation by the ubiquitin-proteasome system via the N-end rule pathway, thereby maintaining low cellular levels of PINK1 [122] [26].
When mitochondria incur damage sufficient to cause ΔΨm dissipation, the import and processing of PINK1 is blocked, leading to its accumulation on the outer mitochondrial membrane (OMM) [26] [121]. There, PINK1 undergoes autophosphorylation at Ser228 (in humans), which triggers a conformational change that stabilizes the kinase as a monomer and activates its catalytic function [26]. This activated PINK1 then phosphorylates ubiquitin at Ser65 and directly phosphorylates Parkin at Ser65 within its ubiquitin-like (Ubl) domain, relieving Parkin's autoinhibited conformation and recruiting it to damaged mitochondria [26].
Once activated and recruited to mitochondria, Parkin – an E3 ubiquitin ligase – initiates a feedforward amplification mechanism by ubiquitinating numerous OMM proteins, including mitofusins (MFN1/2), voltage-dependent anion channel 1 (VDAC1), and TOM20 [45] [26]. These ubiquitinated substrates are further phosphorylated by PINK1, creating more binding sites for Parkin recruitment and resulting in extensive ubiquitin chain formation on the mitochondrial surface [26]. This ubiquitin coating serves as a platform for the recruitment of autophagy receptors – including optineurin (OPTN), NDP52, p62/SQSTM1, and TAX1BP1 – which simultaneously bind ubiquitin through their ubiquitin-binding domains and LC3/GABARAP proteins on developing phagophores through LC3-interacting regions (LIR), thereby facilitating encapsulation of damaged mitochondria into autophagosomes [45] [42] [26].
Diagram 1: PINK1/Parkin Pathway Activation by ΔΨm Loss. Under normal conditions, PINK1 is imported and degraded, but upon ΔΨm dissipation, it accumulates on the OMM, activating Parkin-mediated mitophagy.
Alternative Mitophagy Pathways
While the PINK1/Parkin pathway represents the most extensively studied mechanism of mitophagy, neurons employ several PINK1/Parkin-independent pathways to ensure robust mitochondrial quality control. These alternative routes become particularly important in the context of PINK1 or Parkin mutations and may offer compensatory mechanisms or represent tissue-specific quality control systems [42].
The receptor-mediated mitophagy pathway involves OMM proteins that function as mitophagy receptors, containing LIR motifs that directly bind to LC3/GABARAP family proteins on autophagosomal membranes. Key receptors in this category include BNIP3, NIX/BNIP3L, FUNDC1, and FKBP8 [42]. Under hypoxic conditions or during erythrocyte maturation, BNIP3 and NIX are transcriptionally upregulated and can initiate mitophagy independently of PINK1/Parkin [42]. FUNDC1, another OMM protein, is regulated by phosphorylation: under normal conditions, it is phosphorylated by kinases such as SRC and CK2, which suppresses its activity; during hypoxia or mitochondrial stress, phosphatases such as PGAM5 dephosphorylate FUNDC1, enhancing its interaction with LC3 and promoting mitophagy [42].
Recent research has identified an additional mechanism involving prohibitin 2 (PHB2), an inner mitochondrial membrane protein that functions as a mitophagy receptor when the OMM is ruptured or permeabilized [42] [26]. PHB2 contains an LIR motif that becomes exposed to the cytosol upon OMM damage, enabling direct engagement with LC3 and facilitating mitophagy. This pathway may be particularly relevant for the removal of mitochondria with severe damage that compromises OMM integrity.
A newly identified Mitophagic Stress Response (MitoSR) represents another sophisticated layer of mitochondrial quality control in neurons. In response to increasing mitochondrial stress, neurons activate a graded response that induces the concerted degradation of negative regulators of autophagy, including myotubularin-related phosphatases (MTMR5 and MTMR2) and Rubicon, via the ubiquitin-proteasome pathway and selective proteolysis [123]. MTMR5/MTMR2 normally inhibit autophagosome biogenesis, while Rubicon suppresses lysosomal function. Their degradation in response to mitochondrial stress enhances both early and late stages of mitophagy, representing a compensatory mechanism that operates in parallel to canonical PINK1/Parkin-dependent mitophagy [123].
Table 2: Key Mitophagy Pathways and Their Regulation
| Pathway | Key Components | Activation Signals | Neuronal Relevance |
|---|---|---|---|
| PINK1/Parkin | PINK1, Parkin, ubiquitin, OPTN/NDP52 | ΔΨm loss, oxidative stress | Primary pathway; mutated in familial PD |
| Receptor-Mediated | BNIP3, NIX, FUNDC1, FKBP8 | Hypoxia, metabolic stress | Compensatory pathway in PINK1/Parkin deficiency |
| Lipid-Mediated | Cardiolipin, ceramide | Membrane damage, oxidative stress | Potential backup mechanism |
| MitoSR | MTMR5/MTMR2, Rubicon | Mitochondrial ROS, oxidative damage | Neuronal-specific stress response |
Pathological Consequences of PINK1/Parkin Dysfunction
Mitochondrial Accumulation and Neuronal Vulnerability
The failure of ΔΨm-dependent mitophagy due to PINK1 or Parkin mutations initiates a cascade of pathological events that ultimately lead to selective neuronal vulnerability, particularly affecting dopaminergic neurons in the substantia nigra. The immediate consequence of impaired mitophagy is the accumulation of dysfunctional mitochondria that are unable to maintain adequate ATP production through oxidative phosphorylation [45] [122]. This bioenergetic deficit is particularly detrimental to neurons, which have high energy demands for maintaining ionic gradients, synaptic transmission, and axonal transport.
Dysfunctional mitochondria that escape quality control mechanisms become significant sources of reactive oxygen species (ROS), creating a vicious cycle of oxidative damage that further compromises mitochondrial function and cellular viability [122]. The postmortem analyses of PD patients consistently reveal markers of oxidative stress, including lipid peroxidation, protein carbonylation, and DNA oxidation, particularly in affected brain regions [45]. The susceptibility of dopaminergic neurons to mitochondrial dysfunction may be explained by several factors, including their complex morphology with extensive axonal arborizations, the inherently oxidative environment of dopamine metabolism, and relatively low mitochondrial reserve capacity [122].
Beyond bioenergetic deficits, damaged mitochondria accumulate mutated mitochondrial DNA (mtDNA), exhibit calcium buffering deficiencies, and release pro-apoptotic factors such as cytochrome c, all of which contribute to neuronal dysfunction and eventual death [45] [121]. The accumulation of these defective organelles has been directly observed in PD models and patient tissues, with postmortem analyses revealing abnormal accumulation of mitochondrial proteins such as Miro – an adaptor for mitochondrial transport and a degradation substrate during PINK1/Parkin-mediated mitophagy – in the brains of PD patients but not in age-matched controls [42].
Neuroinflammation and Systemic Pathology
The pathological consequences of defective PINK1/Parkin signaling extend beyond cell-autonomous mitochondrial dysfunction to include significant neuroinflammatory responses that amplify neuronal damage. Damaged mitochondria that evade quality control release damage-associated molecular patterns (DAMPs), including mtDNA, ATP, and cardiolipin, which activate innate immune receptors such as TLR9 and NLRP3 inflammasome in microglia and astrocytes [121]. This triggers the production and release of pro-inflammatory cytokines, including IL-1β, IL-6, and TNF-α, creating a chronic inflammatory environment that exacerbates neurodegeneration [121].
The systemic nature of PINK1/Parkin dysfunction is evidenced by mitochondrial abnormalities observed in peripheral tissues of PD patients, including reduced complex I activity in platelets and increased mitochondrial DNA mutations in skin fibroblasts [45]. These findings suggest that the consequences of impaired mitophagy extend beyond the central nervous system, though neurons appear particularly vulnerable due to their high energy demands and limited regenerative capacity.
The clinical presentation of PINK1/Parkin-linked PD typically differs from idiopathic PD, often presenting as early-onset parkinsonism with slower progression and minimal cognitive decline compared to the later-onset idiopathic form characterized by more rapid cognitive deterioration [45]. This clinical distinction reflects the specific pathophysiology of mitochondrial quality control failure versus the multifactorial etiology of sporadic PD.
Experimental Models and Methodologies
In Vitro Assessment of Mitophagy
Research into PINK1/Parkin-mediated mitophagy employs a diverse array of experimental models and methodological approaches to dissect the molecular mechanisms and pathological consequences of this pathway. Cell culture models, particularly immortalized cell lines such as HeLa, SH-SY5Y, and HEK293, have been instrumental in elucidating the core mechanisms of PINK1/Parkin signaling [120] [124]. These systems allow for precise genetic manipulation, high-resolution imaging, and controlled induction of mitochondrial damage.
The most common method for inducing PINK1/Parkin-mediated mitophagy in vitro involves treatment with mitochondrial uncouplers such as carbonyl cyanide m-chlorophenyl hydrazone (CCCP) or antimycin A, which dissipate ΔΨm and trigger PINK1 stabilization on the OMM [124] [123]. The experimental workflow typically involves:
- Transfection or viral transduction to express fluorescently tagged PINK1, Parkin, or mitochondrial markers
- Treatment with uncouplers (e.g., 10-20 μM CCCP for 1-24 hours) to induce mitochondrial damage
- Fixation or live-cell imaging to monitor Parkin translocation and mitochondrial clearance
- Immunofluorescence or immunoblotting to assess protein localization and degradation
Key readouts include:
- Parkin translocation from cytosol to mitochondria (typically within 1-2 hours after uncoupler treatment)
- Ubiquitination of mitochondrial proteins detected by ubiquitin immunoblotting of mitochondrial fractions
- LC3 recruitment to mitochondria indicating phagophore encapsulation
- Decrease in mitochondrial mass measured by mitochondrial protein levels or fluorescent signals
- Colocalization of mitochondrial markers with lysosomal markers indicating mitochondrial degradation
Table 3: Essential Research Reagents for Studying PINK1/Parkin Mitophagy
| Reagent Category | Specific Examples | Experimental Function | Key Considerations |
|---|---|---|---|
| Mitochondrial Dyes | TMRE, TMRM, JC-1 | Measure ΔΨm | Concentration-dependent uptake; photo-bleaching |
| Chemical Uncouplers | CCCP, FCCP, Antimycin A | Induce ΔΨm loss | Concentration and timing critical for specific effects |
| Genetic Constructs | GFP-Parkin, mCherry-PINK1 | Visualize protein dynamics | Overexpression artifacts; endogenous tagging preferred |
| Autophagy Markers | LC3-GFP, LAMP1-RFP | Track autophagosome/lysosome fusion | Multiple isoforms; processing changes |
| Antibodies | p-S65-ubiquitin, p-S65-Parkin | Detect pathway activation | Phospho-specific antibodies require validation |
| Kinase Inhibitors | Kinetin, BX795 | Modulate PINK1 activity | Off-target effects; dose optimization required |
In Vivo Models and Limitations
While in vitro systems have been invaluable for delineating the molecular mechanisms of PINK1/Parkin-mediated mitophagy, their limitations have prompted the development and characterization of in vivo models [124]. Drosophila models lacking PINK1 or Parkin have been particularly informative, displaying robust mitochondrial defects and dopaminergic neurodegeneration that closely mimics human PD pathology [45] [124]. In contrast, murine knockout models exhibit more subtle phenotypes, with minimal neurodegeneration but measurable deficits in mitochondrial function and stress resistance [45] [124].
The discrepancies between Drosophila and murine models highlight important species-specific differences in mitochondrial biology and neuronal vulnerability, as well as the potential existence of compensatory mechanisms in mammals that obscure the pathological consequences of PINK1/Parkin deficiency [124]. More complex human cellular models, including induced pluripotent stem cell (iPSC)-derived neurons from PD patients with PINK1 or Parkin mutations, have emerged as valuable tools for bridging the gap between simplified in vitro systems and in vivo physiology [123].
Recent methodological advances have enabled more precise assessment of mitophagy in vivo, including:
- Mitophagy reporter mice expressing mitochondrial-targeted fluorescent proteins (e.g., mt-Keima, mt-QC)
- Tissue-specific knockout models to dissect cell-type-specific functions
- Live imaging approaches in transparent model organisms (e.g., zebrafish)
- Proteomic and transcriptomic analyses of patient-derived neurons
A critical consideration in mitophagy research is the distinction between basal mitophagy that occurs under physiological conditions and stress-induced mitophagy triggered by pathological insults or experimental manipulations [124]. The development of more sensitive tools to detect and quantify basal mitophagy has revealed that PINK1/Parkin-independent pathways may play significant roles in physiological mitochondrial turnover, while the PINK1/Parkin pathway appears particularly important for stress-induced quality control [124].
Therapeutic Implications and Research Applications
Current Therapeutic Approaches
The recognition that PINK1/Parkin dysfunction represents a key driver of PD pathogenesis has spurred the development of therapeutic strategies aimed at enhancing mitophagy to promote mitochondrial quality control. These approaches can be broadly categorized into small molecule activators, gene therapy, and non-pharmacological interventions.
Several small molecule compounds designed to activate PINK1 or Parkin have been investigated in preclinical models, showing potential for enhancing mitophagy and ameliorating PD-related pathology [45]. However, a recent study revealed that some candidate drugs initially identified as PINK1/Parkin activators actually function as mitochondrial toxins that damage healthy mitochondria rather than specifically activating the mitophagy pathway [125]. As described in the analogy, "Imagine your microwave was broken, and instead of calling your garbage collector to throw it away, you smashed it up further with a sledgehammer. That would really force you to throw it out, but it's not what we want our drugs to be doing to our cells" [125]. This highlights the critical importance of developing assays that can distinguish true pathway activators from non-specific mitochondrial stressors.
Gene therapy approaches employing lentiviral or adeno-associated viral (AAV) vectors to deliver PINK1 or Parkin genes show promise in animal models, potentially offering a strategy to restore defective mitophagy in familial PD caused by mutations in these genes [121]. However, the translational application of such approaches faces challenges related to delivery efficiency, immune responses, and the potential risks associated with unregulated expression of these proteins.
Non-pharmacological interventions, particularly exercise, have emerged as safe and effective approaches to enhance mitophagy and ameliorate PD symptoms [42]. Physical activity activates mitophagy through key signaling pathways – including AMP-activated protein kinase (AMPK)/Unc-51–like kinase 1 (ULK1) and PINK1/Parkin – thereby enhancing mitochondrial function and antioxidant capacity [42]. The multilayered benefits of exercise, coupled with its safety profile and accessibility, make it an attractive component of comprehensive PD management.
Emerging Research Directions and Clinical Applications
Several emerging research directions hold promise for advancing both our understanding of PINK1/Parkin biology and the development of effective therapies. These include:
Tissue-specific mitophagy enhancers: Developing strategies to selectively enhance mitophagy in neurons without affecting other tissues, potentially through neuron-specific promoters or delivery systems.
Negative regulator targeting: Exploiting endogenous compensatory mechanisms such as the Mitophagic Stress Response by developing compounds that promote the degradation of mitophagy inhibitors like MTMR5, MTMR2, and Rubicon [123].
Pathway-specific biomarkers: Developing reliable biomarkers to assess mitophagy activity in patients, which would facilitate patient stratification and treatment monitoring in clinical trials.
Combination therapies: Integrating mitophagy-enhancing approaches with other neuroprotective strategies targeting complementary pathways implicated in PD pathogenesis.
Diagram 2: Mitochondrial Quality Control Network and Therapeutic Targets. The core PINK1/Parkin pathway is regulated by negative regulators and complemented by alternative pathways, revealing multiple intervention points.
The PINK1/Parkin pathway represents a paradigm of ΔΨm-dependent mitophagy that is essential for neuronal health, with its dysfunction constituting a key mechanism in Parkinson's disease pathogenesis. The precise regulation of this pathway by mitochondrial membrane potential provides a sophisticated quality control system that detects and eliminates damaged mitochondria, preventing the accumulation of dysfunctional organelles that would otherwise trigger oxidative stress, bioenergetic failure, and neuronal death. While significant progress has been made in understanding the molecular mechanisms of this pathway, important challenges remain, including the development of specific mitophagy enhancers that genuinely activate the pathway without causing collateral mitochondrial damage, and the translation of these findings into effective therapies for Parkinson's disease and other neurodegenerative conditions associated with mitochondrial quality control failure. The continued elucidation of PINK1/Parkin biology and its integration with complementary quality control mechanisms will undoubtedly yield new insights and therapeutic opportunities in the coming years.
Mitochondrial membrane potential (ΔΨm) serves as the central regulator of mitochondrial quality control, acting as a key trigger for mitophagy, the selective autophagic clearance of damaged mitochondria. This whitepaper provides a comparative analysis of how ΔΨm collapse and subsequent mitophagy dysregulation manifest distinctly in cardiac and renal pathologies. While both organ systems rely on PINK1/Parkin and receptor-mediated mitophagy pathways, disease-specific variations in ΔΨm sensitivity, mitophagic flux, and downstream consequences reveal fundamental differences in pathological mechanisms. Understanding these organ-specific paradigms is crucial for developing targeted therapeutic interventions for cardiorenal syndromes and other conditions involving mitochondrial dysfunction.
Mitochondrial membrane potential (ΔΨm), generated by the proton gradient across the inner mitochondrial membrane, is essential for ATP production and serves as the primary indicator of mitochondrial health. The collapse of ΔΨm represents the fundamental initiating signal for mitophagy activation across all tissue types. When ΔΨm dissipation occurs, it prevents the import and cleavage of PTEN-induced putative kinase 1 (PINK1), leading to its accumulation on the outer mitochondrial membrane (OMM). This stabilized PINK1 phosphorylates both ubiquitin and the E3 ubiquitin ligase Parkin, initiating a signaling cascade that marks damaged mitochondria for autophagic clearance [126] [127].
The kidney and heart are both high-energy demanding organs rich in mitochondria, making them particularly vulnerable to ΔΨm disruption. However, the temporal patterns, regulatory mechanisms, and pathological consequences of ΔΨm-driven mitophagy differ significantly between these organ systems in disease states. This review systematically compares these differences to inform targeted therapeutic development.
Comparative Pathophysiology: Cardiac vs. Renal ΔΨm and Mitophagy Dysregulation
Table 1: Comparative Analysis of ΔΨm and Mitophagy Dysregulation in Cardiac vs. Renal Pathologies
| Parameter | Cardiac Pathologies | Renal Pathologies |
|---|---|---|
| Primary ΔΨm Disruptors | Chronic pressure overload, ROS/calcium overload, ischemic stress [126] [24] | Uremic toxins, hyperglycemia, ischemia-reperfusion injury [128] [129] |
| Temporal Pattern | Biphasic response - initial upregulation followed by progressive impairment [126] [130] | Generally suppressed mitophagy, though context-dependent upregulation occurs in AKI [128] |
| Key Mitophagy Pathways Affected | PINK1/Parkin, BNIP3/NIX, FUNDC1 [126] [24] | PINK1/Parkin predominantly, with BNIP3 involvement in specific contexts [128] [127] |
| Consequences of ΔΨm Collapse | Bioenergetic crisis, oxidative stress, cardiomyocyte death, pathological remodeling [126] [131] | Tubular cell death, inflammation, fibrosis, progressive nephron loss [128] [129] |
| Unique Pathological Features | Metabolic shift to fatty acid oxidation, dual role of BNIP3 (mitophagy and apoptosis) [126] [130] | Renal osteodystrophy via osteocyte mitophagy blockade [129] |
Table 2: Quantitative Assessment of Mitochondrial Parameters in Disease States
| Parameter | Diabetic Cardiomyopathy | Hypertensive Cardiac Hypertrophy | Diabetic Nephropathy | CKD-MBD |
|---|---|---|---|---|
| ΔΨm | Significant collapse [131] | Progressive dissipation [126] | Not quantified | Not quantified |
| ATP Production | Decreased [131] | Deficient [126] | Not specified | Not specified |
| mtDNA Copy Number | Reduced [131] | Not specified | Reduced [130] | Not specified |
| ROS Production | Elevated [131] | Excessive [126] | Elevated [128] | Increased oxygen-free radicals [129] |
| Mitophagy Status | Activated (early), Impaired (late) [130] | Insufficient clearance [126] | Dysregulated [128] | Blockaded [129] |
Cardiac Pathologies: ΔΨm Sensitivity and Metabolic Implications
In hypertensive cardiac hypertrophy (HCH), chronic pressure overload induces sustained mitochondrial damage characterized by depolarization, ROS overproduction, and calcium overload [126]. The unique metabolic profile of cardiomyocytes creates particular vulnerability to ΔΨm disruption. In diabetic cardiomyopathy, mitochondrial ultrastructural pathology includes cristae dissolution, disorganized arrangements, and vacuolization, directly impairing ΔΨm generation and maintenance [131]. Three-dimensional morphometric analysis reveals significant alterations in mitochondrial architecture under high-glucose conditions, including reduced mitochondrial length and anisotropy with increased thickness, width, flatness, and elongation [131].
The molecular mechanisms of ΔΨm collapse in cardiac pathologies involve:
- Calcium overload: Elevated cytosolic Ca²⁺ activates calpain, which cleaves OMM-stabilized PINK1, preventing Parkin recruitment [126]
- ROS-mediated Parkin inactivation: H₂O₂ oxidizes critical cysteines (Cys431/Cys95) in Parkin's RING domains, abolishing E3 ligase activity [126]
- Dynamical imbalance: Increased DRP1-mediated fission and impaired MFN1/MFN2/OPA1-dependent fusion generate fragmented, depolarized organelles [126] [131]
Renal Pathologies: Distinct ΔΨm Disruptors and Systemic Consequences
In renal diseases, ΔΨm collapse is triggered by unique pathogenic factors, most notably uremic toxins in chronic kidney disease (CKD). In CKD-mineral and bone disorder (CKD-MBD), uremic toxins directly impair mitophagy, leading to dysfunctional mitochondrial accumulation in osteocytes, contributing to renal osteodystrophy [129]. This represents a unique systemic consequence of renal mitochondrial dysfunction not observed in cardiac pathologies.
Diabetic nephropathy involves hyperglycemia-induced ΔΨm disruption through:
- Advanced glycation end products: Promote oxidative stress and mitochondrial damage [128]
- Altered mitochondrial dynamics: Similar to cardiac pathologies but with renal-specific manifestations [128]
- Impaired PINK1/Parkin signaling: Documented in various renal diseases including renal ischemia-reperfusion injury and contrast-induced acute kidney injury [128]
Experimental Methodologies for Assessing ΔΨm and Mitophagy
Protocol: Comprehensive Assessment of Mitochondrial Structure-Function Relationships
Objective: Quantitatively characterize mitochondrial ultrastructure, ΔΨm, and functional parameters in disease models [131].
Sample Preparation:
- Tissue fixation: 2.5% glutaraldehyde + 2.5% paraformaldehyde at 4°C overnight
- Sequential dehydration: Graded ethanol (50%, 70%, 80%, 90%, 100%) and tert-butanol
- Cryovacuum drying with tert-butanol
Imaging and Analysis:
- Scanning electron microscopy (SEM): SU8010, Hitachi at 2 kV accelerating voltage
- 3D reconstruction and morphometric parameters: Length₃D, Thickness₃D, Width₃D, Area₃D, Volume₃D, Anisotropy, Flatness, Elongation
- Cristae analysis: Cristae scores, count, width, gap size, junction widths
Functional Assessment:
- ΔΨm: JC-1 staining or TMRE-based assays [130] [131]
- ATP content: Luciferase-based assays
- mtDNA copy number: Quantitative PCR
- OXPHOS activity: Complexes I-V enzymatic assays
- Oxygen consumption rate: O2k system for basal/maximal respiration, ATP-linked respiration, spare respiratory capacity
Protocol: Dynamic Assessment of Mitophagic Flux
Objective: Quantify mitophagy activation and completion in real-time [130] [129].
Methodologies:
- Mito-Keima fluorescence assay: pH-sensitive fluorescent protein targeted to mitochondria; acidification-resistant Keima signal increases upon lysosomal delivery
- Mito-QC reporter mice: Express mitochondria-targeted tandem mCherry-GFP tag; GFP quenches in acidic lysosomes while mCherry persists
- Western blot analysis: PINK1 stabilization, Parkin recruitment, LC3-I to LC3-II conversion, p62/SQSTM1 degradation
- Immunofluorescence: Co-localization of mitochondrial markers (TOM20, COX IV) with autophagosomal (LC3) and lysosomal (LAMP1) markers
Experimental Workflow for ΔΨm and Mitophagy Assessment
Molecular Mechanisms and Signaling Pathways
PINK1/Parkin-Dependent Mitophagy Pathways
The canonical PINK1/Parkin pathway represents the most extensively studied mechanism for ΔΨm-sensitive mitophagy in both cardiac and renal tissues [126] [127]. Under normal conditions with preserved ΔΨm, PINK1 is continuously imported into mitochondria through the translocase of the outer membrane (TOM) and translocase of the inner membrane (TIM) complexes. Inside mitochondria, PINK1 is cleaved by the inner membrane presenilin-associated rhomboid-like protease (PARL) and ultimately degraded by the proteasome [127].
When ΔΨm collapses, mitochondrial import is impaired, leading to PINK1 accumulation on the OMM. PINK1 then phosphorylates ubiquitin at Ser65 and activates Parkin, which ubiquitinates numerous OMM proteins [126] [127]. These polyubiquitinated proteins are recognized by autophagy receptors such as p62/SQSTM1 and optineurin, which link the damaged mitochondria to the autophagy machinery via LC3 interaction [127].
PINK1/Parkin Pathway in Mitophagy Regulation
Receptor-Mediated Mitophagy Pathways
Beyond the PINK1/Parkin system, cardiac and renal tissues utilize receptor-mediated mitophagy pathways that can respond to ΔΨm collapse independently of ubiquitination [126] [127]. These include:
- BNIP3/NIX pathway: Hypoxia-inducible proteins that homodimerize on the OMM and interact directly with LC3 via their LIR domains [126] [127]
- FUNDC1 pathway: An OMM protein regulated by phosphorylation; dephosphorylation during hypoxia enhances LC3 binding [126]
In hypertensive cardiac hypertrophy, maladaptive alterations occur in these pathways: BNIP3/NIX overexpression shifts from protective mitophagy to apoptosis promotion, while FUNDC1 deficiency impairs hypoxia-responsive clearance [126].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for ΔΨm and Mitophagy Studies
| Reagent/Category | Function/Application | Examples/Specifics |
|---|---|---|
| ΔΨm Indicators | Quantitative measurement of mitochondrial membrane potential | JC-1, TMRE, TMRM [130] [131] |
| Mitophagy Reporters | Live-cell tracking of mitophagic flux | Mito-Keima, Mito-QC (mt-mCherry-GFP) [130] [129] |
| Pathway Modulators | Investigate specific pathway contributions | Mdivi-1 (DRP1 inhibitor), Dynasore (DRP1 inhibitor) [10] |
| Mitochondrial Stress Inducers | Experimental induction of ΔΨm collapse | CCCP (protonophore), PA (palmitic acid) [130] |
| Therapeutic Compounds | Potential mitophagy-targeting interventions | Rapamycin, MitoQ (mitochondrial antioxidant), AST-120 (uremic toxin adsorbent) [129] |
| Antibody Targets | Assessment of pathway components | PINK1, Parkin, BNIP3, NIX, FUNDC1, LC3, p62 [126] [131] |
Therapeutic Implications and Future Directions
The comparative analysis of ΔΨm and mitophagy dysregulation in cardiac versus renal pathologies reveals both shared and distinct therapeutic opportunities. In both organ systems, strategies to modulate mitophagy must be context-specific, considering the paradoxical dual roles of mitophagy as both protective and detrimental depending on disease stage and severity [126] [130].
Cardiac-specific therapeutic approaches:
- Fine-tuning BNIP3/NIX expression: Moderate levels aid clearance while supraphysiological expression drives mitochondrial catastrophe [126]
- FUNDC1 restoration: Enhancing FUNDC1 function protects against hypertensive cardiac hypertrophy by improving mitochondrial clearance and contractility [126]
- Metabolic modulators: Address the unique metabolic shift to fatty acid oxidation in diabetic cardiomyopathy [130] [132]
Renal-specific therapeutic approaches:
- Uremic toxin targeting: AST-120 and mitochondrial-targeted antioxidants (MitoQ) mitigate uremic toxin-induced mitochondrial changes [129]
- PINK1/Parkin augmentation: Enhancing this pathway represents a promising approach for acute kidney injury and diabetic nephropathy [128]
Cross-cutting therapeutic strategies:
- Rapamycin and mTOR inhibition: Shown to reverse uremic toxin effects on mitophagy in renal osteodystrophy [129]
- Lifestyle interventions: Exercise and dietary modifications that enhance mitochondrial quality control in both cardiac and renal tissues [126]
- Gene therapy approaches: Restoration of mitophagy regulators in a tissue-specific manner [126]
Future research should focus on developing more precise methods for monitoring mitophagic flux in human patients, creating tissue-specific delivery systems for mitophagy modulators, and identifying biomarkers that can predict therapeutic response based on individual patterns of ΔΨm and mitophagy dysregulation.
Mitochondrial membrane potential (ΔΨm), the electrochemical gradient across the inner mitochondrial membrane, serves as the fundamental regulator of mitochondrial function beyond its canonical role in ATP production. This potential, typically around -180 mV, facilitates protein import, regulates reactive oxygen species (ROS) production, and acts as a primary sensor for mitochondrial quality control [87]. The dynamic nature of ΔΨm enables spatial and temporal regulation of cellular function, particularly in neuronal adaptation and synaptic plasticity [87]. When ΔΨm becomes compromised through depolarization, it triggers a cascade of quality control mechanisms, most notably mitophagy—the selective autophagic degradation of damaged mitochondria [133]. This process is essential for maintaining cellular homeostasis, and its dysregulation is implicated in numerous diseases, from neurodegeneration to cancer. Therapeutic targeting of ΔΨm represents a promising strategy for modulating mitochondrial quality control, with idebenone and MitoQ emerging as leading investigational agents in this domain.
Molecular Mechanisms of ΔΨm in Mitophagy and Quality Control
ΔΨm as the Primary Signal for Mitophagy Initiation
The loss of ΔΨm serves as the critical initiating signal for mitophagy. In healthy, polarized mitochondria, PTEN-induced putative kinase 1 (PINK1) is continuously imported into the inner membrane and degraded. Upon depolarization, this import is blocked, leading to PINK1 stabilization on the outer mitochondrial membrane (OMM) [133]. Stable PINK1 accumulation recruits and activates the E3 ubiquitin ligase Parkin, which ubiquitinates numerous OMM proteins [133]. This ubiquitination cascade recruits autophagy receptors like optineurin (OPTN) and nuclear dot protein 52 (NDP52), which in turn link the ubiquitinated mitochondria to the core autophagy machinery via LC3 (microtubule-associated protein 1 light chain 3) interaction, culminating in autophagosome engulfment and lysosomal degradation [133].
Integration with Mitochondrial Dynamics
ΔΨm is intrinsically linked to mitochondrial dynamics—the continuous fission and fusion events that remodel the mitochondrial network. Mitochondrial fission generates heterogeneous daughter organelles; those with lower ΔΨm are targeted for mitophagy, while those retaining higher ΔΨm rejoin the network [87]. This quality control mechanism ensures selective elimination of dysfunctional units while preserving functional networks. The proteins mediating these dynamics, including MFN1, MFN2, OPA1, and DRP1, are themselves regulated by ΔΨm-sensitive processes [134].
Table 1: Key Proteins in ΔΨm-Mediated Quality Control
| Protein | Function | Role in ΔΨm Sensing/Quality Control |
|---|---|---|
| PINK1 | Ser/Thr kinase | Accumulates on depolarized mitochondria; initiates mitophagy |
| Parkin | E3 ubiquitin ligase | Amplifies PINK1 signal via ubiquitin chain formation |
| OPA1 | Inner membrane GTPase | Regulates inner membrane fusion; processing regulated by ΔΨm |
| MFN1/MFN2 | Outer membrane GTPases | Mediate outer membrane fusion; targets for Parkin ubiquitination |
| LC3 | Autophagy protein | Decorates autophagosomal membrane; recruits mitochondria via receptors |
| p62/SQSTM1 | Autophagy receptor | Links ubiquitinated cargos to LC3; promotes mitochondrial aggregation |
Clinically Advanced ΔΨm-Targeting Agents
Idebenone: A Synthetic CoQ10 Analog
Idebenone (6-(10-hydroxydecyl)-2,3-dimethoxy-5-methyl-1,4-benzoquinone) is a short-chain synthetic analog of coenzyme Q10 (CoQ10) with enhanced water solubility and superior membrane permeability compared to CoQ10 [135]. Its molecular weight is 338.44 g/mol, and it features a redox-active benzoquinone core that enables electron shuttling in the mitochondrial respiratory chain [135].
Mechanisms of Action:
- Electron Carrier Function: Idebenone effectively transfers electrons from complex II (succinate dehydrogenase) to complex III, bypassing complex I defects [135]. This pathway is particularly therapeutic in mitochondrial disorders with complex I deficiency.
- Antioxidant Activity: As a potent intramitochondrial antioxidant, idebenone scavenges reactive oxygen species (ROS) and reduces lipid peroxidation, protecting against oxidative damage [135].
- ΔΨm Modulation: Idebenone can disrupt mitochondrial membrane potential under certain conditions, particularly in cancer cells, where this effect promotes apoptosis and mitophagy [135] [136]. The reducing environment within cells significantly influences its effects, with the reduced form (idebenol) demonstrating superior efficacy in supporting respiration [136].
Clinical Status and Applications: Idebenone has demonstrated clinical benefits in several neurological conditions and is now being investigated in broader therapeutic areas:
- Leber's Hereditary Optic Neuropathy (LHON): Idebenone is clinically proven for LHON treatment, with an international, multicenter study confirming benefits even in chronic phases [135] [137]. It improves visual acuity and color contrast, particularly for blue-yellow discrimination [137].
- Friedreich's Ataxia: Higher doses of idebenone (up to 2250 mg/day) are well-tolerated and associated with improvements in neurological function and daily activities [135] [137].
- Duchenne Muscular Dystrophy: A Phase 3 trial demonstrated enhanced respiratory function in glucocorticoid non-using patients [135].
- Oncology: Emerging preclinical evidence shows idebenone inhibits triple-negative breast cancer through GADD45/CyclinB/CDK1 signaling, disrupts ΔΨm, promotes mitophagy via AMPK/mTOR pathway, and halts the cell cycle in G2/M phase [135]. Similar effects were observed in melanoma, glioblastoma, neuroblastoma, hepatocellular carcinoma, and pancreatic carcinoma models [135].
Table 2: Clinical Status of Idebenone Across Indications
| Disease Area | Clinical Stage | Key Findings & Mechanisms | References |
|---|---|---|---|
| LHON | Approved/Marketed | Improved visual acuity, color contrast; electron shuttle bypasses CI defects | [135] [137] |
| Friedreich's Ataxia | Phase III (completed) | Stabilized neurological function; reduced oxidative stress markers | [135] [137] |
| Duchenne Muscular Dystrophy | Phase III (completed) | Enhanced respiratory function; reduced cardiac complications | [135] |
| Triple-Negative Breast Cancer | Preclinical | G2/M cell cycle arrest; ΔΨm disruption; AMPK/mTOR-mediated mitophagy | [135] |
| Cardiovascular Diseases | Preclinical/Early Clinical | Ameliorated mitochondrial dysfunction in endothelial cells; anti-atherosclerotic effects | [135] [137] |
MitoQ: A Mitochondria-Targeted Antioxidant
MitoQ is a synthetic mitochondria-targeted derivative of coenzyme Q10, consisting of a ubiquinone moiety covalently linked to a lipophilic triphenylphosphonium (TPP+) cation through a 10-carbon aliphatic chain [138] [139]. The TPP+ cation enables MitoQ to accumulate several hundred-fold within mitochondria, driven by the large ΔΨm [138].
Mechanisms of Action:
- Proton Displacement & Pseudo-ΔΨm Formation: MitoQ adsorbs to the inner mitochondrial membrane with its cationic moiety in the intermembrane space. The positive charges inhibit respiratory chain complexes I, III, and IV, reduce proton production, and decrease oxygen consumption [138]. This creates a "pseudo-ΔΨm" (PMMP) maintained by exogenous positive charges rather than proton gradient, which impairs proton backflow and reduces ATP production [138].
- AMPK/mTOR-Mediated Autophagy Induction: The energy depletion from PMMP increases AMP:ATP ratio, activating AMPK. This inhibits mTOR signaling and induces autophagy, including mitophagy [138].
- Respiratory Inhibition in Oncology: MitoQ completely abrogates oxygen consumption in various human cancer cell lines at nanomolar concentrations (250-500 nM), inducing a glycolytic switch and radiosensitizing hypoxic tumors [140].
Clinical Status and Applications: MitoQ has successfully passed Phase I safety clinical trials and is under investigation for multiple indications:
- Cardiometabolic Diseases: MitoQ reduces mitochondrial oxidative stress, prevents impaired mitochondrial dynamics, and increases mitochondrial turnover by promoting mitophagy and biogenesis [139]. In hypertension, diabetic kidney disease, and alcohol-induced liver damage, it demonstrates protective effects [139] [141].
- Oncology: MitoQ radiosensitizes human breast tumors in mice by inhibiting cancer cell respiration and improving tumor oxygenation [140]. This effect was specific to MitoQ and not observed with other mitochondria-targeted antioxidants (MitoTEMPO, SKQ1) [140].
- Cardiomyocyte Protection: MitoQ protects against oxidative stress-induced mitochondrial dysregulation in human cardiomyocytes, blunting hydrogen peroxide-induced ROS production, mitochondrial hyperpolarization, and cell death while preserving mitochondrial network structure [141].
Experimental Methodologies for Evaluating ΔΨm-Targeting Agents
Assessment of Mitochondrial Membrane Potential
JC-1 Staining Protocol:
- Principle: JC-1 dye exhibits potential-dependent accumulation in mitochondria, forming red fluorescent J-aggregates at high ΔΨm and green fluorescent monomers at low ΔΨm [138].
- Procedure: Cells are loaded with 2-10 μM JC-1 in culture medium at 37°C for 15-30 minutes, washed, and analyzed via fluorescence microscopy or flow cytometry [138].
- Data Interpretation: The red/green fluorescence intensity ratio provides a quantitative measure of ΔΨm. A decreased ratio indicates mitochondrial depolarization. Treatment with carbonyl cyanide m-chlorophenyl hydrazone (CCCP, 10 μM), a proton ionophore, serves as a positive control for depolarization [138].
Tetramethylrhodamine Methyl Ester (TMRM) Staining:
- Principle: TMRM is a cell-permeant cationic dye that accumulates in active mitochondria based on ΔΨm [136].
- Procedure: Cells are incubated with 20-100 nM TMRM for 20-60 minutes at 37°C. For quantitative measurements, use quenched mode with low dye concentrations; for visualization, use non-quenched mode with higher concentrations [136].
- Applications: Ideal for real-time monitoring of ΔΨm changes in response to pharmacological interventions like idebenone or MitoQ [136].
Measurement of Mitochondrial Oxygen Consumption Rate (OCR)
Seahorse XF Analyzer Protocol:
- Principle: Measures oxygen concentration in a microchamber containing live cells, enabling real-time assessment of mitochondrial function [140].
- Cell Preparation: Plate cells at optimal density (20,000-50,000 cells/well for most cell lines) 24 hours before assay in appropriate growth medium.
- Drug Treatment: Treat cells with ΔΨm-targeting agents (e.g., MitoQ at 62.5 nM-1 μM) for predetermined durations (typically 24 hours) [140].
- Mitochondrial Stress Test:
- Basal OCR: Measure baseline oxygen consumption.
- ATP-linked Respiration: Inject oligomycin (1 μM) to inhibit ATP synthase.
- Maximal Respiration: Inject FCCP (0.5-1.5 μM) to uncouple mitochondria and measure maximal OCR.
- Non-mitochondrial Respiration: Inject rotenone (0.5 μM) and antimycin A (0.5 μM) to inhibit complexes I and III [140].
- Data Analysis: Calculate key parameters: basal respiration, ATP production, proton leak, maximal respiration, and spare respiratory capacity.
Assessment of Mitophagy Activation
LC3-I/II Immunoblotting:
- Principle: During autophagy, cytosolic LC3-I is lipidated to form LC3-II, which associates with autophagosomal membranes. The LC3-II/LC3-I ratio indicates autophagic activity [138].
- Procedure: Extract proteins from treated cells, separate via SDS-PAGE (12-15% gels), transfer to PVDF membranes, and probe with anti-LC3 antibodies.
- Interpretation: Increased LC3-II/LC3-I ratio suggests enhanced autophagosome formation. Use chloroquine (10 μM, 4-6 hours) to block autophagosome-lysosome fusion and confirm flux [138].
Cyto-ID Autophagy Assay:
- Principle: Cyto-ID is a cationic amphiphilic tracer that specifically labels autophagic compartments with minimal lysosomal staining [138].
- Procedure: Incubate cells with Cyto-ID dye (1:1000-1:2000 dilution in culture medium) for 30 minutes at 37°C, wash, and analyze via fluorescence microscopy or flow cytometry.
- Applications: Ideal for detecting MitoQ-induced autophagy, with rapamycin (0.5-1 μM, 2-4 hours) serving as a positive control [138].
Research Reagent Solutions
Table 3: Essential Research Reagents for ΔΨm and Mitophagy Studies
| Reagent/Category | Specific Examples | Function/Application | Key Considerations |
|---|---|---|---|
| ΔΨm-Targeting Compounds | Idebenone, MitoQ, MitoTEMPO, SKQ1 | Investigational agents for modulating ΔΨm and mitophagy | MitoQ uniquely inhibits respiration; effects are concentration-dependent (nM-μM range) [135] [138] [140] |
| ΔΨm Indicators | JC-1, TMRM, TMRE | Fluorescent detection of mitochondrial polarization state | JC-1 provides ratio-metric measurements; TMRM suitable for real-time kinetics [138] [136] |
| Mitophagy Modulators | Rapamycin, Chloroquine, CCCP | Inducers and inhibitors of mitophagy pathways | CCCP induces depolarization; chloroquine blocks autophagic flux [138] [133] |
| Respiratory Chain Inhibitors | Rotenone (CI), Antimycin A (CIII), Oligomycin (CV) | Specific inhibition of ETC complexes for functional assays | Used in Seahorse mitochondrial stress tests [136] [140] |
| Autophagy/Mitophagy Markers | LC3 antibodies, p62/SQSTM1 antibodies, Cyto-ID dye | Detection and quantification of autophagic structures | LC3-II conversion and p62 degradation indicate autophagic flux [138] [133] |
Signaling Pathway Visualizations
PINK1/Parkin-Mediated Mitophagy Pathway
Diagram 1: PINK1/Parkin-Mediated Mitophagy Pathway (6 nodes)
MitoQ Mechanism of Action
Diagram 2: MitoQ Mechanism of Action (10 nodes)
Idebenone Mechanisms in Cancer
Diagram 3: Idebenone Anticancer Mechanisms (9 nodes)
Comparative Analysis and Future Directions
Differential Mechanisms and Clinical Applications
While both idebenone and MitoQ target ΔΨm and influence mitophagy, their molecular mechanisms and clinical translation pathways differ significantly. Idebenone primarily functions as an electron carrier and antioxidant, with clinical validation primarily in mitochondrial neurological disorders. Its emerging applications in oncology leverage ΔΨm disruption and cell cycle arrest pathways [135]. In contrast, MitoQ's unique mechanism involves proton displacement and pseudo-ΔΨm formation, leading to energy stress-induced autophagy [138]. Its clinical development focuses on cardiometabolic diseases and oncology, particularly as a radiosensitizer [139] [140].
The contrasting effects on cellular respiration highlight the importance of context-specific application. While MitoQ potently inhibits oxygen consumption across multiple cancer cell lines [140], idebenone can support respiration in complex I-deficient models when properly reduced [136]. This fundamental difference underscores why MitoQ demonstrates strong radiosensitization properties while idebenone shows direct antiproliferative effects in cancer models.
Emerging Research Tools and Technologies
Future research on ΔΨm-targeting agents will benefit from advanced technologies including:
- Super-resolution microscopy for visualizing mitochondrial network dynamics and autophagosome formation
- CRISPR-based screening to identify synthetic lethal interactions with ΔΨm-disrupting agents
- Organoid models for evaluating tissue-specific effects in human-derived systems
- Biosensors for real-time monitoring of ΔΨm and mitophagy flux in live cells
The continued development of these research tools, combined with the structured experimental approaches outlined in this review, will accelerate the translation of ΔΨm-targeting therapies from bench to bedside.
Mitochondrial membrane potential (ΔΨm), the electrochemical gradient across the inner mitochondrial membrane, serves as a fundamental regulator of cellular energy homeostasis and mitochondrial quality control. Generated by proton pumps of the electron transport chain, ΔΨm is essential for ATP production through oxidative phosphorylation and acts as a key sensor for mitochondrial health [142]. In disease states, sustained alterations in ΔΨm trigger quality control mechanisms, particularly mitophagy—the selective autophagic degradation of damaged mitochondria. This whitepaper examines how ΔΨm dysregulation contributes to mitophagy defects in three prevalent diseases: Alzheimer's disease (AD), Type 2 Diabetes Mellitus (T2DM), and cancer. By synthesizing insights from these disease models, we provide a framework for understanding ΔΨm's role in pathological mechanisms and highlight emerging therapeutic strategies targeting mitochondrial quality control.
The centrality of ΔΨm in mitophagy initiation is particularly evident in the PINK1/Parkin pathway. Under normal conditions, PTEN-induced putative kinase 1 (PINK1) is continuously imported into mitochondria and degraded. When ΔΨm collapses, PINK1 import is halted, leading to its accumulation on the outer mitochondrial membrane where it recruits and activates Parkin, an E3 ubiquitin ligase [143] [144]. This pathway represents a critical ΔΨm-sensing mechanism that tags dysfunctional mitochondria for degradation. Beyond this canonical pathway, receptor-mediated mechanisms involving proteins like BNIP3, NIX, and FUNDC1 also contribute to mitochondrial quality control, often in response to more subtle ΔΨm fluctuations [144].
Alzheimer's Disease: Mitophagy Impairment and Neuronal Energetics Failure
Molecular Defects in Alzheimer's-Related Mitophagy
Alzheimer's disease represents a paradigm of mitophagy dysfunction in neurodegenerative disorders. Post-mortem analyses of AD brains consistently reveal impaired mitophagic degradation, evidenced by accumulated mitochondrial DNA, proteins such as COX IV and TOMM20, and structurally damaged mitochondria in the cytoplasmic and autophagic vacuoles [145] [99]. The retrograde transport of damaged mitochondria to neuronal somata for degradation is also compromised, linked to reduced levels of DISC1, a protein regulating axonal mitochondria trafficking that also functions as a mitophagy receptor [145].
Molecular analyses demonstrate altered expression of key mitophagy regulators in AD-affected brain regions. At early AD stages (Braak stage II-III), PINK1 levels increase, while later stages (Braak stage VI) show elevated Parkin alongside persistently high mitochondrial content, suggesting a defective initiation of the PINK1/Parkin cascade [145]. Other studies report decreased mRNA and protein levels of PINK1 in late-stage AD hippocampi (Braak stage V-VI) [146]. Broader downregulation of autophagy and mitophagy proteins occurs, including OPTN, ATG5, ATG12, Beclin-1, BNIP3, BNIP3L, FUNDC1, and VDAC1 [143]. Basal mitophagy levels are reduced by 30-50% in AD hippocampi compared to age-matched controls, with impaired AMPK signaling and defective mitophagosome-lysosome fusion contributing to the pathology [23].
Table 1: Key Mitophagy Alterations in Alzheimer's Disease
| Component | Alteration in AD | Functional Consequence |
|---|---|---|
| PINK1/Parkin | Early increase, later decrease | Failed mitophagy initiation |
| Mitochondrial Content | Increased (COX IV, TOMM20, mtDNA) | Accumulation of damaged organelles |
| LC3-II/I Ratio | Increased | Impaired autophagosome formation/maturation |
| p62/SQSTM1 | Accumulated | Defective autophagic flux |
| AMPK/ULK1/TBK1 | Dysregulated signaling | Reduced mitophagy initiation |
| Lysosomal Fusion | Impaired | Blocked mitochondrial degradation |
| DISC1 | Decreased | Disrupted mitochondrial transport |
Experimental Models and Methodologies
AD Patient-Derived Cellular Models: Fibroblasts from sporadic AD patients exhibit dysfunctional mitophagy with reduced autophagic vacuole formation, decreased lysosomes, and TOMM20-positive mitochondrial accumulation [145]. Parkin recruitment to mitochondria remains deficient even upon CCCP-induced depolarization [145]. Using the MitoTimer probe, researchers have demonstrated that AD fibroblasts lose the spatial maturation gradient (young mitochondria at periphery, old near nucleus) seen in healthy cells, indicating impaired mitochondrial transport and degradation [147].
Induced Pluripotent Stem Cell (iPSC) Models: iPSCs from familial AD patients with PS1(A246E) mutation recapitulate mitophagy defects observed in post-mortem tissue, providing platforms for drug screening [145]. These models allow assessment of how AD-related proteins directly impact mitophagy—APP-CTFs accumulation correlates more strongly with mitophagy failure than Aβ or pTau in human AD brains [148].
Measurement Approaches: ΔΨm can be quantified using fluorescent probes (JC-1, TMRM) in live cells, while mitophagy flux is assessed via mt-Keima reporter systems or Western blot analysis of mitochondrial versus cytosolic LC3-II and PINK1 turnover [23]. Immunofluorescence co-staining of mitochondrial (TOMM20) and autophagosomal (LC3) markers visualizes mitophagosome formation.
Diagram 1: Mitophagy Pathway Disruption in Alzheimer's Disease. In healthy mitochondria, normal ΔΨm facilitates PINK1 import and degradation. In AD, reduced ΔΨm triggers PINK1 accumulation and subsequent Parkin recruitment, but the process is blocked at lysosomal fusion, preventing mitochondrial degradation.
Type 2 Diabetes Mellitus: Metabolic Stress and Mitophagy Adaptation
Mitophagy in Metabolic Regulation and Insulin Resistance
In Type 2 Diabetes Mellitus, mitophagy serves as a crucial adaptive mechanism to metabolic stress characterized by high glucose, elevated free fatty acids, and insulin resistance. Mitochondrial dysfunction in pancreatic β-cells and insulin-responsive tissues (liver, muscle, adipose) contributes significantly to disease pathogenesis [146]. Under physiological conditions, mitophagy maintains a healthy mitochondrial population by eliminating damaged organelles that produce excessive reactive oxygen species (ROS). In T2DM, however, chronic nutrient excess overwhelms this quality control system.
Research demonstrates that patients with mild hyperglycemia show reduced expression of mitophagy-related genes (NIX, PINK1, Parkin) compared to healthy controls, with further declines in established T2DM patients . This impaired mitophagy initiation results in accumulation of dysfunctional mitochondria, evidenced by smaller mitochondrial size and disrupted cristae in hepatocytes from insulin-resistant patients . The resulting oxidative damage promotes β-cell apoptosis and exacerbates insulin resistance .
Table 2: Mitophagy Defects in Type 2 Diabetes Mellitus
| Tissue/Cell Type | Mitophagy Alteration | Functional Consequence |
|---|---|---|
| Pancreatic β-cells | Reduced PINK1/Parkin signaling | Impaired insulin secretion |
| Liver | Downregulated FUNDC1 | Hepatic insulin resistance |
| Skeletal Muscle | Decreased MFN2 expression | Disrupted mitochondrial dynamics |
| Adipose Tissue | Altered ATG5, LC3a/b expression | Obesity-related metabolic dysfunction |
| Retina | Reduced mitophagosomes | Diabetic retinopathy progression |
Mitochondrial Dynamics and Experimental Approaches
T2DM features a shift toward mitochondrial fission, driven by increased Drp1 recruitment and OPAl/MFN degradation . High glucose levels induce excessive ROS production, enhancing the Drp1:Mnf2 ratio and promoting fission . This aberrant mitochondrial dynamics, characterized by fragmented mitochondria, contributes to insulin resistance and β-cell dysfunction .
Experimental Models and Assessment Methods:
In Vivo Models: db/db mice and high-fat diet-fed rodents demonstrate mitophagy impairment in various tissues. These models show reduced Parkin and PINK1 levels in diabetic retinas and increased oxidative stress [146].
Cell Culture Systems: Insulin-resistant hepatocyte models and glucose-treated pancreatic β-cells allow investigation of nutrient overload effects on mitophagy. FUNDC1 knockout models establish its essential role in maintaining metabolic homeostasis [144] [147].
Methodologies: Mitophagy flux measurements use mt-Keima or mito-QC reporters in combination with glucose clamp studies to correlate mitophagy with insulin sensitivity. Mitochondrial morphology is assessed via electron microscopy, while functional assays measure ROS production, ΔΨm (JC-1, TMRM), and oxygen consumption rates.
Cancer: Context-Dependent Mitophagy Roles and Therapeutic Implications
Dual Roles in Tumor Suppression and Promotion
Cancer demonstrates the context-dependent nature of mitophagy, functioning as both tumor suppressor and promoter depending on cancer type, stage, and microenvironment [144]. As a tumor suppressor, mitophagy eliminates damaged mitochondria that would otherwise release pro-death factors, while as a tumor promoter, it maintains mitochondrial health in rapidly dividing cells and supports metabolic adaptation.
In breast cancer, PINK1 demonstrates complex roles—its transcript levels are significantly downregulated in tumors compared to adjacent normal tissue, while protein expression shows higher levels in cancer tissues with distinctive subcellular localization (diffuse cytoplasmic with strong membrane staining in cancer versus granular cytoplasmic in normal) . Functionally, PINK1 suppresses MCF-7 cell growth but promotes MDA-MB-231 triple-negative breast cancer cell proliferation, indicating subtype-specific effects .
Parkin frequently shows downregulated mRNA and protein levels in breast cancer, with high Parkin expression associated with lower histological grade, reduced triple-negative subtypes, decreased lymph node metastasis, and improved prognosis . This tumor-suppressive function contrasts with contexts where mitophagy supports tumor survival under metabolic stress.
Methodologies for Investigating Oncogenic Mitophagy
Cancer Cell Line Models: Breast cancer subtype-representative lines (MCF-7 luminal A, MDA-MB-231 triple-negative, BT-474 HER-2 positive) enable investigation of mitophagy in different oncogenic contexts [144]. PINK1 inhibition enhances paclitaxel-induced apoptosis in BT-474 cells, suggesting therapeutic applications .
Tumor Microenvironment Models: Co-culture systems and 3D tumor spheroids assess how mitophagy in cancer cells interacts with stromal components. Mitophagy influences resistance to hypoxia and nutrient deprivation in tumor cores [143].
Analytical Approaches: Immunohistochemical analysis of PINK1 subcellular distribution provides prognostic information . Western blotting of mitochondrial fractions assesses PINK1/Parkin activation, while proximity ligation assays visualize protein interactions. Metabolic profiling links mitophagy status to cancer cell bioenergetics.
Diagram 2: Comparative Mitophagy Defects Across Disease Models. While all three diseases involve mitophagy dysfunction, the specific points of disruption vary: Alzheimer's primarily affects early PINK1 stabilization, diabetes impacts mitochondrial ubiquitination, and cancer shows altered receptor recruitment with context-dependent outcomes.
The Scientist's Toolkit: Essential Reagents and Methodologies
Table 3: Research Reagent Solutions for Mitophagy Studies
| Reagent/Category | Specific Examples | Research Application | Key Considerations |
|---|---|---|---|
| ΔΨm Indicators | JC-1, TMRM, TMRE | Quantitative measurement of mitochondrial membrane potential | JC-1 shows emission shift (greenred); TMRM suitable for kinetic studies |
| Mitophagy Reporters | mt-Keima, mt-QC, Rosella | Monitoring mitophagy flux in live cells | mt-Keima pH-sensitive; resistant to lysosomal degradation |
| PINK1/Parkin Assays | Phospho-S65 Parkin Ab, Phospho-S65 Ubiquitin Ab | Monitoring pathway activation | Phospho-specific antibodies confirm pathway engagement |
| Mitochondrial Dyes | MitoTracker, TOM20 Ab | Visualizing mitochondrial mass and network | MitoTracker variants for different ΔΨm dependencies |
| Lysosomal Inhibitors | Bafilomycin A1, Chloroquine | Measuring autophagic flux | Block lysosomal degradation to assess cumulative mitophagy |
| Inducers/Inhibitors | CCCP, Oligomycin, Mdivi-1 | Modulating mitophagy experimentally | CCCP dissipates ΔΨm; Mdivi-1 inhibits Drp1-mediated fission |
| Cell Lines | SH-SY5Y, MEFs, patient-derived fibroblasts | Disease modeling | Primary cells maintain patient-specific characteristics |
Standard Experimental Workflow for Mitophagy Assessment
Step 1: Mitochondrial Stress Induction
- Treatment with ΔΨm disruptors (CCCP, valinomycin) or disease-relevant stressors (Aβ oligomers for AD, high glucose/palmitate for T2DM)
- Optimization of dose and duration to avoid excessive cell death
Step 2: Multiparameter Assessment of Mitochondrial Health
- ΔΨm measurement using ratiometric probes (JC-1)
- Simultaneous assessment of ROS production (MitoSOX)
- Mitochondrial morphology analysis (confocal microscopy)
Step 3: Mitophagy Flux Quantification
- Western blot analysis of LC3-II turnover, p62 degradation, PINK1 stabilization
- Immunofluorescence co-localization of mitochondrial and autophagosomal markers
- mt-Keima flow cytometry for quantitative flux measurement
Step 4: Functional Validation
- ATP production assays
- Oxygen consumption rate (Seahorse) measurements
- Assessment of downstream outcomes (insulin secretion, apoptosis, proliferation)
Diagram 3: Comprehensive Workflow for Mitophagy Research. This standardized approach enables systematic investigation of mitophagy across disease models, integrating multiple assessment methods for robust conclusions.
The examination of Alzheimer's disease, Type 2 Diabetes Mellitus, and cancer reveals both shared and distinct patterns of mitophagy dysregulation centered on mitochondrial membrane potential. In AD, ΔΨm reduction fails to adequately initiate quality control, leading to accumulated mitochondrial damage. In T2DM, metabolic stress alters ΔΨm dynamics, promoting fission and overwhelming mitophagic capacity. In cancer, ΔΨm regulation supports context-dependent roles in tumor suppression or promotion. These insights highlight ΔΨm as a critical sensing and signaling hub coordinating mitochondrial quality control.
Emerging therapeutic strategies aim to modulate mitophagy for clinical benefit. In AD models, mitophagy inducers (including NAD+ precursors and urolithin A) reverse pathology and improve cognitive function [145]. Natural compounds like astragaloside IV show promise for diabetic neuropathy by enhancing mitophagy [148]. In cancer, context-specific modulation—inhibiting mitophagy in tumors dependent on mitochondrial quality control versus enhancing it in neurodegenerative conditions—represents a promising frontier. Future research should refine disease-specific mitophagy assessment tools, develop more targeted modulators, and explore combination therapies addressing both mitophagy and complementary quality control pathways.
Mitochondrial membrane potential (ΔΨm) is a critical parameter of mitochondrial health, serving as a fundamental indicator of cellular viability and function. Its integrity is indispensable for ATP production, reactive oxygen species (ROS) regulation, and cellular signaling. Within the mitochondrial quality control (MQC) network, ΔΨm acts as a primary sensor, directly influencing central degradation pathways such as mitophagy. This whitepaper examines the central role of ΔΨm as a biomarker, correlating its dissipation with the progression of neurodegenerative diseases, cancer, and metabolic disorders. We further explore its utility in monitoring therapeutic efficacy, supported by quantitative data, detailed experimental protocols for its assessment, and an analysis of emerging therapeutic strategies targeting its maintenance.
Mitochondrial quality control is a complex, multi-tiered system essential for preserving a healthy mitochondrial population through mechanisms including biogenesis, dynamics (fusion and fission), and mitophagy [10] [52]. At the heart of this regulatory network lies the mitochondrial membrane potential (ΔΨm), an electrochemical gradient across the inner mitochondrial membrane (IMM) that is fundamental for energy transduction and overall organellar fitness [52].
The integrity of ΔΨm is a critical determinant in MQC decision-making. It acts as a key signal for initiating the removal of damaged mitochondria via mitophagy; a collapse in ΔΨm is a recognized "eat-me" signal that triggers the PINK1-Parkin pathway and other mitophagy mechanisms [10] [149]. Furthermore, mitochondrial fusion is energetically demanding and is strongly dependent on an adequate ΔΨm. Notably, a loss of membrane potential in a specific region can lead to uncoupled fusion, where outer membrane fusion proceeds without inner membrane fusion, effectively isolating the damaged component [10]. This positions ΔΨm as a crucial "spatiotemporal-threshold" switch, determining whether a mitochondrion is repaired, recycled, or destined for elimination [52]. Consequently, the loss of ΔΨm integrity is a hallmark of mitochondrial dysfunction and is implicated in the early pathophysiology of a wide spectrum of diseases, offering substantial potential as a dynamic biomarker for disease progression and therapeutic response [150] [25].
Measuring ΔΨm: Experimental Methodologies and Protocols
Accurately determining ΔΨm is challenging due to mitochondrial heterogeneity and the dynamic nature of this potential. A combination of techniques is often necessary to obtain a comprehensive and biologically relevant assessment [151]. The table below summarizes the primary methods used for ΔΨm assessment in research settings.
Table 1: Key Methodologies for Assessing Mitochondrial Membrane Potential (ΔΨm)
| Method | Principle | Key Reagents & Assays | Output Parameters | Advantages | Limitations |
|---|---|---|---|---|---|
| Fluorescent Dye-Based Assays | Cationic, lipophilic dyes accumulate in the mitochondrial matrix in a ΔΨm-dependent manner. | JC-1: Exhibits a fluorescence shift from green (~529 nm) to red (~590 nm) as ΔΨm increases. TMRM, TMRE: Non-ratio-metric dyes whose fluorescence intensity correlates with ΔΨm. Rhodamine 123: Similar to TMRM, used for flow cytometry and imaging. | - JC-1 red/green fluorescence ratio- TMRM/TMRE/Rhodamine 123 fluorescence intensity | - High throughput compatibility (flow cytometry)- Applicable to live cells- Spatial resolution (imaging) | - Sensitive to dye loading conditions- Potential artifacts from plasma membrane potential- Photobleaching |
| Respirometry (O2k-FluoRespirometer) | Couples high-resolution respirometry with fluorometry to measure O2 consumption and ΔΨm simultaneously. | TMRM in combination with substrates (e.g., pyruvate, malate, succinate) and inhibitors (e.g., oligomycin, FCCP) of the electron transport chain. | - Oxygen flow (pmol/(s*ml))- TMRM fluorescence (arbitrary units) over time | - Direct correlation of bioenergetic function with ΔΨm- Provides functional context beyond a static measurement | - Technically complex- Lower throughput- Requires specialized equipment |
| PET Imaging (Emerging) | Uses novel radiotracers that distribute in tissues based on ΔΨm. | [18F]BODIPY-trimethylphosphonium (TPP) and other mitochondrial-targeted radiotracers. | - Standardized Uptake Value (SUV) in target tissues | - Non-invasive, in vivo measurement- Potential for clinical translation and diagnostics | - Limited spatial resolution vs. microscopy- Under development, not yet widespread |
The following workflow details a standardized protocol for evaluating ΔΨm using the JC-1 dye, a widely adopted method for its rationetric properties, which correct for variations in mitochondrial mass, dye loading, and photobleaching.
Protocol: JC-1 Staining for ΔΨm Assessment by Flow Cytometry
- Cell Preparation: Seed cells in a multi-well plate and apply the desired experimental treatments (e.g., compounds, stressors). Include a positive control using a depolarizing agent like Carbonyl cyanide m-chlorophenyl hydrazone (CCCP, 10-50 µM for 10-20 minutes).
- Dye Loading: Carefully remove the culture medium and incubate cells with a pre-warmed JC-1 staining solution (e.g., 5 µM in serum-free media or PBS) for 15-30 minutes at 37°C in the dark.
- Washing: Remove the JC-1 staining solution and gently wash the cells twice with PBS to remove any non-specific dye.
- Cell Harvesting and Analysis: Trypsinize the cells, resuspend in PBS, and immediately analyze via flow cytometry. Use the FL1 (FITC/Green) and FL2 (PE/Red) channels.
- Data Interpretation: A high red/green fluorescence ratio indicates healthy, polarized mitochondria. A decrease in this ratio signifies mitochondrial depolarization. Results should be reported as the mean ratio ± SEM from at least three independent experiments.
ΔΨm as a Quantitative Biomarker in Disease Pathogenesis
The loss of ΔΨm is a common denominator in the pathogenesis of numerous diseases, with the degree of depolarization often correlating with disease severity. The following table synthesizes key findings from recent research linking ΔΨm integrity to specific disease states.
Table 2: Correlations Between ΔΨm Integrity and Disease Progression
| Disease Category | Specific Disease / Context | Key Findings on ΔΨm | Molecular Link to MQC | Reference |
|---|---|---|---|---|
| Neurodegenerative | Alzheimer's Disease (AD) | Synaptic mitochondria in AD models show early ΔΨm dissipation, linked to Aβ accumulation and oxidative stress. | Impaired dynein-Snapin-mediated removal of damaged mitochondria, leading to synaptic mitophagosome accumulation. | [149] |
| Neurodegenerative | Leber Hereditary Optic Neuropathy (LHON) | Mutations in complex I genes (e.g., G11778A) cause ΔΨm collapse, reducing ATP and increasing ROS in retinal ganglion cells. | Direct failure of OXPHOS, triggering apoptosis; inflammation exacerbates damage. | [150] [152] |
| Mitochondrial | MELAS Syndrome | A3243G mutation in mt-tRNA causes cristae disorganization and ETC defects, leading to progressive ΔΨm loss. | Chronic OXPHOS failure induces excessive ROS production and endothelial inflammation. | [152] |
| Cancer | Hematologic Malignancies | Cancer cells hijack mitochondrial dynamics (fission/fusion) to maintain ΔΨm, supporting metabolic flexibility and drug resistance. | DRP1-mediated fission is upregulated, isolating damaged parts and preserving overall network ΔΨm. | [153] |
| Metabolic/Inflammatory | Chronic Obstructive Pulmonary Disease (COPD) | Exposure to cigarette smoke and particulates (e.g., BRPM2.5) directly reduces ΔΨm in bronchial epithelial cells. | ΔΨm loss triggers excessive mito-ROS, creating a vicious cycle of inflammation and dysfunction. | [25] |
| Ischemia-Reperfusion | Intestinal Ischemia-Reperfusion (II/R) Injury | Early II/R injury is characterized by oxidative stress, leading to ΔΨm collapse and activation of pro-apoptotic pathways. | Upregulation of mitochondrial-related hub genes (e.g., Bcl2l11, Pmaip1) involved in apoptosis following ΔΨm loss. | [154] |
The relationship between ΔΨm and key MQC processes like mitophagy can be visualized as a critical regulatory axis. This pathway determines cellular fate based on the severity and persistence of ΔΨm loss.
ΔΨm in Therapeutic Development and Monitoring
The critical role of ΔΨm in cell viability makes it an invaluable tool for drug discovery, both for identifying novel compounds and for monitoring patient response to therapy.
Screening for MQC-Modulating Therapeutics: High-throughput screening platforms using ΔΨm-sensitive dyes can identify small molecules that enhance mitochondrial resilience. Compounds that protect against stress-induced ΔΨm depolarization are potential candidates for treating neurodegenerative and metabolic diseases. For instance, idebenone, approved for LHON, and omaveloxolone, for Friedreich's ataxia, work in part by mitigating oxidative stress that would otherwise collapse ΔΨm [150]. Furthermore, securinine and ABT-737 have been predicted as potential therapeutic agents for intestinal ischemia-reperfusion injury, likely through modulating mitochondrial apoptotic pathways linked to ΔΨm [154].
Monitoring Therapeutic Efficacy: Restoring ΔΨm is a key indicator of successful treatment. In preclinical models of Alzheimer's disease, enhancing the removal of damaged mitochondria (e.g., by overexpressing Snapin to improve retrograde transport) was shown to ameliorate synaptic deficits and mitigate ΔΨm loss [149]. Similarly, in COPD models, interventions that reduce oxidative stress have been shown to improve ΔΨm and ATP levels, indicating a reversal of mitochondrial dysfunction [25].
Emerging Therapy - Mitochondrial Transplantation: A direct approach to restoring ΔΨm is through mitochondrial transplantation and transplantation (MTT). This involves delivering isolated, functional mitochondria into damaged cells or tissues. Studies show that these exogenous mitochondria can integrate into recipient cells, enhance ATP production, restore redox balance, and improve cellular survival [155]. The success of this therapy is intrinsically linked to the ΔΨm of the transplanted organelles, as only mitochondria with intact membrane potential can rescue cellular function.
The Scientist's Toolkit: Essential Research Reagents
This section catalogues critical reagents and tools for investigating ΔΨm and its role in mitochondrial quality control.
Table 3: Key Research Reagent Solutions for ΔΨm and MQC Studies
| Reagent / Tool Category | Specific Examples | Primary Function in Research |
|---|---|---|
| ΔΨm-Indicator Dyes | JC-1, TMRM, TMRE, Rhodamine 123 | Quantitative and visual assessment of ΔΨm status in live cells via fluorescence microscopy or flow cytometry. |
| Chemical Depolarizers | CCCP, FCCP | Positive controls for inducing complete ΔΨm collapse, validating assay sensitivity. |
| MQC Modulators (Small Molecules) | Mdivi-1 (DRP1 inhibitor), Dynasore (DRP1 inhibitor) | Experimental tools to perturb mitochondrial fission and study its effect on ΔΨm and mitophagy. |
| Antioxidants | Vitamin C, Edaravone | Used to investigate the role of oxidative stress in ΔΨm dissipation; shown to reduce inflammation in MELAS iPSC models. |
| Biochemical Assay Kits | ATP Assay Kits, ROS Detection Kits (e.g., MitoSOX) | Correlative measurements to link ΔΨm changes with metabolic output (ATP) and oxidative damage (mito-ROS). |
| Gene Expression Analysis | qRT-PCR primers for MQC genes (e.g., PINK1, Parkin, Pdk4, Bcl2l11) | Validate that ΔΨm changes are associated with transcriptional regulation of key MQC pathways. |
The integrity of the mitochondrial membrane potential (ΔΨm) is unequivocally a master regulator of mitochondrial health and a sensitive, quantitative biomarker with significant utility in both basic research and clinical translation. Its central role in the mitochondrial quality control network, particularly as a trigger for mitophagy, directly links its dissipation to the pathogenesis of a growing list of human diseases. The ability to quantitatively measure ΔΨm using standardized protocols provides a powerful tool for screening novel therapeutics, monitoring disease progression, and validating the efficacy of emerging treatments, including mitochondrial transplantation. Future research focusing on standardizing ΔΨm measurements across platforms and further elucidating its spatiotemporal dynamics in vivo will be crucial for fully unlocking its potential as a cornerstone biomarker in precision medicine.
Mitochondrial membrane potential (ΔΨm) is a critical parameter of mitochondrial health, serving as a key regulator of cellular energy production, quality control mechanisms, and apoptotic signaling. Recent advances in mitochondrial biology have illuminated ΔΨm's fundamental role in coordinating mitophagy and overall mitochondrial quality control, establishing it as a potentially transformative biomarker for precision medicine. This technical guide examines the emerging frontier of incorporating ΔΨm metrics into clinical trial designs, focusing on practical methodologies for ΔΨm quantification, analytical frameworks for data integration, and innovative stratification approaches that leverage ΔΨm dynamics to identify patient subgroups with distinct therapeutic responses. By providing researchers with both theoretical foundations and practical implementation tools, this review aims to facilitate the transition of ΔΨm from a research metric to a clinically actionable biomarker that enhances trial efficiency and therapeutic precision across neurodegenerative, cardiovascular, and oncological indications.
The mitochondrial inner membrane potential (ΔΨm) represents an electrochemical gradient essential for ATP synthesis, protein import, calcium homeostasis, and reactive oxygen species (ROS) regulation. Beyond these established functions, ΔΨm serves as a critical signaling hub within the mitochondrial quality control (MQC) network, directly influencing mitochondrial dynamics, biogenesis, and selective autophagy (mitophagy) [156] [12]. The maintenance of ΔΨm is therefore fundamental to cellular viability, with both depolarization and hyperpolarization carrying significant pathophysiological implications.
Recent research has demonstrated that chronic mitochondrial hyperpolarization triggers profound cellular reprogramming, including nuclear DNA hypermethylation and altered expression of metabolic genes [2]. These epigenetic and transcriptional changes are mediated through phospholipid remodeling rather than traditional redox or metabolic alterations, revealing a novel signaling axis between ΔΨm and nuclear function. Conversely, ΔΨm dissipation represents a key initiating event in PINK1/Parkin-mediated mitophagy, marking dysfunctional organelles for degradation [156]. This dual role—as both indicator of functional capacity and initiator of quality control processes—positions ΔΨm as a master regulator of mitochondrial population health.
In the context of disease, ΔΨm dysregulation manifests across multiple pathological states. Cancer cells frequently exhibit altered ΔΨm profiles, with studies documenting hyperpolarization in glioblastoma, ovarian cancer, and pulmonary hypertension models [2] [157]. Similarly, neurodegenerative conditions like Alzheimer's and Parkinson's disease feature impaired ΔΨm homeostasis linked to defective mitophagy and neuronal vulnerability [156] [158]. These disease-specific alterations in ΔΨm dynamics present unique opportunities for patient stratification based on underlying mitochondrial pathophysiology rather than solely symptomatic or histopathological classifications.
Technical Foundations: ΔΨm Measurement Methodologies
Fluorescent Probe-Based Assays
Fluorescent indicators represent the most widely utilized approach for ΔΨm quantification in both research and preclinical settings. These assays leverage potential-sensitive dyes that accumulate in mitochondria according to the Nernst equation, with fluorescence intensity directly correlating with ΔΨm magnitude.
Table 1: Common Fluorescent Probes for ΔΨm Measurement
| Probe | Excitation/Emission (nm) | Measurement Mode | Key Applications | Considerations |
|---|---|---|---|---|
| TMRE (Tetramethylrhodamine ethyl ester) | 549/575 | Intensity-based | Intact cell imaging, flow cytometry | Potential-dependent accumulation; requires normalization with ΔΨm-independent dyes (e.g., MitoTracker Green) [2] |
| TMRM (Tetramethylrhodamine methyl ester) | 548/573 | Intensity or fluorescence lifetime imaging (FLIM) | Permeabilized cell assays, high-resolution imaging | Reduced phototoxicity; suitable for kinetic measurements [2] |
| JC-1 | 514/529 (monomer); 585/590 (aggregate) | Ratiometric | Apoptosis detection, drug screening | Forms J-aggregates at high ΔΨm; enables qualitative assessment of polarization states |
| Rhodamine 123 | 507/529 | Intensity-based | High-throughput screening | Potential-sensitive accumulation with reversible binding |
Standardized protocols for ΔΨm measurement must account for critical technical considerations. For intensity-based probes like TMRE, normalization to ΔΨm-independent markers (e.g., MitoTracker Green) is essential to control for mitochondrial mass variations [2]. Additionally, careful titration of probe concentrations prevents artifacts from self-quenching or metabolic inhibition. For kinetic assessments, such as calcium uptake capacity in permeabilized cells, TMRM fluorescence can be coupled with calcium indicators (e.g., FuraFF) to simultaneously monitor ΔΨm and mitochondrial function [2].
Advanced Imaging and Analytical Approaches
Beyond conventional fluorescence microscopy, several advanced technologies offer enhanced resolution for ΔΨm monitoring:
- Fluorescence Lifetime Imaging Microscopy (FLIM): Measures nanosecond-scale decay kinetics of ΔΨm-sensitive fluorophores, providing quantitative measurements independent of probe concentration, excitation intensity, or photobleaching.
- High-Content Imaging Platforms: Enable multiplexed ΔΨm assessment alongside other mitochondrial parameters (morphology, mass, ROS production) in automated format suitable for drug screening.
- Correlative Light and Electron Microscopy (CLEM): Integrates dynamic ΔΨm information with ultrastructural analysis of mitochondrial cristae architecture.
Implementation of these methodologies in clinical trial contexts requires rigorous standardization of sample processing, staining protocols, and analytical pipelines to ensure reproducibility across sites and timepoints.
ΔΨm in Mitochondrial Quality Control and Disease Pathogenesis
ΔΨm Regulation of Mitophagy and Dynamics
The integration of ΔΨm into the mitochondrial quality control framework occurs through multiple coordinated mechanisms. As illustrated in Figure 1, ΔΨm dissipation serves as a primary signal for PINK1 stabilization on the outer mitochondrial membrane, initiating Parkin-mediated ubiquitination and autophagic clearance [156]. This quality control pathway ensures the selective removal of depolarized, dysfunctional organelles while preserving the healthy mitochondrial network.
Figure 1: ΔΨm in Mitochondrial Quality Control Signaling
Simultaneously, ΔΨm influences mitochondrial dynamics by modulating the activity of fusion and fission proteins. Hyperpolarized states promote phospholipid remodeling that can alter membrane fluidity and fusion capacity [2], while depolarization often triggers DRP1-mediated fission, facilitating the isolation of damaged components. The interplay between these processes creates a responsive quality control network that maintains mitochondrial integrity through continuous remodeling.
Pathological ΔΨm Alterations in Disease States
Chronic ΔΨm dysregulation manifests differently across pathological contexts, with important implications for therapeutic targeting:
Neurodegenerative Diseases: In Alzheimer's and Parkinson's disease models, progressive ΔΨm impairment correlates with synaptic dysfunction and neuronal loss. The accumulation of depolarized mitochondria resulting from defective PINK1/Parkin signaling exacerbates oxidative stress and promotes neuroinflammation through mitochondrial DNA release and activation of cGAS-STING pathways [156].
Cancer Metabolism: Multiple cancer types exhibit mitochondrial hyperpolarization, which enhances calcium uptake capacity and supports proliferation. In ATP5IF1 (IF1)-deficient cells—a genetic model of hyperpolarization—this ΔΨm elevation triggers nuclear DNA hypermethylation and transcriptional reprogramming of metabolic genes through phospholipid-mediated mechanisms [2].
Cardiac Pathology: Myocardial ischemia-reperfusion injury induces ΔΨm instability that promotes opening of the mitochondrial permeability transition pore (mPTP), culminating in cardiomyocyte death. Extracellular vesicles (EVs) from progenitor cells can restore ΔΨm homeostasis in injured cardiomyocytes by delivering mitochondrial components and modulating fusion/fusion balance through DRP1 and OPA1 regulation [159].
Table 2: ΔΨm Alterations in Pathological States
| Disease Context | ΔΨm Alteration | Functional Consequences | Stratification Potential |
|---|---|---|---|
| Alzheimer's Disease | Progressive depolarization | Impaired synaptic function, increased ROS production | Identify patients with mitochondrial dysfunction predominant subtypes |
| Ovarian Cancer | Chronic hyperpolarization | Enhanced proliferation, chemotherapy resistance | Stratify IF1-deficient tumors for metabolic therapies |
| Cardiac Ischemia | Acute depolarization | mPTP opening, cardiomyocyte apoptosis | Select patients for mitochondrial-protective interventions |
| Pulmonary Hypertension | Elevated ΔΨm in smooth muscle | Hyperproliferation, vascular remodeling | Identify candidates for ΔΨm-normalizing treatments |
These disease-specific alterations highlight the potential of ΔΨm metrics to identify patient subgroups with distinct pathological drivers and therapeutic vulnerabilities.
Integration of ΔΨm Metrics into Clinical Trial Frameworks
Stratification Strategies and Analytical Approaches
The incorporation of ΔΨm into patient stratification paradigms requires careful consideration of analytical frameworks and validation procedures. Building on successful precision medicine approaches in other domains, several strategic models emerge as particularly promising:
AI-Guided Trajectory Classification: Following the paradigm established in Alzheimer's disease trials, where predictive prognostic models (PPMs) stratify patients based on projected progression rates [158], ΔΨm dynamics could be incorporated into similar machine learning algorithms. These models would integrate baseline ΔΨm measurements with other mitochondrial parameters to classify patients into slow versus rapid progression categories for targeted intervention.
Multi-Omic Integration: ΔΨm profiles can be contextualized within broader molecular landscapes through integration with genomic, transcriptomic, and proteomic datasets. This approach aligns with established multi-omics stratification frameworks that have demonstrated utility in oncology and neurology [160]. For instance, ΔΨm hyperpolarization signatures could be combined with ATP5IF1 expression data and phospholipidomic profiles to identify patients with specific metabolic vulnerabilities.
Dynamic Response Assessment: Beyond static measurements, ΔΨm responsivity to pharmacological challenges could serve as a functional stratification metric. Patients might be classified according to mitochondrial resilience based on ΔΨm recovery following exposure to oxidative stress or metabolic inhibitors, creating a functional dimension to stratification.
Practical Implementation and Technical Validation
Successful implementation of ΔΨm stratification requires standardized operationalization across several domains:
Sample Processing Protocols: For clinical trial applications, standardized sample handling is critical. For circulating cells (e.g., platelets, lymphocytes), processing within 2-4 hours of collection with defined anticoagulants preserves ΔΨm integrity. Tissue-based assessments require rapid fixation or fresh processing to prevent artifacts.
Analytical Validation: ΔΨm assays should demonstrate adequate precision (CV <15%), linearity across expected pathological ranges, and stability under expected storage conditions. Reference values should be established using appropriate control materials with defined polarization states.
Multisite Harmonization: In multicenter trials, site-specific differences in instrumentation and technical expertise necessitate rigorous harmonization procedures. This may include centralized batch analysis, standardized instrument calibration, and cross-site technician training with proficiency testing.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for ΔΨm Research and Clinical Assay Development
| Category | Specific Reagents | Function/Application | Considerations for Clinical Translation |
|---|---|---|---|
| ΔΨm Indicators | TMRE, TMRM, JC-1, Rhodamine 123 | Quantitative ΔΨm measurement | TMRE/TMRM preferred for clinical assays due to well-characterized toxicity and accumulation kinetics |
| Validation Reagents | Carbonyl cyanide m-chlorophenyl hydrazone (CCCP), Oligomycin A | Induce controlled depolarization/hyperpolarization for assay calibration | Critical for assay quality control and between-experiment normalization |
| Mitochondrial Mass Markers | MitoTracker Green, Anti-COX4 antibodies | Normalize ΔΨm to mitochondrial content | Essential for distinguishing ΔΨm changes from alterations in mitochondrial mass |
| Quality Control Antibodies | Anti-ATP5IF1, Anti-OPA1, Anti-DRP1 | Assess mitochondrial quality control protein expression | Correlate ΔΨm phenotypes with molecular alterations in MQC pathways |
| Cell Separation Kits | CD235a-/CD45-/CD31- magnetic beads | Isolate specific cell populations from blood/tissue | Enable cell-type-specific ΔΨm assessment in heterogeneous samples |
Experimental Protocols for ΔΨm Assessment
Flow Cytometry-Based ΔΨm Quantification in Peripheral Blood Mononuclear Cells
This protocol provides a standardized approach for ΔΨm measurement in circulating cells, suitable for longitudinal monitoring in clinical trials.
Sample Preparation:
- Collect whole blood in sodium heparin tubes and process within 2 hours.
- Isolate PBMCs using density gradient centrifugation (Ficoll-Paque PLUS).
- Resuspend cells at 1×10^6 cells/mL in pre-warmed assay buffer (HBSS with 10 mM HEPES, pH 7.4).
Staining Procedure:
- Divide cell suspension into experimental and control tubes.
- Load cells with 50 nM TMRE and 100 nM MitoTracker Green for 30 minutes at 37°C in the dark.
- For compensation controls, prepare single-stained tubes for each fluorophore.
- For depolarization control, pre-treat control cells with 20 μM CCCP for 15 minutes before staining.
- Wash cells twice with ice-cold assay buffer and maintain on ice until analysis.
Flow Cytometry Acquisition:
- Acquire data using a flow cytometer equipped with 488 nm excitation and appropriate filter sets (FL2 for TMRE, FL1 for MitoTracker Green).
- Collect a minimum of 10,000 events in the lymphocyte or monocyte gate.
- Use forward and side scatter to exclude debris and aggregates.
Data Analysis:
- Calculate ΔΨm index as the ratio of median TMRE fluorescence to median MitoTracker Green fluorescence.
- Express results relative to CCCP-treated controls to normalize between experiments.
- Include healthy donor samples as reference standards in each experiment.
Imaging-Based ΔΨm Assessment in Cultured Cells
This protocol enables single-cell resolution ΔΨm measurement coupled with morphological analysis.
Cell Preparation:
- Plate cells on glass-bottom dishes at appropriate density 24-48 hours before assay.
- Prior to imaging, replace culture medium with pre-warmed imaging buffer.
Staining and Image Acquisition:
- Load cells with 25 nM TMRM and 50 nM MitoTracker Green for 30 minutes at 37°C.
- For live-cell imaging, maintain temperature at 37°C with 5% CO2 throughout acquisition.
- Acquire images using a confocal microscope with appropriate laser lines and filter sets.
- For each field, capture both TMRM and MitoTracker Green channels with identical acquisition settings across samples.
Image Analysis:
- Segment individual mitochondria using MitoTracker Green signal.
- Calculate mean TMRM intensity for each mitochondrion.
- Normalize TMRM intensity to MitoTracker Green signal on a per-cell basis.
- Compute population statistics including heterogeneity indices.
Regulatory Considerations and Future Directions
The translation of ΔΨm biomarkers from research tools to clinically validated stratification metrics requires careful attention to regulatory frameworks. Analytical validation should follow FDA Bioanalytical Method Validation guidelines, establishing precision, accuracy, linearity, and stability under expected storage conditions. Clinical validation must demonstrate prognostic or predictive value in appropriately powered studies, with defined performance thresholds for stratification decisions.
Future developments will likely focus on non-invasive ΔΨm assessment methodologies, potentially leveraging mitochondrial-specific PET tracers or magnetic resonance spectroscopy approaches. Additionally, the integration of ΔΨm with other mitochondrial parameters into multidimensional quality control indices could provide more comprehensive assessment of mitochondrial health than any single metric alone.
The emerging paradigm of ΔΨm-informed patient stratification represents a convergence of basic mitochondrial biology, technical innovation, and clinical therapeutics. As methodologies mature and validation datasets expand, ΔΨm metrics promise to enhance trial efficiency and therapeutic precision across an expanding spectrum of human diseases.
Conclusion
Mitochondrial membrane potential stands as a central, indispensable regulator of mitochondrial quality control, serving as the critical signal that integrates mitochondrial dynamics with the selective elimination of damaged organelles via mitophagy. The synthesis of knowledge across the four intents confirms that ΔΨm is not merely a bioenergetic parameter but a dynamic signaling hub. Future research must focus on developing more precise tools for in vivo ΔΨm monitoring, unraveling the context-specific outcomes of mitophagy modulation, and advancing the clinical translation of mitochondrial-targeted therapeutics. Bridging the gap between the sophisticated molecular understanding of ΔΨm and its clinical application holds the key to treating a vast spectrum of diseases characterized by mitochondrial dysfunction, from neurodegeneration to metabolic syndromes.
References
- 1. Functional Significance of the Mitochondrial Membrane ... [https://link.springer.com/article/10.1134/S1990747818010129]
- 2. Mitochondrial membrane hyperpolarization modulates ... [https://www.nature.com/articles/s41467-025-59427-5]
- 3. Mechanisms of mitochondria and autophagy crosstalk - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3272286/]
- 4. Mitophagy as a mitochondrial quality control mechanism in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10167083/]
- 5. The pathways of mitophagy for quality control and ... [https://www.nature.com/articles/cdd201281]
- 6. Drug delivery systems for mitochondrial targeting [https://pubs.rsc.org/en/content/articlehtml/2025/nr/d5nr02199e?page=search]
- 7. Best Image Award 2025 Winners Announced [https://wms-site.com/alert-on-mitochondria/1352-best-image-award-2025-winners-announced]
- 8. 2025 Healthcare Trends in Mitochondrial Disease [https://mitocanada.org/2024-healthcare-trends-mitochondrial-disease/]
- 9. Crosstalk between mitochondrial biogenesis and mitophagy to ... [https://jbiomedsci.biomedcentral.com/articles/10.1186/s12929-023-00975-7]
- 10. Molecular mechanisms of mitochondrial quality control - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12400733/]
- 11. Mitochondria at the crossroads: Quality control ... [https://www.sciencedirect.com/science/article/pii/S0969996125000786]
- 12. Mitochondrial Quality Control in Health and Disease - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12356995/]
- 13. Uncoupled turnover disrupts mitochondrial quality control ... [https://insight.jci.org/articles/view/129760]
- 14. Mitochondrial quality control in hematopoietic stem cells [https://stemcellres.biomedcentral.com/articles/10.1186/s13287-025-04304-7]
- 15. Quantitative Analysis of Mitochondrial Morphology and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4289477/]
- 16. Protocol for evaluating mitochondrial morphology changes ... [https://www.sciencedirect.com/science/article/pii/S2666166723007128]
- 17. The molecular mechanisms of mitochondrial dynamics and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12492677/]
- 18. Mitochondrial secrets revealed: UCLA-led team discovers ... [https://cnsi.ucla.edu/september-25-2025-mitochondrial-secrets-revealed-ucla-led-team-discovers-physics-of-how-cells-powerhouse-splits-to-reproduce/]
- 19. ROS-Drp1-mediated mitochondria fission contributes to ... [https://www.sciencedirect.com/science/article/pii/S2213231723001404]
- 20. Mitochondrial fission process 1 protein: a comprehensive ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2025.1646072/full]
- 21. Quantitative analysis of mitochondrial membrane potential ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7871195/]
- 22. Quantitative measurement of mitochondrial membrane ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3448152/]
- 23. Mitophagy and Quality Control Mechanisms in Mitochondrial ... [https://pubmed.ncbi.nlm.nih.gov/29462587/]
- 24. Mitophagy as a mitochondrial quality control mechanism in ... [https://www.sciencedirect.com/science/article/pii/S135581452300038X]
- 25. Mitochondrial quality control: a pathophysiological ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2024.1474310/full]
- 26. The mitophagy pathway and its implications in human ... [https://www.nature.com/articles/s41392-023-01503-7]
- 27. Mitochondrial Quality Control [https://pubmed.ncbi.nlm.nih.gov/40879935/]
- 28. Mechanisms and Functions of Mitophagy and Potential ... [https://www.frontiersin.org/journals/physiology/articles/10.3389/fphys.2020.00935/full]
- 29. Emerging role of mitophagy in human diseases and physiology [https://pmc.ncbi.nlm.nih.gov/articles/PMC5498140/]
- 30. Natural small molecules regulating the mitophagy pathway ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1571767/full]
- 31. Mechanisms of PINK1, ubiquitin and Parkin interactions in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11105328/]
- 32. PINK1- and Parkin-mediated mitophagy at a glance - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3656616/]
- 33. PINK1-mediated phosphorylation of the Parkin ubiquitin- ... [https://www.nature.com/articles/srep01002]
- 34. Review of FUNDC1-mediated mitochondrial autophagy in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12104256/]
- 35. A brief overview of BNIP3L/NIX receptor‐mediated mitophagy - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8634856/]
- 36. Mitophagy mediated by BNIP3 and NIX protects against ferroptosis by downregulating mitochondrial reactive oxygen species - PubMed [https://pubmed.ncbi.nlm.nih.gov/38519771/]
- 37. Reconstitution of BNIP3/NIX-mitophagy initiation reveals ... [https://www.nature.com/articles/s41556-025-01712-y]
- 38. Mitochondrial autophagy: Origins, significance, and role of ... [https://www.sciencedirect.com/science/article/pii/S0167488915000725]
- 39. FUNDC1-induced mitophagy protects spinal cord neurons ... [https://www.nature.com/articles/s41420-023-01780-9]
- 40. Mitochondrial autophagy by Bnip3 involves Drp1-mediated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3213962/]
- 41. Activation of PINK1-Parkin–Mediated Mitophagy Degrades ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6892225/]
- 42. Exercise regulates mitophagy to alleviate parkinsonian ... [https://www.frontiersin.org/journals/aging-neuroscience/articles/10.3389/fnagi.2025.1678460/full]
- 43. Sirtuins in mitophagy: key gatekeepers of mitochondrial quality [https://pmc.ncbi.nlm.nih.gov/articles/PMC12594717/]
- 44. Cellular mitophagy: Mechanism, roles in diseases and ... [https://www.thno.org/v13p0736.htm]
- 45. Mapping the global research landscape of mitophagy in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12632019/]
- 46. New Mitophagy Pathways: Next Therapy Targets? [Sep. 23 ... [https://www.dojindo.com/contents/autophagy_sn20250923.html]
- 47. Researchers find a potential treatment for mitochondrial damage that... [https://www6.slac.stanford.edu/news/2025-05-21-researchers-find-potential-treatment-mitochondrial-damage-causes-disease]
- 48. Advancements in mitochondrial -targeted nanotherapeutics... [https://pubmed.ncbi.nlm.nih.gov/39483479/]
- 49. A Checklist for Successful Quantitative Live Cell Imaging in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3972678/]
- 50. Analysis of mitochondrial membrane potential, ROS, and ... [https://www.sciencedirect.com/science/article/pii/S1016847825000627]
- 51. LDS 698: A highly sensitive probe for tracking ... [https://pubmed.ncbi.nlm.nih.gov/40816051/]
- 52. Mitochondrial Quality Control and Cell Death [https://www.mdpi.com/1422-0067/26/22/11084]
- 53. Role of mitophagy in mitochondrial quality control [https://www.sciencedirect.com/science/article/abs/pii/S1043661821000165]
- 54. Enhancing CNS mitophagy: drug development and ... [https://pubmed.ncbi.nlm.nih.gov/39419743/]
- 55. Chemical mitophagy modulators: Drug development ... [https://www.sciencedirect.com/science/article/pii/S1043661823001913]
- 56. High-Content Analysis (Quantitative Imaging) Information [https://www.thermofisher.com/us/en/home/life-science/cell-analysis/cell-analysis-learning-center/cell-imaging-information/high-content-analysis-information.html]
- 57. Genetically encoded tool for manipulation of ΔΨm identifies its ... [https://pubmed.ncbi.nlm.nih.gov/40250406/]
- 58. Measuring mitochondrial membrane potential | The EMBO ... [https://link.springer.com/article/10.1038/s44318-025-00632-9]
- 59. Genetically encoded tool for manipulation of ΔΨm ... [https://www.sciencedirect.com/science/article/abs/pii/S2451945625000972]
- 60. Genetically encoded tool for manipulation of ΔΨm ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10793441/]
- 61. Electron transport chain inhibitors alter membrane potential ... [https://www.sciencedirect.com/science/article/pii/S0891584918317726]
- 62. Exploring the therapeutic potential of mitochondrial ... [https://www.sciencedirect.com/science/article/pii/S2212877821000661]
- 63. Electron transport chain inhibition increases cellular ... - PubMed [https://pubmed.ncbi.nlm.nih.gov/38876105/]
- 64. Measurement of Mitochondrial Membrane Potential In Vivo ... [https://pubmed.ncbi.nlm.nih.gov/40063520/]
- 65. Toward high-content screening of mitochondrial ... [https://pubmed.ncbi.nlm.nih.gov/25668473/]
- 66. Toward high-content screening of mitochondrial ... [https://www.sciencedirect.com/science/article/abs/pii/S1357272515000308]
- 67. PMI: a ΔΨm independent pharmacological regulator of mitophagy [https://pubmed.ncbi.nlm.nih.gov/25455860/]
- 68. High-Throughput Microscopy Analysis of Mitochondrial ... [https://www.mdpi.com/2073-4409/12/7/1089]
- 69. Frontiers | Effects of fission and mitophagy on mitochondrial... [https://www.frontiersin.org/journals/applied-mathematics-and-statistics/articles/10.3389/fams.2023.1082928/full]
- 70. MitoTox: a comprehensive mitochondrial toxicity database [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-021-04285-3]
- 71. PMI: a ΔΨm independent pharmacological regulator of mitophagy . [https://www.sigmaaldrich.com/SG/en/tech-docs/paper/963000]
- 72. Exploring the Potential of Mitochondria-Targeted Drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11991819/]
- 73. Photo-activation of the delocalized lipophilic cation D112 ... [https://www.nature.com/articles/cddis201719]
- 74. Preparation of mitochondria-targeted camptothecin ... [https://www.sciencedirect.com/science/article/abs/pii/S0968089625002081]
- 75. Mitophagy and Quality Control Mechanisms in ... [https://www.sciencedirect.com/science/article/pii/S096098221830006X]
- 76. Anionic Polymers Promote Mitochondrial Targeting of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7347245/]
- 77. Natural products targeting mitochondria - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC8084865/]
- 78. Natural Compounds Modulating Mitochondrial Functions [https://pmc.ncbi.nlm.nih.gov/articles/PMC4489008/]
- 79. Recent Advances in Marine-Derived Bioactives Towards ... [https://www.mdpi.com/2673-8937/4/4/51]
- 80. Modulators of Mitochondrial Biology Derived from Marine ... [https://encyclopedia.pub/entry/53909]
- 81. Multifunctional Natural Products in Disease Modulation [https://arocjournal.com/journal/multifunctional-natural-products-in-disease-modulation-a-systems-perspective-on-cancer-and-cardiovascular-therapies/]
- 82. Mitochondrial quality control in neurodegenerative diseases [https://pmc.ncbi.nlm.nih.gov/articles/PMC12571580/]
- 83. Elevated mitochondrial membrane potential is a ... [https://www.nature.com/articles/s41467-025-57238-2]
- 84. Identifies Cells with Enhanced... Mitochondrial Membrane Potential [https://pubmed.ncbi.nlm.nih.gov/26674251/]
- 85. Mitochondria in brain diseases: Bridging structural ... [https://www.sciencedirect.com/science/article/pii/S3050562325000078]
- 86. Mitochondrial Transport Proteins in Cardiovascular Diseases [https://pmc.ncbi.nlm.nih.gov/articles/PMC12429345/]
- 87. Mitochondrial membrane potential and compartmentalized ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12454667/]
- 88. Mitochondrial quality control in the brain: The physiological ... [https://www.frontiersin.org/journals/neuroscience/articles/10.3389/fnins.2022.1075141/full]
- 89. Mitophagy: mechanisms, pathophysiological roles, and analysis [https://pmc.ncbi.nlm.nih.gov/articles/PMC3630798/]
- 90. Mitochondrial Quality Control: A Pathophysiological ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8832032/]
- 91. Mitophagy in hepatocytes: Types, initiators and role in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6541449/]
- 92. Molecular mechanisms of mitochondrial quality control [https://translationalneurodegeneration.biomedcentral.com/articles/10.1186/s40035-025-00505-5]
- 93. The Interplay Between Mitochondrial Dynamics and Mitophagy [https://pmc.ncbi.nlm.nih.gov/articles/PMC3078508/]
- 94. DNM1L-mediated fission governs mitophagy & mitochondrial ... [https://biosignaling.biomedcentral.com/articles/10.1186/s12964-025-02142-x]
- 95. Mitochondrial dynamics–fusion, fission, movement, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2758711/]
- 96. Mitochondrial fission and mitophagy are independent ... [https://www.nature.com/articles/s41419-021-03752-2]
- 97. Mitochondrial ROS triggers mitophagy through activating the DNA... [https://pubmed.ncbi.nlm.nih.gov/41026812/]
- 98. Mitophagy: A key regulator in the pathophysiology and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12407537/]
- 99. Mitophagy in Alzheimer's disease and its potential as a ... [https://www.sciencedirect.com/science/article/pii/S0969996125003791]
- 100. Reciprocal Regulation of Mitofusin 2-Mediated Mitophagy ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2022.868465/full]
- 101. The role of mitochondrial dynamics and mitophagy in skeletal ... [https://cmbl.biomedcentral.com/articles/10.1186/s11658-024-00572-y]
- 102. Pharmacokinetic Evaluation of New Drugs Using a Multi ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9953224/]
- 103. Computational approaches to DMPK: A realistic ... [https://www.sciencedirect.com/science/article/pii/S1359644625001357]
- 104. Integrating artificial intelligence into small molecule ... [https://www.nature.com/articles/s44386-025-00029-y]
- 105. Benchmarking compound activity prediction for real-world ... [https://www.nature.com/articles/s42004-024-01204-4]
- 106. Mitochondrial Membrane Potential | AAT Bioquest [https://www.aatbio.com/catalog/mitochondrial-membrane-potential]
- 107. Mitochondrial Quality Control - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3527081/]
- 108. PMI: A ΔΨm Independent Pharmacological Regulator of ... [https://www.sciencedirect.com/science/article/pii/S1074552114003706]
- 109. Mitochondrial Membrane Potential Assay | Sartorius [https://www.sartorius.com/en/applications/life-science-research/live-cell-assays/mitochondrial-membrane-potential]
- 110. Mitochondrial membrane hyperpolarization modulates ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12041266/]
- 111. Mitochondrial Membrane Potential Assay - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9850828/]
- 112. A method to identify and validate mitochondrial modulators ... [https://www.nature.com/articles/srep05285]
- 113. Ferroptosis-related oxidative stress activation in the acute ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1704978/full]
- 114. Calcium and ROS: A mutual interplay - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC4556774/]
- 115. The Complex Interplay between Mitochondria, ROS and ... [https://www.mdpi.com/2076-3921/11/10/1995]
- 116. Mitochondria in oxidative stress, inflammation and aging [https://www.nature.com/articles/s41392-025-02253-4]
- 117. Mitochondrial reactive oxygen species regulate cellular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2933303/]
- 118. Crosstalk between Calcium and Reactive Oxygen Species ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5466514/]
- 119. ROS Function in Redox Signaling and Oxidative Stress - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4055301/]
- 120. Mitophagy and Parkinson's disease: The PINK1–parkin link [https://pmc.ncbi.nlm.nih.gov/articles/PMC3925795/]
- 121. PINK1/PARKIN signalling in neurodegeneration and ... [https://actaneurocomms.biomedcentral.com/articles/10.1186/s40478-020-01062-w]
- 122. Function and Characteristics of PINK1 in Mitochondria - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3600171/]
- 123. Mitochondrial damage triggers the concerted degradation of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12335601/]
- 124. PINK1/Parkin mitophagy and neurodegeneration—what do ... [https://www.sciencedirect.com/science/article/pii/S0959437X16302155]
- 125. Parkinson's Disease Drug Candidates Induce Unexpected ... [https://thisis.caltech.edu/news/parkinsons-disease-drug-candidates-induce-unexpected-damaging-effects]
- 126. Mitophagy in Hypertensive Cardiac Hypertrophy [https://pmc.ncbi.nlm.nih.gov/articles/PMC12358939/]
- 127. Mitophagy: Basic Mechanism and Potential Role in Kidney ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4934814/]
- 128. Regulates Kidney Diseases Mitophagy [https://pubmed.ncbi.nlm.nih.gov/39664332/]
- 129. Mitochondrial dysfunction and mitophagy blockade ... [https://www.sciencedirect.com/science/article/pii/S0085253825000857]
- 130. Dysfunctional Mitochondria Clearance in Situ: Mitophagy in Obesity... [https://www.e-dmj.org/journal/view.php?number=2823]
- 131. Mitochondrial ultrastructural pathology in diabetic cardiomyopathy... [https://cardiab.biomedcentral.com/articles/10.1186/s12933-025-02884-5]
- 132. Mitochondrial Dynamics and Mitophagy in Cardiometabolic ... [https://www.frontiersin.org/journals/cardiovascular-medicine/articles/10.3389/fcvm.2022.917135/full]
- 133. Cellular mitophagy: Mechanism, roles in diseases and small ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9830443/]
- 134. The molecular mechanisms of mitochondrial dynamics ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-025-07078-x]
- 135. Idebenone: Clinical Potential Beyond Neurological Diseases [https://pmc.ncbi.nlm.nih.gov/articles/PMC12433227/]
- 136. The effects of idebenone on mitochondrial bioenergetics [https://www.sciencedirect.com/science/article/pii/S000527281100257X]
- 137. Current Status and Future Prospects of Idebenone in ... [https://clinmedjournals.org/articles/ijsrp/international-journal-of-surgery-research-and-practice-ijsrp-6-102.php]
- 138. MitoQ regulates autophagy by inducing a pseudo ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5388232/]
- 139. Overview of MitoQ on prevention and management of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11934253]
- 140. Mitochondria-targeted antioxidant MitoQ radiosensitizes ... [https://www.nature.com/articles/s41420-024-02277-9]
- 141. MitoQ Protects Against Oxidative Stress-Induced ... [https://www.sciencedirect.com/science/article/pii/S2772976125001886]
- 142. Mitochondrial membrane potential [https://pubmed.ncbi.nlm.nih.gov/28711444/]
- 143. Mitophagy in Cancer: A Tale of Adaptation - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC6562743/]
- 144. Mitophagy: insights into its signaling molecules, biological ... [https://www.nature.com/articles/s41420-024-02226-6]
- 145. Mitophagy in Alzheimer's disease: Molecular defects and ... [https://www.nature.com/articles/s41380-022-01631-6]
- 146. Mitophagy and mitochondrial dynamics in type 2 diabetes ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9004550/]
- 147. in Brain Injuries: Mechanisms, Mitophagy , and Therapeutic... Roles [https://link.springer.com/article/10.1007/s12035-025-04936-z]
- 148. Investigating the interplay between mitophagy and diabetic ... [https://www.sciencedirect.com/science/article/pii/S1043661824003396]
- 149. Mitophagy regulates integrity of mitochondria at synapses and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7507095/]
- 150. Mitochondrial diseases: from molecular mechanisms to ... [https://www.nature.com/articles/s41392-024-02044-3]
- 151. Overview of methods that determine mitochondrial function ... [https://www.sciencedirect.com/science/article/pii/S0026049525001696]
- 152. Pharmacological agents affecting mitophagy and ... [https://www.oaepublish.com/articles/2574-1209.2022.20]
- 153. Mitochondrial Dynamics in Blood Cancer Development and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12553585/]
- 154. Identification and validation of mitochondrial-related genes ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2025.1691749/full]
- 155. Biotechnological approaches and therapeutic potential of ... [https://www.nature.com/articles/s41467-025-61239-6]
- 156. Mitochondrial quality control and transfer communication in ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1542369/full]
- 157. Review of electrophysiological models to study membrane ... [https://www.frontiersin.org/journals/physiology/articles/10.3389/fphys.2025.1536165/full]
- 158. AI-guided patient stratification improves outcomes and ... [https://www.nature.com/articles/s41467-025-61355-3]
- 159. Targeting Mitochondrial Dynamics via EV Delivery in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12609785/]
- 160. The Future of Clinical Trials: Incorporating 'Omics for ... [https://blog.crownbio.com/the-future-of-clinical-trials-incorporating-omics-for-enhanced-stratification]