PARP-1 Cleavage Western Blot vs. DNA Fragmentation Analysis: A Technical Guide for Cancer Research and Drug Development
This article provides researchers and drug development professionals with a comprehensive technical comparison of PARP-1 cleavage detection by western blot and DNA fragmentation analysis.
PARP-1 Cleavage Western Blot vs. DNA Fragmentation Analysis: A Technical Guide for Cancer Research and Drug Development
Abstract
This article provides researchers and drug development professionals with a comprehensive technical comparison of PARP-1 cleavage detection by western blot and DNA fragmentation analysis. We cover the foundational biology of PARP-1 in DNA damage response, including its role in repair, cell death pathways like parthanatos, and its cleavage into signature 89-kDa and 24-kDa fragments. Detailed methodological protocols, troubleshooting guides, and optimization strategies for both techniques are presented. Furthermore, we explore the critical application of these assays in validating PARP inhibitor mechanisms, from catalytic inhibition and DNA trapping to the emerging paradigm of PARP1 degradation via PROTACs, offering a framework for robust experimental validation and data interpretation in preclinical and clinical contexts.
PARP-1 in DNA Damage Response: From Repair to Cleavage and Fragmentation
PARP-1's Multifaceted Role in DNA Repair and Replication
Poly(ADP-ribose) polymerase 1 (PARP1) serves as a critical molecular sensor and coordinator of the DNA damage response, playing essential roles in both DNA repair pathways and replication processes. This guide objectively compares two fundamental techniques for studying PARP1 function: PARP-1 cleavage detection by western blot and DNA fragmentation analysis. While western blot provides specific information about PARP1 protein status and activation during apoptosis, DNA fragmentation analysis offers complementary data about the downstream cellular consequences of DNA damage. Understanding the distinct applications, advantages, and limitations of these methodologies is crucial for researchers investigating PARP1 biology, drug development, and therapeutic responses in cancer and other diseases.
PARP1 is a highly abundant nuclear protein that functions as a primary DNA damage sensor. [1] Upon detecting DNA lesions, PARP1 undergoes rapid activation and catalyzes the transfer of ADP-ribose units from NAD+ to target proteins, a post-translational modification known as PARylation. [2] This process facilitates the recruitment of DNA repair factors to damage sites and coordinates multiple DNA repair pathways, including base excision repair (BER) and single-strand break repair (SSBR). [2]
Beyond its established roles in DNA repair, emerging research has revealed PARP1's critical functions in DNA replication. Recent studies demonstrate that PARP1 auto-modification controls replication fork speed and promotes faithful Okazaki fragment processing. [3] Unligated Okazaki fragments have been identified as major sources of PARP activity during S phase, with perturbations in DNA replication proteins like FEN1 increasing PARP activity independently of exogenous DNA damage or replication stress. [3] [2]
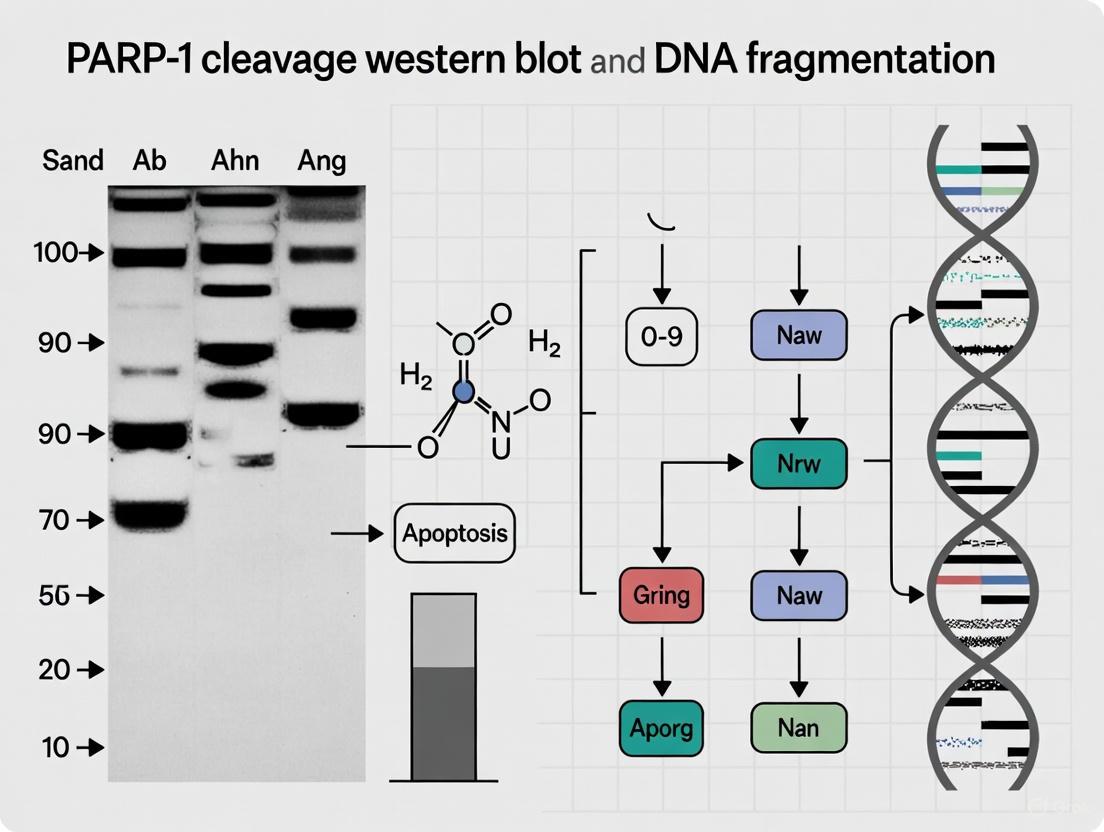
PARP1 also plays a decisive role in determining cell fate in response to severe DNA damage. During apoptosis, PARP1 is cleaved by executioner caspases into specific fragments that contribute to the cell death process. [4] This cleavage serves as a definitive marker for apoptosis and can be effectively detected through western blot analysis. [5]
Methodology Comparison: PARP-1 Cleavage Western Blot vs. DNA Fragmentation Analysis
Table 1: Technical Comparison of PARP-1 Detection Methods
| Parameter | PARP-1 Cleavage Western Blot | DNA Fragmentation Analysis |
|---|---|---|
| Target Detected | PARP1 protein and its cleavage fragments (89 kDa and 24 kDa) | DNA strand breaks and fragmentation patterns |
| Information Provided | Specific PARP1 cleavage status, apoptosis activation, caspase activity | General DNA damage, late-stage apoptosis, necrosis |
| Sensitivity | High (can detect early apoptosis) | Moderate to low (detects mid-late apoptosis) |
| Quantification Approach | Densitometry of protein bands, cleaved to full-length PARP1 ratio | Fragment size distribution, tail moment (comet assay) |
| Stage of Apoptosis Detected | Early to middle phase | Middle to late phase |
| Sample Throughput | Moderate | High |
| Key Experimental Readouts | Presence of 89 kDa and 24 kDa cleavage fragments; cleaved:full-length PARP1 ratio | DNA laddering pattern; comet tail length and intensity |
| Complementary Techniques | Caspase activation assays, viability tests | Annexin V staining, TUNEL assay |
Table 2: Application-Based Method Selection Guide
| Research Context | Recommended Method | Rationale | Key Interpretative Considerations |
|---|---|---|---|
| Therapeutic Screening (PARP inhibitor efficacy) | PARP-1 Cleavage Western Blot | Directly measures apoptosis induction by detecting PARP1 cleavage fragments | Increased cleaved:full-length PARP1 ratio indicates successful apoptosis induction; validates target engagement |
| Mechanistic Studies (DNA repair pathway analysis) | DNA Fragmentation Analysis | Assesses cumulative DNA damage resulting from repair inhibition | Extensive fragmentation suggests repair pathway failure; can indicate synthetic lethality |
| Ferroptosis-Apoptosis Crosstalk | Both techniques recommended | PARP1 cleavage confirms apoptotic commitment; DNA fragmentation assesses genomic integrity | RSL3 induces both PARP1 cleavage and reduced full-length PARP1 via translational suppression [4] |
| Resistance Mechanism Studies | PARP-1 Cleavage Western Blot | Detects altered apoptotic responses in resistant cells | Attenuated cleavage suggests evasion of apoptosis; persistent cleavage indicates maintained sensitivity |
| Clinical Biomarker Development | DNA Fragmentation Analysis with validation | Higher throughput for patient samples; broader damage assessment | Requires validation with specific apoptosis markers to distinguish from necrotic death |
PARP1 Signaling Pathways and Experimental Workflows
PARP1 in DNA Damage Response and Apoptosis
Diagram 1: PARP1's role in DNA damage response and apoptosis. The pathway shows how PARP1 activation leads to either DNA repair or apoptosis, with cleavage serving as a commitment point.
PARP1-DNA Co-condensation in DNA Repair
Diagram 2: PARP1-DNA co-condensation in DNA repair. Recent research reveals that PARP1 and broken DNA ends form co-condensates that maintain spatial connections while facilitating repair. [1]
Detailed Experimental Protocols
PARP-1 Cleavage Western Blot Protocol
Sample Preparation and Protein Extraction
- Cell Lysis: Use RIPA buffer (0.25% Sodium deoxycholate, 50 mM Tris-HCL pH 7.4, 1 mM EDTA, 1% TritonX-100, 1% NP40, 150 mM NaCl) containing protease inhibitor cocktails. [6] Incubate on ice for 30 minutes, then centrifuge at 13,500 rpm for 20 minutes at 4°C to collect supernatant.
- Protein Quantification: Determine protein concentration using BCA assay. [7] Prepare samples with 1× loading buffer, adjusting final protein concentration to 1 mg/mL. [7]
- Recommended Controls: Include untreated cells, apoptosis-induced cells (e.g., with STS or other inducers), and a molecular weight marker for proper fragment identification.
Gel Electrophoresis and Transfer
- Gel Preparation: Prepare 8-12% acrylamide gels using 30% acrylamide/bis solution, Tris-HCl buffers (pH 6.8 and 8.8), SDS, TEMED, and ammonium persulfate. [7]
- Electrophoresis: Load 10-40 μg protein per well. [7] [6] Run at appropriate constant voltage until proper separation is achieved.
- Protein Transfer: Transfer to 0.2 μm nitrocellulose membrane using standard wet or semi-dry transfer systems. Verify transfer efficiency with Ponceau S staining. [7]
Antibody Probing and Detection
- Blocking: Incubate membrane with 5% skim milk in TBST for 1 hour with gentle agitation. [7]
- Antibody Incubation Strategies:
- Primary Antibodies: Use anti-PARP1 antibody (#9532, CST) at appropriate dilution (typically 1:1000-1:2000). [6] [4]
- Detection: Incubate with HRP-conjugated secondary antibodies, develop with chemiluminescent substrate, and image using appropriate detection systems. [7]
Data Analysis and Interpretation
- Band Identification: Full-length PARP1: 116 kDa; Cleavage fragments: 89 kDa and 24 kDa. [8] [5]
- Quantification: Use densitometry software (e.g., ImageJ) to calculate cleaved to full-length PARP1 ratio. [5] Normalize to loading controls (β-actin, GAPDH, or α-tubulin).
- Interpretation: Increased cleaved:full-length ratio indicates apoptosis activation. The 24 kDa fragment irreversibly binds DNA breaks, preventing repair, while the 89 kDa fragment translocates to cytoplasm promoting apoptosis. [4]
DNA Fragmentation Analysis Protocol
Sample Collection and DNA Extraction
- Cell Harvesting: Collect cells by gentle scraping or trypsinization. Pellet by centrifugation.
- DNA Extraction: Use commercial DNA extraction kits or traditional phenol-chloroform extraction methods.
- Quantification: Measure DNA concentration using spectrophotometry or fluorometry.
Fragmentation Analysis Methods
- Gel Electrophoresis: Load 1-2 μg DNA per well on 1.5-2% agarose gels. Include DNA size markers. Run at 5-6 V/cm for 1-2 hours. Visualize with ethidium bromide or SYBR Safe staining.
- Comet Assay (Single Cell Gel Electrophoresis): Embed cells in low-melting-point agarose on slides. Lyse cells in high-salt, detergent-containing buffer. Perform electrophoresis under neutral or alkaline conditions depending on desired damage detection. Stain with DNA-binding fluorescent dyes and analyze by fluorescence microscopy.
- TUNEL Assay: Label 3'-OH ends of fragmented DNA with modified nucleotides using terminal deoxynucleotidyl transferase. Detect with fluorescence or colorimetric methods.
Quantification and Data Analysis
- Gel-Based Methods: Qualitatively assess DNA laddering pattern (approximately 180-200 bp multiples).
- Comet Assay: Quantify tail moment, tail length, and % DNA in tail using specialized software.
- Statistical Analysis: Compare treatment groups with appropriate controls using statistical tests for significance.
Research Reagent Solutions
Table 3: Essential Research Reagents for PARP1 Studies
| Reagent Category | Specific Products/Assays | Research Application | Key Features |
|---|---|---|---|
| PARP1 Antibodies | Anti-PARP1 (#9532, CST) [6] | Western blot, Immunofluorescence | Detects full-length and cleavage fragments; validated for multiple applications |
| Apoptosis Markers | Cleaved Caspase-3, PARP cleavage fragments [5] | Apoptosis detection | Specific markers for early and mid-phase apoptosis |
| PARP Inhibitors | Olaparib, Talazoparib [2] [4] | Mechanism studies, therapeutic screening | Clinical relevance; induce synthetic lethality in HR-deficient cells |
| Detection Systems | HRP-conjugated secondary antibodies, chemiluminescent substrates [7] | Western blot detection | High sensitivity; compatible with quantitative analysis |
| DNA Damage Indicators | γH2AX antibodies [4] [9] | DNA damage assessment | Specific marker for double-strand breaks |
| Cell Death Inducers | RSL3, Staurosporine, Etoposide [4] | Apoptosis induction controls | RSL3 triggers both PARP1 cleavage and reduced full-length PARP1 [4] |
| Specialized Assays | Apoptosis Western Blot Cocktail (ab136812) [5] | Multiplex apoptosis detection | Simultaneous detection of multiple apoptosis markers; improves efficiency |
Emerging Research and Technical Considerations
Recent advances have revealed novel regulatory mechanisms of PARP1, including the USP10-PARP1 positive feedback loop where deubiquitination stabilizes PARP1, and PARP1-mediated PARylation enhances USP10 activity. [6] This regulation promotes DNA damage repair and may influence therapeutic responses.
The discovery of PARP1-DNA co-condensation at double-strand break sites provides new insights into how broken DNA ends are maintained in spatial proximity while allowing repair factor access. [1] This mechanism involves PARP1 forming condensates with DNA through zinc finger domains, with PARylation subsequently remodeling these structures to facilitate repair.
Technical innovations like the sheet protector method for western blotting address reagent conservation concerns, enabling effective antibody distribution with minimal volumes (20-150 μL) while maintaining sensitivity and specificity comparable to conventional methods. [7] This approach offers additional advantages including room temperature incubation without agitation and faster detection timelines.
In therapeutic contexts, PARP1 expression dynamics following DNA damage are clinically relevant. Research demonstrates that sublethal DNA damage can upregulate PARP1 expression, potentially enhancing susceptibility to subsequent PARP-targeted therapies. [9] This principle is being exploited in fractionated radiotherapy approaches to improve treatment efficacy.
Poly(ADP-ribose) polymerase-1 (PARP-1) is a 116-kDa nuclear enzyme that plays a central role in the cellular response to DNA damage, primarily through its involvement in the base excision repair pathway [10] [11]. Beyond its DNA repair functions, PARP-1 has emerged as a critical signaling molecule in cell death pathways, with its cleavage serving as a definitive biochemical marker for apoptosis. The proteolytic cleavage of PARP-1 by caspases represents a decisive step in the commitment to programmed cell death, effectively halting DNA repair while facilitating the dismantling of the cell. This cleavage event generates characteristic fragments, most notably the 89-kDa C-terminal fragment and a 24-kDa N-terminal fragment, which serve as detectable indicators of caspase activation in experimental apoptosis research [12] [10]. Within the context of comparative methodologies for detecting apoptosis, understanding the molecular details of PARP-1 cleavage provides researchers with a specific tool for differentiating between various cell death pathways and assessing the efficacy of apoptotic inducers.
Molecular Mechanisms of PARP-1 Cleavage
Caspase Cleavage Site and Domain Architecture
PARP-1 possesses a modular structure consisting of three primary functional domains: a DNA-binding domain (DBD) at the N-terminus containing two zinc finger motifs, a central automodification domain (AMD), and a C-terminal catalytic domain (CD) responsible for poly(ADP-ribose) polymerization [10] [11]. The caspase cleavage site is located between Asp214 and Gly215 in human PARP-1, strategically positioned between the DNA-binding domain and the automodification domain [12] [10]. This specific location ensures that cleavage separates the N-terminal DNA-binding domain (24-kDa) from the C-terminal portion (89-kDa) containing the automodification and catalytic domains.
Table 1: PARP-1 Domains and Cleavage Fragments
| Domain/Feature | Molecular Weight | Functional Role | Fate After Cleavage |
|---|---|---|---|
| DNA-Binding Domain (DBD) | 24 kDa | Recognizes and binds to DNA strand breaks | Retained in nucleus, irreversibly binds DNA |
| Automodification Domain (AMD) | 22 kDa | Target for poly(ADP-ribosyl)ation | Part of 89-kDa fragment |
| Catalytic Domain (CD) | 54 kDa | Polymerizes ADP-ribose units | Part of 89-kDa fragment |
| Full-length PARP-1 | 116 kDa | DNA damage repair | Cleaved during apoptosis |
| Caspase-cleaved Fragment | 89 kDa | Contains AMD and CD | Translocates to cytoplasm |
The Caspase Cleavage Cascade
The primary caspases responsible for PARP-1 cleavage are the effector caspases-3 and -7, which recognize the DEVD (Asp-Glu-Val-Asp) sequence in PARP-1 [10] [13]. During the initiation of apoptosis, various death signals converge to activate these executioner caspases through either the extrinsic (death receptor) or intrinsic (mitochondrial) pathways. Once activated, caspase-3 and -7 systematically cleave key cellular substrates, with PARP-1 being one of the primary targets. This cleavage event serves two crucial biological functions: first, it inactivates PARP-1's DNA repair activity, preventing futile repair attempts during apoptotic execution; and second, it generates fragments that may actively participate in the cell death process [10] [13].
Caspase-Mediated PARP-1 Cleavage Pathway: This diagram illustrates the sequential process from apoptotic stimulus to the generation and functional consequences of PARP-1 cleavage fragments.
Comparative Analysis of PARP-1 Cleavage Fragments
Signature Fragments Across Cell Death Pathways
Different cell death pathways produce distinct PARP-1 cleavage patterns mediated by specific proteases. While caspase-mediated cleavage during apoptosis generates the characteristic 89-kDa and 24-kDa fragments, other proteases active in alternative cell death pathways create different signature fragments, enabling researchers to differentiate between cell death mechanisms.
Table 2: PARP-1 Cleavage Patterns in Different Cell Death Pathways
| Cell Death Pathway | Primary Proteases | Characteristic Fragments | Functional Consequences |
|---|---|---|---|
| Apoptosis | Caspases-3 and -7 | 89-kDa and 24-kDa | Inactivation of DNA repair; conservation of ATP |
| Necrosis | Lysosomal proteases (cathepsins) | 50-kDa | Non-specific proteolytic degradation |
| Parthanatos | Calpains, cathepsins | Multiple fragments (50-62 kDa) | Energy depletion; AIF-mediated death |
| Granzyme-mediated | Granzyme A | Unknown fragments | Caspase-independent cell death |
The 89-kDa fragment generated by caspase cleavage has recently been shown to serve as a carrier for poly(ADP-ribose) (PAR) polymers, facilitating their translocation from the nucleus to the cytoplasm during certain forms of cell death [13] [14]. Once in the cytoplasm, these PAR polymers can bind to apoptosis-inducing factor (AIF), facilitating its release from mitochondria and subsequent nuclear translocation, where it contributes to caspase-independent DNA fragmentation [13]. This mechanism demonstrates how PARP-1 cleavage fragments can actively participate in amplifying the cell death signal beyond their initial inhibitory function.
Western Blot Analysis of PARP-1 Cleavage
Experimental Protocol for Detection
Western blot analysis remains the gold standard for detecting PARP-1 cleavage due to its ability to differentiate between the full-length protein and its specific cleavage fragments. The following protocol provides a standardized approach for detecting PARP-1 cleavage in cell culture models:
Cell Lysis and Protein Extraction:
- Harvest cells and lyse using RIPA buffer (25 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) supplemented with protease and phosphatase inhibitors.
- Maintain samples at 4°C throughout extraction to prevent protein degradation.
- Centrifuge at 14,000 × g for 15 minutes at 4°C and collect supernatant.
- Quantify protein concentration using BCA or Bradford assay.
Electrophoresis and Immunoblotting:
- Separate 20-50 μg of total protein on 4-12% Bis-Tris polyacrylamide gels.
- Transfer to PVDF membranes using wet or semi-dry transfer systems.
- Block membranes with 5% non-fat dry milk in TBST for 1 hour at room temperature.
- Incubate with primary anti-PARP antibody (e.g., Cell Signaling Technology #9542) at 1:1000 dilution in 5% BSA/TBST overnight at 4°C [12].
- Wash membranes 3× with TBST for 10 minutes each.
- Incubate with HRP-conjugated secondary antibody for 1 hour at room temperature.
- Develop using enhanced chemiluminescence substrate and image.
Key Reagents and Experimental Controls
Table 3: Essential Research Reagents for PARP-1 Cleavage Studies
| Reagent Category | Specific Examples | Function/Application | Validation Parameters |
|---|---|---|---|
| Primary Antibodies | CST #9542, others | Detection of full-length and cleaved PARP-1 | Specificity for 89-kDa fragment; lack of cross-reactivity with other PARP isoforms [12] |
| Apoptosis Inducers | Staurosporine, Actinomycin D | Experimental induction of caspase activation | Dose and time optimization required [13] |
| Caspase Inhibitors | zVAD-fmk | Confirmation of caspase-dependent cleavage | Should prevent 89-kDa fragment formation [15] |
| Positive Control Lysates | Etoposide-treated cell lysates | Assay validation | Should show clear 89-kDa fragment [10] |
| PARP Inhibitors | PJ34, ABT-888 | Investigation of parthanatos pathway | Should not prevent cleavage fragment formation [13] |
Proper validation of antibodies for Western blotting is essential for accurate interpretation of PARP-1 cleavage. According to recent guidelines, antibody specificity should be confirmed using genetic controls such as PARP-1 knockout cells, complemented by orthogonal methods to verify results [16]. The ideal PARP-1 antibody for cleavage detection should recognize both the full-length protein (116-kDa) and the 89-kDa cleavage fragment without cross-reacting with other PARP isoforms or unrelated proteins [12].
PARP-1 Cleavage Western Blot vs. DNA Fragmentation Analysis
Methodological Comparison
When investigating apoptosis, researchers often must choose between detecting PARP-1 cleavage via Western blot or analyzing DNA fragmentation through methods like TUNEL assay or DNA laddering. Each approach offers distinct advantages and limitations that make them suitable for different experimental contexts.
PARP-1 Cleavage vs. DNA Fragmentation Analysis: This diagram compares the strategic advantages and limitations of two principal methods for apoptosis detection in research settings.
Strategic Implementation in Research
For comprehensive apoptosis assessment, particularly in drug development and mechanistic studies, researchers increasingly employ both PARP-1 cleavage analysis and DNA fragmentation methods in parallel. This integrated approach provides complementary information that can delineate the temporal sequence of apoptotic events and offer insights into the specific cell death pathways activated. PARP-1 cleavage analysis offers the distinct advantage of identifying the specific proteases involved in the cell death process based on the fragment signature observed [10] [15]. When comparing different apoptotic inducers or evaluating potential therapeutics, the quantitative nature of Western blot analysis for PARP-1 cleavage provides a reliable metric for assessing the potency and timing of caspase activation.
The caspase-mediated cleavage of PARP-1, generating the characteristic 89-kDa fragment, represents a critical commitment point in the apoptotic pathway that serves both to disable cellular repair mechanisms and potentially amplify cell death signals. Western blot analysis of this event provides researchers with a specific, mechanistic tool for detecting apoptosis that offers complementary information to DNA fragmentation methods. As research continues to elucidate the complex roles of PARP-1 fragments in various cell death pathways, particularly the newly discovered function of the 89-kDa fragment as a PAR carrier, the importance of rigorous detection methodologies becomes increasingly apparent. For drug development professionals, understanding these molecular details enables more precise assessment of therapeutic candidates that either induce or inhibit apoptotic pathways, ultimately contributing to more targeted and effective treatment strategies.
Connecting PARP-1 Activation to Global DNA Fragmentation
Poly(ADP-ribose) polymerase-1 (PARP-1) serves as a critical molecular sensor for DNA damage, with its activation constituting one of the earliest cellular responses to genotoxic stress. Upon detecting DNA strand breaks, PARP-1 catalyzes the transfer of ADP-ribose units from NAD+ to target proteins, including itself—a process known as PARylation or auto-modification [3] [17]. This extensive post-translational modification facilitates DNA repair by recruiting essential repair factors and promoting chromatin relaxation. However, under conditions of severe genotoxic stress, PARP-1 hyperactivation can trigger distinct cellular outcomes, including programmed cell death. A crucial event in this process is the caspase-mediated cleavage of PARP-1 into specific fragments, which serves as both a marker and mediator of apoptosis [18] [8]. This cleavage event generates a recognizable 89 kDa fragment that can be detected via Western blotting, providing researchers with a valuable biochemical tool for monitoring apoptosis. Simultaneously, the apoptotic process activates endonucleases that systematically cleave genomic DNA into characteristic fragments, creating a pattern known as global DNA fragmentation. This article provides a comparative guide to the experimental approaches connecting PARP-1 activation and cleavage to global DNA fragmentation, highlighting key methodologies, their applications, and limitations for researchers and drug development professionals.
PARP-1 Biology and Methodological Principles
PARP-1 Structure, Function, and Cleavage
PARP-1 is a 116 kDa nuclear enzyme comprising several functional domains, including DNA-binding zinc fingers, a BRCT domain, and a C-terminal catalytic domain responsible for PARylation activity [17]. The enzyme operates as an immediate early responder to DNA strand breaks, with its robust activation leading to the synthesis of poly(ADP-ribose) (PAR) chains on itself and other nuclear proteins [3] [17]. This auto-modification facilitates PARP-1's release from DNA, allowing access for repair proteins. However, during apoptosis, executioner caspases (primarily caspase-3) cleave PARP-1 at a specific aspartic acid residue (Asp214), generating two characteristic fragments: a 24 kDa DNA-binding fragment and an 89 kDa catalytic fragment [18] [8]. This cleavage event effectively separates PARP-1's DNA-binding capability from its catalytic activity, inactivating the enzyme and preventing futile ATP consumption during cellular demise.
Detection Principles: Western Blot vs. DNA Fragmentation Analysis
PARP-1 Cleavage Western Blot utilizes antibodies specifically targeting the caspase-cleaved 89 kDa fragment of PARP-1. The Cleaved PARP (Asp214) Antibody (#9541, Cell Signaling Technology) is a well-validated example that detects this endogenous fragment without cross-reacting with full-length PARP-1 or other isoforms [18]. This method provides specific detection of apoptotic signaling with high molecular specificity.
DNA Fragmentation Analysis encompasses several techniques that detect the physical breakdown of genomic DNA during apoptosis. These include:
- Pulsed-Field Gel Electrophoresis (PFGE): Separates large DNA fragments (1 kb to >1 Mb) using alternating electric fields, enabling resolution of chromosomal-sized fragments [19].
- Conventional Gel Electrophoresis: Separates smaller DNA fragments (≤20 kb) using a static electric field, revealing the characteristic "DNA ladder" of oligonucleosomal fragments [19].
- Flow Cytometric (FCM) Sizing: Utilizes intercalating dyes and fluorescence detection to size individual DNA fragments in solution, offering rapid analysis with high sensitivity [19].
- Next-Generation Sequencing (NGS) Approaches: Employ either mechanical or enzymatic fragmentation to assess DNA integrity and fragmentation patterns genome-wide [20].
The following diagram illustrates the core biological relationship between PARP-1 cleavage and DNA fragmentation during apoptosis:
Comparative Experimental Data and Performance Metrics
Technical Performance of DNA Fragmentation Methods
Table 1: Performance Comparison of DNA Fragmentation Detection Methods
| Method | Size Resolution Range | Sample Requirement | Analysis Time | Accuracy | Precision (RSD) | Key Applications |
|---|---|---|---|---|---|---|
| Pulsed-Field GE | 1 kb to >1 Mb | ≥200 ng DNA (~10⁷ cells) | >20 hours/gel | 5% ± 2% | 3% ± 2% | Chromosomal fragmentation, large DNA fragments |
| Conventional GE | ≤20 kb | Varies by protocol | 2-4 hours | N/A | N/A | Apoptotic DNA laddering |
| Flow Cytometry Sizing | 0.125-500 kb | ~1,000 cells | ~30 minutes | 4% ± 4% | 1.2% ± 0.8% | Rapid apoptosis screening, high-throughput |
| Mechanical Shearing (NGS) | Target-specific (e.g., 150-500 bp) | Varies by platform | Library prep + sequencing | High coverage uniformity | High reproducibility | Genome-wide fragmentation mapping |
Table 2: Impact of DNA Fragmentation on Quantitation Methods
| Quantitation Method | Effect of Fragmentation | Sensitivity | Advantages | Limitations |
|---|---|---|---|---|
| Spectrophotometry (A260) | Minimal effect | ~1 ng/μL | Fast, simple, assesses purity | Doesn't distinguish DNA/RNA, low sensitivity |
| Fluorometry (PicoGreen) | Significant underestimation | ~25 pg/μL | Selective for dsDNA, sensitive | Affected by fragments <23 kbp, requires standards |
| qPCR-based | Significant underestimation | ~1 pg human DNA | Highly specific and sensitive | Target-dependent, requires intact primer regions |
PARP-1 Cleavage Detection: Specificity and Validation
The detection of PARP-1 cleavage by Western blot requires rigorous antibody validation to ensure accurate interpretation of apoptotic signaling. The Cleaved PARP (Asp214) Antibody (#9541) exemplifies a well-validated reagent that specifically recognizes the 89 kDa fragment resulting from caspase cleavage at Asp214, without cross-reacting with full-length PARP-1 [18]. For reliable Western blot results, researchers should implement the following validation criteria:
- Specificity Confirmation: Use genetic controls (KO validation) to confirm the absence of non-specific bands [16].
- Selectivity Verification: Test multiple cell lines with known PARP-1 expression patterns to build protein expression profiles [16].
- Band Pattern Interpretation: Recognize that a single distinct band may represent the target protein, cross-reactive species, or protein mixtures, while multiple bands could indicate degradation, splice variants, or post-translational modifications [16].
- Positive Controls: Include lysates from cells undergoing known apoptosis inducers to validate detection capability [16].
Functional studies reveal that the 24 kDa and 89 kDa PARP-1 cleavage products differentially modulate cellular protection, with the 24 kDa fragment conferring protection from oxygen/glucose deprivation damage, while the 89 kDa fragment exhibits cytotoxic properties [8].
Detailed Experimental Protocols
PARP-1 Cleavage Detection by Western Blot
Protocol Overview:
- Sample Preparation: Lyse cells in RIPA buffer supplemented with protease and phosphatase inhibitors. Use approximately 20-30 μg of total protein per sample.
- Gel Electrophoresis: Separate proteins on 4-12% Bis-Tris polyacrylamide gels using MOPS or MES running buffer.
- Protein Transfer: Transfer to PVDF membrane using wet or semi-dry transfer systems.
- Immunoblotting:
- Block membrane with 5% non-fat milk or BSA in TBST for 1 hour.
- Incubate with primary Cleaved PARP (Asp214) Antibody (#9541) at 1:1000 dilution in blocking buffer overnight at 4°C [18].
- Wash membrane 3× with TBST, 10 minutes each.
- Incubate with HRP-conjugated secondary antibody at 1:2000-1:5000 dilution for 1 hour at room temperature.
- Wash membrane 3× with TBST, 10 minutes each.
- Detect using enhanced chemiluminescence substrate.
- Normalization: Probe membrane for housekeeping proteins (e.g., GAPDH, β-actin) to ensure equal loading.
Troubleshooting Notes:
- For cleaner 89 kDa detection, optimize protein loading to avoid signal saturation.
- Include both positive (apoptotic inducer-treated cells) and negative (untreated cells) controls.
- Validate antibody performance in each specific experimental context, as performance can vary based on cell type and treatment conditions [16].
DNA Fragmentation Analysis by Pulsed-Field Gel Electrophoresis
Protocol Overview (based on S. aureus Mu50 analysis [19]):
- Sample Preparation:
- Embed cells in agarose plugs (~1.6×10⁸ cells/100 μL plug) using 1.5% low-melting-point agarose.
- Lyse plugs with lysozyme (1 mg/mL) and lysostaphin (0.1 mg/mL) at 37°C for 105 minutes.
- Treat with proteinase K (1 mg/mL) at 55°C for 60 minutes.
- Inactivate proteinase K with PMSF (1 mM) at 37°C for 30 minutes.
- Wash plugs with H₂O and TE buffer.
Restriction Digestion:
- Digest genomic DNA in plugs with 40 U of appropriate restriction enzyme (e.g., SmaI for Mu50) for 2 hours under manufacturer's conditions.
PFGE Separation:
- Cast 1% agarose gel in 0.5× TBE buffer.
- Load plugs and size markers.
- Run with appropriate pulse conditions for target size range (e.g., 5-30 seconds switch time for 50-500 kb fragments).
- Typical run conditions: 6 V/cm, 14°C, 20 hours.
Visualization:
- Stain gel with ethidium bromide (0.5 μg/mL) or SYBR Safe.
- Image using gel documentation system.
Critical Considerations:
- DNA quality is paramount; avoid excessive handling to prevent mechanical shearing.
- Optimize restriction enzyme choice based on target organism and desired fragment sizes.
- Include appropriate size standards for accurate fragment sizing.
The following workflow diagram illustrates the parallel experimental approaches for detecting PARP-1 cleavage and DNA fragmentation:
Research Reagent Solutions
Table 3: Essential Research Reagents for PARP-1 and DNA Fragmentation Analysis
| Reagent/Category | Specific Examples | Function/Application | Key Characteristics |
|---|---|---|---|
| PARP-1 Cleavage Antibodies | Cleaved PARP (Asp214) Antibody #9541 (Cell Signaling) | Detects caspase-cleaved 89 kDa PARP-1 fragment | Rabbit polyclonal, validated for WB, specific for Asp214 cleavage site [18] |
| DNA Quantitation Kits | PicoGreen dsDNA Assay | Fluorometric DNA quantification | High sensitivity (25 pg/μL), but accuracy affected by fragmentation [21] |
| Restriction Enzymes | SmaI (New England Biolabs) | DNA fragmentation for PFGE fingerprinting | Rare-cutting enzyme, creates manageable fragment numbers [19] |
| Fragmentation Technologies | truCOVER PCR-free Library Prep Kit (Covaris) | Mechanical DNA shearing for NGS | Superior coverage uniformity vs. enzymatic methods [20] |
| Electrophoresis Systems | Pulsed-Field Gel Electrophoresis Systems | Separation of large DNA fragments | Resolves 1 kb to >1 Mb fragments, essential for chromosomal fragmentation analysis [19] |
| Cell Death Inducers | Staurosporine, Etoposide, Camptothecin | Positive controls for apoptosis induction | Trigger caspase activation, PARP-1 cleavage, and DNA fragmentation |
Discussion and Research Implications
The parallel assessment of PARP-1 cleavage and global DNA fragmentation provides complementary insights into apoptotic signaling pathways. PARP-1 cleavage detection offers early, specific evidence of caspase activation, while DNA fragmentation analysis confirms the irreversible commitment to cell death. The methodological comparisons presented here highlight how technique selection should align with specific research objectives:
For high-throughput screening applications, flow cytometric DNA sizing offers rapid analysis with minimal sample requirements (~1,000 cells in 30 minutes) [19]. When high molecular specificity is paramount, PARP-1 cleavage Western blot provides definitive evidence of caspase-3 activation. For comprehensive genome-wide analysis of fragmentation patterns, mechanical shearing coupled with NGS demonstrates superior coverage uniformity, particularly in GC-rich regions [20].
Recent advances in understanding PARP-1 biology continue to refine the interpretation of these assays. The discovery that PARP-1 auto-modification-deficient mutants impact replication fork speed and Okazaki fragment processing reveals connections between PARP-1 function and DNA replication beyond its role in damage response [3]. Furthermore, the differential effects of PARP-1 cleavage fragments—with the 24 kDa fragment conferring protection and the 89 kDa fragment promoting cell death—suggest complex regulatory functions beyond simple enzyme inactivation [8].
These methodologies find particular relevance in cancer research and therapeutic development, where PARP inhibitors exploit synthetic lethality in BRCA-deficient cancers [2]. The accurate assessment of PARP-1 activation and cleavage provides crucial insights into treatment efficacy and mechanisms of resistance. Additionally, the growing recognition of "BRCAness" phenotypes—tumors with homologous recombination deficiencies beyond BRCA mutations—expands the potential applications for these analytical approaches in predicting therapeutic responses [2].
As research progresses, the integration of these traditional methodologies with emerging technologies such as live-cell imaging of PARP-1/2 dynamics [17] and single-molecule analysis will provide increasingly sophisticated tools for connecting PARP-1 activation to global DNA fragmentation outcomes. This continued methodological evolution will enhance our understanding of cell fate decisions in response to genotoxic stress and inform the development of targeted therapeutic interventions.
PARP-1-Dependent Cell Death and its Biomarkers
Parthanatos is a form of programmed cell death that is critically dependent on the hyperactivation of poly(ADP-ribose) polymerase 1 (PARP-1). Unlike apoptosis, parthanatos proceeds independently of caspase activity and is characterized by rapid energy depletion, massive poly(ADP-ribose) (PAR) polymer accumulation, and nuclear translocation of apoptosis-inducing factor (AIF) from mitochondria [22] [23]. This distinct cell death pathway plays a significant role in various pathological conditions, particularly in neurological diseases and acute tissue injury models such as stroke, subarachnoid hemorrhage, and neurodegenerative disorders [24] [23].
The accurate detection of parthanatos is crucial for both basic research and drug development. Among the most reliable biomarkers for identifying this process are PARP-1 cleavage patterns observed via western blot and DNA fragmentation assessed through specialized analyses. This guide provides a comprehensive comparison of these two methodological approaches, offering detailed experimental protocols and data interpretation guidelines tailored for researchers and scientists in the field.
The Molecular Mechanisms of Parthanatos
Parthanatos is initiated by extensive DNA damage, often resulting from oxidative stress, which triggers the hyperactivation of PARP-1 [23]. Overactive PARP-1 consumes large amounts of NAD+ and ATP to synthesize PAR polymers, leading to severe energy depletion within the cell [22] [23]. The accumulated PAR polymers function as a death signal, triggering the release of AIF from mitochondria. Once translocated to the nucleus, AIF recruits macrophage migration inhibitory factor (MIF), which exhibits nuclease activity and drives large-scale DNA fragmentation [22]. This cascade ultimately results in irreversible chromatin condensation and cell death.
Key Signaling Pathway
The diagram below illustrates the core molecular cascade of parthanatos, from the initial DNA damage to the final cell death execution.
Biomarker 1: PARP-1 Cleavage Analysis via Western Blot
Biological Significance and Mechanism
PARP-1 cleavage is a well-established hallmark of cell death, but the specific fragment sizes indicate the activation of different proteases and distinct death pathways. During apoptosis, caspases-3 and -7 cleave PARP-1 at the DEVD214 site, generating characteristic 89 kDa and 24 kDa fragments [25]. In contrast, parthanatos involves PARP-1 hyperactivation but not necessarily its cleavage; however, other proteases activated in cell death contexts can process PARP-1 into different signature fragments. For instance, during necrosis, lysosomal proteases such as cathepsins cleave PARP-1 to produce a 50 kDa fragment [15]. Calpains can generate 55-62 kDa fragments, granzyme A produces a 50 kDa fragment, and matrix metalloproteinases yield 35-45 kDa fragments [25]. Therefore, detecting the full-length PARP-1 (113 kDa) alongside these specific cleavage products provides crucial information about the dominant cell death pathway.
Detailed Experimental Protocol
Sample Preparation:
- Lyse cells or tissue samples in RIPA buffer supplemented with protease inhibitors (e.g., 1 mM PMSF, 10 μg/mL aprotinin, and 10 μg/mL leupeptin).
- For tissues, use a mechanical homogenizer to ensure complete lysis.
- Quantify protein concentration using a BCA or Bradford assay.
- Dilute samples in 4X Laemmli buffer to a final concentration of 1-2 μg/μL and denature at 95°C for 5 minutes.
Gel Electrophoresis and Western Blotting:
- Load 20-40 μg of total protein per lane on a 4-12% Bis-Tris polyacrylamide gel.
- Perform electrophoresis at 120-150 V for 60-90 minutes in MOPS or MES running buffer.
- Transfer proteins to a PVDF membrane using a wet or semi-dry transfer system at 100 V for 60-90 minutes on ice.
Antibody Detection and Visualization:
- Block the membrane with 5% non-fat milk in TBST for 1 hour at room temperature.
- Incubate with primary antibody (e.g., anti-PARP-1 monoclonal antibody, specific for full-length and cleaved fragments) diluted in blocking buffer overnight at 4°C.
- Use appropriate secondary antibody (HRP-conjugated anti-mouse or anti-rabbit) diluted 1:5000 for 1 hour at room temperature.
- Develop using enhanced chemiluminescence (ECL) substrate and image with a digital imaging system.
Data Interpretation Guidelines
- Apoptosis Indicator: The presence of the 89 kDa fragment with corresponding decrease in full-length PARP-1 suggests caspase-dependent apoptosis [25] [26].
- Necrosis/Necroptosis Indicator: Detection of the 50 kDa fragment indicates cathepsin-mediated cleavage, characteristic of necrotic cell death [15].
- Parthanatos Context: In parthanatos, maintain high levels of full-length PARP-1 alongside PAR polymer accumulation; the absence of significant 89 kDa fragment helps distinguish it from apoptosis [23].
Biomarker 2: DNA Fragmentation Analysis
Biological Significance and Mechanism
DNA fragmentation occurs in multiple cell death pathways, but the fragment sizes and patterns differ significantly. In apoptosis, caspase-activated DNase (CAD) produces a ladder of fragments in multiples of 180-200 bp due to cleavage between nucleosomes [25]. In contrast, parthanatos involves MIF nuclease activity downstream of AIF translocation, resulting in large-scale DNA fragmentation (15-50 kb) without the regular nucleosomal pattern [22]. This distinct fragmentation pattern serves as a key diagnostic feature for differentiating parthanatos from other cell death mechanisms.
Detailed Experimental Protocol
DNA Laddering Assay (for Apoptosis Detection):
- Extract genomic DNA using a phenol-chloroform method or commercial DNA extraction kit.
- Quantify DNA concentration using a spectrophotometer or fluorometer.
- Load 500 ng - 1 μg of DNA per lane on a 1.5-2% agarose gel containing 0.5 μg/mL ethidium bromide.
- Perform electrophoresis at 80-100 V for 90-120 minutes in 1X TAE buffer.
- Visualize DNA fragments under UV light; apoptotic samples will show a characteristic ladder pattern.
Pulsed-Field Gel Electrophoresis (for Parthanatos Detection):
- Embed cells in low-melting-point agarose plugs to protect large DNA fragments.
- Lyse cells within plugs using proteinase K-containing buffer (48-72 hours at 50°C with agitation).
- Wash plugs extensively with TE buffer to remove residual detergents and enzymes.
- Load plugs onto a 1% pulsed-field certified agarose gel.
- Run electrophoresis using a CHEF or FIGE system with appropriate settings for separating 15-50 kb fragments (e.g., 6 V/cm, 14°C, with switch times of 1-50 seconds for 18 hours).
- Stain gel with ethidium bromide and visualize under UV light; parthanatos samples will show large fragments (15-50 kb) rather than a nucleosomal ladder.
Comparative Analysis of Biomarkers
The table below provides a direct comparison of the key technical and application characteristics of PARP-1 cleavage analysis and DNA fragmentation analysis.
Table 1: Comparison of PARP-1 Cleavage Western Blot and DNA Fragmentation Analysis
| Parameter | PARP-1 Cleavage Western Blot | DNA Fragmentation Analysis |
|---|---|---|
| Primary Biomarker | Specific proteolytic fragments (89, 50, 35-62 kDa) | Large DNA fragments (15-50 kb) |
| Indicated Process | Protease activation in cell death | Endonuclease activation |
| Key Death Pathway Identified | Apoptosis (89 kDa), necrosis (50 kDa) | Parthanatos (large fragments) |
| Sensitivity | High (can detect ng protein levels) | Moderate (requires significant DNA damage) |
| Time Requirement | 1-2 days | 2-4 days |
| Technical Complexity | Moderate | Moderate to High (especially PFGE) |
| Specialized Equipment | Standard molecular biology equipment | Pulsed-field system for parthanatos |
| Quantification Potential | Densitometry with normalization | Densitometry with standards |
| Key Advantage | Specific protease activity information | Direct evidence of nuclear collapse |
Integrated Experimental Workflow
For comprehensive characterization of parthanatos in research models, the following integrated approach is recommended:
Table 2: Essential Research Reagents for Parthanatos Detection
| Reagent/Category | Specific Examples | Research Function |
|---|---|---|
| PARP-1 Antibodies | Anti-PARP-1 (full-length), Anti-cleaved PARP-1 (89 kDa) [26] | Detect PARP-1 expression and caspase cleavage |
| PAR Detection Reagents | Anti-PAR antibody, PAR ELISA kits | Measure PAR polymer accumulation |
| Cell Death Inducers | MNNG, H₂O₂, Glutamate [22] [23] | Induce parthanatos in experimental models |
| PARP Inhibitors | AG14361, Olaparib analogs [23] | Confirm PARP-1 dependence of cell death |
| DNA Extraction & Analysis | Pulsed-field gel systems, DNA quantification kits | Assess DNA fragmentation patterns |
Both PARP-1 cleavage analysis and DNA fragmentation assessment provide valuable, complementary insights into cell death mechanisms. PARP-1 western blotting excels in identifying the specific proteases activated during cell death, while DNA fragmentation analysis provides direct evidence of the end-stage nuclear events characteristic of parthanatos. For definitive identification of parthanatos, researchers should employ both methodologies in parallel, alongside additional confirmation through PAR polymer detection and AIF translocation assays. This multi-faceted approach ensures accurate discrimination of parthanatos from other cell death pathways, facilitating more precise mechanistic studies in disease models and therapeutic development.
Protocols in Practice: Executing PARP-1 Western Blot and DNA Fragmentation Assays
In the context of DNA fragmentation analysis research, the detection of Poly(ADP-ribose) polymerase-1 (PARP-1) cleavage fragments by western blot stands as a critical methodology for identifying apoptotic cells. PARP-1, a 113 kDa nuclear enzyme involved in DNA repair, becomes a marker for apoptosis when cleaved by caspases into characteristic 89 kDa and 24 kDa fragments [27] [28] [25]. This cleavage separates the DNA-binding domain from the catalytic domain, inactivating the DNA repair function and facilitating cellular disassembly [27] [29]. This guide provides a detailed, experimentally-supported protocol for detecting these signature cleavage fragments, objectively comparing key reagent performance to ensure reliable results in drug development and basic research.
The Biological Foundation of PARP-1 Cleavage
PARP-1 is a primary substrate for caspase-3 and other proteases during programmed cell death. While caspase-mediated cleavage generates the classic 89 kDa fragment during apoptosis, researchers should note that other proteases, including calpains, cathepsins, granzymes, and matrix metalloproteinases (MMPs), can produce different PARP-1 fragments (ranging from 42-89 kDa) in alternative cell death pathways [25] [30]. This protease-specific cleavage makes PARP-1 fragment analysis a valuable tool for discriminating between cell death mechanisms.
The following diagram illustrates the key proteolytic events in PARP-1 cleavage during different cell death pathways:
Research Reagent Solutions: Antibody Comparison
Selecting an appropriate primary antibody is crucial for specific detection of PARP-1 cleavage fragments. The table below summarizes key commercially available antibodies validated for detecting the 89 kDa cleaved PARP-1 fragment:
| Antibody Name | Host & Clonality | Reactivity | Applications | Key Specificity | Catalog Example |
|---|---|---|---|---|---|
| Cleaved PARP (Asp214) | Rabbit Monoclonal | Human, Mouse, Monkey | WB, IHC, IF, FC, ELISA [29] | Detects 89 kDa fragment only; does not recognize full-length PARP1 [29] | #95696 (Cell Signaling) |
| Cleaved PARP (Asp214) | Rabbit Polyclonal | Human, Mouse | WB, Simple Western [27] | Detects 89 kDa fragment produced by caspase cleavage [27] | #9541 (Cell Signaling) |
| Anti-Cleaved PARP1 | Rabbit Polyclonal | Human | WB [31] | Recognizes 85 kDa fragment; specific to cleavage site [31] | ab4830 (Abcam) |
| Cleaved PARP1 | Mouse Monoclonal | Human, Mouse, Rat | WB, IHC, IF/ICC, FC, ELISA [30] | Detects cleaved form only (89 kDa); not full-length [30] | 60555-1-Ig (Proteintech) |
WB: Western Blot; IHC: Immunohistochemistry; IF: Immunofluorescence; FC: Flow Cytometry
Step-by-Step Western Blot Protocol
Sample Preparation from Cultured Cells
Induction of Apoptosis: Treat cells (e.g., Jurkat, HeLa) with a proven apoptotic inducer such as 1 μM Staurosporine for 3-16 hours [31] [30] or 1 μM Etoposide for 16 hours [31]. Include untreated controls.
Cell Lysis: Lyse cells using RIPA or IP lysis buffer (50 mM Tris-HCl pH 7.4, 1% NP-40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA) supplemented with protease and phosphatase inhibitors [6]. Incubate on ice for 30 minutes.
Protein Quantification: Centrifuge lysates at 13,500 rpm for 20 minutes at 4°C. Collect supernatant and determine protein concentration using a Bradford or BCA assay. Prepare samples with 40-50 μg total protein per lane [31].
Gel Electrophoresis and Transfer
SDS-PAGE: Load samples and pre-stained protein ladder onto a 8-12% Tris-Glycine gel. Run electrophoresis at 100-120V until the dye front reaches the bottom. The 89 kDa fragment should run between the 75 and 100 kDa markers [31].
Protein Transfer: Transfer proteins to a PVDF or nitrocellulose membrane using wet or semi-dry transfer systems. Verify transfer efficiency with Ponceau S staining if desired.
Immunoblotting
Blocking: Block membrane with 5% non-fat dry milk or BSA in TBST for 1 hour at room temperature.
Primary Antibody Incubation: Incubate membrane with anti-cleaved PARP1 antibody diluted in blocking buffer overnight at 4°C with gentle agitation. Use optimized dilutions:
Washing: Wash membrane 3 times for 5-10 minutes each with TBST.
Secondary Antibody Incubation: Incubate with appropriate HRP-conjugated secondary antibody (e.g., Goat Anti-Rabbit HRP [31]) for 1 hour at room temperature.
Detection: Develop blots using enhanced chemiluminescence (ECL) substrate and image with a digital imaging system. Expected band at approximately 89 kDa [27] [31].
Membrane Stripping and Reprobing
Strip membrane with mild stripping buffer to remove primary and secondary antibodies.
Reprobe with anti-β-actin or anti-α-tubulin antibody as a loading control [6].
Experimental Data and Antibody Performance Comparison
The table below summarizes experimental data from cited literature demonstrating antibody performance across different cell lines and treatments:
| Cell Line | Treatment | Antibody Used | Result | Reference |
|---|---|---|---|---|
| Jurkat | Etoposide (1 μM, 16 hr) | ab4830 (Abcam) | Strong 85 kDa band in treated cells [31] | abcam |
| HeLa | Staurosporine (3 μM, 16 hr) | ab4830 (Abcam) | Clear 85 kDa band in treated cells [31] | abcam |
| HSC-T6 | Staurosporine (1 μM, 3 hr) | 60555-1-Ig (Proteintech) | Detection of cleaved PARP1 by WB, IF, FC [30] | ptglab |
| A2780 | Staurosporine | 60555-1-Ig (Proteintech) | Cleaved PARP1 detected in treated cells [30] | ptglab |
| Mouse splenocytes | Staurosporine | 60555-1-Ig (Proteintech) | Cleaved PARP1 detected in treated cells [30] | ptglab |
Troubleshooting and Optimization
- No cleaved PARP-1 signal: Ensure apoptosis induction is sufficient; optimize treatment duration and concentration. Include a positive control (Staurosporine-treated cells).
- High background: Increase blocking time, optimize antibody dilution, or increase wash stringency.
- Multiple non-specific bands: Verify antibody specificity and consider using monoclonal antibodies for higher specificity.
- Weak or no signal in positive control: Check antibody expiration, incubation conditions, and ECL substrate activity.
Western blot detection of PARP-1 cleavage fragments provides researchers with a reliable method for apoptosis assessment in the broader context of DNA fragmentation analysis. The protocol outlined here, supported by experimental data from multiple sources, enables specific detection of the characteristic 89 kDa fragment. The antibody performance comparison offers objective guidance for reagent selection based on experimental needs. When properly optimized and controlled, this method serves as a robust approach for evaluating apoptotic responses in basic research and drug development applications.
Essential Controls and Antibody Validation for Specific Detection
In the field of molecular biology research, particularly in studies focusing on cellular stress responses and death pathways, the specific detection of PARP-1 cleavage has emerged as a critical biomarker. As a nuclear enzyme involved in DNA repair, PARP-1 undergoes proteolytic cleavage during various forms of programmed cell death, producing characteristic fragments that serve as signatures for specific protease activities. This comparison guide objectively evaluates two fundamental methodological approaches for detecting PARP-1 cleavage: Western blot analysis and DNA fragmentation analysis. Each technique offers distinct advantages and limitations for researchers investigating apoptosis and other cell death mechanisms in experimental and drug development contexts. Through systematic comparison of their technical requirements, detection capabilities, and experimental considerations, this guide provides scientists with the framework to select appropriate detection strategies based on specific research objectives and resource constraints.
Technical Comparison of Detection Methods
The following table summarizes the core characteristics, advantages, and limitations of Western blot analysis for PARP-1 cleavage versus DNA fragmentation analysis:
| Feature | PARP-1 Cleavage Western Blot | DNA Fragmentation Analysis |
|---|---|---|
| Target Molecule | PARP-1 protein and its cleavage fragments (24 kDa, 89 kDa) [28] [25] | Fragmented DNA molecules [32] |
| Key Detectable Signals | • Full-length PARP-1 (113 kDa)• 89 kDa fragment (catalytic domain)• 24 kDa fragment (DNA-binding domain) [25] [14] | • DNA laddering pattern (apoptosis)• Smear pattern (necrosis)• Specific fragment sizes (e.g., 150-500 bp) [32] |
| Primary Applications | • Apoptosis detection• Caspase/calpain activity assessment• Cell death mechanism differentiation [25] | • Apoptosis confirmation• DNA integrity assessment• Sample quality control [32] [33] |
| Sensitivity | High (can detect nanogram protein amounts) [34] | Varies by method: spectrophotometry (1 ng/μL), PicoGreen (25 pg/μL), qPCR (1 pg) [32] |
| Specificity Control | Knockout validation; cleavage-specific antibodies [34] | Fragment size standardization; reference DNA controls [32] [33] |
| Quantitation Capability | Semi-quantitative (densitometry) | Quantitative with fluorometric/qPCR methods [32] |
| Key Limitations | • Cannot distinguish between different cleavage fragments without specific antibodies• Dependent on antibody quality and specificity [25] [34] | • Accuracy affected by fragmentation degree• Method-dependent variability in results [32] |
| Sample Throughput | Moderate (gel electrophoresis limits parallel processing) | High with microplate-based formats [32] |
| Fragmentation Influence | Not applicable | Significant impact on fluorometric and qPCR quantification [32] |
PARP-1 Cleavage Signaling Pathway
The diagram below illustrates the PARP-1 cleavage pathway and its role in cell death mechanisms:
Experimental Protocols for PARP-1 Cleavage Detection
Western Blot Protocol for PARP-1 Cleavage Fragments
Cell Lysis and Protein Extraction
- Harvest cells and lyse using RIPA buffer supplemented with protease inhibitors
- Centrifuge at 14,000 × g for 15 minutes at 4°C
- Collect supernatant and determine protein concentration using BCA assay [34]
Gel Electrophoresis and Transfer
- Load 20-30 μg protein per lane on 4-12% Bis-Tris polyacrylamide gels
- Run electrophoresis at 120-150V for 60-90 minutes
- Transfer to nitrocellulose or PVDF membranes using standard wet or semi-dry transfer systems [34]
Antibody Incubation and Detection
- Block membranes with 5% non-fat dry milk or BSA in TBST for 1 hour
- Incubate with primary anti-cleaved PARP-1 antibody (e.g., ab32064) at 1:10,000 dilution overnight at 4°C
- Wash membranes 4 times with TBST, 5 minutes each
- Incubate with HRP-conjugated secondary antibody at 1:20,000 dilution for 1 hour at room temperature
- Detect using enhanced chemiluminescence substrate and imaging system [34]
Essential Controls
- Include PARP-1 knockout cell lysates as negative controls
- Use staurosporine-treated (3 μM, 24 hours) cell lysates as positive controls for cleavage
- Include loading controls (e.g., GAPDH, alpha-tubulin) for normalization [34]
DNA Fragmentation Analysis Protocol
DNA Extraction
- Extract DNA using phenol-chloroform method or commercial kits
- Treat with RNase A to remove RNA contamination
- Determine initial concentration using spectrophotometry [32]
Fragment Size Assessment
- For gel electrophoresis: Run 100-500 ng DNA on 1.5-2% agarose gels, stain with ethidium bromide
- For fluorometric quantification: Use PicoGreen dye with standard curve method
- For qPCR-based quantification: Use multi-copy genes (e.g., rDNA, Alu repeats) as targets [32]
Quantitation Methods
- Spectrophotometry: Measure absorbance at 260 nm, minimal fragmentation effect [32]
- Fluorometric (PicoGreen): Prepare standard curve, significant fragmentation effect [32]
- qPCR-based: Use serial dilutions, significantly affected by fragmentation [32]
Research Reagent Solutions
The table below outlines essential reagents and their applications in PARP-1 cleavage and DNA fragmentation studies:
| Reagent Category | Specific Examples | Research Application | Key Characteristics |
|---|---|---|---|
| PARP-1 Cleavage Antibodies | Anti-Cleaved PARP1 [E51] (ab32064) [34] | Specific detection of cleaved PARP1 fragments in Western blot | • Rabbit monoclonal• Recognizes 24-27 kDa fragment• KO-validated specificity |
| Cell Death Inducers | Staurosporine (0.5-3 μM) [34] | Induction of apoptosis and PARP-1 cleavage in positive controls | • Caspase activation• Dose-dependent effect• Treatment: 3-24 hours |
| PARP Inhibitors | Olaparib, Talazoparib [35] [36] | Investigation of PARP-1 function and synthetic lethality | • Catalytic activity inhibition• Research and clinical applications• HR-deficient cancer studies |
| DNA Quantitation Kits | PicoGreen dsDNA Assay [32] | Fluorometric DNA concentration measurement | • High sensitivity (25 pg/μL)• Affected by fragmentation level• Standard curve required |
| Caspase Inhibitors | Z-VAD-FMK [37] | Inhibition of caspase-mediated PARP-1 cleavage | • Pan-caspase inhibitor• Mechanism studies• Apoptosis pathway analysis |
| Protein Extraction Reagents | RIPA Lysis Buffer [34] | Protein extraction for Western blot analysis | • Comprehensive extraction• Protease inhibitors essential• Compatibility with downstream applications |
Methodological Considerations for Accurate Detection
Antibody Validation Strategies
Specific detection of PARP-1 cleavage fragments requires rigorous antibody validation. The anti-cleaved PARP1 antibody [E51] (ab32064) demonstrates specificity through multiple validation approaches, including knockout validation in PARP-1 knockout A549 and HAP1 cells, where no signal is observed at the expected molecular weight (24-27 kDa). Additional validation includes treatment with apoptosis inducers like staurosporine and camptothecin, which enhance cleavage fragment detection. Antibodies should recognize both the 24 kDa DNA-binding domain fragment and the 89 kDa catalytic domain fragment, though most commercial antibodies specifically target the 24 kDa fragment for apoptosis detection [25] [34].
Impact of DNA Fragmentation on Quantitation
The degree of DNA fragmentation significantly influences quantification accuracy in DNA fragmentation analysis. Spectrophotometric methods (e.g., NanoDrop) show minimal effect from fragmentation level, while fluorometric methods (e.g., PicoGreen) and qPCR-based quantification are substantially affected. In 10-fold diluted samples, PicoGreen measurement of DNA fragmented to approximately 150 bp shows approximately 29% reduction in measured concentration compared to unfragmented DNA. Similarly, qPCR-based quantification demonstrates up to 67% reduction in measured concentration for 150 bp fragmented samples compared to intact DNA [32]. These effects must be considered when designing experiments and interpreting results.
Technical Optimization Approaches
For PARP-1 Western blotting, optimal results are achieved with 1:10,000 antibody dilution and 20 μg protein loading. Blocking with 5% non-fat dry milk in TBST and extended washes improve signal-to-noise ratio. For DNA fragmentation studies, fragment size matching between test and reference samples improves aCGH performance, particularly with FFPE samples [33]. The DNA Fragmentation Simulation Method (FSM) allows customized tailoring of fragment sizes, reducing array failure rates from approximately 33% to levels comparable with fresh samples [33].
The selection between PARP-1 cleavage Western blot analysis and DNA fragmentation analysis depends on specific research objectives, with each method offering complementary insights into cell death mechanisms. Western blot provides specific information about protease activity through PARP-1 fragment detection, while DNA fragmentation analysis offers direct evidence of apoptotic progression. Implementation of appropriate controls, validation procedures, and understanding methodological limitations are essential for accurate data interpretation in both techniques. These detection methods continue to play crucial roles in basic research, drug development, and therapeutic response assessment in various disease models, particularly in cancer research and neurodegenerative disorders.
The integrity of genomic DNA is a cornerstone of cellular health and function. In fields ranging from reproductive medicine to cancer research and toxicology, accurately quantifying DNA fragmentation is essential for assessing genotoxicity, diagnosing infertility, and understanding fundamental disease mechanisms. Among the various techniques developed, the Sperm Chromatin Structure Assay (SCSA) and the Comet Assay have emerged as two prominent methodologies. While the SCSA utilizes flow cytometry to measure DNA susceptibility to acid-induced denaturation, the Comet Assay employs single-cell gel electrophoresis to visualize and quantify DNA strand breaks directly. The selection between these methods carries significant implications for research outcomes, particularly in studies investigating cellular responses to stress, chemical agents, or pathological conditions where DNA damage triggers specific molecular pathways such as PARP-1 cleavage.
This guide provides an objective comparison of the SCSA and Comet Assay, supported by experimental data and detailed protocols. It frames this technical comparison within the broader context of DNA damage response research, specifically addressing how these methods complement protein-based techniques like PARP-1 cleavage detection via Western blot in constructing a comprehensive picture of cellular stress and death pathways.
Fundamental Methodological Comparison
The SCSA and Comet Assay differ fundamentally in their underlying principles, with each technique probing different aspects of DNA damage through distinct mechanisms.
The SCSA is an indirect method that assesses DNA fragmentation by measuring the susceptibility of sperm chromatin to acid-induced denaturation in situ [38]. The core principle relies on the metachromatic properties of acridine orange, which fluoresces green when intercalated into double-stranded DNA but shifts to red when associated with single-stranded DNA. The assay involves briefly treating sperm nuclei with a low-pH detergent solution to denature DNA at sites containing strand breaks. Following staining, flow cytometry analysis quantifies the ratio of denatured (red fluorescence) to native (green fluorescence) DNA. The primary metric generated is the DNA Fragmentation Index (DFI), which represents the proportion of cells with fragmented DNA within a sample. This approach enables high-throughput analysis of thousands of cells rapidly, providing population-level statistics with minimal subjective interpretation.
In contrast, the Comet Assay (single-cell gel electrophoresis) directly visualizes and quantifies DNA strand breaks at the individual cell level [39]. Cells are embedded in agarose on a microscope slide, lysed to remove cellular membranes and proteins, and subjected to electrophoresis under neutral or alkaline conditions. Damaged DNA containing strand breaks migrates from the nucleus toward the anode, forming a characteristic "comet" pattern. Under fluorescence microscopy, the intact DNA remains in the "head," while fragmented DNA forms the "tail." Several parameters can be quantified using image analysis software, with % tail DNA (the percentage of total DNA located in the tail) being the most widely accepted and biologically relevant metric [40]. The alkaline version (pH >13) detects single-strand breaks, alkali-labile sites, and cross-linking damage with high sensitivity, while the neutral version primarily detects double-strand breaks.
Table 1: Core Principles and Characteristics of SCSA and Comet Assay
| Feature | SCSA | Comet Assay |
|---|---|---|
| Fundamental Principle | Flow cytometric measurement of DNA denaturation | Electrophoretic separation of DNA fragments |
| Primary Metrics | DNA Fragmentation Index (DFI) | % Tail DNA, Tail Moment |
| Cell Throughput | High (thousands of cells) | Low to medium (50-100 cells typically scored) |
| Level of Analysis | Population-level statistics | Single-cell resolution |
| DNA Damage Detected | Chromatin susceptibility to denaturation | Direct strand breaks, alkali-labile sites |
| Technical Complexity | Moderate (requires flow cytometer) | Low to moderate (requires electrophoresis and imaging) |
Comparative Sensitivity and Performance Data
Recent comparative studies have revealed significant differences in the sensitivity and detection capabilities of the SCSA and Comet Assay across various experimental models and DNA damage induction methods.
Evidence from Sperm Preservation Studies
A direct comparative analysis of equine semen preservation techniques demonstrated a striking disparity in sensitivity between the two assays. While the SCSA revealed no significant increase in DNA damage at any timepoint across various storage conditions, the Comet assay detected substantial damage increases. Specifically, the Comet assay measured a significant increase in % tail DNA after 72 hours of storage in SpermSafe (from 21.1±11.4% to 53.5±0.2%, p≤0.05) and after cryopreservation (from 21.1±11.4% pre-freeze to 67.2±3.5% post-thaw, p≤0.05) [41]. This suggests the Comet Assay possesses superior sensitivity for detecting DNA fragmentation induced by preservation stress in sperm cells.
Multi-Method Comparison in Induced DNA Damage
A comprehensive 2025 study systematically compared four DNA fragmentation detection methods (TUNEL, SCSA, SCD test, and Comet Assay) following DNA damage induction through cryopreservation and in vitro incubation [38]. While all tests detected increased sDF under both experimental conditions, pairwise comparison of fold-increases revealed poor concordance between most methods. The only exception was between the SCD test and Comet Assay, which showed moderate concordance (Lin's concordance correlation coefficients of approximately 0.5). Bland-Altman plot analysis further indicated that TUNEL detects the highest amounts of sDF during cryopreservation. This highlights that despite all methods measuring "DNA fragmentation," they may detect different types of damage or have varying sensitivities to specific lesion classes.
Technical Variability and Standardization Considerations
The Comet Assay demonstrates significant protocol-dependent variability that can affect results and inter-laboratory comparisons. A validation study examining different protocols and image analyzers found that standardizing agarose concentrations, DNA unwinding times, and electrophoresis conditions significantly improved result equivalence [40]. Additionally, the choice of summary measure for single-cell data (median, arithmetic mean, or geometric mean) can substantially influence study outcomes, with median % tail DNA generally providing the most robust statistical properties [42]. These technical considerations are crucial for experimental design and data interpretation when comparing results across studies.
Table 2: Comparative Performance of DNA Fragmentation Assays in Experimental Conditions
| Experimental Condition | SCSA Detection | Comet Assay Detection | Comparative Notes |
|---|---|---|---|
| Equine Semen Cryopreservation | No significant increase in DFI | Significant increase: 21.1% to 67.2% (p≤0.05) | Comet assay showed >3x increase post-thaw [41] |
| 72h Storage in SpermSafe | No significant increase | Significant increase: 21.1% to 53.5% (p≤0.05) [41] | Damage detected only by Comet assay |
| Cryopreservation (Multi-method Study) | Detected increase | Detected increase | Poor concordance with SCSA (CCC <0.5) [38] |
| In Vitro Incubation | Detected increase | Detected increase | Moderate concordance with SCD test only (CCC ~0.5) [38] |
| Tissue-Specific Genotoxicity | Not typically used for tissues | Effectively detects organ-specific damage [39] [40] | Comet adaptable to various tissues |
Integration with PARP-1 Cleavage Research
The relationship between DNA fragmentation and PARP-1 cleavage represents a critical intersection in cell death pathway research, particularly in apoptosis and parthanatos. PARP-1 is a nuclear enzyme that responds to DNA damage by catalyzing poly(ADP-ribosyl)ation of nuclear proteins and itself. During caspase-dependent apoptosis, PARP-1 is cleaved by caspases-3 and -7 into characteristic 24-kDa and 89-kDa fragments [25] [13]. This cleavage event serves as a biochemical hallmark of apoptosis and is frequently detected via Western blot as a complementary method to DNA fragmentation assays.
The 89-kDa PARP-1 fragment generated by caspase cleavage plays a novel role as a carrier of poly(ADP-ribose) (PAR) polymers to the cytoplasm, where it facilitates apoptosis-inducing factor (AIF) release from mitochondria—a crucial step in both apoptosis and parthanatos [13]. This PARP-1 fragment-mediated process directly connects proteolytic cleavage events with nuclear DNA fragmentation, demonstrating the mechanistic relationship between these biomarkers.
In experimental paradigms, researchers can employ DNA fragmentation assays (SCSA or Comet) in parallel with PARP-1 cleavage detection via Western blot to obtain complementary evidence of apoptotic commitment. For instance, in staurosporine-induced apoptosis, both PAR synthesis (indicating PARP-1 activation) and DNA fragmentation are observed, with pharmacological inhibition of either caspases or PARP-1 preventing downstream events including AIF-mediated nuclear shrinkage [13]. This multi-parameter approach provides robust verification of cell death mechanisms and strengthens experimental conclusions.
Detailed Experimental Protocols
SCSA Protocol
Principle: Flow cytometric measurement of DNA denaturation after acid treatment [38].
Reagents:
- Acid detergent solution (0.1% Triton X-100, 0.15 M NaCl, 0.08 N HCl, pH 1.2)
- Staining buffer (0.2 M Na₂HPO₄, 0.1 M citric acid, 1 mM EDTA, 0.15 M NaCl, pH 6.0)
- Acridine orange stock solution (1 mg/mL in distilled water)
Procedure:
- Dilute raw semen to 1-2 × 10⁶ sperm/mL in TNE buffer (0.01 M Tris-HCl, 0.15 M NaCl, 1 mM EDTA, pH 7.4).
- Mix 100 μL of diluted sample with 200 μL of acid detergent solution in a tube.
- After 30 seconds, add 1.2 mL of acridine orange staining solution.
- Analyze samples by flow cytometry within 3-5 minutes of staining.
- Measure fluorescence emission at 515-530 nm (green, double-stranded DNA) and >630 nm (red, denatured single-stranded DNA) after excitation with a 488-nm argon laser.
- Calculate DFI as the ratio of red to total (red + green) fluorescence.
Technical Notes:
- Analyze a minimum of 5,000 events per sample.
- Include reference samples with known DFI for quality control.
- Ensure consistent staining time and temperature across samples.
Comet Assay Protocol
Principle: Single-cell gel electrophoresis to quantify DNA strand breaks [39] [40].
Reagents:
- Lysis solution (2.5 M NaCl, 100 mM EDTA, 10 mM Tris, 1% Triton X-100, 10% DMSO, pH 10)
- Alkaline electrophoresis solution (300 mM NaOH, 1 mM EDTA, pH >13)
- Neutralization buffer (0.4 M Tris-HCl, pH 7.5)
- Fluorescent DNA stain (SYBR Gold, ethidium bromide, or similar)
Procedure:
- Suspend cells in low-melting-point agarose (0.5-0.7% in PBS) at approximately 1 × 10⁴ cells/mL.
- Spread 75-100 μL of cell suspension on pre-coated slides and cover with coverslip.
- Solidify slides at 4°C for 10 minutes, then carefully remove coverslip.
- Immerse slides in freshly prepared lysis solution at 4°C for at least 1 hour (overnight optimal).
- Transfer slides to alkaline unwinding solution for 20 minutes at 4°C in the dark.
- Perform electrophoresis at approximately 1 V/cm for 20-30 minutes (adjust based on cell type).
- Neutralize slides with Tris buffer (pH 7.5) for 5 minutes, then air dry.
- Stain with appropriate DNA dye and visualize using fluorescence microscopy.
- Score 50-100 randomly selected cells per sample using image analysis software.
Technical Notes:
- Include concurrent positive controls (e.g., cells treated with H₂O₂ or ethyl methanesulfonate).
- Standardize electrophoresis conditions (time, voltage, buffer volume) across experiments.
- For specific tissues like urinary bladder, mincing methods provide adequate epithelial cell yield while preserving tissue for histopathology [39].
PARP-1 Cleavage Detection via Western Blot
Principle: Immunodetection of caspase-cleaved PARP-1 fragments [5] [13].
Reagents:
- RIPA lysis buffer with protease inhibitors
- Primary antibodies: anti-PARP-1 (full length and cleaved fragments)
- Secondary antibodies conjugated to HRP or fluorescent dyes
Procedure:
- Prepare cell lysates in RIPA buffer with protease inhibitors.
- Quantify protein concentration and load equal amounts (20-30 μg) per lane on SDS-PAGE gel.
- Transfer proteins to PVDF or nitrocellulose membrane.
- Block membrane with 5% non-fat milk or BSA in TBST.
- Incubate with primary antibody overnight at 4°C.
- Wash and incubate with HRP-conjugated secondary antibody.
- Detect using chemiluminescence or fluorescence imaging.
- Normalize to loading controls (β-actin, GAPDH).
Technical Notes:
- Antibodies should recognize both full-length (116-kDa) and cleaved (89-kDa) PARP-1.
- Include apoptosis-positive controls (e.g., staurosporine-treated cells).
- Use apoptosis antibody cocktails for efficient multi-marker detection [5].
Research Reagent Solutions
Table 3: Essential Reagents for DNA Fragmentation and PARP-1 Analysis
| Reagent/Category | Specific Examples | Research Function |
|---|---|---|
| DNA Staining Dyes | Acridine orange (SCSA), SYBR Gold, Ethidium bromide (Comet) | DNA quantification and visualization |
| Flow Cytometry Reagents | Acid detergent solution, TNE buffer | DNA denaturation and sample preparation for SCSA |
| Electrophoresis Materials | Low-melting-point agarose, alkaline electrophoresis buffer | DNA separation in Comet assay |
| PARP-1 Antibodies | Anti-PARP-1 (full length), anti-cleaved PARP-1 (89 kDa) | Detection of PARP-1 cleavage by Western blot |
| Apoptosis Inducers | Staurosporine, Actinomycin D | Positive controls for DNA damage and PARP-1 cleavage |
| Image Analysis Software | Comet Assay IV, Comet Analyzer, ImageJ | Quantification of DNA damage parameters |
The comparative analysis of SCSA and Comet Assay reveals distinct advantages and limitations for each method in DNA fragmentation research. The Comet Assay demonstrates superior sensitivity for detecting DNA damage induced by various stressors including cryopreservation and chemical exposure, while the SCSA offers higher throughput for population-based screening. The integration of these DNA fragmentation assays with PARP-1 cleavage analysis via Western blot provides complementary evidence in cell death research, offering insights into both upstream DNA damage and downstream proteolytic events in apoptosis and related pathways.
For researchers designing studies on DNA damage response, the selection between SCSA and Comet Assay should be guided by specific research questions, sample types, and required throughput. When maximal sensitivity and single-cell resolution are prioritized, particularly in heterogeneous cell populations, the Comet Assay represents the preferred approach. For large-scale screening studies where population-level statistics are sufficient and equipment access permits, SCSA provides efficient analytical capability. In both contexts, parallel assessment of PARP-1 cleavage fragments strengthens mechanistic conclusions about cell death pathways and enhances the rigor of DNA damage assessment in basic research and drug development applications.
Correlating PARP-1 Cleavage with DNA Damage in Treatment Models
Poly(ADP-ribose) polymerase-1 (PARP-1) is a 113-116 kDa nuclear enzyme that functions as a primary sensor of DNA damage [28] [25]. Upon detecting DNA strand breaks, its catalytic activity is activated, initiating poly(ADP-ribosyl)ation to recruit DNA repair machinery [13] [25]. However, under sustained or severe genotoxic stress, PARP-1 becomes a substrate for proteolytic cleavage by various cell death proteases, generating signature fragments that serve as biochemical hallmarks for specific cell death pathways [25]. The most characterized cleavage occurs at the aspartic acid 214 (Asp214) residue by caspase-3 and -7 during apoptosis, producing 24 kDa and 89 kDa fragments [43] [28] [13]. This proteolytic event inactivates DNA repair capacity and facilitates cellular disassembly, making PARP-1 cleavage a critical nexus between DNA damage response and cell fate determination [43] [13] [25].
This guide objectively compares the experimental approaches for detecting PARP-1 cleavage and DNA fragmentation, two correlated yet distinct biomarkers in cell death research. We provide structured methodological data and comparative analysis to inform researchers and drug development professionals in selecting appropriate assays for their therapeutic model systems.
Methodological Comparison: Western Blot vs. DNA Fragmentation Analysis
Table 1: Comparative Analysis of PARP-1 Cleavage Western Blot and DNA Fragmentation Assays
| Feature | PARP-1 Cleavage Western Blot | DNA Fragmentation Analysis |
|---|---|---|
| Detected Event | Proteolytic cleavage of PARP-1 protein [25] | Physical breakage of genomic DNA [44] |
| Primary Indication | Protease activation (e.g., Caspase-3/7) and apoptosis initiation [43] [25] | Advanced apoptotic execution or severe necrotic death [44] [15] |
| Key Outputs | Fragment size (89 kDa, 24 kDa, 50 kDa), protease specificity [43] [15] [25] | DNA Fragmentation Index (DFI), comet tail moment, ladder pattern [44] |
| Typical Workflow Time | 1-2 days (SDS-PAGE, transfer, immunodetection) [43] [45] | 1 day (SCSA, TUNEL) to 2 days (Comet, gel electrophoresis) [44] |
| Sample Throughput | Medium (typically 10-20 samples per gel) | Variable; high for flow cytometry (SCSA), low for comet assay |
| Quantification | Semi-quantitative (band density ratio of cleaved/full-length) [43] | Quantitative (DFI %, tail moment, % TUNEL-positive cells) [44] |
| Advantages | - Reveals specific protease involved- High specificity with validated antibodies- Provides mechanistic insight [25] | - Direct measure of genotoxicity- Strong correlation with cell death outcomes- Single-cell resolution (SCSA, Comet) [44] |
| Limitations | - Does not directly quantify DNA damage- Semi-quantitative without careful standardization | - Does not identify upstream signaling events- Can miss early apoptosis |
PARP-1 Cleavage Fragments as Signatures of Distinct Cell Death Pathways
PARP-1 is cleaved by different proteases activated in specific cell death contexts, producing characteristic fragments that serve as biochemical signatures.
Table 2: PARP-1 Cleavage Fragments Across Different Cell Death Paradigms
| Cell Death Pathway | Protease Involved | Cleavage Fragments | Functional Consequences | Experimental Inducers |
|---|---|---|---|---|
| Apoptosis | Caspase-3 and Caspase-7 [28] [13] [25] | 89 kDa (C-terminal, catalytic domain) and 24 kDa (N-terminal, DNA-binding domain) [43] [28] | Inactivation of DNA repair, conservation of ATP, facilitation of cellular disassembly [43] [13] | Staurosporine, Actinomycin D [13] |
| Necrosis | Lysosomal Proteases (e.g., Cathepsins B, D, G) [15] | ~50 kDa fragment [15] | Not fully elucidated; may contribute to inflammatory response [15] | 0.1% H₂O₂, 10% Ethanol, 100 μM HgCl₂ [15] |
| Parthanatos | Calpains, other proteases [25] | Fragments ranging from 42-89 kDa [25] [46] | PAR polymer translocation, AIF release, large-scale DNA fragmentation [13] | MNNG (Alkylating agent) [13] |
Experimental Protocol: Detecting PARP-1 Cleavage via Western Blot
Sample Preparation:
- Treat cells (e.g., HeLa, SH-SY5Y, primary neurons) with apoptosis inducers (e.g., 1 μM Staurosporine for 3-6 hours) [13] [46].
- Lyse cells in RIPA buffer supplemented with protease inhibitors.
- Quantify protein concentration using a BCA assay and normalize samples [37].
Gel Electrophoresis and Immunoblotting:
- Load 20-40 μg of total protein per lane on a 4-12% Bis-Tris polyacrylamide gel.
- Separate proteins by electrophoresis and transfer to a PVDF membrane.
- Block membrane with 5% non-fat milk in TBST for 1 hour.
Antibody Incubation and Detection:
- Incubate with primary antibody against cleaved PARP-1 (e.g., Cell Signaling Technology #5625 at 1:1000 or Proteintech 60555-1-Ig at 1:5000-1:50000) overnight at 4°C [43] [46].
- Wash membrane and incubate with HRP-conjugated secondary antibody.
- Detect bands using enhanced chemiluminescence (ECL) substrate.
- Expected results: Full-length PARP-1 at ~116 kDa; cleaved fragment at ~89 kDa [43].
Experimental Protocol: Assessing DNA Fragmentation via SCSA
Sample Collection and Staining:
- Collect semen or cell samples. For clinical studies, collect data on lifestyle factors (age, BMI, smoking status) via structured questionnaires [44].
- Dilute sample in TNE buffer (0.01 M Tris-HCl, 0.15 M NaCl, 0.001 M EDTA, pH 7.4).
- Mix aliquot with acidic solution (pH 1.2) containing 0.1% Triton X-100, 0.15 M NaCl, and 0.08 N HCl for 30 seconds.
- Stain with Acridine Orange (AO) staining solution (6 µg/mL in 0.1 M citric acid, 0.2 M Na2HPO4, 1 mM EDTA, 0.15 M NaCl, pH 6.0) [44].
Flow Cytometry Analysis:
- Analyze samples using a flow cytometer with 488 nm excitation.
- Measure green fluorescence (double-stranded DNA) at 530 ± 30 nm and red fluorescence (denatured, single-stranded DNA) at >630 nm.
- Calculate DNA Fragmentation Index (DFI) as the ratio of red to total (red + green) fluorescence intensity, expressed as a percentage [44].
- Interpretation: DFI >30% is considered abnormal and indicative of significant DNA damage in clinical male fertility studies [44].
Signaling Pathways Integrating PARP-1 Cleavage and DNA Damage
The relationship between PARP-1 cleavage and DNA damage is embedded within complex cell death signaling pathways. The following diagram illustrates the key apoptotic pathway where caspase-mediated PARP-1 cleavage and DNA fragmentation converge.
The Scientist's Toolkit: Key Research Reagents and Materials
Table 3: Essential Reagents for PARP-1 and DNA Damage Research
| Item | Specific Example | Function & Application Note |
|---|---|---|
| Anti-Cleaved PARP-1 Antibodies | Cleaved PARP (Asp214) (D64E10) Rabbit mAb #5625 [43] | Detects endogenous 89 kDa fragment in WB, IP, IHC, IF, FC. High specificity for caspase-cleaved form. |
| Anti-Cleaved PARP-1 Antibodies | Cleaved PARP (Asp214) (19F4) Mouse mAb #9546 [45] | Mouse monoclonal for WB; may detect high levels of full-length PARP. |
| Anti-Cleaved PARP-1 Antibodies | Cleaved PARP1 Monoclonal (60555-1-Ig) [46] | Recognizes only cleaved form, not full-length; works in WB, IHC, IF/ICC, FC, ELISA. |
| Apoptosis Inducers | Staurosporine [13] [46] | Broad-spectrum kinase inhibitor; induces intrinsic apoptosis and caspase-3-mediated PARP-1 cleavage. |
| PARP Inhibitors | PJ34, ABT-888 (Veliparib) [13] | Pharmacological inhibitors used to dissect PARP-1's role in cell death pathways. |
| Caspase Inhibitors | zVAD-fmk [13] [15] | Pan-caspase inhibitor; distinguishes caspase-dependent apoptosis from other death pathways. |
| DNA Damage Inducers | N-methyl-N'-nitro-N-nitrosoguanidine (MNNG) [13] | Alkylating agent; induces PARP-1-dependent, caspase-independent cell death (parthanatos). |
| Flow Cytometry Kits | SCSA Reagents [44] | Used for sperm DNA fragmentation analysis; adaptable for cell line studies. |
The correlative analysis of PARP-1 cleavage and DNA fragmentation provides a powerful dual-axis framework for evaluating treatment efficacy and mechanism of action in experimental models. Western blot analysis of PARP-1 cleavage offers early, mechanistic insight into the specific proteases activated by a therapeutic intervention, while DNA fragmentation assays provide a quantitative, terminal measure of genotoxic impact and cell death. The choice between, or combination of, these techniques should be guided by the specific research question, with PARP-1 cleavage illuminating the initiating mechanisms and DNA fragmentation confirming the irreversible commitment to cell death. This integrated approach is particularly valuable in cancer research and neurotoxicology for profiling cell death pathways activated by novel therapeutics.
Resolving Experimental Challenges: Artifacts, Specificity, and Quantification
Common Pitfalls in PARP-1 Western Blot and How to Avoid Them
In apoptosis research, two key analytical methods provide complementary evidence of programmed cell death: PARP-1 cleavage detection by Western blot and DNA fragmentation analysis. PARP-1, a DNA repair enzyme, is one of the earliest substrates cleaved by caspases during apoptosis, generating characteristic 89 kDa and 24 kDa fragments. While DNA laddering provides confirmation of late-stage apoptotic DNA cleavage, PARP-1 Western blotting detects earlier apoptotic events, offering superior temporal resolution for kinetic studies and drug response evaluation. However, obtaining clean, interpretable PARP-1 blots presents unique challenges due to the protein's abundance, modification states, and cleavage patterns. This guide systematically addresses these pitfalls and provides optimized protocols for reliable detection in pharmaceutical and basic research applications.
Technical Comparison: PARP-1 Western Blot vs. DNA Fragmentation Analysis
Table 1: Methodological Comparison for Apoptosis Detection
| Parameter | PARP-1 Cleavage Western Blot | DNA Fragmentation Analysis |
|---|---|---|
| Biological Process Detected | Early apoptosis (caspase activation) | Late apoptosis (endonuclease activation) |
| Time Resolution | Excellent for kinetic studies | Poor, end-point detection |
| Sample Throughput | Moderate (can multiplex with other targets) | High for sample number, but low for multiplexing |
| Required Expertise | Advanced protein techniques | Standard molecular biology techniques |
| Quantification Potential | High with proper normalization (e.g., Total Protein Normalization) | Semi-quantitative at best |
| Key Limitations | Non-specific bands, high background, protein modifications | Cannot detect early apoptosis, less specific to apoptosis |
| Drug Development Utility | Ideal for mechanism of action studies and pharmacodynamic biomarkers | Confirmation of cell death but limited mechanistic insight |
Common PARP-1 Western Blot Pitfalls and Experimental Solutions
Non-Specific Bands and Multiple Bands
Problem: Multiple bands appear instead of the expected 116 kDa full-length PARP-1 and 89 kDa cleavage fragment, potentially caused by cross-reacting antibodies, protein degradation, or post-translational modifications [47].
Solutions:
- Antibody Optimization: Titrate primary antibody concentration to minimize cross-reactivity; include a secondary-only control to identify non-specific secondary antibody binding [47].
- Pre-adsorption: Pre-adsorb the primary antibody with a lysate that does not contain PARP-1 to remove cross-reactive antibodies [48].
- Protein Integrity: Include protease inhibitors in lysis buffer; store samples at -80°C and avoid freeze-thaw cycles [47].
- Post-translational Modifications: Be aware that PARP-1 undergoes extensive post-translational modification, including ADP-ribosylation, phosphorylation, and ubiquitylation, which can affect migration [17] [49].
Weak or No Signal
Problem: Faint or absent bands for either full-length or cleaved PARP-1, potentially resulting from inefficient transfer, insufficient antigen, or antibody issues [47] [48].
Solutions:
- Transfer Efficiency Verification: Verify complete transfer using reversible staining methods; optimize transfer time and conditions, particularly for the 89 kDa fragment [48].
- Protein Loading: Load at least 20-30 μg of total protein per lane; concentrate samples if studying low-expression systems [47].
- Antigen Retrieval: For PVDF membranes, ensure proper activation by soaking in methanol for 1 minute before transfer [47].
- Antibody Validation: Check antibody datasheet for species reactivity; include a positive control (e.g., apoptotic cell lysate) [47].
High Background and Signal Artifacts
Problem: Excessive background noise obscuring specific bands, often caused by insufficient blocking, inadequate washing, or membrane handling issues [47] [48].
Solutions:
- Blocking Optimization: Use fresh blocking solution (BSA or non-fat dry milk) and extend blocking time; consider rapid blocking solutions for 10-minute incubations [47] [48].
- Enhanced Washing: Increase TBST washing frequency and duration (3-5 washes, 5 minutes each) to remove non-specifically bound antibodies [47].
- Membrane Hydration: Ensure membrane remains fully hydrated during all incubation steps; do not allow it to dry out [47].
- Antibody Concentration: Reduce primary and/or secondary antibody concentration if background persists [47].
PARP-1 Specific Considerations in Apoptosis Signaling
Diagram: PARP-1 in Apoptosis Signaling Pathway
Optimized PARP-1 Western Blot Protocol
Sample Preparation and Electrophoresis
- Cell Lysis: Use RIPA lysis buffer supplemented with protease inhibitors (e.g., PMSF) and PARP-specific inhibitors to prevent degradation and unwanted modification [47].
- Protein Quantification: Perform BCA assay with standard dilution buffer matched to sample buffer [47].
- Gel Electrophoresis: Use 8-10% SDS-PAGE gels for optimal resolution of 116 kDa and 89 kDa fragments; run at 150V until dye front reaches bottom [47].
- Protein Transfer: Activate PVDF membrane in methanol for 1 minute; transfer at 300mA for 60 minutes in ice bath to prevent overheating [47].
Immunodetection
- Blocking: Block with 5% BSA or non-fat dry milk in TBST for 1 hour at room temperature [47].
- Primary Antibody Incubation: Incubate with anti-PARP-1 primary antibody diluted in antibody diluent at 4°C overnight [47].
- Washing: Wash membrane 3 times with TBST for 10 minutes each with agitation [47].
- Secondary Antibody Incubation: Incubate with species-appropriate HRP-conjugated secondary antibody at room temperature for 1-2 hours [47].
- Detection: Develop with enhanced chemiluminescence (ECL) substrate and image with appropriate system [47].
Research Reagent Solutions for PARP-1 Western Blot
Table 2: Essential Reagents for PARP-1 Detection
| Reagent Category | Specific Product/Type | Function in PARP-1 Blot | PARP-Specific Considerations |
|---|---|---|---|
| Lysis Buffer | RIPA Lysis Buffer (strong) | Extracts nuclear and cytoplasmic PARP-1 | Must include protease inhibitors to prevent cleavage during preparation |
| Protease Inhibitors | PMSF or commercial cocktails | Prevents artifactual proteolysis | Critical for maintaining full-length PARP-1 integrity |
| Gel Type | 8-10% Tris-Glycine or Pre-cast Gels | Separates 116 kDa and 89 kDa fragments | Pre-cast gels offer better reproducibility for quantification |
| Membrane | 0.45 μm PVDF | Binds PARP-1 fragments efficiently | Requires methanol activation before transfer |
| Blocking Agent | BSA (5%) or non-fat dry milk (5%) | Reduces non-specific antibody binding | BSA preferred if phospho-specific antibodies are used simultaneously |
| Primary Antibody | Anti-PARP-1 (cleavage specific) | Detects full-length and cleaved fragments | Must be validated for species; monoclonal antibodies preferred for specificity |
| Detection Method | Enhanced Chemiluminescence (ECL) | Visualizes PARP-1 bands | Enhanced ECL provides better sensitivity for low-abundance cleaved fragments |
Data Normalization and Quantification Standards
For publication-quality PARP-1 Western blots, particularly in drug development studies, proper normalization is essential. The field is moving away from housekeeping proteins (HKPs) like GAPDH and β-actin toward Total Protein Normalization (TPN) as the gold standard [50]. TPN accounts for potential variability in protein concentrations, sample loading, and transfer efficiency, providing more accurate quantification of PARP-1 cleavage ratios. This approach is increasingly required by major journals including Journal of Biological Chemistry and Nature [50].
Mastering PARP-1 Western blotting requires careful attention to technical details specific to this apoptosis marker. By understanding the common pitfalls—non-specific bands, weak signals, and high background—and implementing the optimized protocols described, researchers can generate reliable, reproducible data for both basic research and drug development applications. The complementary use of PARP-1 cleavage analysis with DNA fragmentation studies provides a comprehensive approach to apoptosis assessment, with PARP-1 offering the distinct advantage of detecting early apoptotic events crucial for understanding therapeutic mechanisms of action.
Optimizing Antibody Specificity to Minimize Off-Target Detection
In the study of cellular responses to stress, particularly in the context of DNA damage and cell death, PARP-1 cleavage and DNA fragmentation stand as critical biomarkers. For researchers and drug development professionals, accurately detecting these events is paramount for understanding disease mechanisms and treatment efficacy. However, the accuracy of these detections hinges significantly on the specificity of the antibodies used in techniques like western blotting. Off-target detection can lead to misinterpretation of data, potentially derailing research conclusions and drug development pipelines. This guide provides a structured comparison of methodological approaches, detailing experimental protocols and key reagents to empower scientists in optimizing antibody specificity for more reliable and reproducible results in PARP-1 and DNA fragmentation analysis.
PARP-1 Cleavage and DNA Fragmentation as Biomarkers
PARP-1 Cleavage: A Proteolytic Signature
PARP-1 is a nuclear enzyme with a well-established role in DNA repair. Beyond its physiological function, it serves as a key substrate for several proteases activated during different cell death pathways. The cleavage of PARP-1 by these enzymes produces specific signature fragments that act as biochemical fingerprints, revealing the nature of the cell death process.
- Caspase-Dependent Apoptosis: The most well-characterized PARP-1 cleavage occurs during apoptosis, primarily executed by caspase-3 and caspase-7. This proteolysis generates a distinct 89 kDa catalytic fragment and a 24 kDa DNA-binding fragment (DBD). The 24-kD fragment irreversibly binds to damaged DNA, acting as a trans-dominant inhibitor of PARP-1 and other DNA repair enzymes, thereby facilitating the apoptotic process [25]. This cleavage event is considered a hallmark of apoptotic cell death.
- Cleavage by Other Proteases: PARP-1 is also a substrate for other "suicidal" proteases, including calpains, granzymes, cathepsins, and matrix metalloproteinases (MMPs). The activity of these proteases, often associated with alternative cell death pathways like necrosis or neuroinflammation, produces a set of PARP-1 fragments with different molecular weights, serving as biomarkers for these specific pathophysiological conditions [25].
DNA Fragmentation: A Hallmark of Genomic Demise
DNA fragmentation is a fundamental event in cell death, particularly apoptosis. Its analysis provides a window into the integrity of the genome and the cell's fate.
- Sperm DNA Fragmentation Index (DFI): In reproductive medicine, the sperm DFI has emerged as a crucial biomarker for assessing male fertility potential. A DFI value exceeding 30% is considered abnormal and may indicate a higher risk of failed pregnancy and miscarriage, as it potentially exceeds the DNA repair capacity of the oocyte [51] [52].
- Double-Stranded DNA Breaks (dsSDF): Beyond global DNA damage, the assessment of double-stranded sperm DNA fragmentation (dsSDF) via specialized comet assays has shown a particularly strong association with recurrent pregnancy loss, highlighting the importance of measuring specific types of DNA damage [52].
The following diagram illustrates the relationship between different cellular stresses, the proteases they activate, and the specific PARP-1 cleavage fragments that result, which are detectable via western blot.
Experimental Protocols for Key Assays
Western Blot Protocol for Detecting PARP-1 Cleavage
This protocol is optimized for resolving full-length PARP-1 and its cleavage products, a critical requirement for assessing antibody specificity.
Step 1: Protein Extraction and Quantification
- Lyse cells in a suitable IP lysis buffer (e.g., 50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% NP-40, 0.25% Sodium deoxycholate) supplemented with protease inhibitor cocktails [6]. For apoptosis studies, include caspase inhibitors in parallel samples to prevent artifactual cleavage during extraction.
- Centrifuge lysates at 13,500 rpm for 20 minutes at 4°C to remove debris.
- Determine protein concentration using an assay like BCA [6] [37]. Use 20-40 μg of total protein per sample for loading [6].
Step 2: Gel Electrophoresis and Transfer
- Resolve proteins by SDS-PAGE using a 8-12% gradient gel. This is crucial for separating the full-length PARP-1 (116 kDa) from the major apoptotic fragment (89 kDa) and smaller fragments.
- Transfer proteins to a PVDF or nitrocellulose membrane using standard wet or semi-dry transfer systems.
Step 3: Immunoblotting and Antibody Incubation
- Blocking: Incubate the membrane with 5% non-fat milk or BSA in TBST for 1 hour at room temperature to minimize non-specific binding.
- Primary Antibody Incubation: Incubate with the anti-PARP1 primary antibody diluted in blocking buffer overnight at 4°C. Key validation points include:
- Specificity Check: The antibody should clearly distinguish between full-length PARP-1 and the 89 kDa cleavage product. A single, clean band for each is ideal.
- Positive Control: Use lysates from cells treated with a known apoptosis inducer (e.g., Staurosporine) to generate the 89 kDa fragment.
- Negative Control: Use lysates from caspase-inhibited cells to confirm the absence of the 89 kDa band.
- Secondary Antibody Incubation: Incubate with an HRP-conjugated secondary antibody (e.g., goat anti-rabbit) for 1 hour at room temperature [37].
Step 4: Detection and Analysis
- Develop the blot using enhanced chemiluminescence (ECL) reagents and image with a digital imager.
- Probe the same membrane for a loading control (e.g., β-actin or α-Tubulin) to ensure equal protein loading [6].
Sperm DNA Fragmentation Index (DFI) Analysis Protocol
This protocol outlines the assessment of DNA fragmentation using the Sperm Chromatin Structure Assay (SCSA) [51].
Step 1: Sample Collection and Preparation
- Collect semen samples after a recommended period of sexual abstinence.
- Allow samples to liquefy and then analyze within a narrow time window (e.g., 30-60 minutes post-collection) to prevent artifactual DNA damage.
Step 2: Acid Denaturation and Staining
- Dilute a small aliquot of semen (e.g., containing ~2 million sperm) in a buffer.
- Subject the sample to a brief, mild acid treatment (e.g., pH 1.2) to denature DNA at sites of strand breaks.
- Immediately stain with Acridine Orange, a metachromatic dye that fluoresces green when intercalated with double-stranded DNA and red when associated with single-stranded DNA.
Step 3: Flow Cytometry Analysis
- Analyze the stained sample using a flow cytometer.
- Measure the fluorescence of 5,000-10,000 events per sample.
- The DFI is calculated as the ratio of red (denatured, fragmented DNA) to total (red + green) fluorescence intensity. A threshold of >30% DFI is commonly used to define abnormal sperm DNA integrity [51].
Comparative Data and Performance Analysis
Quantitative Comparison of Assay Performance
The table below summarizes the core characteristics, applications, and data outputs of the PARP-1 cleavage western blot versus DNA fragmentation assays, highlighting their complementary roles.
Table 1: Comparative Analysis of PARP-1 Cleavage and DNA Fragmentation Assays
| Feature | PARP-1 Cleavage Western Blot | Sperm DNA Fragmentation (DFI) | Comet Assay (dsSDF) |
|---|---|---|---|
| Biomarker Measured | Proteolytic fragments (e.g., 89 kDa) of PARP-1 protein [25] | Global susceptibility of sperm chromatin to denaturation [51] | Direct double-stranded DNA breaks (dsSDF) [52] |
| Primary Application | Cell death pathway identification (apoptosis vs. other) [25] | Male fertility assessment and prediction of assisted reproduction outcomes [51] | Investigating cause of recurrent pregnancy loss; high sensitivity for male factor contribution [52] |
| Key Output Metric | Presence/absence and intensity of specific protein bands | DNA Fragmentation Index (DFI) percentage [51] | Incidence of Damage (IOD) and tail moment [52] |
| Critical Threshold | Visual detection of 89 kDa fragment | DFI > 30% considered clinically significant [51] | Specific cut-off for dsSDF associated with RPL [52] |
| Diagnostic Performance | Qualitative/Semi-quantitative; gold standard for apoptosis | AUC: ~0.819 for predicting abnormal DFI from lifestyle factors [51] | AUC: 0.909 for association with recurrent pregnancy loss [52] |
Antibody Performance and Specificity Data
When selecting antibodies for PARP-1 detection, it is critical to choose those validated for specific applications. The following table compares common antibodies and their performance in detecting full-length and cleaved PARP-1.
Table 2: Comparison of Anti-PARP1 Antibodies for Western Blot Analysis
| Antibody Clone / Cat. No. | Host Species | Reported Specificity in Western Blot | Key Validation Points | Potential Off-Target Risks |
|---|---|---|---|---|
| CST #9532 [6] [25] | Rabbit | Detects endogenous levels of full-length PARP1 (116 kDa) and the large cleavage fragment (89 kDa) generated by caspase-3/caspase-7 [25]. | Widely cited; used in co-IP and western blot protocols [6]. | May detect other PARP family members if epitope is not unique. Validation with PARP1-knockout cells is recommended. |
| Proteintech 13371-1-AP [6] | Rabbit | Used for immunohistochemistry; western blot performance should be confirmed with apoptosis-induced lysates. | Used alongside CST #9532 in research [6]. | Specificity for cleaved fragments must be empirically determined by the user. |
| Santa Cruz sc-74469 | Mouse | Often reported to detect the 116 kDa and 89 kDa fragments. | Common in older literature. | Higher risk of non-specific bands; less ideal for detecting cleavage in complex samples. |
The Scientist's Toolkit: Essential Research Reagents
A successful experiment relies on high-quality, specific reagents. The following table details essential solutions and tools for optimizing antibody specificity in PARP-1 and DNA damage research.
Table 3: Key Research Reagent Solutions for PARP-1 and DNA Fragmentation Studies
| Reagent / Tool | Function / Application | Example Products / Specifications |
|---|---|---|
| High-Specificity Anti-PARP1 Antibodies | Detection of full-length and cleaved PARP1 in western blot. Critical for minimizing off-target signals. | Rabbit mAb [CST #9532]; Mouse mAb [Santa Cruz sc-74469] (requires rigorous validation). |
| Caspase Inhibitors | Control experiments to confirm that PARP-1 cleavage is caspase-dependent, reducing false positives. | Z-VAD-FMK (pan-caspase inhibitor) [37]. |
| PARP Inhibitors | Tool compounds to study PARP1 function and induce synthetic lethality in HR-deficient models [6] [53]. | Olaparib, Talazoparib [6] [53]. |
| Positive Control Lysates | Essential antibody validation control to ensure proper detection of PARP-1 cleavage fragments. | Lysates from cells treated with Staurosporine or other DNA-damaging agents. |
| Protease Inhibitor Cocktails | Prevent protein degradation during cell lysis and protein preparation, preserving intact PARP1. | Commercial cocktails (e.g., from Roche, Thermo Fisher) containing AEBSF, Aprotinin, etc. [6]. |
| Chemiluminescent Substrates | For sensitive detection of antibody-bound targets in western blotting. | Enhanced Chemiluminescence (ECL) and Super ECL substrates [37]. |
| Flow Cytometry Standards | For calibrating instruments in SCSA to ensure accurate and reproducible DFI measurements [51]. | Fluorescent calibration beads. |
Visualization of Experimental Workflow and Antibody Specificity Check
The following diagram outlines a logical workflow for designing an experiment to detect PARP-1 cleavage, incorporating critical steps to verify antibody specificity and minimize off-target detection.
Strategies to Distinguish Apoptotic Cleavage from Other PARP-1 Fragments
Within the field of cell death research, the cleavage of poly(ADP-ribose) polymerase-1 (PARP-1) serves as a critical diagnostic marker, yet its interpretation requires careful analysis. As a nuclear enzyme central to DNA repair and transcriptional regulation, PARP-1 undergoes proteolytic processing by different proteases activated in distinct cell death pathways [25]. The canonical 89 kDa fragment generated by caspase-3 cleavage at the DEVD214 site has long been recognized as a hallmark of apoptosis [13] [54]. However, emerging research reveals that multiple proteases—including calpains, cathepsins, granzymes, and matrix metalloproteinases—can process PARP-1 into different signature fragments, complicating experimental interpretation [25]. This guide provides a comprehensive comparison of PARP-1 cleavage patterns across cell death modalities, with particular emphasis on distinguishing apoptotic cleavage in western blot analysis versus DNA fragmentation assays within drug development research.
Molecular Signatures of PARP-1 Cleavage Fragments
The definitive identification of cell death pathways relies on recognizing characteristic PARP-1 fragment sizes generated by specific proteases. The table below summarizes the primary cleavage signatures associated with different cell death processes:
Table 1: PARP-1 Cleavage Signatures Across Cell Death Pathways
| Cell Death Pathway | Primary Proteases | Characteristic PARP-1 Fragments | Key Inhibitors | Functional Consequences |
|---|---|---|---|---|
| Apoptosis | Caspase-3, Caspase-7 | 89 kDa + 24 kDa fragments [25] [13] | zVAD-fmk [15] | Inactivation of DNA repair; conservation of cellular energy [25] |
| Necrosis | Cathepsins (B, G), Lysosomal proteases | ~50 kDa fragment [15] | CA-074 (cathepsin B inhibitor) [15] | Potential role in cellular disassembly |
| Parthanatos | Calpains | Multiple fragments (e.g., 55 kDa, 40 kDa) [25] | Calpain inhibitor ALLN [25] | AIF-mediated DNA fragmentation |
| Granzyme-Mediated Death | Granzyme A | ~50 kDa fragment [25] | Not specified | Nuclear-mitochondrial DNA damage |
The 89 kDa apoptotic fragment contains the automodification and catalytic domains but loses nuclear localization capacity, while the 24 kDa fragment retains the DNA-binding domain and nuclear localization signal [13]. In contrast, necrotic cleavage produces a dominant 50 kDa fragment through the action of lysosomal proteases such as cathepsins B and G, a process not inhibited by broad-spectrum caspase inhibitors like zVAD-fmk [15].
Table 2: Comparative Analysis of Apoptotic vs. Necrotic PARP-1 Cleavage
| Characteristic | Apoptotic Cleavage | Necrotic Cleavage |
|---|---|---|
| Primary Inducers | Staurosporine, Etoposide, Anti-FAS [55] | H₂O₂, HgCl₂, Ethanol [15] |
| Protease Family | Caspases | Lysosomal Proteases |
| Key Fragment Sizes | 89 kDa, 24 kDa [25] | 50 kDa [15] |
| Caspase Inhibitor Sensitivity | Sensitive (zVAD-fmk) [15] | Resistant [15] |
| DNA Fragmentation Pattern | Ordered nucleosomal ladder | Random degradation |
| Cellular Energy Requirement | ATP-dependent | ATP-independent |
Experimental Strategies for Distinguishing Cleavage Pathways
Western Blot Methodologies and Reagent Selection
Western blot analysis remains the gold standard for identifying PARP-1 cleavage fragments when optimized with appropriate controls and antibodies. The following workflow and reagent table outline critical methodological considerations:
Table 3: Essential Research Reagents for PARP-1 Cleavage Analysis
| Reagent Category | Specific Examples | Experimental Function | Application Notes |
|---|---|---|---|
| Cleavage-Specific Antibodies | Anti-Cleaved PARP (Asp214) [54] | Detects 89 kDa apoptotic fragment; does not recognize full-length PARP1 | Critical for specific apoptosis detection; 1:1000 dilution for WB [54] |
| Caspase Inhibitors | zVAD-fmk [15] | Broad-spectrum caspase inhibitor; confirms caspase-dependent apoptosis | Prevents 89 kDa fragment formation in apoptosis [15] |
| Lysosomal Protease Inhibitors | CA-074 (cathepsin B inhibitor) [15] | Inhibits cathepsin B-mediated necrotic cleavage | Suppresses 50 kDa fragment formation in necrosis [15] |
| PARP Activity Inhibitors | PJ34, ABT-888 [56] [13] | Inhibits PARP catalytic activity; distinguishes parthanatos | Prevents PAR formation and AIF translocation [13] |
| Apoptosis Inducers | Staurosporine, Anti-FAS [55] | Positive controls for apoptotic cleavage | Generate characteristic 89 kDa fragment within 2-6 hours [55] |
| Necrosis Inducers | H₂O₂ (0.1%), Ethanol (10%) [15] | Positive controls for necrotic cleavage | Produce 50 kDa fragment caspase-independently [15] |
Figure 1: Experimental Workflow for PARP-1 Cleavage Pathway Identification
Protocol for Western Blot-Based Distinction
Sample Preparation: Treat cells with apoptosis inducers (e.g., 1 μM staurosporine for 4 hours) or necrosis inducers (e.g., 0.1% H₂O₂). Include pretreatment groups with 50 μM zVAD-fmk (caspase inhibitor) or 10 μM CA-074 (cathepsin B inhibitor) for 1 hour before inducer application [15] [55].
Protein Extraction and Electrophoresis: Prepare whole-cell lysates using RIPA buffer with protease inhibitors. Load 20-30 μg protein per lane on 4-12% Bis-Tris gels for optimal separation of full-length (116 kDa) and cleaved PARP-1 fragments [55].
Antibody Detection: Transfer to PVDF membranes and probe with:
- Primary antibodies: Anti-cleaved PARP (Asp214) at 1:1000 dilution to specifically detect the 89 kDa apoptotic fragment [54]
- Pan-PARP antibody to detect full-length PARP and all fragments
- Loading control: β-actin or muscle actin [55]
- Secondary antibodies: HRP-conjugated anti-rabbit/anti-mouse at 1:1000-1:5000 dilution
Fragment Analysis: Identify specific cleavage patterns:
- Apoptotic signature: 89 kDa and 24 kDa fragments, inhibited by zVAD-fmk
- Necrotic signature: 50 kDa fragment, resistant to zVAD-fmk but inhibited by cathepsin inhibitors
- Parthanatos: Multiple fragments (55 kDa, 40 kDa), PARP activity-dependent [25]
DNA Fragmentation Analysis Comparative Approach
While western blot detects PARP-1 cleavage directly, DNA fragmentation analysis provides complementary evidence for apoptosis identification:
TUNEL Assay: Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) detects DNA strand breaks characteristic of late apoptosis. Co-localization of TUNEL positivity with PARP-1 cleavage confirms apoptotic progression [57].
DNA Laddering: Extract genomic DNA and separate via agarose gel electrophoresis. Apoptotic cells display ordered nucleosomal DNA fragmentation (∼180-200 bp ladder), while necrotic cells show random DNA degradation [15].
Combined Interpretation: The presence of both 89 kDa PARP-1 fragment and DNA laddering provides conclusive evidence of caspase-dependent apoptosis. Discordant results (e.g., 89 kDa fragment without DNA laddering) may indicate early apoptosis or alternative fragment functions.
Advanced Research Applications and Functional Consequences
Non-Apoptotic Functions of PARP-1 Fragments
Beyond their role as cell death markers, PARP-1 fragments possess distinct biological activities. The 89 kDa fragment translocates to the cytoplasm during apoptosis, where it can mono-ADP-ribosylate RNA polymerase III, facilitating IFN-β production and enhancing antiviral responses [58]. This fragment serves as a PAR carrier to the cytoplasm, inducing AIF release from mitochondria and contributing to nuclear shrinkage [13]. The 24 kDa DNA-binding fragment remains nuclear where it may inhibit DNA repair by blocking access of repair enzymes to damaged DNA [25].
Uncleavable PARP-1 Mutant Studies
Research employing caspase-resistant PARP-1 (PARP-1UNCL) with mutated DEVD214 cleavage site provides insights into fragment-specific functions. Cells expressing PARP-1UNCL show:
- Reduced inflammatory responses to endotoxic shock and ischemia/reperfusion injury
- Impaired NF-κB transcriptional activity despite normal DNA binding
- Protection from oxygen/glucose deprivation damage in neuronal models [28] [59]
These findings demonstrate that PARP-1 cleavage fragments actively regulate inflammatory responses independent of their roles in cell death.
Distinguishing apoptotic PARP-1 cleavage from other fragments requires a multifaceted methodological approach. Western blot analysis with cleavage-specific antibodies provides the most direct detection method, while DNA fragmentation analysis offers complementary evidence of apoptotic progression. Critical to this distinction is the implementation of protease inhibitor studies and recognition of characteristic fragment sizes—the 89 kDa fragment for apoptosis versus 50 kDa for necrosis. As research continues to reveal the diverse biological functions of PARP-1 fragments, these discrimination strategies become increasingly vital for accurate interpretation of cell death mechanisms in basic research and drug development contexts.
Troubleshooting Inconsistent DNA Fragmentation Results
Inconsistent results in DNA fragmentation analysis, particularly in experiments linking PARP-1 cleavage to apoptotic signaling, present significant challenges in biomedical research and drug development. These inconsistencies can stem from various technical and biological factors, including methodological limitations, reagent specificity issues, and the complex interplay of different cell death pathways. This guide objectively compares established and emerging methodologies for detecting DNA fragmentation and its molecular signatures, providing supporting experimental data to help researchers identify and resolve key pain points in their experimental workflows. The content is framed within the broader context of PARP-1 cleavage research, which serves as a critical biomarker connecting DNA damage responses to apoptotic execution.
Technical Comparison of Detection Methodologies
Established Biomarker Analysis: PARP-1 Cleavage Detection
The detection of cleaved PARP-1 fragments via Western blotting remains a gold standard for identifying apoptotic cells in research settings. Multiple commercially available antibodies target specific cleavage sites and fragments, with varying performance characteristics.
Table 1: Comparison of Cleaved PARP-1 Antibodies for Western Blot Analysis
| Product Name | Host Species & Clonality | Reactivity | Specificity | Detected Fragment | Recommended Dilution | Key Applications |
|---|---|---|---|---|---|---|
| Cleaved PARP (Asp214) (D64E10) Rabbit mAb #5625 [60] | Rabbit Monoclonal | Human, Mouse, Monkey | Detects only 89 kDa fragment; not full-length PARP-1 | 89 kDa (catalytic domain) | 1:1000 (WB) | WB, IP, IHC, IF, FC |
| Cleaved PARP1 Antibody #60555-1-Ig [61] | Mouse Monoclonal | Human, Mouse, Rat | Recognizes only cleaved form, not full-length PARP1 | 89 kDa | 1:5000-1:50000 (WB) | WB, IHC, IF/ICC, FC, ELISA |
| Cleaved PARP (Asp214) Antibody #9544 [62] | Rabbit Polyclonal | Human, Mouse | Detects only 89 kDa fragment; not full-length PARP-1 | 89 kDa | 1:1000 (WB) | Western Blotting |
| Anti-Cleaved PARP1 antibody (ab4830) [31] | Rabbit Polyclonal | Human | Specific for 85 kDa fragment at cleavage site Asp214/Gly215 | 85 kDa | 1:1000-1:2000 (WB) | Western Blot |
The cleavage of PARP-1 at Asp214 by caspases separates the 116 kDa full-length protein into 24 kDa DNA-binding and 89 kDa catalytic fragments, serving as a hallmark of apoptosis [60] [25]. This specific cleavage event disrupts DNA repair capabilities and facilitates cellular disassembly. Antibodies such as the Cleaved PARP (Asp214) (D64E10) Rabbit Monoclonal Antibody #5625 provide superior lot-to-lot consistency through recombinant production methods [60].
Emerging Fragmentomics Approaches
Cell-free DNA (cfDNA) fragmentation analysis has emerged as a promising non-invasive biomarker for disease diagnosis and prognosis, particularly in oncology applications. Unlike targeted PARP-1 cleavage detection, fragmentomics analyzes genome-wide patterns of DNA fragmentation.
Table 2: Comparison of DNA Fragmentation Analysis Methods
| Methodology | Biological Target | Sample Type | Key Metrics | Throughput | Applications |
|---|---|---|---|---|---|
| PARP-1 Cleavage Western Blot | Caspase-cleaved PARP-1 protein | Cell lysates, tissues | 89 kDa fragment presence | Medium | Apoptosis confirmation in research |
| Sperm Chromatin Structure Assay (SCSA) [44] | Sperm DNA fragmentation | Semen samples | DNA Fragmentation Index (DFI) | Medium | Male infertility assessment |
| Whole Genome Sequencing Fragmentomics [63] [64] | Genome-wide fragmentation patterns | Plasma cfDNA | Nucleosome positioning, fragment size distribution, end motifs | High | Cancer detection, prenatal testing |
| Targeted Panel Fragmentomics [64] | Fragmentation in specific genomic regions | Plasma cfDNA | Normalized depth, fragment diversity, end motifs | High | Cancer phenotyping, therapy monitoring |
FinaleToolkit represents a significant advancement in cfDNA fragmentation analysis, offering a ~50-fold increase in processing speed for genome-wide fragmentation feature calculation compared to original implementations [63]. This high-speed computational toolkit enables researchers to analyze over 1 billion fragments from cfDNA whole-genome sequencing data in approximately 0.7 hours, making large-scale fragmentomics studies feasible.
Experimental Protocols for Consistent Results
Standardized Western Blot Protocol for Cleaved PARP-1 Detection
Sample Preparation:
Electrophoresis and Transfer:
- Load 20-40 μg of protein per lane on 4-12% Bis-Tris gels.
- Transfer to PVDF membranes using standard wet transfer systems.
Immunoblotting:
- Block membranes with 5% non-fat milk in TBST for 1 hour.
- Incubate with primary antibody (e.g., Cleaved PARP (Asp214) Antibody #9544 at 1:1000 dilution) overnight at 4°C [62].
- Wash and incubate with appropriate HRP-conjugated secondary antibody.
- Detect using enhanced chemiluminescence substrate.
Validation:
- Always include both positive (apoptosis-induced) and negative (untreated) controls.
- Probe for full-length PARP-1 (116 kDa) to confirm specificity of cleavage detection.
- Re-probe membrane with loading control (e.g., GAPDH, β-actin).
Fragmentomics Analysis Workflow for cfDNA
Sample Processing:
- Isolate cfDNA from plasma using validated extraction kits.
- Quality control using Bioanalyzer or TapeStation to confirm fragment size distribution.
Library Preparation and Sequencing:
- Prepare sequencing libraries preserving native fragment ends.
- Sequence using either whole-genome (low-coverage) or targeted approaches.
Bioinformatic Analysis:
- Process raw sequencing data using FinaleToolkit for fragmentation feature extraction [63].
- Calculate key metrics: normalized fragment depth, fragment size distribution, end motif diversity, nucleosome positioning patterns.
- For targeted panels, utilize all exons rather than just first exons for improved performance [64].
PARP-1 Cleavage in Cell Death Signaling Pathways
The following diagram illustrates the central role of PARP-1 cleavage in DNA damage response and apoptotic signaling pathways, explaining its significance as a biomarker in fragmentation analysis:
This pathway highlights how PARP-1 cleavage serves as an irreversible commitment point in apoptotic signaling, with the 24 kDa fragment acting as a trans-dominant inhibitor of DNA repair by irreversibly binding to damaged DNA [25].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for DNA Fragmentation Analysis
| Reagent/Category | Specific Examples | Function & Application | Technical Considerations |
|---|---|---|---|
| Cleaved PARP-1 Antibodies | #5625 (CST), #60555-1-Ig (PTGLab), #9544 (CST), ab4830 (Abcam) | Specific detection of apoptotic cells via Western blot, IHC, IF | Validate specificity with cleavage-site mutants; optimize dilution for each application |
| Apoptosis Inducers | Staurosporine (1-3 μM), Etoposide (1 μM) | Positive controls for PARP-1 cleavage experiments | Titrate concentration and duration to achieve sub-optimal cleavage for assay sensitivity |
| cfDNA Extraction Kits | QIAamp Circulating Nucleic Acid Kit, MagMAX Cell-Free DNA Isolation Kit | Isolation of high-quality cfDNA from plasma/serum | Preserve native fragment ends; minimize contamination with genomic DNA |
| Targeted Sequencing Panels | Tempus xF (105 genes), Guardant360 CDx (55 genes), FoundationOne Liquid CDx (309 genes) [64] | Capture and analysis of cancer-associated genes in cfDNA | FoundationOne panel shows superior performance for fragmentomics analysis [64] |
| Computational Tools | FinaleToolkit [63] | High-speed analysis of cfDNA fragmentation features | 50x faster processing enables genome-wide analysis in large datasets |
Troubleshooting Common Experimental Issues
Inconsistent PARP-1 Cleavage Detection
Unexpected Band Patterns:
- Problem: Multiple bands or smearing on Western blots.
- Solution: Ensure fresh protease inhibitors in lysis buffer; avoid repeated freeze-thaw cycles; titrate antibody concentration (e.g., #60555-1-Ig can be used at 1:5000-1:50000 for WB) [61].
- Advanced Consideration: Recognize that different proteases (calpains, cathepsins, granzymes, MMPs) can generate alternative PARP-1 cleavage fragments (42-89 kDa) beyond the canonical caspase-generated 89 kDa fragment [25].
Weak or No Signal:
- Problem: Failure to detect cleaved PARP-1 in apoptotic samples.
- Solution: Include recommended positive controls (Staurosporine-treated HeLa or Jurkat cells); extend treatment duration; confirm apoptosis via additional markers (caspase-3 activation).
- Experimental Note: PARP-1 cleavage fragments may exhibit different subcellular localization, with the 89 kDa fragment potentially liberating from nucleus to cytosol [25].
Variability in Fragmentomics Data
Low Predictive Performance:
- Problem: Poor discrimination between cancer and non-cancer samples using cfDNA fragmentation.
- Solution: Utilize normalized fragment read depth across all exons rather than just first exons, as this metric demonstrates superior performance (AUROC 0.943-0.964) [64].
- Panel Selection: When using commercial panels, FoundationOne Liquid CDx (309 genes) outperforms smaller panels like Guardant360 CDx (55 genes) for fragmentomics applications [64].
Technical Artifacts in cfDNA Sequencing:
- Problem: Biased representation of fragment patterns.
- Solution: Optimize library preparation to minimize PCR duplicates; use unique molecular identifiers; ensure sufficient sequencing depth (>3000x for targeted panels).
The selection between PARP-1 cleavage analysis and DNA fragmentation approaches depends on specific research questions and experimental contexts. PARP-1 Western blotting provides specific, mechanistically grounded evidence of apoptotic activation in controlled experimental systems, while cfDNA fragmentomics offers non-invasive, multidimensional profiling for clinical applications. Researchers can significantly improve reproducibility by standardizing protocols, validating reagents, and selecting appropriate analytical methods based on well-characterized performance metrics. As both technologies continue to evolve, their integration may provide complementary insights into DNA fragmentation biology across fundamental research and translational applications.
Beyond the Blot: Validating PARP Inhibition and Therapeutic Efficacy
Using Cleavage and Fragmentation as Biomarkers for PARP Inhibitor Efficacy
Poly (ADP-ribose) polymerase inhibitors (PARPi) have transformed cancer treatment, particularly for homologous recombination repair (HRR)-deficient tumors. A critical challenge in their clinical application is identifying robust biomarkers to predict and monitor therapeutic response. Within this context, the analysis of PARP-1 cleavage fragments via western blot and the assessment of DNA fragmentation have emerged as valuable techniques for researching PARP inhibitor efficacy and mechanisms of action. This guide provides a systematic comparison of these methodological approaches, supporting researchers in selecting appropriate techniques for their experimental goals.
Molecular Basis of PARP-1 Cleavage and DNA Fragmentation
PARP-1 Cleavage Fragments as Proteolytic Signatures
PARP-1 is a 116-kDa nuclear enzyme that plays a central role in DNA damage detection and repair. Upon activation by DNA strand breaks, it catalyzes poly(ADP-ribosyl)ation of target proteins [13]. PARP-1 serves as a key substrate for several proteases during distinct cell death pathways:
Caspase-Mediated Cleavage: During apoptosis, caspases-3 and -7 cleave PARP-1 at a specific site (216-Asp-Gly-Val-Asp-219) within the nuclear localization signal, generating 24-kDa and 89-kDa fragments [13] [25]. The 24-kDa fragment contains the DNA-binding domain, while the 89-kDa fragment contains the automodification and catalytic domains.
Functional Consequences: Cleavage separates PARP-1's DNA-binding function from its catalytic activity. The 24-kDa fragment acts as a trans-dominant inhibitor of DNA repair by irreversibly binding to DNA strand breaks, while the 89-kDa fragment can translocate to the cytoplasm [13] [25].
Parthanatos Connection: In caspase-independent parthanatos, PARP-1 overactivation leads to poly(ADP-ribose) (PAR) polymer translocation to the cytoplasm. Caspase activation can generate poly(ADP-ribosyl)ated 89-kDa fragments that serve as PAR carriers to the cytoplasm, inducing apoptosis-inducing factor (AIF) release from mitochondria [13].
DNA Fragmentation as a Cell Death Indicator
DNA fragmentation represents a downstream consequence of PARP inhibition and subsequent DNA damage accumulation:
Replication Stress Mechanism: PARPi impedes maturation of nascent DNA strands during replication, particularly affecting Okazaki fragment processing. This creates post-replicative single-strand nicks or gaps that contribute to genomic instability [65].
Synthetic Lethality Context: In HRR-deficient cells, PARP inhibition causes accumulation of unresolved DNA single-strand breaks that collapse replication forks into double-strand breaks. The inability to repair these lesions through homologous recombination leads to extensive DNA fragmentation and cell death [66] [67].
Cell Death Execution: In later stages of apoptosis and parthanatos, activation of nucleases (including AIF-associated DNAase) causes large-scale DNA fragmentation, which can be detected as a laddering pattern or increased comet tail moments [13] [65].
The relationship between PARP-1 cleavage and DNA fragmentation in cell death pathways can be visualized as follows:
Experimental Approaches: Direct Comparison
PARP-1 Cleavage Western Blot Methodology
Sample Preparation Protocol:
- Harvest cells after PARPi treatment (typically 1-48 hours depending on model system)
- Prepare whole cell extracts using RIPA buffer (25mM Tris-HCl pH 7.6, 150mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) supplemented with protease inhibitors (e.g., PMSF, leupeptin, aprotinin) [13] [68]
- Quantify protein concentration using BCA assay, adjust samples to equal concentration
- Denature samples in Laemmli buffer with XT reducing agent at 90°C for 5 minutes [68]
Electrophoresis and Detection:
- Load 30-50μg protein per lane on 3-8% Criterion XT gels [68]
- Transfer to nitrocellulose membranes at 100V for 1 hour [68]
- Block with 5% non-fat milk in TBST for 1 hour at room temperature
- Incubate with primary antibodies overnight at 4°C:
- Anti-PARP-1 antibody (e.g., sc-53643, Santa Cruz) at 1:500 dilution [68]
- Anti-cleaved PARP (Asp214) specific antibodies for apoptosis detection
- Incubate with HRP-conjugated secondary antibodies (1:2000 dilution) for 1 hour at room temperature [68]
- Develop with ECL substrate and capture chemiluminescence using imaging systems (e.g., ChemiDoc Imager) [68]
Key Quality Controls:
- Include GAPDH (1:1000) or β-actin as loading controls [68]
- Use PARPi-treated and untreated controls as reference points
- Ensure detection of both full-length (116-kDa) and cleaved fragments (89-kDa, 24-kDa)
DNA Fragmentation Analysis Methods
Alkaline Comet Assay Protocol:
- Embed PARPi-treated cells in low-melting-point agarose on microscope slides
- Lyse cells in alkaline lysis buffer (2.5M NaCl, 100mM EDTA, 10mM Tris, 1% Triton X-100, pH 10) for 1 hour at 4°C [65]
- Place slides in alkaline electrophoresis buffer (300mM NaOH, 1mM EDTA, pH >13) for 20-40 minutes to unwind DNA
- Perform electrophoresis at 25V for 20-30 minutes
- Neutralize with Tris buffer and stain with DNA-binding dye (e.g., SYBR Gold, propidium iodide)
- Analyze using fluorescence microscopy; measure tail moment (product of tail length and fraction of total DNA in tail) [65]
BrdU-Labeling for Nascent DNA Strand Assessment:
- Pulse-label cells with 10μM BrdU for 30 minutes prior to PARPi treatment [65]
- Chase with drug-free media for specified periods (0-90 minutes)
- Perform alkaline comet assays as described above
- Detect BrdU-labeled nascent DNA strands using anti-BrdU antibodies [65]
- Quantify tail moments specifically in BrdU-labeled populations
DNA Laddering Assay:
- Extract genomic DNA using phenol-chloroform method
- Load 1-2μg DNA per lane on 1.5-2% agarose gels
- Run electrophoresis at 50-100V for 2-3 hours
- Visualize using ethidium bromide or SYBR Safe staining
- Apoptotic cells show characteristic ~180-200bp DNA laddering pattern
Table 1: Technical Comparison of PARP-1 Cleavage Western Blot vs. DNA Fragmentation Analysis
| Parameter | PARP-1 Cleavage Western Blot | DNA Fragmentation Analysis |
|---|---|---|
| Target | PARP-1 protein and its proteolytic fragments | Genomic DNA integrity |
| Detection Method | Antibody-based immunodetection | DNA staining or antibody-based detection |
| Key Readouts | 89-kDa and 24-kDa fragment presence; full-length:cleaved ratio | Tail moment (comet); oligonucleosomal laddering |
| Time Course | Early apoptosis (hours post-treatment) | Intermediate-late apoptosis (hours-days post-treatment) |
| Sample Throughput | Medium (typically 10-20 samples per gel) | Low-medium (typically 1 sample per slide for comet) |
| Quantification Approach | Densitometry of band intensity | Image analysis of tail moment or ladder intensity |
| Specialized Equipment | Gel electrophoresis system, transfer apparatus, chemiluminescence imager | Fluorescence microscope (comet), gel documentation system |
| Key Advantages | Specific protease activity signature; distinguishes cell death pathways | Direct measure of genomic instability; sensitive detection of early damage |
Table 2: Biological Significance of Detection Events
| Detection Event | Biological Significance | Primary Cell Death Pathway | Upstream Triggers | Downstream Consequences |
|---|---|---|---|---|
| PARP-1 Cleavage (89-kDa/24-kDa) | Caspase activation; commitment to apoptotic program | Apoptosis | PARP trapping, DNA damage, caspase-8/-9 activation | Inactivation of DNA repair, conservation of ATP, facilitation of cellular dismantling |
| PARP-1 Cleavage with PARylated 89-kDa | Cross-talk between apoptosis and parthanatos | Hybrid apoptosis-parthanatos | Extensive DNA damage with caspase activation | AIF-mediated DNA fragmentation, nuclear shrinkage [13] |
| DNA Fragmentation (Comet Assay) | DNA strand break accumulation | Parthanatos; apoptosis; replication stress | PARP inhibition, unresolved Okazaki fragments, replication fork collapse | Genomic instability, cell death through synthetic lethality [65] |
| Oligonucleosomal DNA Laddering | End-stage apoptotic DNA degradation | Late apoptosis | Caspase-activated DNase (CAD) activation | Nuclear condensation, phagocyte recognition |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for PARP-1 Cleavage and DNA Fragmentation Studies
| Reagent Category | Specific Examples | Function/Application | Key Considerations |
|---|---|---|---|
| PARP Inhibitors | Olaparib, Rucaparib, Niraparib, Talazoparib, PJ34 | Induce PARP trapping and synthetic lethality | Varying PARP trapping potency and durability of inhibition [68] |
| PARP-1 Antibodies | Santa Cruz sc-53643; Cleaved PARP (Asp214) antibodies | Detect full-length and cleaved PARP-1 | Specificity for different epitopes; some specifically recognize caspase-cleaved form |
| Apoptosis Inducers | Staurosporine, Actinomycin D, Etoposide | Positive controls for caspase activation and PARP-1 cleavage | Different mechanisms of apoptosis induction |
| Caspase Inhibitors | zVAD-fmk | Confirm caspase-dependent PARP-1 cleavage | Pan-caspase inhibitor establishes mechanism |
| DNA Staining Dyes | SYBR Gold, Propidium Iodide, Ethidium Bromide | Visualize DNA in comet assays and laddering | Sensitivity and compatibility with detection systems |
| BrdU Labeling Kits | Anti-BrdU antibodies, BrdU solution | Label and detect nascent DNA strands | Critical for assessing replication-associated DNA damage [65] |
| Cell Lines | IGROV-1, ES-2 ovarian cancer, DT40 FEN1-/- | Model systems with known PARPi sensitivity | FEN1-deficient cells show enhanced PARP activity and PARPi sensitivity [68] [65] |
Data Interpretation and Integration
Correlation Analysis Between Cleavage and Fragmentation
The temporal relationship between PARP-1 cleavage and DNA fragmentation follows a generally sequential pattern, though significant overlap exists depending on the cell death mechanism:
Early Events (0-6 hours): PARP-1 cleavage fragments typically appear within 1-4 hours of caspase activation, while DNA fragmentation becomes detectable slightly later (4-8 hours) in apoptosis [13].
Parthanatos Timeline: In caspase-independent parthanatos, DNA fragmentation may occur without classical PARP-1 cleavage, instead involving PAR translocation and AIF-mediated DNA degradation [13].
Replication Stress Response: PARPi-induced DNA fragmentation during S-phase may precede significant PARP-1 cleavage, particularly in HR-deficient cells experiencing replication catastrophe [65].
Technical Considerations for Experimental Design
Complementary Approaches:
- Combine western blot and DNA fragmentation analysis for comprehensive cell death characterization
- Include multiple time points to capture dynamic processes
- Use caspase inhibitors to distinguish caspase-dependent and independent mechanisms
Troubleshooting Common Issues:
- Incomplete PARP-1 cleavage detection may require optimization of protein extraction and antibody concentrations
- High background in comet assays may indicate excessive electrophoresis time or voltage
- DNA laddering may be difficult to detect in cell types with low endonuclease activity
The following diagram illustrates a recommended experimental workflow for comprehensive biomarker assessment:
PARP-1 cleavage western blot and DNA fragmentation analysis provide complementary yet distinct information for evaluating PARP inhibitor efficacy. Western blot detection of the characteristic 89-kDa and 24-kDa PARP-1 fragments offers specific evidence of caspase activation and commitment to apoptotic cell death, serving as a precise biomarker for early therapeutic response assessment. DNA fragmentation analysis, particularly through sensitive techniques like the alkaline comet assay, provides direct measurement of genomic instability and replication stress, capturing broader mechanisms of PARPi-induced cytotoxicity including parthanatos and replication catastrophe.
The optimal approach depends on specific research objectives: PARP-1 cleavage analysis excels in mechanism-of-action studies and apoptosis confirmation, while DNA fragmentation assays provide sensitive detection of DNA damage accumulation and are particularly valuable in assessing replication-associated damage in S-phase cells. For comprehensive PARP inhibitor evaluation, implementing both methodologies in a time-course design offers the most complete assessment of therapeutic response and resistance mechanisms, enabling researchers to fully characterize the complex cell death pathways engaged by PARP inhibition.
Poly (ADP-ribose) polymerase (PARP) inhibitors represent a significant advancement in cancer therapy, particularly for tumors with homologous recombination deficiencies. These drugs primarily target PARP1 and PARP2, nuclear enzymes that play a critical role in the DNA damage response (DDR) [69]. The therapeutic effect of PARP inhibitors operates on a dual mechanism: the well-established catalytic inhibition and the more recently discovered PARP trapping effect [69] [36]. Understanding the distinction between these mechanisms is crucial for drug development, as it directly influences compound selection, assay design, and the interpretation of experimental outcomes. This guide objectively compares these mechanisms and the assays used to reveal them, providing a framework for researchers investigating PARP-1 function and inhibition.
Core Concepts: Catalytic Inhibition and PARP Trapping
Catalytic Inhibition: Blocking the Enzymatic Activity
Catalytic inhibition is the classical mechanism by which PARP inhibitors function. These small molecules compete with the cofactor NAD+ for binding to the catalytic domain of PARP1 and PARP2 [69]. Under normal conditions, upon binding to DNA single-strand breaks, PARP enzymes become active and synthesize poly(ADP-ribose) (PAR) chains on themselves (auto-PARylation) and on target proteins. This PARylation serves as a signal to recruit other DNA repair proteins [70] [69]. The auto-PARylation also introduces a strong negative charge on PARP itself, leading to its release from DNA, thereby allowing the repair machinery to access the damage site [69].
PARP Trapping: A More Potent Cytotoxic Mechanism
PARP trapping is a more recently characterized mechanism that explains the significant cytotoxic potency of certain clinical PARP inhibitors. In this scenario, the inhibitor not only blocks PARylation but also "traps" the PARP enzyme on damaged DNA [70] [69]. The trapped PARP-DNA complex creates a physical barrier that blocks replication fork progression and prevents the recruitment of other repair factors. This leads to the stalling and collapse of replication forks, resulting in lethal double-strand breaks during DNA replication [36]. For cancer cells already deficient in homologous recombination repair (e.g., those with BRCA1/2 mutations), this overload of damage is irreparable, leading to synthetic lethality [69] [71].
Table: Fundamental Differences Between Catalytic Inhibition and PARP Trapping
| Feature | Catalytic Inhibition | PARP Trapping |
|---|---|---|
| Primary Effect | Blocks PAR polymer formation | Stabilizes PARP-DNA complexes |
| Consequence | Impairs DNA damage signal amplification | Creates physical replication blocks |
| PARP Release from DNA | Unaffected (initially) | Prevented |
| Cytotoxic Potency | Generally moderate | Highly potent |
| Correlation with Clinical Efficacy | Weak | Strong |
The following diagram illustrates the key differences in the mechanisms of action between a trapped PARP complex and one that is catalytically inhibited.
Quantitative Comparison of PARP Inhibitors
The potency of PARP trapping varies significantly among different clinical PARP inhibitors, and this does not always directly correlate with their catalytic inhibition strength [70] [69]. This differential trapping ability is a key determinant of their cytotoxic potency and clinical efficacy.
Table: Comparison of Clinical PARP Inhibitors by Trapping Potency and Selectivity
| PARP Inhibitor | Relative PARP1Trapping Potency (EC₅₀) | Relative PARP2Trapping Potency (EC₅₀) | PARP1/PARP2Selectivity | Key Clinical Indications |
|---|---|---|---|---|
| Talazoparib | High (Strongest) | High (Strongest) | Non-selective | Recurrent ovarian cancer, BRCA-mutated breast cancer |
| Olaparib | Intermediate | Intermediate | Non-selective | Ovarian, breast, pancreatic, prostate cancer |
| Veliparib | Low (Weakest) | Low (Weakest) | Non-selective | Still under investigation in combinations |
| AZD5305 | High (comparable to Talazoparib) | Very Low | Highly PARP1-Selective | Investigational |
Data derived from PARPtrap assays and cellular viability studies [69]. The trapping efficacy (EC₅₀) is a measure of the concentration required to achieve half-maximal trapping in standardized assays.
The variation in trapping potency is thought to arise from differences in both enzymatic inhibition and allosteric effects that alter the PARP1-DNA interaction in a PARP inhibitor-specific manner [70]. Notably, some inhibitors like AZD5305 demonstrate high selectivity for trapping PARP1 over PARP2, which provides a tool for dissecting the individual roles of these two enzymes in cytotoxicity and may help in designing inhibitors with improved therapeutic windows [69].
Essential Assays and Methodologies
Choosing the appropriate assay is critical for accurately interpreting a compound's mechanism of action. The table below summarizes key methodologies for distinguishing PARP trapping from catalytic inhibition.
Table: Key Assays for Characterizing PARP Inhibition
| Assay Type | What It Measures | Key Readout | Mechanism Revealed |
|---|---|---|---|
| PARPtrap Assay | Compound's ability to trap PARP on DNA | Fluorescence Polarization (FP) | Direct measure of DNA trapping |
| Catalytic Activity Assay | Inhibition of PAR polymer formation | PAR levels (e.g., via Western Blot) | Catalytic inhibition potency |
| Chromatin Fractionation | Amount of PARP retained on chromatin | PARP protein in chromatin fraction | Cellular trapping confirmation |
| Cell Viability Assay | Synthetic lethality in HR-deficient cells | IC₅₀ / EC₅₀ (e.g., HT1080 cells) | Functional cytotoxic outcome |
PARPtrap Assay: Directly Measuring Trapping
The PARPtrap Assay is a homogeneous, high-throughput compatible assay designed specifically to quantify a compound's ability to trap PARP1 or PARP2 onto DNA [69].
Experimental Protocol:
- Principle: The assay is based on Fluorescence Polarization (FP). A small, fluorescently-labeled DNA probe is used. When free in solution, the probe rotates rapidly, resulting in low FP. When bound to a large protein like PARP, its rotation slows, resulting in high FP.
- Procedure:
- Step 1: Incubate purified PARP1 or PARP2 with the fluorescent DNA probe. This forms a complex with high FP.
- Step 2: Add NAD+. In the "no inhibitor" control, PARP becomes auto-PARylated and detaches from the DNA probe due to the added negative charge, causing a drop in FP.
- Step 3: In the test condition, pre-incubate PARP with an inhibitor. If the inhibitor traps PARP, it prevents auto-PARylation and release. The PARP remains bound to the DNA probe, and the FP signal stays high.
- Data Interpretation: An increase in the FP signal relative to the "no inhibitor" control directly indicates PARP trapping. The EC₅₀ for trapping can be calculated from dose-response curves [69].
Chromatin Fractionation: Confirming Trapping in a Cellular Context
This biochemical method assesses the retention of PARP on chromatin after cellular DNA damage, providing a direct measure of trapping within cells.
Experimental Protocol:
- Cell Treatment: Treat cells (e.g., HT1080 fibrosarcoma cells) with PARP inhibitors, optionally combined with a DNA-damaging agent like temozolomide (TMZ) to increase DNA lesions [70].
- Fractionation:
- Harvest cells and lyse with a mild, non-ionic detergent to separate the soluble cytosolic and nucleoplasmic components (Fraction S) from the chromatin-bound components (Fraction C).
- Centrifuge to pellet the chromatin-bound fraction.
- Analysis: Analyze both fractions by Western blotting using anti-PARP1 antibodies.
- Data Interpretation: A strong PARP1 signal in the chromatin-bound fraction (Fraction C) in inhibitor-treated cells, compared to control cells, is a hallmark of PARP trapping. This assay has shown a clear differential trapping potency across PARP inhibitors (Talazoparib > Olaparib > Veliparib) that correlates with cellular sensitivity [70].
Catalytic Inhibition Assays
These assays measure the blockade of PAR polymer synthesis.
Experimental Protocol (Activity-Western Blot):
- Cell Treatment & Lysis: Treat cells with PARP inhibitors and induce DNA damage. Lyse cells.
- Western Blot: Separate proteins by SDS-PAGE and transfer to a membrane.
- Detection: Probe the membrane with an antibody specific for poly(ADP-ribose) (PAR). Strip and re-probe for total PARP1 to confirm equal loading.
- Data Interpretation: A decrease in PAR signal in inhibitor-treated samples, in the presence of DNA damage, indicates effective catalytic inhibition. This can be paired with a PARP-1 cleavage Western blot, as the appearance of the 89 kDa and 24 kDa fragments is a hallmark of apoptosis, while a 50 kDa fragment can indicate necrosis [28] [15].
The Scientist's Toolkit: Key Research Reagents
Table: Essential Reagents for PARP Trapping and Catalytic Inhibition Research
| Reagent / Assay Kit | Function / Application | Key Feature |
|---|---|---|
| PARPtrap Assay Kit (PARP1 & PARP2) | High-throughput screening for PARP trapping ability | Distinguishes trapping from catalytic inhibition; measures selectivity |
| Anti-PAR Antibody | Detection of PAR levels in catalytic inhibition assays (Western Blot) | Essential for quantifying enzymatic block |
| Anti-PARP1 Antibody | Detection of full-length and cleaved PARP1 in Western Blot and chromatin fractionation | Used for apoptosis/necrosis analysis (89/24 kDa; 50 kDa fragments) [28] [15] |
| Fluorescent PARP Inhibitor Probes (e.g., BODIPY FL-Olaparib) | Studying inhibitor binding kinetics (kₒₙ, kₒff) in live cells | Provides insights into binding dynamics [70] |
| HR-Deficient Cell Lines (e.g., BRCA1/2 mutant) | Cell viability assays to determine synthetic lethality | Functional validation of cytotoxicity |
Implications for Research and Drug Development
The distinction between trapping and catalytic inhibition has profound implications. When evaluating PARP inhibitors, scientists must employ both catalytic and trapping assays, as catalytic potency alone is a poor predictor of overall cytotoxic efficacy [70] [69]. The strong correlation between trapping potency and clinical efficacy underscores the importance of this mechanism. Furthermore, the discovery that PARP1, TIMELESS, and TIPIN cooperate to protect the replisome from transcription-replication conflicts (TRCs) in early S phase offers a new perspective on synthetic lethality [36]. Recent evidence suggests that the lethality of PARP inhibitors in HR-deficient cells may be due more to an inability to repair DNA damage caused by these TRCs than solely from the physical blockade of trapped PARPs [36]. This evolving understanding highlights the necessity of context-aware assay selection and data interpretation in PARP-related research.
The therapeutic targeting of poly (ADP-ribose) polymerase 1 (PARP1) has represented a landmark achievement in cancer treatment, particularly for cancers with homologous recombination deficiencies. Traditional PARP inhibitors (PARPis) function primarily by inhibiting catalytic activity and trapping the PARP enzyme on damaged DNA, leading to synthetic lethality in BRCA-deficient cells [72]. However, the clinical efficacy of these inhibitors is often constrained by acquired resistance and on-target toxicities, including hematological side effects linked to PARP2 inhibition and gastrointestinal toxicities associated with tankyrase inhibition [72]. Moreover, the phenomenon of DNA trapping—where PARPis stabilize PARP1 on DNA, creating cytotoxic lesions—while therapeutically beneficial, also contributes to toxicity in normal cells [72].
The emergence of PROteolysis-TArgeting Chimeras (PROTACs) represents a paradigm shift in targeting PARP1. These heterobifunctional molecules recruit the cellular ubiquitin-proteasome system to specifically degrade target proteins, offering potential advantages over inhibition alone. This guide provides an objective comparison of a leading PARP1 degrader against traditional PARPis, detailing the experimental frameworks and Western blot methodologies essential for their rigorous validation within the broader context of PARP1 cleavage research.
PROTACs vs. PARP Inhibitors: A Mechanistic and Functional Comparison
Core Mechanisms of Action
The fundamental distinction between these modalities lies in their mechanism of action. Traditional PARPis, such as Rucaparib, occupy the enzyme's catalytic site, inhibiting its poly(ADP-ribose) polymerase activity but paradoxically enhancing its stable association with DNA—a effect known as DNA trapping [72]. In contrast, PROTACs like 180055 are designed to induce the complete degradation of the PARP1 protein. Compound 180055 consists of the PARPi Rucaparib linked to a ligand for the VHL E3 ubiquitin ligase. This structure simultaneously binds both PARP1 and the ligase, forming a ternary complex that prompts the ubiquitination and subsequent proteasomal degradation of PARP1 [72].
Table 1: Comparative Analysis of PARP1-Targeting Therapeutics
| Feature | Traditional PARP Inhibitors (e.g., Rucaparib) | PARP1 PROTAC Degrader (180055) |
|---|---|---|
| Primary Mechanism | Catalytic inhibition & DNA trapping [72] | Targeted protein degradation via ubiquitin-proteasome system [72] |
| Effect on PARP1 Protein | No change in protein levels | Reduces protein levels (DC50: 180-240 nM) [72] |
| DNA Trapping | Yes, a key cytotoxic mechanism [72] | Not observed [72] |
| Selectivity | Can inhibit PARP2, Tankyrases [72] | Highly specific for PARP1 degradation [72] |
| Kinetics | Rapid, reversible binding | Sustained effect; degradation reversal in ~24h after washout [72] |
| Therapeutic Window | Toxicity from trapping & off-target effects [72] | Potentially wider due to absence of trapping & high specificity [72] |
Quantitative Efficacy and Specificity Data
The degradation efficacy of PROTAC 180055 has been quantitatively assessed across multiple cancer cell lines. It demonstrates a half-maximal degradation concentration (DC50) of 180 nM in T47D (breast cancer) and 240 nM in MDA-MB-231 (breast cancer) cells [72]. Its potency is not confined to breast cancer models; significant degradation of PARP1 protein has been validated in over 12 additional cell lines, including ovarian (IGROV1, A2780), colorectal (RKO), and prostate (DU 145) cancer types [72]. A critical advantage of 180055 is its high specificity for PARP1. Quantitative proteomic analysis revealed minimal off-target degradation, a significant benefit over the broader inhibitory profile of conventional PARPis which can affect PARP2 and tankyrases [72].
Table 2: Quantitative Degradation Profile of PROTAC 180055
| Parameter | Value/Result | Experimental Context |
|---|---|---|
| DC50 | 180 nM (T47D), 240 nM (MDA-MB-231) [72] | 24-hour treatment [72] |
| Degradation Onset | 12 hours post-treatment [72] | Time-course Western blot analysis [72] |
| Reversibility | Protein recovery within 24h of washout [72] | Washout experiment followed by Western blot [72] |
| Key Structural Linker | 8-carbon straight alkyl chain [72] | Structure-activity relationship study [72] |
| Dependency | VHL E3 ligase & Proteasome [72] | Validation with VHL-knockdown cells & MG132 [72] |
Western Blot Validation: A Critical Framework for Degrader Analysis
The Central Role of Western Blotting in PARP1 Research
Western blotting is an indispensable tool for validating PARP1 degraders, as it directly measures the loss of the target protein. This technique has long been central to PARP1 research, not only for detecting full-length PARP1 (∼113 kDa) but also for identifying specific proteolytic cleavage fragments that serve as hallmarks of distinct cell death pathways [25]. For instance, caspase cleavage during apoptosis generates signature fragments of 89 kDa and 24 kDa [25], while necrosis can produce a 50 kDa fragment through lysosomal protease activity [15]. When validating degraders, the objective is to see the diminution or disappearance of these bands, confirming the removal of the substrate itself rather than its cleavage.
Experimental Protocol for Validating PARP1 Degradation
The following protocol is adapted from methodologies used to characterize PROTAC 180055 and incorporates best practices for antibody validation [72] [16].
1. Cell Culture and Treatment:
- Use appropriate PARP1-expressing cell lines (e.g., T47D, MDA-MB-468, OVCAR3).
- Culture cells in recommended media (e.g., RPMI-1640 or DMEM with 10% FBS).
- Seed cells and allow to adhere for 24 hours.
- Treat with a dilution series of the PROTAC compound (e.g., 0.001 µM to 10 µM) for a predetermined period (e.g., 16-24 hours). Include controls: DMSO (vehicle), and the parent PARPi (e.g., Rucaparib) alone.
- To confirm mechanism, include a cohort pre-treated for 1 hour with a proteasome inhibitor (e.g., 10 µM MG132) [72].
2. Cell Lysis and Protein Quantification:
- Lyse cells in a suitable RIPA buffer supplemented with protease and phosphatase inhibitors.
- Centrifuge lysates to clear debris.
- Quantify protein concentration using a standardized assay (e.g., BCA assay).
- Prepare samples with Laemmli buffer, denature at 95°C for 5 minutes.
3. Gel Electrophoresis and Transfer:
- Load 20-30 µg of total protein per well on a 4-12% Bis-Tris polyacrylamide gel.
- Include a pre-stained protein molecular weight marker.
- Run gel at constant voltage until adequate separation is achieved.
- Transfer proteins from gel to a nitrocellulose or PVDF membrane.
4. Immunoblotting:
- Block the membrane with 5% non-fat dry milk or BSA in TBST for 1 hour.
- Incubate with primary antibodies against PARP1 and a loading control (e.g., GAPDH, α-Tubulin) overnight at 4°C.
- Critical Antibody Consideration: Use a PARP1 antibody validated for Western blotting. Antibodies like [E51] (ab32064) are well-characterized and can detect both full-length and cleaved fragments, which is essential for interpreting complex banding patterns [34]. Knockout-validated antibodies are preferred to confirm specificity [16] [34].
- Wash membrane and incubate with appropriate HRP-conjugated secondary antibodies.
- Detect using enhanced chemiluminescence (ECL) reagent and image.
5. Data Analysis:
- Normalize PARP1 band intensity to the loading control.
- Plot normalized density against PROTAC concentration to calculate DC50 values.
- Confirm degradation is reversed by MG132 and is more profound than with PARPi alone.
Troubleshooting Western Blot Complexities
Validating degradation requires careful interpretation of Western blot data. Multiple bands, often perceived as non-specificity, can actually represent biologically relevant states of PARP1, including proteolytic fragments during cell death, alternative splicing isoforms, or post-translationally modified forms [73]. Therefore, it is crucial to:
- Use positive and negative controls. A PARPi control confirms the inhibitor's activity, while a PROTAC+MG132 control confirms the mechanism is proteasome-dependent [72].
- Employ genetic controls where possible. Using PARP1 knockout cell lines (or siRNA knockdown) is the gold standard for confirming antibody specificity, as it demonstrates loss of all specific bands [16] [34] [73].
- Note the expected molecular weight of full-length PARP1 (~113 kDa) and its major cleavage fragments (e.g., 89 kDa, 50 kDa, 24 kDa) to accurately interpret the successful degradation of the full-length protein [25] [15].
Visualizing the Experimental Workflow and Mechanism
The following diagrams illustrate the core concepts and experimental workflow for validating PARP1 degraders.
Diagram Title: PROTAC-Induced PARP1 Degradation Mechanism
Diagram Title: Key Experimental Workflow Steps
The Scientist's Toolkit: Essential Research Reagents
Successful validation hinges on the use of well-characterized reagents. The following table details essential materials and their functions.
Table 3: Key Research Reagent Solutions for PARP1 Degrader Validation
| Reagent / Material | Function / Role | Specific Example / Note |
|---|---|---|
| PARP1 PROTAC | Induces targeted degradation of PARP1 protein | 180055 (Rucaparib-VHL ligand with C8 linker) [72] |
| Parent PARP Inhibitor | Control for catalytic inhibition without degradation | Rucaparib [72] |
| Proteasome Inhibitor | Confirms ubiquitin-proteasome system dependency | MG132 [72] |
| Validated PARP1 Antibody | Detects full-length and cleaved PARP1 in Western blot | Anti-Cleaved PARP1 [E51] (KO-validated) [34] |
| Loading Control Antibody | Ensures equal protein loading across samples | Anti-GAPDH or Anti-α-Tubulin [34] |
| PARP1 Knockout Cell Line | Gold-standard control for antibody specificity [16] | A549 PARP1 KO cells [34] |
| E3 Ligase Ligand | Control for assessing ternary complex formation | VH032 (VHL ligand) [72] |
The advent of PARP1 degraders like PROTAC 180055 offers a powerful new tool for biological research and a promising therapeutic modality. Its key advantages—avoiding DNA trapping and exhibiting high specificity for PARP1—address significant limitations of traditional PARPis [72]. For researchers, robust validation using Western blotting, informed by the deep historical context of PARP1 cleavage analysis, is paramount. By employing rigorous protocols, appropriate controls, and a clear understanding of the complex biochemistry involved, scientists can effectively characterize these novel degraders and advance their potential application in overcoming drug resistance and improving therapeutic outcomes in cancer.
Integrating Multiple Assays for a Comprehensive DDR Profiling
The DNA Damage Response (DDR) is a complex network of signaling pathways that detect and repair DNA lesions, with its failure being a hallmark of cancer and other diseases. Within this network, poly(ADP-ribose) polymerase 1 (PARP1) serves as a critical early responder to DNA damage, and its proteolytic cleavage has emerged as a significant biomarker for different cell death pathways. This guide provides an objective comparison of two fundamental methodologies for DDR assessment: PARP-1 cleavage detection via Western blot and DNA fragmentation analysis. PARP1 is a 116-kDa nuclear enzyme that catalyzes poly(ADP-ribosyl)ation of nuclear acceptor proteins to recruit DNA repair machinery to lesion sites [13] [14]. When DNA damage is excessive, PARP1 becomes a substrate for caspases-3 and -7 during apoptosis, cleaving into characteristic 24-kDa and 89-kDa fragments [10]. This cleavage event inactivates PARP1's DNA repair capability and facilitates cellular disassembly, making it a recognized marker of apoptotic commitment [74]. Meanwhile, DNA fragmentation represents a later downstream event in cell death pathways, providing complementary information about genomic integrity breakdown.
Methodological Principles and Technical Comparison
PARP-1 Cleavage Western Blot
Experimental Principle: This method detects the proteolytic cleavage of full-length PARP1 (116-kDa) into its signature fragments (89-kDa and 24-kDa) using gel electrophoresis and antibody-based detection. The 89-kDa fragment contains the automodification and catalytic domains, while the 24-kDa fragment contains the DNA-binding domain and nuclear localization signal [13]. Caspase-3 and -7 cleave PARP1 at the Asp214-Gly215 site, separating these functional domains [74]. The appearance of the 89-kDa fragment is considered a hallmark of caspase-dependent apoptosis.
Detailed Protocol:
- Cell Lysis: Harvest cells and lyse in RIPA buffer (150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 8.0) supplemented with protease and phosphatase inhibitors
- Protein Quantification: Determine protein concentration using BCA or Bradford assay
- Gel Electrophoresis: Load 20-50 μg protein per lane on 4-12% Bis-Tris polyacrylamide gels; run at 120-150V for 60-90 minutes
- Membrane Transfer: Transfer to PVDF or nitrocellulose membrane at 100V for 60 minutes or 30V overnight at 4°C
- Blocking: Incubate membrane in 5% non-fat dry milk or BSA in TBST for 1 hour
- Antibody Incubation:
- Primary antibody: Anti-cleaved PARP (Asp214) (e.g., Cell Signaling Technology #5625) at 1:1000 dilution in blocking buffer, overnight at 4°C [74]
- Secondary antibody: HRP-conjugated anti-rabbit IgG at 1:2000-1:5000 dilution, 1 hour at room temperature
- Detection: Develop with ECL reagent and image using chemiluminescence detection system
Key Quality Controls: Include positive control (apoptosis-induced cell lysate), negative control (untreated cells), and loading control (β-actin or GAPDH). The cleaved PARP (89-kDa) antibody should not recognize full-length PARP1 or other PARP isoforms [74].
DNA Fragmentation Analysis
Experimental Principle: This method quantifies the percentage of sperm with damaged DNA using the Sperm Chromatin Structure Assay (SCSA) or similar approaches. The DNA Fragmentation Index (DFI) represents the proportion of cells with denaturable DNA that contains single- or double-strand breaks [51]. In somatic cells, DNA fragmentation also occurs during late-stage apoptosis through caspase-activated DNase activity.
Detailed Protocol (SCSA):
- Sample Preparation: Collect semen samples after 2-7 days of sexual abstinence; allow liquefaction for 30 minutes at 37°C
- Acid Denaturation: Mix 100 μL of semen with 200 μL of acid detergent solution (0.1% Triton X-100, 0.15 M NaCl, 0.08 N HCl, pH 1.2) for 30 seconds
- Staining: Add 1.2 mL of acridine orange staining solution (6 μg/mL in 0.1 M citric acid, 0.2 M Na2HPO4, 1 mM EDTA, 0.15 M NaCl, pH 6.0)
- Flow Cytometry Analysis: Analyze samples within 3-10 minutes of staining using flow cytometer with 488 nm excitation; measure green (515-530 nm) and red (>630 nm) fluorescence
- DFI Calculation: DFI = [red fluorescence / (red + green fluorescence)] × 100% [51]
Interpretation Thresholds: DFI ≤ 30% indicates normal DNA integrity, while DFI > 30% suggests abnormal DNA fragmentation that may exceed the repair capacity of sperm and oocytes [51].
Table 1: Technical Comparison of DDR Assessment Methods
| Parameter | PARP-1 Cleavage Western Blot | DNA Fragmentation Analysis |
|---|---|---|
| Biological Process Detected | Early apoptosis initiation via caspase activation | Late-stage apoptosis/DNA degradation |
| Primary Output | Presence/absence of 89-kDa and 24-kDa fragments | DNA Fragmentation Index (DFI) percentage |
| Sample Types | Cell lysates, tissue homogenates | Semen samples, isolated nuclei |
| Time Required | 1-2 days | 2-4 hours |
| Sensitivity | High (can detect <5% apoptotic cells) | Moderate |
| Quantification Capability | Semi-quantitative (densitometry) | Highly quantitative |
| Key Equipment | Gel electrophoresis system, transfer apparatus, imager | Flow cytometer |
| Cost per Sample | Moderate | Low to moderate |
Biological Context and Pathway Integration
PARP1 cleavage and DNA fragmentation occupy distinct positions within the DDR and cell death cascades. The relationship between these events and their broader context in cellular pathways can be visualized as follows:
Pathway Interrelationships: The 89-kDa PARP1 fragment generated by caspase cleavage serves as a poly(ADP-ribose) (PAR) carrier to the cytoplasm, where it facilitates apoptosis-inducing factor (AIF) release from mitochondria [13] [14]. This AIF then translocates to the nucleus and associates with DNAase, resulting in large-scale DNA fragmentation [13]. This cascade demonstrates how PARP1 cleavage acts upstream of DNA fragmentation in certain apoptotic pathways. In the context of parthanatos (a caspase-independent programmed cell death), PAR polymers produced by PARP1 overactivation are translocated to the cytoplasm through a different mechanism involving poly(ADP-ribose) glycohydrolase, subsequently triggering AIF release and DNA fragmentation [13].
Applications and Comparative Performance Data
Predictive Value in Disease Contexts
Male Infertility Assessment: DNA fragmentation analysis has demonstrated significant predictive value in male infertility. A comprehensive study of 746 infertile men revealed that 31.8% exhibited abnormal DFI (>30%) [51]. Six independent predictors were identified: age, body mass index (BMI), smoking, hot spring bathing, stress, and daily exercise duration. The predictive model showed excellent discrimination with an area under the curve (AUC) of 0.819 in the training cohort and 0.814 in the validation cohort [51].
Cancer Research and Therapeutic Monitoring: PARP1 cleavage detection provides critical insights into cancer therapy efficacy. In glioblastoma models, transcriptomic profiling of DDR pathways revealed substantial upregulation of DDR genes after treatment with temozolomide and/or radiation therapy, particularly in radiation-treated cells, peaking within 24 hours after treatment [75]. High expression of ATP23, RAD51C and RPA3 independently associated with poor prognosis in glioblastoma patients [75].
Table 2: Performance Characteristics in Different Biological Contexts
| Application Context | PARP-1 Cleavage Detection | DNA Fragmentation Analysis |
|---|---|---|
| Male Fertility Assessment | Limited application | Primary assessment tool (DFI >30% clinically significant) |
| Cancer Therapy Monitoring | High utility for early apoptosis detection | Moderate utility for late-stage cell death |
| Neurodegenerative Disease Research | Demonstrated value in cerebral ischemia, Alzheimer's, Parkinson's [10] | Less extensively studied |
| Toxicology Studies | Suitable for acute chemical exposure assessment | Appropriate for chronic, cumulative damage evaluation |
| Drug Development Screening | High-throughput compatible with automated Western systems | Moderate throughput with flow cytometry |
Research Reagent Solutions
Table 3: Essential Reagents for DDR Profiling Assays
| Reagent / Resource | Specific Function | Example Specifications |
|---|---|---|
| Anti-Cleaved PARP Antibody | Specifically detects 89-kDa fragment | Rabbit monoclonal, recognizes Asp214 cleavage site [74] |
| HRP-Conjugated Secondary Antibody | Enables chemiluminescent detection | Anti-rabbit IgG, suitable for Western blotting [74] |
| Acridine Orange | Metachromatic nucleic acid dye for SCSA | Excitation 488 nm, green (530 nm) and red (>630 nm) emission [51] |
| Flow Cytometer | Quantifies DNA fragmentation index | 488 nm laser capable, analysis within 3-10 minutes post-staining [51] |
| Protease Inhibitor Cocktail | Preserves protein integrity during lysis | Broad-spectrum, EDTA-free options available |
| Chemiluminescence Substrate | Detects horseradish peroxidase activity | Enhanced sensitivity, prolonged signal duration |
Integrated Workflow Recommendations
For comprehensive DDR profiling, we recommend a sequential approach that leverages the temporal relationship between PARP1 cleavage and DNA fragmentation:
Initial Screening: Implement PARP-1 cleavage Western blot as an early apoptosis detection method, particularly in therapeutic screening applications where early response assessment is valuable. The method's ability to detect initiator caspases-3 and -7 activation provides mechanistic insight into cell death pathways.
Secondary Validation: Follow with DNA fragmentation analysis for quantitative assessment of genomic integrity, particularly in fertility studies or when evaluating cumulative DNA damage. The quantitative nature of DFI provides robust data for statistical analysis and correlation with clinical outcomes.
Specialized Applications: In neurobiology research, PARP-1 cleavage fragments have demonstrated differential effects on cellular viability, with the 24-kDa fragment conferring protection from oxygen/glucose deprivation, while the 89-kDa fragment was cytotoxic [8]. This highlights the importance of detecting specific fragments rather than simply reporting cleavage events.
The complementary nature of these assays provides a more comprehensive DDR profile than either method alone, enabling researchers to capture both early commitment to cell death and final execution phases.
Conclusion
The combined application of PARP-1 cleavage western blot and DNA fragmentation analysis provides a powerful, multi-faceted toolkit for dissecting the DNA damage response in cancer research and therapeutic development. The 89-kDa cleavage fragment serves as a critical, validated marker for apoptotic induction and can act as a carrier for poly(ADP-ribose) in cell death pathways. Meanwhile, DNA fragmentation analysis offers a broader view of genomic instability. Together, these techniques are indispensable for understanding the mechanisms of PARP inhibitors—including catalytic inhibition, DNA trapping, and the emerging strategy of targeted protein degradation with PROTACs, which avoids the DNA trapping associated with traditional inhibitors. As next-generation, PARP1-selective inhibitors and combination therapies with topoisomerase inhibitors advance into the clinic, the rigorous and integrated use of these assays will be paramount for validating drug mechanisms, identifying predictive biomarkers, and ultimately improving patient outcomes by guiding more effective and safer treatment strategies.
References
- 1. PARP1-DNA co-condensation: the driver of broken ... [https://www.nature.com/articles/s41392-024-01832-1]
- 2. Targeting PARP1: A Promising Approach for Next- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12162764/]
- 3. PARP1 auto-modification promotes faithful Okazaki ... [https://www.sciencedirect.com/science/article/pii/S1097276525007476]
- 4. RSL3 promotes PARP1 apoptotic functions by distinct ... [https://cmbl.biomedcentral.com/articles/10.1186/s11658-025-00785-9]
- 5. Apoptosis western blot guide [https://www.abcam.com/en-us/knowledge-center/western-blot/western-blot-analysis-of-apoptosis]
- 6. The deubiquitination-PARylation positive feedback loop of ... [https://www.nature.com/articles/s41388-025-03428-7]
- 7. Sheet Protector Strategy for Western Blot to Reduce ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12462392/]
- 8. Poly(ADP-ribose) polymerase-1 and its cleavage products ... [https://www.sciencedirect.com/science/article/pii/S0167488913004242]
- 9. PARP1-targeted alpha therapy enhances target expression [https://ejnmmires.springeropen.com/articles/10.1186/s13550-025-01256-0]
- 10. PARP-1 cleavage fragments: signatures of cell-death ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3022541/]
- 11. PARP inhibition: PARP1 and beyond - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC2910902/]
- 12. PARP Antibody #9542 [https://www.cellsignal.com/products/primary-antibodies/parp-antibody/9542?srsltid=AfmBOopMgBQljr_56pbm7ievG4EchAMIMD0Zwm4mitOWhFpWiy_ao2OA]
- 13. The 89-kDa PARP1 cleavage fragment serves as a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7948984/]
- 14. The 89-kDa PARP1 cleavage fragment serves as a ... [https://www.sciencedirect.com/science/article/pii/S0021925820000320]
- 15. Characterization of the necrotic cleavage of poly(ADP- ... [https://www.nature.com/articles/4400851]
- 16. Antibody validation for Western blot: By the user, for the user [https://pmc.ncbi.nlm.nih.gov/articles/PMC6983856/]
- 17. The dynamics and Regulation of PARP1 and PARP2 in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12217778/]
- 18. Cleaved PARP (Asp214) Antibody #9541 [https://www.cellsignal.com/products/primary-antibodies/cleaved-parp-asp214-antibody/9541?srsltid=AfmBOoqmT3WtXQBwzpq5K5pvh9iyL-Gno8OHstT-F3pvrnUI5tu-NGqN]
- 19. Performance Assessment of DNA Fragment Sizing by High ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC404634/]
- 20. Optimization of DNA Fragmentation Techniques to ... [https://www.mdpi.com/2075-4418/15/18/2294]
- 21. Fragmentation of DNA affects the accuracy of the DNA ... [https://biologicalproceduresonline.biomedcentral.com/articles/10.1186/1480-9222-15-5]
- 22. PARP-1 and its associated nucleases in DNA damage ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6764844/]
- 23. PARP-1 dependent cell death pathway (Parthanatos) ... [https://www.sciencedirect.com/science/article/abs/pii/S0014299924004539]
- 24. The role of Poly-ADP ribose polymerase (PARP) enzymes in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12183237/]
- 25. PARP-1 cleavage fragments: signatures of cell-death ... [https://biosignaling.biomedcentral.com/articles/10.1186/1478-811X-8-31]
- 26. Cleaved PARP1 antibody (60555-1-PBS) [https://www.ptglab.com/products/Cleaved-PARP1-Antibody-60555-1-PBS.htm?srsltid=AfmBOopFZFm2qQ8rgV0jo-EzePM5Nk0wWWAJ8PnAHB9FyKXkFUH-Aqn-]
- 27. Cleaved PARP (Asp214) Antibody #9541 [https://www.cellsignal.com/products/primary-antibodies/cleaved-parp-asp214-antibody/9541?srsltid=AfmBOoogFoR3BpD0MTTM2jSEaSnmYYiE835bYedB4QhTVLDn9goWgB7F]
- 28. Poly(ADP-ribose) polymerase-1 and its cleavage products ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4013233/]
- 29. Cleaved PARP (Asp214) (D64E10) Rabbit Monoclonal ... [https://www.cellsignal.com/products/primary-antibodies/cleaved-parp-asp214-d64e10-xp-rabbit-mab-bsa-and-azide-free/95696?srsltid=AfmBOooqinhqg4N6Nd_0o3fJ3765ytbLE-qKbRXCdGNP4JO2LkBu4NEO]
- 30. Cleaved PARP1 antibody (60555-1-Ig) [https://www.ptglab.com/products/Cleaved-PARP1-Antibody-60555-1-Ig.htm?srsltid=AfmBOoqKFtrM2ox44YzUq8vemFlDNqDfoYLGkMdMv6oxisU1szixOba2]
- 31. Anti-Cleaved PARP1 antibody (ab4830) [https://www.abcam.com/en-us/products/primary-antibodies/cleaved-parp1-antibody-ab4830]
- 32. Fragmentation of DNA affects the accuracy ... - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC3576356/]
- 33. DNA Fragmentation Simulation Method (FSM) and Fragment ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0038881]
- 34. Anti- Cleaved [E51] (ab32064) | Abcam PARP 1 antibody [https://www.abcam.com/en-us/products/primary-antibodies/cleaved-parp1-antibody-e51-ab32064]
- 35. A Radiotracer Strategy to Quantify PARP-1 Expression In ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5549277/]
- 36. Transcription–replication conflicts underlie sensitivity to ... [https://www.nature.com/articles/s41586-024-07217-2]
- 37. RSL3 promotes PARP1 apoptotic functions by distinct ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12492750/]
- 38. Comparison of TUNEL, SCSA, SCD Test and COMET Assay [https://pubmed.ncbi.nlm.nih.gov/41009547/]
- 39. A comparison of cell-collecting methods for the Comet ... [https://www.sciencedirect.com/science/article/abs/pii/S138357181100341X]
- 40. The effect of different methods and image analyzers on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5801904/]
- 41. Comparative analysis of equine semen preservation ... [https://www.sciencedirect.com/science/article/pii/S0737080624003411]
- 42. In vivo alkaline comet assay: Statistical considerations on ... [https://www.sciencedirect.com/science/article/pii/S0273230024000242]
- 43. Cleaved PARP (Asp214) (D64E10) Rabbit Monoclonal ... [https://www.cellsignal.com/products/primary-antibodies/cleaved-parp-asp214-d64e10-rabbit-monoclonal-antibody/5625?srsltid=AfmBOooUglmO7IR2-jA0Y-UTtAB_oLhkvbF43xE_EzDBJegK5cMh-MR9]
- 44. Predictive model of sperm DNA fragmentation in infertile ... [https://www.frontiersin.org/journals/endocrinology/articles/10.3389/fendo.2025.1675168/full]
- 45. Cleaved PARP (Asp214) (19F4) Mouse Monoclonal Antibody [https://www.cellsignal.com/products/primary-antibodies/cleaved-parp-asp214-19f4-mouse-monoclonal-antibody/9546?srsltid=AfmBOoroeNOsvv_7igBK2QM3X7SgpIlqcVvMPvOlMZVS5jELobsttDo7]
- 46. Cleaved PARP1 antibody (60555-1-Ig) [https://www.ptglab.com/products/Cleaved-PARP1-Antibody-60555-1-Ig.htm?srsltid=AfmBOopfgebXxaetKHLJQM7wO7qd9vNZ8buySCasdmGi4vbmoPhKvEoG]
- 47. Avoiding common mistakes in western blot workflows - Yeasen [https://www.yeasenbio.com/blogs/protein/avoiding-common-mistakes-in-western-blot-workflows-nbsp]
- 48. Troubleshooting Western Blot: Common Problems and Fixes [https://synapse.patsnap.com/article/troubleshooting-western-blot-common-problems-and-fixes]
- 49. Serine ADPr on histones and PARP1 is a cellular target of ... [https://www.nature.com/articles/s41589-025-01974-5]
- 50. 2024 Guide to Quantitative Western Blot Publication [https://www.thermofisher.com/blog/life-in-the-lab/2023-guide-to-quantitative-western-blot-publication/]
- 51. Predictive model of sperm DNA fragmentation in infertile ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12597727/]
- 52. Double-stranded sperm DNA fragmentation assessed ... [https://www.sciencedirect.com/science/article/pii/S1472648325003918]
- 53. FANCA Deficiency Induces Oncogenic R-Loop Dependent ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12236914/]
- 54. Cleaved PARP (Asp214) Antibody #9541 [https://www.cellsignal.com/products/primary-antibodies/cleaved-parp-asp214-antibody/9541?srsltid=AfmBOoquSwPwYD6w2pEtRnVm6rYLbC7n7sWYoMxeGPJJFoLqlrnY8rUM]
- 55. Apoptosis Western Blot Cocktail (pro/cleaved Caspase-3 ... [https://www.abcam.com/en-us/products/panels/apoptosis-western-blot-cocktail-pro-p17-caspase-3-cleaved-parp1-muscle-actin-ab136812]
- 56. PARP-1 Hyperactivation and Reciprocal Elevations in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4081636/]
- 57. Evaluation of poly(ADP-ribose) polymerase cleavage ... [https://www.sciencedirect.com/science/article/pii/S0015028208005621]
- 58. Truncated PARP1 mediates ADP-ribosylation of RNA ... [https://www.nature.com/articles/s41421-021-00355-1]
- 59. Noncleavable poly(ADP-ribose) polymerase-1 regulates ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC522248/]
- 60. Cleaved PARP (Asp214) (D64E10) Rabbit Monoclonal ... [https://www.cellsignal.com/products/primary-antibodies/cleaved-parp-asp214-d64e10-rabbit-monoclonal-antibody/5625?srsltid=AfmBOoox8t2THF0_VNinX689Eooaw3p9g7t8qptnGx8EyJfWAtRrKZ6w]
- 61. Cleaved PARP1 antibody (60555-1-Ig) [https://www.ptglab.com/products/Cleaved-PARP1-Antibody-60555-1-Ig.htm?srsltid=AfmBOor1427ub0xMw3QzH6VpPOhPkUa-RfMYcpkA0Tl9cJbGViuSJVkX]
- 62. Cleaved PARP (Asp214) Antibody #9544 [https://www.cellsignal.com/products/primary-antibodies/cleaved-parp-asp214-antibody/9544?srsltid=AfmBOopEYLfVkw9yYZYn_m8lF6fO7e4ph7VnM5WzFucsrwavelObNVcg]
- 63. FinaleToolkit: accelerating cell-free DNA fragmentation ... [https://pubmed.ncbi.nlm.nih.gov/41221372/?utm_source=FeedFetcher&utm_medium=rss&utm_campaign=None&utm_content=1LQ7EyUJYZpNHCDUyg2JW6KvE1VgkSrIENB_fFDFFpY8bRdoq5&fc=None&ff=20251112070217&v=2.18.0.post22+67771e2]
- 64. Analysis of cfDNA fragmentomics metrics and commercial ... [https://www.nature.com/articles/s41467-025-64153-z]
- 65. PARP inhibition impedes the maturation of nascent DNA ... [https://www.nature.com/articles/s41594-022-00747-1]
- 66. PARP inhibitors: enhancing efficacy through rational ... [https://www.nature.com/articles/s41416-023-02326-7]
- 67. Clinical approaches to overcome PARP inhibitor resistance [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-025-02355-1]
- 68. Differences in Durability of PARP Inhibition by Clinically ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9688250/]
- 69. Setting a Trap for PARP1 and PARP2 [https://bpsbioscience.com/setting-a-trap-for-parp1-and-parp2]
- 70. PARP trapping is governed by the PARP inhibitor ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11259578/]
- 71. Investigating synthetic lethality and PARP inhibitor ... [https://www.nature.com/articles/s41420-025-02382-3]
- 72. Minimizing DNA trapping while maintaining activity ... [https://www.nature.com/articles/s41419-024-07277-2]
- 73. Does a Western blot with multiple bands indicate antibody ... [https://www.genuinbiotech.com/post/antibody-non-specificity]
- 74. Cleaved PARP (Asp214) (D64E10) Rabbit Monoclonal ... [https://www.cellsignal.com/products/primary-antibodies/cleaved-parp-asp214-d64e10-rabbit-monoclonal-antibody/5625?srsltid=AfmBOoo47_zBhyLiMSeYJW0WyK3a5-YvnMEd_tstOtVEWm0W5qr0KyO0]
- 75. Transcriptomic Profiling of DNA Damage Response in Patient ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8997841/]