MOMP: Mitochondrial Outer Membrane Permeabilization in Apoptosis - Mechanisms, Analysis, and Therapeutic Targeting
This article provides a comprehensive analysis of mitochondrial outer membrane permeabilization (MOMP), a pivotal event in the intrinsic apoptotic pathway.
MOMP: Mitochondrial Outer Membrane Permeabilization in Apoptosis - Mechanisms, Analysis, and Therapeutic Targeting
Abstract
This article provides a comprehensive analysis of mitochondrial outer membrane permeabilization (MOMP), a pivotal event in the intrinsic apoptotic pathway. Tailored for researchers, scientists, and drug development professionals, it synthesizes foundational knowledge on the core machinery—primarily the Bcl-2 protein family—with advanced methodological approaches for its detection. The content delves into resolving key controversies in the field, compares established and emerging models of pore formation, and critically evaluates MOMP as a therapeutic target in diseases like cancer. By integrating foundational concepts with current research and technical applications, this review serves as a vital resource for advancing both basic science and therapeutic innovation centered on this 'point of no return' in cell death.
The Core Machinery of MOMP: Bcl-2 Proteins and the Point of No Return
Mitochondrial outer membrane permeabilization (MOMP) is universally recognized as the decisive commitment point in the intrinsic pathway of apoptosis [1] [2]. This event, once triggered, leads to the irreversible release of pro-apoptotic proteins from the mitochondrial intermembrane space into the cytosol, thereby activating the caspase cascade that executes cell death [3]. The regulation of MOMP is primarily governed by the complex interactions between members of the B-cell lymphoma 2 (Bcl-2) protein family [4] [5]. Its pivotal role in cellular fate has made MOMP a significant focus in fundamental apoptosis research and a promising therapeutic target in drug development, particularly for oncology [5]. This whitepaper provides an in-depth technical examination of MOMP, detailing its core mechanisms, regulatory systems, and the experimental approaches used to study it, framed within the broader context of apoptosis research.
The Central Mechanism of MOMP
The Point of No Return in Apoptosis
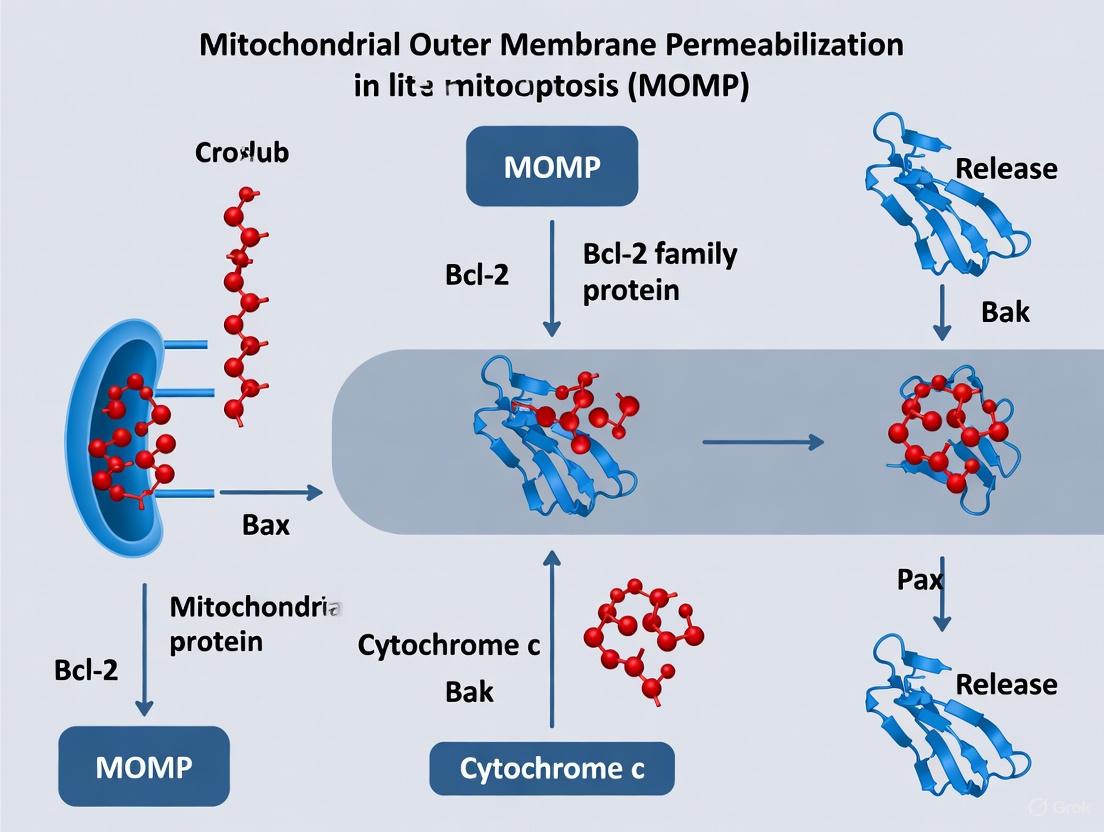
MOMP constitutes a profound change in the permeability of the mitochondrial outer membrane (MOM). Under normal conditions, this membrane is impermeable to proteins larger than approximately 5 kDa [2]. During MOMP, the formation of large pores enables the efflux of soluble proteins from the mitochondrial intermembrane space, such as cytochrome c (∼15 kDa), into the cytosol [3] [2]. This release is rapid and synchronized; time-lapse imaging studies reveal that once initiated, MOMP can permeabilize nearly all mitochondria within a cell within a remarkably short span of 5 to 10 minutes [3].
The release of cytochrome c is the defining biochemical event of MOMP. Once in the cytosol, cytochrome c binds to the protein APAF-1 (apoptotic protease-activating factor-1) in the presence of dATP/ATP. This binding triggers a conformational change in APAF-1, exposing its nucleotide-binding site and oligomerization domain, leading to the assembly of a multi-protein complex known as the apoptosome [3]. The apoptosome serves as a activation platform for the initiator caspase, caspase-9, which in turn activates the executioner caspases (e.g., caspase-3, -6, -7) that systematically dismantle the cell [3] [6].
Other proteins released alongside cytochrome c during MOMP amplify the death signal. A key example is SMAC/DIABLO, which neutralizes a class of cytosolic proteins known as Inhibitor of Apoptosis Proteins (IAPs), such as XIAP. By inhibiting XIAP, SMAC ensures that the activation of caspases proceeds unhindered [3].
The BCL-2 Protein Family: Architects of MOMP
The Bcl-2 family of proteins are the principal arbiters of MOMP, integrating diverse cellular stress signals to determine whether to permeabilize the MOM [4] [5]. This protein family is structurally defined by the presence of Bcl-2 Homology (BH) domains and can be functionally categorized into three groups:
- Multi-domain anti-apoptotic proteins (e.g., BCL-2, BCL-XL, MCL-1): These proteins contain four BH domains (BH1-4) and are characterized by a hydrophobic groove that serves as a binding site for other family members. Their primary function is to preserve mitochondrial integrity and prevent MOMP [4] [5].
- Multi-domain pro-apoptotic effector proteins (e.g., BAX, BAK): These proteins contain BH1-3 domains and are directly responsible for executing MOMP. In response to activation signals, they oligomerize within the MOM, forming pores that facilitate the release of cytochrome c and other proteins [4] [5].
- BH3-only pro-apoptotic proteins (e.g., BIM, BID, PUMA, BAD, NOXA): These proteins sense various intracellular damage signals (e.g., DNA damage, ER stress) and act as molecular messengers that initiate the apoptotic cascade. They function by either directly activating BAX/BAK or by neutralizing the anti-apoptotic proteins [4] [5].
Table 1: Core Components of the BCL-2 Protein Family Regulating MOMP
| Group | Example Proteins | Key Domains | Primary Function in MOMP |
|---|---|---|---|
| Anti-apoptotic | BCL-2, BCL-XL, MCL-1 | BH1, BH2, BH3, BH4 | Inhibit BAX/BAK activation and pore formation; promote cell survival. |
| Pro-apoptotic Effectors | BAX, BAK | BH1, BH2, BH3 | Form permeabilizing pores in the mitochondrial outer membrane upon activation. |
| BH3-only Sensitizers | BAD, NOXA, BIK | BH3 | Neutralize anti-apoptotic proteins, indirectly promoting BAX/BAK activation. |
| BH3-only Activators | BIM, tBID, PUMA | BH3 | Directly bind and activate BAX/BAK to induce MOMP. |
The following diagram illustrates the core interactions within the BCL-2 family that lead to MOMP:
Diagram 1: BCL-2 Family Interactions Leading to MOMP. Cellular stress activates BH3-only proteins, which either directly activate BAX/BAK or neutralize anti-apoptotic proteins, freeing BAX/BAK to form pores in the mitochondrial outer membrane.
Key Models of BCL-2 Family Regulation
The precise mechanisms governing the interactions between BCL-2 family proteins have been the subject of extensive research, leading to several non-mutually exclusive models. These models attempt to explain how the balance between pro- and anti-apoptotic signals is tipped in favor of MOMP.
The Direct Activation and Displacement Models
The Direct Activation Model posits a critical distinction among BH3-only proteins. "Activator" BH3-only proteins (like BIM and tBID) directly bind to and conformationally change BAX and BAK, triggering their activation and oligomerization. "Sensitizer" BH3-only proteins (like BAD and NOXA) promote apoptosis by binding to and sequestering anti-apoptotic proteins, thereby freeing the activators to engage BAX and BAK [4].
In contrast, the Displacement Model suggests that BAX and Bak are constitutively active but are kept in check through constant inhibition by anti-apoptotic proteins. In this model, BH3-only proteins function primarily to displace BAX and BAK from these anti-apoptotic "guardians," thereby unleashing their pore-forming potential [4].
The Embedded Together and Unified Models
More recent models incorporate the critical role of cellular membranes as the central arena for these interactions. The Embedded Together Model emphasizes that the localization and conformation of BCL-2 family proteins at the mitochondrial membrane dictate their interactions and affinities for one another. For instance, the interaction with the membrane can induce conformational changes in proteins like BAX and BCL-XL, altering their binding capabilities and promoting the activation cascade at the site of action [4].
Building on this, the Unified Model proposes that anti-apoptotic proteins suppress MOMP through two distinct modes: by sequestering activator BH3-only proteins (Mode 1) and by directly binding and inhibiting the active forms of BAX and BAK already embedded in the membrane (Mode 2). This model also begins to link the regulation of MOMP with other mitochondrial processes, such as dynamics and fission [4].
Table 2: Comparative Overview of Major BCL-2 Family Interaction Models
| Model | Proposed Mechanism of BAX/BAK Activation | Role of BH3-only Proteins | Key Insight |
|---|---|---|---|
| Direct Activation | Direct binding and activation by "activator" BH3-only proteins. | Activators (e.g., Bim, tBid) directly activate BAX/BAK. Sensitizers (e.g., Bad, Noxa) inhibit anti-apoptotic proteins. | Distinguishes two functional classes of BH3-only proteins. |
| Displacement | Displacement from anti-apoptotic proteins, releasing constitutively active BAX/BAK. | Primarily to displace BAX/BAK from their anti-apoptotic inhibitors. | BAX/BAK are constitutively active and require constant inhibition. |
| Embedded Together | Activation occurs at the membrane, governed by conformational changes and local concentrations. | Can activate BAX/BAK and/or neutralize anti-apoptotic proteins; function is reversible and dependent on membrane context. | The membrane is the active locus; protein conformations and affinities are membrane-dependent. |
| Unified | Relief from inhibition via two modes: release of activators (Mode 1) and release of membrane-embedded BAX/BAK (Mode 2). | Overcome dual inhibition by anti-apoptotic proteins. | Links MOMP regulation to mitochondrial dynamics; proposes two modes of anti-apoptotic action. |
Experimental Approaches for Studying MOMP
Key Methodologies and Workflows
Investigating the dynamic and complex process of MOMP requires a combination of classical biochemical techniques and advanced imaging technologies.
1. Cytochrome c Release Assays: This is a foundational experiment for quantifying MOMP. Isolated mitochondria are incubated with recombinant BCL-2 family proteins (e.g., activated tBID, BAX) or pharmacological agents. After centrifugation, the supernatant (released fraction) and the mitochondrial pellet are analyzed by immunoblotting for cytochrome c. The appearance of cytochrome c in the supernatant directly indicates MOMP has occurred [1] [7].
2. BH3 Profiling: This functional assay evaluates the cellular "priming" for apoptosis by measuring mitochondrial sensitivity to synthetic BH3 peptides. Cells are permeabilized, exposed to different BH3 peptides (e.g., from BIM, BAD, NOXA), and the loss of mitochondrial membrane potential or cytochrome c release is measured. The pattern of response indicates which anti-apoptotic proteins the cell is dependent on for survival and how close it is to the apoptotic threshold [4] [5].
3. Single-Molecule Analysis: Advanced techniques like single-molecule fluorescence are used to dissect the stoichiometry, kinetics, and assembly of BAX/BAK oligomers in artificial membranes or isolated mitochondria. This approach allows researchers to observe the formation of individual pores and measure their properties, providing insights that are masked in ensemble experiments [8].
The generalized workflow for a core MOMP experiment is outlined below:
Diagram 2: Generalized Experimental Workflow for MOMP Analysis. A typical protocol involves isolating mitochondria or treating cells, applying a pro-apoptotic stimulus, and then using fractionation and immunoblotting or other assays to quantify the occurrence of MOMP.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for MOMP and Apoptosis Research
| Reagent / Tool | Category | Primary Function in Research |
|---|---|---|
| Recombinant BCL-2 Proteins (e.g., BAX, BID, BCL-2) | Protein | Used in in vitro reconstitution assays (e.g., with isolated mitochondria) to define specific protein functions and interactions in MOMP. |
| BH3 Peptides (e.g., BIM BH3, BAD BH3) | Peptide | Synthetic peptides used in BH3 profiling to interrogate mitochondrial priming and dependencies on specific anti-apoptotic proteins. |
| BH3-Mimetics (e.g., ABT-199/Venetoclax, ABT-737) | Small Molecule Inhibitor | Potent and specific small molecules that bind the hydrophobic groove of anti-apoptotic BCL-2 proteins, used to probe biological function and as therapeutic agents. |
| Cytochrome c Antibody | Antibody | Essential for immunoblotting and immunofluorescence to detect cytochrome c localization (mitochondrial vs. cytosolic) as a direct readout for MOMP. |
| SMAC/DIABLO Mimetics (e.g., Birinapant) | Small Molecule Inhibitor | Compounds that mimic the N-terminal of SMAC, used to antagonize IAP proteins and study caspase amplification post-MOMP. |
Clinical and Therapeutic Implications
The central role of MOMP and the BCL-2 family in controlling cell death has made them attractive targets for therapeutic intervention, especially in cancer, where apoptosis is often evaded [5].
The most significant success in this field has been the development of BH3-mimetics. These are small, drug-like molecules designed to occupy the hydrophobic groove of anti-apoptotic BCL-2 proteins, thereby mimicking the action of endogenous sensitizer BH3-only proteins [5]. Venetoclax (ABT-199), a highly selective BCL-2 inhibitor, has demonstrated remarkable efficacy in certain hematologic malignancies like chronic lymphocytic leukemia (CLL) and acute myeloid leukemia (AML), leading to its clinical approval [5]. However, targeting other anti-apoptotic members like BCL-XL and MCL-1 has proven more challenging due to on-target toxicities; inhibiting BCL-XL can cause thrombocytopenia, while MCL-1 inhibition has been linked to cardiac complications [5]. Novel strategies such as PROTACs and antibody-drug conjugates are being explored to achieve more tumor-specific targeting and overcome these limitations [5].
MOMP stands as a definitive commitment step in the intrinsic apoptotic pathway, a tightly regulated process governed by the intricate and dynamic interactions of the BCL-2 protein family. Understanding the precise molecular details of how BAX and BAK are activated to form pores, and how this process is inhibited by pro-survival members, has been greatly advanced by sophisticated biochemical, cellular, and single-molecule techniques. This deep mechanistic knowledge has successfully transitioned from basic research to the clinic, with BH3-mimetics like venetoclax validating the BCL-2 family as a druggable target. Future research will continue to refine our models of MOMP regulation, explore its connections to other cellular processes, and develop next-generation therapeutics to manipulate this critical checkpoint in cell fate for the treatment of cancer and other diseases.
The B-cell lymphoma 2 (Bcl-2) family of proteins constitutes the essential regulatory system governing mitochondrial outer membrane permeabilization (MOMP), a decisive event in the intrinsic pathway of apoptosis [9] [10]. This protein family integrates diverse cellular stress signals to determine whether a cell will live or die by controlling the release of cytochrome c and other apoptogenic factors from the mitochondrial intermembrane space [5] [11]. The founding member, BCL2, was first identified in 1984 as the gene translocated in follicular lymphoma, representing the first example of an oncogene that promotes cancer by inhibiting cell death rather than stimulating proliferation [5]. Since this discovery, the Bcl-2 family has expanded to include approximately 25 members in mammals, all characterized by the presence of Bcl-2 homology (BH) domains [12]. These proteins function as a tripartite apoptotic switch that maintains tissue homeostasis, enables developmental tissue sculpting, and eliminates damaged or dangerous cells [5]. Dysregulation of this family contributes fundamentally to cancer pathogenesis and resistance to therapy, making its members compelling targets for pharmaceutical intervention [5] [13].
Bcl-2 Family Structure and Classification
The Bcl-2 family proteins are structurally defined by the presence of up to four α-helical Bcl-2 homology (BH) domains (BH1-BH4), which mediate interactions between family members and regulate their apoptotic functions [12] [13]. These proteins share an evolutionarily conserved structure and are found in most metazoans, highlighting their fundamental role in regulated cell death [12]. Based on their function and domain architecture, the Bcl-2 family is divided into three functional subgroups:
Table 1: Classification of Principal Bcl-2 Family Proteins
| Subgroup | Representative Members | BH Domains Present | Primary Function |
|---|---|---|---|
| Anti-apoptotic | BCL2, BCL-XL, BCL-W, MCL1, BFL-1 | BH1, BH2, BH3, BH4 (all four) | Inhibit MOMP by sequestering pro-apoptotic members |
| Multi-domain Pro-apoptotic | BAX, BAK, BOK | BH1, BH2, BH3 | Directly execute MOMP through oligomerization |
| BH3-only Pro-apoptotic | BIM, BID, BAD, PUMA, NOXA | BH3 only | Initiate apoptosis by sensing stress and antagonizing anti-apoptotic proteins |
The anti-apoptotic proteins, which possess all four BH domains, localize to the outer mitochondrial membrane (OMM) where they prevent membrane permeabilization [5] [12]. Their structure features a characteristic hydrophobic groove formed by the BH1, BH2, and BH3 domains, which serves as the primary interaction site for the BH3 domains of pro-apoptotic partners [5] [12]. The multi-domain pro-apoptotic executioners BAX and BAK normally exist in an inactive conformation but undergo conformational activation during apoptosis, leading to their membrane insertion and oligomerization [10]. The BH3-only proteins function as sentinels that respond to specific cellular damage signals through transcriptional upregulation or post-translational activation, initiating the apoptotic cascade [5] [12].
Molecular Mechanisms of Membrane Permeabilization
The Central Role of MOMP in Apoptosis
Mitochondrial outer membrane permeabilization (MOMP) represents the "point of no return" in the intrinsic apoptotic pathway [10]. Once MOMP occurs, cytochrome c is released from the mitochondrial intermembrane space into the cytosol, where it triggers the formation of the apoptosome complex and subsequent activation of caspase-9 and the downstream caspase cascade [5] [11]. This irreversible commitment to cell death makes MOMP a critical control point regulated by the balanced interactions between pro- and anti-apoptotic Bcl-2 family members [9] [10].
The Bcl-2 family governs MOMP through a complex network of protein-protein interactions that ultimately determine whether the pro-apoptotic executioners BAX and BAK become activated [9]. Current models propose that BH3-only proteins both neutralize anti-apoptotic members and directly activate BAX and BAK, with both functions being necessary for efficient MOMP engagement [9]. Anti-apoptotic proteins maintain mitochondrial integrity by sequestering activated BH3-only proteins and preventing BAX/BAK activation [5].
Mechanisms of Pore Formation
The precise molecular mechanism by which BAX and BAK permeabilize the mitochondrial outer membrane remains an area of active investigation, with several proposed models:
Direct Pore Formation: Activated BAX and BAK undergo conformational changes that enable them to oligomerize and form large pores in the OMM sufficient to allow passage of cytochrome c and other intermembrane space proteins [10] [14]. Structural studies reveal that BAX and BAK share homology with pore-forming bacterial toxins like diphtheria toxin, supporting this direct pore-forming capability [12].
Regulation of Mitochondrial Channels: Bcl-2 family proteins may regulate pre-existing mitochondrial channels, particularly the voltage-dependent anion channel (VDAC) [11] [15]. Anti-apoptotic BCL2 can interact with VDAC to decrease channel conductance and prevent cytochrome c release, while pro-apoptotic members may modulate VDAC to facilitate release [12] [15].
Permeability Transition Pore Involvement: Some evidence suggests Bcl-2 proteins may coordinate permeability of both mitochondrial membranes through the permeability transition (PT) pore, a multi-protein complex that forms at contact sites between the inner and outer membranes [11] [15].
The following diagram illustrates the core regulatory network of Bcl-2 family proteins in controlling MOMP:
Diagram Title: Bcl-2 Protein Regulation of Mitochondrial Apoptosis
Experimental Approaches for Studying Bcl-2 Function
Key Methodologies and Assays
Research into Bcl-2 family function employs a diverse array of biochemical, biophysical, and cell biological techniques to elucidate the complex interactions and mechanisms governing MOMP. The following experimental protocols represent cornerstone methodologies in the field.
BH3 Profiling Assay
Purpose: To measure mitochondrial priming and determine dependence on specific anti-apoptotic Bcl-2 family proteins [5].
Procedure:
- Isolate mitochondria from cells of interest
- Expose mitochondria to synthetic BH3 peptides representing specific BH3-only proteins (e.g., BAD peptide for BCL2/BCL-XL dependence, NOXA peptide for MCL1 dependence)
- Quantify cytochrome c release using ELISA or western blotting
- Measure membrane potential changes using JC-1 or TMRE fluorescent dyes
- Analyze data to determine pattern of anti-apoptotic protein dependence
Applications: Predicting sensitivity to specific BH3-mimetic drugs, identifying mechanisms of resistance, and profiling tumor cell dependencies [5].
Cytochrome c Release Assay
Purpose: To directly measure MOMP in response to Bcl-2 family protein interactions [11].
Procedure:
- Prepare heavy membrane fraction enriched in mitochondria
- Incubate with recombinant Bcl-2 family proteins (e.g., activated BID, BAX)
- Separate mitochondrial and supernatant fractions by centrifugation
- Detect cytochrome c in supernatant by immunoblotting
- Quantify release relative to positive controls (e.g., alamethicin treatment)
Applications: Testing direct effects of Bcl-2 family proteins on MOMP, screening for BH3-mimetic efficacy, and studying regulatory mechanisms [11] [15].
Research Reagent Solutions
The following table summarizes essential experimental tools and reagents used in Bcl-2 family research:
Table 2: Key Research Reagents for Bcl-2 Family Studies
| Reagent Category | Specific Examples | Research Application | Key Function |
|---|---|---|---|
| BH3-Mimetic Compounds | ABT-737, ABT-263 (Navitoclax), ABT-199 (Venetoclax) | Functional inhibition of anti-apoptotic Bcl-2 proteins | Bind hydrophobic groove of anti-apoptotic proteins to displace pro-apoptotic partners |
| Recombinant Proteins | Full-length and truncated Bcl-2 family proteins (e.g., BCL-XLΔTM, BAX) | Structural studies and in vitro reconstitution assays | Enable biochemical characterization of protein-protein interactions |
| Synthetic BH3 Peptides | BAD-like, BIM-like, NOXA-like peptides | BH3 profiling and mitochondrial priming assays | Identify specific anti-apoptotic dependencies in cells |
| Genetic Models | Bcl-2 family knockout mice, CRISPR/Cas9 editing | Study physiological functions and validate drug targets | Establish essential roles in development and tissue homeostasis |
| Structural Biology Tools | X-ray crystallography, NMR spectroscopy, Cryo-EM | Determine atomic-level structures of Bcl-2 family members | Reveal molecular mechanisms of protein interactions and drug binding |
Therapeutic Targeting of Bcl-2 Proteins
BH3-Mimetic Drug Development
The structural characterization of Bcl-2 family interactions has enabled rational drug design targeting the hydrophobic groove of anti-apoptotic proteins. BH3-mimetics are small molecules that structurally mimic the BH3 domain of pro-apoptotic proteins, competitively inhibiting anti-apoptotic family members and promoting apoptosis in cancer cells [5] [13]. The development of these therapeutics represents a milestone in translating basic apoptosis research into clinical applications.
The first generation BH3-mimetic, navitoclax (ABT-263), demonstrated efficacy in hematological malignancies but caused dose-limiting thrombocytopenia due to BCL-XL inhibition [5]. This led to the development of venetoclax (ABT-199), a BCL2-selective inhibitor that received FDA approval in 2016 and has transformed treatment for chronic lymphocytic leukemia and acute myeloid leukemia [5] [13]. Subsequent BH3-mimetics targeting other anti-apoptotic family members, including MCL1 and BCL-XL inhibitors, are undergoing clinical evaluation [5].
Advanced Targeting Strategies
Recent advances in Bcl-2 targeting include novel approaches to overcome limitations of conventional BH3-mimetics:
PROTACs (Proteolysis Targeting Chimeras): Bifunctional molecules designed to selectively degrade target proteins by recruiting ubiquitin ligases, offering potential advantages in efficacy and overcoming resistance [5].
Antibody-Drug Conjugates (ADCs): Enable selective delivery of Bcl-2 inhibitors to tumor cells expressing specific surface markers, potentially mitigating on-target toxicities [5].
BH4 Domain Targeting: Emerging approaches focused on the N-terminal BH4 domain, which is critical for the anti-apoptotic function of BCL2 and BCL-XL [5].
The following diagram illustrates the development timeline and specificity of key BH3-mimetic therapeutics:
Diagram Title: Evolution of BH3-Mimetic Therapeutics
The Bcl-2 family represents a master regulatory system that governs mitochondrial membrane integrity through complex protein interactions that ultimately control MOMP. From its initial discovery as a chromosomal translocation in lymphoma to the current development of sophisticated targeted therapies, research on this protein family exemplifies successful translation of basic molecular mechanisms into clinical applications. While remarkable progress has been made in understanding the structural basis of Bcl-2 family function and developing BH3-mimetic drugs, challenges remain in targeting anti-apoptotic members like MCL1 and BCL-XL without dose-limiting toxicities. Emerging technologies including PROTACs, antibody-drug conjugates, and BH4-domain targeting offer promising approaches to expand the therapeutic potential of Bcl-2 modulation. As research continues to elucidate the nuanced mechanisms of mitochondrial permeabilization and its regulation, targeting the Bcl-2 family holds continued promise for improving cancer therapy and treating other diseases characterized by apoptotic dysregulation.
Mitochondrial outer membrane permeabilization (MOMP) constitutes the pivotal "point of no return" in the intrinsic apoptotic pathway, primarily mediated by the effector proteins Bax and Bak. These pro-apoptotic Bcl-2 family members undergo complex activation and oligomerization processes to form pores in the mitochondrial outer membrane, facilitating the release of cytochrome c and other apoptogenic factors that culminate in cellular destruction. This whitepaper synthesizes current mechanistic understanding of Bax and Bak function, detailing their structural transformations, oligomerization dynamics, and the emerging concept of tunable pore architecture. We present quantitative analyses of their distinct assembly properties, standardized experimental methodologies for studying membrane permeabilization, and visualization of key molecular pathways. For drug development professionals, these insights reveal critical regulatory nodes for therapeutic intervention in cancer and other diseases characterized by apoptotic dysregulation.
Mitochondrial outer membrane permeabilization (MOMP) is the decisive event in the intrinsic apoptotic pathway, irreversibly committing the cell to death [16] [17]. During MOMP, the mitochondrial outer membrane, typically permeable only to molecules smaller than 5 kDa, becomes permeable to proteins larger than 100 kDa [17]. This permeability change allows the release of cytochrome c, Smac/DIABLO, and other intermembrane space proteins into the cytosol, triggering caspase activation and proteolytic cellular dismantlement [16] [18]. The Bcl-2 protein family tightly regulates this process through three functional subgroups: (1) pro-survival proteins (e.g., Bcl-2, Bcl-xL, Mcl-1), (2) initiator BH3-only proteins (e.g., Bid, Bim, Puma), and (3) effector proteins Bax and Bak [16] [18]. Genetic evidence firmly establishes that either Bax or Bak is essential for MOMP, with cells deficient in both proteins exhibiting complete resistance to most intrinsic apoptotic stimuli [16] [19].
Molecular Mechanisms of Bax and Bak Activation
Conformational Activation and Membrane Integration
Bax and Bak undergo substantial conformational changes during activation from inactive forms to membrane-embedded oligomers. Bax primarily resides in the cytosol or loosely associates with mitochondria in healthy cells, while Bak is constitutively integrated into the mitochondrial outer membrane [16] [18]. Activation involves exposure of N-terminal epitopes and the BH3 domain, which becomes accessible for protein-protein interactions [16]. For Bax, activation also entails translocation to mitochondria, where its C-terminal transmembrane domain inserts into the lipid bilayer [16]. This insertion is facilitated by interactions with other Bcl-2 family proteins, particularly activated BH3-only proteins like truncated Bid (tBid) [17].
The BH3:Groove Interaction in Oligomerization
A critical step in Bax and Bak activation involves exposure of the BH3 domain, which subsequently binds to the hydrophobic surface groove of another activated Bax or Bak molecule, forming symmetric homodimers [16]. This BH3:groove interface represents a fundamental mechanism for propagating the oligomerization process. Inhibition of BH3 domain exposure—through mutagenesis or antibody binding—effectively blocks further oligomerization and prevents MOMP [16]. The crystallographic symmetry of this interaction suggests a universal dimerization mechanism that may extend to other Bcl-2 family protein interactions.
Figure 1: Bax and Bak Activation Pathway. Both proteins undergo conformational changes leading to BH3 domain exposure, symmetric dimer formation, and higher-order oligomerization culminating in mitochondrial outer membrane permeabilization (MOMP).
Oligomerization and Pore Formation Dynamics
Distinct Assembly Properties of Bax and Bak
Recent super-resolution microscopy reveals that Bax and Bak form similar line, arc, and ring-shaped oligomeric structures but with distinct assembly characteristics and kinetics [19] [20]. BAK organizes into smaller structures with faster kinetics, while BAX forms larger assemblies that grow more slowly but continue expanding during apoptosis [19]. These structural differences have functional consequences for pore properties and release kinetics of mitochondrial components.
Table 1: Comparative Oligomerization Properties of Bax and Bak
| Property | Bax | Bak |
|---|---|---|
| Initial cellular localization | Cytosolic | Mitochondrial |
| Oligomerization kinetics | Slower | Faster |
| Average ring radius | 34 nm | 18 nm |
| Structure size distribution | Broader | Narrower |
| Assembly maturation | Continues growing | Reaches stable size faster |
Cooperative Assembly in Apoptotic Pores
Bax and Bak do not function in isolation but co-assemble into mixed oligomers during apoptosis [19] [20]. BAK accelerates BAX recruitment and assembly, while BAX incorporation enables continued growth of oligomeric structures [19]. This cooperative interaction creates a regulatory mechanism where the relative abundance of Bax and Bak determines pore growth dynamics and the size selectivity of the permeabilization barrier [19]. The emerging model suggests that BAK nucleates smaller pores that subsequently incorporate BAX to form larger permeabilization structures.
Proteolipidic Pore Nature and Size Regulation
Bax and Bak form proteolipidic pores whose size depends on protein concentration rather than forming fixed proteinaceous channels [21]. At low protein concentrations, pores permit cytochrome c (12 kDa) release, while higher concentrations enable passage of larger proteins like allophycocyanin (104 kDa) [21]. This concentration-dependent pore expansion demonstrates a tunable permeability mechanism rather than a binary open/closed state. Cardiolipin content influences pore formation propensity but does not affect ultimate pore size, which is determined exclusively by Bax/Bak concentration [21].
Quantitative Analysis of Pore Properties
Advanced imaging and biophysical techniques have enabled precise quantification of Bax/Bak oligomeric structures and functional properties. Single-molecule localization microscopy (SMLM) reveals that approximately 40% of BAK assemblies in apoptotic cells form lines, arcs, and rings, with the remainder appearing as dots and aggregates [19]. Atomic force microscopy (AFM) confirms that both arcs and rings associate with membrane pores, directly linking these structures to permeabilization function [19].
Table 2: Structural and Functional Metrics of Bax/Bak Pores
| Parameter | Value | Measurement Technique | Biological Significance |
|---|---|---|---|
| BAK ring radius | 18 nm | SMLM | Determines initial pore size |
| BAX ring radius | 34 nm | SMLM | Enables larger pore formation |
| Pore size range | Cytochrome c (12 kDa) to APC (104 kDa) | GUV permeabilization | Controls release of mitochondrial proteins |
| Pore stability | Long-lived (>45 minutes) | GUV time-course experiments | Ensures irreversible commitment to death |
| MOMP completion time | ~5 minutes per cell | Live-cell imaging | Coordinates cellular self-destruction |
Experimental Methodologies for Studying Bax/Bak Pores
Single Vesicle Permeabilization Assay
Purpose: To visualize pore formation and size selectivity at the individual membrane level [21]. Protocol:
- GUV Preparation: Create giant unilamellar vesicles (GUVs) by electroformation using mitochondrial membrane-mimetic lipid composition (49% PC, 27% PE, 10% PI, 10% PS, 4% cardiolipin) in 300 mM sucrose [21].
- Protein Activation: Incubate Bax or BakΔC21 with cBid (1:10 molar ratio) to generate activated proteins [21].
- Permeabilization Measurement: Mix activated proteins with GUVs in PBS buffer containing fluorescent reporters of different sizes (e.g., Cytochrome c-Alexa488, 12 kDa; allophycocyanin, 104 kDa) [21].
- Image Acquisition and Analysis: Use confocal microscopy to monitor dye influx into individual GUVs over time. Calculate degree of filling using the formula:
Filling (%) = (F_tin - F_tout) / (F_0in - F_0out) × 100, where Ftin and Ftout are fluorescence intensities inside and outside the GUV at time t [21]. Key Applications: Determining pore size selectivity, kinetics of pore formation, and concentration-dependent effects.
Super-Resolution Imaging of Oligomeric Structures
Purpose: To characterize nanoscale organization of Bax and Bak in apoptotic cells [19]. Protocol:
- Cell Culture and Apoptosis Induction: Use BAX/BAK double-knockout HCT116 cells reconstituted with mEGFP-tagged BAK or BAX. Induce apoptosis with 1 μM ABT-737 plus 1 μM S63845 [19].
- Fixation: Fix cells at timepoint when 50% have undergone MOMP (typically 3 hours post-induction) using 4% formaldehyde [19].
- SMLM Imaging: Perform single-molecule localization microscopy with photoactivatable fluorescent proteins. Acquire 10,000-20,000 frames for sufficient localization density [19].
- Cluster Analysis: Use automated structures analysis program (ASAP) to classify and quantify oligomeric architectures (lines, arcs, rings) based on spatial distribution patterns [19]. Key Applications: Comparing oligomer size distributions, structural polymorphisms, and protein-specific assembly properties.
Research Reagent Solutions
Table 3: Essential Research Tools for Bax/Bak Investigation
| Reagent/Cell Line | Function/Application | Key Features |
|---|---|---|
| Bax/Bak DKO HCT116 | Genetic background for reconstitution studies | Resistant to intrinsic apoptotic stimuli |
| ABT-737 | BH3 mimetic inhibitor of Bcl-2/Bcl-xL/Bcl-w | Synergizes with MCL-1 inhibitors for apoptosis induction |
| S63845 | MCL-1-specific inhibitor | Enables complete anti-apoptotic blockade when combined with ABT-737 |
| cBid | Direct activator of Bax and Bak | Generates truncated, active Bid through caspase-8 cleavage |
| BakΔC21 | Recombinant constitutively active Bak | C-terminal truncation facilitates in vitro pore formation studies |
| Cytochrome c-Alexa488 | Reporter for small pore formation (12 kDa) | Fluorescently labeled protein for real-time permeability assessment |
| Allophycocyanin (APC) | Reporter for large pore formation (104 kDa) | Natural fluorescent protein for assessing pore expansion |
Functional Consequences and Pathophysiological Implications
Regulation of Inflammatory Signaling
The size dynamics of Bax/Bak pores directly influence inflammatory outcomes by controlling mitochondrial DNA (mtDNA) release [19] [20]. Larger pores, favored by BAX-dominated oligomers, permit efficient mtDNA efflux, activating the cGAS/STING pathway and promoting paracrine inflammatory signaling [19] [20]. This mechanism connects apoptotic pore properties to immunogenic cell death and has implications for cancer therapy and inflammatory disease.
Therapeutic Targeting Opportunities
The essential role of Bax and Bak in apoptosis execution makes them attractive targets for cancer therapy. BH3-mimetic drugs (e.g., ABT-263/Navitoclax, ABT-199/Venetoclax) indirectly activate Bax/Bak by displacing them from pro-survival Bcl-2 proteins [17] [18]. However, functional or genetic loss of Bax/Bak confers resistance to such therapies, necessitating alternative approaches like Raptinal, which induces MOMP independently of Bax/Bak [22].
Figure 2: Functional Consequences of BAX/BAK Co-Assembly. BAK initiates smaller oligomers that recruit BAX, forming mixed complexes that determine pore size and subsequent inflammatory signaling through differential release of mitochondrial contents.
Bax and Bak function as the essential executioners of MOMP through a sophisticated pore formation process involving coordinated activation, oligomerization, and membrane remodeling. Their distinct but complementary assembly properties enable dynamic regulation of pore size, with implications for both apoptotic efficiency and inflammatory signaling. Quantitative biophysical approaches continue to reveal unexpected complexities in their oligomerization states and functional interactions. For therapeutic development, targeting the Bax/Bak activation pathway remains promising, though bypass strategies may be necessary for cancers with defective effector mechanisms. Future research should focus on structural characterization of full-length pores, single-molecule dynamics in native membranes, and tissue-specific variations in Bax/Bak regulation to fully exploit these proteins for therapeutic benefit.
The BCL-2 protein family constitutes the fundamental regulatory network that controls the mitochondrial pathway of apoptosis, with BH3-only proteins serving as essential initiators that sense and integrate diverse cellular death signals [23] [24]. These specialized proteins, characterized by containing only a single BCL-2 homology 3 (BH3) domain, function as crucial sentinels that propagate both extrinsic and intrinsic cell death signals by engaging with other BCL-2 family members at the mitochondrial outer membrane [23] [24]. The precise mechanism by which BH3-only proteins initiate mitochondrial outer membrane permeabilization (MOMP)—the pivotal event in intrinsic apoptosis—has been the subject of extensive research and debate, culminating in several mechanistic models that explain their activator and sensitizer functions [23] [9] [25].
Within the broader context of MOMP regulation, BH3-only proteins occupy an apical position in the apoptotic cascade, translating various forms of cellular stress into the commitment to cell death [23] [17]. Their activity is counterbalanced by anti-apoptotic BCL-2 family proteins, and the delicate equilibrium between these opposing factions determines cellular fate [5] [25]. This whitepaper examines the classification, mechanisms, and experimental methodologies for investigating the dual activator-sensitizer functions of BH3-only proteins, providing researchers and drug development professionals with a comprehensive technical resource for understanding these critical regulators of programmed cell death.
Classification of BH3-only Proteins by Function
BH3-only proteins are functionally categorized based on their molecular interactions with other BCL-2 family members and their capacity to directly induce MOMP [25] [26] [27]. This classification divides them into direct activators and sensitizers (also known as de-repressors), each with distinct binding profiles and mechanistic roles in apoptosis initiation.
Table 1: Functional Classification of BH3-Only Proteins
| Category | Representative Members | Primary Mechanism | Binding Targets |
|---|---|---|---|
| Direct Activators | Bid, Bim, Puma | Directly activate Bax/Bak; Displace anti-apoptotics | Bcl-2, Bcl-xL, Mcl-1; Bax/Bak |
| Sensitizers | Bad, Noxa, Bik, Bmf, Hrk | Neutralize anti-apoptotic proteins | Selective anti-apoptotics (e.g., Bad→Bcl-2/Bcl-xL; Noxa→Mcl-1) |
The direct activators (including Bid, Bim, and Puma) possess the capability to directly engage and conformationally activate the pro-apoptotic effector proteins Bax and Bak, triggering their oligomerization and integration into the mitochondrial outer membrane [25] [26]. These activators are also capable of binding to anti-apoptotic family members, thereby displacing sequestered pro-apoptotic proteins [23] [24]. In contrast, the sensitizer proteins (including Bad, Noxa, Bmf, Bik, and Hrk) function primarily by selectively engaging and neutralizing specific anti-apoptotic BCL-2 family proteins, thereby liberating direct activators or pre-activated Bax/Bak to initiate MOMP [25] [26] [27].
This functional specialization enables a sophisticated regulatory system where different death signals can be channeled through specific BH3-only proteins to achieve precise control over the apoptotic commitment [23] [24]. The combinatorial interplay between multiple BH3-only proteins allows for graded apoptotic responses and signal integration, as demonstrated by recent genetic studies showing that hepatocyte apoptosis in the absence of key anti-apoptotic proteins requires the collaborative action of several BH3-only members [26].
Molecular Mechanisms of BH3-only Protein Function
The molecular interactions governing BH3-only protein function center on the binding of their amphipathic α-helical BH3 domain to the hydrophobic groove of anti-apoptotic BCL-2 family proteins or to activation sites on Bax/Bak [23] [25]. This molecular recognition event initiates a cascade of protein conformational changes and interactions that ultimately decide cellular fate.
The Direct Activation Model
The direct activation model proposes that a subset of BH3-only proteins (direct activators) can conformationally activate Bax and Bak through physical interaction [26] [27]. According to this model, activator proteins including Bid, Bim, and Puma engage directly with Bax and/or Bak, inducing conformational changes that facilitate their insertion into the mitochondrial outer membrane and subsequent oligomerization into apoptotic pores [25] [26]. Sensitizer proteins in this model function by binding to anti-apoptotic proteins and displacing sequestered activators, which are then free to directly engage Bax/Bak [26].
The Indirect Activation Model
The indirect activation model posits that BH3-only proteins function exclusively by neutralizing anti-apoptotic BCL-2 family members rather than through direct Bax/Bak activation [26]. In this model, all BH3-only proteins are considered sensitizers that displace Bax/Bak from anti-apoptotic sequestration or inhibit anti-apoptotic proteins that normally constrain spontaneously activating Bax/Bak [26]. Recent research supporting this model suggests that the mitochondrial membrane itself may serve as the primary activator of Bax/Bak oligomerization once they are liberated from anti-apoptotic control [26].
Unified Model and Emerging Understanding
Current evidence suggests that both models likely represent aspects of a more complex reality, with BH3-only proteins employing both direct and indirect mechanisms to ensure robust apoptosis induction when appropriate [9] [26]. The emerging unified model proposes that BH3-only proteins must perform both functions—neutralizing anti-apoptotic proteins and directly promoting Bax/Bak activation—to efficiently engage MOMP [9]. This integrated perspective acknowledges that the relative importance of direct versus indirect mechanisms may vary by cell type, death signal, and cellular context [9] [26].
Table 2: Quantitative Apoptosis Induction by BH3-Only Protein Disruption in Hepatocyte-Specific Knockout Mice
| Genetic Model | Serum ALT Reduction | Caspase 3/7 Activity Reduction | TUNEL-Positive Hepatocyte Reduction | Key Findings |
|---|---|---|---|---|
| Mcl-1ΔHep/ΔHep Puma−/− | Significant decrease | Significant decrease | Significant decrease | Puma disruption suppresses hepatocyte apoptosis |
| Bcl-xLΔHep/ΔHep Puma−/− | Significant decrease | Significant decrease | Significant decrease | Puma involved in Bak/Bax activation |
| Mcl-1ΔHep/+ Bcl-xLΔHep/ΔHep Bid−/− Bim−/− | Partial protection | Partial protection | Partial protection | Severe apoptosis persists |
| Mcl-1ΔHep/+ Bcl-xLΔHep/ΔHep Bid−/− Bim−/− Puma−/− | Enhanced protection but incomplete | Enhanced protection but incomplete | Enhanced protection but incomplete | Additional Puma disruption provides incremental benefit |
| Mcl-1iΔHep/iΔHep Bcl-xLiΔHep/iΔHep Bid−/− Bim−/− Puma−/− Noxa−/− | Significant protection | Significant protection | Significant protection | Noxa disruption alleviates hepatocyte apoptosis and prolongs survival |
The unified model is supported by genetic evidence demonstrating that combined disruption of multiple BH3-only proteins—including Bid, Bim, Puma, and Noxa—progressively reduces but does not completely eliminate hepatocyte apoptosis in mice lacking key anti-apoptotic proteins, indicating that these BH3-only proteins collaboratively orchestrate Bak/Bax activation [26]. This functional redundancy ensures robust apoptosis induction while allowing signal-specific responses through different BH3-only protein activation patterns.
Experimental Approaches for Studying BH3-only Protein Functions
Investigating the activator and sensitizer functions of BH3-only proteins requires a multifaceted experimental approach combining genetic manipulation, biochemical analysis, and functional apoptosis assays. The following protocols represent key methodologies for elucidating the specific roles of these proteins in apoptotic signaling.
Genetic Knockout and Knockdown Approaches
Protocol: Genetic Disruption of BH3-only Proteins in Murine Models
- Design targeting vectors for conditional or complete knockout of specific BH3-only genes (e.g., Puma, Noxa, Bid, Bim)
- Generate transgenic mice with floxed alleles for hepatocyte-specific deletion using Alb-Cre or similar tissue-specific promoters
- Cross breeding to create single, double, and triple knockout combinations to assess functional redundancy
- Validate gene disruption through Western blotting of liver tissue lysates to confirm protein ablation
- Monitor phenotypic consequences through serum ALT measurements, caspase 3/7 activity assays, and TUNEL staining to quantify apoptosis
- Assess Bax/Bak activation through immunoprecipitation with conformation-specific antibodies (e.g., Bax 6A7 antibody that recognizes activated Bax)
This approach has revealed that while single BH3-only protein knockouts provide partial protection against apoptosis in specific contexts, combined disruption of multiple BH3-only proteins (Bid, Bim, Puma, and Noxa) is necessary to substantially attenuate hepatocyte apoptosis in the absence of anti-apoptotic proteins [26].
BH3 Profiling and Mitochondrial Assays
Protocol: BH3 Profiling to Assess Mitochondrial Priming
- Isolate mitochondria from target cells or tissues through differential centrifugation
- Permeabilize mitochondrial membranes with digitonin to allow controlled access to intracellular compartments
- Incubate with synthetic BH3 peptides representing different BH3-only proteins (e.g., Bid, Bim, Bad, Noxa peptides)
- Measure mitochondrial response through cytochrome c release by ELISA or Western blotting, or via mitochondrial membrane potential dyes (e.g., JC-1, TMRE)
- Quantify results to determine "mitochondrial priming" - the susceptibility to specific apoptotic signals based on BH3-only protein interactions
BH3 profiling enables researchers to map the dependencies of specific cancer cells on individual anti-apoptotic proteins, predicting sensitivity to BH3-mimetic drugs and providing functional classification of BH3-only proteins as activators or sensitizers based on their ability to directly induce cytochrome c release [17].
Protein Interaction Studies
Protocol: Co-immunoprecipitation for BH3-only Protein Interactions
- Prepare cell lysates from appropriate model systems under non-denaturing conditions
- Incubate lysates with antibodies specific to BH3-only proteins or anti-apoptotic targets
- Capture immune complexes using protein A/G beads
- Wash complexes extensively to remove non-specific interactions
- Elute bound proteins and analyze by Western blotting for co-precipitating BCL-2 family members
- Quantify interactions under different conditions (e.g., with/without apoptotic stimuli)
This approach allows researchers to determine binding specificities between BH3-only proteins and their anti-apoptotic targets, confirming the classification of proteins like Noxa as selective Mcl-1 binders, while Bad preferentially binds Bcl-2 and Bcl-xL [25] [27].
Research Reagent Solutions Toolkit
Table 3: Essential Research Reagents for Investigating BH3-only Protein Functions
| Reagent Category | Specific Examples | Research Application |
|---|---|---|
| Genetic Models | Mcl-1ΔHep/ΔHep mice; Bcl-xLΔHep/ΔHep mice; Puma−/− mice; Bid−/− Bim−/− Puma−/− Noxa−/− multiple KO mice | In vivo analysis of BH3-only protein functions and functional redundancy |
| Cell Lines | Immortalized primary hepatocytes with floxed Mcl-1/Bcl-xL alleles; Bax/Bak DKO cells | Controlled studies of BH3-only protein interactions and mechanisms |
| BH3 Peptides | Synthetic Bid BH3, Bim BH3, Bad BH3, Noxa BH3 peptides | BH3 profiling to determine mitochondrial priming and dependencies |
| Activation-State Antibodies | Bax 6A7 antibody (conformation-specific) | Detection of activated, oligomerized Bax in mitochondrial fractions |
| Apoptosis Detection Kits | TUNEL assay kits; Caspase 3/7 activity assays; Cytochrome c release ELISA kits | Quantification of apoptosis induction in response to BH3-only protein activation |
| BH3-Mimetic Compounds | ABT-737 (Bcl-2/Bcl-xL/Bcl-w inhibitor); ABT-199/Venetoclax (Bcl-2 selective); A-1331852 (Bcl-xL selective); S63845 (Mcl-1 inhibitor) | Experimental tools to mimic sensitizer BH3-only protein function |
Visualizing BH3-only Protein Signaling Pathways
Diagram 1: BH3-only Protein Signaling in MOMP Regulation. This pathway illustrates how cellular stress signals activate BH3-only proteins, which function as either sensitizers that neutralize anti-apoptotic proteins or direct activators that engage effector proteins, leading to mitochondrial outer membrane permeabilization and apoptosis.
The functional classification of BH3-only proteins into activators and sensitizers represents a fundamental framework for understanding the initiation of mitochondrial apoptosis. While mechanistic debates continue regarding the precise molecular events, it is clear that these proteins collaborate to interpret death signals and overcome anti-apoptotic restraints through both direct Bax/Bak activation and indirect displacement mechanisms [9] [26]. The experimental approaches outlined in this technical guide provide researchers with robust methodologies for investigating these proteins in specific physiological and pathological contexts.
The significance of understanding BH3-only protein functions extends to therapeutic applications, particularly in oncology, where BH3-mimetic drugs designed to mimic sensitizer functions have demonstrated remarkable clinical efficacy [5]. As research continues to elucidate the complex interactions and functional redundancies among BH3-only proteins, new opportunities will emerge for targeting specific apoptotic vulnerabilities in cancer and other diseases characterized by dysregulated cell survival.
The mitochondrial outer membrane permeabilization (MOMP) serves as a critical point of no return in intrinsic apoptosis, enabling the release of cytochrome c and other pro-apoptotic factors into the cytosol. While Bcl-2 family proteins have long been recognized as central regulators of MOMP, emerging evidence establishes the voltage-dependent anion channel (VDAC) as an equally crucial player in this process. This technical review synthesizes recent structural and mechanistic insights revealing how VDAC oligomerization contributes to apoptosis induction through novel pathways that both complement and extend beyond traditional Bcl-2-centric models. We examine the conformational changes in VDAC1's N-terminal domain that enable interaction with anti-apoptotic Bcl-2 proteins, the formation of large oligomeric pores capable of mitochondrial membrane permeabilization, and the unanticipated role of VDAC in coordinating inflammatory cell death pathways. The experimental data, quantitative analyses, and methodological frameworks presented herein provide researchers with essential tools for investigating and targeting VDAC-mediated apoptosis in therapeutic contexts.
The voltage-dependent anion channel (VDAC), situated in the outer mitochondrial membrane (OMM), traditionally functions as the primary gateway for metabolic exchange between mitochondria and cytosol, facilitating ATP/ADP flux, Ca²⁺ transport, and metabolite passage [28] [29]. Of the three mammalian isoforms (VDAC1, VDAC2, and VDAC3), VDAC1 has emerged as a critical regulator of mitochondrion-mediated apoptosis, with its oligomerization status serving as a molecular switch between cell survival and death [30] [31].
While the Bcl-2 protein family remains the canonical system for MOMP regulation, VDAC1 oligomerization represents a parallel pathway that responds to mitochondrial stress or damage, including oxidative stress, altered lipid composition, increased Ca²⁺ levels, and low pH [28]. This oligomerization creates large conductance channels (40 nm to 1 µm in diameter) that facilitate MOMP and the subsequent release of apoptogenic proteins such as cytochrome c, AIF, and Smac/Diablo [30] [32]. The discovery that VDAC1 oligomerization can trigger not only apoptosis but also more complex inflammatory cell death pathways like PANoptosis underscores its significance as a therapeutic target in cancer, neurodegenerative diseases, and retinal disorders [33] [34].
Structural Basis of VDAC Oligomerization
VDAC1 Architecture and N-Terminal Dynamics
VDAC1 adopts a β-barrel structure composed of 19 antiparallel β-strands, with an N-terminal α-helix (VDAC1-N) positioned horizontally within the pore interior in its native state [28] [35]. This N-terminal domain exhibits remarkable conformational flexibility, serving as a critical regulatory element that transitions between embedded and exposed states in response to apoptotic stimuli.
Table 1: Structural Elements of VDAC1 and Their Functional Roles
| Structural Element | Composition | Native State | Function in Apoptosis |
|---|---|---|---|
| N-terminal domain | 26 residues, α-helical | Inside pore lumen | Exposes BH3-like motif upon oligomerization; interacts with Bcl-xL |
| β-barrel | 19 antiparallel β-strands | Forms metabolite-passing pore | Oligomerization interface for large pore formation |
| Glycine-rich motif | ²¹GYGFG²⁵ | Connects N-terminus to barrel | Provides flexibility for N-terminal translocation |
| Cysteine residues | C127, C232 | Structural stability | Oxidation promotes oligomerization under oxidative stress |
The mobility of the N-terminal region is facilitated by a conserved glycine-rich sequence (²¹GYGFG²⁵) that connects this domain to the first β-strand of the barrel, providing the structural flexibility necessary for its translocation [35]. Under apoptotic conditions, this N-terminal α-helix undergoes exposure to the pore exterior, becoming available for partner protein binding—a conformational shift that represents a fundamental switch in VDAC1's function from metabolite transport to apoptosis regulation [28].
Oligomerization Triggers and Molecular Interactions
VDAC1 oligomerization is promoted by multiple apoptotic stimuli, including detergents like cholate, negatively charged lipids (e.g., POPG), oxidative stress, increased Ca²⁺ levels, and low pH [28]. The resulting oligomeric assemblies range from dimers and trimers to higher-order structures that form large pores in the OMM, with cross-linking experiments demonstrating the formation of very large VDAC1 oligomers under apoptotic conditions [28] [30].
The structural characterization of VDAC1 in circularized lipid nanodiscs using cryo-EM has revealed distinct conformational states where the N-terminal α-helix is either bound inside the pore or exposed to the exterior, with the latter state enabling interaction with Bcl-2 family proteins [28]. Crystallographic and NMR data have further elucidated how the exposed VDAC1 N-terminal domain forms a complex with the BH3-binding groove of the anti-apoptotic protein Bcl-xL, effectively neutralizing its anti-apoptotic function [28].
Figure 1: Molecular Mechanism of VDAC1 Oligomerization in Apoptosis Induction. The pathway illustrates how apoptotic stimuli trigger VDAC1 oligomerization, leading to N-terminal domain exposure, Bcl-xL binding, and subsequent mitochondrial outer membrane permeabilization (MOMP).
Mechanisms of Apoptosis Induction via VDAC Oligomerization
VDAC1-N as a BH3-Mimetic Sensitizer
The exposed N-terminal domain of oligomerized VDAC1 (VDAC1-N) exhibits functional mimicry of BH3-only sensitizer proteins, directly binding to the BH3-binding groove of Bcl-xL and thereby neutralizing its anti-apoptotic activity [28]. This interaction liberates the pro-apoptotic effector Bak from its inhibitory complex with Bcl-xL, enabling Bak-mediated pore formation and MOMP execution [28].
Biochemical assays demonstrate that VDAC1-N promotes pore formation by Bak through this neutralization mechanism, effectively bypassing the need for traditional BH3-only proteins to initiate apoptosis [28]. This VDAC1-dependent pathway operates alongside canonical Bcl-2 family regulation, providing an alternative route for apoptosis induction under conditions of mitochondrial stress.
Oligomeric Pore Formation and Mitochondrial Membrane Permeabilization
VDAC1 oligomerization creates large pores in the OMM with sufficient diameter to permit the passage of folded proteins such as cytochrome c (diameter ~3.4 nm), which cannot traverse the ~3.0 nm pore of VDAC1 monomers [30]. These oligomeric assemblies form protein-conducting channels that facilitate the release of multiple apoptogenic factors, including cytochrome c, AIF, and Smac/Diablo, from the mitochondrial intermembrane space [32].
Table 2: Quantitative Analysis of VDAC Oligomerization in Apoptosis
| Parameter | Monomeric VDAC | Oligomeric VDAC | Measurement Method |
|---|---|---|---|
| Pore diameter | 1.5-3.0 nm | 40 nm - 1 µm | Electron microscopy, conductance measurements [28] [30] |
| Cytochrome c release | Not permitted | Permitted | Western blotting, immunofluorescence [30] [32] |
| Oligomerization increase during apoptosis | Baseline | Up to 20-fold | Cross-linking + Western blot, BRET [30] |
| Effect of VDAC1-N exposure on Bcl-xL binding | No binding | Kd ~nM range | NMR titration, crystallography [28] |
The formation of these oligomeric channels represents a regulated process that responds to specific apoptotic stimuli rather than nonspecific membrane disruption. Evidence supporting this includes the inhibition of VDAC oligomerization and subsequent cytochrome c release by compounds such as DIDS, ruthenium red, and HK-I, all of which interact directly with VDAC1 [32].
Cross-Talk with Bcl-2 Family Proteins
VDAC1 interacts with multiple Bcl-2 family members beyond Bcl-xL, including Bak, Bax, and Bcl-2 itself [35]. These interactions position VDAC1 at the interface between metabolic regulation and apoptosis execution, with the anti-apoptotic proteins Bcl-2 and Bcl-xL potentially exerting their protective effects partly through modulation of VDAC1 oligomerization [35].
Notably, different VDAC isoforms exhibit distinct roles in apoptosis regulation. While VDAC1 promotes apoptosis, VDAC2 demonstrates anti-apoptotic properties and is crucial for Bak recruitment to mitochondria, highlighting the complex interplay between VDAC isoforms and Bcl-2 family members in determining cellular fate [36] [35].
Experimental Assessment of VDAC Oligomerization
Methodological Approaches
Researchers employ multiple complementary techniques to investigate VDAC oligomerization and its functional consequences in apoptosis:
Chemical Cross-linking: Cell-permeable cross-linkers such as ethylene glycol bis(succinimidylsuccinate) (EGS) or bis(sulfosuccinimidyl)suberate (BS3) stabilize protein-protein interactions in intact cells or isolated mitochondria. Subsequent Western blot analysis using VDAC-specific antibodies reveals dimeric (~72 kDa), trimeric, and higher molecular weight oligomeric species [30] [31]. This approach demonstrated that apoptosis induction by STS, H₂O₂, or selenite augments VDAC oligomerization several-fold [32].
Bioluminescence Resonance Energy Transfer (BRET): BRET technology enables direct monitoring of VDAC1 oligomerization dynamics in living cells. VDAC1 is tagged with either Renilla luciferase (RLuc) as an energy donor or a variant of GFP (GFP2) as an acceptor. Energy transfer between these tags occurs only when VDAC1 molecules are in close proximity (<10 nm), indicating oligomer formation [31]. This method has been validated using known apoptosis inducers (e.g., selenite) and inhibitors (e.g., DNDS) [31].
Cysteine Accessibility Assays: VDAC1 variants with single cysteine substitutions at specific positions (e.g., T6C) enable monitoring of conformational changes through chemical modification with maleimide-polyethyleneglycol reagents (e.g., PM40, 40 kDa). Increased modification efficiency indicates exposure of the N-terminal region during oligomerization [28].
Structural Approaches: Cryo-EM of VDAC1 in lipid nanodiscs of different sizes, NMR spectroscopy, and X-ray crystallography provide high-resolution structural information about VDAC1 oligomers and their interactions with binding partners such as Bcl-xL [28].
Figure 2: Experimental Workflow for Assessing VDAC Oligomerization. The diagram outlines key methodological approaches for investigating VDAC oligomerization, from sample preparation through various detection and analysis techniques.
Research Reagent Solutions
Table 3: Essential Research Reagents for VDAC Oligomerization Studies
| Reagent/Category | Specific Examples | Function/Application | Key Findings Enabled |
|---|---|---|---|
| Chemical Cross-linkers | EGS, BS3, DFDNB | Stabilize protein complexes for oligomer detection | Demonstrated 20-fold increase in VDAC oligomers during apoptosis [30] [32] |
| VDAC1 Mutants | E73V, T6C, L10C | Study structure-function relationships | E73V showed reduced oligomerization; T6C revealed N-terminal exposure [28] |
| Oligomerization Inhibitors | VBIT-3, VBIT-4, VBIT-12, DNDS | Specifically block VDAC1 oligomerization | VBIT compounds prevent cytochrome c release and apoptosis [31] [34] |
| Apoptosis Inducers | Staurosporine, selenite, As₂O₃, H₂O₂ | Trigger VDAC oligomerization | Different inducers all promote VDAC oligomerization [30] [32] |
| BRET Components | VDAC1-Luc, VDAC1-GFP2 | Live-cell monitoring of oligomerization | Real-time quantification of VDAC1 oligomer dynamics [31] |
| Structural Biology | Lipid nanodiscs, cryo-EM, NMR | High-resolution structural analysis | Revealed VDAC1-N terminal exposure mechanism [28] |
Pathophysiological Implications and Therapeutic Targeting
Disease Associations
VDAC1 oligomerization has been implicated in multiple pathological conditions. In cancer, VDAC1 overexpression and enhanced oligomerization contribute to apoptosis resistance, while simultaneously presenting a vulnerability that can be exploited therapeutically [33] [35]. In neurodegenerative diseases such as Alzheimer's and Parkinson's, excessive VDAC1 oligomerization may promote neuronal apoptosis [28] [31]. Recent research has also identified VDAC1 oligomerization as a key driver of PANoptosis (integrated pyroptosis, apoptosis, and necroptosis) in age-related macular degeneration (AMD) via mtDNA release and STING pathway activation [34].
The O-GlcNAcylation of VDAC1 at threonine 165 has been identified as a specific post-translational modification that enhances oligomerization and promotes disease progression in AMD models, revealing a potential regulatory mechanism for controlling VDAC1 oligomerization in pathological conditions [34].
Therapeutic Strategies and Experimental Compounds
Emerging therapeutic approaches target VDAC1 oligomerization to modulate apoptosis in various disease contexts:
VDAC1 Oligomerization Inhibitors: Compounds such as VBIT-3, VBIT-4, and VBIT-12 directly interact with VDAC1 to prevent oligomerization. These agents protect against apoptosis-associated mitochondrial dysfunction by restoring dissipated mitochondrial membrane potential, decreasing ROS production, and preventing cytochrome c release [31] [34]. In AMD models, VBIT-12 treatment preserved mitochondrial integrity, suppressed mtDNA release, and inhibited PANoptosis, restoring RPE function [34].
Metabolic Modulators: Agents like erastin and betulinic acid induce VDAC opening to reverse the Warburg effect in cancer cells, promoting cell death through ferroptosis and apoptosis pathways [33].
VDAC1-Based Peptides: Peptides derived from the VDAC1 N-terminal domain can disrupt interactions with anti-apoptotic proteins like Bcl-xL, showing potential as anti-cancer therapeutics by preventing VDAC1-mediated apoptosis suppression [35].
VDAC oligomerization represents a crucial apoptosis induction mechanism that operates alongside and interacts with the canonical Bcl-2 family protein regulation. The structural insights revealing VDAC1-N terminal exposure upon oligomerization and its subsequent interaction with Bcl-xL provide a mechanistic foundation for understanding how mitochondrial stress transitions to commitment to cell death. The experimental methodologies and research reagents detailed in this review offer researchers comprehensive tools for further investigating this pathway. As therapeutic targeting of VDAC oligomerization progresses, particularly with specific inhibitors like the VBIT compounds, the potential for treating apoptosis-related diseases continues to expand, highlighting VDAC as an essential component of the mitochondrial cell death machinery beyond its traditional metabolic functions.
Advanced Techniques for Monitoring and Quantifying MOMP Dynamics
Biosensors and Live-Cell Imaging for Real-Time MOMP Visualization
Mitochondrial outer membrane permeabilization (MOMP) is a decisive event in the intrinsic apoptotic pathway, serving as a point of no return for programmed cell death. This process is characterized by increased permeability of the mitochondrial outer membrane, allowing proteins such as cytochrome c to escape from the intermembrane space into the cytosol, where they activate caspase proteases and trigger apoptotic execution [37] [25]. As a crucial node in cell death regulation, MOMP represents a promising therapeutic target, particularly in oncology, where its inhibition contributes to cancer cell survival [37]. Understanding the dynamics and regulation of MOMP is therefore essential for both basic cell biology and therapeutic development.
Real-time visualization of MOMP presents significant technical challenges due to the rapid and often heterogeneous nature of the process within cell populations. Recent advances in biosensor design and live-cell imaging methodologies have transformed our ability to monitor MOMP with high spatiotemporal resolution, revealing complex kinetic behaviors and subcellular heterogeneities that were previously obscured in population-level analyses [38] [39]. This technical guide comprehensively outlines the current state of MOMP visualization techniques, providing researchers with practical methodologies for capturing and quantifying this critical cellular event.
Molecular Mechanisms of MOMP
BCL-2 Protein Family Regulation
MOMP is primarily governed by the balanced interactions between pro-apoptotic and anti-apoptotic members of the BCL-2 protein family [25]. The pro-apoptotic effectors BAX and BAK undergo activation upon cellular stress, transitioning from cytosolic monomers to mitochondrial membrane-embedded oligomers that facilitate pore formation [40]. This activation is triggered by BH3-only proteins (such as BID, BIM, and PUMA), while anti-apoptotic members (including BCL-2, BCL-XL, and MCL-1) sequester these activators and effectors to prevent MOMP [37] [25].
The following diagram illustrates the key regulatory steps in BAX activation and pore formation during MOMP:
MOMP Heterogeneity and Sublethal Signaling
MOMP is not a uniform "all-or-nothing" process throughout the mitochondrial network. The concept of minority MOMP (miMOMP) describes instances where only a subset of mitochondria undergo permeabilization under sublethal stress conditions [37]. This incomplete MOMP can lead to non-apoptotic outcomes including cellular senescence, inflammatory responses, and genomic instability due to limited caspase activation and DNA damage [37]. miMOMP represents an important physiological mechanism contributing to tumor development, drug resistance, and other pathological conditions, highlighting the significance of single-cell analysis for detecting these heterogeneous cellular responses.
Biosensors for MOMP Detection
Fluorescent Protein-Based Reporters
Genetically encoded biosensors utilizing fluorescent proteins (FPs) provide powerful tools for visualizing MOMP dynamics in live cells. The most established approach involves tagging cytochrome c with green fluorescent protein (cyt c-GFP), which displays a characteristic transition from punctate mitochondrial patterns to diffuse cytosolic distribution upon MOMP [39]. This release typically occurs within approximately 5 minutes irrespective of the apoptotic stimulus, demonstrating the conserved kinetics of this process [39].
Recent advances in biosensor engineering have substantially improved dynamic range and spectral properties. Chemogenetic FRET pairs combining fluorescent proteins with fluorophore-labeled HaloTag proteins enable near-quantitative FRET efficiencies (≥94%), allowing creation of highly sensitive biosensors with tunable spectral characteristics [41]. This platform, designated "ChemoX," permits multiplexed monitoring of multiple cellular parameters simultaneously by selecting different FP-fluorophore combinations throughout the visible spectrum [41].
Synthetic Fluorophores and Chemical Tags
Synthetic dyes offer complementary approaches for monitoring MOMP-associated changes:
Tetramethylrhodamine esters (TMRE/TMRM) accumulate in active mitochondria based on membrane potential (ΔΨm) and exhibit fluorescence loss upon MOMP due to ΔΨm dissipation [39] [42]. While often used as a proxy for MOMP, careful validation is required as ΔΨm loss can occur through other mechanisms.
Tetracysteine tags (12-amino acid motifs that bind biarsenical dyes FlAsH and ReAsH) provide a minimal genetic tag for monitoring protein localization without the bulk of FPs [39]. This approach has been successfully used to track cytochrome c release and other mitochondrial events, though background staining can present challenges requiring optimized destaining protocols [39].
The table below summarizes key biosensors used for MOMP detection:
Table 1: Biosensors for MOMP Detection and Mitochondrial Function
| Biosensor/Reporter | Detection Method | Target Process | Key Features | Limitations |
|---|---|---|---|---|
| cyt c-GFP | Fluorescence redistribution | Cytochrome c release | Direct MOMP indicator; well-established | Large tag may affect function |
| ChemoX FRET pairs | FRET efficiency | Conformational changes/ proximity | Near-quantitative FRET (≥94%); spectrally tunable | Requires optimized fusion constructs |
| TMRE/TMRM | Fluorescence intensity | Mitochondrial membrane potential (ΔΨm) | ΔΨm-sensitive; widely used | Indirect MOMP indicator; potential phototoxicity |
| Tetracysteine tags (FlAsH/ReAsH) | Fluorescence redistribution | Protein localization | Small genetic tag; suitable for small proteins | Background staining; requires optimization |
Live-Cell Imaging Methodologies
Single-Cell Time-Lapse Imaging
Live-cell imaging of MOMP requires careful experimental design to maintain cell viability while capturing rapid, dynamic processes. The following protocol outlines key considerations for time-lapse imaging of MOMP:
Cell Preparation and Imaging Conditions:
- Plate cells on glass-bottom dishes or multi-well plates pre-coated with attachment factors (collagen, fibronectin, or poly-L-lysine) [39].
- Supplement media with 20mM Hepes for pH stabilization or use controlled CO₂ environment [39].
- Overlay media with mineral oil to prevent evaporation unless using a humidified incubation system [39].
- Maintain constant temperature (37°C) using a stage-top incubator or microscope enclosure to minimize focal drift [39].
- Include control experiments to verify that imaging conditions do not induce phototoxicity or interfere with normal cell functions [39].
Image Acquisition and Analysis:
- For cytochrome c-GFP release, capture images at 30-60 second intervals to resolve the complete release process (typically ~5 minutes) [39].
- Quantify release kinetics using the punctate/diffuse index, calculated as the standard deviation of pixel intensity within individual cells [39].
- High-temporal resolution imaging (e.g., every 3.3 seconds) can resolve BAX recruitment kinetics, revealing that BAX initiates simultaneously throughout the cell but progresses at different rates among individual mitochondrial foci [40].
High-Throughput Single-Cell Analysis
Advanced imaging platforms enable high-throughput MOMP analysis across cell populations. The Live-Cell Imaging on Single-Cell Arrays (LISCA) platform combines micro-patterned cell arrays with automated time-lapse microscopy to extract event times from fluorescence traces of multiple cells in parallel [42]. This approach reveals heterogeneous temporal responses to death stimuli, identifying distinct subpopulations with different event sequences that would be averaged out in bulk analyses.
Event Time Determination:
- For early markers (LysoTracker, TMRM, CellROX), event times (t_breakdown) are defined as the point when fluorescence intensity declines precipitously [42].
- For late markers (Caspase 3/7, phosphatidylserine externalization, membrane permeability), event times (t_onset) are defined as the intersection between basal fluorescence and the tangent at half-maximum intensity [42].
- Mathematical functions (step functions combined with algebraic or exponential terms) are fitted to fluorescence traces to extract precise event times algorithmically [42].
The workflow for high-throughput single-cell analysis is illustrated below:
Integrated Experimental Applications
Multiparameter MOMP Pathway Analysis
Simultaneous monitoring of multiple cell death markers reveals intricate pathway relationships and cell-to-cell heterogeneity. A study investigating nanoparticle-induced cell death combined LysoTracker (lysosomal membrane permeabilization, LMP), TMRM (MOMP), and CellROX (oxidative burst) to establish chronological event sequences [42]. This approach demonstrated that at low nanoparticle doses (25 µg mL⁻¹), A549 and Huh7 cells primarily followed a lysosomal pathway, while at higher doses (100 µg mL⁻¹), A549 cells additionally employed a mitochondrial pathway, revealing previously obscured pathway co-existence [42].
Key findings from multiparameter analysis:
- Temporal heterogeneity: Cells exposed to identical stimuli exhibit marked variations in event timing and sequence [42].
- Pathway cross-talk: Different death pathways can be activated simultaneously within single cells [42].
- Dose-dependent effects: Stimulus intensity can qualitatively alter death mechanism engagement rather than simply accelerating a fixed pathway [42].
BAX Recruitment Kinetics
BAX fusion proteins (e.g., GFP-BAX) enable detailed quantification of recruitment kinetics during apoptosis initiation. High-speed imaging reveals that BAX recruitment begins simultaneously throughout the cell following stress exposure, but progresses at different rates at individual mitochondrial foci [40]. Pro-apoptotic factors are released early during BAX recruitment, with different molecules exhibiting distinct temporal release profiles relative to recruitment initiation [40].
Table 2: Quantitative Parameters of MOMP-Related Processes from Live-Cell Imaging
| Process | Measurement Method | Typical Time Scale | Key Kinetic Features |
|---|---|---|---|
| Cytochrome c release | cyt c-GFP redistribution | ~5 minutes [39] | Complete release; kinetically invariant across stimuli |
| BAX recruitment | GFP-BAX translocation & oligomerization | Variable (minutes) [40] | Simultaneous initiation; heterogeneous rates at foci |
| ΔΨm loss | TMRE/TMRM fluorescence | Coincident with cyto c release [39] | Rapid dissipation following MOMP |
| Early event correlations | LISCA platform [42] | Dose-dependent sequences | Lysosomal & mitochondrial pathway engagement |
Research Reagent Solutions
The following table provides essential reagents and tools for implementing MOMP live-cell imaging assays:
Table 3: Essential Research Reagents for MOMP Live-Cell Imaging
| Category | Specific Reagents | Function/Application | Key Considerations |
|---|---|---|---|
| Fluorescent Proteins | GFP, YFP, RFP variants [41] [39] | Protein tagging; FRET biosensors | Spectral properties; maturation time; photostability |
| Synthetic Fluorophores | TMRE/TMRM [39] [42] | ΔΨm sensing | Concentration optimization; potential phototoxicity |
| Organelle Markers | LysoTracker [42] | Lysosomal integrity assessment | pH-dependent staining; specificity |
| Caspase Reporters | CellEvent Caspase-3/7 [42] | Apoptosis execution phase detection | Specificity for active caspases |
| Membrane Integrity Probes | pSIVA-IANBD, PI/Toto-3 [42] | Phosphatidylserine exposure; membrane permeabilization | Reversible vs. irreversible binding |
| Chemical Inducers | Staurosporine [40] [42] | Apoptosis positive control | Dose optimization for specific cell types |
| Self-Labeling Tags | HaloTag [41] | Chemogenetic FRET biosensors | Fluorophore ligand permeability; labeling efficiency |
Advanced biosensors and live-cell imaging methodologies have transformed our understanding of MOMP from a binary event to a dynamic, heterogeneous process with complex regulation and diverse cellular outcomes. The techniques outlined in this guide enable researchers to capture the temporal and spatial dynamics of MOMP with unprecedented resolution, revealing subtleties in cell death commitment that inform both basic biology and therapeutic development. As biosensor technology continues to evolve, particularly with improvements in dynamic range, spectral tuning, and multiplexing capabilities [41], our ability to dissect the intricate relationships between MOMP and other cell death pathways will further expand, potentially uncovering new therapeutic opportunities for diseases characterized by dysregulated apoptosis.
Single-Molecule Approaches to Decipher Protein Complex Assembly
Mitochondrial outer membrane permeabilization (MOMP) is a pivotal event in the intrinsic apoptotic pathway, serving as a point of no return in the commitment to cell death. Its precise regulation is critical for development, immune function, and tissue homeostasis, while its dysregulation is implicated in diseases ranging from cancer to neurodegeneration [8]. For decades, ensemble averaging techniques have masked the stochasticity and heterogeneity of the protein complex assembly processes that govern MOMP. Single-molecule approaches have revolutionized this field by revealing transient intermediates, dynamic stoichiometries, and spatial distributions that are inaccessible to conventional methods [8] [43].
The central players in MOMP are the Bcl-2 family proteins, which include pro-apoptotic effectors (Bax, Bak), anti-apoptotic guardians (Bcl-2, Bcl-xL), and BH3-only sensors [25]. The critical step is the formation of pores in the mitochondrial outer membrane by activated Bax and Bak proteins. Traditional biochemical methods could identify interacting proteins but failed to reveal the precise sequence of molecular events, the stoichiometry of the resulting complexes, or the fascinating heterogeneity between individual mitochondria within the same cell [44]. This technical guide details how single-molecule methodologies are now illuminating these previously obscure processes, providing unprecedented insights into the molecular mechanisms of apoptosis with direct implications for therapeutic development.
Core Single-Molecule Methodologies and Their Applications to MOMP
Single-Molecule Localization Microscopy (SMLM) and DNA-PAINT
Single-molecule localization microscopy techniques, particularly DNA-PAINT (Points Accumulation for Imaging in Nanoscale Topography), overcome the diffraction limit of light to achieve spatial resolutions in the single-digit nanometer range [45]. DNA-PAINT utilizes transient binding of dye-labeled oligonucleotides to their complementary docking strands attached to proteins of interest, creating a stochastic "blinking" effect that allows precise determination of molecular positions.
Quantitative PAINT (qPAINT) extends this capability to determine copy numbers within protein complexes. By analyzing the binding kinetics of imager strands, researchers can calibrate against standards with known binding sites (e.g., DNA origami structures) to count absolute molecule numbers in cellular complexes [45]. When applied to MOMP regulators, this approach can reveal the stoichiometry of Bax/Bak oligomers at the mitochondrial membrane.
Table 1: Key Single-Molecule Imaging Techniques for MOMP Studies
| Technique | Principle | Spatial Resolution | Key Application in MOMP Research |
|---|---|---|---|
| DNA-PAINT | Transient hybridization of dye-labeled DNA imager strands | ~5-10 nm | Mapping nanoscale organization of Bcl-2 proteins at mitochondria |
| qPAINT | Analysis of binding kinetics for molecular counting | ~10 nm | Determining stoichiometry of Bax/Bak oligomers |
| SiMPull | Single-molecule fluorescence in pull-down format | N/A (not imaging) | Analyzing composition of endogenous Bcl-2 complexes from cell lysates |
| Single-Molecule Tracking | Tracing individual protein trajectories | ~20-40 nm | Monitoring Bax translocation and membrane binding dynamics |
Single-Molecule Pull-Down (SiMPull)
The SiMPull assay combines conventional pull-down principles with single-molecule fluorescence microscopy, enabling direct visualization of individual protein complexes purified from cell extracts or tissues [46]. Proteins are captured on a passivated surface via specific antibodies, and fluorescently tagged components are visualized at the single-complex level.
This technique is particularly valuable for characterizing the composition and stoichiometry of Bcl-2 protein complexes under different apoptotic stimuli. Photobleaching step analysis of fluorescent protein tags can reveal how many regulatory and effector molecules are present in each complex, testing existing models of MOMP regulation [46].
Single-Molecule Diffusion Analysis on Membranes
Supported lipid bilayers provide a simplified model system to investigate how Bcl-2 family proteins interact with membranes and assemble into complexes. By tagging proteins with fluorescent markers and tracking their two-dimensional diffusion using total internal reflection fluorescence (TIRF) microscopy, researchers can detect complex formation through characteristic changes in diffusion rates [47].
When membrane-bound proteins associate, their diffusion typically slows due to increased hydrodynamic drag and potentially increased avidity interactions with the membrane. This approach has demonstrated that complex formation on membrane surfaces can exhibit dramatically altered kinetics compared to solution interactions, with association rates accelerating and dissociation rates slowing, leading to ~100-fold higher apparent affinity in some systems [47].
Experimental Protocols for Key MOMP Applications
Protocol: Quantitative Analysis of Protein Complex Stoichiometry at Mitochondria
This protocol adapts qPAINT methodology [45] for investigating Bcl-2 family proteins:
Sample Preparation:
- Express HaloTag-fused Bax/Bak in appropriate cell lines (e.g., murine embryonic fibroblasts deficient in endogenous Bax/Bak).
- Label with chloroalkane-modified DNA docking strands (2-4 hours, per manufacturer's instructions).
- Induce apoptosis with appropriate stimulus (e.g., 1-5 µM staurosporine or specific BH3 mimetics).
- Fix cells at defined timepoints post-induction (15, 30, 60, 120 minutes) to capture progression of MOMP.
DNA-PAINT Imaging:
- Assemble flow chamber with immobilized cells.
- Image in presence of 1-5 nM Cy3b-labeled imager strands in imaging buffer (500 mM NaCl, 1× PBS, 1 mg/mL glucose oxidase, 0.04 mg/mL catalase, 1% v/v β-mercaptoethanol).
- Acquire 5,000-20,000 frames at 50-200 ms exposure time.
qPAINT Calibration:
- Image DNA origami standards with known docking site numbers (1, 3, 5 sites) under identical conditions.
- Measure binding kinetics (τ_off) for single binding sites.
- Calculate binding site number in cellular complexes using formula: N = τoff(complex) / τoff(single site).
Cluster Analysis:
- Apply DBSCAN algorithm to detect protein clusters.
- Calculate nearest-neighbor distances to assess oligomerization state.
- Correlate stoichiometry with apoptotic progression.
Protocol: Single-Molecule Analysis of Bcl-2 Complex Assembly on Supported Membranes
This protocol modifies established single-molecule diffusion approaches [47] for MOMP studies:
Membrane Preparation:
- Create mitochondrial membrane-mimetic lipid compositions: 65% DOPC, 15% DOPG, 10% DOPS, 5% TOCL, 4.5% cholesterol, 0.5% Biotinyl-PE.
- Form supported lipid bilayers on piranha-cleaned glass coverslips via vesicle fusion.
- Verify membrane fluidity by FRAP or single-particle tracking of control lipids.
Protein Purification and Labeling:
- Express and purify recombinant Bcl-2 proteins with SNAP-tag or HaloTag fusions.
- Label with organic dyes (Alexa Fluor 647, Cy3B) using appropriate labeling protocols.
- Confirm labeling efficiency and protein function after labeling.
Single-Molecule Imaging and Analysis:
- Incubate labeled proteins (10-500 pM) with supported membranes in appropriate buffer (containing 140 mM KCl, 15 mM NaCl, 25 mM HEPES, pH 7.4).
- Image using TIRF microscopy with appropriate excitation lasers and emission filters.
- Track individual proteins with 10-100 ms time resolution.
- Calculate mean squared displacement and diffusion coefficients for each trajectory.
- Identify complex formation by transitions to slower diffusion states.
Diagram 1: Experimental workflow for single-molecule MOMP studies. This generalized pipeline shows the key stages from sample preparation through data analysis, with multiple technique options at the imaging stage.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Essential Research Reagents for Single-Molecule MOMP Studies
| Reagent/Material | Function/Application | Key Considerations |
|---|---|---|
| DNA-PAINT Docking Strands (e.g., chloroalkane-conjugated) | Covalent attachment to HaloTag-fused proteins | Ensure high purity and efficient conjugation to minimize non-specific binding |
| HaloTag/SNAP-tag Plasmids | Genetic encoding for specific protein labeling | Validate fusion protein functionality compared to wild-type |
| DNA Origami Standards | Calibration for qPAINT quantification | Include multiple binding site numbers (1, 3, 5) for standard curve |
| PEG-passivated Surfaces | Minimize non-specific binding in SiMPull | Include biotin-PEG for antibody immobilization |
| Mitochondrial Lipid Mixes | Create biologically relevant membranes for in vitro studies | Include cardiolipin for authentic mitochondrial outer membrane composition |
| Oxygen Scavenging Systems (glucose oxidase/catalase) | Prolong fluorophore longevity during imaging | Optimize concentrations to balance photostability and potential cytotoxicity |
| BH3 Mimetics (e.g., ABT-199, WEHI-539) | Modulate Bcl-2 protein interactions in functional studies | Use specific inhibitors to dissect individual protein contributions |
Visualizing MOMP Regulation by Bcl-2 Proteins
Diagram 2: Bcl-2 protein interactions regulating MOMP. This signaling network shows how apoptotic stimuli trigger a cascade of protein interactions that ultimately lead to pore formation. Single-molecule techniques provide unique insights into previously unobservable aspects of this process.
Single-molecule approaches have fundamentally transformed our understanding of protein complex assembly in general and MOMP regulation in particular. By revealing the stoichiometry, dynamics, and spatial organization of Bcl-2 family proteins at the nanometer scale, these techniques have resolved long-standing controversies and uncovered unexpected complexities in apoptotic signaling. The quantitative frameworks provided by qPAINT, SiMPull, and single-molecule tracking enable researchers to move beyond qualitative models to precise, predictive understanding of how individual molecular interactions culminate in cell fate decisions.
Looking forward, several emerging trends promise to further advance this field. The integration of single-molecule imaging with CRISPR-based genomic editing will enable studies of endogenous proteins under physiological regulation. Advances in label-free detection methods may eventually allow observation of proteins in their native states without potential perturbations from fluorescent tags. Furthermore, the application of machine learning to analyze the vast datasets generated by single-molecule experiments will uncover subtle patterns and correlations that escape conventional analysis. As these methodologies continue to mature, they will undoubtedly reveal new layers of regulation in MOMP and provide innovative targets for therapeutic intervention in the many diseases characterized by apoptotic dysregulation.
Mitochondrial outer membrane permeabilization (MOMP) represents a fundamental commitment point in the execution of apoptotic cell death [48]. Situated at the interface of apoptosis initiation and execution, MOMP functions as a crucial switch that determines cellular life-death decisions [49]. In recent years, advanced live-cell imaging techniques have revealed that MOMP does not occur synchronously throughout all mitochondria within a cell but rather propagates as a wave-like signal [49]. This spatiotemporal propagation ensures efficient and irreversible commitment to apoptosis, particularly in response to extrinsic apoptosis induction [49]. The discovery of MOMP waves in various cell types, including epithelial cells, cardiomyocytes, and syncytiotrophoblasts, highlights the importance of understanding the mechanisms underlying this spatial coordination [49]. This technical guide examines the molecular machinery, computational frameworks, and experimental methodologies for analyzing MOMP propagation, providing researchers with comprehensive tools for investigating this fundamental process in apoptosis research.
Molecular Machinery Governing MOMP Propagation
Core Components of the MOMP Machinery
The spatiotemporal propagation of MOMP is governed by an intricate network of Bcl-2 family proteins and mitochondrial components. At the heart of this process lie the pro-apoptotic effector proteins Bax and Bak, which form pores in the outer mitochondrial membrane through homo-oligomerization [48]. These pores permit the release of pro-apoptotic intermembrane space proteins, including cytochrome c and SMAC, into the cytosol [48]. Cytochrome c then triggers apoptosome formation and caspase activation, while SMAC antagonizes caspase-inhibiting IAP proteins [50]. The activation of Bax and Bak is regulated by a complex interplay with other Bcl-2 family members, including anti-apoptotic proteins such as Bcl-2, Mcl-1, and Bcl-xL, and pro-apoptotic BH3-only proteins [49].
The BH3-only protein Bid serves as a crucial mediator connecting extrinsic apoptosis signals to MOMP. Caspase-8, activated by death receptors, cleaves full-length Bid to generate truncated Bid (tBid), which possesses increased apoptotic activity [49]. tBid both inhibits anti-apoptotic Bcl-2 family members and directly activates Bax/Bak [49]. The high mitochondrial affinity of tBid depends on cardiolipin, a lipid cofactor in the outer mitochondrial membrane, whose mobility is enhanced by peroxidation initiated by reactive oxygen species (ROS) [49].
Integrated tBid-ROS Signaling for Robust MOMP Propagation
Research has revealed that the spatial propagation of MOMP depends on the integrated signaling of both tBid and ROS, rather than either mechanism alone [49]. The interaction between tBid and the outer mitochondrial membrane increases ROS generation, likely by inhibiting state-3 respiration and ATP synthesis, thereby augmenting cardiolipin peroxidation [49]. Conversely, cardiolipin peroxidation facilitates tBid membrane insertion and sensitizes mitochondria to MOMP [49]. This positive feedback loop creates a self-propagating system that ensures complete cellular commitment to apoptosis once the process is initiated.
Table 1: Key Molecular Components in MOMP Propagation
| Component | Type/Class | Primary Function in MOMP Propagation |
|---|---|---|
| Bax/Bak | Pro-apoptotic multi-domain proteins | Form pores in outer mitochondrial membrane through homo-oligomerization |
| tBid | BH3-only protein | Activates Bax/Bak directly and inhibits anti-apoptotic Bcl-2 proteins |
| Cardiolipin | Mitochondrial phospholipid | Serves as membrane cofactor for tBid binding; peroxidation enhances MOMP sensitivity |
| Cytochrome c | Intermembrane space protein | Triggers apoptosome formation and caspase activation upon release |
| SMAC/Diablo | Intermembrane space protein | Antagonizes IAP proteins to enable caspase activity |
| Bcl-2/Bcl-xL | Anti-apoptotic proteins | Antagonize activated Bax/Bak and neutralize BH3-only proteins |
The following diagram illustrates the integrated signaling pathway that facilitates MOMP propagation:
Quantitative Analysis of MOMP Wave Dynamics
Kinetic Parameters of MOMP Propagation
Live-cell imaging studies have yielded crucial quantitative data on the spatiotemporal dynamics of MOMP. In HeLa cells, the entire pool of cytochrome c redistributes throughout the cell body within 2-5 minutes once MOMP is initiated [49]. Cells with a common mitotic history initiate MOMP within a narrow time window of 5-10 minutes [49]. The propagation of MOMP waves exhibits distinct velocities under different experimental conditions. In permeabilized cardiomyocytes, MOMP waves travel with an end-to-end velocity of approximately 0.19 μm/s [49]. The observed wave-like propagation ensures efficient and complete commitment to apoptosis once the process is initiated in a subset of mitochondria.
Table 2: Experimentally Measured MOMP Wave Dynamics
| Parameter | Experimental System | Value | Measurement Technique |
|---|---|---|---|
| MOMP completion time | HeLa cells | 2-5 min | Time-lapse imaging of cytochrome c release |
| MOMP initiation window | HeLa cells with common mitotic history | 5-10 min | High-resolution live-cell imaging |
| Wave propagation velocity | Permeabilized cardiomyocytes | 0.19 μm/s | End-to-end velocity measurement |
| Mitochondrial permeabilization | Individual mitochondria | Seconds | High-resolution imaging |
Mathematical Frameworks for Modeling MOMP Propagation
Computational models of MOMP propagation have evolved from single-mechanism approaches to integrated frameworks that more accurately reflect the biological complexity. Initial models proposed either a reaction-diffusion mechanism governed by anisotropies in tBid production or a process driven by sequential bursts of ROS [49]. However, each individual model could only reproduce a subset of experimental findings, leading to the development of a combined mathematical description that integrates both tBid and ROS signaling [49].
The tBid reaction-diffusion model represents cells as a one-dimensional slab with mesh points reflecting 1 μm distance [49]. tBid is added into the system at mesh position 1 using a sigmoid input function that reflects experimentally determined Bid cleavage kinetics [49]. The ROS reaction-diffusion model implements similar spatial parameterization, with ROS added at a constant input rate and MOMP initiated once a threshold amount of locally absorbed ROS is reached [49]. The integrated model defines tBid input as the near-end master trigger for MOMP, with both tBid and ROS contributing to inducing MOMP in more distant mesh points [49].
Experimental Methodologies for Monitoring MOMP Dynamics
Live-Cell Reporter Systems for Apoptosis Signaling
Advanced fluorescent reporter systems have enabled real-time monitoring of caspase activity and MOMP in individual living cells. These include:
Effector Caspase Reporter Protein (EC-RP): Composed of a FRET donor-acceptor pair (CFP and YFP) connected via a flexible linker containing the caspase cleavage sequence DEVDR, which has 20-fold greater selectivity for caspase-3 relative to caspase-8 compared to previous DEVDG linkers [51]. Cleavage results in loss of energy transfer and increased CFP signal.
Initiator Caspase Reporter Protein (IC-RP): Contains tandem copies of IETD in its linker, efficiently cleaved by caspase-8 but poorly by caspases-3,7, providing a readout of procaspase-3 activation [51].
IMS-RP (Mitochondrial IMS Reporter): Created by fusing RFP to the mitochondrial import sequence of Smac (residues 1-59), serving as a biochemically inert reporter of protein translocation during MOMP [51]. IMS-RP redistribution from punctate mitochondrial to diffuse cytosolic fluorescence indicates MOMP occurrence.
Experimental Workflow for MOMP Wave Analysis
The following diagram outlines a comprehensive experimental workflow for investigating MOMP propagation:
Implementation of Mathematical Models for MOMP Simulation
Computational models of MOMP propagation are typically implemented as MATLAB code for numerical analysis [49]. Partial differential equations are solved using adaptive step Runge-Kutta ODE solvers, with modeling generally performed in one spatial dimension due to computational constraints [49]. For simulating discontinuous mitochondrial patterns, mesh points can be defined as devoid of mitochondria, which consequently do not contribute to tBid adsorption or serve as sources for ROS generation [49]. The MATLAB code for these model variants has been made available as supplemental information in published studies, facilitating adoption and further development by the research community [49].
Research Reagent Solutions for MOMP Investigations
Table 3: Essential Research Reagents for MOMP Studies
| Reagent/Tool | Type | Primary Application | Key Features/Considerations |
|---|---|---|---|
| EC-RP | Fluorescent reporter | Effector caspase activity monitoring | DEVDR linker provides caspase-3 selectivity; CFP/YFP FRET pair |
| IC-RP | Fluorescent reporter | Initiator caspase activity monitoring | Tandem IETD sites for caspase-8 selectivity |
| IMS-RP | Fluorescent reporter | MOMP detection | Smac import sequence (residues 1-59); lacks IAP-binding motif |
| TRAIL/TNF | Apoptosis inducer | Extrinsic pathway activation | Death receptor engagement; caspase-8 activation |
| tBid expression systems | Recombinant protein | Direct MOMP induction | Bypasses upstream signaling; enables localized activation |
| MATLAB with PDE toolbox | Computational software | Mathematical modeling | Numerical solving of reaction-diffusion equations |
| BH3 mimetics (Venetoclax) | Small molecule inhibitors | Bcl-2 family perturbation | Selective Bcl-2 inhibition; research and therapeutic applications |
Discussion and Future Perspectives
The study of MOMP spatiotemporal propagation represents a paradigm shift in understanding cell death decisions, moving beyond biochemical interactions to incorporate spatial and temporal dimensions. The integrated tBid-ROS model provides a robust theoretical framework that accurately reproduces experimental observations and offers mechanistic explanations for the efficiency of MOMP propagation even when mitochondria are spatially separated [49]. This integrated signaling system ensures that once a threshold of apoptotic signaling is reached, the cell commits irreversibly to death, preventing the survival of partially damaged cells that could contribute to genomic instability or oncogenic transformation [51] [48].
Future research directions in MOMP propagation include elucidating how specific Bcl-2 family members coordinate responses across different cell types, investigating the interplay between mitochondrial dynamics and MOMP regulation, and developing more sophisticated multi-scale models that integrate subcellular MOMP dynamics with tissue-level outcomes [48] [50]. The continued refinement of computational models, coupled with advanced imaging techniques, will further enhance our understanding of this fundamental process in apoptosis and its implications for cancer therapy and degenerative diseases.
The tools and methodologies outlined in this technical guide provide researchers with a comprehensive framework for investigating MOMP spatiotemporal dynamics, from experimental design and implementation to computational modeling and data analysis. As these approaches continue to evolve, they will undoubtedly yield new insights into the regulation of cell death and opportunities for therapeutic intervention in diseases characterized by dysregulated apoptosis.
Mitochondrial outer membrane permeabilization (MOMP) is a decisive event in the intrinsic apoptosis pathway, serving as a point-of-no-commitment for cell death execution. This process is primarily regulated by BCL-2 family proteins, which control the release of intermembrane space proteins that activate downstream apoptotic mechanisms [52]. Among these proteins, cytochrome c plays the most characterized role; upon release into the cytosol, it facilitates the formation of the apoptosome complex, leading to caspase activation and systematic cellular dismantlement [53]. Concurrently, mitochondria often undergo dissipation of their transmembrane potential (ΔΨm), an essential component for oxidative phosphorylation and overall organellar health. The functional relationship between cytochrome c release and ΔΨm dissipation is complex and context-dependent, with emerging evidence revealing that these events can be temporally and mechanistically dissociated [53] [54]. This technical guide provides an in-depth examination of the key methodologies for quantifying these critical events, framed within the broader context of MOMP in apoptosis research.
Molecular Mechanisms and Functional Relationships
The Critical yet Dissociable Relationship Between Cytochrome c Release and ΔΨm
The classical model of apoptosis posits that mitochondrial outer membrane permeabilization (MOMP) leads to coordinated cytochrome c release and loss of mitochondrial membrane potential (ΔΨm). However, single-cell analyses have revealed a more complex relationship, demonstrating that these events can be functionally dissociated. During granzyme B-induced apoptosis, cytochrome c and SMAC release occur independently of caspase activity and correlate with the onset of apoptosis, while ΔΨm loss, though also caspase-independent, is transient and can recover if caspase activity is blocked [53]. This recovery occurs because mitochondria can utilize cytoplasmic cytochrome c to maintain ΔΨm and ATP production after MOMP, demonstrating that cytochrome c release does not necessarily cause irreversible mitochondrial dysfunction [54].
The sequence of these molecular events has been experimentally verified using dose and time-dependent strategies with betulinic acid (BetA), confirming that mitochondrial permeability transition pore (mPTP) opening and ΔΨm depolarization sequentially occur prior to cytochrome c release during mPT-mediated mitochondrial dysfunction [55]. This temporal relationship highlights the complex regulatory mechanisms governing mitochondrial function during cell death execution.
BCL-2 Protein Family: Master Regulators of MOMP
The BCL-2 protein family serves as the primary regulatory system controlling MOMP. This family includes pro-apoptotic proteins (such as BAX and BAK), anti-apoptotic proteins (including BCL-2 itself, BCL-XL, and MCL-1), and BH3-only proteins [52]. During apoptosis induction, activated pro-apoptotic members oligomerize to form pores in the mitochondrial outer membrane, facilitating cytochrome c release [56]. The development of BH3 mimetics, such as Venetoclax (a BCL-2 inhibitor), has provided both therapeutic agents and valuable research tools for investigating these regulatory mechanisms, highlighting the translational importance of fundamental apoptosis research [52].
Table 1: Key Proteins in MOMP Regulation and Assessment
| Protein/Factor | Function/Role in Apoptosis | Experimental Utility |
|---|---|---|
| Cytochrome c | Electron transport in respiration; Activates caspase cascade when released to cytosol | Primary marker for MOMP confirmation; measured via immunofluorescence, WB, flow cytometry |
| BAX/BAK | Pro-apoptotic effectors; oligomerize to form pores in OMM during MOMP | Targets for activation monitoring; oligomerization indicates commitment to apoptosis |
| BCL-2/BCL-XL | Anti-apoptotic; sequester pro-apoptotic proteins to prevent MOMP | Inhibition sensitivity (Venetoclax) tests mitochondrial priming & apoptotic predisposition |
| Caspases | Cysteine proteases that execute apoptosis; activated by cytochrome c/Apaf-1 | Activity inhibition (zVAD-fmk) differentiates caspase-dependent/independent death |
| Smac/Diablo | IMS protein released with cytochrome c; antagonizes IAPs to promote apoptosis | Co-measurement with cytochrome c provides comprehensive MOMP assessment |
Diagram 1: MOMP and ΔΨm Loss Signaling Pathways. The core apoptotic pathway involves BAX/BAK activation leading to MOMP and cytochrome c release. The relationship with ΔΨm dissipation can occur through both mPTP opening and MOMP itself.
Quantitative Assays and Methodologies
Cytochrome c Release Detection Methods
Immunofluorescence Microscopy
This technique enables visual confirmation of cytochrome c localization at the single-cell level, allowing researchers to directly observe its release from mitochondria into the cytosol during apoptosis. Cells are stained with antibodies against cytochrome c and a mitochondrial marker (e.g., TOM20), then analyzed using confocal microscopy. Cells with intact mitochondria display a punctate, mitochondrial pattern, while apoptotic cells exhibit a diffuse, cytoplasmic staining pattern. This method provides spatial resolution but is less suitable for high-throughput screening [54].
Subcellular Fractionation and Western Blotting
This biochemical approach involves separating cytosolic and mitochondrial fractions from cell populations, followed by Western blot analysis for cytochrome c. The presence of cytochrome c in the cytosolic fraction indicates MOMP. The protocol involves: (1) harvesting cells by gentle scraping; (2) permeabilizing plasma membrane with digitonin-containing buffer (e.g., CLAMI buffer: 250 mM sucrose, 70 mM KCl, 50 µg/ml digitonin in PBS); (3) centrifuging at low speed (1,000 × g) to collect intact mitochondria; (4) preparing cytosolic (supernatant) and mitochondrial (pellet) fractions; and (5) performing Western blotting with anti-cytochrome c antibodies [54]. This method provides quantitative, population-level data but requires careful optimization to ensure fraction purity.
Flow Cytometric Analysis
Flow cytometry offers a high-throughput, quantitative approach for detecting cytochrome c release in individual cells. The standard protocol utilizes selective plasma membrane permeabilization with digitonin, allowing cytosolic cytochrome c to escape while retaining mitochondria-associated cytochrome c. Cells are stained with anti-cytochrome c antibodies after fixation and permeabilization. A clear fluorescence shift (approximately three-fold difference) distinguishes cells with intact versus released cytochrome c [54]. This method enables rapid analysis of thousands of cells but requires careful titration of digitonin concentration for different cell types.
Nano-Flow Cytometry (nFCM)
An advanced technique, nFCM enables ultra-sensitive, multi-parameter analysis of mitochondrial events at the single-organelle level. Isolated mitochondria are incubated with antibodies against cytochrome c, followed by fluorescent secondary antibodies. nFCM can detect the release of cytochrome c and other cell-death-associated factors (AIF, PNPT1, mtDNA) simultaneously, providing unprecedented resolution of mitochondrial events during MOMP [55]. This method requires specialized instrumentation but offers exceptional sensitivity for studying mitochondrial heterogeneity.
Mitochondrial Membrane Potential (ΔΨm) Measurement Techniques
Fluorescent Probe-Based Assays
Cationic, lipophilic dyes remain the most widely used tools for measuring ΔΨm due to their Nernstian distribution across mitochondrial membranes. These dyes accumulate in the mitochondrial matrix in proportion to ΔΨm, with fluorescence intensity reflecting the potential.
Table 2: Quantitative Measurements of ΔΨm and Cytochrome c Release
| Parameter | Measurement Method | Typical Values in Healthy Cells | Values During Apoptosis | Key Insights from Research |
|---|---|---|---|---|
| ΔΨm | TMRM/TMRE fluorescence (quantitative) | -139 mV (neurons) [57] | -108 mV to -158 mV (regulated range) [57] | Can recover after initial loss if caspases inhibited [53] |
| ΔΨm | DiOC₆(3) fluorescence (semi-quant) | High fluorescence (cell-type dependent) | 40-60% decrease (cell-type dependent) | Dissociable from cytochrome c release timing [55] |
| Cytochrome c Localization | Immunofluorescence microscopy | Punctate mitochondrial pattern | Diffuse cytoplasmic pattern | Direct visual confirmation of MOMP |
| Cytochrome c Release | Subcellular fractionation + WB | Cytochrome c in mitochondrial fraction | Cytochrome c in cytosolic fraction | Biochemical confirmation of MOMP |
| mPTP Opening | Calcein-AM/CoCl₂ quenching (nFCM) | High calcein fluorescence (>90% mitochondria) | ~90% fluorescence decrease [55] | Sequential with ΔΨm loss before cytochrome c release |
TMRM/TMRE (Tetramethylrhodamine Methyl/Ethyl Ester): These potentiometric probes distribute across mitochondrial membranes according to the Nernst equation, enabling quantitative assessment of ΔΨm. For live-cell imaging, cells are loaded with 20-100 nM TMRM/TMRE for 20-30 minutes at 37°C. Fluorescence intensity is measured using fluorescence microscopy or flow cytometry. The absolute ΔΨm values can be calculated in millivolts using specialized calibration protocols accounting for plasma membrane potential, binding, and activity coefficients [57].
JC-1: This unique dye exhibits potential-dependent accumulation in mitochondria, indicated by a fluorescence emission shift from green (~529 nm) to red (~590 nm). The red/green fluorescence ratio provides a semi-quantitative measure of ΔΨm that is relatively independent of mitochondrial mass. However, JC-1's non-equilibrium distribution limits its utility for absolute quantification compared to TMRM [57].
Incucyte MMP Orange Dye: This commercially available reagent enables real-time, kinetic monitoring of ΔΨm in live cells within standard tissue culture incubators. The protocol involves adding the dye to culture media and monitoring fluorescence intensity over time. Control compounds like FCCP (depolarizing agent) and Oligomycin A (hyperpolarizing agent) serve as validation controls [58].
Advanced Quantitative Imaging
Recent methodological advances now enable precise quantification of absolute ΔΨm values in millivolts. This approach incorporates a biophysical model of fluorescent probe compartmentation and dynamics, accounting for plasma membrane potential (ΔΨP), matrix:cell volume ratio, binding coefficients, and optical factors. The calibration protocol involves: (1) simultaneous measurement of TMRM and a plasma membrane potential indicator; (2) determination of cellular volume parameters via confocal microscopy; and (3) application of a mathematical model to deconvolute ΔΨP and ΔΨM from fluorescence intensities [57]. This method provides unprecedented accuracy but requires sophisticated instrumentation and computational analysis.
Integrated Experimental Workflows
Isolated Mitochondria Studies
Analysis of isolated mitochondria provides a reductionist system for studying direct effects on mitochondrial function without confounding cellular processes. The standard isolation protocol involves homogenizing tissues or cells in isotonic sucrose buffer (250 mM sucrose, 10 mM Tris-HCl, 1 mM EDTA, pH 7.4) followed by differential centrifugation. Mitochondrial functionality is validated by measuring respiratory control ratio (RCR > 4) and ADP/O ratio using oxygen electrode analysis [59]. For MOMP-related studies, isolated mitochondria can be treated with pro-apoptotic proteins (tBid, BAX) or pharmacological agents to directly induce permeability transition, followed by simultaneous assessment of ΔΨm (using TMRE or DiOC6(3)), swelling (light scattering at 540 nm), and cytochrome c release (Western blotting or nFCM) [55] [59].
Diagram 2: Experimental Workflow for MOMP Assessment. Complementary approaches using isolated mitochondria and intact cells provide comprehensive understanding of MOMP mechanisms.
Multi-Parameter Single-Cell Analysis
Comprehensive assessment of MOMP requires integrated analysis of multiple apoptotic parameters within the same cell population. A representative workflow for flow cytometric analysis includes: (1) staining cells with TMRM (50 nM, 20 minutes) to measure ΔΨm; (2) processing for cytochrome c immunostaining after digitonin-based permeabilization; (3) counterstaining with Annexin V-FITC and propidium iodide to detect phosphatidylserine externalization and plasma membrane integrity; and (4) analysis by flow cytometry with appropriate fluorescence compensation [54]. This multi-parameter approach enables researchers to correlate ΔΨm loss with cytochrome c release and other apoptotic markers at the single-cell level, revealing population heterogeneity and temporal relationships between events.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for MOMP and ΔΨm Analysis
| Reagent/Category | Specific Examples | Function/Application | Key Considerations |
|---|---|---|---|
| ΔΨm Indicators | TMRM, TMRE, JC-1, DiOC₆(3), Incucyte MMP Orange Dye | Quantitative & semi-quantitative measurement of mitochondrial membrane potential | TMRM/TMRE preferred for quantitative assays; JC-1 for ratio-metric imaging |
| Cytochrome c Detection | Anti-cytochrome c antibodies (e.g., 7H8.2C12), Cytochrome c-GFP constructs | Immunodetection in microscopy, WB, flow cytometry; tracking release in live cells | Digitomin permeabilization critical for specific flow cytometric analysis |
| MOMP Inducers | Staurosporine, Actinomycin D, Betulinic Acid, UV irradiation, recombinant tBid/BAX | Positive controls for inducing MOMP in experimental systems | Different inducers engage apoptotic pathways at different points |
| Pharmacological Inhibitors | zVAD-fmk (pan-caspase inhibitor), Cyclosporin A (mPTP inhibitor), Venetoclax (BCL-2 inhibitor) | Dissecting caspase dependence; testing specific pathway involvement | CsA concentration varies for isolated mitochondria (μM) vs. cells (nM) |
| Mitochondrial Isolation Kits | Commercial kits from various suppliers | Preparing functional mitochondrial fractions for reductionist studies | Validate functionality via RCR measurement after isolation |
| Control Compounds | FCCP (uncoupler), Oligomycin A (ATP synthase inhibitor) | Control for ΔΨm depolarization and hyperpolarization respectively | Essential for assay validation and instrument calibration |
Functional assays for cytochrome c release and ΔΨm loss remain cornerstone methodologies in apoptosis research, providing critical insights into MOMP regulation and execution. The evolving understanding of the complex, often dissociable relationship between these events highlights the importance of multi-parameter, kinetic analysis rather than single time-point assessments. Emerging technologies like nano-flow cytometry and advanced quantitative imaging offer unprecedented resolution for studying these events at the single-organelle level, revealing previously unappreciated heterogeneity in mitochondrial responses [55]. Furthermore, the integration of these functional assays with genetic approaches and pharmacological tools continues to drive both fundamental understanding of cell death mechanisms and the development of novel therapeutic strategies targeting the BCL-2 family for cancer and other diseases [52]. As research progresses, these assays will continue to be refined, offering deeper insights into the mitochondrial regulation of cell death and its translational applications.
The intricate interplay between proteins and lipids is fundamental to cellular life and death. Within the context of mitochondrial outer membrane permeabilization (MOMP)—the "point of no return" in intrinsic apoptosis—elucidating these interactions is paramount for understanding cell fate and developing novel therapeutics [1] [17]. This whitepaper provides an in-depth technical guide on two powerful structural biology methods, cryo-electron microscopy (cryo-EM) and nuclear magnetic resonance (NMR) spectroscopy, for characterizing the protein-lipid interactions that govern MOMP. We detail experimental protocols, summarize key quantitative data, and present essential resources, framing this discussion within the urgent context of apoptosis research and drug development.
Mitochondrial outer membrane permeabilization (MOMP) is a decisive event in the intrinsic apoptotic pathway. During MOMP, the integrity of the outer mitochondrial membrane (OMM) is compromised, leading to the release of pro-apoptotic factors like cytochrome c into the cytosol, which triggers caspase activation and cellular demolition [1] [17]. This process is directly governed by the BCL-2 protein family, whose delicate balance between pro-survival (e.g., BCL-2, MCL-1) and pro-apoptotic members determines cellular fate.
The core executioners of MOMP are the effector proteins BAK and BAX. In their dormant state, BAK is integrated into the OMM, while BAX is largely cytosolic. Upon activation by BH3-only proteins (e.g., tBID, BIM), both undergo profound conformational changes, leading to their homo-oligomerization and insertion into the OMM to form proteolipidic pores [60] [17]. These pores, which allow the egress of cytochrome c, are the physical manifestation of MOMP. Critically, their formation and regulation are not solely dependent on protein-protein interactions within the BCL-2 family but are also intimately dependent on protein-lipid interactions with the OMM itself [60] [61]. The lipid environment is not a passive bystander; it actively participates in the stability, conformation, and function of these apoptotic proteins. Consequently, obtaining high-resolution structural insights into these complexes is essential for a mechanistic understanding of cell death.
Cryo-Electron Microscopy for Membrane Protein Structure Determination
Core Principles and Workflow
Single-particle cryo-electron microscopy (cryo-EM) has emerged as a revolutionary technique for determining high-resolution structures of macromolecular complexes, particularly those resistant to crystallization, such as membrane proteins [62] [63]. The method involves rapidly freezing a thin aqueous sample solution in vitreous (non-crystalline) ice, preserving the particles in a near-native state. The frozen grid is then imaged under a transmission electron microscope, and computational algorithms process thousands of individual particle images to reconstruct a three-dimensional structure [62].
The application of cryo-EM to membrane proteins, however, presents unique challenges, primarily due to the need to solubilize and stabilize the hydrophobic transmembrane domains. The workflow typically involves:
- Protein Extraction and Solubilization: Membrane proteins are extracted from their native lipid bilayers using detergents that form micelles around the hydrophobic domains [62].
- Purification: The target protein is purified using techniques like affinity and size-exclusion chromatography (SEC). Fluorescence-detection SEC (FSEC) is particularly valuable for monitoring the homogeneity of membrane proteins [62].
- Vitrification: The purified sample is applied to a perforated carbon grid, blotted to create a thin film, and plunge-frozen in liquid ethane to form vitreous ice [62].
- Data Collection and Processing: Images are collected with direct electron detectors, and sophisticated software packages perform particle picking, 2D classification, 3D reconstruction, and refinement to generate a final atomic model [62] [64].
Technical Considerations for Studying Protein-Lipid Interactions
A major hurdle in cryo-EM of membrane proteins is the presence of detergent micelles, which can diminish image contrast and obscure underlying protein structure [62]. To overcome this and better mimic the native lipid environment, several advanced sample preparation strategies have been developed:
- Amphipols: Synthetic amphipathic polymers that can replace detergents and stabilize membrane proteins in aqueous solution, often providing a more stable and homogeneous preparation [62].
- Nanodiscs: Lipid bilayers of defined size and composition encircled by a membrane scaffold protein (MSP) or synthetic polymer. Nanodiscs provide a near-native lipid environment for the embedded membrane protein, which is crucial for studying functional protein-lipid interactions [62].
- Lipid Mesophases: Used for crystallizing membrane proteins but can also aid in creating a more native-like environment for certain EM studies.
Recent technological advances, including direct electron detectors and improved image processing software, have propelled cryo-EM to resolve structures at near-atomic resolution (3–4 Å). At this resolution, it is sometimes possible to visualize the electron densities of bound lipids, small molecules, and water molecules, providing direct structural information on protein-lipid interfaces [62].
The following diagram illustrates the key stages of the cryo-EM workflow for a membrane protein, highlighting the sample preparation strategies that enable the study of protein-lipid interactions.
Simulation-Assisted Interpretation of Lipid Densities with LipIDens
A significant challenge in cryo-EM is definitively identifying the chemical nature of low-resolution densities, particularly for lipids that occupy binding sites on membrane proteins. The LipIDens computational pipeline has been developed to address this by integrating molecular dynamics (MD) simulations with cryo-EM data [65].
The LipIDens workflow involves:
- Structure Processing: Preparing the membrane protein structure and embedding it in a model lipid bilayer.
- Coarse-Grained (CG) MD Simulations: Running simulations to extensively sample lipid binding and unbinding events around the protein's transmembrane domain.
- Binding Site Analysis: Using the PyLipID tool to identify lipid binding sites and calculate kinetic parameters (e.g., residence time, occupancy) for different lipid species [65].
- Pose Comparison and Ranking: The top-ranked lipid binding poses from simulations are compared with the experimental cryo-EM density. Lipid poses are ranked based on how well they fit the density and their relative residence times, providing a quantitative assessment for lipid identification [65].
- Pose Refinement: Selected lipid poses can be refined using atomistic MD simulations for higher detail.
This integrative approach allows researchers to move from an ambiguous "lipid-like density" to a molecularly identified and structurally refined lipid binding pose, dramatically enhancing the biological insights gained from a cryo-EM structure.
NMR Spectroscopy for Probing Dynamics and Interactions
Core Principles and Application to MOMP
While cryo-EM excels at determining static, high-resolution structures, nuclear magnetic resonance (NMR) spectroscopy is unparalleled for studying protein dynamics, conformational changes, and weak protein-ligand or protein-lipid interactions in solution [60]. This is particularly relevant for the BCL-2 family proteins, which undergo complex activation transitions.
NMR relies on the magnetic properties of atomic nuclei (e.g., ^1H, ^15N, ^13C). By studying chemical shifts, line widths, and relaxation parameters, researchers can infer on structural and dynamic information at the atomic level. Key applications in MOMP research include:
- Mapping Interaction Sites: Using chemical shift perturbation or paramagnetic relaxation enhancement (PRE) experiments to identify binding interfaces, such as between a BH3 domain and the canonical groove of BCL-2 or between BAX/BAK and membrane mimetics [60].
- Characterizing Conformational Changes: Observing real-time changes in the NMR spectrum as proteins like BAX transition from a dormant to an active state.
- Studying Dynamics: Investigating the intrinsic flexibility of proteins, such as the partially unfolded N-termini of activated BAK and BAX, which is difficult to capture by crystallography or cryo-EM [60].
Technical Considerations and Workflow
A common NMR strategy involves producing isotopically labeled protein (^15N, ^13C) and acquiring multidimensional heteronuclear NMR spectra.
- Sample Preparation: The membrane protein or domain must be stabilized in a membrane-mimetic environment, such as detergent micelles, bicelles, or nanodiscs. For soluble domains (e.g., BAX core), standard aqueous buffers are used.
- Data Collection: A series of NMR experiments (e.g., HSQC, NOESY) are performed. The ^1H-^15N HSQC spectrum serves as a "fingerprint" of the protein's fold.
- Titration Experiments: To map a binding site, a ligand (e.g., a BH3 peptide, a lipid) is titrated into the protein sample, and changes in the chemical shifts of protein residues are monitored. Residues with significant perturbations are part of the binding interface.
- Structure Calculation: For structure determination, distance restraints from NOE experiments and dihedral angle restraints are used to calculate an ensemble of structures that satisfy the experimental data.
NMR has been instrumental in revealing the mechanisms of BCL-2 family protein function. For example, studies using chemically stapled BH3 peptides (SAHBs) and PRE NMR showed that activator BH3 domains bind to a non-canonical trigger site on BAX, comprising helices α1 and α6, initiating a series of allosteric changes that lead to its activation and membrane insertion [60].
Integrated Methodologies: A Practical Guide for MOMP Research
Quantitative Comparison of Structural Techniques
The following table summarizes the key characteristics of cryo-EM and NMR in the context of studying protein-lipid interactions in MOMP.
Table 1: Comparative Analysis of Cryo-EM and NMR for Protein-Lipid Interaction Studies
| Feature | Cryo-Electron Microscopy (Cryo-EM) | Nuclear Magnetic Resonance (NMR) Spectroscopy |
|---|---|---|
| Typical Resolution | Near-atomic to atomic (3–4 Å common) [62] | Atomic (~1-5 Å for structures; site-specific for dynamics) |
| Sample Requirement | ~0.1-1 mg/ml, high homogeneity [62] | ~0.1-1 mM, high solubility & stability |
| Sample State | Vitrified solution (single particles) | Solution in membrane mimetics (micelles, nanodiscs) |
| Key Output | Static 3D density map & atomic model | Ensemble of structures & dynamic parameters |
| Strengths | - Handles large complexes & membranes- Visualizes lipid densities directly- No crystallization needed | - Probes dynamics & kinetics- Identifies weak/transient interactions- Provides atomic-level detail in solution |
| Limitations for MOMP | - Difficulties with heterogeneous samples- Limited time resolution- Lipid identification can be ambiguous | - Size limitation for full-length proteins in membranes- Lower throughput for structure determination |
| Ideal for MOMP Studies | - Oligomeric BAK/BAX pore structures- Visualizing lipid binding sites in OMM proteins | - Mapping BH3 domain binding to BAX/BAK- Characterizing activation-related conformational changes |
Table 2: Key Research Reagents and Resources for MOMP Structural Biology
| Category | Item | Function in Research |
|---|---|---|
| Membrane Mimetics | Detergents (e.g., DDM, LMNG) | Solubilize and stabilize membrane proteins for purification and analysis [62]. |
| Amphipols (e.g., A8-35) | Synthetic polymers that stabilize membrane proteins in the absence of detergents, often improving stability for cryo-EM [62]. | |
| Nanodiscs (MSP/SAP) | Provide a native-like lipid bilayer environment for reconstituting membrane proteins, crucial for functional and structural studies [62]. | |
| Chemical Tools | BH3 Peptides & SAHBs | Stabilized alpha-helical peptides used to activate BAX/BAK and study protein-protein interactions in NMR and biochemical assays [60]. |
| BH3 Mimetics (e.g., Venetoclax) | Small-molecule drugs that inhibit pro-survival BCL-2 proteins; used as tools to study apoptosis and in therapeutic contexts [17]. | |
| Computational Resources | BioDolphin Database | A curated database of over 127,000 lipid-protein interactions, enabling searches and analysis of lipid-binding proteins [66]. |
| LipIDens/PyLipID | MD simulation pipelines that identify lipid binding sites and assist in interpreting lipid-like densities in cryo-EM maps [65]. | |
| Protein Data Bank (PDB) | Central repository for 3D structural data of proteins and nucleic acids, including cryo-EM and NMR structures of BCL-2 family proteins [66]. |
Visualizing the Apoptotic Signaling Pathway and Methodological Integration
The following diagram illustrates the key steps of MOMP within the intrinsic apoptotic pathway and indicates where cryo-EM and NMR provide critical structural and dynamic insights.
The synergistic application of cryo-EM and NMR spectroscopy provides a powerful, multi-faceted approach to dissect the protein-lipid interactions central to MOMP and apoptosis. Cryo-EM delivers high-resolution architectural blueprints of the key molecular machines, like the BAK/BAX pore, within a near-native membrane context. NMR complements this by revealing the dynamic conformational transitions and transient interactions that precede and regulate pore formation. As these techniques continue to advance—driven by better detectors, novel sample preparation methods, and integrative computational tools like LipIDens—our atomic-level understanding of cell death will deepen. This knowledge is not merely academic; it is the foundation for rationally designing next-generation therapeutics, such as BH3 mimetics, to treat cancer and other diseases characterized by dysregulated apoptosis. By providing detailed methodologies and resources, this guide aims to empower researchers in their quest to unravel the structural mysteries of life and death.
Resolving Controversies and Technical Challenges in MOMP Research
Mitochondrial outer membrane permeabilization (MOMP) is a pivotal event in the intrinsic apoptosis pathway, acting as a "point of no return" that leads to the release of cytochrome c and other apoptogenic factors into the cytosol, ultimately triggering cell death [67] [68]. This process is critically implicated in numerous human diseases, including cancer, neurodegenerative disorders, and ischemic injuries of the heart and brain [69] [70]. Despite decades of intensive research, the precise molecular identity of the pores responsible for MOMP remains highly controversial, with three primary candidates emerging: the voltage-dependent anion channel (VDAC) oligomers, the mitochondrial permeability transition pore (mPTP), and Bax/Bak oligomers [71] [72] [73]. This debate is not merely academic; it carries profound implications for understanding fundamental biological processes and developing targeted therapeutic interventions for apoptosis-related pathologies. This review synthesizes current evidence, experimental approaches, and competing models to provide researchers with a comprehensive framework for evaluating these distinct yet potentially interconnected mechanisms of mitochondrial membrane permeabilization.
The Candidates: Molecular Structures and Proposed Mechanisms
Voltage-Dependent Anion Channel (VDAC) Oligomers
VDAC, particularly the VDAC1 isoform, is the most abundant protein in the mitochondrial outer membrane and serves as the primary gatekeeper for metabolite and ion exchange between the cytosol and mitochondria [69]. Under physiological conditions, VDAC regulates mitochondrial metabolism by facilitating the passage of anions such as ATP and ADP in its open state, while showing cation selectivity in its closed state [69]. However, under conditions of oxidative stress, VDAC1 can undergo oligomerization to form large pores in the mitochondrial outer membrane that facilitate the release of mitochondrial DNA (mtDNA) and activate innate immune signaling [71].
The N-terminal domain of VDAC1 contains positively charged residues that interact with mtDNA and promote VDAC oligomerization [71]. This oligomerization appears to be a regulated process, as the highly potent VDAC1 oligomerization inhibitor VBIT-4 can decrease mtDNA release, IFN signaling, and disease severity in a mouse model of systemic lupus erythematosus [71]. Importantly, VDAC-mediated MOMP can occur independently of Bax/Bak, as demonstrated in Bax/Bak-deficient mouse embryonic fibroblasts (MEFs), where a fungal diterpene called cyathin-R was shown to induce VDAC1-dependent apoptosis through promotion of VDAC1 oligomerization [74].
Table 1: Key Evidence Supporting VDAC as a Pore-Forming Structure in MOMP
| Evidence Type | Experimental System | Key Finding | Reference |
|---|---|---|---|
| Genetic Knockout | Vdac1−/−, Vdac3−/−, and Vdac1/3−/− MEFs | Reduced cytosolic mtDNA and IFN-stimulated gene expression | [71] |
| Pharmacological Inhibition | VBIT-4 (VDAC oligomerization inhibitor) | Decreased mtDNA release and ameliorated lupus-like disease in mice | [71] |
| Biochemical Reconstitution | VDAC1-reconstituted liposomes | Increased mtDNA passage across lipid membrane | [71] |
| Chemical Biology Screening | Bax/Bak-deficient MEFs treated with cyathin-R | Induced VDAC1-dependent apoptosis via VDAC1 oligomerization | [74] |
| Antibody Blocking | Anti-VDAC antibodies in HepG2 cells | Prevented superoxide-induced cytochrome c release | [75] |
The Mitochondrial Permeability Transition Pore (mPTP)
The mPTP is conceptualized as a non-selective channel in the inner mitochondrial membrane that opens in response to elevated matrix Ca²⁺ and oxidative stress, permitting the diffusion of molecules up to 1.5 kDa in size [72]. Sustained mPTP opening causes mitochondrial swelling, rupture of the outer mitochondrial membrane, and subsequent apoptotic or necrotic cell death [72] [70]. The mPTP has been extensively studied in the context of ischemia-reperfusion injury, where it serves as a key trigger of pathological cell death [70].
The molecular composition of the mPTP has been intensely debated for decades. While initially proposed to consist of VDAC in the outer membrane, adenine nucleotide translocase (ANT) in the inner membrane, and cyclophilin D (CypD) in the matrix, recent genetic studies have challenged this model [72] [69]. Current evidence increasingly implicates the mitochondrial F₁F₀ (F)-ATP synthase dimers, monomers, or c-subunit ring alone as the core pore-forming structure of the mPTP, with CypD facilitating the transition to the pore-forming conformation [72]. However, this model remains controversial, and it is likely that not all contributing channel-forming complexes have yet been identified [72].
Table 2: Proposed Molecular Components of the mPTP
| Component | Localization | Proposed Role | Current Evidence Status |
|---|---|---|---|
| F-ATP synthase | Inner membrane | Putative pore-forming structure | Strong evidence from genetic and structural studies [72] |
| Adenine nucleotide translocase (ANT) | Inner membrane | Regulator rather than core component | Genetic deletion impairs but does not eliminate mPTP [72] |
| Cyclophilin D (CypD) | Matrix | Regulatory protein that facilitates pore opening | Well-established regulator; genetic deletion desensitizes mPTP [72] [70] |
| Voltage-dependent anion channel (VDAC) | Outer membrane | Not essential for inner membrane pore | mPTP opening occurs in VDAC-deficient mitochondria [72] [67] |
| Bax/Bak | Outer membrane | Regulators of outer membrane permeability during mPTP | Required for mPTP-dependent necrotic cell death [73] |
Bax/Bak Oligomers
Bax and Bak are pro-apoptotic members of the Bcl-2 protein family that are considered central regulators of apoptotic cell death [73] [68]. In response to apoptotic stimuli, these proteins undergo conformational changes, translocate to the mitochondrial outer membrane, and form oligomeric pores that directly mediate MOMP and cytochrome c release [73] [68]. Cryo-electron microscopy studies have revealed that activated Bax mediates the formation and indefinite enlargement of circular pores in membrane vesicles, reaching diameters in the hundreds of nanometers, with Bax molecules densely lining the pore edges [68].
The relative importance of Bax mitochondrial residence versus oligomerization has been investigated using Bax mutants. The G179P variant, which cannot form large oligomers but can still insert into membranes, permeabilizes liposomes but not native mitochondrial outer membrane vesicles (OMVs) [68]. In contrast, the T182I variant inserts poorly into membranes but resides predominantly on mitochondria in cells due to slow retrotranslocation, and mediates apoptosis as efficiently as wild-type Bax [68]. This suggests that Bax's mitochondrial residence in vivo may be more critical for its pro-apoptotic activity than its ability to form higher-order oligomers in model membranes [68].
Beyond their established role in apoptosis, Bax and Bak also function as the outer membrane component of the mitochondrial permeability transition pore (MPTP) in necrotic cell death [73]. Loss of Bax/Bak reduces outer mitochondrial membrane permeability and conductance without altering inner membrane MPTP function, resulting in resistance to mitochondrial calcium overload and necrotic cell death [73].
Comparative Analysis: Key Differences and Experimental Evidence
Structural and Functional Distinctions
The three proposed pore structures differ significantly in their molecular composition, regulation, and functional consequences. VDAC oligomers and Bax/Bak pores form specifically in the outer mitochondrial membrane, while the mPTP is primarily an inner membrane phenomenon [72] [69] [68]. VDAC oligomerization occurs under conditions of oxidative stress and can proceed independently of Bax/Bak, representing a mechanism for MOMP in live cells without immediate commitment to apoptosis [71]. In contrast, Bax/Bak activation typically represents a more definitive step toward apoptotic cell death, though these proteins can also participate in mPTP-dependent necrosis [73].
The mPTP differs fundamentally from both VDAC and Bax/Bak pores in its regulation by matrix calcium and its direct impact on inner membrane potential [72] [70]. While sustained mPTP opening leads to mitochondrial swelling and outer membrane rupture, transient opening at various sub-conductance states may contribute to physiological roles such as alterations in mitochondrial bioenergetics and rapid Ca²⁺ efflux [72].
Experimental Systems and Methodologies
The investigation of these distinct pore types requires different experimental approaches, which has contributed to the ongoing controversies in the field. VDAC oligomerization has been studied using VDAC-reconstituted liposomes to measure mtDNA passage, genetic knockout models, and specific inhibitors like VBIT-4 [71]. Bax/Bak oligomerization is frequently investigated using liposome or outer membrane vesicle (OMV) permeabilization assays, size-exclusion chromatography to examine oligomeric status, and retrotranslocation assays to assess mitochondrial residence [68].
The study of mPTP presents unique technical challenges, as standard practices for plasmalemmal channel proteins do not work well for multi-subunit mitochondrial membrane proteins like the F-ATP synthase [72]. Mitoplast patch-clamp electrophysiology has been used to characterize mPTP channel activity, but linking these measurements to molecular identity has proven difficult [72]. Additionally, the successful preparation of intact F-ATP synthase dimers has only been possible using very mild detergents such as digitonin, glyco-diosgenin (GDN), or lauryl maltose neopentyl glycol (LMNG) [72].
Essential Research Tools and Methodologies
Key Experimental Models and Reagents
The investigation of mitochondrial pores requires specialized research tools that enable the dissection of these complex molecular events. The following table summarizes essential reagents and their applications in MOMP research.
Table 3: Research Reagent Solutions for Investigating Mitochondrial Pores
| Reagent / Model | Type | Primary Application | Key Findings Enabled |
|---|---|---|---|
| VBIT-4 | Small molecule inhibitor | VDAC oligomerization inhibition | Decreased mtDNA release and IFN signaling; ameliorated lupus in mice [71] |
| Bax/Bak DKO MEFs | Genetic cell model | Studying apoptosis-independent MOMP | Revealed VDAC-mediated mtDNA release in absence of Bax/Bak [71] [74] |
| Cyathin-R | Natural product | Inducing VDAC1-dependent apoptosis | Demonstrated Bax/Bak-independent apoptosis via VDAC1 oligomerization [74] |
| Cyclosporine A | Pharmacological inhibitor | mPTP inhibition (via CypD binding) | Established role of mPTP in ischemia-reperfusion injury [70] |
| Liposome/OMV assays | In vitro reconstitution | Studying pore formation mechanisms | Revealed differential requirements for Bax oligomerization in model vs. native membranes [68] |
| Ppif−/− mice | Genetic animal model | Studying mPTP in pathophysiology | Demonstrated role of CypD in necrotic cell death [73] |
Critical Experimental Protocols
Several key methodologies have been instrumental in advancing our understanding of mitochondrial pore formation. The liposome/vesicle permeabilization assay involves loading liposomes or mitochondrial outer membrane vesicles (OMVs) with fluorescent dextrans of various sizes, then incubating them with recombinant proteins (e.g., Bax, VDAC) and activators (e.g., cBID) [68]. Permeabilization is measured by monitoring dextran release through fluorescence detection, providing quantitative data on pore-forming activity and size selectivity [68].
The mitochondrial swelling assay monitors mPTP opening in isolated mitochondria by measuring decreases in light scattering (absorbance at 540 nm) that occur as mitochondria swell due to inner membrane permeabilization [70]. This classical assay is typically triggered by calcium overload and is inhibited by cyclosporine A, confirming mPTP involvement [70].
For detecting VDAC oligomerization, researchers employ cross-linking experiments combined with Western blotting to visualize higher-order complexes, often complemented by genetic or pharmacological inhibition (e.g., VBIT-4) to establish functional significance [71] [74].
The calcium retention capacity (CRC) assay measures the ability of isolated mitochondria to accumulate calcium before triggering mPTP opening, providing a sensitive quantitative assessment of mPTP sensitivity to various pathological stimuli [70].
The debate surrounding the molecular identity of pores mediating MOMP continues to stimulate innovative research and paradigm shifts in cell death biology. Rather than mutually exclusive entities, VDAC oligomers, mPTP, and Bax/Bak oligomers may represent complementary mechanisms that operate under different physiological and pathological contexts. VDAC oligomerization appears particularly important for mtDNA release and innate immune activation under conditions of oxidative stress, while Bax/Bak oligomers serve as the canonical executioners of apoptotic cell death. The mPTP, with its growing connections to the F-ATP synthase, represents a distinct inner membrane trigger that can engage both apoptotic and necrotic pathways.
Future research directions should focus on elucidating the potential crosstalk between these pore systems, particularly how inner membrane mPTP events communicate with outer membrane permeabilization processes. The development of more specific pharmacological tools, advanced structural biology approaches, and conditional genetic models will be essential for resolving ongoing controversies. From a therapeutic perspective, the contextual requirements for each pore type present both challenges and opportunities for selective intervention in specific disease states. As our understanding of these complex mitochondrial pore systems continues to evolve, so too will our ability to target them for therapeutic benefit in the myriad diseases characterized by dysregulated cell death.
Addressing Conflicting Models of Bax/Bak Activation by BH3-Only Proteins
The process of mitochondrial outer membrane permeabilization (MOMP) serves as the pivotal commitment point in intrinsic apoptosis, governed by the interplay between BCL-2 family proteins. For decades, the precise mechanism by which BH3-only proteins activate the core executioners BAX and BAK has been subject to intense debate, giving rise to conflicting molecular models. This whitepaper examines the direct activation and derepression models, synthesizing biochemical, structural, and genetic evidence to arrive at a unified understanding of Bax/Bak activation. Resolving these mechanistic controversies carries profound implications for developing targeted cancer therapeutics, particularly BH3-mimetics, that manipulate this critical cell death switch.
MOMP represents the point of no return in the mitochondrial apoptosis pathway, triggering the release of cytochrome c and other apoptogenic factors that activate caspases and execute cell death [76]. The BCL-2 protein family constitutes the essential regulatory network controlling MOMP, comprising three functional subgroups: (1) multi-domain anti-apoptotic proteins (BCL-2, BCL-XL, MCL-1); (2) multi-domain pro-apoptotic effectors (BAX, BAK); and (3) pro-apoptotic BH3-only proteins (BID, BIM, PUMA, NOXA, etc.) that initiate apoptosis signaling [5]. The founding member BCL-2 was originally identified at the breakpoint of the t(14;18) chromosomal translocation in follicular lymphoma, representing a novel class of oncogenes that promote cell survival rather than proliferation [76] [5]. Genetic studies of Bax-/-Bak-/- double knockout mice definitively established BAX and BAK as the essential gatekeepers of MOMP, without which cells become profoundly resistant to most apoptotic stimuli [76].
Conflicting Models of Bax/Bak Activation
The Direct Activation Model
The direct activation model proposes that certain "activator" BH3-only proteins (BID, BIM, PUMA) directly bind and conformationally activate BAX and BAK, initiating their oligomerization and membrane pore formation [76] [77]. This model distinguishes between "activator" and "sensitizer" BH3-only proteins, with activators capable of directly engaging BAX/BAK. Structural studies reveal that activator BH3 domains bind to a hydrophobic groove on BAX/BAK, inducing profound conformational changes that expose their own BH3 domains and N-terminal regions, enabling homo-oligomerization [76] [78]. The interaction between activator BH3 domains and BAX/BAK is characteristically transient and dynamic ("hit-and-run"), consistent with their low binding affinity, which enables the subsequent critical step of BAX/BAK homo-oligomerization [76].
The Indirect Activation (Derepression) Model
The derepression model posits that BH3-only proteins function primarily by neutralizing anti-apoptotic BCL-2 members, thereby displacing pre-bound BAX/BAK or other pro-apoptotic factors to induce apoptosis indirectly [79]. In this model, BH3-only proteins are classified based on their binding specificities toward different anti-apoptotic members rather than their ability to directly activate BAX/BAK. The model suggests that anti-apoptotic proteins maintain survival by constitutively sequestering active BAX/BAK molecules or their activator proteins [79]. Apoptosis occurs when sufficient BH3-only proteins are unleashed to engage all available anti-apoptotic binding sites, freeing BAX/BAK to oligomerize.
Unified Model
Recent research has converged on a unified model that incorporates elements from both conflicting theories [76] [79]. This synthetic model acknowledges that BH3-only proteins function through dual mechanisms: direct activation of BAX/BAK and indirect activation through inhibition of anti-apoptotic members. The unified model identifies two distinct modes of apoptosis inhibition by pro-survival BCL-2 proteins: MODE 1 (sequestering activator BH3-only proteins) and MODE 2 (directly binding active BAX/BAK) [79]. Importantly, MODE 1 sequestration is less efficient and more easily overcome to promote MOMP compared to MODE 2 inhibition [79]. This hierarchical regulation helps explain the differential apoptotic sensitivities observed across cell types and stress conditions.
Structural Insights into BH3-Mediated Activation
The structural basis of BCL-2 family interactions involves a conserved "BH3-in-groove" mechanism where the amphipathic α-helical BH3 domain of one protein engages the hydrophobic binding groove formed by BH1-3 domains of another [76]. Anti-apoptotic members possess a stable, constitutively exposed binding groove that tightly sequesters BH3 domains, while the grooves of BAX and BAK remain largely inaccessible in their inactive states [76].
BAX activation involves a multi-step process: in viable cells, cytosolic BAX exists as an auto-inhibited monomer with its C-terminal α9 helix engaged in its own dimerization groove [76]. Activator BH3 binding to the α1 helix or α1/α6 trigger site induces mitochondrial targeting and conformational changes that expose the BAX BH3 domain and C-terminal transmembrane anchor [76] [78].
BAK activation follows a somewhat different pathway: BAK is constitutively integrated into the mitochondrial outer membrane via its C-terminal anchor and maintained as an inactive monomer by VDAC2 [76]. Activator BH3 binding induces similar conformational changes that expose the BAK BH3 domain, enabling homo-oligomerization [76].
Structural studies of BAK in complex with the non-canonical BH3-only protein Pxt1 revealed atomic-level details of this activation mechanism, showing how the Pxt1 BH3 helix engages the canonical binding groove of BAK to initiate its pro-apoptotic activity [80].
Table 1: Classification of BH3-Only Proteins Based on Function and Specificity
| Category | BH3-Only Proteins | Anti-apoptotic Targets | BAX/BAK Activation |
|---|---|---|---|
| Activators | BID, BIM, PUMA | BCL-2, BCL-XL, MCL-1 (PUMA) | Direct activation |
| Sensitizers | BAD, NOXA, BIK, BMF, HRK | BCL-2/BCL-XL (BAD), MCL-1 (NOXA) | Indirect only |
| Promiscuous | PUMA, BIM | All major anti-apoptotic members | Direct activation |
Experimental Evidence and Methodologies
Key Experimental Approaches
Multiple complementary approaches have been employed to resolve the mechanism of Bax/Bak activation:
Membrane permeabilization assays using purified components demonstrated that BID and BIM BH3 peptides directly activate BAX to permeabilize liposomal membranes, while other BH3 peptides only sensitize by inhibiting anti-apoptotic proteins [77] [81]. These assays quantified cytochrome c release from mitochondria or fluorescent dextran release from liposomes.
Structural biology techniques including X-ray crystallography, NMR spectroscopy, and cryo-EM have visualized complexes between BH3 peptides and BAX/BAK, revealing the molecular details of the activation mechanism [76] [80]. The crystal structure of BAK bound to the Pxt1 BH3 domain provided direct evidence of BH3-mediated activation [80].
Genetic knockout studies established the essential roles of specific components. While Bax-/-Bak-/- mice demonstrated their absolute requirement for apoptosis, Bid-/-Bim-/-Puma-/-Noxa-/- quadruple knockout mice revealed that BH3-independent autoactivation can occur when anti-apoptotic proteins are downregulated, albeit with slower kinetics [76].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagents for Studying Bax/Bak Activation
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Recombinant Proteins | Purified BAX, BAK, BCL-2, BCL-XL, MCL-1 | In vitro binding and membrane permeabilization assays |
| BH3 Peptides | BID BH3, BIM BH3, BAD BH3, NOXA BH3 | Mapping interactions and specificities |
| BH3 Mimetics | ABT-737 (BCL-2/XL/W), ABT-199/Venetoclax (BCL-2), A-1210477 (MCL-1) | Therapeutic targeting and mechanistic studies |
| Cellular Assays | Cytochrome c release, Caspase activation, Annexin V staining | Monitoring apoptosis in cellular contexts |
| Structural Tools | NMR, X-ray crystallography, Cryo-EM | Determining atomic-level interaction mechanisms |
Figure 1: Unified Model of Bax/Bak Activation by BH3-Only Proteins. This schematic integrates both direct activation and derepression mechanisms, showing how different BH3-only protein classes coordinate to induce MOMP.
Research Methods and Experimental Protocols
Liposomal Membrane Permeabilization Assay
The liposomal membrane permeabilization assay provides a cell-free system to study direct BAX/BAK activation, eliminating complicating cellular factors [77] [81]. The core protocol involves:
Liposome Preparation: Create unilamellar liposomes containing fluorescent markers (e.g., calcein, dextran-conjugated fluorophores) and mitochondrial lipids (phosphatidylcholine, phosphatidylethanolamine, cardiolipin) using extrusion or sonication methods.
Protein Purification: Express and purify recombinant BAX, BAK, and BH3-only proteins from E. coli or insect cells. For BAX, ensure the protein is in its inactive, monomeric form through careful purification and storage conditions.
Activation Measurement: Incubate liposomes with BAX/BAK in the presence or absence of BH3 peptides (typically 0.1-10 µM). Monitor fluorescence dequenching in real-time as the fluorescent marker is released through pores. Include controls with anti-apoptotic proteins to test inhibition.
Data Analysis: Quantify the kinetics and extent of membrane permeabilization. Compare different BH3 peptides to classify them as activators or sensitizers based on their direct membrane-permeabilizing capability.
BH3 Profiling and Competitive Binding Assays
BH3 profiling measures mitochondrial sensitivity to different BH3 peptides as a functional readout of apoptotic priming [77] [81]:
Mitochondrial Isolation: Prepare intact mitochondria from cells or tissues via differential centrifugation.
BH3 Peptide Exposure: Treat mitochondria with synthetic BH3 peptides (typically 0.1-100 µM) representing different specificities. Include positive (e.g., FCCP) and negative controls.
Cytochrome c Release Measurement: After incubation, separate mitochondria from supernatant by centrifugation. Detect cytochrome c in supernatant by immunoblotting or ELISA.
Interpretation: Mitochondria that release cytochrome c in response to direct activator peptides (BID, BIM) but not sensitizer peptides (BAD, NOXA) indicate dependence on specific anti-apoptotic proteins for survival.
Implications for Therapeutic Development
The resolved mechanism of Bax/Bak activation has directly enabled rational design of cancer therapeutics, particularly BH3-mimetics that syntheticially mimic native BH3 domains [5]. Venetoclax (ABT-199), the first FDA-approved BCL-2-specific inhibitor, demonstrates remarkable efficacy in hematologic malignancies by selectively displacing pro-apoptotic proteins from BCL-2 [5]. The unified model explains why resistance can emerge through upregulation of MCL-1 or BCL-XL, which are not targeted by venetoclax, prompting development of combination therapies or broader-spectrum inhibitors [5] [82].
Current challenges in the field include the thrombocytopenia associated with BCL-XL inhibition and cardiac toxicities of MCL-1 inhibitors, driving research into novel delivery approaches such as proteolysis-targeting chimeras (PROTACs) and antibody-drug conjugates to achieve tumor-specific targeting [5]. The structural insights from BH3-groove interactions continue to inform the design of next-generation therapeutics with improved specificity and safety profiles.
The longstanding controversy surrounding Bax/Bak activation mechanisms has progressively resolved into a unified model that incorporates both direct activation and derepression mechanisms. Structural, biochemical, and genetic evidence confirms that specific BH3-only proteins (BID, BIM, PUMA) can directly activate BAX and BAK, while others function primarily by neutralizing specific anti-apoptotic guardians. This hierarchical, multi-layered regulation provides robustness to the apoptotic switch while enabling integration of diverse cellular stress signals. The resolved mechanism continues to guide therapeutic innovation in manipulating cell death for cancer treatment, representing a paradigm for translating basic mechanistic insights into clinical advances.
Mitochondrial outer membrane permeabilization (MOMP) is a decisive event in the intrinsic pathway of apoptosis, serving as a point of no return for cell commitment to death [38] [83]. This process is characterized by the permeabilization of the outer mitochondrial membrane, leading to the release of apoptogenic factors such as cytochrome c from the intermembrane space into the cytosol [84]. Once in the cytosol, cytochrome c facilitates the formation of the apoptosome, which activates caspase proteases that execute the dismantling of the cell [85]. The integrity of the mitochondrial outer membrane is therefore essential for optimal mitochondrial function and cellular survival, as its permeabilization directly couples with the loss of mitochondrial membrane potential (ΔΨm) and disrupts ATP production [38] [83].
Accurate detection and quantification of MOMP are crucial for both fundamental apoptosis research and drug development, particularly in screening compounds that modulate cell death pathways in cancer and other diseases [85]. However, the field faces significant technical challenges, as MOMP is not always a synchronous, all-or-nothing event across the entire mitochondrial network [84] [86]. The emergence of "minority MOMP" – where only a subset of mitochondria undergo permeabilization – introduces substantial complexity for measurement and interpretation [86]. This phenomenon results in sub-lethal caspase activation and can promote genomic instability and carcinogenesis, making its detection both technically challenging and biologically critical [86]. This technical guide examines the current methodologies for MOMP detection, their associated pitfalls, and optimized approaches to overcome these challenges.
Key Methodological Approaches for MOMP Detection
Cytochrome c Release Assays
The release of cytochrome c from the mitochondrial intermembrane space into the cytosol represents a direct biochemical consequence of MOMP and serves as a gold-standard marker for its detection [85]. Multiple methodological approaches exist for monitoring this event.
Immunodetection Methods: Western blot analysis of cytochrome c localization in subcellular fractions (cytosolic vs. mitochondrial) provides a population-level, semi-quantitative assessment of MOMP [85]. While this method offers specificity through antibody-based detection, it involves disruptive cellular fractionation procedures that may artifactually release cytochrome c during sample preparation. Furthermore, it represents an endpoint measurement that destroys spatial information about which specific cells or mitochondria have undergone MOMP.
Imaging Approaches: Immunofluorescence microscopy using anti-cytochrome c antibodies enables the visualization of its redistribution in fixed cells, preserving spatial context [85]. Confocal laser scanning microscopy provides superior resolution for observing cytochrome c release patterns, while live-cell imaging using fluorescent protein-tagged cytochrome c allows real-time monitoring of MOMP dynamics in individual cells [85]. Recent advances include the use of carbon dots for cytochrome c detection, which offer improved photostability and sensitivity [85].
Table 1: Comparison of Cytochrome c Detection Methods
| Method | Spatial Resolution | Temporal Resolution | Quantification Capability | Key Limitations |
|---|---|---|---|---|
| Western Blot | None (population average) | Endpoint only | Semi-quantitative | Disruptive sample preparation; no single-cell data |
| Immunofluorescence | Single-cell | Endpoint only | Quantitative with image analysis | Fixed cells only; no dynamics |
| Live-cell Imaging with FP-tagged Cytochrome c | Single mitochondrion | Real-time (seconds-minutes) | Highly quantitative | Overexpression artifacts; phototoxicity |
| Carbon Dots-based Detection | Single-cell to subcellular | Endpoint or live-cell | Quantitative | Relatively new technology; validation ongoing |
Mitochondrial Membrane Potential (ΔΨm) Measurements
The loss of mitochondrial membrane potential frequently coincides with MOMP and serves as an indirect but readily measurable correlate [38] [83]. This coupling occurs because outer membrane permeabilization can disrupt the inner membrane's proton gradient, though the timing between these events can vary depending on cellular context.
Fluorescent Dyes: Membrane-permeable lipophilic and cationic fluorescent dyes remain the most common approach for assessing ΔΨm [85]. These dyes accumulate in the mitochondrial matrix in a potential-dependent manner, with fluorescence intensity or emission shifts reflecting ΔΨm status. Tetramethylrhodamine esters (e.g., TMRE, TMRM) and JC-1 are widely utilized, with JC-1 offering the advantage of forming J-aggregates that emit at different wavelengths depending on the membrane potential [85]. However, these dyes are susceptible to photobleaching, can exhibit concentration-dependent artifacts, and may themselves influence mitochondrial function through oxidative stress generation.
Biosensors: Genetically-encoded biosensors for ΔΨm represent an emerging alternative to chemical dyes, offering cell-specific targeting and minimal perturbation [38] [83]. These typically consist of fluorescent proteins targeted to the mitochondrial matrix, whose fluorescence properties change with membrane potential. While avoiding dye-related toxicity and providing reproducible expression levels, they generally offer lower dynamic range compared to chemical dyes and require genetic manipulation of the target cells.
Table 2: Comparison of ΔΨm Detection Methods
| Method | Measurement Type | Temporal Resolution | Advantages | Technical Pitfalls |
|---|---|---|---|---|
| TMRE/TMRM | Fluorescence intensity | Medium (minutes) | Reversible; suitable for kinetics | Photobleaching; concentration-dependent artifacts |
| JC-1 | Fluorescence shift (greenred) | Medium (minutes) | Rationetric measurement | Probe aggregation issues; complex loading |
| Genetically-encoded Biosensors | Fluorescence intensity/FRET | Medium (minutes) | Cell-specific expression; minimal toxicity | Lower dynamic range; genetic manipulation required |
| Luminescence Assays | Luminescence intensity | Low (endpoint) | No photobleaching; high throughput | Low spatial information; endpoint measurement |
Bax/Bak Activation and Oligomerization
MOMP execution is primarily mediated by the pro-apoptotic Bcl-2 family proteins Bax and Bak, which undergo conformational activation and oligomerization to form pores in the outer mitochondrial membrane [86]. Monitoring these molecular events provides the most direct assessment of MOMP initiation.
Conformational Changes: Activation-specific antibodies that recognize exposed N-terminal epitopes on Bax or Bak can detect their activation status through immunocytochemistry or flow cytometry [85]. These methods preserve information about the subcellular localization of active proteins but are limited to fixed samples and may miss transient activation states.
Oligomerization Status: Cross-linking experiments followed by immunoblotting can detect Bax/Bak oligomer formation, a key step in pore assembly [85]. While providing biochemical evidence of activation, this approach is disruptive and represents a population average. Advanced techniques such as fluorescence cross-correlation spectroscopy and single-molecule localization microscopy offer superior resolution for studying protein interactions and oligomerization in live or fixed cells, respectively [85].
Critical Technical Pitfalls and Limitations
Challenges in Detecting Heterogeneous and Sub-Lethal MOMP
A fundamental limitation in MOMP detection stems from the assumption that it occurs synchronously throughout the mitochondrial network. In reality, MOMP often exhibits significant heterogeneity, with "minority MOMP" representing a particularly challenging phenomenon to detect and quantify [84] [86]. In this scenario, only a small fraction of mitochondria undergo permeabilization, releasing insufficient cytochrome c to activate robust caspase signaling and commit the cell to death [86]. This sub-lethal event can promote DNA damage and genomic instability through limited caspase-activated DNase (CAD) activity, yet standard population-averaging detection methods frequently miss it entirely [84] [86].
Flow cytometry-based assessments of cytochrome c release or ΔΨm collapse typically gating on cells displaying complete changes will systematically exclude cells undergoing minority MOMP, as they exhibit intermediate signals that are often dismissed as technical noise or incomplete staining [85]. Similarly, western blot analysis of subcellular fractions lacks the sensitivity to detect minor redistributions of cytochrome c affecting only a small mitochondrial subset. Only single-cell imaging approaches possess the requisite spatial resolution to identify minority MOMP events, yet even these methods face sensitivity challenges when the permeabilized minority represents a very small proportion of the total mitochondrial complement [84].
Temporal Resolution and Live-Cell Imaging Challenges
MOMP is a dynamic process that can propagate through the mitochondrial network at variable rates, making temporal resolution a critical factor in accurate detection [85]. Endpoint measurements provide a static snapshot that may miss the initiation, propagation, or reversal of MOMP, potentially leading to incorrect conclusions about its regulation and functional consequences.
Live-cell imaging presents its own technical challenges, particularly regarding phototoxicity and probe limitations [85]. Fluorescent proteins and dyes used for prolonged imaging can generate reactive oxygen species that themselves influence MOMP induction, creating artifacts that confound experimental results. Additionally, the overexpression of fluorescent protein-tagged constructs (e.g., cytochrome c-GFP) may alter natural kinetics and regulation of MOMP, potentially exaggerating or suppressing the phenomenon under investigation [85].
Specificity and Artifact Concerns
Many commonly used MOMP detection methods suffer from significant specificity issues that can lead to misinterpretation. The widely used ΔΨm-sensitive dyes, for instance, respond not only to MOMP but to any process affecting mitochondrial energetics, including uncoupling, inhibition of electron transport, or changes in substrate availability [38] [83]. The assumption that ΔΨm collapse specifically indicates MOMP can therefore be erroneous, particularly in pathological contexts where mitochondrial dysfunction may precede apoptosis.
Sample preparation artifacts represent another major concern. The mechanical stress of cell sorting or the permeability treatments used for antibody access in immunocytochemistry can disrupt mitochondrial integrity, artifactually releasing cytochrome c and generating false positives [85]. Even subtle changes in temperature, pH, or osmotic balance during experimental procedures can induce permeability transitions that mimic MOMP, particularly when working with isolated mitochondria [87].
Optimized Experimental Approaches and Protocols
Multi-Parameter Assessment Strategy
To overcome the limitations of individual methods, a multi-parameter approach combining complementary techniques provides the most robust assessment of MOMP. This strategy should integrate direct measurements (e.g., cytochrome c localization) with correlative indicators (e.g., ΔΨm collapse, Bax activation) to build a comprehensive picture of MOMP induction and consequences.
Recommended workflow:
- Initial screening using plate-based luminescence or fluorescence assays for high-throughput capability
- Validation of positive hits by high-content imaging to assess heterogeneity and single-cell responses
- Mechanistic follow-up using super-resolution microscopy or biochemical approaches to confirm molecular events
This tiered approach balances throughput with mechanistic depth while mitigating the risk of false positives/negatives inherent in any single method.
Protocol: Integrated Cytochrome c Redistribution and ΔΨm Assessment
This protocol combines immunocytochemistry with ΔΨm sensing for correlative single-cell analysis of MOMP in cultured cells.
Materials:
- Cells grown on glass coverslips
- ΔΨm-sensitive dye (e.g., TMRE, 50-100 nM)
- Phosphate-buffered saline (PBS)
- Fixation solution (4% paraformaldehyde in PBS)
- Permeabilization solution (0.1% Triton X-100 in PBS)
- Blocking solution (5% normal serum in PBS)
- Primary antibody against cytochrome c (clone 6H2.B4)
- Fluorescently-labeled secondary antibody
- Mounting medium with DAPI
- Confocal or epifluorescence microscope with high-resolution camera
Procedure:
- Induce apoptosis in cells using the desired stimulus while including appropriate controls.
- 30 minutes before the end of treatment, load cells with TMRE (50 nM in culture medium) at 37°C.
- Rinse cells briefly with pre-warmed PBS to remove extracellular dye.
- Fix cells with 4% PFA for 15 minutes at room temperature.
- Permeabilize with 0.1% Triton X-100 for 5 minutes.
- Block with 5% normal serum for 30 minutes.
- Incubate with anti-cytochrome c primary antibody (1:200 in blocking solution) for 1 hour at room temperature.
- Wash 3× with PBS for 5 minutes each.
- Incubate with fluorescent secondary antibody (1:500) for 45 minutes protected from light.
- Wash 3× with PBS for 5 minutes each.
- Mount coverslips and image using appropriate filter sets.
Interpretation: Cells undergoing MOMP will display diffuse cytochrome c staining throughout the cell (versus punctate mitochondrial pattern in healthy cells) concurrently with diminished TMRE fluorescence. This combined assessment controls for false positives where ΔΨm collapse occurs independently of MOMP.
Protocol: BH3 Profiling for Functional MOMP Priming
BH3 profiling represents a functional assessment of MOMP readiness by measuring mitochondrial sensitivity to pro-apoptotic BH3 peptides, providing predictive information about apoptotic predisposition beyond static molecular measurements [86].
Materials:
- Permeabilization buffer (containing 150 mM KCl, 10 mM HEPES, pH 7.5, 1 mM EGTA, 0.1% BSA)
- Digitonin (5 mg/mL stock)
- BH3 peptides (e.g., BIM, BID, BAD, NOXA)
- ΔΨm-sensitive dye (JC-1 or TMRE)
- Plate reader or flow cytometer
- Isolated mitochondria or intact cells
Procedure for Isolated Mitochondria:
- Isolate mitochondria from fresh tissue or cells using differential centrifugation.
- Load mitochondria with JC-1 (2 μM) for 15 minutes at room temperature.
- Aliquot mitochondria (50 μg protein) into 96-well plates.
- Add BH3 peptides (10-100 μM final concentration) in permeabilization buffer.
- Monitor fluorescence changes over 60-90 minutes (JC-1: 590/530 nm emission ratio; TMRE: fluorescence decrease).
- Calculate percentage ΔΨm loss relative to controls.
Interpretation: Mitochondria primed for MOMP will undergo rapid depolarization in response to specific BH3 domains, with sensitivity patterns indicating which anti-apoptotic Bcl-2 proteins (Bcl-2, Bcl-xL, Mcl-1) are maintaining survival. This functional assay complements structural assessments of MOMP and can identify cells undergoing minority MOMP through their altered sensitivity profiles [86].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for MOMP Detection
| Reagent Category | Specific Examples | Primary Function | Key Considerations |
|---|---|---|---|
| ΔΨm-Sensitive Dyes | TMRE, TMRM, JC-1, Rhodamine 123 | Monitor mitochondrial membrane potential collapse | Concentration optimization critical; potential phototoxicity |
| Cytochrome c Detection | Anti-cytochrome c antibodies (clone 6H2.B4), Cytochrome c-GFP fusion | Direct visualization of cytochrome c release | Fixation/permeabilization conditions affect localization |
| Bax/Bak Activation | Conformation-specific antibodies (6A7 for Bax), Cross-linking agents | Detect activation and oligomerization of core apoptotic proteins | Transient states may be missed; membrane localization important |
| BH3 Peptides | BIM, BID, BAD, PUMA, MS1 | Profile functional priming for MOMP | Peptide purity and concentration critical; specificity patterns inform mechanism |
| Caspase Substrates | DEVD-ase substrates (fluorogenic/colorimetric) | Detect downstream apoptotic signaling | Indicates consequence rather than direct MOMP measurement |
| Permeabilization Agents | Digitonin, Streptolysin O, ALAMAR | Controlled plasma membrane permeabilization for subcellular fractionation | Concentration optimization essential to preserve mitochondrial integrity |
Visualizing MOMP Detection Strategies
The following diagrams illustrate key signaling pathways and methodological approaches for MOMP detection, providing visual guidance for experimental design and interpretation.
Diagram 1: MOMP Signaling Pathway and Detection Nodes
Diagram 2: Integrated MOMP Detection Workflow
Accurate measurement of MOMP remains challenging due to the dynamic and heterogeneous nature of this fundamental apoptotic event. No single method provides a complete picture, necessitating multi-parameter approaches that combine structural, functional, and biochemical assessments. The emergence of sub-lethal MOMP phenomena further complicates detection, requiring specialized methodologies capable of resolving minority events within cellular populations [84] [86].
Future methodological developments should focus on improving spatial and temporal resolution while minimizing perturbation. Biosensor technology continues to advance, with newer generations offering improved dynamic range and reduced artifacts [38] [83]. Super-resolution microscopy techniques are approaching the resolution needed to visualize individual Bax/Bak pores, potentially enabling direct observation of MOMP initiation. Additionally, computational approaches for analyzing the heterogeneity of MOMP responses will enhance our ability to extract meaningful information from complex single-cell data.
For the practicing researcher, maintaining awareness of the limitations inherent in each methodological approach is paramount. Validation through complementary techniques, careful experimental design to minimize artifacts, and appropriate interpretation within the biological context will continue to be essential for advancing our understanding of MOMP in health and disease.
Mitochondrial outer membrane permeabilization (MOMP) represents the pivotal commitment point in the intrinsic apoptosis pathway. Contemporary research has illuminated the profound influence of mitochondrial dynamics—the perpetual cycles of fission and fusion—on the regulation and execution of MOMP. This technical review examines the molecular machinery governing mitochondrial dynamics, with particular emphasis on Dynamin-Related Protein 1 (DRP1) as the central molecular node integrating fission with apoptotic progression. We synthesize current models describing how dysregulated fission facilitates MOMP through BCL-2 family protein activation and cristae remodeling, while fusion acts as a protective mechanism to counteract apoptosis. The document provides detailed experimental methodologies for quantifying these processes and presents emerging therapeutic strategies targeting mitochondrial dynamics to modulate cell death in cancer and neurodegenerative diseases.
Mitochondrial outer membrane permeabilization (MOMP) is considered the 'point of no return' in the intrinsic apoptosis pathway, characterized by the disruption of the mitochondrial outer membrane and subsequent release of pro-apoptotic proteins such as cytochrome c into the cytosol [3] [1]. Once released, cytochrome c activates the APAF-1 protein, triggering the formation of the apoptosome complex and initiating a caspase cascade that ultimately leads to programmed cell death [3]. The efficiency of MOMP is enhanced by proteins like Smac/DIABLO, which counteracts endogenous caspase inhibitors (IAPs), ensuring robust apoptosis execution [3].
Concurrently, mitochondrial dynamics describe the continuous and opposing processes of fission (division) and fusion (merging) that determine mitochondrial morphology, distribution, and quality control [88]. These dynamics are not merely structural phenomena but are fundamentally integrated with mitochondrial function, including their role in apoptosis. The balance between fission and fusion shapes the mitochondrial network, influencing cellular metabolism, stress responses, and ultimately, the decision to undergo MOMP [89] [88]. This review delineates the molecular mechanisms through which mitochondrial fission and fusion converge to regulate MOMP, thereby determining cellular fate.
Molecular Machinery of Mitochondrial Dynamics
Mitochondrial Fission Machinery
The division of mitochondria is a multi-step process primarily mediated by the cytosolic GTPase Dynamin-Related Protein 1 (DRP1). Upon activation, DRP1 is recruited to the mitochondrial outer membrane by specific receptor proteins, where it oligomerizes into helical structures that constrict and ultimately sever the mitochondrial tubule [89] [88].
- DRP1 Structure and Regulation: The DNM1L gene encodes DRP1, which structurally comprises an N-terminal GTPase domain, a middle domain involved in self-assembly, a variable domain (VD) containing most post-translational modification sites, and a C-terminal GTPase effector domain (GED) that regulates GTPase activity [89]. DRP1 function is precisely controlled through various post-translational modifications, particularly phosphorylation. For instance, phosphorylation at Ser616 (e.g., by CDK1, ERK, or CDK5) promotes DRP1 mitochondrial translocation and fission, while phosphorylation at Ser579 inhibits its activity [89].
- Mitochondrial Recruitment: DRP1 recruitment is facilitated by outer membrane receptors, including Mitochondrial Fission Factor (Mff), Mitochondrial Dynamics proteins Mid49 and Mid51, and Fis1 [89]. Although Fis1's role in physiological fission is minor, it participates in stress-induced fission complexes. Mff is considered the dominant receptor, capable of recruiting and activating oligomerized DRP1, while Mids preferentially bind inactive, dimerized DRP1 [89].
- Endoplasmic Reticulum (ER) and Actin Involvement: The initial constriction of mitochondria often occurs at ER-mitochondria contact sites. The ER-associated formin INF2 and Spire1C promote actin polymerization, working alongside myosin II to generate the initial force for membrane constriction before DRP1 recruitment [89].
Mitochondrial Fusion Machinery
Mitochondrial fusion involves the sequential merging of the outer and inner membranes, mediated by a distinct set of GTPases. This process allows for the mixing of mitochondrial contents, promoting complementation and functional homogeneity across the network [90] [88].
- Outer Membrane Fusion: This step is governed by Mitofusin 1 (MFN1) and Mitofusin 2 (MFN2). These proteins are embedded in the outer membrane and contain GTPase domains and heptad repeat regions (HR1 and HR2). Fusion is initiated when MFN proteins on opposing mitochondria form homo- or heterotypic complexes via their HR2 domains, tethering the membranes together. GTP hydrolysis then provides the energy to fuse the outer membranes [90]. MFN1 exhibits higher GTPase activity and is primarily fusogenic, whereas MFN2 also plays a critical role in tethering mitochondria to the endoplasmic reticulum [90].
- Inner Membrane Fusion: Fusion of the inner membrane is exclusively mediated by Optic Atrophy 1 (OPA1). OPA1 exists in multiple isoforms generated by alternative splicing and proteolytic processing. The long isoforms (L-OPA1) are anchored to the inner membrane, while proteolytic cleavage by proteases such as OMA1 and YME1L generates soluble short isoforms (S-OPA1) in the intermembrane space [90]. L-OPA1 is essential for fusion, but S-OPA1 works cooperatively with L-OPA1 to regulate membrane curvature and fusion efficiency. Together, they form higher-order complexes that drive the fusion process [90].
Table 1: Core Protein Machinery of Mitochondrial Dynamics
| Protein | Gene | Localization | Primary Function | Key Regulators |
|---|---|---|---|---|
| DRP1 | DNM1L | Cytosol / OMM | GTPase mediating mitochondrial fission | Phosphorylation (S616, S579), SUMOylation, Mff, Mid49/51 |
| Fis1 | FIS1 | OMM | Atypical receptor; role in stress-induced fission | Protein-protein interactions in complexes |
| Mff | MFF | OMM | Primary receptor for DRP1 recruitment | Post-translational modifications |
| MFN1 | MFN1 | OMM | GTPase mediating outer membrane fusion | Ubiquitination, Proteasomal degradation |
| MFN2 | MFN2 | OMM | GTPase mediating outer membrane & ER tethering | Ubiquitination, PINK1-Parkin |
| OPA1 | OPA1 | IMM | GTPase mediating inner membrane fusion & cristae structure | Proteolytic cleavage (OMA1, YME1L) |
Diagram 1: Mitochondrial Fission and Fusion Pathways
Molecular Interplay Between Dynamics and MOMP
Fission as a Prerequisite for Efficient MOMP
Extensive evidence positions mitochondrial fission as a critical enabling step for efficient MOMP. During apoptosis, mitochondrial networks typically undergo extensive fragmentation, which coincides with MOMP [89]. Several mechanisms underpin this relationship:
- Spatial Restriction of Anti-Apoptotic Proteins: Fission creates smaller, individual mitochondrial units, effectively limiting the diffusion and dilution of pro-apoptotic signals. This compartmentalization can prevent the complementation of damaged components by healthy ones, making individual mitochondria more susceptible to permeabilization [88].
- DRP1 and BCL-2 Family Protein Interaction: DRP1 acts as a central molecular link between the fission machinery and the core apoptotic apparatus. It can directly or indirectly facilitate the activation of BAX and BAK, the pro-apoptotic BCL-2 family proteins responsible for pore formation in the OMM during MOMP [89] [1]. In some cellular contexts, DRP1 is required for the full activation and oligomerization of BAX/BAK at mitochondrial fission sites.
- Cristae Remodeling: The protein OPA1, known for its role in inner membrane fusion, also maintains the tight cristae junctions of the inner membrane. During apoptosis, proteolytic cleavage of OPA1 disrupts cristae structure, a process known as cristae remodeling. This reorganization facilitates the complete release of cytochrome c from the intermembrane space into the cytosol after MOMP has occurred [90] [88]. As OPA1 processing is influenced by the fusion-fission balance, dynamics directly control the efficiency of cytochrome c release.
Fusion as a Mechanism of Apoptotic Resistance
Conversely, mitochondrial fusion acts as a cellular defense mechanism against MOMP. Elongated mitochondrial networks are generally more resistant to apoptosis [88].
- Content Mixing and Dilution: Fusion allows for the homogenization of mitochondrial contents. Damaged components, including oxidized proteins and lipids, can be diluted and neutralized across a larger network. This mixing can also allow mitochondria with damaged DNA to complement each other, maintaining overall function and resisting apoptotic triggers [88].
- Sequestration of Pro-Apoptotic Factors: A hyperfused network may more effectively sequester pro-apoptotic factors like cytochrome c, making their release less efficient even if a localized permeabilization event occurs. The integrity of OPA1-stabilized cristae is crucial for this retention [90].
The dynamic balance between these processes therefore constitutes a critical regulatory node in the cell's decision to live or die. Shifting the balance towards fission predisposes cells to MOMP, while promoting fusion enhances cellular survival.
Diagram 2: Integrated Pathway of Fission and MOMP in Apoptosis
Experimental Analysis: Methodologies and Protocols
Investigating the relationship between mitochondrial dynamics and MOMP requires a multi-faceted approach, combining live-cell imaging to visualize dynamics with biochemical techniques to assess apoptosis.
Live-Cell Imaging of Mitochondrial Dynamics
Visualizing mitochondrial morphology and dynamics in living cells is crucial for understanding their functional state.
- Fluorescent Labeling: Mitochondria can be labeled using:
- Genetically-Encoded Fluorescent Proteins: Stable expression of fluorescent proteins (e.g., mito-GFP, mito-RFP) targeted to the mitochondrial matrix via a targeting sequence provides specific and stable labeling ideal for long-term imaging [91] [92].
- Chemical Dyes: Cell-permeant dyes like MitoTracker系列 (e.g., MitoTracker Green FM, which is relatively independent of membrane potential) can be used for short-term imaging. However, potential dye toxicity and effects on membrane potential must be considered [91].
- Image Acquisition and Analysis:
- Protocol: Cells are plated on imaging-optimized dishes and maintained at 37°C with 5% CO₂ during imaging. Time-lapse sequences are acquired using a high-resolution confocal microscope with a 63x or higher oil-immersion objective [92].
- Quantitative Analysis: Acquired images are analyzed to quantify parameters like:
- Network Morphology: Classifying populations into "fragmented," "intermediate," or "tubular" [92].
- Fission/Fusion Rates: Using photoactivatable probes (e.g., mito-PA-GFP) to track individual mitochondrial events and calculate rates of fission and fusion over time [93].
- Content Mixing: Using FRAP (Fluorescence Recovery After Photobleaching) on a small region of the mitochondrial network to assess connectivity and fusion competence [92].
Assessing MOMP and Apoptotic Commitment
MOMP is a definitive event in apoptosis and can be measured using several key assays.
- Cytochrome c Release Immunofluorescence:
- Cells are transfected with a mitochondrial marker (e.g., mito-RFP).
- After an apoptotic stimulus, cells are fixed, permeabilized, and stained with an anti-cytochrome c antibody and a fluorescent secondary antibody.
- Cells are imaged via confocal microscopy. In healthy cells, cytochrome c staining co-localizes with the mitochondrial marker (punctate pattern). Upon MOMP, cytochrome c diffuses throughout the cytosol, resulting in a weak, diffuse staining pattern, confirming its release [3].
- Monitoring Caspase Activation:
Pharmacological and Genetic Manipulation
To establish causality, researchers manipulate the dynamics machinery and observe the effects on MOMP.
- Genetic Manipulation:
- Knockdown/Knockout: Using siRNA/shRNA or CRISPR/Cas9 to deplete key proteins (e.g., DRP1, MFN1/2, OPA1).
- Dominant-Negative Mutants: Overexpressing mutants like DRP1-K38A (GTPase-deficient) to inhibit fission, or MFN2-HR2 to inhibit fusion.
- Pharmacological Inhibition:
- Fission Inhibitors: Mdivi-1 is a well-characterized small molecule inhibitor that targets DRP1's GTPase activity, preventing its assembly and function [88].
Table 2: Key Reagents for Studying Mitochondrial Dynamics and MOMP
| Reagent / Tool | Category | Primary Function / Target | Example Application in Research |
|---|---|---|---|
| MitoTracker Dyes | Fluorescent Dye | Staining mitochondrial network | Steady-state morphology assessment in live cells [91] |
| Mito-PA-GFP | Photoactivatable Probe | Matrix-localized fluorescent protein | Direct measurement of fission/fusion rates and mitochondrial motility [93] |
| Mdivi-1 | Small Molecule Inhibitor | Allosteric inhibitor of DRP1 GTPase activity | Inhibiting fission to test its requirement for MOMP [88] |
| DRP1 siRNA/shRNA | Genetic Tool | Knockdown of DNM1L gene | Genetic inhibition of fission to study long-term effects on apoptosis sensitivity [89] |
| Dominant-Negative DRP1 (K38A) | Genetic Tool | GTPase-deficient DRP1 mutant | Potent and specific blockade of DRP1-mediated fission [88] |
| Anti-Cytochrome c Antibody | Immunoassay Reagent | Detects cytochrome c localization | Confirming MOMP occurrence via immunofluorescence or cell fractionation [3] |
| Caspase-3/7 Fluorogenic Substrate | Biochemical Assay | Substrate for executioner caspases | Quantifying apoptosis activation downstream of MOMP [94] |
Pathophysiological Implications and Therapeutic Targeting
Dysregulation of mitochondrial dynamics is a hallmark of numerous diseases, making the fission/fusion machinery an attractive therapeutic target.
- Cancer: Many cancer cells exhibit a hyperfused mitochondrial network, which enhances their resistance to chemotherapeutic agents that induce apoptosis via the intrinsic pathway [90]. Conversely, some cancers display excessive fission, promoting cell proliferation. Targeting DRP1 with inhibitors like Mdivi-1 can sensitize resistant cancer cells to chemotherapy by preventing the fission-associated amplification of MOMP [90].
- Neurodegenerative Disorders: Diseases like Alzheimer's, Parkinson's, and Huntington's are characterized by excessive mitochondrial fragmentation and neuronal apoptosis. In Alzheimer's models, CDK5 phosphorylates DRP1 at Ser579, promoting pathological fission and increasing neuronal sensitivity to amyloid-beta (Aβ) toxicity [89]. Strategies to inhibit pathological fission or promote fusion are being explored to protect neurons [89] [88].
- Metabolic and Cardiovascular Diseases: In cardiac ischemia-reperfusion injury, excessive DRP1-mediated fission contributes to cardiomyocyte death. Inhibiting fission has been shown to reduce infarct size and improve cardiac function in animal models [88].
Table 3: Targeting Mitochondrial Dynamics in Disease
| Disease Context | Observed Dynamic Imbalance | Molecular Mechanism | Potential Therapeutic Strategy |
|---|---|---|---|
| Neurodegeneration (e.g., AD, PD) | Excessive Fission | CDK5-mediated DRP1 phosphorylation (S579/S616); Loss of OPA1 function | DRP1 inhibitors (Mdivi-1 analogs); OPA1 stabilizers [89] |
| Chemotherapy Resistance | Excessive Fusion / Reduced Fission | Upregulation of MFN1/2; Altered DRP1 phosphorylation | Pro-fission agents to sensitize to chemo; MFN inhibitors [90] |
| Cardiac Ischemia-Reperfusion | Excessive Fission | Calcineurin-mediated DRP1 dephosphorylation & activation | DRP1 inhibition to reduce cardiomyocyte apoptosis [88] |
| Metabolic Disease (e.g., Insulin Resistance) | Excessive Fission | DRP1 activation impairing fat oxidation | Exercise-mediated reduction of DRP1 activity [88] |
The intricate relationship between mitochondrial dynamics and MOMP represents a fundamental regulatory axis controlling cellular life and death decisions. Fission promotes apoptosis by facilitating BAX/BAK activation and cristae remodeling, thereby enabling efficient MOMP and cytochrome c release. In contrast, fusion acts as a pro-survival mechanism, suppressing MOMP through content mixing and damage dilution. The core fission protein DRP1 serves as a critical molecular integrator of diverse cellular signals, positioning it at the crossroads of dynamics and apoptosis. Understanding these mechanisms at a granular level, facilitated by the advanced experimental methodologies detailed herein, provides a robust framework for developing novel therapeutic interventions. Targeting the mitochondrial dynamics machinery offers promising avenues for treating a spectrum of diseases, from resensitizing chemoresistant cancers to protecting neurons in degenerative conditions, by directly influencing the cell's core apoptotic commitment point.
The precise execution of programmed cell death is critical for multicellular organism homeostasis, with its dysregulation underpinning numerous human diseases. For decades, the intrinsic (mitochondrial) and extrinsic (death receptor) pathways of apoptosis were viewed as largely independent signaling cascades. However, a paradigm shift has occurred with the recognition of sophisticated crosstalk mechanisms that integrate these pathways, ensuring a robust and coordinated cellular response. This convergence is predominantly governed by the pivotal event of mitochondrial outer membrane permeabilization (MOMP), which serves as a central signaling hub. This whitepaper delineates the molecular machinery of intrinsic and extrinsic pathway convergence, emphasizing the role of MOMP as a critical amplifier and integrator of apoptotic signals. We further provide methodological frameworks for investigating this crosstalk and discuss the profound implications for targeted therapeutic strategies in oncology and beyond.
Apoptosis, a genetically regulated form of cell death, is orchestrated through two primary signaling cascades: the intrinsic and extrinsic pathways. The intrinsic pathway is activated by internal cellular stressors—such as DNA damage, oxidative stress, or growth factor deprivation—which ultimately converge on the mitochondria. The extrinsic pathway is initiated externally via the ligation of death receptors (e.g., Fas, TNFR1) on the cell surface [94] [95]. While these pathways are initiated by distinct stimuli and involve unique upstream components, they are not isolated. A critical point of convergence exists, fundamentally centered on the mitochondrial outer membrane permeabilization (MOMP).
MOMP is often considered the 'point of no return' in the apoptotic cascade [1]. It is characterized by the formation of pores in the mitochondrial outer membrane, leading to the release of several pro-apoptotic proteins from the intermembrane space into the cytosol. The most notable of these is cytochrome c, which, once cytosolic, binds to APAF-1 to form the apoptosome. This complex recruits and activates caspase-9, which in turn activates the executioner caspases-3 and -7 [94] [95]. The extrinsic pathway, upon ligand binding, forms a Death-Inducing Signaling Complex (DISC) that directly activates caspase-8. The crucial crosstalk between these two pathways occurs when caspase-8 cleaves the BH3-only protein Bid, generating its active truncated form, tBid, which translocates to the mitochondria to potently induce MOMP [96] [97]. Thus, MOMP transforms from a pathway-specific event into a central amplification step that integrates apoptotic signals from multiple cellular compartments.
Molecular Mechanisms of Pathway Convergence
The Bcl-2 Protein Family and MOMP Regulation
The integrity of the mitochondrial outer membrane is rigorously controlled by the Bcl-2 family of proteins, which are categorized into three functional groups based on their Bcl-2 homology (BH) domains.
- Anti-apoptotic proteins (e.g., Bcl-2, Bcl-xL, Mcl-1) possess four BH domains and preserve mitochondrial integrity by sequestering their pro-apoptotic counterparts [50].
- Multi-domain pro-apoptotic effectors (Bax, Bak) are the direct mediators of MOMP. Upon activation, they oligomerize to form proteolipidic pores in the outer mitochondrial membrane [1] [50].
- BH3-only proteins (e.g., Bid, Bim, Bad, Noxa, PUMA) are the sentinels of cellular stress. They act either by directly activating Bax/Bak (activators like Bim, Puma, tBid) or by neutralizing anti-apoptotic proteins (sensitizers/de-repressors like Bad, Noxa), thereby freeing activators to engage Bax/Bak [98] [50].
The interplay between these factions determines the cell's commitment to MOMP. The "direct activation" model posits that certain BH3-only proteins directly engage and activate Bax and Bak, while others function by inhibiting the anti-apoptotic proteins that constrain them [50].
The Bid-tBid Axis: A Definitive Molecular Bridge
The quintessential molecular bridge interconnecting the extrinsic and intrinsic pathways is the BH3-only protein Bid. The crosstalk mechanism is as follows:
- Extrinsic Pathway Initiation: Binding of a death ligand (e.g., FasL) to its receptor triggers DISC formation, activating caspase-8 [95] [97].
- Bid Cleavage: Activated caspase-8 proteolytically cleaves cytosolic, inactive Bid to generate tBid (truncated Bid) [96].
- Mitochondrial Targeting: tBid translocates to the mitochondria, where it acts as a potent direct activator of the multi-domain pro-apoptotic proteins Bax and/or Bak [96] [50].
- MOMP Induction: tBid-induced activation of Bax/Bak leads to their oligomerization and pore formation in the outer mitochondrial membrane, culminating in MOMP [1].
- Signal Amplification: The release of cytochrome c following MOMP triggers the apoptosome-mediated activation of caspase-9, which massively amplifies the initial death signal by activating executioner caspases [94] [95].
This Bid-mediated cross-talk is particularly critical in so-called Type II cells, where the initial caspase-8 signal from the DISC is insufficient to fully activate executioner caspases and requires mitochondrial amplification for effective apoptosis. In contrast, Type I cells possess a robust DISC and can execute apoptosis with minimal mitochondrial involvement [96] [97].
Contextual Modulation of Crosstalk
The efficiency of this cross-talk is not absolute but is modulated by cellular context. The relative levels of key proteins, such as c-FLIP (which inhibits caspase-8 activation at the DISC) and XIAP (which directly inhibits caspases-3, -7, and -9), can determine a cell's dependence on the mitochondrial amplifier [99] [97]. Furthermore, growth factors and cytokine signaling can influence the "tone" of the intrinsic pathway, thereby altering the threshold for MOMP and the cell's susceptibility to extrinsic death signals [96].
Quantitative Analysis of Crosstalk Dynamics
Table 1: Key Proteins Regulating Apoptotic Crosstalk and Their Functions
| Protein | Family/Type | Primary Function in Crosstalk | Impact of Dysregulation |
|---|---|---|---|
| Bid/tBid | BH3-only | Molecular bridge; cleaved by caspase-8 (extrinsic) to tBid, which activates Bax/Bak at mitochondria (intrinsic). | Reduced cleavage impedes crosstalk, conferring resistance to death receptor stimuli. |
| Bax/Bak | Effector (Pro-apoptotic) | Forms pores in the MOM during MOMP; the convergent target of both pathways. | Loss abolishes MOMP, causing profound apoptosis resistance. |
| Bcl-2/Bcl-xL | Anti-apoptotic | Binds and inactivates BH3-only proteins and Bax/Bak, preventing MOMP. | Overexpression raises apoptotic threshold, a common feature in cancer. |
| Caspase-8 | Initiator Caspase | Key extrinsic pathway protease; activates executioner caspases and cleaves Bid to initiate crosstalk. | Defects block extrinsic initiation and its amplification via mitochondria. |
| c-FLIP | Caspase Homolog | Inhibits caspase-8 activation at the DISC, negatively regulating extrinsic initiation and crosstalk. | Overexpression promotes resistance to death receptor-mediated apoptosis. |
| XIAP | IAP Family | Binds and inhibits caspases-9, -3, and -7; its antagonism by Smac (released during MOMP) is crucial for apoptosis. | Overexpression inhibits downstream execution, even after MOMP. |
Table 2: Characteristics of Type I vs. Type II Cells in Apoptotic Crosstalk
| Feature | Type I Cells | Type II Cells |
|---|---|---|
| DISC Formation | Robust and efficient | Weak or less efficient |
| Mitochondrial Dependency | Low (Caspase-8 directly activates executioners) | High (Requires Bid-mediated MOMP for signal amplification) |
| Sensitivity to Bcl-2 Overexpression | Low (Pathway is largely mitochondrial-independent) | High (Bcl-2 blocks MOMP, aborting the apoptotic signal) |
| Effect of Bid Knockout/Inhibition | Minimal impact | Profound resistance to death receptor-mediated apoptosis |
| Example Cell Types | Some lymphocyte populations | Hepatocytes, pancreatic beta cells |
Experimental Protocols for Investigating Crosstalk
Elucidating the mechanisms of apoptotic crosstalk requires a multidisciplinary approach, combining genetic, biochemical, and live-cell imaging techniques.
Discriminating Type I and Type II Cell Sensitivity
Objective: To determine a cell's dependence on the mitochondrial amplifier (Type I vs. Type II phenotype).
Methodology:
- Treatment: Expose cells to a death receptor agonist (e.g., anti-Fas antibody, TRAIL) in the presence or absence of a specific pharmacological inhibitor of Bcl-2/Bcl-xL (e.g., ABT-263/Navitoclax) or a broad caspase inhibitor (e.g., Z-VAD-FMK).
- Cell Viability Assessment: Quantify apoptosis at various time points (e.g., 6-24 hours) using flow cytometry with Annexin V/PI staining [99].
- Data Interpretation: Type I cells will show significant death upon receptor ligation that is largely unaffected by Bcl-2 inhibition but is blocked by Z-VAD-FMK. Type II cells will show resistance to death receptor ligation alone, but will become highly sensitive when Bcl-2 is co-inhibited, as this lowers the threshold for MOMP [96].
Validating Bid-Mediated Crosstalk
Objective: To confirm the role of Bid in connecting caspase-8 activation to MOMP.
Methodology:
- Genetic Knockdown/Knockout: Use siRNA, shRNA, or CRISPR/Cas9 to generate Bid-deficient cell lines.
- Stimulation and Fractionation: Treat wild-type and Bid-deficient cells with a death receptor agonist. At sequential time points, fractionate cells to isolate cytosolic and heavy membrane (mitochondrial) fractions.
- Western Blot Analysis: Probe the cytosolic fractions for released cytochrome c (marker of MOMP). Probe the mitochondrial fractions for the translocation of tBid. Additionally, analyze whole-cell lysates for caspase-8 activation (cleavage) and effector caspase-3/7 activation (cleavage) [96].
- Expected Outcome: In Bid-deficient cells, death receptor stimulation will lead to normal caspase-8 activation, but will fail to induce cytochrome c release and subsequent effector caspase activation, confirming its essential role in crosstalk in that cell type.
Live-Cell Imaging of MOMP Kinetics
Objective: To kinetically monitor the timing and synchronicity of MOMP in single cells following intrinsic or extrinsic stimulation.
Methodology:
- Transfection: Stably or transiently transduce cells with a fluorescent biosensor, such as cytochrome c-GFP (which localizes to the mitochondrial intermembrane space) or a SMAC-mCherry fusion protein.
- Stimulation and Imaging: Seed cells in an imaging chamber and treat with either an intrinsic stressor (e.g., UV irradiation, etoposide) or an extrinsic stimulus (e.g., TRAIL). Acquire time-lapse confocal microscopy images every 1-5 minutes.
- Data Analysis: MOMP is visualized as a rapid, irreversible transition from a punctate mitochondrial pattern to a diffuse cytosolic fluorescence for the biosensor [50]. This allows for precise quantification of the timing and heterogeneity of MOMP in response to different apoptotic inducers.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Apoptotic Crosstalk Research
| Reagent/Category | Specific Examples | Research Application |
|---|---|---|
| Death Receptor Agonists | Recombinant TRAIL/Apo2L, Anti-Fas (CD95) Agonistic Antibodies (e.g., CH11), Fas Ligand | To specifically activate the extrinsic apoptosis pathway. |
| BH3 Mimetics | ABT-263 (Navitoclax) - targets Bcl-2/Bcl-xL; A-1331852 - Bcl-xL specific; AMG-176 - Mcl-1 specific; ABT-199 (Venetoclax) - Bcl-2 specific. | To sensitize cells to apoptosis by inhibiting anti-apoptotic Bcl-2 proteins, lowering the threshold for MOMP. |
| Caspase Inhibitors | Z-VAD-FMK (pan-caspase inhibitor), Z-IETD-FMK (caspase-8 inhibitor) | To determine caspase-dependency of cell death and dissect specific protease functions. |
| IAP Antagonists | Birinapant, LCL161 | To block XIAP and cIAPs, promoting caspase activation and sensitizing cells to apoptosis [99]. |
| Genetic Tools | siRNA/shRNA against Bid, Bax, Bak, Caspase-8; CRISPR/Cas9 for gene knockout; Bid-deficient mice. | To establish causal roles of specific proteins in the crosstalk mechanism. |
| Detection Antibodies | Anti-cytochrome c, Anti-cleaved caspase-3, Anti-cleaved caspase-8, Anti-Bid/tBid, Anti-Bax | For Western blotting, immunofluorescence, and flow cytometry to monitor pathway activation and protein localization. |
| Viability/Cell Death Assays | Annexin V/Propidium Iodide staining kits, Caspase-Glo 3/7 and 8/9 Assay kits, TMRE/MitoTracker dyes (for mitochondrial health) | To quantitatively assess apoptosis and mitochondrial function. |
Visualizing the Crosstalk: Signaling Pathways and Experimental Workflows
Integrated Apoptotic Signaling Network
Experimental Workflow for Crosstalk Analysis
The intricate crosstalk between the intrinsic and extrinsic apoptotic pathways, with MOMP at its core, represents a fundamental regulatory node in cell fate decisions. The Bid-mediated amplification loop ensures that even weak death receptor signals can be robustly executed, while also providing a mechanism for intracellular stress signals to engage the full apoptotic machinery. Understanding this crosstalk has profound therapeutic implications, particularly in oncology.
Many cancers develop resistance by overexpressing anti-apoptotic proteins like Bcl-2 or Bcl-xL, or by downregulating components of the extrinsic pathway [94] [50]. The development of BH3 mimetics, such as Venetoclax (ABT-199), which specifically inhibits Bcl-2, exemplifies successful therapeutic targeting of the intrinsic pathway. These agents can lower the mitochondrial threshold for apoptosis, thereby sensitizing Type II cells to endogenous death receptor signaling or co-administered extrinsic pathway agonists like TRAIL [99] [50]. Furthermore, IAP antagonists (e.g., Birinapant) can block caspase inhibition, facilitating apoptosis execution once MOMP has occurred [99]. The future of targeting apoptotic crosstalk lies in rational combination therapies that simultaneously engage the extrinsic pathway while lowering the intrinsic threshold via BH3 mimetics, offering a powerful strategy to overcome the apoptotic resistance that plagues many conventional cancer treatments.
Evaluating MOMP Models and Its Validation as a Therapeutic Target
Comparative Analysis of Apoptotic Pore Formation Models
Mitochondrial outer membrane permeabilization (MOMP) represents a pivotal point of no return in the intrinsic apoptotic pathway, serving as the critical event that initiates caspase activation and programmed cell death [25] [1]. This process is directly governed by the BCL-2 family proteins, which converge on mitochondria to disrupt membrane integrity through elusive pore formation mechanisms [100] [25]. The dysregulation of MOMP constitutes a fundamental hallmark in diseases including cancer, making it a prime target for therapeutic intervention [100]. Despite decades of research, the precise structural organization and biophysical properties of apoptotic pores remain poorly understood due to their transient nature and heterogeneous proteolipid composition [100]. This review provides a comprehensive technical analysis of the predominant models of apoptotic pore formation, synthesizing recent structural insights, genetic evidence, and biophysical data to compare the proposed mechanisms through which BCL-2 effector proteins compromise mitochondrial membrane integrity.
Core Models of Apoptotic Pore Formation
BCL-2 Effector Protein Oligomerization Model
The canonical model for MOMP involves the direct oligomerization of pro-apoptotic BCL-2 effector proteins—BAK, BAX, BOK, and truncated BID (tBID)—to form proteolipid pores in the mitochondrial outer membrane [100]. This model posits that upon activation by apoptotic stimuli, these effectors undergo significant conformational changes from dormant monomers to dynamic oligomers that associate with and permeabilize mitochondria [100]. Gene knockout studies have revealed severe developmental phenotypes in mice lacking these effectors, supporting their non-redundant roles in vivo [100]. BAK and BAX double knockout (DKO) mice exhibit the most severe phenotypes among pro-death BCL-2 family DKO mice, including midline defects such as spina bifida, exencephaly, omphalocele, and skeletal abnormalities [100]. The oligomeric structures of these effector proteins observed in super-resolution microscopy typically appear as arcs and rings, suggesting a common organizational principle [100].
The BH3-in-groove homodimer serves as the fundamental building block for BAK and BAX oligomerization [100] [101]. In this configuration, helices α2-α5 form a symmetric homodimer where helices α2 and α3 arrange in an anti-parallel orientation forming the upper hydrophilic face, while helices α4 and α5 assemble in the lower layer presenting a hydrophobic surface that interacts with the membrane [101]. Site-directed spin labeling and electron paramagnetic resonance (EPR) spectroscopy studies have identified a novel α3:α3', α5:α5' oligomerization interface in the BAK oligomeric pore, involving the C-termini of α3 and α5 helices [101]. This interface enables higher-order oligomerization of BH3-in-groove homodimers, ultimately forming pores estimated to be ∼30-60 Å in diameter—sufficient for cytochrome c release [101].
Table 1: Activation Mechanisms of Pore-Forming BCL-2 Effector Proteins
| Effector Protein | Activation Mechanism | Regulatory Partners | Oligomeric Structure |
|---|---|---|---|
| BAK | Direct activation by BH3-only proteins, autoactivation, crossactivation | BH3-only initiators (BID, BIM, PUMA), anti-apoptotic BCL-2 | BH3-in-groove homodimers forming arcs and rings via α3:α3', α5:α5' interface |
| BAX | Direct activation by BH3-only proteins, translocation from cytosol to MOM | BH3-only initiators, anti-apoptotic BCL-2 | BH3-in-groove homodimers similar to BAK |
| BOK | Accumulates upon inhibition of degradation by gp78 E3 ligase | gp78 E3 ligase, IP3 receptors | Uncharacterized oligomeric species |
| tBID | Proteolytic cleavage by caspase-8 | Caspase-8, mitochondrial lipids | Putative monomers, activates BAK/BAX directly |
VDAC1 Oligomerization Model
The voltage-dependent anion channel (VDAC1), the main metabolite transit pore in the mitochondrial outer membrane, has been implicated as an alternative pathway for apoptosis induction [28]. While VDAC1 monomers have an inner diameter of only 1.5-3.0 nm—too small for apoptogenic protein release—oligomerization under apoptotic conditions can form larger pores capable of MOMP [28]. Recent structural studies demonstrate that VDAC1 oligomerization or confinement in small lipid nanodiscs triggers exposure of its N-terminal α-helix (VDAC1-N), which becomes available for partner protein binding [28]. This exposed VDAC1-N forms a complex with the BH3 binding groove of the anti-apoptotic protein Bcl-xL, effectively neutralizing its inhibitory function and promoting Bak pore formation [28].
This VDAC1-mediated mechanism mirrors the function of BH3-only sensitizer proteins that induce Bax/Bak-mediated MOMP [28]. The exposure of VDAC1-N is enhanced by apoptotic stimuli including negatively charged lipids (e.g., POPG), increased Ca²⁺ levels, low pH, and oxidative stress [28]. Cryo-EM and NMR studies have captured VDAC1 in different conformational states where VDAC1-N is either bound inside the pore or exposed to the exterior, providing structural evidence for this regulated mechanism [28]. The crystal structure of the VDAC1-N—Bcl-xL complex confirms this interaction occurs through the canonical BH3-binding groove, representing a structural link between mitochondrial metabolite transport and apoptosis regulation [28].
Lipidic Pore and Proteolipid Models
Beyond purely proteinaceous pores, several models propose that BCL-2 effector proteins function through interactions with membrane lipids to generate proteolipid pores. The "worm hole" model suggests that BAK-induced pores incorporate both protein and lipid elements, with the BH3-in-groove homodimers creating a topography that facilitates membrane curvature and disruption [101]. This model is supported by evidence that BCL-2 proteins interact with specific mitochondrial lipids including cardiolipin [25] [102], and that pore formation depends on membrane composition [25].
In liposome permeabilization assays, BCL-2 family proteins induce content release from model mitochondrial outer membranes, demonstrating their capacity to form pores in lipid bilayers without other protein components [25]. The collaboration between tBID, Bax, and lipids to form supramolecular openings in the mitochondrial outer membrane further supports the involvement of lipid rearrangements in pore formation [1]. Recent studies have also revealed that gasdermin D (GSDMD)—the executioner of pyroptosis—permeabilizes both mitochondrial inner and outer membranes through cardiolipin binding, suggesting conserved mechanisms of proteolipid pore formation across cell death pathways [102].
Table 2: Comparison of Apoptotic Pore Formation Models
| Feature | BCL-2 Effector Oligomerization | VDAC1 Oligomerization | Lipidic Pore Model |
|---|---|---|---|
| Primary Components | BAK, BAX, BOK, tBID | VDAC1 oligomers | BCL-2 proteins + membrane lipids |
| Pore Size | ∼30-60 Å [101] | Larger than monomeric VDAC1 (exact size unknown) | Variable, potentially larger |
| Triggering Stimuli | BH3-only proteins, proteolysis, degradation inhibition [100] | Mitochondrial stress, negative charge, Ca²⁺, low pH [28] | Membrane lipid composition, curvature stress |
| Regulation | Anti-apoptotic BCL-2 proteins [100] | Bcl-xL interaction, VDAC1-N exposure [28] | Lipid composition, membrane properties |
| Structural Evidence | BH3-in-groove dimers, EPR data [101] | Cryo-EM, NMR, crystal structure [28] | Liposome assays, molecular dynamics |
Experimental Approaches for Studying Apoptotic Pores
Structural Biology Techniques
Site-directed spin labeling (SDSL) EPR spectroscopy has been instrumental in mapping conformational changes in BAK during membrane insertion and pore formation [101]. This methodology involves introducing cysteine residues at specific positions in the protein, modifying them with methanethiosulfonate (MTSSL) spin labels, and measuring distance constraints between spin labels through EPR analysis [101]. For BAK, this approach revealed that helices α1 and α6 disengage from the rest of the domain upon membrane targeting, leaving helices α2-α5 as a folded unit that forms the BH3-in-groove homodimer [101]. Distance measurements between 13 pairs of spin-labeled residues in both solution and membrane-inserted states enabled the mapping of oligomerization interfaces.
Cryo-electron microscopy (cryo-EM) has provided recent breakthroughs in understanding VDAC1 oligomerization and its role in apoptosis [28]. Using circularized lipid nanodiscs of different sizes, researchers have structurally characterized VDAC1 in different conformational states, capturing VDAC1-N either bound inside the pore or exposed to the exterior [28]. This technical approach allows for membrane protein structural determination in near-native lipid environments, revealing how apoptotic stimuli trigger VDAC1 conformational changes that enable Bcl-xL binding.
X-ray crystallography of the VDAC1-N—Bcl-xL complex at high resolution (2.1 Å) has provided atomic-level details of the interaction between these key apoptotic regulators [28]. This structure confirmed that VDAC1-N binds to the canonical BH3-binding groove of Bcl-xL in a manner similar to BH3-only proteins, explaining its pro-apoptotic function as a sensitizer.
Biochemical and Biophysical Assays
Liposomal release assays constitute a fundamental methodology for studying membrane permeabilization by BCL-2 proteins in a controlled system [101]. Large unilamellar vesicles (LUVs) with lipid compositions resembling mitochondrial outer membrane contact sites are prepared with encapsulated fluorescent markers such as FITC-dextran [101]. The release of these markers upon incubation with recombinant BCL-2 proteins (e.g., 5 nM BAK with 25 nM tBID) quantifies membrane permeabilization activity under different conditions [101]. This assay allows systematic investigation of how membrane lipid composition, protein mutations, and pharmacological inhibitors affect pore formation.
Chemical crosslinking experiments using amino-selective crosslinkers like bis(sulfosuccinimidyl)suberate (BS3) have been employed to study protein oligomerization under various conditions [28]. For VDAC1, this approach demonstrated that negatively charged detergents (cholate) and lipids (POPG) promote oligomerization, while zwitterionic detergents (LDAO, TritonX-100) do not [28]. Crosslinking combined with SDS-PAGE provides a straightforward method to assess the oligomeric state of proteins in different environments.
PEGylation-based exposure assays utilize cysteine-specific maleimide-polyethyleneglycol reagents (e.g., PM40, 40 kDa) to probe the accessibility of specific protein regions [28]. In VDAC1, a T6C mutation introduced at the N-terminal α-helix enabled monitoring of helix exposure, as the bulky PEG polymer cannot access cysteine residues buried inside the β-barrel pore [28]. The degree of PEG modification under different conditions correlates with structural rearrangements that expose previously buried regions.
Research Reagent Solutions
Table 3: Essential Research Reagents for Apoptotic Pore Studies
| Reagent/Cell Line | Specification/Application | Key Utility in MOMP Research |
|---|---|---|
| BAK/BAX DKO MEFs [100] | Mouse embryonic fibroblasts lacking BAK and BAX | Gold standard for assessing BCL-2 protein function; resistant to mitochondrial apoptosis inducers |
| BCL2allKO HCT116 (AKO) [100] | HCT116 with 17 BCL-2 family genes inactivated | New gold standard for apoptotic studies without BCL-2 family redundancy |
| LUVs mimicking MOM [101] | 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine, phosphatidylinositol, cardiolipin, cholesterol, DOGS-NTA-Ni (36:22:9:8:20:5) | Model membrane system for studying protein-lipid interactions and pore formation |
| Recombinant sBAK/C154S-ΔC-His [101] | Soluble mouse BAK (residues 16-184) with C-terminal His-tag and C154S mutation | Permits site-directed spin labeling for EPR studies of conformational changes |
| N-terminally His-tagged p7/p15 Bid [101] | Caspase-8 cleaved Bid (tBID) activator | Standard activator for BAK/BAX in membrane permeabilization assays |
| MTSSL and MTSSL-d15 [101] | Methanethiosulfonate spin labels | Site-directed spin labeling for EPR distance measurements |
| BS3 crosslinker [28] | Bis(sulfosuccinimidyl)suberate, amine-reactive | Chemical crosslinking to study protein oligomerization states |
| PM40 PEGylation reagent [28] | Maleimide-polyethyleneglycol (40 kDa) | Probing exposure of specific protein regions via cysteine modification |
Integrated Signaling in Apoptotic Pore Formation
MOMP Signaling Pathways
The diagram above illustrates the convergent signaling pathways that regulate mitochondrial outer membrane permeabilization. Both extrinsic (death receptor) and intrinsic (cellular stress) pathways ultimately trigger MOMP through BCL-2 family protein interactions [25]. The extrinsic pathway activates caspase-8, which cleaves BID to generate tBID, while the intrinsic pathway activates BH3-only proteins that either directly activate BAK/BAX or inhibit anti-apoptotic BCL-2 members [100] [25]. Mitochondrial stress can also induce VDAC1 oligomerization, leading to exposure of VDAC1-N, which neutralizes Bcl-xL and promotes BAK activation [28]. These pathways converge on MOMP, resulting in cytochrome c release, caspase activation, and apoptosis execution.
The comparative analysis of apoptotic pore formation models reveals a complex landscape of MOMP regulation with multiple, potentially complementary mechanisms. The BCL-2 effector oligomerization model provides the most extensively characterized pathway, with structural details of the BH3-in-groove homodimers and their oligomerization interfaces increasingly elucidated through advanced biophysical techniques [100] [101]. The VDAC1 oligomerization model offers an alternative pathway that connects mitochondrial metabolite transport with apoptosis regulation through exposure of the N-terminal helix and neutralization of Bcl-xL [28]. Lipidic pore models emphasize the crucial contribution of membrane lipids and proteolipid interactions in membrane disruption [101] [102]. Rather than being mutually exclusive, these models likely represent different facets of a unified MOMP mechanism that incorporates elements of each, with relative contributions potentially varying by cell type, apoptotic stimulus, and physiological context. Future research integrating these models will be essential for developing targeted therapeutic strategies that modulate apoptotic pore formation in disease.
The Voltage-Dependent Anion Channel (VDAC), the most abundant protein in the mitochondrial outer membrane (OMM), serves as the principal gateway governing metabolite and ion exchange between mitochondria and the cytosol [103] [69]. As a critical node in cellular energy metabolism and mitochondria-mediated apoptosis, VDAC has emerged as a promising therapeutic target in cancer research. The channel exists as three isoforms in mammals—VDAC1, VDAC2, and VDAC3—which share approximately 70% sequence similarity but exhibit distinct functional properties [33] [103]. VDAC1, the most extensively studied isoform, functions as a crucial regulator of cellular metabolism and apoptotic signaling, positioning it at the interface between cell survival and death [103] [69]. This technical review examines the molecular mechanisms through which VDAC regulates mitochondrial outer membrane permeabilization (MOMP) and explores the therapeutic potential of VDAC-targeted strategies in oncology, with particular emphasis on their integration with current understanding of apoptotic pathways.
Structural Basis of VDAC Function and Regulation
VDAC Isoforms and Molecular Architecture
VDAC isoforms display distinct tissue distribution patterns and physiological roles. VDAC1 represents the predominant isoform in most tissues, while VDAC3 expression is particularly enriched in testis and spermatozoa [104]. Structural studies using NMR and X-ray crystallography have revealed that VDAC1 forms a 19-strand β-barrel with an N-terminal α-helix (VDAC1-N) located inside the pore [28] [103]. This N-terminal region exhibits significant conformational flexibility, enabling its translocation from the pore interior to the channel surface, a movement critical for channel gating and partner protein interactions [28] [103]. The pore diameter measures approximately 3 nm in the open state, accommodating metabolites up to 5 kDa, but constricts to about 1.5 nm when the N-terminal helix occupies the barrel interior [103].
Table 1: Structural and Functional Properties of VDAC Isoforms
| Isoform | Primary Localization | Expression Pattern | Key Functional Characteristics | Associated Pathology |
|---|---|---|---|---|
| VDAC1 | Mitochondrial Outer Membrane, Plasma Membrane | Highest expression in most tissues | Primary metabolite transport, apoptosis regulation via Bax/Bak, hexokinase binding | Overexpressed in multiple cancers, Alzheimer's disease |
| VDAC2 | Mitochondrial Outer Membrane | High expression in most tissues | Apoptosis regulation, inhibition of Bak activation, immune modulation | Melanoma, lung adenocarcinoma, hepatocellular carcinoma |
| VDAC3 | Mitochondrial Outer Membrane | Lowest expression (except testis) | Redox sensing, mitochondrial quality control | Male sterility when deficient, colorectal cancer |
Conductance States and Metabolic Regulation
VDAC exhibits voltage-dependent gating behavior, transitioning between distinct conductance states that regulate metabolite flux. At low membrane potentials (approximately -20 to +20 mV), VDAC maintains an open conformation with high conductance (~4 nS in 1 M KCl) and preferential permeability to anions, including ATP, ADP, and other metabolic substrates [103] [104]. At higher positive or negative potentials (> 40 mV), the channel transitions to lower conductance states with reduced pore diameter and shifted selectivity toward cations such as K+, Na+, and Ca2+ [103] [104]. This gating behavior enables VDAC to function as a metabolic switch, modulating the cross-talk between glycolytic and oxidative phosphorylation pathways according to cellular energy demands [104].
VDAC in Mitochondrial Outer Membrane Permeabilization and Apoptosis
Oligomerization and MOMP Execution
VDAC oligomerization represents a critical mechanism in mitochondria-mediated apoptosis. Under physiological conditions, VDAC primarily exists in monomeric and dimeric states. However, apoptotic stimuli including oxidative stress, Ca2+ overload, and altered lipid composition induce conformational changes that promote the formation of higher-order oligomers [28]. These oligomeric assemblies undergo substantial structural rearrangements that trigger exposure of the N-terminal α-helix, which becomes accessible for protein-protein interactions [28]. The current model suggests that VDAC oligomers may form large pores in the OMM sufficient to facilitate MOMP and the release of pro-apoptotic proteins such as cytochrome c and AIF from the mitochondrial intermembrane space [103] [105]. This oligomerization process thereby serves as a pivotal switch initiating the intrinsic apoptotic pathway.
Interaction with Bcl-2 Family Proteins
VDAC1 directly engages with both pro-apoptotic and anti-apoptotic members of the Bcl-2 protein family, positioning it as a crucial regulator of apoptotic signaling. Recent structural insights from cryo-EM and NMR spectroscopy demonstrate that the exposed N-terminal α-helix of oligomeric VDAC1 binds to the BH3-binding groove of the anti-apoptotic protein Bcl-xL [28]. This interaction neutralizes Bcl-xL's inhibitory function, thereby liberating pro-apoptotic proteins such as Bak to execute MOMP [28]. The VDAC1-N and Bcl-xL interaction mirrors the mechanism employed by BH3-only sensitizer proteins, effectively priming the apoptotic machinery in response to mitochondrial stress or damage [28]. This molecular pathway establishes VDAC1 as an integral component of the core apoptotic regulatory network.
Metabolic Reprogramming and the Warburg Effect
Cancer cells extensively manipulate VDAC function to support their metabolic reprogramming toward aerobic glycolysis (the Warburg effect). VDAC1 interaction with hexokinase (HK-I and HK-II) at the OMM provides cancer cells with a significant proliferative advantage through dual mechanisms [33] [106]. First, this association grants hexokinase preferential access to mitochondrial ATP, fueling high-rate glycolysis. Second, it sterically hinders VDAC1 interaction with pro-apoptotic factors, thereby increasing the apoptotic threshold and enhancing cellular survival [106] [107]. The closed conformation of VDAC1, induced by tubulin binding, further promotes the Warburg phenotype by restricting mitochondrial metabolite flux and enhancing glycolytic metabolism [33] [104]. Consequently, VDAC occupies a central position in the metabolic adaptations that characterize malignant transformation.
VDAC-Targeted Therapeutic Strategies in Oncology
VDAC Modulation in Cancer Therapy
Pharmacological targeting of VDAC represents a promising avenue for cancer therapy, with multiple compounds developed to modulate channel function through distinct mechanisms. These therapeutic approaches can be broadly categorized into two classes: VDAC openers that promote apoptosis, and VDAC closers that disrupt metabolic adaptations in cancer cells.
Table 2: VDAC-Targeted Therapeutic Compounds and Their Mechanisms
| Compound Class | Representative Agents | Molecular Target | Mechanism of Action | Experimental Evidence |
|---|---|---|---|---|
| VDAC Openers | Erastin, Betulinic Acid | VDAC1/VDAC2 | Induces channel opening, reverses Warburg effect, promotes ferroptosis | Lung cancer, breast cancer models [33] |
| VDAC Closers | Avicin, Acrolein | VDAC1 | Promotes channel closure, disrupts energy metabolism | Preclinical cancer models [33] |
| Hexokinase Disruptors | VDAC1-derived peptides | VDAC1-HK interface | Displaces HK from mitochondria, restores apoptosis sensitivity | In vitro models [107] |
| Small Molecule Antagonists | VA compounds | VDAC1 NADH-binding site | Competes with NADH binding, causes mitochondrial distress | Cholangiocarcinoma organoids [106] |
| Combination Therapy | VBIT-12 + Trametinib | VDAC1 + MEK | Synergistic tumor suppression | Lung adenocarcinoma models [108] |
Isoform-Specific Therapeutic Targeting
Emerging evidence highlights the therapeutic potential of isoform-specific VDAC targeting. Recent research has identified VDAC2 as a promising immune signal-dependent checkpoint that restricts interferon-γ (IFNγ)-mediated tumor destruction [109]. Genetic ablation of VDAC2 in tumor cells unleashes IFNγ-induced BAK activation, mitochondrial damage, and cytosolic release of mitochondrial DNA, which subsequently activates cGAS-STING signaling and type I interferon responses [109]. This integrated mechanism simultaneously engages both intrinsic apoptosis and innate immune activation, resulting potent anti-tumor effects. VDAC2 deficiency significantly enhances responsiveness to immune checkpoint blockade in multiple tumor models, including those resistant to TNF-mediated killing, positioning VDAC2 as a compelling target for combination immunotherapy [109].
Novel Small Molecule Approaches
Innovative drug discovery platforms have identified novel chemical entities that selectively target VDAC1 in cancer cells. Through in silico screening and biochemical validation, VA (VDAC Antagonist) molecules have been characterized as specific VDAC1 binders with a three-ring architectural structure [106]. These compounds compete with NADH for a partially shared binding site on VDAC1, with micromolar affinity interactions that induce mitochondrial distress and preferentially impair cancer cell proliferation [106]. Notably, VA molecules demonstrate significant cytotoxicity against patient-derived cholangiocarcinoma organoids while exhibiting minimal effects on healthy cells, highlighting their potential therapeutic window [106]. This approach exemplifies the feasibility of targeting VDAC1-mediated metabolic regulation for selective cancer therapy.
Experimental Approaches for VDAC Research
Structural and Biophysical Characterization
Advanced structural biology techniques have been instrumental in elucidating VDAC function in apoptosis regulation. The application of cryo-electron microscopy (cryo-EM) to VDAC1 embedded in circularized lipid nanodiscs of varying sizes has enabled structural characterization of different conformational states, particularly the transition from pore-embedded to exposed N-terminal α-helix configurations [28]. This methodological approach has revealed that VDAC1 oligomerization or confinement in small nanodiscs triggers exposure of VDAC1-N, rendering it accessible for interactions with partner proteins such as Bcl-xL [28].
Protocol 1: Assessing VDAC1 Oligomerization and N-terminal Helix Exposure
- Protein Purification: Recombinantly express and purify wild-type VDAC1 and cysteine variants (e.g., T6C, L10C) using detergent extraction and chromatography.
- Cross-linking Analysis: Incubate VDAC1 (0.5-1 mg/mL) with the amino-selective cross-linker bis(sulfosuccinimidyl)suberate (BS3) at 4-25°C for 30 minutes. Terminate reaction with Tris buffer.
- Oligomerization Induction: Utilize negatively charged detergents (e.g., cholate) or lipids (e.g., POPG) to promote VDAC1 oligomerization.
- Cysteine Accessibility Assay: Treat VDAC1-T6C mutant with maleimide-polyethyleneglycol-40kDa (PM40). Resolve via SDS-PAGE to assess modification efficiency.
- Thermal Stability Assessment: Monitor protein unfolding using circular dichroism (CD) spectroscopy or differential scanning fluorimetry.
- Structural Validation: Employ cryo-EM for high-resolution structural analysis of oligomeric states.
Functional Assessment of VDAC in Cell Death
Genetic and pharmacological approaches enable functional characterization of VDAC in apoptotic pathways. CRISPR-Cas9-mediated knockout of VDAC isoforms, particularly VDAC2, combined with stimulation using recombinant IFNγ, provides a robust system for evaluating VDAC-dependent cell death mechanisms [109]. This approach has demonstrated that VDAC2 deficiency potentiates IFNγ-induced caspase-3, caspase-7, and gasdermin E (GSDME) cleavage, indicating activation of secondary necrosis [109].
Protocol 2: Evaluating VDAC-Mediated Cell Death Mechanisms
- Genetic Manipulation: Generate VDAC-deficient cell lines using CRISPR-Cas9 with sgRNAs targeting VDAC1, VDAC2, or VDAC3.
- Immune Cytokine Challenge: Treat cells with IFNγ (10-100 ng/mL) for 24-48 hours. Include controls for TNF and perforin-mediated cytotoxicity.
- Cell Death Assessment: Quantify cell viability using MTT assay, lactate dehydrogenase (LDH) release, and flow cytometry with Annexin V/PI staining.
- Mechanistic Dissection: Employ caspase inhibitors (e.g., emricasan), ferroptosis inhibitors (e.g., ferrostatin-1), or necroptosis inhibitors (e.g., necrostatin-1) to delineate death pathways.
- Downstream Signaling Analysis: Evaluate activation of cGAS-STING pathway and type I interferon responses by immunoblotting for phospho-STING and interferon-stimulated genes.
- In Vivo Validation: Assess therapeutic efficacy in immunocompetent and immunodeficient mouse models with appropriate cancer cell lines.
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Research Reagents for VDAC Investigation
| Reagent/Category | Specific Examples | Experimental Function | Key Applications |
|---|---|---|---|
| VDAC Modulators | Erastin, VBIT-12, Avicin, Betulinic Acid | Pharmacological manipulation of VDAC conformation and function | Assessing VDAC-dependent metabolism and apoptosis |
| Genetic Tools | CRISPR sgRNAs, siRNA, VDAC1-N-terminal peptides | Isoform-specific knockdown or functional interference | Determining isoform-specific functions and mechanisms |
| Structural Biology | Lipid nanodiscs, BS3 crosslinker, Cryo-EM grids | Stabilization of conformational states for structural analysis | Determining VDAC oligomer structure and interactions |
| Apoptosis Assays | Cytochrome c release assays, caspase activity kits, LDH cytotoxicity | Quantification of cell death pathways | Measuring MOMP and downstream apoptotic events |
| Interaction Partners | Recombinant Bcl-xL, Bax, Bak, hexokinase | Binding and competition studies | Characterizing protein-protein interactions with VDAC |
| Metabolic Probes | Seahorse metabolic analyzers, NADH/ATP biosensors | Real-time monitoring of metabolic flux | Assessing VDAC role in metabolic reprogramming |
VDAC represents a multifaceted regulator of mitochondrial function that integrates metabolic and apoptotic signaling pathways in cancer. The channel's dual role in controlling metabolite flux and MOMP positions it as a promising therapeutic target with mechanistic connections to both metabolic reprogramming and immune-mediated tumor destruction. Future research directions should focus on elucidating the structural determinants of isoform-specific functions, developing more selective pharmacological agents with improved therapeutic indices, and exploring combinatorial approaches that leverage VDAC targeting to overcome therapy resistance. The integration of VDAC-targeted strategies with conventional chemotherapy, immunotherapy, and metabolic interventions holds significant promise for advancing cancer treatment paradigms. As our understanding of VDAC biology continues to evolve, so too will opportunities to exploit this critical mitochondrial gatekeeper for therapeutic benefit.
Mitochondrial Outer Membrane Permeabilization (MOMP) represents a point of no return in the intrinsic apoptotic pathway, and its precise regulation by B-cell lymphoma 2 (BCL-2) family proteins is critical for cellular homeostasis. This technical review examines the mechanisms through which anti-apoptotic BCL-2 proteins—including BCL-2, BCL-xL, and MCL-1—inhibit MOMP to promote cell survival. We synthesize current structural and biochemical evidence detailing how these proteins neutralize pro-apoptotic effectors through specific protein-protein interactions. The whitepaper further provides validated experimental methodologies for quantifying MOMP inhibition and discusses the translational implications of these mechanisms for targeted cancer therapeutics, particularly BH3-mimetic compounds. This comprehensive analysis aims to equip researchers with the foundational knowledge and technical protocols necessary for investigating BCL-2-mediated cell survival pathways.
MOMP as the Apoptotic Commitment Point
Mitochondrial Outer Membrane Permeabilization (MOMP) is a decisive event in the intrinsic apoptotic pathway where the mitochondrial outer membrane becomes permeable to proteins normally confined to the intermembrane space [17]. This permeabilization allows the release of cytochrome c and other pro-apoptotic factors into the cytosol, triggering caspase activation and irreversible commitment to cell death [5] [17]. The permeability transition leads to the formation of pores large enough to allow proteins exceeding 100 kDa to pass through [17]. MOMP execution is primarily mediated by the pro-apoptotic BCL-2 effector proteins BAX and BAK, which undergo conformational activation and oligomerization to form these permeability pores in the mitochondrial outer membrane [17].
The BCL-2 Family Tripartite Apoptotic Switch
The BCL-2 protein family constitutes a critical regulatory network that governs MOMP through a delicate balance of pro-survival and pro-death signals [5]. This family consists of three functional subgroups with distinct structural characteristics:
- Multi-domain anti-apoptotic proteins (BCL-2, BCL-xL, MCL-1, BCL-w, BCL-B, and A1/Bfl-1) characterized by four BCL-2 homology (BH) domains (BH1-BH4) that preserve mitochondrial integrity [110] [5]
- Multi-domain pro-apoptotic proteins (BAX, BAK, and BOK) that directly execute MOMP [5]
- BH3-only pro-apoptotic proteins (BIM, BID, BAD, NOXA, PUMA, BMF, HRK) that function as cellular stress sensors and initiate the apoptotic cascade [110] [5]
The founding member, BCL-2, was initially discovered in 1984 as the gene involved in the t(14;18)(q32.3;q21.3) chromosomal translocation found in follicular lymphoma, representing the first example of an oncogene that promotes cancer by blocking cell death rather than stimulating proliferation [5].
Table 1: Core Components of the BCL-2 Protein Family Regulating MOMP
| Protein Category | Representative Members | BH Domains | Primary Function in MOMP Regulation |
|---|---|---|---|
| Anti-apoptotic | BCL-2, BCL-xL, MCL-1 | BH1-BH4 | Maintain mitochondrial membrane integrity, sequester pro-apoptotic proteins |
| Pro-apoptotic Effectors | BAX, BAK | BH1-BH3 | Form permeabilization pores in mitochondrial outer membrane |
| BH3-only Sensitizers | BIM, tBID, PUMA | BH3 only | Directly activate BAX/BAK |
| BH3-only Sensitizers | BAD, NOXA, BMF | BH3 only | Displace activators from anti-apoptotic proteins |
Molecular Mechanisms of MOMP Inhibition by Anti-Apoptotic BCL-2 Proteins
Structural Basis of Pro-Survival Function
Anti-apoptotic BCL-2 proteins share extensive sequence and structural similarity characterized by a globular α-helical fold featuring an eight-helix bundle that forms a hydrophobic surface groove for binding BH3 domains of pro-apoptotic family members [5]. This hydrophobic groove, composed of BH1, BH2, and BH3 domains, serves as the critical interaction site where anti-apoptotic proteins sequester their pro-apoptotic counterparts [110]. The interaction occurs via the binding of the hydrophobic face of the amphipathic BH3 α-helix from pro-apoptotic proteins into the hydrophobic pocket of anti-apoptotic proteins [110].
The anti-apoptotic function primarily manifests through two complementary mechanisms:
- Direct sequestering of activated BH3-only proteins such as BIM and tBID, preventing them from activating BAX and BAK [111]
- Direct inhibition of BAX and BAK at the mitochondrial membrane through complex formation, oligomer disassembly, and/or retrotranslocation to the cytosol [111]
Specific Inhibition Mechanisms by Key Anti-Apoptotic Proteins
BCL-2 itself localizes to the mitochondrial outer membrane via its C-terminal transmembrane domain and demonstrates broad binding capacity for multiple BH3-only proteins [110] [5]. Beyond its canonical role in apoptosis inhibition, BCL-2 regulates additional cellular processes including migration, invasion, autophagy, angiogenesis, and cancer stem-like phenotypes through both mitochondrial and endoplasmic reticulum localization [110].
BCL-xL shares similar structural domains with BCL-2 and employs comparable sequestration mechanisms [110]. Recent research has revealed that BCL-xL interacts with Voltage-Dependent Anion Channel 1 (VDAC1) through its BH4 domain, promoting cell migration by increasing reactive oxygen species production in breast cancer models [110]. BCL-xL exists in balance with its alternatively spliced isoform BCL-xS, which lacks the region with highest homology to BCL-2 and paradoxically promotes apoptosis [110].
MCL-1 was initially discovered during myeloid cell differentiation and demonstrates distinct binding preferences compared to BCL-2 and BCL-xL [110]. While it shares the fundamental mechanism of BH3-domain sequestration, MCL-1 shows particular importance in neutralizing specific BH3-only proteins like NOXA [112]. MCL-1 amplification and overexpression frequently correlate with poor prognosis and resistance to anticancer drugs across various malignancies [110].
Diagram 1: BCL-2 Protein Network Regulating MOMP and Cell Survival. Anti-apoptotic proteins (green) sequester BH3-only proteins and directly inhibit BAX/BAK activation, thereby preventing MOMP and maintaining cell survival.
Alternative Mechanisms and Non-Canonical Functions
Emerging evidence indicates that anti-apoptotic BCL-2 proteins also participate in non-canonical functions beyond direct MOMP regulation. The BH4 domain of BCL-2 and BCL-xL can bind other proteins unrelated to the BCL-2 family, enabling roles in proliferation, autophagy, differentiation, DNA repair, tumor progression, and angiogenesis [110]. Furthermore, these proteins localize not only to mitochondria but also to the endoplasmic reticulum (ER), where they regulate ER Ca²⁺ signaling and storage, indirectly influencing mitochondrial Ca²⁺ uptake and permeability transition [5].
Recent structural studies have revealed novel interaction mechanisms, such as the exposure of the N-terminal α-helix of VDAC1 during oligomerization, which can bind the BH3-binding groove of BCL-xL, effectively acting as a sensitizer BH3-protein to promote BAK-mediated pore formation [28]. This demonstrates the complex interplay between mitochondrial channels and BCL-2 proteins in MOMP regulation.
Experimental Validation of MOMP Inhibition
Methodologies for Assessing MOMP Inhibition
Validating MOMP inhibition by anti-apoptotic BCL-2 proteins requires a multidisciplinary approach combining biochemical, cellular, and structural techniques. The following section details key experimental protocols for investigating these mechanisms.
Mitochondrial Isolation and Cytochrome c Release Assay
This fundamental protocol evaluates the functional capacity of anti-apoptotic proteins to prevent MOMP in isolated mitochondria.
Protocol Steps:
- Mitochondrial Isolation: Homogenize cells in isotonic buffer (250 mM sucrose, 10 mM HEPES, 1 mM EGTA, pH 7.4) followed by differential centrifugation at 800 × g for 10 minutes (nuclear pellet removal) and 10,000 × g for 15 minutes (mitochondrial pellet) [111] [17].
- Treatment Conditions: Resuspend mitochondrial pellets in respiration buffer (125 mM KCl, 10 mM HEPES, 5 mM glutamate, 2.5 mM malate, 1 mM phosphate, pH 7.4) and divide into aliquots for various treatments.
- MOMP Induction: Add recombinant tBID or BIM (5-50 nM) to trigger MOMP in the presence of calcium (10-100 μM) to promote permeability transition [72].
- Inhibition Test: Pre-incubate with recombinant anti-apoptotic BCL-2 proteins (10-100 nM) or transfected mitochondrial fractions overexpressing BCL-2 family members.
- Centrifugation: Pellet mitochondria at 12,000 × g for 10 minutes and collect supernatant.
- Detection: Analyze supernatant for cytochrome c release by Western blot (12-15% SDS-PAGE) using anti-cytochrome c antibodies, or spectrophotometrically at 550 nm following reduction with ascorbate.
Key Controls:
- Positive control: Mitochondria treated with tBID/BIM alone
- Negative control: Untreated mitochondria
- Specificity control: Mitochondria from BAX/BAK double knockout cells
BH3 Profiling and Competitive Binding Assays
BH3 profiling measures mitochondrial sensitivity to specific BH3 peptides, evaluating the protective capacity of anti-apoptotic proteins.
Protocol Steps:
- Mitochondrial Preparation: Isolate mitochondria as described in 3.1.1.
- BH3 Peptide Exposure: Treat with synthetic BH3 peptides (1-10 μM) from specific pro-apoptotic proteins (BAD, NOXA, BIM, HRK) in respiration buffer.
- Membrane Permeability Assessment: Monitor using cytochrome c release (as above) or fluorometric detection of mitochondrial membrane potential with dyes like JC-1 or TMRE.
- Quantification: Measure depolarization kinetics; anti-apoptotic inhibition is indicated by delayed or reduced depolarization in response to specific BH3 peptides.
Interpretation Guide:
- BAD sensitivity: Suggests BCL-2/BCL-xL/BCL-w dependence
- NOXA sensitivity: Indicates MCL-1 dependence
- Resistance to both: Suggests low anti-apoptotic dependence or primed state
Crosslinking and Co-Immunoprecipitation for Protein Complex Analysis
This approach characterizes direct protein-protein interactions between anti-apoptotic and pro-apoptotic BCL-2 family members.
Protocol Steps:
- Cell Lysis: Use mild detergent conditions (1% CHAPS or digitonin in PBS) to preserve native protein complexes.
- Crosslinking (Optional): Treat with membrane-permeable crosslinker (e.g., DSS, 1-5 mM) for 10-30 minutes at room temperature to stabilize transient interactions.
- Immunoprecipitation: Incubate lysates with antibodies specific to anti-apoptotic proteins (anti-BCL-2, anti-BCL-xL, anti-MCL-1) conjugated to protein A/G beads for 2-12 hours at 4°C.
- Washing: Remove non-specifically bound proteins with mild detergent buffers.
- Elution and Analysis: Elute bound complexes with Laemmli buffer and analyze by Western blot for co-precipitated pro-apoptotic proteins (BAX, BAK, BIM, BID).
Table 2: Key Experimental Readouts for MOMP Inhibition Studies
| Method | Primary Readout | Indication of Effective MOMP Inhibition | Complementary Validation |
|---|---|---|---|
| Cytochrome c Release | Cytochrome c in supernatant by Western blot or spectrophotometry | Reduced cytochrome c release in protected mitochondria | Caspase-3 activity assay in cytosolic fractions |
| Mitochondrial Membrane Potential | Fluorescence intensity of JC-1 or TMRE dyes | Maintained red/green (JC-1) or high (TMRE) fluorescence | ATP production assay |
| BAX/BAK Conformational Change | Immunoprecipitation with conformation-specific antibodies | Reduced active BAX/BAK in presence of anti-apoptotic proteins | Size exclusion chromatography of mitochondrial membranes |
| BH3 Profiling | Depolarization kinetics in response to specific BH3 peptides | Distinct protection patterns against specific BH3 peptides | Computational modeling of binding affinities |
Structural Validation Techniques
Surface Plasmon Resonance (SPR) quantitatively measures binding kinetics between anti-apoptotic proteins and BH3 peptides. Immobilize recombinant anti-apoptotic proteins on CMS sensor chips and inject BH3 peptides at varying concentrations (0.1-1000 nM) to determine association/dissociation constants [28].
Nuclear Magnetic Resonance (NMR) Spectroscopy maps interaction surfaces by monitoring chemical shift perturbations of isotopically labeled anti-apoptotic proteins upon titration with BH3 peptides [28]. This approach identified that VDAC1 oligomerization triggers exposure of its N-terminal α-helix, enabling binding to the BH3-binding groove of BCL-xL [28].
X-ray Crystallography provides atomic-resolution structures of anti-apoptotic proteins complexed with BH3 peptides or small-molecule inhibitors. The solved structure of VDAC1-N-terminal peptide complexed with BCL-xL confirmed binding mode similarity to sensitizer BH3 proteins [28].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Investigating MOMP Inhibition
| Reagent Category | Specific Examples | Research Application | Technical Considerations |
|---|---|---|---|
| Recombinant Proteins | BCL-2, BCL-xL, MCL-1, BAX, BAK, tBID | In vitro MOMP assays, binding studies | Verify correct folding by circular dichroism; ensure mitochondrial localization tags for functional assays |
| BH3 Peptides | BIM BH3, BAD BH3, NOXA BH3, PUMA BH3 | BH3 profiling, competitive displacement assays | High-purity synthetic peptides (>95%); confirm α-helical structure by CD spectroscopy |
| Chemical Inhibitors | Venetoclax (BCL-2), A-1331852 (BCL-xL), S63845 (MCL-1) | Specific inhibition of anti-apoptotic proteins in cellular models | Titrate concentration carefully; monitor on-target toxicities (thrombocytopenia for BCL-xL inhibitors) |
| Antibodies for Detection | Anti-cytochrome c, conformation-specific BAX/BAK, anti-BCL-2 family members | Western blot, immunofluorescence, immunoprecipitation | Validate specificity using knockout cell lines; optimize for specific applications (e.g., native conditions for co-IP) |
| Mitochondrial Dyes | JC-1, TMRE, MitoTracker | Live-cell imaging of membrane potential | Calibrate dye concentrations for each cell type; establish baseline fluorescence for quantitative comparisons |
| Expression Plasmids | GFP-tagged BCL-2 mutants, shRNAs for knockdown | Mechanistic studies in cellular models | Use inducible systems for toxic genes; include proper controls for off-target effects |
Translational Applications and Research Perspectives
BH3-Mimetics as Therapeutic Agents
The understanding of MOMP inhibition mechanisms has directly enabled development of BH3-mimetic drugs that selectively antagonize specific anti-apoptotic BCL-2 proteins [110] [5]. Venetoclax (ABT-199), the first FDA-approved selective BCL-2 inhibitor, exemplifies successful translation of this mechanistic knowledge [110] [112]. It binds with high specificity to the BH3-binding groove of BCL-2, displacing pro-apoptotic proteins like BIM to reactivate the apoptotic pathway in cancer cells [112] [113].
The sensitivity to BH3-mimetics depends not only on BCL-2 expression levels but also on the dynamic balance of BCL-2 family proteins, particularly the presence of apoptotic activators bound to BCL-2 and the expression of other anti-apoptotic proteins like MCL-1 that can compensate for BCL-2 inhibition [113]. This understanding has led to combination therapies that target multiple anti-apoptotic proteins simultaneously or sequentially.
Future Research Directions
Despite significant advances, several challenging research questions remain:
- How do specific BCL-2 family members mediate cell-type-dependent stress responses? [17]
- What precise conformational changes enable the transition from F-ATP synthase function to mPTP formation? [72]
- How can we achieve tumor-specific BCL-xL or MCL-1 inhibition to overcome on-target toxicities? [5]
- What role does partial MOMP (minority MOMP or iMOMP) play in non-apoptotic functions and pathophysiological outcomes? [17]
Emerging technologies like proteolysis targeting chimeras (PROTACs) that selectively degrade specific anti-apoptotic proteins, and antibody-drug conjugates for targeted delivery, represent promising approaches to enhance the therapeutic window of MOMP-directed therapies [5].
Diagram 2: Experimental Workflow for Validating MOMP Inhibition. This workflow outlines the key methodological approaches for investigating the inhibitory function of anti-apoptotic BCL-2 proteins on mitochondrial permeabilization.
The inhibition of MOMP by anti-apoptotic BCL-2 proteins represents a fundamental cellular mechanism for maintaining survival under stress conditions. Through precise protein-protein interactions mediated by their hydrophobic BH3-binding grooves, BCL-2, BCL-xL, and MCL-1 effectively neutralize the pro-apoptotic functions of BH3-only proteins and directly restrain the pore-forming activities of BAX and BAK. The experimental methodologies outlined in this review provide a robust framework for validating these mechanisms across biochemical, cellular, and structural domains. As research continues to elucidate the complexity of these regulatory networks, particularly through emerging structural biology techniques and innovative therapeutic approaches, our understanding of MOMP inhibition will undoubtedly expand, offering new opportunities for targeting this critical pathway in disease treatment.
Mitochondrial outer membrane permeabilization (MOMP) is a fundamental biological process often described as the 'point of no return' in the intrinsic pathway of apoptosis [1]. This event is primarily governed by the Bcl-2 family of proteins, where the pro-apoptotic executioners BAX and BAK oligomerize to form pores in the outer mitochondrial membrane [114] [49]. This permeabilization leads to the release of crucial pro-apoptotic factors from the mitochondrial intermembrane space, such as cytochrome c and Smac/DIABLO, into the cytosol [51] [1]. Cytochrome c, in concert with APAF-1, forms the apoptosome, which activates caspase-9 and subsequently the effector caspases-3 and -7, culminating in the organized dismantlement of the cell [114] [115]. Given that the evasion of apoptosis is a recognized hallmark of cancer, the targeted induction of MOMP represents a rational and compelling strategy for anticancer therapy [114]. This whitepaper provides a technical benchmark of MOMP-inducing agents, spanning from pre-clinical tool compounds to agents in clinical development, and details the experimental frameworks for their evaluation.
The Molecular Architecture of MOMP Induction
The regulation of MOMP is centered on a complex interplay between members of the Bcl-2 protein family. The following diagram illustrates the key components and sequence of events in the major apoptotic pathways that converge on MOMP.
The diagram above shows how diverse death signals converge on MOMP. The intrinsic pathway is activated by internal cellular stresses, leading to the upregulation or activation of BH3-only proteins [114]. These proteins either neutralize anti-apoptotic members like Bcl-2 and Bcl-xL ("sensitizing") or directly activate BAX/BAK ("activating") [114] [1]. The extrinsic pathway, initiated by ligands such as TRAIL binding to death receptors, activates caspase-8, which can directly cleave and activate the BH3-only protein Bid to truncated Bid (tBid), thereby engaging the mitochondrial pathway [114] [49]. The oligomerization of BAX and BAK constitutes the final common step, resulting in MOMP and the irreversible commitment to cell death [114] [115] [1].
Benchmarking MOMP-Inducing Agents
MOMP-inducing agents can be categorized based on their molecular targets and mechanisms of action. The following section provides a technical benchmark of key agents, from pre-clinical tools to clinical-stage therapeutics, with their profiles summarized in the accompanying table.
Pre-Clinical Tool Compounds
- Raptinal: This small molecule is a powerful tool for rapidly inducing intrinsic apoptosis. Its mechanism is distinct from other common agents, as it acts downstream of BAX/BAK to trigger MOMP and cytochrome c release [115]. This unique action makes it exceptionally fast and useful for studying apoptosis execution and as a positive control in cell death assays. Raptinal has been shown to induce MOMP and caspase activation independently of BAX/BAK knockout in cellular models, suggesting it may directly target the mitochondrial membrane or other downstream components [115].
- TRAIL/DR Agonists (Pre-clinical variants): Several engineered biologics aim to activate the extrinsic pathway to induce MOMP via caspase-8 and tBid. MEDI3039 is a multivalent DR5 superagonist that has demonstrated efficacy in solid tumor mouse models [114]. Similarly, CPT (circularly permuted TRAIL) is a TRAIL receptor agonist that has been investigated in numerous clinical trials in China for hematologic malignancies and solid tumors [114].
Clinical-Stage Agents
- Venetoclax: A prime example of a successful BH3-mimetic, venetoclax is an FDA-approved oral drug that selectively inhibits the anti-apoptotic protein Bcl-2 [114] [115]. By binding to Bcl-2, it displaces pro-apoptotic BH3-only proteins, allowing them to activate BAX/BAK and trigger MOMP. It is primarily used in hematologic malignancies like chronic lymphocytic leukemia [114].
- ONC201/TIC10: This first-in-class small molecule is a unique clinical-stage agent that transcriptionally upregulates the TRAIL ligand and its death receptor DR5, thereby activating the extrinsic pathway that converges on MOMP via caspase-8 and tBid [114] [116]. It activates the transcription factor Foxo3a through inactivation of Akt and ERK signaling. Its favorable safety profile and induction of apoptosis in tumor cells while sparing normal cells have propelled it into multiple Phase I and II clinical trials for solid tumors and hematologic malignancies [114] [116].
- DR5 Agonistic Antibodies: A class of biologics designed to directly activate the extrinsic pathway. Examples in clinical development include RG7386 (a bispecific antibody) and HexaBody-DR5/DR5 (GEN1029), which are designed to enhance receptor clustering and signaling [114]. While early DR5 agonists like conatumumab showed promise, later development has faced challenges related to efficacy and hepatotoxicity, underscoring the complexity of targeting this pathway [114].
Table 1: Benchmarking Profile of Key MOMP-Inducing Agents
| Agent Name | Class / Target | Mechanism of Action | Key Features / Advantages | Development Status |
|---|---|---|---|---|
| Raptinal [115] | Small Molecule / Downstream of BAX/BAK | Induces MOMP and cytochrome c release via an unknown target downstream of BAX/BAK. | Rapid action (potent in minutes); BAX/BAK-independent; useful as a positive control. | Pre-clinical Tool Compound |
| Venetoclax [114] | BH3-mimetic / Bcl-2 inhibitor | Selectively inhibits anti-apoptotic Bcl-2, displacing BH3-only proteins to activate BAX/BAK. | Oral bioavailability; high selectivity for Bcl-2; proven clinical efficacy in hematologic cancers. | FDA-Approved (Clinical) |
| ONC201/TIC10 [114] [116] | Small Molecule / TRAIL pathway inducer | Upregulates TRAIL and DR5 expression by inactivating Akt/ERK and activating Foxo3a. | Activates both ligand and receptor; p53-independent mechanism; favorable therapeutic window. | Phase II/III Trials |
| TAS266 [114] | Tetravalent Nanobody / DR5 agonist | Multivalent DR5 binding induces receptor clustering and activation of caspase-8. | High avidity and potency due to multivalency. | Clinical Development (Terminated) |
| MEDI3039 [114] | Multivalent Scaffold / DR5 superagonist | Engineered scaffold potently activates DR5 signaling, leading to robust caspase-8 activation. | High potency in vitro and in vivo (mouse models). | Pre-clinical |
The Scientist's Toolkit: Essential Reagents and Protocols
This section details the critical reagents and experimental methodologies for investigating MOMP and the efficacy of inducing agents.
Research Reagent Solutions
Table 2: Essential Reagents for MOMP and Apoptosis Research
| Reagent / Tool | Function / Application | Key Characteristics |
|---|---|---|
| Live-Cell Caspase Reporters (e.g., IC-RP, EC-RP) [51] | FRET-based reporters to monitor initiator (caspase-8) and effector (caspase-3/7) activity in real-time in single cells. | IETD sequence for initiator; DEVDR sequence for effectors (high specificity over caspase-8). |
| IMS-RP (Inter-Membrane Space Reporter) [51] | Fluorescent protein fused to a mitochondrial localization signal (e.g., from Smac) to visually monitor MOMP via its redistribution from mitochondria to cytosol. | Lacks IAP-binding motif (biochemically inert); enables tracking of MOMP dynamics with high temporal resolution. |
| Caspase Inhibitors (e.g., Q-VD-OPh, z-VAD-fmk) [115] | Pan-caspase inhibitors used to confirm the caspase-dependent nature of cell death. | Broad-spectrum, cell-permeable; used to block apoptosis execution downstream of MOMP. |
| siRNA/shRNA Knockdown Kits [115] | For functional validation of protein involvement (e.g., APAF1, caspase-9, BAX/BAK) in the agent's mechanism. | Confirms on-target mechanism; e.g., protection from Raptinal death after APAF1 knockdown [115]. |
| Annexin V / Propidium Iodide (PI) [115] | Flow cytometry assay to detect phosphatidylserine externalization (early apoptosis) and loss of membrane integrity (late apoptosis/necrosis). | Standard quantitative method for assessing apoptosis in cell populations. |
Detailed Experimental Protocol: Quantitative Analysis of MOMP Dynamics
The following workflow, adapted from seminal work, outlines a comprehensive approach to quantitatively analyze the dynamics of MOMP and caspase activation in single cells in response to an inducing agent [51].
Step-by-Step Protocol:
- Stable Cell Line Generation: Generate cell lines (e.g., HeLa, HCT116) stably expressing the IMS-RP, IC-RP, and EC-RP reporters [51]. Validate expression and localization.
- Live-Cell Imaging Setup: Seed cells in glass-bottom dishes. Use a confocal microscope equipped with an environmental chamber to maintain physiologically relevant conditions (37°C, 5% CO₂) throughout the experiment.
- Treatment & Time-Lapse Imaging: Treat cells with the MOMP-inducing agent (e.g., 100 nM TRAIL with cycloheximide, or 10 µM Raptinal). Immediately begin time-lapse imaging, acquiring images for CFP, YFP, and RFP (or other relevant channels) at intervals of 3-5 minutes for a duration of 8-12 hours to capture the highly variable delay to MOMP [51].
- Quantitative Image Analysis:
- MOMP Kinetics: Use image analysis algorithms to quantify the shift of IMS-RP fluorescence from a punctate (mitochondrial) to a diffuse (cytosolic) pattern for each cell. The time of MOMP is defined as the frame where this transition occurs [51].
- Caspase Activation Kinetics: Calculate the CFP/YFP emission ratio for IC-RP and EC-RP. A sharp increase in this ratio indicates cleavage of the linker and, therefore, caspase activity. Determine the timing of initiator and effector caspase activation for each cell [51].
- Data Integration and Modeling: For each cell, align the kinetic trajectories of initiator caspase activity, MOMP, and effector caspase activity. This data can be used to parameterize mathematical models of the apoptotic signaling network, revealing the core regulatory logic and identifying sources of cell-to-cell variability [51] [49]. Key parameters to extract include the delay between stimulus and MOMP, the delay between MOMP and effector caspase activation, and the velocity of MOMP wave propagation [51] [49].
The strategic induction of MOMP remains a cornerstone of anticancer therapeutic development. The successful clinical approval of venetoclax validates Bcl-2 family proteins as druggable targets, while the ongoing clinical investigation of agents like ONC201 and next-generation DR5 agonists highlights the continued pursuit of engaging the extrinsic pathway [114]. The future of this field lies in overcoming the challenges of resistance mechanisms and tumor heterogeneity. This will likely involve the rational combination of MOMP inducers with other targeted agents, such as Bcl-2 inhibitors with DR agonists, to synergistically lower the threshold for apoptosis commitment [114]. Furthermore, the integration of robust predictive biomarkers—such as the levels of anti-apoptotic proteins or the transcriptional profiling of the apoptosis pathway—into clinical trial design will be essential for identifying patient populations most likely to respond to these precision therapeutics [114]. The continued use of sophisticated single-cell analyses and mathematical modeling will further illuminate the dynamics of cell death signaling, guiding the development of more effective and reliable MOMP-targeting strategies.
The study of complex biological processes like apoptosis has been fundamentally transformed by the adoption of systems biology approaches. This interdisciplinary field moves beyond traditional reductionist methods by combining quantitative experimental data with mathematical modeling to understand system-level behaviors [117]. In the specific context of mitochondrial outer membrane permeabilization (MOMP)—the decisive commitment step in the intrinsic apoptosis pathway—systems biology provides powerful tools to decipher the complex regulatory networks that control cellular fate decisions [37] [25]. MOMP represents a critical point of convergence between extracellular death signals and intracellular stress pathways, making it a focal point for both basic research and therapeutic development, particularly in oncology [37].
The validation of mathematical models against experimental data forms the cornerstone of reliable systems biology. This process ensures that computational representations accurately reflect biological reality, enabling researchers to make meaningful predictions about system behavior under novel conditions. For MOMP research, this integration is particularly crucial because the process is regulated by a complex interplay between Bcl-2 family proteins, mitochondrial membrane characteristics, and cellular stress signals [19] [25]. Through iterative cycles of model prediction and experimental validation, researchers can uncover the fundamental design principles of apoptotic control, identify critical regulatory nodes, and develop strategies for therapeutic intervention in diseases characterized by dysregulated cell death.
Mathematical Modeling Frameworks for Biological Systems
Systems biology employs diverse mathematical formalisms to represent and analyze biological networks, each with distinct strengths and applications. The choice of modeling technique depends on the specific research question, available data, and desired level of mechanistic detail. For the analysis of MOMP and apoptosis signaling, several computational frameworks have been successfully applied, ranging from qualitative network representations to quantitative kinetic models [117] [118].
Boolean modeling provides a qualitative approach where proteins and other components are represented as nodes in a network that can exist in either "on" or "off" states (represented by 1 and 0, respectively) [117]. Interactions between nodes are described using logical operators (AND, OR, NOT). While this approach cannot capture the temporal dynamics of protein concentrations, it is valuable for analyzing network topology and identifying stable signaling states, especially when quantitative kinetic data are limited [117] [118].
Ordinary Differential Equations (ODEs) represent the most widely used framework for quantitative dynamic modeling of apoptosis. ODE models assume well-mixed cellular compartments and describe the rate of change for each molecular species based on mass-action kinetics or other rate laws [117]. A simple reversible binding reaction (A + B ⇌ C) would be represented as a system of coupled ODEs: dA/dt = dB/dt = -k₁·A·B + k₂·C and dC/dt = k₁·A·B - k₂·C, where k₁ and k₂ are rate constants [117]. These models excel at capturing the temporal evolution of signaling processes but require estimation of numerous kinetic parameters that are often difficult to measure experimentally [117].
Partial Differential Equations (PDEs) extend ODE frameworks by incorporating spatial information, allowing researchers to model diffusion and compartmentalization effects [117]. While more computationally intensive, PDEs can provide insights into how subcellular localization influences MOMP regulation. Additional approaches include agent-based models that simulate behaviors of individual cells and cellular automata that model spatial interactions, both particularly useful for multi-cellular systems [118].
Specific Modeling Approaches for MOMP and Apoptosis
Mathematical modeling of apoptosis has evolved substantially since pioneering work in the early 2000s. The initial model by Fussenegger et al. captured key aspects of receptor-mediated and stress-induced caspase activation, establishing a foundation for subsequent research [118]. Later models by Bentele et al. provided deeper insights into CD95 signaling pathways, revealing threshold mechanisms whereby c-FLIP proteins inhibit caspase-8 activation at low ligand concentrations [118]. These models successfully explained the binary fate decisions that characterize apoptotic commitment.
For the intrinsic apoptosis pathway culminating in MOMP, several specialized modeling approaches have been developed. One significant study focused on caspase-9 activation within the apoptosome complex, using ODE models to test alternative mechanisms of assembly and activation [119]. The modeling results indicated that activation proceeds through a linear binding model with cooperative interactions rather than through network formation, providing new insights into the regulation of this critical process [119].
Table 1: Mathematical Modeling Approaches in Apoptosis Research
| Modeling Framework | Key Features | Applications in MOMP Research | Limitations |
|---|---|---|---|
| Boolean Networks | Qualitative (on/off states), logic gates | Network topology analysis, stable state identification | No temporal dynamics, no concentration data |
| Ordinary Differential Equations (ODEs) | Quantitative kinetics, temporal dynamics | Caspase activation kinetics, Bcl-2 protein interactions | Requires many parameters, assumes well-mixed system |
| Partial Differential Equations (PDEs) | Incorporates spatial dimensions, diffusion | Subcellular localization effects, gradient formation | Computational complexity, difficult parameter estimation |
| Agent-Based Models | Individual cell behavior, cell-cell interactions | Population heterogeneity, immune cell interactions | Computationally intensive for large systems |
| Hybrid Approaches | Combines multiple frameworks | Multi-scale systems from molecular to tissue level | Implementation complexity |
Recent modeling efforts have increasingly addressed the crosstalk between different cell death modalities. Although current ODE models typically incorporate no more than two regulated cell death types simultaneously, they have revealed critical shared regulators including Bcl-2 family proteins, Ca²⁺ signaling, and p53 [118]. These integrative models highlight the interconnected nature of cell death regulation and represent an important step toward comprehensive computational frameworks for cell fate decisions.
The Biology of MOMP in Apoptosis
Molecular Regulation of MOMP
Mitochondrial outer membrane permeabilization represents the point of no return in the intrinsic apoptosis pathway, characterized by increased permeability of the mitochondrial outer membrane that allows proteins, DNA, and other molecules to pass from the intermembrane space into the cytosol [37]. This process is primarily regulated by the Bcl-2 family of proteins, which can be functionally categorized into three groups: anti-apoptotic proteins (e.g., Bcl-2, Bcl-xL), multi-domain pro-apoptotic effector proteins (BAX, BAK), and BH3-only proteins (Bid, BIM, PUMA) that act as initiators [37] [25].
In healthy cells, anti-apoptotic Bcl-2 proteins maintain mitochondrial integrity by binding and inhibiting pro-apoptotic family members. During apoptosis induction, activated BH3-only proteins either directly engage and activate BAX/BAK or neutralise anti-apoptotic proteins through competitive binding [25]. Once activated, BAX and BAK undergo conformational changes, translocate to the mitochondrial outer membrane, and oligomerize to form pores that facilitate the release of cytochrome c and other pro-apoptotic factors [19] [37]. The release of cytochrome c into the cytosol triggers the formation of the apoptosome complex, which activates caspase-9 and initiates the downstream caspase cascade that executes cell death [119] [25].
Recent research has revealed that BAX and BAK, while functionally redundant in many contexts, exhibit distinct oligomerization properties and assembly kinetics. BAK organizes into smaller structures with faster kinetics than BAX, and the two proteins co-assemble into the same apoptotic pores, reciprocally regulating each other's oligomerization [19]. The relative availability of BAX and BAK molecules determines the growth rate of apoptotic pores and consequently influences the kinetics of mitochondrial content release, including mitochondrial DNA (mtDNA) [19]. This mtDNA release activates the cGAS/STING pathway, connecting MOMP to inflammatory signaling and demonstrating the broader physiological implications of apoptotic pore dynamics [19].
MOMP as a Regulated Process
Contrary to the traditional view of MOMP as a binary "all-or-nothing" process, recent evidence supports a more nuanced model where the extent of MOMP can vary significantly based on the strength and duration of apoptotic stimuli [37]. Completed MOMP occurs when sufficient stress signals trigger widespread BAX/BAK activation across most mitochondria, leading to irreversible commitment to apoptosis. In contrast, minority MOMP (miMOMP) describes a limited permeabilization where only a subset of mitochondria undergo MOMP [37].
This sublethal MOMP has significant pathophysiological consequences. When caspase activity is limited, cells experiencing miMOMP may survive but sustain damage that manifests as genomic instability, cellular senescence, or chronic inflammation [37]. The released mitochondrial content, particularly mtDNA, can activate innate immune pathways even in the absence of complete cell death, contributing to paracrine signaling and microenvironment alterations that influence cancer progression and treatment resistance [19] [37].
Table 2: Key Proteins Regulating MOMP and Their Functions
| Protein | Type | Function in MOMP Regulation | Experimental Detection Methods |
|---|---|---|---|
| BAX | Pro-apoptotic effector | Translocates from cytosol, oligomerizes to form pores | SMLM, AFM, confocal imaging [19] |
| BAK | Pro-apoptotic effector | Mitochondrial resident, oligomerizes with BAX | SMLM, AFM, STED microscopy [19] |
| Bcl-2/Bcl-xL | Anti-apoptotic | Binds and inhibits BAX/BAK activation | Co-immunoprecipitation, FRET |
| Bid, BIM | BH3-only activator | Directly activates BAX/BAK | Western blot, fluorescence polarization |
| Bad, NOXA | BH3-only sensitizer | Neutralizes anti-apoptotic Bcl-2 proteins | Protein interaction assays |
| Cytochrome c | Apoptogenic factor | Released after MOMP, activates apoptosome | ELISA, immunofluorescence, Western blot [119] |
| Caspase-9 | Initiator caspase | Activated in apoptosome, initiates caspase cascade | Fluorogenic substrate assays [119] |
The regulation of MOMP extends beyond the Bcl-2 family to include mitochondrial membrane composition, metabolic status, and interaction with other organellar systems. Lipid components of the mitochondrial outer membrane, including cardiolipin and other phospholipids, influence the recruitment and activation of Bcl-2 proteins [25]. Additionally, cellular stress pathways involving calcium signaling, oxidative stress, and metabolic alterations can modulate the sensitivity to MOMP induction, creating a complex regulatory network that integrates diverse physiological signals [37].
Integrated Validation Workflows
The Model Validation Cycle
Validating mathematical models of MOMP requires a systematic, iterative approach that bridges computational and experimental domains. The core validation cycle begins with model formulation based on existing biological knowledge and preliminary data. This initial model incorporates known molecular interactions, such as BAX/BAK activation kinetics, anti-apoptotic protein binding, and pore formation mechanics [19] [37]. The model structure may be derived from literature curation, prior experimental findings, or hypothesized mechanisms that require testing.
Following formulation, parameter estimation calibrates the model using quantitative experimental data. This process involves determining kinetic constants, binding affinities, and initial concentrations that enable the model to reproduce observed behaviors [117]. Parameter estimation can be challenging due to the limited availability of precise quantitative measurements for many biological processes. Computational tools like COPASI facilitate this process by implementing optimization algorithms that minimize the difference between model predictions and experimental data [120]. For example, in studying temporal evolution of silver nanoparticle toxicity, researchers used COPASI to estimate rate constants from time-course gene expression data, enabling dynamic simulations of cellular stress responses [120].
With parameters established, model simulation generates predictions for system behavior under conditions that may not have been experimentally tested. These simulations can explore a wide range of scenarios, including genetic perturbations (overexpression or knockdown of specific regulators), environmental changes (varying stress intensities), or therapeutic interventions (drug treatments) [118]. The predictive capability of the model is then assessed through experimental validation, where specifically designed experiments test the model's predictions. Discrepancies between predictions and experimental outcomes trigger model refinement, potentially involving structural changes to the network topology or adjustment of kinetic parameters, thus completing the validation cycle [117] [119].
Experimental Methodologies for MOMP Model Validation
Quantitative Imaging Techniques
Advanced microscopy methods provide crucial quantitative data for validating spatial and dynamic aspects of MOMP models. Super-resolution microscopy techniques, particularly single-molecule localization microscopy (SMLM), enable visualization of BAX and BAK oligomeric structures at nanoscale resolution [19]. These approaches have revealed that BAX and BAK form lines, arcs, and rings at the mitochondrial membrane, with distinct size distributions and assembly kinetics [19]. For example, SMLM studies demonstrated that BAK assembles into significantly smaller structures (mean ring radius ~18 nm) compared to BAX (~34 nm), providing quantitative parameters for model refinement [19].
DNA-PAINT (Points Accumulation for Imaging in Nanoscale Topography), a specific SMLM implementation, uses transient DNA hybridization to achieve blinking for super-resolution imaging [121]. This technique allows precise quantification of protein localization and clustering dynamics. Optimal dye selection is critical for DNA-PAINT performance, with studies identifying Cy3B, Atto565, and CF568 as well-performing dyes for green excitation wavelengths, and Atto643, Atto647N, and Cy5B for red excitation [121]. These imaging approaches generate quantitative data on protein clustering kinetics, oligomer size distributions, and spatial relationships that directly inform model parameters.
Atomic force microscopy (AFM) provides complementary structural information by directly imaging membrane-associated protein assemblies under near-physiological conditions. AFM studies have confirmed that both BAX and BAK arcs and rings are associated with membrane pores, validating the fundamental premise of many MOMP models [19]. Correlative microscopy approaches that combine live-cell confocal imaging with fixed-cell SMLM enable researchers to connect dynamic cellular processes with high-resolution structural data, capturing the temporal evolution of MOMP events [19].
Biochemical and Molecular Assays
Caspase activity assays provide crucial kinetic data for validating downstream consequences of MOMP. These assays typically use fluorogenic substrates (e.g., DEVD-AFC for caspase-3) to measure protease activity over time [119]. In studies of caspase-9 activation, researchers measured caspase-3 generation as a proxy for apoptosome activity, establishing linear relationships between substrate cleavage and enzyme concentration that facilitate quantitative modeling [119].
Protein interaction studies using co-immunoprecipitation, surface plasmon resonance, or fluorescence polarization quantify binding affinities between Bcl-2 family members, providing essential parameters for model calibration. Cytochrome c release assays monitor MOMP dynamics directly, using ELISA-based approaches or live-cell imaging with fluorescently tagged cytochrome c [119]. Gene expression analysis through transcriptomics can capture system-wide responses to apoptotic stimuli, as demonstrated in studies of silver nanoparticle toxicity that revealed temporal patterns of stress pathway activation [120].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for MOMP Modeling and Validation
| Category | Specific Reagents | Function/Application | Key Considerations |
|---|---|---|---|
| Recombinant Proteins | Apaf-1, cytochrome c, procaspase-9, Bcl-2 family proteins | Reconstitute apoptosome formation, measure activation kinetics [119] | Maintain proper folding and post-translational modifications |
| Fluorescent Dyes & Labels | Cy3B, Atto643, CF488A, DEVD-AFC substrate | Super-resolution imaging, caspase activity assays [121] [119] | Match dye performance to imaging technique; consider photostability |
| Cell Lines | BAX/BAK DKO HCT116, SKW 6.4 B lymphoblastoid cells | Study specific protein functions, validate model predictions [19] [118] | Verify genetic background and authentication regularly |
| Computational Tools | COPASI, Simpathica, MATLAB | Parameter estimation, model simulation, data analysis [120] [119] | Select tools with appropriate algorithms and visualization capabilities |
| Antibodies | Anti-cytochrome c, anti-BAX/BAK, anti-caspase cleaved forms | Immunodetection, Western blotting, immunoprecipitation | Validate specificity for intended applications |
| Specialized Microscopy Systems | SMLM, STED, AFM, TIRF | Nanoscale imaging of oligomeric structures, pore visualization [19] [121] | Consider resolution limits, sample preparation requirements |
| Apoptosis Inducers | Staurosporine, ABT-737, TRAIL, etoposide | Experimental triggering of MOMP at controlled levels [37] | Titrate concentration for desired MOMP extent (complete vs. minority) |
Case Studies in MOMP Model Validation
Temporal Dynamics of Cellular Stress Responses
A systems biology study on silver nanoparticle (AgNP) toxicity demonstrated the power of integrating time-course transcriptomic data with dynamic simulations [120]. Researchers identified differentially expressed genes (DEGs) at multiple time points (1, 6, and 24 hours) after AgNP exposure, revealing a temporal progression of cellular responses [120]. Early time points showed activation of ribosomal biogenesis and stress pathways, while later stages exhibited shifts toward DNA repair and cell cycle regulation [120].
Protein-protein interaction networks identified key hub genes that changed over time, with ribosomal proteins (RPS27A, RPS11, RPL23A) dominating early responses and cell cycle regulators (CDC20, CDK1, PLK1) emerging later [120]. These experimental findings informed the construction of a pathway map using CellDesigner, which was subsequently translated into a kinetic model using COPASI [120]. Parameter estimation based on time-course gene expression data enabled dynamic simulations that captured the sequential activation of stress response genes, DNA repair attempts, and eventual apoptotic signaling [120]. This approach provided a predictive temporal framework for nanotoxicology research while demonstrating generalizable methods for validating dynamic models of cellular stress responses.
BAX/BAK Oligomerization Dynamics
Research elucidating the distinct oligomerization properties of BAX and BAK provides an exemplary case of how quantitative imaging data can inform and validate mathematical models of MOMP [19]. Through SMLM, AFM, and live-cell imaging, researchers demonstrated that BAK organizes into smaller structures with faster kinetics than BAX, and that these proteins co-assemble into the same apoptotic pores [19]. The quantitative parameters derived from these experiments—including oligomer size distributions, growth rates, and relative abundance effects—provided critical validation data for models of pore assembly kinetics.
This study revealed that BAK recruits and accelerates BAX assembly into oligomers that continue to grow during apoptosis, with the relative availability of BAX and BAK determining pore growth dynamics [19]. These findings challenged simplified models that treated BAX and BAK as functionally identical and led to more sophisticated computational representations that account for their distinct kinetics and cooperative interactions. The functional implications of these dynamics extend to the regulation of mtDNA release and subsequent activation of the cGAS/STING pathway, demonstrating how molecular-scale modeling connects to broader physiological outcomes [19].
Cross-Validation of Apoptosome Assembly Mechanisms
A comparative modeling approach to caspase-9 activation demonstrated how alternative mechanistic hypotheses can be evaluated against experimental data [119]. Researchers used the Simpathica systems biology tool to generate ODE models representing different assembly mechanisms for the apoptosome complex [119]. Model predictions were compared with data from a recombinant system containing purified Apaf-1, cytochrome c, and procaspase-9, allowing direct assessment of caspase activation kinetics under controlled conditions [119].
The modeling results indicated that activation proceeds through a linear binding model with cooperative interactions rather than through network formation, providing mechanistic insights that would be difficult to obtain through experimental approaches alone [119]. This case study highlights the value of model checking approaches in systems biology, where multiple candidate models are systematically evaluated against quantitative data to identify the most plausible mechanisms [119]. The recombinant experimental system provided precisely controlled conditions that reduced biological noise, enabling more rigorous model validation than would be possible in cellular environments.
The integration of mathematical models with experimental data represents a powerful paradigm for advancing our understanding of MOMP regulation. Current challenges in the field include developing models that encompass multiple cell death modalities, incorporating spatial heterogeneity, and bridging scales from molecular interactions to tissue-level outcomes [118]. Future progress will require continued refinement of both computational and experimental methodologies to address these multi-scale challenges.
Emerging opportunities in MOMP modeling include the development of more comprehensive frameworks that simultaneously incorporate three or more regulated cell death pathways, better representation of minority MOMP phenomena and their pathophysiological consequences, and increased attention to the inflammatory and immune consequences of apoptotic cell death [37] [118]. The growing availability of single-cell technologies will enable models that account for cellular heterogeneity in apoptotic responses, potentially revealing mechanisms underlying fractional killing and variable treatment responses in cancer therapy [19].
For researchers pursuing MOMP studies, the successful integration of modeling and experimental approaches requires careful attention to several principles: (1) selecting appropriate modeling frameworks matched to the biological questions and available data, (2) implementing rigorous validation cycles that test model predictions through specifically designed experiments, (3) leveraging advanced imaging and quantification methods to generate high-quality parameter estimates, and (4) maintaining computational models as evolving representations that incorporate new biological insights. As these integrated approaches mature, they promise to accelerate the development of novel therapeutic strategies that target MOMP regulation in cancer and other diseases characterized by dysregulated cell death.
Conclusion
MOMP stands as a decisive, regulated process central to cellular life-and-death decisions, governed by a complex interplay of Bcl-2 family proteins and influenced by mitochondrial lipids and dynamics. The integration of advanced methodologies, from single-molecule analysis to spatiotemporal modeling, is resolving long-standing controversies and providing unprecedented insights into the mechanisms of pore formation. The validation of MOMP's core components, particularly through recent structural studies on proteins like VDAC1, solidifies its promise as a therapeutic target. Future research must focus on translating this mechanistic understanding into novel therapeutic strategies, such as specific BH3-mimetics and VDAC modulators, to overcome apoptosis resistance in cancer and other diseases. The continued refinement of experimental models and the embrace of systems-level approaches will be crucial for harnessing the power of MOMP in biomedical innovation.
References
- 1. Mitochondrial outer membrane permeabilization during ... [https://www.nature.com/articles/4401963]
- 2. Mitochondrial outer membrane permeabilization [https://en.wikipedia.org/wiki/Mitochondrial_outer_membrane_permeabilization]
- 3. The Mitochondrial Pathway of Apoptosis: Part I: MOMP and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9159267/]
- 4. Mechanisms of Action of Bcl-2 Family Proteins - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3683897/]
- 5. The BCL2 family: from apoptosis mechanisms to new ... [https://www.nature.com/articles/s41392-025-02176-0]
- 6. Apoptosis Signaling Pathway: Stages, Types and Key ... [https://geneglobe.qiagen.com/us/knowledge/pathways/apoptosis-signaling]
- 7. Control of mitochondrial permeability by Bcl-2 family members [https://pubmed.ncbi.nlm.nih.gov/14996495/]
- 8. Mitochondrial outer membrane permeabilization at the ... [https://pubmed.ncbi.nlm.nih.gov/33576840/]
- 9. How do BCL-2 proteins induce mitochondrial outer membrane ... [https://pubmed.ncbi.nlm.nih.gov/18314333/]
- 10. The BCL-2 family of proteins and mitochondrial outer ... [https://pubmed.ncbi.nlm.nih.gov/28396106/]
- 11. The role of the Bcl-2 family in the regulation of outer ... [https://pubmed.ncbi.nlm.nih.gov/11175255/]
- 12. Bcl-2 family [https://en.wikipedia.org/wiki/Bcl-2_family]
- 13. The role of BCL-2 family proteins in regulating apoptosis ... [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2022.985363/full]
- 14. Control of mitochondrial permeability by Bcl-2 family ... [https://www.sciencedirect.com/science/article/pii/S0167488903001770]
- 15. The role of the Bcl-2 family in the regulation of outer ... [https://www.nature.com/articles/4400781]
- 16. by which Mechanisms and Bak permeabilise mitochondria during... Bax [https://pmc.ncbi.nlm.nih.gov/articles/PMC2736138/]
- 17. MOMP, cell suicide as a BCL-2 family business [https://www.nature.com/articles/cdd2017179]
- 18. Control of mitochondrial apoptosis by the Bcl-2 family - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2714431/]
- 19. The interplay between BAX and BAK tunes apoptotic pore ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8901441/]
- 20. The interplay between BAX and BAK tunes apoptotic pore ... [https://www.sciencedirect.com/science/article/pii/S1097276522000089]
- 21. Proapoptotic Bax and Bak Proteins Form Stable Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3829170/]
- 22. Raptinal bypasses BAX , BAK , and BOK for mitochondrial outer... [https://www.nature.com/articles/s41419-019-1790-z?error=cookies_not_supported&code=e637a6d2-26ed-402e-aaa1-b59396f8f428]
- 23. BH3-only proteins in apoptosis and beyond: an overview [https://pmc.ncbi.nlm.nih.gov/articles/PMC2928556/]
- 24. BH3-only proteins: Orchestrators of apoptosis [https://www.sciencedirect.com/science/article/pii/S0167488910003101]
- 25. Mitochondrial outer membrane permeabilization: a focus on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5578938/]
- 26. Collaborative orchestration of BH3-only proteins governs Bak ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12162870/]
- 27. BH3-Only Proteins Noxa and Puma Are Key Regulators of ... [https://www.mdpi.com/2075-1729/12/2/256]
- 28. Structural basis of apoptosis induction by the mitochondrial ... [https://www.nature.com/articles/s41467-025-65363-1]
- 29. The Mitochondrial Voltage-Dependent Anion Channel 1, ... [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2017.00060/full]
- 30. Oligomerization of the Mitochondrial Protein Voltage ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3004265/]
- 31. Novel Compounds Targeting the Mitochondrial Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5122769/]
- 32. Apoptosis is regulated by the VDAC1 N-terminal region ... [https://www.sciencedirect.com/science/article/pii/S0005272810000939]
- 33. The dual role of VDAC in cancer: Molecular mechanisms ... [https://www.sciencedirect.com/science/article/pii/S0753332225007243]
- 34. VDAC1 oligomerization activates PANoptosis via mtDNA- ... [https://www.sciencedirect.com/science/article/abs/pii/S0891584925013164]
- 35. Voltage-Dependent Anion Channel 1 As an Emerging ... [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2017.00154/full]
- 36. VDAC1 selectively transfers apoptotic Ca2+ signals to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3263501/]
- 37. MOMP: A critical event in cell death regulation and ... [https://www.sciencedirect.com/science/article/abs/pii/S0304419X25000228]
- 38. Shedding light on mitochondrial outer-membrane ... [https://www.sciencedirect.com/science/article/pii/S1084952123001416]
- 39. Measuring Apoptosis at the Single Cell Level - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2423010/]
- 40. Live-cell imaging to measure BAX recruitment kinetics to ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0184434]
- 41. A general method for the development of multicolor ... [https://www.nature.com/articles/s41589-023-01350-1]
- 42. A high-throughput microscopy method for single-cell ... [https://www.nature.com/articles/s42003-019-0282-0]
- 43. Quantitative single-molecule imaging of protein assembly ... [https://www.sciencedirect.com/science/article/abs/pii/S2451963420300042]
- 44. Dynamics of outer mitochondrial membrane ... [https://www.nature.com/articles/cdd2008187]
- 45. Quantitative single-protein imaging reveals molecular ... [https://www.nature.com/articles/s41467-021-21142-2]
- 46. Probing Cellular Protein Complexes via Single Molecule ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3103084/]
- 47. Assembly of Membrane-Bound Protein Complexes [https://pmc.ncbi.nlm.nih.gov/articles/PMC3318961/]
- 48. MOMP, cell suicide as a BCL-2 family business - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5729535/]
- 49. An Analysis of the Truncated Bid- and ROS-dependent Spatial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4813484/]
- 50. From computational modelling of the intrinsic apoptosis ... [https://www.nature.com/articles/cddis201436]
- 51. Quantitative analysis of pathways controlling extrinsic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2858979/]
- 52. The BCL-2 protein family: from discovery to drug ... [https://www.nature.com/articles/s41418-025-01481-z]
- 53. Functional dissociation of ΔΨm and cytochrome c release ... [https://www.nature.com/articles/4401772]
- 54. Cytochrome C Maintains Mitochondrial Transmembrane ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2169468/]
- 55. Nano-flow cytometry unveils mitochondrial permeability ... [https://www.nature.com/articles/s41420-024-01947-y]
- 56. Mitochondrial dynamics and pore formation in regulated ... [https://www.sciencedirect.com/science/article/pii/S0968000425002191]
- 57. Quantitative measurement of mitochondrial membrane ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3448152/]
- 58. Mitochondrial Membrane Potential Assay [https://www.sartorius.com/en/applications/life-science-research/live-cell-assays/mitochondrial-membrane-potential]
- 59. Flow Cytometric Analysis of Ca2+-Induced Membrane ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2212344/]
- 60. Protein–protein and protein–lipid interactions of pore-forming ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9310348/]
- 61. Lipid-protein interactions in biological membranes [https://pubmed.ncbi.nlm.nih.gov/12729927/]
- 62. Lipid environment of membrane proteins in cryo-EM based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5899730/]
- 63. CryoEM milestones and future directions - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12585528/]
- 64. Extending the reach of single-particle cryoEM [https://www.sciencedirect.com/science/article/pii/S0959440X25000235]
- 65. LipIDens: simulation assisted interpretation of lipid ... [https://www.nature.com/articles/s41467-023-43392-y]
- 66. BioDolphin as a comprehensive database of lipid–protein ... [https://www.nature.com/articles/s42004-024-01384-z]
- 67. Mitochondrial permeability transition pore as a selective ... [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2013.00041/full]
- 68. Mitochondrial residence of the apoptosis inducer BAX is ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7008371/]
- 69. Mitochondrial VDAC1: A Key Gatekeeper as Potential ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5491678/]
- 70. Mitochondrial Permeability Transition Pore-Dependent ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9868081/]
- 71. VDAC oligomers form mitochondrial pores that release ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8325171/]
- 72. Identity, structure, and function of the mitochondrial ... [https://www.nature.com/articles/s41418-023-01187-0]
- 73. Bax and Bak function as the outer membrane component of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3755340/]
- 74. Cell Biology A New Fungal Diterpene Induces VDAC1- ... [https://www.sciencedirect.com/science/article/pii/S0021925820447003]
- 75. VDAC-dependent permeabilization of the outer mitochondrial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2150912/]
- 76. BH3-Dependent and Independent Activation of BAX and BAK ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6186458/]
- 77. BH3 domains of BH3-only proteins differentially regulate Bax ... [https://pubmed.ncbi.nlm.nih.gov/15721256/]
- 78. Building blocks of the apoptotic pore: how Bax and Bak are ... [https://www.nature.com/articles/cdd2013139]
- 79. A Unified Model of Mammalian BCL-2 Protein Family ... [https://www.sciencedirect.com/science/article/pii/S109727651100760X]
- 80. Structural basis for proapoptotic activation of Bak by the ... [https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.3002156]
- 81. BH3 Domains of BH3-Only Proteins Differentially Regulate ... [https://www.sciencedirect.com/science/article/pii/S1097276505010774]
- 82. Harnessing PUMA's lethal potential: BCL-2 family ... [https://www.sciencedirect.com/science/article/pii/S246829422500111X]
- 83. State of the art methods and biosensors [https://pubmed.ncbi.nlm.nih.gov/37438211/]
- 84. Sub-lethal signals in the mitochondrial apoptosis apparatus [https://www.nature.com/articles/s41418-022-01058-0]
- 85. Current trends in luminescence-based assessment of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10613953/]
- 86. Minority MOMP: a toxic, slow demise - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7533071/]
- 87. The Mitochondrial Permeability Transition Pore—Current ... [https://www.mdpi.com/2073-4409/12/9/1273]
- 88. Mitochondrial dynamics in health and disease [https://www.nature.com/articles/s41392-023-01547-9]
- 89. DRP1, fission and apoptosis | Cell Death Discovery [https://www.nature.com/articles/s41420-025-02458-0]
- 90. The molecular mechanisms of mitochondrial dynamics and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12492677/]
- 91. Visualizing Mitochondrial Form and Function within the Cell [https://pmc.ncbi.nlm.nih.gov/articles/PMC6938546/]
- 92. Analysis of mitochondrial dynamics and functions using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3007120/]
- 93. Combining In Vivo 2-Photon Imaging with Photoactivatable ... [https://pubmed.ncbi.nlm.nih.gov/40310022/]
- 94. Apoptosis: A Comprehensive Overview of Signaling Pathways ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11592877/]
- 95. Intrinsic and Extrinsic Pathways of Apoptosis [https://www.thermofisher.com/us/en/home/life-science/antibodies/antibodies-learning-center/antibodies-resource-library/cell-signaling-pathways/cellular-apoptosis-pathway.html]
- 96. Cross-Talk in Cell Death Signaling - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2195865/]
- 97. Apoptosis signaling pathways and lymphocyte homeostasis [https://www.nature.com/articles/cr200752]
- 98. Insights on the crosstalk among different cell death ... [https://www.nature.com/articles/s41420-025-02328-9]
- 99. Convergence of pathway analysis and pattern recognition ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7370234/]
- 100. Apoptotic mitochondrial poration by a growing list of pore ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9975053/]
- 101. Organization of the Mitochondrial Apoptotic BAK Pore [https://pmc.ncbi.nlm.nih.gov/articles/PMC3908389/]
- 102. Gasdermin D permeabilization of mitochondrial inner and ... [https://www.sciencedirect.com/science/article/pii/S1074761323004442]
- 103. VDAC1 at the crossroads of cell metabolism, apoptosis and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6287957/]
- 104. VDAC Modulation of Cancer Metabolism: Advances and ... [https://www.frontiersin.org/journals/physiology/articles/10.3389/fphys.2021.742839/full]
- 105. Permeabilization of the outer mitochondrial membrane [https://www.sciencedirect.com/science/article/pii/S0925443924003107]
- 106. VDAC1-interacting molecules promote cell death in cancer ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11112339/]
- 107. Key regions of VDAC1 functioning in apoptosis induction ... [https://www.sciencedirect.com/science/article/pii/S0005272808007263]
- 108. VDAC1 as prognostic marker and therapeutic target in lung ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12630505/]
- 109. VDAC2 loss elicits tumour destruction and inflammation for ... [https://www.nature.com/articles/s41586-025-08732-6]
- 110. Inhibition of Anti-Apoptotic Bcl-2 Proteins in Preclinical and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7291206/]
- 111. Evolution of the BCL-2-Regulated Apoptotic Pathway - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7123326/]
- 112. Mechanisms of action of the BCL-2 inhibitor venetoclax in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10657905/]
- 113. Understanding the Mechanism and Sensitivity of BCL-2 ... [https://www.targetedonc.com/view/understanding-the-mechanism-and-sensitivity-of-bcl-2-inhibitors]
- 114. Novel Apoptosis-Inducing Agents for the Treatment of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6721450/]
- 115. Raptinal: a powerful tool for rapid induction of apoptotic cell ... [https://www.nature.com/articles/s41420-024-02120-1]
- 116. Identification of TRAIL-inducing compounds highlights small ... [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-015-0346-9]
- 117. Mathematical modeling of apoptosis - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC3699383/]
- 118. Mathematical Modeling of Cell Death and Survival [https://www.mdpi.com/2073-4409/14/22/1792]
- 119. Mathematical modeling of the formation of apoptosome in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2671592/]
- 120. A systems biology approach to understand temporal ... [https://www.nature.com/articles/s41540-025-00561-7]
- 121. a comprehensive performance analysis of fluorescent dyes [https://www.nature.com/articles/s41592-024-02374-8]