Real-Time Imaging of Caspase-3/7 Activation with the ZipGFP Reporter: A Guide for Biomedical Research and Drug Discovery
This article provides a comprehensive resource for researchers and drug development professionals on the ZipGFP-based fluorogenic reporter, a transformative tool for real-time imaging of executioner caspase activation.
Real-Time Imaging of Caspase-3/7 Activation with the ZipGFP Reporter: A Guide for Biomedical Research and Drug Discovery
Abstract
This article provides a comprehensive resource for researchers and drug development professionals on the ZipGFP-based fluorogenic reporter, a transformative tool for real-time imaging of executioner caspase activation. We cover the foundational molecular design of ZipGFP, which offers a 10-fold fluorescence increase upon caspase-3/7 cleavage for superior signal-to-noise ratio. The guide details methodological applications from 2D cultures to physiologically relevant 3D organoid and in vivo models, alongside protocols for integrating apoptosis imaging with assessments of immunogenic cell death and apoptosis-induced proliferation. Critical troubleshooting advice for optimizing signal specificity and kinetic measurements is included, along with a comparative analysis against traditional methods like FRET-based reporters and FLICA probes. This synthesis empowers scientists to robustly apply this technology for high-content screening and the mechanistic dissection of cell death in disease and therapy.
Understanding ZipGFP: The Molecular Design and Mechanism Behind a Superior Caspase Reporter
Apoptosis, or programmed cell death, is a fundamental process in development, tissue homeostasis, and disease pathogenesis, particularly in cancer and neurodegenerative disorders. Traditional methods for detecting apoptosis, including antibody-based assays, viability dyes, and flow cytometry, have provided invaluable insights but suffer from significant limitations including endpoint measurements, extensive sample processing, and inability to capture dynamic, single-cell responses in living systems. This application note explores the transition from classical apoptosis detection methodologies to advanced real-time imaging approaches, with particular emphasis on the ZipGFP caspase reporter technology. We detail the limitations of conventional techniques, present quantitative comparisons of apoptosis detection methods, and provide comprehensive protocols for implementing ZipGFP-based reporters to monitor caspase-3/7 activation in live cells with high spatiotemporal resolution. Framed within broader thesis research on real-time imaging of caspase activation, this note serves as a technical resource for researchers and drug development professionals seeking to implement advanced apoptosis monitoring in their experimental workflows.
Apoptosis is a tightly regulated form of programmed cell death characterized by distinct biochemical and morphological changes, including caspase activation, phosphatidylserine (PS) externalization, DNA fragmentation, and membrane blebbing [1] [2]. Caspases, a family of cysteine-dependent proteases, serve as central regulators and executioners of apoptotic signaling. Among these, caspase-3 functions as a key executioner caspase that cleaves cellular substrates at specific aspartate residues, ultimately leading to the characteristic morphological changes associated with apoptotic cell death [3] [2]. The detection of caspase-3 activation is therefore considered a reliable marker for identifying cells committed to apoptosis.
Traditional methods for apoptosis detection have predominantly relied on techniques such as antibody-based detection (Western blotting, ELISA), flow cytometry with Annexin V staining, and viability dyes that detect late-stage apoptotic events [1] [2] [4]. While these approaches have contributed substantially to our understanding of apoptotic pathways, they share significant limitations for contemporary research needs, particularly in the context of drug discovery and the study of dynamic cellular processes.
Limitations of Traditional Apoptosis Detection Methods
Endpoint Measurements and Temporal Limitations
Classical apoptosis assays provide single timepoint measurements that fail to capture the kinetic progression of cell death within heterogeneous populations. Flow cytometry-based Annexin V assays, considered a gold standard for apoptosis detection, require termination of experiments prior to analysis, thus necessitating tedious optimization for treatment timing and harvesting [4]. These endpoint measurements cannot resolve the temporal sequence of apoptotic events in individual cells, potentially missing critical transitional states and early commitment phases to cell death.
Sample Processing Artifacts
Extensive sample handling in traditional apoptosis assays introduces significant artifacts that compromise data accuracy. Flow cytometry requires mechanical and chemical processing that stresses cells, potentially inducing plasma membrane instability and subsequent false-positive staining of apoptotic markers [4]. Additionally, the requirement for Annexin V binding buffers has been shown to synergize with pro-apoptotic agents, artificially elevating observed apoptosis rates. Studies demonstrate that vehicle-treated cells cultured in Annexin V binding buffer show two-fold increased basal apoptosis rates, while treatment with apoptotic inducers in combination with these buffers reveals eight-fold more apoptosis compared to standard culture conditions [4].
Inability to Resolve Cellular Heterogeneity
Population-level measurements provided by techniques such as Western blotting and spectrophotometric assays mask cell-to-cell variability in apoptotic responses. This limitation is particularly problematic in cancer research, where tumor cell heterogeneity significantly influences treatment responses and the emergence of drug resistance [5] [6]. Traditional methods average responses across cell populations, potentially obscuring critical subpopulations with distinct apoptotic sensitivities.
Limitations in Complex Microenvironments
Conventional apoptosis detection methods perform suboptimally in three-dimensional culture systems and in vivo environments, where light scattering, tissue autofluorescence, and limited reagent penetration complicate analysis [5] [7]. Fluorescence intensity-based measurements are particularly susceptible to these confounding factors, as attenuation varies with wavelength and depth within tissue [5].
Table 1: Comparative Analysis of Apoptosis Detection Methods
| Method | Key Limitations | Temporal Resolution | Cellular Resolution | Applicability to 3D/In Vivo Models |
|---|---|---|---|---|
| Western Blotting | Endpoint measurement; population average; no spatial information | None (endpoint) | None | Limited |
| Flow Cytometry | Extensive processing artifacts; endpoint measurement; suspension requirement | None (endpoint) | Single cell (in suspension) | Limited (requires tissue dissociation) |
| Annexin V Staining | Buffer-induced artifacts; early apoptotic stage detection only | Low (multiple timepoints required) | Single cell (with imaging) | Moderate (with imaging systems) |
| Viability Dyes (PI, DRAQ7) | Late-stage detection only; cannot distinguish death mechanisms | Low to moderate | Single cell | Moderate |
| Caspase Cleavable Peptide Reporters | Potential cleavage by non-caspase proteases; intensity-dependent signals | Moderate to high | Single cell | Moderate |
| FRET-Based Reporters (Intensity) | Susceptible to tissue optical properties; concentration-dependent | High | Single cell | Limited by tissue depth |
| FLIM-FRET Reporters | Technically complex; specialized equipment required | High | Single cell | Excellent |
| ZipGFP Reporters | Requires genetic manipulation; slower activation kinetics (~40 min T½) | High | Single cell | Excellent |
Advanced Real-Time Apoptosis Monitoring Technologies
FRET-Based Caspase Reporters
Förster Resonance Energy Transfer (FRET)-based caspase reporters represent a significant advancement for real-time apoptosis monitoring. These genetically encoded biosensors typically consist of donor and acceptor fluorescent proteins linked by a caspase cleavage sequence (DEVD for caspase-3/7). When the reporter is intact, FRET occurs between the fluorophores; upon caspase activation and cleavage of the linker, the fluorophores separate, resulting in decreased FRET efficiency [5] [6]. While superior to traditional methods, conventional FRET reporters still face limitations for in vivo application, including weak signals due to small fluorescence changes, tissue autofluorescence, and spectral confounding factors in complex tissues [7].
Fluorescence Lifetime Imaging Microscopy (FLIM)
FLIM provides a powerful alternative to intensity-based FRET measurements by quantifying the average time a fluorophore remains in its excited state before emitting a photon. Since fluorescence lifetime is independent of fluorophore concentration, excitation intensity, and imaging depth, FLIM offers significant advantages for imaging in complex 3D environments and in vivo [5] [6]. When combined with FRET reporters, FLIM detects caspase activation through reduction in donor fluorescence lifetime, enabling precise quantification of apoptosis with single-cell resolution in living organisms [5].
The ZipGFP Caspase Reporter Platform
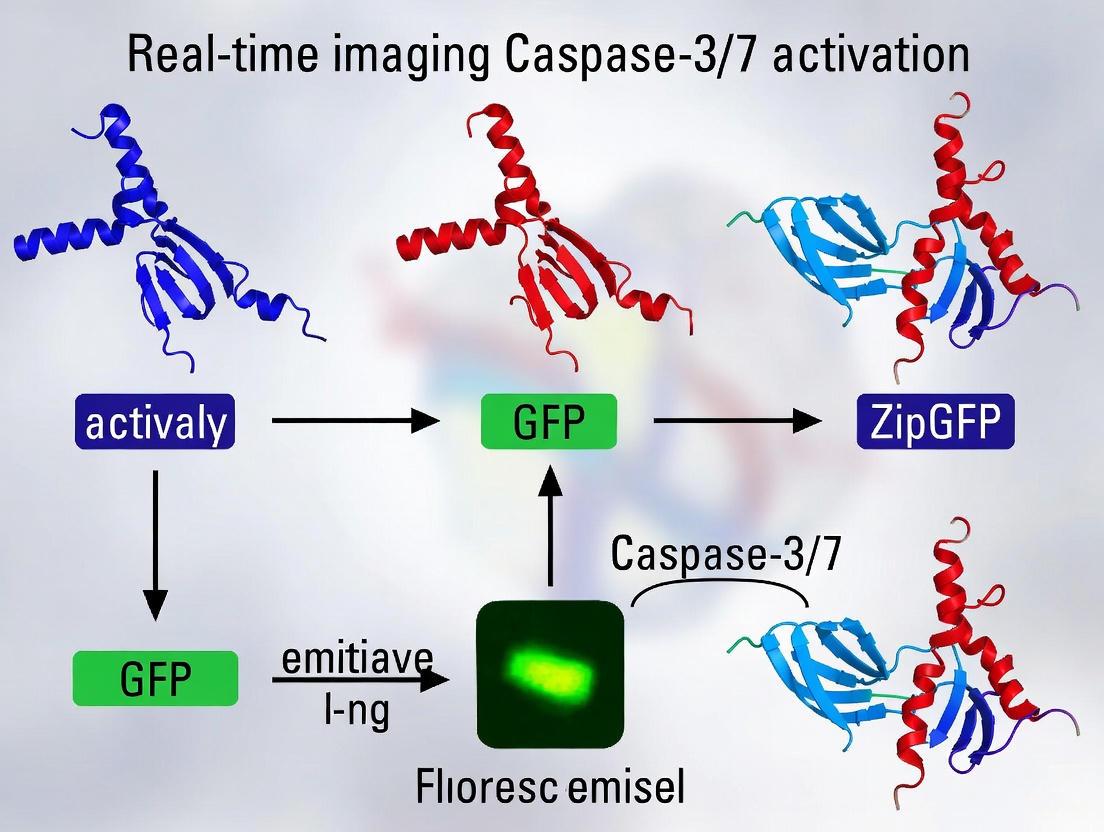
The ZipGFP platform represents a novel approach to fluorogenic caspase reporting that addresses fundamental limitations of FRET-based systems. Unlike FRET reporters that rely on separation of fluorophores, ZipGFP utilizes a "zipped" split GFP system where the eleventh β-strand (β11) of GFP is caged within a heterodimerizing coiled-coil structure (E5/K5) along with the complementary β1-10 fragment [8] [7]. Insertion of caspase cleavage sequences (DEVD) into both "zipped" elements prevents spontaneous GFP reconstitution. Upon caspase activation, cleavage releases both fragments, enabling self-assembly of mature GFP with a 10-fold fluorescence increase [7].
The structural design of ZipGFP offers several advantages over traditional FRET reporters. The fluorogenic signal generation (signal increase upon activation) provides superior signal-to-noise ratios compared to FRET-based systems that rely on signal decrease. Additionally, the single fluorescent output simplifies detection schemes and minimizes potential confounding factors from tissue optical properties [7].
Table 2: Performance Characteristics of Advanced Caspase Reporters
| Parameter | FRET-Based Reporters | FLIM-FRET Reporters | ZipGFP Reporter |
|---|---|---|---|
| Signal Change upon Activation | Decreased FRET efficiency | Decreased donor fluorescence lifetime | 10-fold fluorescence increase |
| Activation Kinetics (T½) | Minutes | Minutes | ~40 minutes (in vitro) ~100 minutes (cellular) |
| Signal-to-Noise Ratio | Moderate (2-3 fold) | High | High (10-fold) |
| Tissue Depth Compatibility | Limited by spectral properties | Excellent | Good |
| Quantification Method | Donor/acceptor ratio | Fluorescence lifetime | Fluorescence intensity |
| Genetic Encoding | Yes | Yes | Yes |
| Co-factor Requirement | None | None | None |
| Demonstrated In Vivo Application | Limited | Yes (mouse xenografts) | Yes (zebrafish embryos) |
Experimental Protocols
ZipGFP Caspase Reporter Implementation Protocol
Background This protocol describes the implementation of ZipGFP-based caspase reporters for real-time apoptosis monitoring in mammalian cells. The ZipGFP design incorporates caspase cleavage sites (DEVD) within both coiled-coil "zipped" elements, preventing spontaneous GFP reconstitution until caspase-mediated cleavage occurs [7].
Materials
- ZipGFP caspase reporter plasmid (Addgene or similar repository)
- HEK293T or other relevant cell lines
- Appropriate cell culture media and supplements
- Transfection reagent (calcium phosphate, FuGENE 6, or lipofectamine)
- Apoptosis inducers (staurosporine, camptothecin, or specific therapeutic agents)
- Fluorescence microscope or live-cell imaging system
- Optional: mCherry reference plasmid for normalization
Procedure
- Cell Culture and Transfection:
- Culture HEK293T or other relevant cells in appropriate medium (DMEM with 10% FBS, penicillin/streptomycin) at 37°C with 5% CO₂.
- Seed cells into 6-well plates at 1.4×10⁵ cells per well one day before transfection.
- Transfect cells with ZipGFP caspase reporter plasmid using calcium phosphate or FuGENE 6 according to manufacturer protocols.
- Include mCherry reference plasmid if normalization for transfection efficiency is required.
Apoptosis Induction and Live-Cell Imaging:
- 24 hours post-transfection, treat cells with apoptosis inducers (e.g., 1 μM staurosporine, 100 μM camptothecin) or vehicle control.
- Transfer cells to fluorescence-compatible imaging medium (phenol red-free DMEM with 1% FBS).
- Acquire time-lapse fluorescence images using appropriate GFP filter sets (excitation ~488 nm, emission ~510 nm) at 15-30 minute intervals for 6-24 hours.
- Maintain cells at 37°C with 5% CO₂ during imaging.
Data Analysis:
- Quantify fluorescence intensity in regions of interest corresponding to individual cells.
- Normalize signals to baseline fluorescence or mCherry reference if included.
- Calculate fold-increase in fluorescence relative to untreated controls.
- Determine apoptosis kinetics by plotting normalized fluorescence versus time.
Technical Notes
- The ZipGFP reporter exhibits a time to half-maximal fluorescence (T½) of approximately 100 minutes in cellular environments following caspase activation [7].
- Optimal signal-to-noise ratio is achieved 4-6 hours after apoptosis induction.
- Include caspase inhibitor (Z-DEVD-FMK) controls to confirm specificity of fluorescence activation.
Multiplexed Apoptosis Assessment with ZipGFP and Annexin V
Background This protocol enables simultaneous detection of caspase activation (via ZipGFP) and phosphatidylserine externalization (via Annexin V) for comprehensive apoptosis monitoring, confirming activation through two distinct biochemical pathways [9] [4].
Materials
- ZipGFP-expressing cells
- Recombinant Annexin V conjugated to Alexa Fluor 594 or similar far-red fluorophore
- Incucyte Live-Cell Analysis System or comparable live-cell imaging platform
- Apoptosis inducers
- Culture media without phenol red
Procedure
- Experimental Setup:
- Establish ZipGFP-expressing cells according to Protocol 4.1.
- Seed cells into 96-well or 384-well imaging-compatible plates.
- Add recombinant Annexin V-594 (0.25-0.5 μg/mL) directly to culture media.
- Treat cells with apoptotic stimuli or controls.
Real-Time Multiplexed Imaging:
- Place plate in live-cell imaging system maintained at 37°C with 5% CO₂.
- Acquire simultaneous images in GFP (ZipGFP) and far-red (Annexin V) channels every 30-60 minutes for 24-48 hours.
- Include phase-contrast images for morphological assessment.
Image Analysis and Quantification:
- Automatically segment cells using phase-contrast or nuclear markers.
- Quantify ZipGFP fluorescence intensity per cell over time.
- Identify Annexin V-positive cells using intensity thresholding.
- Calculate the percentage of cells positive for both markers, each marker individually, and neither marker at each timepoint.
Technical Notes
- Annexin V staining typically precedes ZipGFP activation by 1-2 hours in early apoptosis [4].
- Avoid calcium supplementation beyond standard culture media (DMEM contains 1.8 mM Ca²⁺, sufficient for Annexin V binding) to prevent nonspecific staining artifacts [4].
- This multiplexed approach enables discrimination of apoptotic stages: early (Annexin V positive only), mid (both positive), and late (membrane permeability changes).
Signaling Pathways and Experimental Workflows
Apoptotic Signaling Pathways and Detection Modalities
ZipGFP Experimental Workflow and Mechanism
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Advanced Apoptosis Research
| Reagent/Category | Specific Examples | Function/Application | Key Considerations |
|---|---|---|---|
| Genetic Reporters | ZipGFP caspase reporter | Fluorogenic caspase-3/7 activity reporting | 10-fold signal increase; requires genetic manipulation |
| FRET-based caspase reporters (LSS-mOrange-DEVD-mKate2) | Caspase activity via FRET efficiency change | Compatible with FLIM; ratiometric measurement | |
| Live-Cell Detection Probes | Recombinant Annexin V-488/594 | PS externalization detection | Early apoptosis marker; requires calcium |
| Incucyte Caspase-3/7 Dyes | Cell-permeable caspase substrate | Simple mix-and-read protocol; no washing | |
| Incucyte Annexin V Dyes | PS binding for live-cell imaging | Non-toxic; compatible with long-term imaging | |
| Viability Indicators | YOYO-3, DRAQ7 | Late-stage apoptosis/necrosis detection | Membrane-impermeable DNA dyes |
| SYTOX, Propidium Iodide | Membrane integrity assessment | Traditional viability dyes; potential toxicity | |
| Inhibitors/Inducers | Z-DEVD-FMK | Caspase-3 inhibitor | Specificity controls; 2-4 hour pre-incubation |
| Staurosporine, Camptothecin | Apoptosis induction | Pan-kinase inhibitor; DNA topoisomerase inhibitor | |
| ABT-737 | BCL-2 inhibitor | Intrinsic pathway activation | |
| Cell Lines | HEK293T | Reporter validation and optimization | High transfection efficiency |
| MDA-MB-231 | Cancer apoptosis studies | Breast cancer model; therapeutic screening | |
| Instrumentation | Fluorescence Lifetime Imaging (FLIM) | FRET quantification independent of intensity | Requires specialized equipment |
| Incucyte Live-Cell Analysis System | Kinetic imaging inside incubator | Non-perturbing; automated analysis | |
| Confocal Microscopy with Environmental Control | High-resolution live-cell imaging | Maintains cell viability during imaging |
The evolution from traditional endpoint apoptosis assays to advanced real-time imaging technologies represents a paradigm shift in cell death research. While classical methods including Western blotting, flow cytometry, and early biochemical assays have provided foundational knowledge, their limitations in temporal resolution, cellular heterogeneity assessment, and applicability to complex physiological environments have driven the development of more sophisticated approaches. The ZipGFP caspase reporter platform, with its fluorogenic signal generation, genetic encodability, and compatibility with live-cell imaging in both 2D and 3D contexts, addresses many limitations of traditional FRET-based systems. When integrated with complementary techniques such as Annexin V staining and fluorescence lifetime imaging, ZipGFP technology enables comprehensive, kinetic analysis of apoptotic progression with single-cell resolution. For drug development professionals and basic researchers alike, these advanced apoptosis detection methodologies provide powerful tools to investigate therapeutic efficacy, mechanism of action, and heterogeneous cellular responses in physiologically relevant model systems.
The visualization of biological processes in living systems with high spatiotemporal resolution remains a central challenge in cell biology and drug development. This is particularly true for apoptosis, or programmed cell death, a process where caspase-3 and caspase-7 act as key executioner enzymes [10] [2]. Traditional methods for detecting caspase activation, such as antibody-based assays or fluorescence resonance energy transfer (FRET) reporters, have limitations including poor signal-to-noise ratios, the need for endpoint analyses, and inadequate performance for in vivo imaging [11] [2] [7]. To overcome these hurdles, researchers developed ZipGFP, a fluorogenic caspase reporter that ingeniously combines the principles of split-green fluorescent protein (GFP) and leucine zipper dimerization to create a highly sensitive, specific, and genetically encodable biosensor [12] [8] [7]. This application note details the core architecture of this switch and provides protocols for its use in real-time imaging of caspase-3/7 activation, framed within ongoing thesis research on apoptosis.
Core Architectural Principles
The Self-Assembling Split-GFP Scaffold
The foundational element of the reporter is split GFP. The native GFP molecule is an 11-stranded β-barrel structure with a central α-helix containing the chromophore-forming residues [13]. In split GFP, the protein is divided into two fragments:
- A large fragment (β1-10) containing strands 1 through 10 and the internal chromophore-forming residues (amino acids 65-67).
- A small fragment (β11) containing the 11th β-strand, which includes the highly conserved Glu222, a critical catalytic residue for chromophore maturation [13] [7].
Individually, these fragments are non-fluorescent. However, they spontaneously associate with high affinity to form a folded, fluorescent β-barrel [13] [14]. The chromophore maturation, an autocatalytic process of cyclization and oxidation, can only occur within this reconstituted, properly folded structure [13]. The reconstitution is rapid, with a time to half-maximal fluorescence (T~1/2~) of approximately 40 minutes in vitro [7].
The "Zipping" Mechanism: Leucine Zippers as a Lock
The innovation of ZipGFP lies in preventing the spontaneous reassembly of split GFP until a specific proteolytic event occurs. This is achieved by caging both fragments using leucine zippers [7].
Leucine zippers are dimerization motifs found in a wide array of proteins, notably transcription factors. They are formed by two α-helical monomers, each featuring a characteristic pattern where leucine residues appear at every seventh position. This arrangement creates a hydrophobic stripe along one side of the helix, allowing the two monomers to interdigitate and form a stable, coiled-coil dimer [15] [16].
In the ZipGFP construct, the two fragments of split GFP are each flanked by a different member of a pair of heterodimeric coiled-coil peptides, specifically the well-characterized E5 and K5 leucine zippers [7].
- The large fragment β1-10 is fused to one zipper (e.g., E5).
- The small fragment β11 is fused to the other zipper (e.g., K5).
In the basal state, the high-affinity interaction between the E5 and K5 zippers "zips" the two split-GFP fragments together. This forced proximity sterically occludes the binding cavity, distorting the fragments and preventing their proper association and chromophore formation, thus keeping the reporter dark [7].
The Fluorogenic Switch: Protease-Activated Unlocking
The switch from a dark to a bright state is mediated by the target protease, in this case, executioner caspases.
- A protease cleavage sequence (e.g., DEVD for caspase-3/7) is engineered into the flexible linkers connecting the split-GFP fragments to their respective leucine zippers [10] [7].
- Upon induction of apoptosis, activated caspase-3 or -7 cleaves the DEVD sequence in both linkers.
- Cleavage severs the covalent tether between the leucine zippers and the split-GFP fragments.
- The E5 and K5 zippers dissociate, releasing the β1-10 and β11 fragments.
- Once free, these fragments spontaneously and irreversibly reassemble into the native GFP structure, leading to chromophore maturation and a robust increase in green fluorescence [10] [7].
This design results in a significant signal amplification. The ZipGFP-based caspase reporter has been reported to achieve up to a 10-fold fluorescence increase after protease activation, a substantial improvement over many FRET-based sensors [8] [7].
The diagram below illustrates the logical relationship and mechanism of the ZipGFP fluorogenic switch.
Quantitative Performance Data
The performance of the ZipGFP caspase reporter has been characterized in multiple systems, from live cells to whole organisms. The table below summarizes key quantitative metrics.
Table 1: Performance Metrics of the ZipGFP Caspase Reporter
| Parameter | Performance Value | Experimental Context | Source |
|---|---|---|---|
| Fluorescence Increase | ~10-fold | HEK293 cells, TEV protease reporter | [7] |
| Time to Half-Max (T~1/2~) | ~40 minutes | In vitro, after mixing pre-cleaved fragments | [7] |
| Time to Half-Max (T~1/2~) | ~100 minutes | HEK293 cells, using rapamycin-activatable TEV protease | [7] |
| Application In Vivo | Visualization of physiological apoptosis | Live zebrafish embryos | [12] [8] |
| 3D Model Application | Robust signal detection | Patient-derived organoids (PDAC) & spheroids | [10] |
The reporter's specificity was validated through key control experiments. Treatment with the pan-caspase inhibitor zVAD-FMK almost completely abrogated the fluorescence signal following an apoptotic stimulus [10]. Furthermore, the reporter remained functional in caspase-3 deficient MCF-7 cells, confirming that caspase-7 activation alone is sufficient to generate a signal, as both enzymes share the DEVD cleavage specificity [10].
Experimental Protocols
The following protocols are adapted from methods used in recent literature to apply the ZipGFP caspase reporter in both 2D and 3D cell culture models [10].
Protocol 1: Real-Time Apoptosis Imaging in 2D Monolayers
Purpose: To dynamically track caspase-3/7 activation in adherent cell lines at single-cell resolution.
Materials:
- Stable ZipGFP caspase reporter cell line (e.g., lentivirally transduced)
- Appropriate cell culture medium and reagents
- Apoptosis inducer (e.g., 1–10 µM Carfilzomib, 50–100 µM Oxaliplatin)
- Control: Pan-caspase inhibitor (e.g., 20 µM zVAD-FMK)
- Live-cell imaging compatible multi-well plate
- Confocal microscope or automated live-cell imager (e.g., IncuCyte)
Procedure:
- Seed Reporter Cells: Plate the ZipGFP reporter cells in a multi-well plate to achieve 40-60% confluency at the time of treatment.
- Equilibration: Allow cells to adhere for at least 6 hours or overnight in a 37°C, 5% CO~2~ incubator.
- Treatment:
- Experimental Group: Replace medium with fresh medium containing the apoptosis inducer.
- Inhibition Control: Pre-treat cells with zVAD-FMK for 1 hour, then co-treat with apoptosis inducer and zVAD-FMK.
- Vehicle Control: Treat with vehicle (e.g., DMSO) only.
- Image Acquisition:
- Place the plate in the live-cell imaging system.
- Set imaging parameters: acquire GFP (ex ~475/em ~515 nm) and mCherry (ex ~560/em ~610 nm) channels every 30-60 minutes for 48-120 hours.
- Maintain environmental control at 37°C and 5% CO~2~ throughout.
- Data Analysis:
- Quantify the GFP fluorescence intensity over time, normalized to the mCherry signal to account for cell presence and transduction efficiency.
- Use automated analysis software to count GFP-positive cells and quantify signal kinetics.
Protocol 2: Apoptosis Monitoring in 3D Spheroid/Organoid Models
Purpose: To detect caspase activation within complex, physiologically relevant 3D cultures.
Materials:
- All materials from Protocol 1.
- Cultrex Basement Membrane Extract or Matrigel.
- Patient-derived organoids (PDOs) or spheroid-forming cell lines (e.g., MiaPaCa-2, HUVECs).
Procedure:
- Generate 3D Cultures:
- Spheroids: Seed cells in low-attachment U-bottom plates to allow spheroid formation.
- Organoids: Embed single cells from maintained PDOs in Cultrex/Matrigel droplets according to established protocols.
- Stable Line Generation: If needed, transduce organoid cultures with lentiviral ZipGFP caspase reporter and select with antibiotics to create a stable line.
- Treatment: After 3-5 days of growth, add apoptosis inducer and/or inhibitors directly to the surrounding culture medium.
- Image Acquisition:
- Image 3D structures using a confocal microscope to capture Z-stacks.
- Acquire GFP and mCherry channels at regular intervals (e.g., every 4-6 hours).
- Increase laser power or exposure time relative to 2D cultures to account for light scattering, but monitor for phototoxicity.
- Data Analysis:
- Reconstruct 3D images from Z-stacks.
- Quantify the total GFP fluorescence intensity per spheroid/organoid, normalized to mCherry.
- Alternatively, analyze the volume or number of GFP-positive foci within structures.
The workflow for these experiments, from preparation to analysis, is summarized in the following diagram.
The Scientist's Toolkit: Essential Research Reagents
The following table catalogs key reagents and their functions for implementing ZipGFP-based caspase sensing.
Table 2: Key Research Reagent Solutions for ZipGFP Caspase Studies
| Reagent / Tool | Function / Description | Example Use |
|---|---|---|
| ZipGFP Caspase Reporter Plasmid | Genetically encoded fluorogenic sensor for caspase-3/7. Contains DEVD cleavage site and E5/K5 zippers. | Stable or transient expression in mammalian cells, zebrafish. |
| Executioner Caspase Activators | Small molecules that induce intrinsic/extrinsic apoptosis pathways. | Carfilzomib (proteasome inhibitor), Oxaliplatin (chemotherapeutic), Staurosporine (broad kinase inhibitor). |
| Pan-Caspase Inhibitor (zVAD-FMK) | Irreversible, cell-permeable inhibitor of caspase activity. Essential control for assay specificity. | Pre-incubation/co-treatment to confirm caspase-dependent ZipGFP activation. |
| Fluorescent Cell Viability Marker (e.g., mCherry) | Constitutively expressed fluorescent protein for normalization. | Serves as transfection/transduction control and normalizes for cell number in viability assays. |
| 3D Culture Matrix (e.g., Cultrex) | Extracellular matrix hydrogel to support 3D cell growth. | Used for embedding cells to form spheroids or grow patient-derived organoids. |
The ZipGFP reporter represents a significant advancement in biosensor design, masterfully combining the self-assembly property of split GFP with the tight, regulatable dimerization of leucine zippers. This core architecture creates a specific, sensitive, and fluorogenic switch for monitoring caspase-3/7 activation in real-time, within live cells, and complex 3D environments including patient-derived organoids [10] [7]. Its application provides invaluable insights for drug discovery, enabling the high-content screening of novel chemotherapeutics and the mechanistic dissection of cell death pathways directly in physiologically relevant model systems. Integrating this tool into a thesis research project on apoptosis allows for the dynamic tracking of a key biochemical event, moving beyond static snapshots to a richer understanding of cell fate decisions in response to therapeutic intervention.
Caspases are a family of cysteine proteases that play a central role in executing programmed cell death, or apoptosis. The human caspase family consists of 12 members, which are divided into three main groups based on sequence similarity and biological function [17]. Group II caspases, comprising the executioner caspases-3, -6, and -7, are characterized by short pro-domains and are classically described as the 'executors of apoptosis' [17]. These enzymes have evolved to recognize and cleave specific amino acid sequences in their substrate proteins, with the DEVD motif serving as the primary recognition sequence for caspase-3 and caspase-7.
The nomenclature for caspase substrate recognition sequences describes the cleavage site (↓) located between P1 and P1′ as Pn-P4-P3-P2-P1↓P1′-P2′-Pn′ [17]. The DEVD motif represents the P4-P3-P2-P1 residues, with aspartic acid (D) at the P1 position being absolutely essential for caspase recognition. Among executioner caspases, caspase-3 and caspase-7 share nearly identical substrate specificities, with both enzymes strongly preferring the DEVD sequence [17]. This shared specificity underscores the DEVD motif's critical role as a signature sequence for apoptosis execution.
Table 1: Caspase Substrate Preference Motifs
| Enzyme | Peptide Substrate Motif | Protein Substrate Motif |
|---|---|---|
| Caspase-1 | WEHD | YVHD/FESD |
| Caspase-2 | VDVAD | XDEVD |
| Caspase-3 | DEVD | DEVD |
| Caspase-6 | VQVD | VEVD |
| Caspase-7 | DEVD | DEVD |
| Caspase-8 | LETD | XEXD |
| Caspase-10 | LEHD | LEHD |
Biochemical Basis of DEVD Specificity
Structural Recognition Mechanisms
The exceptional specificity of caspase-3 and caspase-7 for the DEVD motif is rooted in the structural architecture of their active sites. These enzymes contain a defined substrate-binding pocket that sterically and electrostatically accommodates the aspartic acid residues at positions P1 and P4, with glutamic acid (E) at P2 and valine (V) at P3 completing the optimal recognition sequence [17]. This precise arrangement ensures that only proteins containing this specific motif or closely related variants are efficiently cleaved during apoptosis.
Biochemical studies using positional scanning synthetic combinatorial libraries have confirmed that DEVD represents the optimal cleavage sequence for both caspase-3 and caspase-7 [17]. When compared to other caspase family members, caspase-3 demonstrates superior efficiency in cleaving most DEVD-based substrates, often outperforming other caspases to which substrates are reportedly specific [18]. This efficiency contributes to the pivotal role of caspase-3 as the primary executioner caspase in apoptotic progression.
Functional Consequences of DEVD Cleavage
Proteolytic cleavage at DEVD sites leads to critical changes in cellular physiology by altering the function of target proteins. From the hundreds of cellular proteins containing DEVD motifs, only a handful of single cleavage events are capable of sparking apoptosis alone, such as cleavage of caspase-3/-7 and BIMEL [17]. For the most part, cleavage events function cooperatively to generate a proteolytic synthetic lethal outcome [17]. The systematic dismantling of the cell occurs through the combined cleavage of structural proteins, DNA repair enzymes, and signaling molecules, each contributing to the characteristic morphological changes of apoptosis, including membrane blebbing, DNA fragmentation, and formation of apoptotic bodies.
Figure 1: Caspase Activation Pathway Leading to DEVD Cleavage. This simplified pathway illustrates how apoptotic stimuli trigger initiator caspases, which subsequently activate executioner caspases-3 and -7, leading to cleavage of cellular substrates at DEVD motifs and ultimately resulting in the apoptotic phenotype.
Detection Methodologies for DEVD Cleavage
Conventional Caspase Activity Assays
The specificity of the DEVD motif has been exploited to develop sensitive detection methods for caspase-3/7 activity. The Caspase-Glo 3/7 Assay System represents a state-of-the-art bioluminescent approach that provides a proluminescent caspase-3/7 DEVD-aminoluciferin substrate and a proprietary thermostable luciferase in a reagent optimized for caspase-3/7 activity [19]. This homogeneous "add-mix-measure" format results in cell lysis, followed by caspase cleavage of the substrate, liberating free aminoluciferin which is consumed by the luciferase to generate a "glow-type" luminescent signal proportional to caspase-3/7 activity [19].
Similarly, the CellEvent Caspase-3/7 Green Detection Reagent employs a fluorogenic approach where the DEVD peptide sequence is conjugated to a nucleic acid-binding dye [20]. The DEVD peptide inhibits the ability of the dye to bind to DNA, rendering the substrate nonfluorescent until cleavage by activated caspase-3/7 releases the dye, allowing DNA binding and producing a fluorogenic response indicative of apoptosis [20]. This no-wash method is particularly valuable for preserving fragile apoptotic cells typically lost during wash steps and enables real-time monitoring of caspase activation in live cells.
Table 2: Comparison of DEVD-Based Caspase Detection Assays
| Assay Method | Detection Principle | Key Features | Optimal Use Cases |
|---|---|---|---|
| Caspase-Glo 3/7 | Bioluminescent | Homogeneous format, high sensitivity, scalable to 1,536-well plates | High-throughput screening, quantitative activity measurement |
| CellEvent Caspase-3/7 Green | Fluorogenic | No-wash protocol, stains nuclei, compatible with live-cell imaging | Multiplexed apoptosis detection, high-content imaging |
| ZipGFP Reporter | Fluorescence reconstitution | Genetically encoded, irreversible signal, suitable for long-term imaging | Real-time apoptosis tracking in 2D/3D models, single-cell analysis |
| CA-GFP Reporter | Fluorescence dequenching | Genetically encoded, dark state pre-cleavage, 50-fold increase in bacteria | Longitudinal monitoring in living organisms |
Advanced Genetically Encoded Reporters
Recent advances in caspase detection have focused on genetically encoded reporters for real-time imaging applications. The ZipGFP system represents a particularly innovative approach that utilizes a split-GFP architecture where the GFP molecule is divided into two parts tethered via a flexible linker containing the DEVD cleavage motif [7]. Under basal conditions, the forced proximity of the β-strands prevents proper folding and chromophore maturation, resulting in minimal background fluorescence. Upon caspase-3/7 activation, cleavage at the DEVD site separates the β-strands, allowing spontaneous refolding into the native GFP structure with efficient chromophore formation and rapid fluorescence recovery [21] [7].
This ZipGFP design achieves a 10-fold fluorescence increase upon protease cleavage and provides substantial advantages over conventional single-fluorophore or FRET-based caspase reporters by minimizing background noise, enhancing signal stability, and enabling persistent marking of apoptotic events at the single-cell level [7]. The self-assembling properties eliminate the need for external cofactors, making the system particularly well-suited for long-term imaging studies in both 2D monolayers and complex 3D culture environments [21].
ZipGFP Reporter Protocol for Real-Time Caspase Imaging
Reporter Construction and Validation
The ZipGFP executioner caspase reporter is engineered by inserting the DEVD consensus cleavage sequence into both parts of the split GFP, which are "zipped" together using heterodimerizing E5 and K5 coiled coils that prevent self-assembly until proteolytic cleavage occurs [7]. This design maintains the reporter in a dark state until caspase-mediated cleavage at the DEVD motif unzips the fragments, enabling GFP reconstitution.
Validation Steps:
- Transfection Optimization: Generate stable cell lines expressing lentiviral-delivered ZipGFP alongside a constitutive fluorescence marker (e.g., mCherry) for normalization.
- Specificity Confirmation: Treat reporter cells with apoptosis inducers (e.g., 1-10 μM carfilzomib) with or without pan-caspase inhibitors (20-50 μM zVAD-FMK) to demonstrate caspase-dependent signal generation.
- Western Blot Correlation: Confirm cleavage of endogenous caspase-3/7 substrates (e.g., PARP) to validate reporter activation kinetics.
- Caspase-3 Deficient Controls: Utilize MCF-7 cells (caspase-3 deficient) to confirm caspase-7-mediated DEVD cleavage capability [21].
Live-Cell Imaging in 2D and 3D Models
Materials Required:
- ZipGFP reporter cell lines
- Appropriate culture media and supplements
- Apoptosis inducers (e.g., carfilzomib, staurosporine, etoposide)
- Caspase inhibitors (e.g., zVAD-FMK) for controls
- Live-cell imaging chamber with temperature and CO₂ control
- Confocal or widefield fluorescence microscope with time-lapse capability
Experimental Procedure:
- Plate ZipGFP reporter cells in appropriate imaging vessels and culture until desired confluency is reached.
- Pre-incubate control samples with caspase inhibitor for 1-2 hours prior to apoptosis induction.
- Add apoptosis-inducing compounds directly to culture medium and initiate time-lapse imaging.
- Acquire images every 30-60 minutes for 24-120 hours, depending on experimental conditions.
- For 3D models (spheroids/organoids), embed structures in Cultrex or similar matrix prior to treatment.
- Quantify fluorescence intensity using ImageJ or similar software, normalizing ZipGFP signal to constitutive marker.
Figure 2: ZipGFP Caspase Reporter Mechanism. The ZipGFP reporter remains in a dark state until caspase-3/7-mediated cleavage at the DEVD motif separates the constrained GFP fragments, allowing spontaneous reassembly into fluorescent GFP.
Multiplexed Apoptosis Assessment
The ZipGFP platform enables integration with complementary cell death assays for comprehensive phenotyping:
- Annexin V/PI Staining: Correlate reporter activation with phosphatidylserine externalization and membrane integrity.
- Immunogenic Cell Death Markers: Perform endpoint measurements of surface calreticulin exposure via flow cytometry.
- Apoptosis-Induced Proliferation: Combine with proliferation dyes (e.g., CFSE) to detect compensatory proliferation in neighboring cells.
- Mitochondrial Markers: Co-stain with TMRM or JC-1 to monitor mitochondrial membrane potential dynamics.
Research Reagent Solutions
Table 3: Essential Reagents for DEVD-Based Caspase Research
| Reagent/Solution | Manufacturer/Reference | Primary Function | Application Notes |
|---|---|---|---|
| Caspase-Glo 3/7 Assay | Promega [19] | Bioluminescent caspase-3/7 activity quantification | Optimized for 96-, 384-, 1536-well formats; Z'-factor >0.5 |
| CellEvent Caspase-3/7 Green | Thermo Fisher [20] | Fluorogenic live-cell caspase detection | No-wash protocol; compatible with high-content screening |
| ZipGFP Caspase Reporter | To et al. [7] | Genetically encoded caspase activity reporter | 10-fold fluorescence increase; suitable for 2D/3D imaging |
| CA-GFP Reporter | Nicholls et al. [22] | Dark-to-bright caspase reporter | 50-fold fluorescence increase in bacterial systems |
| zVAD-FMK | Various | Pan-caspase inhibitor | Negative control for caspase-dependent processes (20-50 μM) |
| Recombinant Caspase-3 | Various | Enzyme source for in vitro assays | Validation of direct substrate cleavage |
The DEVD cleavage motif remains the definitive signature sequence for executioner caspase-3 and -7 activity, providing a critical readout for apoptotic progression. The development of increasingly sophisticated detection methodologies, particularly the ZipGFP reporter system, has enabled unprecedented spatial and temporal resolution in monitoring caspase activation dynamics. These tools continue to advance our understanding of regulated cell death in physiological and pathological contexts, providing valuable platforms for drug discovery and mechanistic studies of cell fate decisions. The integration of DEVD-based detection in complex model systems, including 3D organoids and in vivo applications, promises to further elucidate the nuanced regulation of apoptosis in development, homeostasis, and disease.
The pursuit of high-fidelity biosensors for real-time imaging of caspase-3/7 activation represents a critical frontier in apoptosis research and drug discovery. Traditional methods, including Western blotting and antibody-based assays, though extensively used, are now recognized as having various shortcomings, particularly their inability to provide dynamic, single-cell resolution data in living systems [2]. The development of the ZipGFP-based caspase reporter addresses these limitations through its engineered molecular architecture that achieves a 10-fold fluorescence increase upon activation while maintaining exceptionally low background signals [12] [8]. This performance breakthrough enables researchers to visualize physiological apoptosis in live zebrafish embryos and complex 3D culture systems with unprecedented spatiotemporal resolution [10] [8]. For scientists and drug development professionals, this technological advancement provides a powerful tool for dissecting apoptotic pathways, screening therapeutic compounds, and investigating emerging biological phenomena such as apoptosis-induced proliferation and immunogenic cell death [10].
Molecular Mechanism of ZipGFP Caspase Reporter
Core Design Principles
The ZipGFP caspase reporter employs a strategically engineered split-GFP architecture that remains dark until activated by caspase-3/7 cleavage [12]. In this design, the GFP molecule is divided into two complementary fragments: β-strands 1–10 and the eleventh β-strand [10]. These fragments are tethered together via a flexible peptide linker containing a specific caspase-3/7 cleavage motif (DEVD) [10]. Under basal conditions (in the absence of caspase activity), the forced proximity of these GFP fragments prevents proper folding and chromophore maturation, resulting in minimal background fluorescence [10]. This ingenious molecular "zipping" effectively suppresses signal generation until the precise biological event of interest occurs.
Table 1: Key Structural Components of the ZipGFP Caspase Reporter
| Component | Structure | Function |
|---|---|---|
| GFP Fragment 1 | β-strands 1–10 | Major portion of GFP β-barrel structure |
| GFP Fragment 2 | Eleventh β-strand | Completes GFP structure upon reassembly |
| Cleavage Linker | DEVD peptide sequence | Specific substrate for caspase-3/7 recognition and cleavage |
| Constitutive Marker | mCherry fluorescent protein | Normalization control for cell presence and transduction efficiency |
Activation Mechanism
Upon induction of apoptosis, executioner caspases (caspase-3 and -7) become activated and recognize the DEVD cleavage motif within the ZipGFP reporter [10]. Proteolytic cleavage at this site separates the two GFP fragments, relieving the forced proximity that inhibited proper folding [10]. Once liberated, the GFP fragments spontaneously refold into the native β-barrel structure, enabling efficient chromophore formation and resulting in rapid fluorescence recovery [10]. This structural reassembly produces a robust, irreversible, and time-accumulating fluorescent signal that provides a highly specific marker for caspase activation at the single-cell level [10]. The 10-fold fluorescence increase represents a significant improvement over previous FRET-based caspase reporters, which struggled with poor signal-to-noise ratios, particularly in in vivo applications [12] [8].
Quantitative Performance Data
Signal Enhancement Metrics
The ZipGFP caspase reporter demonstrates exceptional performance characteristics that make it particularly valuable for sensitive detection of apoptosis. The platform achieves approximately a 10-fold increase in fluorescence signal following caspase-mediated activation, as validated in both live-cell imaging and in vivo models [12] [8]. This substantial signal amplification surpasses the dynamic range of many conventional FRET-based caspase reporters, which typically exhibit more modest signal-to-background ratios [8]. The low background fluorescence of the unactivated reporter is attributed to the effective suppression of GFP maturation through the split-protein architecture, minimizing false-positive signals and enabling clear discrimination between apoptotic and non-apoptotic cells [10] [12].
Table 2: Performance Comparison of Caspase Detection Methods
| Method | Signal Increase | Background Level | Live-Cell Application | In Vivo Utility |
|---|---|---|---|---|
| ZipGFP Reporter | ~10-fold [12] [8] | Very Low [10] [12] | Excellent [10] | Demonstrated in zebrafish embryos [12] [8] |
| FRET-Based Reporters | Moderate (2-4 fold) [23] | Moderate to High [12] | Good [23] | Limited by poor signal-to-noise [12] [8] |
| Immunofluorescence | N/A (endpoint) [24] | Variable (requires optimization) [24] [25] | No (fixed samples only) [24] | Limited |
| Western Blot | N/A (semi-quantitative) [2] | N/A | No (lysate-based) [2] | No |
Specificity and Kinetic Parameters
The ZipGFP reporter exhibits high specificity for executioner caspases (caspase-3 and -7) due to the incorporation of the DEVD recognition motif, which is preferentially cleaved by these enzymes [10]. In caspase-3 deficient MCF-7 cells, the reporter still activates through caspase-7 mediated cleavage, confirming that both executioner caspases can process the DEVD sequence [10]. The kinetic parameters of cleavage are optimized through the split-GFP design, which provides greater substrate accessibility compared to some single-EGFP based sensors [23]. The irreversible nature of the fluorescence signal creates a permanent record of caspase activation, allowing for time-accumulating detection of apoptotic events, which is particularly valuable for tracking asynchronous cell death in heterogeneous populations [10].
Experimental Protocols
Stable Cell Line Generation and Validation
Purpose: To establish reproducible cellular models for real-time imaging of caspase-3/7 activation using the ZipGFP reporter system.
Materials Required:
- ZipGFP caspase reporter construct (DEVD-based) [10]
- Lentiviral packaging system [10]
- Target cells (e.g., MiaPaCa-2, HUVEC, or patient-derived organoids) [10]
- Selection antibiotics (e.g., puromycin)
- Apoptosis inducers (e.g., carfilzomib, oxaliplatin) [10]
- Pan-caspase inhibitor (zVAD-FMK) [10]
- Fluorescence microscope with live-cell imaging capability
Procedure:
- Lentiviral Production: Package the ZipGFP caspase reporter construct (containing the DEVD cleavage site) alongside a constitutive mCherry marker using a lentiviral packaging system [10].
- Cell Transduction: Incubate target cells with lentiviral supernatants in the presence of polybrene (8 μg/mL) to enhance infection efficiency.
- Selection and Expansion: Select successfully transduced cells using appropriate antibiotics (e.g., puromycin) for 7-14 days until a stable polyclonal population is established [10].
- Functional Validation: Treat stable reporter cells with apoptosis inducers (e.g., 10-100 nM carfilzomib or 10-50 μM oxaliplatin) and monitor GFP fluorescence activation over 24-48 hours using time-lapse microscopy [10].
- Specificity Confirmation: Include control treatments with pan-caspase inhibitor zVAD-FMK (20-50 μM) to demonstrate caspase-dependent reporter activation [10].
- Orthogonal Validation: Confirm apoptosis induction using complementary methods such as Annexin V/PI staining by flow cytometry and Western blot analysis for cleaved PARP and cleaved caspase-3 [10].
3D Culture and Organoid Imaging
Purpose: To monitor caspase activation in physiologically relevant 3D model systems that better recapitulate in vivo tissue architecture and complexity.
Materials Required:
- Stable ZipGFP reporter cells [10]
- Cultrex or Matrigel for 3D embedding [10]
- Patient-derived organoids (PDOs) [10]
- Confocal or spinning disk microscope
- Apoptosis-inducing therapeutics
- Proliferation dye (e.g., CellTrace) for detecting apoptosis-induced proliferation [10]
Procedure:
- 3D Model Establishment: Embed stable ZipGFP reporter cells in Cultrex or Matrigel to form 3D spheroids according to standard protocols [10]. For patient-derived organoids, transduce with ZipGFP reporter lentivirus and expand in appropriate culture conditions.
- Treatment Application: Add apoptosis-inducing compounds (e.g., chemotherapeutic agents) to the 3D culture system at clinically relevant concentrations.
- Time-Lapse Imaging: Acquire fluorescence images at regular intervals (e.g., every 2-4 hours) over 72-120 hours using a microscope equipped with environmental control (37°C, 5% CO₂) [10].
- Signal Normalization: Normalize GFP fluorescence intensity to the constitutive mCherry signal to account for potential variations in cell density and imaging depth [10].
- Apoptosis-Induced Proliferation Detection: Label cells with proliferation dye prior to treatment to track compensatory proliferation in neighboring cells following apoptotic stimuli [10].
- Image Analysis: Use automated image analysis software to quantify GFP-positive cells within different regions of the 3D structures and generate kinetic profiles of caspase activation.
Background Reduction and Signal Optimization
Purpose: To minimize non-specific background signals and maximize detection sensitivity for caspase activation.
Materials Required:
- Highly cross-adsorbed secondary antibodies (if using immunofluorescence) [25]
- Serum from host species of secondary antibody for blocking [24] [25]
- Autofluorescence quenching reagents (e.g., TrueBlack Lipofuscin Autofluorescence Quencher) [25]
- Fc receptor blocking reagents (e.g., normal serum or Fab fragment antibodies) [25]
- Optimized fixation and permeabilization reagents (PBS/0.1% Triton X-100) [24]
Procedure:
- Blocking Optimization: Incubate samples with blocking buffer containing 5% serum from the host species of the secondary antibody for 1-2 hours at room temperature to reduce non-specific binding [24] [25].
- Fc Receptor Blocking: For immune cells or samples with high Fc receptor expression, include additional Fc blocking steps using normal serum or Fab fragment antibodies [25].
- Antibody Validation: Use highly cross-adsorbed secondary antibodies to minimize off-target binding, particularly in multiplexed experiments [25].
- Autofluorescence Management: Treat samples with autofluorescence quenching reagents when imaging tissues with inherent autofluorescence (e.g., liver, kidney, or aged tissues) [25].
- Permeabilization Conditions: Optimize permeabilization time and detergent concentration (e.g., PBS/0.1% Triton X-100 for 5 minutes at room temperature) to ensure antibody access while preserving cellular morphology [24].
- Signal-to-Noise Validation: Include appropriate controls (no primary antibody, caspase inhibitor treatments) to establish baseline background levels and validate specific signal detection [10] [24].
Research Reagent Solutions
Table 3: Essential Research Reagents for ZipGFP Caspase Reporter Studies
| Reagent Category | Specific Examples | Function/Application |
|---|---|---|
| Caspase Reporters | ZipGFP DEVD-based biosensor [10] [12] | Specific detection of caspase-3/7 activation via fluorescence reconstitution |
| Constitutive Markers | mCherry fluorescent protein [10] | Normalization control for cell presence and transduction efficiency |
| Apoptosis Inducers | Carfilzomib, Oxaliplatin, Staurosporine [10] [23] | Positive controls for caspase activation pathway validation |
| Caspase Inhibitors | zVAD-FMK (pan-caspase inhibitor) [10] | Specificity controls to confirm caspase-dependent reporter activation |
| Validation Reagents | Annexin V/PI staining kits, Anti-cleaved PARP antibodies [10] | Orthogonal validation of apoptosis induction and caspase activation |
| Background Reduction | Highly cross-adsorbed secondary antibodies [25] | Minimize off-target binding in multiplexed experiments |
| Autofluorescence Quenchers | TrueBlack Lipofuscin Autofluorescence Quencher [25] | Reduce background from endogenous fluorophores in tissues |
| 3D Culture Matrices | Cultrex, Matrigel [10] | Support physiologically relevant 3D spheroid and organoid growth |
Advanced Applications and Integration
Multiparametric Cell Death Analysis
The ZipGFP platform enables integrated analysis of multiple cell death parameters beyond simple caspase activation. Researchers can simultaneously monitor apoptosis-induced proliferation (AIP) - a compensatory process where apoptotic cells stimulate proliferation of neighboring cells - by combining the ZipGFP reporter with proliferation tracking dyes [10]. This capability is particularly valuable for studying tumor repopulation following therapy, a significant clinical challenge in oncology. Additionally, the system can be adapted to detect immunogenic cell death (ICD) by endpoint measurement of surface calreticulin exposure, providing insights into the immunostimulatory potential of cell death modalities [10]. These advanced applications demonstrate how the high-fidelity ZipGFP reporter serves as a core component in comprehensive cell death analysis platforms.
In Vivo and Therapeutic Applications
The significantly improved signal-to-noise ratio of ZipGFP-based caspase reporters enables previously challenging in vivo applications [12] [8]. Researchers have successfully visualized physiological apoptosis during zebrafish embryonic development, demonstrating the utility of this technology for developmental biology research [12] [8]. In therapeutic contexts, the platform facilitates high-content screening of compounds that modulate apoptotic pathways, with direct relevance to drug discovery efforts in cancer, neurodegeneration, and other diseases characterized by dysregulated cell death [2] [10]. The ability to track caspase activation with high spatiotemporal resolution in live animals provides unprecedented opportunities for evaluating therapeutic efficacy and understanding drug mechanisms of action in physiologically relevant contexts.
The study of apoptosis, or programmed cell death, is fundamental to understanding cellular health, disease progression, and therapeutic efficacy. A significant challenge in this field has been the dynamic and irreversible nature of the process, which conventional endpoint assays struggle to capture. This application note details the use of a genetically encoded, ZipGFP-based caspase-3/7 reporter for real-time imaging of apoptotic history. We provide validated protocols for employing this stable, fluorescent reporter system in both 2D and 3D cell culture models, enabling the tracking of irreversible caspase activation with high spatiotemporal resolution. This tool is indispensable for researchers investigating cell death mechanisms, screening novel therapeutics, and studying complex phenomena such as apoptosis-induced proliferation.
Apoptosis is a genetically regulated form of programmed cell death, characterized by distinct morphological changes including cell shrinkage, chromatin condensation, and nuclear fragmentation [26]. It plays a critical role in embryonic development, tissue homeostasis, and the elimination of damaged cells [27]. The process is primarily executed by a family of cysteine proteases known as caspases, with caspase-3 and caspase-7 being the key effector enzymes that systematically dismantle the cell [10].
Traditional methods for detecting apoptosis, such as Annexin V staining, TUNEL assays, or immunoblotting for cleaved substrates, are largely endpoint analyses. They provide a snapshot in time but fail to capture the kinetics and asynchrony of cell death within a population [10]. This is a critical limitation, as apoptosis is a dynamic and irreversible process; once a cell passes a critical point of caspase activation, it is committed to death [26]. There is, therefore, a pressing need for tools that can mark and track this decisive transition irreversibly, allowing researchers to trace the history of apoptotic events within individual cells over time, even after the cell has been cleared.
The ZipGFP Reporter: Mechanism and Irreversible Advantage
The ZipGFP-based caspase-3/7 reporter is a fluorogenic biosensor engineered for specificity and a high signal-to-noise ratio upon irreversible activation [8] [7].
Molecular Design and Mechanism
The reporter is built on a split-GFP architecture where the eleventh β-strand (β11) is physically separated from the rest of the GFP barrel (β1-10). In its inactive state, the self-assembly of these two fragments is prevented by flanking each with heterodimerizing E5 and K5 coiled coils, which "zip" the fragments together and block the binding cavity [7]. A caspase-3/7-specific cleavage sequence (DEVD) is incorporated into the linkers of both fragments.
Upon apoptosis induction and subsequent caspase-3/7 activation, the DEVD sequences are cleaved. This proteolytic event "unzips" the coiled coils, releasing the β11 and β1-10 fragments. These fragments then spontaneously and irreversibly reassemble into a mature, fluorescent GFP β-barrel [10] [7]. The irreversibility of this reassembly is the key feature that allows the system to record and retain the history of caspase activation.
Visualizing the ZipGFP Mechanism
The following diagram illustrates the irreversible activation mechanism of the ZipGFP caspase reporter.
Research Reagent Solutions
The table below catalogues the essential materials and reagents required for implementing the ZipGFP apoptosis reporter system.
Table 1: Key Research Reagents for ZipGFP-Based Apoptosis Tracking
| Reagent / Material | Function / Description | Example/Catalog Consideration |
|---|---|---|
| ZipGFP Caspase-3/7 Reporter | Core biosensor for irreversible detection of executioner caspase activity. | Lentiviral construct encoding the DEVD-zipped split GFP. |
| Constitutive mCherry Marker | Fluorescent marker for normalizing fluorescence and confirming cell presence/transduction. | Co-expressed via IRES or P2A peptide in the reporter construct [10]. |
| Apoptosis Inducers | Positive control agents to validate reporter function. | Carfilzomib, Oxaliplatin, Staurosporine. |
| Pan-Caspase Inhibitor | Control to confirm caspase-specific reporter activation. | zVAD-FMK [10]. |
| Cell Lines | Model systems for validation and experimentation. | HEK293, HeLa, MCF-7 (caspase-3 deficient) [10]. |
| 3D Culture Matrix | For establishing physiologically relevant models. | Cultrex Basement Membrane Extract, Matrigel [10]. |
Detailed Experimental Protocols
Protocol 1: Establishing Stable Reporter Cell Lines
Objective: To generate a stable cell line expressing the ZipGFP caspase-3/7 reporter and the constitutive mCherry marker.
Materials:
- ZipGFP caspase reporter lentiviral particles (commercially sourced or produced in-house)
- Target cell line (e.g., HeLa, HEK293)
- Appropriate cell culture media and supplements
- Polybrene (e.g., 8 µg/mL)
- Puromycin or appropriate selection antibiotic
Procedure:
- Seed cells in a 6-well plate at 30-50% confluence 24 hours before transduction.
- Add viral particles to the culture medium in the presence of polybrene to enhance transduction efficiency.
- After 24 hours, replace the virus-containing medium with fresh complete growth medium.
- Begin antibiotic selection 48 hours post-transduction. Determine the optimal antibiotic concentration by performing a kill curve on non-transduced cells beforehand.
- Maintain selection for at least 5-7 days, until all control (non-transduced) cells are dead.
- Confirm expression using fluorescence microscopy or flow cytometry to verify strong and ubiquitous mCherry expression, indicating successful transduction.
Protocol 2: Real-Time Apoptosis Imaging in 2D Culture
Objective: To dynamically track caspase-3/7 activation in adherent cells in response to a therapeutic agent.
Materials:
- Stable ZipGFP reporter cell line
- Live-cell imaging chamber (e.g., on-stage incubator)
- Confocal or high-content fluorescence microscope
- Apoptosis-inducing agent (e.g., 1 µM Carfilzomib)
- Control vehicle (e.g., DMSO)
Procedure:
- Seed cells in a black-walled, glass-bottom 96-well imaging plate at a density optimized for 24-96 hour growth.
- Pre-scan plates using GFP and mCherry channels to establish baseline fluorescence.
- Treat cells with the inducer (Carfilzomib) or vehicle control. Include a condition co-treated with inducer and 20 µM zVAD-FMK to confirm caspase dependence.
- Initiate time-lapse imaging. Acquire images for both GFP and mCherry channels every 1-2 hours for 48-80 hours.
- Analyze data. Quantify the GFP/mCherry fluorescence ratio for individual cells or the entire well over time. Use image analysis software to count the number of GFP-positive (apoptotic) cells.
Protocol 3: Apoptosis Monitoring in 3D Spheroid/Organoid Models
Objective: To visualize spatially heterogeneous apoptosis within complex 3D structures.
Materials:
- Stable ZipGFP reporter cells or organoids
- 3D culture matrix (e.g., Cultrex)
- Apoptosis-inducing agent
Procedure:
- Generate spheroids/organoids. For spheroids, use low-attachment U-bottom plates. For organoids, embed cells in a droplet of 3D culture matrix and overlay with culture medium.
- Allow structures to form for 3-5 days.
- Treat spheroids/organoids with the apoptotic stimulus or control.
- Image structures at regular intervals using a confocal microscope equipped with environmental control to maintain viability. Acquire z-stacks to capture the entire 3D volume.
- Process images. Generate maximum intensity projections or 3D reconstructions. Quantify the GFP signal intensity normalized to the mCherry signal within the entire structure or specific regions of interest.
Quantitative Data Presentation and Analysis
The following tables summarize typical quantitative outcomes from experiments utilizing the ZipGFP reporter system.
Table 2: Quantitative Analysis of ZipGFP Activation in 2D Culture (e.g., HeLa Cells)
| Treatment Condition | Max Fold Increase in GFP/mCherry Ratio (Mean ± SD) | Time to 50% Max Signal (Hours, Mean ± SD) | % GFP+ Cells at 48h |
|---|---|---|---|
| Vehicle Control (DMSO) | 1.2 ± 0.3 | N/A | < 5% |
| 1 µM Carfilzomib | 8.5 ± 1.2 | 18.5 ± 2.1 | 75% |
| Carfilzomib + zVAD-FMK | 1.5 ± 0.4 | N/A | < 8% |
Table 3: Application of ZipGFP Reporter in Various Experimental Models
| Experimental Model | Key Readout | Utility and Advantage |
|---|---|---|
| 2D Monolayer | Kinetic traces of caspase activation; single-cell fate mapping. | Ideal for high-throughput drug screening and mechanistic studies [10]. |
| 3D Spheroid | Spatial mapping of apoptosis; core vs. periphery cell death kinetics. | Models tumor microenvironments and penetration effects of drugs [10]. |
| Patient-Derived Organoids (PDOs) | Apoptosis heterogeneity in clinically relevant models. | Personalized medicine; predicts patient-specific therapeutic responses [10]. |
Experimental Workflow
The overall workflow for a complete study, from cell line generation to data acquisition and analysis, is summarized below.
From Bench to Bedside: Protocols and Applications for ZipGFP in Diverse Research Models
Regulated cell death, particularly apoptosis, is a fundamental process in tissue homeostasis, disease progression, and therapeutic responses. A key advancement in its study is the development of fluorescent reporters for real-time imaging of caspase activation, the central proteases in apoptosis execution. This application note details a protocol for constructing a stable cell line expressing a ZipGFP-based fluorogenic reporter for caspase-3/7 activity. The ZipGFP reporter represents a significant improvement over traditional FRET-based biosensors, offering minimized background fluorescence and enhanced signal stability through its unique split-GFP architecture that is reconstituted only upon caspase-mediated cleavage [10] [7]. When framed within lentiviral vector technology, this system enables robust, long-term monitoring of apoptotic dynamics in diverse experimental models, including two-dimensional cultures, three-dimensional spheroids, and patient-derived organoids [10]. The following sections provide a comprehensive methodology for generating, transducing, and validating this powerful reporter system, creating an essential tool for high-content screening and mechanistic dissection of cell death pathways in biomedical research and drug development.
Technical Background
The ZipGFP Caspase Reporter System
The ZipGFP reporter is a genetically engineered, caspase-activatable fluorescent biosensor based on an innovative split-GFP design. In this system, the GFP molecule is divided into two parts: β-strands 1–10 and the eleventh β-strand. These fragments are tethered via a flexible linker containing a caspase-3/7-specific DEVD cleavage motif [10] [7]. Under basal conditions, the forced proximity of the β-strands prevents proper folding and chromophore maturation, resulting in minimal background fluorescence—a critical advantage over conventional reporters. Upon activation of caspase-3 or -7 during apoptosis, cleavage at the DEVD site separates the β-strands, allowing spontaneous refolding into the native GFP β-barrel structure. This structural reassembly leads to efficient chromophore formation and rapid fluorescence recovery, providing a highly specific, irreversible, and time-accumulating signal for caspase activation [10]. This design achieves an approximately 10-fold fluorescence increase upon protease activation, enabling sensitive detection of apoptosis even in complex in vivo models [7] [8].
Lentiviral Vector Systems for Stable Expression
Lentiviral vectors (LVs) are the preferred vehicle for stable reporter integration due to their ability to transduce both dividing and non-dividing cells and provide long-term transgene expression. The evolution of LV systems has progressed through multiple generations, with safety improvements achieved by separating viral components across different plasmids. Second-generation systems consolidate essential packaging genes (gag/pol, rev) with the regulatory tat gene in a single plasmid, while third-generation systems further split these components and eliminate tat for enhanced safety [28]. Comparative studies indicate that second-generation systems with specific packaging plasmids like pCMV-dR8.2 dvpr can yield 7.3-fold higher lentiviral production compared to alternative systems, making them highly effective for reporter cell line development [28]. Stable producer cell lines, such as the inducible EuLV System, offer advantages over transient transfection methods by enabling more consistent viral production, better scalability, and reduced batch-to-batch variability [29] [30].
Materials and Reagents
Research Reagent Solutions
Table 1: Essential Reagents for Reporter Cell Line Development
| Reagent Category | Specific Examples | Function | Notes |
|---|---|---|---|
| Lentiviral System | pCMV-dR8.2 dvpr (2nd Gen), psPAX2, pMD2.G | Provides gag/pol, rev, and VSV-G envelope proteins for virus production | pCMV-dR8.2 dvpr shows 7.3-fold higher yield than psPAX2 [28] |
| ZipGFP Reporter Construct | DEVD-ZipGFP transfer vector | Encodes caspase-3/7 sensor with DEVD cleavage site | Contains constitutive mCherry for normalization [10] |
| Transfection Reagent | Lipofectamine 3000 | Delivers plasmid DNA into packaging cells | Shows 4.3-fold higher efficiency than Lipofectamine 2000 [28] |
| Selection Antibiotics | Puromycin, Hygromycin B | Selects for successfully transduced cells | Minimum inhibitory concentration varies by cell type (e.g., 7-10 μg/mL puromycin) [28] |
| Apoptosis Inducers | Carfilzomib, Oxaliplatin | Positive controls for caspase activation | Confirm system functionality [10] |
| Caspase Inhibitor | zVAD-FMK | Pan-caspase inhibitor | Control for caspase-specific signal [10] |
Methodology
Experimental Workflow
The diagram below illustrates the comprehensive workflow for developing a stable ZipGFP reporter cell line, from lentiviral production through functional validation.
Lentiviral Vector Production
Day 1: Plate HEK293T cells at 5×10^5 cells/well in a 6-well plate in complete DMEM medium. Cells should be 70-80% confluent at time of transfection [31].
Day 2: Prepare transfection mixture using Lipofectamine 3000 according to manufacturer's instructions. For a single well, use 1.5 μg total DNA with the following recommended ratios:
- ZipGFP transfer vector: 0.75 μg
- Packaging plasmid (pCMV-dR8.2 dvpr): 0.6 μg
- VSV-G envelope plasmid: 0.15 μg [28]
Add transfection mix dropwise to cells. Ensure even coverage across the well surface.
Day 4: Harvest viral supernatant 48 hours post-transfection. Centrifuge at 500 × g for 10 minutes to remove cell debris. Filter through 0.45 μm membrane.
Concentrate virus by ultracentrifugation at 50,000 × g for 2 hours at 4°C. Resuspend pellet in sterile PBS or serum-free medium (100-1000× concentration) [28]. Aliquot and store at -80°C.
Determine viral titer using Lenti-X GoStix (qualitative) or quantitative methods like p24 ELISA. Titers >5×10^5 IFU/mL are suitable for transduction [28].
Generation of Stable Reporter Cell Lines
Day 1: Plate target cells at 5×10^4 cells/well in 24-well plates. Cells should be 30-40% confluent at time of transduction.
Day 2: Replace medium with fresh medium containing lentiviral supernatant at appropriate multiplicity of infection (MOI). Include 8 μg/mL Polybrene to enhance transduction efficiency. For difficult-to-transfect primary cells, consider spinfection (centrifugation at 600 × g for 60-90 minutes at 32°C) [28].
Day 3: Replace virus-containing medium with fresh complete medium 24 hours post-transduction.
Day 4: Begin antibiotic selection 48 hours post-transduction. Use predetermined minimum inhibitory concentration:
- HEK293T cells: 10 μg/mL puromycin
- Cardiac-derived c-kit expressing cells: 7 μg/mL puromycin [28]
- Other cell types: Determine empirically with kill curve analysis
Maintain selection pressure for 5-7 days, replacing antibiotic-containing medium every 2-3 days until all non-transduced control cells are dead.
Functional Validation and Optimization
Validate reporter functionality using known apoptosis inducers and inhibitors:
- Induction: Treat with 1-10 μM carfilzomib or 10-100 μM oxaliplatin for 4-24 hours [10]
- Inhibition: Pre-treat with 20-50 μM zVAD-FMK (pan-caspase inhibitor) for 1-2 hours before induction [10]
- Specificity: Test in caspase-3-deficient MCF-7 cells to confirm caspase-7-mediated activation [10]
Monitor ZipGFP fluorescence (Ex/Em: 488/510 nm) and mCherry fluorescence (Ex/Em: 587/610 nm) by live-cell imaging over 24-80 hours. Calculate normalized fluorescence as GFP/mCherry ratio to account for cell presence and viability [10].
Table 2: Quantitative Validation Parameters for ZipGFP Reporter Lines
| Validation Parameter | Expected Outcome | Acceptance Criterion |
|---|---|---|
| Basal GFP/mCherry ratio | Low fluorescence signal | <5% of induced signal [10] |
| Carfilzomib response (24h) | Significant fluorescence increase | >10-fold induction [7] |
| zVAD-FMK inhibition | Suppressed GFP signal | >80% signal reduction [10] |
| Caspase-3 deficient cells | Residual caspase-7 activity | Significant signal maintained [10] |
| Time to half-maximal signal (T1/2) | Kinetics of reporter activation | ~40-100 minutes [7] |
| Correlation with Annexin V/PI | Concordance with established methods | >90% agreement [10] |
Applications in Physiologically Relevant Models
The validated ZipGFP reporter cell line enables sophisticated apoptosis imaging in advanced model systems. In 3D spheroid models (e.g., MiaPaCa-2-derived spheroids embedded in Cultrex), the reporter successfully detects heterogeneous caspase activation patterns with temporal resolution, revealing cell death gradients not observable in 2D cultures [10]. For greater physiological relevance, the system can be adapted to patient-derived organoid (PDO) cultures, such as pancreatic ductal adenocarcinoma (PDAC-PDO) models, where it visualizes localized apoptosis within complex tissue-like structures following chemotherapeutic challenge [10].
Beyond apoptosis quantification, this platform supports investigation of emerging cell death phenomena. By incorporating proliferation tracking dyes, researchers can detect apoptosis-induced proliferation (AIP)—a compensatory mechanism where dying cells stimulate division of neighboring survivors, with implications for tumor repopulation after therapy [10]. Furthermore, endpoint measurements of surface calreticulin exposure by flow cytometry enable simultaneous assessment of immunogenic cell death (ICD), bridging apoptosis detection with immune response profiling [10]. These applications demonstrate the system's versatility for studying complex cell death processes in experimental models that closely mimic in vivo pathophysiology.
Troubleshooting Guide
Table 3: Common Issues and Resolution Strategies
| Problem | Potential Causes | Solutions |
|---|---|---|
| Low viral titer | Inefficient transfection, suboptimal plasmid ratios | Use Lipofectamine 3000, optimize DNA:reagent ratio, ensure high-quality plasmids [28] |
| Poor transduction efficiency | Incorrect MOI, no enhancement reagent, target cell refractoriness | Perform MOI curve (0.5-20), include Polybrene (4-8 μg/mL), consider spinfection [28] |
| High background fluorescence | Incomplete reporter "zipping," autofluorescence | Verify construct design includes zipping coils on both fragments, include untransduced controls [7] |
| Weak induced signal | Inadequate apoptosis induction, caspase inhibition | Titrate inducer concentration, check cell line sensitivity, verify serum concentration in media [10] |
| Rapid signal fade | Photobleaching, chromophore instability | Reduce illumination intensity/infrequency, ensure proper environmental control during imaging [10] |
| Incomplete selection | Sublethal antibiotic concentration, resistant cells | Perform kill curve analysis for each cell type, use fresh antibiotic stocks [28] |
The study of dynamic cellular processes requires methodologies that move beyond static snapshots. Live-cell imaging has emerged as a fundamental research tool, enabling the direct visualization and quantitation of biological phenomena in single cells over extended periods [32]. When investigating programmed cell death, or apoptosis, real-time imaging provides a significant advantage by revealing the precise timing and sequence of molecular events, capturing transient intermediates that are often missed in endpoint assays. This application note details a methodology for imaging apoptosis in real-time, specifically framed within the context of a broader thesis focusing on caspase-3/7 activation using an advanced ZipGFP reporter [7]. The protocols are designed for researchers and drug development professionals aiming to generate kinetic, multiparametric data on cell fate decisions in two-dimensional (2D) culture systems. By maintaining cells under optimal physiological conditions throughout the experiment, this approach minimizes artifacts that can arise from cell fixation and provides a more biologically relevant understanding of cell death mechanisms and their modulation by potential therapeutics.
Core Principles of Apoptosis and the ZipGFP Reporter
Molecular Hallmarks of Apoptosis
Apoptosis is a tightly regulated form of cell death characterized by a series of distinct biochemical and morphological changes. Key molecular events include:
- Phosphatidylserine (PS) Externalization: In viable cells, PS is restricted to the inner leaflet of the plasma membrane. Early in apoptosis, PS is translocated to the outer leaflet, creating a specific surface marker for phagocytic cells [33] [34]. This event can be detected using Annexin V conjugates, which bind to PS in a calcium-dependent manner [35] [34].
- Caspase Activation: The execution phase of apoptosis is mediated by the activation of a family of cysteine proteases known as caspases. Caspase-3 and -7 are key effector caspases that cleave a multitude of cellular substrates, leading to the systematic disassembly of the cell [7].
- Nuclear Fragmentation: Caspase-mediated activation of endonucleases results in the cleavage of nuclear DNA into oligonucleosomal fragments, a hallmark of late-stage apoptosis.
The ZipGFP Caspase Reporter System
Traditional Förster resonance energy transfer (FRET)-based caspase reporters are limited by weak signals and are challenging to use in vivo [7]. The ZipGFP-based executioner caspase reporter is a fluorogenic protease reporter designed to overcome these limitations. Its mechanism of action is as follows:
- Rational Design: ZipGFP is based on a self-assembling split GFP. One part contains ten β strands (β1–10), and the other contains the 11th β strand (β11) [7].
- "Zipping" Mechanism: The two fragments are flanked by heterodimerizing E5 and K5 coiled coils, which "zip" the binding cavity shut, preventing the self-assembly of the split GFP and the formation of the fluorophore.
- Protease Activation: The consensus cleavage sequence for caspase-3 (DEVD) is inserted into both parts of the ZipGFP construct. Upon activation of executioner caspases, the linker is cleaved, "unzipping" the coiled coils and allowing β11 to bind to β1–10. This reassembly leads to the maturation of the GFP chromophore and a resultant ~10-fold fluorescence increase [7]. This large signal-to-noise ratio makes ZipGFP ideal for detecting apoptosis with high spatiotemporal resolution in live cells and organisms.
The following diagram illustrates the working mechanism of the ZipGFP caspase reporter system:
Critical Setup for Long-Term Live-Cell Imaging
Maintaining cell viability and function over the course of an extended imaging experiment is paramount to obtaining physiologically relevant data. Failure to properly control the cellular environment is a major source of experimental failure and variability.
Environmental Control
The microscope stage must replicate the conditions of a standard cell culture incubator [36] [37].
- Temperature: Maintain a stable 37°C.
- CO₂: Control 5% CO₂ to regulate media pH.
- Humidity: Maintain high humidity (typically >80%) to prevent media evaporation, which alters osmolarity and can adversely affect cell behavior [36] [37].
This is typically achieved using a stage-top incubator that encloses the culture vessel or an environmental chamber that encloses the entire microscope. The system must be compatible with the microscope stage, objectives, and the cell culture vessels being used [37]. Allowing the microplate to equilibrate on the pre-warmed stage before starting the acquisition helps minimize focus drift caused by thermal expansion and contraction of materials [36].
Imaging Parameters and Phototoxicity
Phototoxicity—cell damage caused by exposure to excitation light—is a major concern in live-cell imaging that can itself induce cell death and confound results [37]. The following strategies are critical for minimization:
- Light Intensity and Exposure: Use the lowest possible intensity of illuminating light and the shortest exposure time necessary to acquire a quality image [36] [37].
- Wavelength: Avoid ultraviolet (UV) light when possible, as it is more phototoxic. Use fluorophores excited by green or red light for longer-term experiments [36] [37].
- Acquisition Frequency: Balance temporal resolution with cell health. Maximize the time intervals between image acquisitions to allow cells to recover [37].
- Hardware: Use objectives with a high numerical aperture (NA) to collect light more efficiently, and sensitive cameras to detect weaker signals [36] [37]. Confocal systems with spinning disk technology can also help reduce photobleaching and phototoxicity [38].
Culture Vessels and Media Formulation
- Vessels: Use thin plastic microplates or glass-bottom dishes with a thickness comparable to a coverslip, especially when using high-NA objectives [36]. Black-walled, clear-bottom plates are preferable to reduce autofluorescence and cross-talk between wells.
- Media: Use phenol red-free media to reduce background fluorescence [36]. For long-term experiments, ensure the media formulation has adequate buffering capacity. While HEPES buffer (e.g., 10-25 mM) can be used for short-term studies to maintain pH outside a CO₂-controlled environment, its compatibility with the cells should be verified, as it can be harmful to some cell types over prolonged periods [36].
The table below summarizes the key parameters for maintaining cell health during imaging:
Table 1: Environmental and Imaging Parameters for Long-Term Live-Cell Imaging
| Parameter | Optimal Setting | Rationale & Considerations |
|---|---|---|
| Temperature | 37°C | Maintains physiological function; stability prevents focus drift. |
| CO₂ | 5% | Maintains physiological pH in bicarbonate-buffered media. |
| Humidity | >80% | Prevents media evaporation and associated osmolarity shifts. |
| Media | Phenol red-free | Reduces background autofluorescence. |
| Imaging Interval | Experiment-dependent | A balance between capturing dynamics and minimizing light exposure. |
| Light Exposure | Lowest possible intensity & duration | Minimizes phototoxicity and photobleaching. |
| Vessel | Black-walled, clear-bottom microplate | Reduces well-to-well cross-talk and autofluorescence. |
Experimental Protocol: Real-Time Apoptosis Imaging with ZipGFP
Reagent and Material Preparation
- Cell Line: Adherent cell line of interest (e.g., HeLa, RPE-1, MEFs).
- Plasmid: ZipGFP-executioner caspase reporter construct (DEVD sequence) [7].
- Culture Vessel: μ-Slide, glass-bottom dish, or black-walled, clear-bottom microplate (e.g., 96-well).
- Microscope System: Inverted microscope equipped with:
- Environmental chamber (37°C, 5% CO₂, humidity control).
- Motorized stage.
- Hardware or software autofocus system.
- Appropriate light source and filters for GFP.
- Transfection Reagent: Suitable for the cell line (e.g., lipofection).
- Apoptosis Inducer: e.g., 1-4 μM Staurosporine, 1-10 μM Paclitaxel, or other relevant agent [36] [39].
- Control Articles:
- Positive Control: Apoptosis inducer (e.g., Staurosporine).
- Negative Control: Caspase inhibitor (e.g., Z-VAD-FMK).
Staining and Treatment Procedure
Cell Seeding and Transfection:
- Seed cells into the culture vessel at an optimized density (e.g., 50-70% confluency for transfection). Allow cells to adhere overnight under standard culture conditions.
- Transfect cells with the ZipGFP-caspase reporter construct according to the manufacturer's protocol. Incubate for 24-48 hours to allow for sufficient reporter expression.
Treatment and Imaging Setup:
- Prepare working dilutions of the apoptosis inducer and any control compounds in pre-warmed, phenol red-free imaging medium.
- If performing a discontinuous time-lapse, replace the medium in the culture vessel with the treatment medium immediately before placing it on the microscope stage.
- For continuous long-term imaging, ensure the environmental chamber is pre-equilibrated to 37°C, 5% CO₂, and high humidity.
Image Acquisition:
- Place the culture vessel onto the microscope stage.
- Using the acquisition software, define multiple fields of view (FOVs) per experimental condition.
- Configure the acquisition settings:
- Focus: Enable the hardware or software autofocus to maintain focus over time. For long-term experiments, autofocus can be applied at every time point or less frequently depending on the stability of the system and the required imaging rate [36].
- Channels: Use a GFP filter set (e.g., Ex/Em ~488/510 nm).
- Timelapse Parameters: Set the total duration (e.g., 24-48 hours) and the imaging interval. For caspase activation kinetics, start with an interval of 15-30 minutes. Adjust based on the expected speed of the biological response.
- Exposure Time: Set the lowest possible exposure time that yields a detectable signal from the ZipGFP reporter to minimize phototoxicity.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for Live-Cell Apoptosis Imaging
| Reagent / Tool | Function / Utility | Example & Notes |
|---|---|---|
| ZipGFP Caspase Reporter | Fluorogenic detection of caspase-3/7 activity; ~10x signal increase upon activation [7]. | Superior signal-to-noise for kinetic assays in live cells and model organisms. |
| Stage-Top Incubator | Maintains physiological temperature, CO₂, and humidity on the microscope stage [37]. | ibidi Stage Top Incubator; must be compatible with microscope and culture vessels. |
| Phenol Red-Free Medium | Imaging medium that minimizes autofluorescence [36]. | Essential for clear fluorescence signal detection. |
| Annexin V Conjugates | Detects phosphatidylserine exposure on the outer membrane leaflet, an early apoptosis marker [35] [33]. | Can be used in multiplexed assays with viability dyes like PI. |
| Fixable Viability Dyes | Distinguishes live from dead cells; fixable versions allow for subsequent intracellular staining [35]. | e.g., eFluor 660, FVD eFluor 780; not recommended: FVD eFluor 450 with Annexin V kits. |
| Propidium Iodide (PI) | Membrane-impermeant DNA dye that stains necrotic/late apoptotic cells [33] [34]. | Do not wash out after adding; analyze cells within 4 hours. |
Data Analysis and Interpretation
Quantitative Kinetic Analysis
The primary data from a ZipGFP timelapse experiment is a series of images tracking fluorescence intensity over time.
- Region of Interest (ROI) Tracking: Use analysis software to define ROIs around individual cells and track them throughout the timelapse.
- Fluorescence Quantification: Measure the mean or integrated fluorescence intensity within each ROI for every time point.
- Data Normalization: Normalize fluorescence data to the initial (t=0) value for each cell to visualize the fold-change in signal.
- Thresholding: Determine a threshold for a positive fluorescence increase (e.g., 2- or 3-fold over baseline) to define the time of caspase activation for each cell.
This single-cell tracking allows for the calculation of key kinetic parameters, such as:
- Time to Activation: The period from treatment addition to the threshold fluorescence increase.
- Activation Synchrony: The distribution of activation times within a population.
- Fraction Responsive: The percentage of cells that undergo caspase activation during the observation period.
Multiparametric Assay Integration
To gain a more comprehensive view of cell fate, the ZipGFP readout can be combined with other probes in a multiplexed assay. However, careful validation is required to avoid spectral overlap and probe interference.
- Morphological Endpoints: Use phase-contrast or brightfield images to score classical apoptotic morphology, such as cell rounding, membrane blebbing, and shrinkage [39].
- Viability Staining: At the end of the timelapse, cells can be stained with a membrane-impermeant dye like propidium iodide (PI) to identify cells that have lost membrane integrity [33] [34]. Note that this is an endpoint measurement.
- Annexin V Staining: For a more detailed analysis of death pathways, a separate experiment can be performed using Annexin V conjugates (to detect PS exposure) combined with PI (to detect loss of membrane integrity) analyzed by flow cytometry. This allows for the discrimination of early apoptotic (Annexin V+/PI-), late apoptotic (Annexin V+/PI+), and necrotic (Annexin V-/PI+) populations [33] [34]. The protocol requires calcium-containing binding buffer and no washing after PI addition.
The following workflow diagram outlines the key steps from experimental setup to data analysis:
Troubleshooting Common Issues
- High Background Fluorescence: Switch to phenol red-free medium and use culture vessels with low autofluorescence. Ensure media and reagents are filtered and free of debris [36] [38].
- Rapid Loss of Cell Health/Phototoxicity: Systematically reduce light intensity, exposure time, and acquisition frequency. Use longer-wavelength fluorophores where possible [36] [37].
- Focus Drift: Ensure the system is thermally equilibrated before starting. Use a robust autofocus system. For hardware autofocus, ensure it is configured for the specific plate type being used [36] [37].
- Weak or No ZipGFP Signal: Optimize transfection efficiency and confirm reporter expression. Titrate the apoptosis inducer to ensure it is effective. Verify that the imaging settings are sufficient to detect the basal fluorescence of the reporter.
Long-term live-cell imaging of apoptosis using genetically encoded reporters like ZipGFP provides an unparalleled window into the dynamics of cell death. The methodology outlined here, emphasizing stringent environmental control, minimization of phototoxicity, and robust quantitative analysis, allows researchers to move beyond population averages and capture the heterogeneity of single-cell responses. When integrated within a broader thesis on caspase-3/7 activation, this approach can yield profound insights into the temporal regulation of cell death, the mechanisms of drug action, and the pathways that allow cells to recover from near-death states—a process known as anastasis [40]. By adhering to these detailed protocols, scientists can generate high-quality, kinetic data to advance our understanding of cell biology and accelerate the development of novel therapeutics.
The study of programmed cell death, particularly apoptosis, is fundamental to understanding tissue homeostasis, disease progression, and therapeutic responses in biomedical research. Executioner caspases-3 and -7 serve as crucial mediators of apoptotic signaling, with their activation representing a committed step toward cellular dismantling [10]. Traditional methods for detecting caspase activation, including immunoblotting, flow cytometry, and endpoint staining, provide valuable snapshots but fail to capture the dynamic, asynchronous nature of apoptosis as it unfolds in complex tissue-like environments [10] [41].
The ZipGFP caspase reporter system represents a significant technological advancement for real-time apoptosis imaging. This genetically encoded biosensor utilizes a split-GFP architecture where two fragments are tethered via a flexible linker containing the caspase-3/7-specific DEVD cleavage motif [10]. Under basal conditions, forced proximity of the β-strands prevents proper GFP folding, minimizing background fluorescence. During apoptosis, caspase-mediated cleavage at DEVD sites allows spontaneous refolding into native GFP structure, generating an irreversible, time-accumulating fluorescent signal that permanently marks apoptotic events [10]. This system substantially outperforms conventional FRET-based caspase reporters by minimizing background noise, enhancing signal stability, and enabling persistent tracking of apoptotic events at single-cell resolution [10].
The transition from two-dimensional (2D) cultures to three-dimensional (3D) models including spheroids and patient-derived organoids (PDOs) represents a critical advancement in cancer research and drug development. These 3D models bridge the gap between conventional cell culture and in vivo physiology by recapitulating essential features of native tissues, including:
- Cellular heterogeneity and organization
- Cell-cell and cell-matrix interactions
- Metabolic and proliferation gradients
- Therapeutic resistance mechanisms relevant to clinical responses [42]
This application note details protocols and experimental workflows for implementing the ZipGFP caspase-3/7 reporter system in 3D spheroid and PDO models, enabling researchers to dynamically track apoptosis execution during therapeutic interventions.
ZipGFP Reporter Mechanism and System Components
Molecular Design and Activation Mechanism
The ZipGFP caspase-3/7 reporter employs an innovative split-GFP design that enables specific, irreversible detection of caspase activation. The molecular mechanism involves:
- Split-GFP Architecture: The GFP molecule is divided into two fragments: β-strands 1–10 and the eleventh β-strand, separated by a flexible peptide linker [10]
- DEVD Cleavage Motif: A caspase-3/7-specific recognition sequence (Asp-Glu-Val-Asp) embedded within the linker region
- Fluorescence Quenching: In the unreconstituted state, forced proximity of the GFP fragments prevents proper chromophore maturation
- Caspase-Dependent Activation: Upon caspase-3/7 activation during apoptosis, cleavage at DEVD sites separates the fragments, allowing spontaneous refolding into native β-barrel structure and fluorescence recovery [10]
Table 1: Key Characteristics of the ZipGFP Caspase-3/7 Reporter System
| Feature | Description | Advantage |
|---|---|---|
| Detection Method | Split-GFP with DEVD cleavage motif | Minimal background, irreversible signal |
| Caspase Specificity | Caspase-3 and caspase-7 | Covers key executioner caspases |
| Signal Properties | Time-accumulating, irreversible | Permanent marking of apoptotic events |
| Co-expression Marker | Constitutive mCherry | Normalization for cell presence and transduction efficiency |
| Spatial Resolution | Single-cell level | Heterogeneous response detection in complex models |
| Temporal Resolution | Real-time, continuous monitoring | Kinetic analysis of apoptosis progression |
Experimental Workflow Visualization
The following diagram illustrates the ZipGFP activation mechanism and experimental workflow for 3D model applications:
Research Reagent Solutions and Essential Materials
Successful implementation of the ZipGFP reporter system in 3D models requires the following key reagents and materials:
Table 2: Essential Research Reagents and Materials for ZipGFP 3D Applications
| Category | Specific Reagent/Material | Function/Application | Example Sources/References |
|---|---|---|---|
| Core Reporter System | Lentiviral ZipGFP-DEVD-mCherry construct | Stable cell line generation | [10] |
| Caspase inhibitor (zVAD-FMK) | Specificity validation | [10] | |
| 3D Culture Components | Extracellular matrix (Cultrex, Matrigel) | 3D scaffold support | [10] [42] |
| U-bottom low-adhesion plates | Spheroid formation | [43] [42] | |
| Patient-derived organoid cultures | Physiologically relevant models | [10] [42] | |
| Apoptosis Inducers | Carfilzomib (proteasome inhibitor) | Apoptosis induction and validation | [10] |
| Oxaliplatin (chemotherapeutic) | Therapy-induced apoptosis modeling | [10] | |
| Staurosporine (broad-spectrum inducer) | Positive control for apoptosis | - | |
| Imaging & Analysis | Live-cell fluorescence microscope | Real-time kinetic imaging | [10] [44] |
| IncuCyte AI Cell Health Module | Automated viability analysis | [10] | |
| Flow cytometer with plate reader | Endpoint validation and ICC | [10] [43] |
Protocol 1: Establishing Stable ZipGFP Reporter Cell Lines
Lentiviral Transduction and Selection
This protocol adapts established methodologies for generating stable caspase reporter cell lines [10] [44]:
Day 1: Cell Plating
- Plate HEK-293T packaging cells at 60-70% confluence in 6-well plates
- Culture in complete DMEM with 10% FBS and 1% penicillin/streptomycin/glutamine
Day 2: Lentiviral Production
- Transfect cells with ZipGFP-DEVD-mCherry lentiviral vector using calcium phosphate or FuGENE 6 transfection reagent [44]
- Use transfer plasmid, packaging plasmid, and envelope plasmid at 4:3:1 ratio
- Change media after 8-12 hours
Day 4-5: Viral Harvest and Transduction
- Collect viral supernatant, filter through 0.45μm membrane
- Add polybrene (8μg/mL) to target cells (MiaPaCa-2, HUVECs, or organoid-derived cells)
- Centrifuge at 800×g for 30-60 minutes (spinfection) to enhance transduction efficiency
Day 6-8: Selection and Expansion
- Begin antibiotic selection with blasticidin (5-10μg/mL) or puromycin (1-2μg/mL) based on vector resistance
- Maintain selection for 7-10 days until control cells (non-transduced) are completely eliminated
- Expand stable pools or isolate single-cell clones by limiting dilution
Validation of Reporter Functionality
Before proceeding to 3D models, validate reporter functionality in 2D culture:
Positive Control Treatment
- Plate ZipGFP reporter cells in 96-well imaging plates
- Treat with carfilzomib (1-10μM) or oxaliplatin (50-200μM) for 24-72 hours
- Include control group with caspase inhibitor zVAD-FMK (20-50μM)
Fluorescence Activation Assessment
- Image using automated live-cell imager (e.g., IncuCyte, ImageXpress)
- Quantify GFP/mCherry fluorescence ratio over 48-72 hours
- Confirm ≥5-fold increase in GFP signal in treated vs. control groups
Orthogonal Validation
- Perform parallel analysis by flow cytometry for Annexin V/PI staining
- Assess caspase-3 cleavage by Western blot (if antibodies available)
- Confirm correlation between GFP activation and established apoptosis markers
Protocol 2: Implementing ZipGFP in 3D Spheroid Models
Spheroid Generation and Culture
Multiple methods exist for generating 3D spheroids, with the choice depending on specific experimental needs and cell types:
Scaffold-Free Spheroid Formation (Liquid Overlay Technique)
- Prepare 96-well U-bottom plates with agarose coating (1.5% in PBS) to prevent attachment
- Seed ZipGFP reporter cells at optimized density (500-5,000 cells/well based on cell type)
- Centrifuge at 300×g for 5 minutes to promote aggregate formation
- Culture in appropriate medium for 3-5 days until compact spheroids form [42]
Extracellular Matrix-Embedded Culture
- Suspend ZipGFP reporter cells in Cultrex or Matrigel (2-5 mg/mL)
- Plate 30-50μL droplets in pre-warmed 24-well plates
- Polymerize for 30 minutes at 37°C before adding culture medium
- Culture for 3-7 days with medium changes every 2-3 days [10]
Agitation-Based Methods
- Seed cells in low-attachment Erlenmeyer flasks or spinner flasks
- Maintain constant agitation (60-80 rpm) to prevent attachment
- Suitable for large-scale spheroid production [42]
Apoptosis Induction and Real-Time Imaging in Spheroids
Experimental Setup
- Select mature spheroids (3-7 days old) with consistent size and morphology
- Treat with therapeutic agents diluted in complete medium
- Include vehicle controls and caspase inhibitor controls for specificity
Image Acquisition Parameters
- Acquire images every 2-6 hours for 72-120 hours using automated microscope
- Use 4× or 10× objectives for full spheroid visualization
- Capture z-stacks (3-5 slices with 20-50μm spacing) for 3D reconstruction
- Maintain physiological conditions (37°C, 5% CO₂) throughout imaging
Quantitative Analysis
- Normalize GFP fluorescence to mCherry signal to account for cell density
- Calculate apoptosis kinetics: time to initiation, half-maximal activation, and rate progression
- Assess spatial distribution patterns: peripheral vs. core apoptosis
Table 3: Quantitative Analysis of ZipGFP Activation in 3D Spheroid Models
| Parameter | Control (DMSO) | Carfilzomib (5μM) | Carfilzomib + zVAD-FMK | Measurement Method |
|---|---|---|---|---|
| GFP/mCherry Ratio (Fold Increase) | 1.0 ± 0.2 | 8.5 ± 1.3 | 1.8 ± 0.4 | Fluorescence intensity ratio |
| Time to Apoptosis Onset (h) | N/A | 18.2 ± 3.1 | N/A | Time to 10% max signal |
| Apoptosis Propagation Rate (%/h) | N/A | 25.7 ± 4.2 | N/A | Slope of activation curve |
| Spatial Heterogeneity Index | 0.05 ± 0.02 | 0.38 ± 0.09 | 0.08 ± 0.03 | Coefficient of variation |
| Viability Reduction (%) | 5.2 ± 2.1 | 78.3 ± 6.5 | 22.4 ± 5.7 | AI-based cell counting |
Protocol 3: Application in Patient-Derived Organoids (PDOs)
Patient-derived organoids preserve the genetic and phenotypic heterogeneity of original tumors, making them particularly valuable for therapeutic response modeling:
PDO Establishment from Tissue Samples
- Mechanically and enzymatically dissociate patient tumor tissue (1-2mm³ pieces)
- Use collagenase/hyaluronidase (1-2 mg/mL) for 30-60 minutes at 37°C
- Filter through 100μm strainer to obtain single cell/small cluster suspension
- Culture in defined medium with niche factors specific to tumor type [10] [42]
ZipGFP Reporter Introduction in PDOs
- Generate single-cell suspension from established PDOs using Accutase
- Transduce with lentiviral ZipGFP reporter using spinfection method
- Re-embed transduced cells in Matrigel droplets (30-50μL) with organoid culture medium
- Expand for 2-3 passages before experimental use to ensure stable reporter expression [10]
Experimental Workflow and Imaging Considerations
The following diagram illustrates the comprehensive workflow for implementing ZipGFP reporter systems in 3D spheroid and organoid models:
Treatment Protocol for PDOs
- Select mature organoids (14-21 days old) with diameter >100μm
- Treat with therapeutic agents across relevant concentration ranges
- Include clinical standard-of-care compounds as reference controls
- Refresh treatment every 2-3 days for longer-term experiments
Advanced Imaging Approaches
- Use confocal or light-sheet microscopy for improved 3D resolution
- Implement fluorescence lifetime imaging (FLIM) for quantitative FRET measurements where applicable [44]
- Correlate ZipGFP activation with additional markers (cell cycle, DNA damage, differentiation)
Multiparametric Endpoint Analysis
- Process organoids for immunohistochemistry to validate caspase cleavage
- Analyze cell death heterogeneity within organoid subregions
- Correlate ZipGFP activation patterns with organoid size, budding morphology, and viability metrics
Data Analysis and Interpretation
Quantitative Metrics for Apoptosis Assessment
The rich data generated from ZipGFP reporter imaging in 3D models enables multiple layers of quantitative analysis:
Temporal Kinetics Analysis
- Time to apoptosis initiation: Duration from treatment to first detectable GFP signal
- Rate of apoptosis propagation: Slope of GFP-positive cell accumulation
- Synchronization index: Degree of coordination in apoptotic response across cell population
Spatial Distribution Assessment
- Radial analysis: Comparison of apoptosis in core vs. peripheral regions
- Heterogeneity mapping: Identification of resistant subpopulations within 3D structures
- Cluster analysis: Detection of coordinated cell death patterns suggesting cell-cell communication
Therapeutic Response Modeling
- Dose-response relationships: EC₅₀ values for apoptosis induction
- Temporal shift analysis: Changes in kinetics across concentration gradients
- Combination index quantification: Synergistic or antagonistic drug interactions
Integration with Complementary Assays
The ZipGFP platform supports integration with additional functional readouts:
Apoptosis-Induced Proliferation (AIP) Detection
- Incorporate proliferation tracking dyes (e.g., CFSE, CellTrace)
- Monitor proliferation bursts in surviving cells following apoptotic events
- Correlate spatiotemporal patterns of apoptosis and compensatory proliferation [10]
Immunogenic Cell Death (ICD) Assessment
- Perform endpoint analysis of calreticulin exposure by flow cytometry
- Correlate caspase activation kinetics with DAMPs release
- Assess phagocytosis by co-culture with dendritic cells or macrophages [10]
Metabolic and Functional Correlates
- Integrate with metabolic assays (ATP, mitochondrial membrane potential)
- Correlate apoptosis timing with cell cycle position using FUCCI reporters
- Assess membrane integrity with time-lapse propidium iodide uptake
Troubleshooting and Technical Considerations
Successful implementation of these protocols requires attention to several technical aspects:
Table 4: Troubleshooting Guide for ZipGFP 3D Applications
| Challenge | Potential Causes | Solutions | Preventive Measures |
|---|---|---|---|
| Poor Spheroid Formation | Incorrect cell density, inadequate coating | Optimize seeding density (10³-10⁴ cells/well), ensure proper U-bottom coating | Test multiple cell densities, use commercial low-attachment plates |
| Weak GFP Signal | Low caspase activity, suboptimal expression | Validate with positive controls, use higher MOI during transduction | Titrate apoptosis inducers, select high-expression clones |
| Background Fluorescence | Incomplete reporter quenching, autofluorescence | Include caspase inhibitor controls, optimize imaging settings | Use minimal exposure settings, include background subtraction |
| Heterogeneous Response | Physiological gradients in 3D models | Implement z-stack imaging, analyze subregions separately | Normalize to viability markers, use smaller spheroids |
| Phototoxicity Effects | Excessive illumination intensity/intensity | Reduce exposure time, increase intervals between imaging | Use low-light sensors, implement adaptive imaging schemes |
| Poor Reporter Penetration | Limited viral transduction in 3D structures | Extend transduction time, use spinfection methods | Transduce before 3D culture, use lentiviral vs. adenoviral vectors |
The integration of ZipGFP caspase-3/7 reporter technology with physiologically relevant 3D models represents a significant advancement in apoptosis research and therapeutic screening. This combination enables dynamic, single-cell resolution tracking of apoptosis execution within tissue-like contexts that preserve critical aspects of tumor microenvironment and cellular heterogeneity.
The protocols outlined here provide researchers with robust methodologies for:
- Establishing functional ZipGFP reporter systems in diverse cellular contexts
- Implementing standardized 3D culture models that bridge the gap between conventional cultures and in vivo physiology
- Extracting quantitative, multiparametric data on apoptosis kinetics, spatial organization, and heterogeneity
- Integrating complementary endpoints including immunogenic cell death markers and proliferation tracking
Future developments in this field will likely focus on expanding the multiplexing capabilities of the platform through incorporation of additional biosensors for parallel monitoring of complementary pathways such as pyroptosis, necroptosis, and autophagy [10]. Additionally, combining this approach with microfluidic organ-on-chip platforms will enable more sophisticated modeling of tissue-tissue interfaces and systemic drug responses [42]. As these technologies mature, their implementation in high-content drug screening and personalized medicine approaches will provide unprecedented insights into therapeutic mechanisms and resistance patterns, ultimately accelerating the development of more effective cancer treatments.
The zebrafish (Danio rerio) has emerged as a premier model organism for monitoring physiological apoptosis in vivo, owing to its optical transparency and rapid ex utero development. This application note details methodologies for real-time imaging of caspase-3/7 activation, contextualized within broader research utilizing ZipGFP reporter systems. We provide a comprehensive framework for researchers investigating developmental apoptosis, leveraging zebrafish's unique advantages for high-resolution, live observation of cell death processes in an intact vertebrate.
Zebrafish as a Model for Apoptosis Research
Zebrafish offer distinctive benefits for apoptosis studies. Embryos are small, optically transparent, and develop rapidly outside the mother, permitting direct observation of developmental processes [45]. A single pair of adults can produce over 100 embryos per week, facilitating high-throughput screening. The molecular machinery of both intrinsic (mitochondrial) and extrinsic (death receptor) apoptotic pathways is highly conserved between zebrafish and mammals [45]. These features, combined with advanced genetic tools, make zebrafish ideal for visualizing apoptotic dynamics in real-time.
Sensor Design and Mechanism for Live Apoptosis Detection
Caspase-Activatable Fluorescent Reporters
The core technology for monitoring caspase-3/7 activation in live embryos centers on genetically encoded fluorescent reporters. These typically utilize a FRET-based design or a fluorescent protein engineered with caspase cleavage sites.
FRET-Based Sensors (e.g., Sensor C3): This design involves a fusion protein of cyan (CFP) and yellow fluorescent protein (YFP), linked by a peptide sequence containing the caspase-3 cleavage motif DEVD. In live cells, excitation of CFP results in FRET to YFP, emitting green light. During apoptosis, activated caspase-3 cleaves the DEVD linker, disrupting FRET and causing a emission shift to blue CFP fluorescence [46]. This allows for ratiometric quantification of caspase activation.
Bright-to-Dark GFP Reporters: An alternative strategy involves direct mutagenesis to insert a caspase-3 cleavage motif (DEVD) into the green fluorescent protein (GFP). In its intact state, the protein fluoresces. Upon caspase-3 activation during apoptosis, cleavage of the scaffold leads to a loss of fluorescence, creating a "bright-to-dark" signal switch [47]. This system is noted for its sensitivity and simpler construction, as it does not require additional peptide linkers [47].
Phosphatidylserine (PS) Exposure Reporters
An alternative method detects the externalization of phosphatidylserine, an early marker of apoptosis. This utilizes a secreted Annexin V protein fused to a fluorescent protein like YFP (secA5-YFP). Annexin V binds with high affinity to PS on the outer leaflet of the plasma membrane in apoptotic cells, labeling them with fluorescence [48]. This method is particularly useful for detecting early apoptosis and has been successfully implemented in transgenic zebrafish lines [48].
The following diagram illustrates the fundamental mechanism of the FRET-based caspase sensor, a core technology in this field:
Diagram 1: Mechanism of a FRET-based caspase-3 sensor. Upon caspase activation, cleavage of the DEVD linker disrupts FRET, shifting emission from green to blue.
Experimental Protocols
Generation of Transgenic Sensor Zebrafish
Objective: Create a stable zebrafish line expressing a caspase sensor (e.g., FRET-based Sensor C3) under a cell-specific promoter (e.g., mnx1 for motor neurons).
Materials:
- Tol2 transposon-based transgenic construct containing
[promoter:sensor C3][46] - Tol2 transposase mRNA
- Microinjection equipment
- Wild-type zebrafish embryos (1-cell stage)
Procedure:
- Construct Preparation: Clone the Sensor C3 sequence downstream of a tissue-specific promoter (e.g., mnx1 for motor neurons) in a Tol2 vector [46].
- mRNA Synthesis: Synthesize Tol2 transposase mRNA in vitro.
- Microinjection: Co-inject approximately 2 nl of a mixture containing the transgenic construct (15-25 ng/µl) and transposase mRNA (25-50 ng/µl) into the cytoplasm of 1-cell stage zebrafish embryos [48].
- Screening (Founder Generation): Raise injected embryos (F0 founders) to adulthood. Outcross F0 fish and screen the F1 progeny for sensor expression using fluorescence microscopy to identify stable transgenic lines [46].
Live-Imaging of Caspase Activation in Embryos
Objective: Capture the spatiotemporal dynamics of caspase-3 activation in real-time at single-cell resolution.
Materials:
- Transgenic zebrafish embryos (e.g.,
Tg(mnx1:sensor C3)) - Low-melting-point agarose
- E3 embryo medium
- Tricaine (MS-222) anesthetic
- Confocal or spinning-disk microscope equipped with temperature control (28.5°C) and a 458 nm laser line [46]
Procedure:
- Sample Preparation: At the desired developmental stage (e.g., 24-48 hours post-fertilization, hpf), dechorionate embryos manually or with pronase treatment [49].
- Mounting: Anesthetize embryos in 0.01 mg/ml tricaine. Embed embryos in a small drop of 0.7-1.2% low-melting-point agarose on a glass-bottom dish, oriented to expose the region of interest [48].
- Image Acquisition:
- Use a 20x-40x water-immersion objective.
- For FRET sensors, acquire time-lapse images using appropriate filter sets for CFP (excitation ~458 nm, emission ~480 nm) and YFP (emission ~535 nm) [46].
- Set an acquisition interval of 2-5 minutes to capture rapid caspase activation, which can occur within 5-6 minutes [46].
- Maintain temperature at 28.5°C throughout imaging.
- Data Analysis:
- Calculate the FRET ratio (CFP emission / YFP emission) pixel-by-pixel.
- A decrease in the FRET ratio (color shift from green to blue) indicates caspase-3 activation [46].
- Track the appearance of apoptotic bodies and correlate with morphological changes like cell shrinkage.
Validation Using Whole-Mount Immunofluorescence (Casp3 Assay)
Objective: Validate sensor activity by detecting activated caspase-3 in fixed embryos, providing a snapshot of apoptosis at a specific time point.
Materials:
- Fixed zebrafish embryos
- Primary antibody: Anti-activated-Caspase-3
- Secondary antibody: Fluorescently-labeled antibody
- Blocking buffer (1x PBST, 10% FBS, 2% BSA)
- 4% Paraformaldehyde (PFA) in PBS
- Methanol
- 1x PDT (1x PBST, 0.3% Triton-X, 1% DMSO) [49]
Procedure:
- Fixation: Fix embryos in 4% PFA at 4°C overnight [49].
- Permeabilization: Dehydrate embryos in 100% methanol at -20°C for at least 2 hours [49].
- Rehydration and Blocking: Rehydrate embryos through a methanol/PBST series. Wash with 1x PDT and block in blocking buffer for 1 hour at room temperature [49].
- Antibody Staining: Incubate with anti-activated-Caspase-3 primary antibody in blocking buffer overnight at 4°C. Wash extensively with 1x PDT, then incubate with fluorescent secondary antibody for 2-4 hours at room temperature [49].
- Imaging: Wash and mount embryos for confocal microscopy. Compare the pattern of antibody staining with the sensor's fluorescence signal from prior live imaging.
Key Quantitative Findings and Data
The application of these live-imaging modalities has yielded critical quantitative insights into physiological apoptosis during zebrafish development.
Table 1: Key Quantitative Findings from Live Apoptosis Imaging in Zebrafish
| Parameter | Finding | Experimental Model | Reference |
|---|---|---|---|
| Caspase-3 Activation Kinetics | Occurs rapidly within 5-6 minutes in a single neuron. | Tg(mnx1:sensor C3) motor neurons |
[46] |
| Spatial Pattern of Activation | Activation occurs simultaneously in the cell body and axon. | Tg(mnx1:sensor C3) motor neurons |
[46] |
| Developmental Death Rate | Only a small percentage of spinal cord motor neurons die during early development. | Tg(mnx1:sensor C3) zebrafish |
[46] |
| Apoptotic Body Clearance | Most apoptotic bodies of motor neurons were not colocalized with macrophages. | Tg(mnx1:sensor C3) zebrafish |
[46] |
| Sensor Sensitivity | Bright-to-dark GFP reporters demonstrated superior sensitivity compared to dark-to-bright systems. | Cultured cells & various models | [47] |
Table 2: Comparison of Primary Live-Cell Apoptosis Detection Methods
| Method | Mechanism | Advantages | Limitations |
|---|---|---|---|
| FRET-Based Caspase Sensor (e.g., Sensor C3) | Caspase-3 cleavage disrupts FRET, changing emission color. | • Ratiometric, quantitative.• Allows kinetic tracking in real-time.• Single-cell resolution. | • Requires genetic engineering.• Signal can be influenced by pH and sensor concentration. |
| Bright-to-Dark GFP Reporter | Caspase-3 cleavage inactivates fluorescence. | • High sensitivity [47].• Simple design without linkers [47].• Applicable across models [47]. | • Signal loss can be confused with photobleaching or cell death. |
| Secreted Annexin V-YFP (secA5-YFP) | Binds phosphatidylserine exposed on the outer membrane. | • Labels early apoptosis.• Can be expressed transgenically.• Does not require cellular internalization. | • Not specific to caspase-driven apoptosis.• Calcium-dependent.• May label stressed but not yet apoptotic cells. |
The following workflow summarizes the integrated process from sensor generation to data analysis:
Diagram 2: Experimental workflow for live apoptosis imaging in zebrafish, from sensor generation to data validation.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Live Apoptosis Imaging
| Reagent / Tool | Function | Example Application / Note |
|---|---|---|
| Tol2 Transposon System | Stable genomic integration of transgenes. | Method of choice for generating stable transgenic zebrafish lines [46] [48]. |
| Cell-Type Specific Promoters | Drives sensor expression in target cells. | e.g., mnx1 for motor neurons [46]; TBP for ubiquitous expression [48]. |
| FRET-Based Sensor C3 | Ratiometric reporting of caspase-3 activation. | Provides a color-based readout (green→blue) for precise kinetic tracking [46]. |
| Secreted Annexin V (secA5-YFP) | Labels phosphatidylserine exposure on apoptotic cells. | Useful for detecting early apoptosis; can be expressed via Gal4-UAS system [48]. |
| Incucyte Caspase-3/7 Dyes | Non-fluorescent, cell-permeable substrates for caspase-3/7. | Yield fluorescent signal upon cleavage; suitable for high-throughput kinetic screening in cell culture [50]. |
| Anti-activated-Caspase-3 Antibody | Immunodetection of cleaved, active caspase-3 in fixed samples. | Gold-standard validation method for fixed embryos (whole-mount immunofluorescence) [49]. |
| Acridine Orange | Vital dye staining apoptotic cells in live embryos. | Rapid staining of live embryos, but dynamic clearance requires immediate imaging [49] [45]. |
Live imaging of physiological apoptosis in zebrafish embryos using caspase-activated reporters provides an unparalleled window into the dynamics of programmed cell death. The protocols and data outlined herein demonstrate the power of this approach to yield quantitative, spatiotemporal insights at single-cell resolution within a living vertebrate. The integration of robust transgenic tools with advanced imaging modalities offers a powerful platform for fundamental developmental biology research and high-throughput applications in drug discovery and toxicology.
Within the field of cell death research, particularly for anticancer therapeutic development, there is a growing need to understand not just if a treatment kills cells, but how it kills them. A critical question is whether cytotoxic agents induce immunogenic cell death (ICD), a process that activates a adaptive immune response against dying cancer cells [51] [52]. A key hallmark of ICD is the pre-apoptotic exposure of calreticulin (CALR) on the cell surface, which acts as an "eat-me" signal to dendritic cells [51] [10] [52]. This application note details a robust methodology, grounded in a real-time imaging caspase-3/7 activation ZipGFP reporter system, for multiplexing the dynamic detection of caspase activation with endpoint validation of surface calreticulin exposure [51] [10]. This integrated approach enables the mechanistic dissection of cell death pathways and their immunogenic potential, providing a powerful tool for high-content screening in both 2D and 3D disease models [51] [53].
The foundational technology for real-time apoptosis imaging is a lentiviral-based, stable reporter system expressing two fluorescent biosensors [51] [10].
- ZipGFP-based Caspase-3/7 Reporter: This biosensor employs a split-GFP architecture where the two fragments are tethered by a flexible linker containing a DEVD motif, the specific cleavage site for executioner caspases-3 and -7. In the absence of caspase activity, the forced proximity of the fragments prevents GFP reconstitution, yielding minimal background fluorescence. Upon caspase-mediated cleavage of the DEVD linker, the GFP fragments spontaneously reassemble into a stable, fluorescent β-barrel structure, providing an irreversible, time-accumulating signal of apoptosis [51] [8].
- Constitutive mCherry Marker: A co-expressed red fluorescent protein serves as a marker for successful transduction and normalizes for cell presence and viability, though its long half-life limits its use for real-time viability assessment [51] [10].
The following workflow diagram illustrates the integrated process of real-time imaging and endpoint flow cytometry within a multiplexed assay.
Key Experimental Protocols
Protocol 1: Generation of Stable Caspase-3/7 Reporter Cell Lines
Objective: To create a stable cell line expressing the ZipGFP-based caspase-3/7 reporter and constitutive mCherry for long-term studies [51] [10].
Materials & Reagents:
- Lentiviral Constructs: pLV-ZipGFP-DEVD (Caspase-3/7 reporter) and pLV-mCherry (constitutive marker) [51].
- Packaging Plasmids: psPAX2 and pMD2.G.
- Cell Line: Relevant cancer cell line (e.g., U2OS, MiaPaCa-2, MCF-7, HUVEC) [51] [10] [52].
- Culture Media: Appropriate complete medium (e.g., DMEM or RPMI-1640 with GlutaMAX, 10% FBS) [54].
- Transfection Reagent: e.g., JetPRIME [54].
- Selection Antibiotic: e.g., Puromycin [54].
Methodology:
- Lentivirus Production: Co-transfect HEK-293T cells with the transfer plasmid (pLV-ZipGFP-DEVD-mCherry) and packaging plasmids (psPAX2, pMD2.G) using a transfection reagent like JetPRIME [54].
- Virus Harvesting: Collect viral supernatant at 48 and 72 hours post-transfection, concentrate by ultracentrifugation if necessary.
- Cell Transduction: Incubate target cells with viral supernatant in the presence of polybrene (e.g., 8 µg/mL) to enhance infection efficiency [54].
- Selection and Expansion: Apply selection pressure with puromycin (e.g., 1-2 µg/mL, concentration requires titration) for 5-7 days to eliminate non-transduced cells.
- Validation: Validate reporter functionality by treating cells with a known apoptosis inducer (e.g., 1 µM carfilzomib) and confirm GFP fluorescence induction via live-cell imaging [51].
Protocol 2: Real-Time Imaging of Caspase Activation & Endpoint Calreticulin Detection
Objective: To dynamically track apoptosis induction and correlate it with surface calreticulin exposure as a key immunogenic marker [51] [52].
Materials & Reagents:
- Stable Reporter Cells: Generated per Protocol 1.
- Inducers/Inhibitors: e.g., Carfilzomib (1 µM), Oxaliplatin (100 µM), Pan-caspase inhibitor Z-VAD-FMK (20 µM) [51].
- Live-Cell Imaging Dyes: e.g., CellTox Green (cytotoxicity), CellEvent Caspase-3/7 Green (alternative caspase sensor), DRAQ7 (viability) [54].
- Flow Cytometry Reagents: Anti-calreticulin antibody (conjugated to e.g., AF647), Annexin V (conjugated to e.g., Pacific Blue), Propidium Iodide (PI) or DAPI [51] [55].
- Equipment: Live-cell imaging system (e.g., IncuCyte), Flow Cytometer, CO₂ incubator.
Methodology:
- Cell Seeding and Treatment: Seed reporter cells in tissue culture-treated multi-well plates (e.g., 96- or 24-well). After adherence, treat with compounds of interest and appropriate controls (vehicle, caspase inhibitor) [51].
- Real-Time Imaging: Place the plate in a live-cell imaging system. Acquire images for both GFP (caspase activation) and mCherry (cell presence) channels every 2-4 hours for 48-120 hours. Maintain environmental control at 37°C with 5% CO₂ [51] [10].
- Image and Data Analysis: Use integrated software (e.g., IncuCyte AI Cell Health Module) to quantify the GFP-positive object count and mCherry area to track apoptosis kinetics and confluency [51].
- Endpoint CALR Staining: Following imaging, harvest cells by trypsinization.
- Data Correlation: Correlate the kinetic parameters of caspase activation (e.g., time to 50% max GFP signal) with the percentage of cells showing surface calreticulin exposure from flow cytometry.
Protocol 3: Adaptation to 3D Spheroid and Organoid Models
Objective: To apply the multiplexed caspase/calreticulin assay to more physiologically relevant 3D culture systems [51] [10].
Materials & Reagents:
- Extracellular Matrix (ECM): Cultrex Basement Membrane Extract or Matrigel.
- 3D Culture Plates: Ultra-low attachment spheroid microplates.
- Patient-Derived Organoids (PDOs): e.g., Pancreatic ductal adenocarcinoma (PDAC) organoids [10].
Methodology:
- 3D Model Generation: For spheroids, seed reporter cells in ultra-low attachment plates. For organoids, embed dissociated reporter cells or transduced PDOs in ECM droplets in culture plates and overlay with media [10].
- Treatment and Imaging: Allow models to form for 3-5 days. Treat with compounds and initiate real-time imaging. For 3D structures, confocal imaging or z-stack acquisition is recommended for better signal resolution [51] [10].
- Endpoint Processing: After imaging, dissociate 3D structures using appropriate enzymes (e.g., Accutase, Dispase) to create single-cell suspensions for subsequent flow cytometric analysis of surface calreticulin [10].
Data Presentation & Analysis
Quantitative Data from Model Studies
The following table summarizes key quantitative findings from validation studies using the ZipGFP caspase reporter system, demonstrating its application across different experimental conditions.
Table 1: Key Quantitative Findings from Caspase-3/7 Reporter System Validation
| Cell Model | Treatment | Key Quantitative Outcome | Citation |
|---|---|---|---|
| Stable Reporter Line (2D) | Carfilzomib (1 µM) | Significant, time-dependent increase in GFP fluorescence over 80 hours; inhibition by Z-VAD-FMK. | [51] |
| MCF-7 (Caspase-3 null) | Carfilzomib (1 µM) | Significant GFP signal induction, confirming caspase-7-mediated DEVD cleavage. | [51] |
| MiaPaCa-2 Spheroids (3D) | Carfilzomib (1 µM) | Time-dependent GFP increase in 3D structure, normalized to mCherry. | [10] |
| Patient-Derived Organoids | Carfilzomib (1 µM) | Localized GFP fluorescence within heterogeneous organoid structures. | [10] |
| HNSCC Patient Tumors | Paclitaxel/Carboplatin/Cetuximab | Increased calreticulin expression in tumor cells post-treatment correlated with lymphoid cell infiltration and no recurrence. | [53] |
Advanced Data Analysis Algorithms
Multiplexed assays generate complex, high-dimensional data. Leveraging advanced computational tools is essential for robust analysis and visualization.
Table 2: Advanced Data Analysis Algorithms for Multiplexed Assay Data
| Algorithm Type | Example Tools | Application in Multiplexed Assays |
|---|---|---|
| Dimensionality Reduction | t-SNE, UMAP [56] [57] | Visualize high-dimensional flow cytometry data in 2D, identifying distinct cell states (e.g., CALR⁺/caspase⁺). |
| Clustering Analysis | FlowSOM, Partitioning Algorithms [56] [57] | Automatically identify and phenotype subpopulations of cells without manual gating. |
| Machine Learning | Predictive Algorithms [52] | Identify physicochemical properties of compounds that predict ICD induction based on caspase/CALR profiles. |
The signaling pathways investigated and the logical relationship between caspase activation and immunogenic marker exposure can be summarized as follows.
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Multiplexed Caspase/Immunogenicity Assays
| Item | Function / Application | Example Reagents & Constructs |
|---|---|---|
| ZipGFP Caspase Reporter | Irreversible, real-time sensor for executioner caspase-3/7 activity. | pLV-ZipGFP-DEVD-mCherry construct [51] [8]. |
| Constitutive Fluorescent Marker | Normalization for cell presence; tracking of cell viability and morphology. | pLV-mCherry, pLV-H2B-RFP (nuclear) [51] [10]. |
| ICD Inducers (Positive Controls) | Induce immunogenic cell death with CALR exposure. | Oxaliplatin, Doxorubicin, Cardiac Glycosides [52]. |
| Caspase Inhibitor (Negative Control) | Confirm caspase-dependence of reporter signal. | Z-VAD-FMK (pan-caspase inhibitor) [51]. |
| Flow Cytometry Antibodies | Detect surface exposure of immunogenic markers. | Anti-Calreticulin Antibody (conjugated), Annexin V (conjugated) [51] [55]. |
| Live-Cell Imaging Dyes | Multiplex with additional viability/death parameters. | CellTox Green (membrane integrity), DRAQ7 (DNA/viability) [54]. |
| 3D Culture Matrix | Enable physiologically relevant model generation. | Cultrex BME, Matrigel [10]. |
Apoptosis-induced proliferation (AiP) is a paradoxical but crucial biological process where dying cells actively release mitogenic signals that stimulate the proliferation of surrounding surviving cells [58]. This mechanism is essential for maintaining tissue homeostasis and driving regeneration following injury. However, in the context of cancer, AiP presents a significant therapeutic challenge, as apoptotic tumor cells following chemo- or radiotherapy can paradoxically stimulate the repopulation of surviving tumor cells, contributing to treatment resistance and disease recurrence [10] [58]. Traditional methods for studying cell death, such as Annexin V staining or terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL), are limited to endpoint analyses and cannot dynamically capture the intricate, temporal relationship between apoptosis and subsequent proliferation [10]. To overcome this limitation, we have developed an integrated experimental platform that combines a stable, genetically encoded ZipGFP reporter for caspase-3/7 activity with proliferation tracking dyes. This Application Note details how this platform enables real-time, single-cell resolution imaging of apoptotic signaling and the consequent proliferative responses within complex cellular populations and 3D models, providing a powerful tool for dissecting AiP mechanisms and screening for potential therapeutic interventions.
The ZipGFP Caspase Reporter System: Mechanism and Advantages
Fundamental Design and Principle
The core of this methodology is the ZipGFP-based executioner caspase reporter, a fluorogenic biosensor engineered for high-sensitivity detection of caspase-3 and caspase-7 activity. The design leverages a split-green fluorescent protein (GFP) architecture where the eleventh β-strand (β11) is physically separated from the remaining 1-10 β-strands (β1-10) [12] [7]. The innovative "zipping" mechanism involves flanking both fragments with heterodimerizing E5 and K5 coiled-coil domains. These domains lock the two GFP fragments in a forced proximity, which prevents their proper association and chromophore formation, resulting in minimal background fluorescence [7]. A critical feature is the incorporation of caspase-specific cleavage motifs (DEVD) into the linkers tethering the GFP fragments to the coiled coils. Upon induction of apoptosis and activation of executioner caspases, cleavage at the DEVD sites releases the GFP fragments from their constraints. The freed β1-10 and β11 fragments then spontaneously self-assemble into a mature, fluorescent GFP β-barrel structure, generating an irreversible, time-accumulating fluorescent signal that marks apoptotic cells [10] [7].
Table 1: Key Characteristics of the ZipGFP Caspase-3/7 Reporter
| Feature | Description | Advantage |
|---|---|---|
| Design Architecture | Split-GFP with caging coiled coils (E5/K5) | Very low background fluorescence (fluorogenic) |
| Activation Mechanism | Cleavage at DEVD motif by caspase-3/7 | High specificity for executioner caspase activity |
| Fluorescence Signal | Irreversible GFP fluorescence upon activation | Permanent marking of apoptotic events; suitable for long-term imaging |
| Dynamic Range | ~10-fold fluorescence increase post-activation | High signal-to-noise ratio for sensitive detection |
| Co-expressed Marker | Constitutive mCherry | Internal control for cell presence and transduction efficiency |
Comparative Advantages over Traditional Reporters
The ZipGFP reporter offers substantial benefits compared to conventional FRET-based caspase sensors. FRET reporters often suffer from poor signal-to-noise ratios due to small fluorescence changes and are hindered by tissue autofluorescence, limiting their effectiveness in vivo [12] [7]. In contrast, the ZipGFP system is fluorogenic, meaning fluorescence is generated de novo upon activation, leading to a high signal-to-noise ratio. This design minimizes background and enables robust detection of apoptosis even in thick, optically challenging samples like 3D organoids and in vivo models [10] [12]. The irreversible nature of the signal ensures that cells which have experienced a caspase activation event are permanently labeled, allowing for accurate tracking of their fate and the dynamics of death propagation within a population over extended time courses.
Integrated Experimental Workflow for AiP Detection
The following diagram and section outline the core workflow for conducting an integrated AiP experiment, from cell preparation to final data analysis.
The integrated AiP assay involves a sequential process designed to capture the entire biological narrative from cell death to compensatory proliferation. The process begins with the generation of a stable cell line expressing the dual fluorescent reporter system. These cells are then pre-labeled with a cytoplasmic proliferation tracking dye before being cultured in the desired format (2D or 3D). Apoptosis is induced with a chosen stimulus, and the cells are immediately transferred to a live-cell imaging system for continuous, automated monitoring. The entire process culminates in the extraction and analysis of multi-parameter data to quantify the relationship between apoptotic cells and subsequent proliferation in their neighbors.
Detailed Experimental Protocols
Protocol 1: Generation of Stable ZipGFP Reporter Cell Lines
Objective: To create a stable cell population constitutively expressing the ZipGFP caspase-3/7 reporter and mCherry.
Materials:
- Lentiviral vector encoding the ZipGFP-DEVD-mCherry construct
- Target cells (e.g., MiaPaCa-2, HUVECs, or patient-derived organoids)
- Appropriate cell culture medium and reagents
- Polybrene (e.g., 8 µg/mL working concentration)
- Puromycin or other suitable selection antibiotic
Method:
- Lentiviral Transduction: Plate target cells at 50-60% confluency in a 6-well plate. The following day, replace the medium with fresh medium containing polybrene to enhance viral infection efficiency.
- Add an appropriate volume of the lentiviral supernatant to the cells. The exact volume should be determined by prior titration.
- Centrifuge the plate at 800 × g for 30-60 minutes at 32°C (spinoculation) to increase transduction efficiency.
- After 24 hours, replace the virus-containing medium with fresh complete growth medium.
- Selection and Expansion: 48 hours post-transduction, begin selection by adding the appropriate concentration of puromycin to the medium. Maintain selection pressure for at least 5-7 days, until all cells in a non-transduced control well have died.
- Expand the resistant cell pool and validate reporter functionality using a known apoptosis inducer (e.g., 1-10 µM Carfilzomib for 24 hours) and live-cell imaging or flow cytometry to detect GFP signal.
Protocol 2: Combined ZipGFP and Proliferation Dye Assay in 2D Culture
Objective: To simultaneously track caspase-3/7 activation and cell proliferation in a 2D monolayer.
Materials:
- Stable ZipGFP reporter cells
- Proliferation dye (e.g., CellTrace Violet, CFSE)
- Apoptosis inducer (e.g., Carfilzomib, Oxaliplatin) and control (DMSO)
- Pan-caspase inhibitor (e.g., zVAD-FMK, 20-50 µM) for specificity controls
- Live-cell imaging-compatible microplate (e.g., 96-well black-walled, clear-bottom plate)
- Live-cell imaging system (e.g., IncuCyte)
Method:
- Proliferation Dye Labeling: Harvest ZipGFP reporter cells and resuspend them in serum-free PBS at a concentration of 1-2 million cells/mL.
- Add the proliferation dye at the manufacturer's recommended working concentration (e.g., 1-5 µM for CellTrace Violet). Incubate for 20 minutes at 37°C.
- Quench the reaction by adding 5 volumes of complete culture medium. Incubate the cells for 5 minutes, then pellet them by centrifugation. Resuspend the labeled cell pellet in fresh complete medium.
- Plating and Treatment: Plate the labeled cells into the live-cell imaging microplate at the desired density (e.g., 10,000 cells/well for a 96-well plate). Allow cells to adhere overnight.
- Pre-treat control wells with the pan-caspase inhibitor zVAD-FMK for 1 hour. Then, treat the experimental wells with the apoptosis inducer and the control wells with an equivalent volume of vehicle (e.g., DMSO).
- Live-Cell Imaging: Immediately place the microplate in the live-cell imaging system. Configure the method to acquire images from all fluorescent channels (ZipGFP, mCherry, proliferation dye) and a brightfield channel every 2-4 hours for 72-120 hours. Maintain standard culture conditions (37°C, 5% CO₂) throughout the imaging period.
Protocol 3: Adaptation to 3D Spheroid and Organoid Models
Objective: To monitor AiP in a more physiologically relevant 3D context.
Materials:
- Stable ZipGFP reporter organoids or cells for spheroid formation
- Cultrex Basement Membrane Extract (BME) or Matrigel
- Advanced 3D culture medium
Method:
- 3D Model Establishment: For spheroids, mix labeled ZipGFP reporter cells with BME/Cultrex according to the manufacturer's instructions and plate droplets in a pre-warmed microplate. For organoids, embed dissociated reporter organoids in BME/Cultrex domes.
- Allow the BME/Cultrex to polymerize at 37°C for 30-60 minutes, then carefully overlay with 3D culture medium.
- After 24-72 hours, when spheroids/organoids have formed, add apoptosis inducers directly to the overlay medium.
- 3D Live-Cell Imaging: Use a confocal or spinning-disk confocal microscope equipped with an environmental chamber for 3D time-lapse imaging. Acquire Z-stacks at each time point to capture the entire structure. The mCherry signal is critical for normalizing the ZipGFP signal against changes in cell density and viability within the 3D mass [10].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagent Solutions for Integrated AiP Analysis
| Reagent / Tool | Function / Description | Example Use Case |
|---|---|---|
| ZipGFP-DEVD Caspase Reporter | Fluorogenic biosensor for caspase-3/7 activity. | Real-time visualization of apoptosis initiation and propagation. |
| Constitutive mCherry | Constitutively expressed red fluorescent protein. | Normalization for cell presence and viability; marker for successful transduction. |
| Cytoplasmic Proliferation Dyes | Cell-permanent dyes that dilute 2-fold with each cell division (e.g., CellTrace Violet, CFSE). | Tracking proliferation history of surviving cells neighboring apoptotic cells. |
| Carfilzomib | Proteasome inhibitor; potent apoptosis inducer. | Used at 1-10 µM to trigger intrinsic apoptosis in reporter validation and AiP assays. |
| zVAD-FMK | Broad-spectrum, cell-permeable caspase inhibitor. | Used at 20-50 µM to confirm caspase-specificity of the ZipGFP signal. |
| LLOMe | Lysosomotropic agent; induces lysosomal cell death. | Alternative cell death inducer; used in studies of cell revival [40]. |
| FlowJo Software | Advanced flow cytometry data analysis platform. | Used for endpoint analysis, clustering (t-SNE, UMAP), and population comparison [56]. |
Data Analysis and Interpretation
Quantitative Metrics and Table Construction
The power of this integrated approach lies in the extraction of quantitative, single-cell data from time-lapse movies. The following metrics should be calculated and compiled for statistical analysis and comparison between experimental conditions.
Table 3: Key Quantitative Metrics for AiP Analysis from Live-Cell Imaging Data
| Metric | Definition | Interpretation in AiP Context |
|---|---|---|
| Apoptosis Kinetics | Time from induction to first detectable ZipGFP signal; rate of new apoptosis events over time. | Characterizes the dynamics and synchrony of cell death initiation. |
| AiP Incidence | Percentage of proliferation dye-labeled cells that have undergone division within a defined radius of a ZipGFP+ cell. | Direct measure of the proliferative output stimulated by apoptotic cells. |
| Proliferation Lag Time | Time delay between the appearance of a ZipGFP+ signal in a founder cell and the first division of a neighboring cell. | Informs on the kinetics of signal production, transmission, and response. |
| Spatial Range of AiP | The maximum distance from a ZipGFP+ cell at which a significant proliferative response is observed. | Provides insight into the diffusion range of the mitogenic factors involved. |
| Caspase Specificity Index | (GFP+ events in treated group) / (GFP+ events in treated + zVAD-FMK group). | Confirms that the observed proliferation is linked to caspase-dependent apoptosis. |
Signaling Pathways in Apoptosis-Induced Proliferation
The following diagram summarizes the key signaling pathways involved in AiP, highlighting how apoptotic caspases can initiate mitogenic signaling.
Validation and Complementary Techniques
To strengthen the findings from live-cell imaging, endpoint analyses are recommended:
- Immunogenic Cell Death (ICD) Assessment: After imaging, cells can be processed for flow cytometric analysis of surface calreticulin exposure, a key damage-associated molecular pattern (DAMP) in ICD [10]. This allows correlation of AiP with immunogenic potential.
- Flow Cytometry Analysis: Cells can be harvested and analyzed using platforms like FlowJo to apply advanced analytical tools such as clustering algorithms (t-SNE, UMAP) to identify distinct cellular states based on ZipGFP, proliferation dye, and other immunostaining markers [56].
- Pathway Inhibition: Using specific inhibitors (e.g., for JNK, NF-κB, or EGFR signaling) during the assay can help delineate the critical pathways required for the observed AiP phenotype [40] [58].
This integrated ZipGFP-proliferation dye platform provides a high-content, dynamic, and mechanistic approach to studying cell death and its paradoxical pro-survival consequences. For drug development professionals, this system is exceptionally valuable for:
- Evaluating On-Target Effects: Confirming that novel chemotherapeutic agents successfully induce caspase-mediated apoptosis in tumor models.
- Identifying Pro-Regenerative Therapies: Screening for compounds that enhance AiP in models of tissue injury and degeneration.
- Mitigating Therapy Resistance: Screening for combination therapies that inhibit or decouple the AiP response, thereby preventing tumor repopulation after initial treatment and improving long-term therapeutic outcomes [10] [58].
By offering a unified method to visualize the entire process from death signal to proliferative response, this application note provides a robust framework for deepening our understanding of tissue homeostasis and developing more effective regenerative and anti-cancer strategies.
Maximizing Performance: A Troubleshooting Guide for Signal, Specificity, and Kinetics
The ZipGFP-based executioner caspase reporter represents a significant advancement in the real-time imaging of apoptosis. Unlike traditional FRET-based reporters, the ZipGFP system is designed as a fluorogenic protease reporter. In its inactive state, the engineered GFP is "zipped" in a way that quenches its fluorescence. Upon cleavage by activated executioner caspases-3 or -7, the reporter undergoes a conformational change that restores its fluorescent function, resulting in a documented 10-fold increase in fluorescence after activation [8]. This high activation ratio is crucial for detecting the subtle, initial stages of apoptosis in real-time assays.
However, leveraging this technology in physiologically relevant environments, such as in live animal models or complex 3D cell cultures, presents two major challenges: achieving a sufficient Signal-to-Noise Ratio (SNR) and managing constitutive fluorescence. A low SNR can obscure genuine caspase activation signals, while excessive background fluorescence can lead to false positives and inaccurate quantification of cell death kinetics. This application note provides detailed methodologies to overcome these pitfalls, ensuring reliable and high-quality data for researchers and drug development professionals.
Understanding and Optimizing Signal-to-Noise Ratio
The Critical Role of SNR in Fluorescence Imaging
In fluorescence microscopy, the Signal-to-Noise Ratio (SNR) quantifies how much the desired fluorescence signal stands out from the background interference. It is mathematically defined as the mean value of the signal (μ) divided by the standard deviation of the noise (σ): SNR = μ / σ [59]. A higher SNR translates to clearer images, better object detection, and improved performance of subsequent analysis algorithms. For the ZipGFP reporter, a high SNR is essential to distinguish the specific fluorescence increase from caspase activation from non-specific background noise.
Strategies to Improve SNR in ZipGFP Imaging
Optimizing SNR requires a holistic approach, addressing both the signal from the activated reporter and the various sources of noise.
| Factor | Goal | Practical Implementation |
|---|---|---|
| Camera Selection | Maximize signal detection while minimizing camera-generated noise. | Use a cooled monochrome CCD camera. Cooling reduces dark current noise, and monochrome sensors typically offer higher sensitivity than color cameras [60]. |
| Optical Components | Maximize light collection efficiency and signal strength. | Use objectives with a high Numerical Aperture (NA). A larger NA provides a brighter image at higher resolution [60]. Ensure fluorescence filters closely match the ZipGFP excitation/emission spectra. |
| Image Acquisition | Balance signal intensity with noise generation and sample health. | Optimize exposure time; longer exposure collects more signal but can increase dark current noise and promote photobleaching. Use binning to increase signal intensity at the cost of spatial resolution. Keep gain low to avoid amplifying noise [60]. |
| Sample Health | Minimize non-specific noise from dying cells. | Maintain healthy cells and use appropriate controls to identify autofluorescence from dead or stressed cells. |
Managing Constitutive Fluorescence
Constitutive fluorescence, or background signal from the unreported probe, is a common pitfall that can mask true caspase activation and lead to false positives.
- Verify Reporter Design and Expression: The ZipGFP reporter is rationally designed to be "zipped," meaning its fluorescence is low in the uncleaved state [8]. However, high overexpression can lead to accumulation of the probe, increasing background. Use inducible or low-copy-number expression systems to find the optimal balance between detectable signal and minimal background.
- Include Rigorous Controls: Always include cells expressing the mutant (non-cleavable) ZipGFP reporter under the same conditions. This control is indispensable for quantifying the level of constitutive fluorescence and setting a baseline for image thresholding.
- Monitor Cell Health: Non-specific stress or alternative cell death pathways can sometimes cause signal independent of caspase-3/7. Ensure cultures are healthy and not over-confluent, and use pharmacological inhibitors (e.g., pan-caspase inhibitors) to confirm the specificity of the signal.
Detailed Experimental Protocol for ZipGFP-based Caspase-3/7 Imaging
Workflow for Apoptosis Imaging with SNR Optimization
The following diagram outlines the key stages of a robust experiment for imaging caspase-3/7 activation with the ZipGFP reporter.
Step-by-Step Protocol
Materials:
- Cells of interest (e.g., stable cell line expressing ZipGFP caspase-3/7 reporter)
- Apoptosis inducer (e.g., Staurosporine, 1 µM)
- Caspase inhibitor control (e.g., Z-VAD-FMK, 20 µM)
- Appropriate cell culture medium and plates (e.g., collagen-coated 8-well µ-slides for imaging)
- Live-cell imaging system equipped with a cooled CCD camera and environmental control (37°C, 5% CO₂)
Procedure:
Cell Preparation:
- Seed cells expressing the ZipGFP caspase reporter into an imaging-optimized plate (e.g., ibidi µ-slide) at a density that will allow ~70% confluency at the time of imaging.
- Incubate for 12-24 hours to allow cells to adhere and fully recover.
Control Setup:
- Experimental Group: Cells treated with apoptosis inducer.
- Negative Control: Cells pre-treated with a broad-spectrum caspase inhibitor (e.g., Z-VAD-FMK, 20 µM) for 1 hour before adding the apoptosis inducer.
- Background Control: Cells expressing a non-cleavable mutant ZipGFP reporter.
Microscope Setup:
- Place the plate on the microscope stage with environmental control set to 37°C and 5% CO₂.
- Select a high-NA objective (e.g., 40x or 60x oil immersion).
- Configure the light source and filter set to match the excitation/emission spectra of ZipGFP.
- Set the focus. For fluorescence microscopes without collision prevention, first position the lens close to the sample, then gradually move it away to adjust the focus [60].
Image Acquisition Optimization:
- Exposure Time: Begin with a moderate exposure time (e.g., 100-500 ms). Increase until a clear signal is obtained from positive control cells, but avoid saturation.
- Gain: Keep the gain as low as possible to prevent amplification of electronic noise.
- Binning: If the signal is too weak, consider 2x2 binning to boost signal intensity at the cost of some resolution.
- Z-stack: For thick samples, acquire a limited Z-stack to ensure the entire cell is in focus.
Time-lapse Imaging:
- Initiate the time-lapse series, acquiring images at regular intervals (e.g., every 10-30 minutes) over the desired duration (e.g., 6-24 hours).
- Add the apoptosis-inducing compound carefully after acquiring 1-2 baseline images.
Data Analysis:
- Use image analysis software (e.g., ImageJ/Fiji, CellProfiler) to quantify the mean fluorescence intensity of individual cells over time.
- Subtract the background fluorescence from each image.
- Normalize the fluorescence intensity (F) to the baseline value (F₀) for each cell to calculate F/F₀.
- A cell is considered apoptotic when its F/F₀ exceeds a threshold defined using the negative control (e.g., mean + 3 standard deviations of the inhibited control).
Research Reagent Solutions and Essential Materials
A carefully selected toolkit is fundamental for the success of live-cell apoptosis imaging.
Table 2: Essential Research Reagents and Materials for ZipGFP Caspase Imaging
| Item | Function/Description | Example/Note |
|---|---|---|
| ZipGFP Caspase-3/7 Reporter | Fluorogenic reporter that increases fluorescence ~10-fold upon cleavage by executioner caspases [8]. | Can be deployed in plasmid or lentiviral vectors for stable cell line generation. |
| Cooled Monochrome CCD Camera | Camera for detection; cooling reduces dark current noise, critical for low-light fluorescence and time-lapse imaging [60]. | Keyence BZ-X; other major brands (Nikon, Olympus) offer similar cooled cameras. |
| High-NA Objective Lens | Microscope lens to collect maximum light; higher NA provides brighter images and better resolution [60]. | e.g., 60x Oil, NA 1.4. |
| Live-Cell Imaging Chamber | System to maintain physiological conditions (37°C, 5% CO₂, humidity) on the microscope stage. | Ibidi µ-Slide; Lab-Tek chambers. |
| Annexin V Probes | Marker for phosphatidylserine externalization, an early apoptosis event. Use to correlate caspase activation with other death markers [54]. | Available conjugated to various fluorophores (e.g., Pacific Blue, FITC). |
| Caspase Inhibitor | Pharmacological control to confirm signal specificity is dependent on caspase activity. | Z-VAD-FMK (pan-caspase inhibitor). |
| CellTox Green / DRAQ7 | Cytoplasmic (CellTox Green) or nuclear (DRAQ7) dyes that stain dead cells due to loss of membrane integrity. Distinguish late-stage death/necrosis [54]. | DRAQ7 is far-red fluorescent and compatible with ZipGFP. |
The ZipGFP caspase reporter is a powerful tool for visualizing programmed cell death with high spatiotemporal resolution. By systematically addressing the common challenges of low SNR and constitutive fluorescence through optimized hardware selection, meticulous experimental design, and rigorous controls, researchers can obtain robust, quantifiable data. These protocols provide a foundation for reliably applying this technology in basic research and drug discovery, enabling the accurate assessment of caspase-3/7 activation in real-time.
Within the context of real-time imaging of caspase-3/7 activation using ZipGFP reporters, a common yet often overlooked challenge is the induction of cellular stress by the reporter system itself. The ZipGFP reporter is a fluorogenic biosensor engineered from a split-green fluorescent protein (GFP), where the two fragments are caged by heterodimerizing coiled-coil peptides (E5 and K5) that prevent their spontaneous reassembly [7]. This system is designed to be irreversibly activated upon cleavage by executioner caspases-3 or -7 at a specific DEVD peptide motif [10] [7]. While this technology enables unprecedented spatiotemporal resolution of apoptotic events in living cells and organisms, high or unbalanced expression of the constituent reporter components can provoke proteostatic stress, disrupt normal cellular functions, and ultimately compromise experimental validity. This document provides detailed application notes and protocols for optimizing ZipGFP reporter expression to minimize cellular stress while maximizing detection sensitivity.
Understanding the ZipGFP Reporter System
Core Mechanism and Design
The ZipGFP caspase-3/7 reporter operates on a unique "zipping" mechanism. The eleventh β-strand (β11) of GFP is fused to one leucine zipper (K5), while the first ten β-strands (β1-10) are fused to the complementary zipper (E5). A caspase-cleavable linker (DEVD) is incorporated into each of these fusion constructs. In the absence of caspase activity, the E5 and K5 coiled coils heterodimerize, physically occluding the binding cavity and preventing the split-GFP fragments from reassembling, thus minimizing background fluorescence [7]. Upon apoptosis induction and subsequent caspase-3/7 activation, the DEVD linkers are cleaved. This liberates the split-GFP fragments from the zipper constraints, allowing them to spontaneously reassociate and form a mature, fluorescent GFP β-barrel structure [10] [7]. The reassembly is irreversible, leading to a cumulative and stable fluorescent signal that permanently marks cells that have undergone caspase activation.
- Proteostatic Overload: Constitutive overexpression of the unreconstituted reporter components (β1-10-E5 and β11-K5) can overwhelm the cellular protein folding and degradation machinery, potentially leading to endoplasmic reticulum stress and activation of the unfolded protein response.
- Imbalanced Stoichiometry: Unequal expression levels of the two zipper-fused GFP fragments can result in unpaired, aggregation-prone proteins. This is a critical source of cytotoxicity and can manifest as punctate, non-specific fluorescent aggregates within cells [7].
- Caspase Cascade Interference: In extreme cases, high levels of the DEVD-containing reporter construct may act as a decoy substrate, potentially sequestering active caspases and inadvertently delaying or modulating the apoptotic process itself.
Quantitative Characterization of the ZipGFP Reporter
Table 1: Performance Characteristics of the ZipGFP Caspase-3/7 Reporter
| Parameter | Value / Description | Experimental Context |
|---|---|---|
| Fluorescence Increase | ~10-fold | Upon caspase activation in HEK293 cells [7] |
| Activation Kinetics (T1/2) | ~40 minutes (in vitro)~100 minutes (in cells) | Measured after TEV protease cleavage; slower cellular kinetics with rapamycin-inducible system [7] |
| Quantum Yield | 0.25 | Of the reassembled ZipGFP complex [7] |
| Key Validation Controls | zVAD-FMK (pan-caspase inhibitor)Carfilzomib & Oxaliplatin (inducers) | Confirmation of caspase-dependent signal [10] |
| Background Fluorescence | Minimal | Due to effective "zipping" of split-GFP fragments [7] |
| Specificity | Activated in Caspase-3 deficient MCF-7 cells | Confirms Caspase-7 can activate the DEVD-based reporter [10] |
Table 2: Stress Indicators and Thresholds in Reporter Cells
| Indicator | Normal Range / Low Stress | Stress Condition / High Expression |
|---|---|---|
| Punctate Fluorescence | Diffuse cytosolic signal | Presence of bright, speckled aggregates [7] |
| Constitutive mCherry Signal | Stable, homogeneous expression | High variance, declining signal over time [10] |
| Cell Growth Rate | >80% of untransduced control | <50% of untransduced control |
| Viability (Annexin V-/PI-) | >90% in untreated cultures | <80% in untreated cultures [10] |
| Basal ZipGFP Signal | <5% of max induced signal | >15% of max induced signal |
Protocols for Optimizing Reporter Expression
Protocol 1: Generating Stable Cell Lines with Balanced Expression
This protocol outlines the creation of stable cell lines using lentiviral vectors, designed to achieve consistent, low-stress expression of the ZipGFP reporter system.
Materials:
- Reporter Construct: Lentiviral transfer plasmid encoding the ZipGFP caspase-3/7 reporter (DEVD sequence in both fragments) and a constitutive fluorescent marker (e.g., mCherry) [10].
- Packaging Plasmids: psPAX2 and pMD2.G.
- Cell Line: HEK293T cells for virus production; target cells (e.g., MiaPaCa-2, HUVECs, patient-derived organoids) [10].
- Culture Reagents: Standard DMEM/FBS medium, polybrene (8 µg/mL), puromycin or appropriate selection antibiotic.
Method:
- Virus Production:
- Co-transfect HEK293T cells with the reporter construct and packaging plasmids using a standard calcium phosphate or PEI method.
- Replace medium after 6-8 hours. Harvest the lentivirus-containing supernatant at 48 and 72 hours post-transfection.
- Concentrate the supernatant using ultracentrifugation or commercial concentration kits. Aliquot and store at -80°C.
Cell Transduction:
- Plate target cells at 40-50% confluence in a 6-well plate.
- The following day, add the lentiviral supernatant supplemented with 8 µg/mL polybrene to the cells.
- Centrifuge the plate at 800 × g for 30 minutes at 32°C (spinoculation) to enhance infection efficiency.
- Replace the virus-containing medium with fresh culture medium after 12-24 hours.
Selection and Clonal Isolation:
- Begin antibiotic selection (e.g., 1-2 µg/mL puromycin) 48 hours post-transduction. Maintain selection for at least 5-7 days until all cells in the non-transduced control well have died.
- Using fluorescence-activated cell sorting (FACS), isolate a population of cells that are positive for the constitutive marker (mCherry). For the highest uniformity, single-cell clone generation is recommended.
- Expand single-cell clones and characterize them as described in Protocol 2.
Protocol 2: Characterizing and Validating Reporter Clones
This protocol details the assessment of stable clones for optimal reporter performance and the absence of significant cellular stress.
Materials:
- Inducers: 1 µM Carfilzomib (proteasome inhibitor) or 100 µM Oxaliplatin [10].
- Inhibitor: 20 µM zVAD-FMK (pan-caspase inhibitor) [10].
- Staining Reagents: Annexin V / Propidium Iodide (PI) apoptosis detection kit.
- Equipment: Live-cell imaging system (e.g., IncuCyte), flow cytometer, western blot apparatus.
Method:
- Baseline Stress Assessment:
- Image the mCherry signal (cell presence) and the baseline GFP signal (apoptotic background) for 24-48 hours in untreated cells using live-cell imaging.
- Calculate the coefficient of variation (CV) of the mCherry signal. A low CV indicates homogeneous expression, a sign of low stress.
- Determine the percentage of cells with a high baseline GFP signal or those showing punctate, aggregated fluorescence.
Functional Validation:
- Treat cells with the following conditions for 24-48 hours: (a) DMSO (vehicle control), (b) inducer (e.g., Carfilzomib), (c) inducer + zVAD-FMK.
- Monitor GFP and mCherry fluorescence every 2-4 hours using live-cell imaging.
- Quantitative Analysis:
- Normalize the GFP signal to the mCherry signal to account for cell loss.
- Plot the kinetics of caspase activation. A robust, time-dependent increase in the GFP/mCherry ratio should be observed in the induced group, which is suppressed by zVAD-FMK co-treatment.
- At the endpoint (e.g., 24 hours), harvest cells and analyze by flow cytometry (Annexin V/PI) and western blot (for cleaved PARP and cleaved Caspase-3) to biochemically confirm apoptosis [10].
Clone Selection:
- Select clones that exhibit: i) low baseline GFP and homogeneous mCherry, ii) a high signal-to-noise ratio (>8-fold induction) upon apoptosis induction, iii) complete inhibition of the GFP signal by zVAD-FMK, and iv) normal morphological appearance and growth rate.
Protocol 3: Application in 3D Culture Systems
The ZipGFP reporter is highly effective in complex 3D models like spheroids and patient-derived organoids (PDOs), which better recapitulate in vivo physiology [10].
Materials:
- Cultrex Basement Membrane Extract or Matrigel.
- Patient-derived organoids (PDOs) [10].
- Confocal or high-content imaging microscope.
Method:
- 3D Model Generation:
- For spheroids, mix reporter cells with Cultrex and seed in a 96-well plate to form domes. Allow to polymerize, then overlay with culture medium.
- For PDOs, transduce organoid fragments as described in Protocol 1 and embed in Matrigel.
- Imaging and Analysis in 3D:
- Treat 3D cultures with apoptosis inducers and image over 80+ hours using a confocal microscope.
- Acquire z-stacks to capture the entire 3D structure.
- Use image analysis software to quantify the total GFP and mCherry fluorescence intensity in each z-stack, normalizing the GFP signal to mCherry to accurately track apoptosis kinetics within the 3D microenvironment [10].
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for ZipGFP Reporter Studies
| Reagent / Material | Function / Description | Example Use Case |
|---|---|---|
| ZipGFP Caspase-3/7 Reporter | Fluorogenic biosensor for real-time apoptosis imaging. | Stable expression in cell lines for live-cell imaging of caspase dynamics [10] [7]. |
| Constitutive mCherry Reporter | Fluorescent marker for cell presence and normalization. | Co-expressed with ZipGFP to normalize for cell number and viability [10]. |
| Carfilzomib | Proteasome inhibitor; induces intrinsic apoptosis pathway. | Positive control for apoptosis induction (e.g., 1 µM) [10]. |
| zVAD-FMK | Irreversible pan-caspase inhibitor. | Control for caspase-specificity of the ZipGFP signal (e.g., 20 µM) [10]. |
| Annexin V / PI Kit | Fluorescent assay for detecting early/late apoptosis and necrosis. | End-point validation of apoptosis by flow cytometry [10]. |
| Lentiviral Packaging System | psPAX2 and pMD2.G plasmids for producing lentiviral particles. | Generation of high-titer virus for creating stable reporter cell lines [10]. |
| Cultrex / Matrigel | Basement membrane extract for 3D cell culture. | Supporting the growth and imaging of reporter cells in spheroid or organoid models [10]. |
Visualizing Workflows and Signaling
The following diagrams, created using Graphviz, illustrate the core concepts and experimental workflows.
ZipGFP Reporter Activation Mechanism
Stable Cell Line Generation & Validation Workflow
Apoptotic Signaling & Caspase Activation Pathways
Within the framework of real-time imaging research utilizing the innovative ZipGFP caspase-3/7 reporter, validating the specific caspase-dependent nature of observed cell death is a fundamental prerequisite for accurate data interpretation. The induction of apoptosis, a complex process involving multiple signaling pathways, can sometimes be triggered by non-caspase-dependent mechanisms. This application note provides detailed protocols for the use of the pan-caspase inhibitor zVAD-FMK, a potent, cell-permeable irreversible inhibitor, to confirm that apoptotic events detected by the ZipGFP reporter are indeed a direct consequence of caspase activation. By integrating this pharmacological validation tool into your research workflow, you significantly enhance the reliability and specificity of your findings in high-content screening and drug discovery applications.
The Principle of Caspase Inhibition with zVAD-FMK
The fluoromethyl ketone (FMK) moiety of zVAD-FMK acts as an irreversible caspase inhibitor that forms a thioether bond with the catalytic cysteine residue in the active site of caspases [61]. Its peptide sequence (Val-Ala-Asp) mimics the natural cleavage site for effector caspases, granting it broad-spectrum activity against a wide range of initiator and executioner caspases. When introduced to live cells, zVAD-FMK passively diffuses across the cell membrane and covalently binds to activated caspases, effectively blocking their proteolytic activity and halting the apoptotic cascade [61]. This mechanism allows researchers to discern whether a particular death stimulus, such as a novel chemotherapeutic agent, operates through a caspase-dependent pathway. In the context of ZipGFP reporter research, where fluorescence emission directly indicates caspase-3/7-mediated cleavage, pre-treatment with zVAD-FMK serves as a critical negative control—specifically, the attenuation or abolition of ZipGFP fluorescence signal upon application of a pro-apoptotic stimulus provides compelling evidence for the caspase-specific nature of the observed cell death.
- Diagram: Logical Workflow for Validating Caspase Dependence
Diagram Title: Logical workflow for validating caspase dependence.
Key Research Reagent Solutions
The following table catalogs essential reagents and tools for conducting caspase dependence validation studies in conjunction with ZipGFP reporter research.
Table 1: Essential Research Reagents for Caspase Validation Studies
| Reagent/Tool | Primary Function & Application | Key Characteristics |
|---|---|---|
| zVAD-FMK | Pan-caspase inhibitor for validating caspase-dependent apoptosis [62] [61]. | Broad-spectrum, irreversible, cell-permeable. |
| ZipGFP Caspase Reporter | Fluorogenic caspase-3/7 reporter for live-cell imaging [7]. | ~10-fold fluorescence increase upon cleavage, high signal-to-noise. |
| DEVD-NucView488 | Fluorogenic substrate for direct caspase-3/7 activity detection [62]. | Homogeneous, live-cell, non-toxic for extended imaging. |
| FAM-VAD-FMK | Fluorescently-labeled caspase inhibitor for direct visualization of active caspases [61]. | Flow cytometry/fluorescence microscopy application. |
| Staurosporine / Camptothecin | Well-characterized inducers of intrinsic apoptosis; used as positive controls [61]. | Reliable and robust apoptosis triggers. |
Quantitative Profiling of zVAD-FMK Efficacy
The effectiveness of zVAD-FMK in inhibiting caspase activity and apoptosis has been quantitatively demonstrated across multiple experimental contexts. The data below summarize key findings from the literature regarding its performance.
Table 2: Quantitative Profile of zVAD-FMK Inhibitory Efficacy
| Experimental Context | Caspase / Apoptosis Readout | Observed Effect of zVAD-FMK | Citation |
|---|---|---|---|
| HeLa Cells treated with Doxorubicin or Etoposide | Caspase activation (DEVD-NucView488) | Attenuated caspase activation [62]. | [62] |
| Jurkat/HeLa Cells treated with Staurosporine | Caspase activity (FAM-VAD-FMK staining) | 3- to 5-fold reduction in fluorescence signal; staining blocked [61]. | [61] |
| HeLa Cells transfected with cell death siRNA | Caspase activation (DEVD-NucView488) | Attenuated caspase activation [62]. | [62] |
| Jurkat Cells treated with Staurosporine | Affinity labeling of Caspase-3, -6, -7; Apoptosis | Inhibition of caspase labeling and apoptosis [61]. | [61] |
Detailed Experimental Protocols
Core Protocol: Validating Caspase Dependence with zVAD-FMK in Live-Cell Imaging
This protocol is designed for researchers using ZipGFP or similar caspase reporters in multi-well plate formats, leveraging automated microscopy for high-content analysis [62].
Materials:
- Cell line of interest stably expressing the ZipGFP caspase-3/7 reporter [7].
- zVAD-FMK (prepared as a 20 mM stock in DMSO, stored at -20°C).
- Pro-apoptotic stimulus (e.g., chemotherapeutic drug, targeted agent).
- Appropriate positive control inducer (e.g., 1 µM Staurosporine).
- Vehicle control (e.g., DMSO, concentration matched to inhibitor stocks).
- Cell culture medium and supplements.
- Black-walled, clear-bottom 96- or 384-well imaging plates.
- Live-cell imaging instrument (e.g., confocal or widefield microscope with environmental control).
Workflow:
- Cell Seeding: Seed ZipGFP-expressing cells into the imaging plate at an optimal density for log-phase growth after the treatment period (e.g., 5,000-10,000 cells/well for a 96-well plate). Incubate for 12-24 hours to allow for cell attachment.
- Pre-treatment with Inhibitor:
- Prepare a 2X working concentration of zVAD-FMK (typically 40-100 µM) in pre-warmed culture medium. The final working concentration usually ranges from 20 to 50 µM [62].
- Gently replace the medium in the designated wells with the 2X zVAD-FMK medium. For vehicle control wells, add medium containing an equal volume of DMSO.
- Incubate the plate for 1-2 hours under standard culture conditions (37°C, 5% CO₂) to allow cellular uptake of the inhibitor.
- Induction of Apoptosis:
- After the pre-incubation, add the pro-apoptotic stimulus directly to the wells at a 2X concentration. In the "stimulus only" control wells, add the stimulus. In the "zVAD-FMK only" control wells, add vehicle. For the "positive control" wells, add Staurosporine.
- Gently mix the plate by swirling.
- Real-Time Live-Cell Imaging:
- Place the imaging plate into the environmentally controlled chamber (maintained at 37°C and 5% CO₂) of the microscope.
- Acquire images using appropriate filters for ZipGFP (Ex/Em ~488/510 nm) at regular intervals (e.g., every 1-4 hours) over a period of 24-48 hours. Include a brightfield or phase-contrast channel to monitor cell morphology and confluence.
- Image and Data Analysis:
- Use image analysis software to quantify the fluorescence intensity of ZipGFP in each well over time.
- Segment cells based on fluorescence to determine the percentage of ZipGFP-positive cells in the population.
- Plot kinetic curves of fluorescence intensity and the percentage of positive cells. Successful caspase inhibition by zVAD-FMK is indicated by a significant reduction in both the rate and extent of ZipGFP fluorescence increase compared to the "stimulus only" condition.
- Diagram: Experimental workflow for caspase dependence validation
Diagram Title: Experimental workflow for caspase dependence validation.
Supplementary Protocol: Validating with Fluorogenic Caspase Substrates
This protocol uses cell-permeable fluorogenic substrates like DEVD-NucView488 as an orthogonal method to confirm ZipGFP reporter findings [62].
Materials:
- DEVD-NucView488 substrate.
- Hoechst 33342 or similar nuclear stain.
- Standard culture medium without phenol red.
Workflow:
- Follow steps 1-3 of the core protocol for cell seeding and pre-treatment with zVAD-FMK.
- Simultaneously with the pro-apoptotic stimulus, add the DEVD-NucView488 substrate (e.g., 1-5 µM final concentration) and a nuclear stain (e.g., 1 µg/mL Hoechst 33342) to all wells.
- Initiate time-lapse imaging immediately. The DEVD-NucView488 substrate becomes fluorescent upon caspase-3/7 cleavage and binds to DNA, providing a complementary measure of caspase activity.
- Analyze the fluorescence in the DEVD-NucView488 channel (Ex/Em ~488/520 nm). Co-localization with the nuclear stain confirms specific caspase activation. The zVAD-FMK pre-treated group should show minimal fluorescence in this channel.
Troubleshooting and Optimization Guide
Table 3: Troubleshooting Common Experimental Issues
| Problem | Potential Cause | Suggested Solution |
|---|---|---|
| No inhibition by zVAD-FMK | Inhibitor concentration too low; cell type with low permeability; predominantly caspase-independent death. | Titrate zVAD-FMK (10-100 µM); use a fluorescent derivative (FAM-VAD-FMK) to confirm uptake [61]; check for other death markers (e.g., propidium iodide). |
| High background in controls | Spontaneous apoptosis due to over-confluence or harsh handling; serum starvation; reagent toxicity. | Ensure cells are in log-phase growth; use fresh medium with serum; titrate all reagents and include a vehicle control. |
| Weak ZipGFP signal | Inefficient transfection/expression; imaging parameters suboptimal; stimulus too weak. | Use a validated stable cell line; optimize exposure time and laser power; perform a stimulus dose-response curve. |
| Cell death despite caspase inhibition | Activation of parallel, non-apoptotic cell death pathways (e.g., necroptosis). | Investigate specific inhibitors for alternative pathways (e.g., Necrostatin-1 for necroptosis) in combination with zVAD-FMK. |
Integrating zVAD-FMK-based caspase inhibition into experimental workflows is an indispensable strategy for validating the specificity of apoptosis induction in real-time imaging studies employing ZipGFP reporters. The detailed protocols and quantitative profiles provided here equip researchers with a robust framework to confidently attribute observed cell death phenotypes to caspase-dependent mechanisms, thereby strengthening the conclusions drawn from high-content screening and basic research in cell death. This approach is particularly critical in drug discovery, where understanding the precise mechanism of action of novel therapeutics is paramount.
In the realm of real-time imaging of biological processes, the development of the ZipGFP caspase reporter has provided researchers with a powerful tool for visualizing caspase-3/7 activation during apoptosis. A critical aspect that governs the application and interpretation of data from this reporter system is its characteristic fluorescence maturation timeline, which spans approximately 40 to 100 minutes. This application note delineates the kinetic considerations essential for employing ZipGFP in drug development research, providing detailed protocols and frameworks to accurately interpret the temporal dynamics of apoptosis.
The ZipGFP Reporter System: Mechanism and Kinetics
The ZipGFP caspase reporter represents a significant advancement in fluorogenic protease reporters, engineered to overcome the limitations of traditional FRET-based systems, particularly their poor signal-to-noise ratio in vivo. The reporter is constructed on a split-GFP architecture where the eleventh β-strand (β11) is physically separated from the first ten β-strands (β1-10). To render the system fluorogenic, both fragments are "zipped" together using heterodimerizing E5 and K5 coiled coils, which prevents their spontaneous reassembly and fluorescence development. This zipping mechanism effectively cages the GFP fragments, minimizing background fluorescence until caspase-mediated cleavage occurs [10] [7].
Upon activation of executioner caspases-3/7 during apoptosis, cleavage at the embedded DEVD sequences liberates the GFP fragments. The subsequent spontaneous reassembly of β1-10 and β11 allows for the refolding of the GFP β-barrel structure and formation of the chromophore, resulting in fluorescence recovery. This structural reassembly is irreversible, enabling persistent marking of apoptotic events at the single-cell level [10].
Table 1: Key Kinetic Parameters of the ZipGFP Caspase Reporter
| Kinetic Parameter | Experimental Value | Experimental Context | Significance |
|---|---|---|---|
| In Vitro Maturation Half-Time (T1/2) | ~40 minutes | Purified components mixed post-TEV protease cleavage [7] | Represents the intrinsic biophysical limit of split-GFP reassembly and chromophore formation. |
| Cellular Activation Half-Time (T1/2) | ~100 minutes | HEK293 cells with rapamycin-activatable TEV system [7] | Reflects the combined timeline of protease cleavage, fragment diffusion, and reassembly in a cellular environment. |
| Fluorescence Increase | 10-fold | Upon TEV protease cleavage in HEK293 cells [7] | Indicates high signal-to-noise ratio and low background fluorescence of the zipped construct. |
The discrepancy between the in vitro and cellular timeframes highlights the significant impact of the intracellular environment on the kinetics of reporter activation. The ~100 minute cellular timeline integrates the processes of caspase cleavage, diffusion of the unzipped fragments, and the subsequent reassembly and chromophore maturation.
Experimental Protocols for Kinetic Validation
Protocol: Validating ZipGFP Kinetics Using a Rapamycin-Inducible System
This protocol allows for the precise measurement of ZipGFP activation kinetics in live cells by using a rapamycin-inducible TEV protease system to simulate caspase activation [7].
Reagents and Materials:
- Plasmid constructs: ZipGFP-TEV, mCherry (transfection control), FKBP-TEV-N, FRB-TEV-C
- Cell line: HEK293T cells
- Culture medium: Appropriate DMEM or RPMI-1640 with 10% FCS
- Rapamycin stock solution (e.g., 1 mM in DMSO)
- Live-cell imaging setup with environmental control (37°C, 5% CO₂)
Procedure:
- Cell Seeding and Transfection: Seed HEK293T cells in a multi-well plate suitable for live-cell imaging. Transfect the cells with the three plasmid constructs using a lipid-based transfection reagent according to manufacturer protocols. Include a constitutive mCherry reporter for normalization.
- Equilibration: 24-48 hours post-transfection, replace the medium with live-cell imaging medium. Place the plate in the imaging system and allow cells to equilibrate for at least 1 hour.
- Baseline Imaging: Acquire baseline images for both GFP and mCherry channels.
- Induction and Time-Lapse Imaging: Add rapamycin to a final concentration of 100 nM. Immediately begin time-lapse imaging, acquiring both GFP and mCherry images at 10-15 minute intervals for 6-8 hours.
- Data Analysis: Quantify the mean GFP and mCherry fluorescence intensity for a population of transfected cells over time. Normalize GFP signal to the mCherry signal to account for potential variations in cell mass or expression level. Plot the normalized GFP fluorescence over time and fit a sigmoidal curve to determine the half-time (T1/2) for reporter activation.
Protocol: Imaging Caspase Dynamics in 3D Spheroid Models
The ZipGFP reporter is adaptable to more physiologically relevant 3D models, such as cancer spheroids or patient-derived organoids [10].
Reagents and Materials:
- Stable ZipGFP reporter cell line (e.g., MiaPaCa-2, HUVEC, or patient-derived organoids)
- Apoptosis-inducing agent (e.g., carfilzomib, oxaliplatin)
- Pan-caspase inhibitor (e.g., zVAD-FMK) for control experiments
- Cultrex Basement Membrane Extract or other 3D culture matrix
- Light-sheet or confocal microscope
Procedure:
- Spheroid/Organoid Generation: Generate spheroids from ZipGFP-expressing cells using hanging drop or ultra-low attachment plates. For organoids, embed cells in Cultrex following established protocols.
- Treatment: After spheroids/organoids have formed, treat with the apoptotic stimulus. Include control groups treated with vehicle alone and a group co-treated with the apoptotic stimulus and 20-50 µM zVAD-FMK.
- Live-Cell Imaging: Transfer the 3D structures to an imaging-compatible chamber. Image using a light-sheet or confocal microscope over 24-80 hours, capturing z-stacks at regular intervals (e.g., every 30-60 minutes).
- Analysis: Quantify the GFP intensity normalized to the constitutive mCherry signal over time. The kinetics of activation can be tracked in different regions of the spheroid/organoid to map the spatiotemporal pattern of apoptosis.
Visualizing the Workflow: From Reporter Design to Data Interpretation
The following diagram illustrates the core mechanism of the ZipGFP reporter and the experimental workflow for its application.
ZipGFP Caspase Activation Mechanism
Experimental Workflow for Kinetic Analysis
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagent Solutions for ZipGFP-Based Apoptosis Imaging
| Reagent / Tool | Function / Application | Key Characteristics |
|---|---|---|
| ZipGFP Caspase Reporter | Core biosensor for detecting caspase-3/7 activity. | DEVD cleavage site, 10-fold fluorescence increase, irreversible activation [12] [7]. |
| Constitutive mCherry Reporter | Internal control for cell presence and normalization. | Long half-life (~24-30 h); normalizes for expression and cell mass [10]. |
| Inducible TEV Protease System | Tool for controlled, non-apoptotic validation of ZipGFP kinetics. | Rapamycin-inducible; validates reporter independent of apoptosis [7]. |
| Apoptosis Inducers | Stimulate caspase activation for experimental setup. | Carfilzomib, oxaliplatin, etoposide, DR5-agonists [10] [63]. |
| Caspase Inhibitors | Specificity controls to confirm caspase-dependent signaling. | zVAD-FMK (pan-caspase inhibitor), Q-VD-OPh [10] [63]. |
| 3D Culture Matrices | Enable physiologically relevant imaging contexts. | Cultrex, Matrigel for spheroid and organoid culture [10]. |
Application in Drug Development: Data Interpretation and Best Practices
The ~40-100 minute maturation timeline of ZipGFP is not a limitation but a defined kinetic parameter that must be incorporated into experimental design and data interpretation. For drug development professionals using this system, several considerations are paramount:
- Temporal Resolution: The kinetics of ZipGFP activation mean that the system reports on caspase activity that occurred within a window prior to imaging. A positive signal indicates that apoptosis was initiated approximately 100 minutes before the observed fluorescence.
- Inhibitor Validation: The specificity of the signal must be confirmed through the use of caspase inhibitors like zVAD-FMK. Abrogation of the GFP signal upon co-treatment confirms that the fluorescence increase is due to caspase activation [10] [7].
- Quantification and Normalization: The constitutive mCherry signal is critical for normalizing the ZipGFP signal, accounting for variations in cell volume, reporter expression level, and overall cell viability, especially in long-term time-lapse experiments [10].
- Context Dependence: Fluorescence maturation times of proteins can be influenced by cellular context, including strain background in microbial systems or cell type in mammalian systems [64] [65] [66]. Therefore, establishing the precise kinetic profile in the specific model system being used is essential for accurate data interpretation.
In conclusion, a thorough understanding of the kinetic profile of the ZipGFP reporter enables its powerful application in monitoring the efficacy of apoptosis-inducing cancer therapeutics, studying apoptosis-induced proliferation, and investigating immunogenic cell death in high-content screening platforms [10].
Application Notes
This document details the application of the ZipGFP caspase-3/7 reporter for monitoring apoptosis in real-time within complex 3D and thick tissue models. The ZipGFP technology addresses the critical limitation of poor signal-to-noise ratio that has historically handicapped the in vivo use of fluorescence resonance energy transfer (FRET)-based caspase reporters [7]. For researchers and drug development professionals, this fluorogenic reporter provides a robust tool for visualizing programmed cell death with high spatiotemporal resolution in challenging biological systems.
The core innovation of ZipGFP lies in its rational design, which "zips" each fragment of split GFP with heterodimerizing E5 and K5 coiled coils, preventing their spontaneous association and fluorophore formation until released by specific caspase-3/7 cleavage at the DEVD sequence [7]. This design achieves an approximately 10-fold fluorescence increase upon protease activation, a significant improvement over traditional FRET-based reporters [8] [7]. The reassembled ZipGFP exhibits a fluorescence quantum yield of 0.25, providing a bright, detectable signal even in thick, scattering tissues [7].
For 3D model systems, this enhanced signal-to-noise ratio is paramount. The fluorogenic nature of ZipGFP means that fluorescence is only generated upon caspase activation, eliminating background signal from uncleaved reporter and enabling clear detection of apoptotic events deep within tissue structures. The reporter's performance kinetics, with a time to half-maximal fluorescence (T1/2) of approximately 40-100 minutes after caspase activation, allows for the temporal tracking of cell death progression in living systems [7].
Quantitative Performance Metrics of ZipGFP Caspase Reporter
Table 1: Key performance characteristics of the ZipGFP caspase-3/7 reporter relevant for 3D and thick tissue imaging
| Parameter | Value | Experimental Context | Significance for 3D/Tissue Models |
|---|---|---|---|
| Fluorescence Increase | ~10-fold | Upon caspase-3/7 cleavage in HEK293 cells [7] | Enables detection against high background autofluorescence |
| Time to Half-Max (T1/2) | ~40-100 minutes | In vitro reassembly and in HEK293 cells with rapamycin-activatable TEV system [7] | Allows tracking of apoptosis progression over relevant biological timescales |
| Quantum Yield | 0.25 | Of reassembled ZipGFP post-cleavage [7] | Provides bright signal necessary for deep tissue penetration |
| Caspase Specificity | DEVD sequence | Consensus cleavage sequence for caspase-3/7 [7] | Ensures specific reporting of apoptosis execution phase |
Beyond its application as a standalone reporter, ZipGFP can be integrated with optogenetic cell death induction systems (optoCDEs) for controlled studies of cell death pathways and their crosstalk [54] [31]. This combination allows researchers to precisely initiate apoptosis and simultaneously visualize the subsequent caspase-3/7 activation in real-time, providing a powerful platform for studying cell death dynamics in 3D microenvironments.
Protocols
Protocol 1: Lentiviral Transduction for Stable ZipGFP Reporter Expression in 3D Culture Models
This protocol establishes a methodology for generating stable cell lines expressing the ZipGFP caspase-3/7 reporter, optimized for subsequent 3D spheroid or thick tissue culture.
Research Reagent Solutions
Table 2: Essential reagents and materials for implementing the ZipGFP caspase reporter system
| Item | Function/Application | Example Catalog Numbers |
|---|---|---|
| Lentiviral Plasmid Encoding ZipGFP | Delivery of caspase reporter construct | N/A |
| HEK293T Cells | Production of lentiviral particles | N/A |
| Polybrene | Enhances viral infection efficiency | TR-1003-G (Merck) [31] |
| Puromycin | Selection of successfully transduced cells | ant-pr-1 (Invivogen) [31] |
| JetPRIME Transfection Reagent | Plasmid transfection for viral production | 101000027 (Polyplus) [31] |
| CellTox Green / DRAQ7 | Real-time viability staining for imaging | G8741 (Promega); 424001 (BioLegend) [31] |
| CellEvent Caspase-3/7 Green | Complementary caspase activity validation | R37111 (Thermo Fisher) [31] |
| 8-well μ-Slides | High-resolution live-cell imaging | 80826 (ibidi) [31] |
Procedure
Lentiviral Particle Production
- Prepare purified lentiviral plasmids encoding the ZipGFP caspase reporter construct using endotoxin-free midi- or maxi-prep kits.
- Plate HEK293T cells in a tissue culture-treated 6-well plate at 5 × 10^5 cells/well in 2 mL of fresh DMEM with GlutaMAX supplement 24 hours prior to transfection [31].
- For each well, prepare transfection mix containing JetPRIME transfection reagent, plasmid DNA, and the necessary packaging plasmids in a 1.5 mL tube. Incubate at room temperature for 15 minutes [31].
- Add the transfection mix dropwise to the cells, ensuring even coverage. Incubate for 24-48 hours until viral particles are produced [31].
- Collect the viral supernatant and filter through a 0.45 μm filter to remove cellular debris [31].
Cell Line Transduction and Selection
- Plate the target cells (e.g., primary fibroblasts, tumor cell lines) for 3D culture at appropriate density.
- Add the filtered viral supernatant to the target cells in the presence of 8 μg/mL Polybrene to enhance infection efficiency [31].
- Centrifuge the plate at 800 × g for 30-60 minutes (spinfection) to increase viral contact with cells.
- After 24-48 hours, replace the viral-containing medium with fresh culture medium containing the appropriate selection antibiotic (e.g., 1-2 μg/mL puromycin). Maintain selection pressure for 5-7 days to establish a stable polyclonal population [31].
Validation of Reporter Function
- Induce apoptosis in a subset of stable cells using a known inducer (e.g., 1-4 μM staurosporine for 2-6 hours).
- Monitor for the characteristic green fluorescence increase using confocal microscopy, confirming proper caspase-dependent activation of the ZipGFP reporter.
Protocol 2: Real-Time Imaging of Caspase-3/7 Activation in 3D Tissue Models
This protocol describes the methodology for inducing apoptosis and capturing real-time ZipGFP activation in thick, complex tissue models using advanced imaging techniques.
Materials and Equipment
- Stable ZipGFP-expressing 3D spheroids or organoids
- Point-scanning confocal microscope (e.g., Zeiss LSM800 or Leica SP8) [31]
- Apoptosis inducer (e.g., chemotherapeutic agent, Trail, staurosporine)
- Opti-MEM reduced serum medium [31]
- Environmental chamber for temperature and CO₂ control
Procedure
Sample Preparation and Mounting
- Transfer mature 3D spheroids/organoids (300-500 μm diameter) into an 8-well μ-Slide suitable for high-resolution imaging [31].
- Allow spheroids to settle for 30-60 minutes before imaging to minimize drift during acquisition.
Image Acquisition Setup
- Mount the slide on the confocal microscope stage with environmental control maintained at 37°C and 5% CO₂.
- Using a 488 nm laser for ZipGFP excitation, set appropriate detection windows (500-550 nm).
- Optimize z-stack parameters to encompass the entire spheroid thickness with minimal step size (e.g., 2-5 μm).
- Set a time-lapse interval of 10-20 minutes over 12-24 hours to capture caspase activation dynamics.
Apoptosis Induction and Data Collection
- Acquire baseline images for 2-3 time points before induction.
- Carefully add apoptosis inducer to the well at the desired final concentration without moving the sample.
- Continue time-lapse acquisition, monitoring for the emergence of ZipGFP fluorescence.
- For complementary viability assessment, include cell-impermeant DNA dyes like DRAQ7 (5 μM final concentration) to mark necrotic cells [31].
Data Analysis and Interpretation
- Process image stacks using Fiji/ImageJ software.
- Quantify fluorescence intensity within regions of interest over time.
- Generate time-course curves of ZipGFP activation for different conditions.
- Correlate caspase activation spatiotemporally with morphological changes of cell death.
Visualizations
ZipGFP Caspase Reporter Mechanism
Experimental Workflow for 3D Tissue Imaging
Proof and Precision: Validating ZipGFP Data and Comparing it to Established Methods
Within apoptosis research, the transition to real-time, single-cell analysis using fluorescent reporters like the ZipGFP caspase-3/-7 biosensor represents a significant technological advancement [10]. However, the validation of these dynamic live-cell findings against established, biochemical endpoint methods remains a critical step for ensuring data integrity and mechanistic understanding. This application note details the methodology for correlating data from the ZipGFP reporter system with the gold-standard technique of Western blotting for cleaved caspase-3 and cleaved PARP. This protocol provides a framework for researchers to confirm that fluorescence activation corresponds precisely to the biochemical hallmarks of apoptosis, thereby bridging live-cell imaging with foundational molecular biology.
Quantitative Correlation of Apoptosis Readouts
The table below summarizes the key characteristics and complementary roles of live-cell reporters and Western blotting in detecting apoptosis.
Table 1: Comparison of Apoptosis Detection Methods
| Method | Key Target(s) | Key Advantage | Primary Limitation | Correlation Role |
|---|---|---|---|---|
| ZipGFP Reporter [10] | Caspase-3/-7 activity | Real-time, single-cell kinetics in live cells (2D & 3D) | Does not directly show protein cleavage | Primary live-cell data source for dynamic activation. |
| Western Blot (Cleaved Caspase-3) [67] [68] | 17/19 kDa caspase-3 fragments | Specific, direct detection of canonical executioner caspase activation [67] | Population-average, endpoint measurement | Validates caspase-3 activation and specific cleavage. |
| Western Blot (Cleaved PARP) [69] | 89 kDa PARP fragment | Direct evidence of downstream apoptotic substrate cleavage [69] | Population-average, endpoint measurement | Confirms downstream apoptotic signaling. |
Experimental Protocol for Correlation
Real-Time Imaging with ZipGFP Caspase-3/7 Reporter
Method Overview: This protocol uses a stable fluorescent reporter cell line expressing a ZipGFP-based biosensor for caspase-3/-7 activity, alongside a constitutive mCherry marker for cell presence and normalization [10].
- Cell Culture: Generate or obtain stable reporter cell lines (e.g., HEK293, HeLa, or patient-derived organoids) using lentiviral transduction. Maintain cells in appropriate medium supplemented with selection antibiotics if required.
- Experimental Setup: Plate cells in collagen-coated 8-well μ-slides or other imaging-optimized dishes. For 3D cultures, embed cells in Cultrex or Matrigel to form spheroids or grow patient-derived organoids [10].
- Live-Cell Imaging:
- Induce Apoptosis: Treat cells with your chosen apoptotic stimulus (e.g., 1 µM staurosporine [69], carfilzomib [10], or oxaliplatin).
- Image Acquisition: Use a live-cell imaging system (e.g., IncuCyte or confocal microscope). Acquire images for GFP (caspase activity) and mCherry (cell presence) channels every 30-60 minutes over 24-80 hours.
- Controls: Include untreated control wells and wells co-treated with a pan-caspase inhibitor (e.g., 20 µM zVAD-FMK) to confirm caspase-specific signal [10].
- Data Analysis: Quantify the GFP/mCherry fluorescence ratio over time using automated image analysis software. This normalized ratio reflects the kinetics of caspase-3/-7 activation at single-cell resolution.
Western Blot Validation for Cleaved Caspase-3 and PARP
Method Overview: This parallel protocol provides biochemical validation of apoptosis by detecting cleaved fragments of caspase-3 and its substrate, PARP, in cell lysates [69] [70] [67].
- Sample Preparation:
- Harvest Lysates: At critical timepoints identified by the live-cell imaging (e.g., during signal initiation, peak, and endpoint), lyse cells directly in RIPA or Laemmli buffer.
- Protein Quantification: Determine protein concentration using a BCA assay and normalize samples for equal loading.
- Gel Electrophoresis and Transfer:
- Immunoblotting:
- Blocking: Block the membrane with 5% non-fat milk in PBS-T (0.1% Tween-20) for 1 hour at room temperature.
- Primary Antibody Incubation: Incubate with specific primary antibodies diluted in blocking buffer overnight at 4°C.
- Cleaved Caspase-3 (Asp175): Use at 1:1000 dilution (Rabbit monoclonal, Cell Signaling Technology #9661) to detect the endogenous 17/19 kDa active fragments [67].
- Cleaved PARP (Asp214): Use at a dilution specified by the manufacturer (e.g., Mouse monoclonal, part of ab136812 cocktail) to detect the 89 kDa fragment [69].
- Loading Control: Use an antibody against GAPDH, β-Actin, or Muscle Actin (42 kDa) [69] [70].
- Secondary Antibody Incubation: Incubate with appropriate HRP-conjugated secondary antibodies (e.g., anti-rabbit and anti-mouse) for 1 hour at room temperature.
- Detection: Develop blots using a chemiluminescent substrate and image with a digital imaging system.
Correlation Workflow
The following diagram illustrates the integrated experimental workflow for correlating real-time imaging with endpoint biochemical validation:
The Apoptotic Signaling Pathway
A clear understanding of the molecular pathway is essential for interpreting data from both ZipGFP reporters and Western blotting. The following diagram outlines the core apoptotic execution pathway, highlighting the key proteins detected in these assays:
The Scientist's Toolkit: Essential Reagents
Table 2: Key Reagents for Apoptosis Detection
| Item | Function/Description | Example Product / Citation |
|---|---|---|
| ZipGFP Caspase-3/7 Reporter | Lentiviral-based stable reporter for real-time caspase activity sensing via DEVD cleavage. | Described in [10] |
| Anti-Cleaved Caspase-3 (Asp175) | Primary antibody for Western Blot; detects active 17/19 kDa fragments. | Cell Signaling #9661 [67] |
| Anti-Cleaved PARP | Primary antibody for Western Blot; detects apoptotic 89 kDa fragment. | Included in Abcam ab136812 [69] |
| Caspase Inhibitor (zVAD-FMK) | Pan-caspase inhibitor used as a negative control to confirm caspase-dependent signals. | Used in [10] |
| Apoptosis Inducers | Chemical agents to trigger apoptosis (e.g., staurosporine, carfilzomib). | Staurosporine [69], Carfilzomib [10] |
| Apoptosis Western Blot Cocktail | Antibody cocktail for simultaneous detection of caspase-3, cleaved PARP, and a loading control. | Abcam ab136812 [69] |
Within the broader context of real-time imaging research utilizing the ZipGFP caspase-3/7 reporter, the cross-validation of apoptotic findings is paramount. The ZipGFP reporter is a rationally designed, GFP-based fluorogenic caspase reporter that allows for the visualization of apoptosis in live cells and organisms with high spatiotemporal resolution [7] [8]. It functions by "zipping" the two fragments of split GFP with heterodimerizing coiled coils, which prevents their reassociation and fluorescence until the linker is cleaved by executioner caspases, resulting in a significant fluorescence increase [7]. While such live-cell imaging tools are invaluable for observing the dynamic processes of apoptosis, flow cytometry-based methods provide a robust, quantitative framework for validating these results at a population level. This application note details the use of Annexin V and Propidium Iodide (PI) staining in flow cytometry as a definitive method to confirm apoptosis, thereby anchoring the data obtained from ZipGFP reporter systems within a established biochemical context.
Principles of Apoptosis Detection
The Biochemical Basis of Annexin V and PI Staining
Apoptosis, or programmed cell death, is characterized by a series of distinct biochemical events. A pivotal early event is the loss of plasma membrane asymmetry, leading to the translocation of phosphatidylserine (PS) from the inner to the outer leaflet of the membrane [71] [33]. Annexin V is a calcium-dependent phospholipid-binding protein with a high affinity for PS, making it a sensitive probe for detecting this externalization [35] [72].
Propidium Iodide (PI) is a vital DNA dye that is excluded from cells with an intact plasma membrane. During the late stages of apoptosis and in necrosis, the integrity of the plasma membrane is compromised, allowing PI to enter the cell, intercalate into nucleic acids, and fluoresce [72] [71].
The simultaneous application of these two markers allows for the discrimination of different cell states within a population, which is foundational for cross-validation.
Correlating Caspase Activation with PS Externalization
The activation of executioner caspases (e.g., caspase-3 and -7) is a central event in the apoptotic cascade, constituting the primary signal detected by the ZipGFP reporter and other caspase substrates like the CellEvent Caspase-3/7 Green reagent [73] [62]. This activation typically precedes the loss of plasma membrane integrity. The externalization of PS, detected by Annexin V, is a downstream event that can be temporally correlated with caspase activation. Cross-validating a ZipGFP signal with Annexin V/PI staining strengthens the conclusion that the observed fluorescence is indeed due to apoptosis, as it confirms the occurrence of two key, sequential apoptotic events [7] [73].
The following diagram illustrates the logical relationship between the detection methods for these key apoptotic events and the resulting cell states.
Experimental Protocols
Detailed Protocol: Annexin V and PI Staining for Flow Cytometry
The following step-by-step protocol is compiled from manufacturer instructions and peer-reviewed methodologies [35] [72] [71].
Materials:
- 1X PBS (cold): Phosphate-buffered saline, without Ca²⁺ or Mg²⁺.
- 1X Binding Buffer: 10 mM HEPES/NaOH (pH 7.4), 140 mM NaCl, 2.5 mM CaCl₂. Prepare fresh from 10X stock by dilution with distilled water. Critical: Avoid buffers containing EDTA, as it chelates calcium and inhibits Annexin V binding [35].
- Fluorochrome-conjugated Annexin V (e.g., Annexin V-FITC, Annexin V-PE).
- Propidium Iodide (PI) Staining Solution or 7-AAD Viability Staining Solution.
- Flow Cytometry Tubes: 12 x 75 mm round-bottom tubes.
Procedure:
- Cell Harvesting and Washing: Harvest cells (adherent cells may require gentle trypsinization) and wash them once with cold 1X PBS. Centrifuge at 400–600 x g for 5 minutes at room temperature. Carefully decant the supernatant.
- Resuspension in Binding Buffer: Resuspend the cell pellet in 1X Binding Buffer at a density of 1–5 x 10⁶ cells/mL.
- Staining with Annexin V: Transfer 100 µL of the cell suspension (approximately 1–5 x 10⁵ cells) to a flow cytometry tube. Add 5 µL of the fluorochrome-conjugated Annexin V. Gently vortex the tube to mix.
- Incubation: Incubate the cells for 10–15 minutes at room temperature (15–25°C). Protect the tubes from light throughout the procedure.
- Washing (Optional): Add 2 mL of 1X Binding Buffer and centrifuge at 400–600 x g for 5 minutes. Discard the supernatant. Note: Some protocols omit this wash to prevent loss of early apoptotic cells [72].
- Resuspension and Viability Staining: Resuspend the cells in 200 µL of 1X Binding Buffer. Add 5 µL of PI Staining Solution (the optimal volume may require titration; 2–10 µL is a common range [72]).
- Analysis: Keep the samples on ice or at 2–8°C and protected from light. Analyze by flow cytometry within 1 hour for optimal results [72] [71]. Do not wash the cells after the addition of PI.
Essential Controls for Validation
Proper controls are non-negotiable for accurate data interpretation and panel setup [72].
- Table: Required Experimental Controls
Control Tube Annexin V Conjugate Vital Dye (PI/7-AAD) Purpose Unstained – – Autofluorescence baseline Annexin V Single Stain + – Compensation & quadrant setting Vital Dye Single Stain – + Compensation & quadrant setting Induced Apoptosis (Experimental) + + Experimental sample Blocking Control (Optional) + (after blocking) + Specificity of Annexin V binding [72]
Integrated Workflow for Cross-Validation with ZipGFP
To directly correlate real-time caspase activity with PS externalization, an integrated experimental workflow is recommended. The following diagram outlines this multi-modal approach.
Data Analysis and Interpretation
Gating Strategy and Population Quantification
Upon acquisition, the flow cytometry data is analyzed by plotting Annexin V fluorescence against PI fluorescence on a bivariate dot plot. This allows for the clear separation of distinct cell populations.
- Viable/Healthy Cells (Annexin V⁻/PI⁻): This population is negative for both stains, indicating intact membranes and no exposed PS.
- Early Apoptotic Cells (Annexin V⁺/PI⁻): These cells are positive for Annexin V but exclude PI, demonstrating exposed PS while maintaining membrane integrity. This is a key population for validating early caspase activation signaled by ZipGFP.
- Late Apoptotic/Necrotic Cells (Annexin V⁺/PI⁺): Positive for both markers, indicating exposed PS and a compromised plasma membrane.
Quantitative Correlation of Methods
The quantitative nature of flow cytometry allows for the direct correlation of the percentage of cells in each apoptotic stage with the fluorescence intensity metrics obtained from ZipGFP imaging. For instance, a time-course experiment should show an increasing percentage of Annexin V⁺/PI⁻ cells that correlates temporally with the increase in ZipGFP fluorescence intensity in the imaged population.
- Table: Expected Correlation Between ZipGFP Signal and Annexin V/PI Staining
ZipGFP Fluorescence (Live Imaging) Corresponding Annexin V/PI Population (Flow Cytometry) Interpretation Low / Baseline Annexin V⁻ / PI⁻ Healthy, viable cells Increasing Signal Annexin V⁺ / PI⁻ Cells in early apoptosis; active caspase-3/7 High / Sustained Signal Annexin V⁺ / PI⁺ Cells in late apoptosis or secondary necrosis
The Scientist's Toolkit: Research Reagent Solutions
The following table details key reagents essential for successfully executing the described cross-validation experiments.
- Table: Essential Research Reagents for Apoptosis Cross-Validation
Item Function & Role in Cross-Validation Example Catalog Numbers / References Annexin V Apoptosis Detection Kits Provides optimized, matched components (Annexin V conjugate, binding buffer, viability dye) for robust and reproducible staining. eFluor 450, FITC, PE, APC formats [35]; BD Biosciences Annexin V-FITC Kit (556420) [72] ZipGFP Caspase Reporter A fluorogenic caspase reporter for live-cell imaging of apoptosis with high signal-to-noise, enabling real-time kinetic studies. [7] [8] CellEvent Caspase-3/7 Green Detection Reagent A cell-permeant, fluorogenic substrate for activated caspase-3/7. An excellent alternative for flow cytometric detection of caspase activity alongside ZipGFP imaging. Catalog # C10427 [73] Propidium Iodide (PI) / 7-AAD Vital dyes used to distinguish cells with intact vs. compromised plasma membranes. Critical for differentiating early and late apoptotic stages. Included in most kits; also sold separately [35] [72] [71] Fixable Viability Dyes (FVD) Amine-reactive dyes that covalently bind to proteins, allowing cell fixation and permeabilization for intracellular staining after viability assessment. eFluor 506, eFluor 660, eFluor 780 [35] Flow Cytometry Staining Buffer A protein-based buffer used to reduce non-specific antibody binding, particularly important in multicolor panels that include surface marker staining. Cat. No. 00-4222 [35]
Troubleshooting and Best Practices
- Calcium is Critical: The binding of Annexin V to PS is absolutely calcium-dependent. Always use calcium-containing binding buffer and avoid the use of EDTA or other calcium chelators during the staining procedure [35].
- Timing is Key: Analyze stained samples promptly (within 1 hour). Prolonged incubation, especially in the presence of PI, can adversely affect cell viability and lead to artifactual results [35] [72].
- Handling Adherent Cells: Exercise caution when harvesting adherent cells. Over-trypsinization can damage the plasma membrane and cause false-positive PI staining. Using enzyme-free cell dissociation buffers is recommended [72].
- Inclusion of Controls: Never omit the single-stained and unstained controls. They are essential for setting appropriate compensation and defining positive and negative populations on the flow cytometry plot [72].
- Method Validation: For clinical research or GLP/GCLP environments, a formal "fit-for-purpose" validation strategy assessing parameters like precision, specificity, and detection limits is recommended to ensure data reliability [74].
The combination of dynamic, real-time imaging tools like the ZipGFP caspase reporter and quantitative, population-based assays like Annexin V/PI flow cytometry provides a powerful, cross-validated approach for studying apoptosis. The protocol detailed herein offers a robust methodology to biochemically confirm the induction of apoptosis signaled by caspase activation. This multi-modal strategy enhances the rigor and reliability of data, which is crucial for fundamental research and high-stakes applications such as drug development, where accurately quantifying cell death is essential for evaluating therapeutic efficacy and mechanism of action.
Within the broader scope of thesis research on real-time imaging of caspase-3/7 activation, selecting an optimal fluorescent reporter is paramount for obtaining reliable, high-quality data in complex physiological environments. For years, Förster Resonance Energy Transfer (FRET)-based reporters have been the cornerstone for visualizing protease activity and other biochemical events in living cells. However, their application in whole-animal studies has been notably challenging. This application note provides a direct comparison between conventional FRET-based caspase reporters and the newer ZipGFP-based fluorogenic reporter, focusing on the critical parameters of signal strength and practical utility in vivo. The analysis aims to equip researchers and drug development professionals with the data needed to select the most appropriate tool for imaging apoptosis in real-time within living animal models such as zebrafish and Drosophila.
Fundamental Mechanisms and Design Principles
FRET-Based Caspase Reporters
FRET (Förster Resonance Energy Transfer) is a non-radiative process where energy is transferred from an excited donor fluorophore to a nearby acceptor fluorophore through long-range dipole-dipole interactions [75]. This transfer is highly efficient when the distance between the two fluorophores is 8 to 10 nanometers or less, making FRET a powerful spectroscopic ruler for probing molecular interactions [75] [76].
In a typical FRET-based caspase reporter, two fluorescent proteins (e.g., CFP and YFP) are linked by a short peptide sequence containing the caspase cleavage site (DEVD) [7] [75]. When the reporter is intact, the close proximity of the two fluorophores enables FRET to occur. Upon caspase activation and cleavage of the linker sequence, the two fluorescent proteins separate, abolishing FRET [75]. This results in a decrease in acceptor emission and an increase in donor emission, which can be measured ratiometrically.
The ZipGFP Fluorogenic Reporter
The ZipGFP reporter operates on a fundamentally different principle. It was rationally designed by modifying a self-assembling split GFP system [7] [77]. One part contains the first ten β-strands (β1–10), which include the chromophore-forming residues. The other part contains the critical 11th β-strand (β11), which includes Glu222, essential for chromophore maturation [7].
The key innovation is the "zipping" of both fragments with heterodimerizing coiled coils (E5 and K5). This zipping physically blocks the binding cavity of β1-10 and occludes β11, preventing their association and consequent fluorescence [7]. A protease cleavage sequence (e.g., DEVD for executioner caspases) is inserted into both "zipped" parts. Upon protease cleavage, the coiled coils are released, allowing β11 to bind to β1-10. This self-assembly leads to the reconstitution of the GFP chromophore and a large increase in fluorescence [7] [77]. The following diagram illustrates this mechanism.
Direct Performance Comparison
Quantitative Performance Metrics
The table below summarizes a direct, quantitative comparison between ZipGFP and FRET-based caspase reporters across key performance indicators.
Table 1: Direct Performance Comparison between ZipGFP and FRET-Based Caspase Reporters
| Performance Parameter | ZipGFP Reporter | FRET-Based Reporters | Experimental Context & Notes |
|---|---|---|---|
| Signal Change Upon Activation | ~10-fold increase [7] | Low signal-to-noise ratio (SNR); small fluorescence change [7] [78] | Measured in HEK293 cells for ZipGFP-TEV. FRET change is often a fractional ratio change. |
| Dynamic Range | Large (Fluorogenic; dark-to-bright) [77] | Limited [78] [77] | ZipGFP's fluorogenic nature provides a superior baseline. |
| Background Fluorescence | Very low when "zipped" [7] | High due to constitutive donor/acceptor fluorescence [7] | Low pre-cleavage background is key for high SNR in vivo. |
| Kinetics (T1/2) | ~40 min (in vitro)~100 min (in cells) [7] | N/A (Rapid, limited by cleavage kinetics) | ZipGFP kinetics limited by GFP fragment reassembly [7]. |
| Quantum Yield (Active State) | 0.25 [7] | Dependent on donor/acceptor pair | For comparison, FlipGFP, a related fluorogenic reporter, has a QY of 0.66 [77]. |
| Cofactor Requirement | None [7] | None | ZipGFP maturation is cofactor-independent, unlike iCasper which requires biliverdin [7]. |
In Vivo Utility and Imaging Performance
Imaging in living animals presents unique challenges, including tissue autofluorescence, cell heterogeneity, and rapid morphological changes [7] [77]. These factors critically impact the performance of fluorescent reporters.
ZipGFP In Vivo Performance: The ZipGFP-based executioner caspase reporter has successfully visualized physiological apoptosis in live zebrafish embryos with high spatiotemporal resolution [7] [8]. Its fluorogenic (dark-to-bright) characteristic is a key advantage here, as the very low background signal before caspase activation minimizes interference from autofluorescence, leading to a high signal-to-noise ratio that is detectable in complex tissue environments [7].
FRET In Vivo Performance: The in vivo application of FRET-based executioner caspase reporters has been "handicapped by poor signal to noise" [7] [8]. The low dynamic range and inherently low SNR of FRET measurements make it difficult to distinguish the genuine signal from background autofluorescence and other noise sources within a living animal [78]. This has significantly limited their use for detailed in vivo studies [7].
Experimental Protocols
Protocol: Using ZipGFP Caspase Reporter in Mammalian Cells
This protocol outlines the methodology for detecting apoptosis in cultured mammalian cells using the ZipGFP-based executioner caspase reporter, as described in the primary literature [7].
Research Reagent Solutions
- ZipGFP Caspase Reporter Construct: Plasmid encoding the ZipGFP reporter with DEVD cleavage sequences in both "zipped" parts.
- mCherry Reference Plasmid: For normalization and transfection control.
- HEK293 Cells: A standard mammalian cell line for transient expression.
- Appropriate Cell Culture Media: DMEM supplemented with FBS.
- Transfection Reagent: Polyethylenimine (PEI) or a commercial equivalent.
- Apoptosis Inducer: e.g., Staurosporine or other relevant agent.
Procedure
- Cell Seeding: Seed HEK293 cells into multi-well imaging plates in complete media and allow them to adhere until ~70% confluent.
- Co-transfection: Transfect cells with a mixture of the ZipGFP caspase reporter plasmid and the mCherry reference plasmid.
- Expression: Incubate cells for 18-24 hours to allow for protein expression.
- Induction (Optional): Induce apoptosis by adding a pro-apoptotic agent (e.g., 1 µM Staurosporine) to the experimental group. Include a negative control (vehicle only).
- Live-Cell Imaging: After an appropriate time (e.g., 2-6 hours post-induction), image cells using a standard fluorescence microscope or conf microscope.
- ZipGFP Channel: Ex/Em ~488 nm/~510 nm.
- mCherry Channel: Ex/Em ~587 nm/~610 nm.
- Data Analysis: Quantify the mean fluorescence intensity for both channels. The green fluorescence (ZipGFP) can be normalized to the red fluorescence (mCherry) to control for expression levels and imaging conditions. A strong increase in the normalized green/red fluorescence ratio indicates caspase activation and apoptosis.
Protocol: Validating ZipGFP Specificity with Rapamycin-Activatable TEV System
This protocol uses a chemically inducible protease system to validate the specificity and kinetics of a ZipGFP-based reporter in cells [7]. The following workflow outlines the key stages of this experiment.
Research Reagent Solutions
- ZipGFP-TEV Reporter Plasmid: ZipGFP with TEV protease cleavage sites.
- Split TEV Protease Plasmids: pC4-FRB-TEV protease (N-terminal fragment) and pC4-FKBP-TEV protease (C-terminal fragment).
- Rapamycin: To induce dimerization of FRB and FKBP.
Procedure
- Co-transfection: Co-express the ZipGFP-TEV reporter, the two split TEV protease fragments, and an mCherry reference in HEK293 cells.
- Baseline Imaging: Acquire baseline green and red fluorescence images.
- Induction: Add rapamycin (e.g., final concentration 100 nM) to the culture medium.
- Time-Lapse Imaging: Perform time-lapse fluorescence imaging over several hours.
- Kinetic Analysis: Plot normalized green fluorescence over time. The time to half-maximal fluorescence (T1/2) for ZipGFP in this cellular context is approximately 100 minutes [7].
The comparative data presented herein strongly supports the superiority of the ZipGFP reporter over traditional FRET-based constructs for detecting caspase activity in living animals. The core advantage of ZipGFP lies in its fluorogenic design, which provides a ~10-fold increase in fluorescence upon activation and an inherently low background [7]. This translates directly to a high signal-to-noise ratio, a critical factor for successful imaging in the autofluorescent and complex environment of a live zebrafish embryo [7] [77].
While FRET-based biosensors remain valuable tools for a range of applications, including some intracellular kinase and protease assays, their inherent limitations in dynamic range and low SNR have consistently hampered their utility for in vivo imaging of caspase activity [78]. The ZipGFP technology effectively overcomes these limitations. For thesis research focused on real-time imaging of caspase-3/7 activation, particularly in an in vivo context, the ZipGFP-based reporter represents a more robust and reliable tool. It enables the visualization of physiological apoptosis with spatiotemporal resolution that was previously difficult to achieve, thereby providing a powerful method to investigate the role of programmed cell death in animal development and disease models.
ZipGFP vs. Fluorogenic Substrates (FLICA) and Bioluminescent Reporters (Z-DEVD-aminoluciferin)
The study of caspase-3/7 activation is fundamental to apoptosis research, with significant implications for understanding development, homeostasis, and disease pathologies. A critical advancement in this field has been the development of genetically encoded reporters and chemical probes that enable real-time monitoring of caspase activity in living systems. This application note provides a detailed comparative analysis of three principal technologies: the ZipGFP caspase reporter, fluorogenic substrates (exemplified by FLICA reagents), and bioluminescent reporters (using Z-DEVD-aminoluciferin). We present quantitative performance data, detailed experimental protocols, and pathway visualizations to guide researchers in selecting the optimal methodology for their specific applications in basic research and drug discovery.
Technology Comparison and Performance Data
The following tables summarize the key characteristics and quantitative performance metrics of the three caspase detection methodologies.
Table 1: Key Characteristics of Caspase-3/7 Detection Technologies
| Feature | ZipGFP Reporter | Fluorogenic Substrates (FLICA) | Bioluminescent Reporters (Z-DEVD-aminoluciferin) |
|---|---|---|---|
| Core Principle | Genetically encoded; protease-activated split GFP reassembly [7] | Cell-permeable, fluorescently-labeled caspase inhibitor probes [79] | Luciferase-catalyzed light emission from caspase-cleaved substrate [80] |
| Signal Type | Fluorescence (Green) | Fluorescence (Varies by dye) | Bioluminescence |
| Live-Cell Capability | Yes (real-time imaging) [7] | Yes (kinetic assays) [79] | Yes (endpoint or kinetic) [80] |
| Temporal Resolution | ~40 min (T1/2 for fluorescence development) [7] | Minutes to hours | Minutes (homogeneous, plate-based) [80] |
| Spatial Resolution | High (single-cell resolution in vivo) [7] | High (single-cell resolution in 2D/3D culture) [79] | Low (population-average, well-level) |
| Throughput | Medium (imaging-based) | Medium (imaging/cytometry-based) | High (plate reader-based) [80] |
Table 2: Quantitative Performance Metrics
| Metric | ZipGFP Reporter | Fluorogenic Substrates | Bioluminescent Assay |
|---|---|---|---|
| Signal-to-Background / Fold-Increase | ~10-fold increase post-activation [7] | Varies with specific reagent | High (low background due to no excitation) [81] |
| Assay Kinetics (T1/2) | ~40 min (in vitro), ~100 min (in cells) [7] | Data not available in search results | Rapid (lytic reagent added directly) [80] |
| Primary Application Demonstrated | Live zebrafish embryos [7] | 2D/3D cell culture [79] | Cell lines (THP-1, J774A.1) and primary cells (BMDMs) [80] |
| Key Advantage | Spatiotemporal imaging in live animals | Compatibility with complex 3D models | Suitability for high-throughput screening [80] |
Mechanism of Action and Experimental Workflows
A. ZipGFP Caspase Reporter
1. Mechanism of Action Diagram
2. Detailed Protocol: Imaging Apoptosis in Mammalian Cells
- Constructs: ZipGFP-Casp3 plasmid (Addgene #81241) [82].
- Cell Culture: HEK293 cells maintained in DMEM with 10% FBS.
- Transfection:
- Seed cells in a multi-well plate or imaging dish.
- Transfect with the ZipGFP-Casp3 plasmid using a suitable transfection reagent (e.g., JetPRIME) [54].
- Include a positive control (e.g., cells treated with 1 µM staurosporine for 4-6 hours) and a negative control (untreated cells).
- Live-Cell Imaging:
- 24-48 hours post-transfection, image cells using a fluorescence microscope equipped with a GFP filter set.
- Maintain cells at 37°C and 5% CO₂ during imaging.
- Acquire time-lapse images every 30-60 minutes to monitor the kinetics of fluorescence development.
- Data Analysis: Quantify the mean fluorescence intensity over time in regions of interest (ROIs) encompassing individual cells. A ≥10-fold increase in fluorescence indicates caspase activation [7].
B. Fluorogenic Substrates (FLICA)
1. Mechanism of Action Diagram
2. Detailed Protocol: ViaStain Live Caspase 3/7 Detection in 2D/3D Culture
- Reagents: ViaStain Live Caspase 3/7 Detection reagent (Revvity, #CS1-V0002-1) [79].
- Cell Preparation:
- Seed cells in a black-walled, clear-bottom 96-well plate. For 3D cultures, establish spheroids or organoids.
- Induce apoptosis in test wells using a chosen stimulus (e.g., chemotherapeutic agent).
- Staining:
- Add the ViaStain reagent directly to the cell culture medium according to the manufacturer's instructions. This is a "no-wash" assay.
- Incubate the plate at 37°C for 30-90 minutes.
- Image Acquisition and Analysis:
- Image the cells using a fluorescence microscope or an image cytometer (e.g., Celigo).
- Quantify the fluorescence intensity from the caspase 3/7 signal. Kinetic assays can be performed by continuous measurement [79].
C. Bioluminescent Caspase Activity Assay
1. Mechanism of Action Diagram
2. Detailed Protocol: Homogeneous Bioluminescent Caspase-3/7 Assay
- Reagents: Caspase-Glo 3/7 Assay System (or similar, based on the referenced principle [80]).
- Cell Preparation:
- Plate cells in a white-walled, clear-bottom 96-well plate.
- Treat cells with apoptotic inducers and include controls.
- Assay Execution:
- Equilibrate the Caspase-Glo 3/7 reagent to room temperature.
- Add an equal volume of reagent to each well containing cells in culture medium.
- Mix contents gently on an orbital shaker for 30 seconds to induce lysis.
- Incubate at room temperature for 30-60 minutes to allow the signal to stabilize.
- Luminescence Measurement:
- Measure luminescence in a plate-reading luminometer.
- The signal is proportional to caspase-3/7 activity. Results can be normalized to cell number using a separate viability assay [80].
Research Reagent Solutions
Table 3: Essential Materials for Caspase-3/7 Research
| Reagent / Material | Function / Description | Example Product / Source |
|---|---|---|
| ZipGFP-Casp3 Plasmid | Genetically encoded reporter for real-time, spatial imaging of caspase-3/7 in live cells and animals. | Addgene Plasmid #81241 [82] |
| ViaStain Live Caspase 3/7 Reagent | Cell-permeable, fluorescent probe for no-wash, kinetic detection of caspase-3/7 in 2D and 3D cultures. | Revvity #CS1-V0002-1 [79] |
| Caspase-Glo 3/7 Assay | Homogeneous, bioluminescent assay for high-throughput, quantitative measurement of caspase-3/7 activity. | Promega (Assay based on principle in [80]) |
| Mammalian Expression Vector | Plasmid backbone for constitutive or inducible expression of genetic reporters like ZipGFP. | pcDNA3.1 [82] |
| Liposomal Transfection Reagent | For efficient delivery of plasmid DNA into mammalian cell lines. | JetPRIME [54] |
| Apoptosis Inducer | Positive control for triggering caspase-3/7 activation. | Staurosporine |
The choice between ZipGFP, FLICA, and bioluminescent reporters is dictated by the specific research question. ZipGFP is unparalleled for studies requiring high spatiotemporal resolution in living organisms, as demonstrated by its successful application in visualizing physiological apoptosis in live zebrafish embryos [7]. Its genetic encoding allows for targeting to specific cell types and long-term tracking. FLICA reagents offer a robust solution for visualizing caspase activation in complex 3D cellular models with single-cell resolution, without the need for genetic manipulation [79]. In contrast, bioluminescent reporters utilizing substrates like Z-DEVD-aminoluciferin provide the highest quantitative throughput and sensitivity for compound screening in drug discovery applications, where population-level data is sufficient and low background is critical [80].
In the context of a thesis focused on real-time imaging of caspase-3/7, ZipGFP represents a powerful tool for mechanistic studies of apoptosis in developing tissues or animal disease models. Its main limitation, the slower kinetics of fluorescence development (~40-minute half-life), is a trade-off for its unique capability to provide spatial information in vivo. For most kinetic and high-throughput applications in cell culture, FLICA and bioluminescent assays remain the more practical and immediate choices.
The study of apoptotic pathways is fundamental to cancer research, providing critical insights into cellular responses to chemotherapeutic agents and potential therapeutic targets. Executioner caspases, particularly caspase-3 and caspase-7, serve as the central proteases responsible for orchestrating the morphological changes associated with programmed cell death. While caspase-3 is often considered the primary executioner caspase, the specific contributions and activation dynamics of caspase-7 remain less characterized, especially in caspase-3-deficient cellular contexts. The human breast carcinoma cell line MCF-7 provides a unique model for investigating caspase-7 activation mechanisms due to its natural deficiency in caspase-3 expression resulting from a 47-base pair deletion within the CASP-3 gene.
This application note details the use of a genetically encoded ZipGFP-based caspase-7 reporter to visualize and quantify the real-time activation dynamics of caspase-7 in MCF-7 cells in response to apoptotic stimuli. The ZipGFP technology represents a significant advancement over traditional FRET-based caspase reporters, offering superior signal-to-noise ratios and greater than 10-fold fluorescence increase upon protease activation, making it particularly suitable for detecting subtle activation kinetics in live cells [7] [77]. This methodology provides researchers with a powerful tool for investigating compensatory caspase activation mechanisms in caspase-3-deficient cancer models, with direct applications in drug discovery and resistance mechanism studies.
Background
Caspase Biology and Apoptotic Signaling
Caspases (cysteine-aspartic proteases) constitute a family of protease enzymes that play essential roles in programmed cell death and inflammation. These enzymes are synthesized as inactive zymogens (pro-caspases) that require proteolytic activation during apoptotic signaling [83]. The executioner caspases-3 and -7 share significant structural homology and substrate recognition preferences, typically cleaving after aspartic acid residues within DEVD peptide sequences [2]. Despite these similarities, emerging evidence suggests non-redundant functions and differential activation kinetics between these two executioner caspases.
Table 1: Caspase Classification and Functions in Programmed Cell Death
| Caspase Type | Members | Activation Mechanism | Primary Functions |
|---|---|---|---|
| Initiator | Caspase-2, -8, -9, -10 | Activation complexes (e.g., DISC, apoptosome) | Initiate apoptotic signaling by processing executioner caspases |
| Executioner | Caspase-3, -6, -7 | Cleaved by initiator caspases | Mediate proteolytic degradation of cellular components |
| Inflammatory | Caspase-1, -4, -5, -11 | Inflammasome complexes | Process pro-inflammatory cytokines and mediate pyroptosis |
The intrinsic apoptotic pathway, triggered by cellular stress and DNA damage, leads to mitochondrial outer membrane permeabilization and cytochrome c release. This facilitates the formation of the apoptosome complex, which activates caspase-9, subsequently processing executioner caspases including caspase-7 [83] [2]. In MCF-7 cells, which lack functional caspase-3, the entire execution phase of apoptosis must be carried out by caspase-7 and caspase-6, creating a unique dependency on these alternative executioner caspases.
ZipGFP Reporter Technology
The ZipGFP caspase reporter represents a novel class of fluorogenic protease reporters that operate on a unique "zipping" mechanism. Unlike FRET-based reporters that rely on energy transfer between two fluorophores, ZipGFP utilizes a specially engineered split GFP system where the two fragments (β1-10 and β11) are flanked by heterodimerizing E5 and K5 coiled coils that prevent their spontaneous association [7] [77].
Upon caspase activation and cleavage of the DEVD recognition sequence incorporated into both fragments, the coiled coil interactions are disrupted, allowing the GFP fragments to reassociate and form a functional fluorescent protein. This design achieves approximately 10-fold fluorescence increase with a activation half-time (T~1/2~) of approximately 40-100 minutes in cellular environments [7]. The quantum yield of the reconstituted ZipGFP is 0.25, providing sufficient brightness for live-cell imaging applications [7].
Table 2: Comparison of Fluorogenic Caspase Reporters
| Reporter | Dynamic Range | Brightness (Quantum Yield) | Activation Kinetics | Key Advantages |
|---|---|---|---|---|
| ZipGFP | ~10-fold | 0.25 | T~1/2~ ~40-100 min | Modular design, simultaneous dual protease detection |
| iCasper | >10-fold | Varies with biliverdin | T~1/2~ ~10 sec | Near-infrared imaging, minimal autofluorescence |
| FlipGFP | ~100-fold | 0.66 | Not specified | Highest brightness, suitable for low-expression systems |
| FRET-based | ~1.5-2 fold | Varies by FP pair | Minutes to hours | Established methodology, ratiometric measurement |
Materials and Methods
Research Reagent Solutions
Table 3: Essential Research Reagents and Materials
| Item | Specification/Supplier | Function/Application |
|---|---|---|
| ZipGFP-Casp7 Reporter | pZipGFP-DEVD plasmid (Addgene) | Caspase-7 activation sensor |
| Cell Line | MCF-7 (Caspase-3 deficient) | Model system for caspase-7 studies |
| Apoptotic Inducers | Doxorubicin (1-5 µM), Etoposide (10-50 µM) | Intrinsic pathway activation |
| Caspase Inhibitor | Z-VAD-FMK (20-50 µM) | Pan-caspase inhibition control |
| Live-Cell Dye | NucView 488 Caspase-3/7 substrate (Biotium) | Independent caspase activity validation |
| Transfection Reagent | Lipofectamine 2000/3000 | Plasmid delivery |
| Imaging Media | Phenol-red free DMEM + 10% FBS | Live-cell imaging compatibility |
| Microscopy System | Confocal or widefield with environmental chamber | Time-lapse imaging |
ZipGFP Caspase-7 Reporter Construct Design
The ZipGFP executioner caspase reporter was engineered by incorporating the canonical DEVD cleavage recognition sequence for executioner caspases into both "zipped" components of the reporter system [7]. The specific construct design includes:
- β1-10 Fragment: Fused to E5 coiled coil domain via N-terminal DEVD cleavage sequence
- β11 Fragment: Fused to K5 coiled coil domain via C-terminal DEVD cleavage sequence
- Expression Vector: CMV promoter-driven mammalian expression vector with antibiotic selection
This symmetric incorporation of DEVD sequences ensures specific detection of executioner caspase activity (caspase-3/7) while maintaining low background fluorescence in the absence of activation [7].
Cell Culture and Transfection Protocol
- Cell Culture: Maintain MCF-7 cells in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, and 1% penicillin-streptomycin at 37°C in a 5% CO~2~ humidified incubator.
- Transfection: Seed cells at 60-70% confluence in 35 mm glass-bottom imaging dishes 24 hours prior to transfection. Transfect with 1-2 µg ZipGFP-Casp7 reporter plasmid using Lipofectamine 2000 according to manufacturer's specifications.
- Recovery: Allow 24-48 hours post-transfection for reporter expression and cellular recovery before initiating apoptosis induction.
- Validation: Confirm transfection efficiency via baseline fluorescence microscopy (>70% efficiency recommended).
Apoptosis Induction and Live-Cell Imaging
- Treatment Preparation: Prepare fresh stock solutions of doxorubicin (1 mM in DMSO) or etoposide (10 mM in DMSO) and dilute to working concentrations in pre-warmed imaging media.
- Apoptosis Induction: Replace culture media with treatment media containing:
- Experimental Group: Doxorubicin (1 µM) or etoposide (20 µM)
- Inhibition Control: Z-VAD-FMK (50 µM) added 1 hour prior to apoptotic inducer
- Vehicle Control: Equivalent DMSO concentration (≤0.1%)
- Imaging Parameters:
- Maintain environmental chamber at 37°C with 5% CO~2~ throughout imaging
- Acquire images every 15-30 minutes for 12-24 hours using 488 nm excitation
- Use 20x objective for population studies or 40-60x oil objective for single-cell analysis
- Maintain consistent exposure settings across all experimental conditions
Image Analysis and Quantification
- Background Subtraction: Apply rolling ball background subtraction to raw images
- Region of Interest (ROI) Definition: Manually outline individual cells or use automated segmentation
- Fluorescence Quantification: Measure mean fluorescence intensity within ROIs for each timepoint
- Normalization: Express data as F/F~0~, where F~0~ represents baseline fluorescence pre-induction
- Kinetic Parameters: Calculate activation half-time (T~1/2~), maximum response (F~max~/F~0~), and area under the curve
Expected Results and Data Interpretation
ZipGFP Activation Kinetics
Successful caspase-7 activation in MCF-7 cells should yield characteristic sigmoidal fluorescence increase curves with the following quantitative expectations:
Table 4: Expected ZipGFP Activation Parameters in MCF-7 Cells
| Parameter | Doxorubicin (1 µM) | Etoposide (20 µM) | Z-VAD-FMK Control |
|---|---|---|---|
| Baseline Fluorescence (a.u.) | 150-300 | 150-300 | 150-300 |
| Maximum Fold-Increase | 8-12x | 6-10x | ≤1.5x |
| Activation Half-Time (T~1/2~) | 4-6 hours | 5-8 hours | N/A |
| Time to Peak Response | 8-12 hours | 10-16 hours | N/A |
The fluorescence increase should initiate 2-4 hours post-treatment, reaching maximum intensity between 8-16 hours depending on the apoptotic stimulus. Z-VAD-FMK pre-treatment should effectively abolish fluorescence development, confirming caspase-dependent activation.
Morphological Correlation with Apoptosis
ZipGFP fluorescence increase should correlate with classical apoptotic morphological changes:
- Early Stage (2-4 hours): Cell shrinkage and membrane blebbing
- Mid Stage (4-8 hours): Chromatin condensation and nuclear fragmentation
- Late Stage (8-16 hours): Formation of apoptotic bodies
These morphological features can be visualized using differential interference contrast (DIC) microscopy or nuclear counterstains (e.g., Hoechst 33342) acquired in parallel with ZipGFP fluorescence.
Applications in Drug Discovery and Development
The ZipGFP caspase-7 reporter system in MCF-7 cells provides a robust platform for several research applications:
- Mechanistic Studies of Apoptotic Pathways: Elucidate compensatory activation mechanisms in caspase-3-deficient cancers
- Drug Screening: Identify novel compounds that restore apoptotic sensitivity in resistant cancer models
- Combination Therapy Development: Test synergistic effects of chemotherapeutic agents
- Resistance Mechanism Investigation: Characterize alterations in caspase-7 activation kinetics in acquired resistance models
The real-time kinetic data provided by this system offers significant advantages over endpoint assays, enabling researchers to capture transient activation events and heterogeneous cellular responses within populations.
Troubleshooting Guide
- Low Fluorescence Signal: Optimize transfection efficiency; confirm plasmid quality; extend expression time; increase laser power/camera exposure
- High Background Fluorescence: Verify DEVD sequence integrity; shorten expression duration; include Z-VAD-FMK control
- No Activation Response: Confirm apoptotic inducer activity via alternative assays (e.g., Western blot for PARP cleavage); test positive control cell line with functional caspase-3
- Rapid Photobleaching: Reduce light exposure; include antioxidant in imaging media; use lower magnification objectives
Visualizations
Diagram 1: Intrinsic Apoptotic Pathway and ZipGFP Caspase-7 Activation in MCF-7 Cells. The diagram illustrates the sequential signaling events from apoptotic stimulus to caspase-7 activation and subsequent ZipGFP reporter cleavage. The intrinsic pathway triggers mitochondrial membrane permeabilization, leading to apoptosome formation and initiator caspase-9 activation, which then processes executioner caspase-7. Active caspase-7 cleaves the ZipGFP reporter, resulting in fluorescence increase, while simultaneously executing the apoptotic program.
Diagram 2: Experimental Workflow for ZipGFP Caspase-7 Activation Monitoring. The flowchart outlines the sequential steps from cell culture and reporter transfection through apoptotic induction, live-cell imaging, and quantitative data analysis. The complete protocol enables real-time tracking of caspase-7 activation kinetics in response to chemotherapeutic agents in caspase-3-deficient MCF-7 cells.
The study of programmed cell death, or apoptosis, is fundamental to biomedical research, spanning cancer biology, neurodegenerative diseases, and regenerative medicine. Executioner caspases-3 and -7 serve as central proteases that irreversibly commit cells to apoptotic death, making them critical biomarkers for detecting and quantifying cell death dynamics [51]. While traditional methods like Annexin V staining and TUNEL assays provide endpoint snapshots of apoptosis, they lack the temporal resolution to capture the dynamic, asynchronous nature of cell death in living systems [51].
Fluorescent caspase reporters have revolutionized apoptosis research by enabling real-time visualization of cell death in live cells and organisms. Among these, ZipGFP-based caspase reporters represent a significant advancement in biosensor technology. Originally described in 2016, ZipGFP utilizes a rational redesign of green fluorescent protein where the molecule is split into two fragments—β-strands 1-10 and the eleventh β-strand—tethered by a flexible linker containing a caspase-3/7-specific DEVD cleavage motif [8]. In its uncleaved state, forced proximity of the β-strands prevents proper folding and chromophore maturation, resulting in minimal background fluorescence. Upon caspase-mediated cleavage at the DEVD site, the fragments separate and spontaneously refold into the native GFP β-barrel structure, leading to efficient chromophore formation and robust fluorescence activation [51].
This technical note provides researchers and drug development professionals with a comprehensive framework for selecting ZipGFP caspase reporters, detailing their optimal applications, performance characteristics, and implementation protocols for advanced apoptosis research.
ZipGFP Mechanism and Key Advantages
Molecular Design and Activation Mechanism
The ZipGFP caspase reporter operates on an elegant molecular principle that combines structural constraints with protease-specific activation. The core innovation lies in the "zippering" of split GFP fragments through leucine zipper domains or direct linkage, maintaining the fluorescent protein in a dark state until caspase cleavage occurs [8]. This design fundamentally differs from FRET-based caspase sensors, which rely on distance changes between donor and acceptor fluorophores.
The activation mechanism proceeds through several precise steps:
- Basal State: In unstimulated cells, the ZipGFP reporter exhibits minimal fluorescence due to prevented chromophore formation.
- Caspase Activation: During apoptosis, executioner caspases-3/7 recognize and cleave the DEVD amino acid sequence within the reporter.
- Structural Liberation: Cleavage releases the structural constraints, allowing the GFP fragments to reconstitute.
- Fluorescence Activation: Proper folding enables chromophore maturation, generating a quantifiable fluorescent signal that accumulates over time [51].
This mechanism provides ZipGFP with distinctive operational advantages, particularly its irreversible activation pattern and signal amplification properties, which enable precise tracking of apoptosis initiation and progression.
Comparative Advantages of ZipGFP Technology
Table 1: Key Advantages of ZipGFP Over Alternative Caspase Sensing Modalities
| Feature | ZipGFP Reporter | FRET-Based Reporters | Small Molecule Probes |
|---|---|---|---|
| Signal-to-Noise Ratio | High (>10-fold increase post-activation) [8] | Moderate (2-3 fold change) | Variable (depends on permeability and retention) |
| Signal Persistence | Irreversible activation; cumulative signal | Reversible; dynamic | Transient; requires continuous presence |
| Spatiotemporal Resolution | Excellent for long-term tracking (hours to days) | Good for acute dynamics | Limited by probe half-life and cell permeability |
| Background Fluorescence | Very low in pre-cleaved state | Constant donor/acceptor background | High non-specific binding in some cases |
| Application Complexity | Suitable for 3D systems and in vivo [8] [51] | Challenging in thick samples due to spectral requirements | Limited penetration in 3D tissues |
| Multiplexing Potential | High (compatible with mCherry and other fluorophores) [51] | Moderate (spectral overlap challenges) | Limited to available probe chemistries |
ZipGFP exhibits particular strength in scenarios requiring high sensitivity and low background. The 10-fold fluorescence increase upon activation significantly outperforms FRET-based systems, enabling detection of subtle caspase activity [8]. Furthermore, the irreversible nature of ZipGFP activation creates a permanent record of apoptotic events, allowing researchers to distinguish cells that have experienced caspase activation even if the event was transient—a valuable feature for studying abortive apoptosis or heterogeneous cell populations.
Experimental Implementation and Workflow
Protocol: Establishing Stable ZipGFP Reporter Cell Lines
Materials and Reagents:
- Lentiviral vector encoding ZipGFP-caspase reporter (commercially available as ZipGFP-Casp3) [84] [85]
- Target cells (mesenchymal stem cells, cancer cell lines, primary fibroblasts) [84]
- Packaging plasmids (psPAX2, pMD2.G) for lentivirus production
- Polybrene (optional, enhances transduction efficiency)
- Apoptosis inducers: Staurosporine, TRAIL, carfilzomib [84] [51]
- Caspase inhibitor: zVAD-FMK (for control experiments) [51]
- Fluorescence-activated cell sorter (FACS) or puromycin for selection
Step-by-Step Procedure:
Vector Preparation:
- Obtain lentiviral vector containing the ZipGFP-Casp3 sensor, which typically includes the DEVD cleavage sequence inserted between split GFP fragments [84] [85].
- For multiplexed experiments, select constructs with constitutive mCherry or other fluorescent markers to normalize for cell presence and transduction efficiency [51].
Lentivirus Production:
- Co-transfect HEK293T cells with the ZipGFP transfer vector and packaging plasmids using standard calcium phosphate or lipid-based methods.
- Collect virus-containing supernatant at 48 and 72 hours post-transfection.
- Concentrate viral particles by ultracentrifugation or PEG precipitation if higher titer is required.
Cell Transduction:
- Plate target cells at 30-50% confluence in appropriate growth medium.
- Add viral supernatant supplemented with 4-8 μg/mL polybrene to enhance infection efficiency.
- Centrifuge plates at 800-1000 × g for 30-60 minutes (spinoculation) to increase transduction efficiency, particularly for difficult-to-transfect cells.
- Replace viral supernatant with fresh medium after 12-24 hours.
Selection and Expansion:
- For puromycin-based selection, begin antibiotic treatment 48 hours post-transduction (optimize concentration for each cell type).
- Alternatively, use FACS to isolate GFP-positive cells after induction with a known apoptosis inducer (e.g., 1μM staurosporine for 4-6 hours).
- Expand sorted/selected cells and validate reporter functionality using established apoptosis inducers.
Quality Control:
- Confirm caspase-specific activation by treating with caspase-3/7 inducer (e.g., 1μM staurosporine) with and without pan-caspase inhibitor zVAD-FMK (20-50μM) [51].
- Verify expected fluorescence increase via flow cytometry or live-cell imaging.
- Assess basal fluorescence in untreated cells to ensure low background signal.
Experimental Workflow for Real-Time Apoptosis Monitoring
The following diagram illustrates a complete experimental workflow from cell preparation to data analysis:
Application Scenarios and Decision Framework
Optimal Use Cases for ZipGFP Technology
Table 2: Recommended Applications of ZipGFP Caspase Reporters
| Research Scenario | Suitability | Key Considerations | Alternative Methods |
|---|---|---|---|
| Long-term apoptosis kinetics | Excellent (signal persistence) | Ideal for tracking over days; irreversible activation | FRET (reversible) may miss transient events |
| 3D culture systems | Excellent (low background) [51] | Superior penetration in organoids/spheroids | Chemical probes limited by penetration |
| In vivo imaging | Good (validated in zebrafish) [8] | Suitable for developmental studies; check expression stability | Bioluminescence may be preferred for deep tissue |
| High-content screening | Excellent [51] | Compatible with automated imaging systems | Plate reader assays faster but less granular |
| Heterogeneous populations | Excellent (single-cell resolution) [51] | Identifies apoptosis-resistant subpopulations | Bulk assays average population response |
| Stem cell research | Validated (MSCs) [84] [85] | Works in primary mesenchymal stem cells | Confirm low basal caspase activity |
| Cancer stem cell studies | Demonstrated (CD133+ cells) [84] | Can isolate apoptosis-resistant cancer stem cells | Combine with surface marker staining |
Decision Framework: When to Select ZipGFP
The following decision pathway provides guidance for selecting appropriate caspase detection methods:
Research Reagent Solutions
Table 3: Essential Reagents for ZipGFP Caspase Reporter Research
| Reagent/Category | Specific Examples | Function/Application | Notes |
|---|---|---|---|
| ZipGFP Reporters | ZipGFP-Casp3 (commercial) [84] [85] | Primary caspase-3/7 sensor | Available as lentiviral constructs |
| Control Inhibitors | zVAD-FMK (pan-caspase inhibitor) [51] | Confirm caspase-specific signal | Use at 20-50μM for validation |
| Apoptosis Inducers | Staurosporine, TRAIL, carfilzomib [84] [51] | Activate executioner caspases | Titrate for optimal dynamic range |
| Validation Antibodies | Anti-cleaved PARP, anti-cleaved caspase-3 [51] | Biochemical confirmation | Essential for protocol validation |
| Secondary Reporters | Constitutive mCherry [51] | Normalization control | Distinguish successful transduction |
| Cell Death Markers | Annexin V, Propidium Iodide [51] | Correlative apoptosis measures | Confirm ZipGFP activation timing |
| Selection Agents | Puromycin, FACS protocols [84] | Establish stable cell lines | Critical for long-term studies |
Technical Considerations and Limitations
While ZipGFP technology offers significant advantages, researchers should consider several technical aspects for optimal experimental design:
Critical Implementation Considerations:
Caspase Specificity Profile: ZipGFP with DEVD cleavage motif is highly specific for executioner caspases-3 and -7, which show strong cleavage activity (+++ rating), but may also be cleaved more weakly (+) by caspase-2, -6, -8, -9, and -10 [51]. This specificity profile is generally suitable for apoptosis research but requires appropriate controls when studying complex cell death pathways involving inflammatory caspases.
Signal Kinetics and Interpretation: The irreversible nature of ZipGFP activation means fluorescence accumulates over time, reflecting cumulative caspase activity rather than instantaneous activity. This is advantageous for identifying cells that have experienced caspase activation but may not capture rapid fluctuations in caspase activity. The signal persists even after caspase activity has ceased, which enables historical tracking but requires careful interpretation in dynamic systems.
Cell Type-Specific Optimization: Successful implementation requires optimization for different cellular contexts. Mesenchymal stem cells, colorectal carcinoma lines (HT-29, Caco2), and dermal fibroblasts have been successfully transduced with ZipGFP reporters [84] [85]. However, transduction efficiency, basal fluorescence, and response to apoptosis inducers should be empirically determined for each new cell type.
Multiplexing Capabilities: ZipGFP's green fluorescence emission (∼510nm) makes it compatible with red fluorescent proteins like mCherry for constitutive expression markers [51]. This enables ratiometric measurements and normalization for cell number and health. However, spectral overlap considerations are essential when combining with other fluorophores in multiplexed experiments.
By understanding these technical considerations and following the implementation guidelines outlined in this document, researchers can effectively leverage ZipGFP technology to advance their apoptosis research with enhanced temporal resolution and sensitivity in complex biological systems.
Conclusion
The ZipGFP reporter represents a significant leap forward in the real-time, dynamic visualization of caspase-3/7 activation, moving beyond static endpoint assays to provide single-cell resolution in everything from simple cultures to complex in vivo models. Its robust design, characterized by high specificity and a strong signal-to-noise ratio, makes it an indispensable tool for dissecting the role of apoptosis in development, homeostasis, and disease pathogenesis. For drug discovery, its application enables the high-content screening of therapeutic efficacy and the study of emergent phenomena like immunogenic cell death and therapy-induced repopulation via apoptosis-induced proliferation. Future directions will see ZipGFP integrated with reporters for other cell death modalities like pyroptosis and necroptosis to deconstruct complex, mixed death scenarios. Furthermore, harnessing this technology to study cell revival processes, such as anastasis, opens new frontiers in understanding cell fate decisions. Ultimately, the adoption of ZipGFP empowers researchers to not only observe cell death with unprecedented clarity but also to translate these insights into novel regenerative and anti-cancer strategies.
References
- 1. Current trends in luminescence-based assessment of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10613953/]
- 2. A Comprehensive Exploration of Caspase Detection Methods [https://pmc.ncbi.nlm.nih.gov/articles/PMC11121653/]
- 3. Evaluation of Caspase‐3 Activity During Apoptosis with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7738394/]
- 4. Robust high-throughput kinetic analysis of apoptosis with ... [https://www.nature.com/articles/cddis2016332]
- 5. Fluorescence Lifetime Imaging of Apoptosis - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4710058/]
- 6. A Caspase-3 Reporter for Fluorescence Lifetime Imaging ... [https://www.mdpi.com/2073-4409/7/6/57]
- 7. Rational design of a GFP-based fluorogenic caspase ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5026494/]
- 8. Rational Design of a GFP-Based Fluorogenic Caspase ... [https://www.sciencedirect.com/science/article/pii/S2451945616302008]
- 9. Incucyte® Apoptosis Assays for Live-Cell Analysis [https://www.sartorius.com/en/applications/life-science-research/live-cell-assays/apoptosis]
- 10. Integrated real-time imaging of executioner caspase ... [https://www.nature.com/articles/s41420-025-02662-y]
- 11. Quantitative analysis of caspase-3 activation by fitting ... [https://www.sciencedirect.com/science/article/abs/pii/S0968432809001103]
- 12. Rational Design of a GFP-Based Fluorogenic Caspase ... [https://pubmed.ncbi.nlm.nih.gov/27447051/]
- 13. Split Green Fluorescent Proteins: Scope, Limitations, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6537611/]
- 14. Engineering an efficient and bright split Corynactis ... [https://www.nature.com/articles/s41598-021-98149-8]
- 15. Leucine zipper [https://en.wikipedia.org/wiki/Leucine_zipper]
- 16. Leucine Zipper - an overview [https://www.sciencedirect.com/topics/agricultural-and-biological-sciences/leucine-zipper]
- 17. Caspases and their substrates - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5520456/]
- 18. Overlapping cleavage motif selectivity of caspases [https://www.nature.com/articles/4402260]
- 19. Caspase-Glo® 3/7 Assay System [https://www.promega.com/products/cell-health-assays/apoptosis-assays/caspase_glo-3_7-assay-systems/]
- 20. No-Wash Imaging of Apoptosis: CellEvent Caspase 3/7 ... [https://www.thermofisher.com/us/en/home/references/newsletters-and-journals/bioprobes-journal-of-cell-biology-applications/bioprobes-issues-2011/bioprobes-64-april-2011/cellevent-caspase-3-7-green-detection-reagent-april-2011.html]
- 21. Integrated real - time of executioner caspase dynamics... imaging [https://www.nature.com/articles/s41420-025-02662-y?error=cookies_not_supported&code=997d5d12-8110-4bfe-afc4-0e9db0552642]
- 22. Structural basis of fluorescence quenching in caspase ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3595455/]
- 23. Designing Caspase-3 Sensors for Imaging of Apoptosis in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3480176/]
- 24. Protocol for detection of caspases using immunofluorescence [https://www.abcam.com/en-us/technical-resources/protocols/immunofluorescence-protocol-to-detect-caspases]
- 25. Amplification and Background Reduction Techniques [https://fluorofinder.com/amplification-and-background-reduction-techniques/]
- 26. Apoptosis: A Review of Programmed Cell Death - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2117903/]
- 27. Apoptosis: A Comprehensive Overview of Signaling ... [https://www.mdpi.com/2073-4409/13/22/1838]
- 28. A guide in lentiviral vector production for hard-to-transfect ... [https://www.nature.com/articles/s41598-021-98657-7]
- 29. The EuLV® System, an inducible stable producer cell line ... [https://www.insights.bio/cell-and-gene-therapy-insights/journal/article/2400/The-EuLV-System-an-inducible-stable-producer-cell-line-for-lentiviral-vector-production]
- 30. Construction of stable packaging cell lines for clinical ... [https://www.nature.com/articles/srep09021]
- 31. Optogenetic Induction of Pyroptosis, Necroptosis, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10366993/]
- 32. Live Cell Imaging, Live Cell Applications, Time-Lapse ... [https://www.moleculardevices.com/applications/live-cell-imaging]
- 33. Protocol for Apoptosis Assay by Flow Cytometry Using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4943750/]
- 34. Apoptosis Protocols [https://health.usf.edu/medicine/corefacilities/flowcytometry/methods-apoptosis]
- 35. Annexin V Staining Protocol for Flow Cytometry [https://www.thermofisher.com/us/en/home/references/protocols/cell-and-tissue-analysis/protocols/annexin-v-staining-flow-cytometry.html]
- 36. Tips for Running a Successful Live Cell Imaging Experiment [https://www.moleculardevices.com/lab-notes/cellular-imaging-systems/tips-running-successful-live-cell-imaging-experiment]
- 37. Live Cell Imaging | Experiment Requirements [https://ibidi.com/content/338-the-live-cell-imaging-experiment]
- 38. Live Cell Imaging & Analysis [https://www.sartorius.com/en/products/live-cell-imaging-analysis]
- 39. Long-term Live-cell Imaging to Assess Cell Fate in Response ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6101178/]
- 40. Programmed cell revival from imminent cell death enhances ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12489119/]
- 41. A Comprehensive Exploration of Caspase Detection Methods [https://www.mdpi.com/1422-0067/25/10/5460]
- 42. Organoid and Spheroid Tumor Models - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC7922036/]
- 43. A three‐dimensional spheroid co‐culture system of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10280145/]
- 44. Fluorescence Lifetime Imaging of a Caspase-3 Apoptosis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5729923/]
- 45. Chapter Sixteen - Studying Apoptosis in the Zebrafish [https://www.sciencedirect.com/science/article/abs/pii/B9780124171589000169]
- 46. Zebrafish live imaging reveals a surprisingly small ... [https://elifesciences.org/reviewed-preprints/95691]
- 47. Designing an apoptosis reporter by mutagenesis-based ... [https://www.sciencedirect.com/science/article/pii/S2090123225004795]
- 48. Live imaging of apoptotic cells in zebrafish - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2974414/]
- 49. Analysis of Apoptosis in Zebrafish Embryos by Whole ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4109746/]
- 50. Kinetic quantification of apoptosis using Incucyte® live-cell ... [https://www.news-medical.net/whitepaper/20251027/Kinetic-quantification-of-apoptosis-using-Incucytec2ae-live-cell-imaging-assays.aspx]
- 51. Integrated real-time imaging of executioner caspase dynamics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12328661/]
- 52. A fluorescent biosensor-based platform for the discovery of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6682369/]
- 53. Neoadjuvant therapy-induced immune dynamics and ... [https://www.nature.com/articles/s41698-025-00954-1]
- 54. Optogenetic Induction of Pyroptosis, Necroptosis, and ... [https://en.bio-protocol.org/en/bpdetail?id=4762&type=0]
- 55. An immunogenic cell injury module for the single- ... [https://www.sciencedirect.com/science/article/pii/S0021925822007426]
- 56. Advanced flow cytometry data analysis ... [https://www.flowjo.com/advanced_analysis.html]
- 57. Multidimensional data analysis algorithms facilitate and ... [https://www.sciencedirect.com/science/article/pii/S0093691X25001190]
- 58. Decoding the divide: what distinguishes apoptosis-induced ... [https://biosignaling.biomedcentral.com/articles/10.1186/s12964-025-02336-3]
- 59. How Signal Noise Ratio Impacts Image Quality in Machine ... [https://www.unitxlabs.com/resources/camera-signal-noise-ratio-machine-vision-system-image-quality/]
- 60. : Causes of Failures and... | KEYENCE America Fluorescence Imaging [https://www.keyence.com/products/microscope/fluorescence-microscope/applications/fluorescence-microscope-applications/fluorescence-imaging.jsp]
- 61. Detection of caspase activation in situ by fluorochrome ... [https://pubmed.ncbi.nlm.nih.gov/11570504/]
- 62. Live Cell Imaging of Caspase Activation for High Content ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3613133/]
- 63. Imaging Caspase-3 Activation as a Marker of Apoptosis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4874215/]
- 64. High Variation of Fluorescence Protein Maturation Times in ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0075991]
- 65. Maturation models of fluorescent proteins are necessary for ... [https://www.sciencedirect.com/science/article/pii/S0006349522007718]
- 66. Limitations of fluorescent timer protein maturation kinetics ... [https://www.sciencedirect.com/science/article/pii/S2589004224011337]
- 67. Cleaved Caspase-3 (Asp175) Antibody #9661 | Cell Signaling Technology [https://www.cellsignal.com/products/primary-antibodies/cleaved-caspase-3-asp175-antibody/9661]
- 68. Caspase 3 (Cleaved Asp175) Polyclonal Antibody (PA5-114687) [https://www.thermofisher.com/antibody/product/Caspase-3-Cleaved-Asp175-Antibody-Polyclonal/PA5-114687]
- 69. Apoptosis Western Blot Cocktail (pro/cleaved Caspase-3, cleaved PARP1, muscle actin) (ab136812) | Abcam [https://www.abcam.com/en-us/products/panels/apoptosis-western-blot-cocktail-pro-p17-caspase-3-cleaved-parp1-muscle-actin-ab136812]
- 70. Caspase Protocols in Mice - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4084876/]
- 71. Annexin V and PI Staining for Apoptosis by Flow Cytometry [https://www.bio-techne.com/resources/protocols-troubleshooting/apoptosis-flow-annexin-v-protocol]
- 72. Annexin V Staining Protocol [https://www.bdbiosciences.com/en-us/resources/protocols/annexin-v-staining-protocol]
- 73. CellEvent™ Caspase-3/7 Green Flow Cytometry Assay Kit [https://www.thermofisher.com/order/catalog/product/C10427]
- 74. Flow cytometry as an integrative method for the evaluation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10160688/]
- 75. Basics of FRET Microscopy [https://www.microscopyu.com/applications/fret/basics-of-fret-microscopy]
- 76. FRET-based dynamic structural biology [https://elifesciences.org/articles/60416]
- 77. Imaging dynamic cell signaling in vivo with new classes of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7131868/]
- 78. Overcoming Limitations of FRET Measurements - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5835960/]
- 79. ViaStain Live Caspase 3/7 Detection for 2D/3D Culture [https://www.revvity.com/product/vs-caspase3-7-for-2d-3d-culture-cs1-v0002-1]
- 80. A bioluminescent caspase-1 activity assay rapidly monitors ... [https://www.sciencedirect.com/science/article/pii/S0022175916302757]
- 81. Illuminating insights into firefly luciferase and other ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2925662/]
- 82. ZipGFP-Casp3 (Plasmid #81241) [https://www.addgene.org/81241/]
- 83. Caspase [https://en.wikipedia.org/wiki/Caspase]
- 84. Generation of Cell Models with Stable Expression of a ... [https://pubmed.ncbi.nlm.nih.gov/40682642/]
- 85. s10517-025-06450-7.pdf [https://link.springer.com/content/pdf/10.1007/s10517-025-06450-7.pdf]