PARP-1 Cleavage Fragments: From Molecular Hallmarks to Therapeutic Targets in Neurodegeneration
This article synthesizes current knowledge on the proteolytic cleavage of PARP-1, a key DNA damage sensor, and the distinct biological activities of its resulting fragments in the context of neurodegenerative...
PARP-1 Cleavage Fragments: From Molecular Hallmarks to Therapeutic Targets in Neurodegeneration
Abstract
This article synthesizes current knowledge on the proteolytic cleavage of PARP-1, a key DNA damage sensor, and the distinct biological activities of its resulting fragments in the context of neurodegenerative diseases. We explore the foundational biology of PARP-1 cleavage by caspases and other proteases, detailing how the generated 24 kDa and 89 kDa fragments can exert opposing effects on neuronal survival, inflammation, and cell death pathways like parthanatos. Methodological approaches for detecting these fragments and modeling their function in vitro and in vivo are reviewed. The article further addresses the central challenge in the field—the paradoxical dual role of PARP-1 in neuroprotection and neurotoxicity—and discusses troubleshooting strategies for therapeutic intervention. Finally, we validate the potential of PARP-1 fragments as biomarkers and compare the therapeutic efficacy of PARP inhibition across various neurodegenerative conditions, providing a comprehensive resource for researchers and drug development professionals aiming to target this pathway.
The Biology of PARP-1 Cleavage: Proteases, Fragments, and Their Roles in Neuronal Health
Poly(ADP-ribose) polymerase 1 (PARP1) is a highly abundant nuclear enzyme that serves as a primary DNA damage sensor in cells. This multifunctional protein plays a critical role in maintaining genomic integrity through its involvement in various DNA repair pathways, including base excision repair (BER), single-strand break repair (SSBR), and double-strand break repair [1] [2]. PARP1's domain architecture enables it to detect DNA damage, initiate poly(ADP-ribose) (PAR) synthesis, and recruit DNA repair factors to damage sites. Beyond its established roles in DNA repair and cancer biology, emerging evidence indicates that PARP1 function—and particularly its cleavage fragments—plays a significant role in the pathogenesis of neurodegenerative diseases [3] [4]. Understanding the precise structure-function relationship of PARP1 domains provides crucial insights for developing targeted therapeutic strategies for both cancer and neurological disorders.
Domain Architecture of PARP1
PARP1 is a multi-domain protein of approximately 116 kDa, consisting of six independently folded domains that orchestrate its DNA damage detection and signaling functions [1] [2]. These domains can be grouped into three primary functional regions: the DNA-binding domain, the auto-modification domain, and the catalytic domain.
Table 1: PARP1 Domains and Their Functions
| Domain Name | Location | Key Functions | Structural Features |
|---|---|---|---|
| Zinc Finger 1 (Zn1) | N-terminal (1-97) | Primary DNA break recognition, cooperative binding with Zn2 | CCHC zinc finger motif, structurally similar to Zn2 |
| Zinc Finger 2 (Zn2) | N-terminal (98-207) | DNA break recognition, key role in SSB detection | CCHC zinc finger motif, shares high structural similarity with Zn1 |
| Zinc Finger 3 (Zn3) | N-terminal (228-368) | Not directly binding DNA breaks; essential for activation | Unique structure unrelated to Zn1/Zn2 |
| BRCT Domain | Central (385-479) | Protein-protein interactions, auto-modification site | BRCT fold (BRCA1 C-terminus-like) |
| WGR Domain | Central (504-525) | DNA binding, allosteric regulation | Tryptophan-Glycine-Arg-rich motif |
| Catalytic Domain | C-terminal (528-1014) | PAR synthesis from NAD+ | Comprises helical subdomain (HD) and ART subdomain |
The N-terminal region contains three zinc finger domains (Zn1, Zn2, and Zn3) that facilitate DNA damage recognition [1] [2]. While Zn1 and Zn2 directly interact with DNA breaks, Zn3 plays a regulatory role in activation rather than direct DNA binding [5]. The central region contains the BRCA1 C-terminus (BRCT) domain, which mediates protein-protein interactions and serves as the primary site for auto-modification, followed by the tryptophan-glycine-arginine (WGR) domain, which also contributes to DNA binding and allosteric regulation [1] [2]. The C-terminal region houses the catalytic domain, which is further divided into the helical subdomain (HD) and the ADP-ribosyl transferase (ART) subdomain [1]. The ART subdomain contains the conserved catalytic pocket that binds NAD+ and facilitates the transfer of ADP-ribose units to target proteins [2].
DNA-Binding Domain: Structure and Mechanism
The DNA-binding domain of PARP1 encompasses Zn1, Zn2, and Zn3, with Zn1 and Zn2 serving as the primary modules for recognizing DNA strand breaks [1] [5]. These zinc fingers belong to a highly unusual class characterized by a CCHC ligand pattern and an unusually long sequence separation (26-37 residues) between ligands 2 and 3 [5].
Structural Basis of DNA Recognition
Biophysical and structural studies reveal that Zn1 and Zn2 are structurally independent in the absence of DNA but cooperate to recognize DNA breaks [5]. The crystal structure of the PARP1 DNA-binding domain in complex with a DNA double-strand break (PDB: 4AV1) shows that Zn1 and Zn2 from separate PARP1 molecules form a strand-break recognition module that facilitates PARP1 dimerization [6]. This dimeric assembly helps activate PARP1 by promoting trans-automodification [6].
Research highlights that Zn2 plays a predominant role in DNA damage recognition, interacting more strongly with nicked or gapped DNA ligands compared to Zn1 [5]. The F1 + F2 fragment (Zn1 + Zn2) recognizes DNA single-strand breaks as a monomer and in a single orientation, contrary to earlier proposals of dimerization [5]. Both fingers contact the DNA phosphate backbone through specific residues—R18 in Zn1 and corresponding residues in Zn2—forming a "phosphate grip" that stabilizes the DNA-protein complex [1].
Damage Recognition Specificity
PARP1 exhibits remarkable versatility in recognizing various DNA lesions, including single-strand breaks, double-strand breaks, gaps, and DNA crosslinks [1] [5]. The DNA-binding domain recognizes different types of DNA single-strand breaks in a highly similar conformation, enabling PARP1 to participate in multiple steps of DNA single-strand break repair and base excision repair [5]. Recent structures of PARP1 domains bound to DNA double-strand breaks (PDB: 7S81) have captured snapshots of PARP1 in active states, providing novel insights into the mechanics of PARP1 allostery and multi-domain interactions with DNA damage [7].
Figure 1: PARP1 DNA Damage Recognition and Activation Mechanism. This diagram illustrates how PARP1 domains cooperate to detect DNA damage and initiate the allosteric activation cascade.
Auto-modification Domain: BRCT and Regulatory Elements
The auto-modification domain (AD) of PARP1 contains the BRCT fold and is flanked by the WGR domain, together forming a critical regulatory unit that controls PARP1 function and dissociation from DNA [1] [2].
Serine Auto-modification Sites
Recent research has identified three key serine residues (Ser499, Ser507, and Ser519) within the auto-modification domain as predominant in vivo PARP1 auto-modification sites [8]. These residues undergo HPF1-dependent serine ADP-ribosylation, which plays a vital role in cellular responses to PARP inhibitors [8]. Efficient modification of these serine residues counters PARP1 trapping on DNA and contributes to inhibitor tolerance [8].
The auto-modification domain serves as a critical regulatory region that controls PARP1's residence time on DNA damage. Auto-modification promotes PARP1 eviction from DNA breaks, which is essential for allowing DNA repair and replication to proceed [9]. Mutation of these serine residues generates an auto-modification-deficient PARP1 that retains catalytic activity but demonstrates prolonged retention at DNA damage sites [9].
Functional Consequences of Auto-modification
Auto-modification directly influences PARP1's functional interactions at DNA damage sites. PARP1 auto-modification promotes faithful Okazaki fragment processing and limits replication fork speed [9]. The negative charge of PAR chains created during auto-modification generates electrostatic repulsion that facilitates PARP1 dissociation from DNA, preventing prolonged occupancy that could impede DNA repair processes [2].
In the context of neurodegenerative diseases, proper regulation of PARP1 auto-modification appears crucial for neuronal health. Dysregulated PAR signaling—whether through overactivation or suppression—can lead to neuronal dysfunction [3]. Huntington's disease exhibits a unique characteristic of reduced PAR levels and impaired PARP1 activity, even in the prodromal phase, suggesting that balanced PARylation is essential for neuronal homeostasis [3].
Catalytic Domain: Structure and Inhibition
The C-terminal catalytic domain of PARP1 comprises two key subdomains: the helical subdomain (HD) and the ADP-ribosyl transferase (ART) subdomain [1] [2]. This domain executes the synthesis of poly(ADP-ribose) chains using NAD+ as a substrate.
Structural Organization
The helical subdomain serves an auto-inhibitory function when PARP1 is in the non-DNA bound state, preventing access to NAD+ [2]. Upon DNA binding and allosteric activation, conformational changes in the HD subdomain relieve this auto-inhibition, allowing the ART subdomain to bind NAD+ and catalyze ADP-ribosylation [1] [10]. The ART subdomain contains the highly conserved catalytic pocket that accommodates NAD+ and facilitates the transfer of ADP-ribose units to target proteins [2].
Structural studies have revealed that the activation of PARP1 involves dynamic changes in the structure of the regulatory helical domain, with captured snapshots of PARP1 in isolated active states displaying specific HD conformations that contribute to PARP1 multi-domain and high-affinity interaction with DNA damage [7].
PARP Inhibition Mechanisms
Clinical PARP inhibitors are essentially NAD+ analogs that contain the nicotinamide moiety and compete with NAD+ for binding to the catalytic pocket [2] [10]. Currently, there are six FDA-approved PARP inhibitors (olaparib, rucaparib, niraparib, talazoparib, fluzoparib, and pamiparib) used in cancer treatment, particularly for BRCA1/2-mutant cancers [2].
PARP inhibitors exert their therapeutic effects through two primary mechanisms: catalytic inhibition and PARP trapping [2] [10]. All current clinical PARP inhibitors block the catalytic activity of PARP1, but their ability to trap PARP1 onto DNA varies and parallels their cytotoxic potency [2]. PARP trapping refers to the prolonged residence of inhibited PARP1 on damaged chromatin, which is more detrimental to cells than PARP1 depletion alone [8].
Table 2: PARP1 Inhibitors and Their Properties
| Inhibitor Name | Trapping Potency | Clinical Status | Primary Applications |
|---|---|---|---|
| Olaparib | Moderate | FDA-approved | Ovarian, breast, pancreatic, prostate cancers |
| Talazoparib | High | FDA-approved | BRCA-mutant breast cancer |
| Niraparib | High | FDA-approved | Ovarian cancer maintenance therapy |
| Rucaparib | Moderate | FDA-approved | Ovarian, prostate cancers |
| Veliparib | Low | Clinical trials | Combination therapy with chemotherapy |
Experimental Approaches for Studying PARP1 Structure-Function
Advancements in structural biology techniques have been instrumental in elucidating PARP1 architecture and activation mechanisms.
Structural Biology Methods
X-ray crystallography has provided high-resolution structures of individual PARP1 domains and multi-domain fragments in complex with DNA [7] [6]. For example, the structure of the human PARP1 DNA-binding domain in complex with DNA (PDB: 4AV1) was solved at 3.10 Å resolution using X-ray crystallography [6]. More recently, cryo-electron microscopy (cryo-EM) has enabled the determination of full-length PARP2 in complex with a nucleosome, providing insights into the structural basis of DNA-dependent PARP activation [1].
Biophysical techniques including NMR spectroscopy, analytical ultracentrifugation, and fluorescence measurements have been employed to characterize PARP1 domain interactions and dynamics in solution [5]. These approaches have revealed that the zinc fingers are structurally independent in the absence of DNA and share highly similar structural folds and dynamics [5].
Functional Assays
Cellular studies using PARP1 mutants, particularly auto-modification-deficient mutants, have been crucial for dissecting the distinct functional roles of PARP1 catalytic activity versus auto-modification [9] [8]. Colony formation assays demonstrate how HPF1 loss enhances PARP1/PARP2 inhibitor sensitivity and PARP-BRCA synthetic lethality [8]. Immunofluorescence-based methods track PARP1 residence on chromatin and recruitment to DNA damage sites, while western blotting detects PAR formation and PARP1 auto-modification states [8].
Table 3: Key Research Reagents and Experimental Tools
| Reagent/Tool | Function/Application | Key Features |
|---|---|---|
| PARP1 domain constructs | Structural and biophysical studies | Individual or combined domains (e.g., Zn1+Zn2, WGR-CAT) |
| Site-directed mutants | Functional dissection | Auto-modification-deficient (S499/507/519A), DNA-binding mutants |
| HPF1 knockout cells | Study serine ADPr role | ~200-fold reduction in DNA damage-induced serine ADPr |
| Clinical PARP inhibitors | Mechanistic studies | Different trapping potencies (talazoparib-high, veliparib-low) |
| DNA damage models | PARP1 activation assays | SSB, DSB, and nucleosome-containing substrates |
PARP1 in Neurodegenerative Disease Context
The role of PARP1 and its cleavage fragments extends beyond DNA repair to encompass significant implications in neurodegenerative diseases. PARP1 is cleaved by caspases during apoptosis, generating fragments that retain potential functional activities [3]. This cleavage occurs in a specific pattern that separates the DNA-binding domain from the catalytic domain, potentially contributing to dysregulated PAR signaling in neuronal cells [3].
Aberrant PAR signaling has been observed in multiple neurodegenerative conditions, including Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis, and cerebellar ataxias [3]. Most of these conditions exhibit increased PAR levels and PARP1 activity, but Huntington's disease presents a unique profile with reduced PAR levels and impaired PARP1 activity, even in prodromal stages [3]. This suggests that PAR signaling homeostasis, rather than simply its absolute level, is critical for neuronal health.
In neurodegenerative disease contexts, PARP1 activation in response to oxidative stress and DNA damage may contribute to pathological processes through multiple mechanisms, including energy depletion due to NAD+ consumption, dysregulation of transcription, and impaired DNA repair [3] [4]. The investigation of PARP1 cleavage fragments in these diseases represents a promising area for future research, potentially offering new biomarkers and therapeutic targets.
PARP1's multi-domain architecture enables its crucial function as a DNA damage sensor and signaling protein. The cooperative action of its zinc fingers, WGR domain, and regulatory regions allows precise detection of DNA strand breaks, while its catalytic domain executes PAR synthesis using NAD+ as substrate. The auto-modification domain, particularly serine residues 499, 507, and 519, serves as a critical regulatory element that controls PARP1's dissociation from DNA damage sites. Understanding these structure-function relationships provides the foundation for comprehending PARP1's roles in both DNA repair and neurodegenerative diseases. The ongoing structural characterization of PARP1 domains and their interdomain communications continues to inform the development of more specific PARP-targeted therapies with potential applications in both oncology and neurodegenerative disorders. Future research focusing on PARP1 cleavage fragments and their activities in neuronal cells may yield important insights into disease mechanisms and therapeutic opportunities.
The maintenance of cellular homeostasis is a fundamental biological process, and the regulated elimination of cells through programmed cell death is one of its critical components. Central to the execution of these death programs are suicidal proteases—enzymes that cleave specific cellular substrates to dismantle the cell in a controlled manner. Among these proteases, caspases, calpains, and granzymes represent three major families with distinct activation mechanisms, substrate specificities, and biological functions. In the context of neurodegenerative disease research, these proteases assume particular significance as their dysregulation can drive the pathological neuronal loss characteristic of conditions such as cerebral ischemia, Alzheimer's disease, and Parkinson's disease [11] [12] [13]. A key substrate that has emerged as a critical biomarker and mediator in these processes is poly(ADP-ribose) polymerase-1 (PARP-1), a nuclear DNA repair enzyme. The cleavage of PARP-1 by different suicidal proteases generates signature fragments that serve as molecular fingerprints, providing researchers with valuable insights into the specific cell death pathways activated in neurological disorders [11] [12] [13]. This technical review examines the roles of caspases, calpains, and granzymes in mediating cell death, with a particular focus on their functions in generating PARP-1 cleavage fragments and the implications for neurodegenerative disease mechanisms and therapeutic development.
Protease Families: Mechanisms and PARP-1 Cleavage Signatures
Caspases: Apoptotic Executioners
Caspases are a family of cysteine-aspartate proteases that serve as primary mediators of apoptosis. They are synthesized as inactive zymogens (procaspases) and undergo proteolytic processing to form active tetrameric enzymes composed of two heterotypic subunits [14]. Caspases are conceptually divided into initiator caspases (e.g., caspase-8, -9) that activate the cell death cascade in response to specific signals, and effector caspases (e.g., caspase-3, -7) that cleave various cellular substrates to execute apoptosis [14]. Adapter protein-mediated oligomerization of procaspases is recognized as a universal mechanism for initiator caspase activation [14].
PARP-1 cleavage by caspases, particularly caspase-3 and -7, is considered a hallmark of apoptosis [11]. These caspases cleave PARP-1 at a specific aspartic acid residue within a nuclear localization signal near the DNA-binding domain, generating signature fragments of 89-kDa and 24-kDa [11] [15]. The 24-kDa fragment, containing two zinc-finger motifs, remains bound to DNA and acts as a trans-dominant inhibitor of DNA repair, while the 89-kDa fragment, containing the auto-modification and catalytic domains, has reduced DNA binding capacity and can translocate to the cytoplasm [11] [15]. Recent research has revealed that the 89-kDa fragment can serve as a carrier for poly(ADP-ribose) (PAR) polymers to the cytoplasm, where it facilitates apoptosis-inducing factor (AIF) release from mitochondria—a critical step in the caspase-independent cell death pathway known as parthanatos [15]. This finding demonstrates an intriguing molecular link between caspase-dependent and caspase-independent cell death pathways.
Calpains: Calcium-Activated Mediators of Necrotic and Apoptotic Death
Calpains constitute a family of calcium-activated cysteine proteases that are localized to the cytosol and mitochondria [16]. They function as key mediators in both apoptotic and necrotic cell death pathways, with their activation triggered by increased intracellular calcium levels [16] [13]. Unlike caspases, which exhibit high substrate specificity for aspartic acid residues, calpains display broader substrate specificity and can cleave a wide range of cellular proteins, including membrane, cytoplasmic, and nuclear substrates [16].
In neurodegenerative contexts such as cerebral ischemia, calpain activation contributes significantly to neuronal injury [13]. Calpains cleave PARP-1 to generate a distinct 50-kDa fragment, which serves as a signature for calpain-mediated cell death [13]. This cleavage event is particularly associated with necrotic cell death, although calpains can also participate in apoptotic pathways. The appearance of the 50-kDa PARP-1 fragment in models of cerebral ischemia provides evidence for the involvement of calpain-mediated proteolysis in the neuronal damage resulting from ischemic insult [13].
Granzymes: Immune-Mediated Cell Death Executors
Granzymes are a set of serine proteases contained within the granules of cytotoxic T lymphocytes and natural killer cells. They function as key mediators of immune-mediated cell death, eliminating virally infected and malignant cells [16]. Granzyme B, the most extensively studied family member, shares functional similarities with caspases by cleaving substrates at aspartic acid residues and can activate caspase-3 directly [16] [13].
In the context of cerebral ischemia, granzyme B has been shown to contribute to neuronal death through both apoptotic and non-apoptotic mechanisms [13]. Research has demonstrated an interaction between granzyme B and PARP-1 in ischemic brain tissue, suggesting that PARP-1 serves as a substrate for granzyme B [13]. While the specific PARP-1 cleavage fragments generated by granzyme B require further characterization, their appearance correlates with immune-mediated cell death in neurological pathologies.
Table 1: Characteristic PARP-1 Cleavage Fragments Generated by Different Suicidal Proteases
| Protease | Protease Class | PARP-1 Fragment Sizes | Primary Cell Death Association | Key Features |
|---|---|---|---|---|
| Caspase-3/7 | Cysteine-aspartase | 89-kDa, 24-kDa | Apoptosis | Hallmark of apoptosis; 24-kDa fragment inhibits DNA repair; 89-kDa fragment can translocate to cytoplasm |
| Calpain | Calcium-activated cysteine protease | 50-kDa | Necrosis/Apoptosis | Associated with calcium dysregulation; observed in cerebral ischemia models |
| Granzyme B | Serine protease | Not fully characterized | Immune-mediated cell death | Released by cytotoxic lymphocytes; can activate caspase-3 |
Table 2: Protease Families and Their Roles in Cell Death
| Protease Family | Activation Mechanism | Primary Intracellular Location | Key Substrates | Role in Neurodegeneration |
|---|---|---|---|---|
| Caspases | Proteolytic processing; adapter-mediated oligomerization | Cytosol, nucleus | PARP-1, cytoskeletal proteins, DNA repair enzymes | Executors of apoptotic neuronal death in cerebral ischemia, Alzheimer's, Parkinson's |
| Calpains | Calcium influx | Cytosol, mitochondria | PARP-1, spectrin, membrane proteins | Mediators of excitotoxicity and ischemic neuronal injury |
| Granzymes | Release from cytotoxic granules | Extracellular, can enter target cells | PARP-1, caspases, ICAD | Contributors to inflammatory component of neurodegeneration |
PARP-1 Cleavage Fragments as Biomarkers in Neurodegeneration
PARP-1 cleavage fragments serve as specific molecular signatures that allow researchers to identify the particular proteases activated and the forms of cell death occurring in neurodegenerative pathologies [11] [12] [13]. The detection of specific PARP-1 fragments in neurological tissues provides a window into the underlying cell death mechanisms, which is crucial for understanding disease pathogenesis and developing targeted therapeutic interventions.
In cerebral ischemia, multiple suicidal proteases are activated, leading to a heterogeneous pattern of cell death that includes both apoptotic and necrotic components [13]. Research using transient focal cerebral ischemia models in rats has demonstrated the concurrent activation of calpain, cathepsin-B, caspase-3, and granzyme-B, with each protease contributing to the generation of distinct PARP-1 cleavage fragments [13]. The appearance of both the 89-kDa (caspase-generated) and 50-kDa (calpain-generated) PARP-1 fragments in ischemic brain tissue indicates the involvement of both apoptotic and necrotic cell death pathways in the pathology of stroke [13]. This heterogeneous cell death response presents challenges for therapeutic intervention but also opportunities for combination therapies targeting multiple proteolytic pathways.
Beyond their value as biomarkers, PARP-1 fragments may actively participate in cell death mechanisms. The 89-kDa PARP-1 fragment generated by caspases has been shown to function as a cytoplasmic PAR carrier that induces AIF-mediated apoptosis, providing a mechanistic link between caspase activation and the parthanatos cell death pathway [15]. This finding is particularly relevant in neurodegenerative diseases where energy failure and DNA damage are prominent features.
Experimental Approaches for Studying Protease Activity and PARP-1 Cleavage
Western Blot Analysis of PARP-1 Cleavage
Objective: To detect and characterize PARP-1 cleavage fragments in experimental models of neurodegeneration.
Methodology:
- Tissue Homogenization: Homogenize brain tissue samples (e.g., from ischemic regions) in RIPA buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1 mM EDTA, 0.4% deoxycholate, 1% Nonidet P-40) containing protease inhibitors (1 mM PMSF) and phosphatase inhibitors (10 mM β-glycerophosphate, 10 mM NaF, 0.3 mM Na₃VO₄) [13].
- Protein Quantification: Determine protein concentration in the supernatant following centrifugation at 14,000 × g for 15 minutes at 4°C.
- SDS-PAGE: Separate equal amounts of protein (20-30 μg) using SDS-polyacrylamide gel electrophoresis (SDS-PAGE) with appropriate percentage gels (e.g., 8-12%) to resolve full-length PARP-1 (113-kDa) and its cleavage fragments (89-kDa, 50-kDa, 24-kDa) [13].
- Western Transfer: Transfer proteins from gels to nitrocellulose membranes using standard wet or semi-dry transfer systems.
- Immunoblotting: Block membranes with 5% non-fat skimmed milk in Tris-buffered saline (TBS) for 1 hour. Incubate with primary antibodies against PARP-1 (diluted according to manufacturer's instructions) for 12-14 hours at 4°C [13]. Use antibodies that recognize both full-length and cleaved PARP-1.
- Detection: Incubate membranes with species-appropriate secondary antibodies conjugated to alkaline phosphatase (AP) or horseradish peroxidase (HRP) for 1 hour at room temperature. Visualize immunoreactivity using BCIP/NBT substrate (for AP) or enhanced chemiluminescence (for HRP) [13].
Interpretation: The presence of specific PARP-1 fragments (89-kDa, 50-kDa, 24-kDa) indicates activation of their respective proteases (caspases, calpains, etc.) and provides insight into the cell death pathways operational in the neurodegenerative model.
Immunohistochemical Localization of Proteases and PARP-1 Fragments
Objective: To determine the spatial distribution and cellular localization of activated proteases and PARP-1 cleavage fragments in neurological tissues.
Methodology:
- Tissue Preparation: Perfuse-fix animals with 4% paraformaldehyde, dissect brains, and prepare paraffin-embedded sections (5-7 μm thickness) [13].
- Deparaffinization and Antigen Retrieval: Deparaffinize sections in xylene, rehydrate through graded alcohol series, and perform antigen retrieval using appropriate methods (e.g., citrate buffer, heat-induced epitope retrieval).
- Immunostaining: Block sections with appropriate blocking serum, then incubate with primary antibodies against target proteases (e.g., cleaved caspase-3, calpain, cathepsin-B, granzyme-B) or PARP-1 fragments overnight at 4°C [13].
- Detection: Use species-appropriate secondary antibodies conjugated to fluorophores (e.g., Alexa Fluor dyes) for fluorescence detection or enzyme conjugates (HRP, AP) for colorimetric detection.
- Counterstaining and Mounting: Counterstain with DAPI (for fluorescence) or cresyl violet (for colorimetry), and mount with appropriate mounting media.
- Microscopy and Analysis: Visualize using fluorescence or brightfield microscopy. For double immunofluorescence, use appropriate filter sets to detect multiple antigens simultaneously [13].
Interpretation: Co-localization of specific proteases with PARP-1 fragments in particular cell types or brain regions provides insight into the spatial organization of cell death pathways in neurodegeneration.
Co-Immunoprecipitation for Protein-Protein Interactions
Objective: To investigate molecular interactions between PARP-1 fragments and cell death effectors such as AIF or granzyme-B.
Methodology:
- Lysate Preparation: Prepare tissue lysates as for Western blotting but using milder lysis buffers without SDS to preserve protein interactions.
- Immunoprecipitation: Pre-clear lysates with protein A/G beads, then incubate with primary antibody against the target protein (e.g., AIF, granzyme-B) or control IgG overnight at 4°C with gentle rotation.
- Bead Capture: Add protein A/G beads and incubate for 2-4 hours at 4°C with rotation.
- Washing and Elution: Wash beads extensively with lysis buffer, then elute bound proteins with Laemmli sample buffer by heating at 95°C for 5 minutes.
- Analysis: Analyze eluates by Western blotting for PARP-1 or its fragments [13].
Interpretation: Successful co-immunoprecipitation of PARP-1 fragments with proteins like AIF demonstrates physical interactions that may mediate their roles in cell death pathways.
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Research Reagents for Studying Suicidal Proteases and PARP-1 Cleavage
| Reagent Category | Specific Examples | Research Application | Key Considerations |
|---|---|---|---|
| PARP-1 Antibodies | Anti-PARP-1 (full length), cleavage-specific antibodies | Detection of PARP-1 and its fragments by Western blot, IHC | Select antibodies that recognize both full-length and cleaved forms; validate for specific applications |
| Protease Antibodies | Anti-caspase-3 (cleaved), anti-calpain, anti-cathepsin-B, anti-granzyme-B | Identifying activated proteases in tissues | Cleaved caspase-3 antibodies detect active form; confirm specificity with positive controls |
| Protease Inhibitors | Z-VAD-FMK (pan-caspase), MDL-28170 (calpain), CA-074 (cathepsin-B) | Determining contribution of specific proteases to cell death | Use appropriate concentrations and pretreatment times; assess potential cross-reactivity |
| Activity Assays | Fluorogenic substrate assays for caspases, calpains | Quantifying protease activity in samples | Use fresh samples; include positive and negative controls; normalize to protein content |
| Animal Models | Transient focal cerebral ischemia (MCAO), neurodegenerative disease models | In vivo study of protease activation in neurodegeneration | Choose appropriate time points for analysis; include sham controls; validate neurological deficits |
Visualization of Protease Activation and PARP-1 Cleavage Pathways
Diagram 1: Suicidal Protease Activation and PARP-1 Cleavage in Neurodegeneration. This diagram illustrates how different neurodegenerative stimuli activate specific suicidal proteases, which cleave PARP-1 to generate signature fragments that mediate distinct cell death pathways. The 89-kDa PARP-1 fragment generated by caspases can promote both apoptosis and parthanatos, while the 50-kDa fragment generated by calpains promotes necrotic death. Granzyme B from immune cells can also cleave PARP-1 and contribute to immune-mediated cell death.
Therapeutic Implications and Future Directions
The central role of suicidal proteases and PARP-1 cleavage in neurodegenerative pathologies makes them attractive targets for therapeutic intervention. Several strategies have emerged for modulating these pathways:
Protease Inhibitors: Developing specific inhibitors for caspases, calpains, and other suicidal proteases represents a logical therapeutic approach. In cerebral ischemia, inhibition of these proteases has shown neuroprotective effects in preclinical models [13]. However, the challenge lies in achieving sufficient specificity to avoid disrupting essential physiological functions of these enzymes, as caspases and other proteases play important roles in normal cellular processes beyond cell death [14] [17].
PARP Inhibition: PARP inhibitors have demonstrated efficacy in attenuating neuronal injury in models of cerebral ischemia, trauma, and excitotoxicity [11] [12]. By preventing PARP-1 overactivation and subsequent energy depletion, these inhibitors can shift the cell death balance toward survival. However, the dual role of PARP-1 in both DNA repair and cell death initiation requires careful dosing to preserve its beneficial functions while inhibiting its detrimental effects.
Combination Therapies: Given the heterogeneity of cell death mechanisms in neurodegeneration, targeting multiple proteolytic pathways simultaneously may yield superior outcomes compared to single-agent approaches [17] [13]. The identification of specific PARP-1 cleavage fragments as biomarkers of particular protease activities enables more rational design of combination therapies tailored to the specific cell death pathways activated in different neurodegenerative conditions or even in individual patients.
The continued investigation of suicidal proteases and their effects on PARP-1 and other substrates will undoubtedly yield new insights into neurodegenerative disease mechanisms and reveal novel therapeutic opportunities for these currently untreatable conditions.
Poly(ADP-ribose) polymerase-1 (PARP-1) is a nuclear enzyme that plays a decisive role in cell fate decisions following DNA damage. Its proteolytic cleavage by executioner caspases during apoptosis generates two signature fragments: a 24-kDa DNA-binding fragment and an 89-kDa catalytic fragment. These fragments are not merely inactive degradation products but actively contribute to the irreversible commitment to cell death. This review provides a comprehensive technical analysis of the structure, function, and experimental methodologies for studying these key fragments. Within the context of neurodegenerative disease research, understanding the dynamics of PARP-1 cleavage is crucial, as it represents a critical molecular switch that can tip the balance between neuronal survival and death.
Structural and Functional Properties of PARP-1 Cleavage Fragments
PARP-1 is a 116-kDa modular protein comprising several functional domains. Caspase-3 and -7 cleave PARP-1 at a specific aspartic acid residue (within the DEVD sequence, amino acids 211-214), separating the DNA-binding function from the catalytic activity [11] [18]. This cleavage produces two major fragments with distinct properties and fates.
Table 1: Characteristics of PARP-1 Cleavage Fragments
| Feature | 24-kDa Fragment (ZnF1–2PARP1) | 89-kDa Fragment (PARP1ΔZnF1–2) |
|---|---|---|
| Domains Contained | Zinc Fingers 1 and 2 (DNA-Binding Domain) | Zinc Finger 3, BRCT, WGR, and Catalytic Domain |
| Primary Function | Binds DNA strand breaks | Retains basal catalytic activity for PAR synthesis |
| Cellular Localization Post-Cleavage | Retained in the nucleus, bound to DNA lesions [19] [15] | Translocates from the nucleus to the cytoplasm [19] [15] |
| Role in Apoptosis | Trans-dominant inhibitor of DNA repair [20] [18] | Serves as a cytoplasmic PAR carrier to induce AIF-mediated apoptosis [19] [15] |
| Regulation by PAR | Acts as an acceptor for PARylation; PAR binding reduces its DNA affinity [18] | Basal activity is inhibited by PAR binding [18] |
The following diagram illustrates the domain architecture of full-length PARP-1 and the consequences of caspase cleavage:
Experimental Protocols for Fragment Analysis
In Vitro PARP-1 Cleavage Assay
This protocol assesses caspase-mediated cleavage of PARP-1 using recombinant proteins.
- Recombinant PARP-1: Purified full-length human PARP-1 (commercially available or expressed in E. coli using pET28-a(+) or pRSFDuet-1 vectors) [18].
- Caspase Enzyme: Active recombinant caspase-3 or caspase-7, diluted in assay buffer.
- Assay Buffer: 50 mM HEPES (pH 7.4), 100 mM NaCl, 0.1% CHAPS, 10% glycerol, 10 mM DTT.
- Procedure:
- Incubate 1 µg of PARP-1 with 20-50 ng of active caspase-3 in a 20 µL reaction volume of assay buffer.
- Incubate at 37°C for 30-60 minutes.
- Terminate the reaction by adding SDS-PAGE loading buffer and boiling for 5 minutes.
- Analysis: The cleavage products (24 kDa and 89 kDa) are resolved by SDS-PAGE (10-12% gel) and visualized by Western blotting using PARP-1 N-terminal-specific (to detect the 24-kDa fragment) or C-terminal-specific antibodies (to detect the 89-kDa fragment) [11] [21].
DNA-Binding Assay for the 24-kDa Fragment
The DNA-binding capacity of the 24-kDa fragment can be analyzed using Electrophoretic Mobility Shift Assay (EMSA).
- Protein: Recombinant 24-kDa fragment (ZnF1–2PARP1, amino acids 1-214) [18].
- DNA Probe: Double-stranded DNA oligonucleotides containing a single-strand break (nick or gap). The DNA is often end-labeled with [γ-³²P] ATP for detection.
- Binding Buffer: 20 mM Tris-HCl (pH 8.0), 50 mM KCl, 1 mM DTT, 5% glycerol, 0.1 mg/mL BSA.
- Procedure:
- Incubate 10-100 nM of the 24-kDa fragment with 1 nM of radiolabeled DNA probe in a 20 µL binding reaction for 20 minutes at room temperature.
- Load the reaction onto a native polyacrylamide gel (4-6%) in 0.5x TBE buffer.
- Run the gel at 100 V for 1-2 hours, then dry and expose to a phosphorimager screen.
- Analysis: The 24-kDa fragment binds DNA breaks with high affinity, forming a stable complex visible as a shifted band on the gel. This binding is central to its role as a trans-dominant inhibitor of DNA repair [5] [20].
PARylation Activity Assay for the 89-kDa Fragment
The basal catalytic activity of the 89-kDa fragment can be measured in vitro.
- Protein: Recombinant 89-kDa catalytic fragment (PARP1ΔZnF1–2, amino acids 215-1014) [18].
- Reaction Buffer: 50 mM Tris-HCl (pH 8.0), 50 mM NaCl, 4 mM MgCl₂.
- Substrate: ²²P-NAD⁺ or biotin-NAD⁺ to allow for detection.
- Procedure:
- Set up a 25 µL reaction containing 0.5-1 µg of the 89-kDa fragment and 100-500 µM NAD⁺ (or labeled NAD⁺).
- Incubate at 25°C for 10-30 minutes.
- Stop the reaction by adding SDS-PAGE loading buffer.
- Analysis:
- SDS-PAGE/Autoradiography: If ²²P-NAD⁺ is used, resolve the proteins by SDS-PAGE, dry the gel, and visualize PAR polymers by autoradiography.
- Western Blot: If biotin-NAD⁺ is used, detect PAR polymers with an anti-PAR antibody or streptavidin-HRP.
- Note: Unlike full-length PARP-1, the 89-kDa fragment's activity is not stimulated by DNA and can be inhibited by free PAR polymers [18].
Table 2: Key Reagent Solutions for PARP-1 Fragment Research
| Reagent / Material | Function / Application | Key Details / Considerations |
|---|---|---|
| Recombinant PARP1 Fragments | In vitro cleavage, binding, and activity assays. | ZnF1–2PARP1 (aa 1-214) for DNA-binding studies; PARP1ΔZnF1–2 (aa 215-1014) for catalytic studies [18]. |
| Active Caspase-3/7 | Executioner protease for generating fragments in vitro. | Commercially available; activity should be verified with fluorogenic substrates (e.g., Ac-DEVD-AFC) [11]. |
| Anti-PARP1 Antibodies | Detection of fragments by Western Blot, Immunoprecipitation. | N-terminal-specific (detects 24-kDa fragment); C-terminal-specific (detects 89-kDa fragment); anti-cleaved PARP1 (Asp214) [19] [21]. |
| DNA Oligonucleotides with Breaks | Substrate for DNA-binding assays (EMSA). | Design double-stranded DNA with single-strand breaks (nicks or 1-nt gaps); can be radioactively or fluorescently labeled [5]. |
| NAD⁺ and Analogues | Substrate for PARylation activity assays. | Use ²²P-NAD⁺ for high sensitivity or biotin-NAD⁺ for compatibility with streptavidin detection [18]. |
| PARP Inhibitors (e.g., PJ34) | Pharmacological control to validate PARP1-dependent effects in cellular models. | Used in cell culture studies to inhibit PARP1 catalytic activity [19]. |
Signaling Pathways in Neurodegeneration
In the context of neurodegenerative diseases, the cleavage of PARP-1 and the subsequent actions of its fragments represent a critical node connecting DNA damage to neuronal death. The following diagram integrates these fragments into the pathological signaling cascades relevant to conditions like Parkinson's and Alzheimer's disease.
Discussion and Research Implications
The 24-kDa and 89-kDa PARP-1 cleavage fragments are more than mere biomarkers of apoptosis; they are active executors of the cell death program. The 24-kDa fragment acts as a trans-dominant inhibitor that actively disrupts DNA repair by sequestering DNA breaks, thereby preventing the recruitment of functional repair complexes and conserving cellular ATP [20] [18]. Meanwhile, the 89-kDa fragment, through its function as a PAR carrier, provides a critical link between nuclear DNA damage and mitochondrial cell death execution via AIF [19] [15]. This pathway, known as parthanatos, is distinct from classical apoptosis and is heavily implicated in neurodegenerative pathologies.
Recent research reveals sophisticated regulatory interplay between the fragments. The 89-kDa fragment's basal activity is inhibited by PAR, but it can be partially complemented for DNA-dependent activation by the 24-kDa fragment, suggesting a potential for functional reassembly post-cleavage under specific conditions [18]. Furthermore, the 24-kDa fragment can exert trans-dominant inhibition over the closely related PARP2, indicating a broader role in shutting down the PARP family's DNA damage response during apoptosis [18].
In neurodegenerative disease research, the study of these fragments provides a mechanistic foundation for the development of novel therapeutic strategies. Inhibiting the initial PARP-1 overactivation or the downstream actions of its cytotoxic fragments could protect vulnerable neurons in conditions such as Parkinson's disease, cerebral ischemia, and Alzheimer's disease, where PARP-1 activation is a recognized contributor to pathology.
Poly(ADP-ribose) polymerase-1 (PARP-1) is a multifunctional nuclear enzyme that plays a pivotal role in determining cellular fate, functioning as a critical regulator of both genomic stability and cell death. As a key DNA damage sensor, PARP-1 exhibits a dual nature in cellular stress responses [22] [23]. Under physiological conditions, it facilitates DNA repair and maintains genomic integrity, while under conditions of severe genotoxic stress, its hyperactivation triggers a programmed necrotic cell death pathway known as parthanatos [24] [25]. This paradoxical nature positions PARP-1 at the crossroads of cell survival and death decisions, with significant implications for cancer therapy, neurodegenerative diseases, and other pathological conditions [23] [3]. The precise mechanistic balance between these opposing functions depends on the extent of DNA damage, the cellular energy status, and the subsequent enzymatic activity of PARP-1, making it a protein of considerable interest for targeted therapeutic interventions.
PARP-1 Structure and Activation Mechanisms
PARP-1 is a 113 kDa nuclear protein composed of several functional domains that coordinate its DNA damage sensing and signaling capabilities [22] [25]. The modular structure includes three DNA-binding zinc finger domains (ZnF1, ZnF2, and ZnF3) at the N-terminus that recognize DNA strand breaks with high specificity and affinity. Following these domains lies a BRCT (BRCA1 C-terminal) domain, which facilitates protein-protein interactions, and a WGR (tryptophan, glycine, arginine) domain that further contributes to DNA binding. The C-terminal region contains the catalytic domain, which is bipartite, consisting of an auto-inhibitory helical subdomain (HD) and the ADP-ribosyl transferase (ART) subdomain that catalyzes poly(ADP-ribose) (PAR) synthesis [25].
The activation mechanism of PARP-1 involves a sophisticated series of conformational changes. Under basal conditions, PARP-1 maintains low intrinsic enzymatic activity [22]. However, upon binding to DNA single-strand or double-strand breaks through its zinc finger domains, PARP-1 undergoes significant conformational changes that relieve autoinhibition and activate the catalytic domain [22] [25]. This activation initiates the synthesis of poly(ADP-ribose) (PAR) chains using NAD+ as a substrate, leading to the modification of PARP-1 itself (automodification) and various nuclear target proteins [22]. The automodification reaction serves as a signal for the recruitment of DNA repair proteins while also facilitating PARP-1's dissociation from DNA, thereby preventing excessive activation that could lead to NAD+ depletion and cell death [22].
PARP-1 in Base Excision Repair (BER)
Mechanistic Role in BER Pathway
PARP-1 serves as a critical initiator and regulator of the Base Excision Repair pathway, which is the primary cellular mechanism for repairing single-strand breaks, oxidized bases, and other forms of base damage [22] [26]. Upon detection of DNA damage through its high affinity for single-strand breaks and apurinic/apyrimidinic (AP) sites, PARP-1 becomes rapidly activated and catalyzes the synthesis of PAR chains on itself and surrounding nuclear proteins [26]. This PARylation acts as a signal for the recruitment of key BER scaffold proteins, particularly XRCC1, which serves as a central platform for assembling the repair machinery [22] [26].
The role of PARP-1 in BER extends beyond simple damage sensing. Research demonstrates that PARP1 can influence the activities of core BER enzymes in the nucleosomal context. In most cases, the presence of PARP1 suppresses the activities of APE1 and DNA polymerase β, while PARylation attenuates this effect to varying degrees depending on the specific enzyme [26]. PARP2, a closely related family member, predominantly affects the final stage of BER—DNA sealing—by DNA ligase IIIα [26]. The interplay between PARP1 and PARP2 in regulating different BER stages highlights the sophisticated coordination of this essential repair pathway.
Chromatin Remodeling and Access to Damage Sites
An essential aspect of PARP-1's function in DNA repair involves chromatin remodeling to facilitate access to DNA damage sites. The compact structure of chromatin presents a significant barrier to DNA repair enzymes, requiring controlled relaxation for efficient repair [3] [26]. PARP-1 contributes to this process through several mechanisms, including the PARylation of all core histone subunits (H2A, H2B, H3, H4) and the linker histone H1 [3]. This extensive modification introduces negative charges through the PAR chains, leading to electrostatic repulsion and chromatin decondensation [22].
Additionally, PARP-1 recruits other chromatin remodeling factors, such as histone PARylation factor 1, which further promotes histone PARylation and chromatin relaxation [3]. The accessibility of DNA damage within nucleosomes is strongly influenced by the spatial orientation of the lesion, with outward-oriented damages being more accessible to repair enzymes than inward-oriented ones [26]. PARP-1 and PARP-2 exhibit differential affinities for damaged nucleosomes depending on this orientation, with PARP2 showing particularly high affinity for gap-containing DNA in the nucleosomal context [26].
Table 1: Quantitative Effects of PARP1 and PARP2 on BER Enzyme Activities in Nucleosomal Context
| BER Enzyme | Effect of PARP1 | Effect of PARP1 PARylation | Effect of PARP2 | Effect of PARP2 PARylation |
|---|---|---|---|---|
| APE1 | Suppression of activity | Attenuation of suppression | Minimal effect | Not determined |
| DNA Polymerase β | Suppression of activity | Partial attenuation | Minor effect | Inhibition |
| DNA Ligase IIIα | Moderate suppression | Variable effects | Influence on DNA sealing | Significant stimulation |
PARP-1 Hyperactivation and Parthanatos
Molecular Mechanisms of Parthanatos
Parthanatos is a programmed necrotic cell death pathway distinguished by its strict dependence on PARP-1 hyperactivation [24] [25]. Unlike apoptosis, parthanatos is caspase-independent and exhibits unique morphological and biochemical features, including dissipation of the inner mitochondrial transmembrane potential, nuclear and chromatin condensation, and DNA fragmentation producing large fragments (15 kb to 50 kb) [24]. The process is initiated when severe DNA damage (caused by alkylating agents, reactive oxygen species, excitotoxicity, or ischemia-reperfusion injury) leads to excessive activation of PARP-1, resulting in catastrophic consumption of cellular NAD+ and ATP pools [24] [25].
The mechanistic pathway of parthanatos involves several critical steps. Following PARP-1 hyperactivation, extensive PAR polymer accumulation occurs, which serves as a death signal [24] [25]. These PAR polymers translocate to the cytoplasm and mitochondria, where they trigger the release of Apoptosis-Inducing Factor (AIF) from mitochondrial membranes [24] [23]. AIF then translocates to the nucleus, where it interacts with Macrophage Migration Inhibitory Factor (MIF), which possesses nuclease activity and acts as the final executioner by cleaving genomic DNA into large fragments [23] [25]. This nuclear action culminates in chromatin condensation and irreversible cell death.
Metabolic Collapse in Parthanatos
A defining feature of parthanatos is the rapid depletion of cellular energy stores, which distinguishes it from other forms of programmed cell death. PARP-1 hyperactivation consumes NAD+ at an accelerated rate, as each ADP-ribose unit added to growing PAR chains requires one NAD+ molecule [24] [25]. The depletion of NAD+ has profound consequences for cellular metabolism, as NAD+ serves as an essential cofactor for critical metabolic pathways including glycolysis, the tricarboxylic acid (TCA) cycle, and oxidative phosphorylation [25].
The energy crisis is further exacerbated by several interconnected mechanisms. NAD+ depletion impairs glycolysis, reducing ATP generation from this primary energy-producing pathway [25]. Additionally, research indicates that PAR polymers released during PAR chain degradation can bind to and inhibit hexokinase-1, the enzyme catalyzing the first committed step of glycolysis [25]. This direct inhibition creates a synergistic effect on energy failure. The resynthesis of NAD+ itself consumes 2-4 ATP molecules per NAD+ molecule, creating a futile cycle that further depletes ATP reserves [24]. The resulting bioenergetic collapse ultimately leads to loss of ion homeostasis, plasma membrane integrity, and cell death.
Table 2: Key Biochemical Events in Parthanatos
| Event | Molecular Components | Consequences | Experimental Evidence |
|---|---|---|---|
| PARP-1 Hyperactivation | DNA strand breaks, PARP1 zinc fingers | Initiation of parthanatic cascade | PARP1 knockout mice are protected [24] |
| PAR Accumulation | PAR polymers, PARG | Mitochondrial depolarization | Detection by anti-PAR antibodies [24] [27] |
| AIF Translocation | Mitochondrial AIF, PAR polymers | DNA fragmentation | AIF knockout studies [24] [23] |
| NAD+ Depletion | NAD+ pools, PARP1 catalytic domain | Bioenergetic failure | NAD+ measurements show >70% depletion [25] |
| Glycolytic Inhibition | Hexokinase-1, PAR polymers | ATP depletion | ATP assays show >80% reduction [25] |
PARP-1 Cleavage Fragments as Cell Death Signatures
Proteolytic Cleavage by Suicide Proteases
PARP-1 serves as a substrate for multiple proteases, often referred to as "suicide proteases," with the resulting cleavage fragments serving as specific biomarkers for different forms of cell death [11]. The cleavage patterns are highly specific to the protease involved and the cell death pathway activated. During apoptosis, PARP-1 is cleaved by caspase-3 and caspase-7 between the second and third zinc-binding domains, producing characteristic fragments of 89 kDa (containing the automodification and catalytic domains) and 24 kDa (containing the DNA-binding domain) [11]. This cleavage inactivates PARP-1, conserving cellular ATP and NAD+ pools during the apoptotic process and preventing unnecessary DNA repair in doomed cells [11].
In contrast to apoptotic cleavage, parthanatos involves minimal PARP-1 proteolysis initially, as the pathway relies on PARP-1's hyperactivation rather than its inactivation [11] [25]. However, other suicide proteases including calpains, cathepsins, granzymes, and matrix metalloproteinases (MMPs) can cleave PARP-1 at distinct sites, generating different signature fragments that indicate alternative cell death programs [11]. For instance, calpain cleavage produces a 55 kDa fragment, while granzyme A generates a 50 kDa fragment, each associated with specific pathological conditions [11].
Functional Consequences of Cleavage Fragments
The proteolytic fragments of PARP-1 are not merely inactive degradation products but can possess distinct biological functions that influence cell death pathways. The 24 kDa DNA-binding fragment generated by caspase cleavage remains tightly bound to DNA strand breaks, where it acts as a trans-dominant inhibitor of intact PARP-1 and other DNA repair enzymes [11]. This irreversible binding prevents DNA repair and conserves cellular energy during apoptosis, facilitating the orderly dismantling of the cell.
Different cleavage fragments exhibit varied cellular localization and protein interactions. The 89 kDa catalytic fragment generated by caspase cleavage is liberated from the nucleus into the cytosol due to its reduced DNA binding capacity [11]. Other fragments, such as those generated by granzyme A during immune cell-mediated killing, display unique subcellular distributions and binding partners [11]. The specific PARP-1 fragments present in a cell can therefore serve as molecular signatures that identify not only the cell death pathway activated but also specific protease activities and potentially the original insult that triggered the death program.
Research Methods and Experimental Approaches
Key Experimental Protocols
Studying PARP-1's dual functions requires specialized methodological approaches that can differentiate between its roles in DNA repair and cell death. Key experimental protocols include:
DNA Repair Assays: To investigate PARP-1's role in BER, researchers employ in vitro repair assays using nucleosome core particles (NCPs) with specifically oriented DNA damage [26]. These assays typically involve reconstructing BER with purified components (APE1, Polβ, XRCC1, LigIIIα) and assessing how PARP1, PARP2, and PARylation influence each enzymatic step. Fluorescence measurements and electrophoretic mobility shift assays (EMSA) are used to determine dissociation constants (Kd) for PARP interactions with damaged DNA and NCPs [26].
Parthanatos Induction and Assessment: Experimental models for inducing parthanatos include treatment with DNA-alkylating agents such as MNNG (N-methyl-N′-nitro-N-nitrosoguanidine) or MMS (methyl methanesulfonate), oxidants like hydrogen peroxide, or excitotoxins including NMDA and glutamate in neuronal models [24] [25]. Key assessment parameters include measuring PAR accumulation using immunodetection, quantifying NAD+ and ATP depletion via commercial assay kits, monitoring AIF translocation through subcellular fractionation and immunofluorescence, and assessing DNA fragmentation using pulse-field gel electrophoresis [24] [25] [27].
Genetic and Pharmacological Manipulation: PARP-1 knockout cells and animals provide essential tools for establishing PARP-1 dependency in observed phenomena [24] [23]. Pharmacological inhibition using PARP inhibitors such as DPQ or clinical compounds (olaparib, niraparib, rucaparib) helps distinguish PARP-1-dependent processes [23] [27]. For in vivo studies, disease models including cerebral ischemia-reperfusion, MPTP-induced Parkinsonism, and streptozotocin-induced diabetes help elucidate PARP-1's pathophysiological roles [23] [25].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Studying PARP-1 in DNA Repair and Parthanatos
| Reagent/Category | Specific Examples | Function/Application | Experimental Notes |
|---|---|---|---|
| PARP Inhibitors | DPQ, Olaparib, Niraparib, Rucaparib, Talazoparib | Inhibit PARP catalytic activity; used to establish PARP-1 dependency | Differential trapping capacities influence outcomes [23] [28] |
| Parthanatos Inducers | MNNG, MMS, H₂O₂, NMDA, Glutamate | Trigger PARP-1 hyperactivation and parthanatos | Concentration-dependent effects; MNNG commonly used [24] [25] |
| NAD+/ATP Assay Kits | Commercial NAD+/NADH and ATP assay kits | Quantify energy depletion in parthanatos | Key for establishing metabolic collapse [25] [27] |
| Antibodies for Detection | Anti-PAR, anti-PARP-1 (full length and fragments), anti-AIF | Detect PAR accumulation, PARP-1 cleavage, AIF translocation | Anti-PAR antibody essential for parthanatos confirmation [24] [27] |
| Genetic Models | PARP1 KO mice, AIF KO models, PARG dysfunction models | Establish genetic evidence for pathway components | PARP1 KO mice are viable but sensitive to DNA damage [22] [24] |
| BER Components | Recombinant APE1, Polβ, XRCC1, LigIIIα | Reconstitute BER in vitro | Used in nucleosomal BER assays [26] |
PARP-1 in Neurodegenerative Diseases: Therapeutic Implications
The dual functions of PARP-1 in DNA repair and cell death have significant implications for neurodegenerative diseases, where the balance between these opposing roles appears disrupted [23] [3]. In conditions such as Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis, increased PARP-1 activity and PAR levels have been commonly observed, suggesting a potential contribution of parthanatos to neuronal loss [3]. For instance, in Alzheimer's disease, brain tissues show increased PARP-1 and PAR, and fibroblasts from patients exhibit elevated PAR levels [3]. Similarly, in Parkinson's disease, cerebrospinal fluid shows increased PAR, indicating potential utility as a biomarker [3].
Interestingly, Huntington's disease presents a unique pattern, with reduced PAR levels and impaired PARP-1 activity even in the prodromal phase [3]. This contrasting finding suggests that dysregulation of PAR signaling in either direction—overactivation or suppression—can lead to neuronal dysfunction. The diminished PARP-1 activity in Huntington's disease may compromise DNA repair capacity, contributing to the accumulation of DNA damage and neuronal vulnerability [3].
Therapeutic strategies targeting PARP-1 must carefully consider this contextual duality. In neurodegenerative diseases where parthanatos contributes to pathology, PARP inhibition may offer neuroprotection [23] [3]. Conversely, in conditions with impaired PARP-1 function, enhancing specific aspects of PARP-1 activity might be beneficial. The development of selective PARP modulators that can inhibit the hyperactivation associated with cell death while preserving DNA repair functions represents an attractive but challenging therapeutic approach.
Visualizing PARP-1 Pathways and Experimental Approaches
PARP-1 Fate Decision Pathway
Experimental Workflow for PARP-1 Studies
PARP-1 embodies a remarkable paradox in cellular stress response, functioning as both guardian of genomic integrity and executioner through parthanatos. The balance between these opposing roles depends on the extent of DNA damage, cellular energy status, and the intricate regulation of PARP-1's enzymatic activity and proteolytic processing. Understanding the molecular switches that determine this fate decision has profound implications for therapeutic interventions in cancer, neurodegenerative diseases, and other conditions characterized by genomic instability or aberrant cell death. The continued investigation of PARP-1 cleavage fragments as biomarkers and functional mediators in cell death pathways promises to yield valuable insights for diagnostic and therapeutic applications, particularly in the context of neurodegenerative diseases where PARP-1 dysregulation appears to be a common but nuanced feature.
PARP-1 as a Cofactor for NF-κB and Regulator of Neuroinflammation
Poly(ADP-ribose) polymerase-1 (PARP-1), also known as ARTD1, is an abundant nuclear protein that functions as a critical regulator of genomic stability, DNA repair, and transcriptional regulation. Beyond its canonical role in DNA damage detection and repair, PARP-1 has emerged as a significant modulator of inflammatory processes in neurological systems, primarily through its functional interactions with the transcription factor NF-κB. In the context of neurodegenerative diseases, understanding the molecular mechanisms governing PARP-1's dual roles in DNA repair and inflammation has become increasingly important. This technical guide examines PARP-1's function as an NF-κB coactivator and its regulation of neuroinflammatory pathways, with particular emphasis on how PARP-1 cleavage fragments generated by proteolytic enzymes may influence inflammatory responses and cellular fate decisions in neurological pathologies.
Molecular Mechanisms of PARP-1 in NF-κB Activation
PARP-1 Domains and Functional Regions
PARP-1 is a modular protein comprising several structurally and functionally distinct domains:
- DNA-binding domain (DBD): Contains two zinc finger motifs that facilitate recognition of and binding to DNA strand breaks
- Automodification domain (AMD): Serves as the primary acceptor site for PARylation, regulating PARP-1's interaction with DNA and other proteins
- Catalytic domain (CD): Mediates the polymerization of ADP-ribose units from NAD+ onto target proteins
The caspase cleavage site (DEVD214) is situated within the DBD, specifically within the nuclear localization signal, and cleavage at this site by executioner caspases generates 24-kDa (DBD fragment) and 89-kDa (AMD+CD fragment) polypeptides [29] [11].
PARP-1 as an NF-κB Coactivator
PARP-1 facilitates NF-κB-mediated transcription through multiple mechanisms that extend beyond its enzymatic activity:
Protein-protein interactions: PARP-1 interacts directly with both p50 and p65 subunits of NF-κB, forming complexes that enhance transcriptional activation [30]. Notably, neither the enzymatic nor DNA-binding activities of PARP-1 are required for this coactivator function [30].
Regulation of nuclear retention: PARylation of p65 by PARP-1 reduces its interaction with nuclear exporting proteins, thereby increasing NF-κB's nuclear retention and transcriptional activity [31].
Chromatin remodeling and facilitator functions: PARP-1 supports the assembly of transcription complexes on NF-κB-responsive promoters, potentially through interactions with histone acetyltransferases like p300 [31] [32].
Table 1: Mechanisms of PARP-1 in NF-κB Activation
| Mechanism | Functional Consequence | Dependence on PARP-1 Enzymatic Activity |
|---|---|---|
| Direct protein-protein interaction with p50/p65 | Enhanced transcription complex assembly | Independent [30] |
| PARylation of NF-κB subunits | Increased nuclear retention | Dependent [31] |
| Interaction with histone acetyltransferases | Chromatin remodeling at target genes | Partially dependent [31] |
| Regulation of NEMO ubiquitination | Enhanced IKK activity and IκB degradation | Dependent [31] |
PARP-1 Cleavage Fragments and Their Functional Consequences
Proteolytic Cleavage of PARP-1
PARP-1 serves as a substrate for multiple proteases, often termed "suicidal proteases," which generate characteristic cleavage fragments that serve as biomarkers for specific cell death pathways [11]:
- Caspase-3 and -7: Cleave PARP-1 at DEVD214↓G to generate 24-kDa (p24) and 89-kDa (p89) fragments, considered a hallmark of apoptosis [11]
- Calpains, cathepsins, granzymes, and MMPs: Generate distinct PARP-1 cleavage fragments associated with alternative cell death programs [11]
Biological Activities of PARP-1 Cleavage Fragments
The cleavage of PARP-1 produces fragments with distinct and often opposing biological functions:
24-kDa fragment (DNA-binding domain): Retains the ability to bind tightly to DNA strand breaks but lacks catalytic activity. This fragment acts as a trans-dominant inhibitor of DNA repair by blocking access of intact PARP-1 and other DNA repair enzymes to damage sites [11].
89-kDa fragment (Automodification + Catalytic domains): Shows reduced DNA binding capacity and can be liberated from the nucleus to the cytosol. This fragment exhibits cytotoxic properties and enhances pro-inflammatory responses [29].
Table 2: PARP-1 Cleavage Fragments and Their Functional Properties
| Fragment | Molecular Weight | Domains Contained | Cellular Localization | Reported Functions |
|---|---|---|---|---|
| p24 | 24 kDa | DNA-binding domain (zinc fingers) | Nuclear | Trans-dominant inhibitor of DNA repair; cytoprotective in ischemia models [29] [11] |
| p89 | 89 kDa | Automodification + Catalytic domains | Nuclear and cytoplasmic | Cytotoxic; enhances NF-κB activity and pro-inflammatory gene expression [29] |
| Uncleavable PARP-1 | 113 kDa | Full-length with mutated caspase site | Nuclear | Cytoprotective; reduces inflammatory gene expression [29] |
PARP-1 in Neuroinflammation and Neurodegenerative Processes
PARP-1 and Neuroinflammatory Signaling
PARP-1 activation contributes significantly to neuroinflammation through multiple pathways:
Regulation of inflammatory gene expression: PARP-1 facilitates the expression of key inflammatory mediators including iNOS, COX-2, cytokines (TNF-α, IL-1β), and chemokines in neural cells [29] [32].
Microglial activation: PARP-1 inhibition suppresses microglial activation and ameliorates post-stroke inflammation in experimental models [33].
Oxidative stress amplification: PARP-1 activation and oxidative stress form a positive feedback loop that sustains inflammatory responses in neurological contexts [31] [34].
PARP-1 Cleavage in Neurological Disease Models
Research using in vitro models of cerebral ischemia has revealed striking differences in how PARP-1 cleavage fragments influence neuronal viability and inflammatory responses:
Cytoprotective fragments: Expression of PARP-1UNCL (uncleavable mutant) or PARP-124 (24-kDa fragment) conferred protection from oxygen/glucose deprivation (OGD) in SH-SY5Y neuroblastoma cells and rat primary cortical neurons [29].
Cytotoxic fragments: Expression of PARP-189 (89-kDa fragment) was cytotoxic and enhanced pro-inflammatory signaling [29].
Differential regulation of NF-κB targets: PARP-1UNCL and PARP-124 decreased iNOS and COX-2 expression while increasing anti-apoptotic Bcl-xL. In contrast, PARP-189 increased iNOS and COX-2 while decreasing Bcl-xL [29].
Table 3: PARP-1 Forms and Their Effects in Ischemia Models
| PARP-1 Form | Effect on Cell Viability | Effect on NF-κB Activity | Effect on Inflammatory Mediators |
|---|---|---|---|
| PARP-1WT (Wild-type) | Baseline toxicity | Baseline activation | Baseline expression |
| PARP-1UNCL (Uncleavable) | Cytoprotective | Similar to PARP-1WT | Decreased iNOS, COX-2; Increased Bcl-xL [29] |
| PARP-124 (24-kDa fragment) | Cytoprotective | Similar to PARP-1WT | Decreased iNOS, COX-2; Increased Bcl-xL [29] |
| PARP-189 (89-kDa fragment) | Cytotoxic | Significantly enhanced | Increased iNOS, COX-2; Decreased Bcl-xL [29] |
Experimental Approaches for PARP-1 Research
In Vitro Models of Neuroinflammation
Oxygen/Glucose Deprivation (OGD) and Restoration Protocol [29]:
- Cell models: SH-SY5Y human neuroblastoma cells and primary rat cortical neurons
- OGD induction: Culture cells in deoxygenated, glucose-free medium in an anaerobic chamber (0.1% O₂) for 2-6 hours
- Restoration phase: Return cells to normal oxygenated medium with glucose (OGD/ROG)
- PARP-1 expression: Generate tetracycline-inducible stable transfectants expressing PARP-1WT, PARP-1UNCL, PARP-124, or PARP-189
- Assessment endpoints: Cell viability assays, NAD+ levels, PAR formation, NF-κB translocation, and expression of inflammatory mediators
PARP-1 Modulation Strategies
Genetic manipulation approaches:
- siRNA-mediated knockdown: Use PARP-1-targeting siRNA (e.g., target sequence: 5'-ACGGTGATCGGTAGCAACAAA-3') at 25 nM concentration [29]
- Expression of cleavage-resistant PARP-1 (PARP-1UNCL) through site-directed mutagenesis of the caspase cleavage site [29]
- Viral vector-mediated expression: Adeno-associated viruses (AAV) for gene delivery in primary neurons [29]
Pharmacological inhibition:
- PARP inhibitors: PJ34 (30 mg/kg in vivo), olaparib, and other clinical-stage inhibitors
- Timing considerations: Delayed PARP-1 inhibition (e.g., 48 hours post-stroke) effectively alleviates neuroinflammation [33]
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Research Reagents for PARP-1/NF-κB Neuroinflammation Studies
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| PARP-1 Expression Constructs | PARP-1WT, PARP-1UNCL (uncleavable), PARP-124 (24-kDa), PARP-189 (89-kDa) [29] | Investigate functional consequences of PARP-1 cleavage |
| Cell Lines | SH-SY5Y human neuroblastoma, Primary rat cortical neurons [29] | In vitro models of neuronal function and stress |
| PARP-1 Modulators | siRNA-PARP-1 (Target: 5'-ACGGTGATCGGTAGCAACAAA-3'), PJ34, Olaparib [29] [33] | Genetic and pharmacological inhibition of PARP-1 |
| Disease Modeling | Oxygen/Glucose Deprivation (OGD) system [29] | Mimic ischemic conditions in neural cells |
| Activity Assays | PAR formation assays, NAD+ quantification [29] | Measure PARP-1 enzymatic activity and cellular energy status |
| NF-κB Assessment | NF-κB translocation assays, iNOS promoter binding activity [29] | Evaluate NF-κB activation and transcriptional function |
| Protease Inhibitors | Caspase inhibitors (Z-VAD-FMK), Calpain inhibitors [11] | Define specific protease pathways in PARP-1 cleavage |
PARP-1 serves as a critical molecular nexus connecting DNA damage response to neuroinflammatory signaling through its multifaceted interactions with NF-κB. The proteolytic cleavage of PARP-1 during cell death programs generates fragments with distinct and often opposing biological activities - the 24-kDa fragment appears to confer cytoprotective effects while the 89-kDa fragment promotes cytotoxicity and enhanced inflammation. Understanding the balance between these cleavage fragments and their differential effects on neuroinflammatory pathways provides valuable insights for developing targeted therapeutic strategies for neurodegenerative diseases, stroke, and other neurological conditions characterized by aberrant neuroinflammation. The experimental approaches outlined in this technical guide provide a framework for investigating these complex molecular relationships in neurological disease models.
Detecting and Deciphering Function: Methods for Fragment Analysis and Experimental Models
The detection of specific poly (ADP-ribose) polymerase-1 (PARP-1) cleavage fragments serves as a critical biomarker for identifying distinct cell death pathways in neurodegenerative diseases. PARP-1, a nuclear enzyme involved in DNA repair, undergoes proteolytic cleavage by various proteases during different forms of cell death, generating signature fragments that can be utilized as diagnostic and therapeutic indicators. This technical guide comprehensively outlines the molecular tools, experimental protocols, and detection methodologies for identifying PARP-1 cleavage products, with particular emphasis on their implications in neurodegenerative pathology. We provide detailed workflows for western blotting, activity assays, and immunohistochemical techniques, along with analytical frameworks for interpreting fragment patterns in the context of caspase-dependent apoptosis, parthanatos, and necrotic cell death pathways relevant to Alzheimer's disease, Parkinson's disease, and other neurological disorders.
PARP-1 is a 113-116 kDa nuclear enzyme that functions as a primary DNA damage sensor and facilitates repair through poly(ADP-ribosyl)ation (PARylation) of target proteins [35] [36]. In neurodegenerative diseases, progressive DNA damage drives PARP-1 activation, but excessive activation leads to distinct proteolytic cleavage events that serve as molecular signatures for specific cell death pathways [35] [36] [34]. The cleavage of PARP-1 by various "suicidal" proteases generates specific fragments with different molecular weights and biological activities, making them valuable biomarkers for discriminating between apoptosis, necrosis, and parthanatos in neurological contexts [35] [21].
The central role of PARP-1 cleavage in neurodegeneration stems from the vulnerability of neuronal cells to oxidative stress and DNA damage accumulation. In conditions like Alzheimer's disease (AD), Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS), and Huntington's disease (HD), the persistence of DNA damage triggers hyperactivation of PARP-1, leading to energy depletion through NAD+ and ATP consumption [36] [34]. This energy crisis activates various proteases that cleave PARP-1 into signature fragments, which subsequently amplify cell death signaling cascades. The detection and characterization of these fragments therefore provides critical insights into the dominant mechanisms of neuronal loss operating in specific neurodegenerative conditions.
PARP-1 Cleavage Fragments: Signatures and Significance
Major PARP-1 Cleavage Fragments
PARP-1 undergoes proteolytic processing by different classes of proteases, generating distinctive fragments that serve as biomarkers for specific cell death pathways. The table below summarizes the key PARP-1 fragments, their origins, and detection significance.
Table 1: Characteristic PARP-1 Cleavage Fragments and Their Significance
| Fragment Size | Generating Proteases | Domain Composition | Cellular Localization | Biological Significance | Associated Cell Death Pathway |
|---|---|---|---|---|---|
| 89 kDa + 24 kDa | Caspases-3 and -7 [35] [29] | 89 kDa: AMD + CD; 24 kDa: DBD [35] [19] | 24 kDa: nuclear; 89 kDa: cytosol [19] | Hallmark of apoptosis; inhibits DNA repair [35] | Caspase-dependent apoptosis [35] |
| 50 kDa | Lysosomal proteases (cathepsins B, D, G) [21] | Not fully characterized | Nuclear | Marker of necrotic cell death [21] | Necrosis [21] |
| 89 kDa (PARylated) | Caspases-3/7 with prior PARP-1 auto-modification [19] | AMD + CD with PAR polymers | Cytosolic translocation [19] | Serves as PAR carrier; induces AIF release [19] | Parthanatos [19] |
Structural Domains and Cleavage Sites
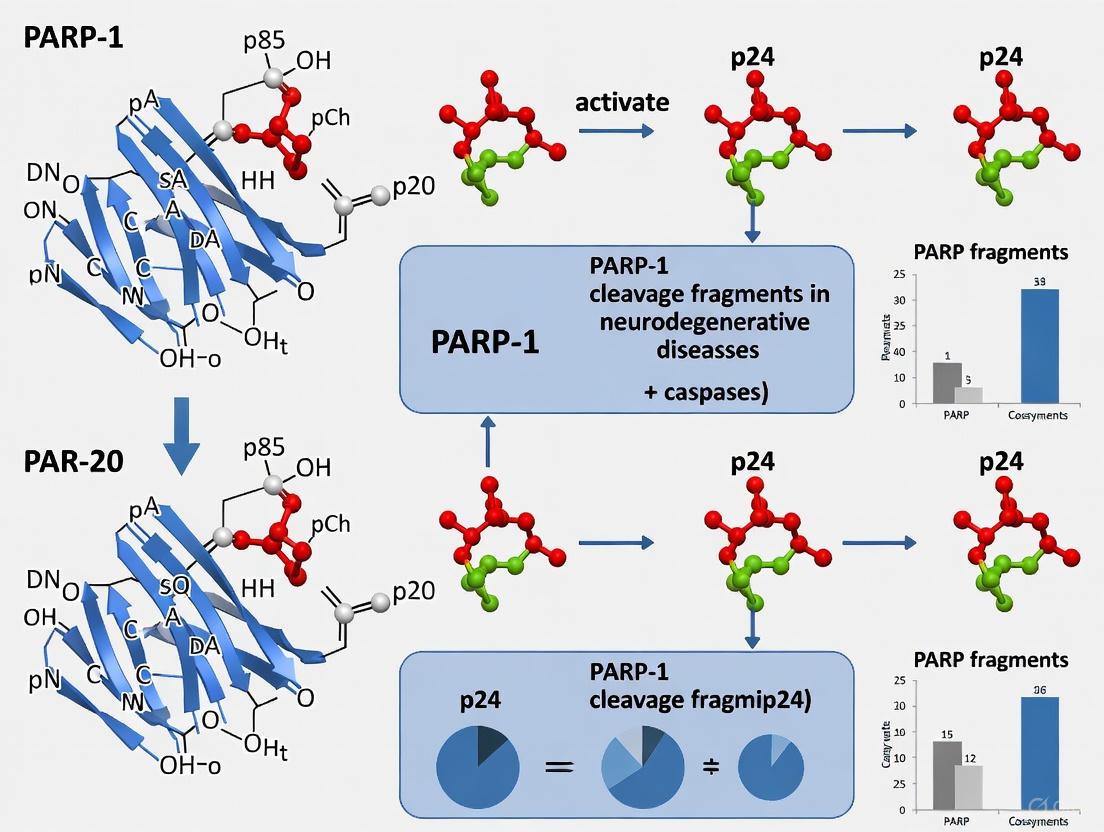
PARP-1 contains three functionally significant domains: a DNA-binding domain (DBD) at the N-terminus containing two zinc finger motifs, an auto-modification domain (AMD) in the central region, and a catalytic domain (CD) at the C-terminus responsible for PAR polymer formation [35]. The caspase cleavage site (DEVD214) is situated within the DBD, specifically between the second zinc finger motif and the nuclear localization signal [29]. This strategic location explains the differential localization of the resulting fragments after cleavage – the 24 kDa fragment containing the DBD remains nuclear, while the 89 kDa fragment containing the AMD and CD translocates to the cytoplasm [19].
The functional consequences of PARP-1 cleavage depend on which domains are liberated. The 24 kDa fragment acts as a trans-dominant inhibitor of intact PARP-1 by irreversibly binding to DNA strand breaks, thereby blocking the DNA repair function of full-length PARP-1 and conserving cellular ATP pools during apoptosis [35]. In contrast, the 89 kDa fragment, particularly when PARylated, can function as a carrier that transports PAR polymers to the cytoplasm where they trigger mitochondrial release of apoptosis-inducing factor (AIF), initiating a cascade of large-scale DNA fragmentation characteristic of parthanatos [19].
PARP-1 Cleavage in Cell Death Pathways Relevant to Neurodegeneration
The following diagram illustrates how different proteolytic events on PARP-1 generate specific fragments that participate in distinct cell death pathways relevant to neurodegenerative diseases:
Experimental Protocols for PARP-1 Fragment Detection
Western Blot Analysis for PARP-1 Fragments
Principle: Western blotting remains the gold standard for detecting and distinguishing PARP-1 cleavage fragments based on molecular weight. This method allows simultaneous detection of full-length PARP-1 (113-116 kDa) and its major cleavage products (89 kDa, 50 kDa, and 24 kDa).
Detailed Protocol:
- Cell Lysis and Protein Extraction: Harvest neuronal cells or tissue samples and lyse in RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) supplemented with protease inhibitors (1 mM PMSF, 10 μg/mL aprotinin, 10 μg/mL leupeptin) and PARP inhibitors (50 μM PJ34 or 10 μM ABT-888) to prevent artifactual cleavage during processing [21] [19].
- Protein Quantification: Determine protein concentration using BCA or Bradford assay. Load 20-50 μg protein per lane on 8-12% SDS-polyacrylamide gels.
- Electrophoresis and Transfer: Separate proteins by SDS-PAGE at 100-120V for 1-2 hours. Transfer to PVDF membranes at 100V for 1 hour at 4°C.
- Blocking and Antibody Incubation: Block membranes with 5% non-fat milk in TBST for 1 hour. Incubate with primary antibodies against PARP-1 (specific for different epitopes) overnight at 4°C.
- Detection: Incubate with HRP-conjugated secondary antibodies for 1 hour at room temperature. Develop using enhanced chemiluminescence substrate and visualize with chemiluminescence imaging systems.
Key Considerations: For optimal fragment resolution, use gradient gels (8-16%) to better separate the 89 kDa and 50 kDa fragments. Include positive controls (e.g., staurosporine-treated cells for apoptotic cleavage, H₂O₂-treated cells for necrotic cleavage) to validate antibody specificity and fragment identification [21] [19].
Activity-Western Blot Technique
Principle: This non-isotopic technique detects PARP-1 fragments based on their enzymatic activity, providing functional information beyond mere immunoreactivity [21].
Protocol Modifications from Standard Western:
- Post-Transfer Membrane Processing: After transfer, incubate PVDF membrane in PARP-1 activity buffer (100 mM Tris-HCl pH 8.0, 10 mM MgCl₂, 1 mM DTT) containing 10 μM NAD⁺ and 0.2 μCi/mL biotin-NAD⁺ for 30 minutes at room temperature.
- PAR Polymer Detection: Block membrane and incubate with streptavidin-HRP conjugate (1:5000) for 1 hour.
- Chemiluminescent Detection: Develop with ECL substrate to visualize PARP-1 fragments that retain catalytic activity [21].
Applications: This technique is particularly valuable for identifying the 89 kDa fragment that retains catalytic activity and becomes auto-poly(ADP-ribosyl)ated, a key feature in parthanatos [19].
Immunohistochemical Detection in Tissue Sections
Principle: Immunohistochemistry enables spatial localization of PARP-1 fragments within specific brain regions and cell types, providing critical pathological context in neurodegenerative samples.
Detailed Protocol:
- Tissue Preparation: Fix brain tissues in 4% paraformaldehyde for 24-48 hours, followed by paraffin embedding and sectioning (5-8 μm thickness).
- Antigen Retrieval: Deparaffinize sections and perform antigen retrieval using citrate buffer (pH 6.0) at 95-100°C for 20 minutes.
- Blocking and Staining: Block endogenous peroxidase with 3% H₂O₂ and non-specific binding with 5% normal goat serum. Incubate with fragment-specific primary antibodies overnight at 4°C.
- Detection and Counterstaining: Apply biotinylated secondary antibodies followed by ABC reagent. Develop with DAB substrate and counterstain with hematoxylin.
- Microscopy and Analysis: Visualize using brightfield microscopy. The 24 kDa fragment shows exclusive nuclear localization, while the 89 kDa fragment may display both nuclear and cytoplasmic distribution depending on cellular state [19].
The Scientist's Toolkit: Key Research Reagents
Table 2: Essential Reagents for PARP-1 Fragment Research
| Reagent Category | Specific Examples | Application/Function | Key Considerations |
|---|---|---|---|
| PARP-1 Antibodies | Anti-PARP-1 (full length) [21], Cleaved PARP-1 (Asp214) [29] | Fragment detection via WB, IHC | Select antibodies targeting different epitopes to distinguish fragments |
| Caspase Inhibitors | zVAD-fmk (pan-caspase) [21] [19] | Inhibit caspase-mediated PARP-1 cleavage | Use to confirm caspase-dependent vs independent cleavage |
| PARP Inhibitors | PJ34, ABT-888 [19], 3-AB [36] | Inhibit PARP-1 catalytic activity | Prevent PAR formation and energy depletion |
| Protease Assays | Ac-DEVD-pNA (caspase substrate) [21] | Measure caspase-3/7 activity | Confirm caspase activation parallel to PARP-1 cleavage |
| Cell Death Inducers | Staurosporine (apoptosis) [19], H₂O₂ (necrosis) [21], MNNG (parthanatos) [19] | Induce specific cell death pathways | Positive controls for PARP-1 cleavage patterns |
| PAR Antibodies | Anti-PAR antibody [19] | Detect PAR polymers | Identify PARylated 89 kDa fragment in parthanatos |
| AIF Antibodies | Anti-AIF [19] | Monitor AIF translocation | Downstream marker of parthanatos activation |
Interpretation of PARP-1 Fragment Patterns in Neurodegenerative Research
Discriminating Cell Death Pathways
The specific pattern of PARP-1 fragments provides crucial information about the dominant cell death mechanism operating in neurodegenerative models or patient samples:
- Apoptotic Signature: Concurrent appearance of 89 kDa and 24 kDa fragments, typically with suppression of full-length PARP-1, indicates caspase-mediated apoptosis. This pattern is observed in models of Alzheimer's disease and Parkinson's disease with caspase activation [35].
- Necrotic Signature: Presence of a 50 kDa fragment in the absence of significant 89/24 kDa fragments suggests necrotic cell death, mediated by lysosomal proteases. This pattern associates with acute neuronal injury models [21].
- Parthanatos Signature: Detection of PARylated 89 kDa fragments, particularly in cytoplasmic fractions, along with mitochondrial AIF release, indicates PARP-1-dependent parthanatos. This pathway is prominent in stroke, Parkinson's disease, and glutamate excitotoxicity models [34] [19].
Technical Considerations and Validation
Fragment Stability: PARP-1 fragments, particularly the 24 kDa DBD fragment, can exhibit rapid turnover. Include protease and PARP inhibitors in all lysis buffers to prevent post-lysis artifacts [21].
Cross-Pathway Interactions: In many neurodegenerative contexts, multiple cell death pathways activate simultaneously. Pharmacological inhibitors (zVAD-fmk for caspases, cathepsin inhibitors for lysosomal proteases, PARP inhibitors for parthanatos) help dissect the relative contributions of each pathway [21] [19].
Subcellular Localization: Fractionation studies (nuclear, cytoplasmic, mitochondrial) provide critical additional information, as fragment localization directly relates to function. The 89 kDa fragment translocates to cytoplasm during parthanatos, while the 24 kDa fragment remains nuclear [19].
The detection and characterization of PARP-1 cleavage fragments represents a powerful biomarker approach for deciphering cell death mechanisms in neurodegenerative diseases. The immunological and molecular tools detailed in this technical guide provide a framework for discriminating between apoptosis, necrosis, and parthanatos - distinct pathways that may require different therapeutic interventions. As research advances, the increasing appreciation of PARP-1 fragment-specific functions, particularly the dual role of the 89 kDa fragment in both apoptosis and parthanatos, highlights the complexity of cell death regulation in neurological contexts. The standardized protocols and analytical approaches outlined here will support consistent application and interpretation of PARP-1 cleavage as a biomarker across neurodegenerative disease models and ultimately in human pathological specimens.
The study of protein cleavage is fundamental to understanding the molecular mechanisms underlying neurodegenerative diseases. Poly (ADP-ribose) polymerase-1 (PARP-1) cleavage, in particular, has been identified as a critical event in cell death pathways relevant to conditions such as Alzheimer's disease, Parkinson's disease, and cerebral ischemia [11] [37] [38]. In vitro, two cellular models stand as pillars for this investigation: the human neuroblastoma cell line SH-SY5Y and primary cortical neurons isolated from rodents. Each model offers unique advantages; SH-SY5Y cells provide a renewable, genetically tractable system for high-throughput screening, while primary cortical neurons offer a more physiologically relevant context, maintaining native neuronal architecture and signaling pathways [29]. This technical guide details the application of these models to study PARP-1 cleavage, providing methodologies, key findings, and essential reagents to advance research in this field.
PARP-1 Cleavage: A Key Event in Cell Death Signaling
PARP-1 is a nuclear enzyme with a well-established role in the detection and repair of DNA single-strand breaks. However, upon excessive activation by severe DNA damage, it becomes a substrate for several proteases, leading to cleavage fragments that can actively regulate cell death and inflammatory responses [11] [38]. The cleavage of PARP-1 by caspases-3 and -7 at the DEVD214 site is a well-documented hallmark of apoptosis, generating a 24-kDa DNA-binding domain (DBD) fragment and an 89-kDa catalytic domain fragment [11] [29]. Notably, these fragments are not merely inert byproducts. Research demonstrates that they differentially modulate cellular viability and the nuclear factor-kappa B (NF-κB) inflammatory signaling pathway, suggesting that the cleavage event itself is a decisive regulatory step in the cellular response to stress [29] [39].
Table 1: Key PARP-1 Fragments and Their Proposed Functions
| Fragment Name | Molecular Weight | Domains Contained | Reported Functions in Neuronal Models |
|---|---|---|---|
| PARP-1 (Full-length) | 113 kDa | DBD, AMD, CD | DNA repair, NF-κB co-activation [11] [29] |
| PARP-124 | 24 kDa | DBD (with 2 zinc fingers) | Acts as a trans-dominant inhibitor of BER; cytoprotective in OGD/ROG models; suppresses iNOS/COX-2 [11] [29] [39] |
| PARP-189 | 89 kDa | AMD, CD | Cytotoxic in OGD/ROG models; enhances NF-κB and iNOS promoter activity [29] [39] |
Abbreviations: DBD (DNA-Binding Domain), AMD (Auto-Modification Domain), CD (Catalytic Domain), BER (Base Excision Repair), OGD/ROG (Oxygen/Glucose Deprivation/Restoration of Oxygen and Glucose), iNOS (inducible Nitric Oxide Synthase), COX-2 (Cyclooxygenase-2).
Experimental Protocols for Cleavage Studies
Differentiation of SH-SY5Y Cells into Neuron-like Cells
The undifferentiated SH-SY5Y neuroblastoma cell line exhibits a proliferative, non-neuronal phenotype. To create a more mature, neuron-like model for neurological research, a well-established two-step differentiation protocol is recommended [40].
- Phase 1 Medium (Days 1-5): Culture SH-SY5Y cells in Dulbecco’s Modified Eagle’s Medium (DMEM) with high glucose (25 mM), L-glutamine (4 mM), 1% Penicillin-Streptomycin (P/S), and 10 µM all-trans Retinoic Acid (RA). This medium lacks sodium pyruvate and fetal bovine serum. Incubate the cells for five days, refreshing the medium every other day [40].
- Phase 2 Medium (Days 6-11): Replace the Phase 1 medium with Neurobasal-A medium (without phenol red), supplemented with 1% L-glutamine, 1% N-2 supplement, 1% P/S, and 50 ng/mL Brain-Derived Neurotrophic Factor (BDNF). Incubate the cells for an additional six days, with medium changes every two to three days [40].
This protocol induces morphological changes, including the extension of long, pronounced neurites, and increases the expression of neuronal markers such as synaptophysin and microtubule-associated protein, thereby yielding a more physiologically relevant model for neuronal studies [40] [41].
Isolation and Culture of Primary Cortical Neurons
For a more direct model of mature neuronal function, primary cortical neurons are isolated from postnatal rodents [29] [42].
- Dissection and Dissociation: Isolate cortices from Sprague-Dawley rats at 2 days of age (P2). Dissociate the cortical tissue enzymatically (e.g., with papain) and triturate mechanically to create a single-cell suspension.
- Plating and Maintenance: Plate the cells on poly-D-lysine or collagen-coated culture vessels at a density of 3.75 x 105 cells/mL. Maintain the neurons in Neurobasal-A medium supplemented with B27 supplement to support survival and maturation. The culture medium is typically replaced after three to four days in vitro (DIV) [29].
- Experimental Use: Neurons are typically ready for experimentation, such as transduction with viral vectors or exposure to insults, at approximately 6-7 DIV, once a mature network has been established [29].
Modeling Cellular Insult and Inducing PARP-1 Cleavage
A standard method to induce PARP-1 cleavage and mimic ischemic-like conditions in vitro is Oxygen/Glucose Deprivation (OGD), often followed by restoration of oxygen and glucose (ROG) to model reperfusion injury [29].
- OGD Protocol: For both differentiated SH-SY5Y cells and primary cortical neurons, replace the standard culture medium with a deoxygenated, glucose-free buffer (e.g., Balanced Salt Solution). Place the culture plates in a hypoxic chamber flushed with 95% N2 and 5% CO2 for a defined period (e.g., 6 hours) at 37°C [29].
- OGD/ROG Protocol: Following the OGD period, replace the glucose-free buffer with complete, oxygenated culture medium and return the cells to a normoxic incubator (5% CO2, 95% air) for a restoration phase (e.g., 15 hours) [29].
This insult triggers DNA damage, calcium dysregulation, and the activation of proteases like caspases and calpains, leading to the cleavage of PARP-1 and other substrates like huntingtin [11] [42].
Key Experimental Findings and Data Interpretation
Research utilizing these models has revealed that PARP-1 cleavage fragments exert opposing effects on neuronal survival and inflammation, as summarized in the table below.
Table 2: Functional Outcomes of PARP-1 Fragment Expression in In Vitro "Ischemia" Models
| PARP-1 Construct Expressed | Effect on Cell Viability (OGD/ROG) | Effect on NF-κB & iNOS Activity | Key Downstream Protein Changes |
|---|---|---|---|
| PARP-1UNCL (Uncleavable) | Cytoprotective [29] [39] | Similar to WT [29] | ↓ iNOS, ↓ COX-2, ↑ Bcl-xL [29] |
| PARP-124 (24 kDa DBD fragment) | Cytoprotective [29] [39] | Lower than PARP-189 [29] | ↓ iNOS, ↓ COX-2, ↑ Bcl-xL [29] |
| PARP-189 (89 kDa Catalytic fragment) | Cytotoxic [29] [39] | Significantly higher than WT [29] | ↑ iNOS, ↑ COX-2, ↓ Bcl-xL [29] |
| PARP-1WT (Wild-type) | Baseline toxicity [29] | Baseline activity [29] | Baseline expression [29] |
A critical insight from these studies is that the protective effect of the PARP-1UNCL and PARP-124 fragments was not accompanied by a reduction in total poly(ADP-ribose) formation or NAD+ depletion, which are hallmarks of PARP-1 overactivation in parthanatos [29]. This suggests that the regulation of cell viability in this context is independent of the canonical enzymatic hyperactivity but is instead mediated through the modulation of NF-κB-dependent inflammatory pathways. The 89-kDa fragment promotes a pro-inflammatory and pro-death environment, while the 24-kDa fragment and the full-length uncleavable protein appear to confer protection by tempering this response [29] [39].
The following diagram illustrates the opposing roles of PARP-1 cleavage fragments in neuronal cell death and survival pathways following an insult like OGD.
The Scientist's Toolkit: Essential Research Reagents
Successful investigation of cleavage events requires a carefully selected toolkit of reagents and methodologies. The table below catalogs essential solutions for studies involving PARP-1 cleavage in neuronal models.
Table 3: Research Reagent Solutions for PARP-1 Cleavage Studies
| Reagent / Method | Function / Target | Application Example |
|---|---|---|
| All-trans Retinoic Acid (RA) | Induces neuronal differentiation [40] | Differentiation of SH-SY5Y cells into neuron-like cells [40] [41]. |
| Brain-Derived Neurotrophic Factor (BDNF) | Enhances neuronal maturation and survival [40] | Second-phase differentiation and maintenance of SH-SY5Y cells and primary neurons [40]. |
| PJ-34 | Potent PARP-1 inhibitor [38] | Studying the effects of PARP-1 enzymatic inhibition on cell death pathways (e.g., parthanatos) [38]. |
| Z-VAD-FMK | Pan-caspase inhibitor [11] | Determining caspase-dependence of PARP-1 cleavage and cell death [11]. |
| Calpain Inhibitors (e.g., MDL-28170) | Inhibits calpain proteases [42] | Investigating calpain-mediated cleavage of substrates like huntingtin and its role in excitotoxicity [42]. |
| siRNA against PARP-1 | Knocks down endogenous PARP-1 expression [29] | Validating functions of exogenously expressed PARP-1 fragments without background interference [29]. |
| Adeno-Associated Virus (AAV) Vectors | Efficient gene delivery to neurons [29] | Transducing primary cortical neurons with PARP-1 constructs (WT, UNCL, 24, 89) [29]. |
| Antibody against 89-kDa PARP-1 fragment | Detects caspase-cleaved PARP-1 [11] [29] | Western blot confirmation of apoptosis and fragment-specific studies. |
| TMRM / JC-1 Dyes | Assesses mitochondrial membrane potential [40] | Measuring mitochondrial health and function in differentiated vs. undifferentiated cells under stress [40]. |
| Neurobasal-A / B27 Medium | Supports survival of post-mitotic neurons [29] | Long-term maintenance of primary cortical neuron cultures [29]. |
SH-SY5Y cells and primary cortical neurons are indispensable and complementary models for dissecting the mechanisms and consequences of protein cleavage, particularly PARP-1 cleavage, in neurodegenerative contexts. The experimental frameworks and reagents detailed in this guide provide a robust foundation for such investigations. The seminal finding that PARP-1 cleavage fragments are not mere biomarkers of cell death but active regulators of viability and inflammation opens a promising avenue for therapeutic intervention. Targeting the specific downstream effects of the cytotoxic 89-kDa fragment or mimicking the protective function of the 24-kDa fragment could lead to novel neuroprotective strategies for a range of neurological diseases.
Oxygen/glucose deprivation (OGD) models serve as a cornerstone ex vivo technique for simulating the ischemic stress that underpins various neurodegenerative pathologies. These models directly replicate the core metabolic crisis of stroke and other neurodegenerative conditions by subjecting brain or spinal cord tissues to controlled environments lacking oxygen and glucose. The fundamental strength of the OGD model lies in its ability to preserve the complex cellular architecture and cell-to-cell interactions of neural tissue while providing precise experimental control over the ischemic environment, a level of control that is often unattainable in in vivo models [43]. This makes OGD an indispensable tool for dissecting the rapid molecular and cellular events that follow ischemic injury.
Critically, the severity of ischemic insult is not a binary state. Reflecting the clinical reality of varying stroke intensities, modern OGD paradigms have evolved beyond simple complete deprivation. Researchers can now induce and meticulously characterize a spectrum of ischemic severities, ranging from mild (partial OGD) to severe (complete OGD) [43]. This gradation is essential, as the cellular response, the type of ensuing cell death, and the activation of specific proteases are highly dependent on the intensity of the initial insult. Framed within the context of PARP-1 biology, OGD models provide a controlled system to investigate how differing levels of metabolic stress trigger the cleavage of PARP-1 by various "suicidal" proteases, producing signature fragments that serve as biomarkers for specific cell-death pathways active in neurodegeneration [11] [44].
Establishing Defined OGD Severity Models
A significant advancement in the field is the move toward standardizing and quantifying ischemic severity. A defined methodology involves the use of an oxygen sensor to establish three distinct conditions, moving beyond the traditional single model of complete deprivation [43].
Table 1: Standardized Conditions for Modeling Ischemic Severity Ex Vivo
| Condition Name | Oxygen Level | Glucose Level | Experimental Purpose |
|---|---|---|---|
| Normoxia (Control) | Normal (~21%) | Normal (e.g., 11 mM) | Healthy baseline control |
| Mild Ischemia | Partial deprivation | Partial deprivation | Models transient ischemic attacks (TIAs) or partial occlusion |
| Severe Ischemia | Complete deprivation | Complete deprivation | Models major stroke with complete arterial occlusion |
The biological response to OGD is remarkably rapid. Validation studies demonstrate that changes in pro-inflammatory cytokine levels, HIF-1α expression, and microglial recruitment to injured neurons can be detected within 30 minutes of inducing OGD, with the magnitude of these changes being directly dependent on the severity of the challenge [43]. The model's pathological relevance is further confirmed by traditional metrics such as TTC staining, which shows a clear increase in infarct volume with increasing ischemic severity [43].
OGD-Induced PARP-1 Activation and Cleavage Fragments
A central pathway activated by OGD is the DNA damage response, orchestrated by the enzyme Poly(ADP-ribose) polymerase-1 (PARP-1). During cerebral ischemia/reperfusion, PARP-1 is hyperactivated in response to oxidative stress and DNA damage [45] [46]. This hyperactivation is a double-edged sword. While PARP-1 is involved in routine DNA repair, its overactivation drives a unique form of programmed cell death known as PARthanatos, which is distinct from apoptosis, necrosis, and other cell death pathways [45].
A key event in PARP-1-related pathology is its cleavage by specific proteases. The pattern of cleavage produces signature fragments that act as biochemical fingerprints, identifying the specific cell-death program activated [11] [44].
Table 2: PARP-1 Cleavage Fragments as Signatures of Protease Activity
| Protease | PARP-1 Fragment Sizes | Associated Cell Death Pathway | Pathological Context |
|---|---|---|---|
| Caspase-3/7 | 89 kDa (Catalytic), 24 kDa (DNA-binding) | Apoptosis | Widespread in various neurological diseases [11] |
| Calpains | Variable (e.g., 55 kDa, 40 kDa) | Excitotoxicity, Necrosis | Linked to calcium-mediated neurotoxicity [11] |
| Cathepsins | Specific fragments not detailed | Lysosomal-mediated death | Implicated in inflammation-associated damage [11] |
| Granzymes | Specific fragments not detailed | Immune-mediated cytotoxicity | Relevant in neuroinflammatory contexts [11] |
| Matrix Metalloproteinases (MMPs) | Specific fragments not detailed | Blood-Brain Barrier disruption | Associated with vascular damage in stroke [11] |
The 89 kDa fragment generated by caspases contains the auto-modification and catalytic domains but has a greatly reduced DNA-binding capacity, while the 24 kDa fragment retains the zinc fingers and binds irreversibly to damaged DNA, acting as a trans-dominant inhibitor of BER and conserving cellular ATP pools [11]. This cleavage is a hallmark of apoptosis. In contrast, PARthanatos is characterized by PARP-1 hyperactivation, massive PAR polymer synthesis, and nuclear translocation of Apoptosis-Inducing Factor (AIF), and it is not blocked by caspase inhibitors [45].
Metabolic Consequences of PARP-1 Hyperactivation
The hyperactivation of PARP-1 during OGD has catastrophic consequences for cellular energy metabolism. PARP-1 uses nicotinamide adenine dinucleotide (NAD+) as a substrate to synthesize poly(ADP-ribose) (PAR) chains. Consequently, excessive PARP-1 activity leads to a rapid and severe depletion of intracellular NAD+ pools. Since NAD+ is an essential cofactor for glycolysis and the mitochondrial tricarboxylic acid (TCA) cycle, its depletion results in a profound bioenergetic collapse and a drop in ATP levels [45] [46].
Recent research has uncovered another critical mechanism: PARP-1 hyperactivation catalyzes the PARylation of key metabolic enzymes. Specifically, hexokinase-1 (HK-1) and the B-subunit of lactate dehydrogenase (LDH-B) have been identified as targets [46]. The PARylation of these enzymes directly inhibits their activity, disrupting glycolysis and the lactate shuttle between astrocytes and neurons. This direct inhibition of metabolic flux contributes to the energy failure and neuronal death observed in OGD and ischemia/reperfusion injury, independent of and concurrent with NAD+ consumption [46].
Detailed Experimental Protocol for Ex Vivo OGD
This protocol outlines the procedure for inducing standardized OGD in organotypic brain or spinal cord slice cultures, based on established methodologies [43] [47].
Reagent and Buffer Preparation
The foundation of a reproducible OGD experiment is the precise preparation of artificial cerebrospinal fluid (aCSF) solutions. Below are the compositions for the three primary conditions.
Table 3: Research Reagent Solutions for OGD Modeling
| Research Reagent | Function in the Model | Specific Example / Concentration |
|---|---|---|
| Normoxic aCSF | Maintains healthy tissue as a control | 11 mM D-glucose, saturated with 95% O₂/5% CO₂ [43] |
| Ischemic aCSF | Creates OGD environment; severity is controlled | Mild: Partial O₂/Glucose; Severe: 0 mM Glucose, saturated with 95% N₂/5% CO₂ [43] |
| Oxygen Sensor | Quantifies and validates ischemic severity | Real-time measurement of dissolved oxygen in the chamber [43] |
| PARP Inhibitors | Tool to probe mechanism of PARthanatos | PJ34, Olaparib, 3-AB (used at various pre-/post-treatment timings) [45] |
| TTC (2,3,5-Triphenyltetrazolium Chloride) | Histological assessment of infarct volume | Standard staining to visualize tissue viability post-OGD [43] |
| Antibodies for PAR / PARP-1 fragments | Detect PARP activation & cleavage | Western Blot/ICC for 89 kDa fragment (apoptosis), PAR polymer (parthanatos) [11] [46] |
- Normoxia (Control) aCSF: 2.5 mM KCl, 1.2 mM NaH₂PO₄, 2.4 mM CaCl₂, 1.2 mM MgCl₂, 126 mM NaCl, 11 mM D-glucose, 4 mM sucrose, 25 mM sodium bicarbonate, 15 mM Tris. The solution must be continuously bubbled with 95% O₂ / 5% CO₂ to maintain oxygenation and physiological pH [43].
- Severe Ischemia aCSF: The composition is identical to the normoxia aCSF but omits D-glucose (replaced with sucrose or an inert osmolyte) and is saturated by bubbling with 95% N₂ / 5% CO₂ to achieve an anoxic environment [43].
- Mild Ischemia aCSF: This is achieved by creating a specific mixture of the normoxic and severe ischemic aCSF solutions, or by controlled bubbling with N₂/CO₂ to achieve a predefined, intermediate level of oxygen tension, verified by an oxygen sensor [43].
OGD Procedure and Tissue Analysis
- Tissue Preparation: Organotypic hippocampal or spinal cord slice cultures (typically 300-400 µm thick) are prepared from postnatal rodents and allowed to stabilize in normoxic aCSF [43] [47].
- OGD Induction: The stabilization aCSF is replaced with pre-equilibrated severe or mild ischemic aCSF. The tissue is incubated in this medium in an airtight chamber maintained at 37°C for a defined period (e.g., 30-60 minutes), with the atmosphere controlled by continuous bubbling of the gas mixture [43].
- Reperfusion/Oxygenation (Optional): To model ischemia-reperfusion injury, the ischemic aCSF is replaced with normoxic aCSF, and the slices are returned to a normal oxygen atmosphere for a variable recovery period.
- Tissue Harvest and Analysis: Following OGD (± reperfusion), slices are harvested for analysis. Key endpoints include:
- Cell Death Assessment: TTC staining for infarct volume, LDH release assay for cytotoxicity, and TUNEL staining for DNA fragmentation [43].
- Molecular Analysis: Protein extraction for Western blotting to detect PARP-1 cleavage fragments (e.g., the 89 kDa caspase-derived fragment) and PAR polymer accumulation [11] [46]. Immunohistochemistry can be used to localize these markers to specific cell types.
- Metabolic Analysis: Measurement of NAD+/NADH levels, ATP assays, and activity assays for PARylated enzymes like HK and LDH [46].
The OGD model remains a highly versatile and physiologically relevant platform for deconstructing the mechanisms of ischemic neuronal stress and death. Its power is greatly enhanced by the ability to precisely control ischemic severity, allowing researchers to model a spectrum of clinical conditions from TIA to major stroke. When integrated with the study of PARP-1 biology, OGD provides critical insights into how metabolic stress is transduced into specific proteolytic signatures and cell fate decisions. The detection of PARP-1 cleavage fragments helps delineate the contribution of apoptosis versus PARthanatos, while the understanding of PAR-mediated metabolic inhibition reveals new therapeutic nodes. Continued refinement of this model, coupled with the investigation of PARP-1's dual roles in neuroprotection and cell death, will undoubtedly accelerate the development of targeted neuroprotective strategies for neurodegenerative diseases.
This technical guide details the rationale and methodologies for the genetic manipulation of poly (ADP-ribose) polymerase-1 (PARP-1), focusing on the generation and application of uncleavable mutants and the expression of specific cleavage fragments. PARP-1 proteolysis by caspases, calpains, and other proteases generates signature fragments that are recognized biomarkers in cell death pathways, with significant implications for neurodegenerative disease pathogenesis [12] [11]. The cleavage of PARP-1, a hallmark of apoptosis, occurs primarily at the DEVD214 site, producing 24-kDa and 89-kDa fragments [11] [29] [48]. Employing uncleavable PARP-1 (PARP-1UNCL) and fragment-specific expression (PARP-124 and PARP-189) provides a powerful experimental paradigm to dissect the non-canonical functions of PARP-1 and its fragments in regulating cell viability, inflammatory responses, and transcriptional programs in neurological disease models [29]. This whitepaper provides an in-depth guide to these genetic tools, framing them within the context of discovering novel therapeutic strategies for disorders such as cerebral ischemia, Alzheimer's disease, and Parkinson's disease.
PARP-1 is a multifunctional nuclear enzyme with well-established roles in DNA repair and the maintenance of genomic stability. In the context of neurodegenerative diseases, PARP-1 overactivation in response to excessive genotoxic or oxidative stress contributes directly to neuronal death [12] [11]. A critical event in this process is the proteolytic cleavage of PARP-1 by a specific subset of "suicidal" proteases. This cleavage is not merely a passive marker of cell death but is increasingly understood to generate fragments with active and opposing biological functions.
The following diagram illustrates the central proteolytic event and the key genetic manipulations covered in this guide.
Diagram 1: PARP-1 Cleavage in Neurodegeneration and Genetic Manipulation Strategies. Proteolytic cleavage of full-length PARP-1 by different proteases generates distinct fragments with specific biological activities. Genetic tools (red boxes) allow researchers to mimic or block these events to determine their functional consequences.
The canonical apoptotic cleavage by caspases-3 and -7 yields a 24-kDa DNA-binding fragment (PARP-124) and an 89-kDa catalytic fragment (PARP-189) [11] [48]. The 24-kDa fragment acts as a dominant-negative inhibitor of DNA repair by occupying DNA strand breaks, thereby conserving cellular energy but potentially promoting genomic instability [11]. The 89-kDa fragment, once considered inert, has been shown to translocate to the cytoplasm, function as a poly(ADP-ribose) (PAR) carrier, and promote apoptosis-inducing factor (AIF)-mediated cell death (parthanatos), a pathway implicated in neurodegeneration [15]. Furthermore, PARP-189 can enhance NF-κB-mediated pro-inflammatory gene expression, creating a toxic feedback loop [29]. In contrast, necrosis induced by agents like H2O2 or HgCl2 results in a distinct ~50-kDa PARP-1 fragment generated by lysosomal proteases such as cathepsins B and G [21].
The strategic expression of an uncleavable PARP-1 mutant (PARP-1UNCL) and the individual fragments (PARP-124 and PARP-189) allows researchers to disentangle this complex web of functions and identify specific pathogenic mechanisms operative in different neurodegenerative contexts.
Quantitative Profiling of PARP-1 Cleavage Fragments
The specific protease and cell death context determines which PARP-1 fragment is generated. These fragments serve as biochemical signatures for diagnosing the mode of cellular demise, a critical factor in understanding neuropathology. The table below summarizes the key characteristics of the major PARP-1 cleavage fragments.
Table 1: Signature PARP-1 Cleavage Fragments in Cell Death Pathways
| Fragment Size | Protease Responsible | Cell Death Context | Primary Localization & Proposed Function | Implication in Neurodegeneration |
|---|---|---|---|---|
| 89 kDa & 24 kDa [11] [48] | Caspase-3 & Caspase-7 [11] [48] | Apoptosis [11] [48] | 89 kDa: Cytoplasmic; PAR carrier, promotes AIF-mediated death (parthanatos) [15]. 24 kDa: Nuclear; dominant-negative inhibitor of DNA repair [11]. | Implicated in cerebral ischemia, Alzheimer's, Parkinson's, and excitotoxicity [12] [11]. |
| ~50 kDa [21] | Cathepsins B & G (Lysosomal Proteases) [21] | Necrosis [21] | Necrotic cleavage; function not fully elucidated, but a marker for lysosomal protease involvement [21]. | Associated with necrotic neuronal death in acute brain injury and possibly chronic degeneration. |
Genetic Engineering of Uncleavable PARP-1 (PARP-1UNCL)
Rationale and Molecular Design
The primary objective of creating an uncleavable PARP-1 mutant is to prevent its proteolytic inactivation and fragment generation during cell death, thereby allowing the investigation of the functional consequences of intact PARP-1 signaling under stress conditions. The design centers on the caspase cleavage site.
- Wild-type Cleavage Site: The canonical caspase-3/7 cleavage site in human PARP-1 is located within the nuclear localization signal (NLS) of the DNA-binding domain, at the amino acid sequence DEVD214↓G [29] [48].
- Mutagenesis Strategy: Site-directed mutagenesis is used to alter the critical aspartic acid (D) residue at position 214. The most common and effective mutation is D214G (asparagine to glycine) or D214N (asparagine to asparagine) [29]. This single amino acid substitution renders the protein resistant to cleavage by caspases without significantly impairing its DNA-binding or catalytic activities under basal conditions.
Experimental Protocol: Generation and Validation of PARP-1UNCL
Step 1: Cloning and Mutagenesis
- Source Vector: A wild-type human PARP-1 (PARP-1WT) cDNA clone (e.g., from the Harvard Institute of Proteomics, ID HsCD00040600) is used as the starting template [29].
- Mutagenesis: Perform site-directed mutagenesis using primers designed to introduce the D214G/N mutation. The resulting construct, PARP-1UNCL, should be cloned into an appropriate expression vector (e.g., pcDNA for transient expression, lentiviral or AAV vectors for stable transduction/neuronal infection) featuring selectable markers (e.g., antibiotic resistance) and/or inducible promoters (e.g., Tetracycline-responsive element) for controlled expression [29].
Step 2: Cell Transfection/Transduction and Model Systems
- In Vitro Models:
- Cell Lines: SH-SY5Y human neuroblastoma cells are a standard model for neuronal studies [29]. Generate stable, inducible tetracycline-regulated cell lines to control PARP-1UNCL expression.
- Primary Neurons: Primary rat cortical neurons provide a more physiologically relevant system. Transduce neurons at days in vitro (DIV) 3-4 using Adeno-Associated Virus (AAV) vectors for high-efficiency gene delivery [29].
- In Vivo Models: AAV vectors expressing PARP-1UNCL can be delivered intracranially into rodent models of neurodegeneration (e.g., middle cerebral artery occlusion for stroke) to assess neuroprotective effects.
Step 3: Functional Validation
- Cleavage Assay: Subject transfected/transduced cells to apoptotic stimuli (e.g., staurosporine 1 μM, 4-6 hours; or oxygen/glucose deprivation (OGD)). Analyze cell lysates by Western blotting.
- Antibodies: Use antibodies recognizing the N-terminus (to detect the 24-kDa fragment) or the C-terminus (to detect full-length PARP-1 and the 89-kDa fragment) [29].
- Expected Result: In PARP-1WT-expressing cells, immunoblotting will show a decrease in full-length PARP-1 and the appearance of the 89-kDa and 24-kDa fragments. In PARP-1UNCL-expressing cells, the full-length protein will persist, and the cleavage fragments will be absent [29].
- Viability Assay: Test the functional outcome of expressing PARP-1UNCL using models of "ischemia" like OGD or OGD followed by restoration of oxygen/glucose (ROG). Assess cell viability 15-24 hours post-OGD using assays like MTT, LDH release, or Annexin V/PI staining [29].
- Expected Result: Expression of PARP-1UNCL confers significant protection against OGD/ROG-induced cell death compared to PARP-1WT or empty vector controls [29].
Fragment-Specific Expression: PARP-124 and PARP-189
Rationale and Molecular Design
Expressing the individual cleavage fragments allows researchers to isolate and study the unique functions of each fragment, independent of the full-length protein and the cleavage event itself.
- PARP-124 (24 kDa Fragment): This fragment comprises the first two zinc finger motifs of the DNA-binding domain. When expressed alone, it is designed to act as a constitutive dominant-negative inhibitor that binds to DNA breaks and blocks the recruitment of repair machinery [11] [29].
- PARP-189 (89 kDa Fragment): This fragment contains the third zinc finger, the BRCT domain, the WGR domain, and the catalytic domain. Its expression mimics the caspase-cleaved product that translocates to the cytoplasm and can engage in non-nuclear signaling, such as enhancing NF-κB activity or acting as a PAR carrier [29] [15].
Experimental Protocol: Cloning and Application
Step 1: Construct Design and Cloning
- PARP-124: Amplify the cDNA sequence corresponding to amino acids 1-214 (or as defined by the caspase cleavage site) of PARP-1. Clone this fragment into an expression vector. Ensure the construct includes a nuclear localization signal (NLS), as this fragment is inherently nuclear [29].
- PARP-189: Amplify the cDNA sequence corresponding to amino acids 215-1014 (or the C-terminus) of PARP-1. Clone this fragment into an expression vector. This fragment may localize to both the nucleus and cytoplasm, but its cytotoxic effects are associated with cytoplasmic functions [29] [15].
Step 2: Functional Characterization in Neuronal Models
- Viability and Cytotoxicity: Express PARP-124 and PARP-189 in SH-SY5Y cells or primary cortical neurons and subject them to OGD/ROG.
- Expected Result: PARP-124 expression is cytoprotective, similar to PARP-1UNCL. In contrast, PARP-189 expression is cytotoxic, even in the absence of stress, and exacerbates cell death following injury [29].
- Mechanistic Studies:
- NF-κB Pathway Analysis: Since PARP-1 is a cofactor for NF-κB, investigate the role of the fragments in inflammation. Use luciferase reporter assays for NF-κB activity and measure the mRNA and protein levels of downstream pro-inflammatory targets like iNOS and COX-2.
- Expected Result: PARP-189 expression significantly increases NF-κB activity and the expression of iNOS and COX-2, while PARP-124 and PARP-1UNCL suppress these pathways [29].
- Cytoplasmic Functions of PARP-189: Investigate the role of PARP-189 in parthanatos. Use immunofluorescence and cellular fractionation to monitor the translocation of AIF from mitochondria to the nucleus in cells expressing PARP-189 [15].
- NF-κB Pathway Analysis: Since PARP-1 is a cofactor for NF-κB, investigate the role of the fragments in inflammation. Use luciferase reporter assays for NF-κB activity and measure the mRNA and protein levels of downstream pro-inflammatory targets like iNOS and COX-2.
The following diagram outlines a core experimental workflow for validating these genetic tools in a neurodegenerative stress model.
Diagram 2: Core Experimental Workflow for Validating PARP-1 Genetic Tools. A generalized pipeline for introducing genetic constructs into model systems, applying disease-relevant stressors, and conducting key downstream analyses.
The Scientist's Toolkit: Essential Research Reagents
The table below catalogues critical reagents and their applications for studies involving PARP-1 cleavage and genetic manipulation.
Table 2: Key Research Reagent Solutions for PARP-1 Studies
| Reagent / Tool | Function / Specificity | Example Application |
|---|---|---|
| Anti-PARP-1 Antibodies | Detect full-length and cleavage fragments. C-terminal antibodies detect 89 kDa; N-terminal antibodies detect 24 kDa. | Western blot validation of cleavage in cellular and tissue lysates [29]. |
| Caspase-3/7 Inhibitor (e.g., z-DEVD-fmk) | Potent, cell-permeable inhibitor of executioner caspases. | Confirming caspase-dependent PARP-1 cleavage in apoptotic models [21]. |
| PARP Inhibitors (e.g., Olaparib) | Small molecules that compete with NAD+ in the catalytic site, inhibiting PARylation. | Studying the impact of catalytic inhibition vs. cleavage; cancer therapy [10] [49]. |
| AAV Vectors for Neuronal Transduction | High-efficiency gene delivery tool for primary neurons and in vivo CNS models. | Stable expression of PARP-1UNCL, PARP-124, or PARP-189 in neurons [29]. |
| Tetracycline-Inducible Expression System | Allows precise temporal control of transgene expression. | Controlling expression of PARP-1 constructs to avoid developmental effects or toxicity [29]. |
| siRNA against Endogenous PARP-1 | Knocks down native PARP-1 expression. | Used in conjunction with mutant expression to study function against a low-background PARP-1 environment [29]. |
The genetic manipulation of PARP-1 through uncleavable mutants and fragment-specific expression has moved the field beyond viewing PARP-1 cleavage merely as a cell death biomarker. These tools have revealed that the 24-kDa and 89-kDa fragments are biologically active players with distinct and often opposing roles in determining cellular fate in the context of neurodegeneration. The consistent findings that PARP-1UNCL and PARP-124 are cytoprotective, while PARP-189 is cytotoxic and pro-inflammatory, highlight the potential of inhibiting PARP-1 cleavage or targeting the downstream pathways of the 89-kDa fragment as novel neuroprotective strategies.
Future research should leverage these genetic tools in more complex, humanized neuronal models and chronic neurodegenerative disease models to fully elucidate the therapeutic potential. Furthermore, exploring the interplay between PARP-1 fragments and other pathogenic processes, such as mitochondrial dysfunction and protein aggregation, will provide a more integrated understanding of disease mechanisms. The methodologies and reagents detailed in this whitepaper provide a robust foundation for such investigations, paving the way for the development of innovative treatments for currently intractable neurodegenerative diseases.
In neurodegenerative disease research, the detection and interpretation of PARP-1 cleavage fragments serve as critical biomarkers for identifying specific cell death pathways and disease mechanisms. These fragments represent more than mere proteolytic artifacts; they signify the activation of specific suicidal proteases and provide a window into the cellular stress response within neurological systems [11]. This technical guide details the subsequent experimental framework for assaying the downstream consequences of PARP-1 activation and cleavage, focusing on the NF-κB signaling pathway, the induction of pro-inflammatory enzymes iNOS and COX-2, and the resultant mitochondrial dysfunction that drives neuronal damage.
The hyperactivation of PARP-1 consumes NAD+ and ATP, leading to energy collapse and direct initiation of parthanatos, a regulated cell death pathway prominent in neurological conditions [34]. Furthermore, PARP-1 functions as a co-activator of NF-κB, creating a vicious cycle of inflammation and metabolic compromise [50]. Therefore, mapping these downstream events is essential for understanding disease pathogenesis and evaluating therapeutic interventions targeting the PARP-1/NF-κB axis.
Core Signaling Pathways and Their Interconnections
The PARP-1/NF-κB/iNOS-COX-2 Axis
The central pathway connecting PARP-1 activation to cellular damage involves the transcription factor NF-κB. Upon activation by cellular stress, PARP-1 facilitates diverse inflammatory responses by promoting the expression of pro-inflammatory genes, including inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) [50]. The following diagram illustrates this core signaling axis and its functional consequences.
This signaling cascade creates a self-amplifying loop in neurodegenerative conditions. For instance, in Alzheimer's disease, increased PARP-1 activity and PAR levels have been observed in brain tissues, correlating with neuroinflammation and disease progression [3]. Similarly, studies on methotrexate-induced intestinal injury confirm the critical role of the "NF-κB-iNOS-COX-2-TNFα inflammatory signaling pathway" in driving tissue damage [51].
Mitochondrial Dysfunction as a Convergence Point
Mitochondria represent a critical convergence point for PARP-1-mediated injury. PARP-1 hyperactivation induces severe NAD+ depletion, which directly impairs mitochondrial respiration and ATP synthesis [52]. Furthermore, NO produced by iNOS can inhibit the mitochondrial electron transport chain, increasing reactive oxygen species (ROS) generation and promoting mitochondrial permeability transition (mPT), a precursor to apoptotic and necrotic cell death [53] [52]. The diagram below outlines the key components of mitochondrial dysfunction relevant to assay design.
The critical role of mitochondrial dysfunction is exemplified in research involving mitochondrial DNA-depleted (ρ0) cells, which demonstrated altered bystander mutagenic responses and decreased NF-κB activity, iNOS, and COX-2 expression, highlighting the essential role of functional mitochondria in sustaining these inflammatory pathways [54].
Experimental Workflow for Assessing Downstream Effects
A comprehensive assessment of these interconnected pathways requires a multi-modal experimental approach. The workflow below integrates key techniques to capture the sequence of events from PARP-1 cleavage to functional mitochondrial decline.
Detailed Methodologies and Protocols
Detecting PARP-1 Cleavage Fragments
Objective: To identify and quantify specific PARP-1 cleavage fragments as indicators of protease activation (e.g., caspases, calpains) in cellular or tissue models of neurodegeneration.
Protocol:
- Sample Preparation: Lyse cells or homogenize tissue samples in RIPA buffer supplemented with protease inhibitors. For subcellular fractionation, use a Nuclear Extraction Kit to separate nuclear and cytoplasmic fractions [55].
- Protein Quantification: Determine protein concentration using a BCA or Bradford assay.
- Western Blotting:
- Load 20-50 μg of protein per lane on a 4-12% Bis-Tris polyacrylamide gel.
- Transfer proteins to a PVDF membrane.
- Block membrane with 5% non-fat milk in TBST for 1 hour.
- Incubate with primary antibodies overnight at 4°C:
- Anti-PARP-1 antibody: To detect full-length (116 kDa) and the 89 kDa caspase-3/7 cleavage fragment [11].
- Caspase-3 (cleaved) antibody: To confirm apoptotic induction.
- Wash and incubate with HRP-conjugated secondary antibody for 1 hour.
- Develop using enhanced chemiluminescence (ECL) substrate.
- Interpretation: Apoptosis is indicated by the appearance of the 89 kDa fragment and a corresponding decrease in full-length PARP-1. The 24 kDa DNA-binding domain fragment is diagnostic but may not be detected by all commercial antibodies [11].
Assessing NF-κB Activation and Nuclear Translocation
Objective: To measure the activation and functional output of the NF-κB pathway.
Protocol (Multiple Approaches):
- Phospho-IκBα and Total IκBα Western Blot:
- Analyze whole cell lysates by Western blot using antibodies against phospho-IκBα (Ser32) and total IκBα. Degradation of IκBα indicates pathway activation [56].
- Nuclear Translocation Assay:
- Perform nuclear and cytoplasmic fractionation [55].
- Probe fractions with an antibody against the p65 subunit of NF-κB. Nuclear accumulation of p65 confirms translocation.
- Alternatively, use immunofluorescence staining in cultured cells with p65 and a nuclear marker (DAPI) to visualize translocation microscopically [51].
- NF-κB Transcriptional Activity Assay:
- Transfert cells with an NF-κB luciferase reporter plasmid.
- After treatments, measure luciferase activity, which correlates directly with NF-κB-driven transcription.
Measuring iNOS and COX-2 Expression and Activity
Objective: To quantify the expression and functional activity of the key inflammatory enzymes iNOS and COX-2.
Protocol:
- Gene Expression:
- Extract total RNA and synthesize cDNA.
- Perform quantitative RT-PCR (qRT-PCR) using primers specific for NOS2 (iNOS) and PTGS2 (COX-2). Normalize to a housekeeping gene (e.g., GAPDH, β-actin) [51].
- Protein Expression:
- Functional Activity:
- iNOS Activity (Nitrite Assay): Measure nitrite (a stable oxidation product of NO) in cell culture supernatants using a commercially available fluorometric or colorimetric assay kit (e.g., OxiSelect In Vitro Nitric Oxide Assay Kit) [55] [53].
- COX-2 Activity (PGE2 ELISA): Quantify Prostaglandin E2 (PGE2) levels in cell culture supernatants using a competitive or sandwich ELISA kit according to the manufacturer's instructions [55].
Evaluating Mitochondrial Dysfunction
Objective: To assess key parameters of mitochondrial health, including membrane potential, ROS production, and content.
Protocol:
- Mitochondrial Membrane Potential (ΔΨm):
- Stain cells with tetramethylrhodamine ethyl ester (TMRE) or JC-1 dye.
- Analyze by flow cytometry or fluorescence microscopy. A decrease in fluorescence intensity (TMRE) or a shift from red to green fluorescence (JC-1) indicates loss of ΔΨm [52].
- Mitochondrial ROS:
- Load cells with MitoSOX Red, a fluorogenic dye specifically targeted to mitochondria that is oxidized by superoxide.
- Quantify fluorescence by flow cytometry or plate reader [53].
- Mitochondrial DNA Content:
- Extract total DNA.
- Perform qPCR to amplify a mitochondrial DNA (mtDNA) gene (e.g., ND1) and a nuclear DNA (nDNA) gene (e.g., 18S rRNA). Calculate the mtDNA/nDNA ratio to estimate mitochondrial content [53].
- Cellular Energetics:
- Measure ATP levels using a commercially available bioluminescence assay kit.
- For a more comprehensive profile, use a Seahorse Analyzer to measure the Oxygen Consumption Rate (OCR) and Extracellular Acidification Rate (ECAR) in live cells.
Quantitative Data from Key Studies
The following tables consolidate quantitative findings from relevant studies, providing reference points for expected experimental outcomes.
Table 1: PARP-1 Dysregulation in Neurodegenerative Diseases (Evidence from Human Samples)
| Disease | Sample Type | Observed Change in PAR Signaling | Reference |
|---|---|---|---|
| Alzheimer Disease (AD) | Brain, Fibroblasts, Lymphoblasts | Increased PARP1 and PAR levels | [3] |
| Parkinson Disease (PD) | Cerebrospinal Fluid (CSF) | Increased PAR levels | [3] |
| Huntington Disease (HD) | CSF, Fibroblasts, iPSC-derived Neurons | Decreased PAR levels and impaired PARP1 activity | [3] |
| Amyotrophic Lateral Sclerosis (ALS) | Spinal Cord (Astroglia) | Increased PARP1 | [3] |
| Cerebellar Ataxias | Fibroblasts, Lymphoblastoids | Increased PAR levels and prolonged PAR response | [3] |
Table 2: Effects of Pharmacological Inhibition on Key Pathways
| Experimental Model | Treatment / Condition | Key Measured Outcome | Effect of Intervention | Reference |
|---|---|---|---|---|
| Human Skin Fibroblasts (ρ+) | Alpha-particle irradiation + Bay 11-7082 (NF-κB inhibitor) | Mutation Frequency (bystander effect) | Significant decrease | [54] |
| Human Skin Fibroblasts (ρ+) | Alpha-particle irradiation + c-PTIO (NO scavenger) | Mutation Frequency (bystander effect) | Significant decrease | [54] |
| 3T3L1 Adipocytes | Palmitate + SMT (iNOS inhibitor) | Adiponectin Secretion, ER Stress (XBP1 splicing) | Near-complete recovery | [53] |
| HepG2 Cells | Amphiregulin stimulation + Bay 11-7082 | iNOS and COX-2 protein expression | Significant reduction | [55] |
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents for Investigating the PARP-1/NF-κB/iNOS Pathway
| Reagent / Tool | Category | Primary Function in Research | Example & Citation |
|---|---|---|---|
| PARP Inhibitors | Small Molecule Inhibitor | Blocks PARP enzymatic activity; used to probe PARP's role in cell death and energy depletion. | Multiple clinical-grade inhibitors (e.g., Olaparib) are used in research. |
| Bay 11-7082 | Small Molecule Inhibitor | Pharmacological inhibitor of IκBα phosphorylation, preventing NF-κB activation. | Used to link NF-κB activity to bystander mutagenesis [54] and iNOS/COX-2 expression [55]. |
| c-PTIO | Nitric Oxide Scavenger | Selectively scavenges nitric oxide (NO), used to dissect the role of NO in signaling and toxicity. | Used to demonstrate NO's role in radiation-induced bystander effects [54]. |
| SMT (S-Methylisothiourea sulfate) | iNOS Inhibitor | A specific inhibitor of inducible NOS (iNOS) activity. | Reversed palmitate-induced ER stress and increased adiponectin synthesis [53]. |
| Anti-PARP-1 (cleaved) Antibodies | Antibody | Detects specific proteolytic fragments (e.g., 89 kDa) of PARP-1, a hallmark of apoptosis. | Critical for identifying caspase-3/7 activation [11]. |
| Phospho-IκBα (Ser32) Antibody | Antibody | Detects the phosphorylated, targeted-for-degradation form of IκBα, indicating canonical NF-κB pathway activation. | Standard readout for NF-κB stimulation [56]. |
| MitoSOX Red / TMRE | Fluorescent Probe | Measures mitochondrial superoxide and membrane potential, respectively; key for assessing mitochondrial health. | Used to quantify palmitate-induced ROS and ΔΨm loss [53] [52]. |
Navigating the PARP Paradox: Therapeutic Challenges and Strategic Inhibition
Poly(ADP-ribose) polymerase-1 (PARP-1) represents a critical regulatory node at the intersection of cellular survival and death decisions in neurological systems. As the predominant member of the PARP family, this nuclear enzyme senses DNA damage and catalyzes the transfer of ADP-ribose units from nicotinamide adenine dinucleotide (NAD+) onto target proteins, a post-translational modification known as PARylation [3] [31]. Under physiological conditions, PARP-1 maintains genomic integrity through its fundamental roles in DNA damage repair, chromatin remodeling, and transcriptional regulation [57] [31]. However, in the context of neurodegenerative diseases and neurological insults, the same enzyme can trigger catastrophic cell death pathways when excessively activated [58] [38].
This review examines the fundamental conundrum of PARP-1 activation in the nervous system, where context-dependent factors—including activation intensity, subcellular localization, proteolytic processing, and metabolic status—determine whether the outcome will be neuroprotective or neurotoxic. Particular emphasis is placed on PARP-1 cleavage fragments as both biomarkers and active participants in neurodegenerative disease pathogenesis, presenting them as potential diagnostic tools and therapeutic targets for researchers and drug development professionals.
PARP-1 Structure, Domains, and Cleavage Fragments
Structural Foundations of PARP-1 Function
PARP-1 is a modular protein comprising three principal functional domains that dictate its cellular functions and proteolytic fate [11] [57]. The DNA-binding domain (DBD), located at the N-terminus, contains two zinc finger motifs that enable PARP-1 to recognize and bind to DNA strand breaks with high affinity [11]. The central auto-modification domain (AMD) serves as a target for covalent PARylation, which regulates PARP-1's dissociation from DNA and interaction with other proteins [11]. The C-terminal catalytic domain (CD) facilitates the transfer of ADP-ribose units from NAD+ to target proteins, generating branched PAR polymers [11] [57]. Recent research has identified that a third zinc finger motif located between the second zinc finger and the AMD plays a crucial role in inter-domain interactions essential for PARP-1 enzymatic activity [11].
Proteolytic Cleavage and Signature Fragments
PARP-1 serves as a preferred substrate for several proteases, generating specific cleavage fragments that serve as biochemical signatures for distinct cell death pathways [11]. The following table summarizes the key PARP-1 fragments, their origins, and their functional significance:
Table 1: PARP-1 Cleavage Fragments and Their Characteristics
| Fragment Size | Protease Responsible | Domains Contained | Cellular Localization | Functional Consequences |
|---|---|---|---|---|
| 89 kDa | Caspases-3 and -7 [11] | AMD + Catalytic Domain [11] | Cytoplasm [15] | Serves as PAR carrier to cytoplasm; induces AIF release [15] |
| 24 kDa | Caspases-3 and -7 [11] | DNA-Binding Domain [11] | Nuclear (retained at DNA lesions) [11] | Acts as trans-dominant inhibitor of PARP-1; blocks DNA repair [11] |
| 55 kDa & 62 kDa | Calpain [11] | Not specified | Nuclear and cytoplasmic [11] | Associated with excitotoxicity and calcium-mediated cell death [11] |
| 40 kDa | Cathepsins [11] | Not specified | Lysosomal/cytoplasmic [11] | Linked to lysosomal-mediated cell death pathways [11] |
The cleavage of PARP-1 by caspases represents a hallmark of apoptosis and produces fragments with dramatically altered functions compared to the full-length protein [11]. The 24-kDa fragment retains the DNA-binding capability but lacks catalytic activity, effectively functioning as a dominant-negative inhibitor that blocks DNA repair by occupying DNA damage sites [11]. Recent research has revealed that the 89-kDa fragment can undergo translocation to the cytoplasm while carrying covalently attached PAR polymers, where it facilitates apoptosis-inducing factor (AIF) release from mitochondria, creating a mechanistic bridge between caspase-dependent apoptosis and PAR-mediated parthanatos [15].
Neuroprotective Functions of PARP-1
DNA Damage Repair and Genomic Maintenance
In response to mild DNA damage, PARP-1 activation serves primarily protective functions through multiple DNA repair pathways [31]. As a first responder to DNA strand breaks, PARP-1 rapidly binds to damaged sites and catalyzes extensive PARylation of itself and surrounding nuclear proteins, including histones [3] [31]. This PARylation creates a negatively charged scaffold that recruits additional DNA repair factors such as XRCC1, DNA ligase III, and other components of the base excision repair (BER) machinery [3] [31]. Beyond BER, PARP-1 contributes to numerous other repair pathways, including nucleotide excision repair (NER), homologous recombination (HR), and non-homologous end joining (NHEJ) [31]. The fundamental role of PARP-1 in maintaining genomic stability is evidenced by the high sensitivity of PARP-1 knockout mice to DNA-damaging agents and the embryonic lethality observed in PARP-1/PARP-2 double knockouts [3].
Transcriptional Regulation and Chromatin Remodeling
Beyond its DNA repair functions, PARP-1 influences gene expression and chromatin architecture through both enzymatic and non-enzymatic mechanisms [57] [31]. PARP-1 can function as a histone chaperone through PARylation of H2A-H2B and H3-H4 histone complexes and has been shown to increase nucleosomal repeat length [57]. The polyanionic nature of PAR polymers can directly destabilize nucleosomes without requiring covalent attachment, indicating multiple mechanisms for chromatin modulation [57]. Through interactions with transcription factors including NF-κB, NFAT, E2F-1, and ELK-1, PARP-1 regulates the expression of numerous genes involved in stress response, inflammation, and cell survival [11] [31]. PARP-1 has been reported to influence approximately 3.5% of the transcriptome in embryonic liver and stem cells, particularly genes controlling cell metabolism, cell cycle progression, and transcription itself [11].
Neurotoxic Consequences of PARP-1 Overactivation
Energy Depletion and Necrotic Cell Death
Under conditions of severe DNA damage, PARP-1 becomes hyperactivated and initiates a cascade of metabolic events leading to cellular energy collapse [58] [38]. The consumption of NAD+ during excessive PAR synthesis depletes cellular NAD+ pools, which in turn impairs glycolysis and mitochondrial respiration, leading to catastrophic ATP depletion [58] [38]. This energy failure mode traditionally represents the primary mechanism of PARP-1-mediated necrotic cell death [58]. The reaction catalyzed by PARP-1 also produces H+ during NAD+ breakdown, directly contributing to intracellular acidification that further promotes necrotic cell death [38]. The table below summarizes the key pathological processes in PARP-1-mediated neurotoxicity:
Table 2: Mechanisms of PARP-1-Mediated Neurotoxicity
| Mechanism | Key Players | Consequences | Associated Diseases | |
|---|---|---|---|---|
| Energy Depletion | NAD+ consumption, ATP depletion [58] [38] | Necrotic cell death [58] | TBI, ischemia [58] | |
| Parthanatos | PAR polymer, AIF release [15] [38] | Caspase-independent programmed death [15] | PD, AD, HD [38] | |
| Neuroinflammation | NF-κB activation, microglial activation, iNOS, TNFα [58] [31] | Chronic inflammation, bystander neuronal damage [58] | PD, AD, ALS [57] [31] | |
| - | Mitochondrial Dysfunction | PARP-1 activation, PAR polymer [58] | Impaired respiration, ROS production [58] | PD, AD [38] |
Parthanatos: A PAR-Dependent Cell Death Pathway
Parthanatos represents a distinct form of programmed cell death that is caspase-independent but mechanistically linked to PARP-1 overactivation [15] [38]. This pathway involves massive PAR synthesis triggered by severe DNA damage, leading to PAR translocation to the cytoplasm where it induces mitochondrial membrane permeabilization and release of AIF [15]. AIF then translocates to the nucleus where it triggers large-scale DNA fragmentation and chromatin condensation [15] [38]. Recent research has demonstrated that caspase-generated 89-kDa PARP-1 fragments can carry PAR polymers to the cytoplasm, serving as PAR carriers that facilitate AIF release, thus creating a mechanistic bridge between apoptotic and parthanatos pathways [15]. Parthanatos has been implicated in the pathogenesis of multiple neurodegenerative disorders, including Parkinson's disease, Alzheimer's disease, and Huntington's disease [38].
Neuroinflammation and Microglial Activation
PARP-1 activation significantly amplifies neuroinflammatory responses through regulation of key transcription factors, particularly NF-κB [58] [31]. PARP-1 promotes NF-κB activation through multiple mechanisms, including facilitating the sumoylation and mono-ubiquitination of NEMO (NF-κB essential modulator), interacting directly with NF-κB family members to enhance transcription complex formation, and sustaining p65 NF-κB nuclear retention by reducing its interaction with nuclear exporting proteins [31]. In microglial cells, PARP-1 activation enhances the production of pro-inflammatory mediators including iNOS, TNF-α, and reactive oxygen species [58]. PARP-1 inhibition with PJ34 has been shown to attenuate microglial activation by limiting NF-κB activity and iNOS expression, thereby decreasing neurotoxic inflammatory responses [58].
PARP-1 in Neurodegenerative Diseases: A Dichotomous Regulatory Role
The role of PARP-1 activation varies significantly across different neurodegenerative conditions, with both overactivation and suppression associated with pathological states [3]. The following table summarizes PARP-1 alterations across major neurodegenerative diseases:
Table 3: PARP-1 Alterations in Human Neurodegenerative Diseases and Models
| Disease | Sample Type | PAR Signaling | References |
|---|---|---|---|
| Alzheimer's Disease | Brain, Fibroblasts, Lymphoblasts | Increased PARP1 and PAR [3] | Love et al., Cecchi et al. [3] |
| Parkinson's Disease | CSF, Fibroblasts | Increased PAR in CSF; Decreased PAR in fibroblasts [3] | Kam et al., Wang et al. [3] |
| Amyotrophic Lateral Sclerosis | Spinal cord astroglia, Motor neurons | Increased PARP1 in astroglia; Decreased in motor neurons [3] | Kim et al., McGurk et al. [3] |
| Huntington's Disease | CSF, Fibroblasts, iPSC-derived neurons | Decreased PAR and impaired PARP1 activity [3] | Maiuri et al. [3] |
| Cerebellar Ataxias | Fibroblasts, Lymphoblastoids | Increased PAR or prolonged PAR response [3] | Hoch et al., Gueven et al. [3] |
Alzheimer's Disease
In Alzheimer's disease, increased PARP-1 activity and PAR levels have been consistently observed in the frontal and temporal lobes of patient brains [3] [59]. Activated PARP-1 has been shown to induce Aβ production and promote Tau tangle formation, thereby worsening cognitive symptoms [60]. PARP-1 overactivation may contribute to AD pathogenesis through multiple mechanisms, including energy depletion, enhanced neuroinflammation, and direct effects on epigenetic regulation [59]. In Drosophila AD models, PARP-1 inhibition with olaparib and MC2050 significantly extended lifespan, improved climbing ability, decreased Aβ42 aggregates, and partially rescued AD-associated epigenetic alterations [59].
Parkinson's Disease
Growing evidence links PARP-1 hyperactivation to Parkinson's disease pathology [57]. Increased PAR levels have been detected in the cerebrospinal fluid of PD patients, suggesting PARP-1 overactivation may contribute to disease progression [3] [57]. Notably, PAR has been shown to promote the fibrillation of α-synuclein, the primary protein component of Lewy bodies, through direct interactions that accelerate the aggregation process [57]. Additionally, PARP-1 activation in microglial cells amplifies neuroinflammatory responses that contribute to the degeneration of dopaminergic neurons in the substantia nigra [58] [57].
Huntington's Disease
In contrast to other neurodegenerative conditions, Huntington's disease exhibits a unique characteristic of reduced PAR levels and impaired PARP1 activity, even during the prodromal phase [3]. This finding challenges the prevailing understanding of PAR's role in neurodegeneration and suggests that dysregulation of PAR signaling in either direction—whether through overactivation or suppression—can lead to neuronal dysfunction [3]. The specific mechanisms through which impaired PARP-1 activity contributes to HD pathogenesis remain under investigation but may involve defective DNA damage repair and altered transcriptional regulation [3].
Experimental Approaches and Research Methodologies
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagents for PARP-1 Studies
| Reagent/Category | Specific Examples | Primary Applications | Key Functions | |
|---|---|---|---|---|
| PARP Inhibitors | PJ34, Olaparib, MC2050 [58] [59] | In vitro and in vivo inhibition studies | Attenuate PARP activity; neuroprotection in models | |
| Cell Death Inducers | MNNG, Staurosporine, Actinomycin D [58] [15] | Inducing parthanatos/apoptosis | Activate specific cell death pathways for study | |
| - | Protease Inhibitors | Caspase, Calpain inhibitors [11] | Cleavage pathway analysis | Block specific PARP-1 cleavage events |
| Model Organisms | PARP-1 KO mice, Drosophila models [58] [59] | Pathophysiology and therapeutic testing | Study PARP-1 function in complex systems | |
| Detection Antibodies | PAR, PARP-1 fragments [11] | Immunoblotting, immunohistochemistry | Identify PARP activation and cleavage |
Standard Experimental Protocols
Assessing PARP-1 Activation and Cleavage In Vitro
Cell Culture and Treatment: Utilize neuronal cell lines (e.g., primary cortical neurons) or microglial cells (e.g., BV2 line) cultured in DMEM supplemented with 10% fetal bovine serum at 37°C with 5% CO₂ [58]. For PARP-1 activation, treat cells with DNA-damaging agents such as N-Methyl-N'-Nitro-N-Nitrosoguanidine (MNNG) or oxidative stress inducers [58]. To induce PARP-1 cleavage, apply apoptotic inducers like staurosporine or actinomycin D [15].
PARP Inhibition Studies: Pre-treat cells with PARP inhibitors (e.g., PJ34 at 1-20 μM concentration) for 1 hour before applying activating stimuli [58]. This approach allows assessment of PARP-dependent phenomena while controlling for potential off-target effects.
Western Blot Analysis: Solubilize cells in RIPA buffer, separate proteins by SDS-PAGE, and transfer to nitrocellulose membranes [58]. Probe with primary antibodies against PARP-1 (to detect full-length and cleavage fragments), PAR polymers (to assess PARP-1 activation), and β-actin (as loading control) [58] [11]. Specific cleavage fragments (e.g., 89-kDa and 24-kDa) serve as biomarkers for particular protease activities and cell death pathways [11].
Evaluating PARP-1-Mediated Cell Death Pathways
Parthanatos Assessment: Monitor PARP-1 overactivation by measuring PAR accumulation using immunoblotting or immunofluorescence [15] [38]. Track AIF translocation from mitochondria to nucleus through subcellular fractionation or immunocytochemistry [15]. Assess mitochondrial membrane potential changes using fluorescent dyes like JC-1 or TMRM [58].
Energy Depletion Measurements: Quantify NAD+ depletion and ATP levels using commercially available kits based on enzymatic cycling assays or luciferase-based methods, respectively [58] [38]. Correlate energy depletion with cell death markers to establish mechanistic connections.
Neuroinflammatory Responses: In microglial cultures, stimulate activation with lipopolysaccharide (LPS) or interferon-γ (IFNγ) with or without PARP inhibitor pre-treatment [58]. Measure pro-inflammatory outputs including NO production (Griess reagent), TNF-α release (ELISA), iNOS expression (Western blot), and NF-κB activity (luciferase reporter assay) [58].
Signaling Pathways and Molecular Mechanisms
The following diagram illustrates the core signaling pathways governing the neuroprotective and neurotoxic outcomes of PARP-1 activation:
PARP-1 Activation Pathways in Neuroprotection and Neurotoxicity
This diagram illustrates how the extent of DNA damage determines PARP-1 activation levels, leading to divergent cellular outcomes. Mild damage triggers moderate activation and neuroprotective responses, while severe damage causes hyperactivation initiating multiple neurotoxic pathways. Proteolytic cleavage by caspases generates fragments that modulate both processes, creating a complex regulatory network.
The dual nature of PARP-1 activation presents both challenges and opportunities for therapeutic development in neurodegenerative diseases. The context-dependent outcomes—where PARP-1 activation can be either protective or detrimental—necessitate sophisticated therapeutic approaches that modulate rather than completely inhibit PARP-1 activity. Future research should focus on several key areas: First, understanding the precise thresholds and signaling contexts that determine the transition from protective to toxic PARP-1 activation. Second, developing context-sensitive PARP inhibitors that can distinguish between basal DNA repair functions and pathological overactivation. Third, exploring the temporal dynamics of PARP-1 activation in disease progression to identify optimal therapeutic windows. Finally, investigating the cell-type specific functions of PARP-1 in the complex landscape of the nervous system, including neurons, microglia, and astrocytes.
The study of PARP-1 cleavage fragments continues to provide valuable insights into the underlying mechanisms of neurodegenerative diseases, serving as both diagnostic biomarkers and potential therapeutic targets. As our understanding of the neuroprotection-neurotoxicity conundrum deepens, the development of precisely targeted PARP-1 modulators holds significant promise for effective interventions across the spectrum of neurodegenerative disorders.
Poly(ADP-ribose) polymerase-1 (PARP-1) is a critical nuclear enzyme involved in DNA damage repair and the maintenance of genomic stability. In the context of neurodegenerative diseases, PARP-1 activation plays a complex, dual role. While its activation is essential for repairing routine DNA damage, persistent overactivation can trigger energy depletion and pathological cell death processes. Emerging evidence reveals a striking dichotomy in PARP-1 activity across different neurodegenerative conditions: it is hyperactive in Alzheimer's disease (AD) and Parkinson's disease (PD) but functionally impaired in Huntington's disease (HD). This contrast not underscores the disease-specific mechanisms at play but also highlights the need for tailored therapeutic strategies. This review synthesizes current evidence on PARP-1 dysregulation in these diseases, framed within the broader context of PARP-1 cleavage fragments and their role in neurodegenerative disease research.
PARP-1 Structure, Function, and Proteolytic Cleavage
Domain Architecture and Primary Functions
PARP-1 is a modular protein comprising three primary functional domains:
- DNA-Binding Domain (DBD): Located at the N-terminus, it contains zinc finger motifs (F1, F2, F3) that recognize and bind to DNA strand breaks. The F1 and F2 domains interact with the DNA backbone and exposed bases, while F3 facilitates interdomain contacts crucial for activation [61] [62].
- Automodification Domain (AMD): This central domain contains a BRCT fold and serves as an acceptor for ADP-ribose units, enabling auto-PARylation. This modification facilitates the release of PARP-1 from DNA after repair is complete [35] [62].
- Catalytic Domain (CAT): Situated at the C-terminus, it includes a WGR motif and the ART (ADP-ribosyltransferase) subdomain. This domain catalyzes the transfer of ADP-ribose units from NAD+ to target proteins, forming poly(ADP-ribose) (PAR) chains [61] [62].
PARP-1's primary role is as a first-line responder to DNA damage. Upon binding to single-strand breaks (SSBs) or double-strand breaks (DSBs), PARP-1 undergoes a dramatic allosteric activation, increasing its catalytic activity up to 1000-fold [61]. This leads to the synthesis of PAR chains, which serve as a signal to recruit other DNA repair proteins, such as XRCC1, to the damage site [61] [3] [63].
PARP-1 as a Substrate for Suicidal Proteases
A critical aspect of PARP-1 biology in neurodegeneration is its role as a substrate for proteases involved in cell death, earning them the name "suicidal proteases" [35]. The cleavage of PARP-1 by these enzymes produces specific signature fragments that serve as biomarkers for different cell death pathways [35].
Table: PARP-1 Cleavage by Suicidal Proteases in Neurodegeneration
| Protease | Cleavage Fragments | Associated Cell Death Pathway | Implication in Neurodegeneration |
|---|---|---|---|
| Caspase-3/7 | 89-kD (AMD + CAT), 24-kD (DBD) | Apoptosis | Hallmark of apoptotic cell death; observed in cerebral ischemia, AD, PD, and brain trauma [35]. |
| Calpains | 55-kD, 40-kD, 35-kD | Excitotoxicity, Necrosis | Associated with calcium-dependent cell death pathways [35]. |
| Granzyme A | 50-kD fragment | Caspase-independent apoptosis | Implicated in immune-mediated neuronal damage [35]. |
| Matrix Metalloproteinases (MMPs) | 55-kD, 40-kD, 35-kD | Inflammatory cell death | Linked to neuroinflammation and blood-brain barrier disruption [35]. |
The most well-characterized cleavage is by caspase-3, which generates an 89-kD fragment (containing the AMD and CAT) and a 24-kD DNA-binding fragment [35]. The 24-kD fragment remains bound to damaged DNA, acting as a trans-dominant inhibitor of PARP-1 and further suppressing DNA repair capacity during apoptosis [35]. This process conserves cellular ATP pools but irrevocably commits the cell to death.
Contrasting PARP-1 Activity in Major Neurodegenerative Diseases
The dysregulation of PARP-1 signaling is a common feature in neurodegeneration, but the direction of this dysregulation is disease-specific.
PARP-1 Overactivation in Alzheimer's and Parkinson's Disease
In both AD and PD, research consistently points to a state of PARP-1 overactivation, which contributes directly to neuropathology.
Alzheimer's Disease (AD): Evidence from human post-mortem brain tissues, fibroblasts, and lymphoblasts shows increased PARP1 levels and PAR signaling in AD [3]. The accumulation of amyloid-beta (Aβ) peptides is a key driver of this overactivation. Aβ induces oxidative stress and DNA damage, triggering PARP-1 activation [64]. For instance, treatment of SH-SY5Y neuroblastoma cells with the Aβ25–35 fragment significantly enhances PARP activity, an effect preventable by PARP inhibition [64]. This overactivation is not merely a consequence but actively participates in pathogenesis by promoting neuroinflammation (via activation of NF-κB) and modulating proteins involved in apoptosis, such as increasing p53 and decreasing Bcl-2 levels [38] [64]. Furthermore, activated PARP1 has been shown to induce Aβ formation and Tau tangles, worsening cognitive symptoms [60].
Parkinson's Disease (PD): Similar to AD, PARP overactivation is a feature of PD. Studies have found elevated PAR levels in the cerebrospinal fluid (CSF) of PD patients [3]. The disease's characteristic mitochondrial dysfunction and oxidative stress generate significant DNA damage, leading to persistent PARP-1 activation. This can deplete cellular NAD+ and ATP pools, ultimately driving a specific form of programmed cell death known as parthanatos [38]. The overactivation of PARP-1 is thus a critical contributor to the loss of dopaminergic neurons in the substantia nigra.
Table: Quantitative Evidence of PARP-1 Dysregulation in Human Samples and Models
| Disease | Sample Type | Observed Change in PARP-1/PAR | Key Supporting Evidence |
|---|---|---|---|
| Alzheimer's Disease (AD) | Human Brain Tissue | Increased PARP1 and PAR | Post-mortem analysis shows elevated PARylation [3]. |
| Human Fibroblasts | Increased PAR | Cellular models from patients confirm dysregulation [3]. | |
| Transgenic Mouse Brain | Increased PARP Activity | TgCRND8 mice overproducing Aβ show elevated activity [64]. | |
| Cell Model | Increased PARP Activity | SH-SY5Y cells treated with Aβ25–35 show enhanced activity [64]. | |
| Parkinson's Disease (PD) | Human CSF | Increased PAR | Biomarker analysis in patient cerebrospinal fluid [3]. |
| Human Fibroblasts | Decreased PAR | Suggests possible cell-type or disease-stage specificity [3]. | |
| Huntington's Disease (HD) | Human CSF | Decreased PAR | A unique finding distinguishing HD from AD and PD [3]. |
| Human Fibroblasts | Decreased PAR response to DNA damage | Impaired PARP-1 function upon challenge [3]. | |
| iPSC-derived Neurons | Decreased PAR | Confirms impairment in relevant human cell types [3]. |
Impaired PARP-1 Activity in Huntington's Disease
Huntington's disease presents a paradoxical picture, characterized by a unique impairment of PARP-1 activity. This is observed even in the prodromal stages of the disease [3].
- Human Evidence: Studies of CSF, fibroblasts, and iPSC-derived neurons from HD patients consistently show reduced PAR levels and a blunted PAR response to DNA-damaging agents [3]. This finding challenges the simplistic view that PARP overactivation is universally detrimental in neurodegeneration and indicates that suppressed PAR signaling can be equally pathological.
- Mechanistic Insights: The mutant huntingtin protein (mHTT), which contains expanded polyglutamine repeats, is central to this dysfunction. mHTT is known to disrupt multiple cellular processes, including transcriptional regulation and mitochondrial function, which may indirectly affect PARP-1. The mHTT protein contributes to the disruption of DNA repair pathways, potentially leading to the observed impairment in PARP-1 function [60]. This failure in DNA damage repair would contribute to the genomic instability and progressive neuronal loss seen in HD.
The diagram below summarizes the divergent pathways driven by PARP-1 dysregulation in these diseases.
Experimental Approaches for PARP-1 Analysis
To investigate the contrasting roles of PARP-1, researchers employ a suite of biochemical, cellular, and molecular techniques.
Key Methodologies and Workflows
The experimental workflow for assessing PARP-1 activity and cleavage typically involves the following key steps, which can be applied to cell cultures, iPSC-derived neurons, or animal brain tissues:
A typical protocol for evaluating Aβ-induced PARP-1 activation, as adapted from Martire et al. (2013), involves the following detailed steps [64]:
- Cell Culture and Treatment:
- Maintain human SH-SY5Y neuroblastoma cells in DMEM/F12 medium supplemented with 10% fetal bovine serum.
- Pre-treat cells with a PARP inhibitor (e.g., 50 µM MC2050) for 1 hour.
- Challenge cells with aggregated Aβ25–35 peptide (e.g., at 10-50 µM concentration) for a defined period (e.g., 24 hours) to induce oxidative stress and DNA damage.
- PARP Activity Assay:
- Lyse cells and extract nuclear proteins.
- Measure PARP activity in a reaction buffer containing 50 mM Tris-HCl (pH 8.0), 25 mM MgCl₂, 1 mM DTT, and labeled NAD+.
- Incorporate tritiated NAD+ ([³H]NAD+) or biotinylated NAD+ into the reaction. The incorporation of radioactive ADP-ribose or the detection of biotin-labeled PAR polymers serves as a direct measure of PARP activity.
- Terminate the reaction and quantify the synthesized PAR. For [³H]NAD+, measure radioactivity via scintillation counting. For biotinylated NAD+, use streptavidin-conjugated detection methods following SDS-PAGE separation and transfer to a membrane.
- Analysis of PARP-1 Cleavage and Downstream Effects:
- Perform Western blotting on total cell lysates.
- Use a primary antibody against PARP-1 to detect the full-length (116 kDa) protein and the signature 89 kDa caspase-derived cleavage fragment.
- Probe for other apoptosis-related proteins like cleaved caspase-3, p53, and Bcl-2 to correlate PARP-1 cleavage with cell death pathways.
- To assess downstream signaling, techniques like Electrophoretic Mobility Shift Assay (EMSA) or reporter gene assays can be used to analyze the activation of transcription factors such as NF-κB.
The Scientist's Toolkit: Key Research Reagents
Table: Essential Reagents for PARP-1 Research in Neurodegeneration
| Reagent / Assay | Specific Example | Primary Function in Research |
|---|---|---|
| PARP Inhibitors | MC2050, PJ34, 3-aminobenzamide (3-ABA), Olaparib | Tool compounds to inhibit PARP catalytic activity and probe its pathological roles in disease models [64] [38]. |
| Anti-PAR Antibody | Multiple commercial clones (e.g., 10H) | Gold-standard for detecting PAR polymer formation (a marker of PARP activation) via Western blot, immunofluorescence, or ELISA [64]. |
| Anti-PARP1 Antibody | Antibodies targeting N-terminal or C-terminal epitopes | Detects full-length PARP-1 and its proteolytic fragments (e.g., 89 kDa) in Western blot, distinguishing activation from cleavage [35]. |
| Caspase-3 Assay | Fluorometric or colorimetric substrate kits | Quantifies caspase-3 activity to confirm apoptotic induction and its link to PARP-1 cleavage [35]. |
| Cell Death Assays TUNEL, Annexin V / PI staining, LDH release | Labels DNA fragmentation (TUNEL) or exposed phosphatidylserine (Annexin V) to quantify and characterize modes of cell death (apoptosis vs. necrosis) [64]. | |
| DNA Damage Inducers | Amyloid-β fragments (Aβ25–35), H₂O₂, Etoposide | Induce oxidative stress or direct DNA damage to trigger and study the PARP-1 response pathway in experimental models [64] [3]. |
Therapeutic Implications and Future Directions
The contrasting dysregulation of PARP-1 necessitates distinct therapeutic approaches.
- For AD and PD: The strategy centers on inhibition of overactivated PARP-1. Multiple PARP inhibitors (e.g., PJ34, olaparib) have shown neuroprotective effects in preclinical models by preventing energy depletion and parthanatos [60] [38]. Notably, PARP inhibition can also attenuate neuroinflammation and mitigate the cognitive complications associated with chemotherapy ("chemo brain"), highlighting its broader potential [60]. The development of selective PARP1 inhibitors (e.g., AZD5305, (S)-G9) aims to provide efficacy while reducing the hematological toxicities associated with first-generation PARP1/2 dual inhibitors [65].
- For HD: The therapeutic challenge is different. Here, the goal is to restore or stimulate the impaired PARP-1 function. This could involve identifying small molecule activators of PARP-1 or targeting the upstream pathways disrupted by mHTT that lead to PARP-1 dysfunction [3]. This approach remains in its early stages but represents a critical frontier.
A promising development is the creation of brain-penetrant PARP inhibitors. Companies are now developing clinical-stage molecules with and without brain penetration, aiming to cover a spectrum of indications from broad-spectrum tumors to central nervous system (CNS) diseases [66]. This opens the possibility that PARP-1 inhibition for AD and PD, or potentially future PARP-1 activators for HD, could be effectively delivered to their primary site of pathology—the brain.
PARP-1 plays a central yet complex role in the pathogenesis of major neurodegenerative diseases. The clear dichotomy—with overactivation in Alzheimer's and Parkinson's disease contributing to energy failure and cell death, and functional impairment in Huntington's disease leading to inadequate DNA repair—underscores the principle that therapeutic strategies must be disease-specific. The analysis of PARP-1 cleavage fragments provides crucial insights into the dominant cell death pathways active in different pathological contexts. Future research must continue to elucidate the precise molecular mechanisms driving this dysregulation and leverage this knowledge to develop targeted interventions, whether through inhibition for AD/PD or restoration of function for HD. The ongoing development of selective and brain-penetrant PARP modulators heralds a new era of precise therapeutic tools to test these hypotheses in clinical settings.
Poly(ADP-ribose) polymerase 1 (PARP1) is a critical nuclear enzyme involved in DNA damage repair, and its inhibition has emerged as a powerful therapeutic strategy. While early research focused primarily on catalytic inhibition, the phenomenon of "PARP trapping"—where PARP1 is stably retained on DNA—has proven to be a pivotal mechanism underlying the efficacy of clinical PARP inhibitors (PARPi). This whitepaper delineates the mechanistic distinctions between enzymatic inhibition and PARP trapping, examining the kinetic and allosteric principles governing these processes. Within the broader context of PARP-1 biology, we explore how PARP cleavage fragments, signatures in neurodegenerative research, connect to the fundamental processes of inactivation and trapping. Understanding these differential mechanisms provides crucial insights for therapeutic development across oncological and neurological contexts.
PARP1 is a multifaceted nuclear enzyme that serves as a primary sensor of DNA damage, with roles spanning DNA repair, chromatin remodeling, transcription regulation, and cell death signaling [67] [48]. The canonical function of PARP1 involves detecting DNA single-strand breaks (SSBs) and catalyzing poly(ADP-ribose) (PAR) chain formation on target proteins using NAD+ as a substrate [67] [48]. This PARylation acts as a signal for the recruitment of additional DNA repair machinery, such as XRCC1, to facilitate efficient DNA repair [67]. The PARP family comprises 17 members, with PARP1 accounting for approximately 80-90% of cellular PARylation activity [48] [68].
Traditional understanding of PARP inhibition centered on catalytic blockade—preventing NAD+ binding and subsequent PAR synthesis. However, a critical breakthrough revealed that PARP inhibitors exert their cytotoxic effects not merely through enzymatic inhibition but predominantly through a phenomenon termed "PARP trapping" [69] [68]. PARP trapping refers to the stable retention of PARP1-DNA complexes in chromatin, which creates physical obstacles to DNA replication and transcription [68]. This trapped state proves particularly deleterious to homologous recombination (HR)-deficient cancer cells, resulting in synthetic lethality [70] [68]. The recognition that different PARPi exhibit varying trapping potencies despite similar catalytic inhibition profiles has fundamentally reshaped drug development and mechanistic understanding [69] [71].
Fundamental Mechanisms: Catalytic Inhibition versus PARP Trapping
Catalytic Inhibition: The Basic Paradigm
Catalytic inhibition of PARP1 occurs when inhibitors competitively bind the NAD+ catalytic pocket, preventing PAR synthesis and subsequent DNA repair processes [68]. In the normal PARP1 enzymatic cycle, DNA damage binding triggers allosteric activation, leading to autoPARylation, which facilitates PARP1 release from DNA and recruitment of repair factors [48] [68]. Pharmacological competition for the NAD+ binding site disrupts this cycle by preventing autoPARylation, thereby extending PARP1's residence time on DNA damage sites [68].
PARP Trapping: A Distinct Cytotoxic Mechanism
PARP trapping represents a more complex phenomenon beyond simple enzymatic blockade. Early models proposed that trapping resulted from extended PARP1-DNA interaction due to inhibited dissociation [69]. However, recent evidence challenges this simplistic view, suggesting instead that trapping occurs "primarily through a kinetic phenomenon at sites of DNA damage that correlates with PARPi koff" [69]. Rather than physical stalling of a single PARP1 molecule, trapping appears to involve a high probability of PARP1 rebinding damaged DNA in the absence of productive repair and recruitment of other DNA binding proteins [69]. This revised model aligns with observations that PARP1 diffusion at DNA damage sites remains unaffected by PARPi presence [69].
Table 1: Key Characteristics of Catalytic Inhibition vs. PARP Trapping
| Characteristic | Catalytic Inhibition | PARP Trapping |
|---|---|---|
| Primary Mechanism | Competition with NAD+ binding | Stable PARP1-DNA complex formation |
| Effect on PARP1-DNA Residence | Moderately increased | Dramatically prolonged |
| Cellular Consequence | Impaired DNA repair signaling | Replication fork collapse & DSB formation |
| Correlation with Cytotoxicity | Moderate | Strong |
| Dependence on PARPi Structure | Low | High |
Allosteric Regulation and Reverse Allostery
The structural basis for differential trapping efficacy among PARPi involves allosteric communication between PARP1 domains. The "reverse allostery" model proposes that inhibitor binding to the catalytic domain transmits conformational changes to the DNA-binding domain, thereby modulating DNA binding affinity [70] [71]. This allosteric coupling explains why inhibitors with similar NAD+-competing potency can exhibit markedly different trapping efficiencies [71]. Recent structural analyses have identified specific PARP1 mutations that disrupt allosteric regulation without affecting catalytic function, further supporting this model and providing insights for targeted inhibitor development [71].
Diagram 1: PARP1 catalytic cycle and trapping mechanism. High koff PARPi promote PARP1 rebinding to DNA damage sites, leading to trapped complexes that cause replication fork collapse.
Quantitative Analysis of PARP Inhibitor Trapping Efficiencies
Differential Trapping Potencies Among Clinical PARPi
PARP inhibitors exhibit a wide spectrum of trapping efficiencies despite targeting the same catalytic domain. Research using HT1080 fibrosarcoma cells demonstrated distinct trapping capabilities across five clinical PARPi, with talazoparib showing the most potent trapping effect, followed by niraparib, olaparib, rucaparib, and veliparib [69]. This differential trapping directly correlates with cellular sensitivity, as evidenced by EC50 values for cell viability spanning three orders of magnitude [69].
Table 2: Comparative Trapping Efficiencies and Cellular Effects of Clinical PARP Inhibitors
| PARP Inhibitor | Relative Trapping Potency | Cellular Sensitivity (EC50) | koff Characteristics | Clinical Status |
|---|---|---|---|---|
| Talazoparib | High | Low nM range | Slow dissociation | FDA-approved |
| Niraparib | Medium-High | nM range | Moderate dissociation | FDA-approved |
| Olaparib | Medium | nM range | Intermediate dissociation | FDA-approved |
| Rucaparib | Medium-Low | nM range | Not specified | FDA-approved |
| Veliparib | Low | μM range | Faster dissociation | Clinical trials |
Kinetic Parameters Governing Trapping Efficiency
The dissociation rate (koff) of PARPi from PARP1 emerges as a critical determinant of trapping potency. Live-cell fluorescence anisotropy measurements revealed significantly different koff rates for veliparib, olaparib, and talazoparib in HT1080 cells, with values similar to those obtained with purified protein [69]. This correlation between cellular koff rates and trapping potency suggests that inhibitor release kinetics, rather than binding affinity alone, drive differential trapping efficacy. Interestingly, PARP activation state influences koff measurements, with apparent dissociation slowing when PARP is activated by DNA damage, though this effect is not unique to high-trapping PARPi [69].
Methodological Approaches for Studying PARP Trapping
Chromatin Fractionation Assay
The chromatin fractionation assay represents a standard methodology for quantifying PARP trapping through biochemical separation of chromatin-bound proteins.
Protocol:
- Cell Treatment and Harvesting: Treat cells with PARPi (e.g., 1μM talazoparib vs. 100μM veliparib) for designated durations. Include controls (DMSO vehicle) and DNA-damaging agents (e.g., temozolomide) as appropriate. Harvest cells by gentle scraping or trypsinization.
- Cellular Fractionation: Resuspend cell pellets in cytosolic extraction buffer (10mM HEPES pH7.9, 10mM KCl, 0.1mM EDTA, 0.1mM EGTA, 1mM DTT, protease inhibitors) with 0.1% NP-40. Incubate 15min on ice, then centrifuge at 5,000×g for 5min at 4°C.
- Nuclear Extraction: Resuspend nuclear pellets in nuclear extraction buffer (20mM HEPES pH7.9, 400mM NaCl, 1mM EDTA, 1mM EGTA, 1mM DTT, protease inhibitors). Incubate with vigorous shaking for 30min at 4°C, then centrifuge at 14,000×g for 15min.
- Chromatin Fraction: Further extract the insoluble chromatin pellet with chromatin extraction buffer (nuclear extraction buffer with 500mM NaCl and 250U/mL Benzonase). Incubate 30min at 4°C with agitation, then centrifuge at 14,000×g for 15min.
- Western Blot Analysis: Resolve fractions by SDS-PAGE and immunoblot for PARP1. Normalize chromatin-bound PARP1 to histone H3 and total PARP1 levels. Compare treatment groups to DMSO controls to calculate fold-increase in chromatin retention [69].
Live-Cell Kinetic Assays for PARP Inhibitor Dissociation
Fluorescence anisotropy-based competitive binding assays enable direct measurement of PARPi koff rates in live cells.
Protocol:
- Fluorescent Tracer Preparation: Utilize covalent BODIPY FL-conjugated olaparib as a fluorescent PARPi tracer [69].
- Cell Preparation: Culture HT1080 or other appropriate cell lines in glass-bottom dishes for live-cell imaging.
- Competition Binding Kinetics: Load cells with fluorescent tracer, then add excess unlabeled competitor PARPi (veliparib, olaparib, or talazoparib) at specified concentrations.
- Anisotropy Measurement: Monitor fluorescence anisotropy over time using appropriate instrumentation. Fit anisotropy decay curves to exponential functions to determine koff rates for each PARPi [69].
- PARP Activation Modulation: Assess koff dependency on PARP activation status by pre-treating cells with DNA-damaging agents (e.g., temozolomide) or NAMPT inhibitors to modulate cellular NAD+ levels [69].
PARP Trapping in the Context of Transcription-Replication Conflicts
Emerging research has revealed an alternative mechanism for PARPi sensitivity in HR-deficient cells that extends beyond traditional trapping models. Recent findings demonstrate that PARP1 functions together with TIMELESS and TIPIN to protect replisomes from transcription-replication conflicts (TRCs) during early S phase [70]. PARP inhibitors induce TRCs specifically in early S phase, and this effect—rather than physical blockage by trapped PARPs—underlies the synthetic lethality with HR deficiency [70]. This paradigm shift is supported by several key observations:
- PARPi-induced DNA damage response is suppressed by transcription elongation inhibitors like DRB [70]
- PARP1 depletion itself is synthetic lethal with HR deficiency, suggesting enzymatic inhibition may suffice for efficacy [70]
- TRC-mediated DNA damage correlates with expressed genes in early-replicating genomic domains [70]
This model reconciles the discrepancy between PARP trapping potency and cellular efficacy for certain inhibitors, as it positions enzymatic inhibition rather than trapping as the primary driver of synthetic lethality in specific contexts.
Diagram 2: PARP1 protection against transcription-replication conflicts. PARP inhibitors disrupt PARP1-TIMELESS-TIPIN complex function, leading to unresolved conflicts that cause R-loop formation, fork stalling, and DSBs lethal to HR-deficient cells.
Research Reagent Solutions for PARP Trapping Studies
Table 3: Essential Research Reagents for PARP Trapping and Inhibition Studies
| Reagent/Category | Specific Examples | Research Application | Key Considerations |
|---|---|---|---|
| Clinical PARP Inhibitors | Talazoparib, Olaparib, Niraparib, Rucaparib, Veliparib | Comparative trapping studies; dose-response analyses | Varying trapping efficiencies at equivalent enzymatic inhibition concentrations |
| Chemical Tools | BODIPY FL-conjugated olaparib | Live-cell binding kinetics; koff measurements | Enables fluorescence anisotropy measurements in cellular contexts |
| DNA Damage Inducers | Temozolomide, Hydrogen peroxide, Etoposide | PARP activation studies; trapping induction | Different agents produce distinct DNA lesion profiles |
| Cellular Models | HT1080 fibrosarcoma, BRCA-deficient vs proficient lines, PARP1 knockout cells | Trapping quantification; synthetic lethality studies | Cell-line specific basal PARP activity affects results |
| Enzymatic Assays | NAD+ consumption assays, PAR immunoblotting | Catalytic inhibition verification | Distinguish catalytic inhibition from trapping effects |
| Chromatin Analysis | Chromatin fractionation, Histone PARylation factor 1 (HPF1) | Trapping quantification; chromatin context studies | HPF1 binding alters PARP1 function and inhibitor effects |
Implications for Neurodegenerative Disease Research
The relationship between PARP1 inactivation mechanisms and neurodegenerative diseases represents a critical interface for therapeutic development. PARP-1 cleavage fragments serve as established biomarkers for specific protease activities in unique cell death programs [11]. Caspase-mediated cleavage of PARP1 generates signature 89-kD and 24-kD fragments, with the 24-kD DNA-binding domain fragment acting as a trans-dominant inhibitor of BER by irreversibly binding to damaged DNA [11]. This cleavage mechanism effectively inactivates PARP1 while conserving cellular ATP pools during apoptosis [11].
In neurodegenerative contexts, PAR signaling demonstrates disease-specific dysregulation. While Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis, and cerebellar ataxias typically show increased PAR levels and PARP1 activity, Huntington's disease exhibits a unique profile with reduced PAR levels and impaired PARP1 activity, even in prodromal stages [3]. This bidirectional dysregulation suggests that both overactivation and suppression of PARP1 can lead to neuronal dysfunction, highlighting the importance of balanced PARylation homeostasis.
The connection between PARP trapping and neurodegenerative processes emerges through several mechanisms:
- PARP overactivation consumes cellular NAD+ pools, leading to energy depletion and necrotic cell death [3] [48]
- PAR polymer itself can stimulate mitochondrial release of apoptosis-inducing factor (AIF), triggering caspase-independent cell death [48]
- Aberrant PARP1 activation contributes to neuroinflammatory gene expression through regulation of transcription factors like NF-κB [3]
Understanding the distinctions between catalytic inhibition and PARP trapping provides a framework for developing targeted therapeutic strategies for neurodegenerative conditions where PARP1 dysfunction contributes to pathology.
The distinction between PARP catalytic inhibition and trapping represents a fundamental advance in understanding PARP inhibitor mechanisms and their therapeutic applications. The kinetic and allosteric principles governing PARP trapping explain why inhibitors with similar enzymatic inhibition profiles exhibit markedly different cellular efficacies and therapeutic indices. The emerging role of PARP1 in preventing transcription-replication conflicts further expands the mechanistic landscape beyond traditional trapping models.
Future research directions should focus on several key areas:
- Elucidating the structural determinants of reverse allostery to guide development of next-generation PARPi with optimized trapping properties
- Investigating cell-type specific variations in PARP trapping consequences, particularly in neuronal contexts relevant to neurodegenerative diseases
- Developing more precise methodologies to distinguish trapped versus catalytically inhibited PARP1 in complex cellular environments
- Exploring the connections between PARP1 cleavage fragments, trapping phenomena, and disease-specific PARylation imbalances
As research continues to unravel the complexities of PARP1 biology, the differentiation between catalytic inhibition and trapping will remain central to therapeutic development across both oncological and neurological contexts.
The blood-brain barrier (BBB) represents one of the most significant obstacles in developing effective treatments for central nervous system (CNS) disorders, including neurodegenerative diseases and primary or metastatic brain tumors [72]. This highly selective cellular barrier sharply restricts paracellular diffusion of polar solutes and actively transports many molecules out of the CNS via efflux transporters such as P-glycoprotein (ABCB1/MDR1) and breast cancer resistance protein (ABCG2) [72]. For Poly(ADP-ribose) polymerase (PARP) inhibitors—a class of drugs that target DNA repair pathways—achieving sufficient brain penetration is particularly crucial for exploiting their potential in treating CNS conditions. The clinical urgency is underscored by the poor prognosis of patients with malignant CNS tumors, where 5-year relative survival for glioblastoma remains only 6.8%, creating a significant unmet need for effective systemic treatments [72]. Within this context, understanding PARP-1 biology, including its cleavage by various proteases into signature fragments, provides critical insights into neurodegenerative pathologies and offers potential biomarkers for monitoring disease progression and therapeutic response [11] [12].
Quantitative Assessment of PARP Inhibitor Brain Penetration
Table 1: Comparative BBB Penetration of PARP Inhibitors in Preclinical Models
| PARP Inhibitor | Experimental Model | Kp, brain | Kp, uu, brain | Brain Concentration | Plasma Concentration | Key Findings |
|---|---|---|---|---|---|---|
| T26 [73] | Mouse intracranial MDA-MB-436 xenograft | N/A | N/A | N/A | N/A | Significant tumor growth inhibition at 0.3 & 3 mg/kg; excellent oral bioavailability (87.74%) |
| Niraparib [72] | Healthy Rhesus Macaques | 3.179 | 0.313 | 378-797 ng/g | 84-436 ng/mL | Distributed across all brain regions; detected in ventricles and choroid plexus |
| Olaparib [72] | Healthy Rhesus Macaques | 0.041 | 0.026 | 11-12 ng/g | 254-322 ng/mL | Brain levels primarily reflect cerebral blood-associated drug; not detected by MALDI-MSI |
| Niraparib [72] | Mouse Metastatic Tumor Model | 0.193 | N/A | 543-658 ng/g | 2535-3847 ng/mL | 5-fold higher Kp, brain than olaparib; accumulated in brain tumor lesions |
| Olaparib [72] | Mouse Metastatic Tumor Model | 0.036 | N/A | 5-6 ng/g | 119-226 ng/mL | Poor BBB penetration; minimal brain accumulation regardless of tumor presence |
Table 2: PARP-1 Cleavage Fragments as Biomarkers in Neurodegeneration
| Cleaving Protease | PARP-1 Fragment Sizes | Biological Significance | Associated Cell Death Pathways | Detection Methods |
|---|---|---|---|---|
| Caspase-3/7 [11] | 89-kD (Catalytic), 24-kD (DBD) | Hallmark of apoptosis; 24-kD fragment acts as trans-dominant inhibitor of PARP-1 | Apoptosis | Western blot, Immunohistochemistry |
| Calpains [74] | Variable (50-62 kD) | Associated with excitotoxic neuronal death | Excitotoxicity, Necrosis | Western blot, Activity assays |
| Cathepsins [11] | Variable | Implicated in lysosomal-mediated cell death | Autophagic cell death | Protease activity assays |
| Granzymes [11] | Variable | Immune-mediated cytotoxicity | Perforin/granzyme-mediated killing | Western blot, Mass spectrometry |
| MMPs [11] | Variable | Associated with inflammatory tissue damage | Inflammation-associated death | Zymography, Western blot |
Methodologies for Assessing BBB Penetration and Neurotoxicity
Parallel Artificial Membrane Permeability Assay (PAMPA-BBB)
The PAMPA-BBB method provides a high-throughput, cost-effective approach for early screening of BBB permeability potential [75]. The stirring Double-Sink PAMPA-BBB technology, patented by Pion Inc., employs a proprietary artificial membrane containing porcine brain lipid extract dissolved in alkane, optimized to predict passive BBB permeability [75].
Experimental Protocol:
- Membrane Preparation: The PAMPA lipid membrane is immobilized on a PVDF matrix of a 96-well "acceptor" filter plate placed on top of a 96-well "donor" plate containing coated magnetic stirrers.
- Compound Preparation: Test compounds are stocked in 10 mM DMSO solutions and diluted to 0.05 mM in aqueous phosphate buffer (final DMSO concentration 0.5%).
- Permeation Study: The assay runs for 60 minutes at room temperature with test samples in the donor compartment stirred using Gutbox technology to reduce the aqueous boundary layer to 60 μm.
- Concentration Measurement: Test article concentrations in both donor and acceptor compartments are measured using a UV plate reader (Nano Quant, Infinite 200 PRO).
- Permeability Calculation: Permeability (Pe) calculations are performed using Pion Inc. software and expressed in units of 10−6 cm/s [75].
Multimodal Imaging for Spatial Drug Distribution
Multimodal imaging approaches, particularly MALDI Mass Spectrometry Imaging (MALDI-MSI) combined with LC-MS/MS bioanalysis, provide comprehensive assessment of spatial distribution and quantitative determination of PARP inhibitors in brain tissue [72].
Experimental Workflow:
- Animal Dosing: NHPs or mice are administered PARP inhibitors (niraparib or olaparib) at clinically relevant doses.
- Tissue Collection: Plasma, cerebrospinal fluid (CSF), and brain tissue are collected at predetermined time points.
- LC-MS/MS Bioanalysis: Bulk drug concentrations in brain homogenates, plasma, and CSF are quantified using validated LC-MS/MS methods.
- MALDI-MSI: Coronal brain tissue sections are prepared and analyzed using MALDI-MSI to visualize spatial distribution of drugs across different brain regions.
- Data Integration: MALDI-MSI data are co-registered with histopathological stains (H&E) to correlate drug distribution with anatomical features [72].
Neurite Outgrowth Inhibition Assay
This assay assesses potential neurotoxicity of BBB-penetrant compounds using GFP-labeled induced pluripotent stem cell (iPSC)-derived human cortical glutamatergic neurons [75].
Protocol Details:
- Cell Culture: GFP-expressing human cortical glutamatergic neurons are thawed with freshly made seeding medium and dispensed at 800 cells/5 μL/well into PDL-coated 1536-well black-wall/clear-bottom assay plates.
- Incubation: Assay plates are incubated at 37°C/5% CO₂ for 42 hours to allow neuronal maturation and neurite outgrowth.
- Compound Treatment: Compounds are added via pintool station with final concentrations ranging from 0.4 nM to 92 μM. Rotenone serves as positive control, DMSO as negative control.
- Imaging and Analysis: After 24-48 hours incubation, fluorescence intensities are measured, and images acquired through High-Content Imaging System (Operetta CLS). Multiple parameters (total neurite length, root number, maximum neurite length) are quantified for neurite outgrowth assessment [75].
Molecular Mechanisms: PARP-1 Cleavage in Neurodegeneration
Diagram 1: PARP-1 Cleavage Pathways in Neurodegeneration
PARP-1 serves as a preferred substrate for several 'suicidal' proteases, and its cleavage generates specific fragments that serve as recognized biomarkers for particular cell death programs [11] [12]. During apoptosis, caspase-3 and caspase-7 cleave PARP-1 into two specific fragments: an 89-kD catalytic fragment containing the auto-modification domain and catalytic domain, and a 24-kD DNA-binding domain (DBD) fragment [11]. The 24-kD fragment retains the zinc-finger motifs and remains tightly bound to damaged DNA in the nucleus, where it acts as a trans-dominant inhibitor of DNA repair by blocking access of additional DNA repair enzymes to strand breaks [11]. This cleavage event is considered a hallmark of apoptosis and has been implicated in multiple neurological diseases, including cerebral ischemia, Alzheimer's disease, and traumatic brain injury [11].
In excitotoxin-induced neuronal death, such as that observed following kainic acid injections in rat visual cortex, PARP-1 cleavage follows a distinct temporal pattern, reaching peak levels 2-3 days after injury and coinciding with increased caspase-3 activity [74]. This delayed cleavage pattern suggests a role for PARP-1 proteolysis in secondary neuronal death mechanisms rather than acute excitotoxic injury. Beyond caspases, PARP-1 is also cleaved by calpains during calcium-mediated excitotoxicity, cathepsins during lysosomal-mediated cell death, granzymes during immune-mediated cytotoxicity, and matrix metalloproteinases (MMPs) in inflammatory tissue damage [11]. Each protease generates distinctive PARP-1 cleavage fragments that can identify specific cell death programs operational in different neurodegenerative contexts.
Research Reagent Solutions for BBB and PARP Studies
Table 3: Essential Research Reagents for BBB Penetration and Neurotoxicity Studies
| Reagent/Cell Line | Supplier | Application | Key Features |
|---|---|---|---|
| PAMPA-BBB Kit | Pion Inc. | BBB permeability screening | Proprietary brain lipid extract; predicts passive BBB penetration |
| GFP-Labeled Human Cortical Glutamatergic Neurons | BrainXell, Inc. | Neurotoxicity assessment | iPSC-derived; express GFP for neurite tracking |
| Primary Human Brain Microvascular Endothelial Cells (BMVEC) | Academic collaborators | BBB in vitro modeling | Isolated from temporal cortex; maintain BBB characteristics |
| PARP Inhibitors (Olaparib, Talazoparib) | Selleck Chemicals | PARP inhibition studies | Clinical-grade inhibitors for translational research |
| Rho GTPase Inhibitors (CT04, NSC23766) | Cytoskeleton Inc. | Cytoskeletal signaling studies | Specifically target RhoA/Rac1 pathways in leukocyte adhesion |
| Selective PARP Inhibitors (AIQ, EB47) | Enzo Life Sciences, Tocris Bioscience | Mechanistic studies | Research-grade inhibitors for pathway analysis |
The development of PARP inhibitors with enhanced BBB penetration represents a promising frontier in neuro-oncology and neurodegenerative disease therapeutics. Recent advances in compound design have yielded molecules like T26, which demonstrates excellent BBB penetrance and significant tumor growth inhibition in intracranial xenograft models [73]. Comparative studies of clinically advanced PARP inhibitors reveal substantial differences in brain penetration capabilities, with niraparib showing superior BBB penetration compared to olaparib across multiple preclinical models [72]. The integration of advanced screening methodologies, including PAMPA-BBB for early permeability assessment and multimodal imaging for spatial distribution analysis, provides robust frameworks for evaluating candidate compounds.
Future directions should focus on identifying predictive biomarkers for PARP inhibitor response, elucidating synergistic effects in combination therapies, addressing resistance mechanisms, and managing potential hematological toxicities [76]. The development of novel PARP inhibitors with improved BBB penetration and enhanced PARP trapping activity, combined with new synergistic treatment approaches, holds significant promise for advancing treatment options for CNS malignancies and neurodegenerative disorders. Continued investigation into PARP-1 cleavage fragments as biomarkers of specific cell death pathways will further enhance our understanding of neurodegenerative processes and facilitate therapeutic monitoring.
Poly(ADP-ribose) polymerase-1 (PARP-1) is a nuclear enzyme that plays a dual role in neuronal survival and death. As a first responder to DNA damage, PARP-1 activation facilitates base excision repair through poly(ADP-ribosyl)ation (PARylation) of nuclear acceptor proteins [77] [11]. However, in neurodegenerative diseases including Alzheimer's disease (AD), Parkinson's disease (PD), and Huntington's disease (HD), excessive DNA damage triggers PARP-1 hyperactivation, culminating in energy depletion and distinct cell death pathways [77] [78]. A critical event in this process is the proteolytic cleavage of PARP-1 by various enzymes, generating fragments with unique biological activities that differentially influence neuronal, astroglial, and microglial responses [11] [39]. These cell-type-specific reactions to PARP-1 cleavage fragments significantly contribute to neuroinflammation, pathological protein aggregation, and ultimately neurodegeneration [77] [79]. Understanding these heterogeneous cellular responses provides crucial insights for developing targeted therapeutic strategies for neurodegenerative diseases.
PARP-1 Cleavage Fragments: Generation and Signatures
PARP-1 is cleaved by specific proteases during different forms of cell death, producing characteristic fragments that serve as biochemical signatures. The table below summarizes the major PARP-1 cleavage fragments, their generating proteases, and their known functions.
Table 1: Major PARP-1 Cleavage Fragments and Their Characteristics
| Fragment Size | Generating Protease(s) | Cell Death Context | Primary Functions and Consequences |
|---|---|---|---|
| 89 kDa + 24 kDa [11] | Caspases-3 and -7 [11] | Apoptosis [11] | |
| ~50 kDa [21] | Cathepsins B and G (Lysosomal proteases) [21] | Necrosis [21] |
|
| PARylated 89 kDa [15] | Caspases-3/7 (with prior PARylation) [15] | Parthanatos [15] |
|
Cell Type-Specific Responses to PARP-1 Cleavage
The cellular response to PARP-1 activation and its cleavage fragments varies significantly among the major cell types of the central nervous system. These differences are driven by distinct receptor expression, metabolic priorities, and innate immune functions.
Neuronal Responses
Neurons, being post-mitotic and highly metabolically active, are particularly vulnerable to PARP-1-mediated energy depletion and the specific toxic effects of its cleavage fragments.
- Parthanatos Execution: Neurons are the primary cell type undergoing PARP-1-dependent parthanatos. The PARylated 89 kDa fragment plays a critical role as a cytoplasmic PAR carrier, triggering AIF release from mitochondria and its translocation to the nucleus, resulting in chromatin condensation and large-scale DNA fragmentation [15] [80].
- Metabolic Catastrophe: PARP-1 hyperactivation depletes neuronal NAD+ and subsequently ATP pools, crippling energy-dependent processes essential for synaptic function and survival [77] [78]. This energy failure is a hallmark of neuronal loss in models of PD and AD.
- Pathological Protein Aggregation: PAR polymers directly interact with and post-translationally modify pathological proteins like α-synuclein. This PARylation accelerates α-synuclein fibril formation and aggregation, creating a toxic feed-forward loop that propagates neurodegeneration in Parkinson's disease models [80].
Astroglial Responses
Astrocytes respond to PARP-1 cleavage products by shifting towards a pro-inflammatory state, which can exacerbate neuronal damage.
- Inflammatory Activation: PARP-1 is a known co-activator of NF-κB-dependent transcription [39]. In astrocytes, this leads to increased expression of pro-inflammatory mediators such as inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) [11] [39]. The expression of the cytotoxic 89 kDa PARP-1 fragment significantly enhances this NF-κB and iNOS promoter activity [39].
- Dysregulated Glutamate Handling: Excessive PARP-1 activation in glial cells, including astrocytes, impairs glutamate uptake, leading to excitotoxic neuronal injuries [78].
Microglial Heterogeneity and Responses
Microglia, the brain's resident immune cells, exhibit high heterogeneity in their response to neurodegeneration, with PARP-1 activation shaping their functional phenotype.
- Phenotypic Switching: Single-cell technologies have revealed that microglia in neurodegenerative diseases adopt multiple reactive states, far beyond the simplistic M1/M2 classification [79]. PARP-1 activation contributes to this shift, promoting a pro-inflammatory phenotype.
- Disease-Associated Microglia (DAM): In Alzheimer's models, a specific microglial state called DAM is identified near Aβ plaques. PARP-1 influences this state by promoting chronic neuroinflammation. These microglia attempt to clear pathological protein aggregates but can become impaired through excessive uptake [78] [79].
- Propagation of Pathology: Activated microglia can contribute to the spread of protein aggregates like α-synuclein and Aβ. PARP-1 activation may exacerbate this process, as microglia can act as a vehicle for propagating protein aggregates, and PARP-1 inhibition has been shown to reduce the cell-to-cell transmission of toxic α-synuclein [79] [80].
Table 2: Summary of Cell Type-Specific Responses to PARP-1 Activation and Cleavage
| Cell Type | Primary Response | Functional Outcome | Contribution to Neurodegeneration |
|---|---|---|---|
| Neurons | Parthanatos, Energy depletion, Pathological protein aggregation | Cell death, Synaptic dysfunction | Direct neuronal loss, Propagation of protein pathology |
| Astrocytes | NF-κB activation, Pro-inflammatory gene expression | Increased NO and prostaglandin production, Dysregulated glutamate handling | Neuroinflammation, Excitotoxicity |
| Microglia | Phenotypic switching to pro-inflammatory states, Phagocytic activation/impairment | Chronic neuroinflammation, Impaired clearance of aggregates | Chronic tissue damage, Propagation of pathology |
Experimental Protocols for Investigating Cell-Type-Specific PARP-1 Biology
Protocol: Assessing PARP-1 Cleavage Fragments by Western Blot
This foundational protocol is used to detect and distinguish between apoptotic and necrotic PARP-1 fragments in cell and tissue lysates.
- Sample Preparation:
- Lyse cells or homogenize tissue samples in RIPA buffer supplemented with protease inhibitors. For suspected necrosis, include inhibitors for lysosomal proteases (e.g., E-64d for cathepsins). For apoptosis, include caspase inhibitors (e.g., z-VAD-fmk) in parallel samples to confirm caspase-dependence [21].
- Quantify protein concentration and normalize samples.
- Gel Electrophoresis and Transfer:
- Load 20-50 μg of protein per lane onto a 7-12% SDS-polyacrylamide gel to resolve proteins from ~20 kDa to 120 kDa.
- Transfer proteins to a PVDF or nitrocellulose membrane.
- Immunoblotting:
- Block membrane with 5% non-fat milk in TBST for 1 hour.
- Incubate with primary antibody overnight at 4°C. Key antibodies include:
- Anti-PARP-1 (full length): Detects full-length ~113 kDa protein.
- Anti-PARP-1 (cleaved): Specific antibodies can detect the apoptotic 89 kDa fragment or the 24 kDa DBD [11].
- Wash membrane and incubate with appropriate HRP-conjugated secondary antibody for 1 hour at room temperature.
- Develop using enhanced chemiluminescence (ECL) substrate.
- Interpretation:
Protocol: Cell-Type-Specific PARP-1 Localization and Activation
This protocol uses immunofluorescence to visualize PARP-1 activation (via PAR) and its fragments in specific neural cell types.
- Cell Culture or Tissue Section Preparation:
- Use primary cultures of neurons, astrocytes, or microglia, or fixed brain tissue sections from disease models.
- For co-localization studies, subject cells/tissue to oxygen/glucose deprivation (OGD) or treat with PARP-1 inducers (e.g., H₂O₂, NO donors) or PARP-1 inhibitors (e.g., Olaparib, Veliparib) [39] [80].
- Immunofluorescence Staining:
- Permeabilize cells/tissue with 0.1% Triton X-100.
- Block with serum from the secondary antibody host.
- Incubate with primary antibodies in the following combinations:
- After washing, incubate with fluorophore-conjugated secondary antibodies (e.g., Alexa Fluor 488, 555, 647).
- Counterstain nuclei with DAPI and mount slides.
- Imaging and Analysis:
- Image using a confocal microscope.
- Analyze co-localization using image analysis software (e.g., ImageJ) to quantify PAR signal intensity specifically within the identified cell types (e.g., Iba1+ microglia vs. MAP2+ neurons).
Signaling Pathways and Molecular Mechanisms
The following diagram illustrates the key cell-type-specific pathways triggered by PARP-1 activation and its cleavage fragments in neurodegeneration.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for PARP-1 and Cell-Type-Specific Studies
| Reagent/Tool | Function and Application | Example Use Case |
|---|---|---|
| PARP Inhibitors (e.g., Veliparib, Olaparib, Talazoparib) [81] [80] | Inhibit PARP-1 catalytic activity; used to probe the role of PARylation in models of neurodegeneration. | Rescuing α-synuclein PFF-mediated PARP activation and cell death in primary neuronal cultures [80]. |
| Anti-PAR Antibody [80] | Detects PAR polymer formation, a direct marker of PARP-1 activation, via Western blot, IF, or ELISA. | Measuring elevated PAR levels in CSF or brain tissues of PD patients and mouse models [80]. |
| Cell-Type-Specific Markers | Identify and isolate specific CNS cell types for downstream analysis. | |
| Caspase Inhibitors (e.g., z-VAD-fmk) [11] [21] | Pan-caspase inhibitor; used to distinguish caspase-dependent apoptosis from other death pathways. | Differentiating apoptotic (caspase-dependent) from necrotic (caspase-independent) PARP-1 cleavage [21]. |
| Primary Cell Cultures (Neurons, Astrocytes, Microglia) [39] [79] | Enable the study of cell-intrinsic responses to PARP-1 activation and cleavage in a controlled environment. | Investigating the differential effects of PARP-1 fragments on NF-κB activation and viability in pure cell populations [39]. |
| PARP-1 Cleavage-Specific Antibodies (e.g., anti-cleaved PARP-1) [11] | Specifically recognize the 89 kDa apoptotic fragment, serving as a definitive apoptosis marker. | Confirming the induction of apoptosis in neuronal cultures treated with DNA-damaging agents [11]. |
From Bench to Biomarker: Validating Fragments and Comparing Therapeutic Strategies
PARP-1 Fragments as Hallmarks of Specific Cell Death Pathways in Human Samples
Poly (ADP-ribose) polymerase-1 (PARP-1) is a nuclear enzyme critically involved in DNA damage response and repair. Beyond its physiological roles, PARP-1 serves as a substrate for various proteases activated in different cell death pathways. The specific proteolytic cleavage fragments generated from these events serve as molecular signatures that distinguish between apoptosis, parthanatos, and other forms of programmed cell death. This technical review comprehensively examines the current understanding of PARP-1 fragments as hallmarks of specific cell death pathways, with particular emphasis on their implications for neurodegenerative disease research and therapeutic development. We provide detailed experimental methodologies for detecting these fragments, summarize key research reagents, and present visual schematics of the underlying molecular mechanisms to serve as a resource for researchers and drug development professionals.
PARP-1 is a multifunctional nuclear enzyme that plays a central role in maintaining genomic stability through its involvement in DNA damage repair. The enzyme consists of three primary functional domains: a DNA-binding domain (DBD) containing two zinc finger motifs, an auto-modification domain (AMD), and a C-terminal catalytic domain (CAT) [82]. Under physiological conditions, PARP-1 facilitates DNA repair through multiple pathways, including base excision repair (BER) and single-strand break repair [35] [3]. However, under conditions of severe cellular stress, PARP-1 becomes a target for proteolytic cleavage by various "suicidal proteases," generating specific fragments that serve as biomarkers for distinct cell death pathways [35].
The cleavage of PARP-1 has emerged as a critical event in cell fate determination, with different proteases generating unique fragment signatures that correspond to specific death programs. Caspases, calpains, cathepsins, granzymes, and matrix metalloproteinases (MMPs) can all cleave PARP-1 at specific sites to produce fragments with characteristic molecular weights and biological activities [35]. These PARP-1 signature fragments are recognized biomarkers for specific patterns of protease activity in unique cell death programs and provide valuable insights into pathological mechanisms, particularly in neurodegenerative diseases where aberrant cell death contributes to disease progression [35] [36].
PARP-1 Domains and Cleavage Sites
Understanding the domain architecture of PARP-1 is fundamental to interpreting the significance of its cleavage fragments. The protein comprises several structurally and functionally distinct domains:
DNA-Binding Domain (DBD): Located at the N-terminus, this domain contains three zinc finger motifs (Zn1, Zn2, and Zn3) that facilitate recognition of and binding to DNA lesions [82]. The Zn1 and Zn2 motifs specifically recognize DNA damage gaps, while Zn3 links structural domains to activate target proteins [82]. This domain also contains a nuclear localization signal (NLS) and an aspartate-glutamate-valine-aspartic acid (DEVD) structure related to apoptosis [82].
Auto-modification Domain (AMD): This central domain contains a BRCT (BRCA1 C terminus) fold that facilitates protein-protein interactions and serves as the target for covalent auto-modification [35] [82]. The AMD is adjacent to the WGR domain, which regulates catalytic activity [81].
Catalytic Domain (CAT): The C-terminal domain contains the active site responsible for poly(ADP-ribose) synthesis [81]. This domain includes an α-helical subdomain (HD) and an ADP-ribosyl transferase (ART) subdomain with the NAD+ binding site and PAR catalytic site [82].
Table 1: PARP-1 Domains and Their Functions
| Domain | Position | Key Features | Function |
|---|---|---|---|
| DNA-Binding Domain (DBD) | N-terminus (1-372) | Three zinc fingers (Zn1, Zn2, Zn3), nuclear localization signal, DEVD motif | Recognizes and binds to DNA lesions |
| Auto-modification Domain (AMD) | Central (373-525) | BRCT fold, WGR domain | Target for auto-modification, protein-protein interactions |
| Catalytic Domain (CAT) | C-terminus (526-1014) | HD and ART subdomains, NAD+ binding site | Catalyzes poly(ADP-ribose) synthesis |
Specific proteases cleave PARP-1 at defined sites within this structure, generating fragments with characteristic sizes and properties that reflect the active cell death pathway and cellular conditions.
PARP-1 Cleavage Fragments as Cell Death Signatures
Caspase-Generated Fragments in Apoptosis
During apoptosis, executioner caspases-3 and -7 cleave PARP-1 at the DEVD²¹⁴↓G²¹⁵ site within the DBD, producing two characteristic fragments: a 24-kDa DNA-binding fragment and an 89-kDa catalytic fragment [35] [15]. The 24-kDa fragment contains the two zinc finger motifs and remains tightly bound to DNA, where it acts as a trans-dominant inhibitor of DNA repair by blocking access to DNA strand breaks for other repair enzymes [35]. The 89-kDa fragment, containing the AMD and CAT domains, has reduced DNA binding capacity and can be liberated from the nucleus into the cytosol [35].
This cleavage event serves as a biochemical hallmark of apoptosis and is thought to prevent futile DNA repair during programmed cell death while conserving cellular ATP pools [35]. Caspase-mediated PARP-1 cleavage has been implicated in numerous neurological conditions, including cerebral ischemia, Alzheimer's disease, multiple sclerosis, Parkinson's disease, traumatic brain injury, NMDA-mediated excitotoxicity, and brain tumors [35].
PARP-1 in Parthanatos
Parthanatos is a caspase-independent programmed cell death pathway distinct from apoptosis and necrosis, characterized by PARP-1 hyperactivation in response to severe DNA damage [24]. Unlike apoptosis, parthanatos involves the translocation of PAR polymers from the nucleus to the cytoplasm, where they trigger mitochondrial release of apoptosis-inducing factor (AIF) [24] [81]. Once released, AIF translocates to the nucleus and induces large-scale DNA fragmentation [24].
Recent research has revealed a fascinating intersection between apoptotic and parthanatos pathways. Studies demonstrate that caspase activation can induce PARP-1 autopoly(ADP-ribosyl)ation and fragmentation, generating poly(ADP-ribosyl)ated 89-kDa and 24-kDa PARP-1 fragments [15]. The 89-kDa fragments with covalently attached PAR polymers translocate to the cytoplasm, where AIF binding to PAR facilitates its translocation to the nucleus [15]. Thus, the 89-kDa PARP-1 fragment can serve as a PAR carrier to the cytoplasm, inducing AIF release from mitochondria and bridging caspase activation with parthanatos execution [15].
Other Protease-Generated Fragments
Beyond caspases, several other proteases cleave PARP-1 to generate signature fragments:
- Calpains: Calcium-activated proteases that generate fragments of 55-kDa and 62-kDa, associated with excitotoxicity and ischemic injury [35].
- Granzymes: Serine proteases delivered by cytotoxic T cells and natural killer cells that generate unique PARP-1 fragments during immune-mediated cell death [35].
- Cathepsins: Lysosomal proteases that contribute to PARP-1 cleavage in specific pathological contexts [35].
- Matrix Metalloproteinases (MMPs): Extracellular matrix proteases that can process PARP-1 in particular microenvironments [35].
Table 2: PARP-1 Cleavage Fragments in Cell Death Pathways
| Protease | Fragments Generated | Cell Death Pathway | Key Features |
|---|---|---|---|
| Caspase-3/7 | 24-kDa (DBD) + 89-kDa (AMD+CAT) | Apoptosis | DEVD²¹⁴↓G²¹⁵ cleavage; 24-kDa fragment inhibits DNA repair |
| Calpain | 55-kDa + 62-kDa | Excitotoxicity, necrosis | Associated with calcium overload |
| PARP-1 hyperactivation | N/A (full-length) | Parthanatos | PAR accumulation, AIF translocation, caspase-independent |
| Caspase + PARP-1 activation | PARylated 89-kDa + 24-kDa | Apoptosis-Parthanatos crossover | 89-kDa fragment serves as cytoplasmic PAR carrier |
Each of these proteolytic events creates a unique molecular signature that can be detected and quantified in human samples to identify the dominant cell death pathway in specific pathological contexts.
Experimental Protocols for PARP-1 Fragment Detection
Immunoblotting for PARP-1 Fragments
Western blot analysis remains the gold standard for detecting and distinguishing PARP-1 fragments based on molecular weight.
Protocol:
- Sample Preparation: Homogenize tissue samples or lyse cells in RIPA buffer supplemented with protease and phosphatase inhibitors. For tissue samples, use a mechanical homogenizer followed by sonication to ensure complete lysis.
- Protein Quantification: Determine protein concentration using BCA or Bradford assay to ensure equal loading.
- Electrophoresis: Separate 20-50 μg of protein on 4-12% Bis-Tris polyacrylamide gels. Include pre-stained molecular weight markers for accurate size determination.
- Membrane Transfer: Transfer proteins to PVDF membranes using wet or semi-dry transfer systems.
- Blocking: Incubate membranes with 5% non-fat dry milk in TBST for 1 hour at room temperature.
- Primary Antibody Incubation: Incubate with anti-PARP-1 antibodies (see Research Reagent Solutions) diluted in blocking buffer overnight at 4°C.
- Secondary Antibody Incubation: Incubate with HRP-conjugated secondary antibodies for 1 hour at room temperature.
- Detection: Develop using enhanced chemiluminescence substrate and image with a digital imaging system.
Key Considerations: Include positive controls (e.g., staurosporine-treated cells for apoptosis, MNNG-treated cells for parthanatos) to ensure proper identification of fragments. Use antibodies targeting different PARP-1 domains (N-terminal for full-length and 24-kDa fragment, C-terminal for full-length and 89-kDa fragment) to confirm fragment identities.
In Situ Fractionation Technique for PARP-1 Localization
This novel technique allows visualization of PARP-1 recruitment to DNA damage sites in vivo by selectively removing unbound nuclear PARP-1 while retaining DNA-bound protein [83].
Protocol:
- Cell Culture and Treatment: Culture cells on glass coverslips and treat with DNA-damaging agents as required.
- Local UV Irradiation: For localized DNA damage, use a 5 μm pore filter to create subnuclear spots of UV damage.
- Extraction: Perform sequential extraction with:
- CSK buffer (10 mM PIPES pH 7.0, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl₂) for 5 minutes
- CSK buffer + 0.5% Triton X-100 for 5 minutes
- CSK buffer + 0.5% Triton X-100 + 0.42 M NaCl for 5 minutes
- Fixation: Fix cells with 4% formaldehyde for 15 minutes.
- Immunostaining: Block with 5% BSA, then incubate with primary antibodies against PARP-1 and DNA damage markers (e.g., DDB2 for UV damage) overnight at 4°C.
- Visualization: Incubate with fluorescent secondary antibodies, counterstain with DAPI, and image by confocal microscopy.
Key Considerations: The C+T+S (CSK+Triton+Salt) protocol offers optimal extraction for visualizing PARP-1 recruitment to DNA damage sites by effectively removing "free" nuclear PARP-1 while retaining chromatin-bound protein [83].
Immunocytochemical Detection of PARP-1 Fragments
For spatial localization of PARP-1 fragments in tissue sections or cultured cells:
Protocol:
- Sample Preparation: Fix cells or tissue sections with 4% paraformaldehyde for 15 minutes at room temperature.
- Permeabilization: Treat with 0.2% Triton X-100 in PBS for 10 minutes.
- Blocking: Incubate with 5% normal serum from the secondary antibody host species for 1 hour.
- Primary Antibody Incubation: Incubate with fragment-specific PARP-1 antibodies overnight at 4°C.
- Secondary Antibody Incubation: Incubate with fluorophore-conjugated secondary antibodies for 1 hour at room temperature.
- Counterstaining: Stain nuclei with DAPI or Hoechst dyes.
- Mounting and Imaging: Mount with anti-fade mounting medium and image using fluorescence or confocal microscopy.
Key Considerations: Include appropriate controls (no primary antibody, isotype controls) to ensure specificity. For co-localization studies with DNA damage markers, use antibodies from different host species with species-specific secondary antibodies.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for PARP-1 Fragment Studies
| Reagent Category | Specific Examples | Function/Application | Key Features |
|---|---|---|---|
| PARP-1 Antibodies | Anti-PARP-1 (N-terminal), Anti-PARP-1 (C-terminal), Anti-cleaved PARP-1 (Asp214) | Detection of full-length and cleaved PARP-1 | Domain-specific antibodies distinguish fragments; cleaved-specific antibodies detect apoptotic fragments |
| Activity Assays | PARP Assay Kit, PAR Detection Kit | Measure PARP enzymatic activity and PAR accumulation | Colorimetric or fluorometric readouts; useful for parthanatos detection |
| Protease Inhibitors | Caspase inhibitors (Z-VAD-FMK), Calpain inhibitors (MDL-28170) | Inhibit specific protease activities to determine involvement in cell death | Selective inhibition helps establish mechanism |
| Cell Death Inducers | Staurosporine (apoptosis), MNNG (parthanatos), H₂O₂ (oxidative stress) | Induce specific cell death pathways for positive controls | MNNG (N-methyl-N′-nitro-N-nitrosoguanidine) is a DNA-alkylating agent that potently initiates parthanatos |
| Model Systems | PARP1 KO cells, AIF KO models, Harlequin mouse (AIF mutant) | Genetic validation of pathways | PARP1 knockout suppresses parthanatos; AIF knockout prevents parthanatos execution |
PARP-1 Fragments in Neurodegenerative Diseases
The detection and quantification of PARP-1 fragments in human samples provides critical insights into the cell death mechanisms operating in neurodegenerative diseases. Different neurodegenerative conditions exhibit distinct PARP-1 activation and cleavage patterns:
Alzheimer's Disease
In Alzheimer's disease, increased PARP-1 activity and PAR accumulation have been observed in brain tissues, fibroblasts, and lymphoblasts from patients [3] [36]. PARP-1 activation contributes to AD pathology through multiple mechanisms, including facilitation of Aβ and tau aggregation, promotion of neuroinflammation, and induction of mitochondrial dysfunction [36] [4]. The detection of caspase-derived PARP-1 fragments in AD brains suggests concurrent apoptotic activation alongside parthanatos-like pathways [35] [36].
Parkinson's Disease
Parkinson's disease demonstrates complex alterations in PAR signaling, with increased PAR levels in cerebrospinal fluid but decreased PAR in fibroblasts [3]. PARP-1 activation in PD models promotes α-synuclein aggregation through PARylation, exacerbating disease pathology [4]. Both caspase-mediated cleavage and parthanatos pathways appear to contribute to dopaminergic neuron loss in PD [24] [35].
Huntington's Disease
Huntington's disease presents a unique profile characterized by reduced PAR levels and impaired PARP-1 activity, even in prodromal stages [3]. This contrasts with other neurodegenerative conditions and suggests that PAR signaling dysregulation, whether through overactivation or suppression, can contribute to neuronal dysfunction [3]. The mechanisms underlying deficient PARP-1 activation in HD remain an active area of investigation.
Amyotrophic Lateral Sclerosis and Ataxias
Amyotrophic lateral sclerosis shows increased PARP-1 expression in spinal cord astroglia but decreased expression in motor neurons, suggesting cell-type-specific regulation [3]. Various forms of cerebellar ataxias, including ataxia with oculomotor apraxia and spinocerebellar ataxia type 7, demonstrate increased PAR levels and PARP-1 activation [3].
Visualization of PARP-1 Cleavage Pathways
Diagram 1: PARP-1 Cleavage in Cell Death Decision. This diagram illustrates how DNA damage severity determines cell fate through PARP-1 activation and cleavage pathways, leading to either apoptosis or parthanatos.
Diagram 2: Molecular Interplay Between Apoptosis and Parthanatos. This diagram details the molecular events following PARP-1 cleavage, showing how the 89-kDa fragment can serve as a PAR carrier to bridge caspase activation with AIF-mediated parthanatos.
PARP-1 cleavage fragments serve as critical biomarkers that distinguish between different cell death pathways in human samples. The specific proteolytic processing of PARP-1 generates unique fragment signatures that reflect the activation of distinct proteases and cellular conditions. Detection of these fragments, particularly through immunoblotting and specialized localization techniques, provides valuable insights into the pathological mechanisms operating in neurodegenerative diseases and other conditions characterized by aberrant cell death.
The intersection between apoptotic and parthanatos pathways, exemplified by the caspase-generated 89-kDa fragment serving as a cytoplasmic PAR carrier, reveals the complexity of cell death regulation and the multifaceted roles of PARP-1 cleavage fragments. As research advances, the detection and characterization of PARP-1 fragments in human samples will continue to provide critical diagnostic and therapeutic insights, particularly for the development of targeted neuroprotective strategies in neurodegenerative diseases.
Poly(ADP-ribose) polymerase-1 (PARP-1) is a nuclear enzyme crucial for DNA damage repair, yet its dysregulation plays a significant role in the pathogenesis of neurodegenerative diseases. Emerging evidence reveals that disease-specific patterns of PARP-1 activation and proteolytic cleavage into signature fragments contribute differentially to neuronal dysfunction across major neurodegenerative disorders. This technical review provides a comprehensive analysis of comparative PARP-1 pathology, highlighting distinct profiles in Alzheimer's disease (AD), Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS), and Huntington's disease (HD). We synthesize quantitative data from human samples and model systems, detail experimental methodologies for detecting PARP-1 fragments, and visualize key signaling pathways. The findings underscore the therapeutic potential of targeting PARP-1 cleavage processes and present essential research tools for investigators in neurodegeneration drug development.
PARP-1 serves as a primary nuclear sensor of DNA damage, coordinating repair through poly(ADP-ribosyl)ation (PARylation) of target proteins. Beyond its repair functions, PARP-1 undergoes proteolytic cleavage by caspases, calpains, and other proteases during cellular stress, generating fragments with distinct biological activities [11]. The 89-kDa fragment retains catalytic activity, while the 24-kDa DNA-binding domain fragment can act as a trans-dominant inhibitor of PARP-1 function [11]. In neurodegenerative diseases, the balance between PARP-1's protective DNA repair functions and its detrimental overactivation appears disrupted in disease-specific patterns. This review systematically compares these pathological signatures across four major neurodegenerative conditions, providing a framework for understanding PARP-1 as a potential therapeutic target and biomarker.
Comparative PARP-1 Pathology Across Neurodegenerative Diseases
Alzheimer's Disease (AD)
In AD, PARP-1 activity is significantly elevated, contributing to the characteristic neuropathology. Postmortem AD brain tissues show increased PARP-1 levels and PARylation compared to controls [3]. This PARP-1 overactivation is linked to multiple AD pathological processes, including the formation of amyloid-β (Aβ) plaques and neurofibrillary tangles [38]. Fibroblasts and lymphoblasts from AD patients similarly demonstrate increased PAR levels, indicating a systemic manifestation of PARP-1 dysregulation [3]. The sustained PARP-1 activation in AD promotes neuroinflammation and contributes to neuronal death through parthanatos, a PARP-1-dependent cell death pathway [38] [34].
Parkinson's Disease (PD)
PD presents a more complex picture of PARP-1 dysregulation, with evidence of both activation and impairment depending on the biological compartment examined. Cerebrospinal fluid (CSF) from PD patients shows increased PAR levels, suggesting elevated PARP-1 activity in the central nervous system [3]. Conversely, fibroblasts from PD patients demonstrate decreased PAR levels, indicating potential tissue-specific differences in PARP-1 regulation [3]. PARP-1 activation in PD models contributes to α-synuclein aggregation, mitochondrial dysfunction, and the loss of dopaminergic neurons in the substantia nigra [38] [60]. This paradoxical pattern highlights the need for compartment-specific analysis when evaluating PARP-1 pathology in PD.
Amyotrophic Lateral Sclerosis (ALS)
ALS exhibits distinct cell-type-specific PARP-1 alterations within the nervous system. In the spinal cord, PARP-1 expression increases in astroglia but decreases in motor neurons, suggesting divergent responses to cellular stress in different neural populations [3]. Overall elevated PAR levels are observed in ALS neural tissues, alongside increased PARP-1 expression in the brain [3]. This PARP-1 overactivation contributes to ALS pathology through multiple mechanisms, including enhanced neuroinflammation, disrupted DNA repair, and promotion of TAR DNA-binding protein-43 (TDP-43) pathology [38].
Huntington's Disease (HD)
HD presents a unique PARP-1 pathology characterized by impaired rather than enhanced activity. Unlike other neurodegenerative conditions, HD patients exhibit reduced PAR levels in CSF, fibroblasts, and induced pluripotent stem cell (iPSC)-derived neurons [3]. Fibroblasts from HD patients show a decreased PAR response to DNA damage, indicating a fundamental defect in PARP-1 activation [3]. This PARP-1 impairment is observed even in the prodromal phase of HD, suggesting it occurs early in disease pathogenesis [3]. The huntingtin protein, mutated in HD, may directly interact with PARP-1, potentially explaining this distinctive pathology [3].
Table 1: PARP-1 Activity Profiles in Human Neurodegenerative Disease Samples
| Disease | Sample Type | PARP-1/PAR Signal | Key References |
|---|---|---|---|
| Alzheimer's Disease | Brain tissue | Increased PARP1 and PAR | [3] |
| Fibroblasts | Increased PAR | [3] | |
| Lymphoblasts | Increased PAR | [3] | |
| Parkinson's Disease | CSF | Increased PAR | [3] |
| Fibroblasts | Decreased PAR | [3] | |
| ALS | Spinal cord astroglia | Increased PARP1 | [3] |
| Spinal cord motor neurons | Decreased PARP1 | [3] | |
| Brain tissue | Increased PARP1 | [3] | |
| Huntington's Disease | CSF | Decreased PAR | [3] |
| Fibroblasts | Decreased PAR response to DNA damage | [3] | |
| iPSC-derived neurons | Decreased PAR | [3] |
Table 2: PARP-1 Cleavage Fragments and Their Functional Consequences
| Fragment | Size | Domains | Functional Consequences | Associated Cell Death Pathways |
|---|---|---|---|---|
| Full-length PARP-1 | 116 kDa | DBD, AMD, CD | DNA damage repair, NAD+ consumption | Survival with mild activation |
| PARP-1 89 kDa | 89 kDa | AMD, CD | Cytotoxic, enhances NF-κB activity, increases iNOS/COX-2 | Apoptosis, parthanatos |
| PARP-1 24 kDa | 24 kDa | DBD | Trans-dominant inhibitor of PARP-1, binds damaged DNA | Apoptosis (inhibition of repair) |
| Caspase-cleaved PARP-1 | 89+24 kDa | Separated domains | Apoptosis biomarker, inhibits DNA repair | Apoptosis |
Experimental Protocols for PARP-1 Fragment Analysis
In Vitro Models of Ischemic Stress (OGD/ROG)
The oxygen/glucose deprivation and restoration of oxygen/glucose (OGD/ROG) model effectively mimics ischemic stress relevant to neurodegenerative pathology [29]. For this protocol, human neuroblastoma cells (SH-SY5Y) or primary rat cortical neurons are cultured under standard conditions. SH-SY5Y cells are maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum, 100 U/ml penicillin, and 100 mg/ml streptomycin at 37°C in 5% CO₂ [29]. Primary cortical neurons are isolated from Sprague-Dawley rats at postnatal day 2 and cultured in Neurobasal Medium-A supplemented with B27 [29].
For OGD exposure, culture medium is replaced with deoxygenated, glucose-free balanced salt solution, and cells are placed in a humidified hypoxia chamber with 85% N₂, 10% H₂, and 5% CO₂ at 37°C for 6 hours [29]. For ROG, the glucose-free medium is replaced with complete medium, and cells are returned to normoxic conditions (95% air, 5% CO₂) for 15-24 hours [29]. PARP-1 cleavage fragments are analyzed by Western blot using antibodies targeting the N-terminal (24 kDa fragment) and C-terminal (89 kDa fragment) regions.
PARP-1 Construct Transfection and siRNA Knockdown
To investigate specific PARP-1 fragments, tetracycline-inducible stable transfectants are generated for PARP-1 wild type (PARP-1WT), uncleavable PARP-1 (PARP-1UNCL), PARP-124 (24 kDa fragment), and PARP-189 (89 kDa fragment) [29]. PARP-1WT plasmid is available from the Harvard Institute of Proteomics (plasmid ID HsCD00040600) [29]. For siRNA-mediated knockdown, cells are transfected with 25 nM PARP-1 siRNA (Target Sequence: 5'-ACGGTGATCGGTAGCAACAAA-3') using Lipofectamine RNAi max [29]. Control experiments utilize scrambled siRNA (AllStars Negative Control siRNA). Transfection efficiency is measured 48-72 hours post-transfection before subjecting cells to experimental conditions.
PARP-1 Cleavage Analysis by Western Blot
Cells are lysed in RIPA buffer supplemented with protease and phosphatase inhibitors. Protein extracts (20-50 μg) are separated by 4-12% Bis-Tris SDS-PAGE and transferred to PVDF membranes [29]. Membranes are blocked with 5% non-fat milk and incubated with primary antibodies against PARP-1 (to detect full-length and fragments), PAR polymer (for PARylation activity), and loading controls (β-actin or GAPDH) overnight at 4°C [29]. After incubation with HRP-conjugated secondary antibodies, signals are detected by enhanced chemiluminescence. The 89 kDa and 24 kDa fragments are quantified densitometrically and normalized to full-length PARP-1.
Immunofluorescence Analysis of PARP-1 Localization
Cells grown on coverslips are fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and blocked with 5% normal goat serum [29]. Primary antibodies against PARP-1 fragments are applied overnight at 4°C, followed by incubation with fluorophore-conjugated secondary antibodies. Nuclei are counterstained with DAPI, and images are captured by confocal microscopy. Subcellular localization of PARP-1 fragments (nuclear vs. cytoplasmic) is analyzed quantitatively across experimental conditions.
Diagram 1: PARP-1 Cleavage and Fragment Signaling Pathway
PARP-1-Dependent Cell Death Mechanisms
Parthanatos
Parthanatos is a programmed cell death pathway distinct from apoptosis, characterized by PARP-1 overactivation [38] [34]. This process begins with severe DNA damage that triggers excessive PARP-1 activation, leading to massive PAR polymer synthesis. The resulting PAR polymers translocate from the nucleus to the cytoplasm, where they bind apoptosis-inducing factor (AIF) in mitochondria [38] [34]. AIF is then released and translocates to the nucleus with macrophage migration inhibitory factor (MIF), where it triggers large-scale DNA fragmentation [34]. Parthanatos is particularly relevant in AD and PD, where oxidative stress and DNA damage are prominent features.
Apoptosis
PARP-1 cleavage by caspases is a hallmark of apoptotic cell death. During apoptosis, executioner caspases-3 and -7 cleave PARP-1 at the DEVD214 site, separating the 24 kDa DNA-binding domain from the 89 kDa catalytic domain [11]. This cleavage inhibits PARP-1's DNA repair activity, conserving cellular ATP for the apoptotic process while preventing DNA repair in doomed cells [11]. The 24 kDa fragment remains bound to DNA breaks, acting as a trans-dominant inhibitor of PARP-1 and other DNA repair enzymes [11].
Necrosis
In severe energy stress conditions, PARP-1 overactivation can lead to necrotic cell death through NAD+ and ATP depletion [38]. As PARP-1 consumes NAD+ for PAR synthesis, NAD+ reservoirs become depleted, disrupting mitochondrial respiration and ATP production [38]. This energy collapse leads to loss of ion homeostasis, cellular swelling, and necrotic death. The breakdown of NAD+ also produces H+, contributing to intracellular acidification that further promotes necrotic pathways [38].
Diagram 2: Experimental Workflow for PARP-1 Fragment Analysis
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for PARP-1 Fragment Studies
| Reagent/Category | Specific Examples | Research Application | Technical Notes |
|---|---|---|---|
| Cell Models | SH-SY5Y human neuroblastoma cells, Primary rat cortical neurons | In vitro neurodegeneration models | Primary neurons better represent physiological responses; SH-SY5Y suitable for high-throughput screens |
| PARP-1 Constructs | PARP-1WT, PARP-1UNCL (uncleavable), PARP-124, PARP-189 | Functional analysis of specific fragments | Tetracycline-inducible systems provide temporal control; AAV vectors enable neuronal transduction |
| PARP-1 siRNA | Target sequence: 5'-ACGGTGATCGGTAGCAACAAA-3' | PARP-1 knockdown studies | Use scrambled siRNA as negative control; transfect with Lipofectamine RNAi max |
| PARP Inhibitors | PJ-34, 3-aminobenzamide, Olaparib | Therapeutic mechanism studies | PJ-34 widely used in preclinical models; concentrations typically 1-10 μM |
| Antibodies | Anti-PARP-1 (full length), Anti-PARP-1 (cleaved fragments), Anti-PAR | Detection by Western blot, immunofluorescence | Validate antibodies for specific fragment recognition; anti-PAR for activity assessment |
| Activity Assays | PAR ELISA kits, NAD+ detection assays | PARP-1 functional assessment | Correlate PAR levels with NAD+ depletion for complete activity profile |
| Cell Death Assays | LDH release, MTT, Annexin V/PI staining | Viability and death mechanism analysis | Combine with PARP-1 cleavage analysis to link fragments to specific death pathways |
Discussion and Therapeutic Implications
The comparative analysis of PARP-1 pathology across neurodegenerative diseases reveals both common themes and disease-specific patterns. While AD, PD, and ALS generally exhibit PARP-1 overactivation, HD presents a unique profile of impaired PARP-1 function [3]. This dichotomy suggests that therapeutic strategies must be disease-specific, with PARP inhibition potentially beneficial in AD, PD, and ALS, while PARP activation might be more appropriate for HD [3] [38].
The proteolytic cleavage of PARP-1 into specific fragments represents a critical regulatory mechanism across these conditions. The 89 kDa fragment appears predominantly cytotoxic, enhancing inflammatory responses through NF-κB activation and increasing expression of pro-inflammatory mediators like iNOS and COX-2 [29]. In contrast, the 24 kDa fragment may serve adaptive functions in some contexts by conserving cellular energy during apoptosis [11].
Several PARP inhibitors show promise in preclinical models of neurodegeneration. First-generation inhibitors like 3-aminobenzamide and second-generation compounds like PJ-34 have demonstrated neuroprotective effects in models of AD, PD, and ALS [38] [77]. Third-generation FDA-approved PARP inhibitors, including olaparib and niraparib, originally developed for cancer therapy, are now being investigated for neurodegenerative applications [38] [60]. These inhibitors may be particularly valuable for mitigating the neurotoxic effects of chemotherapy, which shares mechanistic similarities with neurodegeneration [60].
Future research should focus on developing more sensitive detection methods for PARP-1 fragments in biological fluids, establishing fragment profiles as biomarkers for disease progression, and designing therapeutic strategies that specifically target the detrimental fragments while preserving PARP-1's beneficial DNA repair functions. The distinct PARP-1 pathology in HD warrants particular attention, as it challenges the prevailing paradigm of PARP-1 overactivation in neurodegeneration and suggests a more complex relationship between PAR signaling and neuronal survival.
PARP-1 and its cleavage fragments represent important players in the pathological landscape of neurodegenerative diseases, with distinct profiles emerging across AD, PD, ALS, and HD. The disease-specific patterns of PARP-1 activation and fragment formation highlight the complex, dual nature of this enzyme in neuronal homeostasis and dysfunction. The experimental methodologies and research tools outlined in this review provide a foundation for further investigation into PARP-1-targeted therapeutic strategies. As our understanding of PARP-1 fragment biology advances, these insights may yield novel diagnostic approaches and treatment modalities for these currently incurable conditions.
The development of Poly (ADP-ribose) polymerase (PARP) inhibitors represents a landmark achievement in precision oncology, demonstrating the profound therapeutic potential of targeting DNA damage response pathways. These agents, including olaparib and rucaparib, exploit synthetic lethality in homologous recombination-deficient cancers, offering a powerful therapeutic strategy validated through extensive clinical trials. This whitepaper delineates the core mechanisms of PARP inhibition, summarizes clinical validation data, and provides detailed experimental methodologies. Furthermore, it explores the critical implications of PARP-1 biology, including its proteolytic cleavage, for ongoing research in neurodegenerative diseases, where PARP-1 overactivation is increasingly recognized as a key pathological driver.
PARP enzymes, particularly PARP-1, function as critical DNA damage sensors and facilitate DNA repair through multiple pathways, primarily base excision repair (BER) for single-strand breaks (SSBs) [67]. The synthetic lethality between PARP inhibition and deficiencies in homologous recombination (HR) repair, such as in BRCA1/2 mutated tumors, provides the fundamental rationale for their cancer specificity [67] [84]. PARP inhibitors (PARPi) trap PARP enzymes on DNA, preventing repair and causing replication fork collapse into lethal double-strand breaks (DSBs) in HR-deficient cells [60] [67].
Beyond oncology, PARP-1 overactivation is implicated in neuronal cell death pathways, including parthanatos, a programmed necrosis mechanism characterized by PAR polymer-induced mitochondrial collapse and nuclear translocation of Apoptosis-Inducing Factor (AIF) [38] [34]. PARP-1 is also a known substrate for caspase-3 and other proteases, generating specific cleavage fragments that serve as biomarkers for different cell death programs [11]. Understanding the mechanisms validated in oncology provides a critical framework for investigating PARP-1's role in neurodegenerative diseases and exploring the therapeutic repurposing of PARPi.
Mechanism of Action: Core Pathways and Experimental Validation
Primary Mechanisms of PARP Inhibitors
PARP inhibitors exert their effects through multiple, interconnected mechanisms:
- Catalytic Inhibition: PARPi competitively bind the NAD+ site of PARP's catalytic domain, inhibiting auto-PARylation and the subsequent recruitment of repair proteins like XRCC1 to DNA damage sites. This prevents the repair of single-strand breaks (SSBs) [67].
- PARP Trapping: This clinically dominant mechanism involves the stabilization of PARP-DNA complexes at stalled replication forks, which are more cytotoxic than unrepaired SSBs alone. Different PARPi have varying trapping potencies [85].
- Synthetic Lethality in HR-Deficient Cells: In healthy cells, SSBs that persist into S-phase are repaired via HR. In HR-deficient cells (e.g., BRCA1/2 mutations), these breaks lead to irreparable DSBs, genomic instability, and cell death, while sparing healthy cells with functional HR [67] [84].
Signaling Pathway and Synthetic Lethality
The diagram below illustrates the core mechanism of PARP inhibition and its relationship with homologous recombination repair, leading to synthetic lethality.
PARP-1 Cleavage as a Cell Death Signature
In neurodegenerative contexts, PARP-1 activation and cleavage are critical events. PARP-1 is a primary substrate for "suicidal proteases," and its cleavage generates signature fragments that serve as biomarkers for specific cell death pathways [11].
- Apoptosis: Executioner caspases (e.g., caspase-3 and -7) cleave PARP-1 into a characteristic 89-kD catalytic fragment and a 24-kD DNA-binding fragment (DBD). The 24-kD fragment acts as a trans-dominant inhibitor of BER by irreversibly binding to DNA strand breaks [11].
- Other Proteases: PARP-1 is also cleaved by calpains, cathepsins, granzymes, and matrix metalloproteinases (MMPs) in other cell death programs, producing fragments of distinct molecular weights (e.g., 50-kD, 40-kD) [11].
The detection of these specific fragments in neuronal tissues provides critical evidence for the type of cell death occurring in neurodegenerative pathologies.
Clinical Validation and Quantitative Data
FDA-Approved PARP Inhibitors and Indications
PARP inhibitors are approved for maintenance therapy and treatment of specific cancers with HR deficiencies [86].
Table 1: FDA-Approved PARP Inhibitors and Their Key Indications (as of March 2024) [86]
| Drug Name | Approved Indications (Cancer Types) | Primary Biomarker Requirements |
|---|---|---|
| Olaparib | Ovarian, Breast, Pancreatic, Prostate | Germline or somatic BRCA1/2 mutations; Other HRR gene variants (prostate) |
| Rucaparib | Ovarian, Prostate | Germline or somatic BRCA1/2 mutations; Other HRR gene variants (prostate) |
| Niraparib | Ovarian | Biomarker-independent for maintenance in advanced cancer; gBRCA for recurrent cancer |
| Talazoparib | Breast | Germline BRCA1/2 mutations |
Pivotal trials demonstrate the significant efficacy of PARPi in prolonging progression-free survival (PFS), particularly in patients with BRCA mutations.
Table 2: Selected Clinical Trial Data for PARP Inhibitors in Ovarian Cancer [84]
| Trial | Phase | Eligibility | Arms | Number of Patients | Median PFS (Months) |
|---|---|---|---|---|---|
| SOLO1 | 3 | Newly diagnosed, gBRCAmut | Olaparib Maintenance | 260 | Not Reached (HR=0.30) |
| Placebo | 131 | 13.8 | |||
| SOLO2 | 3 | Platinum-sensitive, gBRCAmut | Olaparib Maintenance | 196 | 19.1 |
| Placebo | 99 | 5.5 | |||
| ARIEL3 | 3 | Platinum-sensitive | Rucaparib Maintenance | 375 | 10.8 |
| Placebo | 189 | 5.4 |
Preclinical Activity in Pediatric Solid Tumors
Preclinical data supports the investigation of PARPi in other malignancies. A study of olaparib in pediatric solid tumor models showed growth inhibition and synergy with DNA-damaging agents [85].
Table 3: In Vitro Growth Inhibition (IC50) of Olaparib in Pediatric Solid Tumor Cell Lines [85]
| Tumor Origin | Cell Line | Olaparib IC50 (μM) Mean (SD) |
|---|---|---|
| Ewing Sarcoma | TC-71 | 1.5 (0.9) |
| Ewing Sarcoma | RD-ES | 1.0 (0.3) |
| Medulloblastoma | HTB-185 | 2.1 (0.9) |
| Medulloblastoma | HTB-186 | 2.4 (0.7) |
| Neuroblastoma | SK-NAS | 33.8 (8.7) |
| Neuroblastoma | NGP | 2.5 (0.8) |
| Rhabdomyosarcoma | SJCRH-30 | 3.6 (1.2) |
Experimental Protocols and Research Toolkit
This section outlines key methodologies for validating PARP inhibitor mechanisms and detecting PARP-1 cleavage in research.
Protocol: In Vitro Cytotoxicity and Combination Synergy Assay
This protocol is adapted from studies evaluating olaparib in pediatric solid tumor cell lines [85].
- Cell Seeding: Plate cells in 96-well plates at a density determined by growth kinetics (e.g., 3,000-5,000 cells/well) and allow to adhere overnight.
- Drug Treatment:
- Prepare serial dilutions of olaparib, rucaparib, and cytotoxic chemotherapies (e.g., temozolomide, topotecan) in complete medium.
- Treat cells with single agents or fixed-ratio combinations for a defined period (e.g., 72-96 hours).
- Viability Assessment: Use a standardized cell viability assay (e.g., MTT, CellTiter-Glo).
- Data Analysis:
- Calculate IC50 values for single agents using non-linear regression.
- Analyze drug interactions using the Chou-Talalay Combination Index (CI) method. CI < 1 indicates synergy, CI = 1 additivity, and CI > 1 antagonism.
Protocol: Immunoblotting for PARP-1 Cleavage Fragments
This method is critical for detecting apoptosis and other protease-mediated PARP-1 cleavage in cellular and tissue samples [11].
- Sample Preparation: Lyse cells or homogenize tissue samples in RIPA buffer supplemented with protease and phosphatase inhibitors. Quantify protein concentration.
- Gel Electrophoresis: Load 20-50 μg of protein per lane onto a 7-12% SDS-PAGE gel and separate proteins by molecular weight.
- Membrane Transfer: Transfer proteins from the gel to a PVDF or nitrocellulose membrane.
- Immunoblotting:
- Blocking: Incubate membrane in 5% non-fat milk in TBST for 1 hour.
- Primary Antibody Incubation: Incubate with anti-PARP-1 antibody overnight at 4°C.
- Key Cleavage Signatures:
- Apoptosis: Probe for loss of full-length PARP-1 (116-kD) and appearance of the 89-kD fragment.
- Other Cell Death: Antibodies targeting specific neo-epitopes can detect other fragments (e.g., 50-kD, 40-kD).
- Key Cleavage Signatures:
- Washing: Wash membrane 3x with TBST.
- Secondary Antibody Incubation: Incubate with HRP-conjugated secondary antibody for 1 hour at room temperature.
- Detection: Use enhanced chemiluminescence (ECL) substrate and visualize bands using a digital imager. Use a loading control (e.g., GAPDH, β-Actin) for normalization.
Protocol: In Vivo PARP Inhibition and Efficacy Study
This protocol is based on xenograft models used to evaluate olaparib [85].
- Xenograft Establishment: Subcutaneously implant human cancer cells (e.g., Ewing sarcoma, neuroblastoma) into immunodeficient mice.
- Randomization and Dosing: When tumors reach a predefined volume (e.g., 150-200 mm³), randomize mice into treatment groups:
- Vehicle control
- PARPi alone (e.g., olaparib 50 mg/kg orally, daily)
- Chemotherapy alone (e.g., cyclophosphamide/topotecan)
- PARPi + Chemotherapy combination
- Monitoring:
- Measure tumor dimensions 2-3 times weekly to calculate volume.
- Monitor animal body weight as an indicator of toxicity.
- Endpoint Analysis:
- At study endpoint, harvest tumors and snap-freeze for pharmacodynamic analysis.
- Assess PARP activity in tumor lysates using a PAR ELISA to confirm target engagement.
The Scientist's Toolkit: Key Research Reagents
Table 4: Essential Reagents for PARP Inhibition and Cleavage Studies
| Reagent / Assay | Function / Application | Key Details |
|---|---|---|
| PARP Inhibitors (Olaparib, Rucaparib) | Tool compounds for in vitro and in vivo mechanistic studies. | Varying potencies for catalytic inhibition vs. PARP trapping [85]. |
| Anti-PARP-1 Antibody | Detection of full-length and cleaved PARP-1 by immunoblotting. | Critical for identifying the 89-kD apoptotic fragment and other signature fragments [11]. |
| PAR ELISA Kit | Quantification of poly(ADP-ribose) levels in cell/tissue lysates. | Gold standard for measuring pharmacodynamic PARP inhibition in vivo [85]. |
| Caspase-3/7 Assay | Fluorometric or luminescent measurement of caspase activity. | Confirms apoptotic activation concurrent with PARP-1 cleavage [11]. |
| HR-Deficient Cell Lines | Models for synthetic lethality (e.g., BRCA1/2 mutant). | Include isogenic HR-proficient pairs for controlled experiments. |
| Chou-Talalay Assay Software | Statistical analysis of drug combination effects (e.g., CompuSyn). | Quantifies synergy (Combination Index < 1) [85]. |
PARP-1 in Neurodegeneration: Connecting Cleavage to Pathogenesis
The mechanistic insights from oncology directly illuminate PARP-1's role in neurodegenerative diseases like Alzheimer's (AD), Parkinson's (PD), and Huntington's (HD).
PARP-1 Overactivation and Parthanatos
In neurodegeneration, persistent oxidative stress and DNA damage lead to PARP-1 overactivation. This depletes cellular NAD+ and ATP pools, leading to energy failure and triggering parthanatos [38] [34]. This cell death pathway is distinct from apoptosis and involves:
- Massive PAR polymer synthesis.
- PAR translocation to the mitochondria.
- Release and nuclear translocation of AIF.
- AIF/MIF-mediated large-scale DNA fragmentation [38] [34].
PARP-1 Cleavage as a Diagnostic and Mechanistic Biomarker
The cleavage of PARP-1 by different proteases creates a molecular signature of the dominant cell death pathway active in neuronal tissue.
- The presence of the caspase-3-derived 89-kD fragment indicates concurrent apoptosis [11].
- Cleavage by other proteases like calpain suggests alternative cell death mechanisms relevant in excitotoxicity and ischemia [11].
- Detecting these fragments in cerebrospinal fluid (CSF) or post-mortem brain tissue (e.g., in AD, PD, ALS) can provide insights into disease progression and the balance between different neuronal death pathways [11] [38].
Experimental Workflow for PARP-1 Analysis
The following diagram outlines a logical workflow for analyzing PARP-1 activation and cleavage in a research context, such as a neurodegenerative disease model.
The validation of PARP inhibitor mechanisms in oncology provides a robust foundation for exploring their application in neurodegenerative diseases. The concepts of synthetic lethality, PARP trapping, and biomarker-driven therapy are now being translated to neurological research. Critical to this effort is the understanding of PARP-1 cleavage fragments as specific signatures of active cell death pathways. Future work should focus on detecting these fragments in patient biofluids, developing more brain-penetrant PARP inhibitors, and designing clinical trials that target PARP-1 overactivation in defined neurodegenerative populations. The lessons from oncology underscore the power of targeting DNA repair pathways and offer a promising strategic framework for confronting the challenge of neuronal cell death.
Poly (ADP-ribose) polymerase-1 (PARP-1) is a nuclear enzyme central to maintaining genome integrity through its role in DNA damage repair [35]. Beyond this fundamental function, PARP-1 has emerged as a critical sensor of cellular stress and a key executioner in various cell death pathways relevant to neurodegenerative diseases [35] [87]. The proteolytic cleavage of PARP-1 by a specific set of 'suicidal' proteases generates stable, characteristic fragments that serve as molecular signatures of ongoing pathological processes [35] [12]. These fragments are not merely inactive degradation products but often possess distinct biological activities that can actively influence cell fate decisions. This technical review examines the biomarker potential of PARP-1 cleavage fragments, focusing specifically on correlating their presence and abundance with disease stage and progression trajectories in neurodegenerative conditions. The emerging understanding of these relationships provides researchers and drug development professionals with valuable tools for diagnosing disease stages, monitoring progression, and evaluating therapeutic efficacy in clinical and preclinical settings.
PARP-1 Cleavage Fragments: Protease-Specific Signatures in Cell Death
PARP-1 serves as a preferred substrate for several proteases activated in distinct cell death pathways, with each protease generating characteristic cleavage fragments that can be detected using specific antibodies and molecular techniques [35].
Major Proteases and Their Characteristic PARP-1 Fragments
Table 1: Protease-specific PARP-1 cleavage fragments and their characteristics
| Protease | Cleavage Fragments | Cell Death Pathway | Primary Detection Methods |
|---|---|---|---|
| Caspase-3/7 | 89 kDa (AMD + Catalytic Domain) and 24 kDa (DBD) | Apoptosis | Western Blot (anti-PARP-1 antibody) |
| Calpain | 55-62 kDa fragments | Necrosis, Excitotoxicity | Western Blot, Immunohistochemistry |
| Cathepsins | Varied fragments (e.g., 50 kDa) | Lysosomal-mediated death | Activity-based probes, Western Blot |
| Granzyme A | 50 kDa fragment | Immune-mediated cytotoxicity | Western Blot, Mass Spectrometry |
| MMPs | Varied fragments | Inflammation-associated death | Zymography, Western Blot |
Caspase-3/7-mediated cleavage represents the most extensively characterized PARP-1 proteolytic event, generating an 89-kDa fragment containing the auto-modification and catalytic domains, and a 24-kDa DNA-binding fragment [35]. This cleavage event is considered a biochemical hallmark of apoptosis and serves to inactivate DNA repair capacity while conserving cellular ATP [35]. The 24-kDa fragment remains nuclear-localized and acts as a trans-dominant inhibitor of BER by irreversibly binding to DNA strand breaks, thereby preventing recruitment of functional repair complexes [35].
Calpain-mediated cleavage generates distinct 55-62 kDa fragments typically associated with non-apoptotic cell death pathways, including necrosis and excitotoxicity, which are particularly relevant in acute neuronal injury and chronic neurodegeneration [35]. The differential fragment sizes result from calpain's recognition of distinct cleavage sites within the PARP-1 structure compared to caspases.
Other proteases including cathepsins, granzymes, and matrix metalloproteinases (MMPs) also target PARP-1, generating unique signature fragments that provide a molecular readout of the specific cell death program activated [35]. These cleavage events often occur in disease-specific contexts, such as granzyme-mediated cleavage in immune-mediated neurodegeneration and cathepsin-mediated cleavage in lysosomal dysfunction pathologies.
PARP-1 Domains and Cleavage Fragment Functions
Table 2: Functional domains of PARP-1 and their roles in cleavage fragments
| Domain | Size | Function | Location in Fragments |
|---|---|---|---|
| DNA-Binding Domain (DBD) | 46 kDa | Contains two zinc finger motifs; binds DNA damage | 24 kDa caspase fragment |
| Auto-Modification Domain (AMD) | 22 kDa | Target for poly(ADP-ribosyl)ation; protein interactions | 89 kDa caspase fragment |
| Catalytic Domain (CD) | 54 kDa | Polymerizes ADP-ribose units from NAD+ | 89 kDa caspase fragment |
| Third Zinc Finger | - | Facilitates inter-domain interactions; enzymatic regulation | Varies by cleavage site |
The 24-kDa DBD fragment generated by caspase cleavage deserves particular attention as a potential biomarker. This fragment retains the ability to bind DNA damage sites with high affinity but lacks catalytic function, effectively acting as a dominant-negative inhibitor of both PARP-1 and DNA repair processes [35]. Its nuclear retention and stability make it an excellent indicator of prior or ongoing apoptotic activity in neuronal populations.
Quantitative Correlations in Neurodegenerative Diseases
The presence and abundance of specific PARP-1 cleavage fragments show distinct patterns across neurodegenerative conditions, reflecting differences in underlying disease mechanisms and progression timelines.
Disease-Specific PARP-1 Cleavage Patterns
Table 3: PARP-1 cleavage patterns across neurodegenerative diseases
| Disease | Observed PARP-1 Changes | Associated Fragments | Correlation with Disease Stage |
|---|---|---|---|
| Alzheimer's Disease | Increased PARP1 and PAR levels; decreased nucleolar PARP-1 in hippocampus [3] [88] | Caspase-derived fragments (89 kDa) [35] | Nucleolar PARP-1 decreases in MCI stage [88] |
| Parkinson's Disease | Elevated PAR in CSF; PARP-1 activation in PARthanatos [3] [87] | Caspase and calpain fragments [87] | Correlates with motor deficit severity in models [87] |
| Huntington's Disease | Reduced PAR levels; impaired PARP1 activity in prodromal phase [3] | Altered cleavage pattern | Present even in pre-symptomatic stages [3] |
| Amyotrophic Lateral Sclerosis | Increased PARP1 in spinal cord astroglia; decreased in motor neurons [3] | Cell-type specific fragmentation | Varies by cell type and disease stage [3] |
| Cerebellar Ataxias | Increased PAR levels [3] | Caspase-derived fragments | Correlates with neuronal loss [3] |
In Alzheimer's disease, research has demonstrated a significant decrease of nucleolar PARP-1 in hippocampal pyramidal cells in both MCI and AD cases, with the most pronounced decrease observed in MCI cases in the CA1 region [88]. This surprising finding suggests that nucleolar PARP-1 displacement occurs early in disease progression, potentially contributing to cognitive impairment through disrupted ribosome biogenesis before widespread neuronal loss occurs [88]. Concurrently, overall PAR levels and caspase-mediated PARP-1 cleavage increase as the disease progresses, indicating a complex regulation of PARP-1 function across different cellular compartments [3].
In Parkinson's disease, PARP-1 plays a central role in PARthanatos, a programmed cell death pathway distinct from apoptosis [87]. In this pathway, extensive DNA damage triggers hyperactivation of PARP-1, leading to NAD+ and ATP depletion, followed by apoptosis-inducing factor (AIF) release from mitochondria and translocation to the nucleus, where it triggers chromatin condensation and large-scale DNA fragmentation [87]. The correlation between PARP-1 activation, AIF nuclear translocation, and motor deficits in MPTP models underscores the potential of PARP-1 cleavage fragments as biomarkers for disease progression [87].
Huntington's disease presents a unique pattern, with consistently observed reductions in PAR levels and impaired PARP-1 activity, even during prodromal stages [3]. This contrasts with most other neurodegenerative conditions where PARP-1 overactivation is typically observed, suggesting that the balance of PAR signaling—whether excessive or insufficient—may be critical for neuronal health [3].
Experimental Protocols for Detection and Quantification
Standardized Western Blot Protocol for PARP-1 Fragment Detection
Sample Preparation: Cells or tissue samples should be lysed using IP lysis buffer (0.25% Sodium deoxycholate, 50 mM Tris-HCL pH 7.4, 1 mM EDTA, 1% Triton X-100, 1% NP40, and 150 mM NaCl) containing protease inhibitor cocktails [89]. Lysates should be centrifuged at 13,500 rpm for 20 minutes at 4°C to collect supernatant containing soluble proteins [89].
Electrophoresis and Transfer: Load 20-50 μg of protein per lane on 4-12% Bis-Tris polyacrylamide gels for optimal separation of PARP-1 fragments. Transfer to PVDF membranes using standard wet or semi-dry transfer systems.
Antibody Detection: Incubate with primary antibodies against PARP-1 (recommended: #9532, Cell Signaling Technology) at appropriate dilutions (typically 1:1000) [89]. Anti-PAR antibodies (e.g., #83732, CST) can be used in parallel to detect PAR polymer accumulation [89]. Species-appropriate HRP-conjugated secondary antibodies should be used for chemiluminescent detection.
Quantification: Densitometric analysis of full-length PARP-1 (116 kDa) and cleavage fragments (89 kDa for caspase-derived) should be performed using image analysis software. The ratio of cleaved to full-length PARP-1 provides a quantitative measure of protease activity.
Immunohistochemical Protocol for Tissue Localization
Tissue Preparation: Formalin-fixed, paraffin-embedded tissue sections should be deparaffinized with xylene and rehydrated through graded ethanol series [88].
Antigen Retrieval: Microwave irradiation in 10 mM citrate buffer (pH 6.0) for 15 minutes effectively retrieves PARP-1 epitopes [88].
Staining Procedure: Incubate with PARP-1 monoclonal antibody (1:200 dilution; Cat # 1522G, AbD Serotec) followed by biotinylated secondary antibody (horse anti-mouse, 1:200, Vector Laboratories) [88]. Develop using ABC system (Vector Laboratories) and appropriate chromogens.
Scoring Method: For nucleolar PARP-1 assessment, score cells for presence or absence of PARP-1 in the nucleolus across hippocampal subfields (CA1-CA4) [88]. Nucleolar diameter measurements can provide additional quantitative parameters.
Figure 1: PARP-1 Cleavage Fragment Generation and Detection Pathway
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key research reagents for PARP-1 fragment studies
| Reagent Category | Specific Examples | Application | Technical Notes |
|---|---|---|---|
| PARP-1 Antibodies | #9532 (CST), 13371-1-AP (Proteintech) | Western Blot, IHC, Immunofluorescence | Validate for fragment detection |
| PAR Antibodies | #83732 (CST) | Detection of PAR polymer formation | Indicator of PARP-1 activity |
| USP10 Antibodies | #8501 (CST), sc-365828 (Santa Cruz) | Studying PARP-1 stabilization | Co-immunoprecipitation applications |
| PARP Inhibitors | Olaparib (Beyotime, SC9118) | Functional studies of PARP-1 | Therapeutic and experimental use |
| Protease Inhibitors | Caspase inhibitors (Z-VAD-FMK), Calpain inhibitors | Pathway manipulation | Determine protease specificity |
| Activity Assays | PARP-1 Activity Assay Kits | Quantitative enzymatic activity | Measure catalytic function |
| Cell Death Assays | MTT, LDH release, Annexin V | Correlation with viability | Link fragments to cell fate |
Experimental Considerations: When designing studies to investigate PARP-1 cleavage fragments, researchers should consider several technical aspects. First, the subcellular localization of fragments impacts their detection—nuclear extraction protocols may enhance detection of the 24-kDa DBD fragment [35]. Second, the timing of sample collection is critical, as cleavage fragments may be transient in acute models but accumulate in chronic conditions [35] [87]. Third, species-specific antibody reactivity should be validated, particularly when working with animal models of neurodegeneration.
Advanced Techniques: For comprehensive characterization of PARP-1 cleavage events, liquid chromatography-tandem mass spectrometry (LC-MS/MS) following immunoprecipitation provides unbiased identification of cleavage sites and fragment-specific post-translational modifications [89]. Proximity Ligation Assays (PLA) can visualize interactions between PARP-1 fragments and their binding partners in fixed tissues, providing spatial context to fragment biology [89].
The systematic characterization of PARP-1 cleavage fragments and their correlation with disease stages provides a powerful framework for understanding and quantifying neurodegenerative disease progression. The distinct protease-specific fragments serve as molecular footprints of the specific cell death pathways activated in different disease contexts and stages. As research advances, the integration of fragment detection with other biomarkers of neurodegeneration—including α-synuclein aggregates in Parkinson's disease, Aβ and tau in Alzheimer's disease, and emerging markers of neuroinflammation—will enable more comprehensive molecular staging of disease progression. For drug development professionals, PARP-1 cleavage fragments offer promising pharmacodynamic biomarkers for evaluating target engagement and treatment efficacy, particularly for therapeutics targeting specific cell death pathways. The ongoing development of standardized detection protocols and validated reagent systems will be essential for translating these research tools into clinically applicable biomarkers for the next generation of neurodegenerative disease therapeutics.
The hyperactivation of poly (ADP-ribose) polymerase-1 (PARP-1) and the subsequent generation of specific PARP-1 cleavage fragments are established hallmarks of cellular stress responses in neurodegenerative diseases. While PARP-1's primary role involves DNA damage repair, its dysregulation triggers multiple pathogenic cascades, including persistent neuroinflammation, mitochondrial dysfunction, and distinct forms of regulated cell death such as parthanatos. This whitepaper explores the therapeutic rationale for combining PARP inhibitors (PARPi) with anti-inflammatory agents and DNA repair-targeting strategies. We detail the underlying molecular mechanisms, present structured quantitative data, and provide validated experimental protocols for evaluating these synergistic approaches. The content is framed within the context of neurodegenerative disease research, offering drug development professionals a comprehensive technical guide for advancing novel combination therapies.
PARP-1 is a nuclear enzyme that functions as a primary sensor of DNA damage. Upon activation by DNA strand breaks, it catalyzes the transfer of ADP-ribose polymers (PAR) onto target proteins, facilitating DNA repair [11]. In neurological diseases, however, excessive PARP-1 activation depletes cellular NAD+ and ATP pools, leading to energy failure and a specialized form of programmed cell death known as parthanatos [34]. This cell death cascade involves the accumulation of PAR polymers, binding to apoptosis-inducing factor (AIF), and its translocation from mitochondria to the nucleus, resulting in large-scale DNA fragmentation [34].
Critically, PARP-1 is also a substrate for several 'suicidal' proteases, including caspases, calpains, and cathepsins. The proteolytic cleavage of PARP-1 produces specific signature fragments that serve as biomarkers for particular protease activities and cell death programs [11]. For instance, caspase-3 cleavage generates a characteristic 89-kD catalytic fragment and a 24-kD DNA-binding domain (DBD) fragment. The 24-kD fragment remains bound to damaged DNA, acting as a trans-dominant inhibitor of further PARP-1 activity and thereby conserving cellular ATP during apoptosis [11]. In neurodegenerative contexts, the persistence of these fragments contributes to the pathophysiological signature, offering both diagnostic and therapeutic insights.
Beyond direct DNA repair, PARP-1 activation influences a substantial portion of the transcriptome and modulates key transcription factors like NF-κB, thereby regulating the expression of pro-inflammatory cytokines and adhesion molecules [11]. This establishes a critical link between DNA damage, PARP-1 activation, and chronic neuroinflammation—a common feature across neurodegenerative conditions, including Alzheimer's disease (AD), Parkinson's disease (PD), and amyotrophic lateral sclerosis (ALS).
Table 1: PARP-1 Cleavage Fragments as Cell Death Signatures
| Protease | PARP-1 Fragment Sizes | Associated Cell Death Process | Functional Consequences |
|---|---|---|---|
| Caspase-3/-7 | 89-kD (Catalytic), 24-kD (DBD) | Apoptosis | 24-kD fragment irreversibly binds DNA, inhibiting repair; conserves ATP [11] |
| Calpain | 55-kD, 62-kD, etc. | Excitotoxicity, Necrosis | Fragments lack defined function; serve as disease biomarkers [11] |
| Cathepsins | Varies | Lysosomal-mediated death | Contributes to proteolytic signature in unique death programs [11] |
| Granzyme A | ~50-kD (N-terminal) | Immune-mediated cytotoxicity | Induces caspase-independent cell death [11] |
Molecular Rationale for Combination Therapy
PARP Inhibition and Neuroinflammation
PARP-1 activation and PAR polymer accumulation directly exacerbate neuroinflammation. Activated PARP-1 can promote the expression of pro-inflammatory cytokines and chemokines, such as TNF-α, IL-1β, IL-6, and CCL2, via its interaction with NF-κB [11] [60]. This creates a vicious cycle in the neurodegenerative microenvironment, where inflammation induces DNA damage, leading to further PARP-1 activation. In PD models, PARP-1 activation was shown to be critically involved in a feed-forward loop that drives pathologic α-synuclein neurodegeneration [80]. The resulting PAR polymers directly modify α-synuclein, accelerating its aggregation and cell-to-cell transmission [80]. Combining PARPi with anti-inflammatory agents thus targets a core pathogenic feedback loop.
PARP Inhibition and DNA Repair Targeting
The concept of synthetic lethality with PARPi is well-established in oncology, particularly in BRCA-mutated cancers, where inhibiting the base excision repair (BER) pathway in cells already deficient in homologous recombination repair (HRR) leads to cell death [90]. While not classically synthetic lethal in neurodegeneration, a analogous principle applies. Neurons are post-mitotic cells with high metabolic rates and are particularly vulnerable to persistent DNA damage. Combining PARPi with agents that induce low levels of DNA stress or target alternative repair pathways can selectively enhance death of vulnerable, stressed neurons while preserving healthy ones, a strategy that requires careful calibration.
Mitochondrial Dysfunction and Energetic Crisis
PARP-1 hyperactivation consumes NAD+, leading to a depletion of this critical cofactor and a subsequent drop in ATP production [91]. This bioenergetic collapse is a hallmark of parthanatos and contributes significantly to neuronal loss. Research in AD models has shown that increasing NAD+ bioavailability through supplementation with nicotinamide (NAM, a form of vitamin B3) or through PARP inhibition can rescue mitochondrial function, protect against neurodegeneration, and improve behavioral outcomes [91]. Therefore, combining PARPi with mitochondrial protectors or NAD+ precursors represents a compelling synergistic strategy to address the energetic deficit in neurodegenerative brains.
Quantitative Data and Clinical Evidence
The therapeutic potential of PARP inhibition is supported by preclinical data across multiple neurodegenerative disease models. The following table summarizes key findings.
Table 2: Preclinical Evidence for PARP Inhibition in Neurodegeneration
| Disease Model | Intervention | Key Outcomes | Reference |
|---|---|---|---|
| Alzheimer's Disease (AD) Fly Model | Parp mutation (genetic inhibition) | Increased NAD+ levels; improved mitochondrial function (Δψm); reduced neurodegeneration; improved mid-life survival [91] | Cell Death & Disease (2021) |
| Alzheimer's Disease (AD) Fly Model | Nicotinamide (NAM) supplementation | Increased NAD+ pools; enhanced mitochondrial membrane potential (Δψm); significant decrease in photoreceptor neurodegeneration [91] | Cell Death & Disease (2021) |
| Parkinson's Disease (PD) In Vitro/In Vivo | PARP inhibitors (Veliparib, Rucaparib, Talazoparib) or PARP-1 KO | Prevented α-synuclein PFF-mediated PARP activation and cell death; reduced phosphorylation and aggregation of α-synuclein; reduced transmission of toxic α-synuclein [80] | Science (2018) |
| Human Cohort (UK Biobank) | PARP1 polymorphisms (rs3219134) | Major allele for SNP rs3219134 confers significantly higher risk of AD (OR 1.42; 95% CI 1.06–1.96) [91] | Cell Death & Disease (2021) |
Proposed Combination Strategies and Mechanisms
Based on the established molecular pathways, three primary combination strategies are proposed.
- PARPi + Anti-inflammatory Agents: Combining a PARPi (e.g., Olaparib, Rucaparib) with anti-inflammatory drugs, such as a selective NF-κB inhibitor or a cytokine receptor antagonist (e.g., Anakinra for IL-1), to simultaneously disrupt the DNA damage response and break the cycle of chronic neuroinflammation.
- PARPi + DNA Damage Response Modulators: Using a PARPi alongside a low-dose agent that introduces specific types of DNA lesions (e.g., a topoisomerase inhibitor) or inhibits a complementary DNA repair pathway (e.g., ATM/ATR inhibitors). This approach must be carefully titrated to avoid excessive toxicity.
- PARPi + NAD+ Precursors / Mitochondrial Boosters: Administering a PARPi together with NAD+ precursors like Nicotinamide Riboside (NR) or Nicotinamide Mononucleotide (NMN) to correct the NAD+ deficit and support mitochondrial bioenergetics, thereby preventing parthanatos.
The following diagram illustrates the core signaling pathways involved in PARP-1 activation and the points of intervention for these combination therapies.
Figure 1: PARP-1 Activation Pathway and Combination Therapy Targets. This diagram illustrates the core signaling cascade from initial stress to cell death (parthanatos) and chronic neuroinflammation. Key intervention points for PARP inhibitors, anti-inflammatory agents, and NAD+ precursors are shown in green, highlighting the synergistic strategy.
Experimental Protocols for Validation
Protocol 1: Evaluating PARP-1 Cleavage and Cell Death In Vitro
Aim: To assess the efficacy of combination treatments in reducing PARP-1 cleavage fragments and cell death in neuronal cultures challenged with a neurodegenerative stressor (e.g., α-synuclein PFFs or Oxidative Stress).
Materials:
- Primary Cortical Neurons from rodent embryos (E17-18).
- α-Synuclein Preformed Fibrils (PFFs) or H₂O₂ (for oxidative stress).
- PARP Inhibitors: Talazoparib (BMN 673, 100 nM) or Rucaparib (AG-014699, 1 µM) [80].
- Anti-inflammatory Agent: e.g., NF-κB inhibitor BAY 11-7082 (5 µM).
- NAD+ Precursor: Nicotinamide (NAM, 1-5 mM) [91].
- Antibodies: Anti-PAR monoclonal antibody (for total PARylation), Anti-PARP-1 (for full-length and cleavage fragments), Anti-α-synuclein (phospho S129), Anti-Iba1 (microglial activation), Anti-GFAP (astrocyte activation).
Methodology:
- Cell Culture & Treatment: Plate primary neurons in neurobasal medium. At DIV 7-10, pre-treat cultures with PARPi, anti-inflammatory agent, or combination for 2 hours.
- Disease Insult: Challenge neurons with α-synuclein PFFs (5 µg/mL) or H₂O₂ (100 µM) for 24-48 hours.
- Cell Lysate Collection: Harvest cells in RIPA buffer containing protease and phosphatase inhibitors.
- Western Blot Analysis:
- Separate proteins (20-30 µg) via SDS-PAGE (8-12% gel).
- Transfer to PVDF membrane.
- Probe with primary antibodies: Anti-PAR (1:1000), Anti-PARP-1 (1:1000), Anti-phospho-S129 α-synuclein (1:1000).
- Use appropriate HRP-conjugated secondary antibodies.
- Visualize using chemiluminescence and quantify band intensity. The 89-kD caspase-derived fragment and the 24-kD DBD fragment are key markers [11].
- Immunocytochemistry: Fix neurons and stain for PAR, cell death markers (e.g., Propidium Iodide), and cell-type-specific markers. Analyze using confocal microscopy.
- Viability Assay: Perform MTT or LDH assay to quantify cell death.
Protocol 2: Assessing Neuroinflammation and Microglial Activation
Aim: To quantify the synergistic effect of PARPi and anti-inflammatory agents on glial activation and cytokine release.
Materials:
- BV-2 Microglial Cell Line or primary microglia.
- PARP Inhibitor and Anti-inflammatory Agent (as in Protocol 1).
- ELISA Kits: For TNF-α, IL-1β, IL-6.
- qPCR Reagents: Primers for Tnf, Il1b, Il6, Ccl2.
Methodology:
- Cell Stimulation: Activate BV-2 cells with LPS (100 ng/mL) or α-synuclein PFFs in the presence of vehicle, PARPi, anti-inflammatory agent, or combination.
- Cytokine Measurement: Collect culture supernatant after 6-24 hours. Analyze cytokine levels using specific ELISAs according to manufacturer protocols.
- Gene Expression Analysis: Extract total RNA from cells. Synthesize cDNA and perform qPCR with cytokine-specific primers. Normalize to Gapdh or Actb.
- Statistical Analysis: Use ANOVA with post-hoc tests to compare combination therapy against monotherapies.
The following workflow provides a logical overview of the key experimental steps for validating the combination therapy.
Figure 2: Experimental Workflow for Combination Therapy Validation. This flowchart outlines the key steps for in vitro assessment of synergistic effects, from model establishment to data integration.
The Scientist's Toolkit: Essential Reagents and Models
Table 3: Key Research Reagent Solutions for PARP Combination Studies
| Reagent / Model | Specific Example | Function/Utility in Research |
|---|---|---|
| PARP Inhibitors | Talazoparib (BMN 673), Rucaparib (AG-014699), Veliparib (ABT-888) | Tool compounds to inhibit PARP catalytic activity; Talazoparib is a potent PARP trapper [80]. |
| NAD+ Precursors | Nicotinamide (NAM), Nicotinamide Riboside (NR) | Boost intracellular NAD+ levels, rescue mitochondrial function, and counteract PARP-1-induced NAD+ depletion [91]. |
| Neurodegeneration Inducers | α-Synuclein Preformed Fibrils (PFFs), Amyloid-β Oligomers | Induce protein aggregation, PARP activation, and downstream pathologies relevant to PD and AD [80]. |
| Antibodies for Detection | Anti-PAR Monoclonal Antibody, Anti-PARP-1 (cleavage specific), Anti-phospho-S129 α-synuclein | Detect PAR polymer accumulation, PARP-1 cleavage fragments (e.g., 89-kD, 24-kD), and disease-specific protein phosphorylation [11] [80]. |
| In Vitro Models | Primary Cortical/Hippocampal Neurons, BV-2 Microglial Cell Line | Physiologically relevant systems for studying neuron-specific vulnerability and neuroinflammatory responses [80]. |
| In Vivo Models | Drosophila Aβ-Arc, Mouse PFF Injection Model | Genetically tractable fly model for AD; mouse model for studying α-synuclein pathology and transmission in PD [91] [80]. |
The synergistic combination of PARP inhibition with anti-inflammatory and DNA repair-targeting agents represents a promising, mechanistically grounded strategy for treating neurodegenerative diseases. This approach addresses multiple, interconnected pathological cascades—PARP-1 hyperactivation, parthanatos, bioenergetic collapse, and chronic neuroinflammation—simultaneously. The experimental frameworks and tools provided here offer a roadmap for researchers to rigorously validate these combinations in preclinical models. Future work should focus on optimizing dosing regimens to minimize toxicity, developing biomarkers (such as PAR levels in CSF or specific PARP-1 cleavage fragments) for patient stratification, and advancing brain-penetrant PARP inhibitors to improve clinical translation. By moving beyond single-target paradigms, the field can accelerate the development of truly disease-modifying therapies for currently intractable neurodegenerative conditions.
Conclusion
The cleavage of PARP-1 into specific fragments is not merely a bystander effect of cellular stress but a critical regulatory node determining neuronal fate in neurodegenerative diseases. The 24 kDa and 89 kDa fragments possess distinct and often opposing functions, influencing DNA repair, transcriptional regulation, and the initiation of parthanatos. The central challenge for therapeutic development lies in strategically modulating this pathway—whether by inhibiting the enzyme's catalytic activity, preventing the formation of toxic fragments, or exploiting the protective functions of specific cleavage products. Future research must prioritize the development of brain-penetrant therapeutics and define patient subgroups most likely to benefit from PARP-targeted strategies based on their specific PARylation signature. Unlocking the precise regulatory mechanisms governing PARP-1 cleavage and fragment action will open new avenues for precision medicine in a spectrum of currently untreatable neurodegenerative disorders.
References
- 1. PARP Power: A Structural Perspective on PARP1, PARP2 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8150716/]
- 2. Human PARP1 substrates and regulators of its catalytic activity [https://pmc.ncbi.nlm.nih.gov/articles/PMC9995695/]
- 3. Tipping the PARylation scale: Dysregulation of PAR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12602731/]
- 4. Adenosine diphosphate-ribosylation greatly affects ... [https://www.frontiersin.org/journals/aging-neuroscience/articles/10.3389/fnagi.2025.1575204/full]
- 5. The DNA-Binding Domain of Human PARP-1 Interacts with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3094755/]
- 6. 4AV1: Crystal structure of the human PARP-1 DNA binding ... [https://www.rcsb.org/structure/4av1]
- 7. 7S81: Structure of human PARP1 domains (Zn1, Zn3, WGR ... [https://www.rcsb.org/structure/7S81]
- 8. Serine-linked PARP1 auto-modification controls PARP ... [https://www.nature.com/articles/s41467-021-24361-9]
- 9. PARP1 auto-modification promotes faithful Okazaki ... [https://www.sciencedirect.com/science/article/pii/S1097276525007476]
- 10. PARP1: Structural insights and pharmacological targets for ... [https://www.sciencedirect.com/science/article/pii/S1568786421000811]
- 11. PARP-1 cleavage fragments: signatures of cell-death ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3022541/]
- 12. PARP-1 cleavage fragments: signatures of cell-death ... [https://pubmed.ncbi.nlm.nih.gov/21176168/]
- 13. Differential PARP Cleavage: An Indication of Heterogeneous Forms of... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11506027/]
- 14. Proteases for cell suicide: functions and regulation of ... [https://pubmed.ncbi.nlm.nih.gov/11104820/]
- 15. The 89-kDa PARP1 cleavage fragment serves as a ... [https://www.sciencedirect.com/science/article/pii/S0021925820000320]
- 16. and Calpains : Non- Granzymes in... | SpringerLink caspase Proteases [https://link.springer.com/chapter/10.1007/978-3-319-19497-4_3]
- 17. Inhibitors of Proteases: A Well-Grounded Strategy in Drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12298585/]
- 18. Regulatory apoptotic fragment of PARP1 complements ... [https://www.sciencedirect.com/science/article/abs/pii/S1568786423001477]
- 19. The 89-kDa PARP1 cleavage fragment serves as a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7948984/]
- 20. Functional Competition between Poly(ADP-ribose ... [https://www.sciencedirect.com/science/article/pii/S0021925819345764]
- 21. Characterization of the necrotic cleavage of poly(ADP- ... [https://www.nature.com/articles/4400851]
- 22. Functional Aspects of PARP1 in DNA Repair and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4030864/]
- 23. Poly(ADP-ribose) polymerase-1 and its ambiguous role in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9877585/]
- 24. Molecular Mechanisms of Parthanatos and Its Role in Diverse ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9266317/]
- 25. Molecular mechanisms of cell death by parthanatos [https://pmc.ncbi.nlm.nih.gov/articles/PMC11445734/]
- 26. The contribution of PARP1, PARP2 and poly(ADP-ribosyl) ... [https://www.nature.com/articles/s41598-021-84351-1]
- 27. PARP-1 overexpression contributes to Cadmium-induced ... [https://www.nature.com/articles/s41598-017-04555-2]
- 28. The basi(c)s of PARP inhibitor efficacy and resistance [https://www.sciencedirect.com/science/article/pii/S0093775423000611]
- 29. Poly(ADP-ribose) polymerase-1 and its cleavage products ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4013233/]
- 30. The Enzymatic and DNA Binding Activity of PARP-1 Are ... [https://www.sciencedirect.com/science/article/pii/S0021925819373417]
- 31. Multifaceted Role of PARP-1 in DNA Repair and Inflammation [https://pmc.ncbi.nlm.nih.gov/articles/PMC7017201/]
- 32. Signaling Mechanism of Poly(ADP-Ribose) Polymerase-1 ... [https://www.sciencedirect.com/science/article/pii/S0002944010002142]
- 33. Delayed PARP-1 Inhibition Alleviates Post-stroke ... [https://www.frontiersin.org/journals/cellular-neuroscience/articles/10.3389/fncel.2020.00077/full]
- 34. Poly (ADP-Ribose) polymerase 1 and parthanatos in ... [https://www.sciencedirect.com/science/article/pii/S0969996123003303]
- 35. PARP-1 cleavage fragments: signatures of cell-death ... [https://biosignaling.biomedcentral.com/articles/10.1186/1478-811X-8-31]
- 36. Poly (ADP-ribose) polymerase 1 and neurodegenerative ... [https://www.sciencedirect.com/science/article/abs/pii/S1568163723002374]
- 37. The role of PARP1 in neurodegenerative diseases and aging [https://pubmed.ncbi.nlm.nih.gov/33460497/]
- 38. Roles and therapeutic potential of PARP-1 in ... [https://www.sciencedirect.com/science/article/abs/pii/S0006295225006380]
- 39. Poly(ADP-ribose) polymerase-1 and its cleavage products ... [https://www.sciencedirect.com/science/article/pii/S0167488913004242]
- 40. Characterization of Differentiated SH-SY5Y as Neuronal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4904349/]
- 41. Quantitative proteomics and in-cell cross-linking reveal ... [https://www.nature.com/articles/s42003-022-03478-7]
- 42. Calcium-dependent Cleavage of Endogenous Wild-type ... [https://www.sciencedirect.com/science/article/pii/S0021925819723230]
- 43. New approach to control ischemic severity ex vivo [https://www.sciencedirect.com/science/article/abs/pii/S0165027024002668]
- 44. PARP-1 cleavage fragments: Signatures of cell-death ... [https://augusta.elsevierpure.com/en/publications/parp-1-cleavage-fragments-signatures-of-cell-death-proteases-in-n]
- 45. Emerging role of PARP-1 and PARthanatos in ischemic stroke [https://pmc.ncbi.nlm.nih.gov/articles/PMC9010225/]
- 46. PARP-1 inhibitor alleviates cerebral ischemia/reperfusion ... [https://www.sciencedirect.com/science/article/abs/pii/S0014299924000657]
- 47. The role of H3K27 acetylation in oxygen-glucose ... [https://pubmed.ncbi.nlm.nih.gov/39643249]
- 48. Poly (ADP-ribose) polymerase [https://en.wikipedia.org/wiki/Poly_(ADP-ribose)_polymerase]
- 49. PARP Inhibitors: Clinical Relevance, Mechanisms of Action ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2020.564601/full]
- 50. Signaling Mechanism of Poly(ADP-Ribose) Polymerase-1 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3069822/]
- 51. NF-κB-iNOS-COX2-TNF α inflammatory signaling pathway ... [https://pubmed.ncbi.nlm.nih.gov/29935243/]
- 52. Mitochondrial Protection by PARP Inhibition [https://www.mdpi.com/1422-0067/21/8/2767]
- 53. Mitochondrial dysfunction and activation of iNOS are ... [https://www.nature.com/articles/emm201264]
- 54. Mitochondrial Function and NF-κB Mediated Signaling in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3715144/]
- 55. Amphiregulin Induces iNOS and COX-2 Expression through ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10586434/]
- 56. NF-κB in biology and targeted therapy - Nature [https://www.nature.com/articles/s41392-024-01757-9]
- 57. PARkinson's: from cellular mechanisms to potential therapeutics [https://pmc.ncbi.nlm.nih.gov/articles/PMC8821123/]
- 58. PARP-1 Inhibition Attenuates Neuronal Loss, Microglia ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3967421/]
- 59. Neuroprotective Effects of PARP Inhibitors in Drosophila ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9027574/]
- 60. The role of Poly-ADP ribose polymerase (PARP) enzymes in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12183237/]
- 61. Rapid Detection and Signaling of DNA Damage by PARP - 1 - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8364484/]
- 62. Transcriptional regulation mechanism of PARP and its application in... 1 [https://epigeneticsandchromatin.biomedcentral.com/articles/10.1186/s13072-024-00550-w]
- 63. 揭秘!PARP抑制剂临床应用大突破,耐药机制大起底 - ByDrug [https://bydrug.pharmcube.com/news/detail/55f6da637b77984b1a29589a7546df87]
- 64. - PARP Modulates Amyloid Beta Peptide-Induced... | PLOS One 1 [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0072169]
- 65. 选择性PARP1抑制剂在癌症治疗中的潜力 [https://www.ebiotrade.com/newsf/2025-10/20251022164101983.htm]
- 66. 合成致死2.0:PARP之后,谁是下一个百亿靶点? [https://www.phirda.com/artilce_40642.html?module=trackingCodeGenerator]
- 67. PARP Inhibitors: Clinical Relevance, Mechanisms of Action ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7509090/]
- 68. Alternate therapeutic pathways for PARP inhibitors and ... [https://www.nature.com/articles/s12276-021-00557-3]
- 69. PARP trapping is governed by the PARP inhibitor ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11259578/]
- 70. Transcription–replication conflicts underlie sensitivity to ... [https://www.nature.com/articles/s41586-024-07217-2]
- 71. Review The complex universe of inactive PARP1 [https://www.sciencedirect.com/science/article/abs/pii/S016895252400204X]
- 72. Assessment of brain penetration and tumor accumulation ... [https://www.nature.com/articles/s41598-025-18255-9]
- 73. Design, Synthesis, and Pharmacodynamic Evaluation of Highly... [https://pubmed.ncbi.nlm.nih.gov/39789975/]
- 74. PARP cleavage, DNA fragmentation, and pyknosis during ... [https://www.sciencedirect.com/science/article/abs/pii/S0014488603003662]
- 75. Investigating blood–brain barrier penetration and ... [https://www.nature.com/articles/s41598-025-90888-2]
- 76. PARP inhibitors in gliomas: Mechanisms of action, current ... [https://www.sciencedirect.com/science/article/pii/S0305737224001798]
- 77. Roles and therapeutic potential of PARP-1 in ... [https://pubmed.ncbi.nlm.nih.gov/41022359/]
- 78. The role of Poly-ADP ribose polymerase (PARP) enzymes ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1615843/full]
- 79. Microglia in neurodegenerative diseases: mechanism and ... [https://www.nature.com/articles/s41392-023-01588-0]
- 80. PARP Pathway May Hold Promise for Parkinson Disease ... [https://www.neurologylive.com/view/parp-pathway-may-hold-promise-for-parkinson-disease-treatment]
- 81. Existing Evidence for the Repurposing of PARP-1 Inhibitors in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8833351/]
- 82. Biological function, mediate cell death pathway and their ... [https://www.frontiersin.org/journals/nutrition/articles/10.3389/fnut.2022.1093939/full]
- 83. Characterization of the interactions of PARP-1 with UV- ... [https://www.nature.com/articles/srep19020]
- 84. PARP inhibitors: clinical development, emerging differences ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8992523/]
- 85. Preclinical Evaluation of the PARP Inhibitor, Olaparib, in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3849815/]
- 86. PARP inhibitors: Overview and indications [https://www.jax.org/education-and-learning/clinical-and-continuing-education/clinical-topics/tumor-testing/parpi-detail]
- 87. Decoding Parkinson's Disease: The interplay of cell death ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12312120/]
- 88. Evidence for Decreased Nucleolar PARP-1 as an Early ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6885846/]
- 89. The deubiquitination-PARylation positive feedback loop of the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12256264/]
- 90. - Wikipedia PARP inhibitor [https://en.wikipedia.org/wiki/PARP_inhibitor]
- 91. Parp mutations protect from mitochondrial toxicity in ... [https://www.nature.com/articles/s41419-021-03926-y]