Structural Biology of Caspases: Decoding Initiator and Executioner Mechanisms for Therapeutic Development
This article provides a comprehensive analysis of the structural differences between initiator and executioner caspases, crucial proteases regulating cell death and inflammation.
Structural Biology of Caspases: Decoding Initiator and Executioner Mechanisms for Therapeutic Development
Abstract
This article provides a comprehensive analysis of the structural differences between initiator and executioner caspases, crucial proteases regulating cell death and inflammation. Targeting researchers and drug development professionals, we explore foundational domain architectures, activation mechanisms, and quaternary structures that dictate functional specialization. The content extends to methodological applications in research and therapy, addresses common experimental challenges, and offers comparative validation of structural features. By synthesizing recent structural biology advances, this review aims to inform the rational design of caspase-targeted therapeutics for cancer, neurodegenerative disorders, and inflammatory diseases.
Core Structural Architecture: Domains, Activation Mechanisms, and Quaternary Organization
Caspases (cysteine-aspartic proteases) are central regulators of programmed cell death and inflammation, functioning as inactive zymogens that require precise activation mechanisms. Their pro-domain structures serve as critical molecular signatures that determine activation pathways, hierarchical positioning within signaling cascades, and ultimate biological function. Within the broader context of initiator versus executioner caspase structural research, pro-domain organization provides the fundamental classification framework that dictates how these proteases integrate into multiprotein complexes and respond to cellular stimuli [1] [2]. This technical guide examines the structural and functional characteristics of caspase recruitment domains (CARD), death effector domains (DED), and short pro-domains, establishing how these signatures define caspase activation mechanisms and functional specialization in cell death pathways.
The classification of caspases based on pro-domain architecture has evolved beyond the traditional apoptotic versus inflammatory dichotomy, recognizing that apoptotic caspases can drive inflammatory lytic cell death under specific conditions [1]. This more nuanced understanding emphasizes the importance of structural biology in predicting caspase function across diverse physiological and pathological contexts. Given the clinical relevance of caspases across cancer, neurodegenerative disorders, and inflammatory diseases, comprehensive understanding of pro-domain organization and its relationship to function provides critical insights for therapeutic targeting [1] [3].
Caspase Pro-domain Architectures: Structural Features and Classification
Caspase pro-domains represent the primary structural determinants that regulate zymogen activation through specific protein-protein interactions. These N-terminal domains precede the conserved catalytic subunit comprising large (p20) and small (p10) subunits and exhibit significant length and compositional variation that defines caspase hierarchy within signaling pathways [2] [4].
Table 1: Caspase Pro-domain Classification and Functional Correlations
| Pro-domain Type | Representative Caspases | Domain Length | Adapter/Complex | Primary Functions |
|---|---|---|---|---|
| CARD | Caspase-1, -2, -4, -5, -9, -11, -12 | ~90-100 amino acids | Apoptosome, Inflammasome, PIDDosome | Innate immunity, intrinsic apoptosis, inflammatory cell death |
| DED | Caspase-8, -10 | ~70-80 amino acids | DISC (FADDosome) | Extrinsic apoptosis, necroptosis regulation, embryonic development |
| Short/None | Caspase-3, -6, -7 | <30 amino acids | Activated by initiator caspases | Apoptotic execution, substrate cleavage, pyroptosis induction |
The CARD domain represents a compact protein interaction module comprising six antiparallel amphipathic α-helices that form a conserved fold facilitating homotypic CARD-CARD interactions [4]. This structure enables recruitment of CARD-containing caspases to corresponding adapter proteins such as Apaf-1 in the apoptosome (caspase-9) or ASC in the inflammasome (caspase-1) [5]. The DED domain adopts a similar α-helical structure but facilitates distinct interaction networks, primarily recruiting caspases-8 and -10 to the death-inducing signaling complex (DISC) through homotypic DED-DED interactions with adapter proteins like FADD [4] [3]. Short pro-domains, characteristic of executioner caspases, lack structured interaction domains and instead maintain the latent state through intrasteric regulation until cleaved by upstream initiator caspases [2] [6].
Table 2: Structural and Biophysical Properties of Caspase Pro-domains
| Property | CARD Domains | DED Domains | Short Pro-domains |
|---|---|---|---|
| Secondary Structure | 6-7 antiparallel α-helices | 6 antiparallel α-helices | Unstructured/Loosely folded |
| Molecular Surface | Acidic and basic patches for complementary binding | Hydrophobic grooves for specific DED interactions | Minimal surface features |
| Interaction Type | Homotypic CARD-CARD | Homotypic DED-DED | Proteolytic cleavage sites |
| Representative Structure | Caspase-9 CARD (1JXQ) | Caspase-8 DED (not specified) | Caspase-3 pro-domain (1GFW) |
| Binding Affinity (Kd) | Low micromolar range for optimal complex assembly | Low micromolar range for optimal complex assembly | N/A |
Molecular Mechanisms of Pro-domain-Mediated Caspase Activation
CARD Domain Activation Mechanisms
CARD-containing caspases employ their pro-domains as recruitment modules that localize inactive zymogens to specific activation platforms through complementary CARD-CARD interactions. Caspase-9 activation exemplifies this mechanism, wherein the CARD domain binds reciprocally to the CARD domain of Apaf-1 within the heptameric apoptosome complex [4]. This interaction occurs through electrostatic complementarity between basic residues on the caspase-9 CARD and acidic residues on the Apaf-1 CARD, with key interactions involving conserved residues that facilitate high-affinity binding and caspase-9 dimerization [4]. Similarly, inflammatory caspases such as caspase-1 utilize CARD domains to recruit to inflammasome complexes via adapter proteins like ASC, which contains both a PYD domain for sensor interaction and a CARD domain for caspase recruitment [3].
The induced proximity model explains subsequent activation events, whereby caspase monomer dimerization at the activation platform facilitates conformational changes that reorganize active sites into catalytically competent configurations [2] [4]. For caspase-9, this occurs through formation of a proteolytic-based molecular timer wherein autocleavage between large and small subunits unmasks a neo-epitope for XIAP binding while promoting apoptosome dissociation [2]. CARD-mediated interactions thus serve dual functions: initial recruitment and subsequent regulation of activated caspases through controlled retention or release from activation platforms.
DED Domain Activation Mechanisms
DED-containing caspases-8 and -10 employ their pro-domains to recruit to death receptor signaling complexes through serial DED-DED interactions that form filamentous structures [4] [3]. At the DISC, Fas-associated death domain (FADD) serves as an adapter protein containing both a death domain (DD) for receptor interaction and a DED for caspase recruitment. The caspase-8 pro-domain engages in hierarchical interactions with FADD-DED, initiating filament formation that facilitates caspase-8 dimerization and activation [3]. This mechanism demonstrates remarkable regulation through cFLIP proteins, which contain DEDs but lack catalytic activity; cFLIPL heterodimerizes with caspase-8 to generate a single active site, while cFLIPS acts as a dominant-negative inhibitor by occupying DED binding sites without facilitating activation [2].
DED-mediated activation exemplifies how pro-domain interactions can be modulated by regulatory proteins to determine cell fate decisions. The composition of DED filaments—whether homomeric caspase-8 or heteromeric caspase-8/cFLIP—directs signaling outcomes toward apoptosis, necroptosis, or survival pathways [2] [5]. This regulatory sophistication underscores the importance of DED interactions as control points for extrinsic cell death signaling and explains why caspase-8 deficiency causes embryonic lethality due to disrupted development and hematopoiesis [4].
Activation of Short Pro-domain Executioner Caspases
Executioner caspases-3, -6, and -7 contain short pro-domains (<30 amino acids) that lack protein interaction capability, rendering them dependent on proteolytic activation by initiator caspases [2] [6]. These caspases exist as latent dimers in healthy cells, with activity restrained by the intersubunit linker that separates large and small catalytic subunits. Cleavage by initiator caspases at specific aspartic acid residues within this linker enables conformational rearrangement that assembles the active site and releases enzymatic activity [2] [3].
This activation mechanism creates hierarchical amplification within caspase signaling pathways, as a single initiator caspase can activate multiple executioner caspases that subsequently engage in feedback amplification loops [6]. The short pro-domains of executioner caspases thus represent evolutionary adaptations that prevent inadvertent activation while enabling rapid, explosive protease cascades once initiator caspase thresholds are surpassed. Recent evidence indicates that executioner caspases can also participate in non-apoptotic processes including differentiation, suggesting these short pro-domains may facilitate regulated subactivation without full commitment to cell death [6] [7].
Experimental Approaches for Pro-domain Characterization
Structural Determination Methodologies
X-ray Crystallography of Pro-domain Complexes: Successful structural determination of CARD and DED interactions requires expression and purification of isolated pro-domains followed by co-crystallization with binding partners. For CARD-CARD interactions, the caspase-9 CARD/Apaf-1 CARD complex (PDB: 1JXQ) revealed the electrostatic principles governing homotypic interactions, with basic residues (K15, K18, R56) on caspase-9 complementary to acidic residues (E66, D27) on Apaf-1 [4]. Protocol: Clone human caspase-9 CARD (residues 1-104) and Apaf-1 CARD (residues 1-97) into pET vectors with N-terminal His-tags. Express in E. coli BL21(DE3) at 18°C overnight with 0.5mM IPTG. Purify using Ni-NTA affinity chromatography followed by size exclusion chromatography (Superdex 75). Mix purified proteins at 1:1 molar ratio and crystallize using sitting drop vapor diffusion with 25% PEG 3350, 0.1M Bis-Tris pH 5.5, 0.2M ammonium acetate. Collect diffraction data at synchrotron sources and solve structure using molecular replacement.
Cryo-electron Microscopy of Activation Complexes: For large caspase activation platforms like the apoptosome or DISC, cryo-EM provides structural insights into pro-domain function in context. The apoptosome structure (PDB: 3J2T) reveals how caspase-9 CARD interactions position the catalytic domains for activation. Protocol: Express and purify full-length caspase-9 and Apaf-1 from baculovirus-infected insect cells. Assemble apoptosome by incubating with dATP and cytochrome c for 1 hour at 25°C. Apply 3μL aliquots to glow-discharged Quantifoil grids, blot, and plunge-freeze in liquid ethane. Collect data using Titan Krios microscope with Gatan K3 detector at nominal magnification of 105,000x. Process data using cryoSPARC with non-uniform refinement to achieve ~3.5Å resolution.
Biophysical Interaction Analyses
Surface Plasmon Resonance (SPR) for Binding Kinetics: SPR quantifies pro-domain interaction affinities and kinetics. Protocol: Immobilize GST-tagged Apaf-1 CARD on CMS chip using amine coupling. Flow purified caspase-9 CARD at concentrations from 10nM to 10μM in HBS-EP buffer (10mM HEPES pH 7.4, 150mM NaCl, 3mM EDTA, 0.005% surfactant P20) at 30μL/min. Monitor association for 180s and dissociation for 300s. Fit data to 1:1 Langmuir binding model to determine KD, kon, and koff values. Expected results: CARD-CARD interactions typically exhibit KD values of 0.1-10μM, optimized for reversible but specific complex formation.
Isothermal Titration Calorimetry (ITC) for Thermodynamics: ITC measures the enthalpy, entropy, and stoichiometry of pro-domain interactions. Protocol: Dialyze both interaction partners (caspase pro-domain and adapter) extensively against PBS pH 7.4. Load 200μM caspase-9 CARD into syringe and 20μM Apaf-1 CARD into sample cell. Perform 25 injections of 1.5μL at 180s intervals while maintaining constant stirring at 750rpm. Fit data to single-site binding model to determine ΔH, ΔS, and binding stoichiometry. Expected results: CARD-CARD interactions typically show favorable enthalpy (negative ΔH) with 1:1 binding stoichiometry.
Functional Cellular Assays
Fluorescence Resonance Energy Transfer (FRET) Caspase Activation Reporters: FRET-based biosensors monitor real-time caspase activation in live cells. Protocol: Transfect HeLa cells with CFP-YFP FRET reporter containing caspase cleavage site (DEVD for executioner caspases, IETD for initiator caspases). Image cells every 30s using confocal microscopy with 458nm excitation and collect emissions at 475-495nm (CFP) and 525-550nm (YFP). Calculate FRET ratio and monitor decrease upon caspase activation. For specific pathway analysis, pre-treat with pathway inhibitors: Z-VAD-FMK (pan-caspase), Z-IETD-FMK (caspase-8), Z-LEHD-FMK (caspase-9).
Co-immunoprecipitation of Activation Complexes: Co-IP validates pro-domain interactions in physiological contexts. Protocol: Lyse cells in mild lysis buffer (1% CHAPS, 150mM NaCl, 10mM HEPES pH 7.4) with protease inhibitors. Incubate lysate with anti-caspase-9 antibody overnight at 4°C. Add protein A/G beads for 2h, wash extensively, and elute with SDS sample buffer. Analyze by Western blotting for Apaf-1, caspase-9, and cytochrome c. For DISC IP, use anti-FADD antibody and blot for caspase-8, FADD, and receptor components.
Research Reagents and Methodologies Toolkit
Table 3: Essential Research Reagents for Caspase Pro-domain Studies
| Reagent/Category | Specific Examples | Function/Application | Key Features |
|---|---|---|---|
| Structural Biology | Apaf-1 CARD (1CY5), Caspase-9 CARD (1JXQ) | Structure determination of pro-domain complexes | High-purity, isotopically labeled for NMR |
| Chemical Inhibitors | Z-VAD-FMK (pan-caspase), Z-IETD-FMK (caspase-8), Z-LEHD-FMK (caspase-9) | Mechanistic studies of specific caspase functions | Irreversible, cell-permeable inhibitors |
| Activity Assays | Ac-DEVD-pNA (caspase-3), Ac-IETD-pNA (caspase-8), Ac-LEHD-pNA (caspase-9) | Quantitative enzyme activity measurements | Colorimetric or fluorogenic substrates |
| Cellular Reporters | CFP-YFP-DEVD, CFP-YFP-IETD FRET constructs | Live-cell monitoring of caspase activation | Real-time kinetic measurements |
| Antibodies | Anti-caspase-8 (DED domain specific), Anti-CARD domain (caspase-9), Anti-cleaved caspase-3 | Detection in Western blot, IP, immunofluorescence | Domain-specific, cleavage-sensitive |
| Expression Systems | Baculovirus (insect cells), pET vectors (E. coli) | Recombinant protein production | Post-translational modifications, high yield |
Visualization of Caspase Pro-domain Signaling Pathways
Caspase Pro-domain Activation Pathways - This diagram illustrates how CARD, DED, and short pro-domain caspases integrate into distinct activation pathways, highlighting the protein interaction networks that define initiator versus executioner caspase functions.
Caspase Pro-domain Structural Organization - This structural diagram compares the domain architectures of representative caspases, highlighting the relationship between pro-domain length, organization, and hierarchical positioning within activation cascades.
The structural signatures embedded within caspase pro-domains represent fundamental determinants of function that extend beyond simple classification schemes. The CARD, DED, and short pro-domain organizations establish precise activation mechanisms that integrate caspases into specific signaling networks while maintaining regulatory control over potent proteolytic activity. Understanding these structural principles provides critical insights for therapeutic development, particularly for diseases characterized by dysregulated cell death such as cancer, neurodegenerative conditions, and autoimmune disorders [1] [3] [5].
Future research directions include elucidating the structural basis for cross-talk between different pro-domain types, understanding how post-translational modifications regulate pro-domain interactions, and developing targeted therapeutics that specifically modulate pro-domain interactions rather than catalytic activity. The emerging roles of caspases in non-apoptotic processes including cellular differentiation and tissue remodeling further emphasize the importance of understanding how sublethal caspase activation is controlled through pro-domain-mediated localization and complex formation [6] [7]. As structural biology techniques advance, particularly cryo-EM and computational approaches like AlphaFold2, our understanding of caspase pro-domain function in native cellular environments will continue to expand, revealing new opportunities for therapeutic intervention in caspase-mediated diseases.
Caspases, a family of cysteine-aspartic proteases, are the core effectors of programmed cell death and inflammation. They are synthesized as inactive precursors, or zymogens, that require activation to gain full proteolytic functionality [8] [9]. A fundamental distinction within this enzyme family lies in the quaternary structure of their zymogen states and their subsequent activation mechanisms. Initiator caspases (caspase-1, -2, -4, -5, -8, -9, -10, -11, -12), which act apically in signaling cascades, exist predominantly as monomers in their latent forms. In contrast, executioner caspases (caspase-3, -6, -7), which carry out the dismantling of the cell, exist as preformed, stable dimers even before activation [8] [9]. This structural difference is not merely incidental; it forms the cornerstone of the hierarchical regulation of caspase activity, ensuring that cell death proceeds in a controlled and orderly fashion. This whitepaper delves into the structural and biochemical principles underlying these distinct zymogen states, framing them within the context of initiator versus executioner caspase function and their implications for therapeutic targeting.
Structural and Functional Classification of Caspases
Caspases are traditionally classified based on their primary roles in apoptosis, pyroptosis, and inflammation. Table 1 summarizes this functional classification and the inherent zymogen states.
Table 1: Functional Classification of Caspases and Their Zymogen States
| Programmed Cell Death Pathway | Type of Caspase | Enzyme | Native Zymogen State |
|---|---|---|---|
| Apoptosis | Initiator | Caspase-2, -8, -9, -10 | Monomer [8] |
| Apoptosis | Executioner | Caspase-3, -6, -7 | Dimer [8] |
| Pyroptosis | Inflammatory / Initiator | Caspase-1, -4, -5, -11, -12 | Monomer [9] |
Beyond this functional classification, a more structurally informed system groups caspases by their pro-domain architecture, which directly correlates with their zymogen state and activation mechanism. Initiator and inflammatory caspases possess long pro-domains containing protein-protein interaction motifs, such as the Caspase Recruitment Domain (CARD) or Death Effector Domain (DED). These domains are critical for their activation via induced proximity. Executioner caspases, however, have short pro-domains lacking these motifs and are activated through direct cleavage by initiator caspases [8] [9].
Zymogen Structures and Activation Mechanisms
The Monomeric State of Initiator Caspases
Initiator caspases, such as caspase-8 and caspase-9, are monomeric in their latent state. Their activation is triggered by recruitment to large activation platforms. For example, procaspase-8 is recruited to the Death-Inducing Signaling Complex (DISC) via interactions between its DEDs and the adaptor protein FADD [10]. Similarly, procaspase-9 is recruited to the apoptosome via CARD-CARD interactions with the adaptor protein Apaf-1 [8]. This recruitment leads to the high-localized concentration of the zymogens, facilitating their dimerization.
This process is known as "induced proximity" or "proximity-induced dimerization" [8] [11]. The dimerization event itself is the primary step that activates the initiator caspases, enabling them to undergo autocatalytic processing [11]. The crystal structure of the procaspase-1 zymogen domain (an inflammatory initiator caspase) revealed that while the isolated domain is monomeric in solution, it forms a dimer in the crystal, with the dimer interface providing insight into the first autoproteolytic events [12].
Diagram: Activation of Initiator Caspases via Induced Proximity
The Dimeric State of Executioner Caspases
Executioner caspases, such as caspase-3 and -7, adopt a stable dimeric conformation even in their latent, inactive state [8]. The activation of these zymogens does not require dimerization but is instead dependent on proteolytic cleavage by an upstream initiator caspase. This cleavage occurs at specific aspartic acid residues in the inter-domain linker region, separating the large and small subunits. Following cleavage, the subunits reassociate within the pre-existing dimer to form the active enzyme, which is a heterotetramer comprising two large and two small subunits [9]. This cleavage event allows the active-site loops to reorganize into a productive conformation, dramatically enhancing enzymatic activity.
Diagram: Activation of Executioner Caspases via Proteolytic Cleavage
Comparative Structural and Kinetic Analysis
The divergent activation mechanisms for initiator and executioner caspases have profound implications on their kinetic parameters and regulatory logic. Table 2 provides a quantitative comparison of these properties.
Table 2: Structural and Kinetic Properties of Caspase Zymogens
| Property | Initiator Caspases (e.g., -8, -9) | Executioner Caspases (e.g., -3, -7) | Experimental Evidence |
|---|---|---|---|
| Native Zymogen State | Monomer [8] | Dimer [8] | Size-exclusion chromatography, Multi-angle light scattering (MALS) [10] |
| Primary Activation Trigger | Induced proximity / Dimerization [11] | Proteolytic cleavage [8] | In vitro reconstitution assays, Analysis of cleavage-site mutants [12] [8] |
| Key Regulatory Motif | Long pro-domain (CARD/DED) [9] | Short pro-domain [9] | Crystal structures (e.g., PDB: 1IBC, 1ICE for caspase-1; 3R5J for caspase-2) [1] |
| Activation Complex | DISC (caspase-8), Apoptosome (caspase-9), Inflammasome (caspase-1) [8] [9] | N/A (Activated directly by initiator caspases) | Co-immunoprecipitation, Structural studies of complexes [8] [10] |
| Consequence of Activation | Auto-proteolytic processing [11] | Increase in kcat (e.g., 130-fold for caspase-1) [12] | Kinetic enzymology, Activity assays with fluorogenic substrates [12] |
The stability of the active dimer can be dependent on specific cleavage events. For instance, studies on procaspase-1 demonstrated that autoproteolysis at a specific aspartic acid (Asp316) is necessary for its conversion to a stable dimer in solution. This dimer stabilization was concurrent with a 130-fold increase in kcat, which was the sole kinetic factor contributing to its full activation [12].
Detailed Experimental Analysis of Caspase Activation
Experimental Protocol: Analyzing Initiator Caspase Dimerization
The study of initiator caspase activation often involves biochemical and structural methods to probe dimerization. The following protocol, based on research into caspase-8 activation, outlines a key approach [10].
- Objective: To purify and determine the oligomeric state of the caspase-8 tandem DEDs and elucidate its dimerization interface via X-ray crystallography.
- Expression and Purification:
- Cloning: The gene for the pro-domain of caspase-8 (residues 1-178) is cloned into an expression vector with a C-terminal hexa-histidine tag.
- Transformation: The plasmid is transformed into E. coli BL21(DE3) competent cells.
- Expression: Protein expression is induced with 0.25 mM IPTG at 18°C for ~18 hours.
- Purification: The bacterial lysate is applied to a nickel-nitrilotriacetic acid (Ni-NTA) affinity column. The target protein is eluted with an imidazole gradient and further purified by size-exclusion chromatography (Superdex 200 column) in a buffer containing 20 mM Tris-HCl pH 8.0 and 150 mM NaCl [10].
- Oligomeric State Analysis:
- Multi-angle Light Scattering (MALS): The absolute molar mass of the purified tandem DEDs is determined by coupling the size-exclusion column to a MALS detector. This confirms whether the protein exists as a monomer, dimer, or higher oligomer in solution [10].
- Crystallization and Structure Determination:
- Crystallization: Crystals are grown using the hanging-drop vapor-diffusion method with a reservoir solution containing 2.0 M ammonium sulfate and 0.1 M cacodylate pH 6.5.
- Data Collection: X-ray diffraction data is collected at a synchrotron beamline and processed.
- Structure Solution: The structure is solved by molecular replacement using a known caspase-8 DED mutant structure as a search model. The final model is refined through iterative cycles of manual building and computational refinement [10].
- Key Finding: The crystal structure of caspase-8 tandem DEDs revealed a novel domain-swapped dimer, providing a molecular basis for DED-mediated dimerization during DISC assembly [10].
Experimental Protocol: Analyzing Executioner Caspase Activation and Kinetics
The activation of executioner caspases is typically studied by monitoring their cleavage and the resultant kinetic enhancement. The following protocol is derived from studies on procaspase-1 and -7 [12].
- Objective: To activate an executioner caspase zymogen and characterize the kinetic consequences of proteolytic cleavage.
- Protein Expression and Refolding:
- Expression: The p20 and p10 subunits of the caspase (e.g., caspase-1) are expressed individually in E. coli, often forming inclusion bodies.
- Solubilization and Refolding: The insoluble pellets are solubilized in 6 M Guanidine-HCl. The denatured p20 and p10 subunits are mixed at a 1:1 ratio and refolded by dialysis into a refold buffer (e.g., 50 mM HEPES pH 8.0, 100 mM malonate, 1 M NDSB-201, 10% sucrose, 20 mM DTT) [12].
- Purification: The refolded, active caspase heterotetramer is purified using dialysis and additional chromatography steps.
- Analysis of Processing and Dimerization (SEC-MALS):
- The procaspase zymogen (e.g., a catalytic mutant) is mixed with the active enzyme.
- Time-point aliquots are analyzed by Size-Exclusion Chromatography coupled to Multi-Angle Light Scattering (SEC-MALS). This technique separates protein complexes based on size and provides an absolute measurement of their molecular weight in solution, confirming the oligomeric state.
- Simultaneously, samples are analyzed by SDS-PAGE to visualize proteolytic cleavage [12].
- Kinetic Analysis:
- Enzymatic activity is measured using fluorogenic peptide substrates that contain a caspase cleavage site (e.g., DEVD).
- The catalytic efficiency (kcat/Km) of the uncleaved zymogen is compared to that of the fully processed enzyme. As seen with caspase-1, activation can result in a massive increase in kcat, with no significant change in Km, leading to a much more efficient enzyme [12].
Diagram: Workflow for Analyzing Executioner Caspase Activation
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents for Studying Caspase Zymogens and Activation
| Reagent / Method | Function in Research | Specific Examples / Notes |
|---|---|---|
| C-terminal His-tag Vectors | Facilitates purification of recombinant caspases via immobilized metal affinity chromatography (IMAC). | pRSET, pET24a vectors; used with Ni-NTA affinity resin [12] [10]. |
| Size-Exclusion Chromatography (SEC) | Separates proteins by hydrodynamic radius; used to analyze oligomeric state and purity. | Superdex 200 column; often coupled with MALS for absolute molecular weight determination [12] [10]. |
| Multi-Angle Light Scattering (MALS) | Determines the absolute molar mass of a protein in solution, critical for confirming monomeric vs. dimeric states. | Wyatt Technology miniDAWN Treos system; coupled with SEC and refractive index detection [12] [10]. |
| Fluorogenic Peptide Substrates | Quantify caspase enzymatic activity. The substrate specificity varies between caspases. | Ac-DEVD-AFC for caspase-3; Ac-WEHD-AFC for caspase-1; cleavage releases fluorescent AFC [13]. |
| Refold Buffer Systems | Enable in vitro reconstitution of active caspase from bacterially expressed subunits. | Contains HEPES pH 8.0, malonate, nondetergent sulfobetaine (NDSB-201), sucrose, and DTT [12]. |
| X-ray Crystallography | Provides high-resolution atomic structures of zymogens and active caspases. | Revealed domain-swapped dimer of caspase-8 DEDs [10] and dimeric procaspase-1 [12]. |
The dichotomy between monomeric initiator caspases and dimeric executioner caspases represents a fundamental architectural and regulatory strategy in cell death signaling. Initiator caspases are tightly controlled through localization and induced-proximity dimerization, acting as signal integrators at the apex of pathways. In contrast, executioner caspases, pre-formed as dimers, are kept in check until a proteolytic event unleashes their potent catalytic activity, allowing for the rapid and irreversible execution of the cell death program. Understanding these mechanisms at a structural and kinetic level, as detailed in this whitepaper, is paramount for the development of novel therapeutics. Targeting the unique activation interfaces of initiator caspases or the allosteric networks of executioner caspases presents promising avenues for treating diseases ranging from cancer and autoimmunity to neurodegenerative disorders.
Caspases, a family of cysteine-aspartic proteases, are the central executioners of programmed cell death or apoptosis. These enzymes are synthesized as inactive zymogens (pro-caspases) and must undergo precise molecular events to become activated, culminating in the controlled dismantling of the cell. The fundamental distinction in caspase activation mechanisms lies between the initiator caspases (caspase-8, -9, and -10), which are activated primarily by induced proximity, and the executioner caspases (caspase-3, -6, and -7), which are activated principally by proteolytic cleavage. This dichotomy is not merely sequential but is rooted in profound structural differences that dictate their roles in the apoptotic cascade. Initiator caspases possess long pro-domains that facilitate recruitment to and activation within large multi-protein complexes, whereas executioner caspases have short pro-domains and rely on initiator caspases for their proteolytic activation [1] [5] [6]. Understanding these mechanisms is paramount for researchers and drug development professionals aiming to modulate cell death in diseases such as cancer and neurodegenerative disorders.
Core Activation Mechanisms
The Induced Proximity Model for Initiator Caspases
The induced proximity model proposes that the zymogens of initiator caspases possess low, intrinsic enzymatic activity. Their activation is triggered by their recruitment into high-order signaling complexes, which brings multiple procaspase molecules into close proximity. This forced dimerization is the critical event that drives their activation [14].
- Molecular Architecture: Initiator caspases (caspase-8, -9, -10) are monomers in their inactive state. They contain long N-terminal pro-domains, either Death Effector Domains (DEDs) in caspases-8 and -10 or a Caspase Activation and Recruitment Domain (CARD) in caspase-9 [5] [15]. These domains mediate homotypic interactions with adapter proteins in activation platforms.
- Activation Platforms: Caspase-8 and -10 are recruited to the Death-Inducing Signaling Complex (DISC) via interactions between their DEDs and the adapter protein FADD. Caspase-9 is recruited to the apoptosome through CARD-CARD interactions with Apaf-1 [5] [15].
- The Role of Dimerization: Oligomerization within these platforms forces the initiator caspase zymogens to dimerize. Dimerization alone is sufficient to generate catalytic activity by stabilizing a productive conformation of the active site [16] [15]. This is followed by autoprocessing of the intersubunit linker, which further stabilizes the active dimer but is not strictly required for initial activity [17] [15].
Proteolytic Cleavage for Executioner Caspase Activation
In contrast to initiator caspases, executioner caspases are activated through precise proteolytic cleavage by already-active initiator caspases.
- Molecular Architecture: Executioner caspases (caspase-3, -6, -7) exist as stable homodimers even in their inactive zymogen state. Their pro-domains are short, lacking the ability to recruit to large complexes [6] [15].
- The Activation Trigger: The key activation event is cleavage at specific aspartic residues within the intersubunit linker that separates the large and small subunits of the catalytic domain. This cleavage is performed by an upstream initiator caspase (e.g., caspase-8 or -9 cleaving pro-caspase-3) [13] [6].
- Conformational Change: Proteolytic cleavage triggers a conformational rearrangement that allows surface loops to stabilize the active site, resulting in a massive increase (several orders of magnitude) in catalytic activity. For executioner caspases, cleavage is both necessary and sufficient for full activation [15].
Table 1: Comparative Features of Initiator and Executioner Caspase Activation
| Feature | Initiator Caspases (e.g., -8, -9, -10) | Executioner Caspases (e.g., -3, -6, -7) |
|---|---|---|
| Primary Activation Mechanism | Induced Proximity / Dimerization | Proteolytic Cleavage |
| Native State in Cell | Monomeric | Dimeric |
| Pro-domain | Long (DED or CARD) | Short |
| Activation Platform | DISC (caspase-8/-10) or Apoptosome (caspase-9) | N/A |
| Role of Cleavage | Stabilizes the active dimer; can modify specificity [17] [15] | Essential for activating catalytic activity [15] |
| Catalytic Activity Enhancement | Upon dimerization [16] | >10,000-fold after cleavage [15] |
Key Experimental Evidence and Protocols
The distinct activation models are supported by foundational biochemical and structural experiments.
Experimental Evidence for Induced Proximity
Research on caspase-9 provided critical insights challenging a simplistic dimerization model. A landmark study engineered a constitutively dimeric caspase-9 by mutating residues at the dimer interface (e.g., Phe404) that created steric hindrance [16]. This engineered dimer exhibited higher catalytic activity and pro-apoptotic potential than wild-type monomeric caspase-9. However, its activity was only a small fraction of the activity achieved when caspase-9 was activated by the Apaf-1 apoptosome. This demonstrated that while dimerization is crucial, the activation platform provides a qualitative enhancement beyond mere dimerization, leading to a refined "induced conformation" model [16].
Protocol: In Vitro Dimerization and Activation Assay This protocol is used to study initiator caspase activation artificially, as performed in studies on caspase-10 [17].
- Protein Engineering: Create a fusion protein of the initiator caspase (e.g., lacking its pro-domain) with a synthetic dimerization domain, such as FK506-binding protein (Fv).
- Expression and Purification: Express the recombinant fusion protein in E. coli and purify via affinity chromatography (e.g., Ni-NTA for His-tagged proteins).
- Induced Dimerization: Incubate the purified protein with a cell-permeable, bivalent dimerizer drug (e.g., AP20187) that cross-links the Fv domains. A typical reaction uses a 1:1 molar ratio of protein to dimerizer for 30 minutes at 25°C [17].
- Activity Measurement: Assess caspase activation by adding a fluorogenic peptide substrate (e.g., Ac-IETD-AFC). Cleavage releases the fluorophore (AFC), which is measured by excitation at 405 nm and emission at 510 nm.
Experimental Evidence for Proteolytic Activation
The activation of executioner caspases is readily demonstrated by incubating the purified zymogen with an upstream protease. For example, cleaving pro-caspase-3 with caspase-8 or -9 results in its activation. The cleaved, active caspase-3 can then be characterized using peptide libraries or natural substrates to define its specificity, which often diverges from that of the initiator caspases [13]. Studies using positional scanning substrate combinatorial libraries (PS-SCL) have quantified the dramatic shift in substrate specificity and catalytic efficiency that occurs upon cleavage and activation of executioner caspases [17] [13].
Protocol: Analyzing Executioner Caspase Activation and Specificity
- Cleavage Reaction: Incubate purified executioner caspase zymogen (e.g., pro-caspase-3) with a purified, active initiator caspase (e.g., caspase-8) in standard caspase assay buffer (e.g., 20 mM PIPES, 100 mM NaCl, 10% sucrose, 10 mM DTT, 0.05% CHAPS, pH 7.4).
- Activation Confirmation: Monitor successful cleavage by SDS-PAGE, observing the appearance of large and small subunit fragments.
- Specificity Profiling: Use the activated caspase in a PS-SCL assay. The library consists of tetrapeptide substrates with fixed and variable amino acids. Incubate the caspase with the library and measure the fluorescence for each combination to define the optimal substrate sequence (e.g., DEVD for caspase-3) [17] [13].
- Kinetic Analysis: Determine catalytic parameters (kcat, KM) for optimal substrates using a range of substrate concentrations and measuring initial reaction velocities.
Table 2: Quantitative Catalytic Parameters of Selected Caspases
| Caspase | Activation State | Key Substrate | Reported KM (μM) | Catalytic Enhancement |
|---|---|---|---|---|
| Caspase-9 | Monomeric (zymogen) | LEHD | Very high | Low basal activity [16] |
| Caspase-9 | Apoptosome-bound | LEHD | Not explicitly stated | Activity enhanced >1000x vs. monomer [16] |
| Caspase-3 | Uncleaved zymogen | DEVD | Not explicitly stated | Low activity [15] |
| Caspase-3 | Cleaved (active) | DEVD | Not explicitly stated | >10,000-fold increase in activity [15] |
Visualization of Activation Pathways
The following diagrams illustrate the core concepts and experimental workflows for caspase activation mechanisms.
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents for Caspase Activation Research
| Reagent / Tool | Function / Application | Example Use Case |
|---|---|---|
| Synthetic Dimerizers (e.g., AP20187) | Artificially induces dimerization of engineered caspase fusion proteins. | Probing the induced proximity mechanism without the need for full activation platforms [17]. |
| Fluorogenic Peptide Substrates (e.g., Ac-DEVD-AFC) | Quantifying caspase activity. Caspase cleavage releases a fluorescent group (AFC). | Measuring the kinetic parameters (KM, kcat) of activated caspases in vitro [17] [13]. |
| Positional Scanning Substrate Combinatorial Library (PS-SCL) | Defining the comprehensive substrate specificity profile of a caspase. | Identifying optimal tetrapeptide cleavage motifs and comparing specificity between caspases [17] [13]. |
| Caspase Inhibitors (e.g., z-VAD-fmk) | Broad-spectrum, irreversible caspase inhibitor. | Active site titration to determine concentration of active enzyme; confirming caspase-dependent phenotypes [17]. |
| Kosmotropic Salts (e.g., Sodium Citrate) | Promotes protein-protein interactions and dimerization in solution. | Activating initiator caspases like caspase-10 in a cell-free system for biochemical studies [17]. |
| Recombinant Caspase Zymogens | Highly purified, inactive procaspases. | Serving as substrates in cleavage assays or for in vitro activation studies [17] [16]. |
Caspases, or cysteine-dependent aspartate-specific proteases, are a family of clan CD cysteine proteases that serve as critical effectors in apoptosis, inflammation, and cellular homeostasis. Their catalytic activity and stringent specificity for aspartate residues are governed by a unique structural fold that is conserved across evolution. This review examines the structural architecture of the clan CD fold that underpins caspase catalysis, detailing the molecular mechanisms of substrate recognition, activation, and inhibition. Framed within broader research on initiator versus executioner caspase structural differences, we synthesize recent structural biology findings to elucidate how conserved features enable catalytic function while variations confer specialized roles. The clinical relevance of caspase structures for targeted drug development is also discussed, providing a comprehensive technical resource for researchers and drug development professionals.
Caspases are a family of intracellular cysteine proteases that play central roles in programmed cell death (apoptosis), inflammation, and cellular differentiation [18] [9]. Their name derives from their unique catalytic mechanism: they are cysteine-dependent aspartate-specific proteases that cleave target proteins specifically after aspartic acid residues [19] [9]. In humans, 12 caspases have been identified, each sharing a conserved structural framework known as the clan CD fold [19] [18].
The clan CD fold represents a structurally related group of proteases that also includes metacaspases in plants, legumains, and bacterial proteases such as gingipains and clostripain [19]. Common to all clan CD proteases is a stringent specificity for a particular amino acid at the P1 position in substrates; for caspases, this is an absolute requirement for aspartic acid [19]. This review will explore the structural basis of caspase catalysis, focusing on the conserved elements of the clan CD fold that enable both their shared catalytic mechanism and their functional diversification into initiator and executioner roles in apoptotic signaling.
Table: Classification of Human Caspases
| Role Category | Caspase | Pro-domain Type | Primary Function |
|---|---|---|---|
| Initiator | Caspase-2 | CARD | Stress-induced apoptosis, cell cycle regulation |
| Caspase-8 | DED | Extrinsic apoptosis pathway, necroptosis inhibition | |
| Caspase-9 | CARD | Intrinsic apoptosis pathway | |
| Caspase-10 | DED | Extrinsic apoptosis pathway (human only) | |
| Executioner | Caspase-3 | Short | Primary apoptosis executioner, cleaves numerous substrates |
| Caspase-6 | Short | Apoptosis executioner, cleaves structural proteins | |
| Caspase-7 | Short | Apoptosis executioner, overlaps with caspase-3 substrates | |
| Inflammatory | Caspase-1 | CARD | Pyroptosis, IL-1β and IL-18 processing |
| Caspase-4 | CARD | Non-canonical inflammasome, LPS sensing | |
| Caspase-5 | CARD | Non-canonical inflammasome, LPS sensing | |
| Caspase-12 | CARD | Inflammatory response modulator (inactive in most humans) | |
| Other | Caspase-14 | Short | Epithelial cell differentiation, skin barrier formation |
The Conserved Clan CD Fold: Architectural Principles
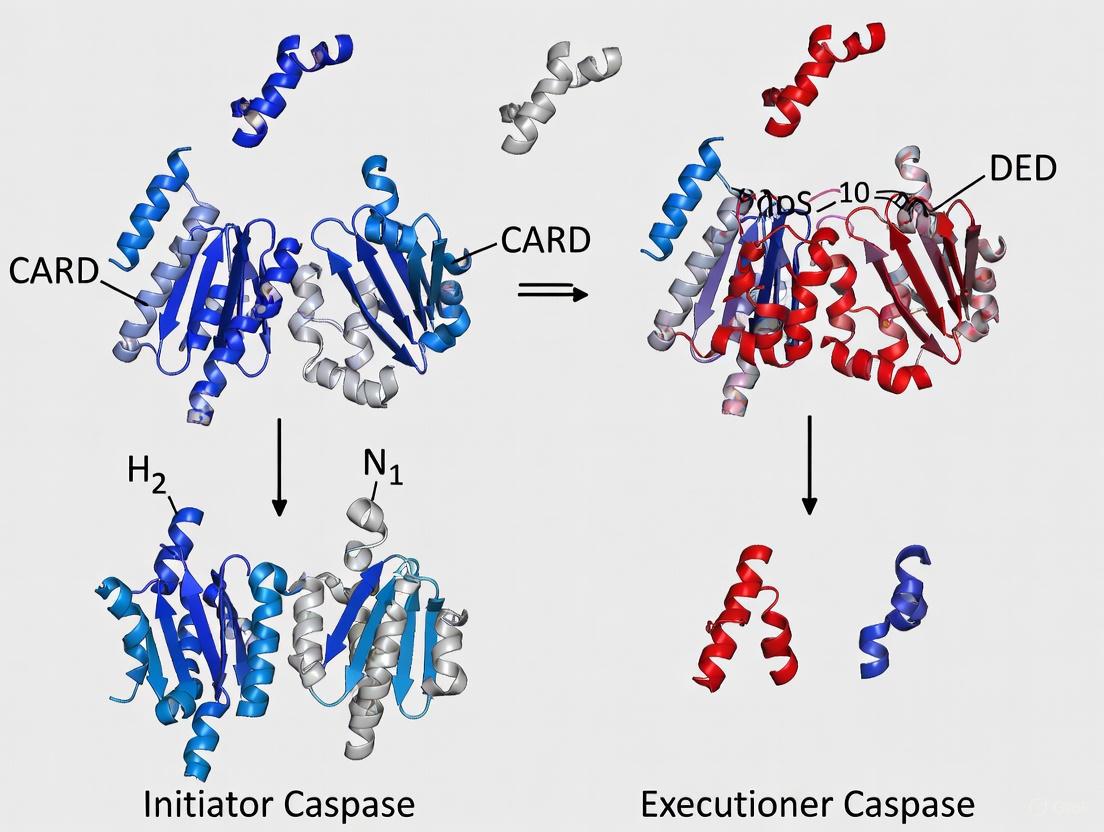
The clan CD fold common to all caspases forms a compact cylinder consisting of a central β-sheet surrounded by α-helices [20]. The active caspase is an obligate dimer, with each monomer contributing one catalytic domain [21]. Each catalytic domain in the mature enzyme is composed of two subunits - a large subunit (p17/p18) and a small subunit (p10/p12) - that are derived from the same precursor molecule through proteolytic cleavage at specific aspartic acid residues [20] [21].
The canonical active caspase exists as a heterotetramer with the composition (p17/p10)₂, forming a symmetric dimer of heterodimers [20] [9]. In this quaternary structure, the two active sites are formed at the dimer interface, with residues from both monomers contributing to each catalytic center [20]. This arrangement is conserved across both initiator and executioner caspases, though their activation mechanisms differ significantly.
The Catalytic Dyad and Active Site Architecture
The catalytic mechanism of all caspases depends on a conserved cysteine-histidine dyad [20] [18]. The nucleophilic cysteine residue (Cys285 in caspase-1 numbering) is positioned in the active site to attack the carbonyl carbon of the scissile peptide bond in substrate proteins [19]. This cysteine is stabilized by a neighboring histidine residue (His237), which acts as a general base to enhance the nucleophilicity of the catalytic cysteine [19].
The primary specificity pocket that confers aspartate selectivity is formed by three strictly conserved residues: Arg179, Arg341, and Gln283 (caspase-1 numbering) [19]. This deep, highly basic pocket is perfectly shaped to accommodate the negatively charged aspartic acid side chain of substrates, accounting for the up to four orders of magnitude lower catalytic efficiency for cleavage of peptides with a P1 glutamic acid residue in most caspases [19].
Diagram Title: Caspase Zymogen Activation and Quaternary Structure
Structural Distinctions Between Initiator and Executioner Caspases
Zymogen Activation Mechanisms
A fundamental structural difference between initiator and executioner caspases lies in their activation mechanisms. Initiator caspases (caspase-2, -8, -9, -10) exist as inactive monomers in their zymogen form and require dimerization for activation [22] [21]. This dimerization is facilitated by binding to adaptor proteins through protein-protein interaction motifs in their large pro-domains (CARD or DED domains) [22]. In contrast, executioner caspases (caspase-3, -6, -7) exist as inactive dimers in their zymogen form and are activated by proteolytic cleavage between their large and small subunits [22].
For initiator caspases, the induced proximity model explains their activation: when initiator caspase zymogens are brought into close proximity through adapter protein complexes, they dimerize and become activated [22]. Cleavage of initiator caspases occurs after dimerization but is not required for initial activity; rather, it serves to stabilize the active dimer [22]. Executioner caspases, however, exist as latent dimers with obstructed active sites; proteolytic cleavage at the inter-subunit linker induces a conformational change that repositions the loops to form a competent active site [22].
Pro-domain Architecture and Recruitment Complexes
The pro-domain structure represents another key distinction between initiator and executioner caspases. Initiator caspases contain long pro-domains with either CARD (caspase-2, -9) or DED (caspase-8, -10) domains that mediate protein-protein interactions [22] [18]. These domains belong to the death fold superfamily, which share a common structural motif of six or seven antiparallel amphipathic α-helices arranged in a characteristic fold [22].
Executioner caspases have only short pro-domains that lack protein interaction motifs [22]. The presence of large pro-domains in initiator caspases enables their recruitment to specific activation complexes: caspase-8 to the Death-Inducing Signaling Complex (DISC), caspase-9 to the Apoptosome, and caspase-1 to the Inflammasome [9]. These multiprotein complexes facilitate the dimerization and activation of the initiator caspases, which then propagate the death or inflammatory signal by cleaving and activating downstream executioner caspases.
Table: Structural and Activation Differences Between Initiator and Executioner Caspases
| Feature | Initiator Caspases | Executioner Caspases |
|---|---|---|
| Zymogen Form | Inactive monomers | Inactive dimers |
| Activation Mechanism | Dimerization induced by adapter proteins | Proteolytic cleavage by initiator caspases |
| Pro-domain | Long (CARD or DED domains) | Short |
| Primary Function | Signal initiation and amplification | Substrate cleavage and cellular dismantling |
| Activating Complexes | DISC (caspase-8), Apoptosome (caspase-9), Inflammasome (caspase-1) | Activated directly by initiator caspases |
| Representative Members | Caspase-2, -8, -9, -10 | Caspase-3, -6, -7 |
Substrate Recognition and Specificity Determinants
Extended Substrate Binding Groove
Caspases recognize short peptide sequences in their substrates through an extended binding groove that accommodates approximately four to six amino acid residues N-terminal to the cleavage site (P4-P1 positions) [19]. The groove comprises several substrate-binding pockets (S4-S1) that confer sequence specificity [19] [13]. While the S1 pocket is virtually identical across all caspases and strictly recognizes aspartic acid, the other subsites show variation that underlies the differing substrate specificities among caspase family members [19].
The S4 pocket shows the most variation among caspases and is a primary determinant of substrate specificity [19]. For example, caspase-1 prefers bulky hydrophobic residues (Trp, Tyr) in the P4 position; caspase-3 has a nearly absolute requirement for aspartic acid at P4; while caspase-8 preferentially accommodates branched aliphatic residues (Leu, Val) [19]. These differences in the S4 pocket structure and composition enable the functional specialization of different caspases for specific substrate cohorts.
Structural Basis of Caspase-2 Pentapeptide Specificity
Caspase-2 is unique among caspases in its strong preference for pentapeptide substrates (VDVAD) rather than tetrapeptides [23]. Structural studies have revealed that two residues, Thr380 and Tyr420, are critical for P5 residue recognition [23]. Mutation of these residues reduces catalytic efficiency by approximately 4-fold and 40-fold, respectively, demonstrating their importance in substrate binding [23]. The requirement for a P5 residue in caspase-2 substrates may contribute to its unique functions in stress-induced apoptosis and cell cycle regulation.
Diagram Title: Caspase Substrate Recognition and Specificity Pockets
Experimental Approaches for Caspase Structure Determination
Homology Modeling of Caspase Structures
Homology modeling has been extensively used to predict caspase three-dimensional structures when experimental structures are unavailable [24] [20]. This approach is based on the principle that evolutionary related proteins share similar structures, and that structural conformation is more highly conserved than amino acid sequence [24]. The methodology involves several key steps:
- Template identification using sequence similarity search tools such as BLAST against the Protein Data Bank [24] [20]
- Target-template alignment using programs such as CLUSTALW or CLUSTALX [24] [20]
- Model building using methods such as rigid-body assembly, segment matching, or satisfaction of spatial restraints [24]
- Model refinement through energy minimization and molecular dynamics simulations [24]
- Model validation using geometric verification and energy profiling [24]
For example, the three-dimensional structure of caspase-6 was successfully predicted through homology modeling using caspase-3 and caspase-8 as templates [20]. The resulting model revealed the characteristic caspase fold with a central β-sheet surrounded by α-helices and correctly predicted the Cys/His catalytic dyad and substrate-binding sites [20].
X-ray Crystallography of Caspase-Inhibitor Complexes
X-ray crystallography has provided most of the high-resolution structural information on caspases [20] [23]. This experimental approach involves:
- Protein expression and purification: Recombinant caspase proteins are typically expressed in E. coli and purified using affinity chromatography [23]
- Crystallization: Using vapor diffusion methods with solutions containing precipitans such as PEG [23]
- Data collection: Measuring X-ray diffraction at synchrotron sources [23]
- Structure determination: Using molecular replacement with known caspase structures as search models [23]
Structural studies of caspase-inhibitor complexes have been particularly informative for understanding substrate recognition. For example, crystal structures of caspase-2 in complex with peptide aldehyde inhibitors (VDVAD-CHO, ADVAD-CHO, DVAD-CHO) have revealed the molecular basis for its unique pentapeptide specificity and identified key residues involved in P5 recognition [23].
Table: Key Research Reagents for Caspase Structural Studies
| Reagent Type | Specific Examples | Function/Application |
|---|---|---|
| Expression Vectors | pET23b with C-terminal His6 tag | Recombinant protein expression in E. coli |
| Purification Systems | HisTrap FF column, Hi-Trap Q anion exchange | Protein purification using affinity and ion exchange chromatography |
| Fluorogenic Substrates | Ac-DEVD-AFC, Ac-VDVAD-AFC, Ac-IETD-AFC | Enzyme activity assays and kinetic measurements |
| Inhibitors | Z-VAD-FMK (pan-caspase), Ac-DEVD-CHO | Active site titration and specificity studies |
| Crystallization Reagents | HEPES buffer, PEG 3350 | Protein crystallization for X-ray studies |
Clinical Implications and Therapeutic Targeting
Understanding the structural basis of caspase catalysis has significant clinical implications, particularly for drug development. Caspases are implicated in numerous diseases, including cancer, neurodegenerative disorders, and inflammatory conditions [18] [9]. In cancer, caspase deficiency can contribute to tumor development by reducing apoptotic cell death, while in neurodegenerative diseases like Alzheimer's, excessive caspase activation can lead to inappropriate neuronal loss [9].
The structural insights into caspase substrate specificity and active site architecture have enabled the development of targeted caspase inhibitors [19] [18]. For example, the tetrapeptide sequence DEVD corresponds to the optimal cleavage site for caspase-3 and has been used to develop specific inhibitors and detection reagents [19]. Similarly, understanding the unique pentapeptide specificity of caspase-2 has enabled the design of selective inhibitors that may be useful for dissecting its biological functions [23].
Emerging therapeutic approaches include the use of caspase gene therapies and direct protein delivery systems [25]. For instance, recent studies have explored the exogenous introduction of specific caspases using redox-responsive polymeric nanogels as a potential cancer therapeutic strategy [25]. Surprisingly, while caspase-3 has the highest catalytic efficiency among executioner caspases, delivery studies have found caspase-7 and caspase-9 to be more effective at inducing apoptotic cell death in certain cellular contexts, highlighting the importance of the cellular regulatory environment in determining caspase efficacy [25].
The conserved clan CD fold provides the fundamental structural framework that enables caspase catalysis while allowing functional specialization through variations in key structural elements. The stringent specificity for aspartic acid at the P1 position is maintained across all caspases through a highly conserved basic pocket, while differences in other substrate-binding pockets enable recognition of distinct substrate cohorts. The structural distinctions between initiator and executioner caspases—particularly in their pro-domains and activation mechanisms—underlie their specialized roles in apoptotic and inflammatory signaling pathways. Continued structural studies of caspases, using both experimental and computational approaches, will further elucidate the molecular details of their regulation and function, providing foundations for targeted therapeutic interventions in caspase-mediated diseases.
Quaternary Structure Transitions During Activation
Caspases, cysteine-dependent aspartate-specific proteases, are central regulators of programmed cell death and inflammation. A critical distinction within this enzyme family lies between initiator caspases (e.g., caspase-2, -8, -9, -10) and executioner caspases (e.g., caspase-3, -6, -7), a classification defined by their unique positions in the apoptotic signaling cascade and, fundamentally, their divergent structural mechanisms of activation [26] [27]. The transition from an inactive zymogen to an active protease is governed by precise and distinct quaternary structure transitions for each class. Initiator caspases are characterized by long prodomains that facilitate the formation of large multimeric activation platforms, such as the Death-Inducing Signaling Complex (DISC) for caspase-8 or the apoptosome for caspase-9. This recruitment leads to their dimerization and subsequent activation [26]. In contrast, executioner caspases typically exist as stable dimers in their inactive state and require proteolytic cleavage by initiator caspases to achieve a conformational rearrangement into their active form [1] [26]. This article provides an in-depth technical examination of these quaternary structure transitions, framing them within the broader context of research aimed at elucidating the fundamental structural differences between initiator and executioner caspases, with significant implications for targeted drug development.
Structural Basis of Caspase Activation
Canonical Caspase Structure and the Activation Paradigm
All caspases are synthesized as inactive proenzymes (zymogens) that share a common structural organization. The canonical structure includes an N-terminal prodomain, a large subunit (p20), and a small subunit (p10) [26]. A conserved pentapeptide active-site motif, QACXG, is located within the large catalytic subunit and is essential for proteolytic function [26]. The activation of these zymogens involves proteolytic cleavage at specific aspartic acid residues, which separates the domains and allows for their reassembly into the active enzyme [26]. However, the pathways leading to this cleavage and the resultant quaternary structures differ markedly between initiator and executioner caspases.
Table 1: Core Structural Domains of Caspase Zymogens
| Structural Domain | Description | Role in Activation |
|---|---|---|
| Prodomain | N-terminal region; length varies significantly between initiator and executioner caspases. | Contains protein-protein interaction motifs (CARD, DED) for recruitment to activation platforms (initiators). |
| Large Subunit (p20) | Contains the catalytic cysteine residue within the QACXG motif. | Forms one part of the active site; cleavage from the prodomain and small subunit is required for activity. |
| Small Subunit (p10) | -- | Contributes to the formation of the active site heterodimer. |
Quaternary Transitions in Initiator Caspases: Induced Proximity and Dimerization
Initiator caspases, such as caspase-8 and -9, possess long prodomains featuring Death Effector Domains (DED) or Caspase Recruitment Domains (CARD). These domains are not directly involved in catalysis but are essential for homotypic protein-protein interactions [26]. The activation mechanism for initiator caspases is primarily driven by dimerization rather than proteolytic cleavage.
The process begins when specific death signals trigger the assembly of large multi-protein complexes. For the extrinsic pathway, death ligands like Fas and TNF bind to their receptors, leading to the formation of the Death-Inducing Signaling Complex (DISC), which recruits procaspase-8 via DED interactions [27]. For the intrinsic (mitochondrial) pathway, cellular stress signals induce the formation of the apoptosome, a complex involving Apaf-1 and cytochrome c, which recruits procaspase-9 via CARD interactions [26] [27].
Within these confined activation platforms, the local concentration of initiator caspase zymogens increases dramatically. This "induced proximity" forces the inactive monomers to dimerize [26]. The dimerization event itself is sufficient to generate a low level of enzymatic activity, a phenomenon known as the induced-proximity model. This initial activity allows the caspases to cleave each other in an interchain reaction, which stabilizes the active dimer but is not the primary trigger for activation. The core quaternary transition is therefore from an inactive monomer to an active homodimer.
Diagram 1: Initiator caspase activation pathway.
Quaternary Transitions in Executioner Caspases: Cleavage-Induced Conformational Change
Executioner caspases, such as caspase-3 and -7, have short prodomains and exist as pre-formed homodimers in their inactive state [26]. The activation mechanism for these caspases is not driven by dimerization but by proteolytic cleavage that induces a conformational shift.
In the inactive state, the executioner caspase dimer is intact, but a flexible loop region occupies the active site, rendering the enzyme catalytically incompetent. Activation occurs when an upstream initiator caspase (e.g., caspase-8 or -9) cleaves the executioner zymogen at specific inter-subunit linker aspartic acid residues. This cleavage allows the large and small subunits to separate from the prodomain and re-associate [26].
The critical quaternary structure transition involves a significant conformational rearrangement of the dimer. The cleaved subunits realign to form two mature and structured active sites, each capable of binding and cleaving substrate. Therefore, while the dimeric state is maintained, the transition is from an inactive, cleavage-incompetent dimer to an active, cleavage-competent dimer. This cleavage event is essential for executioner caspase activity; it is not merely a stabilizing factor but the definitive activation switch.
Diagram 2: Executioner caspase activation pathway.
Table 2: Comparative Analysis of Quaternary Structure Transitions
| Feature | Initiator Caspases (e.g., Casp-8, -9) | Executioner Caspases (e.g., Casp-3, -7) |
|---|---|---|
| Prodomain | Long (contains CARD or DED) | Short |
| Baseline State | Inactive Monomer | Inactive Homodimer |
| Primary Activation Trigger | Induced Proximity / Dimerization | Proteolytic Cleavage |
| Key Activation Complex | DISC (Casp-8), Apoptosome (Casp-9) | -- |
| Core Quaternary Transition | Monomer → Stable Homodimer | Inactive Dimer → Active Dimer |
| Role of Interchain Cleavage | Stabilizes the active dimer | Induces active conformation; essential for activity |
Experimental Methodologies for Studying Quaternary Transitions
Structural Biology and Biophysical Approaches
Determining the high-resolution structures of caspase intermediates is fundamental to understanding quaternary transitions.
- X-ray Crystallography: This technique has been instrumental in providing atomic-level snapshots of both inactive and active states of various caspases. By solving the structures of monomeric initiator caspase mutants and comparing them to their dimeric, active forms, researchers can pinpoint the conformational changes induced by dimerization [1]. Similarly, structures of executioner caspases before and after cleavage reveal the precise realignment of loops and subunits.
- Size-Exclusion Chromatography with Multi-Angle Light Scattering (SEC-MALS): This method is critical for empirically determining the absolute molecular weight and oligomeric state (monomer vs. dimer) of caspases in solution under different conditions. It can be used to confirm that initiator caspases are monomeric at low concentrations and dimerize upon inclusion in activation platforms or at high concentrations.
Table 3: Key Structural Data from the Protein Data Bank (PDB)
| Caspase | PDB Entry (Example) | Oligomeric State | Description / Relevance to Activation |
|---|---|---|---|
| Caspase-1 | 1IBC, 1ICE | -- | Inflammatory caspase; early structural insights. |
| Caspase-2 | 3R5J, 3R6G | -- | Initiator caspase structure. |
| Caspase-3 | 1GFW, 1PAU | Dimer | Structures of inactive and active executioner caspase. |
| Caspase-8 | (Multiple entries) | Monomer/Dimer | Structures illustrating both monomeric zymogen and dimeric active states. |
| Caspase-9 | (Multiple entries) | Monomer/Dimer | Structures within the apoptosome context and as isolated dimer. |
Functional and Cell-Based Activity Assays
While structural methods provide static pictures, activity assays are necessary to correlate structure with function in a dynamic context.
- Immunofluorescence (IF) for Caspase Detection: This antibody-based protocol allows for the spatial visualization of caspase activation within fixed cells or tissue sections [28]. The procedure involves:
- Sample Preparation and Fixation: Cells are grown on slides and fixed to preserve cellular architecture.
- Permeabilization: Treatment with a detergent like Triton X-100 allows antibodies to access intracellular caspases.
- Blocking: Incubation with a serum (e.g., goat serum) to reduce non-specific antibody binding.
- Primary Antibody Incubation: Application of a caspase-specific primary antibody (e.g., anti-active Caspase-3) overnight at 4°C.
- Secondary Antibody Incubation: Application of a fluorescently-labeled secondary antibody for detection.
- Mounting and Imaging: Slides are mounted and visualized using a fluorescence microscope [28]. This method is ideal for co-localization studies with other apoptotic markers but is limited to fixed samples.
- Genetically Encoded Fluorescent Biosensors: These tools, such as the Venus-based Caspase-3-like Activity Indicator (VC3AI), enable real-time, non-invasive monitoring of executioner caspase activity in live cells [29]. The VC3AI sensor is a cyclized, non-fluorescent chimera containing a caspase-3/7 cleavage site (DEVD). Upon cleavage by active caspase-3/7 during apoptosis, the sensor undergoes a conformational change that restores fluorescence, providing a direct, switch-on readout of caspase activity at the single-cell level [29]. This is superior for kinetic studies and monitoring apoptosis in 3D culture models.
Diagram 3: Experimental workflow for studying caspase activation.
The Scientist's Toolkit: Key Research Reagents
Table 4: Essential Reagents for Caspase Structure and Function Research
| Reagent / Tool | Function and Application | Example / Note |
|---|---|---|
| Caspase-Specific Antibodies | Detect expression, cleavage, and localization of caspases in techniques like Western Blot and Immunofluorescence (IF). | Anti-active Caspase-3 antibody for IF [28]. |
| Fluorogenic Caspase Substrates | Measure caspase activity in cell lysates or live cells. The substrate emits fluorescence upon cleavage. | Ac-DEVD-AFC (for caspase-3/7); the release of AFC is quantified. |
| Pharmacologic Caspase Inhibitors | Probe the functional role of specific caspases in a pathway. Can be pan-caspase or specific. | Z-VAD-fmk (pan-caspase inhibitor); Z-DEVD-fmk (caspase-3/7 inhibitor) [29]. |
| Genetically Encoded Biosensors | Monitor caspase activity in real-time within live cells, allowing for kinetic single-cell analysis. | VC3AI: A cyclized Venus-based sensor that becomes fluorescent upon caspase-3/7 cleavage [29]. |
| Recombinant Caspase Proteins | Used for in vitro biochemical studies, structural biology (crystallography), and high-throughput inhibitor screening. | Purified, inactive caspase-6 or -7 for studying activation kinetics. |
The fundamental difference in the quaternary structure transitions during initiator and executioner caspase activation—dimerization-driven versus cleavage-driven—underscores a sophisticated evolutionary adaptation for ensuring tight and irreversible control over cell death pathways. The precise structural understanding of these mechanisms, enabled by a suite of biophysical and cell-biological tools, provides an invaluable foundation for drug discovery. Targeting the specific protein-protein interfaces required for initiator caspase dimerization (e.g., at the DISC or apoptosome) or designing allosteric inhibitors that lock executioner caspases in their inactive dimeric state represent promising therapeutic strategies. As structural biology techniques continue to advance, offering deeper insights into these dynamic transitions, the potential for developing highly specific caspase-modulating drugs for cancer, neurodegenerative disorders, and autoimmune diseases will grow exponentially.
Research Tools and Therapeutic Translation: From Structural Insights to Clinical Applications
Caspases, a family of cysteine-aspartate proteases, are fundamental regulators of programmed cell death (apoptosis) and inflammation. They are broadly classified into initiator caspases (including caspase-8, -9, and -10), which initiate apoptotic signaling cascades, and executioner caspases (including caspase-3, -6, and -7), which carry out the proteolytic cleavage of cellular targets [18]. Understanding the distinct structural properties and activation mechanisms of these two classes is a central goal in cell biology and drug development. Structural biology techniques, primarily X-ray crystallography and cryo-electron microscopy (cryo-EM), have been indispensable in elucidating these molecular details. These techniques reveal the atomic-level three-dimensional structures of caspase complexes, providing insights into their domain organization, activation mechanisms, and substrate specificity. This knowledge is crucial for understanding the molecular basis of diseases characterized by dysregulated cell death, such as cancer and neurodegenerative disorders, and for informing the rational design of therapeutic compounds.
The following table summarizes key structural differences between initiator and executioner caspases, which form the foundation for their specialized roles in apoptosis.
Table 1: Key Structural and Functional Differences Between Initiator and Executioner Caspases
| Feature | Initiator Caspases (e.g., caspase-8, -9) | Executioner Caspases (e.g., caspase-3, -6, -7) |
|---|---|---|
| Primary Function | Initiate apoptosis signaling cascades | Execute apoptosis by cleaving cellular substrates |
| Activation Mechanism | Induced proximity, dimerization on activation platforms (e.g., apoptosome, DISC) | Proteolytic cleavage by initiator caspases |
| Pro-Domain | Long pro-domains containing DED (caspase-8) or CARD (caspase-9) domains for recruitment | Short pro-domains |
| Characteristic Domains | Death Effector Domain (DED) or Caspase Activation and Recruitment Domain (CARD) | Lacks long recruitment domains |
| Structural Flexibility | Often exist as monomers (caspase-9) or monomer/dimer mixtures (caspase-8) prior to activation [25] | Constitutively dimeric in their active forms |
Elucidating Caspase Activation and Specificity Through X-ray Crystallography
X-ray crystallography has been a cornerstone technique for determining the high-resolution atomic structures of caspases, providing foundational knowledge about their active sites, the conformational changes during activation, and the molecular basis for substrate selection.
Molecular Basis for Caspase-9 Selectivity
A prime example of how crystallography illuminates functional specificity is the investigation into why caspase-9 directly activates procaspase-3 but not procaspase-6. Research has shown that this selectivity is governed by both the sequence and the local structural context of the substrate cleavage site. Caspase-9 cleaves procaspase-3 at its intersubunit linker (ISL) site, 172IETD↓S. In contrast, procaspase-6 possesses two ISL cleavage sites (176DVVD↓N and 190TEVD↓A), neither of which is directly cleaved by caspase-9 [30].
Engineered constructs revealed that the P4-P1' sequence of the procaspase-6 ISL site 1 (DVVDN) is accessible yet uncleavable by caspase-9. While caspase-9 can recognize the sequence of the procaspase-6 ISL site 2 (TEVDA), the local structural context surrounding this site prevents proteolytic cleavage [30]. This demonstrates that substrate selection is not solely based on a short linear motif but is also critically dependent on the three-dimensional structural environment accessible through crystallographic analysis.
Experimental Protocol: Determining a Caspase Structure by X-ray Crystallography
The following workflow outlines the standard methodology for determining a caspase structure using X-ray crystallography, integrating specific examples from caspase studies.
Diagram 1: Caspase X-ray Crystallography Workflow
Protein Expression and Purification: The gene for the target caspase (e.g., wild-type or engineered construct) is cloned into an expression plasmid, such as pET23b or pET11a. The plasmid is transformed into E. coli (e.g., BL21(DE3) strain). Protein expression is induced with IPTG, typically at lower temperatures (25-30°C) to improve protein folding and solubility. Cells are then lysed, and the recombinant caspase is purified using a combination of Ni²⁺-affinity chromatography (exploiting an engineered His-tag) followed by anion exchange chromatography to achieve high purity [30] [31].
Crystallization and Data Collection: Purified caspase is concentrated to high levels (e.g., 5-20 mg/mL) and subjected to sparse matrix screening to identify conditions that promote crystal formation. This involves vapor diffusion methods where the protein solution is mixed with a precipitant solution and allowed to equilibrate. Successfully grown crystals are harvested and cryo-cooled in liquid nitrogen for data collection. X-ray diffraction data are collected at synchrotron facilities, generating a set of structure factor intensities [32].
Structure Determination: The diffraction data are processed (indexed, integrated, and scaled) to determine the crystal's symmetry and unit cell parameters. Phasing methods, such as molecular replacement (using a related caspase structure as a search model), are used to determine the phases of the structure factors. An initial atomic model is built into the experimental electron density map and undergoes iterative cycles of refinement and manual model adjustment to improve the fit to the data [32].
Advancing Caspase Structural Studies with Cryo-Electron Microscopy
Cryo-EM has emerged as a powerful complementary technique to X-ray crystallography, particularly for studying larger, more complex, or heterogeneous caspase complexes that are difficult to crystallize.
Technical Considerations and a Novel Tagging System
A significant challenge in single-particle cryo-EM is studying proteins smaller than 50-100 kDa, as they produce low signal-to-noise ratios and lack distinct features for particle alignment. While caspase homo-dimers are often on the lower end of this size range, their incorporation into larger activation complexes makes them amenable to cryo-EM study. Recent research has investigated the small, tetrameric, metal-binding protein Csp1 as a potential "bio-tag" to overcome these challenges. Csp1 is compact, stable, and exhibits enhanced electron scattering, providing excellent particle contrast in cryo-EM micrographs. In a proof-of-concept study, the structure of Csp1 alone was determined to 2.98 Å resolution. Furthermore, a complex between an epitope-tagged Csp1 and a ~40 kDa Fab fragment yielded a medium-resolution structure (5.40 Å for the Fab), demonstrating its potential as a fiducial marker for structural studies of smaller proteins and complexes [33].
Sample Preparation Strategies for Near-Native Conditions
Cryo-EM allows for structural analysis under closer-to-native conditions. Strategies are evolving from studying purified proteins in isolation to analyzing complexes with minimal perturbation. One approach involves using crude cell lysates to identify cellular composition and complexes, though this loses some native environmental information. A more elegant method is cryo-electron tomography (cryo-ET) combined with cryo-focused ion beam (cryo-FIB) milling, which enables the imaging of caspases and other proteins within vitrified cells, preserving their native context. However, this technique currently has limitations in throughput and achievable resolution, often requiring the target to be naturally abundant [34]. Hybrid strategies that combine the high resolution of SPA with the native context of cellular samples are an area of active development, effectively blurring the lines between these techniques [34].
Table 2: Key Research Reagent Solutions for Caspase Structural Biology
| Research Reagent / Material | Function in Experiment | Specific Example from Literature |
|---|---|---|
| pET Vectors (e.g., pET23b, pET11a) | Bacterial expression plasmids for high-yield recombinant caspase production. | Used for expressing caspase-3, -6, and -9 constructs [30]. |
| Ni²⁺-Affinity Resin | Purification of recombinant His-tagged caspases via immobilized metal affinity chromatography (IMAC). | Standard step in purification protocols for caspase-3 and -6 [30] [31]. |
| Profinity eXact Purification Tag | A fusion tag system that allows for highly specific, protease-free cleavage and purification. | Used for purifying the Csp1 bio-tag candidate for cryo-EM [33]. |
| Csp1 "Bio-Tag" | An electron-dense, metal-binding protein tag to improve contrast and particle alignment in cryo-EM of small proteins. | Tested as a fiducial marker for determining the structure of a ~40 kDa Fab fragment [33]. |
| Cross-linking Agents (e.g., DTT) | Stabilizes self-assembled nanomaterials for caspase encapsulation and delivery; used in protein complex stabilization. | Used to cross-link PEG-PDS polymers for caspase nanogel formation [25]. |
Integrated Structural Workflows and Caspase-Specific Protocols
Combining multiple biophysical and structural techniques often provides the most comprehensive understanding of caspase structure and function, especially when studying dynamic processes like protein metalation or inhibitor binding.
Complementary Techniques for a Holistic View
The investigation of metal-based drug interactions with proteins showcases the power of integrated approaches. X-ray crystallography can pinpoint the metal coordinates and protein residues involved in binding. However, it provides a static, time- and space-averaged view. Mass spectrometry techniques, particularly electrospray ionization (ESI-MS), are invaluable for characterizing metal/protein adducts, identifying binding stoichiometry, and studying interactions in solution, thus complementing and validating crystallographic data [32]. Other techniques like vibrational spectroscopy, electron paramagnetic resonance, and computational methods provide additional insights into the dynamics and energetics of these interactions, offering a solution-state perspective that crystallography alone cannot [32].
Specialized Protocol for Challenging Caspase Constructs
Some caspases, such as caspase-6, present unique challenges for structural biology due to low expression yields, propensity for aggregation, and self-cleavage. Specialized protocols are required, particularly for sensitive techniques like NMR spectroscopy, which demands high concentrations of stable, monodisperse protein [31].
Optimized Expression and Purification for Caspase-6:
- Media Optimization: For isotopic labeling required for NMR, caspase-6 is expressed in minimal media (e.g., M9). Using 2x M9 minimal media, which has a higher buffering capacity, allows E. coli to reach a higher optical density, improving protein yield [31].
- Concentration and Buffer Exchange: Standard spin-filter concentration is often unreliable for caspase-6, leading to significant losses. A more robust strategy involves using a gravity flow column for buffer exchange into a deuterated NMR buffer, followed by concentration under a controlled gas pressure, significantly improving sample recovery and homogeneity [31].
Diagram 2: Caspase-6 NMR Sample Challenges
X-ray crystallography and cryo-EM have provided profound insights into the structural distinctions between initiator and executioner caspases. Crystallography has delivered high-resolution details of active sites, conformational changes, and the principles of substrate specificity, such as the critical role of both sequence and local context in caspase-9's selectivity [30]. Meanwhile, cryo-EM is opening new frontiers by enabling the study of larger activation complexes, like the apoptosome, and offering pathways to visualize caspases in more native-like environments [34]. The continued development of integrated structural workflows, including the use of complementary biophysical techniques and novel tools like the Csp1 bio-tag [33], promises to further deepen our understanding of caspase regulation and function. This expanding structural knowledge is fundamental for elucidating the mechanisms of cell death in health and disease and for paving the way toward targeted therapeutic interventions.
Design of Caspase-Specific Substrates and Activity Probes
The design of specific substrates and activity-based probes for caspases is fundamentally grounded in the distinct structural and biochemical differences between initiator and executioner caspases. Caspases, an evolutionarily conserved family of cysteine-dependent aspartate-specific proteases, are synthesized as inactive zymogens (procaspases) that require proteolytic activation for full function [35] [26]. The human caspase family consists of 12 members that have historically been classified based on their structural domains, substrate preferences, and roles in biological processes including apoptosis, pyroptosis, and inflammation [35] [1] [5].
Table 1: Structural and Functional Classification of Human Caspases
| Caspase Type | Members | Pro-Domain | Primary Activation Mechanism | Biological Functions |
|---|---|---|---|---|
| Initiator | Caspase-2, -8, -9, -10 | Long (CARD or DED) | Induced proximity & dimerization | Initiate apoptotic signaling; non-apoptotic functions |
| Executioner | Caspase-3, -6, -7 | Short | Cleavage by initiator caspases | Cleave cellular substrates to execute cell death |
| Inflammatory | Caspase-1, -4, -5, -11 | Long (CARD) | Inflammasome assembly | cytokine maturation; pyroptosis |
The critical structural distinction lies in their pro-domains: initiator caspases possess long pro-domains containing protein-protein interaction motifs such as the caspase activation and recruitment domain (CARD) or death effector domain (DED), which facilitate their recruitment to and activation within large signaling complexes [26] [1]. In contrast, executioner caspases contain short pro-domains and exist as constitutive dimers, requiring cleavage by initiator caspases for activation [35]. This structural divide directly informs the differential substrate specificity and catalytic efficiency that researchers must exploit when designing specific probes.
Structural Principles Governing Caspase Specificity
Active Site Architecture and Substrate Recognition
Caspase substrate recognition occurs through interaction with a groove that accommodates approximately four amino acids (P4-P1) N-terminal to the cleavage site [35]. The enzymes display a nearly absolute requirement for aspartic acid at the P1 position, with additional specificity determined by residues at P2-P4 positions [35] [36]. Structural analyses reveal that the S4 pocket, which accommodates the P4 residue of substrates, constitutes the primary determinant of specificity differences between caspases [37].
Executioner caspases-3 and -7 display a strong preference for aspartic acid at the P4 position (DEVD motif), whereas initiator caspase-8 prefers hydrophobic residues at P4 (L/VEXD motif) [35] [36]. Caspase-6 represents an exception with its preference for valine at P4 (VEHD motif), despite its classification as an executioner caspase based on domain architecture [37]. These specificity profiles directly reflect the physicochemical properties of their respective S4 binding pockets.
Structural Basis of Zymogen Activation
The activation mechanisms of initiator versus executioner caspases present additional opportunities for specific targeting. Initiator caspases exist as monomers that dimerize upon activation, often through recruitment to activation platforms such as the Death-Inducing Signaling Complex (DISC) for caspase-8 or the apoptosome for caspase-9 [35] [5]. This induced proximity model contrasts with executioner caspases, which exist as dimers in their zymogen form and require interdomain cleavage to achieve full catalytic competence [35].
The crystal structure of caspase-7 illustrates the canonical executioner caspase fold: a head-to-tail dimer with each monomer consisting of large (~20 kDa) and small (~10 kDa) subunits [35]. The active site contains a conserved pentapeptide motif QACXG, with the cysteine residue serving as the catalytic nucleophile [26]. These structural features create distinct conformational landscapes that can be exploited for specific probe design.
Figure 1: Hierarchical Caspase Activation Cascade. Initiator caspases (e.g., caspase-8) undergo dimerization at activation platforms, while executioner caspases (e.g., caspase-3) require proteolytic cleavage.
Quantitative Profiling of Caspase Substrate Specificity
Proteomic Approaches to Substrate Identification
Modern proteomic technologies, particularly N-terminomics, have enabled global identification of native caspase substrates in complex biological systems [35] [37]. The subtiligase-based N-terminomics methodology allows selective enrichment and identification of protein N-termini generated by caspase cleavage [37]. This approach involves labeling newly generated N-terminal with biotin tags following caspase activation, affinity purification using avidin beads, tryptic digestion, and identification by LC-MS/MS [37].
Application of these methods has revealed that individual caspases cleave hundreds to thousands of cellular substrates, with striking differences in catalytic efficiency (kcat/KM) spanning over 500-fold for each caspase [37]. Quantitative mass spectrometry studies demonstrate virtually no correlation in catalytic efficiencies among caspases-2, -3, -6, -7, and -8 when comparing common substrates, highlighting their specialized cellular functions and the importance of quantitative profiling for probe design [37].
Table 2: Caspase Substrate Preference Motifs from Proteomic Studies
| Caspase | Optimal Peptide Motif | Protein Substrate Consensus | Catalytic Efficiency Range (kcat/KM) |
|---|---|---|---|
| Caspase-1 | WEHD | YVHD/FESD | 5.0×10² - 1.2×10⁵ M⁻¹·s⁻¹ |
| Caspase-2 | VDVAD | XDEVD | >500-fold variation |
| Caspase-3 | DEVD | DEVD | 10⁴ - 10⁶ M⁻¹·s⁻¹ |
| Caspase-6 | VEID | VEVD | >500-fold variation |
| Caspase-7 | DEVD | DEVD | Similar to caspase-3 |
| Caspase-8 | LETD | XEXD | >500-fold variation |
Kinetic Characterization of Natural Substrates
Quantitative MS-based enzymology has transformed our understanding of caspase substrate hierarchies. By measuring catalytic efficiencies for hundreds of natural protein substrates within cell extracts, researchers have established that cleavage rates are dominated more by local primary sequence than tertiary protein structure [37]. This critical insight validates the use of peptide-based probes for monitoring caspase activity, as linear recognition sequences largely determine cleavage efficiency.
Studies comparing protein substrates with corresponding synthetic peptides have found similar catalytic efficiencies, confirming the primacy of local sequence context [37]. For example, caspase-2 cleaves substrates with a consensus motif of XDEVD, remarkably similar to the executioner caspase-3 motif despite its classification as an initiator caspase [37]. These findings enable rational design of optimized substrates and probes based on primary sequence information.
Design Strategies for Caspase-Specific Substrates
Peptide-Based Substrate Design
The foundation of caspase substrate design involves four-residue peptide sequences (P4-P1) based on natural cleavage preferences, coupled to various reporting systems [35] [36]. The classic approach utilizes fluorogenic substrates such as Ac-DEVD-AMC for caspase-3, where cleavage releases the fluorescent 7-amino-4-methylcoumarin (AMC) group [26]. Similarly, Ac-IETD-AMC and Ac-LEHD-AMC target caspase-8 and -9, respectively [26].
Recent advances have focused on improving specificity between highly homologous caspases, particularly distinguishing caspase-3 from caspase-7. While both executioner caspases share a preference for DEVD sequences, subtle differences in their S2 and S3 pockets can be exploited [38]. Incorporation of non-natural amino acids and strategic substitutions have yielded substrates with significantly improved selectivity profiles.
Structural Optimization Approaches
Several strategies have emerged to enhance caspase substrate specificity and utility:
Prime-Side Modifications: Extending recognition elements to the P1' position and beyond can improve selectivity, as demonstrated by the development of caspase-3-specific probes containing ketoester groups at the prime side that enhance selectivity over caspase-7 [38].
Non-Natural Amino Acid Incorporation: Substitution with amino acid analogs such as 3-palmitoyl-lysine (3Pal) at P5 and pentafluorophenylalanine at P3 in the ATS010-KE inhibitor significantly improved caspase-3 selectivity [38].
Conformational Constraint: Cyclic peptides and peptidomimetics that pre-organize the recognition sequence can enhance binding affinity and specificity by reducing the entropic penalty of binding.
These design principles enable researchers to create substrates with tailored specificity for individual caspases, addressing the challenge of overlapping substrate preferences within the caspase family.
Activity-Based Probe Design for Caspase Monitoring
Fundamentals of Activity-Based Profiling
Activity-based probes (ABPs) represent powerful tools for monitoring caspase activation in biological systems. These reagents typically consist of three key elements: a caspase recognition sequence, an electrophilic warhead that covalently modifies the catalytic cysteine, and a detection tag (fluorescent, biotin, or radiolabel) [38]. Unlike substrate-based probes, ABPs form irreversible covalent complexes with their target enzymes, enabling enrichment and direct identification of active caspases.
The electrophilic warhead is a critical determinant of ABP specificity and reactivity. Common warheads include fluoromethyl ketones (FMK), acyloxymethyl ketones (AOMK), and more recently, ketoester (KE) groups that target the prime side of the caspase active site [38]. ABPs such as Ac-ATS010-KE demonstrate dramatically improved caspase-3 selectivity (9-fold over caspase-7) compared to earlier designs [38].
Advanced Probe Designs and Applications
Recent innovations in caspase ABP design have addressed several challenges:
Caspase-3-Selective PET Tracers: Development of [¹⁸F]MICA-316 for apoptosis imaging represents a significant advance in caspase detection, though clinical translation remains challenging due to limited tumor uptake [38].
Multimodal Probes: Incorporation of both fluorescent and affinity tags enables parallel visualization and enrichment of active caspases from complex samples.
Cell-Permeable Designs: Structural modifications such as per-methylation can enhance cell permeability while maintaining target engagement, though this approach requires careful optimization as over-methylation can impair function [38].
Figure 2: Activity-Based Probe Architecture. Caspase ABPs contain three essential elements: recognition sequence, electrophilic warhead, and detection tag.
Experimental Protocols for Caspase Profiling
N-Terminomics for Global Substrate Identification
The subtiligase-based N-terminomics protocol provides a comprehensive method for identifying native caspase substrates [37]:
Sample Preparation: Prepare cell extracts from Jurkat cells or other relevant cell lines as a source of physiological caspase substrates.
Caspase Treatment: Incubate extracts with active recombinant caspase (e.g., caspase-2 or -6) at physiological concentrations for defined timepoints.
N-Terminal Labeling: Employ subtiligase to biotinylate newly generated N-terminal following caspase cleavage, selectively labeling cleavage products over naturally blocked N-terminal.
Affinity Enrichment: Capture biotinylated peptides using streptavidin beads, followed by extensive washing to remove non-specific binders.
TEV Protease Release: Cleave captured peptides with TEV protease to release them from beads while retaining an aminobutyric acid (Abu) tag as a cleavage signature.
LC-MS/MS Analysis: Identify released peptides by liquid chromatography tandem mass spectrometry, with database searching to map cleavage sites.
Quantitative Kinetics: Perform time-course experiments with multiple caspase concentrations to determine catalytic efficiencies (kcat/KM) for individual substrates.
This methodology enables identification of hundreds to thousands of cleavage events with quantitative kinetic information, providing a comprehensive view of caspase substrate preferences in near-physiological conditions [37].
Characterization of Probe Binding Kinetics
Rigorous kinetic characterization is essential for validating caspase probe specificity:
Progress Curve Analysis: Monitor reaction progress over time using fluorogenic substrates to determine initial velocities at varying substrate concentrations.
Inhibition Constants: For ABPs, measure the apparent second-order rate constant (kinact/KI) by pre-incubating caspase with varying probe concentrations before adding substrate.
Selectivity Profiling: Test probes against a panel of recombinant caspases to determine specificity ratios, with particular attention to discriminating between highly homologous caspases (e.g., caspase-3 vs. -7).
Cellular Validation: Confirm probe functionality in cell-based models of apoptosis, comparing results with established caspase activation markers such as PARP cleavage.
These protocols ensure comprehensive characterization of novel caspase substrates and probes, validating their utility for biological research and therapeutic development.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Caspase Studies
| Reagent Category | Specific Examples | Primary Application | Considerations |
|---|---|---|---|
| Broad-Spectrum Inhibitors | Z-VAD-FMK, Q-VD-OPh | Pan-caspase inhibition; apoptosis confirmation | Q-VD-OPh shows reduced toxicity at high concentrations |
| Selective Peptide Inhibitors | Ac-DEVD-CHO (caspase-3), Ac-IETD-CHO (caspase-8) | Specific caspase inhibition; mechanistic studies | Aldehyde-based inhibitors have limited cell permeability |
| Activity-Based Probes | Ac-ATS010-KE, [¹⁸F]MICA-316 | Direct detection of active caspases; molecular imaging | Warhead chemistry critically determines specificity |
| Fluorogenic Substrates | Ac-DEVD-AMC (caspase-3/7), Ac-WEHD-AMC (caspase-1) | Continuous activity monitoring; high-throughput screening | Verify specificity against related caspases |
| Antibody-Based Reagents | Anti-cleaved caspase-3, anti-PARP | Western blot detection of caspase activation | Provides indirect evidence of activity |
| Expression Systems | Recombinant caspases, caspase-GFP fusions | Biochemical studies; subcellular localization | Proper folding and activity validation required |
The design of caspase-specific substrates and activity probes continues to evolve with advancing understanding of caspase structure and function. Emerging strategies include the development of conformation-specific inhibitors that selectively target pathological caspase activation states while preserving physiological functions [39], and microenvironment-responsive delivery systems that exploit features of the apoptotic cell environment [39]. The integration of structural biology insights with quantitative proteomic profiling promises to yield increasingly specific tools for dissecting caspase functions in complex biological systems, ultimately supporting therapeutic development for conditions ranging from cancer to neurodegenerative diseases.
The ability to induce programmed cell death in cancerous cells represents a cornerstone of oncology research. Caspases, a family of cysteine-aspartic proteases, are central executioners of apoptosis and other forms of programmed cell death, making them attractive candidates for protein-based cancer therapies [5] [9]. However, their delivery into specific cellular compartments remains a significant challenge due to their fragile tertiary structure, large molecular size, and poor membrane permeability [40]. Exogenous caspase delivery systems aim to overcome these barriers by packaging active caspase enzymes into nanoscale carriers that facilitate intracellular delivery, thereby directly triggering apoptotic pathways in target cells [25]. This approach is particularly valuable for addressing cancers that have developed resistance to conventional small-molecule drugs through mutations in apoptotic signaling pathways [25].
The structural and functional differences between initiator and executioner caspases fundamentally influence their therapeutic application. Initiator caspases (including caspase-2, -8, -9, and -10) typically contain long prodomains with protein-protein interaction motifs such as death effector domains (DEDs) or caspase activation and recruitment domains (CARDs), which enable their assembly into activating complexes like the death-inducing signaling complex (DISC) or apoptosome [41] [9]. In contrast, executioner caspases (including caspase-3, -6, and -7) possess short prodomains and exist primarily as dimers that require proteolytic activation by initiator caspases [41]. These structural distinctions dictate their placement within apoptotic cascades and determine the strategic approach for their therapeutic delivery.
Caspase Biology and Structural Foundations
Classification and Molecular Structures
Caspases are synthesized as inactive zymogens (pro-caspases) that require proteolytic processing for activation. The activation mechanism differs significantly between initiator and executioner caspases, with initiator caspases undergoing induced-proximity-mediated dimerization while executioner caspases require cleavage by upstream caspases [42] [9]. The active form of most caspases is a heterotetramer composed of two large and two small subunits that form two active sites positioned at opposite ends of the molecule [41].
Table 1: Functional Classification of Mammalian Caspases
| Role in Cell Death | Caspase Type | Members | Activation Complex | Primary Substrates |
|---|---|---|---|---|
| Apoptosis | Initiator | Caspase-2, -8, -9, -10 | DISC (caspase-8/10), Apoptosome (caspase-9) | Executioner caspases, BID, various proteins |
| Executioner | Caspase-3, -6, -7 | Cleavage by initiator caspases | PARP, lamin, ICAD, >600 cellular targets | |
| Pyroptosis | Inflammatory | Caspase-1, -4, -5, -11 | Inflammasome | Pro-IL-1β, Pro-IL-18, gasdermin D |
| Differentiation | Other | Caspase-14 | Not applicable | Filaggrin, other structural proteins |
The substrate specificity of caspases is largely determined by residues in the active site that form distinct pockets (S4-S1) accommodating the substrate amino acids (P4-P1) [41]. While caspases generally recognize tetrapeptide sequences terminating in aspartic acid, individual caspases exhibit distinct preferences. For example, caspase-3 and -8 both recognize the sequence DEVD but with different catalytic efficiencies, while caspase-9 shows preference for LEHD [13]. These specificity profiles have important implications for designing delivery systems that maintain caspase activity and for understanding potential off-target effects.
Structural Differences Between Initiator and Executioner Caspases
The fundamental structural differences between initiator and executioner caspases dictate their activation mechanisms and functional roles:
Initiator Caspases (caspase-8, -9, -10):
- Contain long N-terminal prodomains with death folds (CARD or DED domains) that facilitate recruitment to activation platforms [41] [9]
- Exist primarily as monomers in their inactive state and require dimerization for activation [42]
- Undergo autoproteolysis following dimerization, but catalytic activity can precede complete processing [42]
- Examples: Caspase-8 contains two N-terminal DED domains that mediate recruitment to the DISC, while caspase-9 contains a CARD domain that facilitates interaction with Apaf-1 in the apoptosome [9]
Executioner Caspases (caspase-3, -6, -7):
- Feature short prodomains without extensive protein interaction motifs [41]
- Reside as constitutive dimers even in their zymogen forms [41]
- Require proteolytic cleavage between the large and small subunits by initiator caspases for full activation [9]
- Examples: Caspase-3 and -7 share similar tetrapeptide specificities but have non-overlapping substrate pools and cellular functions [13]
These structural distinctions are crucial when designing delivery systems, as initiator caspases may require conditions that facilitate proper dimerization, while executioner caspases need protection from premature activation during the encapsulation process.
Diagram 1: Caspase Activation Pathways in Apoptosis. The intrinsic pathway responds to cellular stress, while the extrinsic pathway is triggered by death ligands. Both converge on executioner caspase activation.
Nanocarrier Platforms for Caspase Delivery
Design Considerations for Intracellular Protein Delivery
Effective caspase delivery systems must overcome multiple biological barriers, including stability in circulation, cellular uptake, endosomal escape, and intracellular release while maintaining caspase activity [40]. Key design parameters include:
- Stability: Protection from proteolytic degradation and maintenance of structural integrity during circulation [40]
- Cellular Uptake: Efficient internalization via endocytosis or other mechanisms [25]
- Endosomal Escape: Capacity to disrupt endosomal membranes before lysosomal degradation [40]
- Controlled Release: Intracellular activation mechanisms (e.g., redox-responsive, pH-sensitive) for precise cargo release [25] [43]
- Bioactivity Preservation: Maintenance of caspase conformation and enzymatic activity throughout delivery process [25]
Nanocarrier Platforms and Their Applications
Various nanocarrier systems have been developed to address these challenges, each with distinct advantages for caspase delivery:
Table 2: Nanocarrier Platforms for Exogenous Caspase Delivery
| Nanocarrier Type | Composition | Loading Method | Caspases Delivered | Key Features |
|---|---|---|---|---|
| Polymeric Nanogels | PEG-PDS copolymers | Thiol-disulfide exchange with solvent-exposed cysteines | Caspase-3, -6, -7, -8, -9 [25] | Redox-responsive release, ~10 nm size, high encapsulation efficiency |
| Lipid-Based Systems | Lipid-calcium carbonate nanoparticles | Electrostatic/hydrophobic interactions | Caspase-8 (implicit) [40] [44] | Co-delivery with small molecules, mitochondrial targeting via cardiolipin |
| Polymeric Nanoparticles | PEG-PLGA | Physical encapsulation | Activity-based probes for caspase-3 [45] | Controlled release kinetics, FDA-approved materials |
| Inorganic Nanoparticles | Mesoporous silica | Physical adsorption/covalent conjugation | Caspase-3, -9 [40] | High surface area, tunable pore size, functionalizable surface |
| Polymer-Protein Complexes | Triblock copolymers | Covalent conjugation/hydrophobic interaction | Caspase-3/7 analogues (RNase A) [40] | Dual cargo loading, synergistic therapy |
Redox-Responsive Nanogels: A Case Study
Redox-responsive polymeric nanogels represent one of the most extensively characterized platforms for caspase delivery. These systems typically employ amphiphilic random copolymers composed of polyethylene glycol (PEG) and pyridyl disulfide (PDS) units that self-assemble in aqueous media [25]. The delivery process involves:
- Formulation: Caspases are encapsulated via reaction of solvent-exposed cysteine residues with polymer PDS units at a typical caspase/polymer ratio of 1:25 [25]
- Stabilization: Cross-linking with reducing agents (e.g., DTT) stabilizes the nanostructure, yielding monodisperse particles of approximately 10 nm [25]
- Cellular Uptake: Caspase-loaded nanogels (casp-NGs) are internalized via endocytosis in serum-containing cell cultures [25]
- Intracellular Release: Glutathione (GSH) reduces disulfide cross-links, releasing native caspase into the cytoplasm [25]
This platform has successfully delivered caspase-3, -6, -7, -8, and -9, with studies revealing unexpected findings about their relative potency. Contrary to expectations that caspase-3 would be most effective due to its central role in apoptosis and high catalytic efficiency, caspase-7 and caspase-9 demonstrated superior efficacy in inducing apoptotic cell death in certain cellular contexts [25].
Diagram 2: Nanogel-Mediated Caspase Delivery Workflow. Redox-responsive nanogels facilitate caspase encapsulation, cellular uptake, and intracellular release via glutathione-mediated reduction.
Experimental Protocols and Methodologies
Preparation of Caspase-Loaded Nanogels
Materials:
- PEG-PDS copolymer (synthesized as previously described [25])
- Recombinant active caspase (expressed and purified using standard methods)
- Dithiothreitol (DTT) for cross-linking
- Purification columns (e.g., desalting columns)
Protocol:
- Polymer Preparation: Dissolve PEG-PDS copolymer in aqueous buffer at 1-5 mg/mL concentration
- Caspase Encapsulation: Add caspase solution to polymer at 1:25 caspase/polymer molar ratio, incubate for 2 hours at 4°C with gentle agitation
- Cross-linking: Add DTT to final concentration of 5 mM, incubate for 30 minutes at room temperature to stabilize nanogel structure
- Purification: Remove unencapsulated caspase and excess DTT using desalting columns or dialysis
- Characterization: Determine encapsulation efficiency by measuring pyridine-2-thiol release at 343 nm, assess particle size by dynamic light scattering (target ~10 nm), confirm monodispersity [25]
In Vitro Assessment of Caspase Delivery and Activity
Cell Culture and Treatment:
- Culture appropriate cell lines (e.g., PC3, HCT116, MCF-7) in standard conditions
- Treat cells with caspase-loaded nanogels at optimized concentrations in serum-containing media
- Incubate for 4-24 hours depending on experimental endpoint
Apoptosis Assessment Methods:
- Activity-Based Probing: Use caspase-selective activity-based probes (ABPs) like CS1 for caspase-3 to detect active enzyme [45]
- Western Blot Analysis: Detect caspase activation by monitoring cleavage fragments (e.g., p17/p19 for caspase-3) and substrate cleavage (e.g., PARP-1)
- Cell Viability Assays: Measure apoptotic cell death using MTT, Annexin V/propidium iodide staining, or similar methods
- Substrate Cleavage Profiling: Use global proteomic methods to identify specific cleavage events characteristic of different caspases [13]
Validation of Caspase Specificity and Function
Specificity Controls:
- Selective ABPs: Employ hybrid combinatorial substrate library (HyCoSuL)-derived probes like CS1 that distinguish caspase-3 from caspase-7 [45]
- Knockout Cell Lines: Utilize caspase-deficient cells (e.g., MCF-7 lacking caspase-3) to confirm specificity [45]
- Inhibitor Studies: Include pan-caspase inhibitors (e.g., Z-VAD-FMK) or specific inhibitors to confirm caspase-dependent effects
Functional Assessment:
- Mitochondrial Membrane Potential: Monitor ΔΨm collapse using JC-1 or TMRE dyes
- Morphological Analysis: Assess apoptotic morphology (membrane blebbing, chromatin condensation)
- Caspase Cascade Activation: Measure downstream caspase activation to confirm engagement of apoptotic machinery
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Exogenous Caspase Delivery Research
| Reagent Category | Specific Examples | Function/Application | Key Characteristics |
|---|---|---|---|
| Nanocarrier Materials | PEG-PDS copolymers [25] | Redox-responsive nanogel formation | Amphiphilic, thiol-reactive, self-assembling |
| PEG-PLGA [45] | Polymeric nanoparticle matrix | Biocompatible, controlled release, FDA-approved | |
| Lipid-calcium carbonate hybrids [40] | Co-delivery systems | pH-responsive, high loading capacity | |
| Caspase Detection Tools | CS1 activity-based probe [45] | Selective caspase-3 labeling | HyCoSuL-optimized sequence, AOMK warhead, biotin tag |
| DEVD-AMC substrate [45] | Caspase-3/7 activity measurement | Fluorogenic, continuous monitoring capability | |
| Analytical Reagents | Streptavidin-HRP [45] | ABP detection in Western blot | High sensitivity, compatible with standard protocols |
| Cell Culture Reagents | Entinostat + rTRAIL [45] | Apoptosis induction in PC3 cells | Synergistic combination for caspase activation |
| Camptothecin [45] | Intrinsic pathway activation in MCF-7 cells | Topoisomerase inhibitor, caspase-7 activation | |
| Molecular Biology Tools | PARP-1 antibodies [45] | Apoptosis marker detection | Early cleavage target, indicates executioner caspase activity |
Comparative Efficacy and Therapeutic Implications
Relative Potency of Different Caspases
Systematic comparison of caspase efficacy following exogenous delivery has yielded surprising insights. While caspase-3 is often considered the primary executioner caspase due to its vast substrate pool and high catalytic efficiency, studies using nanogel delivery revealed that caspase-7 and caspase-9 demonstrated superior efficacy in inducing apoptotic cell death in certain cellular contexts [25]. This finding suggests that the optimal caspase for therapeutic application may depend on the specific apoptotic blockades present in different cancers.
The differential efficacy appears to correlate with levels of pro-survival factors that both directly and indirectly impact the introduced caspase, highlighting the importance of matching the delivered caspase to the specific apoptotic dysregulation in target cells [25]. For example, caspase-9 may bypass upstream blocks in the intrinsic pathway, while caspase-7 may overcome inhibition that specifically targets caspase-3.
Synergistic Approaches: Co-delivery Systems
Combining caspases with other therapeutic agents represents a promising strategy to enhance efficacy and address therapeutic resistance. Several co-delivery approaches have been developed:
- Caspase-Small Molecule Combinations: Co-delivery of paclitaxel with caspase-3 using nanocapsules that utilize electrostatic interactions for protein loading and physical encapsulation for small molecules [40]
- Dual Protein Delivery: Integration of RNase A with doxorubicin via covalent conjugation in dual-cargo nanocapsules [40]
- Gene-Protein Combinations: CRISPR-Cas9 ribonucleoprotein with photosensitizers for combination therapy [40]
These approaches demonstrate the potential of nanocarrier systems to unify the pharmacokinetics of multiple therapeutic agents with distinct physicochemical properties, enhancing tumor accumulation while minimizing systemic toxicity.
Exogenous caspase delivery via nanocarrier systems represents a promising therapeutic strategy that directly targets the apoptotic machinery in cancer cells. The structural differences between initiator and executioner caspases significantly influence delivery system design and therapeutic application. Redox-responsive nanogels and other advanced nanocarriers have demonstrated efficient intracellular delivery of functional caspases, with surprising findings regarding the relative efficacy of different caspase family members.
Future research directions should focus on optimizing carrier systems for specific caspase types based on their structural characteristics, developing more sophisticated co-delivery approaches that address multiple nodes in apoptotic signaling networks, and advancing understanding of how cancer-specific mutations in cell death pathways influence therapeutic response to exogenous caspase delivery. As these technologies mature, they hold significant potential for addressing the challenge of apoptotic resistance in advanced cancers, potentially offering new treatment options for patients with limited therapeutic alternatives.
Caspases, a family of cysteine-dependent aspartate-specific proteases, are pivotal regulators of programmed cell death and inflammation. Their activation cascades drive critical cellular processes, and dysregulation underpins numerous diseases, making them attractive therapeutic targets. The structural distinction between initiator caspases (caspase-2, -8, -9, -10) and executioner caspases (caspase-3, -6, -7) is fundamental to their function and, consequently, to therapeutic strategies aimed at their modulation [18]. Initiator caspases possess long prodomains containing death folds such as CARD (caspase activation and recruitment domain) or DED (death effector domain) that facilitate recruitment to and activation within large signaling complexes like the apoptosome or DISC (death-inducing signaling complex) [22] [46]. They exist as inactive monomers that are activated by induced proximity and dimerization [22]. In contrast, executioner caspases have short prodomains and pre-exist as inactive dimers, becoming activated through cleavage by initiator caspases [22] [6]. This structural dichotomy defines their roles in apoptosis and informs the development of targeted therapies for cancer, inflammatory, and neurodegenerative diseases [46] [47] [18].
Molecular Mechanisms of Caspase Activation
Structural Basis of Initiator and Executioner Caspase Activation
The activation mechanisms of initiator and executioner caspases are fundamentally different, governed by their distinct structural compositions.
Initiator Caspase Activation: Initiator caspases (caspase-8, -9, -10) are single-polypeptide chains featuring long N-terminal prodomains. These prodomains contain protein-protein interaction motifs, either CARD or DED, that are essential for their recruitment to specific activation platforms [22] [18]. For example, caspase-8 is recruited to the DISC via DED interactions in the extrinsic apoptotic pathway, while caspase-9 is recruited to the apoptosome via CARD interactions in the intrinsic pathway [46]. This recruitment brings multiple caspase monomers into close proximity, facilitating dimerization and auto-activation through the "induced proximity" model [22]. Cleavage between the large and small subunits then occurs, which stabilizes the active dimer but is not strictly required for initial activation [22]. An exception is caspase-9, where cleavage does not stabilize the dimer but alters its regulatory properties [22].
Executioner Caspase Activation: Executioner caspases (caspase-3, -6, -7) possess very short prodomains and exist as dormant dimers in healthy cells [22] [6]. These zymogens contain the potential to form two active sites, but these sites are structurally constrained. Activation occurs when initiator caspases cleave at specific aspartic acid residues located between the large and small subunits [22] [6]. This cleavage triggers a conformational change that realigns the active site loops, forming the mature and maximally functional protease capable of cleaving hundreds of cellular substrates to orchestrate apoptotic cell death [22].
Figure 1: Molecular Pathways of Caspase Activation. Initiator caspases are activated by dimerization at multiprotein complexes, while executioner caspases are activated by proteolytic cleavage. Positive feedback loops amplify the death signal.
Non-Apoptotic Functions and Survival Signaling
Beyond their classical role in cell death, caspases participate in diverse non-apoptotic processes. Executioner caspases can be activated at sub-lethal levels to drive cellular processes including cell differentiation, proliferation, and immune modulation [48] [6]. In differentiation, caspase activity remodels cells during erythropoiesis, lens cell development, and skeletal muscle differentiation without triggering death [48]. Furthermore, cells can survive transient executioner caspase activation through a process called anastasis (Greek for "rising to life") [6]. Survival in this context can have dual consequences: promoting tissue regeneration or, concerningly, potentially enabling cancer cells to recover, acquire mutations, and exhibit increased aggressiveness [6] [49]. Proteomic studies reveal that under non-lethal stress, the proteolytic landscape is entirely shaped by caspase-3 and -7 activity, cleaving a specific subset of proteins distinct from the broad-scale cleavage observed in apoptosis [49]. This illustrates that cellular fate is determined by the magnitude, duration, and context of caspase activation rather than its mere occurrence.
Caspase-Targeted Therapeutic Agents
Therapeutic strategies targeting caspases fall into two main categories: inhibitors to block excessive cell death in degenerative and inflammatory diseases, and activators to induce cell death in pathologies like cancer.
Caspase Inhibitors in Clinical Development
Caspase inhibitors have been investigated for a range of conditions where unchecked cell death or inflammation drives pathology. The table below summarizes key caspase inhibitors that have progressed to clinical trials.
Table 1: Selected Caspase Inhibitors in Clinical Development
| Inhibitor Name | Target Caspase(s) | Class/Type | Therapeutic Indication | Clinical Status | Key Findings/Challenges |
|---|---|---|---|---|---|
| VX-740 (Pralnacasan) | Caspase-1 | Peptidomimetic, Irreversible | Rheumatoid Arthritis, Osteoarthritis | Trials Terminated | Significant efficacy in trials; development halted due to liver toxicity in animal models [47]. |
| VX-765 (Belnacasan) | Caspase-1 | Peptidomimetic, Reversible | Epilepsy, Psoriasis | Trials Terminated | Potent anti-inflammatory effect; clinical trials terminated due to liver toxicity concerns [47]. |
| IDN-6556 (Emricasan) | Pan-caspase | Peptidomimetic, Irreversible | Liver Diseases (NASH, HCV) | Trials Terminated | Efficacy in preclinical and early clinical studies; development terminated after phase II/III due to undisclosed reasons [47]. |
| Q-VD-OPh | Pan-caspase | Peptide-based (FMK), Irreversible | Neurodegeneration, Sepsis (Preclinical) | Preclinical | Broad-spectrum inhibitor with low cellular toxicity at high concentrations; used extensively in research [47]. |
| NSAIDs (e.g., Ibuprofen) | Multiple (Caspase-4, etc.) | Non-peptide small molecule | General Anti-inflammation | FDA-Approved (for other indications) | Identified as a COX-independent caspase inhibitor at physiologic concentrations; represents drug repurposing opportunity [50]. |
The development of caspase inhibitors has faced significant challenges, primarily due to inadequate efficacy, poor target specificity, and adverse side effects, particularly hepatotoxicity [47]. The failure of several promising candidates highlights the complexity of caspase biology, including their multifaceted roles in non-apoptotic processes and the activation of compensatory cell death pathways upon caspase inhibition [47].
Caspase Activators and Delivery Strategies
Inducing apoptosis in cancer cells by activating caspases is a compelling therapeutic strategy. A direct approach involves the exogenous introduction of active caspases into cells. Research using redox-responsive polymeric nanogels (NGs) has demonstrated the feasibility of delivering active caspase proteins intracellularly [25]. Contrary to expectations that caspase-3 would be most effective due to its high catalytic efficiency, studies found that caspase-7 and caspase-9 were the most effective at inducing apoptotic cell death in tested cancer cell lines [25]. This suggests that the efficacy of a caspase is context-dependent, influenced by the unique repertoire of pro-survival and anti-apoptotic proteins in different cancers. Gene therapies delivering constitutively active caspase genes (e.g., caspase-3, -9) have also been explored but often require additional apoptosis inducers or tissue-specific promoters for efficacy [25].
Table 2: Efficacy of Exogenously Delivered Caspases in Inducing Apoptosis
| Caspase Delivered | Role/Type | Relative Efficacy in Apoptosis Induction | Key Notes |
|---|---|---|---|
| Caspase-3 | Executioner | Moderate | High catalytic efficiency but efficacy limited by cellular regulatory blocks [25]. |
| Caspase-6 | Executioner | Lower | Canonical executioner, but its removal has minimal impact on apoptosis [25]. |
| Caspase-7 | Executioner | High | Functionally distinct from caspase-3 despite similar cleavage motif; effective in exogenous delivery [25]. |
| Caspase-8 | Initiator (Extrinsic) | Variable | Activated by DED-mediated complex formation; delivery of a soluble, less-aggregating form (ΔDED) is feasible [25]. |
| Caspase-9 | Initiator (Intrinsic) | High | Activated by CARD-mediated apoptosome formation; effective in exogenous delivery studies [25]. |
Experimental Approaches for Caspase Research
Protocol: Exogenous Caspase Delivery via Nanogels
The following detailed methodology is adapted from studies that successfully introduced active caspases into cells using redox-responsive nanogels [25].
Principle: Cysteine residues on the caspase surface form disulfide bonds with pyridyl disulfide (PDS) groups on an amphiphilic PEG–PDS polymer, enabling encapsulation. Upon cellular uptake and exposure to high intracellular glutathione (GSH), the nanogel is reduced, releasing the active caspase.
Materials:
- Recombinant Caspase: Active, full-length wild-type or constitutively active mutant (e.g., caspase-3, -7, -9).
- PEG–PDS Polymer: Amphiphilic random copolymer of polyethylene glycol (PEG) and pyridyl disulfide (PDS).
- Cross-linker: Dithiothreitol (DTT).
- Cell Culture Reagents: Appropriate cell line (e.g., cancer cell lines like HCT116), serum-containing medium.
Procedure:
- Polymer Assembly and Caspase Encapsulation: Incubate the PEG–PDS polymer with the recombinant caspase at a optimized weight ratio (e.g., 1:25 caspase/polymer) in an aqueous buffer. The cysteine residues on the caspase will react with the PDS units on the polymer, leading to encapsulation during self-assembly.
- Nanogel Cross-linking: Add DTT to the caspase-polymer mixture to cross-link and stabilize the nanomaterial. Cross-linking can be monitored by measuring the release of pyridine-2-thiol via its absorbance at 343 nm.
- Characterization: Determine the size and dispersity of the resulting caspase-loaded nanogels (casp-NGs) using dynamic light scattering. Monodisperse particles of ~10 nm are typically obtained. Measure encapsulation efficiency.
- Cell Treatment: Add the purified casp-NGs directly to serum-containing cell cultures. The nanogels are internalized by cells.
- Intracellular Release: Inside the cell, following endosomal escape, the high concentration of intracellular GSH reduces the disulfide cross-links, degrading the nanogel and releasing the native, active caspase.
- Viability Assessment: Monitor apoptotic cell death using standard assays (e.g., flow cytometry with Annexin V/PI staining, caspase activity assays, or clonogenic survival assays) 24-72 hours post-treatment.
Protocol: Profiling Caspase-Mediated Proteolysis
To map the proteolytic events downstream of caspase activation, quantitative proteomics can be employed [49] [13].
Principle: Mass spectrometry (MS)-based proteomics identifies and quantifies protein fragments generated by cleavage. Comparing stressed cells (non-lethal and apoptotic) to caspase-3/-7 double knock-out (DKO) cells reveals the caspase-dependent proteolytic landscape.
Materials:
- Cell Lines: Wild-type and CASP3/CASP7 DKO isogenic cell lines (e.g., HCT116).
- Inducers: Apoptotic inducer (e.g., high-dose cisplatin) and non-lethal stressor (e.g., low-dose cisplatin).
- Lysis Buffer: MS-compatible denaturing buffer (e.g., containing SDS or urea).
- Protease Inhibitors: Include where appropriate to capture steady-state cleavage.
- Trypsin/Lys-C: for protein digestion.
- Tandem Mass Spectrometer: e.g., Q-Exactive series.
- Database: e.g., The Cleavage Profiling database (https://cleaved-apoptosis.unil.ch).
Procedure:
- Cell Treatment and Lysis: Treat wild-type and DKO cells with either a non-lethal stress, an apoptotic stress, or a vehicle control. Harvest cells at appropriate time points and lyse in denaturing buffer.
- Protein Digestion and Peptide Preparation: Digest the extracted proteins with trypsin/Lys-C. For N-terminal peptide enrichment, use the TAILS (Terminal Amine Isotopic Labeling of Substrates) method or similar, which involves blocking primary amines, digesting proteins, and enriching for internal (neo-N-terminal) peptides.
- Mass Spectrometry Analysis: Analyze the peptides by liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS).
- Data Processing: Use search engines (e.g., MaxQuant) against a human protein database to identify peptides and proteins. Crucially, disable the "specific protease" setting to allow identification of non-tryptic cleavage events.
- Bioinformatic Analysis:
- Identify proteins with significant changes in peptide abundance that indicate cleavage.
- Map the cleavage sites and determine the sequence specificity.
- Compare the proteolytic profiles between wild-type and DKO cells to assign caspase-3/7 dependence.
- Validate key cleavage events by western blot.
Figure 2: Experimental Workflow for Profiling Caspase-Dependent Proteolysis. Quantitative proteomics comparing wild-type and caspase-3/7 double knock-out (DKO) cells under stress identifies the specific proteolytic events mediated by these executioner caspases.
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents for Caspase-Targeted Therapeutic Research
| Reagent / Tool | Function/Description | Example Use Case | Key Considerations |
|---|---|---|---|
| Caspase Knock-out Cell Lines | Isogenic cell lines (e.g., DKO for caspase-3/-7) to define specific caspase functions. | Determining caspase-dependent proteolytic events and phenotypes in stress responses [49]. | Confirms on-target effects and identifies non-apoptotic functions. |
| Redox-Responsive Nanogels (PEG-PDS) | Intracellular protein delivery vehicle for exogenous caspase introduction. | Directly testing the apoptotic potential of specific caspases in different cancer backgrounds [25]. | Allows delivery of active enzyme, bypassing upstream regulatory blocks. |
| Broad-Spectrum Caspase Inhibitor (Q-VD-OPh) | Irreversible, cell-permeable pan-caspase inhibitor with low toxicity. | Determining if a cellular phenotype is caspase-dependent; neuroprotection studies [47]. | Preferred over Z-VAD-FMK due to higher specificity and lower cellular toxicity. |
| Selective Peptide Substrates/Inhibitors | Fluorogenic substrates (e.g., DEVD-AFC for caspase-3/7) or aldehyde inhibitors (Ac-DEVD-CHO). | Measuring enzymatic activity of specific caspases in cell lysates or purified systems [13]. | Beware of cross-reactivity; e.g., DEVD is cleaved by both caspase-3 and -7. |
| Quantitative Proteomics Platform (LC-MS/MS) | Global, unbiased identification of protein cleavage sites and neo-N-termini. | Mapping the proteolytic landscape in apoptosis and non-lethal stress [49] [13]. | Requires specialized equipment and bioinformatic expertise for data analysis. |
The therapeutic targeting of caspases remains a field of high promise and significant challenge. The structural dichotomy between initiator and executioner caspases provides a rational framework for designing specific drugs, yet the complexity of caspase biology, with its extensive crosstalk, feedback loops, and non-apoptotic functions, demands highly sophisticated approaches [6] [18]. Future success will likely depend on strategies that move beyond broad inhibition or activation. These may include context-specific activation in cancers by leveraging the unique apoptotic blocks in different tumors, highly selective inhibitors that spare caspases involved in homeostatic functions, or combination therapies that modulate caspases in concert with other regulatory pathways [25] [47]. A deeper understanding of the structural and molecular mechanisms of caspase activation and substrate recognition, particularly through the lens of initiator versus executioner caspases, will be fundamental to translating these powerful proteases into effective and safe therapeutics for a wide range of human diseases.
Exploiting Structural Differences for Selective Drug Design
Caspases, a family of cysteine-aspartic proteases, are central regulators of programmed cell death (PCD) and inflammation [9]. These enzymes specifically cleave their substrates after aspartic acid residues and are synthesized as inactive zymogens (pro-caspases) that require proteolytic activation for function [9] [6]. The caspase family is broadly categorized into initiator caspases (caspase-2, -8, -9, -10) and executioner caspases (caspase-3, -6, -7) based on their position in apoptotic signaling cascades, as well as inflammatory caspases (caspase-1, -4, -5, -11) that primarily regulate immune responses [9] [5]. Dysregulation of caspase activity is implicated in a wide spectrum of diseases, including cancer, neurodegenerative disorders, and autoimmune conditions, making them promising targets for therapeutic intervention [1] [9] [5].
The structural biology of caspases reveals fundamental differences between initiator and executioner caspases that dictate their activation mechanisms and functions. Initiator caspases possess long pro-domains containing protein-protein interaction motifs such as death effector domains (DEDs) in caspase-8 and -10 or caspase activation and recruitment domains (CARDs) in caspase-9, which facilitate their recruitment to and activation within large multiprotein complexes like the apoptosome or death-inducing signaling complex (DISC) [51] [9] [5]. In contrast, executioner caspases have short pro-domains and are activated primarily through cleavage by initiator caspases [6]. This review examines the structural distinctions between initiator and executioner caspases and explores how these differences can be exploited for selective drug design in disease contexts.
Structural Biology of Caspases: A Comparative Analysis
Fundamental Architectural Differences
All caspases share a conserved catalytic domain comprised of large (~20 kDa) and small (~10 kDa) subunits, but critically differ in their N-terminal pro-domain architecture and activation mechanisms [6]. The table below summarizes the key structural and functional differences between initiator and executioner caspases.
Table 1: Structural and Functional Classification of Human Caspases
| Category | Caspase | Pro-domain Feature | Activation Complex | Primary Functions |
|---|---|---|---|---|
| Initiator | Caspase-8 | Two DEDs | DISC, FADDosome | Extrinsic apoptosis, necroptosis regulation, pyroptosis [9] [5] |
| Caspase-9 | CARD | Apoptosome | Intrinsic apoptosis [51] [9] | |
| Caspase-10 | Two DEDs | DISC | Extrinsic apoptosis, negatively regulates caspase-8 [9] [5] | |
| Caspase-2 | CARD | PIDDosome | Cell cycle, tumorigenesis, apoptosis [1] [5] | |
| Executioner | Caspase-3 | Short pro-domain | Cleaved by initiator caspases | Apoptosis execution, pyroptosis via GSDME cleavage [5] [6] |
| Caspase-6 | Short pro-domain | Cleaved by initiator caspases | Apoptosis execution, activates caspase-8 [5] | |
| Caspase-7 | Short pro-domain | Cleaved by initiator caspases | Apoptosis execution, suppresses pyroptosis [5] | |
| Inflammatory | Caspase-1 | CARD | Inflammasome | Pyroptosis via GSDMD cleavage, cytokine maturation [9] [5] |
| Caspase-4/5 | CARD | Non-canonical inflammasome | Pyroptosis via GSDMD cleavage [9] [5] | |
| Caspase-11 | CARD | Non-canonical inflammasome | Pyroptosis via GSDMD cleavage (mouse) [9] |
Activation Mechanisms: Dimerization Versus Cleavage
The activation mechanisms of initiator and executioner caspases represent a fundamental structural difference with profound implications for drug design. Initiator caspases are activated primarily by induced proximity dimerization [52] [53]. For example, caspase-8 activation occurs through DED-mediated filament formation within the Death-Inducing Signaling Complex (DISC), where dimerization drives its activation [52] [53]. Similarly, caspase-9 is activated through CARD-mediated recruitment to the Apaf-1 apoptosome, forming a heptameric wheel-like structure that facilitates dimerization and activation [51] [54]. Crucially, initiator caspases can exhibit catalytic activity without proteolytic processing, as dimerization alone is sufficient for activation [51] [53].
In contrast, executioner caspases exist as inactive dimers in healthy cells and require cleavage by initiator caspases between the large and small subunits to undergo conformational changes that form the active site [6] [55]. This creates a hierarchical activation cascade where initiator caspases activate executioner caspases, which then cleave hundreds of cellular substrates to execute cell death [13] [6].
Table 2: Quantitative Analysis of Caspase Substrate Specificity and Cleavage
| Caspase | Approximate Number of Substrates | Preferred Cleavage Motif | Cleavage Efficiency Variation | Key Functional Substrates |
|---|---|---|---|---|
| Caspase-8 | Hundreds [13] | (I/L/V/E)ETD [13] | >500-fold [13] | Caspase-3, -7, Bid, GSDMC [5] [6] |
| Caspase-9 | Few dozen [13] | (I/L/V/E)EHD [13] | Limited data | Caspase-3, -7, Bid [55] |
| Caspase-3 | Hundreds [13] | DEVxD [13] | >500-fold [13] | PARP, ICAD, GSDME [5] [6] |
| Caspase-7 | Hundreds [13] | DEVxD [13] | >500-fold [13] | PARP, GSDMB, GSDMD [5] |
| Caspase-2 | Hundreds [13] | VDxxD [13] | >500-fold [13] | Bid, Golgin-160 [5] |
Experimental Approaches for Studying Caspase Structure and Function
Structural Biology Methodologies
X-ray Crystallography has been instrumental in elucidating caspase structures. For example, structural studies of caspase-8 DEDs revealed a novel domain-swapped dimerization mechanism with both open and closed conformations that differ in their solvent exposure of hydrophobic patches critical for protein-protein interactions [52]. Similarly, crystallography of caspase-9 in complex with Apaf-1 CARD domains demonstrated a 1:1 complementary interface essential for caspase-9 activation [51]. Recent advances have revealed a multimeric interaction requiring three distinct interfaces for full caspase-9 activation [51].
Cryo-Electron Microscopy (cryo-EM) has enabled visualization of large caspase activation complexes. The apoptosome structure reveals a wheel-shaped heptameric assembly of Apaf-1 and cytochrome c that serves as the activation platform for caspase-9 [54]. More recently, cryo-EM has determined the structure of caspase-8 tandem DED filaments, providing insights into DED interactions within the DISC and with regulatory proteins like cFLIP and vFLIP [52].
Biochemical and Cellular Assays
Activity-Based Protease Profiling utilizes mass spectrometry-based proteomics to globally identify caspase substrates and characterize their cleavage specificities in live cells and cell extracts [13]. These studies reveal that each caspase has a preferred substrate cohort with cleavage rates varying over 500-fold, and that the number of substrate targets varies widely—from few dozen for caspases-4, -5, -9, and -14 to hundreds for caspases-1, -2, -3, -6, -7, and -8 [13].
Genetic Knockout Models have been essential for defining non-redundant functions of individual caspases. For instance, caspase-9 knockout mice die perinatally with severe brain abnormalities due to suppressed apoptosis during development [51]. Similarly, caspase-3 and caspase-7 knockout mice display distinct phenotypes, indicating non-overlapping functions despite their similar substrate specificities [55]. Studies with these models have revealed that caspase-3 inhibits ROS production and is required for efficient apoptosis execution, while caspase-7 is required for apoptotic cell detachment [55].
Table 3: Research Reagent Solutions for Caspase Studies
| Reagent/Category | Specific Examples | Function/Application | Experimental Context |
|---|---|---|---|
| Caspase Inhibitors | Z-VAD-FMK (pan-caspase), VX-765 (caspase-1), Emricasan (caspase-2, -3, -9) [1] | Mechanism-based inhibitors for functional studies and therapeutic development | Determining caspase-specific contributions to cell death pathways [1] |
| Activity Reporters | DEVD-AFC (caspase-3/7), LEHD-AFC (caspase-9), IETD-AFC (caspase-8) [13] | Fluorescent substrates for kinetic analysis of caspase activity | Measuring activation kinetics and inhibitor potency in vitro [13] |
| Structural Biology Tools | Caspase-8 DED mutants (F122A, Q125C) [52], Caspase-9 CARD-Apaf-1 complex [51] | Engineered proteins for crystallization and structural studies | Elucidating dimerization interfaces and activation mechanisms [51] [52] |
| Cellular Models | Caspase-3, -7, -9 deficient MEFs [55], Caspase-9 null embryonic stem cells [51] | Genetically defined systems for functional studies | Dissecting distinct roles in apoptosis execution and mitochondrial events [51] [55] |
| FRET Biosensors | SCAT3, SCAT9 [6] | Live-cell imaging of caspase activation kinetics | Monitoring spatiotemporal dynamics of caspase activation in single cells [6] |
Visualization of Caspase Activation Pathways
The following diagrams illustrate key structural and functional relationships in caspase activation pathways, created using Graphviz DOT language with sufficient color contrast for readability.
Caspase Activation Hierarchies in Cell Death Pathways
Diagram 1: Caspase activation hierarchies in apoptotic pathways. Initiator caspases (blue) activate executioner caspases (green) in both extrinsic and intrinsic apoptosis pathways. Caspase-8 cleaves Bid to connect the pathways via mitochondrial permeabilization.
Structural Domains and Activation Complexes
Diagram 2: Structural domains and activation mechanisms of initiator versus executioner caspases. Initiator caspases feature long pro-domains (DED or CARD) and activate by dimerization, while executioner caspases have short pro-domains and require cleavage for activation.
Strategic Framework for Targeted Drug Development
Targeting Initiator Caspase Activation Complexes
The multiprotein complexes that activate initiator caspases present unique opportunities for selective intervention. For caspase-8, the DED-mediated filament formation within the DISC offers potential targets for disrupting protein-protein interactions without directly inhibiting the catalytic site [52]. Structural studies have identified conserved hydrophobic patches (Phe122/Leu123) that are critical for DED dimerization and could be targeted by small molecules or peptides [52]. Similarly, for caspase-9, the CARD-CARD interaction with Apaf-1 in the apoptosome represents a specific target for modulating intrinsic apoptosis [51]. Drugs targeting these protein interaction interfaces could achieve greater selectivity than active-site inhibitors.
Exploiting Differential Substrate Specificities
Although caspases share a preference for aspartic acid in the P1 position, they exhibit distinct preferences for the P2-P4 positions, enabling the development of selective substrates and inhibitors [13]. For example, caspase-8 prefers (I/L/V/E)ETD motifs, while caspase-9 cleaves (I/L/V/E)EHD sequences, and executioner caspases-3 and -7 favor DEVxD motifs [13]. These subtle differences in substrate specificity can be leveraged to design drugs that discriminate between caspases with overlapping functions. The development of caspase-3 selective inhibitors that don't affect caspase-7, despite their similar cleavage preferences, demonstrates the feasibility of this approach [55].
Allosteric Regulation and Exosite Targeting
Beyond the active site, caspases contain secondary interaction sites (exosites) that influence substrate recognition and catalytic efficiency. For instance, caspase-7 uses an exosite to promote poly(ADP ribose) polymerase 1 proteolysis [13]. Similarly, phosphorylation at specific residues (e.g., Thr125 in caspase-9) can regulate activity without direct active site blockade [51]. Allosteric inhibitors that target these regulatory sites could provide enhanced selectivity compared to active-site directed compounds that often show cross-reactivity among caspase family members.
Context-Dependent Therapeutic Strategies
The multifaceted roles of caspases in different cell death pathways necessitate context-dependent therapeutic approaches. In cancer, where caspase-8 expression is often reduced, promoting its activation could restore apoptosis, while in neurodegenerative diseases, caspase inhibition might protect neurons [1] [9]. The emerging understanding that cells can survive executioner caspase activation under certain conditions further complicates therapeutic strategies [6]. For inflammatory conditions, selective caspase-1 inhibition shows promise for autoimmune diseases, while caspase-4/5/11 inhibition might be beneficial for sepsis [9] [5].
The structural differences between initiator and executioner caspases provide a robust foundation for developing selective therapeutic agents. Initiator caspases, with their long pro-domains and dependence on dimerization within multiprotein complexes, present unique targeting opportunities distinct from executioner caspases with their short pro-domains and cleavage-dependent activation. Advances in structural biology, particularly in understanding caspase activation complexes and substrate specificity profiles, continue to reveal new avenues for selective intervention. The ongoing characterization of caspase functions beyond apoptosis, including roles in differentiation, inflammation, and cellular remodeling, further expands the therapeutic potential of caspase-targeted drugs. As our understanding of caspase biology deepens, so too will our ability to design increasingly selective modulators of these critical cell death regulators.
Experimental Challenges and Strategic Solutions in Caspase Research
Caspases, a family of cysteine-aspartate proteases, are central regulators of programmed cell death, including apoptosis and pyroptosis. Their traditional classification as initiators (e.g., caspase-8, -9) and executioners (e.g., caspase-3, -6, -7) is defined by their position in the proteolytic cascade and their structural features. A significant challenge in targeting these enzymes for therapeutic purposes is the substantial cross-reactivity observed between different caspase types, stemming from their highly conserved active sites and overlapping substrate specificities. This whitepaper delves into the structural underpinnings of this cross-reactivity, presents quantitative data on shared substrate pools, and discusses advanced experimental strategies to overcome these specificity challenges in research and drug development. Understanding these mechanisms is critical for developing precise caspase-modulating therapies for cancer, neurodegenerative disorders, and inflammatory diseases.
Caspases (cysteine-dependent aspartate-specific proteases) are a family of proteases that play critical roles in programmed cell death (PCD), inflammation, and cellular homeostasis [9] [5]. In humans, the 12 identified caspases are historically categorized by function: inflammatory caspases (caspase-1, -4, -5, -11), apoptotic initiators (caspase-2, -8, -9, -10), and apoptotic executioners (caspase-3, -6, -7) [18] [9]. Initiator caspases are characterized by long N-terminal pro-domains containing death folds (either CARD or DED domains) that facilitate recruitment to and activation within large multimolecular signaling complexes like the apoptosome or DISC [9] [5]. In contrast, executioner caspases typically have short pro-domains and are activated by initiator caspases through proteolytic cleavage [9].
The central challenge in both understanding caspase biology and developing targeted therapeutics is the significant cross-reactivity between different caspase types. This cross-reactivity arises because all caspases share a common fold and a catalytic mechanism that stringently requires aspartic acid in the P1 position of the substrate [19]. Although each caspase has a theoretically optimal substrate recognition sequence, the reality within a living cell is a complex network of overlapping specificities. This means that an executioner caspase like caspase-3 can sometimes process substrates typically associated with initiator caspases, and vice-versa, leading to potential non-canonical pathway activation and blurred functional boundaries [25] [13]. This review dissects the structural and molecular basis of this cross-reactivity within the context of initiator versus executioner caspase differences.
Structural Basis of Cross-reactivity
Conserved Catalytic Architecture
The fundamental similarity driving cross-reactivity is the highly conserved three-dimensional structure of the caspase catalytic domain. All caspases exist as homodimers, with each monomer contributing to one active site. The active site itself contains a deep, highly basic pocket formed by the conserved residues Arg-179, Arg-341, and Gln-283 (caspase-1 numbering) that perfectly accommodates the P1 aspartic acid side chain of the substrate [19]. This architecture makes specificity for aspartate a non-negotiable feature but also means the primary pocket offers little opportunity for discriminating between different caspases.
Specificity is instead determined by interactions with the extended substrate recognition groove, which accommodates the P4-P2-P3-P1-P1' residues of the substrate [19]. While each caspase has preferences for specific amino acids at these positions, these preferences are not absolute. The most striking differences are often observed in the S4 pocket. For instance, caspase-1 prefers bulky hydrophobic residues (Trp, Tyr), caspase-3 has a near-absolute requirement for aspartic acid (Asp), and caspase-8 accommodates branched aliphatic residues (Leu, Val) [19]. However, the plasticity of these binding pockets allows for the recognition of a wider range of sequences than their "optimal" motif would suggest.
Key Structural Differences: Initiator vs. Executioner
Despite the conserved core, critical structural differences exist between initiator and executioner caspases that influence their activation and function, yet also contribute to potential cross-talk.
Table 1: Structural and Activation Differences Between Initiator and Executioner Caspases
| Feature | Initiator Caspases (e.g., Casp-8, -9) | Executioner Caspases (e.g., Casp-3, -6, -7) |
|---|---|---|
| Pro-domain | Long pro-domain containing death folds (CARD or DED) [9] | Short pro-domain [9] |
| Activation Mechanism | Induced proximity; dimerization driven by large complexes (DISC, apoptosome) [9] | Proteolytic cleavage by initiator caspases [9] |
| Basal State | Monomeric (caspase-9) or monomer/dimer mixture (caspase-8) in zymogen form [25] | Dimeric zymogen requiring cleavage for activity [9] |
| Representative Optimal Peptide Substrate Motif | Caspase-8: (L/V/I)EXD [18] | Caspase-3: DEXD [18] |
| Catalytic Efficiency | Generally lower | Generally higher (e.g., caspase-3 has high catalytic efficiency) [25] |
The following diagram illustrates the canonical activation pathways and structural features of initiator and executioner caspases, highlighting points where cross-reactivity can occur.
Quantitative Analysis of Caspase Cross-reactivity
Global proteomic studies have been instrumental in quantifying the extent of substrate overlap between different caspases, moving beyond idealized peptide substrates to reveal the complex reality within cells.
Overlapping Substrate Pools
Proteomic analyses show that the number of cellular substrates identified for individual caspases varies widely, from a few dozen for caspases-4, -5, -9, and -14 to hundreds for caspases-1, -2, -3, -6, -7, and -8 [13]. This immediately suggests significant functional overlap, particularly among the executioners and some initiators. While each caspase has a preferred substrate cohort, the rates of cleavage for these substrates can vary by over 500-fold within a single caspase's target pool, and there is considerable sharing between caspases [13]. For example, caspase-3 and -7, despite being paralogs with high sequence identity, display both overlapping and non-overlapping substrate pools, and the removal of both is required to confer significant resistance to apoptosis [25] [13].
Efficiency of Substrate Cleavage
The kinetic parameters of caspase activity further illuminate the cross-reactivity landscape. The table below summarizes the catalytic efficiency (kcat/KM) of different caspases against common and optimal peptide substrates, demonstrating both their inherent preferences and the potential for overlap.
Table 2: Catalytic Efficiency of Selected Caspases Against Fluorogenic Substrates
| Caspase | Type | Optimal Tetrapeptide Motif | Example Substrate | kcat/KM (M⁻¹s⁻¹) | Reference |
|---|---|---|---|---|---|
| Caspase-1 | Inflammatory | WEHD | Ac-WEHD-AMC | 3.34 x 10⁶ | [19] |
| Caspase-2 | Initiator | VDVAD* | Ac-VDVAD-AFC | ~10⁵ (Requires P5 residue) | [19] |
| Caspase-3 | Executioner | DEVD | Ac-DEVD-AMC | 1.4 x 10⁶ | [19] |
| Caspase-6 | Executioner | VEID | Ac-VEID-AMC | Data not fully quantified | [13] |
| Caspase-7 | Executioner | DEVD | Ac-DEVD-AMC | Lower than caspase-3 | [13] |
| Caspase-8 | Initiator | IETD | Ac-IETD-AMC | Liberal accommodation at S4 | [19] |
| Caspase-9 | Initiator | LEHD | Ac-LEHD-AMC | Data not fully quantified | [13] |
*Caspase-2 exhibits very low activity on tetrapeptides and cleaves substrates efficiently only when a P5 residue is present [19].
A key experimental demonstration of functional cross-reactivity comes from studies involving the exogenous introduction of caspases. Contrary to expectations, while caspase-3 has high catalytic efficiency, studies found that exogenously introduced caspase-7 and caspase-9 were more effective at inducing apoptotic cell death in certain cellular contexts [25]. This highlights that the efficacy of a caspase is not determined by catalytic potency alone but is also shaped by the cellular network of pro-survival factors and inhibitors that differentially regulate each caspase [25].
Experimental Strategies to Overcome Specificity Challenges
The Researcher's Toolkit: Reagents and Methodologies
To dissect the complex web of caspase cross-reactivity, researchers employ a suite of specific reagents and experimental strategies.
Table 3: Essential Research Reagents for Studying Caspase Specificity
| Reagent / Method | Function & Rationale | Key Considerations |
|---|---|---|
| Positional Scanning Synthetic Combinatorial Libraries (PS-SCL) | Defines the inherent subsite preference (P4-P1) of a caspase using libraries of fluorogenic tetrapeptides [19]. | Provides a consensus optimal motif but does not always predict cleavage of full-length protein substrates. |
| Fluorogenic/Luminescent Peptide Substrates (e.g., Ac-DEVD-AMC) | Measures caspase activity in cell lysates, live cells, or in vivo. AMC release increases fluorescence; aminoluciferin release produces luminescence [19]. | DEVD is cleaved by caspase-3 and -7, and also by other caspases like caspase-8, leading to potential overinterpretation of specificity. |
| Active-site Labeling Probes (Biotin-/Fluorophore-conjugated) | Irreversibly binds to the active-site cysteine, allowing visualization and pull-down of active caspases. | Can distinguish active enzymes from zymogens but does not provide information on substrate specificity. |
| Genetic Knockout/Knockdown Models | Removing a specific caspase gene allows for the assessment of its non-redundant functions and identification of its unique substrates. | Compensation by other caspases can occur, masking the true phenotype. |
| Global Proteomics (N-terminomics/Degradomics) | Identifies natural caspase substrates on a proteome-wide scale in live cells or extracts by labeling and sequencing neo-N-terminal after cleavage [13]. | The gold standard for identifying the true substrate repertoire of a caspase in a physiological context. |
Detailed Protocol: Using Degradomics to Map Cross-reactive Substrates
The following workflow outlines a key proteomic method for empirically identifying the full range of substrates cleaved by a specific caspase, thereby directly characterizing cross-reactivity.
Protocol Steps:
- Cell Lysis and Fractionation: Harvest cells undergoing a specific death stimulus (e.g., treated with a chemotherapeutic agent to activate intrinsic apoptosis). Prepare a cytosolic extract and fractionate it using liquid chromatography (e.g., ion exchange). This simplifies the complex proteome and removes endogenous caspases [13].
- In Vitro Proteolysis: Incubate individual fractions with a highly active, recombinant caspase of interest (e.g., caspase-9). A control reaction without caspase addition is crucial. This step allows the exogenous caspase to cleave its potential substrates within the cellular lysate.
- N-terminal Labeling (TAILS Method): Use the Terminal Amine Isotopic Labeling of Substrates (TAILS) method. All primary amines (α-amines of original N-termini and ε-amines of lysines) are blocked by reductive dimethylation. The neo-N-termini created by caspase cleavage are then specifically labeled with a polymer that enables their enrichment [13].
- Mass Spectrometry and Data Analysis: Digest the samples with trypsin. The polymer-containing peptides (neo-N-terminal peptides) are enriched and analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS). Bioinformatics pipelines are used to map the cleavage sites back to the proteome, identifying the specific substrates and the exact aspartic acid residue cleaved by the caspase [13].
This protocol directly reveals the substrate landscape of a caspase, highlighting shared substrates (cross-reactivity) and unique substrates, providing a data-driven understanding of specificity beyond theoretical motifs.
Discussion and Future Perspectives
The cross-reactivity between caspase types is not merely a biochemical nuisance; it is an embedded feature of a robust and fail-safe cellular death network. This redundancy ensures that if one pathway is blocked, as often happens in cancer, another can potentially be activated [25]. From a therapeutic standpoint, this presents both a challenge and an opportunity.
The traditional goal of developing exquisitely specific caspase inhibitors has proven difficult due to the conserved active sites. However, new strategies are emerging. One approach involves targeting exosites—secondary substrate binding sites distinct from the catalytic cleft—which can be more variable and offer greater specificity for modulating the cleavage of specific key substrates [13]. Another promising avenue is the development of conformation-specific inhibitors that stabilize specific enzyme conformations, potentially allowing selective blockade of pro-metastatic functions without affecting other roles, as explored for caspase-6 [39].
Furthermore, the exogenous introduction of specific caspases using advanced delivery systems like redox-responsive nanogels represents a novel therapeutic strategy to bypass endogenous apoptotic blocks in cancer cells [25]. The finding that caspase-7 or -9 can be more effective than caspase-3 in certain contexts underscores the need to profile the apoptotic network of a tumor to select the most effective caspase therapeutic [25]. As our understanding of caspase biology evolves beyond a simple "apoptosis executioner" model to encompass a "functional continuum" of roles in sublethal signaling and cellular remodeling [39], the need for precision tools to dissect and manipulate their specific functions becomes ever more critical for advancing therapeutic applications.
Optimizing Active Caspase Production and Stability
Caspases are a family of cysteine-aspartic proteases that serve as critical mediators of programmed cell death (PCD), including apoptosis and pyroptosis, and play essential roles in cellular homeostasis, development, and immunity [5] [9] [18]. These enzymes are synthesized as inactive zymogens (pro-caspases) that require proteolytic cleavage for activation, typically after aspartic acid residues, forming active heterotetramers consisting of two large and two small subunits [9]. The caspase family is broadly categorized into initiator caspases (including caspase-2, -8, -9, and -10), which initiate cell death cascades, and executioner caspases (including caspase-3, -6, and -7), which carry out the proteolytic dismantling of cellular structures [9] [18]. A third subgroup, inflammatory caspases (including caspase-1, -4, -5, and -11), primarily regulates inflammatory responses and pyroptosis [18].
Understanding the structural and functional distinctions between initiator and executioner caspases is fundamental to advancing caspase-related research and therapeutic development. Initiator caspases possess long pro-domains containing death folds such as caspase activation and recruitment domains (CARD) or death effector domains (DED) that facilitate their recruitment to and activation within large multiprotein complexes like the death-inducing signaling complex (DISC) or apoptosome [5] [9]. In contrast, executioner caspases typically have short pro-domains and are activated through cleavage by initiator caspases, creating a proteolytic cascade that amplifies the cell death signal [9]. These structural differences directly impact production strategies, activation mechanisms, and stability profiles for different caspase types, necessitating tailored approaches for working with specific caspases in research and drug discovery contexts.
Structural and Functional Differences Between Initiator and Executioner Caspases
The fundamental structural distinctions between initiator and executioner caspases dictate their activation mechanisms, substrate specificities, and functional roles in cellular signaling pathways. Understanding these differences is paramount for developing optimized production and stabilization protocols.
Domain Architecture and Activation Mechanisms
Initiator caspases, including caspase-2, -8, -9, and -10, are characterized by extended N-terminal pro-domains that contain protein-protein interaction motifs, either CARD (in caspase-2 and -9) or DED (in caspase-8 and -10) domains [5] [9]. These domains mediate homotypic interactions with adaptor proteins in activation platforms such as the DISC (for caspase-8 and -10) or the apoptosome (for caspase-9) [5]. Activation occurs through proximity-induced dimerization, where bringing multiple pro-caspase molecules into close contact promotes their autoproteolytic processing and activation [9]. This mechanism allows initiator caspases to function as signal integrators that respond to specific death stimuli.
Executioner caspases (-3, -6, and -7) possess only short pro-domains and exist predominantly as stable dimers in their zymogen forms [9]. They are not activated by proximity-induced dimerization but rather by precise proteolytic cleavage between the large and small subunits by initiator caspases. Once cleaved, executioner caspases undergo conformational changes that reorganize the active site into a catalytically competent state [9]. This hierarchical activation cascade allows for tremendous signal amplification, where a small number of activated initiator caspase molecules can trigger the processing of numerous executioner caspase zymogens.
Substrate Specificity and Biological Functions
The substrate preferences of caspases are largely determined by residues in the S1-S4 binding pockets, with each caspase demonstrating distinct sequence specificities [13] [18]. Initiator caspases typically recognize tetra-peptide motifs with the general sequence (L/V/I)EXD, while executioner caspases prefer DEXD motifs, and inflammatory caspases favor (W/L/Y)EHD sequences [18]. Beyond their primary roles in apoptosis initiation and execution, individual caspases participate in diverse cellular processes. For instance, caspase-8 serves as a molecular switch between apoptosis, necroptosis, and pyroptosis [5], while caspase-3 can drive both apoptotic and pyroptotic cell death through cleavage of different gasdermin family members [5] [18].
Table 1: Key Structural and Functional Properties of Major Caspases
| Caspase | Type | Pro-domain | Activation Complex | Preferred Substrate Motif | Key Functions |
|---|---|---|---|---|---|
| Caspase-8 | Initiator | DED | DISC, RIPoptosome | (L/V/I)EXD | Extrinsic apoptosis, necroptosis inhibition, pyroptosis switch [5] |
| Caspase-9 | Initiator | CARD | Apoptosome | (L/V/I)EXD | Intrinsic apoptosis, cleaves caspase-3/7 [5] [9] |
| Caspase-10 | Initiator | DED | DISC | (L/V/I)EXD | Extrinsic apoptosis, immune regulation [56] |
| Caspase-3 | Executioner | Short | N/A | DEXD | Apoptosis execution, GSDME cleavage for pyroptosis [5] [18] |
| Caspase-7 | Executioner | Short | N/A | DEXD | Apoptosis execution, PARP cleavage [5] |
| Caspase-1 | Inflammatory | CARD | Inflammasome | (W/L/Y)EHD | Pyroptosis, cytokine maturation (IL-1β, IL-18) [57] [9] |
Production Strategies for Active Caspases
Producing high-quality, active caspases with minimal background contamination or autoproteolysis presents significant technical challenges. Both traditional and innovative engineering approaches have been developed to address these issues.
Traditional Recombinant Expression and Purification
Conventional methods for caspase production involve expressing pro-caspases in bacterial systems such as E. coli, followed by purification and in vitro activation. The standard protocol involves cloning the caspase gene into an expression vector, transforming into an appropriate E. coli strain (e.g., BL21(DE3)), inducing expression with IPTG, and purifying the zymogen using affinity chromatography (typically nickel-NTA for His-tagged constructs) [56]. The purified pro-caspase is then activated by incubation with high concentrations of other proteases (e.g., caspase-8 for executioner caspases) or through auto-activation under specific buffer conditions.
While this approach can yield functional enzymes, it often suffers from several limitations: (1) low yields due to caspase toxicity in bacterial systems, (2) heterogeneous cleavage products from incomplete activation, (3) premature activation during purification, and (4) difficulty controlling the activation process precisely. These challenges are particularly pronounced for initiator caspases, which require specific adaptor proteins and activation platforms not present in bacterial expression systems.
Innovative Engineering Approaches
Recent advances in protein engineering have yielded innovative solutions to overcome the limitations of traditional caspase production methods. One particularly promising approach involves engineering caspases to replace their native activation sites with sequences recognized by exogenous proteases, such as tobacco etch virus (TEV) protease [56].
Castellón et al. (2025) demonstrated this strategy by developing a TEV-activatable caspase-10 construct (proCASP10TEV Linker) in which the endogenous cleavage sites were replaced with TEV recognition sequences [56]. This engineered caspase exhibited minimal background activity in the absence of TEV protease but could be robustly activated upon TEV addition, showing comparable activity to recombinant active caspase-10. The optimized construct displayed improved stability and reduced autoproteolysis, making it suitable for high-throughput screening applications [56].
Table 2: Comparison of Caspase Production Methods
| Method | Procedure | Advantages | Limitations | Ideal Applications |
|---|---|---|---|---|
| Traditional Recombinant | Express pro-caspase in E. coli, purify, activate with proteases | Well-established protocol, no specialized reagents needed | Low yield, premature activation, heterogeneous products | Small-scale studies, initial characterization |
| TEV-Activatable Engineering | Replace caspase cleavage sites with TEV recognition sequence, activate with TEV protease | Low background, controlled activation, high stability, homogeneous product | Requires protein engineering, additional purification step | High-throughput screening, structural studies, drug discovery |
| Cell-Free Systems | In vitro transcription/translation of caspase constructs | Bypasses toxicity issues, rapid production | Low yield, high cost, scaling challenges | Functional assays, incorporation of unnatural amino acids |
Stabilization Strategies and Buffer Optimization
Maintaining caspase stability and activity after production represents a critical challenge in experimental workflows. Multiple factors including buffer composition, pH, ionic strength, and temperature must be carefully optimized for each caspase type.
Buffer Composition and Additives
The canonical storage buffer for caspases typically contains 20-50 mM HEPES (pH 7.2-7.5), 100-150 mM NaCl, 10% glycerol, 0.1% CHAPS, and 1-10 mM DTT [56]. However, specific caspases may require tailored optimization. Key considerations include:
Reducing Agents: DTT (1-10 mM) or β-mercaptoethanol (2-5 mM) are essential for maintaining the catalytic cysteine in a reduced state. However, excessive concentrations may promote non-specific reduction or protein destabilization.
Kosmotropes and Chaotropes: Sodium citrate (a kosmotrope) has been shown to promote caspase autoproteolysis and increase background activity in caspase-10, suggesting that kosmotropic salts should be used with caution [56]. Conversely, mild chaotropes may help stabilize certain caspase conformations.
Detergents: CHAPS (0.1%) helps maintain caspase solubility and prevents aggregation without significantly inhibiting enzymatic activity. Other non-ionic detergents like Triton X-100 (0.01-0.1%) or Tween-20 (0.05%) may also be suitable alternatives.
Osmolytes and Stabilizers: Glycerol (10-20%) serves as a cryoprotectant and stabilizer. Other compatible osmolytes such as sucrose (100-200 mM) or trehalose (50-100 mM) may provide additional stabilization, particularly for long-term storage.
Temperature and Storage Considerations
Active caspases are generally stored at -80°C in small aliquots to avoid freeze-thaw cycles, which dramatically accelerate inactivation. For short-term use (hours to days), caspases may be maintained on ice or at 4°C, though significant activity loss can occur within 24 hours at these temperatures. The implementation of single-use aliquots is strongly recommended to preserve activity across multiple experiments.
Executioner caspases typically demonstrate greater stability than initiator caspases, reflecting their evolutionary adaptation to function downstream in proteolytic cascades. Inflammatory caspases, particularly caspase-1, often present intermediate stability profiles. These differences highlight the need for caspase-specific optimization of storage conditions rather than applying a universal standard to all family members.
Table 3: Optimized Stabilization Conditions for Different Caspase Types
| Caspase | Optimal pH | Recommended [NaCl] | Critical Additives | Storage Temperature | Special Considerations |
|---|---|---|---|---|---|
| Caspase-8 | 7.2-7.4 | 100-150 mM | 5-10 mM DTT, 0.1% CHAPS | -80°C (aliquots) | Prone to aggregation, avoid repeated freezing/thawing |
| Caspase-9 | 7.4-7.6 | 50-100 mM | 1-5 mM DTT, 20% glycerol | -80°C (aliquots) | CARD domain interactions may require specific stabilizers |
| Caspase-3 | 7.2-7.5 | 100-200 mM | 1-5 mM DTT, 10% glycerol | -80°C (aliquots) | Relatively stable, tolerates wider pH range |
| Caspase-7 | 7.2-7.5 | 100-200 mM | 1-5 mM DTT, 0.1% CHAPS | -80°C (aliquots) | Similar to caspase-3 but slightly less stable |
| Caspase-1 | 7.0-7.4 | 150-200 mM | 2-5 mM DTT, 10% glycerol | -80°C (aliquots) | Inflammasome activation requires specific conditions |
Activity Assessment and Quality Control
Rigorous assessment of caspase activity and purity is essential for ensuring experimental reproducibility and interpreting results accurately. Multiple complementary methods should be employed to fully characterize caspase preparations.
Kinetic Assays and Specific Activity Determination
Fluorogenic substrates containing the preferred cleavage sequences for specific caspases, conjugated to fluorophores like 7-amino-4-trifluoromethylcoumarin (AFC) or 7-amino-4-methylcoumarin (AMC), provide a sensitive method for quantifying caspase activity [56] [58]. The general protocol involves incubating caspase with substrate (typically 10-200 μM) in reaction buffer (20-50 mM HEPES pH 7.4, 100 mM NaCl, 0.1% CHAPS, 10% glycerol, 1-5 mM DTT) and monitoring fluorescence increase over time (AFC: excitation 400 nm, emission 505 nm; AMC: excitation 380 nm, emission 460 nm).
Specific activity should be calculated using the initial linear rate of fluorescence increase, converted to product formation using a standard curve of free fluorophore. Quality preparations typically exhibit Michaelis-Menten kinetics with Km values in the low micromolar range for preferred substrates. For example, engineered TEV-activated caspase-10 showed comparable kinetics to recombinant active caspase-10 with Ac-VDVAD-AFC as substrate [56].
Orthogonal Validation Methods
Beyond kinetic assays, multiple validation methods should confirm caspase quality:
Gel Electrophoresis and Western Blotting: SDS-PAGE under reducing conditions should show the appropriate subunit pattern (large and small subunits for active caspase) with minimal degradation products or pro-form contamination. Western blotting with caspase-specific antibodies provides additional confirmation of identity and processing state [56].
Inhibitor Profiling: Incubation with pan-caspase inhibitors (e.g., zVAD-FMK, 10-50 μM) should abolish >90% of activity, confirming that observed activity is caspase-mediated [58]. Caspase-selective inhibitors can further verify identity through characteristic inhibition profiles.
Activity-Based Probes: Fluorescent or biotinylated caspase inhibitors (e.g., Rho-DEVD-AOMK) enable direct visualization of active enzyme and can detect contamination with other proteases or background activation in zymogen preparations [56].
The Scientist's Toolkit: Essential Research Reagents
Successful caspase research requires a comprehensive set of specialized reagents and tools. The following table outlines essential materials for production, stabilization, and assessment of caspase activity.
Table 4: Essential Research Reagents for Caspase Studies
| Reagent Category | Specific Examples | Function/Application | Technical Considerations |
|---|---|---|---|
| Expression Systems | pET vectors, BL21(DE3) E. coli | Recombinant caspase production | Codon optimization may be needed for human caspases |
| Purification Tools | Ni-NTA resin, His-tag antibodies, size exclusion columns | Isolation of recombinant caspase | Imidazole must be thoroughly removed before activity assays |
| Activation Reagents | TEV protease, active caspase-8, high salt buffers | Conversion of zymogen to active caspase | TEV protease offers controlled activation with low background [56] |
| Stabilization Additives | DTT, glycerol, CHAPS, HEPES buffer | Maintain caspase activity during storage | Optimize concentration for each caspase type |
| Fluorogenic Substrates | Ac-DEVD-AFC (caspase-3/7), Ac-VDVAD-AFC (caspase-2), Ac-IETD-AFC (caspase-8) | Kinetic activity measurements | Verify specificity with caspase-selective inhibitors |
| Caspase Inhibitors | zVAD-FMK (pan-caspase), DEVD-CHO (caspase-3/7), VX-765 (caspase-1) | Specificity controls, mechanistic studies | Cell-permeable forms available for cellular assays |
| Activity-Based Probes | Biotin-/fluorophore-DEVD-AOMK, -VEID-AOMK | Direct labeling of active caspases | Can detect multiple active caspases in complex mixtures |
| Detection Antibodies | Anti-cleaved caspase-3, anti-caspase-8, anti-PARP | Western blot validation of activity | Confirm cleavage of native substrates |
| Cellular Reporters | DEVD-ZipGFP, DEVD-based FRET reporters | Live-cell caspase activity monitoring | Enables real-time kinetics in physiological contexts [58] |
Troubleshooting Common Challenges
Despite optimized protocols, researchers frequently encounter specific challenges when working with caspases. The following section addresses common issues and provides evidence-based solutions.
Low Yield and Purity Issues
Low expression yields often result from caspase toxicity in bacterial systems. Strategies to mitigate this include using lower induction temperatures (18-25°C), reduced IPTG concentrations (0.1-0.5 mM), and shorter induction times (4-16 hours). Alternatively, caspase toxicity can be addressed through the use of engineered TEV-activatable constructs that minimize background activity during expression [56]. For purity concerns, tandem affinity purification strategies or inclusion of additional purification steps (e.g., ion exchange chromatography after initial His-tag purification) can significantly improve sample homogeneity.
Premature Activation and Autoproteolysis
Uncontrolled activation during expression or purification remains a major challenge, particularly for executioner caspases. This can be minimized by including caspase inhibitors in lysis and initial purification buffers (except when purifying active enzyme), working quickly at 4°C, and using engineered caspase variants with reduced autoproteolysis susceptibility [56]. The TEV-activation system represents a particularly effective solution, as it physically separates the activation step from expression and purification, ensuring a homogeneous population of properly activated enzyme [56].
Rapid Activity Loss
Significant activity loss during storage or experiments typically stems from oxidation of the catalytic cysteine, proteolytic degradation, or protein aggregation. Maintaining fresh reducing agents (DTT should be added immediately before use), using single-use aliquots, including protease inhibitor cocktails (except when assaying activity), and optimizing stabilizer concentrations can dramatically improve stability. For particularly labile caspases, consider adding non-hydrolyzable substrate analogs (at nanomolar concentrations) that stabilize the active conformation without permanently inhibiting activity.
Optimizing the production and stability of active caspases requires careful consideration of their structural and functional differences, particularly the distinction between initiator and executioner caspases. Traditional recombinant methods, while useful for some applications, face significant limitations in yield, homogeneity, and control over activation. Innovative approaches, such as TEV-protease activatable caspase engineering, offer promising solutions with improved control, reduced background activity, and enhanced stability [56].
As caspase research continues to evolve, several emerging areas present opportunities for further advancement. The development of more robust caspase expression systems, including mammalian and cell-free platforms, could better recapitulate native post-translational modifications and complex formation. Additionally, the creation of caspase variants with enhanced stability through rational design or directed evolution would address a major limitation in both basic research and therapeutic development. Finally, advanced delivery systems for active caspases in therapeutic contexts, particularly in cancer treatment, represent an exciting frontier where production and stabilization optimizations may directly translate to clinical benefits [59].
The critical role of caspases in health and disease, from cancer to neurodegenerative disorders to inflammatory conditions, continues to drive methodological innovations [5] [1] [18]. By applying the optimized strategies outlined in this technical guide, researchers can overcome historical challenges in caspase production and stabilization, accelerating both basic mechanistic studies and the development of caspase-targeted therapeutics.
Addressing Challenges in Measuring Caspase Activation Kinetics
Caspases, a family of cysteine-aspartic proteases, are central regulators of programmed cell death (PCD) and inflammation. Their activation kinetics are critical determinants of cellular fate, dictating whether a cell undergoes apoptosis, pyroptosis, necroptosis, or survival. A fundamental challenge in caspase research lies in accurately measuring these activation kinetics, which are profoundly influenced by the distinct structural and activation mechanisms of initiator versus executioner caspases. Initiator caspases (caspase-2, -8, -9, -10) exist as inactive monomers that require dimerization for activation, typically through adapter proteins in large multiprotein complexes. In contrast, executioner caspases (caspase-3, -6, -7) preexist as inactive dimers and are activated through cleavage between their large and small subunits [22]. This structural dichotomy creates significant methodological challenges for researchers investigating the temporal dynamics of caspase activation in various biological contexts, from basic research to drug discovery.
Structural and Mechanistic Basis of Caspase Activation
Fundamental Differences Between Initiator and Executioner Caspases
The activation mechanisms of caspases diverge significantly based on their classification as initiators or executioners, with profound implications for kinetic measurements.
Table 1: Structural and Activation Differences Between Caspase Types
| Feature | Initiator Caspases | Executioner Caspases |
|---|---|---|
| Representative Members | Caspase-8, -9, -10, -2 [26] [22] | Caspase-3, -6, -7 [26] [22] |
| Native State in Cell | Inactive monomers [22] | Inactive dimers [22] |
| Primary Activation Mechanism | Dimerization via induced proximity [22] | Cleavage between large and small subunits [22] |
| Prodomain Structure | Long prodomains (∼150–200 amino acids) with DED or CARD interaction domains [6] [22] | Short prodomains (less than 30 amino acids) lacking interaction domains [6] |
| Key Activation Complexes | DISC (caspase-8), apoptosome (caspase-9), PIDDosome (caspase-2) [5] | Activated by initiator caspases or granzyme B [22] |
| Cleavage Role in Activation | Stabilizes active dimer but not required for initial activation [22] | Essential for formation of active enzyme [22] |
| Activation Kinetics | Rapid, all-or-none pattern once initiated [6] | Feed-forward amplification; peaks within 15 minutes in apoptosis [6] |
Visualizing Caspase Activation Pathways
The following diagram illustrates the fundamental structural and activation differences between initiator and executioner caspases that underpin the kinetic measurement challenges:
Figure 1: Fundamental activation pathways of initiator versus executioner caspases, highlighting structural differences that complicate kinetic measurements.
Technical Challenges in Kinetic Measurement
Temporal and Spatial Resolution Limitations
Traditional endpoint assays like Western blotting provide limited insights into caspase activation kinetics because they capture a single time point and lack temporal resolution [26]. However, caspase activation occurs in rapid, often irreversible bursts - once initiated, executioner caspase activation peaks within 15 minutes during apoptosis [6]. This rapid kinetics necessitates real-time monitoring approaches to capture meaningful data. Spatially, caspase activation is compartmentalized within specific subcellular locations and macromolecular complexes, creating microdomains of activity that bulk measurement techniques often miss. For instance, executioner caspases can be proximal to cell membrane proteins like Fasciclin 3 in Drosophila, creating spatially restricted activation pools that facilitate non-lethal functions [60].
Specificity and Sensitivity Issues
The high sequence and structural homology among caspase family members presents significant challenges for developing specific detection reagents [56]. Most commercially available antibodies and fluorescent substrates show considerable cross-reactivity, complicating the interpretation of kinetic data for individual caspases. Additionally, the dynamic range required to detect both basal non-lethal caspase activity (in cellular remodeling) and lethal activation during apoptosis spans several orders of magnitude, pushing the limits of conventional detection methods [60]. The "all-or-none" pattern of executioner caspase activation further complicates population-level measurements, as averaging across cells can obscure single-cell heterogeneity in activation timing and extent [6] [61].
Live-Cell Monitoring Challenges
While real-time monitoring is ideal for capturing caspase kinetics, several technical hurdles persist:
- Phototoxicity: Extended live-cell imaging can artificially induce cell death pathways [61]
- Reporter perturbation: Overexpression of fluorescent biosensors may alter natural caspase activation dynamics
- Microenvironment effects: The intracellular environment can quench fluorescence or affect substrate accessibility
- Multi-parameter tracking: Simultaneously monitoring multiple caspases with different spectral properties remains technically challenging
Advanced Methodologies and Experimental Approaches
Real-Time Kinetic Monitoring Systems
SPARKL (Single-cell and Population-level Analyses Using Real-time Kinetic Labeling): This advanced workflow integrates high-content live-cell imaging with automated detection of fluorescent caspase reporters, enabling zero-handling, non-disruptive monitoring of caspase activation kinetics [61]. The system captures single-cell resolution data across entire populations over extended time periods, allowing researchers to correlate caspase activation with morphological changes and cell fate decisions. Implementation requires:
- Environmental control systems for maintained viability during extended imaging
- Automated image acquisition systems with appropriate fluorescent filter sets
- Computational pipelines for single-cell tracking and signal quantification
- Validation with established caspase inhibitors and activators
FRET (Fluorescence Resonance Energy Transfer)-Based Sensors: Genetically encoded FRET reporters undergo conformational changes upon caspase cleavage, altering energy transfer between donor and acceptor fluorophores [26] [6]. These sensors provide ratiometric measurements that are less susceptible to expression level variations, making them ideal for quantitative kinetic analyses. Optimal experimental setup includes:
- Selection of appropriate caspase-specific cleavage sequences (DEVD for executioner caspases, IETD for caspase-8)
- Control experiments with caspase-resistant mutant reporters
- Calibration with known caspase activators and inhibitors
- Multiplexing with cell viability markers for correlation with cell fate
Proximity Labeling for Caspase Interactome Mapping
TurboID-Based Proximity Proteomics: This cutting-edge approach identifies proteins proximal to caspases in their native cellular environment, revealing compartment-specific activation complexes [60]. The methodology involves:
- Generation of caspase-TurboID fusion proteins with preserved function
- In vivo biotin labeling with precise temporal control
- Streptavidin-based purification of biotinylated proteins
- Mass spectrometric identification and quantification
This technique has revealed that the Drosophila executioner caspase Drice exists as an inactive proform proximal to cell membrane proteins, including specific isoforms of the cell adhesion molecule Fasciclin 3 [60]. This spatial restriction likely facilitates non-lethal caspase activation for neuronal function modulation without triggering apoptosis.
Engineered Activatable Caspases for Screening
TEV Protease-Activatable Caspase Systems: To address the challenge of spontaneous activation in recombinant caspase preparations, researchers have developed engineered caspases where native cleavage sites are replaced with tobacco etch virus (TEV) protease recognition sequences [56]. This system enables:
- Low-background screening assays by eliminating pre-activation
- Precise temporal control over caspase activation
- Identification of zymogen-selective inhibitors
- High-throughput screening with improved Z'-factors (>0.5)
Implementation requires careful protein engineering to replace aspartate cleavage sites with ENLYFQG sequences without compromising caspase activity upon activation [56]. This approach is particularly valuable for identifying selective inhibitors that target caspase zymogens rather than active enzymes, potentially improving therapeutic specificity.
Research Reagent Solutions for Caspase Kinetic Studies
Table 2: Essential Reagents for Caspase Activation Kinetic Studies
| Reagent Category | Specific Examples | Function and Application | Technical Considerations |
|---|---|---|---|
| Fluorogenic Substrates | Ac-DEVD-AFC (caspase-3/7), Ac-VDVAD-AFC (caspase-2), Ac-IETD-AFC (caspase-8) [62] [56] | Enzyme activity measurement via fluorescence release after cleavage | Substrate selectivity is relative; confirm with specific inhibitors |
| FRET Reporters | SCAT3, SCAT9, CFP-YFP DEVD constructs [6] | Real-time caspase activity monitoring in live cells | Requires genetic manipulation; photobleaching concerns |
| Covalent Inhibitors | Z-VAD-fmk (pan-caspase), Ac-DEVD-CHO (caspase-3) [1] [62] | Mechanism-based inhibition for validation studies | Irreversible binding may alter natural kinetics |
| Activity-Based Probes | Rho-DEVD-AOMK [56] | Direct labeling and detection of active caspases | Can be used for subcellular localization |
| Expression Constructs | TEV-activatable caspases [56], Gal4-Manipulated Area-Specific CaspaseTracker (MASCaT) [60] | Controlled activation and compartment-specific monitoring | Requires verification of native-like activity |
| Live-Cell Compatible Dyes | Propidium iodide, SYTOX, Annexin V conjugates [61] | Multiplexing cell death markers with caspase activation | Timing of addition critical to avoid toxicity |
Experimental Protocols for Key Applications
Stopped-Flow Kinetics for Caspase Inhibition Studies
This protocol enables determination of individual kinetic parameters for caspase inhibition, providing insights into inhibition mechanisms and potency [62].
Materials:
- Highly pure, recombinant caspase (≥95% purity)
- Fluorogenic substrate (e.g., Ac-DEVD-AMC for caspase-3)
- Inhibitor compounds dissolved in DMSO
- Stopped-flow fluorescence spectrometer
- Assay buffer (20 mM HEPES, pH 7.5, 100 mM NaCl, 10% sucrose, 0.1% CHAPS)
Procedure:
- Prepare caspase solution in assay buffer at 2× final concentration
- Mix substrate and inhibitor at 2× final concentration in assay buffer
- Load solutions into stopped-flow instrument syringes
- Rapidly mix equal volumes (typically 50-100 μL each) and monitor fluorescence
- Collect data for 5-10 half-lives of the reaction
- Fit data to appropriate kinetic models (e.g., three-step mechanism for irreversible inhibitors)
- Determine individual kinetic parameters (k₁, k₂, k₃) for inhibition mechanism
Technical Notes:
- Maintain final DMSO concentration ≤1% to avoid enzyme inhibition
- Include control reactions without inhibitor for baseline activity
- For irreversible inhibitors, expect progressive decrease in reaction rate over time
- Account for substrate depletion in extended measurements
MASCaT System for Compartment-Specific Caspase Monitoring
The Gal4-Manipulated Area-Specific CaspaseTracker/CasExpress (MASCaT) system enables sensitive monitoring of caspase activity near specific subcellular locations, particularly the plasma membrane [60].
Materials:
- Tissue-specific Gal4 driver lines
- UAS-Fas3G constructs for membrane targeting
- CaspaseTracker reporter (caspase-sensitive GFP variant)
- Standard Drosophila culture materials
- Confocal microscopy setup with environmental control
Procedure:
- Cross appropriate Gal4 driver lines with UAS-Fas3G and CaspaseTracker lines
- Age-match adult progeny for consistent analysis
- Prepare whole-mount brain dissections in physiological buffer
- Image using confocal microscopy with minimal laser power to prevent phototoxicity
- Quantify CaspaseTracker signal intensity in specific neuronal populations
- Correlate caspase activation with behavioral outputs (e.g., olfactory attraction assays)
Validation Steps:
- Verify Fas3G expression via immunostaining
- Confirm non-lethal status through cell viability markers
- Test specificity with caspase inhibitor treatments
- Demonstrate physiological relevance through behavioral assays
Data Interpretation and Analytical Frameworks
Kinetic Modeling Approaches
Proper interpretation of caspase activation kinetics requires fitting data to appropriate mechanistic models. For initiator caspases, the induced proximity model predicts sigmoidal activation curves due to cooperative dimerization [22]. Executioner caspases typically follow hyperbolic kinetics with a rapid exponential phase due to feed-forward amplification [6]. When studying inhibition, irreversible caspase inhibitors like Z-VAD-fmk typically follow a three-step kinetic mechanism with two rapid equilibrium steps followed by a slower inactivation step [62].
Single-Cell versus Population Analysis
The "all-or-none" characteristic of executioner caspase activation necessitates single-cell analysis approaches [6] [61]. Population averaging can obscure critical heterogeneities in activation timing and extent, potentially missing biologically significant subpopulations. Advanced analytical frameworks should incorporate:
- Threshold determination for defining activation events
- Timing variability analysis (onset time, time to peak)
- Correlation with cell cycle status or differentiation state
- Spatial patterning within tissues or culture systems
Accurately measuring caspase activation kinetics remains challenging due to the fundamental structural and mechanistic differences between initiator and executioner caspases, their rapid activation dynamics, and spatial compartmentalization. However, emerging technologies like SPARKL, TurboID proximity labeling, TEV-activatable caspases, and compartment-specific reporters are progressively overcoming these limitations. The future of caspase kinetic research lies in developing increasingly sophisticated tools that can capture the full complexity of caspase activation in real-time, at single-cell resolution, and within relevant physiological contexts. These advances will be crucial for understanding the nuanced roles of caspases in both cell death and non-apoptotic processes, ultimately accelerating the development of caspase-targeted therapeutics for cancer, neurodegenerative diseases, and inflammatory disorders.
Strategies for Differentiating Apoptotic vs. Inflammatory Caspase Functions
Caspases are cysteine-dependent aspartate-specific proteases that serve as critical regulators of cell death and inflammation. Historically, caspases were classified into two distinct categories: apoptotic caspases (caspase-2, -3, -6, -7, -8, -9, -10), which oversee programmed cell death, and inflammatory caspases (caspase-1, -4, -5, -11), which drive inflammatory responses [18] [63]. However, emerging research over the past decade has revealed extensive functional overlap and crosstalk between these groups, challenging this traditional binary classification [18] [64]. For instance, apoptotic caspases such as caspase-3 and -8 can participate in lytic, inflammatory cell death pathways like pyroptosis under certain conditions [18] [64]. This complexity necessitates refined strategies for accurately differentiating caspase functions, a crucial endeavor for understanding disease mechanisms and developing targeted therapies. This guide outlines a multi-faceted approach—encompassing structural analysis, functional assessment, and advanced experimental protocols—to unequivocally distinguish apoptotic and inflammatory caspase functions within the broader context of initiator and executioner caspase structural differences.
Structural and Molecular Determinants
The fundamental strategy for differentiating caspase functions begins with a detailed analysis of their structural and molecular features. These inherent characteristics dictate their activation mechanisms, interaction partners, and ultimate biological roles.
Domain Architecture and Activation Mechanisms
Caspases are synthesized as inactive zymogens (procaspases) whose domain organization provides the first clue to their function. The presence and type of protein-protein interaction domains in the prodomain are key discriminators between initiator caspases (which trigger pathways) and executioner caspases (which implement death) [63] [3].
- Inflammatory Caspases and Apoptotic Initiators with CARD Domains: Caspase-1, -4, -5, -9, and -11 possess a Caspase Activation and Recruitment Domain (CARD) in their prodomains [63] [64]. This domain facilitates their recruitment to and activation within large multiprotein complexes—inflammasomes for inflammatory caspases and the apoptosome for caspase-9 [63] [3]. Activation proceeds via the induced proximity model, where dimerization of caspase zymogens brought into close contact leads to their auto-proteolytic cleavage and full activation [65].
- Apoptotic Initiators with DED Domains: Caspase-8 and -10 contain Death Effector Domains (DEDs) in their prodomains [63] [6]. These domains mediate their recruitment to the Death-Inducing Signaling Complex (DISC) formed by activated death receptors, such as those in the TNF family [3] [65]. Similar to CARD-containing caspases, activation follows the induced proximity model [65].
- Executioner Caspases (Short/No Pro-domain): Caspase-3, -6, and -7 have very short prodomains lacking recognizable protein interaction motifs [63] [6]. They exist as inactive dimers in healthy cells and cannot auto-activate. Their activation is strictly dependent on cleavage by upstream initiator caspases (e.g., caspase-8, -9, or -10), which results in a conformational change that creates the active protease [3] [6].
Table 1: Caspase Classification by Pro-domain and Activation Mechanism
| Classification | Caspases | Pro-domain Type | Activation Complex | Activation Mechanism |
|---|---|---|---|---|
| Inflammatory Initiators | Caspase-1, -4, -5, -11 | CARD | Inflammasome | Induced Proximity / Dimerization |
| Apoptotic Initiators | Caspase-2, -9 | CARD | Apoptosome (Caspase-9) | Induced Proximity / Dimerization |
| Apoptotic Initiators | Caspase-8, -10 | DED | DISC | Induced Proximity / Dimerization |
| Apoptotic Executioners | Caspase-3, -6, -7 | Short/None | N/A | Cleavage by Initiator Caspases |
Diagram 1: Caspase Activation Pathways. Caspase activation mechanisms are determined by pro-domain structure, directing recruitment to specific signaling complexes.
Substrate Specificity and Recognition
Beyond activation, caspases are functionally defined by their substrate specificity. Caspases cleave their targets after specific aspartic acid residues, with primary substrate selectivity determined by the four amino acids N-terminal to the cleavage site (P4-P1 positions) [18].
- Group I (Inflammatory Caspases): Caspase-1, -4, and -5 prefer the tetrapeptide sequence (W/L/Y)EHD [18]. A classic and defining substrate for caspase-1 is pro-IL-1β, which it processes to generate the active inflammatory cytokine [63].
- Group II (Apoptotic Executioners): Caspase-3 and -7 show a strong preference for the sequence DEXD [18]. A well-characterized substrate for these caspases is Poly (ADP-ribose) polymerase (PARP), whose cleavage inactivates DNA repair and facilitates cellular dismantling during apoptosis [64].
- Group III (Apoptotic Initiators): Caspase-6, -8, and -9 prefer the sequence (L/V/I)EXD [18]. For example, caspase-8 cleaves and activates caspase-3 and -7, and also cleaves the protein Bid to link the extrinsic and intrinsic apoptotic pathways [3] [6].
The critical functional differentiator in the inflammatory versus apoptotic debate is the cleavage of gasdermin proteins. Inflammatory caspases (caspase-1, -4, -5, -11) directly cleave gasdermin D (GSDMD) to trigger pyroptosis [63] [64]. In contrast, apoptotic caspase-3 cleaves gasdermin E (GSDME), which can also result in pyroptosis if GSDME is expressed [18] [64]. This illustrates how apoptotic caspases can drive inflammatory lytic death, blurring functional lines.
Table 2: Caspase Substrate Specificity and Key Downstream Effectors
| Caspase Group | Representative Caspases | Preferred Tetrapeptide Motif | Prototypic Substrates & Effectors | Primary Functional Association |
|---|---|---|---|---|
| Group I | Caspase-1, -4, -5 | (W/L/Y)EHD | pro-IL-1β, pro-IL-18, GSDMD | Inflammation, Pyroptosis |
| Group II | Caspase-3, -7 | DEXD | PARP, ICAD, GSDME | Apoptosis, (Pyroptosis via GSDME) |
| Group III | Caspase-6, -8, -9 | (L/V/I)EXD | Caspase-3, -7, Bid | Apoptosis Initiation |
Functional Characterization in Cell Death Pathways
The ultimate classification of a caspase often depends on its functional role within specific cell death pathways. Observing the morphological and biochemical outcomes of caspase activation provides a definitive strategy for differentiation.
Associated Cell Death Pathways
- Apoptosis (Traditionally "Apoptotic Caspases"): This is a non-lytic, non-inflammatory form of cell death. It is characterized by cell shrinkage, membrane blebbing, chromatin condensation, and the formation of apoptotic bodies that are efficiently cleared by phagocytes [63] [3].
- Pyroptosis (Traditionally "Inflammatory Caspases"): This is a lytic, pro-inflammatory form of cell death driven by plasma membrane pore formation, leading to cell swelling, rupture, and release of pro-inflammatory cytokines and DAMPs [18] [63].
- Necroptosis: This caspase-independent lytic cell death is paradoxically inhibited by caspase-8. When caspase-8 activity is absent or inhibited, RIPK1 and RIPK3 activate MLKL to execute necroptosis [18] [64].
- PANoptosis: This is an emerging concept describing an inflammatory, lytic cell death pathway that integrates components from apoptosis, pyroptosis, and necroptosis. PANoptosomes can simultaneously activate caspases like caspase-1, -3, -7, and -8, making simple classification impossible and highlighting the need for a pathway-based functional analysis [18].
Key Readouts for Functional Differentiation
To distinguish caspase functions experimentally, researchers should measure the following parameters:
- Membrane Integrity: Pyroptosis and necroptosis result in rapid plasma membrane rupture, which can be measured by the release of Lactate Dehydrogenase (LDH) or uptake of dyes like Propidium Iodide (PI). Apoptotic cells maintain membrane integrity until late stages [64].
- Inflammatory Cytokine Secretion: The processing and release of IL-1β and IL-18 is a hallmark of inflammatory caspase activity (particularly caspase-1) and pyroptosis [63].
- Effector Molecule Cleavage: Immunoblotting for the cleavage of specific effector proteins is a direct method. This includes:
- Morphological Analysis: Imaging techniques (e.g., time-lapse microscopy, electron microscopy) can reveal characteristic shapes of cell death: apoptotic bodies (apoptosis) versus cell swelling and lysis (pyroptosis/necroptosis) [3].
Experimental Strategies and Methodologies
A robust strategy for differentiating caspase functions relies on a combination of genetic, pharmacological, and biochemical approaches.
Core Experimental Workflow
A systematic workflow for characterizing an unknown caspase function or a cell death trigger involves the sequential application of specific tools and assays.
Diagram 2: Experimental Workflow for Caspase Function Characterization. A stepwise approach to definitively identify cell death pathways and responsible caspases.
Detailed Methodologies
1. Pharmacological Inhibition:
- Protocol: Pre-treat cells with caspase inhibitors 1-2 hours before applying the death-inducing stimulus.
- Pan-caspase Inhibitor: z-VAD-fmk (e.g., 20-50 µM). Abrogation of cell death confirms a caspase-dependent process.
- Specific Inhibitors: Use selective inhibitors for individual caspases (e.g., caspase-1 inhibitor Z-YVAD-fmk; caspase-3 inhibitor Z-DEVD-fmk) to pinpoint involvement. Residual cell death upon specific inhibition suggests involvement of other caspases or alternative pathways [66].
- Data Interpretation: If z-VAD-fmk blocks death, but a caspase-1 specific inhibitor does not, the pathway likely depends on apoptotic caspases. Conversely, if both block death, caspase-1 is involved.
2. Genetic Manipulation:
- CRISPR/Cas9 or siRNA Knockdown: Generate knockout cell lines or transiently knock down expression of specific caspases (e.g., CASP1, CASP3, CASP8, GSDMD).
- Protocol: Transfert cells with specific siRNA or perform CRISPR editing. Validate knockout/knockdown efficiency by Western blot. Apply death stimulus and assess viability and phenotypic readouts (LDH, morphology) [66].
- Data Interpretation: Resistance to death in CASP1 or GSDMD KO cells points to pyroptosis. Resistance in CASP3/7 KO cells points to apoptosis. This provides more specific evidence than pharmacology alone.
3. Biochemical Analysis of Cleavage Events:
- Western Blot Protocol:
- Harvest stimulated and control cells at various time points.
- Perform SDS-PAGE and immunoblotting with antibodies against:
- Data Interpretation: Detection of cleaved GSDMD is a signature of inflammatory caspase or caspase-8 activity. Cleaved PARP and caspase-3 indicate apoptotic executioner activity.
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents for Differentiating Caspase Functions
| Reagent Category | Specific Examples | Function & Application |
|---|---|---|
| Pharmacological Inhibitors | z-VAD-fmk (pan-caspase), Z-YVAD-fmk (caspase-1), Z-DEVD-fmk (caspase-3) | Chemical tool to probe dependency on specific caspase activities in a pathway. |
| Genetic Tools | siRNA, CRISPR/Cas9 knockout constructs | To definitively ablate gene function of specific caspases (e.g., CASP1, CASP3, CASP8) or gasdermins (e.g., GSDMD, GSDME). |
| Antibodies for Immunoblotting | Anti-cleaved Caspase-3, Anti-cleaved PARP, Anti-GSDMD (Full length & N-terminal), Anti-GSDME, Anti-IL-1β | To detect proteolytic processing and activation of key caspases and their substrates. |
| Cell Death Assays | Propidium Iodide (PI), Lactate Dehydrogenase (LDH) Release Assay | To quantify lytic cell death (pyroptosis/necroptosis) by measuring membrane integrity. |
| Cytokine Detection | ELISA Kits for IL-1β, IL-18 | To measure the specific output of inflammatory caspase (especially caspase-1) activation. |
Integration and Therapeutic Implications
Successfully differentiating caspase functions requires integrating data from all the above strategies. The field is moving beyond the rigid apoptotic/inflammatory dichotomy toward a context-dependent model where the cellular outcome is determined by the specific caspase activated, the stimulus, the cellular environment, and the repertoire of available substrates (like gasdermins) [18] [64].
This refined understanding has profound implications for drug discovery. Dysregulated caspase activity is implicated in cancer, neurodegenerative diseases, and autoimmune and inflammatory disorders [18] [3]. For example, targeting caspase-1 with specific inhibitors may be a strategy for autoinflammatory diseases, while promoting caspase-3 activation could be desirable in cancer therapy. However, the discovery that caspase-3 can also drive inflammatory lytic death via GSDME underscores the need for highly specific therapeutic strategies that consider this functional complexity [18] [6]. A multifaceted differentiation strategy is, therefore, not just an academic exercise but a critical prerequisite for developing safe and effective caspase-targeted therapies.
Technical Considerations for Studying Caspases in Complex Biological Systems
Caspases (cysteine-aspartic proteases) are a family of evolutionarily conserved cysteine proteases that cleave their substrates after aspartic acid residues, serving as critical mediators of programmed cell death (PCD) and cellular homeostasis [5] [68]. In the context of initiator versus executioner caspase structural biology research, understanding their distinct activation mechanisms and domain architectures is fundamental. These enzymes are synthesized as inactive zymogens (procaspases) that require proteolytic activation, typically through cleavage at specific aspartic acid residues themselves [68]. The central role of caspases extends beyond apoptosis to include pyroptosis, necroptosis, and various non-apoptotic cellular remodeling events, making their study in complex biological systems particularly challenging [1] [13].
The classification of caspases has traditionally separated them into apoptotic and inflammatory categories; however, emerging evidence reveals significant functional overlap, with apoptotic caspases participating in inflammatory lytic cell death pathways [1]. A more structurally informed classification system categorizes caspases based on their pro-domain architecture into three groups: CARD-domain containing, DED-domain containing, and short/no pro-domain caspases [1]. This structural distinction forms the basis for understanding their differential activation mechanisms and signaling pathways in complex biological systems.
Structural Biology of Caspases: Initiator versus Executioner Configurations
Domain Architecture and Activation Mechanisms
All procaspases share a common structural organization comprising an N-terminal pro-domain, a large subunit (~20 kDa), and a small subunit (~10 kDa) [68]. The critical structural distinction between initiator and executioner caspases lies in their pro-domain length and composition, which dictates their activation mechanisms and places them within specific signaling networks.
Initiator caspases (caspases-2, -8, -9, -10) contain long pro-domains with protein-protein interaction motifs, either CARD (caspase activation and recruitment domain) or DED (death effector domain) [1] [5]. These domains enable recruitment to and activation within large multiprotein signaling complexes such as the FADDosome (extrinsic apoptosis), apoptosome (intrinsic apoptosis), or inflammasome (pyroptosis) [5]. This proximity-induced dimerization model allows initiator caspases to undergo autocatalytic activation without requiring cleavage by upstream proteases.
Executioner caspases (caspases-3, -6, -7) typically contain short pro-domains and are activated through direct proteolytic cleavage by initiator caspases or other executioner caspases [68]. This cleavage between the large and small subunits enables the formation of the active heterotetramer consisting of two large and two small subunits [68]. The active site of mature caspases features a catalytic dyad of cysteine and histidine residues positioned within a substrate specificity pocket that contains an arginine residue critical for recognizing the P1 aspartic acid in substrates [68].
Table 1: Structural and Functional Classification of Mammalian Caspases
| Caspase | Pro-domain Type | Category | Activation Complex | Primary Functions |
|---|---|---|---|---|
| Caspase-1 | CARD | Inflammatory | Inflammasome | Pyroptosis, cytokine maturation |
| Caspase-2 | CARD | Initiator | PIDDosome | Cell cycle, DNA damage response |
| Caspase-3 | Short | Executioner | Cleaved by initiators | Apoptosis execution, substrate cleavage |
| Caspase-4/5/11 | CARD | Inflammatory | Non-canonical inflammasome | Pyroptosis via GSDMD cleavage |
| Caspase-6 | Short | Executioner | Cleaved by initiators | Apoptosis, lamin cleavage |
| Caspase-7 | Short | Executioner | Cleaved by initiators | Apoptosis, PARP cleavage |
| Caspase-8 | DED | Initiator | FADDosome | Extrinsic apoptosis, necroptosis regulation |
| Caspase-9 | CARD | Initiator | Apoptosome | Intrinsic apoptosis |
| Caspase-10 | DED | Initiator | FADDosome | Extrinsic apoptosis, immune regulation |
| Caspase-12 | CARD | Inflammatory | ER stress complex | ER stress-induced apoptosis |
Active Site Architecture and Substrate Recognition
The enzymatic activity of caspases depends on a structurally conserved active site that recognizes tetra-peptide sequences on substrates with high specificity. The substrate binding pocket accommodates four amino acids (P4-P3-P2-P1) with cleavage occurring after the P1 aspartic acid residue [68]. Different caspases exhibit distinct preferences for these positions, creating a hierarchy of substrate recognition that determines their specific roles in cellular pathways.
Structural studies using X-ray crystallography (with numerous PDB entries available for caspases-1, -2, -3, -7, -8, and -9) have revealed how exosites outside the primary active site contribute to substrate selection and catalytic efficiency [1]. For example, caspase-7 utilizes an exosite to promote poly(ADP ribose) polymerase 1 (PARP-1) proteolysis [13]. These structural nuances explain how executioner caspases with similar active site architectures can have distinct substrate profiles and functional outputs in complex biological systems.
Methodological Approaches for Caspase Research
Monitoring Caspase Activity in Live Cells
Advanced biosensor technologies enable real-time monitoring of caspase activation dynamics in live cells. Fluorescence anisotropy-based FRET biosensors allow simultaneous measurement of extrinsic (caspase-8), intrinsic (caspase-9), and effector (caspase-3/7) caspase activities within single living cells, overcoming intercellular variability [69]. These biosensors utilize spectrally separated fluorophores and measure changes in fluorescence anisotropy resulting from caspase-mediated cleavage of specific peptide linkers.
Experimental Protocol: Co-imaging Caspase Activity by Fluorescence Anisotropy Microscopy
- Biosensor Design: Engineer three spectrally distinct biosensors containing caspase-specific cleavage sequences (DEVD for caspase-3/7, IETD for caspase-8, LEHD for caspase-9) flanked by FRET donor-acceptor pairs.
- Cell Transfection: Introduce biosensor constructs into target cells using appropriate transfection methods (lipofection, electroporation, or viral transduction).
- Stimulation and Imaging: Expose cells to apoptotic stimuli while performing time-lapse anisotropy microscopy using appropriate excitation/emission filters for each biosensor.
- Data Analysis: Calculate anisotropy values (r) using the formula: r = (I∥ - I⟂)/(I∥ + 2I⟂), where I∥ and I⟂ represent parallel and perpendicular emission intensities relative to polarized excitation.
- Kinetic Modeling: Derive time of maximum activity for each caspase from anisotropy measurements and correlate relative activation times to refine signaling network models.
This approach provides valuable insights into signal propagation kinetics and reveals cell-to-cell heterogeneity in caspase activation patterns that would be obscured in population-level assays.
Proteomic Approaches for Substrate Identification
Global proteomic methods have revolutionized the identification of caspase substrates, revealing hundreds to thousands of cleavage events during apoptosis [13]. These approaches include:
- N-terminal TAILS (Terminal Amine Isotopic Labeling of Substrates): Enriches for N-terminal peptides to identify cleavage sites.
- COFRADIC (Combined FRActional Diagonal Chromatography): Separates protein fragments based on chromatographic properties.
- Substrate Phage Display: Libraries of random peptides displayed on phage particles to determine cleavage specificity.
Experimental Protocol: Global Mapping of Caspase Cleavage Events
- Sample Preparation: Treat cells with apoptotic stimuli or directly add active caspases to cell extracts. Include control samples with caspase inhibitors.
- Protein Extraction and Digestion: Lyse cells and process proteins for mass spectrometry analysis.
- N-terminal Enrichment: Use positive selection methods (e.g., charge-based) or negative selection (e.g., blocking internal amines) to enrich for native and caspase-generated N-termini.
- Mass Spectrometry Analysis: Perform LC-MS/MS on enriched samples using high-resolution instruments.
- Bioinformatic Analysis: Identify cleavage sites by searching spectra against protein databases and apply algorithms to distinguish caspase-specific cleavage events from other processing.
These proteomic studies have revealed that the number of substrate targets varies widely between caspases, from few dozen for caspases-4, -5, -9, and -14 to hundreds for caspases-1, -2, -3, -6, -7, and -8 [13]. Furthermore, cleavage rates vary over 500-fold within substrate cohorts for individual caspases, indicating a hierarchy of preferred targets.
Table 2: Caspase Substrate Specificity and Proteomic Profiling
| Caspase | Preferred Cleavage Motif | Approximate Number of Identified Substrates | Key Physiological Substrates |
|---|---|---|---|
| Caspase-1 | WEHD | ~100+ | pro-IL-1β, pro-IL-18, GSDMD |
| Caspase-2 | VDVAD | ~200+ | Bid, Golgin-160, RIPK1 |
| Caspase-3 | DEVD | ~500+ | PARP-1, ICAD, Lamin A, GSDME |
| Caspase-6 | VEID | ~200+ | Lamin A/C, Caspase-8, GSDMB |
| Caspase-7 | DEVD | ~200+ | PARP-1, Caspase-6, GSDMD |
| Caspase-8 | LETD | ~300+ | Bid, Caspase-3, -7, RIPK1, GSDMC |
| Caspase-9 | LEHD | Few dozen | Caspase-3, -7 |
| Caspase-10 | IEAD | Limited data | Caspase-3, -4, -7 |
Technical Challenges in Complex Biological Systems
Specificity and Compensation in Caspase Networks
The overlapping substrate specificities among caspases present significant challenges in determining individual caspase functions in complex systems. For example, caspases-3 and -7 share preferences for DEXD motifs, while caspases-8 and -10 have similar recognition patterns [13] [68]. This redundancy can lead to compensatory mechanisms where knocking out one caspase results in upregulation of another, obscuring phenotypic analysis.
Mitigation Strategies:
- Use multiple, specific caspase inhibitors in combination rather than single gene knockouts.
- Employ caspase-specific substrates with distinct optimal cleavage motifs.
- Implement activity-based protein profiling (ABPP) using specific active-site probes.
- Analyze temporal activation patterns to establish hierarchy rather than relying solely on endpoint measurements.
Context-Dependent Caspase Functions
Caspases display remarkable context-dependent functionality, particularly evident in their roles across different cell death modalities. For instance, caspase-8 serves as a molecular switch between apoptosis, necroptosis, and pyroptosis depending on cellular conditions and stimulus [5]. Similarly, caspase-3 can promote either non-inflammatory apoptosis or highly inflammatory pyroptosis depending on which gasdermin protein it cleaves (GSDME versus GSDMB/GSDMD) [1] [5].
Experimental Considerations:
- Characterize caspase functions in multiple cell types and under varying stimulation conditions.
- Monitor multiple cell death parameters simultaneously (membrane integrity, caspase activation, cytokine release).
- Consider species-specific differences, particularly for inflammatory caspases (human caspases-4/5 versus rodent caspase-11).
Visualization of Caspase Signaling Pathways
Caspase Signaling Pathways Cross-Talk
Research Reagent Solutions for Caspase Studies
Table 3: Essential Research Reagents for Caspase Experimental Workflows
| Reagent Category | Specific Examples | Technical Function | Application Context |
|---|---|---|---|
| Fluorescent Biosensors | DEVD-, IETD-, LEHD-based FRET reporters | Caspase activity sensing via cleavage-induced fluorescence dequenching | Live-cell caspase activity kinetics and multiplexed imaging [69] |
| Active Recombinant Caspases | Human caspases-1, -3, -8, -9 | Standardized enzyme preparations for in vitro assays | Substrate specificity profiling and biochemical characterization [13] |
| Caspase Inhibitors | Z-VAD-FMK (pan-caspase), VX-765 (caspase-1), Ac-DEVD-CHO (caspase-3) | Irreversible (FMK) or reversible (aldehyde) active site blockade | Pathway dissection and validation of caspase-dependent phenotypes [1] |
| Activity-Based Probes | Biotin- or fluorophore-conjugated ABPs | Covalent active site labeling for enrichment/detection | Identification of active caspases in complex mixtures and in situ visualization |
| Antibody Panels | Cleavage-specific (e.g., anti-cleaved caspase-3), active conformations | Selective recognition of activated caspases or cleavage products | Immunodetection, Western blotting, and immunohistochemistry |
| Proteomic Kits | TAILS, COFRADIC, N-terminal enrichment kits | System-wide identification of protease cleavage sites | Global substrate discovery and degradomics [13] |
The structural and functional complexity of caspase networks demands integrated methodological approaches that account for their overlapping specificities, context-dependent functions, and dynamic activation patterns. Successful experimental design must incorporate multiple complementary techniques—from structural biology and live-cell imaging to global proteomics—to overcome the limitations of any single approach. The continuing development of more specific biosensors, inhibitors, and analytical methods will enable deeper understanding of how initiator and executioner caspase structural differences translate to their distinct yet interconnected functional roles in health and disease. As caspase-targeted therapeutics advance, these technical considerations will become increasingly critical for translating basic research into clinical applications.
Comparative Structural Analysis and Functional Validation Across Caspase Families
Within the broader context of research on initiator versus executioner caspase structural differences, a critical focal point is the direct comparison of their active sites. This structural divergence is the fundamental determinant of their distinct roles in the apoptotic cascade. Initiator caspases (e.g., caspase-8, -9) function as upstream signaling integrators that launch the cell death pathway, while executioner caspases (e.g., caspase-3, -6, -7) act as downstream effectors that carry out the systematic dismantling of the cell by cleaving hundreds of cellular substrates [22] [68] [9]. This in-depth technical guide delineates the structural and molecular mechanisms underlying these functions, focusing on active site architecture, substrate recognition, and activation kinetics, which are pivotal for informed drug development strategies targeting specific caspases.
Structural Biochemistry and Activation Mechanisms
The activation mechanisms of initiator and executioner caspases represent one of the most fundamental structural distinctions between these two classes.
Activation States and Quaternary Structures
Initiator Caspases (Monomer to Dimer): Initiator caspases such as caspase-8 and -9 preexist in the cytosol as inactive monomers [22]. Their activation is governed by the "induced proximity" model, where adapter proteins (e.g., FADD for caspase-8, Apaf-1 for caspase-9) facilitate their dimerization into active enzymes [22] [70]. This dimerization occurs via homophilic interactions between death folds—CARD domains in caspase-9 or DED domains in caspase-8—located in their long prodomains [22] [18] [9]. A critical rule is that cleavage between their large and small subunits is not required for initial enzymatic activity but serves to stabilize the active dimer, particularly for caspases like caspase-8 [22]. Caspase-9 is an exception, where cleavage does not confer stability [22].
Executioner Caspases (Dimer to Cleaved Dimer): In stark contrast, executioner caspases such as caspase-3 and -7 preexist as inactive dimers [22]. These zymogens are functionally inert because their catalytic sites are structurally constrained. Activation requires proteolytic cleavage at specific aspartate sites between the large and small subunits, a step almost invariably catalyzed by an active initiator caspase [22] [71] [9]. This cleavage event triggers a conformational change that allows the loops forming the substrate-specificity pocket to snap into their correct positions, creating the mature, maximally functional protease [22].
The diagram below illustrates these distinct activation pathways.
Active Site Architecture and Substrate Specificity
While all caspases share a canonical fold and a cysteine-histidine catalytic dyad that confers specificity for aspartic acid at the P1 position of substrates, key variations in their active site pockets determine distinct substrate preferences [68] [72] [18].
The substrate binding cleft is commonly described using the Schechter and Berger nomenclature, where the substrate residues are designated P1, P2, P3, P4, etc., N-terminal to the scissile bond, and the enzyme's corresponding binding subsites are labeled S1, S2, S3, S4, etc. [68]. Structural analyses, including homology modeling, reveal that while the S1 pocket and catalytic dyad are nearly identical across caspases, the S2-S4 subsites display considerable diversity [72].
S4 Subsite: The S4 pocket is a primary determinant of specificity.
- Caspase-1, -4, -5, -11 possess a large hydrophobic S4 pocket that prefers bulky hydrophobic residues like Tryptophan (W) [72] [18].
- Caspase-3 and -7 have a highly specific S4 conformation that favors Aspartate (D) [72]. This is the basis for their shared preference for the DEVD sequence.
- Caspase-6, -8, -9, -10 have hybrid S4 structures with little inherent specificity, generally accommodating Valine (V) or Leucine (L) [72] [73].
S2 and S3 Subsites: The S2 pockets can be categorized into two groups: a smaller group (caspase-3, -6, -7) limited to small P2 residues like Valine (V) or Alanine (A), and a larger group (all other caspases) that can accommodate bulkier residues [72]. A conserved Arginine in the S3 subsite suggests a universal preference for Glutamate (E) at P3, though caspase-2 is a notable exception capable of accommodating charged residues [72].
P5 Recognition: Recent studies have extended recognition to the P5 position. Caspase-3 and -6 can recognize a P5 residue, which enhances substrate hydrolysis, while caspase-7 and -8 show no such preference [73]. In caspase-3, the P5 main-chain is anchored by Ser209, and the P5 leucine side-chain interacts with Phe250 and Phe252 [73].
Table 1: Comparative Substrate Specificity Profiles of Key Human Caspases
| Caspase | Classification | Preferred Tetrapeptide Motif (P4-P1) | P5 Preference | Structural Basis for Specificity |
|---|---|---|---|---|
| Caspase-1 | Inflammatory | WEHD [18] | N/D | Large hydrophobic S4 pocket [72] |
| Caspase-2 | Initiator | DEXD [73] | Essential [73] | Unique S3 that accommodates charged residues [72] |
| Caspase-3 | Executioner | DEXD (DEVD) [68] [73] | Hydrophobic (e.g., L) [73] | S4 specific for Asp; S5 binding via Ser209, Phe250/252 [72] [73] |
| Caspase-6 | Executioner | L/VEXD (VEID) [73] | Polar (e.g., Q) [73] | Hybrid S4; Polar S5 interactions with Lys265 [73] |
| Caspase-7 | Executioner | DEXD (DEVD) [68] [73] | None [73] | S4 specific for Asp; Lacks P5-binding residues [73] |
| Caspase-8 | Initiator | L/VEXD (IETD) [73] | None [73] | Hybrid S4; Lacks P5-binding residues and loop-4 [73] |
| Caspase-9 | Initiator | L/VEXD (LEHD) [73] | N/D | Hybrid S4; Additional Arg enhances P3 Glu binding [72] |
Table 2: Summary of Fundamental Structural and Functional Differences
| Feature | Initiator Caspases | Executioner Caspases |
|---|---|---|
| Examples | Caspase-8, -9, -10 [70] [9] | Caspase-3, -6, -7 [70] [9] |
| Physiological Role | Signal initiation and amplification [22] | Mass substrate proteolysis [68] |
| Native State | Inactive monomers [22] | Inactive dimers [22] |
| Primary Activation Trigger | Dimerization (Induced Proximity) [22] | Cleavage between large and small subunits [22] |
| Prodomain | Long (contains CARD or DED) [22] [9] | Short [22] [9] |
| Cleavage for Activity | Not required for initial activity; stabilizes dimer [22] | Absolutely required for activity [22] |
| Representative Activating Complex | DISC (Casp-8), Apoptosome (Casp-9) [9] | N/A (Activated directly by initiators) |
Experimental Analysis of Caspase Structure and Function
A multi-faceted approach is required to delineate the structural and functional properties of caspases. The following section outlines key methodologies and reagents central to this field.
Key Experimental Protocols
1. Determination of Enzyme Kinetics and Specificity:
- Objective: To quantitatively measure the activity and substrate preference of purified caspases.
- Methodology: Recombinant caspases are expressed in E. coli and purified via affinity chromatography (e.g., C-terminal His-tag using Ni-NTA) [73]. Enzymatic activity is measured using a colorimetric assay. Caspases are incubated in an assay buffer (e.g., 50 mM HEPES, pH 7.5, 100 mM NaCl, 0.1% CHAPS, 10% glycerol, 10 mM DTT) with para-nitroanilide (pNA)-conjugated peptide substrates (e.g., Ac-DEVD-pNA for caspase-3) [73]. Upon cleavage, the release of free pNA is monitored spectrophotometrically at 405 nm. The Michaelis-Menten constants (K~M~) and catalytic turnover (k~cat~) are calculated from initial velocity measurements at varying substrate concentrations to define specificity constants (k~cat~/K~M~) [73].
2. Structural Elucidation via X-ray Crystallography:
- Objective: To obtain high-resolution, three-dimensional structures of caspases, often in complex with substrates or inhibitors.
- Methodology: Purified caspase proteins are crystallized. The crystals are then exposed to X-rays, and the resulting diffraction patterns are used to solve the protein's atomic structure [72] [73]. This technique has been instrumental in visualizing the active site conformations of both inactive procaspases (e.g., procaspase-7) and their active forms, revealing the critical conformational changes upon activation [22]. Co-crystallization with peptide-based inhibitors (e.g., Ac-LDESD-CHO) allows for the direct mapping of substrate-enzyme interactions within the active site cleft [73].
3. Global Substrate Identification using Proteomics:
- Objective: To identify the full repertoire of cellular proteins cleaved during apoptosis (the caspase "degradome").
- Methodology: Apoptosis is induced in cell cultures, and the proteome is analyzed at various time points using mass spectrometry (MS)-based techniques. Methods like N-terminal COFRADIC (Combined FRActional Diagonal Chromatography) selectively label and isolate protein N-terminal, allowing for the systematic identification of in vivo caspase cleavage sites [13]. This approach has revealed that executioner caspases like caspase-3 can cleave hundreds to over a thousand cellular substrates, whereas initiator caspases have a more limited substrate pool [13].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Caspase Research
| Research Reagent | Function and Application | Example Use-Case |
|---|---|---|
| Recombinant Caspases | Purified enzymes for in vitro biochemical studies, kinetics, and structural biology. | Expression in E. coli followed by Ni-NTA purification for kinetic assays with synthetic substrates [73]. |
| Synthetic Peptide Substrates | Chromogenic or fluorogenic tetrapeptide/pentapeptide conjugates to probe enzyme activity and specificity. | Ac-DEVD-pNA (colorimetric) for caspase-3/7; Ac-VEID-pNA for caspase-6; used to determine kinetic parameters [73]. |
| Activity-Based Probes (ABPs) | Biotin- or fluorophore-labeled irreversible inhibitors that covalently tag active caspases in complex proteomes. | Identifying active caspases in cell lysates; monitoring caspase activation during apoptosis in live cells. |
| Peptide Inhibitors | Reversible (aldehyde, e.g., Ac-DEVD-CHO) or irreversible (fluoromethyl ketone, e.g., Z-VAD-FMK) inhibitors for functional validation. | Confirming the specific role of a caspase in a proteolytic event by inhibiting its activity in cellulo [73]. |
| Crystallography Reagents | Chemicals for protein crystallization and peptide inhibitors for co-crystallization. | Solving the 3D structure of caspase-3 in complex with the pentapeptide inhibitor Ac-LDESD-CHO [73]. |
The diagram below summarizes the logical workflow for a structural and functional study of a caspase.
Implications for Drug Discovery and Therapeutic Targeting
The precise understanding of caspase active site differences provides a rational foundation for therapeutic intervention. Dysregulated caspase activity is implicated in a spectrum of diseases, including neurodegeneration, cancer, and inflammatory disorders [18] [73] [9].
- Specificity is Paramount: The challenge in developing caspase-targeting drugs lies in achieving high specificity to avoid unintended on-target effects. For example, a pan-caspase inhibitor could simultaneously block desired apoptosis in cancer cells and detrimental inflammation in surrounding tissue. The structural insights detailed in this guide offer a roadmap.
- Exploiting Active Site Differences: The distinct S4 and S5 subsites of different caspases present unique chemical environments that can be targeted. For instance, designing a compound that exploits the P5-binding pocket of caspase-3 (involving Ser209, Phe250, Phe252) could yield a highly selective inhibitor for this executioner caspase, potentially useful in conditions of excessive apoptosis like spinal muscular atrophy [73].
- Targeting Non-Conserved Exosites: Beyond the conserved active site, some caspases utilize exosites—secondary substrate-binding sites remote from the catalytic cleft—to enhance specificity and catalysis. Caspase-7, for example, uses an exosite to promote the efficient cleavage of PARP-1 [13] [71]. These exosites represent promising targets for allosteric inhibitors that could modulate caspase activity with exceptional specificity, offering a new avenue for drug development that moves beyond active-site competition [13].
In conclusion, the direct structural comparison of initiator and executioner caspase active sites reveals a sophisticated evolutionary adaptation that ensures the fidelity and amplification of the cell death signal. The distinct activation mechanisms, quaternary structures, and substrate recognition profiles are all directly encoded in their three-dimensional architectures. A deep understanding of these differences is not only critical for fundamental biochemical knowledge but also serves as an indispensable guide for the rational design of next-generation, highly specific caspase-targeted therapeutics for a wide range of human diseases.
Within the intricate machinery of programmed cell death and inflammation, caspases function as central conductors, translating proteolytic activity into decisive cellular outcomes. The functional consequences of caspase activation are inextricably linked to their precise cleavage of hundreds of cellular protein substrates [35]. This review delves into the molecular logic governing caspase substrate specificity, contrasting the profiles of initiator and executioner caspases. Understanding these profiles—defined by recognition motifs and catalytic efficiencies—is fundamental to elucidating how these enzymes drive distinct biological processes, from non-lytic apoptosis to highly inflammatory pyroptosis [1] [18]. The burgeoning field of caspase biology continues to reveal that substrate cleavage is not a monolithic event but a nuanced and coordinated process, the details of which are critical for therapeutic intervention in cancer, neurodegenerative diseases, and inflammatory disorders [1].
Caspase Classification and Physiological Roles
Caspases are a family of cysteine-dependent aspartate-specific proteases that are expressed as inactive zymogens (procaspases) and undergo activation through dimerization and proteolytic cleavage [35]. Traditionally, caspases have been classified based on their primary physiological functions.
- Inflammatory Caspases: This group includes caspase-1, -4, -5, and -11 (in mice). They are primarily involved in the maturation of pro-inflammatory cytokines such as IL-1β and in the cleavage of gasdermin D (GSDMD) to trigger the lytic cell death pathway of pyroptosis [1] [18].
- Initiator Caspases (Apoptotic): Comprising caspase-2, -8, -9, and -10, these caspases are characterized by long pro-domains (either CARD or DED) that enable recruitment to and activation within large multimolecular signaling complexes like the apoptosome or the Death-Inducing Signaling Complex (DISC) [35] [74]. They function upstream to initiate the apoptotic cascade.
- Executioner Caspases (Apoptotic): This group includes caspase-3, -6, and -7, which possess short pro-domains. They are activated by initiator caspases and are responsible for the systematic cleavage of a broad repertoire of structural and regulatory proteins, leading to the characteristic morphological changes of apoptosis [35] [25].
However, recent research underscores the limitations of a rigid functional classification. For instance, the apoptotic executioner caspase-3 can cleave gasdermin E (GSDME) to drive pyroptosis, and caspase-8 is a critical component in inflammatory PANoptosis [1] [18]. Consequently, alternative classification systems based on substrate specificity or pro-domain structure are increasingly valuable for understanding the nuanced roles of these enzymes.
Substrate Recognition Motifs Across the Caspase Family
Caspase substrate recognition is typically described using the Schechter and Berger nomenclature, where the substrate residues N-terminal to the scissile bond are designated P4-P3-P2-P1↓ and the cleavage occurs C-terminal to an aspartic acid in the P1 position [35]. Early characterization using fluorogenic tetrapeptide libraries established baseline specificity profiles for each caspase [35] [13]. Subsequent proteomic studies profiling cleavage in native protein contexts have refined these motifs, revealing that while short peptide motifs are informative, native protein folding and the presence of exosites can profoundly influence cleavage efficiency and site accessibility [35].
Table 1: Caspase Substrate Preference Motifs from Peptide and Proteomic Studies
| Caspase | Historical Peptide Substrate Motif | Proteomic-Derived Protein Substrate Motif | Primary Role |
|---|---|---|---|
| Caspase-1 | WEHD | YVHD / FESD | Inflammatory |
| Caspase-2 | VDVAD | XDEVD | Initiator |
| Caspase-3 | DEVD | DEVD | Executioner |
| Caspase-4 | (W/L)EHD | — | Inflammatory |
| Caspase-5 | (W/L)EHD | — | Inflammatory |
| Caspase-6 | VQVD | VEVD | Executioner |
| Caspase-7 | DEVD | DEVD | Executioner |
| Caspase-8 | LETD | XEXD | Initiator |
| Caspase-9 | (W/L)EHD | — | Initiator |
| Caspase-10 | LEHD | LEHD | Initiator |
| Caspase-14 | WEHD | VSQD / HSED | Epithelial Differentiation |
The data in Table 1 highlights several key points. First, the executioner caspases-3 and -7 share a strong preference for DEVD motifs, which is reflected in both peptide and proteomic analyses [35]. Second, initiator caspases like -8 and -10 prefer a bulky hydrophobic residue (Leu, Val) at the P4 position (e.g., LETD, LEHD) [35] [18]. Third, inflammatory caspases like -1 prefer an aromatic residue at P4 (WEHD) [35]. The "X" in proteomic-derived motifs indicates a variable residue, underscoring the greater diversity of sequences cleaved in native proteins compared to optimized peptide substrates.
Structural Basis for Specificity and Efficiency
The distinct substrate preferences of initiator and executioner caspases are rooted in their three-dimensional structures, particularly the architecture of their active sites.
All mature caspases form head-to-tail homodimers, with each monomer contributing one active site [35]. The catalytic site contains a conserved cysteine-histidine dyad. The S1 pocket, which accommodates the P1 aspartate, is a defining and deeply conserved feature across all caspases [18]. The key structural differences that dictate specificity lie in the S2-S4 subsites.
Executioner caspases like caspase-3 and -7 possess a shallow, hydrophilic S4 pocket that optimally accommodates the negatively charged side chain of aspartic acid in the P4 position of the DEVD motif [35]. In contrast, initiator caspases such as -8 and -9 have a deeper, hydrophobic S4 pocket that preferentially binds large aliphatic or aromatic residues like leucine or valine [35] [18]. This fundamental difference in the S4 pocket is a major determinant for the substrate cohort targeted by each caspase type.
Beyond the core active site, exosites—secondary substrate-binding sites distinct from the catalytic cleft—play a crucial role in enhancing cleavage efficiency and specificity for certain native protein substrates. For example, caspase-7 uses an exosite to promote the efficient proteolysis of poly(ADP-ribose) polymerase 1 (PARP-1) [35]. This mechanism allows caspases to selectively cleave critical substrates with high efficiency amidst a pool of hundreds of potential targets, adding a layer of regulation that cannot be predicted from short peptide sequences alone.
Quantitative Profiling of Cleavage Efficiencies
Modern proteomic technologies, such as N-terminomics, have enabled the global identification of native caspase substrates in complex cellular lysates or live cells, moving beyond inferred specificities from peptide libraries [35] [74]. These studies reveal that each caspase has a preferred substrate cohort, with cleavage rates varying by over 500-fold within the group of targets for a single caspase [35]. This indicates a hierarchy of substrate cleavage, where a subset of critical proteins is processed rapidly and efficiently to execute cell death.
A striking example of the power of these approaches is the recent deorphanization of caspase-9 substrates. While caspase-3 has a vast substrate pool of over 900 proteins, caspase-9 was long thought to have very few substrates beyond caspase-3 and -7 [74]. However, deep profiling via subtiligase N-terminomics identified 124 protein substrates for caspase-9, more than half of which are shared with caspase-3, but often cleaved at distinct sites [74]. This suggests a broader and more complex role for caspase-9, involving both the activation of executioners and the direct proteolysis of key apoptotic substrates.
Table 2: Comparative Substrate Pool and Efficiencies of Select Caspases
| Caspase | Type | Number of Identified Substrates | Cleavage Rate Variation | Key High-Efficiency Substrates |
|---|---|---|---|---|
| Caspase-3 | Executioner | ~900+ [74] | >500-fold [35] | PARP-1, Caspase-6, Caspase-9, ICAD |
| Caspase-9 | Initiator | ~124 [74] | Not Quantified | Caspase-3, Caspase-7, Vimentin, HDAC7 |
| Caspase-7 | Executioner | Hundreds [35] | >500-fold [35] | PARP-1, Caspase-6, Lamin A |
| Caspase-8 | Initiator | Hundreds [35] | >500-fold [35] | Caspase-3, Caspase-7, BID, RIPK1 |
Methodologies for Profiling Substrate Specificity
Experimental Workflow for Global Substrate Identification
The "reverse N-terminomics" workflow is a powerful method for unequivocally attributing cleavage events to a specific, exogenously added caspase under controlled conditions [74]. The following diagram and protocol outline this key technique.
Diagram Title: Reverse N-Terminomics Workflow
Detailed Protocol: Reverse N-terminomics [74]
- Lysate Preparation: Cells (e.g., Jurkat T-cells) are lysed in a buffer containing broad-spectrum protease inhibitors and iodoacetamide to alkylate and inhibit endogenous cysteine proteases.
- Caspase Incubation: Purified, active caspase is added to the clarified lysate. For initiator caspases like caspase-9, caspase-deficient cell lines (e.g., Jurkat JMR) may be used, and executioner caspase activity is blocked with inhibitors like Ac-DEVD-fmk to isolate the activity of the caspase of interest.
- Proteolytic Reaction: The reaction is incubated to allow substrate cleavage, generating new, neo-N-termini with free amine groups.
- Subtiligase Labeling: The engineered ligase, subtiligase, is used to specifically biotinylate these newly generated N-termini using a biotinylated peptide ester tag. This step selectively labels cleavage products over naturally acetylated N-termini.
- Affinity Capture: The biotinylated protein fragments are captured using NeutrAvidin or streptavidin beads.
- On-Bead Digestion and Elution: Captured proteins are digested on-bead with trypsin. Peptides that were originally neo-N-termini (and thus biotinylated) remain bound. These are specifically released using Tobacco Etch Virus (TEV) protease, which cleaves a sequence engineered into the biotin tag.
- Mass Spectrometry Analysis: The eluted peptides are analyzed by Liquid Chromatography with Tandem Mass Spectrometry (LC-MS/MS). The TEV elution leaves a non-native aminobutyric acid (abu) residue on the peptide, allowing unambiguous identification of the original cleavage site.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Caspase Substrate Specificity Research
| Reagent / Tool | Function / Application |
|---|---|
| Fluorogenic Tetrapeptide Substrates (e.g., Ac-DEVD-AFC) | Kinetic analysis of caspase activity and initial specificity profiling in a high-throughput format. |
| Positional Scanning Synthetic Combinatorial Libraries (PS-SCL) | Unbiased determination of optimal tetrapeptide recognition motifs for purified caspases. |
| Active, Recombinant Caspases | Essential for in vitro cleavage assays, structural studies, and reverse N-terminomics experiments. |
| Caspase-Specific Inhibitors (e.g., Ac-DEVD-fmk, Z-VAD-FMK) | To validate caspase-dependent cleavage and to inhibit specific caspases in complex lysates. |
| Subtiligase Enzyme | Engineered ligase critical for the N-terminomics workflow to label and isolate neo-N-termini. |
| Biotin-Ester Peptide Tag | The labeling reagent used in conjunction with subtiligase for affinity enrichment. |
| Caspase-Deficient Cell Lines (e.g., Caspase-9 deficient JMR) | To create lysates devoid of specific endogenous caspase activity, ensuring clean attribution of cleavage events. |
The substrate specificity profiles of caspases, defined by recognition motifs and cleavage efficiencies, are a fundamental property that dictates their biological function. The structural differences between initiator and executioner caspases, particularly in the S4 binding pocket, direct them toward distinct substrate cohorts. However, the modern view, powered by global proteomic analyses, reveals a landscape of surprising complexity—characterized by vast substrate pools, hierarchical cleavage, functional redundancy, and unique roles for underappreciated enzymes like caspase-9. Moving forward, the challenge lies in moving from cataloguing cleavage events to understanding the functional consequence of each proteolytic step within the integrated network of cell death and differentiation. A deeper grasp of these mechanisms, including the role of exosites and the contextual regulation of caspase activity, will be indispensable for developing effective caspase-targeted therapies for cancer and inflammatory diseases.
Validation Through Mutational Analysis and Disease-Associated Mutations
Within the broader research on the structural differences between initiator and executioner caspases, validation through mutational analysis and the study of disease-associated mutations provide critical functional insights. Caspases, a family of cysteine-aspartic proteases, are central regulators of programmed cell death (apoptosis) and inflammation [9]. They are synthesized as inactive zymogens (pro-caspases) and undergo activation through dimerization and proteolytic cleavage [3] [9]. The classification of caspases into initiators (e.g., caspase-8, -9, -10) and executioners (e.g., caspase-3, -6, -7) is based on their position and role in the apoptotic cascade, which is also reflected in their distinct structural features, particularly within their N-terminal pro-domains [3] [18]. Initiator caspases possess long pro-domains containing death folds, such as the Death Effector Domain (DED) in caspase-8 and -10, or the Caspase Recruitment Domain (CARD) in caspase-9, which facilitate recruitment to and activation within large signaling complexes like the DISC (Death-Inducing Signaling Complex) or the apoptosome [3] [41] [9]. In contrast, executioner caspases contain short pro-domains and are activated by direct cleavage by initiator caspases [3]. This review details how modern mutational analysis, including deep mutational scanning and the investigation of natural variants, validates the functional consequences of these structural differences and informs our understanding of human disease.
Key Structural and Functional Differences Between Initiator and Executioner Caspases
The functional distinction between initiator and executioner caspases is underpinned by specific structural elements, as summarized in Table 1. A key difference lies in their N-terminal pro-domains. Initiator caspases use their long pro-domains, which feature protein-protein interaction domains like CARD or DED, to be recruited to large activation platforms such as the apoptosome (caspase-9) or the DISC (caspase-8 and -10) [3] [41] [9]. This recruitment brings multiple initiator caspase zymogens into close proximity, leading to their activation through dimerization, a mechanism described by the "induced proximity" model [3] [75]. Once activated, initiator caspases then cleave and activate the downstream executioner caspases.
Executioner caspases, such as caspase-3 and -7, have short pro-domains and exist as inactive dimers in their zymogen form [3]. Their activation is not driven by recruitment to large complexes but by proteolytic cleavage by initiator caspases at specific internal aspartic acid residues. This cleavage event separates the large and small subunits, resulting in a conformational change that assembles the active site and confers enzymatic activity [3] [41].
Table 1: Core Structural and Functional Differences Between Initiator and Executioner Caspases
| Feature | Initiator Caspases (e.g., Caspase-8, -9, -10) | Executioner Caspases (e.g., Caspase-3, -6, -7) |
|---|---|---|
| Pro-domain | Long, contains CARD or DED death folds [3] [18] | Short, lacks death folds [3] |
| Activation Mechanism | Induced proximity & dimerization at activation platforms (e.g., DISC, apoptosome) [3] [75] | Proteolytic cleavage by initiator caspases [3] |
| Primary Function | Initiate apoptotic cascade by activating executioner caspases [3] [9] | Execute cell death by cleaving hundreds of cellular substrates [9] |
| Representative Activation Complex | Death-Inducing Signaling Complex (DISC), Apoptosome [9] | N/A |
The following diagram illustrates the hierarchical relationship and distinct activation pathways of initiator and executioner caspases.
Modern Mutational Analysis Techniques
Deep Mutational Scanning of Executioner Caspases
Recent technological advances have enabled high-throughput, systematic analysis of caspase function. Deep mutational scanning (DMS) represents a powerful approach for comprehensively mapping the relationship between protein sequence and function. As detailed in a 2022 study, a microfluidic platform was developed to screen hundreds of thousands of variants of the executioner caspases-3 and -7 (CASP3 and CASP7) [76]. This platform encapsulated single E. coli cells, each expressing a unique caspase variant, into picoliter droplets containing a cell lysis reagent and a fluorogenic peptide substrate. Following incubation, droplets with high fluorescence, indicating proteolytically active caspase variants, were sorted for downstream sequencing [76]. This method allowed for the kinetic analysis of over 360,000 variants per hour, generating extensive functional data [76].
The DMS data confirmed known critical residues, showing that mutations to the catalytic cysteine and histidine residues were highly deleterious. Furthermore, it identified positions critical for structural integrity, such as residues in the hydrophobic core, and validated the essential nature of internal cleavage sites (D175 in CASP3, D198 in CASP7) for zymogen maturation [76]. Beyond validation, this unbiased approach revealed new insights, such as identifying the G177R mutation in CASP3, which was experimentally validated to increase catalytic efficiency (k~cat~/K~M~) over two-fold compared to the wild-type enzyme, despite its location distant from the active site [76].
Table 2: Key Findings from Deep Mutational Scanning of CASP3 and CASP7 [76]
| Mutational Impact | Identified Residues/Regions | Functional Consequence |
|---|---|---|
| Highly Deleterious | Catalytic Cysteine & Histidine | Abolished enzymatic activity |
| Intolerant to Mutation | Hydrophobic core residues | Disrupted protein folding and stability |
| Intolerant to Mutation | Internal cleavage sites (D175 in CASP3, D198 in CASP7) | Prevented zymogen maturation and activation |
| Activating Mutation | G177R (adjacent to D175 in CASP3) | >2-fold increase in catalytic efficiency (k~cat~/K~M~) |
Experimental Protocol: Microfluidic Deep Mutational Scanning
The following workflow outlines the key steps in performing a deep mutational scan for caspases, based on the methodology from the cited study [76].
Detailed Methodology:
- Library Generation: Create a diverse library of caspase variants (e.g., CASP3 or CASP7) using error-prone PCR. The cited library contained 2–4 amino acid substitutions per variant on average [76].
- Cell Transformation: Transform the plasmid library into an appropriate expression host, typically E. coli.
- Microfluidic Encapsulation: Use a droplet microfluidic device to co-encapsulate single cells, expressing a unique caspase variant, with cell lysis reagents and a fluorogenic caspase substrate (e.g., containing the DEVD sequence) [76] [77]. Droplet volume is approximately 10 picoliters.
- On-chip Incubation: Incubate the droplets in an on-chip continuous flow reactor for a short period (e.g., 3 minutes) to allow cell lysis and the enzymatic reaction to proceed.
- Fluorescence-activated Droplet Sorting: Analyze droplets with an integrated laser fluorimeter. Sort droplets displaying high fluorescence signals, which correspond to cells expressing active caspase variants.
- High-throughput Sequencing: Isolate DNA from sorted droplets and subject it to high-throughput sequencing (e.g., Illumina) to identify the sequences of functional caspase variants.
- Functional Validation: Retransform sorted sequences and validate the kinetic properties (k~cat~, K~M~) of individual candidate variants using traditional plate reader-based enzyme assays [76].
Disease-Associated Mutations and Functional Validation
Caspase-10 Mutations in Autoimmune Lymphoproliferative Syndrome (ALPS)
The analysis of naturally occurring human mutations provides a critical "experiment of nature" for validating protein function in a physiological context. The role of caspase-10 (CASP10), an initiator caspase of the extrinsic apoptotic pathway, has been debated in the context of Autoimmune Lymphoproliferative Syndrome (ALPS). ALPS is typically caused by defects in the FAS-mediated apoptosis pathway [78].
A 2024 study systematically evaluated the impact of three CASP10 variants (p.C401LfsX15, p.V410I, and p.Y446C) identified in patients without canonical ALPS or in healthy individuals [78]. The study employed a combination of genetic analysis, protein biochemistry, and functional cellular assays to validate the pathological relevance of these mutations.
Key Experimental Findings and Workflow [78]:
- Genetic Identification: Subjects carrying heterozygous or homozygous CASP10 variants were identified from a genetic database. Clinical presentation was highly variable and did not consistently meet ALPS diagnostic criteria.
- Protein Expression Analysis (Western Blot):
- The frameshift mutation p.C401LfsX15, when homozygous, led to a complete loss of CASP10 protein expression due to RNA instability. The heterozygous state resulted in reduced protein levels.
- The missense variants p.V410I and p.Y446C did not disrupt CASP10 protein expression, with levels comparable to healthy controls.
- Functional Apoptosis Assay (FAS-mediated apoptosis): T-cell blasts from subjects with either homozygous or heterozygous CASP10 variants were stimulated through the FAS receptor. The assay revealed that none of the variants, including the homozygous p.C401LfsX15 that abolished protein expression, impaired FAS-mediated apoptosis compared to healthy controls. In contrast, control cells from ALPS patients with FAS mutations showed a profound apoptosis defect.
Conclusion: This comprehensive validation demonstrated that CASP10 is dispensable for FAS-mediated apoptosis in humans, challenging the notion that CASP10 mutations are a significant contributor to ALPS pathogenesis [78]. This highlights the importance of functional validation for interpreting genetic variants and understanding functional redundancies, such as the potential compensation by the homologous initiator caspase, caspase-8.
Experimental Protocol: Validating Apoptosis Function in Patient Cells
The following protocol summarizes the key steps for validating the functional impact of caspase mutations derived from patient samples, as used in the cited ALPS study [78].
Detailed Methodology:
- Patient and Control Sample Collection: Isolate Peripheral Blood Mononuclear Cells (PBMCs) from subjects carrying the caspase variants of interest, healthy controls, and positive disease controls (e.g., ALPS patients with confirmed FAS mutations) [78].
- Generation of Immortalized Cell Lines: Generate T lymphoblastoid cell lines (T-LCLs) or T-blasts from the PBMCs to establish a long-lasting source of biological material [78].
- Protein Expression Analysis:
- Functional Apoptosis Assay:
- Stimulate T-blasts or other relevant cell lines with an apoptotic inducer, such as an antibody against FAS (CD95), to trigger the extrinsic apoptosis pathway [78].
- Quantify apoptosis using methods like flow cytometry to measure cell death markers (e.g., Annexin V/propidium iodide staining) or by monitoring the cleavage of downstream substrates [77].
- Data Comparison and Interpretation: Compare the apoptosis rates and protein expression levels in variant-carrying cells directly to those from healthy controls and known positive controls. A key outcome is determining if the mutation causes a loss-of-function that aligns with the disease phenotype [78].
The Scientist's Toolkit: Key Research Reagents and Solutions
The following table compiles essential reagents and materials used in the mutational analysis and functional validation of caspases, as derived from the cited experimental protocols.
Table 3: Research Reagent Solutions for Caspase Mutational Analysis
| Reagent / Material | Specification / Example | Primary Function in Experimental Protocol |
|---|---|---|
| Caspase Substrates | Fluorogenic peptides (e.g., DEVD-AMC/AFC for caspase-3/7, IETD-AFC for caspase-8, LEHD-AMC for caspase-9) [77] | Synthetic substrates for measuring caspase enzymatic activity in kinetic assays and high-throughput screens [76] [77]. |
| Antibodies for Immunoblotting | Anti-cleaved caspase-3, anti-CASP10, anti-PARP, anti-GAPDH [78] [77] | Detect protein expression, proteolytic maturation (cleavage), and loading controls in Western Blot analysis [78] [77]. |
| Cell Culture & Assay Components | PBMCs, T-blasts/Lymphoblastoid Cell Lines (B-LCL/T-LCL), FAS Ligand (FasL) [78] | Provide cellular models for ex vivo functional validation of apoptosis and protein expression in a relevant physiological context [78]. |
| Microfluidic Platform | Droplet generator, on-chip incubator, fluorescence-activated droplet sorter [76] | Enables ultra-high-throughput screening of caspase variant libraries by performing and analyzing millions of isolated enzymatic reactions [76]. |
| Lysis & Assay Buffers | HEPES-based lysis buffer (e.g., 50 mM HEPES, pH 7.5, 0.1% CHAPS, 1 mM DTT) [77] | Extract and stabilize proteins from cells or tissues while maintaining caspase activity for subsequent biochemical assays. |
Cross-species Conservation of Structural and Functional Features
Caspases, a family of cysteine-aspartic proteases, are central regulators of programmed cell death and inflammation [9]. They are broadly classified by function into initiator caspases (e.g., caspase-2, -8, -9, -10 in humans), executioner caspases (e.g., caspase-3, -6, -7), and inflammatory caspases (e.g., caspase-1, -4, -5, -11) [1] [9]. A critical understanding of biology and therapeutic targeting of these proteases requires a deep knowledge of the structural and functional features that are conserved across diverse species. These evolutionarily preserved characteristics—from protein domains and activation mechanisms to substrate specificities—underline the fundamental roles caspases play in maintaining cellular and organismal homeostasis. This whitepaper synthesizes current research to provide a detailed technical guide on the cross-species conservation of caspase biology, with a specific focus on the structural distinctions between initiator and executioner caspases.
Structural Conservation of Caspases
Universal Catalytic Domain and Specific Cleavage
All caspases share a conserved catalytic domain that defines their function. The active site contains a quintessential QACXG motif (where X is R, Q, or G) and is uniquely configured to cleave substrate proteins specifically after aspartic acid residues, a characteristic that gives the caspase family its name (cysteine-aspartic proteases) [79] [9]. This specific catalytic mechanism is a universal feature observed from invertebrates to mammals.
Classification and Variation of Pro-Domains
A primary structural difference between initiator and executioner caspases lies in their N-terminal pro-domain regions. These domains are crucial for the upstream regulation and activation of the caspases. Based on pro-domain structure, caspases can be categorized into three main groups, a classification system that holds across species [1].
- CARD-Domain Containing: This group includes initiator caspases such as caspase-1, -2, -4, -5, -9, -11, and -12. The Caspase Activation and Recruitment Domain (CARD) enables protein-protein interactions with adaptor molecules in large activating complexes like the apoptosome or inflammasome [79] [9].
- DED-Domain Containing: This group includes the initiator caspases-8 and -10, which contain two Death Effector Domains (DED). These domains facilitate recruitment to activation complexes such as the Death-Inducing Signaling Complex (DISC) [9].
- Short/No Pro-Domain: This group consists of the executioner caspases-3, -6, and -7. They possess only a short pro-domain, which is consistent with their role as downstream effectors that are activated by proteolytic cleavage by initiator caspases [1] [9].
Table 1: Structural and Functional Classification of Caspases Across Species
| Functional Role | Caspase | Pro-Domain | Activation Complex | Conservation |
|---|---|---|---|---|
| Initiator | Caspase-2, -9 | CARD | Apoptosome | Human, Mouse, Ticks (RhCaspase9) [79] [9] |
| Initiator | Caspase-8, -10 | DED | DISC | Human, Mouse, Ticks (RhCaspase8) [79] [9] |
| Executioner | Caspase-3, -6, -7 | Short/None | Cleavage by Initiators | Human, Mouse, Ticks (RhCaspase7) [79] [9] |
| Inflammatory | Caspase-1, -4, -5, -11 | CARD | Inflammasome | Human (Casp-1, -4, -5), Mouse (Casp-1, -11) [9] |
Dimerization and Activation Mechanism
The fundamental activation mechanism is conserved for initiator caspases. They exist as inactive monomers (zymogens) and require dimerization to become activated. This dimerization is facilitated by the binding of their pro-domains (CARD or DED) to multi-protein complexes such as the apoptosome (caspase-9), DISC (caspase-8), or inflammasome (caspase-1) [9]. Once dimerized, initiator caspases undergo auto-proteolytic cleavage to stabilize the active enzyme.
In contrast, executioner caspases are already present as stable dimers in their inactive form. Their activation is triggered by proteolytic cleavage by initiator caspases. This cleavage event removes the pro-domain and separates the large and small subunits, which then reassociate to form the active protease [9]. This creates an amplifying cascade where initiator caspases activate the more abundant executioner caspases.
Functional Conservation in Apoptotic Pathways
The hierarchical caspase cascade is a functionally conserved feature of apoptotic cell death. Research in the tick Rhipicephalus haemaphysaloides provides compelling evidence for this conservation in invertebrates. The study identified and characterized homologs of initiator and executioner caspases—RhCaspase8/9 (initiators) and RhCaspase7 (executioner)—demonstrating that their functional interactions are preserved [79].
Experimental Evidence of Functional Conservation
Experimental Protocol: Co-transfection Assay for Caspase Interaction [79]
- Objective: To determine the hierarchical relationship between putative initiator and executioner caspases.
- Cloning: Full-length cDNA of tick caspases (RhCaspase7, -8, and -9) are cloned into expression vectors, often with tags (e.g., GFP, HA) for detection.
- Cell Transfection: Mammalian cell lines (e.g., HEK293T) are co-transfected with combinations of plasmids:
- RhCaspase7 (potential executioner) + RhCaspase8 (potential initiator)
- RhCaspase7 + RhCaspase9 (potential initiator)
- Control vectors.
- Detection Method: After 24-48 hours, cell lysates are analyzed by Western Blot using tag-specific or caspase-specific antibodies.
- Outcome Measurement: Cleavage of the executioner pro-caspase (RhCaspase7) is assessed. Cleavage indicates that the initiator caspase (RhCaspase8 or -9) is active and can process the downstream executioner, thereby defining the functional hierarchy.
Results: The co-transfection assays in the tick study confirmed that RhCaspase7 was cleaved when co-expressed with either RhCaspase8 or RhCaspase9. This provided direct molecular evidence that RhCaspases 8 and 9 are initiator caspases, and RhCaspase7 is an executioner caspase, confirming the conservation of the caspase cascade in ticks [79].
Diagram 1: Conserved Caspase Activation Cascade. This pathway illustrates the fundamental, cross-species hierarchy where initiator caspases activate executioner caspases, leading to substrate cleavage and cell death.
Quantitative Analysis of Caspase Features
Substrate Specificity and Proteolytic Activity
While the core catalytic mechanism is conserved, caspases have distinct substrate specificities. Proteomic studies have revealed that the number of substrate targets for individual caspases varies widely, from a few dozen for caspases-4, -5, and -9 to hundreds for caspases-3, -6, -7, and -8 [13]. Furthermore, the rates of substrate cleavage can vary over 500-fold within a caspase's substrate cohort [13].
A detailed comparative study of the highly similar executioner caspases-3 and -7 in human cells revealed significant functional differences. Despite their structural similarity, caspase-3 exhibited significantly stronger protease activity against cellular substrates than caspase-7 [80]. This difference was mapped to specific amino acid regions that form distinct three-dimensional structures at the homodimer interface, which are critical for both protease activity and homodimer-forming activity within cells [80].
Table 2: Quantitative and Functional Differences in Key Executioner Caspases
| Feature | Caspase-3 | Caspase-7 | Experimental Context |
|---|---|---|---|
| Protease Activity | Stronger | Weaker | In vitro and within cells; against low molecular weight and cellular substrates [80] |
| Critical Regions | 7 specific amino acid regions required for high activity in cells | Corresponding regions confer lower activity | Identified using chimeric constructs in human cells [80] |
| Homodimer Formation | Specific homodimer-forming activity dependent on 5 amino acid regions | Different homodimer-forming affinity | Within human cells [80] |
| Key Substrates | Poly(ADP-ribose) polymerase 1 (PARP1) [1] | Poly(ADP-ribose) polymerase 1 (PARP1) [1] | Cleaved during apoptosis in multiple species |
| Non-Apoptotic Role | PANoptosis, Pyroptosis, Stem cell and neural differentiation [1] | Not as widely implicated in lytic cell death pathways | Roles in innate immunity and development [1] |
The Scientist's Toolkit: Key Research Reagents and Methodologies
Advancing research on caspase conservation relies on a suite of standard experimental protocols and reagents.
Table 3: Essential Research Reagents and Methods for Caspase Studies
| Reagent / Method | Function / Purpose | Example from Research |
|---|---|---|
| RNA Interference (RNAi) | Gene silencing to determine caspase function in vivo. | RhCaspase9-RNAi inhibited tick feeding, while RhCaspase7/8 RNAi reduced salivary gland apoptosis [79]. |
| Site-Directed Mutagenesis | To identify critical amino acid regions defining caspase activity. | Used to map 7 regions critical for caspase-3's superior activity over caspase-7 [80]. |
| Reverse-Transcription qPCR (RT-qPCR) | Quantify transcriptional levels of caspase genes across tissues and conditions. | Demonstrated increased RhCaspase 7, 8, 9 transcription in engorged tick salivary glands [79]. |
| Western Blot | Detect protein expression, cleavage, and activation. | Confirmed cleavage of RhCaspase7 when co-transfected with RhCaspase8 or -9 [79]. |
| TUNEL Assay | Label and quantify DNA fragmentation, a hallmark of apoptosis. | Measured apoptosis levels in tick salivary glands after caspase gene silencing [79]. |
| Global Proteomics / Degradomics | Systematically identify and quantify caspase substrates in live cells or extracts. | Revealed that caspase substrate cohorts range from dozens to hundreds of targets [13]. |
| Chimeric Caspase Constructs | Swapping domains between caspases to dissect function. | Used to identify regions conferring high activity to caspase-3 [80]. |
Diagram 2: Experimental Workflow for Characterizing Caspases. This workflow outlines a standard pipeline for identifying and functionally characterizing novel or homolog caspases in a model organism.
Implications for Drug Development
The profound conservation of caspase structures and functions underscores their validity as therapeutic targets. The differential roles of caspases in various cell death pathways (apoptosis, pyroptosis, PANoptosis) and diseases, from cancer to neurodegeneration, provide a strategic roadmap for intervention [1] [9]. For instance, caspase-1 inhibitors like VX-765 (Belnacasan) have been investigated for autoimmune diseases, while the over-activation of caspase-3 in neurodegenerative contexts makes it a target for inhibitory compounds such as Minocycline and Emricasan [1]. The nuanced differences between highly similar caspases, such as the starkly different intracellular activity of caspase-3 and -7 despite similar in vitro activity, highlight the critical importance of targeting specific functional regions and understanding cellular context for effective and precise therapeutic development [80].
Integration of Structural Data with Proteomic Substrate Profiling
In the field of caspase biology, a fundamental challenge lies in connecting the intricate three-dimensional structures of these proteases to their specific functions in mediating programmed cell death. Caspases, the cysteine-dependent aspartate-specific proteases, are traditionally classified as either initiators (caspase-2, -8, -9, -10) or executioners (caspase-3, -6, -7) based on their position in the apoptotic cascade and their structural features [6] [26]. Initiator caspases possess long pro-domains containing either death effector domains (DED) or caspase activation and recruitment domains (CARD), which facilitate their recruitment to and activation within large multiprotein complexes such as the apoptosome or death-inducing signaling complex [5] [6]. In contrast, executioner caspases contain short pro-domains and are activated through cleavage by initiator caspases, subsequently carrying out the proteolytic dismantling of the cell by cleaving hundreds of cellular substrates [13] [6].
The integration of structural biology techniques with proteomic approaches for substrate profiling has emerged as a powerful paradigm for elucidating the molecular mechanisms that govern caspase specificity and function. This integrated approach enables researchers to move beyond static structural snapshots to understand how dynamic conformational changes, protein-protein interactions, and cellular context influence substrate selection and cleavage kinetics. Such insights are critical for understanding the precise roles of caspases in both apoptotic and non-apoptotic processes, and for developing targeted therapeutic interventions for diseases characterized by dysregulated cell death, including cancer, neurodegenerative disorders, and autoimmune conditions [1] [5] [26].
Structural Biology Techniques for Caspase Characterization
Mass Spectrometry-Based Structural Proteomics
Recent advances in mass spectrometry (MS)-based structural proteomics have revolutionized our ability to study caspase structures and interactions under near-physiological conditions. Three principal methods—cross-linking MS (XL-MS), hydrogen-deuterium exchange MS (HDX-MS), and limited proteolysis MS (LiP-MS)—provide complementary insights into caspase topology, dynamics, and protein-protein interactions [81] [82].
Cross-linking Mass Spectrometry (XL-MS) utilizes bifunctional chemical cross-linkers to covalently link amino acid side chains in spatial proximity within caspase complexes. Following enzymatic digestion and MS analysis, the identified cross-linked peptides provide distance constraints that are invaluable for modeling protein structures and mapping interaction interfaces [81]. XL-MS can be applied across various biological scales, from purified caspase proteins to intact cellular environments, enabling the characterization of caspase interactions in situ. The spatial proximity information derived from XL-MS, determined by the cross-linker's spacer arm length, helps define both intra- and intermolecular interactions critical for caspase activation and function [81].
Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS) measures the rate at which backbone amide hydrogens in caspases exchange with deuterium atoms from the solvent. Regions with rapid exchange typically represent flexible or solvent-accessible regions, while slow-exchanging regions often correspond to structured elements or protein interaction interfaces [81]. By comparing the HDX profiles of inactive zymogens and active caspases, researchers can identify conformational changes associated with caspase activation. Similarly, comparing caspase alone versus in complex with inhibitors or binding partners reveals interfaces involved in molecular recognition [81].
Limited Proteolysis Mass Spectrometry (LiP-MS) employs nonspecific proteases under native conditions to selectively cleave accessible, flexible regions of caspase proteins. The resulting proteolytic patterns provide information on surface accessibility and conformational changes [81]. When combined with quantitative proteomics, LiP-MS can identify structural changes in caspases across different cellular states or in response to perturbations.
Integration with Complementary Structural Techniques
The true power of MS-based structural proteomics emerges when these approaches are integrated with traditional structural biology techniques and computational modeling. Electron cryo-microscopy (cryo-EM) and cryo-electron tomography (cryo-ET) can resolve the structures of large caspase-containing complexes, such as the apoptosome or inflammasome, that are often challenging to study by crystallography [81] [82]. Nuclear magnetic resonance (NMR) spectroscopy provides atomic-resolution information on caspase dynamics and interactions in solution, complementing the static snapshots provided by crystallography [81].
The recent integration of artificial intelligence (AI)-based structure prediction tools, such as AlphaFold and RoseTTAFold, has further transformed structural caspase biology [81] [82]. These tools generate high-accuracy models of caspase structures and complexes, which can be refined and validated using experimental data from the aforementioned techniques. This integrative approach enables the modeling of caspase complexes that are difficult to characterize experimentally, bridging the gap between static structures and dynamic cellular environments [81].
Table 1: Structural Biology Techniques for Caspase Characterization
| Technique | Key Information Obtained | Applications in Caspase Research | Sample Requirements |
|---|---|---|---|
| XL-MS | Distance restraints, protein-protein interfaces, protein complex architecture | Mapping caspase activation complexes, identifying interaction partners | Purified proteins to intact cells (1-100 μg protein) |
| HDX-MS | Protein dynamics, conformational changes, binding interfaces | Characterizing zymogen activation, inhibitor binding, allosteric regulation | Purified protein complexes |
| LiP-MS | Surface accessibility, conformational states, structural changes | Identifying caspase activation states, conformational changes upon binding | Cell lysates to purified proteins |
| Cryo-EM/ET | High-resolution 3D structures of large complexes | Visualizing apoptosome, inflammasome structures | Purified complexes, cellular sections |
| NMR | Atomic-resolution dynamics, interactions in solution | Mapping binding interfaces, studying protein dynamics | Purified, isotopically labeled proteins |
| AI Modeling | High-accuracy protein structure predictions | Modeling caspase complexes, predicting functional impacts of mutations | Sequence information |
Proteomic Methods for Caspase Substrate Profiling
Global Proteomic Approaches for Substrate Identification
Proteomic technologies have enabled the systematic identification and quantification of caspase cleavage events on a global scale, revealing that individual caspases can cleave dozens to hundreds of cellular substrates with remarkable specificity [13]. These approaches typically utilize bottom-up proteomics, where proteins from caspase-active cell extracts or live cells are digested into peptides and analyzed by tandem mass spectrometry (MS/MS) to identify cleavage sites [83] [81].
Several specialized proteomic strategies have been developed for comprehensive caspase substrate profiling:
N-terminal TAILS (Terminal Amine Isotopic Labeling of Substrates) and other N-terminal enrichment techniques selectively label and isolate protein N-terminal, enabling the identification of protease cleavage sites by distinguishing natural N-terminal from neo-N-terminal generated by caspase cleavage [13]. This approach allows for the global mapping of caspase cleavage events and has revealed that caspases exhibit distinct substrate specificities and hierarchies, with cleavage rates varying over 500-fold within each caspase's substrate cohort [13].
Multiplex Substrate Profiling utilizes synthetic peptide libraries to systematically characterize caspase substrate specificity [13]. By incubating caspases with diverse peptide sequences and quantifying cleavage rates using MS, researchers can define the optimal cleavage motifs for individual caspases. This approach has revealed that while caspases share a preference for aspartic acid in the P1 position, they exhibit distinct preferences for amino acids in the P2-P4 positions, contributing to their functional specialization [13].
Quantitative MS-based Enzymology combines recombinant caspase incubation with cellular proteomes and quantitative proteomics to simultaneously determine catalytic efficiencies (kcat/KM) for hundreds of cellular substrates [13]. This approach has established clear hierarchies of substrate cleavage, identifying which cellular proteins are most efficiently cleaved by each caspase and providing insights into the functional consequences of these cleavage events.
Functional Classification of Caspase Substrates
Proteomic studies have revealed that caspases target diverse functional classes of proteins, with the number of identified substrates varying widely between different caspases [13]. Caspases-3, -6, -7, and -8 typically cleave hundreds of substrates, while caspases-4, -5, -9, and -14 cleave only a few dozen targets [13]. These substrates can be broadly categorized based on the functional consequences of their cleavage:
Apoptosis Amplification Substrates include proteins whose cleavage enhances caspase activation itself, such as the cleavage of procaspase-3, -6, and -7 by initiator caspases, creating positive feedback loops that ensure rapid and complete caspase activation [6]. Similarly, cleavage of Bcl-2 family members like BID amplifies the apoptotic signal by promoting mitochondrial outer membrane permeabilization [6].
Structural Protein Substrates encompass cytoskeletal and nuclear envelope proteins whose cleavage facilitates the morphological changes characteristic of apoptosis. These include lamins, whose cleavage disrupts the nuclear envelope, and gelsoin, whose cleavage activates it to mediate actin fragmentation [6]. The dismantling of cellular architecture through these cleavage events contributes to the formation of apoptotic bodies and facilitates phagocytic clearance [6].
Signaling Modulator Substrates include proteins involved in cell survival, DNA repair, and other signaling pathways. Cleavage of these proteins typically inactivates pro-survival signals and activates pro-apoptotic factors. For example, cleavage of PARP-1 inactivates its DNA repair function and promotes apoptotic dismantling [6]. Similarly, cleavage of various kinase and phosphatase components alters signaling networks to favor cell death execution.
Immune Response Modulator Substrates comprise proteins that, when cleaved, generate "find-me" and "eat-me" signals to recruit phagocytes and promote clearance of apoptotic cells. Caspase-mediated cleavage of certain cellular proteins can release chemotactic factors like fractalkine, which attracts macrophages to sites of apoptosis [6].
Table 2: Proteomic Methods for Caspase Substrate Identification
| Method | Principle | Key Insights | Advantages |
|---|---|---|---|
| N-terminal Enrichment (TAILS) | Isolation and identification of neo-N-termini generated by caspase cleavage | Global mapping of caspase cleavage sites in complex proteomes | Identifies endogenous cleavage events in live cells |
| Multiplex Substrate Profiling | Incubation of caspases with diverse synthetic peptide libraries | Definition of optimal cleavage motifs and substrate specificity profiles | Systematic characterization of sequence preferences |
| Quantitative MS-based Enzymology | Measurement of catalytic efficiencies for hundreds of natural substrates | Establishment of substrate hierarchies and cleavage kinetics | Provides biochemical relevance to identified substrates |
| Stable Isotope Labeling (SILAC, TMT) | Quantitative comparison of protein abundance between caspase-active and control samples | Identification of substrate stability changes and cleavage dynamics | Enables multiplexed analysis of multiple conditions |
| Activity-Based Protein Profiling | Use of mechanism-based probes to label active caspases | Identification of active caspase populations and their interactors | Distinguishes active enzymes from zymogens |
Integrated Experimental Workflows
Workflow for Integrated Structural and Substrate Profiling
The most powerful insights into caspase biology emerge from workflows that seamlessly integrate structural proteomics with global substrate identification. The following diagram illustrates a comprehensive workflow for such an integrated approach:
Diagram Title: Integrated Structural and Substrate Profiling Workflow
This integrated workflow begins with parallel sample processing for structural proteomics and substrate profiling analyses. For structural characterization, caspase-containing samples are subjected to XL-MS, HDX-MS, and/or LiP-MS to obtain structural constraints and interaction maps. Simultaneously, substrate profiling approaches identify caspase cleavage sites and quantify cleavage kinetics. The integration of these datasets enables the correlation of structural features with substrate specificity, ultimately leading to comprehensive models of caspase function that can be validated through biochemical and cellular assays.
Caspase Signaling Pathways and Complexes
Caspases function within complex signaling networks that integrate signals from multiple programmed cell death pathways. The following diagram illustrates the key caspase-containing complexes and their roles in apoptosis, pyroptosis, and necroptosis:
Diagram Title: Caspase Signaling Complexes and Pathways
Caspases are activated within specific multiprotein complexes that determine their substrate specificity and functional outcomes. The FADDosome, containing caspase-8, is central to extrinsic apoptosis initiated by death receptor engagement [5]. The apoptosome, containing caspase-9, mediates intrinsic apoptosis triggered by mitochondrial outer membrane permeabilization [5]. The inflammasome activates caspase-1, which drives pyroptosis through cleavage of gasdermin D [5]. Recent evidence indicates significant crosstalk between these pathways, with certain caspases functioning in multiple complexes. For example, caspase-8 can serve as a molecular switch between apoptosis, necroptosis, and pyroptosis depending on cellular context and activation conditions [5].
Experimental Protocols
Cross-Linking Mass Spectrometry for Caspase Complexes
Objective: To identify protein-protein interactions and spatial proximities within caspase complexes using XL-MS.
Materials:
- Purified caspase complex or caspase-expressing cells
- DSSO (disuccinimidyl sulfoxide) or DSBU (disuccinimidyl dibutyric urea) cross-linker
- Ammonium bicarbonate buffer (50 mM, pH 8.0)
- Quenching solution (1 M ammonium bicarbonate)
- Trypsin/Lys-C mix for digestion
- Strong cation exchange (SCX) chromatography materials
- LC-MS/MS system capable of MS2 and MS3 fragmentation
Procedure:
- Sample Preparation: For purified caspase complexes, dilute to 1 mg/mL in ammonium bicarbonate buffer. For cells, wash with PBS and resuspend at 10×10^6 cells/mL.
- Cross-Linking: Add cross-linker to a final concentration of 1-2 mM and incubate at room temperature for 30-60 minutes.
- Quenching: Add quenching solution to a final concentration of 100 mM and incubate for 15 minutes.
- Digestion: Recover proteins by precipitation or directly digest with trypsin/Lys-C (1:50 enzyme:substrate ratio) overnight at 37°C.
- Peptide Fractionation: Desalt peptides and fractionate using SCX chromatography to reduce complexity.
- LC-MS/MS Analysis: Analyze fractions using LC-MS/MS with methods optimized for cross-link identification, including stepped collision energy and MS3 fragmentation for cross-link validation.
- Data Analysis: Process data using specialized XL-MS software (e.g., XlinkX, plink) to identify cross-linked peptides. Use identified cross-links as distance constraints for integrative modeling of caspase complexes.
Key Considerations: Cross-linker concentration and reaction time should be optimized to maximize cross-linking efficiency while minimizing non-specific cross-links. Include appropriate controls without cross-linker to distinguish cross-linked peptides from non-cross-linked peptides [81].
Global Caspase Substrate Identification Using N-terminal Enrichment
Objective: To comprehensively identify caspase cleavage sites in complex cellular proteomes using N-terminal enrichment strategies.
Materials:
- Cell culture system with appropriate caspase activation conditions
- Lysis buffer (e.g., 50 mM HEPES, 150 mM NaCl, 1% NP-40, protease inhibitors without EDTA)
- Protein quantification assay
- Amine-blocking reagents (e.g., sulfo-NHS-acetate)
- Trypsin
- Polymer-based amine-reactive resins (for TAILS)
- LC-MS/MS system
- Database search software (e.g., MaxQuant, FragPipe)
Procedure:
- Caspase Activation: Treat cells with appropriate apoptotic stimuli (e.g., staurosporine, TRAIL) or transduce with active caspases. Include control untreated cells.
- Cell Lysis: Harvest cells at appropriate time points and lyse in suitable buffer. Confirm caspase activation by western blotting for caspase cleavage or activity assays.
- Protein Processing: Reduce, alkylate, and digest proteins with trypsin.
- N-terminal Enrichment: Block primary amines of internal peptides using amine-reactive reagents (e.g., dimethylation or acetylation). Enrich for N-terminal peptides using negative selection with amine-reactive polymers (TAILS) or positive selection using charge-based methods.
- LC-MS/MS Analysis: Analyze enriched peptides by LC-MS/MS using data-dependent acquisition or data-independent acquisition methods.
- Data Analysis: Search MS data against appropriate protein databases, allowing for variable modification of N-terminal. Identify caspase cleavage sites by detecting neo-N-terminal with semi-specific search parameters. Validate identifications using decoy database searches and filter results based on false discovery rate.
Key Considerations: The timing of caspase activation is critical—harvest cells at multiple time points to capture early and late cleavage events. Include caspase inhibitor controls to distinguish caspase-specific cleavage from other proteolytic events [13].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Research Reagent Solutions for Caspase Studies
| Reagent/Category | Specific Examples | Function/Application | Key Features |
|---|---|---|---|
| Caspase Activity Probes | CellEvent Caspase-3/7 Green, Image-iT LIVE Poly Caspase Assays | Detection of active caspases in live or fixed cells | Fluorogenic substrates (DEVD-based for caspase-3/7; VAD-based for pan-caspase) |
| Cross-linking Reagents | DSSO, DSBU, BS3 | Covalent linking of proximal amino acids for XL-MS | MS-cleavable or non-cleavable linkers with specific spacer arm lengths |
| Activity-Based Probes | Biotin- or fluorophore-labeled caspase inhibitors | Labeling active caspase populations for pull-down or imaging | Covalently binds active site cysteine, enables enrichment of active caspases |
| Structural Biology Reagents | Cryo-EM grids, NMR isotopes, crystallization screens | Sample preparation for structural studies | Optimized for specific techniques (e.g., graphene oxide grids for cryo-EM) |
| Mass Spectrometry Standards | TMT/Isobaric tags, SILAC amino acids, retention time standards | Quantitative proteomics and instrument calibration | Enable multiplexed quantification and analytical reproducibility |
| Caspase Inhibitors | Z-VAD-FMK (pan-caspase), DEVD-CHO (caspase-3/7) | Specific caspase inhibition for control experiments | Irreversible (FMK) or reversible (CHO) inhibitors with varying specificity |
| AI/Modeling Software | AlphaFold, RoseTTAFold, Coot, PyMOL | Protein structure prediction and visualization | Integration with experimental data for model building and refinement |
The integration of structural data with proteomic substrate profiling represents a powerful paradigm for advancing our understanding of caspase biology. By combining insights from XL-MS, HDX-MS, and LiP-MS with global substrate identification approaches, researchers can establish direct links between caspase structure, dynamics, and function. This integrated approach has revealed the molecular determinants of caspase specificity, the structural basis for initiator versus executioner caspase functions, and the dynamic remodeling of caspase complexes during cell death signaling.
Future directions in this field will likely focus on extending these integrated approaches to more physiological contexts, including the characterization of caspase complexes and functions in intact cells and tissues. The continued development of MS-based structural proteomics methods, particularly those capable of analyzing endogenous protein complexes in native environments, will further enhance our ability to connect caspase structure with function. Similarly, advances in single-cell proteomics may enable the correlation of caspase activation states with substrate cleavage in heterogeneous cell populations, providing new insights into cell fate decisions in response to apoptotic stimuli.
For researchers investigating initiator versus executioner caspase functions, the integrated workflow presented here provides a comprehensive roadmap for connecting structural features with functional outcomes. By applying these approaches, the scientific community can continue to unravel the complex regulatory mechanisms that govern caspase activity and substrate selection, ultimately informing the development of novel therapeutic strategies for diseases characterized by dysregulated cell death.
Conclusion
The structural distinctions between initiator and executioner caspases—from their pro-domain architectures and zymogen states to their activation mechanisms and quaternary organizations—form the fundamental basis for their specialized roles in cell death pathways. These differences create unique therapeutic opportunities for targeted intervention in diseases ranging from cancer to neurodegeneration. Future research must focus on elucidating full-length caspase structures within multiprotein complexes like apoptosomes and inflammasomes, developing isoform-specific modulators with minimal off-target effects, and exploring the therapeutic potential of exogenous caspase delivery systems. The integration of structural biology with chemical biology and disease models will be crucial for translating our understanding of caspase mechanisms into effective clinical therapies that can precisely modulate cell death in pathological conditions.
References
- 1. Caspases: structural and molecular mechanisms and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12052993/]
- 2. A Primer on Caspase Mechanisms - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6043420/]
- 3. Caspase Functions in Cell Death and Disease - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3683896/]
- 4. Initiator Caspase - an overview [https://www.sciencedirect.com/topics/pharmacology-toxicology-and-pharmaceutical-science/initiator-caspase]
- 5. Caspases as master regulators of programmed cell death [https://www.nature.com/articles/s12276-025-01470-9]
- 6. Death and survival from executioner caspase activation [https://www.sciencedirect.com/science/article/abs/pii/S108495212300143X]
- 7. Making the head: Caspases in life and death [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2022.1075751/full]
- 8. Cellular Mechanisms Controlling Caspase Activation and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3660825/]
- 9. Caspase [https://en.wikipedia.org/wiki/Caspase]
- 10. Molecular basis of dimerization of initiator caspase was ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6748139/]
- 11. Mechanisms of caspase activation - PubMed - NIH [https://pubmed.ncbi.nlm.nih.gov/14644197/]
- 12. Crystal Structure of Procaspase-1 Zymogen Domain Reveals ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2649088/]
- 13. Caspases and their substrates | Cell Death & Differentiation [https://www.nature.com/articles/cdd201744]
- 14. (PDF) Caspase : The activation - induced model proximity [https://www.academia.edu/15496576/Caspase_activation_The_induced_proximity_model]
- 15. Evolution of Caspases and the Invention of Pyroptosis [https://www.mdpi.com/1422-0067/25/10/5270]
- 16. Engineering a Dimeric Caspase -9: A Re-evaluation of the Induced ... [https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.0030183]
- 17. and Specificity of human Activation -10 - PMC Caspase [https://pmc.ncbi.nlm.nih.gov/articles/PMC2943529/]
- 18. structural and molecular mechanisms and functions in cell ... [https://www.nature.com/articles/s41421-025-00791-3]
- 19. Caspase Substrates and Inhibitors - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3721276/]
- 20. prediction of three-dimensional structure of caspase-6 and ... [https://www.sciencedirect.com/science/article/pii/S0006291X03013949]
- 21. Mechanisms of caspase activation [https://www.sciencedirect.com/science/article/abs/pii/S0955067403001364]
- 22. Caspase Activation and Inhibition - PMC - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC9341465/]
- 23. Structural and Enzymatic Insights into Caspase-2 Protein Substrate Recognition and Catalysis - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3190824/]
- 24. Homology Modeling a Fast Tool for Drug Discovery [https://pmc.ncbi.nlm.nih.gov/articles/PMC3507339/]
- 25. Exogenous Introduction of Initiator and Executioner Caspases ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8385707/]
- 26. A Comprehensive Exploration of Caspase Detection Methods [https://pmc.ncbi.nlm.nih.gov/articles/PMC11121653/]
- 27. Apoptosis: A Comprehensive Overview of Signaling ... [https://www.mdpi.com/2073-4409/13/22/1838]
- 28. Protocol for detection of caspases using immunofluorescence [https://www.abcam.com/en-us/technical-resources/protocols/immunofluorescence-protocol-to-detect-caspases]
- 29. Visualization of caspase-3-like activity in cells using a ... [https://www.nature.com/articles/ncomms3157]
- 30. Caspase-9 Activation of Procaspase-3 but not ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8489496/]
- 31. Considerations for the self-destructive protease caspase-6 [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0337291]
- 32. Investigation of metallodrug/protein interaction by X-ray ... [https://pubs.rsc.org/en/content/articlehtml/2025/qi/d4qi03277b]
- 33. A novel "bio-tag" for cryo-EM studies based on the small, ... [https://www.sciencedirect.com/science/article/abs/pii/S0006349525001729]
- 34. (IUCr) Cryo-EM single-particle analysis expanding towards ... [https://journals.iucr.org/d/issues/2025/11/00/zha5001/]
- 35. Caspases and their substrates - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5520456/]
- 36. Substrate Specificities of Caspase Family Proteases [https://www.sciencedirect.com/science/article/pii/S0021925818408010]
- 37. Quantitative MS-based enzymology of caspases reveals ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4833263/]
- 38. Development of caspase-3-selective activity-based probes for ... [https://ejnmmipharmchem.springeropen.com/articles/10.1186/s41181-024-00291-x]
- 39. The multifunctionality of the caspase family of proteases [https://www.sciencedirect.com/science/article/abs/pii/S0098299725000755]
- 40. Nanocarriers for intracellular co-delivery of proteins and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9485877/]
- 41. The protein structures that shape caspase activity, specificity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1134104/]
- 42. The intricate biophysical puzzle of caspase-1 activation [https://www.sciencedirect.com/science/article/abs/pii/S0003986121000035]
- 43. Intracellular aggregation of exogenous molecules for ... [https://pubs.rsc.org/en/content/articlehtml/2025/cs/d5cs00141b]
- 44. Caspase-8 and BID Caught in the Act with Cardiolipin [https://pmc.ncbi.nlm.nih.gov/articles/PMC12609577/]
- 45. Development of an advanced nanoformulation for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7331821/]
- 46. Caspases as therapeutic targets - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3918066/]
- 47. A long way to go: caspase inhibitors in clinical use [https://www.nature.com/articles/s41419-021-04240-3]
- 48. The concealed side of caspases: beyond a killer of cells - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11615176/]
- 49. The proteolytic landscape of cells exposed to non-lethal ... [https://www.nature.com/articles/s41420-021-00539-4]
- 50. NSAIDs are Caspase Inhibitors - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC5357154/]
- 51. Caspase-9: structure, mechanisms and clinical application [https://pmc.ncbi.nlm.nih.gov/articles/PMC5410359/]
- 52. Structural basis for dimerization of the death effector ... [https://www.nature.com/articles/s41598-018-35153-5]
- 53. Insights into the Regulatory Mechanism for Caspase-8 ... [https://www.sciencedirect.com/science/article/pii/S1097276503000595]
- 54. The Cellular Engine for the Activation of Caspase-9 [https://www.sciencedirect.com/science/article/pii/S0969212602007323]
- 55. Caspase-9, caspase-3 and caspase-7 have distinct roles ... [https://bmcmolcellbiol.biomedcentral.com/articles/10.1186/1471-2121-14-32]
- 56. An activation-based high throughput screen identifies caspase ... [https://pubs.rsc.org/en/content/articlehtml/2025/cb/d5cb00017c]
- 57. Design and optimization of caspase-1-responsive fluorescent ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12598616/]
- 58. Integrated real-time imaging of executioner caspase ... [https://www.nature.com/articles/s41420-025-02662-y]
- 59. From mechanism to application: programmed cell death ... [https://www.sciencedirect.com/science/article/pii/S2452199X25002889]
- 60. Executioner caspase is proximal to Fasciclin 3 which ... [https://elifesciences.org/articles/99650]
- 61. Single-Cell and Population-Level Analyses Using Real ... [https://www.sciencedirect.com/science/article/pii/S1534580719306999]
- 62. Kinetic and structural characterization of caspase-3 and ... [https://www.sciencedirect.com/science/article/abs/pii/S1570963910001329]
- 63. Caspases in cell death, inflammation and disease - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6611727/]
- 64. Caspases as master regulators of programmed cell death [https://pmc.ncbi.nlm.nih.gov/articles/PMC12229594/]
- 65. Caspase Activation Pathways: an Overview - NCBI - NIH [https://www.ncbi.nlm.nih.gov/books/NBK14011/]
- 66. Caspase activation pathways: some recent progress [https://www.nature.com/articles/cdd200959]
- 67. Research Article Apoptotic and proinflammatory caspases ... [https://www.sciencedirect.com/science/article/pii/S2772735125000526]
- 68. Caspases and Their Substrates - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8886984/]
- 69. Co-imaging extrinsic, intrinsic and effector caspase activity ... [https://www.sciencedirect.com/science/article/pii/S2213231718305524]
- 70. Initiator Caspase - an overview [https://www.sciencedirect.com/topics/medicine-and-dentistry/initiator-caspase]
- 71. Caspases play in traffic | Cell Death & Disease [https://www.nature.com/articles/cddis201755]
- 72. Structural and functional analysis of caspase active sites [https://pubmed.ncbi.nlm.nih.gov/12680769/]
- 73. Structural Basis for Executioner Caspase Recognition of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2782447/]
- 74. Deorphanizing Caspase-3 and Caspase-9 Substrates In ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9116730/]
- 75. Caspase Activation: Revisiting the Induced Proximity Model [https://www.sciencedirect.com/science/article/pii/S0092867404005756]
- 76. Microfluidic deep mutational scanning of the human ... [https://www.nature.com/articles/s41420-021-00799-0]
- 77. Caspase Protocols in Mice - PMC - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC4084876/]
- 78. Study of the potential role of CASPASE-10 mutations in ... [https://www.nature.com/articles/s41419-024-06679-6]
- 79. Initiator and executioner caspases in salivary gland apoptosis of Rhipicephalus haemaphysaloides | Parasites & Vectors | Full Text [https://parasitesandvectors.biomedcentral.com/articles/10.1186/s13071-020-04164-5]
- 80. Cell Biology Identification of Functional Regions Defining ... [https://www.sciencedirect.com/science/article/pii/S0021925820599262]
- 81. State-of-the-Art and Future Directions in Structural Proteomics [https://pmc.ncbi.nlm.nih.gov/articles/PMC12546877/]
- 82. Structural Proteomics State-of-the-Art and Future Directions ... [https://www.sciencedirect.com/science/article/pii/S1535947625001641]
- 83. Application of Machine Learning to Proteomics Data [https://pmc.ncbi.nlm.nih.gov/articles/PMC3837439/]