PARP-1 Cleavage: A Critical Switch Between Cell Death Pathways in Health and Disease
This article provides a comprehensive analysis of the mechanisms and functional consequences of poly(ADP-ribose) polymerase-1 (PARP-1) cleavage during programmed cell death (PCD).
PARP-1 Cleavage: A Critical Switch Between Cell Death Pathways in Health and Disease
Abstract
This article provides a comprehensive analysis of the mechanisms and functional consequences of poly(ADP-ribose) polymerase-1 (PARP-1) cleavage during programmed cell death (PCD). We explore how specific proteolytic cleavage of PARP-1 by caspases, calpains, and other proteases generates signature fragments that serve as biomarkers for distinct cell death modalities, including apoptosis, parthanatos, and necrosis. The content details methodological approaches for detecting PARP-1 cleavage fragments and their applications in basic research and drug development. We examine how PARP-1 cleavage functions as a molecular switch directing cellular fate decisions, with particular emphasis on its implications for cancer therapy, neurodegenerative diseases, and the development of PARP-targeted therapeutics. This resource is tailored for researchers, scientists, and drug development professionals seeking to understand and manipulate PARP-1-mediated cell death pathways.
The Molecular Architecture and Proteolytic Landscape of PARP-1
Poly(ADP-ribose) polymerase-1 (PARP-1) is a highly abundant nuclear enzyme that serves as a critical molecular sensor for DNA damage and plays a fundamental role in maintaining genomic integrity. As the founding member of the PARP superfamily, which comprises 17 distinct proteins, PARP-1 accounts for approximately 85% of total cellular PARP activity [1]. This multifunctional enzyme participates in various cellular processes, including DNA repair, chromatin remodeling, transcriptional regulation, and cell death signaling. The diverse functions of PARP-1 are facilitated by its modular domain architecture, which enables the protein to detect DNA damage, undergo auto-modification, and catalyze poly(ADP-ribose) formation. Within the context of programmed cell death research, PARP-1 serves as a key substrate for several suicidal proteases, including caspases, calpains, and granzymes, generating specific cleavage fragments that serve as signature biomarkers for different cell death pathways [1]. Understanding the domain structure of PARP-1 provides critical insights into its mechanism of action and its role as a central node in cell fate decisions.
PARP-1 is organized into three primary functional domains that work in concert to detect DNA damage and initiate appropriate cellular responses: an N-terminal DNA-binding domain (DBD), a central auto-modification domain (AMD), and a C-terminal catalytic domain (CAT) [1]. The full-length protein contains approximately 1-2 million copies per cell, reflecting its importance as a first responder to genomic insults [1].
Table 1: Primary Domains of Human PARP-1
| Domain | Location | Molecular Weight | Key Functions | Structural Features |
|---|---|---|---|---|
| DNA-Binding Domain (DBD) | N-terminal (residues 1-214) | 24 kDa | Recognizes DNA strand breaks, facilitates chromatin binding | Contains two zinc finger motifs (F1 & F2) [2] |
| Auto-Modification Domain (AMD) | Central (residues 214-524) | 22 kDa | Accepts PAR polymers; mediates protein-protein interactions | Contains BRCT fold and third zinc finger (F3) [1] |
| Catalytic Domain (CAT) | C-terminal (residues 525-1014) | 54 kDa | Catalyzes PAR polymerization using NAD+ | ADP-ribosyl transferase (ART) fold with helical subdomain [3] |
The functional integration of these domains allows PARP-1 to transition between different functional states in response to cellular signals, particularly during DNA damage response and programmed cell death processes.
DNA-Binding Domain (DBD)
Structural Composition and Zinc Finger Motifs
The DNA-binding domain of PARP-1 encompasses the first 214 amino acids and contains two zinc finger motifs (F1 and F2) that are structurally independent in the absence of DNA [2]. These zinc fingers belong to a highly unusual class characterized by a CCHC ligand pattern and an extended sequence separation (26-37 residues) between ligands 2 and 3 [2]. This distinctive structural arrangement enables specific recognition of DNA lesions. Both fingers share remarkably similar structural folds and dynamics, with finger 2 (F2) demonstrating significantly stronger binding to nicked or gapped DNA ligands compared to finger 1 (F1) [2]. The DBD fragment (residues 1-214) corresponds to the apoptotic fragment released through cleavage by caspase-3 and -7 during programmed cell death [2].
DNA Recognition Mechanism
The DBD recognizes DNA single-strand breaks as a monomer and in a single orientation, with recognition primarily achieved by F2 [2]. This interaction occurs in an essentially identical manner whether F2 is present in isolation or within the two-finger fragment. The DBD engages in at least two high-affinity binding modes with chromatin, one of which does not involve free DNA ends, consistent with PARP-1's role as a chromatin architectural protein [4]. Upon binding to DNA damage sites, the DBD facilitates the activation of the catalytic domain through an allosteric mechanism.
Table 2: DNA-Binding Affinities of PARP-1 Domains
| Binding Substrate | PARP-1 (Kd, nM) | N-parp (Kd, nM) | AM-PARP-1 (Kd, nM) |
|---|---|---|---|
| 30Nick | 23.4 ± 4.8 | 27.8 ± 5.6 | 33.2 ± 23.5 |
| Nuc207 | 1.0 ± 0.2 | 48.8 ± 21.2 | 13.2 ± 2 |
| LE-Tri | 12.7 ± 6.4 | 20.3 ± 2.6 | 10 ± 2 |
| NLE-Tri | 4.8 ± 2.1 | 22.8 ± 4.8 | 101 ± 23 |
| H2A-H2B | >500 | >500 | 2.3 ± 0.8 |
| H3-H4 | >500 | >500 | >500 |
Data adapted from PMC4156740 showing dissociation constants for various PARP-1 constructs with different chromatin substrates [4].
Experimental Analysis of DNA Binding
Protocol 1: Electrophoretic Mobility Shift Assay (EMSA) for PARP-1 DNA Binding
- Principle: Measure protein-DNA complex formation based on reduced mobility in polyacrylamide gel.
- Procedure:
- Incubate 0-100 nM purified PARP-1 DBD fragment with 5 nM fluorescently-labeled DNA containing single-strand breaks.
- Use DNA ligands with nicks, gaps, or double-strand breaks.
- Resolve complexes on 6% non-denaturing polyacrylamide gel.
- Visualize using fluorescence imaging or autoradiography.
- Key Finding: F1+F2 fragment forms 1:1 monomeric complex with DNA single-strand breaks [2].
Diagram Title: PARP-1 DNA Damage Recognition Mechanism
Auto-Modification Domain (AMD)
Structural Features and Function
The central auto-modification domain (approximately 22 kDa) contains a BRCT (BRCA1 C-terminal) fold, a motif commonly found in DNA repair proteins that facilitates protein-protein interactions [1]. This domain also contains a third zinc finger motif (F3) that is structurally unrelated to F1 and F2 and is not directly involved in DNA binding but is essential for catalytic activation [1]. The AMD serves as the primary acceptor site for PAR polymers during auto-modification, a process that dramatically alters PARP-1's functions and interactions.
Auto-Modification and Functional Consequences
Auto-modification represents a critical regulatory mechanism that switches PARP-1 from a chromatin architectural protein to a transcription factor and DNA damage responder [4]. Automodified PARP-1 (AM-PARP-1) demonstrates reduced affinity for intact chromatin but maintains binding capability for nucleosomes with exposed DNA ends [4]. This modification confers unexpected functionalities, including the ability to sequester histones and efficiently assemble nucleosomes, suggesting a model where DNA damage or transcription events trigger transient histone chaperone activity [4].
Experimental Analysis of Auto-Modification
Protocol 2: In Vitro Auto-Modification Assay
- Principle: Monitor incorporation of radiolabeled ADP-ribose units from NAD+ onto PARP-1.
- Procedure:
- Incubate 100 nM purified full-length PARP-1 with activated DNA (1 µg/mL) in reaction buffer.
- Add 50 µM NAD+ containing 1 µCi [32P]-NAD+.
- Terminate reactions at 0, 1, 2, 5, and 10 minutes.
- Resolve proteins by SDS-PAGE and visualize automodified PARP-1 by autoradiography.
- Quantify PAR incorporation using phosphorimaging.
- Modification: Include 3-aminobenzamide (PARP inhibitor) in control reactions.
Catalytic Domain (CAT)
Structural Organization and Mechanism
The C-terminal catalytic domain (54 kDa) contains the ADP-ribosyl transferase (ART) fold that catalyzes the polymerization of ADP-ribose units from NAD+ onto acceptor proteins [3]. A key structural feature is the helical subdomain (HD), which acts as an autoinhibitory domain in the folded state [3]. The catalytic mechanism involves cleaving NAD+ and transferring the resulting ADP-ribose moiety onto target proteins, with subsequent addition of ADP-ribose units to form linear or branched polymers of ADP-ribose.
Allosteric Regulation and Activation
PARP-1 exhibits a low level of basal catalytic activity that increases up to 1000-fold upon DNA strand break binding [3]. This activation involves an allosteric network connecting PARP-1 multi-domain detection of DNA damage to catalytic domain structural changes that relieve autoinhibition. The autoinhibitory HD selectively restricts access to the NAD+-binding site through a steric block mechanism, where small compounds like benzamide can access the active site but larger molecules like benzamide adenine dinucleotide (BAD) cannot bind to the unactivated enzyme [3].
Experimental Analysis of Catalytic Activity
Protocol 3: Catalytic Activity Measurement Using NAD+ Analogs
- Principle: Utilize non-hydrolyzable NAD+ analogs to probe active site accessibility.
- Procedure:
- Express and purify PARP-1 CAT ΔHD (constitutively active) and CAT WT.
- Perform differential scanning fluorimetry with NAD+ analogs.
- Measure apparent melting temperature (Tₘ) changes.
- Use benzamide adenine dinucleotide (BAD) to assess binding.
- Key Finding: HD presents selective steric block to PARP-1 active site [3].
Table 3: PARP-1 Catalytic Domain Interactions with NAD+ Analogs
| Compound | Effect on CAT WT Tₘ | Effect on CAT ΔHD Tₘ | Inhibition of PARP-1 Activity |
|---|---|---|---|
| BAD | No change | +10 °C increase | Strong inhibition |
| Carba-NAD+ | No change | <+1 °C increase | Minimal inhibition |
| Benzamide | +6 °C increase | +8 °C increase | Strong inhibition |
| ADP-ribose | No change | No change | No inhibition |
Data adapted from Nature Communications (2018) showing PARP-1 interactions with NAD+ analogs [3].
Diagram Title: PARP-1 Catalytic Activation Pathway
PARP-1 Cleavage in Programmed Cell Death
Proteolytic Cleavage by Suicidal Proteases
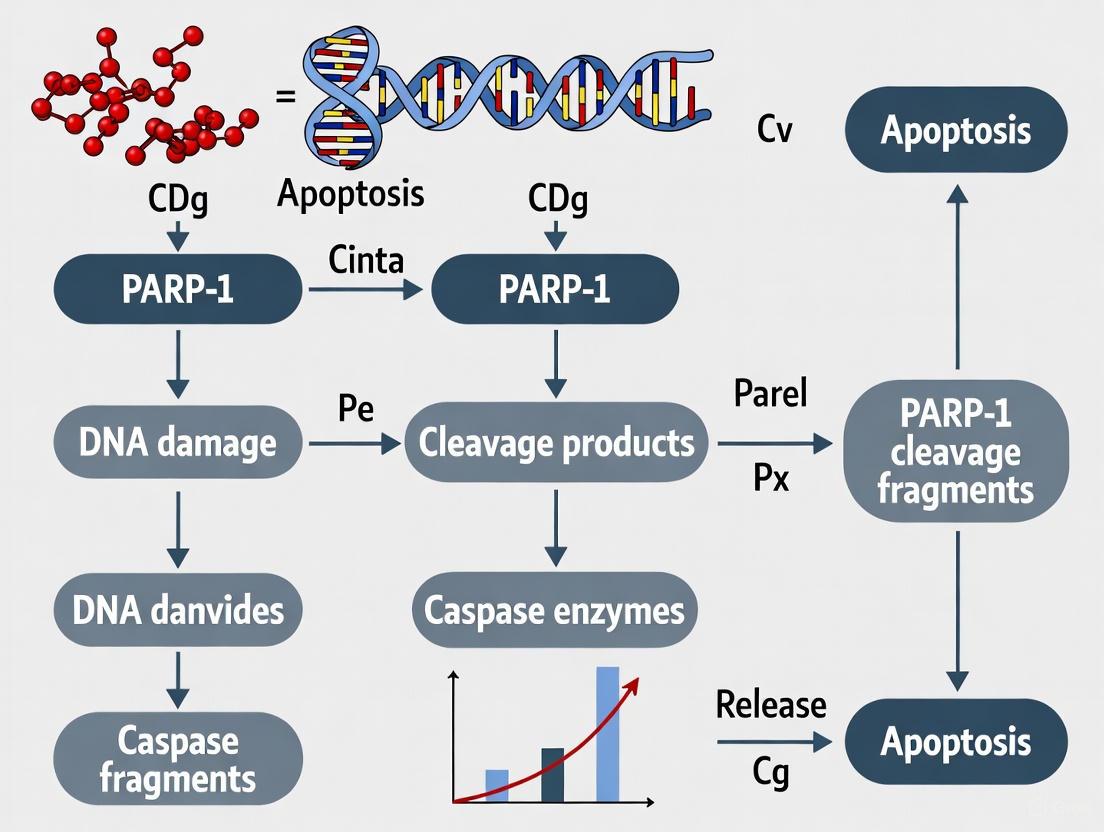
PARP-1 serves as a preferred substrate for several suicidal proteases during programmed cell death, generating specific proteolytic fragments that serve as signature biomarkers for different cell death pathways [1]. Caspases, particularly caspase-3 and caspase-7, cleave PARP-1 after aspartate residues within the sequence DEVD, producing characteristic 89-kD and 24-kD fragments [1]. The 24-kD fragment corresponds to the DNA-binding domain, which retains the ability to bind tightly to DNA strand breaks but lacks catalytic activity, thereby acting as a trans-dominant inhibitor of DNA repair [1].
Functional Consequences of Cleavage
PARP-1 cleavage represents a strategic cellular process to redirect energy resources during cell death. The 24-kD DBD fragment irreversibly binds to nicked DNA, inhibiting DNA repair enzymes and conserving cellular ATP pools that would otherwise be depleted by excessive PAR synthesis [1]. The 89-kD fragment containing the auto-modification and catalytic domains has greatly reduced DNA binding capacity and is liberated from the nucleus into the cytosol [1]. This cleavage event is considered a hallmark of apoptosis and has been implicated in numerous pathological conditions, including cerebral ischemia, neurodegenerative diseases, and brain tumors.
Experimental Detection of PARP-1 Cleavage
Protocol 4: Western Blot Analysis of PARP-1 Cleavage
- Principle: Detect signature PARP-1 fragments using domain-specific antibodies.
- Procedure:
- Prepare cell lysates from treated and control samples.
- Separate proteins using 7.5% SDS-PAGE.
- Transfer to PVDF membrane and block with 5% non-fat milk.
- Incubate with anti-PARP-1 antibodies targeting:
- N-terminal epitopes (detects 24-kD fragment)
- C-terminal epitopes (detects 89-kD fragment)
- Visualize using chemiluminescence detection.
- Interpretation: Ratio of cleaved to full-length PARP-1 indicates apoptosis extent.
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Research Reagents for PARP-1 Studies
| Reagent | Function/Application | Key Features |
|---|---|---|
| Benzamide adenine dinucleotide (BAD) | Non-hydrolyzable NAD+ analog for active site studies | Reveals PARP-1 substrate-blocking mechanism [3] |
| Caspase-3 | Protease for generating signature PARP-1 cleavage fragments | Produces 24-kD DBD and 89-kD CAT fragments [1] |
| Tri-nucleosome substrates | Chromatin models for binding studies | NLE-Tri and LE-Tri for quantitative affinity measurements [4] |
| HI-FI FRET system | Quantitative binding affinity measurements | Determines Kd values for PARP-1-chromatin interactions [4] |
| Anti-PARP-1 domain antibodies | Detection of specific fragments in cell death | Domain-specific epitopes for cleavage analysis [1] |
| 3-aminobenzamide | PARP catalytic inhibitor | Control for auto-modification experiments [3] |
The modular domain architecture of PARP-1 enables this multifunctional enzyme to integrate DNA damage detection with appropriate cellular responses, including DNA repair, chromatin remodeling, and participation in programmed cell death pathways. The DNA-binding domain provides specific recognition of DNA lesions, the auto-modification domain serves as a regulatory hub and protein interaction platform, and the catalytic domain executes PAR synthesis with sophisticated allosteric control. In the context of programmed cell death research, PARP-1 cleavage by suicidal proteases generates signature fragments that not only serve as biomarkers but also actively participate in cell death execution by inhibiting DNA repair and conserving cellular energy. The quantitative methodologies and research tools outlined in this review provide scientists with robust approaches for investigating PARP-1 structure and function in both physiological and pathological contexts, with particular relevance for cancer therapy development and understanding cell fate decisions.
The poly(ADP-ribose) polymerase (PARP) family represents a crucial group of enzymes that orchestrate cellular responses to stress, maintain genomic integrity, and regulate programmed cell death. Comprising 17 members in humans, these NAD⁺-dependent enzymes catalyze the transfer of ADP-ribose units to target proteins through either poly(ADP-ribosyl)ation (PARylation) or mono(ADP-ribosyl)ation (MARylation) processes [5]. While PARP1 has been extensively studied for its role in DNA repair and as a substrate for caspases during apoptosis, understanding the functional specialization across the entire PARP family provides critical insights for targeted therapeutic development. The distinct structural domains and specialized functions of individual PARP members create a sophisticated regulatory network that extends far beyond DNA damage repair, encompassing telomere maintenance, metabolic regulation, immune responses, and multiple forms of programmed cell death [5] [6]. This review systematically examines the PARP family's classification, functional specialization, and emerging roles in cellular fate decisions, with particular emphasis on PARP-1 cleavage as a signature event in programmed cell death pathways.
Classification and Structural Characteristics of the PARP Family
The PARP family is classified into five major subcategories based on structural domains and functional specializations. All PARPs share a conserved C-terminal catalytic domain (CAT) that mediates NAD⁺-dependent ADP-ribose polymerization, while their N-terminal domains (NTDs) exhibit considerable variation, determining substrate specificity and subcellular localization [5].
Table 1: Classification of the Human PARP Family
| Subcategory | Family Members | Key Structural Domains | Primary Functions |
|---|---|---|---|
| DNA-Dependent PARPs | PARP1, PARP2, PARP3 | Zinc finger DNA-binding domains, WGR domain, BRCT domain | DNA damage detection and repair, base excision repair |
| Tankyrases | PARP5A (TNKS1), PARP5B (TNKS2) | Ankyrin repeat domains (ARDs), SAM domain, HPS domain | Telomere maintenance, Wnt signaling regulation |
| CCCH-Type PARPs | PARP7, PARP12, PARP13 | CCCH-type zinc finger motifs, WWE domain | RNA binding, antiviral defense, post-transcriptional regulation |
| MacroPARPs | PARP9, PARP14, PARP15 | Macrodomains (ADP-ribose binding), WWE domains | Immune response regulation, interferon signaling |
| Atypical PARPs | PARP4, PARP6, PARP8, PARP10, PARP11, PARP16 | Varied domain structures including: VWA, CAM, RRM, TM | Specialized functions in vesicle trafficking, cell adhesion, endoplasmic reticulum stress response |
Structural Basis for Functional Specialization
The functional specialization among PARP family members is dictated by their distinct structural architectures. PARP1, the canonical family member, contains three zinc finger domains at its N-terminus that facilitate DNA damage recognition, a central BRCT (BRCA1 C Terminus) auto-modification domain, and a C-terminal catalytic domain that executes ADP-ribose polymerization [5] [7]. The Zn1 and Zn2 domains recognize DNA strand breaks, while the Zn3 domain orchestrates conformational rearrangement during activation [5]. Tankyrases (PARP5A/5B) feature extended ankyrin repeat domains that mediate protein-protein interactions, enabling their roles in telomere maintenance and Wnt signaling pathway regulation [5]. CCCH-type PARPs contain zinc finger motifs specialized for RNA binding, positioning them as key regulators of post-transcriptional gene expression and antiviral defense [5]. The structural diversity across the PARP family enables their participation in a remarkably broad spectrum of cellular processes while maintaining specificity in their respective functions.
Functional Specialization Among PARP Family Members
DNA Damage Response and Repair Specialists
PARP1, PARP2, and PARP3 constitute the DNA-dependent PARP subfamily that specializes in genomic maintenance. PARP1 serves as the primary sensor of single-strand DNA breaks, initiating base excision repair (BER) through its zinc finger domains that rapidly detect DNA lesions [5] [7]. Upon binding to DNA damage sites, PARP1 undergoes conformational activation, synthesizing extensive PAR chains that serve as recruitment platforms for DNA repair factors including XRCC1, DNA ligase III, and other BER components [7] [1]. PARP2 shares approximately 69% structural homology with PARP1 and plays complementary roles in DNA repair, particularly in BER and the resolution of single-strand and double-strand breaks [7]. While PARP1 accounts for approximately 85% of total cellular PARylation activity, PARP2 contributes an additional 10-15% of this activity, with both enzymes serving as primary targets for PARP inhibitor therapeutics in oncology [7].
Telomere and Signaling Regulation
Tankyrases (PARP5A and PARP5B) specialize in telomere length maintenance through their interactions with telomeric repeat-binding factor 1 (TRF1) [5]. Their ankyrin repeat domains enable assembly of multi-protein complexes that regulate telomere accessibility. Beyond telomere biology, tankyrases control Wnt/β-catenin signaling pathway activity by promoting axin degradation, thereby influencing cell proliferation and differentiation programs [5]. The distinctive HPS domain in Tankyrase-1 and its absence in Tankyrase-2 illustrate how structural variations within subfamilies enable functional diversification.
RNA Regulation and Antiviral Defense
CCCH-type PARPs (PARP7, PARP12, PARP13) represent a specialized subgroup that coordinates post-transcriptional regulation and antiviral immunity. These PARPs contain CCCH-type zinc finger motifs with high affinity for RNA molecules, enabling their roles in mRNA stability and translation regulation [5]. PARP13 (ZAP/ZC3HAV1) has emerged as a critical antiviral factor that binds viral mRNAs and targets them for degradation, while PARP7 and PARP12 similarly restrict viral replication through RNA-mediated mechanisms [5]. Interestingly, PARP13 exists as two isoforms (PARP13.1 and PARP13.2), with PARP13.2 completely lacking the catalytic domain while retaining antiviral activity, demonstrating that non-enzymatic functions can be equally crucial for biological specialization [5].
Immune Response Modulation
MacroPARPs (PARP9, PARP14, PARP15) specialize in immune regulation through their macrodomains that exhibit high affinity for ADP-ribose moieties. PARP14 functions as a key coordinator of inflammatory signaling, dampening NF-κB activation and promoting anti-inflammatory macrophage polarization [5]. PARP9 forms a complex with DTX3L that regulates DNA damage responses in immune contexts, while PARP15 modulates interferon signaling pathways [5]. The specialization of macroPARPs in immunoregulation highlights how the PARP family has evolved to control diverse physiological processes beyond DNA metabolism.
PARP-1 Cleavage in Programmed Cell Death
PARP-1 as a Protease Substrate in Cell Death Pathways
PARP-1 serves as a preferred substrate for multiple proteases activated during programmed cell death, with specific cleavage fragments serving as signature biomarkers for distinct cell death pathways [1]. The protease-specific cleavage patterns of PARP-1 provide a molecular record of the cell death mechanism in operation, with implications for both pathological processes and therapeutic interventions.
Table 2: PARP-1 Cleavage Fragments in Programmed Cell Death
| Protease | Cleavage Sites | PARP-1 Fragments | Cell Death Pathway | Functional Consequences |
|---|---|---|---|---|
| Caspase-3/7 | Asp214/Gly215 (within the nuclear localization signal) | 24-kDa DNA-binding fragment + 89-kDa catalytic fragment | Apoptosis | 24-kDa fragment irreversibly binds DNA breaks, inhibiting repair; 89-kDa fragment translocates to cytoplasm promoting apoptosis |
| Calpain | Multiple sites within the N-terminal DNA-binding domain | 55-kDa and 62-kDa fragments | Necrosis, excitotoxicity | Incomplete cleavage preserves catalytic activity while reducing DNA binding capacity |
| Granzyme A | Lys202, Arg203, Arg207 | 50-kDa and 64-kDa fragments | Lymphocyte-mediated cytotoxicity | Unique cleavage pattern distinct from caspases |
| Granzyme B | Similar to caspase-3 | 24-kDa and 89-kDa fragments | Lymphocyte-mediated cytotoxicity | Mimics caspase-3 cleavage pattern |
| Cathepsins | Varied sites | 35-kDa, 40-kDa, 50-kDa fragments | Lysosome-mediated cell death | Cleavage pattern dependent on cathepsin type and cellular context |
| MMP-2/9 | Not fully characterized | 55-kDa fragment | Extracellular matrix remodeling, inflammation | Limited information on functional consequences |
Caspase-Mediated PARP-1 Cleavage in Apoptosis
During apoptosis, executioner caspases-3 and -7 cleave PARP-1 at Asp214-Gly215 within the nuclear localization signal, generating characteristic 24-kDa DNA-binding and 89-kDa catalytic fragments [1]. The 24-kDa fragment contains the two zinc finger domains and irreversibly binds to DNA strand breaks, acting as a trans-dominant inhibitor of DNA repair by blocking access of intact PARP-1 and other repair factors to damage sites [1]. Simultaneously, the 89-kDa fragment translocates from the nucleus to the cytoplasm where it acquires pro-apoptotic functions, directly promoting caspase-mediated DNA fragmentation and apoptotic execution [1]. This cleavage event serves as a biochemical hallmark of apoptosis and represents a commitment point in the cell death pathway, ensuring irreversible progression of the dismantling process.
Cross-Talk Between PARP-1 and Ferroptosis
Emerging evidence reveals complex cross-talk between PARP-1 and ferroptosis, an iron-dependent form of programmed cell death characterized by lipid peroxidation. The ferroptosis activator RSL3 triggers PARP-1-dependent apoptotic signaling through dual mechanisms: caspase-dependent PARP-1 cleavage and epitranscriptomic regulation of PARP-1 expression via m6A RNA modification [8]. RSL3 inhibits METTL3-mediated m6A modification of PARP-1 mRNA, reducing its stability and translational efficiency, thereby depleting full-length PARP-1 and sensitizing cells to DNA damage-induced apoptosis [8]. This pathway operates in parallel to RSL3-induced caspase-3 activation and PARP-1 cleavage, demonstrating how PARP-1 serves as an integration node for ferroptosis-apoptosis cross-talk.
Diagram 1: PARP-1 Cleavage Pathways in Programmed Cell Death. This diagram illustrates the dual pathways through which PARP-1 regulates and executes programmed cell death, integrating both caspase-dependent cleavage and epitranscriptomic regulation mechanisms.
Experimental Approaches for PARP Research
TurboID Proximity Labeling for PARP Interactome Mapping
Recent advances in proteomic technologies have enabled comprehensive mapping of PARP family interactomes under standardized conditions. The TurboID proximity labeling technique has emerged as a powerful method for capturing both stable and transient interactions of PARP family members that evade detection by conventional co-immunoprecipitation approaches [9].
Protocol: TurboID Proximity Labeling for PARP Interactors
- Plasmid Construction: Clone PARP family genes into pDONR221 vectors and transfer to destination vectors (pKO187) containing N-terminal V5-TurboID or EGFP tags using Gateway LR Clonase enzyme [9].
- Cell Culture and Transfection: Culture HEK293T cells in DMEM supplemented with 10% FBS and penicillin-streptomycin. Transfect cells at 90% confluency in 150mm dishes with 15μg V5-TurboID-tagged PARP plasmid using polyethylenimine (75μl) [9].
- Expression Optimization: Express high-abundance PARPs (PARP5A, PARP6, PARP8, PARP10, PARP13, PARP15, PARP16) for 24 hours; low-abundance PARPs (PARP1, PARP2, PARP3, PARP4, PARP5B, PARP7, PARP9, PARP11, PARP12, PARP14) for 48 hours [9].
- Biotin Labeling: Incubate cells with 50μM biotin in DMEM for 1 hour at 37°C to enable proximity-dependent protein labeling [9].
- Protein Extraction and Denaturation: Lyse cells in SDS-lysis buffer (50mM Tris-HCl pH 8, 1% SDS, 40mM DTT, 5% glycerol, protease inhibitors) at 95°C for 15 minutes [9].
- Affinity Purification: Dilute lysate 10-fold with NP40-RIPA buffer and incubate with streptavidin magnetic beads overnight at room temperature [9].
- On-bead Digestion: Wash beads extensively, then digest with trypsin (1μg/μL) in 50mM ammonium bicarbonate with shaking at 37°C for 12-18 hours [9].
- LC-MS/MS Analysis: Desalt peptides using C18 microcolumns, resuspend in mobile phase A, and analyze by data-dependent acquisition LC-MS/MS [9].
This approach has identified 6,314 high-confidence PARP-interacting proteins, revealing both shared and unique interaction networks across the PARP family and providing unprecedented insights into the functional cooperativity and specialization among PARP members [9].
Analysis of PARP-1 Cleavage Fragments
Detecting and quantifying PARP-1 cleavage fragments provides critical information about activated cell death pathways in physiological and pathological contexts.
Protocol: PARP-1 Cleavage Fragment Analysis
- Cell Lysis and Protein Extraction: Lyse cells in RIPA buffer (50mM Tris-HCl pH 8.0, 150mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) supplemented with protease inhibitors [1] [8].
- Western Blotting: Separate proteins by SDS-PAGE (8-12% gradient gels) and transfer to PVDF membranes [8].
- Antibody Detection: Probe membranes with PARP-1 antibodies targeting different domains:
- Full-length PARP-1: ~116 kDa
- Caspase-cleaved PARP-1: 89-kDa fragment (catalytic domain) and 24-kDa fragment (DNA-binding domain)
- Calpain-cleaved PARP-1: 55-kDa and 62-kDa fragments [1]
- Protease Inhibition Studies: Pre-treat cells with specific protease inhibitors to identify cleavage mechanisms:
- Caspase inhibition: Z-VAD-FMK (20-50μM)
- Calpain inhibition: Calpeptin (10-50μM)
- Cathepsin inhibition: E64d (10-30μM) [1]
- Functional Assays: Assess DNA repair capacity after PARP-1 cleavage using comet assays, γH2AX immunofluorescence, or host cell reactivation assays [1] [8].
Table 3: Research Reagent Solutions for PARP Studies
| Reagent/Category | Specific Examples | Primary Applications | Key Functions |
|---|---|---|---|
| PARP Inhibitors | Olaparib, Niraparib, Talazoparib, Rucaparib, Veliparib | Oncology research, synthetic lethality studies | Inhibit PARP catalytic activity; induce PARP trapping on DNA |
| Activity Assays | PARylation immunoassays, NAD⁺ consumption assays | PARP functional characterization | Quantify PARP enzymatic activity and inhibition |
| Cell Death Inducers | RSL3, Erastin, Staurosporine, Etoposide | Cell death pathway studies | Activate specific programmed cell death pathways |
| Protease Inhibitors | Z-VAD-FMK (caspase), Calpeptin (calpain), E64d (cathepsin) | Protease activity characterization | Identify specific proteases responsible for PARP cleavage |
| Proximity Labeling Tools | TurboID, APEX2 | Interactome mapping | Capture transient and stable protein interactions |
| PARP Cleavage Antibodies | Anti-PARP1 (cleaved Asp214), Anti-PARP1 (C-terminal) | Cell death mechanism analysis | Detect and quantify specific PARP cleavage fragments |
The PARP protein family exemplifies functional specialization through structural diversification, with individual members evolving distinct domains that target them to specific cellular processes. While PARP1 remains the most characterized family member, serving as both a DNA damage sensor and a key substrate in programmed cell death pathways, the broader PARP family coordinates an extensive regulatory network encompassing DNA repair, telomere maintenance, RNA biology, immune signaling, and metabolic regulation. The protease-specific cleavage of PARP-1 generates signature fragments that not only serve as biomarkers for distinct cell death pathways but also actively participate in apoptotic execution through both dominant-negative inhibition of DNA repair and gain-of-function pro-apoptotic activities. Emerging research continues to reveal unexpected connections between PARP family members and novel cell death modalities such as ferroptosis, highlighting the complexity of their regulatory networks. As technological advances like TurboID proximity labeling provide increasingly comprehensive maps of PARP interactions, and as structural biology reveals the molecular basis for their functional specialization, new opportunities emerge for developing isoform-selective PARP-targeted therapies with enhanced efficacy and reduced toxicity profiles across diverse human diseases.
Poly (ADP-ribose) polymerase-1 (PARP-1) cleavage into specific 24-kD and 89-kD fragments represents a critical biochemical hallmark of apoptosis and serves as a recognized biomarker for caspase activation in programmed cell death research. This proteolytic event, primarily executed by caspase-3 and caspase-7, effectively inactivates PARP-1's DNA repair capacity while generating fragments with distinct cellular functions. Beyond its traditional role as an apoptosis marker, emerging evidence indicates that the 89-kD fragment may actively participate in cell death signaling by facilitating poly(ADP-ribose) (PAR) translocation to the cytoplasm. This technical guide comprehensively examines the molecular mechanisms, experimental methodologies, and functional consequences of PARP-1 cleavage, providing researchers and drug development professionals with essential frameworks for investigating this crucial event in cell death pathways.
PARP-1 is a nuclear enzyme with well-established roles in DNA repair, genome stability, and transcriptional regulation. As a primary sensor of DNA damage, PARP-1 becomes activated upon binding to DNA strand breaks and catalyzes the synthesis of poly(ADP-ribose) (PAR) chains using NAD+ as a substrate [10] [11]. During apoptosis, PARP-1 undergoes specific proteolytic cleavage that serves as a biochemical signature of caspase activation, generating characteristic 24-kD and 89-kD fragments [11]. This cleavage event represents a molecular switch that redirects cellular fate from DNA repair toward programmed demolition, making it a critical mechanism in cell death research and a potential therapeutic target in cancer and other pathologies [10].
The cleavage of PARP-1 not only inactivates its DNA repair function but may also generate bioactive fragments with distinct roles in cell death pathways. Recent research has revealed that the 89-kD fragment can serve as a cytoplasmic PAR carrier, potentially bridging caspase-dependent apoptosis with PAR-thanatos, a caspase-independent programmed cell death pathway [12] [13]. This whitepaper provides an in-depth technical examination of PARP-1 cleavage mechanisms, detection methodologies, and functional implications within the broader context of programmed cell death research.
Molecular Architecture of PARP-1 and Cleavage Sites
Structural Domains of PARP-1
PARP-1 is a modular protein of 1,014 amino acids organized into three primary functional domains that dictate its cellular functions and cleavage patterns [11] [14]:
DNA-Binding Domain (DBD): Located at the N-terminus, this domain contains three zinc finger motifs (Zn1, Zn2, Zn3) that recognize and bind to DNA strand breaks. Zn1 and Zn2 specifically recognize DNA damage gaps by binding to the 5' and 3' ends respectively, while Zn3 links the structural domains to activate the target protein [14]. This domain also contains a nuclear localization signal (NLS) and the aspartate-glutamate-valine-aspartic acid (DEVD) motif that serves as the primary caspase cleavage site [14].
Automodification Domain (AMD): This central domain contains a BRCT (BRCA1 C-terminus) fold that facilitates protein-protein interactions and serves as the primary target for PARP-1 automodification. The AMD is crucial for recruiting DNA repair machinery to damage sites and regulates PARP-1's release from DNA following poly(ADP-ribosyl)ation [11].
Catalytic Domain (CAT): Located at the C-terminus, this domain contains the NAD+ binding site and catalyzes PAR synthesis. It consists of the α-helical subdomain (HD) and ADP-ribosyl transferase (ART) subdomain, which are responsible for transferring ADP-ribose units to target proteins [14]. A critical WGR segment within this domain interacts with DNA and other domains to form an inter-regional network that activates catalysis upon DNA binding [14].
Caspase Cleavage Site and Fragment Generation
The primary caspase cleavage site in human PARP-1 is located between amino acids Asp214 and Gly215 within the DEVD motif (residues 211-214) of the DBD [10] [11]. Cleavage at this site produces two signature fragments:
- 24-kD Fragment: Comprises the N-terminal DBD containing two zinc finger motifs (Zn1 and Zn2)
- 89-kD Fragment: Contains the third zinc finger (Zn3), the AMD, and the CAT [11]
This cleavage event separates the DNA-binding capability from the catalytic activity, effectively eliminating PARP-1's functionality in DNA repair [10]. The 24-kD fragment retains the ability to bind DNA strand breaks but lacks catalytic function, while the 89-kD fragment contains the catalytic domain but cannot be recruited to DNA damage sites [11].
Table 1: PARP-1 Fragments Generated by Caspase-Mediated Cleavage
| Fragment | Molecular Weight | Domains Contained | Cellular Localization | Primary Functions |
|---|---|---|---|---|
| 24-kD Fragment | 24 kDa | DNA-Binding Domain (Zn1, Zn2) | Nuclear | Binds irreversibly to DNA strand breaks; acts as trans-dominant inhibitor of PARP-1; blocks DNA repair |
| 89-kD Fragment | 89 kDa | Zn3, Automodification Domain, Catalytic Domain | Cytoplasmic (after cleavage) | Serves as PAR carrier to cytoplasm; may facilitate AIF release; contains residual catalytic activity |
| Full-length PARP-1 | 116 kDa | All domains (DBD, AMD, CAT) | Nuclear | DNA damage sensing and repair; transcriptional regulation; energy metabolism |
Experimental Methodologies for Detecting PARP-1 Cleavage
Induction of Apoptosis and PARP-1 Cleavage
Multiple experimental approaches can induce apoptosis and subsequent PARP-1 cleavage for research purposes:
Chemical Inducers:
- Staurosporine: A broad-spectrum protein kinase inhibitor that induces intrinsic apoptosis (typically used at 0.5-2 μM for 4-24 hours) [12] [15]
- Paclitaxel: Microtubule-stabilizing agent that activates the intrinsic apoptotic pathway (commonly used at 0.1 μM for 48 hours) [16] [15]
- Etoposide: Topoisomerase II inhibitor that causes DNA damage and apoptosis [15]
- Actinomycin D: Transcription inhibitor that induces apoptosis through DNA damage [12]
Treatment Protocols: For in vitro studies using cell lines (e.g., HeLa, HT29, MEFs), researchers typically treat cells at 60-80% confluency with apoptosis inducers for varying durations based on the agent and cell type. For example, paclitaxel treatment is commonly administered for 48 hours at 0.1 μM concentration, while staurosporine treatments may range from 4-24 hours at 0.5-2 μM [16] [15]. Serum withdrawal from dependent cell lines (e.g., FL5.12 cells) for 12-24 hours provides an alternative method for inducing intrinsic apoptosis [17].
Detection and Analysis Methods
Western Blotting: The most common method for detecting PARP-1 cleavage utilizes specific antibodies targeting different PARP-1 epitopes:
- Antibody Selection: Use antibodies recognizing the N-terminal region (detects full-length and 24-kD fragment) or C-terminal region (detects full-length and 89-kD fragment)
- Sample Preparation: Prepare whole cell extracts using RIPA buffer with protease inhibitors to prevent post-lysis degradation
- Electrophoresis: Separate proteins using 8-12% SDS-PAGE gels to resolve the size difference between full-length PARP-1 (116 kDa) and cleavage fragments (89 kDa and 24 kDa)
- Validation: Include positive controls (apoptotic cell extracts) and caspase inhibitor controls (e.g., zVAD-fmk) to confirm specificity
Immunohistochemistry (IHC): For tissue samples or fixed cells, IHC using antibodies specific to cleaved PARP-1 (c-PARP) enables spatial detection of apoptosis:
- Fixation: Use 4% formaldehyde (pH 7.4) for 16 hours for optimal antigen preservation [16]
- Antibody Validation: Confirm specificity using caspase-deficient cells or caspase inhibitor pretreatments
- Limitations: c-PARP IHC may be less efficient in ethanol-fixed tissues and requires validation with additional apoptosis markers [16]
Caspase Activity Assays: Parallel measurement of caspase-3 and caspase-7 activities strengthens the interpretation of PARP-1 cleavage data:
- Fluorogenic Substrates: Use DEVD-based substrates (e.g., Ac-DEVD-AFC) to measure caspase-3/7 activity
- Inhibitor Controls: Include pan-caspase inhibitors (e.g., zVAD-fmk, IDN-6556 at 20-50 μM) or specific caspase inhibitors to confirm caspase-dependent cleavage [15]
Functional Consequences of PARP-1 Cleavage
Inactivation of DNA Repair and Conservation of Cellular Energy
The primary consequence of PARP-1 cleavage is the termination of its DNA repair functions through multiple mechanisms:
Separation of Functional Domains: Cleavage between the DBD and catalytic domains prevents PARP-1 from simultaneously binding DNA damage sites and performing PAR synthesis [10] [11]
Trans-dominant Inhibition: The 24-kD fragment remains tightly bound to DNA strand breaks, physically blocking access for other DNA repair proteins and creating a dominant-negative effect that further inhibits DNA repair [11]
ATP Conservation: By preventing PARP-1 overactivation, caspase-mediated cleavage conserves cellular NAD+ and ATP pools that would otherwise be depleted by excessive PAR synthesis, thereby maintaining energy-dependent apoptotic execution [10]
This cleavage event represents a strategic cellular decision to abandon DNA repair in favor of programmed cell death when damage is irreparable. Studies in L929 cells have demonstrated that preventing PARP-1 cleavage (e.g., through caspase inhibition) can shift the mode of cell death from apoptosis to necrosis due to ATP depletion [10].
Emerging Roles of PARP-1 Fragments in Cell Death Signaling
Beyond the simple inactivation of DNA repair, PARP-1 fragments may actively participate in cell death signaling:
Cytoplasmic Translocation of 89-kD Fragment: Recent research indicates that the 89-kD fragment, particularly when poly(ADP-ribosyl)ated, can translocate to the cytoplasm where it may serve as a PAR carrier [12] [13]
Cross-talk with Parthanatos: The 89-kD fragment facilitates the translocation of PAR polymers to the cytoplasm, where they can bind to apoptosis-inducing factor (AIF), potentially bridging caspase-mediated apoptosis with PAR-thanatos [12] [13]
Modulation of Mitochondrial Function: PARP-1 cleavage fragments may influence mitochondrial membrane permeability and the release of pro-apoptotic factors, though these mechanisms require further elucidation [17]
Table 2: Research Reagent Solutions for PARP-1 Cleavage Studies
| Reagent Category | Specific Examples | Concentration/Usage | Primary Function | Experimental Considerations |
|---|---|---|---|---|
| Caspase Inhibitors | zVAD-fmk (pan-caspase) | 20-50 μM | Blocks PARP-1 cleavage; confirms caspase dependence | Potential off-target effects; use multiple inhibitors for validation |
| PARP Inhibitors | 3-aminobenzamide (3AB) | 1-5 mM | Suppresses PARP activity; prevents energy depletion | May shift cell death from necrosis to apoptosis |
| Apoptosis Inducers | Staurosporine, Paclitaxel, Etoposide | Varies by agent | Activates caspase cascade; induces PARP-1 cleavage | Optimal concentration varies by cell type; time-course recommended |
| Detection Antibodies | Anti-PARP-1, c-PARP specific | Manufacturer's dilution | Identify full-length and cleaved fragments | Validate specificity with knockout cells or siRNA |
| Activity Assays | Fluorogenic caspase substrates (DEVD-AFC) | 50-200 μM | Measure caspase-3/7 activation | Correlate activity with cleavage extent |
| Cell Lines | PARP-1(-/-) MEFs, Caspase-3/7 deficient | N/A | Provide genetic controls for specificity | Confirm absence of target protein |
PARP-1 Cleavage in Broader Cell Death Contexts
Integration with Apoptotic Signaling Networks
PARP-1 cleavage does not occur in isolation but is integrated within complex apoptotic signaling networks:
Caspase Hierarchy: PARP-1 is primarily cleaved by executioner caspases-3 and -7, which are themselves activated by initiator caspases (e.g., caspase-9 in intrinsic pathway) [17]. Each caspase appears to have distinct roles, with caspase-3 being particularly important for efficient apoptotic execution, while caspase-7 may contribute to cell detachment [17]
Mitochondrial Regulation: Caspase-9 can cleave Bid to generate tBid, which promotes mitochondrial remodeling and ROS production, creating feedback amplification loops that enhance apoptotic signaling [17]
Energy-Dependent Cell Fate Decisions: The interplay between PARP-1 activation and cleavage serves as a metabolic switch between apoptosis and necrosis - intact PARP-1 activity depletes ATP promoting necrosis, while PARP-1 cleavage conserves ATP supporting apoptosis [10]
Therapeutic Implications and Research Applications
Understanding PARP-1 cleavage has significant implications for therapeutic development and disease research:
Cancer Therapeutics: PARP inhibitors are used in BRCA-deficient cancers through synthetic lethality approaches. The relationship between PARP inhibition and PARP-1 cleavage may influence treatment efficacy and resistance mechanisms [18]
Neurodegenerative Diseases: Excessive PARP-1 activation contributes to neuronal death in stroke, Parkinson's, and Alzheimer's disease, making the regulation of PARP-1 cleavage a potential therapeutic target [11]
Inflammation and Ischemic Injury: PARP-1 deficient mice show protection against inflammatory and ischemic injury, highlighting the pathophysiological importance of PARP-1 regulation [10]
PARP-1 cleavage into 24-kD and 89-kD fragments represents a critical commitment point in programmed cell death, serving both as a biomarker of caspase activation and an active regulatory event that influences cellular fate decisions. The well-established role of this cleavage event in terminating DNA repair and conserving cellular energy during apoptosis continues to be refined with emerging evidence suggesting additional signaling functions for the cleavage fragments, particularly in mediating cross-talk between different cell death pathways. For researchers and drug development professionals, robust detection methodologies and appropriate experimental controls are essential for accurate interpretation of PARP-1 cleavage data. As our understanding of PARP-1's multifaceted roles in cell death continues to evolve, so too will opportunities for targeting this pathway in therapeutic applications across oncology, neurodegeneration, and inflammatory diseases.
Poly(ADP-ribose) polymerase-1 (PARP-1) cleavage is a established hallmark of programmed cell death, serving as a critical signaling event that determines cellular fate. While caspase-mediated PARP-1 cleavage is well-characterized in apoptosis, emerging research highlights significant roles for alternative proteolytic pathways in regulating PARP-1 function and contributing to diverse cell death modalities. This technical guide examines the mechanisms and consequences of PARP-1 cleavage by calpain, cathepsin, and granzyme proteases, providing researchers with advanced experimental frameworks and analytical tools for investigating these pathways in physiological and pathological contexts, including neurodegeneration, cancer, and viral infections.
PARP-1 is a nuclear enzyme with multifaceted roles in DNA repair, transcriptional regulation, and cell death decision-making. As an abundant nuclear protein with approximately 1-2 million copies per cell, PARP-1 accounts for approximately 85% of total cellular PARP activity [11]. Its domain architecture features a 46-kDa DNA-binding domain (DBD) containing zinc finger motifs at the N-terminus, a 22-kDa auto-modification domain (AMD), and a 54-kDa catalytic domain (CD) at the C-terminus [11]. Beyond its canonical DNA repair function, PARP-1 serves as a preferred substrate for multiple proteases, earning its designation as a "suicidal protease" substrate whose cleavage fragments serve as biomarkers for specific cell death pathways [11].
The cleavage of PARP-1 represents a molecular switch that can either promote or suppress cell death depending on the cellular context and the specific protease involved. While caspase-mediated cleavage generates characteristic 24-kDa and 89-kDa fragments during apoptosis, alternative proteases including calpains, cathepsins, and granzymes produce distinct PARP-1 cleavage signatures with unique functional consequences [11]. These proteolytic events occur in specific subcellular compartments and physiological contexts, offering new avenues for therapeutic intervention in diseases characterized by dysregulated cell death.
Calpain-Mediated PARP-1 Cleavage
Calpain Family Characteristics and Activation Mechanisms
Calpains constitute a family of calcium-activated neutral cysteine proteases that function at neutral pH in the cytosol [19]. The ubiquitously expressed calpain isoforms, μ-calpain (calpain I) and m-calpain (calpain II), are heterodimers composed of a large 80-kDa catalytic subunit and a small 30-kDa regulatory subunit. These isoforms are distinguished by their in vitro calcium requirements: 2-80 μM for calpain I and 0.2-0.8 mM for calpain II [19]. A third isoform, calpain-2, has been implicated in neurodegeneration, inflammation, and cancer, though its substrates remain incompletely characterized [20].
Calpain activation represents a critical regulatory node in calcium-mediated cell death pathways. Under physiological conditions, calpain exists as an inactive proenzyme in the cytosol, where resting free calcium concentrations range from 50-100 nM [19]. Activation occurs through multiple mechanisms: (1) calcium-induced autolysis of N-terminal propeptides from both subunits, leading to conformational change and subunit dissociation; (2) calcium-triggered translocation from cytosol to membrane, where phospholipids facilitate activation; and (3) phosphorylation at specific residues such as Ser-369 by protein kinase A [19]. Calpain activity is tightly regulated by its endogenous inhibitor, calpastatin, which exists in molar excess under normal conditions [19].
Table 1: Calpain Family Isoforms and Characteristics
| Isoform | Calcium Requirement | Structure | Primary Localization | Key Functions |
|---|---|---|---|---|
| Calpain I (μ-calpain) | 2-80 μM | Heterodimer (80kD + 30kD) | Cytosol, membrane | Apoptosis, signal transduction |
| Calpain II (m-calpain) | 0.2-0.8 mM | Heterodimer (80kD + 30kD) | Cytosol, membrane | Cell proliferation, differentiation |
| Calpain-2 | Millimolar range | Heterodimer with CAPNS1 | Cytosol, nucleus | Neurodegeneration, inflammation, cancer |
Calpain Substrate Specificity and PARP-1 Cleavage
Unlike caspases with their specific recognition motifs, calpains lack a strict consensus sequence, instead favoring specific structural contexts and accessible loops in their substrates [20]. This broad specificity allows calpains to target numerous cellular proteins, including cytoskeletal components, membrane proteins, transcription factors, and enzymes involved in apoptosis. Recent N-terminomics and proteomics approaches have identified over 51 putative calpain-2 substrates in THP-1 human monocyte-like cells, expanding the known repertoire of calpain targets [20].
In the context of PARP-1 cleavage, calpain activation has been documented in neuronal apoptosis following spinal cord injury and in neurodegenerative diseases including Alzheimer's, Parkinson's, and amyotrophic lateral sclerosis [19]. Calpain-mediated PARP-1 cleavage generates distinct fragments that differ from the canonical caspase-generated portions, though the exact molecular weights of these fragments require further characterization. The functional consequences of calpain-mediated PARP-1 cleavage include amplification of calcium-mediated cell death signals and contribution to the pathological processes in neurological disorders.
Experimental Protocols for Investigating Calpain-Mediated PARP-1 Cleavage
Calpain Activation and PARP-1 Cleavage Assay
Principle: This protocol induces calpain activation using calcium ionophores in monocyte-like cells and detects resultant PARP-1 cleavage fragments through Western blotting with specific antibodies.
Reagents and Solutions:
- THP-1 human monocyte-like cell line
- Phorbol 12-myristate 13-acetate (PMA) for macrophage differentiation
- Calcium ionophore (e.g., A23187 or ionomycin)
- Calpain-specific inhibitors (e.g., MDL-28170)
- Lysis buffer: 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM EDTA, plus protease inhibitors
- Antibodies: Anti-PARP-1 (full-length and cleavage-specific), anti-calpain-2, anti-calpastatin
Procedure:
- Culture THP-1 cells in RPMI-1640 medium with 10% FBS and maintain at 37°C in 5% CO₂.
- Differentiate THP-1 cells into macrophages by treating with 100 nM PMA for 48 hours.
- Activate calpain by treating differentiated cells with 2-5 μM calcium ionophore for 1-4 hours.
- For inhibition controls, pre-treat cells with 20-50 μM calpain inhibitor for 1 hour before ionophore addition.
- Harvest cells by centrifugation and lyse in ice-cold lysis buffer for 30 minutes.
- Clarify lysates by centrifugation at 14,000 × g for 15 minutes at 4°C.
- Determine protein concentration using BCA assay.
- Separate 20-30 μg of protein by SDS-PAGE (8-12% gradient gel) and transfer to PVDF membrane.
- Perform Western blotting with PARP-1 antibodies to detect full-length and cleavage fragments.
- Probe with calpain-2 and calpastatin antibodies to confirm activation status.
Technical Notes: Calpain activation is transient and calcium-dependent, requiring precise timing of treatments. Include calpastatin Western blots to monitor inhibitor cleavage, which indicates calpain activation. Use caspase inhibitors (e.g., Z-VAD-FMK) to distinguish calpain-mediated cleavage from caspase-mediated cleavage [19] [20].
N-terminomics/TAILS Analysis for Calpain Substrate Identification
Principle: Terminal Amine Isotopic Labeling of Substrates (TAILS) identifies natural protein N-termini and protease-generated cleavage products on a proteome-wide scale, enabling discovery of novel calpain substrates including potential PARP-1 cleavage fragments.
Reagents and Solutions:
- WT and CAPN2⁻¹⁻ THP-1 cells
- PMA for differentiation
- Calcium ionophore
- Hyperplex TAILS kit
- Formaldehyde (light [¹²CH₂O] and heavy [¹³CD₂O])
- Sodium cyanoborohydride
- HPG-ALD polymer
- Mass spectrometry-grade trypsin/Lys-C
Procedure:
- Differentiate WT and CAPN2⁻¹⁻ THP-1 cells with PMA as described above.
- Activate calpain with calcium ionophore in experimental groups.
- Harvest cells and lyse in denaturing buffer.
- Reduce and alkylate cysteine residues.
- Label primary amines with formaldehyde isotopes (light for control, heavy for treated samples).
- Combine equal protein amounts from light- and heavy-labeled samples.
- Trypsinize combined samples.
- Remove internal peptides by HPG-ALD polymer enrichment.
- Elute and analyze N-terminal peptides by LC-MS/MS.
- Process data using TopFIND database and bioinformatic tools to identify calpain-specific cleavage events.
Technical Notes: This approach identified 51 potential calpain-2 substrates in THP-1 cells, including known and novel targets [20]. For PARP-1-specific analysis, combine with immunoprecipitation using PARP-1 antibodies prior to TAILS analysis.
Cathepsin-Mediated PARP-1 Cleavage
Cathepsin Family Characteristics and Activation Mechanisms
Cathepsins represent a group of lysosomal proteases that include serine (cathepsins A and G), aspartic (cathepsins D and E), and cysteine (cathepsins B, C, F, H, K, L, O, S, V, W, and X) proteases. These enzymes normally function in the acidic environment of lysosomes but can contribute to cell death when released into the cytosol or extracellular space during lysosomal membrane permeabilization (LMP). In the context of viral infections, cathepsins L and B facilitate SARS-CoV-2 invasion by cleaving viral spike protein within endosomes and lysosomes [21].
The role of cathepsins in PARP-1 cleavage is less characterized than caspase or calpain pathways, but emerging evidence suggests their involvement in specific cell death contexts. Cathepsins can initiate or amplify death signals that ultimately converge on PARP-1 cleavage, either directly or through activation of other proteases. Nuclear localization of cathepsins has been reported in certain cancer cells, suggesting potential direct nuclear substrates including PARP-1 [22].
Experimental Protocols for Cathepsin-Mediated PARP-1 Cleavage
Lysosomal Membrane Permeabilization and PARP-1 Cleavage Assay
Principle: This protocol induces lysosomal membrane permeabilization using specific agents, leading to cathepsin release into the cytosol and subsequent PARP-1 cleavage.
Reagents and Solutions:
- Appropriate cell line (e.g., primary macrophages or cancer cell lines)
- Lysosomotropic agents (e.g., L-Leucyl-L-leucine methyl ester, siramesine)
- Cathepsin inhibitors (E-64d for cysteine cathepsins, pepstatin A for aspartyl cathepsins)
- Lysotracker Red for lysosomal integrity assessment
- Antibodies: Anti-PARP-1, anti-cathepsin B, anti-cathepsin L, anti-LAMP1
Procedure:
- Culture cells in appropriate medium and plate at optimal density.
- Treat cells with lysosomotropic agents (e.g., 100-500 μM L-Leucyl-L-leucine methyl ester) for 2-8 hours.
- For inhibition controls, pre-treat with cathepsin inhibitors (10 μM E-64d, 1 μM pepstatin A) for 1 hour.
- Monitor lysosomal integrity using Lysotracker Red staining according to manufacturer's protocol.
- Harvest cells and prepare cytosolic and nuclear fractions using differential centrifugation.
- Confirm cathepsin release to cytosol by Western blotting of fractions.
- Analyze PARP-1 cleavage by Western blotting of nuclear fractions.
- Correlate PARP-1 cleavage patterns with cathepsin activation.
Technical Notes: Cathepsin-mediated PARP-1 cleavage often occurs in the context of oxidative stress or toxic insults. Combine with caspase inhibitors to distinguish caspase-independent pathways. Use fractionation to confirm subcellular localization of cathepsins during cell death [21].
Granzyme-Mediated PARP-1 Cleavage
Granzyme Characteristics and Cytotoxic Functions
Granzymes are serine proteases stored in the granules of cytotoxic T lymphocytes and natural killer cells that induce apoptosis in target cells upon granule release. Among the several granzyme isoforms, granzyme A and granzyme B represent the most abundant and best-characterized members. Granzyme B cleaves substrates after aspartate residues, similar to caspase-3, and can directly process caspase-3, -7, -8, and -10 to initiate apoptosis [11]. Granzyme A utilizes a distinct cleavage mechanism with preference for basic residues (Arg or Lys) and can trigger caspase-independent cell death pathways.
Both granzyme A and B have been demonstrated to cleave PARP-1, generating signature fragments that serve as biomarkers for immune-mediated cell death [11]. Granzyme B cleavage of PARP-1 produces fragments similar to those generated by caspase-3, while granzyme A produces a distinct cleavage pattern that may have unique functional consequences.
Experimental Protocols for Granzyme-Mediated PARP-1 Cleavage
Granzyme Delivery and PARP-1 Cleavage Assay
Principle: This protocol utilizes purified granzymes with perforin to introduce these proteases into target cells, mimicking immune-mediated cytotoxicity and enabling analysis of PARP-1 cleavage.
Reagents and Solutions:
- Target cells appropriate for cytotoxicity assays (e.g., Jurkat, HeLa)
- Purified granzyme A and granzyme B
- Perform (or streptolysin O for sublytic delivery)
- Granzyme inhibitors: 3,4-dichloroisocoumarin (general serine protease inhibitor)
- Caspase inhibitors (Z-VAD-FMK) to distinguish pathways
- Antibodies: Anti-PARP-1, anti-granzyme B, cleaved caspase-3
Procedure:
- Culture target cells in appropriate medium and plate at 70-80% confluence.
- Wash cells with serum-free medium.
- Deliver granzymes using sublytic perforin (50-100 ng/mL) or streptolysin O (100-500 ng/mL) with 100-500 nM granzyme for 1-4 hours.
- For inhibition controls, pre-treat cells with 20 μM 3,4-dichloroisocoumarin or 50 μM Z-VAD-FMK for 1 hour.
- Harvest cells at specific time points and prepare whole cell lysates.
- Analyze PARP-1 cleavage by Western blotting using antibodies recognizing full-length and cleaved forms.
- Confirm granzyme activity using specific fluorogenic substrates.
- Correlate PARP-1 cleavage patterns with other cell death markers (phosphatidylserine exposure, DNA fragmentation).
Technical Notes: Granzyme B cleavage of PARP-1 generates fragments similar to caspase-3 (89-kDa and 24-kDa), while granzyme A produces different fragments. Use granzyme-specific inhibitors and caspase inhibitors to distinguish these pathways. Include controls for perforin/permeabilization agent alone [11].
Comparative Analysis of PARP-1 Cleavage Fragments
The functional consequences of PARP-1 cleavage vary significantly depending on the protease involved and the specific cleavage sites utilized. These differential cleavage patterns produce fragments with distinct biological activities that can either promote or suppress cell death pathways.
Table 2: PARP-1 Cleavage Fragments by Different Proteases
| Protease | Cleavage Fragments | Fragment Localization | Functional Consequences |
|---|---|---|---|
| Caspase-3/7 | 24-kDa (DBD) + 89-kDa (AMD+CD) | Nuclear (24-kDa) Cytoplasmic (89-kDa) | Inhibition of DNA repair, energy conservation, promotion of apoptosis |
| Calpain | Not fully characterized | Nuclear and cytoplasmic | Contribution to calcium-mediated cell death, neurodegenerative diseases |
| Granzyme B | Similar to caspase-3 | Nuclear and cytoplasmic | Immune-mediated cytotoxicity, apoptosis induction |
| Granzyme A | Distinct from caspase-3 | Nuclear and cytoplasmic | Caspase-independent cell death, unique biological functions |
| MMPs | Various fragments | Dependent on fragment | Potential signaling functions, incomplete characterization |
The 89-kDa PARP-1 fragment generated by caspase cleavage has recently been shown to serve as a cytoplasmic poly(ADP-ribose) (PAR) carrier that induces apoptosis-inducing factor (AIF)-mediated parthanatos, a caspase-independent programmed cell death pathway [13]. This fragment, when modified with PAR polymers, translocates to the cytoplasm where it facilitates AIF release from mitochondria, ultimately leading to cell death. This finding demonstrates how PARP-1 cleavage fragments can actively participate in cell death execution rather than simply representing inactivation of the enzyme.
Similarly, research has revealed that truncated PARP1 (tPARP1) generated during apoptosis recognizes the RNA polymerase III (Pol III) complex in the cytosol, mono-ADP-ribosylates Pol III, and facilitates IFN-β production during cytosolic DNA-induced apoptosis [23]. This represents a novel biological function connecting PARP-1 cleavage to innate immune responses during cell death.
Research Reagent Solutions
Table 3: Essential Research Reagents for Studying Alternative Proteolytic Pathways
| Reagent Category | Specific Examples | Research Application | Key Considerations |
|---|---|---|---|
| Calpain Activators | Calcium ionophores (A23187, ionomycin) | Induce calpain activation in cellular models | Requires precise concentration optimization to avoid necrosis |
| Calpain Inhibitors | MDL-28170, calpeptin, ALLN | Validate calpain-specific substrate cleavage | Limited specificity; may inhibit other cysteine proteases |
| Cathepsin Inhibitors | E-64d (cysteine cathepsins), pepstatin A (aspartyl cathepsins) | Distinguish cathepsin-mediated pathways | Cell permeability varies between inhibitors |
| Granzyme Preparations | Purified granzyme A and B | Study immune-mediated cytotoxicity | Requires perforin or other delivery systems for cellular uptake |
| PARP-1 Antibodies | Cleavage-specific, full-length, fragment-specific | Detect PARP-1 cleavage patterns | Validation needed for specific fragments in different models |
| Activity-Based Probes | DCG-04 (cysteine proteases) | Profile active protease populations | Enables monitoring of protease activation in complex mixtures |
| Caspase Inhibitors | Z-VAD-FMK (pan-caspase) | Distinguish caspase-independent pathways | Use in combination with other protease inhibitors |
| N-terminomics Tools | TAILS kits, isobaric tags | Proteome-wide cleavage site discovery | Requires specialized mass spectrometry expertise |
Signaling Pathway Visualizations
Calpain Activation and PARP-1 Cleavage Pathway
Comparative PARP-1 Cleavage by Proteases
The investigation of alternative proteolytic pathways targeting PARP-1 has revealed sophisticated regulatory mechanisms that extend beyond canonical caspase-mediated apoptosis. Calpain, cathepsin, and granzyme proteases contribute to PARP-1 cleavage in specific physiological and pathological contexts, generating distinct cleavage fragments with unique biological activities. The emerging roles of these cleavage fragments in processes such as parthanatos, innate immune activation, and neurodegenerative pathways highlight the complexity of PARP-1 as a signaling node in cell death decision-making.
Future research directions should focus on characterizing the exact cleavage sites utilized by these alternative proteases, identifying the full spectrum of biological activities associated with the resulting fragments, and exploring therapeutic opportunities for modulating these pathways in disease contexts. The development of more specific protease inhibitors and activity-based probes will facilitate these investigations, potentially leading to novel treatment strategies for conditions ranging from cancer to neurodegenerative disorders where dysregulated proteolysis contributes to pathology.
Poly(ADP-ribose) polymerase-1 (PARP-1) serves as a primary nuclear sensor for DNA damage, playing a pivotal role in maintaining genomic integrity through its involvement in DNA repair pathways and cell death signaling. As the most abundant member of the PARP family, PARP-1 accounts for approximately 85% of cellular PARP activity and possesses a unique ability to detect DNA strand breaks within seconds of their formation [24] [25]. This rapid response mechanism initiates a complex signaling cascade that determines cellular fate, ranging from successful DNA repair to programmed cell death. The critical positioning of PARP-1 at the intersection of DNA repair and cell death pathways has made it a compelling therapeutic target, particularly in oncology, where PARP inhibitors are now approved for treating homologous recombination-deficient cancers [24] [26].
Beyond its established role in DNA damage response, PARP-1 participates in various physiological processes, including transcription regulation, chromatin remodeling, and cell death programs [1] [25]. This multifunctional enzyme catalyzes the transfer of ADP-ribose units from nicotinamide adenine dinucleotide (NAD+) to target proteins, forming linear or branched poly(ADP-ribose) (PAR) polymers. This PARylation process serves as a critical post-translational modification that regulates protein function and facilitates the assembly of DNA repair complexes at damage sites [24] [27]. Understanding the molecular mechanisms governing PARP-1 activation, polymer synthesis, and its role in cell death pathways provides crucial insights for developing novel therapeutic strategies for cancer and other human diseases.
Molecular Architecture of PARP-1
PARP-1 is a multifunctional enzyme composed of 1,014 amino acids with a molecular weight of approximately 116 kDa [25]. Its structural organization features three primary domains that enable its DNA damage sensing and signaling capabilities, though finer structural analyses reveal six distinct functional domains that facilitate its complex regulation.
Table 1: Domain Structure of PARP-1
| Domain Name | Molecular Weight | Key Functions | Structural Features |
|---|---|---|---|
| DNA-Binding Domain (DBD) | 46 kDa | Recognizes DNA breaks via zinc fingers | Contains three zinc-finger motifs (F1, F2, F3) |
| Automodification Domain (AMD) | 22 kDa | Serves as target for auto-PARylation | BRCT fold facilitating protein-protein interactions |
| Catalytic Domain (CAT) | 54 kDa | Catalyzes PAR synthesis from NAD+ | Comprises helical (HD) and ART subdomains |
The N-terminal DNA-binding domain (DBD) contains three zinc-finger motifs (F1, F2, and F3) that recognize and bind to various DNA structures, including single-strand breaks (SSBs), double-strand breaks (DSBs), hairpins, and cruciforms [24] [25]. The F1 and F2 domains utilize a "base-stacking loop" and "backbone grip" to interact with exposed nucleotide bases and the DNA phosphate backbone, respectively [24] [28]. While both F1 and F2 bind DNA, they exhibit different affinities and functions—F1 has lower DNA affinity but is essential for PARP-1 activation, whereas F2 contributes to DNA localization and retention but is dispensable for activation [24]. The F3 domain does not directly bind DNA but facilitates interdomain contacts essential for PARP-1 assembly upon DNA damage recognition [24] [28].
The central automodification domain (AMD) contains a BRCT fold, a motif found in many DNA repair proteins that mediates protein-protein interactions [1] [25]. This domain serves as the primary target for auto-PARylation, with several glutamic acid residues functioning as acceptor sites for covalent PAR attachment [25]. Automodification represents a key regulatory mechanism that modulates PARP-1 activity and facilitates the recruitment of DNA repair machinery to damage sites.
The C-terminal catalytic domain (CAT) is the most conserved region across PARP family members and contains the NAD+-binding site that executes PAR synthesis [25]. This domain comprises two critical subdomains: the helical domain (HD), which maintains autoinhibition in the absence of DNA damage, and the ADP-ribosyltransferase (ART) domain, which catalyzes PAR formation [24] [27] [28]. The CAT domain also includes a highly conserved 50-amino acid "PARP signature" motif essential for NAD+ binding and catalytic activity [25].
DNA Damage Detection and Allosteric Activation
DNA Damage Recognition Mechanism
PARP-1 functions as a first-line responder to DNA damage, capable of detecting DNA strand breaks within 1-3 seconds of their formation [24] [28]. This exceptional rapidity enables PARP-1 to initiate the DNA damage response (DDR) before other repair factors arrive at the lesion site. PARP-1 exhibits remarkable versatility in recognizing various DNA lesions, with particularly high affinity for single-strand breaks (SSBs)—the most common form of DNA damage [24]. However, it also effectively binds and signals double-strand breaks (DSBs) and other DNA structural abnormalities [24] [29].
The mechanism of DNA damage recognition involves coordinated action of PARP-1's zinc finger domains. The F1 and F2 domains recognize specialized DNA structures rather than specific sequences, interacting with exposed nucleotide bases through a "base-stacking loop" and with the phosphate backbone through a "backbone grip" [24] [28]. The F3 domain, while not directly binding DNA, establishes essential interdomain contacts with F1 and DNA that facilitate the assembly of PARP-1 into its active conformation [24]. This DNA binding triggers a dramatic conformational change in PARP-1 from a "beads on a string" architecture to a collapsed structure where the zinc fingers, WGR domain, and CAT domain collectively engage damaged DNA, creating an extensive network of interdomain contacts [24] [28].
Allosteric Activation Switch
The transition from inactive to fully active PARP-1 represents a remarkable example of allosteric regulation in response to DNA damage. In the absence of DNA damage, PARP-1 maintains minimal basal activity due to autoinhibition mediated by the folded helical domain (HD), which sterically blocks NAD+ access to the catalytic active site [24] [28]. Binding to DNA breaks induces local unfolding within three of the seven helices comprising the HD, as revealed by hydrogen/deuterium exchange coupled with mass spectrometry (HXMS) [24]. This DNA-induced destabilization of the HD activates PARP-1 by relieving enzymatic autoinhibition, allowing full NAD+ access to the catalytic site and resulting in a 1000-fold increase in enzymatic activity [24] [28].
The allosteric communication between DNA binding sites and the catalytic center occurs over a considerable distance of approximately 40 Å [24]. NMR studies have further demonstrated that PARP-1 interaction with a SSB, followed by NAD+ binding, results in stepwise and additive destabilization of the HD during activation [24]. This allosteric switch represents a sophisticated regulatory mechanism that ensures PARP-1 remains largely inactive under normal physiological conditions but becomes rapidly and robustly activated in response to genomic insults.
Figure 1: PARP-1 Allosteric Activation Pathway. DNA damage binding induces helical domain (HD) unfolding, relieving autoinhibition and enabling NAD+ access for PAR synthesis.
PAR Synthesis Mechanisms and Signaling
Canonical Protein PARylation
PARP-1 catalyzes the transfer of ADP-ribose units from NAD+ to target proteins, initiating with the attachment of a single ADP-ribose unit (mono-ADP-ribosylation) followed by elongation to form linear or branched PAR polymers (poly-ADP-ribosylation) [25]. The highly negatively charged PAR chains dramatically alter the physicochemical properties of modified proteins, affecting their DNA-binding capacity, protein-protein interactions, and subcellular localization [25]. PARP-1 itself represents the most abundant auto-PARylation target, with automodification serving as a key regulatory mechanism that modulates PARP-1 affinity for DNA and facilitates its release from DNA break sites after signaling [24] [25].
The PARylation process occurs through a coordinated mechanism involving two sites within the ART domain: a donor site that binds NAD+, and an acceptor site that extends the ADP-ribose chain [27]. Recent research has identified histone PARylation factor 1 (HPF1) as a critical binding partner that modulates PARP-1 enzymatic activity to target serine residues rather than the traditional aspartate/glutamate residues [27] [28]. This HPF1-mediated serine-directed PARylation represents the predominant modification in response to DNA damage and significantly influences PARP-1 function in DNA repair [27].
The cellular functions of PARylation extend beyond DNA repair to include chromatin relaxation through PAR-mediated histone modification, recruitment of DNA repair factors such as XRCC1 through specific PAR-binding motifs, and regulation of various nuclear processes including transcription and replication [24] [25] [29]. The massive PAR synthesis at DNA damage sites creates a dense matrix that facilitates the assembly of repair complexes and promotes chromatin decondensation to allow repair machinery access to damaged DNA.
Novel Protein-Free PAR Synthesis
Recent groundbreaking research has revealed that PARP-1 possesses the capacity for de novo synthesis of protein-free PAR molecules—a previously unrecognized catalytic activity that expands the signaling potential of PARP enzymes [27]. This novel mechanism occurs when a molecule of NAD+ or ADP-ribose docks in the PARP-1 acceptor site and attaches to an NAD+ molecule bound to the donor site, initiating ADP-ribose chains that emanate from NAD+/ADP-ribose rather than protein residues [27].
This protein-free PAR synthesis occurs alongside canonical protein modification activity and is stimulated by PARP-1 interaction with DNA breaks [27]. The isolated ART domain of PARP-1 demonstrates constitutive activity in producing free PAR, suggesting that DNA binding primarily relieves autoinhibition rather than directly activating the catalytic mechanism [27]. HPF1 regulates the balance between free PAR and protein-linked PAR production by favoring synthesis of protein-linked PAR [27].
Table 2: Comparison of PARP-1 Catalytic Activities
| Feature | Canonical Protein PARylation | Protein-Free PAR Synthesis |
|---|---|---|
| Initial Substrate | Protein glutamate/aspartate/serine residues | NAD+ or free ADP-ribose molecules |
| Product | Protein-attached PAR chains | Free PAR molecules not attached to proteins |
| HPF1 Regulation | Switches specificity to serine residues | Reduces free PAR production |
| Cellular Function | Post-translational modification of target proteins | Proposed role in parthanatos signaling |
| Dependency on DNA | Stimulated by DNA damage | Similarly stimulated by DNA damage |
Cellular studies demonstrate that DNA damage stimulates free PAR production, and this free PAR originates primarily from PARP-1 de novo synthesis rather than through PAR degradation by glycohydrolases such as PARG, ARH3, or TARG1 [27]. The discovery of direct protein-free PAR synthesis represents a paradigm shift in understanding PAR signaling and broadens the scope of PARP enzyme signaling capacity in cellular physiology and pathology.
PARP-1 in Programmed Cell Death
PARP-1 Cleavage as a Cell Death Signature
PARP-1 serves as a preferred substrate for multiple cell death proteases, and its proteolytic cleavage generates specific fragments that serve as recognized biomarkers for distinct cell death programs [1]. Different proteases target PARP-1 at specific cleavage sites, producing characteristic fragment patterns that identify the active protease and the particular form of cell death occurring in pathological conditions [1].
The most extensively characterized PARP-1 cleavage occurs during caspase-dependent apoptosis, where executioner caspases-3 and -7 cleave PARP-1 at a specific aspartate residue (DEVD214↓G) within the nuclear localization signal near the DNA-binding domain [1] [12]. This cleavage produces two signature fragments: a 24-kDa DNA-binding fragment containing the two zinc-finger motifs, and an 89-kDa fragment containing the automodification and catalytic domains [1]. The 24-kDa fragment remains tightly bound to DNA breaks, acting as a trans-dominant inhibitor of PARP-1 activity and other DNA repair enzymes, thereby conserving cellular ATP pools during apoptosis [1]. The 89-kDa fragment exhibits greatly reduced DNA binding capacity and translocates to the cytoplasm [1] [12].
Beyond caspases, PARP-1 serves as a substrate for other "suicidal" proteases including calpains, cathepsins, granzymes, and matrix metalloproteinases (MMPs), each generating distinct PARP-1 cleavage fragments associated with specific cell death pathways [1]. These PARP-1 signature fragments provide valuable diagnostic tools for identifying active proteases and particular forms of cell death in various pathological contexts, including neurodegeneration, cerebral ischemia, trauma, and excitotoxicity [1].
Parthanatos: PAR-Mediated Cell Death
Under conditions of excessive DNA damage, PARP-1 hyperactivation triggers a specialized cell death pathway termed parthanatos, which represents a caspase-independent programmed necrosis distinct from both apoptosis and conventional necrosis [12] [29]. Parthanatos features characteristic nuclear shrinkage, chromatin condensation, and large-scale DNA fragmentation (ranging from 15-50 kb) [29]. This cell death pathway depends specifically on PARP-1 hyperactivation and PAR accumulation, as PARP-1 inhibitors or genetic deletion completely block parthanatos, while caspase inhibitors are ineffective [29].
The molecular mechanism of parthanatos involves deadly crosstalk between the nucleus and mitochondria mediated by PAR polymer [29]. Following extreme DNA damage, PARP-1 hyperactivation generates substantial PAR accumulation, which functions as a cell death signal [12] [29]. Recent research has demonstrated that caspase-mediated cleavage of PARP-1 during apoptosis can also contribute to parthanatos through an unexpected mechanism: the 89-kDa PARP-1 fragment with covalently attached PAR polymers translocates to the cytoplasm, where it serves as a PAR carrier that facilitates apoptosis-inducing factor (AIF) release from mitochondria [12] [13]. AIF then recruits macrophage migration inhibitory factor (MIF), a specific 3'-exonuclease, to the nucleus, where MIF executes large-scale DNA fragmentation, culminating in cell death [29].
This caspase-mediated interaction between apoptosis and parthanatos pathways extends our current understanding of programmed cell death mechanisms and reveals potential new therapeutic targets for conditions where parthanatos contributes to pathology, including stroke, neurodegenerative diseases, and other forms of cellular injury [12] [29].
Figure 2: PARP-1-Mediated Parthanatos Cell Death Pathway. Excessive DNA damage triggers PARP-1 hyperactivation and PAR accumulation, leading to AIF-mediated large-scale DNA fragmentation.
Experimental Approaches and Research Tools
Key Methodologies for PARP-1 Research
Advancing our understanding of PARP-1 biology has relied on sophisticated experimental approaches that probe its structure, function, and cellular dynamics. Structural biology techniques including X-ray crystallography and hydrogen/deuterium exchange coupled with mass spectrometry (HXMS) have been instrumental in elucidating the allosteric activation mechanism of PARP-1 [24] [28]. Crystallographic studies of PARP-1 bound to DNA breaks have provided high-resolution snapshots of the enzyme's collapsed active conformation, revealing interdomain contacts and structural distortions in the helical domain that relieve autoinhibition [24]. HXMS has complemented these static structures by capturing the dynamic changes in PARP-1 backbone dynamics upon DNA binding, particularly the local unfolding within the HD that enables NAD+ access to the catalytic site [24] [28].
Biochemical assays monitoring PAR synthesis remain fundamental for studying PARP-1 catalytic activity. These include in vitro PARylation assays using purified PARP-1 components with NAD+ as substrate, often incorporating activating DNA oligonucleotides to stimulate PARP-1 activity [27]. Recent investigations of protein-free PAR synthesis have employed systematic variations of standard protocols, including the use of isolated ART domains that exhibit constitutive activity without requiring DNA activation [27]. Advanced mass spectrometry techniques enable precise mapping of PARylation sites on target proteins and quantification of PAR chain length and branching patterns [27] [30].
Cellular studies utilize DNA-damaging agents such as hydrogen peroxide, alkylating agents (e.g., MNNG), and chemotherapeutic drugs (e.g., etoposide) to activate PARP-1 in physiological contexts [1] [29]. Immunofluorescence microscopy with PAR-specific antibodies allows visualization of PAR accumulation at DNA damage sites and tracking of PARP-1 translocation during cell death processes [12] [29]. Genetic approaches including PARP-1 knockout cells and RNAi-mediated knockdown provide essential tools for establishing PARP-1-specific functions versus those compensated by other PARP family members [24] [29].
Essential Research Reagents
Table 3: Key Research Reagents for PARP-1 Studies
| Reagent/Category | Specific Examples | Research Applications | Key Functions |
|---|---|---|---|
| PARP Inhibitors | Olaparib, Veliparib, Talazoparib | Cancer research, mechanistic studies | Inhibit PARP catalytic activity; induce PARP trapping |
| DNA-Damaging Agents | Hydrogen peroxide, MNNG, Etoposide, Hydroxyurea | PARP activation studies | Induce SSBs, DSBs, replication stress |
| Activity Assays | NAD+ consumption, PAR immunoblotting, HXMS | Enzymatic characterization | Quantify PAR synthesis, structural dynamics |
| Cell Death Inducers | Staurosporine, Actinomycin D | Apoptosis/parthanatos research | Activate caspases, trigger programmed cell death |
| Detection Antibodies | Anti-PAR, Anti-PARP-1 cleavage fragments | Cell death pathway analysis | Identify specific proteolytic fragments |
| Genetic Tools | PARP-1 KO cells, siRNA/shRNA | Functional studies | Establish PARP-1-specific phenotypes |
The expanding toolkit for PARP-1 research continues to drive discoveries in DNA damage response and cell death signaling. Particularly valuable are reagents that specifically detect PARP-1 cleavage fragments, which serve as precise biomarkers for different cell death pathways [1]. The development of increasingly specific PARP inhibitors, including those that allosterically tune PARP-1 release from DNA breaks, represents an active area of pharmaceutical research with implications for cancer therapy and beyond [24] [26] [28].
PARP-1 stands as a paramount DNA damage sensor and signaling molecule that integrates DNA repair with cell fate decisions through its sophisticated activation mechanism and versatile PAR synthesis capabilities. The allosteric switch mechanism—whereby DNA binding induces helical domain unfolding to relieve catalytic autoinhibition—represents an elegant molecular solution for rapid damage detection and response [24] [28]. The recent discovery of protein-free PAR synthesis significantly expands the signaling potential of PARP-1 beyond traditional protein modification, suggesting more diverse roles in cellular physiology and pathology [27].
The critical positioning of PARP-1 at the intersection of DNA repair and cell death pathways, particularly through its role in parthanatos, underscores its importance as a therapeutic target [12] [29]. The detailed understanding of PARP-1 cleavage by various cell death proteases and the specific functions of the resulting fragments provide valuable insights for diagnostic and therapeutic applications [1]. As research continues to unravel the complexity of PARP-1 biology, including its regulation by accessory factors such as HPF1 and its coordination with associated nucleases, new opportunities will emerge for targeted interventions in cancer, neurodegenerative diseases, and other conditions where PARP-1-mediated processes contribute to pathology.
Post-translational modifications (PTMs) represent crucial biochemical processes that rapidly diversify protein function, regulate activity, and control complex cellular signaling networks. In the context of programmed cell death (PCD) research, PTMs serve as fundamental regulatory mechanisms that determine cellular fate. Among the hundreds of known PTMs, phosphorylation, acetylation, and ADP-ribosylation stand out as particularly significant for their roles in modulating key cell death pathways. These modifications regulate critical processes including DNA damage response, chromatin remodeling, metabolic adaptation, and the direct activation of cell death executioners. The intricate interplay between these PTMs creates a sophisticated control system that integrates survival and death signals, with profound implications for therapeutic interventions in cancer and other diseases.
Within PCD research, the cleavage and regulation of poly(ADP-ribose) polymerase-1 (PARP-1) serves as a paradigm for understanding how PTMs coordinate cellular fate decisions. PARP-1, a prominent nuclear enzyme responsible for the majority of ADP-ribosylation activity in cells, becomes activated in response to DNA damage and participates in a complex network of PTM-mediated signaling that ultimately determines whether a cell undergoes repair or initiates suicide programs. This whitepaper provides a comprehensive technical examination of these three pivotal PTMs, with particular emphasis on their mechanisms, cross-regulatory relationships, and indispensable functions in PARP-1-mediated cell death pathways.
Core Mechanisms of Key Post-Translational Modifications
Phosphorylation: The Ubiquitinary Regulatory Switch
Phosphorylation, catalyzed by kinases and reversed by phosphatases, represents the most extensively studied PTM. This reversible process involves the transfer of a phosphate group from ATP to specific amino acid side chains, primarily serine, threonine, and tyrosine residues. The addition of a negatively charged phosphate group induces conformational changes that regulate protein activity, stability, subcellular localization, and protein-protein interactions.
In apoptotic signaling, phosphorylation events serve as critical control points at multiple levels of death pathways. The BCL-2 family proteins, which govern mitochondrial outer membrane permeabilization (MOMP) - a pivotal event in intrinsic apoptosis - are extensively regulated by phosphorylation. Specific phosphorylation events can either promote or inhibit the pro-apoptotic activities of BCL-2 family members, thereby modulating cellular susceptibility to death signals [31]. Additionally, caspase enzymes, the ultimate executioners of apoptosis, are subject to complex phosphorylation regulation that either activates or suppresses their proteolytic functions, creating multiple layers of control throughout the cell death process [32].
Acetylation: Metabolic Integration and Histone Regulation
Lysine acetylation, mediated by acetyltransferases (HATs) and deacetylases (HDACs), involves the transfer of an acetyl group from acetyl-CoA to the ε-amino group of lysine residues. This modification neutralizes the positive charge of lysine, potentially altering protein conformation, DNA-binding affinity, and protein interactions. Originally characterized in the context of histone modification and chromatin organization, acetylation is now recognized as a widespread regulatory mechanism affecting numerous non-histone proteins, particularly those involved in cell death and metabolism.
The functional consequences of acetylation extend to the direct regulation of apoptotic machinery. Acetylation of specific lysine residues on caspases can influence their activation status and catalytic efficiency, thereby modulating the threshold for apoptosis initiation [32]. Furthermore, key transcription factors such as p53 undergo acetylation that enhances their sequence-specific DNA binding, promoting the expression of pro-apoptotic genes in response to cellular stress. The intimate connection between acetylation and central metabolism (through acetyl-CoA availability) positions this PTM as a crucial integrator of metabolic status and cell death decisions.
ADP-Ribosylation: DNA Damage Sensing and PARP-1 Function
ADP-ribosylation entails the transfer of ADP-ribose units from NAD+ to target proteins, with PARP-1 accounting for approximately 90% of the NAD+ consumption by this enzyme family in cells [33]. This modification occurs as either mono-ADP-ribosylation (MARylation) or poly-ADP-ribosylation (PARylation), which involves the formation of branched polymers. PARP-1 contains a modular domain architecture that includes zinc finger domains (Zn1, Zn2, Zn3), a WGR domain, and a catalytic domain (CAT) that mediates the poly(ADP-ribose) synthesis activity [34].
The activation mechanism of PARP-1 involves direct binding to DNA lesions through its zinc finger domains. Structural studies reveal that PARP-1 engages DNA as a monomer, with the Zn1, Zn3, and WGR domains collectively binding to DNA and forming a network of interdomain contacts that links the DNA damage interface to the catalytic domain [35]. This DNA-induced organization of PARP-1 domains into a collapsed conformation triggers structural distortions that destabilize the CAT domain, resulting in dramatically enhanced automodification and substrate modification activities [34] [35].
Table 1: Key Characteristics of Major Post-Translational Modifications in Cell Death
| Feature | Phosphorylation | Acetylation | ADP-ribosylation |
|---|---|---|---|
| Chemical Group | Phosphate (PO₄) | Acetyl group (COCH₃) | ADP-ribose unit(s) |
| Donor Molecule | ATP | Acetyl-CoA | NAD+ |
| Target Residues | Ser, Thr, Tyr | Lys | Asp, Glu, Lys, Arg |
| Enzymes Adding | Kinases | HATs/KATs | PARPs/ARTDs |
| Enzymes Removing | Phosphatases | HDACs/KDACs | PARG, ARH3 |
| Primary Functions in Cell Death | Signal transduction cascades, protein activation/inactivation | Chromatin remodeling, metabolic integration, transcription | DNA damage response, chromatin modulation, energy depletion |
PARP-1 Cleavage in Programmed Cell Death
PARP-1 as a Caspase Substrate in Apoptosis
During the execution phase of apoptosis, PARP-1 serves as a primary substrate for caspase proteases, particularly effector caspases-3 and -7. These enzymes cleave PARP-1 at a specific aspartic acid residue (DEVD²¹⁴↓G²¹⁵ in human PARP-1), separating the N-terminal DNA-binding domain from the C-terminal catalytic domain [32]. This proteolytic event serves as both a biochemical marker of apoptosis and a functionally significant step in the cell death process. The cleavage of PARP-1 prevents excessive NAD+ and ATP consumption that would otherwise occur due to persistent PARP-1 activation, thereby conserving cellular energy for the orderly execution of the apoptotic program [33]. This cleavage also inactivates PARP-1's DNA repair functions, facilitating the dismantling of the nucleus and contributing to the irreversibility of the cell death process.
The detection of PARP-1 cleavage has emerged as a standard biochemical assay for apoptosis in experimental systems. Western blot analysis typically reveals the intact 116-kDa full-length PARP-1 and the characteristic 89-kDa cleavage fragment when apoptosis occurs. This cleavage event represents a pivotal point of crosstalk between ADP-ribosylation and proteolytic signaling in cell death regulation, with the 89-kDa fragment losing its catalytic activity while retaining DNA-binding capacity [32].
PARP-1 in Alternative Cell Death Modalities
Beyond its established role in apoptosis, PARP-1 activation contributes significantly to other forms of programmed cell death, including parthanatos - a PAR-dependent cell death pathway. In parthanatos, extensive PARP-1 activation leads to the generation of extensive PAR polymers that trigger mitochondrial release of apoptosis-inducing factor (AIF) [36]. AIF translocates to the nucleus where it facilitates chromatin condensation and large-scale DNA fragmentation, independent of caspase activity. This PARP-1-dependent cell death pathway becomes particularly important under conditions of severe genotoxic stress or oxidative damage.
In sepsis and other inflammatory conditions, endothelial cell dysfunction involves PARP-1 activation contributing to various programmed cell death modalities. The septic milieu triggers endothelial injury through coordinated activation of multiple death mechanisms, including apoptosis, pyroptosis, and ferroptosis, with PARP-1 playing a significant role in this pathological process [36]. The involvement of PARP-1 in these alternative cell death pathways highlights its broader significance beyond DNA repair and apoptosis, establishing it as a central node in multiple cell fate decisions.
Table 2: PARP-1 in Different Programmed Cell Death Modalities
| Cell Death Type | Key Initiators | Role of PARP-1 | Functional Consequences |
|---|---|---|---|
| Apoptosis | Caspases-3/7, DNA damage | Caspase substrate; cleavage inactivates DNA repair | Prevents energy depletion; facilitates nuclear dismantling |
| Parthanatos | Severe DNA damage, oxidative stress | Hyperactivation produces PAR polymers; AIF release | Caspase-independent death; large-scale DNA fragmentation |
| Pyroptosis | Inflammatory caspases, GSDMD cleavage | Inflammatory gene expression via NF-κB | Promotes inflammation; endothelial barrier disruption |
| Sepsis-associated EC Death | Multiple PAMPs/DAMPs, inflammatory mediators | Contributes to endothelial barrier dysfunction | Microvascular leakage, coagulopathy, organ failure |
Experimental Methodologies for Studying PTMs in Cell Death
Analyzing PARP-1 Cleavage in Apoptosis
The detection and quantification of PARP-1 cleavage serves as a fundamental methodology in cell death research. The standard experimental approach involves:
Cell Lysis and Protein Extraction: Prepare whole-cell lysates using RIPA buffer supplemented with protease inhibitors (e.g., PMSF, complete protease inhibitor cocktail) and caspase inhibitors (e.g., Z-VAD-FMK) when appropriate to prevent post-lysis cleavage. For subcellular localization studies, perform nuclear-cytoplasmic fractionation using differential centrifugation with non-ionic detergents.
Western Blot Analysis: Resolve proteins (20-30 μg per lane) by SDS-PAGE (8-10% gels) and transfer to PVDF membranes. Block with 5% non-fat milk or BSA in TBST for 1 hour. Incubate with primary antibodies against PARP-1 (specific for the N-terminal region to detect both full-length and cleaved fragments) overnight at 4°C. Common antibodies include rabbit anti-PARP-1 (Abcam) used at 1:1000 dilution [37]. After washing, incubate with HRP-conjugated secondary antibodies (1:5000) for 1 hour at room temperature. Detect using enhanced chemiluminescence substrate and image with a digital imaging system.
Data Interpretation: Quantify band intensities using densitometry software. The characteristic cleavage pattern shows full-length PARP-1 at 116 kDa and the large fragment at 89 kDa. The ratio of cleaved to full-length PARP-1 provides a quantitative measure of apoptosis extent. Include controls such as untreated cells and cells treated with known apoptosis inducers (e.g., staurosporine) as positive controls.
Assessing PARP-1 Enzymatic Activity
Measuring PARP-1 enzymatic activity provides complementary information to cleavage assays:
NAD+ Consumption Assay: Monitor NAD+ depletion using colorimetric or fluorometric methods. Incubate cell lysates or immunoprecipitated PARP-1 with NAD+ and activated DNA (sonicated or oligonucleotide-treated) in reaction buffer. Measure NAD+ levels at different timepoints using enzymatic cycling assays.
PAR Polymer Detection: Use anti-PAR antibodies (e.g., 10H) for immunofluorescence or Western blot to detect PAR formation. For immunofluorescence, fix cells with 4% paraformaldehyde, permeabilize with 0.2% Triton X-100, and incubate with anti-PAR primary antibody followed by fluorophore-conjugated secondary antibody. Counterstain with DAPI and image using confocal microscopy.
Activity-Modifying Treatments: To study PARP-1 inhibition, use specific PARP inhibitors (e.g., olaparib, veliparib) at concentrations ranging from 1-10 μM. To induce PARP-1 activation, treat cells with DNA-damaging agents such as H₂O₂ (0.5-2 mM for 1 hour) [37] or methyl methanesulfonate.
Research Reagent Solutions for PTM Studies
Table 3: Essential Research Reagents for PARP-1 and Cell Death Studies
| Reagent Category | Specific Examples | Function/Application | Experimental Notes |
|---|---|---|---|
| PARP-1 Antibodies | Rabbit anti-PARP-1 (Abcam) [37] | Detection of full-length and cleaved PARP-1 in Western blot | Use N-terminal targeting antibodies to detect cleavage fragments |
| Caspase Inhibitors | Z-VAD-FMK (pan-caspase inhibitor) | Inhibition of caspase-mediated PARP-1 cleavage | Validate specificity with specific caspase inhibitors |
| PARP Inhibitors | Olaparib, Veliparib, 3-AB | Inhibition of PARP enzymatic activity | Use at appropriate concentrations (typically 1-10 μM) for specific inhibition |
| Apoptosis Inducers | Staurosporine, H₂O₂ [37], Etoposide | Positive controls for PARP-1 cleavage | Titrate for appropriate response in specific cell models |
| Activity Assays | Anti-PAR antibody (10H), NAD/NADH assay kits | Detection of PAR formation and NAD+ consumption | Combine with inhibition controls for specificity |
| Cell Death Detection | Annexin V-FITC [37], DAPI, PI | Apoptosis quantification by flow cytometry | Use in combination with PARP-1 cleavage for correlation |
| Caspase Activity Assays | Caspase-3/7 luminescent assays [38] | Quantification of executioner caspase activity | Correlate with PARP-1 cleavage timing and extent |
Signaling Pathway Visualizations
PARP-1 Cleavage and Cell Death Pathways
PTM Crosstalk in Cell Death Regulation
Concluding Perspectives and Therapeutic Implications
The intricate interplay between phosphorylation, acetylation, and ADP-ribosylation creates a sophisticated regulatory network that determines cellular fate in response to stress and damage. PARP-1 stands at the crossroads of these PTM pathways, functioning as both a sensor of genomic integrity and a mediator of cell death decisions. The cleavage of PARP-1 by caspases represents a committed step in apoptosis that prevents energy depletion and facilitates orderly cellular dismantling, while excessive PARP-1 activation can trigger alternative death pathways such as parthanatos.
The therapeutic targeting of PTM pathways, particularly PARP-1, has yielded significant clinical advances in cancer treatment. PARP inhibitors exploit synthetic lethality in BRCA-deficient tumors by blocking both PARP-mediated DNA repair and homologous recombination repair, leading to selective tumor cell death [33]. Additionally, the modulation of phosphorylation and acetylation pathways through kinase inhibitors and HDAC inhibitors represents promising strategies for overcoming apoptosis resistance in cancer. Understanding the complex crosstalk between these PTM systems will enable the development of more effective combination therapies that simultaneously target multiple nodes within cell death networks.
As research continues to unravel the complexity of PTM networks in cell death regulation, new opportunities will emerge for therapeutic intervention in cancer, neurodegenerative disorders, and inflammatory conditions. The integration of structural biology, chemical biology, and systems approaches will be essential for mapping the dynamic landscape of PTM crosstalk and identifying optimal targets for precision medicine.
Detection Methods and Research Applications of PARP-1 Cleavage
Poly(ADP-ribose) polymerase-1 (PARP-1) is a 113 kDa nuclear enzyme that plays a fundamental role in DNA repair and maintenance of genomic integrity. Beyond its physiological functions, PARP-1 has emerged as a critical signaling molecule in programmed cell death pathways. The proteolytic cleavage of PARP-1 by various cell death proteases generates specific signature fragments that serve as recognizable biomarkers for distinguishing between different forms of cell death [11]. These cleavage events not only inactivate the DNA repair function of PARP-1 but also generate fragments with distinct biological activities that can actively participate in and amplify cell death processes [39].
The analysis of PARP-1 cleavage fragments via Western blotting has become an essential technique in cell death research, particularly in the context of neurodegeneration, cancer, and drug development [40]. This technical guide provides comprehensive methodologies for identifying these signature fragments and interpreting their significance within the broader mechanism of PARP-1 cleavage during programmed cell death research.
PARP-1 Domains and Cleavage Sites
PARP-1 contains three primary functional domains: a 46-kDa DNA-binding domain (DBD) at the N-terminus containing two zinc finger motifs, a 22-kDa automodification domain (AMD) in the central region, and a 54-kDa catalytic domain (CD) at the C-terminus [11]. The DBD facilitates tight binding to DNA damage sites, while the CD catalyzes the polymerization of ADP-ribose units from donor NAD+ molecules onto target proteins [11]. The AMD contains a BRCT fold that facilitates protein-protein interactions and recruitment of DNA repair enzymes [11].
Different proteases cleave PARP-1 at specific sites within these domains, generating characteristic fragments with distinct molecular weights and biological functions. The most well-characterized cleavage occurs at the DEVD214 site within the nuclear localization signal (NLS) of the DBD [39]. This site is preferentially targeted by caspase family proteases during apoptosis.
Signature Cleavage Fragments and Their Biological Significance
Apoptotic Versus Necrotic Cleavage Patterns
The cleavage pattern of PARP-1 serves as a biochemical signature that distinguishes between different cell death pathways. During apoptosis, caspase-mediated cleavage generates characteristic 89 kDa and 24 kDa fragments, whereas during necrosis, lysosomal proteases produce a dominant 50 kDa fragment [41].
Table 1: PARP-1 Cleavage Fragments in Different Cell Death Pathways
| Cell Death Pathway | Cleavage Fragments | Responsible Proteases | Molecular Weight | Biological Consequences |
|---|---|---|---|---|
| Apoptosis | 89 kDa + 24 kDa | Caspase-3, Caspase-7 | 89 kDa (AMD+CD), 24 kDa (DBD) | Inactivation of DNA repair, conservation of cellular energy |
| Necrosis | 50 kDa | Cathepsin B, Cathepsin G | 50 kDa | Lysosomal protease-mediated cleavage |
| Parthanatos | PAR-modified 89 kDa | Caspase-3/7 (with PARylation) | 89 kDa (PAR-modified) | AIF-mediated cell death |
Functional Consequences of PARP-1 Cleavage
The biological consequences of PARP-1 cleavage extend beyond mere inactivation of the enzyme. The 24 kDa fragment containing the zinc-finger motifs remains tightly bound to DNA strand breaks, acting as a trans-dominant inhibitor of DNA repair by blocking access of other repair enzymes to damage sites [11]. This irreversible binding conserves cellular ATP pools by preventing PARP-1 activation and excessive NAD+ consumption [11].
The 89 kDa fragment, comprising the automodification and catalytic domains, has reduced DNA binding capacity and can be liberated from the nucleus into the cytosol [11]. Recent research has revealed that this fragment can serve as a cytoplasmic PAR carrier that induces apoptosis-inducing factor (AIF)-mediated apoptosis, creating a link between caspase-dependent apoptosis and parthanatos [13].
In necrosis, the 50 kDa fragment generated by cathepsins represents a distinct proteolytic pattern that is not inhibited by broad-spectrum caspase inhibitors like zVAD-fmk, confirming the involvement of different protease families in this process [41].
Experimental Methodology for PARP-1 Cleavage Detection
Sample Preparation and Protein Extraction
Proper sample preparation is critical for accurate detection of PARP-1 cleavage fragments. The following protocol ensures preservation of these proteolytic fragments:
Homogenization Buffer: Use RIPA buffer (1% NP-40 or Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 150 mM NaCl, 50 mM Tris-HCl, pH 7.8, 1 mM EDTA) supplemented with complete mini protease inhibitor cocktail tablets [42].
Tissue Processing: Snap-freeze tissue in liquid nitrogen and dice into 1 mm pieces on dry ice. Add to ice-cold RIPA buffer and homogenize using a Dounce tissue homogenizer (25 strokes on ice) [42].
Cell Culture Processing: For pelleted cells, add ice-cold RIPA buffer directly. For plated cells, wash cells first, then add RIPA buffer and scrape [42].
Sonication and Clarification: Sonicate homogenized samples on ice for 5 × 20 seconds at 50% power. Clear extracts by centrifugation at 34,000 ×g at 4°C for 30 minutes [42].
Protein Quantification: Measure total protein concentration using a detergent-compatible assay (e.g., RC DC protein assay from Bio-Rad). Dilute homogenates to at least 2 mg/mL to enable loading of 10-80 μg per lane [42].
Western Blotting and Detection
For optimal detection of PARP-1 cleavage fragments, follow these quantitative Western blotting procedures:
Gel Electrophoresis: Use TGX stain-free SDS-gels for better resolution of protein separation. Include molecular weight markers in all gels [43].
Protein Transfer: Transfer to low-fluorescent PVDF membrane using semi-dry or wet transfer systems [42].
Antibody Incubation: Use validated primary antibodies against PARP-1 and appropriate fluorescent or HRP-conjugated secondary antibodies with at least four 3-minute wash steps between incubations [42].
Signal Detection: Develop immunochemical signal using imager-compatible chemiluminescence substrate (e.g., Clarity from Bio-Rad) and capture signal using a CCD-camera-based imager (e.g., ChemiDoc MP) [42].
Data Normalization and Quantification
For publication-quality quantitative Western blot data, implement proper normalization techniques:
Total Protein Normalization (TPN): Normalize target protein signals to the total amount of protein in each lane rather than using housekeeping proteins alone. TPN is not affected by experimental manipulations and provides a larger dynamic range for detection [44].
Linear Dynamic Range Determination: Create a 1/2 dilution series of a pooled sample starting from 100 μg protein load over at least 12 dilutions. Plot relative density versus fold dilution for each primary antibody to determine the linear dynamic range [42].
Validation: Select the protein load for each antibody that corresponds to the middle of the linear dynamic range to avoid membrane saturation [42].
Table 2: Research Reagent Solutions for PARP-1 Cleavage Studies
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Cell Death Inducers | Staurosporine, Etoposide (VP-16), H₂O₂, Actinomycin D | Induce specific cell death pathways for PARP-1 cleavage studies |
| Protease Inhibitors | zVAD-fmk (caspase inhibitor), E64d (cysteine protease inhibitor) | Determine protease families involved in PARP-1 cleavage |
| PARP-1 Antibodies | Monoclonal anti-PARP-1, Cleavage-specific antibodies | Detect full-length and cleaved PARP-1 fragments |
| Detection Systems | Chemiluminescent substrates, Fluorescent secondaries | Visualize and quantify PARP-1 cleavage fragments |
| Normalization Reagents | No-Stain Protein Labeling Reagents, Housekeeping protein antibodies | Ensure accurate quantification of Western blot data |
Troubleshooting and Technical Considerations
Common Experimental Challenges
Several technical challenges can affect the accurate detection and interpretation of PARP-1 cleavage fragments:
Incomplete Cleavage Detection: Use antibodies that recognize both full-length PARP-1 and cleavage fragments to ensure comprehensive detection of all proteolytic products.
Non-Specific Bands: Optimize antibody concentrations and blocking conditions to minimize non-specific signals that may be confused with genuine cleavage fragments.
Sample Degradation: Implement strict cold chain maintenance and protease inhibition during sample preparation to prevent artifactual proteolysis.
Quantification Errors: Employ total protein normalization rather than relying solely on housekeeping proteins, which may vary under experimental conditions [44].
Publication Standards
For publication of Western blot data featuring PARP-1 cleavage fragments, adhere to current journal requirements:
- Maintain original, unprocessed images of all blots as supplementary information [43]
- Avoid excessive cropping that removes molecular weight markers or important bands [44]
- Disclose all image processing procedures including brightness/contrast adjustments [43]
- Include appropriate positive and negative controls for cell death induction and protease activity
Western blot analysis of PARP-1 cleavage fragments provides critical insights into the mechanisms of programmed cell death. The distinct signature fragments generated by different proteases serve not only as biomarkers for specific cell death pathways but also as active participants in cell death execution. The methodologies outlined in this technical guide enable researchers to accurately detect and quantify these fragments, advancing our understanding of PARP-1's multifaceted roles in cell death and supporting drug development efforts targeting these pathways.
Immunohistochemical Detection in Tissue Sections and Disease Models
Poly(ADP-ribose) polymerase-1 (PARP-1) is a nuclear enzyme with a well-established role as a DNA damage sensor and a key mediator of DNA repair pathways. This 113-116 kDa protein catalyzes the synthesis of poly(ADP-ribose) (PAR) chains on target proteins using NAD+ as a substrate [1] [25]. Beyond its DNA repair functions, PARP-1 participates in diverse cellular processes including transcription regulation, inflammation, and most notably, programmed cell death [1] [45]. During apoptosis, PARP-1 serves as a primary substrate for executioner caspases-3 and -7, which cleave the protein at the conserved sequence DEVD214↓G, generating signature fragments of 24 kDa and 89 kDa [1] [12]. The detection of these cleavage fragments, particularly through immunohistochemical (IHC) methods, has become a fundamental biomarker for identifying and characterizing programmed cell death in both research and clinical contexts, providing critical insights into disease mechanisms and treatment responses across numerous pathological conditions.
The 24-kDa fragment encompasses the DNA-binding domain containing two zinc-finger motifs and remains tightly bound to DNA breaks, acting as a trans-dominant inhibitor of DNA repair by blocking access of intact PARP-1 and other repair factors to damaged DNA [1] [39]. Conversely, the 89-kDa fragment contains the auto-modification domain and catalytic domain, and recent research has revealed it can translocate to the cytoplasm where it functions as a PAR carrier to induce AIF-mediated apoptosis (parthanatos) or interact with cytoplasmic proteins including the RNA polymerase III complex during innate immune responses [12] [23]. This cleavage event is not merely a passive marker of cell death but actively contributes to the apoptotic process by preventing wasteful NAD+ consumption while facilitating the dismantling of the cell.
PARP-1 Biology and Technical Detection Principles
PARP-1 Domains and Cleavage Fragments
Understanding the domain architecture of PARP-1 is essential for interpreting IHC detection patterns. The protein consists of three primary functional domains:
- DNA-binding domain (DBD): An N-terminal 46-kDa domain containing three zinc-finger motifs that recognize DNA strand breaks [25] [46].
- Auto-modification domain (AMD): A central 22-kDa domain with a BRCT fold that serves as the primary target for PARP-1 automodification and facilitates protein-protein interactions [1] [25].
- Catalytic domain: A C-terminal 54-kDa domain that catalyzes PAR synthesis from NAD+ [1] [25].
During caspase-mediated apoptosis, cleavage occurs within the nuclear localization signal near the DBD, producing the 24-kDa fragment (containing the DBD) and the 89-kDa fragment (containing the AMD and catalytic domain) [1] [12]. It is crucial to note that multiple proteases beyond caspases—including calpains, cathepsins, granzymes, and matrix metalloproteinases—can cleave PARP-1, generating fragments of varying sizes (42-89 kDa) that may indicate different cell death programs [1] [46].
Detection Methodologies and Reagent Selection
The cornerstone of PARP-1 IHC detection lies in appropriate antibody selection and antigen retrieval optimization. Antibodies targeting different PARP-1 epitopes provide distinct experimental capabilities:
Table 1: Key Antibodies for PARP-1 Immunohistochemical Detection
| Antibody Target | Clone/Reference | Recommended Dilution | Detects Full-Length | Detects Cleaved Fragments | Primary Applications |
|---|---|---|---|---|---|
| C-terminal region | 13371-1-AP [46] | 1:1000-1:4000 | Yes (113-116 kDa) | Yes (89 kDa) | WB, IHC, IF/ICC, IP, FC |
| Not specified | ab6079 [47] | 1:500 | Yes | Not specified | IHC (tissue scanning) |
| Not specified | 46D11 [48] | 0.4 μg/mL | Yes | Not specified | IHC |
For comprehensive detection of both full-length and cleaved PARP-1, the polyclonal antibody 13371-1-AP represents an optimal choice as it targets the C-terminal region (amino acids 667-1014), enabling recognition of both the full-length protein (113-116 kDa) and the 89-kDa cleavage fragment that retains this epitope [46]. This antibody has been successfully validated in Western blot (WB), IHC, and immunofluorescence (IF) applications across human, mouse, and rat specimens.
Optimized IHC Protocol for PARP-1 Detection
The following protocol has been established based on validated methodologies from multiple research studies:
Tissue Preparation and Sectioning:
- Fix tissues in 4% formaldehyde for 24 hours maximum to prevent epitope masking
- Embed in paraffin using standard dehydration protocols
- Section at 4μm thickness and mount on charged slides
Antigen Retrieval and Staining:
- Deparaffinize and rehydrate sections through xylene and graded ethanol series
- Perform heat-induced epitope retrieval using:
- Block endogenous peroxidase activity with 3% H₂O₂
- Apply primary antibody at optimized dilution (1:1000-1:4000 for 13371-1-AP) in appropriate blocking buffer
- Incubate with species-specific secondary antibody conjugated to HRP
- Develop with 3,3'-diaminobenzidine (DAB) chromogen [48] [47]
- Counterstain with hematoxylin, dehydrate, and mount
Critical Controls and Validation:
- Include positive control tissues known to express PARP-1 (e.g., breast cancer, ovarian cancer)
- Use negative controls with rabbit IgG instead of primary antibody [48]
- Validate cleavage detection with apoptosis-induced tissue sections
- Correlate with Western blot analysis to confirm fragment sizes
PARP-1 Cleavage in Disease Models and Signaling Pathways
PARP-1 in Neurodegenerative Diseases
PARP-1 cleavage plays a significant role in the pathogenesis of various neurodegenerative conditions. In cerebral ischemia, traumatic brain injury, and excitotoxicity, PARP inhibition attenuates neuronal damage, demonstrating the enzyme's central role in these pathologies [1]. The cleavage fragments generated during neurodegenerative processes actively participate in cell death signaling rather than merely serving as bystander markers.
The diagram below illustrates the central role of PARP-1 cleavage in coordinating different cell death pathways:
PARP-1 in Cancer Biology and Therapy Response
PARP-1 expression and cleavage have significant prognostic implications in oncology. In high-grade epithelial ovarian cancer, PARP-1 expression serves as a negative prognostic indicator, with PARP-positive patients showing significantly worse progression-free survival (median 12 months vs. 16 months in PARP-negative patients) and overall survival (median 52 months vs. 65 months) following platinum-based chemotherapy [47]. Similarly, in operable breast cancer, nuclear PARP-1 overexpression independently predicts poor prognosis, with hazard ratios of 10.05 for disease-free survival and 1.82 for overall survival [49].
The relationship between PARP-1 expression and cancer outcomes can be summarized as follows:
Table 2: PARP-1 Expression as a Prognostic Indicator in Human Cancers
| Cancer Type | Detection Method | PARP-1 Expression Pattern | Clinical Correlation | Reference |
|---|---|---|---|---|
| High-grade epithelial ovarian cancer | IHC (ab6079 antibody) | 52.32% positive (45/86 cases) | Reduced PFS (12 vs 16 months) and OS (52 vs 65 months) | [47] |
| Operable breast cancer | IHC signal intensity scanning | ~33% overexpression in DCIS and invasive carcinoma | Independent prognostic factor; HR 10.05 for DFS, HR 1.82 for OS | [49] |
| Cervical cancer | PARPi-FL fluorescence imaging | Overexpressed in cancer vs. normal tissue | Potential for tumor delineation during colposcopy | [48] |
Technical Considerations for Different Disease Models
The accurate interpretation of PARP-1 IHC across disease models requires attention to model-specific considerations:
Ischemia-Reperfusion Models:
- In oxygen/glucose deprivation (OGD) models, PARP-1 cleavage fragments regulate cellular viability and inflammatory responses differentially [39]
- The 89-kDa fragment exhibits cytotoxic effects while the 24-kDa fragment is protective
- Uncleavable PARP-1 (PARP-1UNCL) mutants provide protection from ischemic damage
Cancer Models:
- PARP-1 overexpression reflects genomic instability and increased DNA repair activity
- Cleavage fragments indicate apoptotic response to genotoxic therapies
- PARP-1 expression in triple-negative breast cancer associates with aggressive phenotype [49]
Neurodegenerative Disease Models:
- PARP-1 activation occurs early in neurodegenerative processes
- Different cleavage fragments may activate distinct cell death pathways (apoptosis vs. parthanatos)
- PARP inhibition shows therapeutic potential across multiple neurodegenerative conditions [45]
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for PARP-1 Immunohistochemical Detection
| Reagent Category | Specific Product/Method | Application Purpose | Technical Notes |
|---|---|---|---|
| Primary Antibodies | 13371-1-AP (Proteintech) | Detection of full-length and 89-kDa cleaved PARP-1 | C-terminal epitope (667-1014 aa); works in WB, IHC, IF |
| Primary Antibodies | ab6079 (Abcam) | PARP-1 protein expression scanning | Used at 1:500 dilution; validated in ovarian cancer |
| Primary Antibodies | 46D11 (Cell Signaling) | PARP-1 immunohistochemistry | 0.4 μg/mL concentration; binds human and mouse PARP1 |
| Detection System | Bond Refine-HRP (Leica) | Signal amplification | Compatible with automated stainers |
| Detection System | DAB detection kit (Ventana) | Chromogenic development | Standard IHC visualization |
| Antigen Retrieval | TE buffer (pH 9.0) | Epitope unmasking | 10-minute retrieval recommended for 13371-1-AP |
| Antigen Retrieval | Citrate buffer (pH 6.0) | Alternative epitope retrieval | Can be used as alternative to TE buffer |
| Positive Controls | Breast cancer tissue | Assay validation | Known PARP-1 overexpression |
| Positive Controls | Ovarian cancer tissue | Assay validation | High PARP-1 expression common |
Data Interpretation and Analytical Considerations
Quantification and Scoring Methods
Standardized scoring systems are essential for reproducible PARP-1 IHC interpretation:
Intensity-Based Scoring:
- 0: No detectable staining
- 1+: Weak nuclear staining
- 2+: Moderate nuclear positivity
- 3+: Strong, uniform nuclear staining [47]
Distribution Assessment:
- Percentage of positive tumor cells
- Heterogeneity of staining within tissue architecture
- Subcellular localization (nuclear vs. cytoplasmic)
For the 89-kDa cleavage fragment, cytoplasmic localization may be observed in addition to nuclear staining, reflecting its translocation during apoptosis [12] [23]. This spatial information provides valuable insights into the activation status of cell death pathways.
Troubleshooting Common Technical Issues
Poor Signal Intensity:
- Optimize antigen retrieval duration and pH
- Validate antibody dilution series
- Check reagent expiration and storage conditions
High Background Staining:
- Titrate primary antibody concentration
- Optimize blocking conditions (serum, BSA, or commercial blockers)
- Increase wash stringency between steps
Inconsistent Cleavage Fragment Detection:
- Validate with positive control tissues known to undergoing apoptosis
- Correlate with Western blot analysis of tissue lysates
- Ensure proper tissue preservation and fixation timing
Immunohistochemical detection of PARP-1 and its cleavage fragments provides powerful insights into cell death pathways across diverse disease models. The 24-kDa and 89-kDa fragments generated during apoptosis serve not merely as passive biomarkers but as active participants in cell death execution through mechanisms including DNA repair inhibition, cytoplasmic signaling, and activation of parthanatos. The optimized protocols and analytical frameworks presented herein enable researchers to reliably detect and quantify these molecular events, advancing our understanding of PARP-1's functional roles in neurodegeneration, cancer, and other pathological conditions. As research progresses, the integration of PARP-1 cleavage detection with spatial transcriptomics and single-cell analysis approaches will further elucidate the complex relationships between DNA damage, cell death execution, and tissue pathophysiology.
Live-Cell Imaging and FRET-Based Cleavage Reporters
The detection of programmed cell death (PCD) is fundamental to cancer research, neurobiology, and therapeutic development. Among the most reliable hallmarks of apoptosis is the caspase-mediated cleavage of specific cellular substrates, with poly(ADP-ribose) polymerase-1 (PARP-1) standing as a quintessential marker. This technical guide details the implementation of Fluorescence Resonance Energy Transfer (FRET)-based cleavage reporters for real-time monitoring of PARP-1 proteolysis in live cells. We explore the molecular design principles of these biosensors, provide validated experimental protocols, and contextualize their application within mechanistic studies of PCD. By enabling quantitative, single-cell analysis of caspase-3/7 activity—the primary executioner proteases that cleave PARP-1—these reporters provide unparalleled insights into the dynamics of cell death, facilitating drug discovery and the functional characterization of cell death pathways.
Programmed cell death (PCD), particularly apoptosis, is a tightly regulated process essential for development, tissue homeostasis, and the elimination of damaged cells [50]. The evasion of apoptosis is a recognized hallmark of cancer, making the induction of cell death a desired endpoint for many anticancer therapies [51]. A critical biochemical event in the execution phase of apoptosis is the activation of a cascade of cysteine-aspartate proteases, or caspases [50] [1]. Caspase-3, a key executioner caspase, is activated during the early stages of both the intrinsic and extrinsic apoptotic pathways and is responsible for the proteolytic cleavage of numerous cellular proteins, the inactivation of which leads to the morphological hallmarks of apoptosis [51].
Among the primary and most well-characterized substrates of caspase-3 is PARP-1, a nuclear enzyme involved in DNA repair and other nuclear processes [1]. During apoptosis, caspase-3 cleaves PARP-1 at a specific aspartic acid residue within the DEVD(_{214}) amino acid sequence, separating its N-terminal DNA-binding domain (DBD) from its C-terminal catalytic domain [39] [1]. This cleavage event produces two signature fragments: a 24-kD DBD fragment and an 89-kD catalytic fragment [1]. The 24-kD fragment retains the ability to bind tightly to DNA strand breaks, where it acts as a trans-dominant inhibitor of DNA repair, thereby conserving cellular ATP and facilitating the apoptotic process [1]. The appearance of these cleavage fragments is widely accepted as a biochemical hallmark of apoptosis, distinguishing it from other forms of cell death like necroptosis or pyroptosis [50] [39].
The Principle of FRET-Based Cleavage Reporters
Fluorescence Resonance Energy Transfer (FRET) is a distance-dependent physical process where energy is transferred from an excited donor fluorophore to a nearby acceptor fluorophore. This principle can be harnessed to create highly sensitive molecular biosensors for protease activity.
Molecular Design of PARP-1-Inspired Reporters
FRET-based caspase reporters are typically engineered by fusing two compatible fluorescent proteins (e.g., CFP/YFP or GFP/mCherry) via a flexible peptide linker that contains the specific caspase cleavage sequence DEVD [51] [52]. In the intact, uncleaved reporter, the two fluorophores are in close proximity, enabling efficient FRET. Upon induction of apoptosis and activation of caspase-3, the reporter is cleaved within the DEVD sequence. This cleavage leads to the physical separation of the donor and acceptor fluorophores, causing a loss of FRET efficiency that can be quantified as a change in the emission ratio (Table 1).
Table 1: Quantitative Changes in FRET-Based Caspase Reporter Upon Cleavage
| Parameter | Uncleaved Reporter (High FRET) | Cleaved Reporter (Low FRET) | Measurement Method |
|---|---|---|---|
| Emission Ratio (Acceptor/Donor) | High | Low | Fluorescence microscopy or plate reader |
| Donor Emission Intensity | Lower | Higher | Fluorescence microscopy or plate reader |
| Acceptor Emission Intensity | Higher | Lower | Fluorescence microscopy or plate reader |
| FRET Efficiency | High (e.g., 20-30%) | Low (e.g., <5%) | Acceptor photobleaching or lifetime imaging (FLIM) |
This design mirrors the natural cleavage of PARP-1, where the separation of functional domains abrogates its normal activity. The reporter thus serves as a biomimetic synthetic substrate that provides a quantifiable, real-time signal proportional to caspase-3 activity within single, live cells [52].
Signaling Pathway and Reporter Activation
The following diagram illustrates the caspase-3 activation pathway during apoptosis and the subsequent mechanism of the FRET-based reporter cleavage.
Experimental Protocols for FRET Reporter Assays
This section provides detailed methodologies for implementing FRET-based cleavage reporters, from initial cellular transduction to final data analysis.
Reporter Construct Generation and Cell Line Engineering
The first step is to create a stable cell line that reliably expresses the FRET-based caspase reporter.
Materials:
- Reporter Plasmid: pBABEpuro-based retroviral vector containing the FRET construct (e.g., eGFP-DEVD-mCherry) [51].
- Cell Lines: Adherent cells such as human SH-SY5Y neuroblastoma cells, murine 4T1 mammary carcinoma cells, or RPE1hTERT immortalized retinal epithelial cells [39] [51] [53].
- Culture Reagents: Standard Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin, and streptomycin.
- Transfection/Transduction Reagents: Lipofectamine 2000 for transient transfection; polybrene for retroviral transduction [51].
- Selection Antibiotic: Puromycin for selecting stably transduced cells.
Procedure:
- Plasmid Preparation: Propagate and purify the reporter plasmid using a standard maxi-prep kit.
- Transient Transfection (for initial validation):
- Seed cells at 50-70% confluency in a 12-well plate.
- The following day, transfect with 1-2 µg of plasmid DNA using Lipofectamine 2000 according to the manufacturer's protocol.
- Incubate for 24-48 hours before imaging.
- Generation of Stable Cell Lines (for high-throughput work):
- Use a retroviral transduction system. Transfect Phoenix eco packaging cells with the retroviral reporter construct.
- 24-48 hours post-transfection, collect the viral supernatant, filter it through a 0.45 µm membrane, and add it to target cells in the presence of 4 µg/ml polybrene.
- Repeat the transduction 2-3 times for higher efficiency.
- 48 hours after the final transduction, begin selection with 1 µg/ml puromycin.
- Maintain selection pressure for at least one week, then use fluorescence-activated cell sorting (FACS) to isolate a population of cells with high and uniform fluorescence [51].
Live-Cell Imaging and Apoptosis Induction
Once a stable reporter cell line is established, it can be used to monitor apoptosis in real-time.
Materials:
- Imaging Equipment: Confocal microscope or high-content imaging system with environmental control (37°C, 5% CO₂).
- Fluorescence Filter Sets: Filters appropriate for the donor (e.g., eGFP: Ex 488nm, Em 500-550nm) and acceptor (e.g., mCherry: Ex 560nm, Em 570-620nm).
- Apoptosis Inducers: Staurosporine (1 µM), Etoposide (VP-16, 50-100 µM), or other cytotoxic agents relevant to the research context [1] [51].
- Caspase Inhibitor Control: Z-VAD-FMK (20-50 µM) to confirm caspase-specific cleavage.
Procedure:
- Plate cells: Seed stably expressing cells onto collagen-coated glass-bottom dishes or 96-well imaging plates at a density of 1x10⁴ cells per well.
- Acquire baseline images: Place the plate on the pre-warmed microscope stage and acquire baseline FRET images for both donor and acceptor channels.
- Induce apoptosis: Add the apoptotic stimulus directly to the medium. Include control wells with vehicle (e.g., DMSO) and wells pre-treated with Z-VAD-FMK for one hour.
- Time-lapse imaging: Collect images at regular intervals (e.g., every 30-60 minutes) over a period of 6-24 hours to capture the dynamics of caspase activation.
Data Analysis and FRET Quantification
The raw imaging data must be processed to quantify the loss of FRET, which serves as a metric for caspase-3 activity.
Key Calculations:
- FRET Ratio (Sensitized Emission): The most common method for quantification involves calculating the background-subtracted acceptor-to-donor emission intensity ratio.
( \text{FRET Ratio} = \frac{\text{Intensity}{\text{Acceptor Channel}}}{\text{Intensity}{\text{Donor Channel}}} )
- A decrease in this ratio over time indicates reporter cleavage and ongoing apoptosis (Table 1).
- % Apoptotic Cells: A cell can be scored as apoptotic once its FRET ratio drops below a predefined threshold (e.g., 50% of its baseline value) [51]. The percentage of apoptotic cells in a population can then be determined and compared across different treatment conditions.
Table 2: Key Research Reagent Solutions for FRET-Based Apoptosis Detection
| Reagent / Material | Function / Role | Example / Specification |
|---|---|---|
| FRET Reporter Construct | Genetically encoded caspase sensor | pBABEpuro vector with eGFP-DEVD-mCherry cassette [51] |
| Caspase-3/7 Substrate | Core cleavage site within reporter | Amino acid sequence: DEVDG [51] |
| Fluorescent Protein Pair | FRET donor and acceptor | eGFP (Donor) and mCherry (Acceptor) [51] |
| Apoptosis Inducer | Positive control for caspase activation | Staurosporine (1 µM) or Etoposide (50-100 µM) [1] |
| Pan-Caspase Inhibitor | Negative control to confirm specificity | Z-VAD-FMK (20-50 µM) |
| Retroviral System | For stable cell line generation | Phoenix eco packaging cells + Polybrene (4 µg/ml) [51] |
| Selection Antibiotic | Maintains reporter expression in population | Puromycin (1 µg/ml) [51] |
Advanced Applications and Technical Considerations
Integration with PARP-1 Specific Research
FRET reporters provide a dynamic readout of general caspase activity, which can be correlated with direct endpoints of PARP-1 cleavage. To validate the reporter's readout in the context of PARP-1 biology, researchers can employ complementary techniques:
- Western Blotting: Confirming the appearance of the signature PARP-1 cleavage fragments (89 kDa and 24 kDa) in cell lysates from the same experiment [1].
- Inhibitor Studies: Using PARP inhibitors (e.g., Olaparib) can help dissect the relationship between PARP-1's role in DNA damage response and the initiation of apoptosis [53]. For instance, cells with deficiencies in DNA repair pathways like CHD6 show heightened sensitivity to PARP-1/2 trapping inhibitors, leading to increased apoptosis that can be sensitively detected by the FRET reporter [53].
Troubleshooting and Optimization
- Low FRET Efficiency: Ensure the linker between fluorophores is not too rigid or long. Verify the health and expression level of the fluorescent proteins.
- High Background Signal: Optimize cell density and imaging parameters. Ensure thorough washing after any reagent additions.
- Variable Response: Use low-passage cells and maintain consistent culture conditions. A FACS-sorted homogeneous population is preferable for quantitative studies.
FRET-based cleavage reporters represent a powerful methodology for the real-time, quantitative analysis of caspase activity in live cells. By modeling the central apoptotic event of PARP-1 cleavage, these biosensors bridge the gap between traditional biochemical endpoint assays and dynamic single-cell biology. The protocols and considerations outlined in this guide provide a robust framework for implementing this technology in diverse research settings, from basic investigations into the mechanisms of PCD to high-throughput drug discovery campaigns aimed at inducing or inhibiting cell death. The ability to track the fate of individual cells within a heterogeneous population makes these reporters an indispensable tool for the modern cell death researcher.
PARP-1 Cleavage as a Biomarker in Preclinical Drug Screening
Poly (ADP-ribose) polymerase-1 (PARP-1) is a nuclear enzyme with well-established roles in DNA damage repair and maintenance of genome integrity. Beyond its DNA repair functions, PARP-1 participates in diverse physiological and pathological processes, including multiple forms of programmed cell death [1]. The cleavage of PARP-1 by various cell death proteases generates specific, stable fragments that serve as recognizable biomarkers for particular protease activities and cell death pathways [1]. During apoptosis, PARP-1 is cleaved by caspases, while in other cell death pathways, it is processed by calpains, cathepsins, granzymes, and matrix metalloproteinases (MMPs), each generating distinctive signature fragments [1]. This article explores the molecular mechanisms of PARP-1 cleavage during programmed cell death and its application as a specific, mechanistically informative biomarker in preclinical drug screening.
Molecular Mechanisms of PARP-1 Cleavage
Domain Architecture and Cleavage Sites
PARP-1 is a modular protein comprising several functional domains: a DNA-binding domain (DBD) containing two zinc finger motifs at the N-terminus, an auto-modification domain (AMD) in the central region, and a catalytic domain (CD) at the C-terminus [1]. The caspase cleavage site DEVD214 is situated within the DBD and is part of the nuclear localization signal (NLS) [39]. Cleavage at this site by effector caspases separates the DNA-binding domain from the catalytic domain [39].
Proteases and Their Characteristic Cleavage Fragments
Different proteases cleave PARP-1 at specific sites, generating signature fragments that serve as biomarkers for specific cell death pathways:
Table 1: PARP-1 Cleavage Fragments by Different Proteases
| Protease | Cleavage Sites | Characteristic Fragments | Associated Cell Death Pathway |
|---|---|---|---|
| Caspase-3/7 | DEVD214↓G [39] [1] | 24 kDa (DBD) + 89 kDa (AMD+CD) [54] [1] | Apoptosis [1] |
| Calpain | Unknown | 55 kDa + 62 kDa (alternative fragments) [1] | Ca²⁺-mediated necrosis [1] |
| Granzyme A | Unknown | 50 kDa + 66 kDa (alternative fragments) [1] | Lymphocyte-mediated cytotoxicity [1] |
| MMPs | Unknown | 35-42 kDa (alternative fragments) [1] | Inflammation-associated cell death [1] |
Functional Consequences of Cleavage
Caspase-mediated cleavage of PARP-1 produces two primary fragments with distinct functions:
The 24-kDa fragment contains the DNA-binding domain and nuclear localization signal. This fragment remains tightly bound to DNA strand breaks, acting as a trans-dominant inhibitor of DNA repair by blocking access of intact PARP-1 and other repair enzymes to DNA damage sites [54] [1].
The 89-kDa fragment contains the auto-modification and catalytic domains. During caspase-dependent apoptosis, this fragment can be translocated to the cytoplasm where it may function as a carrier of poly(ADP-ribose) (PAR) polymers [54]. PAR polymers attached to the 89-kDa fragment can bind apoptosis-inducing factor (AIF) in the cytoplasm, facilitating AIF release from mitochondria and its translocation to the nucleus, resulting in large-scale DNA fragmentation [54].
The functional significance of this cleavage is context-dependent. In some scenarios, cleavage conserves cellular energy by inactivating PARP-1's NAD+-consuming catalytic activity [39]. However, emerging evidence suggests the fragments themselves may actively regulate cell death and inflammatory responses [39].
Detection Methods and Experimental Protocols
Western Blot Analysis for PARP-1 Cleavage
Protocol Overview:
- Cell Treatment and Lysis: Treat cells with experimental compounds (e.g., 1-10 μM staurosporine for 6h as positive control for apoptosis) [54]. Harvest cells and lyse in RIPA buffer supplemented with protease inhibitors.
- Protein Quantification and Separation: Determine protein concentration using BCA assay. Load 20-30 μg protein per lane and separate by SDS-PAGE (8-12% gradient gels recommended) [54].
- Membrane Transfer and Blocking: Transfer to PVDF membrane, block with 5% non-fat milk in TBST for 1 hour.
- Antibody Incubation: Incubate with primary antibodies (anti-PARP-1 that detects both full-length and cleavage fragments) overnight at 4°C [54]. Recommended dilutions: 1:1000 in TBST with 1% BSA.
- Detection: Use HRP-conjugated secondary antibodies (1:2000-1:5000) and chemiluminescent substrate. Image using digital imaging system.
Expected Results: Apoptotic samples show simultaneous decrease in full-length PARP-1 (116 kDa) and appearance of 89 kDa fragment [54]. The 24 kDa fragment may be less consistently detected due to its tight binding to chromatin [1].
Immunofluorescence and Cellular Localization
Protocol Overview:
- Cell Culture and Treatment: Culture cells on glass coverslips. Treat with experimental compounds.
- Fixation and Permeabilization: Fix with 4% paraformaldehyde for 15 minutes, permeabilize with 0.1% Triton X-100 for 10 minutes.
- Antibody Staining: Incubate with primary antibodies against PARP-1 (to detect fragments) and AIF (to monitor parthanatos) [54]. Use species-appropriate fluorescent secondary antibodies.
- Counterstaining and Imaging: Counterstain with DAPI for nuclei. Image using confocal microscopy.
Expected Results: During caspase-mediated apoptosis, the 89-kDa PARP-1 fragment translocates to the cytoplasm [54]. In parthanatos, AIF translocates from mitochondria to the nucleus, leading to nuclear shrinkage [54].
Pharmacological Inhibition Controls
Include specific inhibitors in experimental designs:
- Caspase inhibitors: zVAD-fmk (20-50 μM) to inhibit caspase-dependent cleavage [54]
- PARP inhibitors: PJ34 or ABT-888 (1-10 μM) to inhibit PARP catalytic activity [54]
- Calpain inhibitors: MDL-28170 (10-50 μM) to inhibit calpain-mediated cleavage
Validation: PARP inhibition should prevent PAR synthesis and AIF-mediated nuclear shrinkage in parthanatos models [54].
Quantitative Analysis of PARP-1 Cleavage in Drug Screening
Benchmarking Cleavage Patterns Against Reference Compounds
Table 2: PARP-1 Cleavage Response to Reference Compounds
| Compound/Stimulus | PARP-1 Cleavage Pattern | Cell Death Pathway | Time Course | Key Accompanying Markers |
|---|---|---|---|---|
| Staurosporine (1-2 μM) [54] | 89 kDa fragment appearance [54] | Caspase-dependent apoptosis with parthanatos features [54] | PAR detection: 1-4h; AIF translocation: 4-6h [54] | Caspase-3 activation, PAR accumulation, AIF nuclear translocation [54] |
| NMDA excitotoxicity [1] | 89 kDa fragment appearance [1] | Excitotoxicity with parthanatos | Variable (hours) | AIF release, large-scale DNA fragmentation [1] |
| Actinomycin D [54] | 89 kDa fragment appearance [54] | Caspase-dependent apoptosis | 6-24 hours | Caspase-3 activation, PARP-1 fragmentation [54] |
| Oxygen/Glucose Deprivation (OGD) [39] | 89 kDa fragment appearance [39] | Ischemia-reperfusion injury | 6h OGD + 15h ROG [39] | NF-κB activation, iNOS and COX-2 expression [39] |
Quantification Approaches for High-Throughput Screening
- Densitometric Analysis: Quantify band intensities from Western blots. Calculate cleavage index as (89 kDa fragment) / (full-length + 89 kDa fragment) × 100%.
- High-Content Imaging: Use automated microscopy to quantify fragment localization and AIF translocation in multi-well formats.
- Plate-Based Assays: Develop ELISA-based methods for quantifying specific fragments in cell lysates.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for PARP-1 Cleavage Studies
| Reagent/Category | Specific Examples | Function/Application | Considerations |
|---|---|---|---|
| PARP-1 Antibodies | Anti-PARP-1 (full length and fragment specific) | Detection of full-length and cleavage fragments by Western blot, IF | Validate for specific fragments (24 kDa, 89 kDa) [54] [1] |
| Caspase Inhibitors | zVAD-fmk (pan-caspase inhibitor) [54] | Inhibition of caspase-mediated PARP-1 cleavage | Use 20-50 μM for effective inhibition [54] |
| PARP Inhibitors | PJ34, ABT-888 (veliparib) [54] | Inhibition of PARP catalytic activity | Confirm specificity for PARP-1 vs. other PARP family members [54] |
| Apoptosis Inducers | Staurosporine, Actinomycin D [54] | Positive controls for caspase-mediated PARP-1 cleavage | Titrate concentration for optimal cleavage detection [54] |
| Cell Death Assays | Propidium iodide, Annexin V, LDH release | Correlation of PARP-1 cleavage with cell death | Combine with PARP-1 cleavage detection for mechanism [54] |
| AIF Antibodies | Anti-AIF [54] | Detection of AIF translocation in parthanatos | Monitor mitochondrial vs. nuclear localization [54] |
PARP-1 Cleavage in Different Cell Death Contexts
Apoptosis versus Parthanatos
PARP-1 cleavage fragments play distinct roles in different cell death pathways:
- In caspase-dependent apoptosis, PARP-1 cleavage primarily inactivates DNA repair function and conserves cellular energy, facilitating apoptotic progression [1].
- In parthanatos, PARP-1 overactivation leads to PAR polymer formation and AIF release, with cleavage occurring downstream or as a parallel event [54].
- The 89-kDa fragment generated during caspase-mediated apoptosis can serve as a PAR carrier to the cytoplasm, where it facilitates AIF release from mitochondria, creating a bridge between apoptotic and parthanatos pathways [54].
Functional Studies with PARP-1 Mutants
Studies expressing specific PARP-1 fragments reveal their functional significance:
- Expression of PARP-1UNCL (uncleavable mutant) or the 24-kDa fragment protects from oxygen/glucose deprivation damage in neuronal models [39].
- Expression of the 89-kDa fragment is cytotoxic and increases NF-κB activity and expression of pro-inflammatory proteins (iNOS, COX-2) while decreasing anti-apoptotic Bcl-xL [39].
- These findings suggest PARP-1 cleavage products actively regulate cellular viability and inflammatory responses beyond merely marking protease activation [39].
Application in Preclinical Drug Screening
Mechanism of Action Profiling
PARP-1 cleavage analysis provides critical information for drug mechanism characterization:
- Cytotoxic anticancer agents: PARP-1 cleavage pattern indicates whether cell death occurs through apoptosis (classic 89 kDa fragment) or alternative pathways.
- Neuroprotective agents: Reduction of PARP-1 cleavage in models of cerebral ischemia, excitotoxicity, or neurodegenerative diseases indicates therapeutic potential [1].
- Inflammation modulators: PARP-1 cleavage fragments regulate NF-κB activity and inflammatory gene expression, making them relevant biomarkers for anti-inflammatory drug screening [39].
Predictive Biomarker Development
Specific PARP-1 cleavage patterns may predict therapeutic responses:
- Tumors with specific DNA repair deficiencies (e.g., BRCA mutations) may show altered PARP-1 cleavage in response to PARP inhibitors [55] [56].
- In prostate cancer, PARP1 loss and PARP2 gain are associated with aggressive disease and may influence PARP-1 processing and function [57] [58].
- PARP-1 cleavage signatures can guide patient stratification for targeted therapies, particularly in determining susceptibility to PARP inhibitor treatments [55] [56].
PARP-1 cleavage represents a sophisticated biomarker system that provides mechanistically rich information in preclinical drug screening. The specific cleavage fragments generated reveal not only that cell death is occurring but also the specific protease pathways activated and the functional consequences for cellular survival and inflammation. As drug discovery increasingly focuses on targeted therapies with specific mechanisms of action, PARP-1 cleavage analysis offers a window into compound mechanism that extends beyond simple cytotoxicity assessment. Integration of PARP-1 cleavage profiling into standardized preclinical screening workflows will enhance mechanistic understanding of drug candidates and support the development of more targeted, effective therapeutic strategies.
The cleavage of poly(ADP-ribose) polymerase-1 (PARP-1) during programmed cell death is a critical event that transcends its traditional role as a mere apoptotic biomarker. This in-depth technical guide explores how PARP-1 cleavage fragments and their specific signatures serve as predictive indicators for chemotherapeutic response. We examine the molecular mechanisms through which distinct cleavage products regulate cell fate decisions—including apoptosis, necrosis, and parthanatos—and how these pathways influence cancer treatment outcomes. The document provides a comprehensive framework for researchers and drug development professionals to quantify and interpret PARP-1 cleavage patterns, along with detailed experimental protocols for assessing chemosensitivity in preclinical models. By integrating current understanding of PARP-1 biology with emerging clinical data, this review establishes a foundation for developing PARP-1-based biomarkers to personalize cancer therapy.
Poly(ADP-ribose) polymerase-1 (PARP-1) is a nuclear enzyme with pivotal functions in DNA damage repair, transcriptional regulation, and cell death signaling. As a DNA damage sensor, PARP-1 binds to DNA single-strand and double-strand breaks through its zinc finger motifs, activating its catalytic domain to transfer ADP-ribose units from NAD+ to target proteins, including itself—a process known as PARylation [59] [11]. This post-translational modification facilitates the recruitment of DNA repair machinery and plays a crucial role in maintaining genomic integrity. However, beyond its DNA repair functions, PARP-1 participates in multiple cell death pathways, making it a critical determinant of cellular response to genotoxic stress, including chemotherapy.
In the context of cancer therapeutics, PARP-1's role is multifaceted. While PARP-1 activation initially promotes DNA repair and cell survival, excessive DNA damage triggers specific cleavage patterns that commit cells to different death pathways [11] [14]. The development of PARP inhibitors (PARPis) represents a significant advancement in targeted cancer therapy, particularly for BRCA-mutated cancers where synthetic lethality approaches have shown clinical success [60] [61]. However, predicting response to these therapies remains challenging, necessitating a deeper understanding of PARP-1's cleavage mechanisms and their functional consequences.
PARP-1 Cleavage as a Mechanism in Programmed Cell Death
Molecular Architecture and Cleavage Sites
PARP-1 is a modular protein comprising several functional domains: a DNA-binding domain (DBD) containing three zinc finger motifs, an automodification domain (AMD), and a C-terminal catalytic domain (CAT) [11] [14]. The DBD recognizes DNA damage, while the CAT domain catalyzes PAR synthesis using NAD+ as a substrate. Between these domains lies a nuclear localization signal (NLS) and the aspartate-glutamate-valine-aspartic acid (DEVD) motif, which serves as the primary cleavage site for caspases during apoptosis [11] [39].
Table 1: PARP-1 Domains and Their Functions
| Domain | Structural Features | Function | Cleavage Proteases |
|---|---|---|---|
| DNA-Binding Domain (DBD) | Three zinc finger motifs (Zn1, Zn2, Zn3), Nuclear Localization Signal (NLS) | Recognizes and binds to DNA strand breaks | Caspases-3/7 (at DEVD214), Lysosomal proteases |
| Automodification Domain (AMD) | BRCT fold, glutamate and serine residues for PAR attachment | Target for auto-PARylation; mediates protein-protein interactions | Calpains, Cathepsins |
| Catalytic Domain (CAT) | WGR domain, HD and ART subdomains, NAD+ binding site | Catalyzes poly(ADP-ribose) formation from NAD+ | Caspase-independent proteases |
During programmed cell death, PARP-1 undergoes proteolytic cleavage at specific sites, generating fragments with distinct biological activities. The best-characterized cleavage occurs at DEVD214 within the DBD, executed by activated caspases-3 and -7 during apoptosis [11] [39]. This cleavage produces a 24-kDa N-terminal fragment containing the DBD and a 89-kDa C-terminal fragment comprising the AMD and CAT domains. In contrast, necrosis induces a different cleavage pattern, generating a prominent 50-kDa fragment through lysosomal proteases such as cathepsins B and G [41].
Cell Death Pathways and PARP-1 Cleavage Signatures
The fate of cells exposed to genotoxic stress is determined by the intensity and duration of DNA damage, which directly influences PARP-1 activation and cleavage patterns. Three primary cell death pathways involve PARP-1:
Apoptosis: Moderate DNA damage activates caspases, which cleave PARP-1 at DEVD214, generating 24-kDa and 89-kDa fragments. The 24-kDa fragment retains DNA-binding capacity but lacks catalytic activity, potentially acting as a trans-dominant inhibitor of intact PARP-1 [11] [39]. This cleavage conserves cellular ATP by preventing excessive PARP-1 activation and NAD+ depletion, facilitating orderly apoptotic execution.
Necrosis: Severe oxidative stress or DNA damage triggers PARP-1 overactivation, leading to catastrophic NAD+ and ATP depletion. Under these conditions, lysosomal proteases (cathepsins) cleave PARP-1, generating a characteristic 50-kDa fragment and other cleavage products distinct from apoptotic fragments [41].
Parthanatos: This caspase-independent programmed cell death pathway involves massive PARP-1 activation, PAR polymer synthesis, and translocation of PAR polymers and PARP-1 fragments to the cytoplasm. Here, they interact with mitochondrial proteins such as apoptosis-inducing factor (AIF), triggering chromatin condensation and large-scale DNA fragmentation [59] [13].
Table 2: PARP-1 Cleavage Fragments in Cell Death Pathways
| Cell Death Pathway | Cleavage Fragments | Proteases Involved | Functional Consequences |
|---|---|---|---|
| Apoptosis | 24-kDa (DBD) and 89-kDa (AMD+CAT) | Caspases-3/7 | Inactivation of DNA repair, conservation of cellular energy |
| Necrosis | 50-kDa and other fragments | Cathepsins B, D, G (lysosomal) | Cellular energy depletion, loss of membrane integrity |
| Parthanatos | PAR polymers, 89-kDa fragment | PARP-1 overactivation | AIF translocation, large-scale DNA fragmentation |
The 89-kDa fragment generated during apoptosis has recently been shown to translocate to the cytoplasm, where it can function as a carrier for PAR polymers, facilitating AIF release from mitochondria and potentially bridging apoptotic and parthanatos pathways [13]. This finding reveals unexpected complexity in PARP-1's role in cell fate decisions.
PARP-1 Cleavage in Predicting Chemotherapeutic Response
PARP-1 Cleavage Patterns as Biomarkers
The detection of specific PARP-1 cleavage fragments provides valuable insights into the mode and extent of cell death induced by chemotherapeutic agents. Apoptotic cleavage (24-kDa and 89-kDa fragments) typically indicates a controlled response to moderate DNA damage, while necrotic cleavage (50-kDa fragment) or parthanatos-associated PAR accumulation suggests more severe cellular injury [11] [41]. These patterns can serve as biomarkers to predict therapeutic response and patient outcomes.
In clinical contexts, the presence of apoptotic PARP-1 fragments in tumor samples following initial chemotherapy may indicate successful activation of cell death pathways and predict favorable treatment response. Conversely, the persistence of full-length PARP-1 or the emergence of necrotic fragments might signify resistance or excessive toxicity, respectively [59] [61]. Furthermore, the ratio of PARP-1 fragments to full-length protein could provide a quantitative measure of therapeutic efficacy.
PARP Inhibitors and Synthetic Lethality
PARP inhibitors (PARPis) such as olaparib, talazoparib, and veliparib exploit the concept of synthetic lethality in DNA repair-deficient cancers, particularly those with BRCA1/2 mutations [60] [61]. These inhibitors trap PARP-1 on DNA, preventing its dissociation and generating persistent DNA lesions that require homologous recombination for repair. In BRCA-deficient cells lacking functional homologous recombination, PARPi-induced DNA damage proves lethal.
Recent evidence suggests that PARP1 cleavage patterns may influence response to PARP inhibitors. A single nucleotide polymorphism (rs1805414) in PARP1, while synonymous, affects PARP1 mRNA secondary structure, expression levels, and potentially protein function [60]. Patients with the GCC variant (SNP) exhibit lower PARP1 expression and may respond differently to PARPi therapy than those with the GCT variant (WT). This finding highlights the potential of PARP1 genotyping as a companion diagnostic for PARPi therapy.
PARP-1 in Differential Chemosensitivity
The cellular context of PARP-1 expression and activation significantly influences chemosensitivity. For example:
- HER2-positive breast cancers demonstrate sensitivity to PARP inhibition independent of BRCA status, potentially through attenuation of NF-κB signaling [59].
- Acute lymphoblastic leukemia (ALL) cells show enhanced response to antibody-drug conjugates (e.g., Inotuzumab ozogamicin) when combined with PARP inhibitors like talazoparib, while acute myeloid leukemia (AML) cells exhibit more heterogeneous responses [61].
- PARP-1 cleavage fragments differentially regulate NF-κB activity—the 89-kDa fragment enhances pro-inflammatory signaling, while the 24-kDa fragment and uncleavable PARP-1 exert protective effects in neuronal models of ischemia [39].
These context-dependent effects underscore the importance of understanding PARP-1 biology in specific cancer types to optimize therapeutic strategies.
Experimental Protocols for Assessing PARP-1-Mediated Chemotherapeutic Response
Detection and Quantification of PARP-1 Cleavage
Protocol 1: Western Blot Analysis of PARP-1 Cleavage Fragments
Materials:
- Cell lysates from treated and untreated cells
- PARP-1 antibodies: N-terminal specific (for 24-kDa fragment), C-terminal specific (for 89-kDa fragment), and full-length specific
- SDS-PAGE gel (4-20% gradient recommended)
- Enhanced chemiluminescence detection system
Procedure:
- Treat cells with chemotherapeutic agents (e.g., 50 μM etoposide for 24 hours) or PARP inhibitors at appropriate concentrations.
- Harvest cells and prepare lysates using RIPA buffer supplemented with protease inhibitors.
- Resolve 20-50 μg of protein lysate by SDS-PAGE and transfer to PVDF membrane.
- Incubate with PARP-1 antibodies (1:1000 dilution) overnight at 4°C.
- Detect bound antibodies using HRP-conjugated secondary antibodies and ECL reagent.
- Quantify band intensities using densitometry software; calculate ratios of cleavage fragments to full-length PARP-1.
Interpretation: Apoptotic induction is confirmed by detection of 89-kDa and 24-kDa fragments. Necrotic cleavage may be indicated by a 50-kDa fragment. The timing and extent of cleavage provide insights into the dominant cell death pathway activated.
Protocol 2: Immunofluorescence Staining for Subcellular Localization of PARP-1 Fragments
Materials:
- Cells grown on chamber slides
- Paraformaldehyde (4%) for fixation
- Triton X-100 (0.1%) for permeabilization
- PARP-1 antibodies with fluorescently-labeled secondary antibodies
- DAPI for nuclear counterstaining
- Confocal microscope
Procedure:
- Culture cells on chamber slides and treat with test compounds.
- Fix cells with 4% PFA for 15 minutes, permeabilize with 0.1% Triton X-100 for 10 minutes.
- Block with 5% BSA for 1 hour, then incubate with PARP-1 antibodies overnight at 4°C.
- Incubate with fluorescent secondary antibodies (1:500) for 1 hour at room temperature.
- Counterstain with DAPI, mount slides, and image using confocal microscopy.
Interpretation: Cytoplasmic translocation of the 89-kDa fragment or PAR polymers indicates parthanatos, while nuclear retention of fragments suggests apoptotic signaling.
Functional Assays for PARP-1 Activity in Cell Death
Protocol 3: NAD+ Depletion Assay for PARP-1 Overactivation
Materials:
- NAD/NADH quantification kit
- Cell culture plates (96-well format recommended)
- Microplate reader
Procedure:
- Seed cells in 96-well plates and treat with DNA-damaging agents with or without PARP inhibitors.
- At various time points (2, 4, 8, 16, 24 hours), harvest cells and extract NAD+ according to kit instructions.
- Measure NAD+ levels colorimetrically or fluorometrically.
- Normalize NAD+ values to protein content or cell number.
Interpretation: Rapid NAD+ depletion indicates PARP-1 overactivation and predicts necrotic cell death or parthanatos, while moderate NAD+ decline suggests controlled PARP-1 activity compatible with apoptosis.
Protocol 4: Clonogenic Survival Assay with PARP Inhibition
Materials:
- Six-well tissue culture plates
- Crystal violet staining solution
- PARP inhibitors (e.g., talazoparib, olaparib)
Procedure:
- Seed cells at low density (200-1000 cells/well) in six-well plates.
- After 24 hours, treat cells with chemotherapeutic agents alone, PARP inhibitors alone, or combinations.
- Incubate for 10-14 days to allow colony formation.
- Fix cells with methanol, stain with crystal violet, and count colonies (>50 cells).
- Calculate survival fractions relative to untreated controls.
Interpretation: Synergistic reduction in clonogenic survival with combination treatment indicates potential therapeutic benefit, particularly in DNA repair-deficient models.
Research Reagent Solutions
Table 3: Essential Research Reagents for PARP-1 Cleavage Studies
| Reagent/Category | Specific Examples | Function/Application | Key Considerations |
|---|---|---|---|
| PARP Inhibitors | Talazoparib, Olaparib, Veliparib | Induce synthetic lethality in HR-deficient cells; chemosensitizers | Different trapping potentials; variable effects on PARP-1 cleavage |
| Antibodies | Anti-PARP-1 (N-terminal), Anti-PARP-1 (C-terminal), Anti-PARP (cleaved) | Detect full-length and cleavage fragments; subcellular localization | Specificity for fragments vs. full-length critical for interpretation |
| Cell Lines | BRCA-mutated vs. WT; isogenic pairs | Model genetic backgrounds; test synthetic lethality | Verify mutation status and PARP-1 expression periodically |
| Apoptosis Inducers | Staurosporine, Etoposide, Actinomycin D | Positive controls for apoptotic PARP-1 cleavage | Concentration and timing optimize for specific cell types |
| Necrosis Inducers | Hydrogen peroxide, Ethanol, Mercuric chloride | Induce necrotic PARP-1 cleavage patterns | Confirm necrosis by additional markers (e.g., LDH release) |
| Activity Assays | NAD+ quantification kits, PAR ELISA kits | Measure PARP-1 enzymatic activity | Normalize to cell number; kinetic measurements recommended |
| Protease Inhibitors | zVAD-fmk (caspase inhibitor), E64d (cathepsin inhibitor) | Distinguish cleavage pathways | Use combination inhibitors to confirm protease specificity |
Signaling Pathways and Experimental Workflows
Diagram 1: PARP-1 Cleavage Pathways in Cell Death Decisions. This diagram illustrates how DNA damage intensity determines PARP-1 activation and subsequent cleavage by different proteases, leading to distinct cell death outcomes.
Diagram 2: Experimental Workflow for PARP-1 Cleavage Analysis. This workflow outlines the key steps in assessing PARP-1 cleavage patterns and their functional implications for predicting therapeutic response.
The analysis of PARP-1 cleavage provides a powerful window into cellular responses to genotoxic stress, offering predictive insights for chemotherapeutic efficacy. The distinct cleavage signatures associated with different cell death pathways serve as biomarkers that can guide treatment selection and optimization. As research continues to unravel the complex functions of PARP-1 fragments beyond their traditional roles, new opportunities emerge for therapeutic intervention.
Future directions in this field include developing standardized assays for PARP-1 cleavage fragments in clinical samples, validating the prognostic value of PARP-1 cleavage patterns in prospective trials, and exploring the therapeutic potential of modulating specific cleavage events. The integration of PARP-1 genotyping, particularly for functional SNPs like rs1805414, may further refine patient stratification for PARP inhibitor therapy. Additionally, understanding the non-canonical functions of PARP-1 fragments in inflammatory signaling and gene regulation may reveal novel targets for combination therapies.
As we deepen our understanding of PARP-1's multifaceted roles in cell death decisions, we move closer to personalized cancer treatments that leverage the predictive power of PARP-1 cleavage to maximize therapeutic efficacy while minimizing adverse effects.
Poly(ADP-ribose) polymerase-1 (PARP-1) represents a critical nuclear enzyme involved in DNA damage repair and cell fate decisions. Beyond its canonical role in base excision repair, PARP-1 has emerged as a central executioner in multiple regulated cell death pathways, particularly through its proteolytic cleavage patterns. During programmed cell death, PARP-1 undergoes specific cleavage by various proteases, generating signature fragments that serve as biomarkers for particular cell death modalities and potential therapeutic targets. This technical guide examines how distinct PARP-1 cleavage patterns correlate with neuroprotective outcomes across various neurological disease models, providing researchers with methodological frameworks and analytical approaches for investigating these mechanisms in drug development contexts.
The cleavage of PARP-1 represents a molecular switch point that determines cellular fate in response to stress signals. In neurological contexts, where irreversible neuronal loss underpins disease progression, understanding how to modulate PARP-1 cleavage fragments offers promising therapeutic avenues. This review synthesizes current evidence linking specific cleavage patterns to neuroprotection, detailing experimental methodologies for quantifying these events, and providing visual frameworks for understanding the complex signaling networks involved.
PARP-1 Cleavage Mechanisms and Signature Fragments
Protease-Specific Cleavage Patterns
PARP-1 serves as a substrate for multiple cell death proteases, each generating characteristic cleavage fragments that serve as signatures for specific cell death pathways:
Caspase-3/7 cleavage: Generates 24-kDa (DNA-binding domain) and 89-kDa (catalytic domain) fragments, considered hallmarks of apoptosis [1]. The 24-kDa fragment contains two zinc finger motifs (ZnF1-2) that remain bound to DNA breaks, acting as a trans-dominant inhibitor of DNA repair, while the 89-kDa fragment translocates to the cytoplasm [62] [1].
Calpain cleavage: Produces 55-kDa and 62-kDa fragments, associated with excitotoxicity and calcium-mediated cell death pathways relevant in stroke and traumatic brain injury [1].
Cathepsin cleavage: Generates 50-kDa and 42-kDa fragments, linked to lysosomal-mediated cell death pathways [1].
Granzyme A cleavage: Creates a 70-kDa fragment, while Granzyme B generates 64-kDa and 50-kDa fragments, important in immune-mediated neuronal damage [1].
Matrix Metalloproteinases (MMPs): Produce various fragments including 55-kDa, 42-kDa, and 36-kDa fragments, potentially relevant in neuroinflammatory contexts [1].
Structural and Functional Consequences of Cleavage
The functional consequences of PARP-1 cleavage depend on both the specific protease involved and the cellular context:
Caspase-mediated cleavage disables PARP-1's DNA repair function while conserving cellular energy (NAD+, ATP) by preventing PARP-1 overactivation [1]. The 24-kDa fragment (ZnF1-2PARP1) irreversibly binds to DNA strand breaks, competitively inhibiting repair processes and facilitating apoptotic progression [62].
The 89-kDa catalytic fragment (PARP1ΔZnF1-2) retains basal enzymatic activity but cannot be stimulated by DNA damage. This fragment can be inhibited by poly(ADP-ribose) (PAR) polymers and, when complemented with the regulatory ZnF1-2PARP1 fragment, can partially restore DNA-dependent activation [62].
Uncleavable PARP-1 mutants (PARP-1UNCL), where the caspase cleavage site DEVD214 has been mutated, demonstrate significant neuroprotective effects in ischemia models, highlighting the therapeutic potential of modulating PARP-1 cleavage [39].
Table 1: PARP-1 Cleavage Fragments and Their Characteristics
| Protease | Cleavage Fragments | Cell Death Pathway | Functional Consequences |
|---|---|---|---|
| Caspase-3/7 | 24-kDa + 89-kDa | Apoptosis | Inhibition of DNA repair; energy conservation |
| Calpain | 55-kDa + 62-kDa | Excitotoxicity | Calcium-associated cell death |
| Cathepsins | 50-kDa + 42-kDa | Lysosomal cell death | Lysosomal membrane permeabilization |
| Granzyme A | 70-kDa | Immune-mediated cytotoxicity | T-cell mediated neuronal damage |
| Granzyme B | 64-kDa + 50-kDa | Immune-mediated cytotoxicity | T-cell mediated neuronal damage |
| MMPs | 55-kDa, 42-kDa, 36-kDa | Neuroinflammation | Extracellular matrix remodeling |
Diagram 1: Protease-specific cleavage of PARP-1 generates signature fragments associated with distinct cell death pathways. The 24-kDa and 89-kDa fragments from caspase cleavage represent the most characterized apoptotic signature.
PARP-1 Cleavage in Neurological Disease Models
Ischemic Stroke Models
In vitro ischemia models utilizing oxygen/glucose deprivation (OGD) have demonstrated compelling correlations between PARP-1 cleavage patterns and neuroprotective outcomes:
Uncleavable PARP-1 (PARP-1UNCL) and the 24-kDa fragment (PARP-124) significantly enhance neuronal survival in both neuroblastoma cells (SH-SY5Y) and primary rat cortical neurons subjected to OGD. Conversely, the 89-kDa fragment (PARP-189) exhibits cytotoxic effects [39].
The neuroprotection conferred by PARP-1UNCL and PARP-124 occurs independently of PAR formation or NAD+ levels, suggesting alternative mechanisms beyond energy conservation [39].
PARP-1 cleavage fragments differentially regulate NF-κB signaling: PARP-189 increases NF-κB activity and upregulates pro-inflammatory proteins (iNOS, COX-2), while PARP-1UNCL and PARP-124 decrease these inflammatory mediators and increase anti-apoptotic Bcl-xL expression [39].
Neurodegenerative Disease Models
Alzheimer's disease models show increased PARP-1 activity and PAR levels in patient brains, fibroblasts, and lymphoblasts, suggesting potential therapeutic applications for PARP inhibition [63] [64].
Parkinson's disease demonstrates elevated PAR levels in cerebrospinal fluid, though fibroblasts show decreased PAR, indicating tissue-specific PARP-1 dysregulation [63].
Huntington's disease presents a unique pattern with reduced PAR levels and impaired PARP-1 activity even in prodromal stages, challenging the prevailing understanding of PARP-1 overactivation in neurodegeneration [63].
Cerebellar ataxias including ataxia with oculomotor apraxia type 1 (AOA1) show increased PARP-1 cleavage and PAR accumulation in neural progenitor cells, contributing to defective neural differentiation [65].
Heavy Metal Neurotoxicity Models
Cadmium-induced toxicity in rat proximal tubular cells (NRK-52E) and primary rat proximal tubular cells demonstrates PARP-1 overactivation leading to parthanatos, a PARP-1-dependent programmed cell death pathway. This process involves oxidative stress, mitochondrial damage, and interplay with MAPK signaling pathways (JNK1/2 and p38) [66].
Table 2: PARP-1 Cleavage Patterns in Neurological Disease Models
| Disease Model | PARP-1 Cleavage/Observation | Experimental System | Neuroprotective Correlation |
|---|---|---|---|
| Cerebral Ischemia | Increased caspase cleavage | OGD in SH-SY5Y and primary neurons | PARP-1UNCL and 24-kDa fragment protective |
| Alzheimer's Disease | Increased PARP-1 activity and PAR levels | Patient brains, fibroblasts, lymphoblasts | PARP inhibition protective |
| Parkinson's Disease | Elevated PAR in CSF | Patient cerebrospinal fluid | PARP inhibition protective |
| Huntington's Disease | Reduced PAR levels and PARP-1 activity | Patient fibroblasts, iPSC-derived neurons | Distinct mechanism from other neurodegenerative diseases |
| AOA1 | Increased cleaved PARP-1/total PARP-1 ratio | APTX-mutant iPSC-derived neural cells | Defective neural differentiation |
| Cadmium Neurotoxicity | PARP-1 overactivation, parthanatos | NRK-52E cells, primary rPT cells | PARP inhibitors and antioxidant treatment protective |
Experimental Methodologies for Assessing PARP-1 Cleavage
In Vitro PARylation Assay
This protocol enables quantitative assessment of PARP-1 enzymatic activity and fragment functionality:
Reagents Required:
- Purified PARP1, PARP1 variants (PARP1ΔZnF1-2, ZnF1-2PARP1) and PARP2 [62]
- SSB-DNA (single-strand break DNA) or DSB-DNA (double-strand break DNA)
- NAD+ solution (working concentration: 50-200 μM)
- Reaction buffer: 50 mM Tris-HCl (pH 8.0), 50 mM NaCl, 5 mM MgCl2, 1 mM DTT
- SDS-PAGE components and Western blot apparatus
- Anti-PAR antibody (e.g., mouse anti-PAR polymer antibody)
Procedure:
- Incubate PARP-1 or its fragments (0.5-2 μM) with DNA breaks (SSB or DSB, 0.1-1 μM) in reaction buffer at 25°C for 5 minutes.
- Initiate PARylation reaction by adding NAD+ to a final concentration of 50-200 μM.
- Allow reaction to proceed for 10-30 minutes at 25°C.
- Terminate reaction by adding SDS-PAGE loading buffer.
- Analyze PARylation by SDS-PAGE followed by Western blotting with anti-PAR antibody.
- Quantify band intensities using densitometry software [62].
Cell Culture Models of Ischemic Injury
Oxygen/Glucose Deprivation (OGD) and Restoration of Oxygen/Glucose (ROG):
Cell Lines:
- Human neuroblastoma SH-SY5Y cells
- Primary rat cortical neurons
Protocol:
- Culture SH-SY5Y cells in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 mg/ml streptomycin at 37°C in 5% CO2.
- For primary cortical neurons, isolate from Sprague-Dawley rats at postnatal day 2 and culture in Neurobasal Medium-A supplemented with B27.
- Generate tetracycline-inducible stable transfectants expressing PARP-1WT, PARP-1UNCL, PARP-124, or PARP-189 using Lipofectamine RNAi max.
- For OGD, replace culture medium with deoxygenated, glucose-free balanced salt solution and place cells in a hypoxic chamber (1% O2, 5% CO2, 94% N2) for 2-6 hours at 37°C.
- For ROG, return cells to normal oxygenated, glucose-containing medium and incubate under normal conditions for 15-24 hours.
- Assess cell viability using MTT assay, Annexin V/propidium iodide staining, or LDH release assay [39].
Induced Pluripotent Stem Cell (iPSC) Neural Differentiation Model
Protocol for Generating Neural Progenitor Cells (NPCs) from iPSCs:
- Culture control and APTX-mutant iPSCs in essential 8 medium on vitronectin-coated plates.
- Induce neural differentiation using a modified dual SMAD inhibitor protocol with 10 μM SB431542 (TGF-β inhibitor) and 100 nM LDN193189 (BMP inhibitor) for 10-12 days.
- Passage neural rosettes using dispase to isolate NPCs.
- Maintain NPCs in neural maintenance medium containing DMEM/F-12, N2 supplement, B27 supplement, and growth factors (20 ng/mL EGF and 20 ng/mL FGF2).
- Differentiate NPCs to early immature neurons (EiNs) by withdrawing growth factors and adding brain-derived neurotrophic factor (BDNF) and glial cell line-derived neurotrophic factor (GDNF) for 2 weeks.
- Assess DNA damage using comet assay and PAR immunostaining [65].
Diagram 2: Experimental workflow for correlating PARP-1 cleavage patterns with neuroprotection. Multiple model systems and analytical approaches enable comprehensive assessment of PARP-1 fragment functions.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for PARP-1 Cleavage Studies
| Reagent/Category | Specific Examples | Function/Application | Experimental Context |
|---|---|---|---|
| PARP-1 Constructs | PARP-1WT, PARP-1UNCL (uncleavable), PARP-124 (24-kDa), PARP-189 (89-kDa) | Functional analysis of cleavage fragments; determination of neuroprotective potential | In vitro ischemia models (OGD/ROG); transfection studies [39] |
| Cell Lines | SH-SY5Y (human neuroblastoma), NRK-52E (rat kidney epithelial), Primary cortical neurons, iPSC-derived neural cells | Disease modeling; mechanistic studies in relevant cellular contexts | Neurodegeneration, ischemia, toxicology studies [39] [66] [65] |
| PARP Inhibitors | DPQ, Olaparib, 3-AB | Inhibition of PARP enzymatic activity; investigation of parthanatos pathways | Cadmium toxicity, ischemia-reperfusion injury [66] [67] |
| Protease Inhibitors | Z-VAD-FMK (caspase), MG132 (proteasome), Calpain inhibitors | Specific inhibition of proteolytic pathways; determination of protease-specific PARP-1 cleavage | Apoptosis, excitotoxicity models [67] |
| Antibodies for Detection | Anti-PARP-1 (full length), Anti-cleaved PARP-1 (Asp214), Anti-PAR polymer, Anti-β-actin (loading control) | Detection and quantification of PARP-1 and its cleavage fragments; assessment of PARP activity | Western blot, immunofluorescence, immunoprecipitation [39] [66] [65] |
| DNA Damage Inducers | Hydrogen peroxide, Tert-butyl hydroperoxide, Etoposide, Cadmium acetate | Induction of DNA strand breaks; activation of PARP-1 and cleavage pathways | Oxidative stress models, genotoxicity assays [66] [65] |
| Signaling Pathway Modulators | SP600125 (JNK inhibitor), SB203580 (p38 inhibitor), NAC (antioxidant) | Investigation of crosstalk between PARP-1 cleavage and signaling pathways | MAPK pathway analysis, oxidative stress studies [66] |
Data Interpretation and Therapeutic Implications
Correlation Analysis of Cleavage Patterns and Neuroprotection
The relationship between specific PARP-1 cleavage fragments and neuronal survival outcomes reveals important therapeutic insights:
The 24-kDa fragment demonstrates consistent neuroprotective properties across multiple ischemia models, associated with reduced inflammatory signaling (decreased iNOS, COX-2) and enhanced anti-apoptotic factor expression (increased Bcl-xL) [39].
The 89-kDa fragment generally correlates with cytotoxic outcomes, potentially through sustained inflammatory activation and possible cytoplasmic signaling functions [39].
PARP-1 cleavage inhibition (via uncleavable PARP-1UNCL) provides neuroprotection, suggesting therapeutic potential of strategies that prevent specific cleavage events rather than broadly inhibiting PARP-1 enzymatic activity [39].
Emerging Therapeutic Approaches
PARP inhibitor repurposing: Several PARP inhibitors developed for oncology applications show promise for neurological disorders, though the unique PARP-1 dysregulation in certain diseases like Huntington's requires careful targeting strategies [68] [64].
Fragment-specific therapeutics: Developing approaches to enhance protective fragments (24-kDa) while inhibiting detrimental fragments (89-kDa) represents a more nuanced strategy than pan-PARP inhibition [39].
Combination therapies: Targeting both PARP-1 cleavage and interconnected pathways (MAPK, NF-κB) may provide enhanced neuroprotection, as demonstrated in cadmium toxicity models where JNK1/2 and p38 inhibition reduced parthanatos [66].
PARP-1 cleavage patterns serve as critical determinants of cell fate in neurological disease models, with specific fragments exhibiting distinct and sometimes opposing effects on neuronal survival. The caspase-generated 24-kDa fragment consistently correlates with neuroprotective outcomes, while the 89-kDa fragment and various calpain-generated fragments associate with detrimental effects. The experimental methodologies outlined herein provide robust frameworks for investigating these relationships, enabling drug development professionals to identify novel therapeutic targets that selectively modulate specific PARP-1 cleavage events rather than employing broad PARP inhibition. As research advances, the correlation between PARP-1 cleavage signatures and neuroprotection promises to yield more precise therapeutic interventions for complex neurological disorders.
Technical Challenges and Interpretation of PARP-1 Cleavage Data
Distinguishing Apoptotic vs. Non-Apoptotic Cleavage Fragments
Poly (ADP-ribose) polymerase-1 (PARP-1) is a nuclear enzyme with a well-established role in the routine repair of DNA damage. Beyond this fundamental function, PARP-1 participates in diverse physiological and pathological processes, including gene transcription, immune responses, and several forms of cell death [11]. The cleavage of PARP-1 by a specific set of proteases is a recognized biomarker for cell death. However, the specific proteolytic fragments generated serve as signatures that can distinguish between different cell death programs, such as apoptosis and various forms of lytic death [11]. This technical guide details the mechanisms behind PARP-1 cleavage, the signature fragments produced in different contexts, and the methodologies used for their discrimination, providing a critical resource for research in cell death and therapeutic development.
PARP-1 Domain Architecture and Cleavage Sites
PARP-1 contains several critical structural domains: a DNA-Binding Domain (DBD) with zinc finger motifs at the NH2 terminus, a central Auto-Modification Domain (AMD) with a BRCT fold, and a C-terminal Catalytic Domain (CD) [11]. During cell death, PARP-1 becomes a substrate for various "suicidal" proteases, which cleave the protein at specific sites, resulting in fragments with distinct sizes and functions [11].
Domain Organization and Cleavage Map
The following diagram illustrates the domain structure of full-length PARP-1 and the cleavage sites targeted by different proteases during apoptotic and non-apoptotic cell death.
Protease-Specific Cleavage of PARP-1 and Resulting Fragments
Different proteases activated in distinct cell death pathways cleave PARP-1 at unique sites, generating signature fragments that can be used for mechanistic identification.
Apoptotic Cleavage by Caspases
During apoptosis, executioner caspases-3 and -7 cleave PARP-1 at the Asp214 residue, located between the DBD and AMD [11] [23]. This proteolysis produces two well-characterized fragments:
- A 24 kDa fragment containing the first two zinc finger motifs (DBD). This fragment is retained in the nucleus, where it irreversibly binds to DNA strand breaks, acting as a trans-dominant inhibitor of DNA repair processes and conserving cellular ATP [11].
- An 89 kDa fragment (tPARP1) containing the third zinc finger, the BRCT domain, the WGR domain, and the catalytic domain. This fragment translocates from the nucleus to the cytosol [11] [23]. Recent research has revealed that tPARP1 is not merely an inactive byproduct; it can recognize the RNA Polymerase III (Pol III) complex in the cytosol via its BRCT domain and mediate its ADP-ribosylation. This activity facilitates innate immune responses, such as IFN-β production, during poly(dA-dT)-stimulated apoptosis [23].
Non-Apoptotic Cleavage by Other Proteases
In non-apoptotic cell death pathways, PARP-1 is cleaved by other proteases, generating different signature fragments.
- Calpains, calcium-activated proteases associated with necrosis and other lytic cell death forms, cleave PARP-1 to generate fragments of 55-62 kDa [11]. These fragments typically contain the WGR and catalytic domains but lack the N-terminal DNA-binding domain, resulting in a distinct pattern from caspase cleavage.
- Other Proteases, including cathepsins, granzymes, and matrix metalloproteinases (MMPs), can also process PARP-1, yielding specific fragments that serve as biomarkers for their respective protease activities and associated cell death programs [11].
The table below provides a consolidated overview of the key PARP-1 cleavage fragments.
Table 1: Signature PARP-1 Cleavage Fragments in Cell Death
| Cleaving Protease | Cell Death Context | Fragment Sizes | Domain Composition | Key Functional Consequences |
|---|---|---|---|---|
| Caspase-3/7 | Apoptosis | 24 kDa | ZnF1, ZnF2 (DBD) | Binds DNA irreversibly; inhibits DNA repair [11] |
| 89 kDa (tPARP1) | ZnF3, BRCT, WGR, CD | Translociates to cytosol; mediates ADP-ribosylation of Pol III to potentiate immune response [23] | ||
| Calpain | Necrosis, Lytic Death | 55-62 kDa | WGR, CD | Distinct catalytic activity and localization; biomarker for non-apoptotic death [11] |
| Other (Cathepsins, Granzymes, MMPs) | Various | Specific fragments | Variable | Recognized biomarkers for specific patterns of protease activity [11] |
Experimental Methodologies for Discriminating Cell Death
Accurately determining the mode of cell death requires methodologies that can differentiate between apoptosis and necrosis based on specific biochemical events.
Live-Cell Imaging with FRET-Based Caspase Sensors
A highly sensitive, real-time method for discriminating apoptosis from necrosis uses live cells stably expressing a genetically encoded FRET (Förster Resonance Energy Transfer)-based caspase sensor alongside a stable fluorescent marker targeted to mitochondria (e.g., Mito-DsRed) [69].
- Principle: The FRET probe consists of ECFP (donor) and EYFP (acceptor) linked by a DEVD sequence, a substrate for executioner caspases. Caspase cleavage separates the fluorophores, resulting in a loss of FRET, detectable as a change in the ECFP/EYFP emission ratio.
- Discrimination Workflow: The following diagram outlines the decision process for classifying cell death events using this system.
- Protocol Details: Cells are treated with the agent of interest and imaged in real-time using wide-field, confocal, or high-throughput microscopy. The following parameters are tracked simultaneously:
- Caspase Activation: Measured as a loss of FRET (increase in ECFP/EYFP ratio).
- Membrane Integrity: Assessed by the retention or loss of the soluble cytosolic FRET probe.
- Mitochondrial Retention: The Mito-DsRed marker, being tethered to an organelle, is retained longer in necrotic cells, allowing differentiation from apoptotic cells that retain both probes until late stages [69].
- Data Analysis: This method allows quantification of three distinct populations: 1) Live cells (no FRET loss, probes retained), 2) Apoptotic cells (FRET loss, Mito-DsRed retained), and 3) Necrotic cells (no FRET loss, FRET probe lost, Mito-DsRed retained) [69].
Quantitative Phase Imaging (QPI) for Label-Free Analysis
Quantitative Phase Imaging (QPI) is a label-free technique that analyzes time-dependent changes in cellular morphology and mass distribution to identify cell death subroutines [70].
- Principle: QPI measures the optical path length delay induced by cellular components, providing quantitative data on biomass distribution and density in real-time without requiring stains or labels.
- Key Parameters:
- Cell Density: Measured in picograms per pixel (pg/pixel), often decreases during lytic death due to swelling, while showing distinct dynamics during apoptosis.
- Cell Dynamic Score (CDS): A measure of the average intensity change of cell pixels, reflecting morphological activity like membrane blebbing (apoptosis) or swelling (necrosis).
- Protocol: Cells are treated and imaged over time. Machine learning algorithms can be trained on parameters like cell density and CDS to classify cell death modalities, such as caspase-dependent apoptosis versus caspase-independent lytic death, with reported accuracy exceeding 75% [70].
Flow Cytometry with Annexin V/PI Staining and Protein Analysis
Flow cytometry using annexin V and propidium iodide (PI) provides a robust, quantitative method for analyzing apoptosis induction and tracking protein expression in defined subpopulations [71].
- Principle:
- Annexin V-FITC binds to phosphatidylserine (PS), which is externalized to the outer leaflet of the plasma membrane during early apoptosis.
- Propidium Iodide (PI) is a DNA dye that only enters cells upon loss of plasma membrane integrity, a hallmark of late apoptosis and necrosis.
- Protocol Enhancement with Protein Staining: This protocol can be enhanced by simultaneously staining cells with fluorochrome-conjugated antibodies (e.g., APC) against specific proteins of interest. This multiparametric approach enables the tracking of protein expression changes from viable to apoptotic cells within the same sample [71].
- Gating Strategy: Cells can be differentiated into:
- Viable cells: Annexin V-negative, PI-negative.
- Early apoptotic cells: Annexin V-positive, PI-negative.
- Late apoptotic/necrotic cells: Annexin V-positive, PI-positive.
The table below summarizes the core techniques for distinguishing apoptotic and non-apoptotic cell death.
Table 2: Key Experimental Protocols for Discriminating Cell Death
| Method | Key Readouts | Apoptotic Signature | Necrotic/Lytic Signature | Key Advantages |
|---|---|---|---|---|
| FRET-Based Caspase Sensing [69] | Caspase activation (FRET loss), Membrane integrity (probe retention), Mitochondrial mass | FRET loss, probes retained | No FRET loss, soluble probe lost, Mito-DsRed retained | Real-time, single-cell resolution, distinguishes primary/secondary necrosis |
| Quantitative Phase Imaging (QPI) [70] | Cell density, Cell Dynamic Score (CDS), Morphology | Specific dynamics of density and CDS ("Dance of Death") | Swelling, membrane rupture (decreased density) | Label-free, non-invasive, provides rich biophysical data |
| Flow Cytometry (Annexin V/PI) [71] | PS exposure, Membrane integrity, Protein expression | Annexin V+/PI- (early); Annexin V+/PI+ (late) | Can be Annexin V+/PI+ (primary necrosis) | Quantitative, high-throughput, enables multiparametric protein analysis |
The Scientist's Toolkit: Essential Research Reagents
The following table lists key reagents and tools essential for conducting research on PARP-1 cleavage and cell death mechanisms.
Table 3: Essential Research Reagents for PARP-1 and Cell Death Analysis
| Research Reagent / Tool | Function and Application | Specific Example / Target |
|---|---|---|
| PARP-1 Cleavage-Specific Antibodies | Detect full-length and specific cleavage fragments (e.g., 89 kDa) via Western Blot [23] | Anti-PARP1 (cleaved) antibody |
| Caspase Inhibitor | Pharmacologically inhibit caspase activity to confirm caspase-dependent apoptosis [70] | z-VAD-FMK (pan-caspase inhibitor) |
| FRET-Based Caspase Sensor | Real-time visualization of caspase activation in live cells [69] | Stable cell line expressing ECFP-DEVD-EYFP |
| Mitochondrial Marker | Label mitochondria to assess cellular integrity and stage of death in live-cell imaging [69] | Mito-DsRed, MitoTracker |
| Annexin V-FITC / PI Kit | Standard flow cytometry assay to distinguish viable, early apoptotic, and late apoptotic/necrotic populations [71] | Commercial apoptosis detection kit |
| Fluorochrome-Conjugated Antibodies | Multiparametric flow cytometry to track protein expression changes in specific cell death subpopulations [71] | APC-conjugated anti-CD44 antibody |
| Chemical Inducers of Cell Death | Induce specific cell death pathways for experimental studies [69] [70] | Doxorubicin, Staurosporine, H₂O₂, Valinomycin |
Optimizing Antibody Selection for Specific Fragment Detection
In programmed cell death research, the cleavage of poly(ADP-ribose) polymerase-1 (PARP-1) serves as a critical biochemical marker distinguishing between different cell death modalities. PARP-1, a 113-116 kDa nuclear enzyme, functions as a primary DNA damage sensor and facilitates DNA repair through its catalytic activity. During apoptosis, PARP-1 undergoes specific proteolytic cleavage at the Asp214-Gly215 bond by executioner caspases-3 and -7, generating characteristic 24 kDa and 89 kDa fragments [72] [1]. This cleavage event separates the N-terminal DNA-binding domain (DBD) from the C-terminal catalytic domain, effectively inactivating DNA repair capabilities and facilitating cellular disassembly. Beyond caspase-mediated apoptosis, emerging research reveals that PARP-1 cleavage fragments also serve as signatures for other suicidal proteases including calpains, cathepsins, granzymes, and matrix metalloproteinases, each producing distinctive fragment patterns that identify specific cell death pathways [1]. The accurate detection of these specific fragments requires sophisticated antibody selection and experimental optimization, which this technical guide addresses for researchers and drug development professionals working in cell death mechanisms.
PARP-1 Cleavage Mechanisms in Cell Death Pathways
Molecular Architecture of PARP-1 and Cleavage Sites
PARP-1 contains three functionally distinct domains: two zinc-finger motifs constituting the 46-kDa DNA-binding domain (DBD) at the N-terminus, a 22-kDa automodification domain (AMD) in the central region, and a 54-kDa catalytic domain (CD) at the C-terminus [1]. The caspase cleavage site DEVD214G is situated within the DBD, specifically interrupting the nuclear localization signal [39] [54]. Cleavage at this aspartic acid residue produces a 24-kDa fragment containing the DBD and a separate 89-kDa fragment containing both the automodification and catalytic domains [72]. Recent studies have identified that the 89-kDa PARP-1 fragment, when modified with poly(ADP-ribose) (PAR) polymers, can translocate to the cytoplasm during caspase-mediated apoptosis, where it facilitates apoptosis-inducing factor (AIF) release from mitochondria, creating a novel bridge between caspase-dependent apoptosis and parthanatos [54].
Protease-Specific Cleavage Patterns
Different cell death proteases generate characteristic PARP-1 cleavage fragments that serve as signature biomarkers for specific death pathways:
- Caspases-3 and -7: Generate 24-kDa and 89-kDa fragments during apoptosis [1]
- Calpains: Produce 55-kDa and 62-kDa fragments during calcium-mediated cell death [1]
- Granzyme A: Creates a 50-kDa fragment during cytotoxic T-cell-mediated killing [1]
- Cathepsins: Generate various fragments during lysosomal-mediated cell death [1]
- Matrix Metalloproteinases: Produce distinct fragments in extracellular matrix remodeling and associated cell death [1]
The presence of these specific fragments provides researchers with critical information about the dominant cell death pathway activated under experimental conditions or in pathological contexts.
Figure 1: PARP-1 Cleavage Signaling Pathway during Programmed Cell Death. This diagram illustrates the molecular cascade from DNA damage to caspase-mediated PARP-1 cleavage and the functional consequences of fragment generation, including links to parthanatos.
Antibody Selection Guide for PARP-1 Fragment Detection
Key Antibody Characteristics for Specific Fragment Detection
Selecting appropriate antibodies for cleaved PARP-1 detection requires careful consideration of several parameters to ensure specificity and reproducibility. Antibodies targeting the cleavage site at Asp214 provide the highest specificity for apoptosis detection, as they recognize the neoepitope created by caspase cleavage [72]. These antibodies specifically detect the 89-kDa fragment without cross-reacting with full-length PARP-1 or other PARP isoforms, making them ideal for distinguishing apoptosis from other cell death forms. The clone F21-852, for instance, demonstrates specific reactivity with the 89-kDa fragment containing the automodification and catalytic domains downstream of the caspase-3 cleavage site at Asp214, without recognizing intact PARP-1 [73].
For comprehensive death pathway analysis, researchers may employ multiple antibodies targeting different PARP-1 epitopes. Antibodies recognizing the N-terminal DBD can detect the 24-kDa fragment, while those targeting the C-terminal catalytic domain can identify the 89-kDa fragment. The combination provides verification of complete PARP-1 cleavage. Furthermore, antibodies that recognize full-length PARP-1 alongside cleavage-specific antibodies enable quantification of the cleavage ratio, offering insights into the extent of apoptotic engagement within experimental systems.
Commercial Antibody Comparison
Table 1: Commercial Antibodies for Cleaved PARP-1 Detection
| Supplier | Clone/Catalog | Specificity | Applications | Reactivity | Observed Band Size |
|---|---|---|---|---|---|
| Cell Signaling Technology | #9541 (Polyclonal) | 89 kDa fragment (Asp214) | WB, Simple Western | Human, Mouse | 89 kDa |
| Cell Signaling Technology | #60068 (D64E10) | 89 kDa fragment (Asp214) | Flow Cytometry | Human, Mouse, Monkey | Not specified |
| Abcam | ab32064 [E51] | Cleaved PARP1 (27 kDa observed) | WB, IHC-P | Human, Mouse, Rat | 27 kDa (observed) |
| BD Biosciences | 552933 (F21-852) | 89 kDa fragment (Asp214) | Flow Cytometry | Human | 89 kDa |
Technical Considerations for Antibody Validation
Rigorous validation is essential for reliable cleaved PARP-1 detection. Researchers should confirm antibody specificity using:
- PARP-1 knockout cell lines to verify absence of non-specific binding [74]
- Induced apoptosis controls (e.g., camptothecin-treated Jurkat cells) to confirm expected cleavage detection [73]
- Caspase inhibition to demonstrate cleavage reduction in zVAD-fmk treated samples [54]
- Multi-platform validation where antibodies are tested across Western blot, flow cytometry, and immunohistochemistry applications
Batch-to-batch consistency represents another critical consideration, particularly for long-term studies. Recombinant monoclonal antibodies typically offer superior lot-to-lot consistency compared to traditional monoclonal or polyclonal antibodies [75]. For quantitative studies, antibodies with demonstrated linear detection ranges across experimental conditions should be selected, and normalization to housekeeping proteins is essential for accurate inter-experiment comparisons.
Experimental Protocols for Cleaved PARP-1 Detection
Western Blot Protocol for PARP-1 Cleavage Detection
Materials:
- RIPA Lysis Buffer (25 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS)
- Protease inhibitor cocktail
- Phosphatase inhibitor cocktail
- BCA Protein Assay Kit
- 4-12% Bis-Tris Protein Gels
- PVDF or Nitrocellulose Membranes
- Cleaved PARP (Asp214) Antibody (e.g., Cell Signaling #9541)
- HRP-conjugated Secondary Antibody
- Chemiluminescent Substrate
Method:
- Cell Treatment and Lysis: Induce apoptosis in cells (e.g., 1×10^6 Jurkat cells) using 4-6 µM camptothecin for 4 hours at 37°C [73]. Wash cells twice with cold PBS and lyse in RIPA buffer containing protease and phosphatase inhibitors on ice for 30 minutes. Centrifuge at 14,000 × g for 15 minutes at 4°C and collect supernatant.
Protein Quantification and Preparation: Determine protein concentration using BCA assay. Dilute samples with Laemmli buffer to equal concentrations (20-40 µg per lane) and denature at 95°C for 5 minutes.
Gel Electrophoresis: Load samples onto 4-12% Bis-Tris gradient gels alongside pre-stained protein markers. Run at constant voltage (120-150V) until dye front reaches bottom.
Membrane Transfer: Transfer proteins to PVDF membrane using wet or semi-dry transfer systems according to manufacturer's protocols.
Blocking and Antibody Incubation: Block membrane with 5% non-fat dry milk in TBST for 1 hour at room temperature. Incubate with primary cleaved PARP antibody (1:1000 dilution in 5% BSA/TBST) overnight at 4°C with gentle agitation [72]. Wash membrane 3×10 minutes with TBST. Incubate with species-appropriate HRP-conjugated secondary antibody (1:2000-1:5000) for 1 hour at room temperature. Wash 3×10 minutes with TBST.
Detection: Develop blots using enhanced chemiluminescence substrate according to manufacturer's instructions. Image using chemiluminescence detection system with multiple exposure times to ensure linear signal detection.
Expected Results: Successful apoptosis induction should yield a clear 89 kDa band in treated samples, with minimal detection in untreated controls. Full-length PARP-1 (113-116 kDa) should be visible in untreated samples and diminish in apoptotic samples.
Flow Cytometry Protocol for Intracellular Cleaved PARP Detection
Materials:
- Cytofix/Cytoperm Solution (BD Biosciences #554722)
- Perm/Wash Buffer (BD Biosciences #554723)
- PE Mouse Anti-Cleaved PARP (Asp214) (BD Biosciences #552933)
- Isotype Control Antibody
- Flow Cytometry Staining Buffer (PBS with 1% BSA)
Method:
- Apoptosis Induction and Cell Preparation: Induce apoptosis in 1×10^6 proliferating Jurkat cells using 4-6 µM camptothecin for 4 hours at 37°C [73]. Include untreated control cells. Wash cells twice with cold PBS.
Fixation and Permeabilization: Resuspend cells in Cytofix/Cytoperm solution at 2×10^6 cells/ml. Incubate for 20 minutes on ice. Pellet cells and discard supernatant. Wash cells twice with 0.5 ml Perm/Wash buffer per 1×10^6 cells, discarding supernatants.
Antibody Staining: Resuspend cells in Perm/Wash buffer at 10×10^6/ml. Aliquot 1×10^6 cells per 100-µl test. Add 20 µl antibody per test, incubate for 30 minutes at room temperature protected from light.
Washing and Analysis: Wash each test in 1.0 ml Perm/Wash Buffer, discard supernatant. Resuspend in 0.5 ml Perm/Wash Buffer and analyze by flow cytometry using appropriate channels for PE detection (excitation 496/566 nm, emission 576 nm) [73].
Expected Results: Camptothecin-treated cells should show distinct positive population for cleaved PARP compared to isotype control and untreated cells, enabling quantification of apoptotic population percentage.
Figure 2: Flow Cytometry Workflow for Cleaved PARP Detection. This experimental workflow outlines the key steps for intracellular detection of cleaved PARP using camptothecin-induced Jurkat cells as an apoptosis model system.
Research Reagent Solutions for PARP-1 Cleavage Studies
Table 2: Essential Research Reagents for PARP-1 Cleavage Studies
| Reagent Category | Specific Examples | Function/Application | Key Features |
|---|---|---|---|
| Apoptosis Inducers | Camptothecin (4-6 µM) [73], Staurosporine (1 µM) [74] | Induce caspase-dependent apoptosis and PARP-1 cleavage | Dose-dependent apoptosis induction; validated in multiple cell lines |
| Caspase Inhibitors | zVAD-fmk (pan-caspase inhibitor) [54] | Confirm caspase-dependence of PARP-1 cleavage | Broad-spectrum caspase inhibition; establishes mechanism |
| Fixation/Permeabilization Kits | Cytofix/Cytoperm Kit (BD #554714) [73] | Intracellular antibody staining for flow cytometry | Preserves epitopes while allowing antibody penetration |
| PARP Inhibitors | PJ34, ABT-888 (Veliparib) [54] | Investigate PARP activity in cell death pathways | Tool compounds for mechanistic studies |
| Validated Cell Lines | Jurkat (ATCC TIB-152), HeLa, A549 [74] [73] [54] | Model systems for PARP cleavage studies | Well-characterized apoptosis responses; reference controls |
| Detection Systems | HRP-conjugated secondaries, Chemiluminescent substrates [72] | Signal detection in immunoblotting | Sensitive detection of low-abundance fragments |
Troubleshooting and Optimization Strategies
Common Detection Issues and Solutions
Weak or No Signal:
- Cause: Insufficient apoptosis induction or suboptimal antibody concentration
- Solution: Titrate apoptosis inducer (camptothecin 1-10 µM) using extended treatment times (4-6 hours). Perform antibody titration (typically 1:500-1:2000 for Western blot) to determine optimal concentration [73] [72].
Non-Specific Bands:
- Cause: Antibody cross-reactivity with unrelated proteins or degraded PARP-1
- Solution: Include PARP-1 knockout controls [74], optimize blocking conditions (5% BSA or non-fat milk), increase stringency washes (high salt, detergent).
High Background in Flow Cytometry:
- Cause: Inadequate washing or non-specific antibody binding
- Solution: Increase Perm/Wash buffer volumes and wash cycles, include isotype controls, titrate antibody to optimal concentration, use Fc receptor blocking if applicable.
Discrepant Band Sizes:
- Cause: Variability in observed versus predicted molecular weights
- Solution: Note that observed band sizes may vary - the 89-kDa fragment may run at 85-90 kDa, and some antibodies detect smaller fragments (e.g., 27 kDa with ab32064) [74]. Always include positive controls and appropriate molecular weight markers.
Validation Strategies for Specific Fragment Detection
Robust validation ensures accurate interpretation of cleaved PARP-1 detection experiments:
- Genetic Validation: Use PARP-1 knockout cell lines to confirm antibody specificity [74]
- Pharmacological Validation: Employ caspase inhibitors (zVAD-fmk) to demonstrate caspase-dependence of cleavage [54]
- Multi-Antibody Confirmation: Use antibodies targeting different epitopes to verify complete cleavage (both 24-kDa and 89-kDa fragments)
- Time-Course Experiments: Establish kinetic profile of cleavage following apoptosis induction
- Platform Correlation: Compare results across Western blot, flow cytometry, and immunohistochemistry platforms
Optimizing antibody selection for specific PARP-1 fragment detection requires systematic consideration of multiple factors, including the specific cell death pathway under investigation, the required application platform, and the necessary validation strategies. The 89-kDa fragment generated by caspase cleavage at Asp214 remains the best-characterized cleavage product and serves as a robust apoptosis marker across multiple detection platforms. As research continues to elucidate the complex roles of PARP-1 fragments in different cell death modalities, antibody-based detection methods will remain essential tools for mechanistic studies and drug development targeting cell death pathways. The protocols and guidelines presented here provide researchers with a framework for implementing reliable cleaved PARP-1 detection in programmed cell death research.
Poly(ADP-ribose) polymerase-1 (PARP-1) is a nuclear enzyme with well-established roles in DNA repair, transcriptional regulation, and the maintenance of genomic integrity [11] [76]. Beyond these homeostatic functions, PARP-1 has emerged as a central executioner and biomarker in multiple programmed cell death (PCD) pathways. The proteolytic cleavage of PARP-1 by a specific set of cellular proteases generates distinctive fragments that serve as molecular signatures, providing critical insights into the particular death pathway activated within a cell population [11] [77]. The canonical 89 kDa and 24 kDa fragments generated by caspase cleavage are established hallmarks of apoptosis. However, the reality within stressed tissues and experimental models is often far more complex, characterized by heterogeneous or "mixed" cell death populations where multiple PCD pathways are activated simultaneously or sequentially [11] [78].
Interpreting this incomplete or mixed cleavage signature is a significant challenge in cell death research. The presence of multiple PARP-1 fragments indicates the concurrent activity of different proteases (e.g., caspases, calpains, cathepsins, granzymes, matrix metalloproteinases), each activated within a specific PCD context [11]. This technical guide provides an in-depth analysis of PARP-1 cleavage patterns as indicators of mixed cell death populations. It details the molecular signatures, quantitative assessments, and experimental protocols necessary to decipher these complex biological signals within the broader framework of PCD research and drug development.
PARP-1 Cleavage Fragments as Biomarkers of Specific Cell Death Pathways
PARP-1 serves as a preferred substrate for several "suicidal" proteases activated during different forms of PCD. The cleavage of PARP-1's 113 kDa full-length structure produces specific fragments with defined molecular weights, which can be detected via western blotting and serve as reliable biomarkers [11].
Table 1: PARP-1 Cleavage Fragments and Their Associated Cell Death Pathways
| PARP-1 Fragment | Molecular Weight | Generating Protease(s) | Cell Death Pathway | Key Functional Consequences |
|---|---|---|---|---|
| Full-length PARP-1 | 113 kDa | N/A | Homeostasis / DNA Repair | Active in DNA damage repair and gene transcription [11]. |
| Apoptotic Fragments | 89 kDa & 24 kDa | Caspase-3 & Caspase-7 | Apoptosis (Type I PCD) | Inactivation of DNA repair; 24 kDa fragment acts as trans-dominant inhibitor of BER; 89 kDa fragment can translocate to cytosol [11] [39] [13]. |
| tPARP-1 (Truncated) | 89 kDa (can be modified) | Caspase-3 | Apoptosis (Cytosolic Role) | Cytosolic tPARP1 can mediate ADP-ribosylation of RNA Pol III, facilitating innate immune response [23]. |
| PARP-1 p35 | ~35 kDa | Calpain, Cathepsins | Necrosis / Parthanatos / Autophagy (Type II/III PCD) | Associated with caspase-independent cell death; can promote AIF release [11]. |
The 89 kDa fragment, when modified by poly(ADP-ribose) (PAR) polymers, can translocate to the cytoplasm during parthanatos, a caspase-independent cell death pathway. Here, it acts as a carrier for PAR, facilitating the binding to and release of Apoptosis-Inducing Factor (AIF) from mitochondria, which ultimately leads to DNA degradation [13]. The 24 kDa DNA-binding domain fragment remains nuclear and can act as a trans-dominant inhibitor of DNA repair by irreversibly binding to DNA strand breaks, thereby conserving cellular ATP and preventing aberrant DNA repair during apoptosis [11].
Table 2: Key Proteases in Cell Death and Their PARP-1 Targets
| Protease | Class | Primary Cell Death Pathway | PARP-1 Cleavage Signature | Additional Notes |
|---|---|---|---|---|
| Caspase-3/7 | Cysteine-aspartic protease | Apoptosis (Type I PCD) | 89 kDa + 24 kDa fragments | Gold-standard apoptosis biomarker; cleaves at DEVD214↓G motif [11] [39]. |
| Calpain | Calcium-dependent cysteine protease | Necrosis, Parthanatos | ~35 kDa fragment | Links calcium toxicity to cell death; implicated in neurodegeneration [11]. |
| Cathepsins | Lysosomal proteases | Lysosomal Cell Death | ~35 kDa fragment | Released upon lysosomal membrane permeabilization [11]. |
| Granzyme A | Serine protease | Immune-mediated killing | Not specified (distinct from caspase) | Activated in cytotoxic T-lymphocytes and NK cells [11]. |
| MMPs | Matrix Metalloproteinases | Necrosis-associated | Not specified | Associated with tissue remodeling and inflammatory cell death [11]. |
Quantitative Data: Fragment Ratios and Cell Fate Decisions
The relative abundance of different PARP-1 fragments provides quantitative insights into the dominant cell death mechanism and the cellular consequences. Experimental data from models of cerebral ischemia and other pathologies demonstrate that the fate of a cell is influenced by the balance between these cleavage events.
Table 3: Quantitative Cell Viability and NF-κB Activity Based on PARP-1 Status
| PARP-1 Construct Expressed | Cell Viability Post-OGD/ROG | NF-κB Activation Level | Downstream Protein Expression | Interpretation |
|---|---|---|---|---|
| PARP-1WT (Wild-type) | Baseline | Baseline | Baseline iNOS, COX-2, Bcl-xL | Standard apoptotic and inflammatory response [39]. |
| PARP-1UNCL (Uncleavable) | Increased | Similar to PARP-1WT | ↓ iNOS, ↓ COX-2, ↑ Bcl-xL | Cleavage is cytotoxic; prevention favors survival and anti-inflammatory state [39]. |
| PARP-124 (24 kDa Fragment) | Increased | Similar to PARP-1WT | ↓ iNOS, ↓ COX-2, ↑ Bcl-xL | The 24 kDa fragment is cytoprotective, potentially by blocking excessive DNA repair [39]. |
| PARP-189 (89 kDa Fragment) | Decreased ↓ | Significantly Higher ↑ | ↑ iNOS, ↑ COX-2, ↓ Bcl-xL | The 89 kDa fragment is cytotoxic and pro-inflammatory [39]. |
The data from these controlled experiments suggest that the 89 kDa fragment is not merely an inactive byproduct of caspase cleavage but possesses inherent cytotoxic and pro-inflammatory properties, likely through its role in activating NF-κB and its downstream targets like iNOS and COX-2 [39]. Conversely, the 24 kDa fragment and the uncleavable PARP-1 mutant appear to promote cell survival under ischemic stress.
Experimental Protocols for Detecting and Interpreting Mixed Cleavage
Standard Western Blot Protocol for PARP-1 Cleavage Detection
Sample Preparation:
- Cell Lysis: Harvest cells and lyse in RIPA buffer (25 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) supplemented with protease inhibitors (e.g., 1 mM PMSF, 10 μg/mL aprotinin, 10 μg/mL leupeptin) and a pan-caspase inhibitor (e.g., Z-VAD-FMK, 20 μM) if aiming to specifically inhibit apoptotic cleavage. For detecting calpain-mediated cleavage, include calpain inhibitors (e.g., MDL-28170, 25 μM) [11] [39].
- Protein Quantification: Determine protein concentration using a Bradford or BCA assay. Prepare samples with Laemmli buffer and denature at 95°C for 5 minutes.
Gel Electrophoresis and Immunoblotting:
- Load 20-30 μg of total protein per lane on a 4-20% gradient SDS-PAGE gel to ensure optimal separation of full-length PARP-1 (113 kDa) and its major fragments (89 kDa, 24 kDa, and ~35 kDa).
- Transfer proteins to a PVDF membrane.
- Blocking: Incubate membrane with 5% non-fat milk in TBST (Tris-Buffered Saline with 0.1% Tween-20) for 1 hour at room temperature.
- Primary Antibody Incubation: Incubate with specific primary antibodies diluted in blocking buffer overnight at 4°C.
- Anti-PARP-1 Antibody (e.g., recognizing the C-terminal catalytic domain): Detects full-length (113 kDa) and the 89 kDa fragment [13].
- Anti-Cleaved PARP-1 (Asp214) Antibody: Specifically detects the 89 kDa fragment generated by caspase cleavage [39].
- Antibody against the N-terminal DNA-binding domain: Can detect the 24 kDa fragment [11].
- Secondary Antibody Incubation: Incubate with an appropriate HRP-conjugated secondary antibody for 1 hour at room temperature.
- Detection: Use enhanced chemiluminescence (ECL) substrate and image with a digital gel documentation system.
Protocol for Poly(dA-dT)-Stimulated Apoptosis and tPARP-1 Analysis
This protocol is used to study the non-canonical role of the truncated 89 kDa PARP-1 (tPARP1) in cytosolic DNA-induced apoptosis and innate immune signaling [23].
- Cell Transfection: Seed human embryonic kidney 293T (HEK293T) cells or other relevant cell lines (e.g., PARP1-deficient 293T for rescue experiments) in 6-well plates.
- Induction of Apoptosis: Transfert cells with poly(deoxyadenylic-deoxythymidylic) acid [poly(dA-dT)] (1 μg/well) using a standard transfection reagent (e.g., Lipofectamine 2000). Poly(dA-dT) mimics pathogenic DNA and stimulates the cytosolic DNA-sensing pathway.
- Validation of Apoptosis (6-24 hours post-transfection):
- Western Blotting: Confirm PARP-1 cleavage using the protocol in section 4.1.
- Flow Cytometry: Harvest cells and stain with Annexin V-FITC and Propidium Iodide (PI) according to manufacturer's instructions. Analyze using flow cytometry to quantify the percentage of cells in early (Annexin V+/PI-) and late (Annexin V+/PI+) apoptosis [23].
- Morphological Assessment: Observe cells under a phase-contrast microscope for characteristic apoptotic morphology (cell shrinkage, rounding, membrane blebbing).
- Co-immunoprecipitation (Co-IP) to Study tPARP1-Pol III Interaction:
- Lyse cells from step 2 in a mild, non-denaturing lysis buffer (e.g., 20 mM Tris-HCl pH 8.0, 137 mM NaCl, 1% Triton X-100, 2 mM EDTA, plus protease inhibitors).
- Incubate the clarified lysate with an antibody against PARP-1 or a tag on expressed tPARP1, with Protein A/G agarose beads overnight at 4°C.
- Wash beads extensively with lysis buffer, elute proteins with Laemmli buffer, and perform western blotting to probe for subunits of the RNA Polymerase III (Pol III) complex (e.g., POLR3A, POLR3B, POLR3F) [23].
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Reagents for PARP-1 and Cell Death Research
| Reagent / Tool | Category | Specific Example / Catalog Number | Primary Function in Research |
|---|---|---|---|
| Anti-PARP-1 Antibody | Antibody | Rabbit mAb #9532 (Cell Signaling) | Detects full-length PARP-1 (113 kDa) and the 89 kDa cleavage fragment [39]. |
| Anti-Cleaved PARP-1 (Asp214) Antibody | Antibody | Mouse mAb #9541 (Cell Signaling) | Specifically detects the 89 kDa fragment resulting from caspase cleavage; superior for confirming apoptosis [39] [23]. |
| Caspase Inhibitor | Pharmacological Inhibitor | Z-VAD-FMK (pan-caspase inhibitor) | Inhibits caspase-mediated cleavage, used to dissect caspase-dependent vs. independent death [11]. |
| Calpain Inhibitor | Pharmacological Inhibitor | MDL-28170 | Inhibits calpain activity, used to investigate calpain's role in PARP-1 cleavage and parthanatos [11]. |
| PARP-1 siRNA | Genetic Tool | Target Sequence: 5'-ACGGTGATCGGTAGCAACAAA-3' (Qiagen) | Silences endogenous PARP-1 expression, allowing for functional studies of PARP-1 variants or fragments [39]. |
| PARP-1 Expression Constructs | Molecular Biology | PARP-1WT, PARP-1UNCL (D214A), PARP-124, PARP-189 | Used to express wild-type, uncleavable, or individual fragments to study their specific functions [39] [23]. |
| Poly(dA-dT) | Biochemical Inducer | e.g., Sigma-Aldrich P0883 | Mimics cytosolic pathogenic DNA, stimulating the Pol III/tPARP1 pathway and inducing innate immune apoptosis [23]. |
| Annexin V-FITC / PI Apoptosis Kit | Detection Kit | e.g., BioLegend 640914 | Allows for quantification of apoptotic cells by flow cytometry, a standard correlate for PARP-1 cleavage [23]. |
Integrated Signaling in Mixed Cell Death
The interplay between different cell death pathways is complex. A key intersection exists between apoptosis and parthanatos. Caspase activation, a hallmark of apoptosis, can also trigger events characteristic of parthanatos. Research shows that caspase cleavage of PARP-1 generates the 89 kDa tPARP1 fragment, which can be poly(ADP-ribosyl)ated. This modified tPARP1 fragment can then translocate to the cytoplasm, bind to AIF, and facilitate its nuclear translocation, culminating in DNA degradation—a defining feature of parthanatos [13]. This demonstrates how a proteolytic event in one pathway (apoptosis) can actively initiate or amplify another (parthanatos), leading to a mixed death phenotype.
Interpreting incomplete PARP-1 cleavage patterns is a critical skill for researchers investigating cell death in complex biological systems, from neurodegenerative diseases to cancer therapy. The presence of multiple PARP-1 fragments is not merely a sign of messy biology but a precise molecular record of the proteolytic activities within a cell population. By systematically applying the methodologies outlined in this guide—rigorous western blotting with specific antibodies, quantitative assessment of fragment ratios, the use of selective inhibitors, and functional assays—scientists can move beyond simple apoptosis detection to a more nuanced understanding of mixed cell death populations. This advanced diagnostic capability is essential for accurately profiling drug mechanisms, understanding disease pathogenesis, and developing targeted therapeutic strategies that account for the intricate interplay between cell death pathways.
Addressing Cross-Reactivity with Other PARP Family Members
In the context of programmed cell death (PCD) research, poly(ADP-ribose) polymerase 1 (PARP-1) has emerged as a critical molecular switch, with its cleavage serving as a definitive biomarker for specific cell death pathways. PARP-1, a 116 kDa nuclear enzyme, is a primary DNA damage sensor and facilitator of DNA repair through its poly(ADP-ribosyl)ation activity [1] [79]. However, the human PARP family comprises 17 members with structural similarities, creating significant challenges for research specificity and therapeutic development [80]. This technical guide addresses the critical issue of cross-reactivity within the PARP family, providing researchers with methodologies to ensure precise PARP-1 investigation, particularly within the complex landscape of PCD where PARP-1 cleavage fragments serve as signature biomarkers for specific suicidal proteases [1].
The challenge of cross-reactivity extends beyond basic research into therapeutic development. While current clinically approved PARP inhibitors target both PARP1 and PARP2, evidence suggests that PARP2 inhibition contributes to hematological toxicity without being essential for synthetic lethality in BRCA-mutated cancers [81]. This understanding has driven the development of next-generation PARP1-selective inhibitors with improved safety profiles, highlighting the clinical significance of distinguishing between PARP family members [81].
The PARP Family: Structural Relationships and Potential for Cross-Reactivity
PARP Family Classification and Characteristics
The PARP superfamily, recently reclassified under a proposed nomenclature by Hottiger et al. as ADP-ribosyltransferases (ARTDs), is categorized based on structural motifs and catalytic functions [80]. PARP-1 remains the most extensively studied member, accounting for approximately 85% of total cellular PARP activity [1]. Understanding the structural relationships within this family is fundamental to addressing cross-reactivity challenges.
Table 1: Key PARP Family Members and Their Characteristics
| PARP Member | Primary Functions | Structural Domains | DNA Damage Response | Potential Cross-Reactivity Concerns |
|---|---|---|---|---|
| PARP-1 (ARTD1) | DNA repair, transcriptional regulation, programmed cell death | Three zinc fingers, BRCT, WGR, catalytic domain | Primary responder to single-strand breaks | Primary target of investigation; reference for specificity |
| PARP-2 (ARTD2) | DNA repair, spermatogenesis, adipogenesis | WGR, catalytic domain | Back-up for PARP-1 in SSB repair | ~69% homology in catalytic domain with PARP-1 [80] |
| PARP-3 (ARTD3) | Mitosis, centromere organization, DNA double-strand break repair | WGR, catalytic domain | Roles in NHEJ and cellular resistance to ionizing radiation | Often co-expressed with PARP-1; similar molecular weight |
| Tankyrase-1 (PARP-5a) | Telomere homeostasis, Wnt signaling, GLUT4 vesicle trafficking | Ankyrin repeats, SAM, catalytic domain | Not primarily involved in DNA damage response | Distinct structure but may cross-react with poor-quality antibodies |
Structural Basis for Cross-Reactivity
The potential for cross-reactivity arises primarily from conserved domains across PARP family members. PARP-1 and PARP-2 share approximately 69% homology in the catalytic domain, presenting the most significant challenge for specificity [80]. This homology is particularly problematic when investigating PARP-1 cleavage during PCD, as detection methods must distinguish between PARP-1 fragments and full-length or cleaved forms of other PARP members.
PARP-1 contains several distinctive domains: a 46-kD DNA-binding domain (DBD) with two zinc finger motifs at the NH2 terminus, a 22-kD auto-modification domain (AMD) containing a BRCT fold, and a 54-kD catalytic domain (CD) at the carboxyl terminus [1]. The presence of multiple zinc finger motifs in the DBD distinguishes PARP-1 from other family members and provides potential targets for specific detection strategies.
PARP-1 Cleavage in Programmed Cell Death: Protease-Specific Signatures
PARP-1 as a Molecular Marker for Cell Death Pathways
During programmed cell death, PARP-1 serves as a preferred substrate for multiple "suicidal" proteases, with each protease generating specific cleavage fragments that serve as biomarkers for distinct cell death pathways [1]. The detection of these specific fragments is crucial for accurate characterization of cell death mechanisms, but requires reagents capable of distinguishing them from other PARP family members.
Table 2: PARP-1 Cleavage Fragments in Different Programmed Cell Death Pathways
| Cell Death Pathway | Primary Protease(s) | Cleavage Site | PARP-1 Fragments Generated | Biological Consequences |
|---|---|---|---|---|
| Apoptosis | Caspase-3 and Caspase-7 | Asp214-Gly215 [79] | 24-kD DBD + 89-kD fragment (AMD+CD) [1] | Inactivation of DNA repair; conservation of ATP; dismantling of cellular structures |
| Necroptosis | Not established | Not established | Not established | Potential cross-talk with parthanatos |
| Parthanatos | Calpains, Cathepsins | Multiple sites | 55-kD, 40-kD, 35-kD fragments [1] | AIF-mediated chromatinolysis; PAR polymer accumulation |
| Cytotoxic Lymphocyte Killing | Granzyme A | Not aspartate-specific | 50-kD fragment [1] | Unique signature of immune-mediated destruction |
| Inflammatory Cell Death | MMPs (e.g., MMP-2, -9) | Not established | 55-kD, 40-kD, 35-kD fragments | Potential role in blood-brain barrier disruption |
The most well-characterized PARP-1 cleavage occurs during apoptosis, where caspase-3 and caspase-7 cleave PARP-1 between Asp214 and Gly215, separating the DNA-binding domain (24 kDa) from the catalytic domain (89 kDa) [79]. This cleavage event serves as a gold standard biomarker for apoptotic cell death and has been extensively utilized in cell death research.
PARP-1 in Parthanatos: A PARP-1-Dependent Cell Death Pathway
Parthanatos represents a distinct form of programmed cell death that is explicitly PARP-1-dependent, characterized by excessive PARP-1 activation leading to poly(ADP-ribose) (PAR) polymer accumulation, mitochondrial permeability changes, and massive energy depletion [66]. Unlike apoptosis, parthanatos involves calpain and cathepsin-mediated PARP-1 cleavage, generating different signature fragments (55-kD, 40-kD, and 35-kD) that can be distinguished from apoptotic cleavage products [1]. This pathway has been demonstrated in cadmium-induced renal tubular cell death, where PARP-1 overexpression contributes to toxicity through mechanisms involving oxidative stress and mitochondrial damage [66].
Diagram 1: PARP-1 in Parthanatos. This PARP-1-dependent cell death pathway involves hyperactivation, PAR polymer accumulation, and calpain-mediated cleavage.
Experimental Strategies to Address Cross-Reactivity
Antibody-Based Detection Methods
Western blotting remains the primary method for detecting PARP-1 cleavage, but requires rigorous validation to ensure specificity. The (46D11) Rabbit Monoclonal Antibody (#9532, Cell Signaling Technology) provides a validated solution that detects endogenous levels of total full-length PARP-1 (116 kDa) and the large fragment (89 kDa) produced by caspase cleavage, without cross-reactivity with PARP-2 and PARP-3 [79]. This specificity is achieved through immunization with a synthetic peptide corresponding to residues surrounding Gly623 of human PARP-1, which lies within a non-conserved region of the catalytic domain.
For researchers investigating parthanatos, anti-PAR polymer antibodies are essential for detecting PAR accumulation, a hallmark of this pathway. These antibodies should be validated against PARP-1 specific PARylation, as other PARP family members may produce PAR chains under certain conditions.
Genetic Approaches for PARP-1 Specificity
Genetic techniques provide powerful alternatives to address cross-reactivity concerns:
siRNA Knockdown: PARP-1-specific siRNAs can selectively reduce PARP-1 expression without affecting other PARP family members. In cadmium toxicity studies, PARP-1 siRNA effectively reduced PARP-1 expression and attenuated cell death, confirming PARP-1's specific role in parthanatos [66].
CRISPR-Cas9 Knockout: Complete PARP-1 knockout cells provide definitive controls for antibody specificity and functional studies, allowing researchers to confirm whether observed signals or phenotypes are truly PARP-1-dependent.
Selective Inhibitors: Next-generation PARP-1 selective inhibitors with greater than 100-fold selectivity for PARP-1 over PARP-2 provide pharmacological tools to distinguish PARP-1-specific functions [81].
Diagram 2: Experimental Workflow for Specific PARP-1 Detection. This workflow integrates multiple validation strategies to ensure specificity.
The Scientist's Toolkit: Essential Reagents for PARP-1 Research
Table 3: Research Reagent Solutions for PARP-1 Specific Investigation
| Reagent Category | Specific Product Examples | Key Features and Applications | Validation for Specificity |
|---|---|---|---|
| PARP-1 Specific Antibodies | PARP (46D11) Rabbit mAb #9532 [79] | Detects full-length (116 kDa) and caspase-cleaved (89 kDa) PARP-1; no cross-reactivity with PARP-2/3 | Validated in human, mouse, rat, monkey; knockout cell confirmation recommended |
| PAR Polymer Detection | Mouse anti-PAR polymer antibody [66] | Detects PAR accumulation in parthanatos; essential for distinguishing from apoptosis | Should show reduced signal with PARP-1 knockdown |
| Selective Inhibitors | Next-generation PARP1-selective inhibitors [81] | >100-fold selectivity for PARP-1 over PARP-2; functional specificity validation | Confirm reduced cytotoxicity compared to pan-PARP inhibitors |
| Genetic Tools | PARP-1 specific siRNAs [66] | Selective PARP-1 knockdown without affecting other PARP members; ideal for functional studies | Validate knockdown efficiency and specificity via Western blot |
| Activity Assays | NAD+/NADH Assay Kits [66] | Measures PARP activity through NAD+ consumption; use with selective inhibitors for specificity | Combine with PARP-1 knockdown for confirmation |
Advanced Technical Protocols for PARP-1 Specific Detection
Western Blot Protocol for Discriminating PARP-1 Cleavage
Sample Preparation:
- Harvest cells using gentle lysis without detergents that might activate PARP-1 artificially (RIPA buffer is suitable)
- Include PARP inhibitors in lysis buffer only if measuring basal PARP-1 levels, not cleavage
- Process samples quickly on ice to prevent post-lysis proteolysis
Electrophoresis and Transfer:
- Use 6-15% SDS-polyacrylamide gradient gels to resolve full-length PARP-1 (116 kDa) and cleavage fragments (89 kDa, 24 kDa)
- Transfer to PVDF membranes using standard Western blot protocols
- Confirm transfer efficiency with Ponceau S staining
Antibody Detection:
- Primary antibody: PARP (46D11) Rabbit mAb at 1:1000 dilution in 5% BSA/TBST overnight at 4°C [79]
- Secondary antibody: HRP-conjugated anti-rabbit IgG at 1:5000 dilution for 2 hours at room temperature
- Develop with enhanced chemiluminescence reagents
Specificity Controls:
- Include PARP-1 knockout cell lysates as negative controls
- Test antibody against recombinant PARP-2 and PARP-3 proteins
- Use caspase inhibitor (Z-VAD-FMK) to confirm apoptosis-specific cleavage
- Employ PARP inhibitor (DPQ) to validate parthanatos-related cleavage [66]
Immunofluorescence Protocol for Spatial Localization of PARP-1 Cleavage
Cell Fixation and Permeabilization:
- Fix cells with 4% paraformaldehyde for 15 minutes at room temperature
- Permeabilize with 0.1% Triton X-100 in PBS for 10 minutes
- Block with 5% normal goat serum for 1 hour
Antibody Staining:
- Incubate with PARP (46D11) Rabbit mAb at 1:400 dilution overnight at 4°C
- Use species-appropriate fluorescent secondary antibody (e.g., Alexa Fluor 488) at 1:1000 dilution for 1 hour
- Counterstain nuclei with DAPI
Specificity Validation:
- Include PARP-1 knockout cells as negative controls
- Pre-absorb antibody with immunizing peptide to confirm signal specificity
- Use compartment-specific markers to confirm nuclear localization of full-length PARP-1 and potential cytoplasmic translocation of cleavage fragments
Implications for Therapeutic Development and Disease Research
The development of PARP-1 selective inhibitors represents a significant advancement in cancer therapy, addressing toxicity concerns associated with pan-PARP inhibition [81]. PARP2 inhibition has been linked to hematological toxicity, while synthetic lethality in BRCA-mutated cancers depends primarily on PARP1 inhibition [81]. This therapeutic rationale underscores the importance of distinguishing PARP-1 specific functions in both basic research and drug development.
In neurodegenerative diseases, PARP-1 cleavage fragments serve as specific biomarkers for different cell death pathways [1]. The accurate detection of these fragments helps distinguish between apoptotic and parthanatic cell death, with significant implications for understanding disease mechanisms and developing targeted neuroprotective strategies. Research in cerebral ischemia, traumatic brain injury, and Parkinson's disease has demonstrated the value of specific PARP-1 cleavage detection in understanding pathological processes [1].
Addressing cross-reactivity with other PARP family members is essential for accurate PARP-1 research in programmed cell death. Through the implementation of specific antibodies, genetic controls, selective inhibitors, and validated experimental protocols, researchers can confidently distinguish PARP-1-specific functions and cleavage events from those of other PARP family members. As the field advances toward more precise therapeutic interventions, the tools and methodologies outlined in this technical guide will remain fundamental to rigorous PARP-1 investigation and the development of targeted therapies that maximize efficacy while minimizing off-target effects.
Troubleshooting Sample Preparation for Labile Fragments
Poly(ADP-ribose) polymerase-1 (PARP-1) is a nuclear enzyme that serves as a critical sensor of DNA damage and plays a decisive role in cell fate decisions. Its cleavage by various proteases generates specific fragments that serve as biomarkers for different programmed cell death (PCD) pathways [11]. During massive DNA damage, PARP-1 overactivation can trigger parthanatos, a caspase-independent PCD pathway characterized by poly(ADP-ribose) (PAR) polymer-induced mitochondrial membrane permeabilization and apoptosis-inducing factor (AIF) release [76] [13]. In contrast, caspase-mediated cleavage of PARP-1 during apoptosis generates signature fragments that inhibit DNA repair and facilitate orderly cellular dismantling [11].
The labile nature of PARP-1 fragments presents significant technical challenges for researchers. This guide provides comprehensive troubleshooting methodologies for preparing and analyzing these proteolytically sensitive fragments within the context of PCD research.
PARP-1 Cleavage Fragments: Significance and Characteristics
PARP-1 serves as a substrate for multiple proteases activated in different cell death pathways, yielding characteristic fragments with distinct biological activities [11].
Table 1: PARP-1 Cleavage Fragments in Cell Death Pathways
| Protease | Cleavage Site | Fragments Generated | Cell Death Pathway | Biological Consequences |
|---|---|---|---|---|
| Caspase-3/7 | DEVD²¹⁴ | 24 kDa (DBD) + 89 kDa (CAT) | Apoptosis | Inhibition of DNA repair; 24 kDa fragment acts as trans-dominant inhibitor of PARP-1 [11] |
| Calpain | Multiple sites | 50 kDa + 40 kDa + 24 kDa | Necrosis, excitotoxicity | Alternative cleavage pattern; contributes to bioenergetic collapse [11] |
| Cathepsins | Not specified | 50 kDa + 40 kDa | Lysosomal cell death | Context-dependent fragmentation [11] |
| Granzyme A | Not specified | ~50 kDa | Immune-mediated killing | Unique fragment generation [11] |
| MMPs | Not specified | 50-55 kDa | Extracellular remodeling | Limited information available [11] |
The 89 kDa fragment generated by caspase cleavage can serve as a cytoplasmic PAR carrier, inducing AIF release from mitochondria and mediating crosstalk between apoptosis and parthanatos pathways [13]. Conversely, the 24 kDa DNA-binding domain fragment remains nuclear and irreversibly binds to damaged DNA, blocking repair enzyme access [11].
Critical Challenges in Labile Fragment Preparation
Fragment Instability Factors
- Post-translational modifications: PARP-1 undergoes auto-poly(ADP-ribosyl)ation, which alters fragment mobility and antibody recognition [13]
- Rapid degradation: Cleavage fragments are susceptible to secondary proteolysis by other activated proteases
- Experimental artifacts: Fragments can degrade during sample preparation if protease inhibitors are inadequate
- Modification-dependent migration: PARylated fragments exhibit altered electrophoretic mobility [13]
Pathway Cross-talk Complications
PARP-1 cleavage occurs in multiple cell death contexts, creating interpretation challenges:
- Caspase-dependent apoptosis: Generates classic 89 kDa and 24 kDa fragments [11]
- Parthanatos: Features PARP-1 overactivation and PAR polymer formation prior to cleavage [76]
- Hybrid cell death: May involve simultaneous activation of multiple proteases [11]
Optimized Experimental Protocols
Lysis Buffer Formulation for Fragment Preservation
Recommended Composition:
- 50 mM Tris-HCl (pH 7.4)
- 150 mM NaCl
- 1% NP-40 or Triton X-100
- 0.5% Sodium deoxycholate
- 0.1% SDS
- 1 mM EDTA
- Protease inhibitors: 1 mM PMSF, 10 µg/mL aprotinin, 10 µg/mL leupeptin, 1 µg/mL pepstatin A
- PARP activity modulators: 10 µM PJ34 (PARP inhibitor) or 1 mM NAD+ (PARP substrate)
- Phosphatase inhibitors: 1 mM NaF, 1 mM Na₃VO₄ for phosphorylation studies
Critical Notes:
- Avoid repeated freeze-thaw cycles of lysates
- Process samples immediately on ice
- Pre-chill all centrifuges to 4°C
- Consider adding PARP inhibitors to prevent auto-modification during processing
PARP-1 Cleavage Detection by Western Blotting
Electrophoresis Conditions:
- Gradient gel: 4-20% SDS-PAGE for optimal separation of 24 kDa and 89 kDa fragments
- Loading: 20-50 µg protein per lane
- Transfer: Low-molecular-weight optimized conditions (low methanol, extended transfer time)
Antibody Selection Strategy:
- N-terminal antibodies: Detect 24 kDa fragment (DNA-binding domain)
- C-terminal antibodies: Detect 89 kDa fragment (catalytic domain)
- PAR antibodies: Detect PARylated fragments (parthanatos indicator) [76]
Visualization Enhancement:
- Use high-sensitivity chemiluminescent substrates
- Include positive controls (etoposide-treated cells for apoptosis, MNNG-treated cells for parthanatos)
- Run full-length PARP-1 standard for reference
Cell Death Induction Models for PARP-1 Cleavage Studies
Table 2: Experimental Models for PARP-1 Cleavage Studies
| Inducer | Concentration | Exposure Time | Primary Cell Death Pathway | Expected Fragments | Validation Markers |
|---|---|---|---|---|---|
| Staurosporine | 0.5-2 µM | 4-8 hours | Apoptosis [13] | 89 kDa + 24 kDa | Caspase-3 activation, PS externalization |
| MNNG | 50-200 µM | 15-60 minutes | Parthanatos [76] | PAR polymers > fragments | AIF translocation, PAR accumulation |
| H₂O₂ | 0.5-2 mM | 30-120 minutes | Oxidative stress-dependent | Variable | 8-OHdG, lipid peroxidation |
| TNF-α + CHX | 10-50 ng/mL + 10 µg/mL | 6-18 hours | Necroptosis/apoptosis | Context-dependent | RIPK1 phosphorylation, MLKL activation |
Protocol for Differentiating Apoptosis from Parthanatos
Sequential Assessment Method:
- Measure caspase activation (caspase-3/7 activity assays)
- Detect PAR accumulation (immunofluorescence with PAR antibodies)
- Monitor AIF localization (cell fractionation + Western blotting)
- Assess mitochondrial membrane potential (JC-1 or TMRM staining)
Interpretation Guide:
- Apoptosis: Caspase-3 activation + PARP-1 cleavage (89/24 kDa) + no AIF translocation
- Parthanatos: Minimal caspase activation + PAR accumulation + AIF nuclear translocation
- Hybrid death: Mixed characteristics with both caspase activation and PAR accumulation
Research Reagent Solutions
Table 3: Essential Reagents for PARP-1 Fragment Studies
| Reagent Category | Specific Examples | Function/Application | Technical Notes |
|---|---|---|---|
| PARP inhibitors | PJ34, Olaparib, Niraparib [76] [82] | Inhibit PARP activity; prevent auto-modification | Use varying concentrations based on purpose (low for preservation, high for inhibition studies) |
| Caspase inhibitors | Z-VAD-FMK, Q-VD-OPh | Block apoptosis-mediated cleavage | Use pan-caspase or specific inhibitors depending on research question |
| Protease inhibitors | Complete Mini tablets, PMSF, leupeptin | Prevent artifactual proteolysis | Include in all lysis and storage buffers |
| PAR antibodies | 10H, polyclonal anti-PAR | Detect PAR polymer formation | Essential for parthanatos identification [76] |
| PARP-1 antibodies | N-terminal specific, C-terminal specific, full-length | Detect specific fragments | Validate antibody epitopes for fragment identification |
| Activity assays | NAD+ consumption, PAR synthesis | Measure PARP activation | Correlate with fragmentation patterns |
| Cell death inducers | Staurosporine, MNNG, H₂O₂ [13] [76] | Activate specific cell death pathways | Use multiple inducers to confirm pathway specificity |
Visualization of PARP-1 Cleavage Pathways
Diagram 1: PARP-1 Cleavage in Cell Death Pathways
Diagram 2: Experimental Workflow for Fragment Analysis
Troubleshooting Common Technical Issues
Problem 1: Missing or Faint Fragment Detection
Potential Causes:
- Inadequate protease inhibition during sample preparation
- Over- or under-expression of PARP-1 due to cell type variability
- Antibody epitope masking by PAR modifications
Solutions:
- Validate protease inhibitor efficacy using activity assays
- Include positive control cells with known PARP-1 expression
- Treat blots with PARG to remove PAR chains before antibody probing
Problem 2: Non-Specific Banding Patterns
Potential Causes:
- Cross-reactivity with PARP-1 isoforms or unrelated proteins
- Partial degradation generating intermediate fragments
- Post-translational modifications altering mobility
Solutions:
- Use multiple antibodies targeting different epitopes
- Include PARP-1 knockout cells as negative controls
- Optimize gel conditions for better fragment separation
Problem 3: Inconsistent Results Between Experiments
Potential Causes:
- Variations in cell confluence at treatment time
- Subtle differences in inducer preparation or storage
- Environmental factors affecting cell stress levels
Solutions:
- Standardize cell culture conditions precisely
- Prepare fresh inducer stocks and use consistent concentrations
- Include internal controls in each experiment
Successful preparation and analysis of labile PARP-1 fragments requires meticulous attention to experimental conditions and understanding of the cell death context. The protocols outlined herein provide a framework for obtaining reproducible, interpretable data that can distinguish between apoptosis, parthanatos, and hybrid cell death pathways. As research in programmed cell death advances, these methodologies will enable more precise dissection of PARP-1's multifaceted roles in cellular fate decisions and contribute to therapeutic development targeting specific cell death pathways.
In programmed cell death research, the cleavage of Poly(ADP-ribose) polymerase 1 (PARP-1) serves as a critical biochemical marker, facilitating the distinction between apoptosis and other cell death pathways. During apoptosis, executioner caspases-3 and -7 specifically cleave PARP-1 at the DEVD214↓G215 site, generating characteristic 24 kDa and 89 kDa fragments [39] [11] [83]. This proteolytic event separates the N-terminal DNA-binding domain (DBD) from the C-terminal catalytic domain, effectively inactivating PARP-1's DNA repair function and committing the cell to apoptotic death [11] [23]. Accurate quantification of this cleavage process provides researchers with a powerful tool for assessing apoptosis efficiency in response to various stimuli, including chemotherapeutic agents, DNA damage, and developmental cues. This technical guide outlines standardized methodologies and normalization strategies for precise quantification of PARP-1 cleavage efficiency, enabling robust and reproducible measurement in diverse experimental systems.
Biological Significance of PARP-1 Cleavage
Functional Consequences of Cleavage
PARP-1 cleavage represents more than merely a biomarker; it constitutes a decisive biochemical switch that regulates cell fate through multiple mechanisms:
Inactivation of DNA Repair: The 24 kDa fragment containing the DBD remains nuclear and acts as a trans-dominant inhibitor of DNA repair by irreversibly binding to DNA strand breaks, thereby blocking recruitment of intact PARP-1 and other repair factors [11].
Altered Signaling Pathways: The 89 kDa fragment, containing the automodification and catalytic domains, translocates to the cytoplasm where it can participate in novel signaling functions [13] [23]. Recent research indicates this fragment may serve as a cytoplasmic PAR carrier that induces AIF-mediated apoptosis (parthanatos) and can ADP-ribosylate RNA Polymerase III, potentially linking apoptosis to innate immune responses [13] [23].
Differential Biological Activities: Studies expressing individual cleavage fragments demonstrate their opposing biological roles. The 24 kDa fragment confers protection from ischemic damage, while the 89 kDa fragment exhibits cytotoxic properties and enhances pro-inflammatory NF-κB activity [39].
PARP-1 Cleavage in Different Cell Death Modalities
The detection of PARP-1 cleavage fragments helps distinguish between apoptosis and alternative cell death pathways:
Apoptosis: Characterized by caspase-dependent cleavage producing the classic 89 kDa and 24 kDa fragments [11] [83].
Necrosis: Typically involves PARP-1 hyperactivation leading to NAD+ and ATP depletion rather than specific cleavage [84] [14].
Parthanatos: May involve caspase-independent PARP-1 activation and PAR-mediated cell death, though cleavage fragments may still form through secondary caspase activation [13].
Table 1: PARP-1 Proteolytic Fragments in Research
| Fragment Size | Domains Contained | Localization | Function |
|---|---|---|---|
| 24 kDa | Zinc fingers 1 & 2, NLS [39] [11] | Nuclear | Dominant-negative inhibitor of DNA repair [11] |
| 89 kDa | Zinc finger 3, BRCT, WGR, CAT [39] [13] | Cytoplasmic | Cytotoxic; induces AIF release; ADP-ribosylates Pol III [39] [13] [23] |
Quantitative Approaches for PARP-1 Cleavage Assessment
Immunoblotting and Densitometric Analysis
Western blotting remains the gold standard for PARP-1 cleavage quantification, providing both fragment size confirmation and relative abundance measurement:
Antibody Selection: Utilize antibodies specific for either the cleavage site (e.g., Asp214) detecting only the 89 kDa fragment, or antibodies recognizing epitopes in both full-length and cleaved PARP-1 for comprehensive assessment [83].
Gel Electrophoresis: Employ 4-12% Bis-Tris gradient gels with precise molecular weight markers to resolve the 116 kDa full-length PARP-1 from the 89 kDa fragment [39] [83].
Detection Method: Prefer chemiluminescent substrates with wide linear dynamic range, or advanced digital imaging systems for superior quantitation [39].
Calculation Methods for Cleavage Efficiency
Multiple calculation approaches enable researchers to express cleavage efficiency according to experimental requirements:
Cleavage Fragment Ratio:
[89 kDa band intensity] / [116 kDa band intensity]- Provides direct measure of cleavage extent [39].Percentage Cleavage:
[89 kDa intensity] / [116 kDa + 89 kDa intensities] × 100- Intuitive percentage metric [39].Normalized Cleavage Index:
[89 kDa intensity] / [Loading Control Intensity]- Controls for sample-to-sample variation [39].
Table 2: Normalization Strategies for PARP-1 Cleavage Quantification
| Normalization Approach | Calculation Method | Advantages | Limitations |
|---|---|---|---|
| Total Protein | Cleavage signal/total protein stain | Accounts for total protein loading; unaffected by biological variability [39] | May not reflect cellularity; requires additional staining step |
| Housekeeping Proteins | Cleavage signal/GAPDH, β-actin, etc. | Widely adopted; minimal optimization [39] | Housekeeping protein levels may vary under experimental conditions |
| Internal Reference Control | Cleavage signal in treated/untreated control | Direct fold-change calculation; ideal for dose-response [39] | Requires parallel untreated controls |
| DNA Content | Cleavage signal/cell number or DNA content | Normalizes to cellularity; useful in cell death studies [39] | Requires additional quantification method |
Experimental Protocols for PARP-1 Cleavage Analysis
Cell Culture and Apoptosis Induction
Materials:
- SH-SY5Y human neuroblastoma cells or primary rat cortical neurons [39]
- Staurosporine (0.5-1 μM, 4-16 hours) or Actinomycin D (0.5-2 μM, 8-24 hours) [13]
- Oxygen/Glucose Deprivation (OGD) system for ischemia models [39]
Procedure:
- Culture cells in appropriate medium (DMEM complete for SH-SY5Y; Neurobasal-A with B27 for cortical neurons) [39]
- At 70-80% confluence, treat with apoptosis inducer for optimized duration
- Include vehicle-treated controls for baseline PARP-1 assessment
- Harvest cells at predetermined time points post-treatment
Protein Extraction and Western Blotting
Materials:
- RIPA lysis buffer with protease inhibitors [39]
- BCA or Bradford protein assay kit
- 4-12% Bis-Tris gradient gels
- PVDF or nitrocellulose membranes
- Cleaved PARP (Asp214) antibody (e.g., Cell Signaling Technology #5625) [83]
- HRP-conjugated secondary antibodies
- Enhanced chemiluminescence substrate
Procedure:
- Lyse cells in RIPA buffer (20 min, 4°C), centrifuge (14,000 × g, 15 min), collect supernatant [39]
- Quantify protein concentration, adjust samples to equal concentration
- Load 20-40 μg protein per lane, separate by SDS-PAGE (120V, 90 min)
- Transfer to membrane (100V, 60 min or 30V overnight)
- Block with 5% non-fat milk in TBST (60 min, RT)
- Incubate with primary antibody (1:1000 dilution, 4°C overnight) [83]
- Wash (3 × 10 min TBST), incubate with HRP-secondary (1:2000, 60 min RT)
- Develop with ECL substrate, image with digital documentation system
In Situ Fractionation for PARP-1 Localization Studies
For subcellular localization of PARP-1 fragments during apoptosis:
Materials:
- CSK buffer: 10 mM PIPES (pH 6.8), 100 mM NaCl, 300 mM sucrose, 3 mM MgCl₂ [85]
- CSK+T buffer: CSK + 0.5% Triton X-100 [85]
- CSK+T+S buffer: CSK + 0.5% Triton X-100 + 0.42 M NaCl [85]
Procedure:
- Gently permeabilize cells with CSK buffer (5 min, 4°C)
- Extract with CSK+T buffer (5 min, 4°C) to remove soluble proteins
- Extract with CSK+T+S buffer (5 min, 4°C) to remove loosely-bound nuclear proteins
- Fix remaining chromatin-associated proteins with 4% formaldehyde [85]
- Proceed with immunostaining for PARP-1 fragments and microscopy
Visualization of PARP-1 Cleavage Signaling Pathways
Diagram 1: PARP-1 Cleavage Apoptotic Pathway
Experimental Workflow for Cleavage Efficiency Quantification
Diagram 2: Cleavage Efficiency Quantification Workflow
Research Reagent Solutions for PARP-1 Cleavage Studies
Table 3: Essential Research Reagents for PARP-1 Cleavage Analysis
| Reagent Category | Specific Examples | Function/Application | Key Features |
|---|---|---|---|
| Cleavage-Specific Antibodies | Cleaved PARP (Asp214) (D64E10) Rabbit mAb #5625 [83] | Detects 89 kDa fragment; apoptosis-specific | Does not recognize full-length PARP-1; suitable for WB, IHC, IF, FC [83] |
| PARP-1 Expression Constructs | PARP-1WT, PARP-1UNCL (uncleavable), PARP-124, PARP-189 [39] | Functional studies of cleavage fragments | Tetracycline-inducible systems; AAV delivery for primary neurons [39] |
| Apoptosis Inducers | Staurosporine, Actinomycin D [13]; Oxygen/Glucose Deprivation [39] | Activate caspases to initiate PARP-1 cleavage | Staurosporine: broad-spectrum kinase inhibitor; OGD: physiological ischemia model [39] [13] |
| Caspase Inhibitors | Z-VAD-FMK (pan-caspase inhibitor) | Confirm caspase-dependence of cleavage | Validates specific cleavage mechanism [11] |
| Protease Inhibitor Cocktails | PMSF, leupeptin, aprotinin [39] | Prevent post-lysis proteolysis | Maintain fragment integrity during processing [39] |
| Cell Fractionation Reagents | CSK buffers with Triton X-100 and NaCl [85] | Subcellular localization of fragments | Sequential extraction to distinguish free vs. chromatin-bound PARP-1 [85] |
Advanced Applications and Technical Considerations
PARP-1 Cleavage in Disease Models and Drug Development
The quantification of PARP-1 cleavage efficiency finds critical applications across multiple research domains:
Oncology Research: Assessment of chemotherapeutic efficacy through apoptosis induction in cancer cells; evaluation of PARP inhibitor compounds that modulate cleavage dynamics [86].
Neurodegenerative Disease Models: Analysis of PARP-1 cleavage patterns in cerebral ischemia, excitotoxicity, and other neurological insults [39] [11].
Inflammatory Pathways: Investigation of connections between PARP-1 cleavage and NF-κB-mediated inflammation, particularly with the 89 kDa fragment enhancing pro-inflammatory responses [39].
Drug Development: Screening for novel PROTAC molecules (e.g., 180055) that degrade PARP-1 without DNA trapping effects, representing next-generation therapeutic approaches [86].
Troubleshooting and Method Validation
Ensure reliable PARP-1 cleavage quantification through these quality control measures:
Fragment Specificity Confirmation: Include caspase inhibitor controls to verify cleavage is apoptosis-specific [11].
Antibody Validation: Confirm antibody specificity using PARP-1 knockout cells or siRNA knockdown approaches [39].
Linearity Assessment: Perform serial dilutions to ensure band intensities fall within the linear detection range [39].
Multiple Detection Methods: Correlate immunoblot findings with complementary techniques such as immunofluorescence for subcellular localization or flow cytometry for single-cell analysis [83].
Standardized quantification of PARP-1 cleavage efficiency provides invaluable insights into apoptotic signaling across diverse research contexts. The methodologies outlined in this technical guide—from experimental design through data normalization and interpretation—enable precise assessment of this critical cell death marker. As research continues to reveal novel functions for PARP-1 fragments beyond their traditional roles, particularly in inflammatory signaling and cytoplasmic functions, accurate cleavage quantification becomes increasingly important for understanding the full biological significance of PARP-1 proteolysis in programmed cell death.
Comparative Analysis of PARP-1 Cleavage Across Cell Death Pathways
Poly(ADP-ribose) polymerase-1 (PARP-1) is a multifunctional nuclear enzyme that plays a decisive role in determining cellular fate in response to stress. As a key DNA damage sensor, PARP-1's activity can guide cells toward survival or death through distinct pathways. Within programmed cell death research, PARP-1 functions as a critical molecular switch whose specific activation mechanism directs cells toward two principal death pathways: caspase-mediated apoptosis or PAR-mediated parthanatos. The dichotomy between PARP-1 cleavage and overactivation represents a fundamental regulatory mechanism that determines the mode of cell death. This technical guide examines the molecular mechanisms, signaling pathways, and experimental approaches for investigating PARP-1's dual roles in these distinct cell death processes, providing a framework for researchers exploring therapeutic interventions in cancer, neurodegeneration, and other diseases involving dysregulated cell death.
PARP-1 Structure and Functional Domains
PARP-1 is a 116-kDa protein consisting of 1,014 amino acids with three primary functional domains that dictate its activity in cell death pathways [25] [14]. Understanding this structural organization is essential for interpreting its differential roles in apoptosis and parthanatos.
DNA-Binding Domain (DBD): The N-terminal 46-kDa domain contains three zinc-finger motifs that recognize DNA damage. Zinc fingers 1 and 2 function as "nick sensors" that bind to DNA strand breaks, while zinc finger 3 facilitates interdomain contact and assembly of the DNA-activated conformation [25]. This domain also contains the nuclear localization signal (NLS) and the aspartate-glutamate-valine-aspartic acid (DEVD) motif that serves as the caspase cleavage site [14].
Automodification Domain (AMD): The central 22-kDa domain contains a BRCT (breast cancer C-terminal) motif found in DNA repair proteins and serves as a regulatory segment. It includes glutamate residues (Glu 488 and 491) and serine residues (Ser 499, 507, and 519) that function as auto-poly(ADP-ribosyl)ation sites [25] [14]. This domain facilitates protein-protein interactions and PARP-1 homodimerization or heterodimerization with PARP-2 [25].
Catalytic Domain (CAT): The C-terminal 54-kDa domain executes the enzyme's primary function by synthesizing poly(ADP-ribose) (PAR) polymers using NAD+ as substrate. It contains the highly conserved "PARP signature" motif present in all PARP family members and includes the α-helical subdomain (HD) and ADP-ribosyl transferase (ART) subdomain [25] [14]. The WGR domain within this region interacts with DNA and other domains to form an inter-regional network linking damaged DNA to catalytic activity [14].
Table 1: PARP-1 Structural Domains and Their Functions
| Domain | Molecular Weight | Key Structural Features | Primary Functions |
|---|---|---|---|
| DNA-Binding Domain (DBD) | 46 kDa | Three zinc-finger motifs, NLS, DEVD caspase cleavage site | DNA damage recognition, nuclear localization, caspase targeting |
| Automodification Domain (AMD) | 22 kDa | BRCT motif, glutamate and serine modification sites | Protein-protein interactions, dimerization, auto-modification |
| Catalytic Domain (CAT) | 54 kDa | WGR domain, HD and ART subdomains, PARP signature motif | PAR synthesis from NAD+, catalytic activity execution |
PARP-1 in Apoptosis: Cleavage and Inactivation
Caspase-Mediated Cleavage Mechanism
In caspase-dependent apoptosis, PARP-1 undergoes proteolytic cleavage that inactivates its DNA repair function and facilitates programmed cell death. Executioner caspases-3 and -7 recognize the DEVD motif (amino acid sequence 211-DEVD-214) located in the DBD, cleaving PARP-1 into specific signature fragments [11] [13]. This cleavage event produces two predominant fragments: a 24-kDa fragment containing the DBD with two zinc-finger motifs, and an 89-kDa fragment containing the AMD and CAT domains [11]. The 24-kD fragment retains DNA binding capacity but lacks catalytic function, while the 89-kD fragment has diminished DNA binding ability due to separation from the DBD [11].
Functional Consequences of Cleavage
The biological significance of PARP-1 cleavage in apoptosis serves multiple purposes. The 24-kDa fragment remains tightly bound to DNA strand breaks, acting as a trans-dominant inhibitor of DNA repair by blocking access of intact PARP-1 and other repair enzymes to damage sites [11]. This irreversible binding conserves cellular ATP pools that would otherwise be depleted by PARP-1 activation and subsequent PAR synthesis [11]. Simultaneously, the inactivation of PARP-1's DNA repair function prevents futile repair attempts in committed apoptotic cells, allowing the apoptotic program to proceed efficiently. Recent research has also revealed that the 89-kDa fragment may serve as a cytoplasmic PAR carrier under certain conditions, potentially inducing AIF release from mitochondria and creating crosstalk between apoptotic and parthanatos pathways [13].
PARP-1 in Parthanatos: Overactivation and PAR-Mediated Cell Death
Hyperactivation and Energy Depletion
Parthanatos represents a distinct form of programmed cell death characterized by PARP-1 overactivation following severe DNA damage. Unlike apoptosis, parthanatos occurs independently of caspases and involves massive synthesis of PAR polymers that initiate a specific death signaling cascade [66]. When DNA damage exceeds repairable levels, PARP-1 undergoes hyperactivation, consuming NAD+ at an accelerated rate to synthesize extensive PAR chains on itself and target proteins [25] [87]. This excessive PAR synthesis depletes intracellular NAD+ pools, forcing the cell to consume ATP to regenerate NAD+, resulting in severe energy depletion [87]. The consequent collapse of cellular energy status ultimately leads to loss of ionic homeostasis and membrane integrity, characteristic of necrotic cell death.
PAR Signaling and Mitochondrial Disruption
The key mediator in parthanatos is the PAR polymer itself, which functions as a death signaling molecule. Following massive PAR synthesis, PAR polymers are released from nuclear proteins and translocate to the cytoplasm [13] [66]. Within the cytoplasm, PAR directly interacts with apoptosis-inducing factor (AIF), facilitating its release from mitochondria [13] [66]. Mitochondrial AIF translocation to the nucleus triggers large-scale DNA fragmentation and chromatin condensation, culminating in cell death that displays morphological features distinct from both apoptosis and necrosis [13] [66]. This PAR-mediated death pathway represents a self-amplifying cycle where DNA damage initiates PARP-1 activation, leading to PAR-induced mitochondrial disruption that potentially causes additional DNA damage.
Table 2: Comparative Analysis of PARP-1 in Apoptosis vs. Parthanatos
| Parameter | Apoptosis | Parthanatos |
|---|---|---|
| Initial Trigger | Mild to moderate DNA damage, developmental cues | Severe genotoxic stress, excessive ROS |
| PARP-1 Activity | Transient activation followed by cleavage and inactivation | Sustained overactivation and hyperactivity |
| Key Proteases | Caspases-3 and -7 | Calpains (caspase-independent) |
| PARP-1 Fragments | 24-kDa (DBD) + 89-kDa (AMD+CAT) | Not systematically characterized |
| Energy Status | ATP preservation | NAD+/ATP depletion |
| PAR Synthesis | Limited and transient | Massive and sustained |
| Mitochondrial Involvement | Cytochrome c release, caspase activation | AIF release and nuclear translocation |
| Morphological Features | Membrane blebbing, chromatin condensation, apoptotic bodies | Loss of membrane integrity, necrotic features |
Quantitative Assessment of Metabolic Changes
The metabolic consequences of PARP-1 activation differ dramatically between apoptosis and parthanatos, particularly regarding cellular energy status. Quantitative analysis of nucleotide levels following PARP-1 activation reveals distinct patterns that differentiate these death pathways.
Table 3: Temporal Changes in Energy Metabolites Following PARP-1 Activation
| Time Post-MNNG | NAD+ Levels | ATP Levels | AMP Levels | AMP/ATP Ratio | PARP-1 Inhibitor Effect |
|---|---|---|---|---|---|
| 5 minutes | ~80% of control | ~95% of control | ~110% of control | 0.006 | Partial NAD+ protection |
| 15 minutes | ~20% of control | ~30% of control | 2,000% of control | 0.128 | Complete prevention of ATP loss |
| 30 minutes | ~15% of control | ~10% of control | ~1,500% of control | 0.320 | Maintains normal AMP/ATP ratio |
| 60 minutes | ~10% of control | ~5% of control | ~800% of control | 0.128 | Complete metabolic protection |
Data derived from HEK293 cells treated with 100 μM MNNG (alkylating agent) with or without PARP inhibitor AG14361 [87]. The dramatic increase in AMP levels is attributed to PAR degradation by PARG and NUDIX enzymes following massive PAR synthesis [87].
Experimental Approaches and Methodologies
Detecting PARP-1 Cleavage and Activation
Western Blot Analysis for Cleavage Fragments: The canonical method for detecting apoptotic PARP-1 cleavage involves Western blotting using antibodies against specific PARP-1 domains. The appearance of the 89-kDa fragment (detected with antibodies against the CAT domain) and the 24-kDa fragment (detected with antibodies against the DBD) serves as a biochemical marker of apoptosis [11] [13]. Protocol: Extract proteins using RIPA lysis buffer with protease inhibitors, separate 20-40 μg total protein on 6-15% SDS-polyacrylamide gels, transfer to PVDF membranes, block with 5% non-fat milk, and incubate with primary antibodies (e.g., anti-PARP-1, Santa Cruz sc-7150) overnight at 4°C [66]. Detect using appropriate HRP-conjugated secondary antibodies and enhanced chemiluminescence reagents.
PAR Immunodetection: For parthanatos assessment, monitor PAR formation using anti-PAR antibodies (e.g., mouse anti-PAR polymer, USBio 045159) [66] [87]. Immunofluorescence staining reveals nuclear PAR accumulation followed by cytoplasmic translocation, while Western blotting detects high-molecular-weight PAR polymers.
Cell Viability Assays: Distinguish death modalities using specific inhibitors combined with viability measurements. Employ PARP inhibitors (e.g., AG14361, DPQ) for parthanatos, and caspase inhibitors (e.g., Z-VAD-FMK) for apoptosis [66] [87]. Use CCK-8 assays for viability quantification and Annexin V/PI staining for death modality characterization by flow cytometry.
Metabolic and Energetic Assessment
Nucleotide Measurements: Quantify NAD+/NADH levels using specific assay kits (e.g., Suzhou Ered Biological Technology Co. Ltd) following manufacturer protocols [66] [87]. Measure ATP levels using luciferase-based ATP assay kits (e.g., Beyotime Biotechnology). Monitor AMP accumulation as an indicator of PAR turnover and energy stress.
Mitochondrial Assessment: Evaluate mitochondrial membrane potential using JC-1 dye (Beyotime Biotechnology) with fluorescence shift from red (aggregates) to green (monomers) indicating depolarization [66]. Detect AIF translocation by subcellular fractionation followed by Western blotting with anti-AIF antibodies (Abcam ab110327) or by immunofluorescence microscopy.
Research Reagent Solutions
Table 4: Essential Research Reagents for PARP-1 Cell Death Studies
| Reagent Category | Specific Examples | Application and Function |
|---|---|---|
| PARP Inhibitors | AG14361, DPQ, 3-AB | Inhibit PARP catalytic activity, prevent parthanatos |
| Caspase Inhibitors | Z-VAD-FMK (pan-caspase), DEVD-CHO (caspase-3/7) | Block apoptotic PARP-1 cleavage, confirm caspase-dependence |
| DNA Damaging Agents | MNNG (alkylator), H₂O₂ (oxidative stress), Cadmium acetate | Activate PARP-1, induce specific death pathways |
| Antibodies | Anti-PARP-1 (Santa Cruz sc-7150), anti-PAR (USBio 045159), anti-AIF (Abcam ab110327) | Detect cleavage, activation, and subcellular localization |
| Viability/Cytotoxicity Assays | CCK-8, Annexin V-FITC/PI, LDH release | Quantify cell death and distinguish modalities |
| Metabolic Assay Kits | NAD+/NADH Assay Kit, ATP Assay Kit, JC-1 Mitochondrial Potential Kit | Assess bioenergetic status and mitochondrial function |
| siRNA/shRNA | PARP-1-specific siRNA (Invitrogen) | Genetically validate PARP-1 role in death pathways |
Signaling Pathway Visualization
Discussion and Research Implications
The dichotomy between PARP-1 cleavage in apoptosis and overactivation in parthanatos represents a critical regulatory mechanism in cell fate determination. The decision between these pathways appears to depend on the intensity and duration of DNA damage, with moderate damage favoring apoptosis and severe, irreparable damage triggering parthanatos [25] [87]. Recent research has revealed unexpected complexity in these pathways, including potential crosstalk mechanisms. The discovery that caspase-mediated cleavage can generate poly(ADP-ribosyl)ated 89-kDa fragments that translocate to the cytoplasm and potentially contribute to AIF release suggests overlapping mechanisms between the two death pathways [13].
From a therapeutic perspective, PARP-1 represents a promising target for various pathological conditions. In cancer therapy, PARP inhibitors are already employed to induce synthetic lethality in BRCA-deficient tumors, but understanding their context-specific effects on cell death pathways remains crucial [18]. In neurodegenerative diseases and acute injury models, where parthanatos contributes to pathology, developing inhibitors that specifically block the cell death-promoting functions of PARP-1 without compromising its DNA repair capabilities represents an important research direction. The emerging understanding of PARP-1's role in different cell death modalities continues to provide new insights for therapeutic interventions across a spectrum of human diseases.
Programmed cell death (PCD) is a fundamental biological process essential for maintaining cellular homeostasis, embryonic development, and host defense. Among the various regulated cell death pathways, apoptosis, necroptosis, and pyroptosis represent the most extensively characterized mechanisms, each with distinct molecular signatures and functional consequences [88]. These pathways function as crucial defense mechanisms against pathogens and cellular stress, with their dysregulation contributing to numerous diseases, including cancer, neurodegenerative disorders, and inflammatory conditions [89] [90].
The intricate interplay between these cell death modalities is orchestrated by specialized molecular machinery, with caspases serving as central regulators that determine cellular fate [89]. A critical biochemical event intersecting these pathways is the cleavage of poly(ADP-ribose) polymerase-1 (PARP-1), a nuclear DNA repair enzyme whose proteolytic inactivation serves as a hallmark of apoptotic commitment and a point of crosstalk between different cell death modalities [14] [76]. This whitepaper provides a comprehensive technical comparison of apoptosis, necroptosis, and pyroptosis, with particular emphasis on their molecular regulators, experimental methodologies, and the contextual role of PARP-1 cleavage within the broader landscape of PCD research.
Molecular Mechanisms and Key Regulators
Apoptosis: The Silent Pathway
Apoptosis is a non-lytic, immunologically silent form of PCD characterized by cell shrinkage, chromatin condensation, DNA fragmentation, and formation of apoptotic bodies that are efficiently cleared by phagocytes [88] [91]. This pathway proceeds through two main molecular cascades:
- Extrinsic Pathway: Initiated by ligand binding to death receptors (e.g., Fas, TNFR1) leading to formation of the death-inducing signaling complex (DISC) and activation of caspase-8 [92] [91].
- Intrinsic Pathway: Triggered by intracellular stressors (e.g., DNA damage, oxidative stress) that cause mitochondrial outer membrane permeabilization (MOMP), cytochrome c release, and formation of the apoptosome complex, resulting in caspase-9 activation [92].
Both pathways converge on the activation of executioner caspases-3 and -7, which systematically cleave cellular substrates, including PARP-1 [76] [91]. Caspase-mediated cleavage of PARP-1 serves to conserve cellular energy by preventing NAD+ depletion and shifts cellular fate from survival to death [14] [76].
Necroptosis: Programmed Necrosis
Necroptosis represents a lytic, inflammatory form of PCD that typically occurs when caspase activity, particularly caspase-8, is inhibited [88] [90]. This pathway is characterized by cellular swelling, plasma membrane rupture, and release of damage-associated molecular patterns (DAMPs) that trigger robust inflammatory responses [88].
The core molecular machinery of necroptosis centers on the kinase cascade involving RIPK1, RIPK3, and the terminal effector MLKL [88] [90]. Phosphorylated MLKL undergoes oligomerization and translocates to the plasma membrane, where it forms pores that disrupt ionic homeostasis and ultimately cause membrane rupture [88]. Unlike apoptosis, necroptosis does not typically involve PARP-1 cleavage; rather, PARP-1 overactivation can contribute to a distinct cell death pathway known as parthanatos [76].
Pyroptosis: Inflammatory Cell Death
Pyroptosis is a highly inflammatory form of lytic cell death primarily executed by members of the gasdermin protein family, particularly GSDMD [89] [90]. This pathway is characterized by plasma membrane pore formation, cellular swelling, osmotic lysis, and release of proinflammatory cytokines (IL-1β, IL-18) and cellular contents [90].
The molecular triggers for pyroptosis include:
- Canonical Inflammasome Activation: Pathogen-associated molecular patterns (PAMPs) or DAMPs activate inflammasome sensors (e.g., NLRP3, AIM2), leading to caspase-1 activation, which cleaves and activates GSDMD and pro-inflammatory cytokines [90].
- Non-canonical Inflammasome Activation: Intracellular lipopolysaccharide (LPS) directly activates human caspases-4/5 or murine caspase-11, which subsequently cleave GSDMD [90].
Recent evidence indicates that certain caspases, particularly caspase-3, can cleave other gasdermin family members (e.g., GSDME) to initiate pyroptosis, demonstrating intriguing crossover between apoptotic and pyroptotic signaling [89].
Table 1: Comparative Analysis of Programmed Cell Death Pathways
| Feature | Apoptosis | Necroptosis | Pyroptosis |
|---|---|---|---|
| Morphology | Cell shrinkage, membrane blebbing, apoptotic bodies | Cellular swelling, organelle edema, membrane rupture | Cellular swelling, plasma membrane pore formation, lysis |
| Inflammatory Response | Non-inflammatory | Highly inflammatory | Highly inflammatory |
| Key Initiators | Caspase-8 (extrinsic), Caspase-9 (intrinsic) | RIPK1, RIPK3 (upon caspase inhibition) | Caspase-1/4/5/11, GSDMD |
| Key Effectors | Caspase-3/7, BAX/BAK | MLKL | GSDMD-N terminal pores |
| PARP-1 Cleavage | Yes (by caspase-3/7) | No | Context-dependent |
| Physiological Role | Development, tissue homeostasis, immune tolerance | Host defense, tissue repair | Anti-pathogen defense, inflammation |
PARP-1 Cleavage: A Strategic Nexus in Cell Death Decision
PARP-1 is a multifunctional nuclear enzyme comprising three primary domains: an N-terminal DNA-binding domain (DBD), a central automodification domain (AMD), and a C-terminal catalytic domain (CAT) [25] [14]. Upon detecting DNA damage, PARP-1 becomes activated and catalyzes the synthesis of poly(ADP-ribose) (PAR) chains using NAD+ as a substrate, facilitating DNA repair through recruitment of repair machinery [25] [18].
The cleavage of PARP-1 by executioner caspases-3 and -7 at specific aspartic acid residues (within the DEVD motif) serves as a definitive biochemical marker of apoptosis [14] [76]. This proteolytic event separates the N-terminal DNA-binding domain from the C-terminal catalytic domain, effectively inactivating the enzyme and preventing futile DNA repair attempts and NAD+ depletion during apoptotic execution [76].
The functional consequences of PARP-1 cleavage include:
- Energy Conservation: Prevents excessive NAD+ and ATP consumption in doomed cells [76]
- Apoptosis Promotion: Facilitates the dismantling of cellular infrastructure [14]
- Necrosis Prevention: Redirects cell death toward the non-inflammatory apoptotic pathway [76]
In contrast to apoptosis, PARP-1 remains intact during canonical necroptosis. However, PARP-1 overactivation due to severe DNA damage can trigger an alternative cell death pathway known as parthanatos, which involves PAR-mediated translocation of apoptosis-inducing factor (AIF) from mitochondria to the nucleus, culminating in chromatin condensation and large-scale DNA fragmentation [76]. The role of PARP-1 in pyroptosis remains less defined but may involve contextual crosstalk between different cell death modalities.
Experimental Methodologies for Cell Death Research
Detection of PARP-1 Cleavage
Western Blot Analysis represents the gold standard for detecting PARP-1 cleavage. This methodology involves:
- Sample Preparation: Lysate preparation from treated cells using RIPA buffer supplemented with protease inhibitors [14]
- Electrophoresis: Separation of proteins (20-50 μg per lane) on 8-10% SDS-PAGE gels
- Transfer: Electroblotting to PVDF or nitrocellulose membranes
- Immunoblotting: Incubation with anti-PARP-1 antibodies that recognize both full-length (116 kDa) and cleaved (89 kDa) fragments
- Quantification: Densitometric analysis to calculate cleavage ratio
Key Technical Considerations:
- Include positive controls (e.g., staurosporine-treated cells) to validate apoptosis induction
- Normalize protein loading using housekeeping proteins (e.g., GAPDH, β-actin)
- Optimize antibody concentrations for specific detection of cleavage fragments
Pathway-Specific Assessment Techniques
Table 2: Experimental Approaches for Cell Death Pathway Characterization
| Methodology | Application | Key Readouts | Technical Notes |
|---|---|---|---|
| Caspase Activity Assays | Apoptosis quantification | Fluorometric/colorimetric measurement of caspase-3/7, -8, or -9 activity | Use specific substrates (e.g., DEVD for caspase-3); combine with inhibition studies |
| Annexin V/PI Staining | Apoptosis vs. necrosis discrimination | Phosphatidylserine externalization (early apoptosis) vs. membrane integrity (necrosis) | Time-course analysis critical for distinguishing primary vs. secondary necrosis |
| MLKL Phosphorylation Detection | Necroptosis confirmation | Western blot with phospho-specific MLKL antibodies (Thr357/Ser358) | Combine with RIPK1 inhibition (necrostatin-1) for pathway validation |
| GSDMD Cleavage Analysis | Pyroptosis detection | Immunoblotting for GSDMD-N terminal fragment | Inflammasome activators (e.g., nigericin) serve as positive controls |
| LDH Release Assay | Lytic cell death quantification | Measurement of lactate dehydrogenase in supernatant | Correlates with plasma membrane rupture in necroptosis/pyroptosis |
| IL-1β/IL-18 Measurement | Inflammasome activation | ELISA or Western blot for processed cytokines | Specific indicator of inflammasome-mediated pyroptosis |
Pharmacological Modulation Strategies
Strategic use of pathway-specific inhibitors and activators enables precise dissection of cell death mechanisms:
- Apoptosis Induction: Staurosporine (protein kinase inhibitor), Actinomycin D (transcription inhibitor)
- Apoptosis Inhibition: Z-VAD-FMK (pan-caspase inhibitor), specific caspase inhibitors (e.g., Z-DEVD-FMK for caspase-3)
- Necroptosis Induction: TNFα + SMAC mimetic + Z-VAD-FMK (TSZ combination)
- Necroptosis Inhibition: Necrostatin-1 (RIPK1 inhibitor), GSK'872 (RIPK3 inhibitor)
- Pyroptosis Induction: Nigericin (NLRP3 activator), intracellular LPS delivery
- Pyroptosis Inhibition: Disulfiram (GSDMD inhibitor), VX-765 (caspase-1 inhibitor)
- PARP-1 Inhibition: Olaparib, veliparib (clinical PARP inhibitors)
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Cell Death Pathway Investigation
| Reagent Category | Specific Examples | Research Application | Mechanistic Insight |
|---|---|---|---|
| Caspase Inhibitors | Z-VAD-FMK (pan-caspase), Z-DEVD-FMK (caspase-3) | Apoptosis inhibition, necroptosis induction | Discerns caspase-dependent vs independent death |
| Kinase Inhibitors | Necrostatin-1 (RIPK1), GSK'872 (RIPK3) | Necroptosis pathway dissection | Validates RIPK1/RIPK3/MLKL axis requirement |
| PARP Inhibitors | Olaparib, veliparib, PJ34 | PARP activity blockade | Distinguishes PARP-mediated cell death (parthanatos) |
| Gasdermin Inhibitors | Disulfiram | Pyroptosis inhibition | Confirms gasdermin-dependent lysis |
| Inflammasome Activators | Nigericin, ATP, monosodium urate crystals | Pyroptosis induction | NLRP3 inflammasome-specific triggering |
| Death Receptor Ligands | TNFα, FasL, TRAIL | Extrinsic apoptosis/necroptosis initiation | Death receptor pathway activation |
| Detection Antibodies | Anti-cleaved PARP-1, anti-pMLKL, anti-GSDMD | Specific pathway marker detection | Molecular confirmation of activation states |
Therapeutic Implications and Concluding Perspectives
The strategic manipulation of cell death pathways represents a promising frontier in therapeutic development, particularly for cancer, neurodegenerative diseases, and inflammatory disorders. PARP inhibitors have already demonstrated clinical success in oncology, particularly for BRCA-deficient cancers, by exploiting synthetic lethality and preventing DNA repair in malignant cells [76]. Concurrently, inhibiting PARP-1 activation shows neuroprotective potential in models of Parkinson's disease and cerebral ischemia by preventing parthanatos [76].
Emerging therapeutic strategies include:
- Contextual Pathway Modulation: Selective inhibition of necroptosis or pyroptosis in inflammatory diseases while preserving apoptotic capacity [88] [90]
- Combination Therapies: PARP inhibitors with DNA-damaging agents for enhanced anticancer efficacy [76]
- Cell Death Pathway Switching: Redirecting lytic death (necroptosis/pyroptosis) to non-inflammatory apoptosis in chronic inflammatory conditions
The intricate crosstalk between apoptosis, necroptosis, and pyroptosis, exemplified by molecular sensors like caspase-8 that can participate in all three pathways, underscores the complexity of cellular fate decisions [89]. PARP-1 cleavage serves as both a definitive apoptotic marker and a strategic point of therapeutic intervention. As research continues to unravel the sophisticated regulatory networks governing these pathways, opportunities will expand for developing precise, context-dependent therapies that manipulate cell death signaling for therapeutic benefit.
Future research directions should focus on elucidating the spatiotemporal dynamics of cell death pathway crosstalk, identifying novel biomarkers for pathway activation in human diseases, and developing more specific inhibitors that can distinguish between closely related death modalities. The integration of systems biology approaches with traditional biochemical methods will be essential for mapping the complex decision-making networks that determine cellular survival and death.
Cell-Type Specific Variations in Cleavage Patterns and Consequences
This technical guide examines the mechanisms and implications of poly (ADP-ribose) polymerase-1 (PARP-1) cleavage during programmed cell death (PCD), with specific emphasis on how these processes exhibit remarkable cell-type specificity. PARP-1, a fundamental nuclear enzyme involved in DNA repair and cellular homeostasis, undergoes proteolytic cleavage by various "suicidal" proteases during different PCD pathways, generating distinctive fragments that serve as molecular signatures for specific cell death programs. The cleavage patterns, kinetics, and functional consequences of PARP-1 fragmentation vary significantly across cell types, influencing cellular fate decisions, differentiation pathways, and pathological outcomes. Understanding these cell-type specific variations provides critical insights for targeted therapeutic interventions in cancer, neurodegenerative disorders, and other diseases characterized by dysregulated cell death.
PARP-1 is an abundant nuclear enzyme with approximately 1-2 million copies per cell, accounting for roughly 85% of total cellular PARP activity [11]. This multifunctional protein plays crucial roles beyond its canonical DNA repair functions, including participation in transcription regulation, immune responses, inflammation, learning, memory, synaptic functions, angiogenesis, and aging [11] [40].
The PARP-1 protein contains several functionally distinct domains [11]:
- A 46-kD DNA binding domain (DBD) containing two zinc finger motifs at the NH2 terminus that facilitates tight binding to specific DNA motifs
- A 22-kD auto-modification domain (AMD) that functions as a target for direct covalent auto-modification
- A 54-kD catalytic domain (CD) at the carboxyl terminus that polymerizes linear or branched poly-ADP ribose units from NAD+ on target proteins
PARP-1's normal function involves routine repair of DNA damage through addition of poly (ADP ribose) polymers in response to various cellular stresses [11] [40]. However, during programmed cell death, PARP-1 becomes a preferred substrate for several proteases, leading to specific cleavage fragments that serve as biomarkers for particular cell death pathways and exhibit significant variation across different cell types.
PARP-1 Cleavage by Specific Proteases: Molecular Signatures
Table 1: Proteases Generating Specific PARP-1 Cleavage Fragments
| Protease | PARP-1 Fragment Sizes | Primary Cell Death Pathway | Key Features of Fragments |
|---|---|---|---|
| Caspase-3/7 | 89-kD (AMD + CD) and 24-kD (DBD) | Apoptosis | 24-kD fragment irreversibly binds to nicked DNA, inhibiting DNA repair; hallmark of apoptosis [11] |
| Caspase-1 | Specific cleavage fragments | Inflammatory pathways | Associated with interleukin converting enzyme activity [11] |
| Calpains | Variable fragments | Calcium-dependent cell death | Associated with pathological conditions including cerebral ischemia and excitotoxicity [11] [40] |
| Granzymes | Cell-type specific fragments | Immune-mediated cell death | Induced during cytotoxic T-cell mediated elimination of target cells [11] |
| Matrix Metalloproteinases (MMPs) | Distinct fragmentation patterns | Tissue remodeling pathologies | Associated with extracellular matrix degradation contexts [11] |
| Cathepsins | Lysosomal cell death signatures | Lysosome-dependent cell death | Released during lysosomal membrane permeabilization [93] [11] |
Cell-Type Specific Variations in PARP-1 Cleavage Patterns
Neural Cells
In the central nervous system, PARP-1 cleavage plays a significant role in neurodegeneration. PARP inhibition attenuates injury in pathologies including cerebral ischemia, trauma, and excitotoxicity, demonstrating the central role of PARP-1 cleavage in these conditions [11] [40]. Caspase-3 mediated cleavage of PARP-1 has been specifically implicated in several neurological diseases including cerebral ischemia, Alzheimer's disease, multiple sclerosis, Parkinson's disease, traumatic brain injury, NMDA-mediated excitotoxicity, and brain tumors, particularly gliomas [11].
The 89-kD catalytic fragment generated by caspase cleavage has a greatly reduced DNA binding capacity and is liberated from the nucleus into the cytosol in neural cells, while the 24-kD cleaved fragment with two zinc-finger motifs is retained in the nucleus, irreversibly binding to nicked DNA where it acts as a trans-dominant inhibitor of active PARP-1 [11]. This irreversible binding of the 24-kD PARP-1 fragment to DNA strand breaks inhibits DNA repair enzymes and attenuates DNA repair, simultaneously conserving cellular ATP pools in energy-sensitive neural cells [11].
Pancreatic β-Cells and Islet Development
PARP-1 plays a critical regulatory role in stem cell differentiation and control of gene expression during the development and function of β-cells in pancreatic islets [94]. Studies with PARP-1 null mice reveal distinct pancreatic and islet architectural differences, with KO mice showing extensive ductal fibrosis not evident in wild-type mice [94].
Table 2: PARP-1 Dependent Variations in Pancreatic Islet Development
| Parameter | PARP-1 Wild-Type Mice | PARP-1 Knockout Mice | Biological Significance |
|---|---|---|---|
| Islet Size | Standard islet dimensions | Approximately half the size of wild-type | Indicates immature islets in KO mice [94] |
| Islet Frequency | Baseline distribution | 2× more abundant than wild-type | Compensatory increase in islet number [94] |
| Endocrine Fraction | ~3% of pancreas mass | ~1% of pancreas mass | Reduced endocrine capacity in KO mice [94] |
| Insulin/Pdx1 Co-localization | Present within islets | Insulin present without Pdx1 staining | Disrupted differentiation signaling [94] |
| Progenitor Cell Morphology | Tightly packed epithelioid morphology | Elongated, fibroblastic morphology with large nuclei | Suggests genetic instability in KO cells [94] |
PARP-1 depletion in human pancreatic progenitor PANC-1 cells completely abrogates new β-cell formation, while pharmacological inhibition using PJ34 promotes endocrine β-cell differentiation and maturation [94]. This demonstrates the cell-type specific requirement of PARP-1 (though not necessarily its enzymatic activity) for proper development and differentiation of human islets.
Embryonic Stem Cells and Differentiation
In embryonic stem cells (ESCs), PARP-1 promotes differentiation by antagonizing the DNA binding transcription factor Sox2 to stimulate expression of the gene encoding fibroblast growth factor 4 (FGF4) that promotes differentiation [94]. The ESCs from PARP-1−/− mice exhibit altered expression of about 10% of genes compared to only 3% of genes in the liver, highlighting the cell-type specific impact of PARP-1 deletion [94].
Caspase activity plays a novel role in mediating ESC differentiation through cleavage of key transcription factors including Nanog [95]. Stem cells lacking the Casp3 gene show marked defects in differentiation, while forced expression of a caspase cleavage-resistant Nanog mutant in ESCs strongly promotes self-renewal [95]. This links a major component of the programmed cell-death pathway to the regulation of ESC development, demonstrating how cleavage events typically associated with cell death can be repurposed for differentiation programs in specific cell types.
Cancer Cells and Therapeutic Implications
In cancer cells, particularly triple-negative breast cancer (TNBC), HERPUD1 (an ER stress response protein) mediates palmitic acid-induced unfolded protein response, sustaining TNBC aggressiveness [96]. HERPUD1 deletion enhances TNBC cell sensitivity to chemotherapy drugs like doxorubicin,表现为细胞存活率下降、caspase-3剪切和PARP1 cleavage增加 [96]. This demonstrates the cell-type specific regulation of PARP-1 cleavage in response to different stress pathways in cancer cells.
The development of PARP-1 degraders such as PROTAC 180055 represents a novel approach to target PARP-1 in cancer cells while minimizing DNA trapping effects associated with traditional PARP inhibitors [97]. This compound effectively degrades PARP-1 and inhibits its enzyme activity, avoiding DNA capture effect, and shows significant cytotoxicity against BRCA1-mutated MOLT4 cells while having lower toxicity to normal myocardial cell lines [97].
Experimental Protocols for Assessing PARP-1 Cleavage
Detection and Quantification of PARP-1 Cleavage Fragments
Protocol 1: Western Blot Analysis for PARP-1 Cleavage Fragments
Materials:
- Cell lysates from treated and control conditions
- PARP-1 primary antibodies (specific for different domains)
- Secondary antibodies conjugated with HRP
- Enhanced chemiluminescence detection system
- SDS-PAGE gel electrophoresis system
- Nitrocellulose or PVDF membranes
Procedure:
- Prepare cell lysates using RIPA buffer supplemented with protease inhibitors
- Quantify protein concentration using BCA assay
- Separate 20-50 μg of total protein on 8-12% SDS-PAGE gels
- Transfer proteins to nitrocellulose or PVDF membranes
- Block membranes with 5% non-fat milk in TBST for 1 hour
- Incubate with primary antibodies against PARP-1 (specific for N-terminal, C-terminal, or full-length epitopes) overnight at 4°C
- Wash membranes and incubate with HRP-conjugated secondary antibodies for 1 hour at room temperature
- Develop using ECL substrate and image with chemiluminescence detection system
- Quantify band intensities using image analysis software, focusing on full-length PARP-1 (116-kD) and cleavage fragments (89-kD for caspase cleavage)
Protocol 2: Immunofluorescence Assessment of PARP-1 Localization
Materials:
- Cells grown on chamber slides or coverslips
- Paraformaldehyde fixation solution
- Permeabilization buffer (Triton X-100)
- Blocking solution (BSA or serum)
- PARP-1 primary antibodies with different epitope specificities
- Fluorescently-labeled secondary antibodies
- Nuclear counterstains (DAPI or Hoechst)
- Fluorescence microscope with high-resolution imaging capabilities
Procedure:
- Culture cells on sterile coverslips until desired confluence
- Treat cells with appropriate experimental conditions
- Fix cells with 4% paraformaldehyde for 15 minutes at room temperature
- Permeabilize with 0.1% Triton X-100 for 10 minutes
- Block with 3% BSA or serum from secondary antibody host for 1 hour
- Incubate with primary antibodies against PARP-1 overnight at 4°C
- Wash and incubate with fluorophore-conjugated secondary antibodies for 1 hour in darkness
- Counterstain nuclei with DAPI for 5 minutes
- Mount coverslips and image using fluorescence microscopy
- Analyze subcellular localization patterns of PARP-1 and cleavage fragments
Functional Assays for PARP-1 Cleavage Consequences
Protocol 3: DNA Repair Capacity Assessment Post-Cleavage
Materials:
- Comet assay reagents (low-melting point agarose, lysis solution, electrophoresis apparatus)
- γH2AX immunofluorescence materials
- Host cell reactivation assay components
- DNA damage-inducing agents (H2O2, etoposide, irradiation source)
Procedure:
- Induce PARP-1 cleavage through appropriate stimuli (e.g., death receptor activation, cytotoxic compounds)
- Apply controlled DNA damage using sublethal doses of H2O2 or irradiation
- Assess DNA repair capacity using:
- Alkaline Comet Assay: Measure single-strand break repair at various timepoints
- γH2AX Foci Formation: Quantify double-strand break markers via immunofluorescence
- Host Cell Reactivation: Transfer damaged reporter plasmids and measure recovery
- Compare repair kinetics between cells with intact vs. cleaved PARP-1
- Correlate specific PARP-1 cleavage fragments with repair deficiencies
Protocol 4: Cell Fate Determination After PARP-1 Cleavage Initiation
Materials:
- Annexin V/propidium iodide staining kit
- Caspase activity assays (fluorometric or colorimetric)
- Mitochondrial membrane potential dyes (JC-1, TMRM)
- Clonogenic survival assay materials
- Live-cell imaging system with environmental control
Procedure:
- Induce sublethal PARP-1 cleavage using calibrated stimuli
- Monitor real-time cell fate using:
- Annexin V/PI staining: Track phosphatidylserine exposure and membrane integrity
- Caspase activity assays: Measure executioner caspase activation
- Mitochondrial potential: Assess cytochrome c release capacity
- Morphological analysis: Document classical apoptosis markers
- For surviving cells, assess long-term outcomes:
- Clonogenic assays: Measure proliferative capacity after cleavage event
- Senescence-associated β-galactosidase staining: Detect premature senescence
- Differentiation markers: Evaluate altered differentiation pathways
- Correlate specific PARP-1 cleavage patterns with eventual cell fate decisions
Research Reagent Solutions for PARP-1 Cleavage Studies
Table 3: Essential Research Reagents for PARP-1 Cleavage Analysis
| Reagent/Category | Specific Examples | Function/Application | Cell-Type Specific Considerations |
|---|---|---|---|
| PARP-1 Antibodies | Anti-PARP-1 (N-terminal), Anti-PARP-1 (C-terminal), Cleavage-specific antibodies | Detection of full-length and cleaved PARP-1 fragments | Antibody affinity may vary by cell type due to post-translational modifications [11] [40] |
| Caspase Inhibitors | Z-VAD-FMK (pan-caspase), DEVD-CHO (caspase-3 specific) | Inhibition of caspase-mediated PARP-1 cleavage | Effectiveness varies by cell type due to differential caspase expression [11] |
| PARP Inhibitors | PJ34, Olaparib, Rucaparib | Inhibition of PARP-1 enzymatic activity | Cell-type specific toxicity profiles; differential effects on cleavage [94] [97] |
| PARP-1 Degraders | PROTAC 180055 | Selective degradation of PARP-1 protein | BRCA-mutant cells show heightened sensitivity [97] |
| Cell Death Inducers | LLOMe, GPN, sphingosine, TNF-α, staurosporine | Induction of specific PCD pathways activating different proteases | Cell-type specific responses to different inducers [93] [78] |
| Detection Kits | Caspase-3 activity kits, Annexin V apoptosis kits, Comet assay kits | Assessment of functional consequences of PARP-1 cleavage | Optimization required for different cell types [11] [78] |
Signaling Pathways and Molecular Mechanisms
The following diagrams illustrate key signaling pathways involving PARP-1 cleavage and its cell-type specific consequences:
Diagram 1: PARP-1 Cleavage Pathways and Cell-Type Specific Consequences. This diagram illustrates how different death signals activate specific proteases that cleave PARP-1 into distinct fragments, leading to diverse functional outcomes in different cell types.
Diagram 2: Experimental Workflow for PARP-1 Cleavage Studies. This diagram outlines a comprehensive experimental approach for investigating PARP-1 cleavage patterns and their functional consequences across different cell types.
The cell-type specific variations in PARP-1 cleavage patterns and their consequences represent a critical layer of complexity in programmed cell death research. The specific cleavage fragments generated in different cellular contexts serve as molecular signatures that not only indicate the activation of particular cell death pathways but also influence functional outcomes including DNA repair capacity, differentiation potential, and susceptibility to therapeutic interventions.
Understanding these cell-type specific variations has profound implications for targeted therapeutic development. The emergence of PARP-1 degraders like PROTAC 180055 that can selectively target PARP-1 without DNA trapping effects represents a promising advancement in cancer therapy, particularly for BRCA-mutant cancers [97]. Similarly, the role of PARP-1 in pancreatic β-cell differentiation highlights its importance in diabetes research and potential regenerative medicine applications [94].
Future research directions should focus on:
- Comprehensive mapping of PARP-1 cleavage patterns across diverse cell types and states
- Elucidation of how specific cleavage fragments influence gene expression programs in different cellular contexts
- Development of cell-type specific therapeutic strategies that leverage PARP-1 cleavage mechanisms
- Investigation of PARP-1 cleavage in the context of novel programmed cell death pathways including necroptosis, pyroptosis, and ferroptosis
The integration of these findings will advance our understanding of PARP-1's multifaceted roles in cellular homeostasis and death, ultimately enabling more precise therapeutic interventions tailored to specific cellular contexts and disease states.
Poly(ADP-ribose) polymerase-1 (PARP-1) is a nuclear enzyme that functions as a primary responder to DNA damage, playing a well-established role in DNA repair mechanisms including base excision repair (BER) and alternative non-homologous end joining [1]. Beyond its DNA repair functions, PARP-1 has emerged as a central regulator of multiple programmed cell death (PCD) pathways, with its proteolytic cleavage serving as a critical decision point in cellular fate. During caspase-dependent apoptosis, PARP-1 is cleaved by caspases-3 and -7 at the DEVD214 site, located within its nuclear localization signal, generating 24-kDa and 89-kDa fragments [1] [39]. This cleavage event was traditionally viewed as a mechanism to inactivate PARP-1 and conserve cellular energy during apoptosis. However, emerging evidence indicates that the cleavage fragments themselves possess distinct biological activities that actively regulate cell death pathways and inflammatory responses [39] [12] [98].
The development of CRISPR/Cas9 models of non-cleavable PARP-1 represents a transformative approach for dissecting the functional significance of PARP-1 cleavage in PCD. These engineered cellular systems allow researchers to isolate the specific contributions of PARP-1 cleavage fragments from the full-length protein, providing unprecedented insight into the molecular mechanisms governing cell fate decisions in response to genotoxic stress. This technical guide comprehensively outlines the strategies, methodologies, and applications of non-cleavable PARP-1 models for advanced mechanistic studies in cell death research and therapeutic development.
PARP-1 Cleavage Fragments: Mechanisms and Consequences
Proteolytic Cleavage of PARP-1 by Cell Death Proteases
PARP-1 serves as a substrate for multiple "suicidal" proteases activated during different cell death programs, with each protease generating characteristic cleavage fragments that serve as biomarkers for specific death pathways [1].
- Caspase-Mediated Cleavage: During apoptosis, executioner caspases-3 and -7 cleave PARP-1 at the DEVD↓G motif (amino acids 211-214), producing p24 (24-kDa) and p89 (89-kDa) fragments [1] [39]. The p24 fragment contains the DNA-binding domain (DBD) with two zinc fingers, while the p89 fragment contains the auto-modification domain (AMD) and catalytic domain (CD) [1].
- Necrotic Cleavage: In necrosis, PARP-1 is cleaved by lysosomal proteases (cathepsins B and G), generating a dominant 50-kDa fragment [41]. This cleavage is not inhibited by broad-spectrum caspase inhibitors like zVAD-fmk.
- Other Proteases: Additional proteases including calpains, granzymes, and matrix metalloproteinases (MMPs) can also cleave PARP-1, producing distinctive signature fragments [1].
Table 1: PARP-1 Cleavage Fragments in Different Cell Death Pathways
| Cleavage Fragment | Molecular Weight | Generating Protease | Cell Death Pathway | Functional Consequences |
|---|---|---|---|---|
| p24 | 24 kDa | Caspases-3/7 | Apoptosis | Contains DBD; remains nuclear-bound; acts as trans-dominant inhibitor of PARP-1 [1] |
| p89 | 89 kDa | Caspases-3/7 | Apoptosis | Contains AMD and CD; translocates to cytoplasm; serves as PAR carrier inducing AIF-mediated death [12] [13] |
| p50 | 50 kDa | Cathepsins B/G | Necrosis | Not well characterized; serves as biomarker for lysosomal protease activation [41] |
Biological Activities of PARP-1 Cleavage Fragments
The traditional view that PARP-1 cleavage simply inactivates the enzyme has been superseded by evidence that the fragments themselves are biologically active and can differentially influence cell fate.
p24 Fragment Activities: The 24-kDa DBD fragment remains tightly bound to DNA strand breaks, where it acts as a trans-dominant inhibitor of DNA repair by blocking access of repair machinery to damage sites [1]. This function may promote the conservation of cellular ATP during the execution phase of apoptosis.
p89 Fragment Activities: The 89-kDa fragment can be poly(ADP-ribosyl)ated and translocates to the cytoplasm, where it serves as a PAR carrier that facilitates apoptosis-inducing factor (AIF) release from mitochondria, promoting the caspase-independent cell death pathway known as parthanatos [12] [13]. This fragment also demonstrates pro-inflammatory properties by enhancing NF-κB transcriptional activity and increasing expression of inflammatory mediators like iNOS and COX-2 [39] [98].
Functional Opposition of Fragments: Research demonstrates that the p24 and p89 fragments can exert opposing effects on cell viability. In models of oxygen/glucose deprivation (in vitro ischemia), expression of the p24 fragment was cytoprotective, while expression of the p89 fragment was cytotoxic [39] [98].
Engineering Non-Cleavable PARP-1 Models Using CRISPR/Cas9
Molecular Design of Non-Cleavable PARP-1
The creation of non-cleavable PARP-1 (PARP-1UNCL) involves introducing mutations at the caspase cleavage site (DEVD214) to prevent proteolytic processing while preserving the normal functions of the protein.
- Target Site Identification: The canonical caspase cleavage site in human PARP-1 is located between amino acids Asp214 and Gly215 within the nuclear localization signal of the DNA-binding domain [39].
- Mutation Strategy: The most common approach involves mutating the critical aspartic acid residue (D214) to alanine (D214A), which prevents caspase recognition and cleavage while minimizing structural perturbations [39].
- Functional Preservation: The mutation strategy is designed to maintain normal PARP-1 functions including DNA damage binding, catalytic activity, and protein-protein interactions, allowing specific investigation of cleavage-dependent phenomena.
CRISPR/Cas9 Genome Editing Protocol
The following protocol details the generation of non-cleavable PARP-1 models using CRISPR/Cas9-mediated homology-directed repair (HDR).
Reagent Preparation
- gRNA Design: Design a single-guide RNA (sgRNA) targeting sequences proximal to the PARP-1 caspase cleavage site (DEVD214) in exon X (verify exact exon for your model system). Select a target with high on-target efficiency and minimal off-target potential using established algorithms (e.g., CRISPOR, ChopChop) [99].
- Donor Template Construction: Generate a single-stranded oligodeoxynucleotide (ssODN) or double-stranded DNA (dsDNA) donor template containing the D214A mutation flanked by homologous arms (80-100 bp each). Incorporate silent mutations in the PAM sequence to prevent Cas9 re-cleavage after HDR [99].
- CRISPR/Cas9 Delivery System: Utilize a ribonucleoprotein (RNP) complex comprising purified Cas9 protein and synthetic sgRNA for maximal editing efficiency and reduced off-target effects. Alternatively, plasmid or viral vector systems can be employed for difficult-to-transfect cells [99].
Cell Transfection and Screening
- Delivery Optimization: Transfect the RNP complex and donor template into target cells using appropriate methods (electroporation for immune cells, lipofection for adherent lines). Determine optimal ratios of RNP:donor through pilot experiments.
- Clonal Isolation: 48-72 hours post-transfection, isolate single cells by fluorescence-activated cell sorting (FACS) or limiting dilution into 96-well plates. Expand clonal populations for 2-3 weeks.
- Genotypic Validation: Screen clones by PCR amplification of the targeted region followed by Sanger sequencing or next-generation sequencing to identify homozygous D214A mutants. Confirm the absence of random integrations or large deletions.
Table 2: Research Reagent Solutions for PARP-1 CRISPR Modeling
| Reagent Category | Specific Examples | Function/Application | Technical Notes |
|---|---|---|---|
| CRISPR/Cas9 System | Synthetic sgRNA, purified Cas9 protein | Forms RNP complex for precise genome editing | RNP complexes reduce off-target effects and increase HDR efficiency [99] |
| Donor Template | ssODN with D214A mutation | Provides repair template for HDR | Include silent PAM-disruption mutations to prevent re-cleavage [99] |
| Delivery Method | Electroporation, lipofection | Introduces editing components into cells | Electroporation typically provides highest efficiency for RNP delivery [99] |
| Selection System | FACS, antibiotic resistance | Enriches for successfully edited cells | Fluorescent co-markers (GFP) enable tracking of transfected cells [99] |
| Validation Tools | PARP-1 antibodies, caspase substrates | Confirms successful mutation and functional characterization | Anti-PARP-1 antibodies that recognize cleavage fragments are essential [1] |
Validation of Non-Cleavable PARP-1 Models
Comprehensive validation ensures the PARP-1UNCL model accurately recapitulates the intended genetic and functional properties.
- Genomic DNA Analysis: Confirm precise incorporation of the D214A mutation without additional sequence alterations through Sanger sequencing and T7 endonuclease I or TIDE analysis to quantify editing efficiency.
- Protein-Level Validation:
- Cleavage Resistance: Treat PARP-1UNCL and wild-type cells with caspase inducers (e.g., staurosporine, actinomycin D) and assess PARP-1 cleavage by immunoblotting using antibodies recognizing the p89 and p24 fragments. PARP-1UNCL should demonstrate complete resistance to caspase-mediated cleavage [39].
- Expression and Localization: Verify that PARP-1UNCL maintains normal nuclear localization patterns using immunocytochemistry with PARP-1 antibodies.
- Functional Validation:
- DNA Repair Capacity: Evaluate the ability of PARP-1UNCL to recruit to DNA damage sites and catalyze PAR synthesis following genotoxic stress (e.g., H2O2, UV irradiation) using immunofluorescence for PAR and laser microirradiation assays [85].
- Catalytic Activity: Measure enzymatic activity through in vitro PARP assays comparing PAR synthesis between PARP-1UNCL and wild-type protein.
Experimental Applications and Methodologies
Assessing Cell Death Pathways in PARP-1UNCL Models
The PARP-1UNCL system enables precise dissection of PARP-1's role in different PCD pathways through well-established assays.
Apoptosis Induction and Assessment
- Protocol: Treat PARP-1WT and PARP-1UNCL cells with classical apoptosis inducers (staurosporine 0.5-1 μM, actinomycin D 0.5-1 μg/mL) for 4-24 hours.
- Assessment Methods:
- PARP-1 Cleavage Immunoblotting: Resolve cell lysates by SDS-PAGE and immunoblot with antibodies recognizing the p89 PARP-1 fragment. PARP-1UNCL should show complete absence of cleavage fragments [39].
- Caspase-3/7 Activity: Measure using fluorogenic substrates (DEVD-AFC) or cleaved caspase-3 immunoblotting.
- Phosphatidylserine Externalization: Detect via Annexin V-FITC/propidium iodide staining and flow cytometry.
- Expected Outcomes: PARP-1UNCL cells typically demonstrate delayed apoptosis execution and altered kinetics of cell death, revealing the contribution of PARP-1 cleavage to apoptotic progression [39].
Parthanatos Induction and Assessment
- Protocol: Indce parthanatos using MNNG (50-200 μM) or H2O2 (0.5-2 mM) treatment for 15 minutes to 2 hours.
- Assessment Methods:
- AIF Translocation: Monitor by immunofluorescence for AIF; parthanatos features AIF translocation from mitochondria to nucleus.
- PAR Accumulation: Detect poly(ADP-ribose) polymer formation using anti-PAR antibodies.
- Mitochondrial Membrane Potential: Assess using JC-1 or TMRM dyes.
- Expected Outcomes: PARP-1UNCL cells typically show resistance to parthanatos, with reduced AIF translocation and cell death, confirming the role of PARP-1 cleavage in this pathway [12] [13].
Necrosis and Inflammatory Response Assessment
- Protocol: Indce inflammatory responses with TNF-α (10-50 ng/mL) + cycloheximide (10 μg/mL) or LPS (100 ng/mL-1 μg/mL) treatment for 6-24 hours.
- Assessment Methods:
- NF-κB Activation: Measure by nuclear translocation of p65 (immunofluorescence) or NF-κB luciferase reporter assays.
- Inflammatory Mediator Expression: Quantify iNOS, COX-2, and cytokine expression by immunoblotting and qRT-PCR.
- Cell Viability: Assess using MTT, ATP-based, or LDH release assays.
- Expected Outcomes: PARP-1UNCL typically shows reduced NF-κB activation and decreased expression of inflammatory mediators compared to wild-type, demonstrating the pro-inflammatory function of PARP-1 cleavage fragments [39] [98].
Quantitative Analysis of PARP-1UNCL Phenotypes
Rigorous quantification of cellular responses in PARP-1UNCL models enables precise characterization of PARP-1 cleavage-dependent phenomena.
Table 3: Quantitative Assessment of PARP-1UNCL in Cell Death Models
| Experimental Paradigm | Key Readout Parameters | PARP-1WT Response | PARP-1UNCL Response | Biological Interpretation |
|---|---|---|---|---|
| Staurosporine-Induced Apoptosis | % Cleaved PARP-1; Caspase-3 Activity; % Annexin V+ Cells | Robust PARP-1 cleavage; High caspase activity; Rapid apoptosis execution | No PARP-1 cleavage; Delayed caspase activation; Attenuated apoptosis kinetics | PARP-1 cleavage accelerates apoptotic progression [39] |
| MNNG-Induced Parthanatos | AIF Nuclear Translocation; PAR Accumulation; % Cell Death | Significant AIF translocation; Extensive PAR formation; High cell death | Reduced AIF translocation; Limited PAR accumulation; Protection from death | PARP-1 cleavage is required for AIF-mediated death [12] [13] |
| Inflammatory Stimulation | NF-κB Luciferase Activity; iNOS Protein Levels; IL-6 Secretion | Enhanced NF-κB activation; Increased iNOS expression; High cytokine production | Reduced NF-κB activation; Decreased iNOS expression; Lower cytokine production | p89 fragment promotes inflammatory responses [39] [98] |
| Oxygen/Glucose Deprivation | Cell Viability (% control); NAD+ Levels; PAR Formation | Reduced viability; NAD+ depletion; Transient PAR formation | Enhanced protection; Preserved NAD+ levels; Minimal PAR changes | PARP-1 cleavage contributes to ischemic damage [39] [98] |
Discussion and Research Implications
Interpreting Results from Non-Cleavable PARP-1 Models
The integration of data from PARP-1UNCL models reveals the multifaceted role of PARP-1 cleavage in cell death regulation:
- Dual Roles in Apoptosis: While PARP-1 cleavage was historically viewed as an inactivation mechanism, evidence from PARP-1UNCL models demonstrates that it actively facilitates apoptotic execution, potentially through the actions of the p89 fragment in promoting mitochondrial dysfunction [12].
- Cross-Talk Between Cell Death Pathways: PARP-1UNCL studies highlight the intersection between apoptosis and parthanatos, demonstrating that caspase-mediated cleavage can unexpectedly promote AIF-mediated death through p89-PAR complex formation and cytoplasmic translocation [12] [13].
- Transcriptional and Inflammatory Regulation: The protective effects of PARP-1UNCL in inflammatory models underscore the significant role of PARP-1 cleavage in modulating NF-κB activity and subsequent inflammatory gene expression, independent of its DNA repair functions [39] [98].
Therapeutic Implications and Future Directions
The insights gained from non-cleavable PARP-1 models have significant implications for therapeutic development:
- PARP Inhibitor Applications: Understanding the distinct roles of PARP-1 cleavage fragments may inform the use of PARP inhibitors in different pathological contexts, particularly in balancing DNA repair inhibition against cell death promotion.
- Ischemic Injury Interventions: The robust protection afforded by PARP-1UNCL in models of cerebral ischemia and myocardial infarction suggests that strategies targeting PARP-1 cleavage specifically may yield therapeutic benefits without compromising genomic stability [39].
- Inflammatory Disease Modulation: The ability of PARP-1UNCL to suppress NF-κB-driven inflammation suggests novel approaches for inflammatory and autoimmune conditions through selective inhibition of PARP-1 cleavage.
CRISPR/Cas9-generated non-cleavable PARP-1 models represent a sophisticated toolset for dissecting the complex relationship between PARP-1 proteolysis and programmed cell death. These validated experimental systems enable researchers to isolate the specific contributions of PARP-1 cleavage fragments from the full-length protein, revealing their distinct and sometimes opposing biological activities. The methodologies outlined in this technical guide provide a comprehensive framework for implementing PARP-1UNCL models in diverse research contexts, from basic mechanisms of cell death to therapeutic development for cancer, neurodegenerative diseases, and inflammatory conditions. As research in this field advances, these models will continue to illuminate the intricate regulation of cellular fate decisions and identify novel targets for therapeutic intervention in PARP-1-associated pathologies.
Poly (ADP-ribose) polymerase (PARP) inhibitors represent a cornerstone of targeted cancer therapy, primarily known for exploiting synthetic lethality in homologous recombination (HR)-deficient cancers. While their traditional mechanism involves disrupting DNA repair to induce apoptotic cell death, emerging research reveals a complex interplay between PARP inhibition and diverse regulated cell death (RCD) pathways. The efficacy of PARP inhibitors (PARPi) extends beyond BRCA-mutant cancers, with therapeutic implications in various malignancy contexts. This review systematically examines the therapeutic implications of PARP inhibitors across different cell death modalities, focusing on mechanistic insights, experimental approaches, and translational applications. Understanding these multifaceted death contexts is critical for optimizing PARPi-based therapies and overcoming resistance.
PARP-1 in Programmed Cell Death: Molecular Foundations
PARP-1, the most abundant and well-characterized PARP family member, functions as a critical molecular switch directing cellular fate in response to stress. Its role in cell death is context-dependent, influenced by the intensity and duration of activation.
- DNA Repair and Cell Survival: Under mild genotoxic stress, PARP-1 acts as a key DNA damage sensor. It detects DNA strand breaks and catalyzes the synthesis of poly(ADP-ribose) (PAR) chains using NAD+ as a substrate. This PARylation facilitates the recruitment of DNA repair machinery, promoting cell survival and genomic integrity [100] [101].
- Parthanatos: PARP-1-Dependent Cell Death: Under severe oxidative or genotoxic stress, PARP-1 becomes hyperactivated, leading to catastrophic NAD+ and ATP depletion. This bioenergetic crisis triggers a distinct form of regulated necrosis termed parthanatos. A key executioner in this pathway is the mitochondria-derived protein Apoptosis Inducing Factor (AIF), which, upon release, translocates to the nucleus and works in concert with Macrophage Migration Inhibitory Factor (MIF) to drive DNA fragmentation [76]. This pathway is implicated in neurological diseases and ischemic injuries, where PARP inhibition shows protective effects.
- Apoptosis and PARP-1 Inactivation: During apoptosis, PARP-1 is cleaved and inactivated by executioner caspases-3 and -7. This cleavage is considered a hallmark of apoptosis and serves to prevent futile PARP-1 activation and energy depletion, thereby ensuring the apoptotic process proceeds efficiently [76].
Table 1: PARP-1 Functions in Different Cell Death Contexts
| Cell Death Context | PARP-1 Role | Key Effectors | Functional Outcome |
|---|---|---|---|
| DNA Repair & Survival | Pro-survival | PAR polymers, DNA repair proteins | Genomic integrity maintenance |
| Parthanatos | Pro-death | Hyperactivation, PAR polymer, AIF, MIF | NAD+/ATP depletion, DNA degradation |
| Apoptosis | Substrate | Caspase-3/7 cleavage | Inactivation to conserve energy |
The following diagram illustrates the central role of PARP-1 in directing cell fate towards survival, parthanatos, or apoptosis based on the intensity of the stress signal.
PARP Inhibitors and Ferroptosis
Ferroptosis is an iron-dependent form of cell death characterized by the lethal accumulation of lipid peroxides. Recent evidence indicates a significant mechanistic crosstalk between PARP inhibition and ferroptosis induction, offering a promising avenue to overcome PARPi resistance.
Mechanisms of Synergy
The synergistic effect of PARP inhibitors and ferroptosis primarily involves the regulation of key antioxidant defenses:
- p53-Dependent SLC7A11 Suppression: Olaparib has been shown to downregulate the expression of SLC7A11, the core component of the System Xc- cystine/glutamate antiporter. This downregulation is often dependent on the tumor suppressor p53. Reduced SLC7A11 impairs cystine uptake, leading to depletion of the antioxidant glutathione (GSH) and subsequent inactivation of GPX4, the central enzyme responsible for detoxifying lipid peroxides [102].
- BRCA1-Mediated GPX4 Regulation: A non-canonical role of BRCA1 in promoting ferroptosis has been identified. BRCA1 facilitates the ubiquitination and proteasomal degradation of GPX4. Consequently, BRCA1-deficient cells exhibit GPX4 accumulation and are resistant to ferroptosis. This creates a unique vulnerability: combining PARP inhibitors with GPX4 inhibitors can synergistically induce ferroptosis in BRCA1-deficient cancer cells, bypassing traditional DNA repair-based synthetic lethality [102].
- Metabolic Reprogramming via CD36: Niraparib can trigger ferroptosis independently of p53 and BRCA status by transcriptionally upregulating the fatty acid transporter CD36. Enhanced CD36-mediated fatty acid uptake leads to dysregulated lipid metabolism and peroxidation, sensitizing ovarian cancer cells to ferroptotic death and suppressing peritoneal metastasis in preclinical models [102].
Table 2: Key Mechanisms Linking PARP Inhibition to Ferroptosis
| Mechanism | Key Players | Molecular Consequence | Therapeutic Context |
|---|---|---|---|
| SLC7A11 Suppression | Olaparib, p53 | Glutathione depletion, GPX4 inactivation | BRCA wild-type cancers |
| GPX4 Stabilization | BRCA1 loss | Increased GPX4 protein, Ferroptosis resistance | BRCA1-deficient cancers |
| Lipid Metabolic Rewiring | Niraparib, CD36 | Fatty acid overload, Lipid peroxidation | Peritoneal metastasis |
Experimental Protocol: Assessing Ferroptosis Induction by PARP Inhibitors
Objective: To evaluate the induction of ferroptosis in cancer cells treated with a PARP inhibitor (e.g., Olaparib).
Materials and Reagents:
- Cell lines of interest (e.g., Ovarian cancer cell lines with varying BRCA and p53 status)
- PARP inhibitor (e.g., Olaparib, dissolved in DMSO)
- Ferroptosis inducer (e.g., Erastin, RSL3) and inhibitor (e.g., Ferrostatin-1, Liproxstatin-1)
- C11-BODIPY 581/591 probe (for lipid ROS detection)
- Commercial GSH and GPX4 activity assay kits
- Antibodies for SLC7A11, GPX4, and p53
Methodology: 1. Treatment Groups: Seed cells and treat with: - Vehicle control (DMSO) - Olaparib alone - Ferroptosis inhibitor alone - Olaparib + Ferroptosis inhibitor - Positive control (e.g., Erastin) 2. Viability Assay: After 48-72 hours, measure cell viability using assays like CCK-8 or CellTiter-Glo to determine IC50 values and combination index. 3. Lipid Peroxidation Measurement: Treat cells with Olaparib for 24-48 hours. Load cells with C11-BODIPY dye. The shift from red to green fluorescence, measured by flow cytometry or fluorescence microscopy, indicates lipid peroxidation. 4. GSH and GPX4 Activity Analysis: Harvest Olaparib-treated cells and use commercial kits to quantify intracellular GSH levels and GPX4 enzymatic activity. 5. Western Blot Analysis: Analyze protein lysates from treated cells to assess changes in SLC7A11 and GPX4 expression levels.
Data Interpretation: A synergistic reduction in cell viability with Olaparib and ferroptosis inducers, coupled with increased lipid ROS, decreased GSH, and downregulation of SLC7A11, confirms ferroptosis as a contributing death mechanism.
PARP Inhibitors and Immunogenic Cell Death (ICD)/Pyroptosis
PARP inhibitors can stimulate antitumor immunity by inducing immunogenic cell death (ICD), particularly through the activation of pyroptosis, a highly inflammatory form of RCD.
Mechanisms of PARPi-Induced ICD
The induction of ICD by PARP inhibitors involves a defined biochemical cascade:
- cGAS-STING Pathway Activation: PARP inhibitor-induced DNA damage leads to the accumulation of cytosolic DNA fragments. These fragments are sensed by the cyclic GMP-AMP synthase (cGAS), which synthesizes a second messenger that activates the Stimulator of Interferon Genes (STING) pathway. STING activation promotes the production of type I interferons and proinflammatory cytokines, enhancing dendritic cell maturation and CD8+ T cell priming [103].
- Gasdermin-Mediated Pyroptosis: Research has delineated a specific pathway where PARP inhibitor-induced DNA damage initiates an NF-κB-TNFα-TNFR signaling axis, leading to the activation of caspase 8 and caspase 3. These caspases then cleave Gasdermin D (GSDMD) and/or Gasdermin E (GSDME). The cleaved Gasdermin fragments form pores in the plasma membrane, leading to pyroptosis [103]. This lytic cell release damage-associated molecular patterns (DAMPs) such as ATP, HMGB1, and calreticulin, which are hallmark signals of ICD that recruit and activate immune cells.
The following diagram summarizes this dual-pathway mechanism leading to enhanced antitumor immunity.
Experimental Protocol: Evaluating PARPi-Induced Pyroptosis and ICD
Objective: To confirm the induction of pyroptosis and assess the immunogenic potential of cancer cells treated with PARP inhibitors.
Materials and Reagents:
- Murine cancer cell lines (syngeneic models)
- PARP inhibitor (e.g., Niraparib)
- Caspase inhibitors (e.g., Z-VAD-FMK for pan-caspase, Z-DEVD-FMK for caspase-3)
- GSDMD/E knockout cells (genetic or CRISPR/Cas9-mediated)
- Antibodies for GSDMD/GSDME (cleaved and full-length), HMGB1, Calreticulin
- ELISA kits for ATP, IFN-β, HMGB1
- Dendritic cells (DCs) and CD8+ T cells for co-culture assays
Methodology:
- Cell Death Analysis: Treat cells with a PARP inhibitor and measure cell death via LDH release assay, a hallmark of lytic cell death like pyroptosis. Confirm the role of pyroptosis by pre-treating with caspase inhibitors or using GSDMD/E-deficient cells.
- Gasdermin Cleavage Assay: Detect the cleavage of GSDMD/GSDME by Western blot in PARPi-treated cells. The appearance of cleaved fragments indicates activation of the pyroptosis executioner.
- DAMP Detection:
- ATP Release: Measure extracellular ATP in the supernatant of treated cells using a luminescent ATP detection assay.
- HMGB1 and Calreticulin Translocation: Use immunofluorescence or Western blot of cell supernatant and fractions to detect the release of HMGB1 and the translocation of calreticulin to the cell surface.
- DC Phagocytosis and T Cell Activation Assay:
- Co-culture conditioned media from PARPi-treated cancer cells with bone marrow-derived DCs. Assess DC maturation by flow cytometry (CD80, CD86, MHC-II).
- Load matured DCs with tumor antigen and co-culture with naïve or antigen-specific CD8+ T cells. Measure T cell activation (e.g., IFN-γ production by ELISA, CD69 expression by flow cytometry) and cytotoxic activity.
Data Interpretation: Successful PARPi-induced ICD is confirmed by caspase/GSDMD-dependent LDH release, elevated DAMP secretion, and the subsequent ability of treated cell debris to promote DC maturation and antigen-specific T cell activation.
Research Reagent Solutions
The following table provides a curated list of essential research tools for investigating the interplay between PARP inhibitors and different cell death pathways.
Table 3: Key Research Reagents for Studying PARP Inhibitors in Cell Death
| Reagent / Tool | Primary Function | Application Context | Example Product/Assay |
|---|---|---|---|
| PARP Inhibitors | Inhibit PARP enzymatic activity, trap PARP on DNA. | Core agents for all mechanistic and therapeutic studies. | Olaparib, Niraparib, Rucaparib |
| Ferroptosis Modulators | Induce or inhibit ferroptosis. | Elucidating PARPi-ferroptosis crosstalk. | Inducers: Erastin, RSL3Inhibitors: Ferrostatin-1, Liproxstatin-1 |
| C11-BODIPY 581/591 | Fluorescent lipid peroxidation sensor. | Quantifying lipid ROS in ferroptosis. | Flow cytometry, fluorescence microscopy |
| GSH/GSSG Assay Kit | Quantify intracellular glutathione levels. | Measuring antioxidant capacity in ferroptosis. | Colorimetric/Fluorometric kits |
| cGAS/STING Antibodies | Detect pathway activation. | Confirming innate immune activation by PARPi. | Phospho-STING, TBK1 Western blot |
| Gasdermin D/E Antibodies | Detect full-length and cleaved forms. | Confirming pyroptosis execution. | Cleaved GSDMD (Asp275) IHC/Western |
| Caspase Inhibitors | Pharmacologically inhibit specific caspases. | Defining caspase-dependency in cell death pathways. | Z-VAD-FMK (pan), Z-DEVD-FMK (casp-3) |
| LDH Release Assay Kit | Measure lactate dehydrogenase release. | Quantifying plasma membrane rupture (pyroptosis/necrosis). | Colorimetric cytotoxicity kits |
| DAMP Detection Assays | Measure release of immunogenic signals. | Evaluating ICD potential. | ATP: Luminescence assayHMGB1: ELISA |
The therapeutic landscape of PARP inhibitors is rapidly expanding beyond their canonical role in synthetic lethality. As this review delineates, PARPIs engage a network of cell death pathways, including ferroptosis and immunogenic pyroptosis, offering combinatorial strategies to broaden their efficacy and overcome resistance. Targeting the ferroptosis axis is a promising approach for BRCA-wild-type and PARPi-resistant cancers. Simultaneously, the ability of PARPIs to trigger pyroptosis and ignite antitumor immunity provides a strong rationale for their combination with immunotherapies. Future research should focus on refining patient stratification biomarkers based on these non-apoptotic death mechanisms and developing next-generation PARP-1 selective inhibitors to improve therapeutic windows. Understanding the complex interplay between PARP inhibition and diverse cell death contexts is paramount for unlocking the full potential of this pivotal drug class in oncology.
Evolutionary Conservation of PARP-1 Cleavage Across Species
Poly(ADP-ribose) polymerase-1 (PARP-1) cleavage is a highly conserved event in programmed cell death that serves as a critical biochemical hallmark of apoptosis across species. This whitepaper examines the evolutionary conservation of PARP-1 cleavage, detailing the molecular mechanisms, functional consequences of cleavage fragments, and implications for therapeutic development. We demonstrate that the caspase-mediated cleavage of PARP-1 generates signature fragments with distinct biological activities that have been maintained through evolution, highlighting their fundamental importance in cellular homeostasis and disease pathogenesis. The conservation of these mechanisms provides valuable insights for drug development, particularly in targeting resistant cancers and neurodegenerative disorders.
PARP-1 is a nuclear enzyme that plays a dual role in cellular homeostasis, functioning primarily in DNA damage repair under mild genotoxic stress while transitioning to a mediator of cell death under extensive damage conditions [104]. As a key substrate for caspases during apoptosis, PARP-1 cleavage serves as a biochemical hallmark of programmed cell death execution [11] [23]. The cleavage of PARP-1 by caspases represents a crucial regulatory point that determines cellular fate, either promoting survival through DNA repair or facilitating elimination through apoptosis.
The evolutionary conservation of PARP-1 cleavage across species underscores its fundamental importance in maintaining genomic integrity and regulating cell death pathways. Understanding these conserved mechanisms provides critical insights for developing targeted therapies for cancer, neurodegenerative disorders, and other conditions characterized by dysregulated cell death.
Molecular Architecture of PARP-1 and Cleavage Sites
Domain Organization
PARP-1 is a modular protein composed of several functional domains that dictate its cellular functions:
- DNA-binding domain (DBD): Contains three zinc finger motifs (Zn1, Zn2, Zn3) that recognize various DNA structures including single-strand breaks, double-strand breaks, and cruciforms [105]
- Nuclear localization signal (NLS): Situated near the DBD, facilitates nuclear import
- Automodification domain (AMD): Also known as the BRCT domain, serves as a target for auto-poly(ADP-ribosyl)ation and mediates protein-protein interactions
- WGR domain: Named after conserved amino acids (Trp, Gly, Arg), contributes to DNA binding
- Catalytic domain (CD): Mediates poly(ADP-ribose) polymerization using NAD+ as a substrate [104]
Caspase Cleavage Site
The primary caspase cleavage site in human PARP-1 is located at aspartic acid residue 214 (DEVD↓G) within the nuclear localization signal, situated between the DNA-binding domain and the automodification domain [39] [11]. This site is preferentially cleaved by executioner caspases-3 and -7 during apoptosis, generating two characteristic fragments: a 24-kDa N-terminal fragment containing the DNA-binding domain, and an 89-kDa C-terminal fragment containing the automodification and catalytic domains [11] [54].
Table 1: PARP-1 Domains and Cleavage Fragments
| Domain/Region | Position | Function | Location in Fragments |
|---|---|---|---|
| Zinc Finger 1 (Zn1) | ~1-100 | DNA damage recognition | 24-kDa fragment |
| Zinc Finger 2 (Zn2) | ~101-200 | DNA damage recognition | 24-kDa fragment |
| Zinc Finger 3 (Zn3) | ~201-233 | DNA binding modulation | 24-kDa fragment |
| Nuclear Localization Signal | ~214-226 | Nuclear import | 24-kDa fragment |
| Caspase Cleavage Site | D214 | Caspase-3/7 recognition | Cleavage site |
| BRCT Domain | ~389-486 | Protein-protein interactions | 89-kDa fragment |
| WGR Domain | ~487-586 | DNA binding | 89-kDa fragment |
| Catalytic Domain | ~662-1014 | PAR synthesis | 89-kDa fragment |
Evolutionary Conservation of PARP-1 Cleavage
Conservation Across Species
Comparative analysis of PARP-1 orthologs reveals significant evolutionary conservation of the caspase cleavage site and the resulting fragment structures. Notably, several lower eukaryotes naturally express PARP-1 variants that resemble the truncated form generated by caspase cleavage in higher organisms [23]. For instance, PARP-1 orthologs in certain lower organisms lack the first two zinc finger motifs or even the third zinc finger, structurally mirroring the 89-kDa tPARP1 fragment produced during apoptosis in humans [23].
This evolutionary pattern suggests that the truncated PARP-1 form has biological functions independent of its role in DNA damage detection. The conservation of these truncated forms indicates positive evolutionary selection for the functional activities of PARP-1 cleavage fragments across diverse species.
Functional Conservation
The functional consequences of PARP-1 cleavage are conserved across species, with similar biological outcomes observed in mammalian, avian, and invertebrate models:
- Inactivation of DNA repair: The separation of DNA-binding domains from catalytic domains conserves cellular energy and prevents futile repair during apoptosis [11]
- Generation of pro-apoptotic signals: Cleavage fragments acquire new functions that promote cell death execution [39] [54]
- Subcellular redistribution: Fragments translocate to different cellular compartments to execute their functions [23] [54]
Table 2: Evolutionary Conservation of PARP-1 Cleavage
| Species/Organism Group | Cleavage Conservation | Fragment Sizes | Key Functional Similarities |
|---|---|---|---|
| Mammals (Human, Mouse) | Complete conservation of caspase site | 24-kDa & 89-kDa | DNA repair inhibition, cytoplasmic translocation |
| Birds (Chicken) | High conservation | Similar fragments | Caspase-dependent cleavage |
| Lower Vertebrates | Conservation | Similar fragments | Apoptosis execution |
| Insects (Drosophila) | Conservation with variations | Variable sizes | Cell death regulation |
| Lower Eukaryotes | Natural truncation similar to cleavage | Truncated forms | Alternative functions |
Biochemical Mechanisms and Consequences of Cleavage
Fragment-Specific Functions
The cleavage of PARP-1 generates two primary fragments with distinct and evolutionarily conserved functions:
24-kDa Fragment (N-terminal)
- Contains the first two zinc fingers and nuclear localization signal [11]
- Remains tightly bound to DNA strand breaks, acting as a trans-dominant inhibitor of DNA repair by blocking access of other DNA repair proteins [11] [54]
- Serves as a signature biomarker for caspase-mediated apoptosis [11]
89-kDa Fragment (C-terminal)
- Contains the automodification domain, WGR domain, and catalytic domain [54]
- Translocates from the nucleus to the cytoplasm due to loss of the nuclear localization signal [23] [54]
- Serves as a carrier of poly(ADP-ribose) (PAR) polymers to the cytoplasm [54] [13]
- Binds to apoptosis-inducing factor (AIF) via PAR polymers, facilitating AIF release from mitochondria and subsequent nuclear translocation [54] [13]
- In lower eukaryotes, similar truncated forms catalyze ADP-ribosylation of novel substrates like RNA polymerase III [23]
Structural Basis for Altered Function
The cleavage-induced separation of domains fundamentally alters PARP-1 function through several mechanisms:
- Loss of DNA binding coordination: Without the N-terminal zinc fingers, the 89-kDa fragment cannot efficiently localize to DNA damage sites [23]
- Exposure of cryptic functional domains: The BRCT domain in the 89-kDa fragment becomes accessible for interaction with new protein partners, such as RNA polymerase III [23]
- Altered subcellular localization: The separate fragments partition to different cellular compartments, enabling spatially distinct functions [54]
Diagram 1: PARP-1 Cleavage and Apoptosis Signaling Pathway
Experimental Analysis of PARP-1 Cleavage
Detection Methodologies
Western Blot Analysis
- Primary Antibodies: Use antibodies recognizing either full-length PARP-1 and the 89-kDa fragment, or specific antibodies detecting only the 89-kDa fragment [54]
- Characteristic Band Pattern: Full-length PARP-1 (116-kDa) with cleavage products (89-kDa and 24-kDa) [39] [54]
- Validation: Confirm apoptosis induction with additional markers (caspase-3 activation, Annexin V staining) [8] [23]
Immunofluorescence and Subcellular Localization
- Track fragment translocation using antibodies specific to different PARP-1 domains [54]
- Demonstrate nuclear-to-cytoplasmic shift of 89-kDa fragment while 24-kDa fragment remains nuclear [54]
Co-immunoprecipitation assays
- Identify novel binding partners of cleavage fragments [23]
- Map interaction domains using truncation mutants [23]
Functional Assays
Apoptosis Induction and Inhibition
- Inducers: Staurosporine (0.1-1 μM), actinomycin D (0.5-5 μM), poly(dA-dT) transfection [23] [54]
- Inhibitors: zVAD-fmk (pan-caspase inhibitor, 20-50 μM), PJ34 (PARP inhibitor, 1-10 μM) [54]
- Cell Viability Assessment: MTT assay, Annexin V/PI staining, morphological analysis [8] [23]
Protein-Protein Interaction Mapping
- Generate internal truncation mutants to identify interaction domains [23]
- Use point mutations (e.g., F473A in BRCT domain) to disrupt specific interactions [23]
Diagram 2: Experimental Workflow for PARP-1 Cleavage Analysis
Research Reagent Solutions
Table 3: Essential Research Reagents for PARP-1 Cleavage Studies
| Reagent/Category | Specific Examples | Function/Application | Key Research Findings |
|---|---|---|---|
| PARP-1 Antibodies | Anti-PARP-1 (full-length and fragment-specific) | Detection and quantification of cleavage | 89-kDa fragment translocation to cytoplasm [54] |
| Caspase Inhibitors | zVAD-fmk (pan-caspase) | Inhibit PARP-1 cleavage | Complete suppression of cleavage and cell death [54] |
| PARP Inhibitors | PJ34, ABT-888, Olaparib | Block PARP-1 catalytic activity | Reduced PAR synthesis and AIF-mediated death [54] |
| Apoptosis Inducers | Staurosporine, Actinomycin D, poly(dA-dT) | Activate caspases and induce cleavage | PARP-1 fragmentation and apoptotic signaling [23] [54] |
| Expression Constructs | PARP-1WT, PARP-1UNCL (uncleavable), PARP-124, PARP-189 | Functional studies of cleavage fragments | Cytoprotective effect of PARP-1UNCL and PARP-124 [39] |
| Cell Lines | PARP-1-deficient 293T, HeLa with PARP-1 shRNA | Loss-of-function studies | Reduced PAR synthesis and AIF translocation in knockdown [23] [54] |
Therapeutic Implications and Future Directions
Cancer Therapeutics
PARP inhibitors (PARPi) have emerged as powerful targeted therapies for cancers with DNA repair deficiencies, particularly BRCA-mutant ovarian and breast cancers [104] [8]. The conserved nature of PARP-1 cleavage mechanisms informs therapeutic strategies:
- Synthetic lethality: PARP inhibition in DNA repair-deficient cancers causes accumulation of unrepaired DNA damage leading to cell death [104]
- Overcoming resistance: RSL3 (ferroptosis inducer) promotes apoptosis in PARPi-resistant cells by reducing full-length PARP-1 through METTL3-mediated m6A modification and inducing caspase-dependent cleavage [8]
- Combination therapies: Targeting both PARP-1 and related pathways (ferroptosis, apoptosis) shows promise against resistant malignancies [8]
Neurodegenerative Disorders
In neurological conditions such as cerebral ischemia, Parkinson's disease, and Alzheimer's disease, PARP-1 overactivation contributes to neuronal death [39] [11]. Inhibition of PARP-1 cleavage or activity provides neuroprotection in experimental models:
- Uncleavable PARP-1 (PARP-1UNCL) confers protection from oxygen/glucose deprivation damage in neuronal models [39]
- The 24-kDa fragment shows cytoprotective effects while the 89-kDa fragment is cytotoxic in ischemic challenges [39]
- PARP inhibitors attenuate brain injury in models of stroke, trauma, and excitotoxicity [11]
Emerging Research Directions
Future research on PARP-1 cleavage conservation should focus on:
- Exploiting fragment-specific functions for targeted therapeutics
- Developing biomarkers based on cleavage fragments for treatment monitoring
- Exploring the role of PARP-1 cleavage in non-apoptotic processes and inflammatory responses
- Investigating tissue-specific and species-specific differences in cleavage consequences
The evolutionary conservation of PARP-1 cleavage across species underscores its fundamental role as a regulatory switch in programmed cell death. The consistent generation of 24-kDa and 89-kDa fragments with specialized functions highlights the evolutionary optimization of this mechanism for precise control of cell fate decisions. Understanding these conserved processes provides a robust foundation for developing novel therapeutic strategies that target PARP-1 cleavage in cancer, neurodegenerative diseases, and other conditions characterized by dysregulated cell death. The continued investigation of PARP-1 cleavage fragments and their conserved functions will undoubtedly yield valuable insights for both basic biology and clinical applications.
Conclusion
PARP-1 cleavage serves as a decisive molecular switch that directs cellular fate between survival, apoptosis, and alternative cell death pathways like parthanatos. The specific proteolytic processing of PARP-1 by different enzymes generates signature fragments that not only serve as biomarkers but actively participate in cell death execution. In apoptosis, caspase-mediated cleavage inactivates PARP-1's DNA repair function and conserves cellular energy, while in parthanatos, PARP-1 overactivation triggers AIF-mediated death. The dual nature of PARP-1 - as both a DNA guardian and cell death executioner - presents unique therapeutic opportunities, evidenced by the clinical success of PARP inhibitors in oncology and their investigational use in neurodegenerative conditions. Future research should focus on developing cleavage-specific inhibitors, understanding tissue-specific regulation of PARP-1 processing, and exploring the non-canonical functions of PARP-1 fragments in immune signaling and transcriptional regulation. The continued elucidation of PARP-1's multifaceted roles in cell death will undoubtedly yield novel therapeutic strategies for cancer, neurodegenerative diseases, and other conditions characterized by dysregulated cell death.
References
- 1. PARP-1 cleavage fragments: signatures of cell-death ... [https://biosignaling.biomedcentral.com/articles/10.1186/1478-811X-8-31]
- 2. The DNA-Binding Domain of Human PARP-1 Interacts with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3094755/]
- 3. NAD+ analog reveals PARP-1 substrate-blocking ... [https://www.nature.com/articles/s41467-018-03234-8]
- 4. Automodification switches PARP-1 function from chromatin ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4156740/]
- 5. PARP (Poly ADP‐ribose Polymerase) Family in Health and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12402623/]
- 6. PARP (Poly ADP-ribose Polymerase) Family in Health and ... [https://pubmed.ncbi.nlm.nih.gov/40904702/]
- 7. Poly(ADP-ribose) polymerase-1 (PARP1)-based dual ... [https://www.sciencedirect.com/science/article/abs/pii/S0223523425007020]
- 8. RSL3 promotes PARP1 apoptotic functions by distinct ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12492750/]
- 9. Comprehensive dataset of interactors for the entire PARP ... [https://www.nature.com/articles/s41597-025-04722-5]
- 10. Activation and Caspase-mediated Inhibition of PARP - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC99613/]
- 11. PARP-1 cleavage fragments: signatures of cell-death ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3022541/]
- 12. The 89-kDa PARP1 cleavage fragment serves as a ... - PubMed [https://pubmed.ncbi.nlm.nih.gov/33168626/]
- 13. The 89-kDa PARP1 cleavage fragment serves as a ... [https://www.sciencedirect.com/science/article/pii/S0021925820000320]
- 14. Biological function, mediate cell death pathway and their ... [https://www.frontiersin.org/journals/nutrition/articles/10.3389/fnut.2022.1093939/full]
- 15. Caspase-mediated cleavage of the centrosomal proteins ... [https://www.nature.com/articles/s41419-018-0632-8]
- 16. Assessment of Apoptosis by Immunohistochemistry to Active... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2664986/]
- 17. - Caspase , 9 - caspase and 3 - caspase have distinct roles during... 7 [https://bmcmolcellbiol.biomedcentral.com/articles/10.1186/1471-2121-14-32]
- 18. PARP-1, a determinant of cell survival in response to DNA ... [https://www.sciencedirect.com/science/article/pii/S0301472X03000833]
- 19. Role of Calpain in Apoptosis - PMC - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC3584455/]
- 20. N‐terminomics and proteomics analysis of Calpain‐2 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12023407/]
- 21. The roles of cellular protease interactions in viral infections ... [https://link.springer.com/article/10.1007/s43440-022-00394-9]
- 22. New Perspectives on the Role of Nuclear Proteases in Cell ... [https://www.mdpi.com/2079-7737/12/6/797]
- 23. Truncated PARP1 mediates ADP-ribosylation of RNA ... [https://www.nature.com/articles/s41421-021-00355-1]
- 24. Rapid Detection and Signaling of DNA Damage by PARP-1 [https://pmc.ncbi.nlm.nih.gov/articles/PMC8364484/]
- 25. Signaling Mechanism of Poly(ADP-Ribose) Polymerase-1 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3069822/]
- 26. The basi(c)s of PARP inhibitor efficacy and resistance [https://www.sciencedirect.com/science/article/pii/S0093775423000611]
- 27. PARP enzyme de novo synthesis of protein-free poly(ADP ... [https://www.sciencedirect.com/science/article/abs/pii/S1097276524008645]
- 28. Rapid Detection and Signaling of DNA Damage by PARP-1 [https://www.sciencedirect.com/science/article/abs/pii/S0968000421000281]
- 29. PARP-1 and its associated nucleases in DNA damage ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6764844/]
- 30. The deubiquitination-PARylation positive feedback loop of ... [https://www.nature.com/articles/s41388-025-03428-7]
- 31. Comprehensive landscape of cell death mechanisms [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2025.1611055/full]
- 32. Caspases as master regulators of programmed cell death [https://pmc.ncbi.nlm.nih.gov/articles/PMC12229594/]
- 33. PARP1 [https://en.wikipedia.org/wiki/PARP1]
- 34. Crystal structures of poly(ADP-ribose) polymerase-1 (PARP-1 ... [https://pubmed.ncbi.nlm.nih.gov/21233213/]
- 35. 4DQY: Structure of Human PARP-1 bound to a DNA ... [https://www.rcsb.org/structure/4DQY]
- 36. Research advances on the role of programmed endothelial ... [https://www.nature.com/articles/s41420-025-02728-x]
- 37. - PARP negatively regulates nucleolar protein pool and mitochondrial... 1 [https://genesenvironment.biomedcentral.com/articles/10.1186/s41021-024-00312-w]
- 38. The BCL - 2 family protein BCL-RAMBO interacts and cooperates with... [https://www.nature.com/articles/s41598-023-41196-0?error=cookies_not_supported]
- 39. Poly(ADP-ribose) polymerase-1 and its cleavage products ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4013233/]
- 40. PARP-1 cleavage fragments: signatures of cell-death ... [https://pubmed.ncbi.nlm.nih.gov/21176168/]
- 41. Characterization of the necrotic cleavage of poly(ADP- ... [https://www.nature.com/articles/4400851]
- 42. The Design of a Quantitative Western Blot Experiment [https://pmc.ncbi.nlm.nih.gov/articles/PMC3971489/]
- 43. 2024 Scientific Publication Requirements for Western Blots ... [https://azurebiosystems.com/blog/publication-requirements-for-western-blots-and-gels/]
- 44. 2024 Guide to Quantitative Western Blot Publication [https://www.thermofisher.com/blog/life-in-the-lab/2023-guide-to-quantitative-western-blot-publication/]
- 45. Roles and therapeutic potential of PARP-1 in ... [https://www.sciencedirect.com/science/article/abs/pii/S0006295225006380]
- 46. PARP1 antibody (13371-1-AP) [https://www.ptglab.com/products/PARP1-Antibody-13371-1-AP.htm?srsltid=AfmBOorJLo5Ukb0IbRnWkoHPbB4P-OnaWZVFgDyF_J2I3EgmBMKNEpym]
- 47. The Prognostic Value of PARP Expression in High-Grade ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7471102/]
- 48. PARP1: A Potential Molecular Marker to Identify Cancer ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8882878/]
- 49. Nuclear PARP-1 protein overexpression is associated with ... [https://www.sciencedirect.com/science/article/pii/S092375341934671X]
- 50. Programmed cell death detection methods - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC9308588/]
- 51. Development of a fluorescence-based cellular apoptosis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6372133/]
- 52. Quantitative analysis of fluorescent caspase substrate ... [https://www.nature.com/articles/4400769]
- 53. CHD6 has poly(ADP-ribose)- and DNA-binding domains ... [https://www.nature.com/articles/s41467-025-56085-5]
- 54. The 89-kDa PARP1 cleavage fragment serves as a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7948984/]
- 55. PARP1-Dependent and Independent Pathways for ... [https://pubmed.ncbi.nlm.nih.gov/40955140/]
- 56. Biomarker-Guided Development of DNA Repair Inhibitors [https://www.sciencedirect.com/science/article/pii/S109727652030277X]
- 57. Prognostic Value of PARP1 and PARP2 Copy Number ... [https://pubmed.ncbi.nlm.nih.gov/40210166/]
- 58. Prognostic Value of PARP1 and PARP2 Copy Number ... [https://www.sciencedirect.com/science/article/abs/pii/S0023683725000819]
- 59. Beyond DNA Repair: Additional Functions of PARP-1 in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3841914/]
- 60. A single nucleotide variant of human PARP1 determines ... [https://www.nature.com/articles/s41698-020-0113-2]
- 61. Exploring the role of PARP1 inhibition in enhancing antibody ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-024-05838-9]
- 62. Regulatory apoptotic fragment of PARP1 complements ... [https://www.sciencedirect.com/science/article/abs/pii/S1568786423001477]
- 63. Tipping the PARylation scale: Dysregulation of PAR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12602731/]
- 64. Roles and therapeutic potential of PARP-1 in ... [https://pubmed.ncbi.nlm.nih.gov/41022359/]
- 65. Induced pluripotent stem cells carrying novel APTX ... [https://www.nature.com/articles/s41420-025-02723-2]
- 66. PARP-1 overexpression contributes to Cadmium-induced ... [https://www.nature.com/articles/s41598-017-04555-2]
- 67. RSL3 promotes PARP1 apoptotic functions by distinct ... [https://cmbl.biomedcentral.com/articles/10.1186/s11658-025-00785-9]
- 68. Poly (ADP-Ribose) polymerase 1 and parthanatos in ... [https://www.sciencedirect.com/science/article/pii/S0969996123003303]
- 69. A quantitative real-time approach for discriminating ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5253725/]
- 70. The Quantitative-Phase Dynamics of Apoptosis and Lytic ... [https://www.nature.com/articles/s41598-020-58474-w]
- 71. Flow cytometry-based quantitative analysis of cellular ... [https://www.sciencedirect.com/science/article/pii/S2405844024096968]
- 72. Cleaved PARP (Asp214) Antibody #9541 [https://www.cellsignal.com/products/primary-antibodies/cleaved-parp-asp214-antibody/9541?srsltid=AfmBOorRu6vnb62IGUpyiNp9fp9qg99P4Hqnto4NYLFknPiyw6Dc86lp]
- 73. BD Pharmingen™ PE Mouse Anti-Cleaved PARP (Asp214) [https://www.bdbiosciences.com/en-us/products/reagents/flow-cytometry-reagents/research-reagents/single-color-antibodies-ruo/pe-mouse-anti-cleaved-parp-asp214.552933]
- 74. Anti-Cleaved PARP1 antibody [E51] (ab32064) [https://www.abcam.com/en-us/products/primary-antibodies/cleaved-parp1-antibody-e51-ab32064]
- 75. Cleaved PARP (Asp214) (D64E10) Rabbit Monoclonal ... [https://www.cellsignal.com/products/antibody-conjugates/cleaved-parp-asp214-d64e10-xp-rabbit-mab-pacific-blue-conjugate/60068?srsltid=AfmBOorZjhM9qjyKpf-6niSk7wmOlSsbZGdOKifQoqVdek7Eyt9_zNv9]
- 76. Poly(ADP-ribose) polymerase-1 and its ambiguous role in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9877585/]
- 77. - PARP fragments: signatures of 1 - cleavage ... | Sigma-Aldrich cell death [https://www.sigmaaldrich.com/DE/en/tech-docs/paper/115479]
- 78. Research progress on morphology and mechanism of ... [https://www.nature.com/articles/s41419-024-06712-8]
- 79. (46D11) Rabbit mAb | Cell Signaling Technology PARP [https://www.cellsignal.com/products/primary-antibodies/parp-46d11-rabbit-mab/9532]
- 80. The PARP family: insights into functional aspects of poly ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6496725/]
- 81. Targeting PARP1: A Promising Approach for Next- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12162764/]
- 82. Human PARP1 substrates and regulators of its catalytic activity [https://pmc.ncbi.nlm.nih.gov/articles/PMC9995695/]
- 83. Cleaved PARP (Asp214) (D64E10) Rabbit Monoclonal ... [https://www.cellsignal.com/products/primary-antibodies/cleaved-parp-asp214-d64e10-rabbit-monoclonal-antibody/5625?srsltid=AfmBOor8RUbE1PpIIUOXxFg15yiJ7Edzz9KJVwEFvFSaifLswhfCh0Rm]
- 84. PARP inhibition: PARP1 and beyond - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC2910902/]
- 85. Characterization of the interactions of PARP-1 with UV- ... [https://www.nature.com/articles/srep19020]
- 86. Minimizing DNA trapping while maintaining activity ... [https://www.nature.com/articles/s41419-024-07277-2]
- 87. PARP-1 Modulation of mTOR Signaling in Response to a DNA ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0047978]
- 88. Necroptosis, pyroptosis and apoptosis: an intricate game of ... [https://www.nature.com/articles/s41423-020-00630-3]
- 89. Caspases as master regulators of programmed cell death [https://www.nature.com/articles/s12276-025-01470-9]
- 90. Pyroptosis versus necroptosis: similarities, differences, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6294779/]
- 91. Apoptosis, Pyroptosis, and Necroptosis—Oh My! The Many ... [https://www.sciencedirect.com/science/article/abs/pii/S002228362100615X]
- 92. Regulated Cell Death in Traumatic Brain Injury [https://www.mdpi.com/2073-4409/14/23/1878]
- 93. Programmed cell revival from imminent cell death enhances ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12489119/]
- 94. Role of poly(ADP-ribose) polymerase-1 in regulating ... [https://www.nature.com/articles/s41598-022-25405-w]
- 95. Caspase Activity Mediates the Differentiation of Embryonic ... [https://www.sciencedirect.com/science/article/pii/S1934590908001628]
- 96. CK2调控HERPUD1稳定性介导棕榈酸诱导的未折叠蛋白 ... [https://www.ebiotrade.com/newsf/2025-11/20251107001940896.htm]
- 97. PARP1降解新突破:180055精准打击肿瘤细胞且无DNA捕获 ... [https://news.bioon.com/article/3b7985e3378d.html]
- 98. Poly(ADP-ribose) polymerase-1 and its cleavage products ... [https://www.sciencedirect.com/science/article/pii/S0167488913004242]
- 99. DNA repair pathway choices in CRISPR-Cas9 mediated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8187289/]
- 100. PARP‑1 in liver diseases: Molecular mechanisms ... [https://www.spandidos-publications.com/10.3892/mmr.2025.13689]
- 101. cellular stress signaling through poly(ADP-ribose) and PARP-1 [https://genesdev.cshlp.org/content/26/5/417]
- 102. Research progress on ferroptosis and PARP inhibitors in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12058875/]
- 103. Potentiation of cancer immunogenicity by targeting PARP [https://pmc.ncbi.nlm.nih.gov/articles/PMC12198780/]
- 104. The multifaceted roles of PARP1 in DNA repair and chromatin ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6591728/]
- 105. Structural and Biophysical Studies of Human PARP-1 in ... [https://www.sciencedirect.com/science/article/abs/pii/S0022283609014569]