The Apoptosome Complex: Decoding Caspase-9 Activation for Therapeutic Innovation
This article provides a comprehensive analysis of the caspase-9 activation mechanism mediated by the apoptosome complex, a critical control point in the intrinsic apoptosis pathway.
The Apoptosome Complex: Decoding Caspase-9 Activation for Therapeutic Innovation
Abstract
This article provides a comprehensive analysis of the caspase-9 activation mechanism mediated by the apoptosome complex, a critical control point in the intrinsic apoptosis pathway. Tailored for researchers and drug development professionals, we synthesize foundational knowledge with cutting-edge methodological advances, resolving long-standing mechanistic debates between induced proximity and allosteric activation models. The content explores the pathophysiological consequences of dysregulated apoptosome activity in cancer and degenerative diseases, evaluates current and emerging strategies for therapeutic targeting, and critically assesses model systems for studying this complex. By integrating structural biology, biochemical regulation, and clinical insights, this resource aims to bridge fundamental research with translational applications for apoptosis-related pathologies.
The Apoptosome Blueprint: Architecture and Core Activation Mechanism
The apoptosome is a quintessential signaling platform in the intrinsic apoptotic pathway, serving as the molecular epicenter for the initiation of programmed cell death in response to cellular stress and damage [1] [2]. This multi-protein complex achieves the critical activation of caspase-9, the apical protease that sets in motion a proteolytic cascade culminating in cellular dismantling [3] [4]. The formation, architecture, and function of the mammalian apoptosome are governed by the heptameric scaffold of Apoptotic Protease-Activating Factor 1 (Apaf-1) and its regulatory cofactor, cytochrome c [5] [6]. Within the context of broader research on caspase-9 activation mechanisms, understanding the precise composition and stoichiometry of this scaffold is fundamental. This whitepaper provides an in-depth technical analysis of the Apaf-1-cytochrome c scaffold, integrating high-resolution structural data to delineate the molecular mechanism underpinning its function, a subject of profound significance for researchers and drug development professionals targeting apoptotic pathways in diseases such as cancer and neurodegeneration.
Structural Architecture of the Apoptosome
The mature mammalian apoptosome is a wheel-shaped complex with seven-fold rotational symmetry, approximately 145 Å in height and comprising seven copies each of Apaf-1 and cytochrome c [5] [6] [2]. The assembly of this 1.3 MDa complex marks the "point of no return" for the intrinsic apoptotic pathway [2]. Cryo-electron microscopy (cryo-EM) structures, particularly the landmark 3.8 Å resolution structure (PDB ID 3JBT), have provided an atomic-level view of its intricate organization [5] [6].
The overall architecture can be divided into four major structural regions:
- The Central Hub: A ring formed by the oligomerized nucleotide-binding and oligomerization domains (NOD) of the seven Apaf-1 molecules.
- The Spokes: Each consisting of the C-terminal regulatory region of an Apaf-1 monomer, which extends outward from the hub.
- The CARD Disk: A flexibly tethered, disk-like spiral composed of Caspase Activation and Recruitment Domains (CARDs) from Apaf-1 and procaspase-9, situated above the central hub.
- The Cytochrome c Binding Platform: The site where cytochrome c molecules are sandwiched within the Apaf-1 spokes.
Table 1: Core Quantitative Parameters of the Human Apoptosome
| Parameter | Specification | Structural Basis / Notes |
|---|---|---|
| Symmetry | Heptameric (7-fold) | Seven Apaf-1 molecules form the core ring [5] [6] |
| Molecular Weight | ~1.3 Megadaltons (MDa) | Calculated mass of the full complex [2] |
| Apaf-1:Cytochrome c Stoichiometry | 7:7 | One cytochrome c molecule binds per Apaf-1 subunit [6] |
| Apaf-1:Procaspase-9 Stoichiometry | 7:(3-4) | The CARD disk typically incorporates 3-4 procaspase-9 molecules [7] [2] |
| Overall Dimensions | ~145 Å (height) | Measured from cryo-EM density maps [5] |
Domain Organization of Apaf-1 and Cytochrome c Binding
Apaf-1 is a modular protein comprising several functional domains that undergo dramatic conformational changes during activation. A single Apaf-1 monomer has an estimated molecular weight of ~140 kDa and contains three major regions [5] [1] [2]:
- Caspase Recruitment Domain (CARD): The N-terminal domain responsible for homotypic interaction with the CARD of procaspase-9.
- Nucleotide-Binding and Oligomerization Domain (NOD or NB-ARC): This central domain includes:
- WD40 Repeat Region: The C-terminal regulatory region composed of 15 WD40 repeats that fold into two distinct β-propeller domains—a 7-bladed propeller (WD1) and an 8-bladed propeller (WD2) [5]. This region is connected to the NOD via a second helical domain (HD2).
In the autoinhibited, monomeric state, Apaf-1 exists in a compact, "closed" conformation where the WD40 repeats pack against the rest of the protein, preventing oligomerization [5]. The critical trigger for apoptosome assembly is the binding of cytochrome c, which is released from the mitochondrial intermembrane space, to the cleft between the two β-propellers of the WD40 region [5] [2]. This binding, coupled with dATP/ATP exchange, releases the autoinhibition, allowing Apaf-1 to adopt an extended, "open" conformation competent for oligomerization [5].
Table 2: Key Domains of Apaf-1 and Their Functions
| Domain | Location in Protein | Primary Function | Key Structural Features |
|---|---|---|---|
| CARD | N-terminus | Recruits procaspase-9 via CARD-CARD interactions [2] | Forms an acentric spiral disk in the active apoptosome [7] |
| NBD | Within NOD | Binds dATP/ATP; drives conformational change [5] [1] | Contains Walker A and B motifs; part of the central hub [2] |
| HD1 | Within NOD | Mediates oligomerization contacts [5] | Part of the central hub |
| WHD | Within NOD | Contributes to nucleotide binding and interface stability [5] | Part of the central hub; stacks against NBD/HD1 |
| HD2 | Links NOD to WD40 | Connects the hub to the regulatory region; provides flexibility [5] [2] | Acts as a structural arm |
| WD40 Repeats | C-terminus | Binds cytochrome c; maintains autoinhibition in monomer [5] [2] | Forms two β-propellers (7- and 8-bladed) |
Diagram 1: The Apoptosome Assembly and Caspase Activation Pathway. This diagram illustrates the sequence of molecular events from the initial cellular stress signal to the execution of apoptosis, highlighting the central role of the heptameric Apaf-1-cytochrome c scaffold.
Detailed Experimental Protocol for Apoptosome Reconstitution and Analysis
The following section outlines a standardized methodology for the in vitro reconstitution and structural analysis of the human apoptosome, as derived from key biochemical and cryo-EM studies [5].
Expression and Purification of Full-Length Human Apaf-1
- Objective: To obtain high-purity, monomeric Apaf-1 protein.
- Protocol:
- Expression System: Utilize a baculovirus expression system in insect cells (e.g., Sf9) to ensure proper post-translational folding of the large, multi-domain Apaf-1 protein.
- Cell Lysis and Clarification: Lyse cells in a mild, non-denaturing buffer (e.g., 20 mM Tris-HCl pH 8.0, 150 mM NaCl, 5% Glycerol) supplemented with protease inhibitors. Clarify the lysate by high-speed centrifugation.
- Affinity Chromatography: Purify the protein using affinity chromatography tailored to the tag used (e.g., Ni-NTA for His-tagged Apaf-1).
- Size-Exclusion Chromatography (SEC): As a final polishing step, subject the protein to SEC (e.g., Superose 6 column). This separates the monomeric, autoinhibited Apaf-1 from higher-order aggregates and ensures sample homogeneity. The peak corresponding to monomeric Apaf-1 is collected for downstream experiments.
In Vitro Reconstitution of the Apoptosome
- Objective: To assemble the functional apoptosome complex from its purified components.
- Protocol:
- Reaction Mixture: Combine the following components in a molar excess to drive the reaction to completion:
- Purified Apaf-1 monomer (e.g., 1 µM)
- Horse cytochrome c (e.g., 5-10 µM)
- dATP (1 mM)
- MgCl₂ (e.g., 2-5 mM, as a cofactor for nucleotide binding)
- Incubation: Incubate the reaction mixture at a physiologically relevant temperature (e.g., 25-30°C) for 1-2 hours to allow for cytochrome c binding, nucleotide exchange, and oligomerization.
- Validation of Assembly: Analyze the assembly efficiency using analytical SEC. A successful assembly is indicated by a clear shift in the elution volume from a peak corresponding to the Apaf-1 monomer to a peak corresponding to the high-molecular-weight (~1.3 MDa) apoptosome complex [5].
- Reaction Mixture: Combine the following components in a molar excess to drive the reaction to completion:
Functional Validation: Caspase-9 Activation Assay
- Objective: To confirm the biological activity of the reconstituted apoptosome.
- Protocol:
- Incubation with Procaspase-9: Incubate the reconstituted apoptosome with recombinant, inactive procaspase-9 zymogen.
- Proteolytic Activity Measurement: Use a fluorogenic or colorimetric substrate that is specifically cleaved by activated caspase-9 (e.g., LEHD-pNA). The rate of substrate cleavage, measured by the increase in fluorescence or absorbance over time, provides a quantitative measure of caspase-9 activity.
- Control: Compare the activity to negative controls containing only Apaf-1 or procaspase-9 alone. A significant increase in substrate cleavage confirms that the apoptosome is functionally active.
Structural Analysis by Single-Particle Cryo-Electron Microscopy
- Objective: To determine the high-resolution structure of the apoptosome.
- Protocol:
- Sample Vitrification: Apply 3-4 µL of the purified apoptosome complex to a freshly plasma-cleaned cryo-EM grid. Blot excess liquid and plunge-freeze the grid in liquid ethane to embed the particles in a thin layer of vitreous ice.
- Data Collection: Image the vitrified sample using a high-end cryo-electron microscope (e.g., FEI Titan Krios) operating at 300 kV. Collect a large dataset of micrographs (e.g., 900+ micrographs) with a high-quality direct electron detector.
- Image Processing:
- Particle Picking: Automatically select ~200,000 individual apoptosome particles from the micrographs.
- 2D Classification: Perform reference-free 2D classification to separate well-defined, intact apoptosome particles from junk aggregates or ice contamination.
- 3D Reconstruction: Use an initial low-resolution model as a reference for 3D classification and refinement. Iterative cycles of 3D classification and auto-refinement (e.g., using RELION software) yield a final high-resolution 3D reconstruction.
- Model Building: The final cryo-EM map, at a resolution of 3.8 Å, allows for the atomic model of Apaf-1 and cytochrome c to be built and refined, revealing specific amino acid side chains and interactions [5] [6].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagent Solutions for Apoptosome Research
| Reagent / Material | Specification / Example | Critical Function in Experimentation |
|---|---|---|
| Recombinant Apaf-1 | Full-length human, His-tagged, from baculovirus system [5] | The core scaffold protein for in vitro reconstitution; ensures proper folding. |
| Cytochrome c | Horse heart cytochrome c (commercially available) [5] [6] | The critical trigger for apoptosome assembly; binds Apaf-1 WD40 repeats. |
| Nucleotides | dATP (or ATP), high-purity, Mg²⁺ salt | The energy source and allosteric regulator driving conformational change and oligomerization [5]. |
| Procaspase-9 | Recombinant human, untagged or tagged | The substrate for the apoptosome; used to validate complex functionality via activation assays. |
| Caspase Substrate | Fluorogenic peptide (e.g., Ac-LEHD-AFC) | Allows quantitative measurement of caspase-9 enzymatic activity upon activation by the apoptosome. |
| Chromatography Media | Ni-NTA Agarose (Affinity), Superose 6 (SEC) | For purification of recombinant Apaf-1 and separation of monomeric vs. oligomeric complexes [5]. |
| Cryo-EM Grids | Quantifoil or C-flat, 300 mesh gold | The support structure for vitrifying the protein sample for high-resolution imaging. |
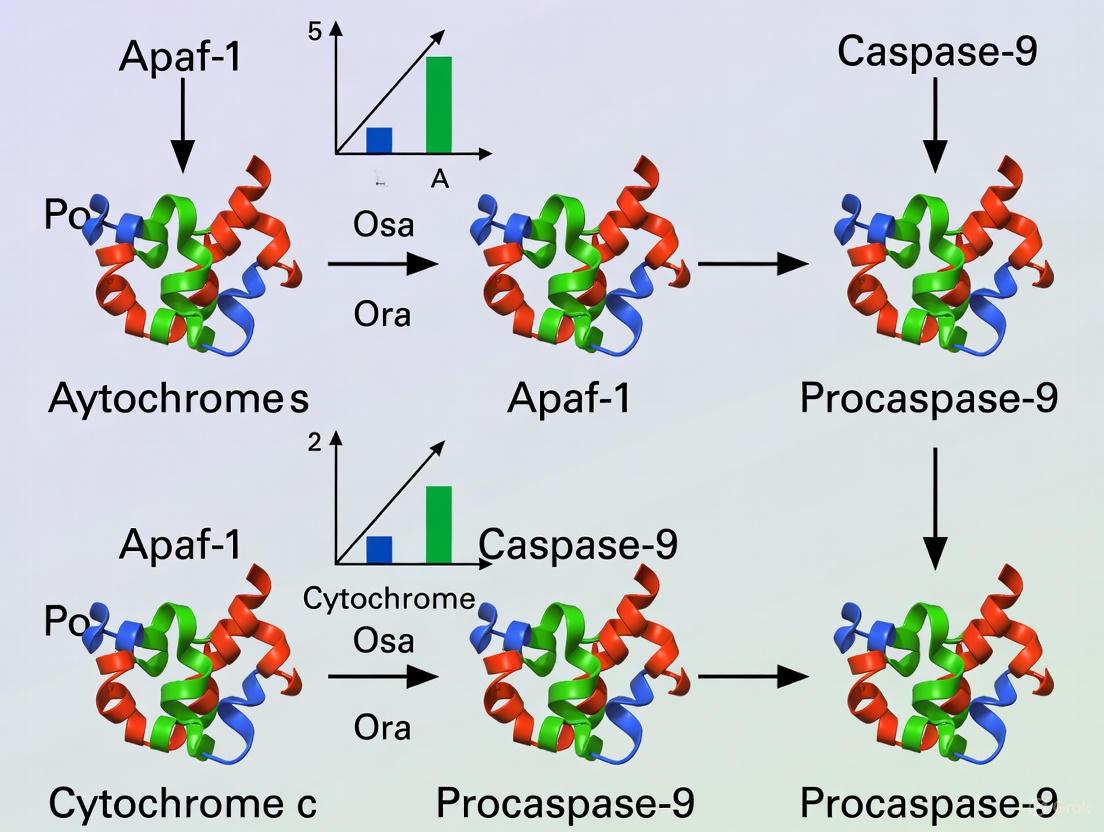
Mechanism of Procaspase-9 Activation on the Scaffold
The heptameric Apaf-1-cytochrome c scaffold activates procaspase-9 through a mechanism known as proximity-induced dimerization, rather than proteolytic cleavage [4]. The current model, supported by structural and biochemical data, involves the following steps:
- Recruitment: The CARD domains of the Apaf-1 scaffold recruit the CARD domains of procaspase-9 molecules via homotypic interactions. These CARDs assemble into a flexible, disk-like spiral structure above the central hub, recruiting an estimated three to four procaspase-9 molecules per heptameric apoptosome [7] [2].
- Dimerization and Activation: The catalytic domains of the recruited procaspase-9 molecules are flexibly tethered to their CARDs. The scaffold acts as a platform to dramatically increase the local concentration of procaspase-9, facilitating their homodimerization. Dimerization is sufficient to align the catalytic clefts and create the active enzyme conformation [4]. This model is further supported by the formation of specific heterodimers between the catalytic domains of procaspase-9 and the Apaf-1 platform, which may also contribute to stabilizing the active state [7].
- Execution Phase: Once activated, caspase-9 remains bound to the apoptosome, where it efficiently cleaves and activates the downstream effector caspases-3 and -7, initiating the execution phase of apoptosis [3].
Diagram 2: Mechanism of Procaspase-9 Activation on the Apoptosome Scaffold. The model shows recruitment of procaspase-9 CARDs to the platform, leading to proximity-induced homodimerization of their catalytic domains and subsequent activation of effector caspases.
The apoptosome, as a precisely assembled heptameric Apaf-1-cytochrome c scaffold, represents a master regulatory node in intrinsic apoptosis. Its formation is a masterclass in allosteric regulation, where cytochrome c binding and nucleotide exchange unlock a pre-programmed oligomerization sequence. The resulting structure serves as a proteolytic activation platform, not through direct catalysis, but by concentrating and aligning procaspase-9 zymogens to facilitate their activation by induced proximity and dimerization. The high-resolution structural data now available provides an unparalleled roadmap for understanding this process at an atomic level. For the research and drug development community, targeting the specific protein-protein interfaces within the apoptosome—such as the cytochrome c binding cleft, the oligomerization interface, or the CARD-CARD disk—offers promising, albeit challenging, therapeutic avenues for modulating cell death in human disease.
Caspase-9 serves as the essential initiator caspase within the intrinsic apoptotic pathway, with its activation and regulation fundamentally governed by a precise modular architecture. This whitepaper delineates the tripartite domain structure of caspase-9—comprising the Caspase Activation and Recruitment Domain (CARD), a flexible linker region, and the catalytic protease domain—and examines how this structure underpins its function within the apoptosome complex. We synthesize current mechanistic models of activation, present quantitative biophysical data on domain-specific interactions, and provide detailed methodologies for studying these processes. The structural insights discussed herein provide a critical foundation for therapeutic intervention in apoptosis-related diseases, including cancer and degenerative disorders.
Caspase-9 is an initiator caspase that plays an indispensable role in the intrinsic (mitochondrial) apoptotic pathway [8] [9]. Unlike effector caspases, its activation is tightly regulated by integration into a multimolecular signaling platform known as the apoptosome. This complex forms in response to cellular stress signals that trigger mitochondrial outer membrane permeabilization and cytochrome c release [1]. The apoptosome is a wheel-shaped heptameric assembly of Apaf-1 molecules, which, upon binding cytochrome c and dATP/ATP, oligomerizes to create a platform for caspase-9 recruitment and activation [10] [11]. The domains of caspase-9 mediate critical, specific interactions required for its recruitment, activation, and control of the downstream apoptotic cascade. A profound understanding of this domain architecture is therefore essential for elucidating the molecular basis of caspase-9 function and its role in cell fate decisions.
Domain Architecture of Caspase-9
The functional caspase-9 protein is organized into three primary structural regions: an N-terminal prodomain, a central catalytic domain, and a short linker that connects them.
Table 1: Core Domains of Human Caspase-9
| Domain | Residue Location | Key Structural Features | Primary Function |
|---|---|---|---|
| CARD (Pro-Domain) | N-terminus (approx. 1-96) | Death-domain fold, six-helix bundle [12] | Mediates homotypic interaction with Apaf-1 CARD for apoptosome recruitment [8] [13] |
| Linker Domain | Connects CARD to protease domain (residues 97-138) | Flexible peptide loop [8] | Facilitates access to the active site without obligatory cleavage; presumed to allow movement upon recruitment [8] [14] |
| Protease Domain | C-terminus (residues 139-416) | Heterodimer of large (p35) and small (p12) subunits; contains catalytic cysteine residue [13] | Executes proteolytic activity; cleaves and activates downstream effector caspases-3, -6, and -7 [9] [13] |
The CARD domain belongs to the death-domain fold (DDF) superfamily, all of which fold into a conserved six-helix bundle and mediate specific protein-protein interactions [12]. This domain is responsible for the highly specific homotypic interaction with the CARD of Apaf-1, tethering the inactive caspase-9 zymogen to the apoptosome [8] [1]. The catalytic protease domain itself is subdivided into a large and a small subunit. A defining feature of caspase-9's active site is the QACGG motif, which differs from the QACRG motif conserved in most other caspases [13]. Furthermore, the caspase-9 dimer is asymmetric, featuring only one functional catalytic site per dimer, which has significant implications for its activation mechanism [13] [11].
Diagram 1: Hierarchical domain structure of procaspase-9.
The CARD Domain: Structure and Function in Apoptosome Recruitment
The CARD domain is the linchpin for integrating caspase-9 into the apoptotic machinery. Structural studies, particularly by NMR spectroscopy, confirm that the CARD domain of caspase-9 (C9CARD) is a folded, stable six-helix bundle in solution and exists as a monomer under physiological conditions [12]. Its primary function is to engage in a homotypic interaction with the CARD domain of Apaf-1 within the assembled apoptosome.
Recent cryo-electron microscopy structures reveal that this interaction is not a simple 1:1 binding event but involves multiple asymmetric interfaces between Apaf-1 and caspase-9 CARDs, forming a flexibly tethered "disk" above the central hub of the apoptosome [12] [1]. This multimeric interaction is crucial for stabilizing the active conformation of caspase-9. Intriguingly, the C9CARD itself possesses an intrinsic capacity to self-assemble into helical filaments under specific conditions, such as at mildly acidic pH or low salt concentrations [12]. This polymerization is governed by electrostatic interactions and is critically regulated by a pH-sensitive histidine switch at residue H38. The protonation of H38 at lower pH enhances filament formation, a process that can be mimicked by a H38R mutation to introduce a constitutive positive charge [12]. This suggests that H38 may act as a physiological pH sensor, potentially regulating caspase-9 activation in specific cellular microenvironments.
Table 2: Key Residues and Mutations in the CARD Domain
| Residue/Feature | Functional Role | Experimental Effect of Mutation |
|---|---|---|
| Histidine 38 (H38) | pH sensor; protonation enhances filament formation [12] | H38R: Enhances filament propensity (positive charge mimic). H38D/H38N: Decreases filament propensity [12] |
| Charged Residues | High density of Arg, Lys, Asp, Glu; mediates electrostatic interactions [12] | Low salt conditions promote aggregation/filament formation; high salt (500 mM NaCl) increases solubility [12] |
| CARD-CARD Interface | Binds Apaf-1 CARD via multiple interfaces [12] | Disruption prevents apoptosome recruitment and caspase-9 activation [8] |
The Protease Domain: Catalytic Mechanism and Activation
The protease domain is responsible for the proteolytic activity of caspase-9. It cleaves downstream effector caspases, primarily caspase-3 and -7, to initiate the execution phase of apoptosis [9] [13]. The activation mechanism of this domain is a subject of ongoing research and debate, with two principal models prevailing.
The induced proximity/dimerization model posits that the apoptosome serves primarily as a platform to concentrate caspase-9 zymogens, facilitating their homodimerization and subsequent activation [8] [14]. In support of this, artificial dimerization of caspase-9 is sufficient to induce its activity [11]. In contrast, the allosteric regulation model proposes that binding to the apoptosome backbone induces a conformational change in caspase-9 that unlocks its catalytic activity. Evidence for this includes the finding that caspase-9 is active only when bound to the apoptosome and reverts to an inactive monomeric state upon dissociation [11]. Mathematical modeling of apoptosis execution kinetics suggests that a mechanism of allosteric activation more accurately reproduces experimental data than one based solely on homodimerization [11]. It is likely that both mechanisms—conformational change and increased local concentration—contribute to the full activation of caspase-9 within the physiological context of the apoptosome.
Experimental Analysis of Caspase-9 Domain Function
NMR Spectroscopy for CARD Domain Characterization
Objective: To determine the solution-state structure, dynamics, and oligomeric status of the isolated C9CARD. Methodology:
- Sample Preparation: Recombinant (^{15}\text{N})-labeled and (^{13}\text{C},^{15}\text{N})-labeled C9CARD is expressed and purified. The sample is buffer-exchanged into a low-salt, pH 6.5 buffer for NMR experiments [12].
- Data Collection: A series of NMR experiments are performed:
- (^1\text{H})-(^{15}\text{N}) HSQC: To assess protein folding and monodispersity.
- Triple-resonance experiments (HNCA, HNCOCA, etc.): For sequential backbone resonance assignment.
- (^{15}\text{N}) spin relaxation (T1, T2): To determine the rotational correlation time and confirm the domain is monomeric in solution.
- (^{15}\text{N}) CPMG relaxation dispersion: To probe for millisecond-timescale dynamics or transient oligomerization [12].
- pKa Determination: The pKa of H38 is determined by monitoring the chemical shift changes of its side chain imidazole rings in (^1\text{H})-(^{15}\text{N}) HSQC spectra as a function of pH [12]. Key Findings: C9CARD is a stable, folded monomer with a rotational correlation time of ~6.5 ns. No millisecond-exchange dynamics are detected. The pKa of H38 supports its role in pH-dependent filament formation [12].
Cryo-Electron Microscopy for Filament and Apoptosome Structure
Objective: To determine the high-resolution structure of C9CARD filaments and the caspase-9/apoptosome holo-complex. Methodology:
- Sample Vitrification: C9CARD (wild-type and H38R mutant) is incubated under filament-forming conditions (e.g., low salt, pH ~6.5). The sample is applied to EM grids, blotted, and plunge-frozen in liquid ethane [12].
- Imaging and Processing: Micrographs are collected on a cryo-electron microscope. Image processing workflows (particle picking, 2D classification, 3D reconstruction) are used to generate helical reconstructions for filaments [12]. For the apoptosome, samples of Apaf-1, cytochrome c, and caspase-9 are assembled and processed using single-particle analysis [1] [10].
- Model Building: Atomic models are built and refined into the resulting electron density maps [12]. Key Findings: Cryo-EM has revealed the wheel-shaped heptameric structure of the apoptosome and the flexibly tethered CARD disk [10]. For C9CARD, it provided 3.2-Å and 3.4-Å structures of the wild-type and H38R filaments, respectively, revealing the molecular basis of polymerization and the role of H38 [12].
Diagram 2: Workflow for structural study of caspase-9 domains.
In Vitro Filamentation Assay
Objective: To investigate the conditions that promote C9CARD self-assembly into filaments. Methodology:
- Condition Screening: Purified C9CARD is dialyzed into buffers of varying pH (5.5 to 7.5) and salt concentrations (0 to 500 mM NaCl) [12].
- Aggregation Assessment: Protein solubility and aggregation are monitored by visual inspection, light scattering, or centrifugation assays.
- Visualization: The structural nature of the aggregates is confirmed using negative-stain electron microscopy, where samples are applied to grids, stained with uranyl acetate, and imaged to identify filamentous structures [12]. Key Findings: C9CARD filament formation is enhanced under low-salt or mildly acidic conditions, indicating a significant role for electrostatic interactions and histidine protonation [12].
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents for Caspase-9 Domain Research
| Reagent / Material | Function and Application | Key Characteristics / Targets |
|---|---|---|
| Recombinant C9CARD | Structural studies (NMR, Cryo-EM); in vitro filamentation assays [12] | Isolated CARD domain (approx. residues 1-96); can be isotopically labeled ((^{15}\text{N}), (^{13}\text{C})) |
| Caspase-9 Mutants (e.g., H38R, H38D) | To probe the role of specific residues in activation and regulation [12] | H38R mimics protonated state; H38D introduces negative charge to disrupt pH sensing |
| Z-LEHD-FMK | Cell-permeable, irreversible caspase-9 inhibitor [15] | Used for functional studies in cells and animal models to probe caspase-9's role |
| Apaf-1 CARD / Apoptosome | For reconstitution studies of the activation platform [1] [11] | Used in co-immunoprecipitation, in vitro apoptosome assembly, and binding assays |
| Inducible Caspase-9 (iCasp9) | A safety switch in adoptive cell therapies (e.g., CAR-T) [13] | Caspase-9 fused to FK506-binding protein; dimerizes upon addition of small molecule drug, triggering apoptosis |
The functional capacity of caspase-9 as the apex protease of the intrinsic apoptotic pathway is an emergent property of its precise domain architecture. The CARD domain provides specificity in recruitment, the linker affords necessary flexibility, and the protease domain holds the catalytic potential, which is unleashed through a complex process of allosteric activation and dimerization on the apoptosome. The discovery that the CARD domain can form pH-regulated filaments adds a novel layer of complexity to its regulation. Continued high-resolution structural and biophysical dissection of these domains and their interactions within the full-length protein and the apoptosome complex will be vital for fully elucidating the activation mechanism. This knowledge is a prerequisite for the rational design of novel therapeutics that can modulate caspase-9 activity to treat cancer, autoimmune diseases, and degenerative disorders by targeting the very engine of the intrinsic apoptotic pathway.
The apoptosome complex is a critical signaling platform in the intrinsic apoptosis pathway, serving as the molecular bridge between cellular stress signals and the execution of programmed cell death. Its formation is initiated by the release of cytochrome c from the mitochondrial intermembrane space into the cytosol in response to various apoptotic stimuli, including DNA damage, radiation, and chemotherapeutic agents [16] [17]. In the cytosol, cytochrome c binds to the key adaptor protein, Apoptotic Protease-Activating Factor 1 (Apaf-1), triggering a sequence of nucleotide-dependent events that culminate in the assembly of the wheel-like heptameric apoptosome [18]. This complex then recruits and activates the initiator caspase, procaspase-9, which subsequently activates downstream effector caspases, such as caspase-3 and -7, leading to the controlled dismantling of the cell [19] [16].
The precise molecular events governing apoptosome assembly, particularly the roles of cytochrome c binding and nucleotide exchange on Apaf-1, have been the focus of extensive biochemical research. This technical guide synthesizes current understanding of these mechanisms, framing them within the broader context of apoptosome complex and caspase-9 activation mechanism research, and provides detailed methodologies for its experimental investigation.
Core Molecular Mechanism
The Key Players: Apaf-1, Cytochrome c, and Nucleotides
Apaf-1 is a multi-domain protein comprising three fundamental regions:
- An N-terminal Caspase Recruitment Domain (CARD) that mediates homotypic interactions with the CARD of procaspase-9.
- A central Nucleotide-Binding and Oligomerization Domain (NOD).
- A C-terminal regulatory region composed of WD-40 repeats that binds cytochrome c and maintains Apaf-1 in an auto-inhibited state in the absence of an apoptotic signal [18] [8].
The binding of cytochrome c to the WD-40 repeats of Apaf-1 is the pivotal event that relieves this auto-inhibition. Research using reconstituted systems with purified components has demonstrated that this binding induces significant conformational changes, enabling Apaf-1 to engage nucleotides [18] [20]. Notably, Apaf-1 co-purifies with dATP as an endogenous cofactor. The binding of cytochrome c induces the hydrolysis of this bound dATP to dADP, a crucial first step that primes the protein for activation [18].
Following hydrolysis, the resulting dADP is replaced by exogenous dATP or ATP. This exchange is the definitive trigger for Apaf-1 oligomerization. A non-hydrolyzable ATP analog, β,γ-methylene adenosine 5′-triphosphate (ADPCP), can also support apoptosome formation and caspase activation, indicating that nucleotide binding, rather than subsequent hydrolysis, is the key requirement for this step [20]. The energy from dATP/ATP binding drives the assembly of seven Apaf-1/cytochrome c complexes into the active apoptosome, a wheel-like structure with a central hub formed by the CARD and NOD domains and seven radiating spokes formed by the WD-40 repeats, each bound to a single cytochrome c molecule [18].
Diagram: Cytochrome c and (d)ATP Induced Apoptosome Assembly
Quantitative Analysis of Nucleotide Dynamics
The following table summarizes key quantitative findings on nucleotide dynamics during apoptosome formation, as revealed by biochemical assays.
Table 1: Quantitative Data on Nucleotide Dynamics in Apoptosome Formation
| Parameter | Experimental Finding | Significance | Citation |
|---|---|---|---|
| Apaf-1 Bound Cofactor | dATP found as endogenous cofactor | Primes Apaf-1 for activation upon cytochrome c binding | [18] |
| Nucleotide Binding Stimulation | Cytochrome c significantly stimulates dATP binding to Apaf-1 | Relieves auto-inhibition, allowing Apaf-1 to engage nucleotide | [20] |
| Nucleotide Hydrolysis | Cytochrome c binding induces dATP hydrolysis to dADP on Apaf-1 | First required step for apoptosome formation; primes for exchange | [18] |
| Nucleotide State in Active Complex | dATP remains as dATP (not dADP) in bound state of active apoptosome | Nucleotide binding, not hydrolysis, drives oligomerization | [20] |
| Nucleotide Analog Support | Non-hydrolyzable ATP analog (ADPCP) supports apoptosome formation | Confirms that hydrolysis is not required for oligomerization after the initial step | [20] |
Experimental Analysis & Methodologies
Reconstitution of the Caspase Activation Pathway
A foundational experimental approach for studying apoptosome formation involves the reconstitution of the caspase activation pathway using highly purified components. This method allows for precise control over each element and direct observation of interactions.
Table 2: Essential Reagents for Reconstituting Apoptosome Activity
| Research Reagent | Source / Production Method | Critical Function in the Experiment |
|---|---|---|
| Recombinant Apaf-1 | Baculovirus expression system in Sf21 insect cells, purified via nickel-affinity and ion-exchange chromatography [18] | Core scaffold protein for apoptosome assembly; often engineered with N- and C-terminal histidine tags for purification. |
| Cytochrome c | Purified from horse heart; further purified via Mono S cation-exchange chromatography [18] | Apoptotic signal; binds Apaf-1 WD-40 repeats to initiate the activation sequence. |
| Recombinant Procaspase-9 | Baculovirus expression system, purified via nickel-affinity and ion-exchange chromatography [18] | Initiator caspase; substrate for apoptosome activity measurement. |
| Recombinant Procaspase-3 | Baculovirus expression system, purified via nickel-affinity and ion-exchange chromatography [18] | Effector caspase; activation by caspase-9 is a readout for functional apoptosome formation. |
| Nucleotides (dATP/dADP) | Commercially sourced purified nucleotides; radiolabeled [α-³³P]dATP for binding assays [18] | Essential cofactors for Apaf-1 activation; used to track binding and hydrolysis. |
| Fluorogenic Caspase Substrate | e.g., DEVD substrate (cleaved by caspase-3) [18] | Allows quantitative measurement of caspase-3 activity as a final output of the pathway. |
Detailed Protocol:
- Protein Incubation: Incubate purified Apaf-1 (e.g., 20-35 ng) with cytochrome c (e.g., 175 pmol) and dATP/ATP (e.g., 10 µM) in an appropriate reaction buffer (e.g., 20 mM HEPES-KOH pH 7.5, 10 mM KCl, 1.5 mM MgCl₂, 1 mM DTT) at 30°C for several hours to allow complex formation [18].
- Complex Assembly Analysis:
- Glycerol Gradient Centrifugation: Layer the reaction mixture onto a 10-30% glycerol gradient and centrifuge at high speed (e.g., 256,000 × g for 3 hours). Fractionate the gradient and analyze fractions for Apaf-1 (via Western blot) to distinguish monomeric Apaf-1 from high-molecular-weight apoptosome complexes [18].
- Size-Exclusion Chromatography: As an alternative, fractionate the reaction mixture using a Superose 6 column to separate complexes based on size [18].
- Functional Assay: To test the activity of formed complexes (e.g., individual glycerol gradient fractions), incubate them with procaspase-9 and procaspase-3 in the presence of dATP and a fluorogenic DEVD caspase-3 substrate. Measure the cleavage of the substrate over time using a fluorescence plate reader to quantify functional caspase activation [18].
Diagram: Experimental Workflow for Apoptosome Reconstitution
Analyzing Nucleotide Binding and Hydrolysis
Directly probing the nucleotide status of Apaf-1 is key to understanding the activation mechanism.
Detailed Protocols:
- Liquid Chromatography-Mass Spectrometry (LC-MS) for Nucleotide Identification:
- Incubate Apaf-1 with or without cytochrome c under appropriate buffer conditions.
- Dialyze the samples to remove salts and unbound nucleotides.
- Extract nucleotides from the protein complex using phenol/chloroform/isoamyl alcohol.
- Analyze the aqueous phase containing the extracted nucleotides using LC-MS to identify and quantify the specific nucleotide (dATP vs. dADP) bound to Apaf-1 at different stages [18].
- Thin-Layer Chromatography (TLC) for Nucleotide Hydrolysis:
- Incubate Apaf-1 with cytochrome c and radiolabeled [α-³³P]dATP.
- Purify the apoptosome complex (e.g., via size-exclusion chromatography).
- Spot the complex or supernatant onto a PEI-cellulose TLC plate.
- Separate nucleotides using a suitable solvent (e.g., 1 M formic acid / 0.5 M LiCl).
- Visualize and quantify the ratio of dATP to dADP using a phosphorimager to determine the extent of hydrolysis [18].
- Malachite Green Phosphate Assay:
- This colorimetric assay can be used to directly measure the inorganic phosphate (Pi) released during dATP hydrolysis.
- Incubate Apaf-1 with cytochrome c and dATP, and measure the free Pi concentration over time using the Malachite Green reagent, which changes color in the presence of Pi [18].
Research Context and Therapeutic Implications
Integration with Broader Apoptosis Research
The cytochrome c/(d)ATP-dependent activation of the apoptosome represents the core of the intrinsic apoptosis pathway. This pathway integrates signals from cellular stress, DNA damage, and oncogenic insults. The Bcl-2 family of proteins acts as a crucial upstream regulator by controlling mitochondrial outer membrane permeabilization (MOMP), the event that determines cytochrome c release [16] [17]. Once released, cytochrome c converts Apaf-1 from a silent monomer into an active caspase-activating machine. The activated caspase-9 within the apoptosome then cleaves and activates the executioner caspases-3 and -7, leading to the systematic dismantling of the cell [19] [16]. Failing to activate caspase-9 has profound pathophysiological outcomes, most notably contributing to cancer development and resistance to chemotherapy [8]. Tumor cells often exhibit downregulation of Apaf-1 or caspase-9, or overexpression of endogenous inhibitors like XIAP, allowing them to evade cell death [8].
The Scientist's Toolkit: Key Research Reagent Solutions
The following table compiles essential materials and reagents for experimental research in this field.
Table 3: Research Reagent Solutions for Apoptosome Studies
| Reagent / Material | Key Function & Application | Experimental Notes |
|---|---|---|
| Recombinant Human Apaf-1 | Core structural component for in vitro reconstitution studies; allows study of mutants. | Often His-tagged for purification; baculovirus system produces functional protein [18]. |
| Purified Cytochrome c | Apoptotic trigger; used to initiate apoptosome assembly in vitro. | Commercial horse heart cytochrome c is commonly used and effective [18]. |
| dATP / ATP Analogs | To dissect the role of nucleotide binding vs. hydrolysis in assembly. | Non-hydrolyzable analogs (e.g., ADPCP) confirm binding is sufficient for oligomerization [20]. |
| Caspase-Specific Antibodies | Western blot analysis of caspase-9 and caspase-3 processing and activation. | Critical for confirming the downstream proteolytic cascade in cell-based or in vitro assays. |
| Fluorogenic / Chromogenic Caspase Substrates | Quantitative measurement of caspase-9 and caspase-3 enzyme activity. | DEVD-based substrates for caspase-3/7; LEHD-based for caspase-9 [18]. |
| XIAP Protein | Key endogenous inhibitor of caspases; used to study regulation of the pathway. | Used in experiments to understand how effector caspase activity is restrained [21]. |
The release of cytochrome c and the subsequent (d)ATP exchange on Apaf-1 constitute the definitive biochemical trigger for the assembly of the caspase-9 activating apoptosome complex. The precise, multi-step mechanism—involving cytochrome c binding, dATP hydrolysis to dADP, and exchange for exogenous dATP—ensures that this potent cell death pathway is tightly regulated. Continued biochemical and structural dissection of this process, utilizing the experimental methodologies outlined, is fundamental to advancing our understanding of cell death in health and disease. This knowledge provides the essential foundation for developing novel therapeutic strategies aimed at modulating apoptosis in conditions such as cancer and degenerative disorders.
Programmed cell death (PCD) is an evolutionarily conserved process essential for development, tissue homeostasis, and host defense in multicellular organisms [22]. The core components of the apoptotic machinery were first identified through genetic studies in the nematode Caenorhabditis elegans (C. elegans), providing a foundational model for understanding equivalent pathways in mammalian systems [3] [23]. This whitepaper examines the profound evolutionary conservation between the C. elegans cell death pathway and mammalian apoptosis, with specific focus on the activation mechanisms of the key initiator caspases—CED-3 in nematodes and caspase-9 in mammals. The molecular architecture comprising caspase activators (CED-4/Apaf-1), inhibitors (CED-9/Bcl-2 family), and executioners (CED-3/caspase-9) represents one of the best-characterized examples of evolutionary conservation in metazoan biology [24] [23]. Understanding these conserved mechanisms provides crucial insights for drug development targeting apoptotic pathways in human diseases including cancer, neurodegenerative disorders, and fibrotic conditions [8] [15].
Core Components: A Comparative Analysis
The apoptotic pathway in C. elegans is governed by three core interacting components: CED-3, CED-4, and CED-9 [23]. Mammalian systems have evolved homologs for each of these components, forming analogous but more complex regulatory networks. The following table summarizes the key components and their evolutionary relationships:
Table 1: Evolutionary Conservation of Core Apoptotic Components
| Function | C. elegans Component | Mammalian Component | Domain Structure | Key Features |
|---|---|---|---|---|
| Initiator Caspase | CED-3 | Caspase-9 | CARD → p20 → p10 | Activated within multiprotein complexes; requires cleavage for full activity [22] [25] |
| Caspase Activator | CED-4 | Apaf-1 | CARD → NB-ARC → WD40 | Nucleotide-binding; forms oligomeric activation platform [25] [23] |
| Caspase Inhibitor | CED-9 | Bcl-XL/Bcl-2 | BH4 → BH3 → BH1 → BH2 | Mitochondrial membrane localization; regulates activator function [23] |
| Regulatory Molecule | EGL-1 | BH3-only proteins | BH3 domain only | Disrupts activator-inhibitor interaction [23] |
Genetic and biochemical analyses demonstrate that CED-9 directly binds and inhibits CED-4, preventing CED-3 activation in living cells [24]. During apoptosis, EGL-1 is transcriptionally activated and binds to CED-9, disrupting the CED-9/CED-4 complex and releasing CED-4 to activate CED-3 [23]. An equivalent ternary complex exists in mammalian cells involving Apaf-1, caspase-9, and Bcl-XL, confirming the functional conservation of this regulatory mechanism [23].
Activation Mechanisms: From Nematodes to Mammals
The C. elegans Pathway
In C. elegans, the CED-4 protein exists in an asymmetric dimer that is specifically recognized and inhibited by one molecule of CED-9 [23]. This specific interaction prevents CED-4 from activating CED-3. Upon induction of apoptosis, EGL-1 binding induces pronounced conformational changes in CED-9 that result in the dissociation of the CED-4 dimer [23]. The released CED-4 dimer further dimerizes to form a tetramer, which facilitates the autoactivation of CED-3 [23]. This mechanism ensures tight regulation of the cell death pathway, with CED-4 serving as the crucial activation platform.
The Mammalian Apoptosome
In mammalian systems, caspase-9 activation occurs through the formation of a multiprotein complex known as the apoptosome, composed of apoptotic protease-activating factor 1 (Apaf-1), cytochrome c, and caspase-9 [8] [25]. Similar to CED-4, Apaf-1 contains a caspase recruitment domain (CARD) that selectively binds to the CARD motif in caspase-9 through homotypic interactions [8] [25]. Following apoptotic stimuli, cytochrome c is released from mitochondria and binds to Apaf-1, promoting oligomerization into a heptameric apoptosome complex [25]. This complex then recruits and activates procaspase-9.
Two primary models have been proposed for caspase-9 activation. The "induced proximity model" suggests that the apoptosome serves as a platform to bring caspase-9 molecules into close proximity, promoting dimerization and activation [8] [25]. The "induced conformation model" proposes that binding to the apoptosome induces conformational changes in caspase-9 that activate the protease [8]. Recent evidence indicates that procaspase-9 has higher affinity for the apoptosome than its cleaved form, and that procaspase-9 autoprocessing acts as a molecular timer that regulates the duration of apoptosome activity [8].
Table 2: Caspase-9 Activation Mechanisms and Evidence
| Activation Model | Key Mechanism | Supporting Evidence | C. elegans Parallel |
|---|---|---|---|
| Induced Proximity | Apoptosome platforms accumulate local procaspase-9 concentration to promote dimer-driven activation [8] | Procaspase-9 dimerization leads to rapid autocatalytic cleavage; uncleaved caspase-9 retains activity [8] [25] | CED-4 tetramerization facilitates CED-3 autoactivation [23] |
| Induced Conformation | Direct Apaf-1 binding alters caspase-9 conformation to create active protease [8] | Crystal structure reveals complementary CARD-CARD interface essential for activation [8] | EGL-1 induces conformational changes in CED-9 to release CED-4 [23] |
| Proximity-Induced Dimerization | Hybrid model emphasizing both local concentration and structural rearrangement [8] | Procaspase-9 more affinitive to apoptosome than cleaved form; autocleavage regulates activity duration [8] | CED-4 oligomerization state controls CED-3 activation efficiency [23] |
Experimental Approaches and Methodologies
Yeast-Based Reconstitution Systems
Several key insights into caspase activation mechanisms have been obtained using yeast-based reconstitution systems. These approaches exploit the fact that yeast lack endogenous caspase pathways, providing a null background for functional studies [24]. The typical workflow involves:
- Transformation: Introduction of mammalian or nematode caspase pathway components into Saccharomyces cerevisiae [24]
- Selection: Growth under selective conditions to maintain plasmid expression
- Functional Assay: Measurement of caspase activity using fluorescent substrates (e.g., AFC-based detection) or viability assays [24]
This system was instrumental in demonstrating that CED-9 does not directly inhibit CED-3, but rather functions through regulation of CED-4 [24]. Similarly, yeast models have been used to study the mammalian Apaf-1/caspase-9 activation mechanism and to screen for caspase inhibitors [24].
Biochemical Reconstitution and Structural Studies
The C. elegans apoptotic pathway has been fully reconstituted in vitro using homogeneous recombinant proteins, allowing detailed biochemical characterization [23]. Key methodologies include:
- Protein complex purification: Recombinant CED-4/CED-9 complex purification using affinity chromatography [23]
- Crystallography: Determination of the CED-4/CED-9 complex structure at 2.6 Å resolution [23]
- Site-directed mutagenesis: Functional analysis of specific residues in CED-9 (e.g., G169E) that disrupt EGL-1 mediated release of CED-4 [23]
These studies revealed that one molecule of CED-9 binds to an asymmetric dimer of CED-4, specifically recognizing only one of the two CED-4 molecules, which prevents CED-4 from activating CED-3 [23].
Visualization of Conserved Activation Pathways
The following diagram illustrates the evolutionary conservation of caspase activation pathways from C. elegans to mammalian systems:
Research Reagents and Experimental Tools
The following table outlines essential research reagents and their applications for studying conserved caspase activation mechanisms:
Table 3: Essential Research Reagents for Caspase Activation Studies
| Reagent/Catalog Number | Application | Experimental Function | Research Context |
|---|---|---|---|
| Z-LEHD-FMK (Selleck) [15] | Caspase-9 inhibition | Irreversible caspase-9 inhibitor; blocks intrinsic apoptosis | In vivo studies of pulmonary fibrosis [15] |
| Recombinant CED-4/CED-9 complex [23] | Biochemical reconstitution | Platform for structural studies of caspase activation mechanism | Crystallography of ternary complex [23] |
| Yeast caspase expression system [24] | Functional screening | Null background for caspase pathway reconstitution | Validation of CED-9/CED-3 interaction [24] |
| Anti-caspase-9 antibodies [15] | Protein detection | IHC, WB for caspase-9 expression and cleavage | Analysis of fibrotic lung tissues [15] |
| AFC-based caspase substrates [24] | Enzymatic activity | Fluorogenic measurement of caspase activity | Kinetic studies in yeast and mammalian systems [24] |
The evolutionary conservation from C. elegans to mammalian systems provides a powerful framework for understanding the fundamental mechanisms of caspase activation and apoptotic regulation. The core architecture comprising CED-3/caspase-9, CED-4/Apaf-1, and CED-9/Bcl-XL has been maintained throughout metazoan evolution, with increasing complexity in mammalian systems enabling more nuanced regulation [23]. The conserved mechanism of caspase activation through oligomeric platforms (CED-4 tetramer/Apaf-1 apoptosome) represents a fundamental principle in cell death biology [25] [23].
These insights have direct implications for drug development, particularly for diseases involving dysregulated apoptosis. The conservation of these pathways means that findings in model organisms like C. elegans continue to provide valuable insights for understanding human disease mechanisms and developing therapeutic strategies [8] [15]. Continued investigation of these evolutionarily conserved pathways will undoubtedly yield new targets for therapeutic intervention in cancer, neurodegenerative diseases, and fibrotic disorders.
Physiological Roles in Development and Tissue Homeostasis
The apoptosome complex and its associated initiator caspase, caspase-9, constitute the core engine of the intrinsic apoptotic pathway. Within the context of broader research on caspase-9 activation mechanisms, it is crucial to understand that this system functions not merely as a death switch but as a master regulator of cellular and tissue homeostasis. The apoptosome is a large multimeric protein complex that forms in response to cellular stress signals, particularly those involving mitochondrial outer membrane permeabilization and cytochrome c release [2]. This complex, primarily composed of Apoptotic Protease Activating Factor 1 (Apaf-1), cytochrome c, and the initiator caspase-9, serves as an activation platform that precisely controls the initiation of the caspase cascade [7] [2]. Beyond its well-established role in apoptosis, emerging evidence reveals that caspase-9 activity is integral to normal development, tissue remodeling, and cellular differentiation processes [8] [26]. This whitepaper synthesizes current understanding of caspase-9's physiological functions, framed within ongoing research into the molecular mechanics of apoptosome-mediated caspase activation, providing researchers and drug development professionals with a comprehensive technical resource.
Molecular Mechanisms of Caspase-9 Activation
Apoptosome Structure and Assembly
The apoptosome is a quaternary protein structure with a calculated mass of approximately 1 megadalton, exhibiting a characteristic wheel-like architecture with seven-fold rotational symmetry in humans [2]. Its assembly represents a critical control point in the intrinsic apoptotic pathway, triggered by the release of cytochrome c from the mitochondrial intermembrane space into the cytosol in response to various cellular stresses [2]. The core component Apaf-1 exists in an autoinhibited state until cytochrome c binding, which occurs in a cleft between two β-propeller domains formed by WD40 repeats at the C-terminal region [7] [2]. This binding, coupled with nucleotide exchange from ADP to ATP/dATP, induces extensive conformational changes that transition Apaf-1 to an extended, assembly-competent state [7] [2].
The fully assembled apoptosome consists of seven Apaf-1 molecules arranged in a circular configuration, with each Apaf-1 subunit comprising three major domains: (1) an N-terminal Caspase Recruitment Domain (CARD), (2) a central Nucleotide-Binding and Oligomerization Domain (NOD) belonging to the AAA+ ATPase family, and (3) a C-terminal regulatory region composed of WD40 repeats that form β-propeller structures [2]. The CARD domains of Apaf-1 are flexibly attached above the central hub and organize into a disk-like, acentric spiral structure that serves as the recruitment platform for procaspase-9 [7] [2]. This spiral configuration typically accommodates three to four procaspase-9 molecules rather than a full complement of seven, due to constraints in linker length and specific binding interfaces [7].
Models of Caspase-9 Activation
The precise mechanism by which the apoptosome activates caspase-9 remains an active area of investigation, with two primary models proposed and substantiated by experimental evidence:
Induced Proximity/Dimerization Model: This model posits that the apoptosome serves primarily as a platform to concentrate procaspase-9 molecules, facilitating their proximity-induced homodimerization [8]. According to this view, caspase-9 activation is driven by dimerization rather than proteolytic cleavage, with the apoptosome functioning to increase the local concentration of procaspase-9 zymogens [8]. Procaspase-9 is recruited to the apoptosome through homotypic CARD-CARD interactions between Apaf-1 and procaspase-9 [8] [2]. The catalytic activity of caspase-9 is thought to be maintained through its continued association with the apoptosome complex [8].
Induced Conformation Model: This alternative model suggests that binding to the apoptosome induces specific conformational changes in procaspase-9 that activate its proteolytic function [8]. Structural studies have revealed that Apaf-1-caspase-9 interactions involve multiple binding interfaces rather than a simple 1:1 interaction [8]. Recent high-resolution structural data indicates that caspase-9 activation involves the formation of both homodimers (with other caspase-9 molecules) and heterodimers with Apaf-1 subunits on the apoptosome platform [7]. These interactions create distinct active states that may determine cell fate decisions, exemplifying potential sequential stages in the intrinsic cell death pathway [7].
Table 1: Key Structural Features of the Human Apoptosome
| Component | Domain Architecture | Function | Structural Features |
|---|---|---|---|
| Apaf-1 | N-terminal CARD | Procaspase-9 recruitment | Forms spiral disk with 3-4 caspase-9 CARDs |
| Central NOD (NBARC) | Oligomerization & nucleotide binding | AAA+ ATPase family; Walker A/B motifs | |
| C-terminal WD40 repeats | Cytochrome c binding & regulation | Forms two β-propellers (7 & 8 blades) | |
| Caspase-9 | N-terminal CARD | Apaf-1 binding | Homotypic interactions with Apaf-1 CARD |
| Catalytic domain | Protease activity | Heterodimer of large & small subunits | |
| Cytochrome c | Heme-binding protein | Apoptosome activation | Binds between β-propellers of Apaf-1 |
Following activation at the apoptosome, caspase-9 demonstrates selective substrate specificity. It directly cleaves and activates executioner caspase-3 but does not efficiently activate procaspase-6, which requires processing by caspase-3 [27]. This selectivity appears governed by both the sequence context and local structural environment surrounding the cleavage sites in target proteins [27].
Figure 1: Caspase-9 Activation Pathway via the Apoptosome Complex. The intrinsic apoptosis pathway initiates with mitochondrial stress, leading to cytochrome c release and apoptosome assembly, ultimately resulting in caspase-9 activation and downstream physiological outcomes.
Physiological Functions in Development and Homeostasis
Roles in Developmental Processes
Caspase-9-mediated apoptosis serves as a fundamental mechanism shaping tissue and organ development through selective elimination of specific cell populations. Genetic knockout studies demonstrate that mice lacking caspase-9 die perinatally with severe brain abnormalities, including exencephaly and reduced apoptosis in neural tissues, highlighting its non-redundant role in central nervous system development [8] [26]. During brain development, caspase-9 activation eliminates excess neuronal populations, with its absence resulting in neuronal overgrowth and disrupted cortical architecture [8]. Beyond the nervous system, caspase-9 contributes to the massive cell death of immature hematopoietic cells and neurons, with deficiencies leading to accumulation of superfluous cells in developing tissues [28] [26].
Recent evidence indicates that caspase-9 also participates in non-lethal processes essential for proper neural circuit formation. Studies reveal that caspase-9 is essential for postnatal motor circuit reorganization, with deficient activation causing corticospinal circuit defects and impaired skilled movement [26]. This function appears to operate through non-apoptotic mechanisms, as corticospinal axon elimination depends on caspase-9 activity without engaging effector caspases-3, -6, or -7 [26]. Additionally, caspase-9-mediated cleavage of semaphorin7a is required for proper axonal projection during olfactory development, further illustrating its non-lethal functions in neural wiring [26].
Tissue Homeostasis and Turnover
In mature tissues, caspase-9 activity maintains homeostasis by eliminating damaged, senescent, or potentially harmful cells. This homeostatic function extends across multiple tissue types, with dysregulation contributing to various pathological states. The apoptosome complex integrates diverse cellular stress signals, including DNA damage, oxidative stress, and growth factor deprivation, to determine cell fate decisions [2] [26]. This regulatory function ensures tissue integrity by removing compromised cells while preserving healthy counterparts.
The critical importance of caspase-9 in homeostatic maintenance is evidenced by its involvement in cellular differentiation pathways. Surprisingly, caspase-9 and caspase-3 activities participate in determining myoblast differentiation fate, indicating that apoptotic components are co-opted for specialized cellular functions beyond cell death [8] [26]. Similarly, caspase-9 plays a role in hematopoietic development, where it contributes to the balanced production and elimination of blood cell lineages [26]. These findings collectively demonstrate that caspase-9 activity is not exclusively dedicated to cell elimination but also contributes to cellular differentiation and functional specialization programs.
Non-Apoptotic Functions
Emerging research has unveiled several non-apoptotic functions of caspase-9 that contribute to cellular homeostasis. Caspase-9 activity is essential for mitochondrial homeostasis, with genetic or pharmacological ablation resulting in depolarized mitochondrial membrane potential, reduced reactive oxygen species production, aberrant accumulation of mitochondrial fusion-fission proteins, and impaired autophagy flux [26]. This mitochondrial regulation function depends on caspase-9 proteolytic activity, though its relevant substrates in this pathway remain incompletely characterized.
Additionally, non-catalytic caspase-9 regulates endosomal sorting and lysosomal biogenesis by facilitating retrograde transport of the insulin-like growth factor receptor 2 (IGFR2) from endosomes to the trans-Golgi network [26]. An endogenous alternatively-spliced short isoform, caspase-9b, which lacks the large catalytic subunit, inhibits apoptosis and promotes cell proliferation through NF-κB pathway activation [26]. These diverse non-apoptotic functions expand the physiological repertoire of caspase-9 beyond its traditional role in cell death execution.
Table 2: Physiological Roles of Caspase-9 in Development and Homeostasis
| Physiological Process | Caspase-9 Function | Consequence of Dysregulation | Experimental Evidence |
|---|---|---|---|
| Neural Development | Elimination of excess neurons; neural circuit refinement | Brain abnormalities; perinatal lethality in knockouts | Caspase-9 null mice exhibit exencephaly [8] |
| Hematopoietic Development | Regulation of hematopoietic cell populations | Disrupted blood cell homeostasis | Caspase-9 deficiency affects hematopoietic cells [26] |
| Muscle Differentiation | Regulation of myoblast differentiation | Impaired muscle development | Caspase-9 activity in myoblast differentiation [8] |
| Tissue Homeostasis | Elimination of damaged/senescent cells | Tissue degeneration or hyperplasia | Caspase-9 polymorphisms linked to homeostasis defects [26] |
| Mitochondrial Quality Control | Regulation of mitochondrial function | Impaired energy metabolism; reduced autophagy | Caspase-9 ablation disrupts mitochondrial parameters [26] |
Regulatory Mechanisms
Endogenous Regulators
Caspase-9 activity is tightly controlled through multiple endogenous regulatory mechanisms to ensure appropriate apoptotic responses. Phosphorylation represents a key regulatory strategy, with several protein kinases targeting caspase-9 at specific residues to modulate its function. Phosphorylation at Thr125 by multiple kinases, including ERK1/2, DYRK1A, CDK1-cyclinB1, and p38α, inhibits caspase-9 processing and activation [8] [29]. This phosphorylation site resides in the hinge region near the N-terminus of the large subunit and appears to function without preventing caspase-9 recruitment to Apaf-1 [8]. Instead, phosphorylated caspase-9 may serve as a dominant-negative regulator that modulates the recruitment of non-phosphorylated caspase-9 to the apoptosome platform [8].
Several endogenous proteins directly interact with and inhibit caspase-9 activity. X-linked Inhibitor of Apoptosis Protein (XIAP) represents a potent endogenous caspase-9 inhibitor, with its Bir3 domain selectively targeting the D315 neoepitope of cleaved caspase-9 [26]. Additionally, proteins including ATG7, the BIRC5/LAMTOR5 complex, and HAX-1 inhibit caspase-9 activation through various mechanisms [26]. Alternative splicing generates caspase-9b, a naturally occurring isoform lacking catalytic activity that functions as an endogenous dominant-negative inhibitor by competing with full-length caspase-9 for binding to the apoptosome [26].
Alternative Splicing and Isoform Regulation
Alternative splicing represents a critical mechanism regulating caspase-9 function and generating functional diversity. The caspase-9 gene produces multiple mRNA variants through alternative splicing of exons and introns during pre-mRNA processing [28]. This splicing-mediated regulation modulates cell and tissue homeostasis and is implicated in both developmental and pathological processes [28]. Splicing factors such as SRSF1 and hnRNP L regulate the alternative splicing of caspase-9 via specific intronic splicing enhancers, affecting the chemotherapeutic sensitivity of non-small cell lung cancer cells [28]. This regulatory layer demonstrates how caspase-9 activity is integrated with broader cellular signaling networks to determine cell fate decisions.
Experimental Approaches and Research Tools
Methodologies for Studying Caspase-9 Function
Research into caspase-9 physiology employs diverse methodological approaches to interrogate its activation, regulation, and functional outcomes. Structural biology techniques, particularly cryogenic electron microscopy (cryo-EM), have provided high-resolution structures of apoptosomes from C. elegans (CED-4), D. melanogaster (Dark), and H. sapiens (Apaf-1) [7]. These structural studies define critical protein interfaces, including intra- and interdomain interactions, and reveal interactions between apoptosomes and their respective initiator caspases [7]. Biochemical approaches, including expression and purification of recombinant caspase constructs, site-directed mutagenesis, and in vitro cleavage assays, enable detailed investigation of caspase-9 specificity and activation requirements [27].
Genetic manipulation techniques, including knockout mouse models and RNA interference, have been instrumental in establishing caspase-9's essential functions in development and tissue homeostasis [8] [26]. Caspase-9 null embryonic stem cells and embryonic fibroblasts demonstrate resistance to apoptotic stimuli including UV irradiation, γ-irradiation, and dexamethasone treatment [8]. Human genetic studies identifying CASP9 polymorphisms associated with various cancers, neurological disorders, and other pathologies provide clinical correlates for experimental findings [26].
Research Reagent Solutions
Table 3: Essential Research Reagents for Caspase-9 and Apoptosome Studies
| Reagent/Category | Specific Examples | Function/Application | Research Context |
|---|---|---|---|
| Expression Plasmids | pET23b-Casp3-His; pET23b-Casp9-His; pET11a-Casp6 | Recombinant protein expression | In vitro studies of caspase activation and specificity [27] |
| Cell Lines | MLE-12 (alveolar epithelial); HeLa; Caspase-9 null ES cells | Cellular models for functional studies | Investigation of cell-type specific functions [30] [26] |
| Animal Models | Caspase-9 knockout mice; Bleomycin-induced fibrosis models | In vivo functional analysis | Developmental studies; disease modeling [8] [30] |
| Pharmacological Inhibitors | Z-LEHD-FMK (caspase-9 inhibitor); XIAP Bir3 domain | Selective caspase-9 inhibition | Functional validation; therapeutic exploration [30] [26] |
| Antibodies | Anti-cleaved-caspase-9 (D315/D330 neoepitopes) | Detection of activated caspase-9 | Assessing caspase-9 activation status [26] |
Figure 2: Experimental Approaches for Studying Caspase-9 Physiology. Multiple complementary methodologies provide mechanistic insights into caspase-9 activation, physiological functions, and pathological roles.
The apoptosome complex and its activation of caspase-9 represent a sophisticated molecular machinery essential for physiological processes beyond mere cell elimination. Caspase-9 functions as a crucial regulator of development, tissue homeostasis, and cellular differentiation through both apoptotic and non-apoptotic mechanisms. The emerging understanding of caspase-9's multimodal functions reveals a complex regulatory network integrating multiple signaling pathways, post-translational modifications, and alternative splicing events to determine cellular outcomes. Future research directions should focus on elucidating the structural determinants of caspase-9 substrate specificity, the molecular mechanisms underlying its non-apoptotic functions, and the therapeutic potential of modulating caspase-9 activity in pathological conditions. As research continues to unravel the complexities of caspase-9 regulation and function, new opportunities will emerge for targeting this pathway in diseases characterized by dysregulated cell survival and death, including cancer, neurodegenerative disorders, and autoimmune conditions.
Advanced Techniques for Apoptosome Analysis and Therapeutic Targeting
The intricately regulated process of programmed cell death, or apoptosis, is fundamental to development and tissue homeostasis in multicellular organisms. The apoptosome complex, a central signaling platform in the intrinsic apoptotic pathway, is responsible for the activation of the initiator protease, caspase-9. For decades, the precise structural mechanism underlying caspase-9 activation remained one of the most elusive questions in cell death research. The resolution of this mystery has been primarily driven by advances in two complementary structural biology techniques: cryo-electron microscopy (cryo-EM) and methyl-TROSY NMR spectroscopy. This whitepaper provides an in-depth technical guide on the application of these methods within the context of apoptosome research, detailing how their synergistic use has decoded the complex activation dynamics of caspase-9, offering invaluable insights for drug discovery targeting apoptotic disorders such as cancer and neurodegenerative diseases.
The Apoptosome Complex: A Central Apoptotic Signaling Platform
Assembly and Function
The intrinsic apoptotic pathway is triggered by diverse cellular stressors, including DNA damage and growth factor withdrawal, leading to mitochondrial outer membrane permeabilization (MOMP). This event results in the release of cytochrome c from the mitochondrial intermembrane space into the cytosol [1] [31]. Cytochrome c binds to the adapter protein Apoptotic Protease-Activating Factor 1 (Apaf-1), which exists in an inactive, monomeric state bound to dATP or ATP [1]. Upon cytochrome c binding, Apaf-1 undergoes a nucleotide exchange (dATP/ATP exchange) and subsequent oligomerization into a wheel-like apoptosome complex comprising seven Apaf-1 subunits [1] [25]. This platform then recruits the initiator caspase, procaspase-9, via homotypic interactions between the caspase recruitment domains (CARDs) present in both Apaf-1 and caspase-9 [8] [32].
Table 1: Core Components of the Human Apoptosome Complex
| Component | Role in Apoptosome | Key Domains |
|---|---|---|
| Apaf-1 | Scaffold Protein | CARD, NOD/NB-ARC (ATPase domain), WD40 Repeats |
| Cytochrome c | Activating Signal | Heme-binding protein |
| Caspase-9 | Initiator Caspase | CARD, Large Catalytic Subunit, Small Catalytic Subunit |
| (d)ATP | Essential Cofactor | Energy source for conformational change |
The Mechanistic Debate: Caspase-9 Activation Models
The mechanism by which the apoptosome activates caspase-9 has been the subject of intense debate, giving rise to two primary, competing hypotheses:
- The Induced Proximity/Dimerization Model: This model posits that the apoptosome serves primarily as a platform to increase the local concentration of procaspase-9 molecules, facilitating their proximity-induced homodimerization, which is the key event for activation [32] [4] [25]. In this model, dimerization of caspase-9's protease domains drives its activation.
- The Holoenzyme/Allosteric Regulation Model: This model argues that binding to the apoptosome induces allosteric conformational changes within a monomeric caspase-9, resulting in its activation. Here, caspase-9 and the apoptosome form a active holoenzyme where Apaf-1 acts as an allosteric regulator [33] [25].
Recent evidence, particularly from hybrid methodologies, suggests that the actual mechanism is nuanced and may integrate aspects of both models [32].
Cryo-Electron Microscopy in Apoptosome Research
Methodology and Workflow
Cryo-EM has been instrumental in visualizing the architecture of large complexes like the apoptosome. The standard workflow involves:
- Sample Preparation: The native apoptosome complex (∼1.1 MDa) is reconstituted in vitro from purified Apaf-1, cytochrome c, and (d)ATP. The sample is then vitrified in liquid ethane to preserve its native state in a thin layer of amorphous ice [1].
- Data Acquisition: Images of the frozen-hydrated complexes are collected using a transmission electron microscope under low-dose conditions to minimize radiation damage. Thousands of micrographs are recorded.
- Image Processing: Computational algorithms are used to select and align millions of individual particle images. These are then classified to isolate homogeneous populations and reconstructed into a three-dimensional density map.
- Model Building and Refinement: Atomic models are built and refined into the cryo-EM density map to interpret the structural details.
Key Structural Insights from Cryo-EM
Modeling of cryo-EM images at ~9.5 Å resolution revealed the apoptosome as a wheel-shaped particle with seven-fold symmetry [1]. The structure features a central hub formed by the oligomerized NOD domains of Apaf-1, with seven bent spokes radiating outwards, each composed of the Apaf-1's helical and WD40 domains. The CARD domains of Apaf-1 form a flexibly tethered "disk" above the central hub, which is responsible for recruiting procaspase-9. This flexible tethering is crucial, as it allows the CARDs, and by extension the catalytic domains of caspase-9, significant conformational freedom, a observation that proved critical for understanding the activation mechanism [1].
Figure 1: A generalized workflow for structural determination of the apoptosome complex using single-particle cryo-EM.
NMR Spectroscopy for Studying Dynamics and Mechanism
Methyl-TROSY NMR: A Tool for Mega-Dalton Complexes
While cryo-EM provides static snapshots, NMR spectroscopy offers unique insights into protein dynamics and weak interactions in solution. Traditional NMR is limited by the size of the complex, but the development of methyl-TROSY NMR has enabled the study of complexes exceeding 1 MDa, such as the native apoptosome [34]. This technique focuses on methyl groups in key amino acids like isoleucine, leucine, and valine, which serve as sensitive probes of structure and dynamics even in very large systems.
A seminal application involved studying caspase-9 within the 1.3-MDa native apoptosome complex and a smaller 480-kDa engineered apoptosome mimic using methyl-TROSY NMR [34]. The key technical achievement was observing the NMR signals of isotopically labeled caspase-9 while it was bound to these massive scaffolds.
Revealing a Monomeric, Substrate-Triggered Activation Mechanism
The methyl-TROSY NMR data provided a critical breakthrough. It revealed that the protease domain (PD) of caspase-9 remains predominantly monomeric after recruitment to the apoptosome, with dimerization dissociation constants in the millimolar range [34]. This finding challenged the pure induced-proximity model. The data supported a refined substrate-triggered dimerization model: the apoptosome organizes caspase-9 protease domains in a way that they are monomeric and inactive until a peptide substrate binds. Substrate binding then acts as a linchpin, rapidly inducing the dimerization of caspase-9 PDs and triggering their full catalytic activity [34]. This provides a crucial regulatory layer to prevent spurious activation.
Table 2: Key Quantitative Findings from Methyl-TROSY NMR Study [34]
| Parameter Studied | Experimental Finding | Interpretation |
|---|---|---|
| Caspase-9 Protease Domain (PD) Dimerization (free in solution) | Dissociation constant (K~d~) in the millimolar range | Very weak intrinsic tendency to dimerize |
| Caspase-9 PD State within the Apoptosome | NMR spectra consistent with a monomeric state | Apoptosome binding alone does not induce dimerization |
| Proposed Activation Trigger | Rapid and extensive dimerization upon substrate presence | Activation is substrate-triggered, not platform-induced |
Integrated Structural Models and a Synthesis of Activation Mechanisms
The combined data from cryo-EM, NMR, and biochemical assays has led to a sophisticated, integrated model of caspase-9 activation that moves beyond the simple dichotomy of earlier hypotheses.
A Hybrid Activation Model
The current model synthesizes elements from both induced proximity and allosteric regulation:
- Recruitment and Positioning: The apoptosome first recruits procaspase-9 via CARD-CARD interactions. Cryo-EM shows the CARD disk is flexibly tethered, allowing the caspase-9 protease domains to sample different orientations [1].
- Allosteric Priming: Binding to the apoptosome may induce subtle conformational changes that "prime" caspase-9, making it more susceptible to activation, though it remains a monomer as per NMR data [34] [33].
- Substrate-Triggered Dimerization: The organized arrangement of primed, monomeric caspase-9 protease domains on the apoptosome surface creates a high local concentration. When a substrate (like procaspase-3) enters the active site, it acts as a nucleating factor, stabilizing the formation of a transient, active caspase-9 homodimer that processes the substrate with high efficiency [34] [32].
- Regulation by Cleavage: The model also incorporates a "molecular timer" mechanism. Uncleaved procaspase-9 has a higher affinity for the apoptosome and homodimerizes more readily. Autocatalytic cleavage at Asp-315 produces caspase-9-p35/p12, which inhibits homodimerization and leads to its release from the complex, thus timing the activity of the apoptosome [32]. Feedback cleavage by caspase-3 at Asp-330 can partially restore this activity.
Figure 2: Integrated model of caspase-9 activation on the apoptosome, synthesizing structural data from Cryo-EM and NMR.
The Role of Heterodimerization
Beyond homodimerization, research using site-specific crosslinking has revealed that procaspase-9 can also form a heterodimer with Apaf-1, where the small subunit of caspase-9 binds to the NOD domain of Apaf-1 [32]. This heterodimer appears to be particularly efficient at activating the downstream effector, procaspase-3. The formation of both homo- and heterodimers on the apoptosome highlights the complex regulatory landscape and contributes to the overall proteolytic activity of the complex.
Experimental Protocols for Key Investigations
Protocol: Analyzing Caspase-9 Dimerization via SEC-MALS
Aim: To determine the oligomeric state of recombinant caspase-9 constructs (e.g., non-cleavable ProC9-TM vs. processed C9-p35/p12) in solution [32].
- Protein Purification: Express and purify recombinant human caspase-9 constructs (e.g., in E. coli or insect cells) using affinity and size-exclusion chromatography.
- Sample Preparation: Concentrate proteins to a high concentration (e.g., 40 µM) in a suitable buffer.
- SEC-MALS Analysis:
- Inject the protein sample onto a high-performance size-exclusion chromatography column equilibrated with running buffer.
- Connect the outlet of the column in-line to a multi-angle light scattering (MALS) detector and a refractometer.
- The MALS detector measures the absolute molar mass of the eluting particles independently of their shape, while the UV and refractive index signals provide concentration.
- Data Interpretation: Analyze the data using dedicated software. A measured molar mass approximately twice that of the caspase-9 monomer indicates stable homodimer formation under the conditions tested.
Protocol: Studying Caspase-9 within the Apoptosome by Methyl-TROSY NMR
Aim: To characterize the dynamics and oligomeric state of caspase-9's protease domain while bound to the apoptosome [34].
- Isotope Labeling: Produce caspase-9 with specific isotopic labeling (e.g., (^{13})CH(_3)- on Ile, Leu, Val methyl groups) in a recombinant expression system.
- Apoptosome Reconstitution: Form the native apoptosome complex by incubating purified, unlabeled Apaf-1 with cytochrome c and (d)ATP. Then, add the isotopically labeled caspase-9 to form the holo-complex.
- NMR Data Collection:
- Use a high-field NMR spectrometer (e.g., 800 MHz or higher) equipped with a cryogenic probe.
- Acquire methyl-TROSY spectra on two samples: i) the free, isotopically labeled caspase-9, and ii) the caspase-9–apoptosome complex.
- Maintain constant temperature (e.g., 25-37°C) and use optimized pulse sequences for large complexes.
- Spectra Analysis: Compare the NMR spectra of free and bound caspase-9. Chemical shift perturbations indicate binding. The line shape and number of cross-peaks can be used to infer the oligomeric state (e.g., a spectrum consistent with a monomeric state).
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents for Apoptosome and Caspase-9 Structural Studies
| Reagent / Material | Function in Research | Example Application |
|---|---|---|
| Recombinant Apaf-1 | Core scaffold protein for in vitro reconstitution of the apoptosome. | Cryo-EM sample preparation; biochemical activity assays [32] [1]. |
| Recombinant Caspase-9 (Wild-type & Mutants) | The initiator caspase under investigation. Key mutants include catalytically inactive (C287A), non-cleavable (D315A, "ProC9-TM"), and dimerization-deficient (F404D). | Studying activation mechanisms, cleavage effects, and dimerization interfaces [32]. |
| Cytochrome c | Apoptogenic signal that triggers Apaf-1 oligomerization. | Essential component for initiating apoptosome assembly in all experimental setups [1] [31]. |
| (d)ATP / ATP | Essential nucleotide cofactor for Apaf-1 conformational change and oligomerization. | Required for functional reconstitution of the apoptosome complex [1]. |
| Caspase-Specific Peptide Substrates | Synthetic fluorogenic or chromogenic peptides (e.g., LEHD-amc) to measure enzymatic activity. | Quantifying caspase-9 activity in spectrophotometric assays [32]. |
| Site-Specific Crosslinkers | Chemical reagents (e.g., BS3, formaldehyde) to covalently trap transient protein-protein interactions. | Providing direct biochemical evidence for caspase-9 homo- and heterodimerization on the apoptosome [32]. |
| Isotope-Labeled Nutrients | (^{15})NH(4)Cl, (^{13})C-glucose, precursors for (^{13})CH(3)- labeling for NMR sample preparation. | Production of isotopically labeled caspase-9 for NMR-based structural and dynamics studies [34]. |
| Cryo-EM Grids | Ultrathin, perforated carbon films on metal grids (e.g., Quantifoil, C-flat) for sample vitrification. | Supporting the thin layer of ice containing the apoptosome complex for cryo-EM imaging [1]. |
The activation of caspase-9 (Casp9) is a pivotal event in the intrinsic apoptotic pathway, a programmed cell death process essential for development, tissue homeostasis, and the elimination of damaged cells [35] [8]. This pathway is triggered by intracellular stressors such as DNA damage, leading to mitochondrial outer membrane permeabilization and the release of cytochrome c into the cytosol. Cytochrome c then binds to Apoptotic Protease-Activating Factor 1 (Apaf-1), prompting its oligomerization into a wheel-like signaling platform known as the apoptosome [35] [36]. The apoptosome recruits initiator caspase-9 via homotypic interactions between the caspase activation domains (CARDs) of Apaf-1 and caspase-9 [8]. Once activated on the apoptosome, caspase-9 cleaves and activates downstream effector caspases (e.g., caspase-3 and -7), initiating a proteolytic cascade that culminates in cell death [35] [37].
Understanding the precise structural mechanism by which the apoptosome activates caspase-9 has been a central challenge in apoptosis research. Two primary models have been proposed: the "induced proximity" or "proximity-induced dimerization" model, which posits that the apoptosome serves as a platform to concentrate caspase-9 monomers, facilitating their dimerization and activation; and the "induced conformation" or "holoenzyme" model, which suggests that the apoptosome induces activating conformational changes in monomeric caspase-9 [8] [32]. Recent scientific investigations have yielded data that both challenge and refine these models, highlighting the necessity of robust biochemical assays to monitor caspase-9 dimerization and activity within this critical complex. This guide provides an in-depth technical overview of the methods enabling this research, framed within the context of elucidating the caspase-9 activation mechanism.
Key Mechanistic Insights: A Foundation for Assay Design
The following diagram illustrates the core signaling pathway and molecular interactions central to caspase-9 activation, integrating key recent findings.
Figure 1. The Caspase-9 Activation Pathway within the Apoptosome. Recent mechanistic studies reveal that binding to the apoptosome primes caspase-9, but extensive dimerization occurs predominantly upon substrate presence [35]. This substrate-driven step is critical for full activation and subsequent triggering of the apoptotic cascade.
Central to the debate on caspase-9 activation has been the role of dimerization. Caspase-9 is an initiator caspase that, like other family members, depends on dimerization for its activity [35] [38]. However, unlike effector caspases, it exists as an inactive monomer in the cytosol and only gains significant proteolytic activity upon binding the apoptosome [35]. A groundbreaking 2023 study using methyl-TROSY NMR spectroscopy demonstrated that the protease domains (PDs) of caspase-9 remain predominantly monomeric after recruitment to the apoptosome [35]. This finding challenges the simple induced proximity model. Instead, the data support a mechanism where the apoptosome primes and organizes caspase-9 monomers, enabling them to rapidly and extensively dimerize only when a substrate is available [35]. This provides a crucial additional layer of regulation, ensuring that caspase-9 is not fully activated prematurely.
Furthermore, research has shown that the affinity of caspase-9 for the apoptosome and its propensity to dimerize are influenced by its cleavage status. The uncleaved procaspase-9 (proCasp9) has a higher affinity for the apoptosome than the cleaved, mature form (Casp9-p35/p12) [32]. Autocleavage within the apoptosome does not directly activate caspase-9 but instead initiates a "molecular timer," regulating the duration of apoptosome activity by facilitating the release of cleaved caspase-9, which has a reduced dimerization affinity [32] [8].
Quantitative Biochemical Profiling of Caspase-9
The investigation of caspase-9's behavior relies on quantitative methods that provide insights into its oligomeric state and enzymatic function. The table below summarizes key quantitative findings from recent research.
Table 1. Quantitative Profiling of Caspase-9 Dimerization and Activity
| Parameter Measured | Experimental Condition | Key Finding | Technique Used | Citation |
|---|---|---|---|---|
| Dissociation Constant (Kd) for Dimerization | Caspase-9 Protease Domain (PD), substrate-free | Weak dimerization, Kd in the millimolar range | SEC-MALS, NMR | [35] |
| Oligomeric State | Caspase-9 PD within native 1.3 MDa apoptosome | PD remains monomeric on the scaffold | Methyl-TROSY NMR | [35] |
| Oligomeric State | Caspase-9 PD with substrate mimic (Z-LEHD-fmk) | Forms stable dimer | SEC-MALS | [35] |
| Catalytic Activity | Uncleaved Procaspase-9 (ProC9-TM) vs. Cleaved (C9-p35/p12) in apoptosome | ProC9-TM exhibits more robust activity | In vitro caspase-3 cleavage assay | [32] |
| Cell Death Efficiency | iCasp9 HeLa cells + 0.25 nM AP20187 (CID) | ~68% cell death | Live-cell imaging & FRET | [38] |
| Cell Death Efficiency | iCasp9 HeLa cells + CID + XIAP inhibitor (AT406) | Significant increase in death percentage | Live-cell imaging & FRET | [38] |
Core Experimental Protocols and Workflows
This section details the methodologies essential for studying caspase-9 dimerization and activity, from analyzing bulk solutions to probing single cells within complex assemblies.
Analyzing Oligomeric State: Size-Exclusion Chromatography with Multi-Angle Light Scattering (SEC-MALS)
SEC-MALS is a gold-standard technique for determining the absolute molecular weight and oligomeric state of proteins in solution, independent of elution volume.
Detailed Protocol:
- Protein Preparation: Purify recombinant caspase-9 protease domain (Casp9 PD) or full-length protein. For the substrate-bound state, pre-incubate the protein with a molar excess of an irreversible inhibitor like the tetrapeptide Z-LEHD-fmk, which covalently traps the active conformation [35].
- Chromatography System: Equilibrate a high-quality size-exclusion column (e.g., Superdex 200 Increase) with a suitable buffer (e.g., 20 mM HEPES, 150 mM NaCl, pH 7.5).
- In-Line Detection: The HPLC system should be connected in series to three detectors: a UV spectrophotometer, a Multi-Angle Light Scattering (MALS) detector, and a differential refractometer.
- Sample Injection and Run: Inject a concentrated sample (e.g., 50-100 µL at ~1-5 mg/mL) onto the column. Isocratically elute the protein at a slow, constant flow rate (e.g., 0.5 mL/min).
- Data Analysis: Use the manufacturer's software (e.g., Astra for Wyatt systems) to analyze the data. The software combines the light scattering and refractive index signals to calculate the absolute molecular weight across the entire elution peak.
- Interpretation: A calculated molecular weight of ~30 kDa indicates a Casp9 PD monomer, while a value of ~60 kDa confirms a dimer [35]. This provides direct, quantitative evidence of dimerization propensity.
Probing Structure and Dynamics in Mega-Dalton Complexes: Methyl-TROSY NMR
Solution-state NMR spectroscopy, particularly methyl-TROSY, is uniquely powerful for characterizing flexible, dynamic domains within very large complexes like the apoptosome, which can exceed 1 MDa in size [35].
Detailed Protocol:
- Isotope Labeling: Produce caspase-9 with specific isotopic labeling. A common strategy is to express the protein in deuterated medium with protonated, carbon-13-labeled methyl groups on isoleucine, leucine, and valine (ILV) residues. This reduces signal complexity and relaxation, allowing observation in large complexes.
- Complex Reconstitution: Reconstitute the native apoptosome by incubating Apaf-1, cytochrome c, and (d)ATP with the isotopically labeled caspase-9. Alternatively, an engineered apoptosome mimic (e.g., using a heptameric proteasome scaffold) can be used [35].
- NMR Data Acquisition: Acquire two-dimensional (^1H)-(^{13}C) correlation spectra on a high-field NMR spectrometer (≥ 600 MHz) using methyl-TROSY pulse sequences. These sequences are optimized to maintain signal for high-molecular-weight systems.
- Spectral Analysis:
- Compare the NMR spectrum of caspase-9 within the apoptosome to its spectrum as an isolated monomer and a salt-induced dimer.
- The presence of distinct peak patterns or chemical shifts can report on the oligomeric state and conformational changes.
- Key Insight: If the spectrum of apoptosome-bound caspase-9 closely resembles that of the monomer and does not change upon binding, it indicates that the protease domains remain monomeric and highly dynamic while tethered to the scaffold [35].
Monitoring Activation Dynamics in Single Cells: FRET-Based Live-Cell Imaging
For studies in a more physiological context, such as with the inducible caspase-9 (iCasp9) system used in cell therapies, FRET-based reporters allow real-time monitoring of caspase activity in single living cells.
Detailed Protocol:
- Reporter Design: Construct a FRET-based caspase-3/7 reporter, such as a plasmid encoding CFP and Venus fluorescent proteins linked by the caspase-3 cleavage sequence DEVD [38].
- Cell Line Engineering: Stably transduce the cell line of interest (e.g., HeLa) with the FRET reporter and the iCasp9 construct (a fusion of caspase-9 to a dimerizer domain like FKBP12(F36V)).
- Live-Cell Imaging:
- Plate cells on an imaging-optimized dish and place on a confocal or widefield fluorescence microscope with an environmental chamber (37°C, 5% CO₂).
- Acquire time-lapse images of CFP and Venus channels before and after adding the dimerizer drug (AP20187 or AP1903).
- Data Quantification:
- For each cell, track the fluorescence intensity over time in both channels.
- Calculate the FRET ratio (CFP/Venus or Venus/CFP, depending on the construct design).
- Activation Signature: Cleavage of the linker by effector caspases separates CFP and Venus, leading to a loss of FRET and a measurable change in the FRET ratio. A sharp increase (or decrease) in this ratio indicates caspase-3 activation and commitment to cell death [38]. This reveals cell-to-cell heterogeneity in the timing and probability of apoptosis.
The following workflow chart integrates these core protocols into a cohesive strategy for investigating caspase-9.
Figure 2. Experimental Workflow for Investigating Caspase-9. A combined approach using in vitro reconstitution and multiple analytical techniques is essential to build a complete mechanistic picture. Research objectives guide the selection of specific protocols.
The Scientist's Toolkit: Essential Research Reagents
Table 2. Key Reagent Solutions for Caspase-9 Research
| Reagent / Tool | Function & Application | Example & Notes |
|---|---|---|
| Recombinant Caspase-9 Proteins | In vitro reconstitution assays; studying purified protein biochemistry. | Full-length, protease domain (PD), cleavage mutants (e.g., ProC9-TM), dimerization-deficient mutants (e.g., F404D) [32]. |
| Apoptosome Components | Reconstruct the native activation platform. | Recombinant Apaf-1, cytochrome c, (d)ATP. Can be reconstituted from insect cell systems [35] [32]. |
| Activity-Based Probes / Inhibitors | Trapping active conformations; measuring enzyme kinetics. | Irreversible inhibitors: e.g., Z-LEHD-fmk (traps active dimer for SEC-MALS) [35]. Peptide substrates: e.g., LEHD-amc (for fluorometric activity assays) [32]. |
| Inducible Caspase-9 (iCasp9) System | Model for studying dimerization and death kinetics in live cells. | iCasp9 construct (Casp9-FKBP12 fusion); Chemical Dimerizer (CID): AP20187 or AP1903 [39] [38]. |
| FRET-Based Caspase Reporters | Real-time, single-cell monitoring of downstream caspase activation. | Plasmid encoding CFP-DEVDR-Venus. Cleavage by caspase-3 alters FRET ratio, signaling apoptosis commitment [38]. |
| XIAP Inhibitors | Investigating regulation of caspase-9 activity; boosting killing efficiency. | e.g., AT406. Used in combination with iCasp9 inducer to overcome endogenous inhibition [38]. |
Discussion and Research Implications
The integrated use of the assays and reagents detailed herein has profoundly advanced our understanding of caspase-9 activation. The model emerging from recent data is nuanced: the apoptosome acts as a regulated activation platform that primes caspase-9 monomers. The critical step of full dimerization is gated by substrate availability, adding a crucial checkpoint to prevent inadvertent cell death [35]. Furthermore, the cleavage of caspase-9 serves not to activate it but to install a molecular timer that controls the duration of apoptosome signaling [32] [8].
These insights have direct implications for drug discovery and therapeutic applications. The iCasp9 system is already a promising "safety switch" in adoptive cell therapies [39] [38]. However, heterogeneous cell responses and the development of drug resistance remain challenges. Research using the single-cell FRET assays has shown that resistance is linked to low initial iCasp9 expression and high XIAP/Caspase-3 ratios in surviving cells [38]. This knowledge directly informs combination therapy strategies, such as using a XIAP inhibitor like AT406 after initial iCasp9 activation to significantly enhance killing efficiency and overcome resistance [38].
For researchers, the path forward involves leveraging these sophisticated tools to answer remaining questions. How do specific oncogenic mutations impair the dimerization priming step? Can small molecules be identified that allosterically promote or stabilize the active caspase-9 dimer on the apoptosome? The continuous refinement of biochemical, biophysical, and cellular assays, as outlined in this guide, is essential for translating the complex mechanism of caspase-9 activation into novel and effective therapeutic strategies for cancer and other diseases.
Engineering Apoptosome Mimics for Controlled Activation Studies
The apoptosome is a critical signaling platform in the intrinsic apoptotic pathway, serving as the molecular engine for the activation of caspase-9 and the subsequent commitment to programmed cell death. This wheel-shaped heptameric complex is assembled upon the release of cytochrome c from mitochondria in response to cellular stress signals, including DNA damage and oxidative stress [40] [36]. The formation of this complex represents a pivotal control point in the life-or-death decision of cells, with its dysregulation contributing to various pathologies, including cancer, neurodegenerative diseases, and autoimmune disorders [40] [41]. Research into the fundamental mechanisms of apoptosome formation and function provides the necessary foundation for engineering apoptosome mimics, which serve as valuable tools for dissecting the molecular events of caspase activation and for screening potential therapeutic compounds that modulate apoptotic signaling [40] [42].
The core component of the apoptosome is Apoptotic Protease-Activating Factor 1 (Apaf-1), which exists as a monomer in an inactive conformation under normal physiological conditions. Apaf-1 is characterized by three functional domains: an N-terminal Caspase Recruitment Domain (CARD), a central Nucleotide-Binding Domain (NBD), and C-terminal WD40 repeats [40]. The CARD mediates interactions with the CARD of procaspase-9, the NBD binds dATP/ATP, and the WD40 domain is responsible for cytochrome c binding [40]. Recent research has revealed that Apaf-1 exhibits evolutionary conservation in DNA sensing functionality, binding cytoplasmic DNA via a positively charged surface between its NB-ARC and WD1 domains, which competes with cytochrome c binding and potentially switches cell fate between apoptosis and inflammation [43].
Structural and Mechanistic Principles of the Apoptosome
Domain Architecture and Assembly Mechanism
The assembly of the apoptosome follows a carefully orchestrated molecular process initiated when cytochrome c, released from mitochondria during cellular stress, binds to the WD40 repeats of monomeric Apaf-1. This binding event triggers dATP hydrolysis and nucleotide exchange, inducing a conformational change that exposes the CARD domain and enables Apaf-1 oligomerization into a heptameric, wheel-like structure approximately 1.4 MDa in size [40] [36]. This structural transition is essential for creating a platform that can recruit and activate procaspase-9.
The activation mechanism of caspase-9 following its recruitment to the apoptosome remains an area of active investigation, with several competing models proposed. The induced proximity model suggests that the apoptosome serves primarily as a platform to bring caspase-9 molecules into close proximity, enabling autoactivation through monomer clustering [40]. The proximity-driven dimerization model posits that the apoptosome promotes the dimerization of caspase-9 monomers, which is sufficient for activation [8]. In contrast, the induced conformation model proposes that binding to the apoptosome induces specific conformational changes in caspase-9 that enhance its catalytic activity [8]. Recent structural studies have revealed that caspase-9 activation involves multimeric interactions between the CARD domains of Apaf-1 and caspase-9 requiring three distinct interfaces, rather than simple 1:1 binding [8].
Table 1: Key Domains of Apaf-1 and Their Functional Roles
| Domain | Position | Primary Function | Interacting Partners |
|---|---|---|---|
| CARD | N-terminal | Protein-protein interaction | Caspase-9 CARD domain |
| NBD/NB-ARC | Central | Nucleotide binding (dATP/ATP) and oligomerization | dATP, ATP, cytochrome c |
| WD40 Repeats | C-terminal | Cytochrome c binding | Cytochrome c, DNA [43] |
Regulatory Influences on Apoptosome Function
Apoptosome activity is subject to multiple layers of cellular regulation, with the balanced expression and activity of pro-apoptotic and anti-apoptotic factors determining the sensitivity of cells to apoptotic stimuli. Inhibitor of Apoptosis Proteins (IAPs), particularly XIAP, function as crucial negative regulators by directly binding to and inhibiting active caspases, including caspase-9 [44]. This inhibition is counteracted by mitochondrial proteins such as SMAC/DIABLO, which are released along with cytochrome c during mitochondrial outer membrane permeabilization and function as endogenous IAP antagonists [44] [42].
Post-translational modifications further fine-tune apoptosome activity. Phosphorylation of caspase-9 at Thr125 by various kinases, including ERK1/2, DYRK1A, CDK1-cyclinB1, and p38α, inhibits its proteolytic processing without necessarily preventing recruitment to Apaf-1 [8]. The phosphorylated caspase-9 may act as a dominant-negative regulator by competing with non-phosphorylated caspase-9 for apoptosome binding [8]. Understanding these regulatory mechanisms is essential for engineering functional apoptosome mimics that accurately recapitulate the controlled activation characteristics of the native complex.
Engineering Apoptosome Mimics: Design Strategies and Implementation
Structural Scaffolds for Complex Assembly
The engineering of functional apoptosome mimics requires the identification or design of appropriate structural scaffolds that can support the oligomerization state and protein interaction surfaces of the native complex. One promising approach utilizes truncated Apaf-1 constructs containing the CARD and NBD domains but lacking the WD40 region, which may retain the ability to oligomerize in a cytochrome c-independent manner when combined with nucleotide analogs [40]. These minimal apoptosomes can provide insights into the core structural requirements for caspase-9 activation while offering a more tractable system for biophysical characterization.
Alternative scaffolding strategies include the use of synthetic protein assemblies based on symmetrical oligomeric proteins that can be engineered to display Apaf-1 CARD domains in defined geometric patterns. Computational protein design approaches can generate de novo protein assemblies with specified symmetry that mimic the heptameric architecture of the native apoptosome, potentially enabling more precise control over the spatial arrangement of caspase-9 binding sites than achieved with truncated Apaf-1 constructs alone.
Table 2: Quantitative Parameters for Apoptosome Mimic Engineering
| Parameter | Native Apoptosome | Minimal Apoptosome Mimic | Measurement Technique |
|---|---|---|---|
| Molecular Weight | ~1.4 MDa [36] | 500-700 kDa | Size exclusion chromatography with multi-angle light scattering |
| Stoichiometry (Apaf-1:Caspase-9) | 7:3-4 [40] | Adjustable (1:1 to 7:7) | Analytical ultracentrifugation |
| Caspase-9 Activation Rate | 0.1-0.5 min⁻¹ [40] | Variable (0.01-0.3 min⁻¹) | Fluorometric caspase activity assay |
| dATP/ATP Requirement | 1-10 µM [40] | 1-100 µM (concentration-dependent) | Nucleotide binding assays |
| Cytochrome c EC₅₀ | 0.1-1 µM [40] | Not applicable (constitutive activity) | Cytochrome c titration studies |
Reconstitution of Functional Apoptosome Mimics
Protocol 3.2.1: Expression and Purification of Recombinant Apaf-1 and Caspase-9
Construct Design: Clone cDNA encoding human Apaf-1 (residues 1-1080, lacking WD40 repeats) and full-length caspase-9 into bacterial expression vectors (e.g., pET series) containing N-terminal His₆-tags with TEV protease cleavage sites.
Protein Expression: Transform constructs into E. coli BL21(DE3) cells. Grow cultures in LB medium at 37°C to OD₆₀₀ ≈ 0.6-0.8. Induce protein expression with 0.5 mM IPTG and incubate for 16-20 hours at 18°C.
Protein Purification: Harvest cells by centrifugation (4,000 × g, 20 min) and resuspend in lysis buffer (50 mM Tris-HCl pH 8.0, 500 mM NaCl, 10 mM imidazole, 10% glycerol, 1 mM PMSF). Lyse cells by sonication and clarify lysate by centrifugation (20,000 × g, 45 min). Purify proteins using Ni²⁺-NTA affinity chromatography with elution buffer (50 mM Tris-HCl pH 8.0, 500 mM NaCl, 250 mM imidazole). Remove His₆-tags by TEV protease cleavage during dialysis overnight. Further purify by ion-exchange and size-exclusion chromatography.
Protocol 3.2.2:In VitroAssembly of Minimal Apoptosome Mimics
Assembly Reaction: Combine purified Apaf-1ΔWD40 (5-10 µM) with caspase-9 (5-15 µM) in assembly buffer (20 mM HEPES-KOH pH 7.5, 100 mM KCl, 1.5 mM MgCl₂, 1 mM EDTA, 1 mM DTT) in the presence of 1 mM dATP.
Incubation Conditions: Incubate the reaction mixture for 60-90 minutes at 30°C to allow complex formation.
Complex Isolation: Separate assembled complexes from unincorporated components using size-exclusion chromatography (Superose 6 Increase 10/300 GL column) equilibrated with assembly buffer without dATP.
Quality Assessment: Analyze complex formation by native PAGE, dynamic light scattering, and negative stain electron microscopy to verify proper oligomerization.
Diagram 1: Native Apoptosome Assembly Pathway
Characterization of Engineered Apoptosome Mimics
Protocol 3.3.1: Caspase Activation Assay
Substrate Preparation: Prepare 50 µM of the fluorogenic caspase-9 substrate Ac-LEHD-AFC (or Ac-LEHD-AMC) in assay buffer (20 mM HEPES-KOH pH 7.5, 100 mM KCl, 1.5 mM MgCl₂, 1 mM EDTA, 1 mM DTT, 0.1% CHAPS).
Reaction Setup: Incubate assembled apoptosome mimics (10-50 nM) with substrate in a 96-well plate at 37°C. Include controls with unassembled Apaf-1 and caspase-9 alone.
Kinetic Measurement: Monitor fluorescence emission (AFC: λex = 400 nm, λem = 505 nm; AMC: λex = 380 nm, λem = 460 nm) every 2-5 minutes for 60-120 minutes using a plate reader.
Data Analysis: Calculate initial velocities and determine catalytic efficiency (kcat/Km) from substrate titration experiments. Compare activities with native apoptosome complexes reconstituted with full-length Apaf-1 and cytochrome c.
Protocol 3.3.2: Oligomerization State Analysis by Analytical Ultracentrifugation
Sample Preparation: Dilute apoptosome mimics to appropriate concentrations (0.5-1.0 mg/mL) in assembly buffer without DTT.
Centrifugation Parameters: Conduct sedimentation velocity experiments at 40,000 rpm and 20°C using an An-50 Ti rotor. Monitor sedimentation using absorbance (280 nm) or interference optics.
Data Analysis: Fit sedimentation data using continuous size distribution models to determine sedimentation coefficients and molecular weights. Compare with values obtained for native apoptosome (approximately 11S).
Research Reagent Solutions for Apoptosome Studies
Table 3: Essential Research Reagents for Apoptosome Mimic Engineering
| Reagent Category | Specific Examples | Function/Application | Commercial Sources |
|---|---|---|---|
| Recombinant Proteins | Apaf-1 (full-length and ΔWD40), Caspase-9 (full-length and variants) | Structural and functional studies of apoptosome assembly | Novus Biologicals, Abcam, R&D Systems |
| Caspase Substrates | Ac-LEHD-AFC, Ac-LEHD-AMC, Ac-DEVD-AFC | Fluorometric measurement of caspase-9 and caspase-3 activity | Enzo Life Sciences, BioVision, Cayman Chemical |
| Chemical Activators | Cytochrome c (equine heart), dATP, Bid cleavage products | Triggering apoptosome assembly in reconstitution experiments | Sigma-Aldrich, Millipore |
| SMAC Mimetics | JP1201, LCL161, Birinapant | Antagonizing IAP-mediated caspase inhibition to enhance apoptosis [44] | MedChemExpress, Selleck Chemicals |
| BH3 Mimetics | ABT-263 (Navitoclax), ABT-199 (Venetoclax) | Inhibiting anti-apoptotic Bcl-2 proteins to promote MOMP [42] | Cayman Chemical, AdooQ BioScience |
| Antibodies | Anti-Apaf-1, Anti-Caspase-9, Anti-Cleaved Caspase-3, Anti-Cytochrome c | Detection and quantification of apoptosome components | Cell Signaling Technology, Santa Cruz Biotechnology |
Applications in Drug Discovery and Therapeutic Development
Engineered apoptosome mimics serve as valuable tools for screening compounds that modulate the intrinsic apoptosis pathway, with particular relevance for cancer therapeutics. These systems enable the identification of small molecules that either promote or inhibit apoptosome formation and caspase activation, offering potential strategies for overcoming apoptotic resistance in cancer cells [44] [42]. Smac mimetics such as JP1201 have demonstrated promising results in enhancing chemotherapy efficacy in robust mouse models of pancreatic cancer when combined with gemcitabine, reducing primary and metastatic tumor burden and prolonging survival in both xenograft and transgenic models [44]. The effect of JP1201 was phenocopied by XIAP siRNA in vitro and correlated with elevated levels of TNFα protein in vivo, confirming the role of IAP inhibition in restoring apoptotic sensitivity [44].
The development of BH3 mimetics represents another strategic application targeting the upstream regulation of apoptosome formation. These small molecule compounds function by saturating the binding capacity of pro-survival Bcl-2 family members (e.g., Bcl-2, Bcl-XL, MCL-1), thereby blocking their anti-apoptotic activity and promoting mitochondrial outer membrane permeabilization [42]. This leads to cytochrome c release and subsequent apoptosome assembly, effectively bypassing common resistance mechanisms in cancer cells. Engineered apoptosome mimics provide a controlled system for evaluating the efficacy of BH3 mimetics and their combinations with other therapeutic agents, enabling mechanistic studies without the complexity of intact cellular systems.
Diagram 2: Drug Screening Using Apoptosome Mimics
Technical Challenges and Future Directions
Despite significant advances in apoptosome mimic engineering, several technical challenges remain. A primary limitation concerns recapitulating the full regulatory complexity of the native apoptosome, including post-translational modifications such as phosphorylation at Thr125 of caspase-9, which inhibits its processing without necessarily preventing recruitment to Apaf-1 [8]. Future iterations of engineered apoptosomes may incorporate regulatory components such as kinases (ERK2, DYRK1A, CDK1-cyclinB1) and phosphatases that modulate caspase-9 activity through phosphorylation-dephosphorylation cycles, enabling more physiologically relevant models of apoptosome regulation [8].
The recent discovery of Apaf-1 as an evolutionarily conserved DNA sensor that competes with cytochrome c binding presents both challenges and opportunities for apoptosome mimic engineering [43]. This finding suggests that Apaf-1 may serve as a cell fate checkpoint, determining whether cells initiate inflammation or undergo apoptosis based on distinct ligand binding. Future apoptosome mimics could be engineered to explore this dual functionality, potentially leading to novel insights into the interconnectedness of apoptotic and inflammatory signaling pathways. Such systems would need to accommodate both cytochrome c and DNA as competing ligands, with the capacity to measure pathway selection outcomes in response to different molecular triggers.
Emerging technologies in protein engineering, including computational design and directed evolution, offer promising approaches for creating next-generation apoptosome mimics with enhanced stability, tunable activity, and additional reporter functionalities. The incorporation of non-natural amino acids with unique chemical properties could enable site-specific labeling and cross-linking studies to elucidate dynamic aspects of apoptosome assembly and caspase activation. As these engineered systems become increasingly sophisticated, they will continue to provide fundamental insights into the molecular mechanisms of apoptosis while serving as valuable platforms for therapeutic discovery and development.
The apoptosome is a central signaling platform in the intrinsic pathway of apoptosis, a programmed cell death process crucial for development, tissue homeostasis, and disease prevention in multicellular organisms [35] [1]. This large molecular complex, with a molecular weight exceeding 1.1 MDa, forms when cytosolic Apaf-1 (Apoptotic protease-activating factor 1) binds to cytochrome c released from mitochondria and oligomerizes into a heptameric ring structure [35] [45] [1]. The apoptosome recruits and activates the initiator protease caspase-9 (Casp9), which then cleaves and activates downstream effector caspases, such as caspase-3 and caspase-7, ultimately leading to cell dismantling [35] [1].
The precise mechanism of caspase-9 activation has been the subject of extensive research and debate. Two primary models have emerged: the proximity-induced dimerization model and the holoenzyme model [25] [32]. The former posits that apoptosome assembly increases the local concentration of caspase-9 monomers, facilitating their homodimerization into the active form [32]. The latter suggests that the apoptosome induces allosteric changes in monomeric caspase-9, leading to its activation [32]. Recent evidence, including NMR spectroscopy studies, indicates that the caspase-9 protease domain remains monomeric upon apoptosome recruitment but is primed for rapid, substrate-induced dimerization, integrating aspects of both models [35]. Small molecule regulators have been instrumental in dissecting these complex activation mechanisms, offering both research tools and potential therapeutic leads.
PETCM: Mechanism and Experimental Applications
PETCM (alpha-(trichloromethyl)-4-pyridineethanol) is a identified small molecule activator of the apoptosome. Its primary mechanism of action is to relieve the inhibitory effect of Prothymosin α (ProT), a nuclear oncoprotein, on apoptosome formation [46].
Mechanism of Action
Prothymosin α negatively regulates apoptosis by directly interacting with Apaf-1 and inhibiting the formation of the functional apoptosome complex [46]. PETCM directly binds to Apaf-1, thereby blocking its interaction with ProTα. This disruption releases Apaf-1 from inhibition, permitting cytochrome c/dATP-mediated oligomerization into the active apoptosome and subsequent caspase-9 activation [46]. The interaction between PETCM and Apaf-1 can be studied using binding assays with biotinylated small molecule analogs and recombinant proteins.
Table 1: Key Characteristics of PETCM
| Property | Description |
|---|---|
| Target Protein | Apaf-1 [46] |
| Primary Mechanism | Relieves Prothymosin α-mediated inhibition of apoptosome formation [46] |
| Functional Outcome | Activates caspase-3 in cell extracts; promotes cell death [46] |
| Binding Study Method | Streptavidin bead pull-down with biotinylated analogs (e.g., Biotin-BETT) and recombinant Apaf-1 [46] |
Experimental Protocols
In Vitro Binding Assay for Target Identification
This protocol identifies cellular targets of small molecules like PETCM.
- Compound Immobilization: Incubate the biotinylated small molecule (e.g., Biotin-BETT) with streptavidin-coated magnetic beads for 30 minutes at 4°C [46].
- Blocking: Block beads with 5 mg/mL BSA to prevent non-specific binding [46].
- Binding Incubation: Incubate the compound-coated beads with a protein source (e.g., HeLa S100 cell extract or purified recombinant Apaf-1) overnight at 4°C in buffer (e.g., 20 mM HEPES, 10 mM KCl, 1.5 mM MgCl₂, 1 mM EDTA, 150 mM NaCl, 0.1% NP-40) [46].
- Wash and Elute: Wash beads extensively with buffer to remove unbound proteins. Elute bound proteins by boiling in SDS-PAGE loading buffer [46].
- Analysis: Analyze eluted proteins by SDS-PAGE, followed by western blotting for specific proteins or mass spectrometry for identification [46].
Reconstitution of Caspase Activation Pathway
This assay tests the functional effect of PETCM on a fully reconstituted system.
- Component Mixing: Combine purified recombinant proteins including Apaf-1, cytochrome c, procaspase-9, procaspase-3, Hsp70, PHAPI, CAS, and ProT in an appropriate buffer (e.g., 20 mM HEPES pH 7.5, 10 mM KCl, 1.5 mM MgCl₂, 1 mM DTT) with dATP/ATP [46].
- Compound Addition: Add PETCM or DMSO vehicle control to the reaction mixture [46].
- Incubation: Incubate the reaction at 30°C for 1 hour to allow for apoptosome formation and caspase activation [46].
- Activity Measurement: Measure caspase-3 or caspase-9 activity using fluorogenic substrates (e.g., Ac-DEVD-AFC for caspase-3) by monitoring fluorescence release over time [46].
Diagram 1: PETCM relieves ProT-mediated inhibition of apoptosome formation.
Other Pharmacological Modulators of the Apoptosome
Beyond PETCM, other small molecules have been identified that modulate the apoptosome complex through diverse mechanisms. These compounds serve as valuable tools for probing apoptotic signaling and represent potential starting points for drug development.
Table 2: Other Small Molecule Modulators of Apoptosome Function
| Modulator | Target/Effect | Mechanistic Class | Key Experimental Finding |
|---|---|---|---|
| BETT [46] | Apaf-1 | Activator | Relieves Prothymosin α inhibition of apoptosome formation, similar to PETCM [46]. |
| Z-LEHD-fmk [35] | Caspase-9 | Irreversible Inhibitor | Acts as a substrate mimic, covalently binding the catalytic cysteine (C287) and trapping caspase-9 in an active conformation [35]. |
The Scientist's Toolkit: Essential Research Reagents
Successful experimental investigation of the apoptosome and its small molecule modulators relies on a suite of specialized reagents and methodologies.
Table 3: Key Research Reagent Solutions for Apoptosome Studies
| Reagent / Assay | Function in Research | Key Utility |
|---|---|---|
| Recombinant Apaf-1 | Purified full-length protein, often from baculovirus/insect cell systems [46]. | Essential for in vitro reconstitution of apoptosome and binding studies [46]. |
| Fluorogenic Caspase Substrates (e.g., Ac-LEHD-AFC for caspase-9) [35] [45] | Peptides linked to a fluorophore (e.g., AFC); cleavage releases fluorescence. | Quantitative measurement of caspase enzyme activity in real-time [35] [45]. |
| SEC-MALS (Size-Exclusion Chromatography with Multi-Angle Light Scattering) [35] [32] | Separates biomolecules by size while determining absolute molecular weight. | Directly assesses oligomeric state (monomer vs. dimer) of proteins like caspase-9 in solution [35] [32]. |
| Methyl-TROSY NMR | An NMR technique optimized for large complexes using deuterated, methyl-labeled proteins [35]. | Provides atomic-level insights into dynamics and structure of caspase-9 within the mega-dalton apoptosome complex [35]. |
| Site-Specific Crosslinking | Genetically introducing crosslinking sites (e.g., cysteine pairs) into proteins [32]. | Provides direct biochemical evidence for protein-protein interactions (e.g., caspase-9 homodimerization) within the apoptosome [32]. |
Diagram 2: Apoptosome-mediated caspase-9 activation and activity detection.
Small molecule regulators like PETCM are powerful tools for deconstructing the intricate activation mechanism of the apoptosome and caspase-9. By specifically targeting key regulatory nodes, such as the Apaf-1/Prothymosin α interaction, these compounds provide definitive evidence for biochemical pathways and have solidified our understanding of the proximity-induced dimerization model. The experimental frameworks established for their study—including in vitro reconstitution assays, binding studies, and advanced structural techniques—provide a roadmap for the discovery and characterization of next-generation modulators. As research continues, these small molecules may not only illuminate fundamental cell death biology but also evolve into novel therapeutics for diseases characterized by dysregulated apoptosis, such as cancer and neurodegenerative disorders.
High-Throughput Screening Platforms for Drug Discovery
High-Throughput Screening (HTS) is an automated, rapid assessment method central to modern drug discovery and biological research. It is designed to quickly evaluate the biological or biochemical activity of hundreds of thousands of chemically or genetically diverse compounds against specific therapeutic targets [47] [48]. The primary objective of HTS is to identify initial "hit" compounds that show potential therapeutic effects, which can then be validated and optimized into lead candidates for further development [47]. A key advantage of HTS over rational drug design is its ability to rapidly deliver diverse drug leads, especially when little is known about the pharmacological target's structure [48]. This makes it particularly valuable for exploring novel biological mechanisms, such as those involving the apoptosome complex and caspase-9 activation.
The core principle involves using automated, miniaturized assays—typically run in 96-, 384-, or 1536-well microplates—alongside robust data analysis to process large compound libraries efficiently [48]. While HTS focuses on speed and throughput, often using single-parameter readouts, it is distinct from High-Content Screening (HCS), which provides a more detailed, multi-parameter analysis of cellular responses using automated fluorescence microscopy and image analysis [47]. In practice, HTS is often employed for primary screening of vast compound libraries, while HCS is utilized in secondary and tertiary screening phases to gain deeper insight into a compound's mechanism of action and cellular effects [47].
HTS Technologies and Methodologies
Core Components of an HTS Platform
A functional HTS platform integrates several automated and standardized components to ensure reliability, reproducibility, and speed.
- Sample and Library Preparation: HTS relies on combinatorial chemical libraries prepared in a standardized, automation-friendly manner, typically stored and processed on microplates [48]. The quality and diversity of these libraries are critical for success. Modern compound management involves highly automated procedures for storage, retrieval, nanoliter liquid dispensing, solubilization, and quality control [48].
- Automation and Robotics: Automated liquid-handling robots are indispensable. They enable low-volume dispensing of nanoliter aliquots, minimizing assay setup times and providing the accuracy and reproducibility required for miniaturized assays [48]. This automation is key to achieving the high throughput that defines HTS.
- Detection Technologies: HTS assays are broadly categorized as biochemical or cell-based [48].
- Biochemical Assays often target purified enzymes or proteins. Fluorescence-based methods are common due to their sensitivity and adaptability [48]. Mass spectrometry (MS)-based methods for unlabeled biomolecules are also gaining traction [48].
- Cell-Based Assays utilize whole cells to provide a more physiologically relevant context. These can range from simple viability readouts to more complex reporter assays.
- Data Management and Analysis: The massive datasets generated by HTS pose a significant challenge. A major issue is the prevalence of false positives, which can arise from assay interference, chemical reactivity, autofluorescence, or colloidal aggregation [48]. Triage strategies, including expert rule-based filters and machine learning models trained on historical HTS data, are used to rank compounds by their probability of success [48].
Advanced HTS: Ultra-High-Throughput Screening (uHTS)
Ultra-High-Throughput Screening (uHTS) pushes the boundaries of scale, allowing for the testing of millions of compounds per day [48]. This requires even greater miniaturization, often using 1536-well plates or higher densities with volumes of 1–2 µL, and advanced microfluidics [48]. A key limitation for uHTS has been the ability to directly monitor each microwell and perform multiplexed measurements of multiple analytes in parallel, though the development of miniaturized, multiplexed sensor systems is helping to address this challenge [48].
Table 1: Comparison of HTS and uHTS Core Attributes
| Attribute | HTS | uHTS |
|---|---|---|
| Throughput | Up to ~100,000 compounds per day [48] | >300,000 compounds per day [48] |
| Well Format | 96-, 384-, 1536-well [48] | 1536-well and higher [48] |
| Liquid Handling | Automated robotic dispensers [48] | Advanced microfluidics [48] |
| Multiplexing Capability | Limited | Requires miniaturized, multiplexed sensor systems [48] |
| Technical Complexity & Cost | High | Significantly greater [48] |
HTS in Apoptosome and Caspase-9 Activation Research
Biological Context: The Apoptosome and Caspase-9
The intrinsic apoptosis pathway is a tightly regulated process crucial for eliminating damaged or unwanted cells. A key event in this pathway is the formation of the apoptosome, a large multi-protein complex that acts as a caspase activation platform [8] [1]. Cellular stressors, such as DNA damage, trigger mitochondrial outer membrane permeabilization, leading to the release of cytochrome c into the cytoplasm. Cytochrome c binds to and activates Apaf-1, which then oligomerizes into a heptameric apoptosome complex [1].
This complex recruits the initiator caspase-9 through homotypic interactions between the CARD domains of Apaf-1 and caspase-9 [8] [1]. Two non-mutually exclusive models explain caspase-9 activation on the apoptosome:
- Proximity-Induced Dimerization: The apoptosome serves as a platform to concentrate procaspase-9 monomers, facilitating their homodimerization and subsequent activation [32].
- Induced Conformation: Binding to the apoptosome induces allosteric conformational changes in caspase-9 that activate it [8].
Notably, caspase-9 must remain bound to the apoptosome to exhibit significant catalytic activity, and its autoprocessing at Asp-315 initiates a "molecular timer" that regulates the duration of apoptosome activity [32]. Failure to properly activate caspase-9 is linked to cancer and developmental disorders, making this pathway a compelling therapeutic target [8].
Application of HTS to the Pathway
HTS provides a powerful tool for interrogating this complex biological system. It can be used to:
- Identify small molecules that modulate apoptosome formation.
- Discover direct activators or inhibitors of caspase-9 activity.
- Uncover regulators of upstream pathway components (e.g., Bcl-2 family proteins).
- Perform functional genomic screens to identify novel genes critical for intrinsic apoptosis.
Experimental Protocol: An HTS Campaign for Caspase-9 Activators
A typical HTS campaign targeting caspase-9 activation involves a multi-stage process:
Assay Development and Validation:
- Format Selection: A biochemical assay using purified components (Apaf-1, caspase-9, cytochrome c) or a cell-based assay with a caspase-9 activity reporter (e.g., a cleavable fluorescent substrate) is developed.
- Miniaturization: The assay is optimized and validated in a 384-well or 1536-well microplate format. Key parameters like Z'-factor (>0.5) are calculated to ensure assay robustness and reproducibility [48].
- Readout: A fluorescence-based readout is often used. For example, a peptide substrate conjugated to a fluorophore (e.g., LEHD-amc) is cleaved by active caspase-9, generating a fluorescent signal [32].
Primary Screening:
- A large combinatorial library of small molecules is screened against the target assay.
- Automated liquid handlers dispense nanoliter volumes of compounds and reagents into the microplates.
- Plates are incubated and read using a high-throughput plate reader.
Hit Triage and Data Analysis:
- Raw data is processed to identify "hits"—compounds that show activity beyond a set threshold (e.g., >3 standard deviations from the mean).
- Hit lists are triaged using cheminformatic filters to remove known pan-assay interferents and compounds with undesirable properties [48].
Confirmatory and Counter-Screening:
- Primary hits are re-tested in dose-response to confirm activity and calculate potency (IC50/EC50).
- Counter-screens are run to rule out non-specific activity (e.g., testing for fluorescence interference or general protease activation).
Secondary Screening:
- Confirmed hits are advanced to more physiologically relevant secondary assays. This is where High-Content Screening (HCS) becomes highly valuable. An HCS assay could be used to visualize caspase-9 recruitment to the apoptosome, mitochondrial membrane potential, and downstream caspase-3 activation in cells, providing a multi-parameter, mechanistic profile of the hit compound [47].
Successful HTS requires a suite of reliable reagents, tools, and data resources. The table below details key components for establishing an HTS campaign focused on caspase-9 and apoptosome research.
Table 2: Research Reagent Solutions for HTS on Caspase-9/Apoptosome
| Category / Item | Function / Application in HTS |
|---|---|
| Compound Libraries | Diverse collections of small molecules (e.g., 100,000+ compounds) for primary screening; the source of potential hits [48]. |
| Recombinant Proteins | Purified, active Apaf-1, caspase-9, and cytochrome c for setting up in vitro biochemical reconstitution assays [32] [1]. |
| Caspase Activity Assays | Fluorogenic or luminogenic peptide substrates (e.g., Ac-LEHD-amc); cleaved by active caspase-9 to generate a quantifiable signal [32]. |
| Cell Lines | Engineered cell lines (e.g., with caspase-9 reporters or specific genetic knockouts) for cell-based primary or secondary screening. |
| Public Data Repositories | PubChem BioAssay: Contains HTS data from various contributors, searchable by Assay ID (AID). ChEMBL, BindingDB: Provide additional bioactivity data [49]. |
| Automation & Data Analysis | PubChem PUG-REST: A web service API for programmatically retrieving large-scale HTS data for computational analysis [49]. |
Visualization of the Target Pathway
Understanding the biological pathway targeted by the HTS campaign is essential for rational assay design and data interpretation. The following diagram illustrates the intrinsic apoptosis pathway and the central role of the apoptosome in activating caspase-9.
High-Throughput Screening remains a cornerstone technology for initiating drug discovery campaigns against complex biological targets like the apoptosome-caspase-9 axis. Its power lies in its ability to empirically and rapidly test vast chemical space, generating crucial starting points for therapeutic development. The integration of robust assay design, advanced automation, and sophisticated data analysis is critical for a successful HTS campaign. Furthermore, the strategic use of public data repositories and the complementary application of High-Content Screening for hit validation can significantly de-risk the early discovery process. As technologies advance towards uHTS and integrate more deeply with AI and machine learning, the potential to unravel and target intricate mechanisms such as caspase-9 activation with greater speed and precision will only continue to grow.
Resolving Mechanistic Conflicts and Regulatory Challenges
The apoptosome complex serves as the central signaling hub in the intrinsic pathway of apoptosis, functioning as a molecular platform that activates caspase-9 following cellular stress signals. This activation represents a critical commitment point in programmed cell death, making its mechanistic understanding fundamental to both basic biology and therapeutic development [9] [14]. For decades, two primary models have framed the scientific discourse regarding how the apoptosome achieves caspase-9 activation: the induced proximity model and the induced conformation model [50] [25].
The induced proximity model posits that the apoptosome primarily serves as a scaffold to bring caspase-9 molecules into close proximity, facilitating their dimerization and subsequent autoactivation through increased local concentration [50]. In contrast, the induced conformation model argues that binding to the apoptosome induces specific structural changes in caspase-9 that directly create its active conformation, with dimerization being a secondary consequence or qualitatively different from artificially induced dimerization [50] [14].
This whitepaper synthesizes historical and emerging evidence to reconcile these seemingly divergent models, presenting a unified mechanistic understanding of caspase-9 activation for researchers and drug development professionals. We integrate quantitative biochemical data, structural biology insights, and recent NMR spectroscopy findings to provide a comprehensive technical guide to the apoptosome's function.
The Apoptosome: Structure and Assembly
The apoptosome is a large signaling platform that forms in response to mitochondrial outer membrane permeabilization and cytochrome c release into the cytosol. In humans, this ~1.3 MDa complex consists of seven apoptotic protease activating factor-1 (Apaf-1) molecules, each bound to cytochrome c and a nucleotide (dATP/ATP) [7] [2].
Structural Organization
Each Apaf-1 subunit contains three major domains:
- CARD (Caspase Activation and Recruitment Domain): An N-terminal domain that recruits procaspase-9 through homotypic CARD-CARD interactions [2] [14].
- NB-ARC/NOD (Nucleotide-Binding Apaf-1/R-proteins/CED-4 or Nucleotide-Oligomerization Domain): A central AAA+ ATPase family domain that mediates oligomerization [2].
- WD40 repeats: A C-terminal regulatory region that forms two β-propeller domains which bind cytochrome c, maintaining Apaf-1 in an autoinhibited state prior to activation [7] [2].
Upon cytochrome c binding and nucleotide exchange (ADP to dATP/ATP), Apaf-1 undergoes a dramatic conformational change from a closed, monomeric form to an extended conformation that oligomerizes into a wheel-like structure with 7-fold symmetry [2] [51]. The CARD domains of Apaf-1 form a flexibly tethered, disk-like spiral above the central platform, creating a binding surface for caspase-9 recruitment [7] [2].
Table 1: Core Components of the Human Apoptosome
| Component | Structure/Type | Function in Apoptosome Assembly |
|---|---|---|
| Apaf-1 | 140 kDa multi-domain protein | Scaffold protein that oligomerizes to form the apoptosome platform |
| Cytochrome c | Heme protein (12.5 kDa) | Triggers conformational change in Apaf-1, relieving autoinhibition |
| dATP/ATP | Nucleotide | Energy source and allosteric regulator; required for oligomerization |
| Procaspase-9 | 46 kDa zymogen (monomer) | Initiator caspase recruited to and activated by the apoptosome |
The Dueling Models: Historical Evidence and Experimental Foundations
The Induced Proximity Model
First proposed by Salvesen and Dixit, the induced proximity model suggests that initiator caspases like caspase-9 autoactivate simply by being brought into close proximity on activation platforms [25]. This model posits that caspase-9 exists as an inactive monomer in solution and that the apoptosome's primary function is to concentrate these monomers, facilitating dimerization through statistical chance [50].
Key evidence supporting this model includes:
- Caspase-9 exhibits significantly enhanced catalytic activity when artificially dimerized [50]
- The apoptosome creates a multivalent surface that can recruit multiple caspase-9 molecules simultaneously [2]
- Engineered dimeric forms of caspase-9 show increased activity compared to wild-type monomers [50]
The Induced Conformation Model
The induced conformation model argues that mere proximity is insufficient for full caspase-9 activation and proposes that specific interactions with the apoptosome induce conformational changes that directly create the active enzyme [50] [14]. This model emerged from observations that artificially dimerized caspase-9 failed to achieve the same level of activity as apoptosome-bound caspase-9 [50].
Supporting evidence includes:
- Engineered dimeric caspase-9 exhibits only a fraction of the activity of Apaf-1-activated wild-type caspase-9 [50]
- The activity of engineered dimeric caspase-9 is not stimulated by Apaf-1, unlike wild-type enzyme [50]
- Structural studies suggest specific interfaces between Apaf-1 and caspase-9 CARD domains are essential for activation [14]
Key Experimental Evidence and Methodologies
Seminal Caspase-9 Dimerization Studies
A pivotal 2005 study by Shi and colleagues provided critical evidence challenging the pure induced proximity model [50]. The researchers engineered a constitutively dimeric form of caspase-9 and compared its activity to wild-type caspase-9 activated by the apoptosome.
Table 2: Comparison of Caspase-9 Activation States and Activity
| Caspase-9 Form | Oligomeric State | Catalytic Activity | Stimulation by Apaf-1 | Cell Death Induction |
|---|---|---|---|---|
| Wild-type monomer | Monomeric | Basal level | Yes | Moderate |
| Engineered dimer | Constitutive dimer | Enhanced over monomer, but significantly lower than apoptosome-bound | No | Increased over monomer |
| Apoptosome-bound | Scaffold-organized | Maximum activity | N/A | High |
Experimental Protocol:
- Protein Engineering: Created constitutively dimeric caspase-9 using genetic fusion strategies
- In Vitro Activity Assays: Measured enzymatic activity of dimeric vs. wild-type caspase-9 using fluorogenic substrate cleavage
- Apoptosome Reconstitution: Activated wild-type caspase-9 with purified Apaf-1, cytochrome c, and dATP
- Cellular Assays: Expressed dimeric caspase-9 in cells and measured apoptosis induction
- Structural Validation: Confirmed engineered dimer structure resembled wild-type using crystallography
The critical finding was that while dimeric caspase-9 showed enhanced activity over the monomer, it never reached the activity level of apoptosome-activated wild-type enzyme, suggesting the apoptosome provides more than just a concentration effect [50].
Recent NMR Spectroscopy Insights
A groundbreaking 2023 study used methyl-TROSY NMR spectroscopy to examine caspase-9 activation within the native ~1.3 MDa apoptosome complex [35]. This approach allowed researchers to observe the dynamic behavior of caspase-9 in near-physiological conditions.
Experimental Protocol:
- Isotope Labeling: Produced ¹³CH₃-labeled caspase-9 with deuterated background
- Apoptosome Reconstitution: Assembled native apoptosome complex with labeled caspase-9
- NMR Spectroscopy: Acquired methyl-TROSY spectra of caspase-9 within the complex
- Dimerization Assessment: Measured chemical shifts and line shapes to determine oligomeric state
- Substrate Titration: Added caspase-9 substrate to observe conformational changes
The NMR data revealed that caspase-9 protease domains remain predominantly monomeric when bound to the apoptosome in the absence of substrate, challenging both pure proximity and conformation models [35]. Only upon addition of peptide substrate did significant dimerization occur, suggesting a more sophisticated activation mechanism where the apoptosome "primes" caspase-9 for activation, with substrate binding driving the final dimerization step.
Diagram 1: Integrated caspase-9 activation pathway
Reconciliation: A Unified Model of Caspase-9 Activation
Current evidence supports a hybrid model where the apoptosome provides both a concentrating function and specific allosteric effects that together optimize caspase-9 activation. The unified mechanism can be described as follows:
Sequential Activation Process
Recruitment and Priming: The apoptosome recruits caspase-9 monomers via CARD-CARD interactions, creating a primed state where the protease domains are optimally positioned but not yet fully active [35].
Conformational Optimization: Specific interactions between caspase-9 and the apoptosome platform induce subtle conformational changes that optimize the active site, lowering the energy barrier for activation [50] [14].
Substrate-Driven Dimerization: Upon substrate binding, the primed caspase-9 molecules rapidly dimerize, achieving full catalytic activity through a combination of proximity and optimized conformation [35].
This unified model explains why artificial dimerization produces partially active enzyme (lacking apoptosome-specific optimization), while also accounting for the substrate dependence of dimerization observed in recent structural studies.
Diagram 2: Model reconciliation and supporting evidence
Research Reagent Solutions and Technical Tools
Table 3: Essential Research Reagents for Apoptosome and Caspase-9 Studies
| Reagent/Tool | Type/Composition | Research Application | Key Features |
|---|---|---|---|
| Recombinant Apaf-1 | Full-length human protein | Apoptosome reconstitution | WD40 domain for cytochrome c binding, CARD domain for caspase-9 recruitment |
| Z-LEHD-fmk | Tetrapeptide fluoromethyl ketone | Caspase-9 inhibition | Irreversible active-site binder; traps active conformation for structural studies |
| Methyl-TROSY NMR | ¹³CH₃-labeled, deuterated proteins | Studying large complexes in solution | Enables observation of caspase-9 within intact 1.3 MDa apoptosome |
| Engineered Caspase-9 Dimer | Constitutively dimeric mutant | Proximity model testing | Bypasses apoptosome requirement; tests sufficiency of dimerization |
| Cryo-EM Apoptosome | Frozen hydrated complexes | Structural characterization | Reveals spatial organization of Apaf-1, cytochrome c, and caspase-9 |
| Caspase-9 Activity Assays | Fluorogenic substrates (LEHD-afc) | Enzymatic activity measurement | Quantifies activation under different conditions; inhibitor screening |
The historical dichotomy between induced proximity and induced conformation models has progressively dissolved as more sophisticated structural and biophysical techniques have revealed the nuanced mechanism of caspase-9 activation. The emerging unified model presents the apoptosome as a sophisticated activation platform that uses both geometric organization and specific allosteric effects to prime caspase-9 for substrate-driven activation.
This reconciliation has important implications for drug discovery, particularly in cancer therapeutics where resistance to apoptosis is a common feature. Rather than targeting either dimerization interfaces or Apaf-1 binding alone, effective therapeutic strategies might need to address the entire activation cycle or exploit the substrate-dependence of the final activation step. Future research should focus on dynamic visualization of the entire activation process and identification of small molecules that can modulate specific steps in this carefully orchestrated process.
The substrate-driven dimerization paradigm describes a biological mechanism where the binding of a specific molecular entity induces or stabilizes the dimerization of a protein or protein complex, thereby regulating its function. This review examines recent structural and biochemical evidence that solidifies this concept as a fundamental regulatory mechanism across diverse biological systems, with a particular focus on its implications for apoptosome complex and caspase-9 activation mechanisms. Growing evidence indicates that diverse molecular actors—including peptides, small molecule degraders, lipids, and endogenous substrates—can function as molecular glues that drive functional dimerization with significant consequences for cellular signaling, metabolic pathways, and therapeutic development.
Mechanistic Insights from Recent Structural Studies
Molecular Glue-Driven Dimerization in Targeted Protein Degradation
Recent cryo-EM structures have revealed unprecedented mechanisms of substrate-driven dimerization. The molecular glue degrader MRT-31619 induces CRBN homo-dimerization by mimicking a natural substrate degron. Structural analysis shows that two MRT-31619 molecules assemble into a helix-like structure that bridges two CRBN molecules, with the spirocyclic linker creating a conformationally restricted interface that stabilizes the dimer [52].
Key features of this mechanism include:
- Degron Mimicry: The molecular glue copies the structural features of a native G-loop degron, tricking the ubiquitination system.
- Complementary Architecture: The two compound copies adopt virtually identical conformations (all-atom RMSD of 0.064 Å) with a fixed dihedral angle of 81° between ring systems.
- Dimer Stability: Hydrophobic contacts between spirocyclic linkers and non-classical C–H···O hydrogen bonds create a stable dimeric complex that buries 1528 Ų of solvent-accessible surface area [52].
This mechanism represents a significant advancement in targeted protein degradation, demonstrating how small molecules can co-opt natural dimerization mechanisms for therapeutic purposes.
Lipid-Mediated Dimerization in Membrane Transport Systems
Structural studies of the taurine transporter (TauT) have revealed that cholesterol molecules act as "molecular glue" to facilitate functional dimerization. In the TauT homodimer, two cholesterol molecules wedge between TM5 from one protomer and TM7 and EL3 from the other, creating a stable dimer interface [53].
The functional significance was demonstrated through mutagenesis studies:
- Hydrophobic Interactions: Residues Val262 and Leu265 in TM5 mediate critical intermolecular contacts.
- Functional Consequences: Mutations Val262Arg, Val262Glu, Leu265Arg, and Leu265Glu completely abolished TauT activity, while alanine mutations retained partial function [53].
- Cholesterol Dependence: Cholesterol removal during purification eliminated dimer formation, confirming its essential role in the dimerization process [53].
This represents a paradigm where membrane lipid composition directly regulates transporter oligomerization and function through substrate-driven mechanisms.
Apoptosome-Mediated Caspase-9 Dimerization
The apoptosome complex activates caspase-9 through a sophisticated dimerization mechanism that integrates both homo- and heterodimerization events. Research has demonstrated that the apoptosome induces formation of both caspase-9 homodimers and Apaf-1:caspase-9 heterodimers, each with distinct functional properties [32].
Table 1: Caspase-9 Dimer Types and Their Functional Roles in the Apoptosome
| Dimer Type | Formation Trigger | Primary Function | Cleavage Specificity |
|---|---|---|---|
| Caspase-9 Homodimer | Proximity-induced on apoptosome platform | Stable recruitment & autocatalysis | Intramolecular cleavage at Asp-315 |
| Apaf-1:Caspase-9 Heterodimer | Binding to Apaf-1 NOD domain | Downstream substrate activation | procaspase-3 cleavage |
| Processed Caspase-9 Homodimer | Kosmotropic salts (in vitro) | Reduced activity due to linker interference | Diminished procaspase-3 cleavage |
The dimerization mechanism follows a precise sequence:
- Initial Recruitment: Apaf-1 oligomerization creates the apoptosome platform.
- Homodimer Formation: Procaspase-9 homodimerizes on the platform, increasing avidity through multivalent interactions.
- Autocatalytic Processing: Homodimers undergo intramolecular cleavage at Asp-315.
- Heterodimer Formation: Processed caspase-9 forms heterodimers with Apaf-1 NOD domains.
- Substrate Activation: Heterodimers efficiently activate downstream caspase-3 [54] [32].
This intricate mechanism represents a molecular timer that regulates the duration of apoptosome activity through controlled dimerization events.
Diagram 1: Caspase-9 activation through sequential homo- and heterodimerization in the apoptosome. The mechanism functions as a molecular timer that regulates cell death signaling.
Quantitative Analysis of Dimerization Effects
Recent studies have provided quantitative insights into how dimerization impacts protein function, stability, and catalytic efficiency across different biological systems.
Table 2: Quantitative Effects of Dimerization on Protein Function Across Biological Systems
| Protein System | Dimerization Effect | Functional Impact | Experimental Evidence |
|---|---|---|---|
| CYP121A1 | ~75% decrease in product formation in monomeric mutant | Compromised substrate specificity for cYY | Chromatographic analysis of product peaks [55] |
| Caspase-9 | Higher avidity of procaspase-9 for apoptosome | Enhanced molecular timer function | SEC-MALS, surface plasmon resonance [32] |
| SULT1A1 | Increased catalytic efficiency and stability | Potential half-site reactivity mechanism | Molecular dynamics simulations [56] |
| TauT | Complete loss of transport in interface mutants | Essential for cholesterol-mediated function | Uptake assays with Val262/Leu265 mutants [53] |
| CRBN | Potent degradation (DC50 < 1µM) without hook effect | Selective CRBN degradation | Proteomic profiling, ternary complex formation [52] |
The data reveal a consistent pattern where dimerization enhances functional efficiency across diverse protein families, though the magnitude of effect varies significantly based on the biological context and specific mechanism involved.
Experimental Approaches for Studying Dimerization
Structural Biology Techniques
Cryo-electron microscopy has emerged as a powerful tool for visualizing dimer interfaces in complex biological systems. For TauT structural determination, researchers employed detergent purification (LMNG) and nanodisc reconstitution with POPC/POPE/POPG lipids or brain total lipids, enabling visualization of both monomeric and dimeric states in near-native conditions [53].
Key methodological considerations:
- Sample Preparation: Nanodisc reconstitution was crucial for identifying the cholesterol-mediated TauT dimer, which was not observed in detergent alone.
- Resolution: Structures were determined at 3.20-3.26 Å resolution, sufficient for identifying side-chain interactions at dimer interfaces.
- Conformational Flexibility: Multiple cryo-EM maps (2.9 Å and 3.1 Å) were obtained for CRBN dimerization, capturing substantial conformational flexibility in the complex [52] [53].
Biophysical and Biochemical Assays
Site-specific crosslinking with unnatural amino acids provides direct evidence of dimer interfaces within native complexes. The incorporation of 3,4-dihydroxy-L-phenylalanine (L-DOPA) at position F406 in caspase-9 enabled periodate-mediated crosslinking, confirming both caspase-9 homodimerization and Apaf-1:caspase-9 heterodimer formation within the apoptosome [32].
Complementary approaches include:
- SEC-MALS: Quantified molecular weights of complexes in solution, demonstrating concentration-dependent ProC9 homodimerization.
- Spectral Binding Assays: Revealed fourfold weakened interaction between monomeric CYP121A1 and its cYY substrate.
- NanoBRET Assays: Confirmed CRBN homo-dimerization driven by MRT-31619 and its abrogation by W386A mutation [52] [55] [32].
Functional Assays
Reconstituted enzyme assays using purified components remain essential for establishing causal relationships between dimerization and function. For CYP121A1, incubation of WT and dimer-disrupted (I166A_I180A) enzymes with cYY substrate followed by reverse-phase liquid chromatography revealed a 75% decrease in product formation in the monomeric mutant [55].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Studying Substrate-Driven Dimerization
| Reagent / Method | Primary Function | Example Application |
|---|---|---|
| Site-specific crosslinking (L-DOPA) | Covalent capture of transient dimers | Caspase-9 homo- and heterodimer identification [32] |
| SEC-MALS | Solution molecular weight determination | ProC9 homodimer quantification [32] |
| NanoBRET | Monitoring protein-protein interactions in cells | CRBN-CRBN engagement [52] |
| Glutaraldehyde crosslinking + MS | Identification of dimer interfaces | CYP121A1 interface mapping [55] |
| Nanodisc reconstitution | Membrane protein stabilization in native lipids | TauT dimer visualization [53] |
| Kosmotropic salts (e.g., ammonium citrate) | Enforcing homodimerization in solution | Caspase-9 activation studies [32] |
| HaloTag/ NanoLuc constructs | Monitoring ternary complex formation | CRBN dimerization assays [52] |
Implications for Therapeutic Development
The substrate-driven dimerization paradigm offers novel approaches for therapeutic intervention, particularly in targeted protein degradation and apoptosis modulation. Molecular glues like MRT-31619 demonstrate how small molecules can induce homo-dimerization of E3 ligases, leading to selective degradation of previously "undruggable" targets [52].
Key therapeutic implications:
- Cancer Therapy: Regulating caspase-9 dimerization in the apoptosome could fine-tune apoptosis in cancer cells.
- Anti-infectives: Targeting essential dimeric enzymes like CYP121A1 in M. tuberculosis.
- Neurodegenerative Disorders: Modulating TauT dimerization could influence taurine homeostasis in neurological conditions.
The emerging understanding that many biological receptors dimerize upon ligand interaction has inspired the development of dimeric drug compounds designed to simultaneously engage two binding sites, creating thermodynamically stronger interactions than monomeric drugs [57].
Diagram 2: Therapeutic advantage of dimeric drugs. Dimeric compounds can simultaneously engage two binding sites on a receptor dimer, creating stronger interactions and enhanced biological responses compared to monomeric drugs.
The substrate-driven dimerization paradigm represents a fundamental mechanism in cellular regulation, with recent evidence illuminating diverse implementations across biological systems. From apoptosome-mediated caspase activation to molecular glue-induced degradation, this paradigm continues to reveal sophisticated regulatory strategies employed in nature. The convergence of structural biology, biophysical techniques, and functional studies has been instrumental in deciphering these mechanisms, providing new avenues for therapeutic intervention and a deeper understanding of cellular signaling networks. As research progresses, the deliberate targeting of dimerization interfaces promises to yield novel therapeutic strategies with enhanced specificity and efficacy.
Caspase-9 is an initiator caspase that plays an indispensable role in the intrinsic apoptosis pathway. It is activated within a multi-protein signaling platform known as the apoptosome, which forms in response to cellular stress signals such as DNA damage or growth factor withdrawal [1] [9]. The apoptosome complex consists of apoptotic protease-activating factor 1 (Apaf-1), cytochrome c, and procaspase-9 [1]. Once activated, caspase-9 proteolytically activates downstream effector caspases (caspase-3 and -7), initiating a cascade that leads to controlled cell death [9] [14]. The activity of caspase-9 is not left unchecked; it is precisely regulated by endogenous cellular mechanisms to ensure apoptotic commitment occurs appropriately. The two most critical regulatory mechanisms are post-translational modification, notably phosphorylation, and protein-protein interaction with inhibitors like the X-linked Inhibitor of Apoptosis Protein (XIAP) [58] [8]. Understanding these regulatory networks is crucial for elucidating cell fate decisions in both physiological and pathological contexts, including cancer and neurodegenerative diseases.
Phosphorylation: A Reversible Mechanism for Controlling Caspase-9 Activity
Phosphorylation serves as a rapid and reversible mechanism to fine-tune caspase-9 activity. Several kinases have been identified that phosphorylate specific residues on caspase-9, primarily leading to its inhibition.
Key Phosphorylation Sites and Regulatory Kinases
The most well-characterized inhibitory phosphorylation site on caspase-9 is Thr125, which is located in the hinge region near the N-terminus of the large subunit [8]. Phosphorylation at this site inhibits the proteolytic processing of procaspase-9. Multiple kinases target this residue, including ERK1/2, DYRK1A, CDK1-cyclinB1, and p38α [8]. The phosphorylated caspase-9 is thought to act as a dominant-negative regulator by potentially competing with non-phosphorylated procaspase-9 for recruitment to the apoptosome platform, although the exact mechanistic details remain an area of investigation [8].
Table 1: Endogenous Regulators of Caspase-9 Activity
| Regulator | Type | Target/Mechanism | Effect on Caspase-9 |
|---|---|---|---|
| ERK2 | Kinase | Phosphorylation at Thr125 | Inhibition |
| CDK1-Cyclin B1 | Kinase Complex | Phosphorylation at Thr125 | Inhibition |
| DYRK1A | Kinase | Phosphorylation at Thr125 | Inhibition |
| p38α | Kinase | Phosphorylation at Thr125 | Inhibition |
| XIAP | Inhibitory Protein | BIR3 domain binding to D315 neoepitope | Inhibition |
| cIAP1 | Inhibitory Protein | BIR domain binding & ubiquitin-mediated degradation | Inhibition |
| Caspase-9b | Splice Variant | Competes with full-length caspase-9 for apoptosome binding | Inhibition |
| HAX-1 | Protein | Binds and inhibits apoptosome-mediated activation | Inhibition |
| Survivin/BIRC5 | Protein Complex | Complexes with LAMTOR5 to inhibit activation | Inhibition |
Experimental Protocols for Studying Phosphorylation
To investigate the role of phosphorylation in caspase-9 regulation, researchers often employ a combination of molecular biology and biochemistry techniques.
Site-Directed Mutagenesis and Functional Apoptosis Assays: A common approach involves generating phosphorylation-deficient and phosphorylation-mimetic mutants of caspase-9. For Thr125, this typically entails mutating the codon to alanine (T125A) to prevent phosphorylation, or to aspartic acid (T125D) to mimic constitutive phosphorylation [8]. These mutant constructs are then transfected into cell lines (e.g., caspase-9 null embryonic fibroblasts or HeLa cells). The sensitivity of these cells to intrinsic apoptosis inducers (e.g., UV irradiation, etoposide, or staurosporine) is assessed and compared to cells expressing wild-type caspase-9. Assays include:
- Cell Viability Assays: MTT, XTT, or CellTiter-Glo assays to measure overall cell survival.
- Western Blotting: To monitor caspase-9 processing (cleavage into p35/p12 fragments), activation of downstream caspases (e.g., cleavage of caspase-3 and PARP), and the phosphorylation status of caspase-9 using phospho-specific antibodies.
- In Vitro Kinase Assays: Recombinant caspase-9 is incubated with purified active kinases (e.g., ERK2) in the presence of ATP. Phosphorylation is detected via Western blot or by measuring a reduction in caspase-9 enzymatic activity using fluorogenic substrates like LEHD-amc [8].
XIAP Inhibition: Direct Targeting of Active Caspase-9
XIAP is a potent and direct endogenous inhibitor of caspase-9. It belongs to the inhibitor of apoptosis (IAP) family of proteins, which are characterized by the presence of baculoviral IAP repeat (BIR) domains [59].
Molecular Mechanism of XIAP-Mediated Inhibition
The primary mechanism of XIAP inhibition involves its BIR3 domain binding specifically to the neoepitope of caspase-9 that is exposed after autocatalytic cleavage at Asp315 [58] [60]. This cleavage event generates the caspase-9 (p35/p12) form and creates a short peptide sequence that is essential for high-affinity interaction with the BIR3 domain of XIAP [58] [59]. This interaction physically blocks the active site of caspase-9, thereby inhibiting its activity and its ability to activate downstream effector caspases like caspase-3 [59]. It is important to note that XIAP has a significantly lower affinity for the uncleaved, apoptosome-bound procaspase-9, which allows the initial activation to proceed before being reined in by XIAP [32].
Key Experimental Evidence and Methodologies
The critical role of XIAP's BIR3 domain in caspase-9 inhibition has been solidified by elegant point-mutation studies.
Point Mutation Analysis and Binding Assays: Key residues within XIAP's BIR3 domain, such as Tryptophan 310 (W310) and Glutamic Acid 314 (E314), are essential for forming a groove that binds the N-terminus of processed caspase-9 [59]. Mutagenesis of these residues (e.g., W310A or E314S) disrupts this interaction without affecting the overall structure of XIAP.
- Co-immunoprecipitation (Co-IP): 293T cells are co-transfected with plasmids encoding caspase-9 and either wild-type or mutant XIAP. The XIAP protein is immunoprecipitated using an antibody (e.g., against a FLAG tag), and the precipitates are probed for the presence of bound caspase-9 by Western blotting. Mutants like W310A and E314S show a marked reduction in caspase-9 binding [59].
- Functional Rescue Experiments: Cell lines are transfected with mutant XIAP constructs that are defective in binding caspase-3, caspase-9, or both, and then subjected to apoptotic stimuli (e.g., UV irradiation). The ability of each mutant to inhibit cell death is quantified. Mutants that have lost the ability to bind both caspase-3 and caspase-9 are completely ineffectual, demonstrating that caspase inhibition is central to XIAP's anti-apoptotic function [59].
Diagram 1: Regulatory Network of Caspase-9. This diagram illustrates the activation of caspase-9 on the apoptosome and its subsequent inhibition by XIAP (via BIR3 domain binding to the D315 neoepitope) and kinase-mediated phosphorylation at Thr125.
Integrated Regulation and Functional Cross-Talk
The phosphorylation and XIAP regulatory pathways are not isolated; they function in an integrated manner to provide robust control over caspase-9 activity. This cross-talk is embedded within the broader "molecular timer" model of the apoptosome.
The Synergy of the Molecular Timer and XIAP
The molecular timer is a model wherein the autocleavage of procaspase-9 at Asp315 not only creates a XIAP-binding site but also reduces the affinity of the activated caspase-9 (p35/p12) for the apoptosome, leading to its eventual dissociation and the cessation of apoptosome activity [60] [32]. Systems modeling has demonstrated that this molecular timer synergizes with XIAP to suppress apoptosis execution effectively. The timer ensures that caspase-9 activity is transient, while XIAP acts as a failsafe to inhibit any remaining active caspase-9 that dissociates from the complex. This synergy is particularly important in scenarios of "minority MOMP" (miMOMP), where only a subset of mitochondria undergo permeabilization, leading to sub-lethal caspase activation. In such cases, the combined action of the molecular timer and XIAP can prevent full-blown apoptosis, potentially allowing the cell to survive and initiate inflammatory or other pro-tumourigenic responses [60].
Experimental Workflow for Studying Integrated Regulation
Diagram 2: Experimental Workflow for Caspase-9 Regulation Studies. A generalized protocol for investigating the integrated regulation of caspase-9, incorporating genetic mutants, biochemical perturbations, and multi-faceted readouts.
The Scientist's Toolkit: Key Research Reagents and Models
Table 2: Essential Research Tools for Studying Caspase-9 Regulation
| Reagent / Model | Function/Description | Key Application |
|---|---|---|
| Non-cleavable Caspase-9 (D315A) | Caspase-9 mutant resistant to autocleavage. | Disables the molecular timer and high-affinity XIAP binding; used to study prolonged apoptosome activity [60] [32]. |
| Caspase-9 Phospho-Mutants (T125A, T125D) | Alanine (non-phosphorylatable) or aspartic acid (phospho-mimetic) mutants. | Used to dissect the functional consequences of phosphorylation at Thr125 [8]. |
| XIAP BIR3 Domain Point Mutants (W310A, E314S) | XIAP mutants with disrupted caspase-9 binding. | Critical for validating the specificity of XIAP-caspase-9 interaction in Co-IP and functional assays [59]. |
| Caspase-9 Dimerization Mutant (F404D) | Mutant within the GCFNF dimerization motif. | Used to probe the role of homodimerization in caspase-9 activation within the apoptosome [32]. |
| Recombinant Apaf-1/Apoptosome | Purified components for in vitro reconstitution. | Allows for controlled biochemical study of caspase-9 activation and regulation without confounding cellular factors [32]. |
| SMAC Mimetics (e.g., AT-406, Birinapant) | Small molecules that antagonize IAP proteins. | Used to probe the physiological role of XIAP and to sensitize cells to apoptosis by relieving caspase inhibition [58]. |
| Caspase-9b Expression Construct | Plasmid encoding a naturally occurring dominant-negative splice variant. | Used to study competitive inhibition of full-length caspase-9 at the apoptosome [58]. |
The precise regulation of caspase-9 through phosphorylation and XIAP inhibition represents a critical juncture in the control of intrinsic apoptosis. Phosphorylation provides a rapid, kinase-mediated switch to preemptively inhibit caspase-9 activation, while XIAP acts as a direct, high-affinity damper on its catalytic activity. These pathways are not redundant but are deeply integrated, cooperating with the intrinsic molecular timer of the apoptosome to fine-tune the apoptotic signal. Disruption of this delicate balance has profound implications, contributing to diseases ranging from cancer to neurodegeneration. Continued research into these regulatory mechanisms, leveraging the sophisticated tools and models available, is paramount for developing targeted therapeutic strategies that can modulate caspase-9 activity for clinical benefit.
Caspase-9 is a critical initiator caspase in the intrinsic (mitochondrial) apoptotic pathway, which is activated through its recruitment to a multi-protein scaffolding complex known as the apoptosome [8] [9]. The apoptosome is a 1.1-MDa to 1.3-MDa heptameric complex composed of apoptotic protease-activating factor 1 (Apaf-1), which forms upon cytochrome c release from mitochondria [35] [25]. This complex serves as a platform for the activation of caspase-9, which subsequently cleaves and activates downstream effector caspases, such as caspase-3 and caspase-7, leading to the execution phase of apoptosis [35] [9]. The precise mechanism of caspase-9 activation on the apoptosome has been a subject of extensive research, with models ranging from induced proximity-driven dimerization to direct conformational activation by the complex itself [35] [8].
A critical layer of regulation in this process occurs at the pre-mRNA level through alternative splicing, which generates distinct caspase-9 isoforms with opposing functions in cell death [61] [62]. The caspase-9 gene can be processed to include or exclude a cassette of exons 3, 4, 5, and 6, giving rise to two primary variants: the full-length, pro-apoptotic caspase-9a and a shorter, anti-apoptotic isoform known as caspase-9b [61] [62] [63]. The balance between these isoforms is a crucial determinant of cellular susceptibility to apoptotic stimuli, and its dysregulation is implicated in diseases such as cancer [62] [64]. This review provides an in-depth technical examination of caspase-9b, its mechanistic role as a dominant-negative regulator within the apoptosome system, the molecular mechanisms governing its splicing, and the experimental and therapeutic implications of this regulatory node.
Molecular Identity and Function of Caspase-9b
Structural Characteristics of Caspase-9b
Caspase-9b was first identified as an endogenous, alternatively spliced isoform of caspase-9 that functions as a key regulatory molecule in apoptosis [61]. Structurally, caspase-9b lacks a significant portion of the central large subunit that constitutes the canonical caspase domain present in full-length caspase-9a [61]. Despite this truncation, the caspase-9b isoform retains the N-terminal Caspase Recruitment Domain (CARD), which is essential for protein-protein interactions [61] [8]. This structural composition allows caspase-9b to engage with specific components of the apoptotic machinery while being catalytically inactive, positioning it as a potent endogenous inhibitor.
Mechanism of Apoptosis Inhibition
Caspase-9b exerts its anti-apoptotic function primarily by acting as a dominant-negative regulator that interferes with the core activation mechanism of caspase-9a at the apoptosome.
- Interaction with Apaf-1: Through its intact CARD domain, caspase-9b binds to the CARD domain of Apaf-1 on the apoptosome complex [61]. This interaction allows caspase-9b to compete with pro-caspase-9a for binding sites on the Apaf-1 scaffold [61] [64].
- Inhibition of Caspase-9 Activation: By occupying the recruitment sites on the apoptosome, caspase-9b physically prevents the binding and subsequent activation of pro-caspase-9a [61]. This competition disrupts the formation of a functional Apaf-1–caspase-9a activation complex.
- Suppression of Downstream Signaling: The sequestration of the apoptosome by caspase-9b effectively blocks the proteolytic activation of downstream effector caspases, such as caspase-3. In vitro assays have demonstrated that caspase-9b can inhibit cytochrome c-dependent caspase-9 and caspase-3 activation mediated by Apaf-1 [61].
The functional consequence of this mechanism is a significant increase in the cellular threshold for undergoing apoptosis. Ectopic expression of caspase-9b confers resistance to diverse apoptotic stimuli, including death receptor oligomerization and chemotherapeutic agents [61] [62].
Table 1: Key Characteristics of Caspase-9a and Caspase-9b
| Feature | Caspase-9a (Pro-apoptotic) | Caspase-9b (Anti-apoptotic) |
|---|---|---|
| Structure | Full-length, contains CARD, large, and small subunit domains | Lacks most of the large subunit domain; retains CARD domain |
| Expression | Constitutively expressed in many tissues | Detected in many cell lines at mRNA and protein levels [61] |
| Primary Function | Initiator caspase in the intrinsic apoptotic pathway | Dominant-negative inhibitor of apoptosis [61] |
| Apoptosome Binding | Binds Apaf-1 via CARD for activation [35] | Binds Apaf-1 via CARD, competing with caspase-9a [61] |
| Catalytic Activity | Active upon dimerization on apoptosome | Inactive |
| Net Effect on Cell Death | Promotes apoptosis | Inhibits apoptosis, induces chemoresistance [62] |
Regulation of Caspase-9 Alternative Splicing
The decision to include or exclude the exon 3-4-5-6 cassette in the caspase-9 pre-mRNA is a tightly regulated process controlled by cis-acting RNA elements and trans-acting splicing factors, which in turn can be modulated by specific cellular signaling pathways.
Key Splicing Factors: SRSF1 and hnRNPs
- SRSF1 (SRp30a) as a Splicing Enhancer: The serine/arginine-rich splicing factor SRSF1 is a critical enhancer for the inclusion of the four-exon cassette, favoring the production of the pro-apoptotic caspase-9a isoform [62] [63]. It mediates this effect by binding to a specific intronic splicing enhancer located in intron 6 (C9-I6/ISE) of the caspase-9 pre-mRNA. Downregulation of SRSF1 leads to a significant decrease in the caspase-9a/9b ratio, shifting the balance towards the anti-apoptotic isoform [62] [63].
- hnRNP L and hnRNP U as Competitive Regulators: Heterogeneous nuclear ribonucleoproteins (hnRNPs) also play a crucial role. hnRNP L acts as a splicing repressor that binds to an exonic element in exon 3, promoting the exclusion of the cassette and the generation of caspase-9b [64]. Conversely, hnRNP U functions as a splicing enhancer that competes with hnRNP L for binding to the same cis-element in exon 3. The outcome of this competition is determined by the phosphorylation status of hnRNP L, which is regulated by the AKT signaling pathway [64].
Upstream Signaling Pathways
The phospho-status of SR proteins, including SRSF1, is a primary mechanism by as which external signals control alternative splicing.
- Ceramide-Protein Phosphatase 1 (PP1) Pathway: The lipid second messenger ceramide, generated in response to cellular stress and chemotherapeutic agents, activates PP1 [62] [63]. PP1 dephosphorylates SRSF1 on specific serine residues (e.g., Ser199, 210, 227, 234), which is required for ceramide to promote the inclusion of the exon cassette and increase the caspase-9a/9b ratio [62].
- AKT Pathway: The pro-survival AKT kinase pathway phosphorylates hnRNP L. Phosphorylated hnRNP L maintains its binding to the repressive element in exon 3, thereby blocking the access of the enhancer hnRNP U and favoring the generation of caspase-9b [64]. This mechanism directly links oncogenic survival signaling to the suppression of pro-apoptotic splicing.
Figure 1: Signaling pathways regulating caspase-9 alternative splicing. Cellular stresses promote pro-apoptotic caspase-9a splicing via the ceramide-PP1 pathway, while survival signals promote anti-apoptotic caspase-9b splicing via the AKT pathway.
Experimental Analysis of Caspase-9 Splicing and Function
Key Methodologies and Reagents
Studying the regulation and function of caspase-9 isoforms requires a combination of molecular, biochemical, and cellular techniques.
Table 2: Research Reagent Solutions for Caspase-9 Splicing and Function Studies
| Reagent / Method | Function / Purpose | Key Experimental Details |
|---|---|---|
| SRSF1 SMARTpool siRNA | Silences SRSF1 expression to assess its role as a splicing enhancer [62]. | Transfect A549 (NSCLC) cells using Dharmafect 1; 100 nM concentration; analyze RNA/protein after 48h [62]. |
| Caspase-9b-specific siRNA | Targets caspase-9b transcript for degradation to study its functional roles [62]. | Sequence: GATTTGGTGATGTCGAGCATT. Used to demonstrate increased chemosensitivity in NSCLC cells [62]. |
| Anti-Sense RNA Oligos (ASRO) | Redirects splicing towards caspase-9b production [62]. | 2'-O-(2-methoxy) ethyl 23-mer MOE ASRO targeted to 5' splice site of exon 4 (e.g., 5'-GAGTGTACCTTGGCAGTCAGGTC-3') [62]. |
| In vitro Caspase Activation Assay | Measures cytochrome c/Apaf-1 dependent caspase-9 and -3 activity [61]. | Used to demonstrate caspase-9b inhibition of apoptosome function [61]. |
| Co-Immunoprecipitation (Co-IP) | Validates protein-protein interactions (e.g., caspase-9b with Apaf-1 or cIAP1) [64]. | Critical for confirming dominant-negative mechanism and identifying novel interactions. |
| RT-PCR / Western Blot | Detects and quantifies caspase-9a and 9b mRNA and protein ratios [62]. | Standard method for evaluating splicing outcomes and isoform expression. |
Illustrative Experimental Workflow
The following diagram outlines a generalized protocol for investigating the functional consequences of caspase-9 alternative splicing, integrating many of the reagents listed above.
Figure 2: Generalized workflow for functional studies of caspase-9 splicing. Experiments begin by modulating the splicing machinery, validating the resulting isoform shift, and then assessing the functional consequences on apoptosis and underlying molecular mechanisms.
Functional Consequences and Therapeutic Implications
Role in Cancer Pathogenesis and Chemoresistance
The dysregulation of caspase-9 alternative splicing is a significant mechanism in cancer biology, particularly in the development of resistance to chemotherapy.
- Chemoresistance in NSCLC: Manipulation of the caspase-9a/9b ratio directly impacts the sensitivity of non-small cell lung cancer (NSCLC) cells to chemotherapeutics. Forced skewing towards caspase-9b expression via antisense RNA oligonucleotides (ASROs) increases the IC₅₀ of NSCLC cells to daunorubicin, cisplatinum, and paclitaxel. Conversely, knockdown of caspase-9b sensitizes cells to these drugs [62].
- Synergistic Drug Effects: The alternative splicing of caspase-9 is a documented mechanism for the synergistic effect observed in combination therapies. For instance, the efficacy of combining daunorubicin with the EGFR inhibitor erlotinib involves modulation of the caspase-9 splice variant ratio [62].
- Beyond the Apoptosome: Interaction with NF-κB Pathway: Recent research indicates that caspase-9b has functions independent of its role in apoptosome inhibition. It can directly bind to cellular inhibitor of apoptosis protein 1 (cIAP1), leading to the activation of the canonical NF-κB survival pathway and further enhancing cell survival, anchorage-independent growth, and tumorigenicity in NSCLC models [64].
Caspase-9b in the Broader Context of Apoptosome Research
The study of caspase-9b provides critical insights into the regulation of the apoptosome. Recent structural biology studies, using techniques like cryo-EM and methyl-TROSY NMR, have revealed that the protease domains of apoptosome-bound caspase-9 are flexibly tethered and remain monomeric until substrate binding drives their dimerization and full activation [35]. The presence of caspase-9b, competing for CARD-binding sites, likely disrupts this carefully orchestrated sequence by reducing the local concentration of procaspase-9a monomers available for productive dimerization on the scaffold. This underscores the apoptosome not just as a static activator, but as a dynamic platform whose composition and output are finely tuned by regulatory isoforms like caspase-9b.
Caspase-9b is a pivotal endogenous regulator of the intrinsic apoptotic pathway, functioning through a dominant-negative mechanism that disrupts the activation of caspase-9a at the apoptosome. Its generation is controlled by a sophisticated network of splicing factors, including SRSF1, hnRNP L, and hnRNP U, which are in turn regulated by key cellular signaling pathways like ceramide-PP1 and AKT. The resulting balance between pro- and anti-apoptotic isoforms significantly influences cellular fate in response to stress and damage. The documented role of caspase-9b in promoting chemoresistance in cancers like NSCLC highlights its therapeutic relevance. Understanding the molecular mechanics of caspase-9 alternative splicing and the function of its isoforms provides a solid foundation for developing novel strategies to overcome apoptosis evasion in human diseases, particularly by targeting the splicing machinery or the specific interactions of caspase-9b itself.
Overcoming Apoptosome Defects in Chemotherapy Resistance
The apoptosome, a critical signaling platform that activates caspase-9 in the intrinsic apoptosis pathway, represents a pivotal control point in cellular life-or-death decisions. Defects in apoptosome formation and function constitute a major mechanism by which cancer cells develop resistance to chemotherapy. This whitepaper examines the molecular architecture of the apoptosome, quantitative parameters governing its assembly, and the mechanistic basis of its dysregulation in malignancies. Within the broader context of caspase-9 activation mechanism research, we detail emerging therapeutic strategies to bypass apoptosome defects, including BH3 mimetics, SMAC mimetics, and novel protein degradation technologies. The integration of quantitative mathematical modeling with experimental validation provides a powerful framework for developing targeted interventions to restore apoptotic signaling in treatment-resistant cancers.
The apoptosome is a multiprotein complex that functions as the cellular engine for caspase-9 activation, serving as the pivotal control point in the intrinsic apoptosis pathway [36]. This wheel-shaped heptameric assembly forms when apoptotic protease-activating factor 1 (Apaf-1) oligomerizes in the presence of cytochrome c and dATP/ATP [11] [36]. The fully assembled complex consists of seven Apaf-1 molecules arranged in a symmetric ring structure, with their caspase recruitment domains (CARDs) forming a central hub that recruits and activates procaspase-9 [11].
The molecular mechanism of caspase-9 activation has been extensively debated, with two primary hypotheses emerging: the dimerization hypothesis suggests that apoptosome binding facilitates caspase-9 activation through homodimerization, while the allosteric activation hypothesis proposes that binding induces a conformational change that activates caspase-9 [11]. Recent systems biology approaches utilizing mathematical simulations have challenged the prevailing dogma, demonstrating that only scenarios assuming allosteric caspase-9 activation can accurately reproduce experimental data on apoptosis execution kinetics [11]. This allosteric mechanism enables the apoptosome to function as a proteolytic molecular timer that provides transient caspase-9 activities, with processed caspase-9 being released from the complex and subsequently inactivated [11].
Within the broader landscape of caspase activation mechanisms, the apoptosome represents a specialized adaptor platform that specifically regulates the intrinsic apoptosis pathway initiated by mitochondrial outer membrane permeabilization (MOMP). This positions the apoptosome as a critical integration point for cellular stress signals and a key determinant of cell fate decisions in response to chemotherapeutic agents.
Molecular Architecture and Assembly Mechanisms
Structural Components and Assembly Kinetics
The apoptosome backbone is composed of Apaf-1, a member of the STAND (signal transduction ATPases with numerous domains) family, which contains three functional domains: an NH2-terminal CARD domain, a central nucleotide-binding and oligomerization domain, and a COOH-terminal WD40 repeat domain [11]. Apoptosome assembly initiates when cytochrome c, released during MOMP, binds to Apaf-1 along with dATP/ATP, inducing a conformational change that enables heptamerization [11].
Table 1: Quantitative Parameters of Apoptosome Assembly
| Parameter | Value | Experimental Context | Citation |
|---|---|---|---|
| Cytochrome c cytosolic accumulation t½ | 1.5 min | HeLa cells | [11] |
| Kd for procaspase-9 binding to Apaf-1 | 0.7 μm | In vitro determination from IC50 | [11] |
| IC50 for procaspase-9 binding | 0.8 μm | In vitro competition assay | [11] |
| Km of caspase-9 toward LEHD-afc substrate | 686 μm | Fluorigenic substrate assay | [11] |
The assembly process proceeds through precisely regulated molecular interactions. Cytochrome c and dATP/ATP bind to Apaf-1 in an interchangeable order [11], with activated Apaf-1 monomers capable of reverting to auto-inhibited states, adding a regulatory checkpoint to apoptosome formation. Mathematical modeling of these interactions has revealed that Apaf-1 activation is a rapid process that reaches completion quickly following cytochrome c release [11].
Caspase-9 Activation Mechanisms
The central question in apoptosome function concerns the mechanism of caspase-9 activation. Systems biology approaches comparing dimerization-based versus allosteric activation models have demonstrated superior performance of the allosteric model in replicating experimental observations [11]. Key evidence supporting allosteric activation includes:
- Caspase-9 bound to the apoptosome processes procaspase-3 significantly more efficiently than force-dimerized free caspase-9 [11] [19]
- Processed caspase-9 released from the apoptosome is inactive and monomeric in the cytosol [11]
- Mathematical simulations implementing homodimerization as a prerequisite for activation fail to replicate experimental kinetics of apoptosis execution [11]
The allosteric regulation model positions the apoptosome as both an activation platform and a regulatory constraint that maintains precise control over caspase-9 activity, functioning as a proteolytic molecular timer [11].
Apoptosome Defects in Chemotherapy Resistance
Molecular Mechanisms of Defective Apoptosome Function
Cancer cells employ multiple strategies to disrupt apoptosome function, thereby evading chemotherapy-induced cell death. These mechanisms operate at various levels of the apoptosome assembly and activation cascade:
- Insufficient cytochrome c release: Despite MOMP, inadequate release of cytochrome c or mutation of critical lysine residues (especially K72) can abrogate apoptosome formation [65]
- Apaf-1 dysregulation: Reduced expression or functional impairment of Apaf-1 prevents proper apoptosome assembly, observed in various chemoresistant cancers [66] [67]
- Caspase-9 mutations: Acquired mutations in caspase-9 can inhibit its function even when properly recruited to the apoptosome [65]
- IAP overexpression: Elevated levels of XIAP and other inhibitor of apoptosis proteins directly bind to and inhibit active caspase-9 [65] [21]
The integrity of apoptosome-mediated caspase activation is particularly crucial for chemotherapy response, as many chemotherapeutic agents act through the intrinsic pathway by inducing DNA damage or other cellular stresses that ultimately converge on mitochondrial cytochrome c release [67].
Quantitative Impact on Apoptosis Thresholds
Mathematical modeling of apoptosome function has revealed precise regulatory features that become dysregulated in chemotherapy resistance. Systems biology approaches have identified a XIAP threshold concentration at which apoptosis execution is suppressed in cancer cells [11]. When XIAP levels exceed this threshold, they effectively neutralize activated caspase-9, preventing downstream caspase-3/7 activation despite successful apoptosome assembly.
Additionally, the proteasome-dependent degradation of effector caspases has been identified as an important restraint mechanism during the pre-MOMP delay phase [21]. When this restraint is impaired, cells can enter a physiologically indeterminate state of partial cell death with potential to generate genomic instability [21]. This finding highlights how apoptosome dysregulation contributes not only to chemotherapy resistance but also to tumor evolution and progression.
Therapeutic Strategies to Overcome Apoptosome Defects
Direct Targeting of Apoptosis Regulators
Table 2: Therapeutic Approaches Targeting Apoptosome-Related Pathways
| Therapeutic Class | Representative Agents | Molecular Target | Mechanism of Action | Development Status |
|---|---|---|---|---|
| BH3 Mimetics | Venetoclax, Navitoclax | BCL-2, BCL-XL, BCL-w | Inhibit anti-apoptotic BCL-2 proteins to promote MOMP | FDA-approved (Venetoclax) [65] [68] |
| SMAC Mimetics | Birinapant, LCL161 | XIAP, cIAP1 | Antagonize IAP inhibition of caspases | Clinical trials [65] |
| PROTACs | DT2216, MCL-1 degraders | BCL-2 family, IAPs | Targeted protein degradation | Preclinical/early clinical [69] |
| TRAIL Agonists | TLY012, Eftozanermin alfa | DR4/5 | Activate extrinsic apoptosis pathway | Clinical development [65] |
BH3 mimetics represent a paradigm shift in targeting the intrinsic apoptosis pathway. Venetoclax, the first FDA-approved BCL-2-selective BH3 mimetic, has demonstrated remarkable efficacy in hematologic malignancies by binding to BCL-2 and displacing pro-apoptotic proteins like BIM, which in turn directly activates BAX and BAK to induce MOMP [65] [68]. This bypasses upstream defects in apoptosome assembly by ensuring sufficient cytochrome c release to activate remaining functional apoptosome complexes.
Emerging Technologies: PROTACs and Targeted Protein Degradation
Proteolysis Targeting Chimeras (PROTACs) represent a novel therapeutic modality with particular relevance for overcoming apoptosome defects. These heterobifunctional molecules consist of three elements: a target protein-binding ligand, an E3 ubiquitin ligase binding ligand, and a linker connecting both [69]. PROTACs targeting anti-apoptotic proteins (BCL-2, BCL-XL, MCL-1, IAPs) induce their ubiquitination and proteasomal degradation, effectively removing key barriers to apoptosis execution [69].
Specific and nongenetic IAP-dependent Protein Erasers (SNIPERs) constitute a related technology that simultaneously degrades cIAP1/2 or XIAP together with target proteins [69]. This dual degradation approach is particularly promising for overcoming the high XIAP thresholds that suppress caspase-9 activity in chemoresistant cancers [11].
Combination Strategies to Restore Apoptotic Sensitivity
Rational combination therapies represent the most promising approach to address the multifactorial nature of apoptosome-related chemoresistance:
- BH3 mimetics with TRAIL agonists: BH3 mimetics promote MOMP while TRAIL agonists activate complementary extrinsic pathways, creating synergistic apoptosis induction [65]
- IAP antagonists with conventional chemotherapy: SMAC mimetics lower the threshold for caspase-9 activation, sensitizing cells to chemotherapy-induced apoptosome formation [65]
- PROTACs with targeted inhibitors: Simultaneous degradation of anti-apoptotic proteins and inhibition of survival pathways addresses multiple resistance mechanisms [69]
These combinations leverage the growing understanding of cross-talk between apoptosis pathways and the quantitative nature of caspase regulation to design effective strategies against chemoresistant malignancies.
Experimental Approaches and Research Methodologies
Quantitative Analysis of Apoptosome Function
Advanced methodologies have enabled precise quantification of apoptosome dynamics in single cells:
Live-cell caspase activity reporters permit real-time tracking of initiator and effector caspase activities throughout apoptosis progression. When combined with flow cytometry and immunoblotting, these approaches have revealed that initiator caspases are active during the prolonged and variable delay that precedes MOMP and effector caspase activation [21].
Mathematical modeling of core apoptosis pathways has identified key regulatory features, including the importance of XIAP and proteasome-dependent degradation of effector caspases in restraining activity during the pre-MOMP delay [21]. These models typically employ ordinary differential equations solved using numerical integration methods (e.g., ODE15s solver implementing backward differentiation formula) [11].
Molecular Interaction Mapping
Molecular interaction maps provide comprehensive visual representations of signaling networks regulating apoptosome function. These conventions have been developed to summarize complex networks including AKT, NF-κB, p53, Chk2, and c-Abl pathways [66]. Such maps facilitate the identification of critical nodes for therapeutic intervention and help visualize cross-talk between apoptosis regulatory pathways.
Structural biology techniques including cryo-EM have revealed the wheel-shaped heptameric architecture of the apoptosome, providing a structural framework for understanding caspase-9 activation mechanisms [36]. These structural insights inform the rational design of compounds targeting specific protein-protein interactions within the apoptosome complex.
Research Reagent Solutions
Table 3: Essential Research Reagents for Apoptosome Studies
| Reagent/Category | Specific Examples | Research Application | Key Features |
|---|---|---|---|
| Caspase Activity Reporters | LEHD-afc, DEVD-afc | Fluorigenic substrate cleavage assays | Km for LEHD-afc = 686 μm; specific for caspase-9 [11] |
| Recombinant Proteins | Apaf-1, cytochrome c, procaspase-9 | In vitro apoptosome reconstitution | Enables biochemical analysis of assembly requirements |
| Cell Line Models | HeLa, various cancer lines with Apaf-1 defects | Study of apoptosome dysfunction | HeLa used for XIAP threshold determination [11] |
| BH3 Mimetics | ABT-737, ABT-263 (Navitoclax), ABT-199 (Venetoclax) | BCL-2 family inhibition research | Tool compounds for intrinsic pathway modulation [68] |
| Mathematical Modeling Environments | MATLAB with ODE15s solver | Systems biology simulations | Backward differentiation formula for numerical integration [11] |
Future Directions and Clinical Translation
The evolving understanding of apoptosome structure and function continues to inform novel therapeutic approaches. Promising research directions include:
Precision targeting of caspase-9 allosteric sites: Structural insights into the caspase-9 activation mechanism may enable development of compounds that mimic the apoptosome-induced conformational change, directly activating caspase-9 independent of apoptosome assembly [11] [19].
Dynamic BH3 profiling: This functional assay measures apoptotic priming in cancer cells and has been used to identify optimal BH3 mimetic combinations in resistant xenograft models [67]. Clinical implementation could guide personalized therapy selection based on individual tumor dependencies.
Expanded PROTAC applications: Advances in E3 ubiquitin ligase recruitment and linker optimization are creating opportunities to target previously "undruggable" components of the apoptosome regulatory network [69].
The successful clinical development of venetoclax provides a roadmap for translating fundamental insights into apoptosome biology into effective cancer therapies. Future efforts must focus on identifying predictive biomarkers for apoptosome-dependent therapies and developing rational combinations that address the complex redundancy in apoptosis regulation.
As caspase-9 activation mechanism research continues to evolve, the apoptosome remains a compelling therapeutic target for overcoming chemotherapy resistance. Integrating structural biology, systems-level modeling, and innovative therapeutic modalities holds promise for restoring apoptotic competence in treatment-refractory malignancies.
Pathophysiological Validation and Cross-Pathway Integration
The apoptosome, a multiprotein complex central to the intrinsic apoptosis pathway, functions as a critical platform for initiator caspase-9 activation. Its impairment represents a fundamental mechanism enabling tumorigenesis and cancer cell survival. This whitepaper synthesizes current structural and mechanistic insights into apoptosome-mediated caspase-9 activation and delineates how its dysfunction contributes to carcinogenesis. We provide detailed experimental methodologies for investigating apoptosome function and present emerging therapeutic strategies targeting this complex. Within the broader context of caspase-9 activation research, understanding apoptosome impairment reveals pivotal molecular disruptions in cell death signaling that offer promising targets for oncological therapeutic development.
The apoptosome is a quaternary protein structure that forms in the cytosol in response to mitochondrial outer membrane permeabilization and cytochrome c release [2] [70]. This wheel-shaped complex consists of apoptotic protease-activating factor 1 (Apaf-1), cytochrome c, and the initiator caspase, procaspase-9 [71]. The assembly of this complex marks the point of no return in the intrinsic apoptosis pathway [2].
In mammalian systems, the core apoptosome is a heptameric complex with sevenfold rotational symmetry, with a calculated mass of approximately 1 megadalton [2]. Each Apaf-1 subunit contains three major regions: (1) an N-terminal caspase recruitment domain (CARD) that recruits procaspase-9; (2) a central nucleotide-binding and oligomerization domain (NB-ARC/NOD) that enables Apaf-1 oligomerization; and (3) a C-terminal WD40 region that forms β-propeller domains for cytochrome c binding and regulation of apoptosome assembly [2]. The formation of this complex is triggered when cytochrome c, released from mitochondria, binds to Apaf-1, facilitating the exchange of bound ADP for ATP/dATP and inducing conformational changes that enable oligomerization [7] [2].
The primary function of the apoptosome is to serve as an activation platform for procaspase-9. Through CARD-CARD interactions, procaspase-9 is recruited to the complex, where it undergoes activation through proximity-induced homodimerization [32]. Once activated, caspase-9 cleaves and activates downstream effector caspases, including caspase-3 and caspase-7, initiating the proteolytic cascade that leads to apoptotic cell death [8] [70]. Recent research has revealed that the apoptosome induces the formation of both caspase-9 homodimers and caspase-9/Apaf-1 heterodimers with distinct activities, adding complexity to the activation mechanism [32].
Table 1: Core Components of the Mammalian Apoptosome Complex
| Component | Structure/Features | Function in Apoptosome |
|---|---|---|
| Apaf-1 | Multi-domain protein (CARD, NOD, WD40 repeats) | Scaffold protein that oligomerizes to form the apoptosome platform |
| Cytochrome c | Heme-containing protein | Initiates apoptosome assembly by binding Apaf-1 WD40 domains |
| Procaspase-9 | Initiator caspase with CARD domain | Executioner protease activated on the apoptosome platform |
| Nucleotide (dATP/ATP) | Purine nucleotides | Cofactor required for oligomerization and activation |
Molecular Architecture and Activation Mechanisms
Structural Organization of the Apoptosome
High-resolution structural studies have revealed critical details of apoptosome organization. The human apoptosome forms a wheel-shaped complex with a central hub comprised of the NOD domains of Apaf-1, from which the V-shaped regulatory regions extend outward [2]. These regulatory regions are formed by two β-propeller domains (one with seven blades and one with eight) created by 15 WD40 repeats, with cytochrome c binding within the cleft between them [2].
The CARD domains of Apaf-1 are flexibly attached above the central hub. Upon binding procaspase-9, these domains organize into a disk-like, acentric spiral structure on top of the platform [7] [2]. This disk typically consists of four Apaf-1 and three to four procaspase-9 CARDs, meaning not all Apaf-1 CARDs participate in the spiral due to linker length constraints [2]. This architecture creates a specialized environment for caspase-9 activation through multiple mechanisms.
Mechanisms of Caspase-9 Activation
Two primary models have been proposed for caspase-9 activation on the apoptosome: the "induced proximity" model and the "induced conformation" model [8]. Recent evidence suggests both mechanisms contribute to activation:
Proximity-Induced Homodimerization: The apoptosome serves as a platform to concentrate procaspase-9 molecules, promoting their homodimerization and subsequent activation [8] [32]. Procaspase-9 possesses a higher affinity for itself compared to processed caspase-9, facilitating stable homodimer formation within the apoptosome [32].
Allosteric Regulation: Oligomerized Apaf-1 induces conformational changes in monomeric caspase-9 that result in its activation [32]. This is facilitated by the formation of heterodimers between procaspase-9 and Apaf-1, where procaspase-9 binds via its small subunit to the NOD domain in Apaf-1 [32].
The catalytic activity of caspase-9 is regulated by proteolytic processing. Autocleavage at Asp-315 produces caspase-9 (p35/p12), while cleavage by caspase-3 at Asp-330 generates caspase-9 (p35/p10) [26]. These cleaved forms exhibit reduced affinity for the apoptosome but maintain catalytic activity, implementing a "molecular timer" that regulates the duration of apoptosome activity [8] [32].
Diagram 1: Apoptosome-Mediated Apoptosis Activation Pathway. This diagram illustrates the sequential events from mitochondrial stress to caspase activation, highlighting the central role of apoptosome assembly.
Apoptosome Dysfunction in Human Cancers
Disruption of normal apoptosome formation and function constitutes a significant mechanism in tumor development and progression [72] [71]. Cancer cells employ multiple strategies to impair apoptosome function, thereby evading programmed cell death and increasing resistance to therapeutic agents.
Mechanisms of Apoptosome Impairment
Table 2: Mechanisms of Apoptosome Dysfunction in Human Cancers
| Dysfunction Mechanism | Molecular Basis | Cancer Associations |
|---|---|---|
| Reduced Apaf-1 Expression | Epigenetic silencing, loss of heterozygosity, transcriptional regulation | Leukemia, melanoma, colorectal, bladder, renal cell carcinoma |
| Altered Apaf-1 Splicing | Expression of alternative isoforms with missing WD40 repeats | Impaired cytochrome c binding and procaspase-9 activation |
| Inhibitory Protein Binding | Hsp70, Hsp90, and XIAP binding to apoptosome components | Blockade of procaspase-9 recruitment and activation |
| Post-Translational Modifications | Phosphorylation of caspase-9 at Thr125 | Inhibition of caspase-9 processing and activity |
| Cytochrome c Release Defects | Altered Bax/Bak function or increased Bcl-2 expression | Failure to initiate apoptosome assembly |
Specific Cancer Associations
Apoptosome impairment has been documented across diverse cancer types. In leukemia cells, reduced Apaf-1 expression contributes to disease pathogenesis and treatment resistance [2]. Ovarian cancers frequently exhibit downregulation of Apaf-1, correlating with poor response to chemotherapy [2] [71]. Additionally, CASP9 polymorphisms have been linked to increased susceptibility to lung, bladder, pancreatic, colorectal, and gastric cancers [8].
The physiological consequence of apoptosome dysfunction is particularly evident in treatment resistance. For example, head and neck squamous cell carcinoma cells exhibiting reduced caspase-9 activity and Apaf-1 expression demonstrate resistance to cisplatin treatment [8]. Similarly, testicular cancer cells with failed caspase-9 activation show increased apoptotic thresholds to chemotherapeutic agents [8].
Experimental Approaches for Apoptosome Research
Methodologies for Apoptosome Assembly and Analysis
In Vitro Apoptosome Reconstitution: Cell-free apoptosome assembly can be achieved by incubating recombinant Apaf-1 with cytochrome c and dATP/ATP in buffer containing 20 mM HEPES-KOH (pH 7.5), 10 mM KCl, 1.5 mM MgCl₂, 1 mM EDTA, 1 mM EGTA, and 1 mM DTT [32]. The assembly reaction typically proceeds for 60 minutes at 30°C, after which the complex can be isolated by gel filtration chromatography or glycerol gradient centrifugation [32].
Caspase Activation Assays: Apoptosome-mediated caspase activation can be quantified using fluorogenic substrates. The standard reaction includes reconstituted apoptosome, procaspase-9, and the substrate LEHD-amc (50 µM) in assay buffer [32]. Cleavage is monitored continuously by fluorescence emission at 460 nm with excitation at 380 nm. Alternatively, caspase-3 activation can be assessed using DEVD-amc substrate following addition of procaspase-3 to the reaction mixture [32].
Crosslinking Studies for Dimerization Analysis: Site-specific crosslinking techniques enable detection of caspase-9 homo- and heterodimers within the apoptosome. Cysteine mutants of caspase-9 are generated at strategic positions in the dimer interface, and apoptosome complexes are reconstituted with these mutants [32]. The complexes are then treated with crosslinkers such as BMH (bismaleimidohexane) or BMOE (bismaleimidoethane), followed by non-reducing SDS-PAGE and immunoblotting to detect crosslinked species [32].
Advanced Imaging Techniques
Recent advances in microscopy have enabled visualization of apoptosome formation in living cells. A 2025 study employed Apaf1-GFP expressing HeLa cells to monitor Apaf-1 dynamics during apoptosis induction [73]. This approach revealed that Apaf-1 molecules accumulate into multiple cytoplasmic foci upon apoptosis induction, and their disassembly correlates with cell survival [73].
Correlative light and electron microscopy (CLEM) and cryo-electron tomography (cryo-ET) have provided unprecedented views of apoptosome ultrastructure in near-native states [73]. These techniques reveal that Apaf-1 foci consist of cloud-like irregular meshwork structures rather than discrete wheel-shaped complexes, suggesting a more dynamic and pleiomorphic organization than previously understood [73].
Table 3: Key Research Reagents for Apoptosome Studies
| Reagent/Tool | Application | Experimental Function |
|---|---|---|
| Recombinant Apaf-1 | In vitro reconstitution | Core structural component for apoptosome assembly |
| Cytochrome c | Apoptosome activation | Trigger for Apaf-1 oligomerization |
| Fluorogenic Caspase Substrates (LEHD-amc, DEVD-amc) | Activity assays | Quantification of caspase-9 and caspase-3 activation |
| Site-Specific Crosslinkers (BMH, BMOE) | Dimerization studies | Stabilization of protein interactions for analysis |
| Apaf1-GFP Constructs | Live-cell imaging | Visualization of apoptosome dynamics in cells |
| Caspase-9 Mutants (F404D, ProC9-TM) | Mechanistic studies | Disruption or modulation of dimerization and processing |
Therapeutic Targeting and Research Perspectives
Therapeutic Strategies Targeting Apoptosome Dysfunction
Several therapeutic approaches aim to overcome apoptosome impairment in cancer:
Small Molecule Activators: Compounds that promote apoptosome assembly or enhance caspase-9 activation represent promising therapeutic avenues. These include agents that mimic cytochrome c function or facilitate Apaf-1 oligomerization independently of cytochrome c.
XIAP Antagonists: As XIAP (X-linked Inhibitor of Apoptosis Protein) directly inhibits caspase-9 through its Bir3 domain, small-molecule XIAP antagonists can restore apoptosis in cancer cells with intact apoptosome machinery [26]. The Bir3 domain of XIAP serves as an endogenous highly selective caspase-9 inhibitor, making it a compelling therapeutic target [26].
Gene Therapy Approaches: Restoration of Apaf-1 expression in deficient tumors via gene delivery systems represents another strategy under investigation. This approach aims to resensitize cancer cells to intrinsic apoptosis signals.
Emerging Research Directions
Recent studies have revealed novel dimensions of apoptosome biology with therapeutic implications:
Non-Apoptotic Functions: Caspase-9 exhibits functions beyond cell death execution, including regulation of cellular differentiation, mitochondrial homeostasis, and autophagy [26]. These non-apoptotic roles may influence tumor behavior and treatment responses.
Dynamic Assembly Regulation: The discovery that Apaf-1 forms transient, cloud-like assemblies in cells rather than stable wheel-shaped complexes suggests a more dynamic regulation of apoptosome activity than previously recognized [73]. The disassembly of these foci correlates with cell survival, indicating potential regulatory mechanisms that could be therapeutically targeted.
Context-Specific Activation Mechanisms: Emerging evidence suggests alternative caspase-9 activation pathways that operate independently of Apaf-1, particularly in specific cellular contexts such as autophagy induction or metabolic stress [26]. Understanding these alternative pathways may reveal new opportunities for therapeutic intervention.
Diagram 2: Apoptosome Impairment Mechanisms and Therapeutic Strategies. This diagram outlines primary dysfunction mechanisms in cancer and corresponding therapeutic approaches to restore apoptotic function.
Apoptosome impairment represents a critical mechanism in tumorigenesis, enabling cancer cells to evade programmed cell death and resist conventional therapies. The intricate molecular architecture of the apoptosome and its central role in caspase-9 activation make it a compelling focus for both basic research and therapeutic development. Ongoing research continues to elucidate the structural basis of apoptosome assembly, the mechanisms of caspase-9 activation, and the diverse strategies cancer cells employ to disrupt this pathway.
Future directions in apoptosome research include developing more precise structural models of the complex in cellular environments, understanding context-specific regulation of its activity, and designing targeted therapies that can overcome specific impairment mechanisms. As our knowledge of apoptosome biology deepens, so too will our ability to manipulate this critical complex for therapeutic benefit in cancer and other diseases characterized by apoptotic dysregulation.
Caspase-9 in Neuronal Death Pathways
Caspase-9 functions as the essential initiator caspase within the intrinsic apoptotic pathway, playing a critical role in both physiological neuronal development and pathological neuronal death. Its activation is mediated through the apoptosome complex, a process regulated by multiple molecular mechanisms. Dysregulation of caspase-9 signaling contributes significantly to various neurodegenerative diseases, making it a promising therapeutic target. This whitepaper provides an in-depth examination of caspase-9 activation mechanisms, pathological implications in neurological disorders, experimental methodologies for study, and emerging therapeutic strategies targeting the apoptosome complex for drug development in neurological diseases.
Caspase-9 belongs to the cysteine-aspartic protease family and serves as the apical protease in the mitochondrial stress-induced apoptosis pathway [8]. As an initiator caspase, it exists as an inactive monomeric zymogen (procaspase-9) within cells until activated through a highly regulated process [8] [9]. The constitutive expression of caspase-9 is ubiquitous across mammalian tissues, with particular importance in the central nervous system, where it plays crucial roles in developmental apoptosis and neuronal homeostasis [8]. Genetic knockout studies demonstrate that mice lacking caspase-9 die perinatally with severe brain abnormalities due to impaired apoptosis during brain development, underscoring its essential function in neural development [8].
The activation of caspase-9 occurs primarily within a multiprotein activation platform known as the apoptosome, which forms in response to various cellular stressors including DNA damage, oxidative stress, and excitotoxicity [25] [26]. This complex represents a critical control point in the intrinsic apoptotic pathway, integrating death signals from the mitochondria and initiating the caspase cascade that leads to cellular demolition [40]. The apoptosome core consists of apoptotic protease-activating factor 1 (Apaf-1), cytochrome c released from mitochondria, and procaspase-9, forming in the presence of dATP/ATP [25] [40]. Understanding the precise molecular mechanisms governing caspase-9 activation within this complex provides the foundation for comprehending its role in neuronal death pathways and developing targeted therapeutic interventions.
Molecular Mechanisms of Caspase-9 Activation
Apoptosome-Mediated Activation
The molecular activation of caspase-9 occurs through a meticulously orchestrated process centered on the apoptosome complex. Apaf-1, the core component of the apoptosome, features three critical domains: an N-terminal caspase recruitment domain (CARD), a central nucleotide-binding domain (NBD), and C-terminal WD-40 repeats [40]. In its inactive state, Apaf-1 exists as a monomer, with its CARD domain interacting intramolecularly with the WD-40 region, preventing premature caspase-9 binding [40]. The activation cascade initiates when cytochrome c, released from mitochondria following apoptotic stimuli, binds to the WD-40 repeats of Apaf-1, inducing a conformational change that facilitates dATP/ATP binding and hydrolysis [40]. This triggers Apaf-1 oligomerization into a heptameric wheel-like structure that serves as the activation platform for procaspase-9 [25].
The current understanding of caspase-9 activation encompasses several mechanistic models, each supported by experimental evidence. The induced proximity model posits that the apoptosome serves primarily as a platform to bring caspase-9 zymogens into close proximity, enabling autocatalytic activation [8] [25]. An evolved version, the proximity-driven dimerization model, suggests that the apoptosome accumulates local concentrations of procaspase-9 to promote dimer-driven activation [8] [4]. Experimental evidence supporting this model demonstrates that both Hofmeister salts and a reconstituted mini-apoptosome activate caspase-9 through a second-order process consistent with dimerization [4]. In contrast, the induced conformation model proposes that the apoptosome actively alters caspase-9 conformation through direct binding, creating an active catalytic site [8] [25]. This model is supported by structural studies showing that caspase-9 in complex with Apaf-1 exhibits proteolytic activity several orders of magnitude higher than the free enzyme, functioning as a holoenzyme with caspase-9 as the catalytic subunit and Apaf-1 as its allosteric regulator [33].
Table 1: Models of Caspase-9 Activation
| Model | Key Mechanism | Experimental Evidence |
|---|---|---|
| Induced Proximity | Apoptosome brings caspase-9 molecules into close proximity for autocatalysis | Based on original caspase activation studies; supported by induced proximity concept for initiator caspases [25] |
| Proximity-Driven Dimerization | Apoptosome increases local concentration to promote dimer formation | Second-order activation observed with mini-apoptosome; caspase-8/caspase-9 hybrid activated by apoptosome [4] |
| Induced Conformation | Direct interaction with apoptosome induces active conformation in caspase-9 | Caspase-9/Apaf-1 complex shows significantly higher activity than free enzyme [33] |
The resulting active caspase-9, whether through dimerization or conformational change, remains bound to the apoptosome as part of a holoenzyme complex that recruits and activates downstream effector caspases, particularly caspase-3 and caspase-7 [8] [9]. This holoenzyme complex demonstrates significantly enhanced catalytic efficiency compared to free caspase-9, highlighting the critical regulatory function of the apoptosome in amplifying the apoptotic signal [33]. Recent structural insights have revealed that the interaction between Apaf-1 and caspase-9 involves multiple interfaces beyond simple 1:1 binding, including a multimeric interaction between CARD domains that requires three distinct interfaces for effective caspase-9 activation [8].
Structural Features and Regulatory Domains
Caspase-9 possesses characteristic structural domains that mediate its activation and function. The N-terminal region contains a long prodomain featuring a CARD motif that selectively binds to the complementary CARD domain in Apaf-1 through homotypic interactions [8] [9]. This CARD-CARD interaction is essential for recruiting procaspase-9 to the apoptosome complex. Following the prodomain, a linker loop connects to the catalytic domain, which consists of large (p35) and small (p12) subunits [8]. Unlike effector caspases, caspase-9 demonstrates activity in its uncleaved form, suggesting that the extended linker loop provides sufficient flexibility for active site formation without proteolytic processing [8]. However, upon activation, caspase-9 undergoes autocatalytic cleavage at specific aspartic acid residues, producing the p35/p12 fragments that remain associated within the holoenzyme complex [8] [26].
Figure 1: Caspase-9 Activation Pathway via the Apoptosome Complex
Caspase-9 in Neurological Diseases
Neurodegenerative Disorders
Dysregulation of caspase-9-mediated apoptosis represents a common pathological mechanism across multiple neurodegenerative conditions. In Alzheimer's disease (AD), caspase-9 activation contributes to the progressive neuronal loss characteristic of the disease pathology [74]. Experimental evidence indicates that caspase-9 is activated in response to amyloid-beta toxicity and tau pathology, initiating the apoptotic cascade that leads to neuronal demolition [74] [26]. The observed correlation between caspase-9 inhibition and p53 expression has implications for AD pathophysiology, as this interaction may increase neuronal resistance to apoptotic stimuli [8].
In Parkinson's disease (PD), caspase-9 activation occurs in response to mitochondrial dysfunction, a hallmark of PD pathogenesis [74] [26]. The vulnerability of dopaminergic neurons in the substantia nigra to caspase-9-mediated apoptosis is exacerbated by oxidative stress and complex I impairment, which promote cytochrome c release and apoptosome formation [74]. Similarly, in Huntington's disease (HD), activated caspase-9 and caspase-3 are present at end-stage disease, suggesting that apoptosis contributes significantly to neuronal death in advanced pathology [8]. Mutant huntingtin protein directly sensitizes neurons to caspase-9 activation through mechanisms involving mitochondrial dysfunction and impaired energy metabolism [74].
Amyotrophic lateral sclerosis (ALS) represents another neurodegenerative condition where caspase-9 plays a crucial role. Studies in SOD1 mutant mouse models of ALS demonstrate abnormally reduced levels of anti-apoptotic BCL-2 and increased expression of pro-apoptotic BAX in spinal cord motor neurons [74]. Notably, BIM deficiency extends lifespan in SOD1G93A mutant mice, while administration of broad-spectrum caspase inhibitors delays symptom onset and terminal illness [74]. The absence of apoptosis effectors BAX and BAK similarly produces protective effects in ALS models, confirming the significance of the intrinsic apoptotic pathway in ALS pathogenesis [74].
Table 2: Caspase-9 in Neurodegenerative Diseases
| Disease | Key Pathological Features | Caspase-9 Involvement |
|---|---|---|
| Alzheimer's Disease | Amyloid-beta plaques, neurofibrillary tangles, neuronal loss | Activated in response to Aβ toxicity and tau pathology; contributes to synaptic dysfunction and neuronal death [74] [26] |
| Parkinson's Disease | Dopaminergic neuron loss, Lewy bodies, mitochondrial dysfunction | Activated by mitochondrial dysfunction and oxidative stress; mediates death of substantia nigra neurons [74] [26] |
| Huntington's Disease | Mutant huntingtin, striatal neuron loss, motor dysfunction | Activated at endstage disease; contributes to neuronal death alongside caspase-3 [8] [74] |
| Amyotrophic Lateral Sclerosis | Motor neuron degeneration, muscle atrophy, SOD1 mutations | Correlated with BCL-2 reduction and BAX increase; caspase inhibition delays disease progression in models [74] |
Non-Apoptotic Functions and Alternative Death Pathways
Beyond its canonical role in apoptosis, caspase-9 participates in diverse non-apoptotic neuronal functions and alternative cell death pathways. Recent research has revealed that caspase-9 regulates corticospinal circuit organization through non-apoptotic mechanisms, with caspase-9 deficiency leading to skilled movement deficits despite the absence of overt cell death [26]. This function appears independent of effector caspases-3, -6, and -7, suggesting novel signaling mechanisms [26]. Additionally, caspase-9 contributes to mitochondrial homeostasis, with genetic or pharmacological ablation resulting in mitochondrial membrane depolarization, reduced reactive oxygen species production, and aberrant accumulation of mitochondrial dynamics proteins [26].
In cases where caspase activity is inhibited, neurons may undergo alternative caspase-independent death pathways. Studies demonstrate that cortical neurons treated with camptothecin and the pan-caspase inhibitor BAF avoid apoptosis initially but subsequently undergo delayed caspase-independent death characterized by mitochondrial dysfunction [75]. This alternative death pathway involves reduced mitochondrially generated ATP and increased reactive oxygen species generation, both contributing to neuronal demise despite caspase inhibition [75]. These findings highlight the complex interplay between caspase-dependent and independent death mechanisms in neurological contexts and suggest that effective neuroprotective strategies may need to target multiple parallel pathways.
Experimental Analysis of Caspase-9 Activity
Research Reagent Solutions
The investigation of caspase-9 function in neuronal death pathways requires a specialized toolkit of research reagents and methodological approaches. The table below summarizes essential experimental resources for studying caspase-9 activation and function:
Table 3: Research Reagent Solutions for Caspase-9 Investigation
| Reagent/Category | Specific Examples | Research Application | Mechanistic Function |
|---|---|---|---|
| Caspase Inhibitors | zVAD-fmk (broad-spectrum), Boc-aspartyl(OMe)-fluoromethylketone (BAF) | Inhibition of apoptotic death to study caspase-independent pathways [74] [75] | Irreversible binding to active site cysteine; prevents substrate cleavage |
| Pharmacological Activators | Camptothecin (topoisomerase-I inhibitor), ABT-263 (BCL-2 inhibitor) | Induction of intrinsic apoptotic pathway in neuronal cultures [75] [8] | DNA damage stress; displacement of pro-apoptotic proteins from anti-apoptotic BCL-2 |
| Genetic Models | Caspase-9 knockout mice, BAX/BAK deficient neurons, SOD1 mutant mice (ALS model) | Study of developmental apoptosis and disease-specific mechanisms [8] [74] | Elimination of caspase function; modeling human neurodegenerative diseases |
| Activity Assays | D315 neoepitope antibodies, D330 neoepitope antibodies, fluorogenic substrate cleavage | Detection of specific caspase-9 activation forms and proteolytic activity [26] | Distinguish autoactivation (D315) vs. caspase-3-mediated cleavage (D330) |
| Mitochondrial Probes | TMRM (ΔΨm), DHE (ROS), LysoTracker Red (lysosomal function) | Assessment of mitochondrial function in caspase-dependent and independent death [75] | Monitor membrane potential, reactive oxygen species, and organelle function |
| Protein Interaction Tools | XIAP Bir3 domain, dominant-negative caspase-9 mutants, Apaf-1 constructs | Study of regulation mechanisms and apoptosome formation [26] | Selective caspase-9 inhibition; disruption of complex formation |
Methodological Approaches
The experimental analysis of caspase-9 in neuronal systems employs diverse methodological approaches. Primary neuronal cultures from embryonic rodents provide a physiologically relevant system for investigating caspase-9 activation mechanisms. These cultures, typically prepared from E18 rat cortical neurons and maintained in neurobasal medium with B-27 supplement, allow for controlled manipulation of apoptotic stimuli and pharmacological inhibition [75]. In such systems, caspase-9 activation can be induced using DNA-damaging agents like camptothecin (10 μM), which triggers the intrinsic pathway through p53 activation and Bax-mediated cytochrome c release [75].
Assessment of caspase-9 activation employs multiple complementary techniques. Immunochemical detection of specific cleavage neoepitopes distinguishes between autoactivation (D315 cleavage site) and caspase-3-mediated cleavage (D330 site), providing insight into activation mechanisms [26]. Fluorogenic substrate assays measure catalytic activity directly, while Western blot analysis monitors processing of caspase-9 and its downstream targets [26] [75]. Additionally, live-cell imaging with fluorescent dyes such as TMRM (for mitochondrial membrane potential), DHE (for reactive oxygen species), and calcein AM (for viability) enables real-time tracking of apoptotic events in neuronal populations [75].
To dissect the specific contribution of caspase-9 to neuronal death, researchers employ genetic manipulation approaches including siRNA knockdown, CRISPR/Cas9 gene editing, and analysis of neurons from caspase-9 deficient animals [8] [26]. These tools allow for specific interrogation of caspase-9 function separate from other initiator caspases. Furthermore, biochemical reconstitution experiments using purified components (Apaf-1, cytochrome c, caspase-9) enable detailed analysis of apoptosome structure and function, as demonstrated in studies of mini-apoptosome complexes [4].
Figure 2: Experimental Workflow for Studying Caspase-9 in Neuronal Death
Therapeutic Targeting and Clinical Implications
Pharmacological Strategies
The central role of caspase-9 in neuronal death pathways has motivated the development of targeted therapeutic strategies. Pharmacological caspase-9 inhibitors represent a promising approach for neurodegenerative conditions where excessive apoptosis contributes to pathology. Several inhibitor classes have shown efficacy in preclinical models, including small-molecule active site inhibitors that directly bind the catalytic cleft, protein-based inhibitors derived from endogenous caspase regulators like the XIAP Bir3 domain, and dominant-negative caspase-9 mutants that disrupt apoptosome function [26]. In ALS models, the broad-spectrum caspase inhibitor zVAD-fmk delays disease onset and extends survival, while in models of acute brain injury, caspase inhibition provides neuroprotection and functional improvement [74].
Alternative approaches target regulatory nodes upstream of caspase-9 activation. BCL-2 family modulators such as ABT-263 can influence caspase-9 activation by altering mitochondrial apoptotic susceptibility, though interestingly, caspase-9 inhibition sometimes enhances ABT-263-induced apoptosis through compensatory caspase-8 activation, revealing complex regulatory networks [8]. Cytochrome c release inhibitors provide another strategic approach, preventing the initial apoptosome assembly signal, while compounds that modulate Apaf-1 conformation or oligomerization offer more direct targeting of the caspase-9 activation platform itself [40].
Emerging strategies also aim to exploit non-apoptotic caspase-9 functions for therapeutic benefit. The identification of caspase-9 roles in neuronal differentiation, circuit refinement, and mitochondrial homeostasis suggests that modulators of caspase-9 activity (rather than complete inhibitors) might provide optimal outcomes in chronic neurodegenerative conditions [26]. Additionally, the development of neoepitope-specific antibodies that distinguish between different caspase-9 activation states (D315 vs D330 cleavage sites) enables more precise monitoring of therapeutic responses and disease progression in both preclinical and clinical settings [26].
Clinical Translation and Challenges
The translation of caspase-9-targeted therapies from preclinical models to clinical applications faces several challenges. The dual roles of caspase-9 in both physiological processes (developmental apoptosis, tissue homeostasis) and pathological neurodegeneration create a therapeutic window concern, as systemic caspase-9 inhibition might disrupt normal cellular turnover or promote tumorigenesis [8] [26]. Additionally, the evidence for caspase-independent death pathways in neurons suggests that caspase inhibition alone may provide only transient protection, necessitating combination approaches that address parallel degeneration mechanisms [75].
Clinical reports have identified CASP9 gene polymorphisms associated with neurological conditions, including the Ex5+32G/A polymorphism linked to multiple sclerosis susceptibility and the -1263A/G polymorphism associated with discogenic low back pain [8]. These genetic associations highlight the relevance of caspase-9 regulation in human disease but also indicate population variability that might influence therapeutic responses. Furthermore, tissue-specific expression patterns and alternative splicing of caspase-9 (generating isoforms like caspase-9b with anti-apoptotic functions) add layers of complexity to therapeutic targeting [26].
Despite these challenges, the compelling preclinical evidence supporting caspase-9's role in neuronal death pathways continues to drive therapeutic innovation. Future directions include the development of CNS-penetrant selective inhibitors, gene therapy approaches targeting caspase-9 expression, and biomarker strategies using caspase-9 cleavage products to monitor disease progression and treatment response. As our understanding of caspase-9 biology in neurological contexts continues to evolve, particularly regarding its non-apoptotic functions and interactions with other cell death pathways, new therapeutic opportunities will likely emerge for addressing the profound medical need in neurodegenerative diseases.
The apoptosome complex, a central signaling platform in the intrinsic apoptotic pathway, executes cellular suicide in response to internal damage signals. This whitepaper delineates the critical molecular cross-talk that connects extrinsic death receptor signaling to core apoptosome function through caspase-8-mediated Bid integration. We examine how the cleavage of Bid generates tBid, which propagates death signals to mitochondria, resulting in cytochrome c release and subsequent caspase-9 activation via the apoptosome. This integration mechanism enables signal amplification in cell types with limited initial caspase-8 activation and represents a pivotal regulatory node in cellular fate determination. Understanding these connections provides novel insights for therapeutic intervention in cancer and other diseases characterized by apoptotic dysregulation.
Apoptosis, a programmed cell death mechanism essential for development and homeostasis, proceeds through two principal signaling pathways: the extrinsic (death receptor) pathway and the intrinsic (mitochondrial) pathway. The apoptosome complex, formed upon cytochrome c release from mitochondria, activates caspase-9 as the initiator caspase of the intrinsic pathway [8] [9]. While these pathways were initially characterized as distinct entities, emerging evidence demonstrates significant molecular cross-talk that integrates their signaling networks [76] [77].
The critical intersection point between these pathways involves caspase-8-mediated cleavage of Bid, a BH3-only Bcl-2 family protein, which translates death receptor activation into mitochondrial engagement [76] [78]. This cross-talk mechanism enables cells with limited direct caspase-8 activation to amplify death signals through the intrinsic pathway, ensuring efficient apoptosis execution. The resulting apoptosome formation and caspase-9 activation represent the final common pathway for many apoptotic stimuli, making this cross-talk a fundamental regulatory checkpoint in cell fate determination [8] [32].
Molecular Mechanisms of Caspase-8 and Bid Integration
Caspase-8 Activation and Function
Caspase-8 serves as the initiator caspase in the extrinsic apoptotic pathway. Its activation occurs through a well-defined mechanism:
- Death-Inducing Signaling Complex (DISC) Formation: Upon ligand binding to death receptors (e.g., Fas, TRAIL receptors), the adaptor protein FADD (Fas-associated death domain) recruits procaspase-8 to form the DISC [78] [9].
- Proximity-Induced Activation: Within the DISC, caspase-8 zymogens undergo proximity-induced dimerization and autocatalytic processing [78]. The initial cleavage between the p18 and p10 domains generates a heterodimer (p43-p10/p43-p10) essential for substrate recognition [78].
- Catalytic Activity: Active caspase-8 exists as a heterotetramer (p18-p10/p18-p10) that cleaves downstream substrates, including effector caspases and the BH3-only protein Bid [78] [9].
Bid Cleavage and tBid Generation
Bid serves as the critical molecular link connecting caspase-8 activation to mitochondrial apoptosis:
- Cleavage Mechanism: Caspase-8 cleaves full-length Bid (22 kDa) at a specific aspartic acid residue (Asp-59) to generate truncated Bid (tBid, 15 kDa) [78] [77].
- Mitochondrial Targeting: tBid translocates to the mitochondrial outer membrane, where it undergoes conformational activation and membrane insertion [79] [78].
- BCL-2 Family Interactions: At the mitochondria, tBid directly activates the pro-apoptotic effector proteins Bax and Bak, while simultaneously inhibiting anti-apoptotic Bcl-2 family members [77].
Mitochondrial Integration and Apoptosome Activation
The integration of tBid into mitochondrial signaling triggers key events leading to apoptosome formation:
- MOMP Induction: tBid-mediated Bax/Bak activation induces mitochondrial outer membrane permeabilization (MOMP), enabling cytochrome c release into the cytosol [79] [77].
- Apoptosome Assembly: Cytochrome c binds to Apaf-1, promoting ATP-dependent oligomerization into the heptameric apoptosome complex [8] [32].
- Caspase-9 Activation: The apoptosome recruits and activates caspase-9 through CARD-CARD domain interactions, initiating the caspase cascade that executes apoptosis [8] [32].
Figure 1: Molecular pathway of caspase-8 and Bid integration connecting extrinsic apoptosis to apoptosome-mediated caspase-9 activation. Dashed lines indicate alternative pathways.
Experimental Analysis of Caspase-8/Bid Cross-Talk
Key Methodologies for Studying Cross-Talk
Research into caspase-8 and Bid integration employs multiple technical approaches to elucidate the molecular mechanisms:
3.1.1 Protein Localization Studies
- Confocal Microscopy: GFP-tagged caspase-8 mutants and mito-dsRed2 enable visualization of caspase-8 mitochondrial translocation [78].
- Subcellular Fractionation: Biochemical separation of mitochondrial and cytosolic fractions confirms protein redistribution during apoptosis [78].
- Immunofluorescence: Antibodies against active caspase-8 and tBid visualize their co-localization at mitochondrial membranes [78].
3.1.2 Complex Identification
- Co-Immunoprecipitation: Identifies native complexes containing caspase-8 and Bid on mitochondrial membranes [78].
- Crosslinking Techniques: Site-specific crosslinking stabilizes transient interactions for complex analysis [32].
- Blue Native PAGE: Resolves native protein complexes in their functional states [78].
3.1.3 Functional Assessments
- Caspase Activity Assays: Fluorogenic substrates (e.g., LEHD-afc for caspase-9) quantify enzymatic activity [8] [32].
- Cytochrome c Release Assays: Isolated mitochondria or permeabilized cells measure MOMP induction [78].
- Cell Viability Assays: Annexin V/PI staining, MTT assays, and colony formation determine apoptotic outcomes [78] [80].
Critical Experimental Findings
Research using these methodologies has yielded fundamental insights into cross-talk mechanisms:
- Mitochondrial Caspase-8 Localization: Caspase-8 stably inserts into mitochondrial outer membranes during extrinsic apoptosis, forming a platform for Bid cleavage [78].
- Bid Cleavage Specificity: Caspase-8 cleaves Bid within a native mitochondrial complex, ensuring spatial specificity of tBid generation [78].
- Type I/Type II Distinction: Cells are classified based on their dependence on mitochondrial amplification (type II) versus direct caspase-8 signaling (type I) [76].
- Molecular Timer Regulation: Caspase-9 autoprocessing at Asp-315 initiates a molecular timer that regulates apoptosome activity duration [32].
Table 1: Key Experimental Findings in Caspase-8/Bid Cross-Talk Research
| Finding | Experimental Approach | Biological Significance | Reference |
|---|---|---|---|
| Caspase-8 mitochondrial translocation | Confocal microscopy with GFP-tagged constructs | Identifies specialized platform for Bid cleavage | [78] |
| Native caspase-8/Bid complex formation | Co-immunoprecipitation and crosslinking | Demonstrates spatial organization of cleavage event | [78] |
| Type I/Type II cell classification | Bcl-2 overexpression and caspase inhibition | Explains differential apoptotic signaling requirements | [76] |
| Apoptosome molecular timer mechanism | Caspase-9 cleavage mutants and activity assays | Reveals regulation of apoptosome activity duration | [32] |
Research Reagent Solutions for Cross-Talk Studies
Table 2: Essential Research Reagents for Studying Caspase-8/Bid/Apoptosome Cross-Talk
| Reagent Category | Specific Examples | Research Application | Key Function | |
|---|---|---|---|---|
| Caspase-8 Tools | Caspase-8-GFP constructs (wild-type and mutants C360S, DM1, DM2) | Localization and functional studies | Tracking caspase-8 processing and mitochondrial translocation | [78] |
| Caspase-8 inhibitory peptides (Z-IETD-FMK) | Pathway inhibition studies | Specific blockade of caspase-8 activity | ||
| Bid Reagents | Anti-Bid antibodies (full-length and tBid specific) | Immunodetection and quantification | Monitoring Bid cleavage and tBid generation | [78] [77] |
| Recombinant Bid and tBid proteins | In vitro reconstitution assays | Direct testing of mitochondrial effects | ||
| Apoptosome Components | Recombinant caspase-9 (wild-type and mutants F404D, ProC9-TM) | Apoptosome activation studies | Investigating caspase-9 dimerization and activity | [32] |
| Anti-Apaf-1 antibodies | Complex detection | Apoptosome formation analysis | ||
| Activity Reporters | Fluorogenic substrates (LEHD-afc for caspase-9, DEVD-afc for caspase-3) | Enzymatic activity quantification | Real-time measurement of caspase activation | [32] |
| VC3AI (Venus-based caspase-3 activity indicator) | Live-cell imaging | Visualizing caspase-3 activation in living cells | [80] | |
| Cell Death Modulators | Bcl-2/Bcl-xL expression vectors | Mitochondrial resistance studies | Blocking intrinsic pathway activation | [76] [80] |
| Caspase-9 inhibitor (Z-LEHD-FMK) | Specific pathway inhibition | Testing apoptosome dependence | [30] |
Type I/Type II Cell Paradigm and Cross-Talk Regulation
Fundamental Distinctions in Apoptotic Signaling
The classification of cells into type I and type II categories represents a critical framework for understanding caspase-8/Bid cross-talk:
Type I Cell Characteristics:
- Robust DISC Formation: Generate large amounts of active caspase-8 at the DISC [76].
- Mitochondrial Independence: Undergo apoptosis without cytochrome c release or Bcl-2 inhibition [76].
- Direct Activation Pathway: Caspase-8 directly activates effector caspases (caspase-3/7) sufficiently for apoptosis execution [76] [78].
Type II Cell Characteristics:
- Limited DISC Formation: Produce insufficient active caspase-8 for direct effector caspase activation [76].
- Mitochondrial Dependence: Require cytochrome c release and apoptosome formation for apoptosis amplification [76] [78].
- Bid-Mediated Amplification: Caspase-8 cleaves Bid to engage the mitochondrial pathway for signal amplification [76] [78].
Molecular Determinants of Cell Type Specification
Several factors determine whether a cell follows type I or type II apoptosis pathways:
- DISC Formation Efficiency: The abundance of death receptors and adapter proteins influences initial caspase-8 activation levels [76].
- Cellular Context and Signaling Tone: Growth factors and cytokines can modulate mitochondrial sensitivity to tBid through MAPK pathway activation [76].
- Bcl-2 Family Expression Ratios: The balance between pro- and anti-apoptotic Bcl-2 family members regulates mitochondrial priming [76] [77].
- XIAP Levels: Differential expression of XIAP, which inhibits caspases-3, -7, and -9, influences apoptotic requirements [79].
Figure 2: Comparative signaling pathways in type I and type II cells, highlighting the role of Bid cleavage and mitochondrial amplification in type II apoptosis.
Therapeutic Implications and Research Applications
Cancer Therapeutic Opportunities
The caspase-8/Bid/apoptosome axis presents multiple therapeutic targets for cancer treatment:
- Chemosensitization Strategies: Enhancing caspase-8 activation or Bid cleavage can overcome resistance in type II tumor cells [8].
- Bcl-2 Family Targeting: Small molecule inhibitors (e.g., ABT-263) counteracting anti-apoptotic Bcl-2 proteins can facilitate tBid-mediated apoptosis [8].
- IAP Antagonists: Smac mimetics that neutralize XIAP can lower the threshold for apoptosome-mediated caspase activation [79].
Disease Pathogenesis Connections
Dysregulation of caspase-8/Bid cross-talk contributes to various disease states:
- Cancer Development: Reduced caspase-8 expression or Bid mutation enables tumor cells to evade extrinsic apoptosis [8].
- Liver Pathologies: Hepatocytes predominantly exhibit type II characteristics, explaining the protective effect of Bid deficiency in Fas-mediated liver failure [76].
- Neurodegenerative Disorders: Impaired apoptosome function contributes to defective apoptosis in neurological diseases [8].
Experimental and Technical Considerations
Research in this field requires careful attention to methodological details:
- Cell Type Validation: Researchers must empirically determine whether specific cell lines utilize type I or type II pathways under experimental conditions [76].
- Simultaneous Pathway Assessment: Comprehensive studies should monitor both caspase-8 activation and mitochondrial events to fully characterize apoptotic signaling [78].
- Physiological Relevance: The form of death ligand (multimeric vs. trimeric) influences signaling outcomes and should approximate physiological conditions [76].
The integration of caspase-8 and Bid signaling represents a fundamental mechanism that connects extrinsic apoptosis to the core apoptosome complex and caspase-9 activation. This cross-talk enables apoptotic signal amplification in cell types with limited initial caspase-8 activation and provides critical regulatory checkpoints for cell fate determination. The type I/type II cell paradigm offers a framework for understanding how cellular context influences apoptotic pathway utilization, with significant implications for therapeutic development in cancer and other diseases. Future research elucidating the precise molecular regulation of these integration points will continue to advance both fundamental knowledge and clinical applications in programmed cell death.
The apoptosome complex, a heptameric scaffold formed by Apaf-1, represents a critical control point in intrinsic apoptosis by serving as the activation platform for caspase-9 [35] [7]. This canonical pathway involves cytochrome c release from mitochondria, Apaf-1 oligomerization, and recruitment of procaspase-9, leading to its activation through proximity-induced dimerization [8] [2]. While this apoptotic function has been extensively studied, emerging research reveals that components of this mitochondrial-regulated system participate in fundamental non-apoptotic processes, particularly in cellular differentiation and homeostasis. This paradigm shift establishes mitochondria not merely as executioners of cell death but as master regulators of cell fate decisions, integrating metabolic and signaling cues to direct developmental and differentiation programs [81] [82]. This whitepaper examines the mechanistic basis of mitochondrial regulation in cellular differentiation, providing technical insights and methodologies for researchers investigating mitochondrial functions beyond apoptosis.
Mitochondrial Dynamics: The Structural Basis of Functional Plasticity
Mitochondria exist in a dynamic equilibrium between fission and fusion states, processes governed by conserved GTPase proteins. This continuous remodeling underpins mitochondrial functional plasticity and quality control, forming the structural foundation for their role in cellular differentiation.
Core Machinery Governing Mitochondrial Morphology
Table 1: Core Proteins Regulating Mitochondrial Dynamics
| Protein | Primary Function | Mechanism of Action | Impact on Differentiation |
|---|---|---|---|
| DRP1 (Drp1/DNM1L) | Mitochondrial fission | GTPase that oligomerizes at OMM constriction sites; recruited by MFF, Fis1, MiD49/51 [83] [84] [85] | Essential for embryonic development; regulates stem cell fate decisions [82] |
| MFN1/2 (Mitofusin 1/2) | Outer membrane fusion | Forms homo/hetero-dimers; GTP hydrolysis mediates OMM tethering [83] [84] [85] | MFN2 required for trophoblast differentiation; maintains stem cell populations [82] |
| OPA1 | Inner membrane fusion | Regulates cristae architecture; exists in long and short forms [83] [84] [85] | Controls steroidogenesis in trophoblasts; essential for neuronal differentiation [82] |
| FIS1 | Fission adapter | Tethers DRP1 to OMM; regulates mitochondrial fragmentation [83] [84] | Modulates mitochondrial network during metabolic transitions in differentiation |
The fission process is initiated by recruitment of cytosolic DRP1 to the outer mitochondrial membrane (OMM) at endoplasmic reticulum (ER)-contact sites, where it oligomerizes into spiral structures that constrict and ultimately divide the organelle [85]. Fusion mechanisms are more complex, requiring coordinated merging of both outer and inner membranes: MFN1 and MFN2 mediate OMM fusion, while OPA1 facilitates inner mitochondrial membrane (IMM) fusion and maintains cristae architecture [84] [85]. These dynamics are not merely structural but are intimately coupled to functional outputs—fission generates discrete mitochondrial units that can be selectively removed via mitophagy, while fusion enables content mixing to maintain functional homogeneity across the network.
Experimental Assessment of Mitochondrial Dynamics
Protocol 1: Evaluating Mitochondrial Morphology and Dynamics
Live-Cell Imaging of Mitochondrial Networks: Transfect cells with mito-DsRed or mt-GFP and culture in glass-bottom dishes. Capture time-lapse images using confocal microscopy at 5-15 second intervals over 15-30 minutes. Analyze morphology using classification systems (punctate, tubular, interconnected) or quantitative parameters (aspect ratio, form factor, branch length) with specialized software like ImageJ MiNa plugin [82].
FRAP (Fluorescence Recovery After Photobleaching) for Fusion Assays: Label mitochondria with MitoTracker Green. Photobleach a defined region using high-intensity laser and monitor fluorescence recovery over time. Calculate fusion rates from recovery kinetics, as fusion allows unbleached proteins to diffuse into bleached areas [84].
Mitochondrial Tracking in Migrating Cells: Use GFP-tagged mitochondrial proteins (e.g., TOM20) for live imaging in wound healing assays or microfluidic devices. Track individual mitochondrial movement using automated tracking algorithms to quantify velocity, directionality, and confinement ratio [82].
Transmission Electron Microscopy for Ultrastructural Analysis: Fix cells with glutaraldehyde and post-fix with osmium tetroxide. Embed in resin, section, and stain with uranyl acetate and lead citrate. Image cristae structure at high magnification (≥20,000X). Quantify cristae density, width, and junction size [85].
Metabolic Regulation of Cell Fate Decisions
Mitochondria regulate differentiation through metabolic reprogramming, shifting energy production pathways and generating metabolites that influence epigenetic landscapes and signaling cascades.
Bioenergetic Transitions in Differentiation
The transition from pluripotency to differentiated states involves a fundamental metabolic shift from glycolytic to oxidative metabolism. Pluripotent stem cells maintain limited mitochondrial oxidative capacity and rely predominantly on glycolysis, despite having functional mitochondria [82]. Upon differentiation commitment, cells undergo mitochondrial maturation characterized by increased mitochondrial mass, expanded cristae, enhanced membrane potential, and increased oxidative phosphorylation (OXPHOS) activity. This bioenergetic transition is not merely a consequence but a driver of differentiation, as inhibition of OXPHOS or mitochondrial biogenesis impairs differentiation across multiple lineages [81] [82].
Metabolites as Signaling Molecules
Table 2: Mitochondrial Metabolites Regulating Differentiation
| Metabolite | Metabolic Pathway | Signaling Role | Differentiation Context |
|---|---|---|---|
| Acetyl-CoA | TCA cycle, Pyruvate dehydrogenase | Substrate for histone acetylation; regulates epigenetic state | Modulates chromatin accessibility during lineage specification [82] |
| α-ketoglutarate | TCA cycle | Co-factor for dioxygenases (TET proteins, JmjC-domain histone demethylases) | Regulates DNA/histone methylation status; maintains pluripotency [82] |
| Reactive Oxygen Species (ROS) | Electron transport chain | Second messengers; modify redox-sensitive signaling proteins | Moderate levels promote differentiation; high levels induce senescence [83] [82] |
| Calcium | Mitochondrial calcium uniporter (MCU) | Regulates calcium-sensitive enzymes and transcription factors | Modulates NFAT signaling in T-cell differentiation [85] |
| ATP | OXPHOS | Energy currency; substrate for kinases; extracellular signaling via purinergic receptors | Alters energy sensors (AMPK); influences mechanical properties via cytoskeleton [85] |
Mitochondria-derived metabolites function as critical signaling molecules that influence cell fate. For instance, mitochondrial ROS at moderate concentrations activate signaling pathways such as HIF-1α and NRF2 that promote differentiation, while excessive ROS can trigger senescence or apoptosis [83] [82]. Similarly, calcium shuttling between mitochondria and ER regulates calcium-sensitive transcription factors like NFAT that control lineage-specific differentiation programs.
Mitochondrial Integration of Developmental Signaling Pathways
Mitochondria serve as signaling hubs that integrate and amplify developmental cues through direct interactions with core signaling pathways, creating feedback loops that reinforce differentiation commitments.
Molecular Interfaces with Signaling Cascades
Diagram 1: Mitochondrial Integration of Signaling Pathways in Cell Fate Determination
Mitochondria engage in bidirectional communication with major developmental signaling pathways:
Notch Signaling: Mitochondrial fusion proteins (MFN2) and fission regulators (DRP1) physically interact with Notch intracellular domain (NICD), influencing its stability and transcriptional activity. Conversely, Notch activation can modulate mitochondrial biogenesis through c-MYC regulation [82].
Wnt/β-catenin Signaling: Wnt activation stimulates DRP1-dependent mitochondrial fission, while mitochondrial ROS can enhance Wnt signaling through oxidation-sensitive disheveled proteins. β-catenin can localize to mitochondria and influence metabolism [82].
Hippo-YAP/TAZ Pathway: Mechanical cues transmitted via YAP/TAZ influence mitochondrial dynamics and metabolism. Reciprocally, mitochondrial ROS and metabolic status regulate LATS kinases, creating a feedback loop that influences nuclear YAP/TAZ localization [82].
These interactions create regulatory circuits that reinforce differentiation decisions, positioning mitochondria as signal integrators that consolidate intracellular and extracellular information to guide appropriate cell fate choices.
Experimental Approaches for Pathway Analysis
Protocol 2: Investigating Mitochondria-Signaling Pathway Interactions
Subcellular Fractionation and Western Analysis: Separate cytoplasmic, nuclear, and mitochondrial fractions using differential centrifugation. Validate purity with compartment-specific markers (TOM20 for mitochondria, Lamin A/C for nucleus). Probe for transcription factors (β-catenin, YAP, NICD) across fractions to assess localization changes during differentiation [82].
Proximity Ligation Assay (PLA) for Protein Interactions: Use primary antibodies against mitochondrial proteins (e.g., MFN2) and signaling components (e.g., β-catenin). Apply PLA probes and amplify fluorescent signals. Image using confocal microscopy and quantify puncta per cell to detect in situ protein interactions [82].
Metabolite Manipulation and Pathway Readouts: Treat differentiating cells with membrane-permeable metabolites (cell-permeable α-KG, dimethyl-succinate) or inhibitors of mitochondrial enzymes (rotenone, antimycin A). Assess signaling pathway activity via Western blot for phosphorylation states or qPCR for target genes [81].
CRISPR Interference for Pathway Components: Use dCas9-KRAB with sgRNAs targeting promoters of mitochondrial dynamics genes in stem cells. Induce differentiation and monitor effects on signaling pathway activation using reporter constructs and transcriptional assays [82].
Mitochondrial Control of Differentiation: Lineage-Specific Mechanisms
The regulatory principles of mitochondrial control of differentiation manifest in lineage-specific mechanisms across diverse tissues and developmental contexts.
Mitochondrial Regulation of Stem Cell Compartments
In hematopoietic stem cells (HSCs), mitochondrial fragmentation through DRP1 activation promotes quiescence maintenance, while inhibition of fission drives HSCs into cycle and depletes the stem cell pool [82]. Similarly, in neural stem cells (NSCs), a shift from glycolytic to oxidative metabolism coupled with mitochondrial fusion drives differentiation into neurons, while fission maintains the proliferative NSC state [82]. These patterns demonstrate how mitochondrial dynamics are tailored to specific stem cell niches.
Experimental Model Systems
Diagram 2: Mitochondrial Transitions During Cellular Differentiation
Several model systems have proven invaluable for investigating mitochondrial regulation of differentiation:
Dictyostelium discoideum: This social amoeba exhibits starvation-induced differentiation with well-defined transitions. Studies demonstrate that mitochondrial ribosomal protein S4 (mt-RPS4) is essential for initiation of differentiation, with partial disruption causing complete failure of aggregation and multicellular development [81].
Murine Embryonic Stem Cells (mESCs): Provide a manipulable system for studying lineage specification. Research shows that DRP1 deletion impairs cardiac mesoderm differentiation, while promoting neural ectoderm fate through altered ROS signaling and metabolic rewiring [82].
Human iPSC Differentiation Systems: Enable study of human-specific differentiation processes. Mitochondrial morphology transitions from fragmented in iPSCs to highly networked in differentiated lineages, with dynamics proteins regulating efficiency of specific lineage commitments [82].
Drosophila and C. elegans: Offer genetic tools for in vivo studies of development. In Drosophila, mitochondrial fusion is required for proper germline differentiation, while in C. elegans, mitochondrial dynamics influence lifespan and developmental timing [84] [86].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Investigating Mitochondrial Functions in Differentiation
| Reagent/Category | Specific Examples | Research Application | Technical Notes |
|---|---|---|---|
| Inhibitors of Dynamics | Mdivi-1 (DRP1 inhibitor), Dynasore (dynamin inhibitor), FCCP (uncoupler) | Acute perturbation of mitochondrial functions; testing necessity of dynamics | Confirm specificity with genetic approaches; monitor off-target effects [84] [85] |
| Metabolic Modulators | Rotenone (Complex I inhibitor), Oligomycin (ATP synthase inhibitor), DMOG (PHD inhibitor) | Dissecting metabolic requirements in differentiation; mimicking physiological transitions | Use dose-response curves; combine with rescue experiments [81] [82] |
| Fluorescent Reporters | MitoTimer, mt-cpYFP, Mito-roGFP, MitoTracker variants | Live imaging of mitochondrial properties (redox, pH, membrane potential) | Validate specificity with controls; consider phototoxicity in live imaging [85] |
| CRISPR Tools | sgRNAs for dynamics proteins (DRP1, MFN1/2, OPA1), metabolic enzymes | Genetic perturbation of specific mitochondrial components | Use inducible systems for temporal control; monitor compensatory mechanisms [82] |
| Antibodies | Anti-TOM20 (mitochondrial mass), Anti-COX IV (OXPHOS), Anti-H3K9ac (epigenetic) | Assessing mitochondrial content, function, and downstream effects | Validate for specific applications (WB, IF, EM); use compartment-specific loading controls [81] |
The emerging paradigm of mitochondrial regulation of cellular differentiation represents a significant expansion beyond their canonical apoptotic functions. Through integrated control of metabolism, signaling pathway modulation, and dynamic morphological remodeling, mitochondria establish a central regulatory hub that coordinates cell fate decisions. The molecular mechanisms governing these processes—from metabolite-mediated epigenetic modifications to physical interactions with developmental signaling cascades—provide new insights into fundamental biology with profound implications for regenerative medicine, cancer biology, and developmental disorders.
Future research directions should focus on elucidating the precise molecular interfaces between mitochondrial components and differentiation machinery, developing technologies for real-time monitoring of mitochondrial signaling in developing tissues, and exploiting these mechanisms for therapeutic applications in differentiation-related pathologies. As our understanding of these non-apoptotic mitochondrial functions deepens, we anticipate new opportunities for manipulating cell fate decisions in clinical contexts, from enhancing regenerative medicine approaches to targeting cancer stem cell populations.
Within the realm of cellular biology and immunology, supramolecular signaling complexes serve as critical platforms for the regulation of programmed cell death and inflammation. This whitepaper provides a comparative analysis of three such complexes—the apoptosome, the Death-Inducing Signaling Complex (DISC), and the inflammasome—with a particular focus on the mechanistic insights into apoptosome-mediated caspase-9 activation. Understanding the distinct assembly mechanisms, structural architectures, and functional outcomes of these complexes is paramount for developing targeted therapeutics for cancer, autoimmune disorders, and neurodegenerative diseases [9].
The core function of these complexes is the activation of specific caspases, which are cysteine proteases that act as master regulators of programmed cell death. The apoptosome activates caspase-9 to initiate intrinsic apoptosis, the DISC activates caspase-8 to initiate extrinsic apoptosis, and the inflammasome activates caspase-1 to drive pyroptosis and cytokine maturation [87] [88] [9]. Despite this common theme of caspase activation, the triggering stimuli, molecular components, and regulatory mechanisms differ substantially.
Complex-Specific Assembly and Activation Mechanisms
The Apoptosome: A Caspase-9 Activation Platform
The apoptosome is a central signaling platform in the intrinsic apoptotic pathway, which is activated in response to cellular stress signals such as DNA damage or oxidative stress. Its primary role is to dimerize and activate the initiator caspase, caspase-9.
- Activation Trigger: Intracellular stress signals, particularly mitochondrial outer membrane permeabilization (MOMP), lead to the release of cytochrome c into the cytosol [9].
- Core Components: The complex is assembled from cytochrome c, the apoptotic protease-activating factor 1 (Apaf-1), and the inactive procaspase-9 zymogen [9].
- Structural Assembly: Apaf-1 oligomerizes into a symmetric, wheel-like structure approximately 1.1 to 1.3 MDa in size in the presence of cytochrome c and dATP/ATP. This heptameric scaffold provides multiple binding sites for caspase-9 [34].
- Caspase Activation Mechanism: A pivotal 2024 study using methyl-TROSY NMR spectroscopy revealed that the protease domains (PDs) of caspase-9 molecules recruited to the apoptosome remain predominantly monomeric. The apoptosome organizes these caspase-9 PDs, priming them for rapid and extensive dimerization only upon binding to a peptide substrate. This indicates that the apoptosome does not directly induce caspase-9 dimerization but instead acts as an allosteric regulator that dramatically lowers the concentration threshold for its activation. The dissociation constant for caspase-9 dimerization in the absence of the apoptosome is remarkably weak, in the millimolar range [34]. Once activated, caspase-9 cleaves and activates the effector caspases-3 and -7, triggering the apoptotic cascade [9].
The Death-Inducing Signaling Complex (DISC): Extrinsic Apoptosis Initiator
The DISC is formed in response to extracellular death signals, such as Fas ligand (FasL), binding to their cognate death receptors on the cell surface.
- Activation Trigger: Engagement of death receptors of the tumor necrosis factor (TNF) receptor superfamily (e.g., Fas, TRAIL receptors) by their specific ligands [89] [90].
- Core Components: The complex comprises the trimerized death receptor, the adapter protein FADD (Fas-associated death domain), and the initiator procaspase-8 (or -10) [89] [90] [9].
- Structural Assembly: The elucidation of the Fas/FADD death domain complex structure revealed a tetrameric arrangement comprising four Fas DDs and four FADD DDs. A crucial step in DISC formation is the "opening" of the Fas death domain, which exposes a hydrophobic patch for FADD binding and simultaneously generates a Fas/Fas bridge. This opening acts as a mechanistic switch, preventing accidental activation yet allowing for highly processive DISC formation and clustering upon a sufficient stimulus [90]. Quantitative mass spectrometry analysis of the native TRAIL DISC showed a non-stoichiometric composition, with caspase-8 being present in up to 9-fold excess over FADD. This finding led to the proposal of a DED chain model, where procaspase-8 molecules interact sequentially via their death effector domains (DEDs) to form a caspase-activating filament or chain [89].
- Caspase Activation Mechanism: The DED chain facilitates the dimerization and autoproteolysis of caspase-8. Once active, caspase-8 can directly cleave and activate effector caspases-3 and -7. It also cleaves the protein BID to generate tBID, which amplifies the death signal by engaging the intrinsic apoptotic pathway via the mitochondria [9].
The Inflammasome: Inflammatory Caspase Activator
Inflammasomes are cytosolic multiprotein complexes that form in response to pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs), serving as a cornerstone of innate immunity.
- Activation Trigger: A wide array of stimuli, including bacterial components (e.g., flagellin, LPS), viral dsDNA, and host-derived crystals (e.g., uric acid) [91] [92] [87].
- Core Components: A typical inflammasome consists of a sensor protein (e.g., NLRP3, NLRC4, AIM2), an adapter protein ASC (apoptosis-associated speck-like protein containing a CARD), and the inflammatory procaspase-1 [91] [92].
- Structural Assembly: Inflammasomes assemble through a hierarchical mechanism. In the resting state, sensor molecules like NLRC4 are autoinhibited through intricate intramolecular interactions. Ligand binding to the sensor (e.g., NAIPs sensing bacterial ligands for NLRC4, or dsDNA binding to the HIN domain of AIM2) relieves this autoinhibition. The activated sensors then oligomerize, recruiting ASC via homotypic PYD-PYD interactions. ASC, in turn, nucleates the formation of filamentous structures through its CARD domain, recruiting multiple procaspase-1 molecules [92]. This nucleated polymerization mechanism allows for signal amplification and operates on an "all-or-none" cooperative principle [92].
- Caspase Activation Mechanism: The clustering of procaspase-1 on the ASC filament leads to its dimerization, autoprocessing, and activation. Active caspase-1 then proteolytically matures the pro-inflammatory cytokines IL-1β and IL-18. Concurrently, it cleaves gasdermin D (GSDMD), whose N-terminal fragment oligomerizes to form pores in the plasma membrane, leading to a lytic form of cell death called pyroptosis [91] [87].
Comparative Analysis
The following tables provide a direct comparison of the key features, quantitative data, and research tools associated with the apoptosome, DISC, and inflammasome.
Table 1: Core Characteristics and Functional Outcomes of the Three Signaling Complexes
| Feature | Apoptosome | DISC | Inflammasome |
|---|---|---|---|
| Primary Function | Initiation of intrinsic apoptosis | Initiation of extrinsic apoptosis | Inflammation and pyroptosis |
| Key Initiator Caspase | Caspase-9 | Caspase-8 | Caspase-1 (canonical) |
| Activation Trigger | Cytochrome c release (cellular stress) | Death receptor ligation (extracellular signal) | PAMPs/DAMPs (pathogen/ danger signals) |
| Core Scaffold Protein | Apaf-1 | FADD | ASC, NLRC4 |
| Molecular Size | ~1.1 - 1.3 MDa [34] | Not explicitly stated; forms large clusters | Micron-sized puncta [92] |
| Caspase Activation Mechanism | Apoptosome-organized monomer priming for substrate-induced dimerization [34] | DED-mediated procaspase-8 chain formation-induced dimerization [89] | CARD-mediated filament-induced dimerization [92] |
| Key Downstream Effectors | Caspases-3/7, DNA fragmentation | Caspases-3/7, tBID (links to intrinsic pathway) | Mature IL-1β, IL-18, GSDMD pores |
| Cell Death Fate | Apoptosis (non-inflammatory) | Apoptosis (non-inflammatory) | Pyroptosis (highly inflammatory) |
Table 2: Key Research Reagent Solutions for Experimental Study
| Reagent / Solution | Complex | Function in Experimentation |
|---|---|---|
| Methyl-TROSY NMR | Apoptosome | Enables high-resolution structural studies of very large (>1 MDa) complexes like the native apoptosome under near-physiological conditions [34]. |
| Engineered Apoptosome Mimic | Apoptosome | A 480-kDa engineered scaffold used to simplify structural studies while retaining native-like activation properties [34]. |
| Quantitative Mass Spectrometry | DISC | Used to determine the precise stoichiometry of native complexes, revealing the excess of caspase-8 over FADD [89]. |
| Reconstitution Model (DED Chain) | DISC | A system to functionally test the role of specific DED interactions in procaspase-8 activation and cell death induction [89]. |
| Cryo-Electron Microscopy (Cryo-EM) | Inflammasome | Visualizes the filamentous architecture of inflammasome components (e.g., ASC specks, AIM2 filaments) [92]. |
Experimental Protocols for Key Studies
Protocol 1: Studying Caspase-9 Activation using Methyl-TROSY NMR
This protocol is based on the recent study that provided mechanistic insight into caspase-9 activation on the apoptosome [34].
- Complex Formation: Recombinantly express and purify full-length caspase-9 and its isolated protease domain (PD). Form the native apoptosome complex (∼1.3 MDa) from cell lysates or assemble an engineered 480-kDa apoptosome mimic in vitro.
- Isotope Labeling: Employ bacterial expression systems to produce caspase-9 with specific isotopic labeling (e.g., (^{13})C-methyl labeling of isoleucine, leucine, and valine residues) for NMR sensitivity.
- NMR Sample Preparation: Incorporate the isotope-labeled caspase-9 into the pre-formed apoptosome complex. The complex is stabilized in a suitable NMR buffer.
- Data Acquisition: Acquire methyl-TROSY NMR spectra on a high-field NMR spectrometer. Compare spectra of:
- Free, monomeric caspase-9 PD.
- Caspase-9 PD within the apoptosome complex.
- Caspase-9 PD within the apoptosome in the presence of a peptide substrate.
- Biochemical Validation: Perform complementary biochemical assays, such as caspase activity assays, to correlate structural observations with enzymatic function.
- Data Analysis: Analyze chemical shift perturbations and line shapes to determine the oligomeric state (monomer vs. dimer) of caspase-9 in each condition.
Protocol 2: Elucidating DISC Stoichiometry and Assembly
This protocol outlines the approach used to develop the DED chain model for DISC assembly [89].
- Native Complex Isolation: Stimulate cells (e.g., lymphocytes) with a relevant death ligand (e.g., TRAIL) to induce native DISC formation. Use immunoprecipitation with an antibody against the death receptor (e.g., TRAIL-R) to isolate the intact DISC from cell lysates.
- Quantitative Mass Spectrometry: Subject the purified DISC proteins to digestion with trypsin. Analyze the resulting peptides using high-resolution mass spectrometry with a label-free quantitative (LFQ) approach. Use the intensity of the peptide signals to calculate the relative stoichiometry of all core DISC components (receptor, FADD, caspase-8).
- Structural Modeling: Use computational modeling to propose a three-dimensional structure of the DED chain based on known domain structures and the quantified stoichiometry.
- Functional Reconstitution: Introduce point mutations into key interacting residues of the procaspase-8 DED2 domain, as predicted by the model.
- Validation: Transfect the wild-type and mutant procaspase-8 constructs into a suitable cell line. Measure the ability of the mutant to undergo activation and induce cell death in response to DISC formation, compared to the wild-type control.
Visualizing Signaling Pathways and Complex Assembly
The following diagrams, generated using DOT language, illustrate the core signaling pathways and structural relationships of each complex.
Apoptosome-Mediated Caspase-9 Activation Pathway
DISC Assembly via the DED Chain Model
Hierarchical Inflammasome Assembly
Discussion and Future Perspectives
The comparative analysis underscores that while the apoptosome, DISC, and inflammasome are all caspase-activating platforms, they employ distinct strategies. The apoptosome functions as a regulatory scaffold that allosterically primes caspase-9 for activation, a mechanism recently clarified by advanced NMR techniques [34]. In contrast, the DISC and inflammasome rely on induced proximity and helical filament formation (DED or CARD chains) to drive caspase dimerization [89] [92].
From a therapeutic standpoint, these complexes present attractive drug targets. The precise understanding of the weak, millimolar caspase-9 dimerization and its regulation by the apoptosome opens avenues for developing small molecules that could either inhibit or promote this interaction in cancers where apoptosis is evaded [34]. Similarly, the hyperactive Fas mutant (I313D) that promotes Fas opening demonstrates that targeting the conformational state of death receptors could modulate DISC activity [90]. For inflammasomes, the detailed structural knowledge of sensor autoinhibition and ASC polymerization is driving the search for specific inhibitors for autoinflammatory diseases [91] [87].
Future research will continue to leverage structural biology techniques like cryo-EM and NMR, complemented by quantitative cell biology, to further unravel the dynamic regulation of these complexes in health and disease, paving the way for a new generation of highly specific therapeutics.
Conclusion
The apoptosome represents a sophisticated molecular machine whose activation mechanism, once hotly contested, is now understood as a substrate-primed, dimerization-driven process that integrates multiple regulatory inputs. The convergence of structural biology and biochemical evidence has largely resolved the historical debate, favoring a model where the apoptosome serves as a platform that organizes caspase-9 monomers for rapid, substrate-dependent dimerization rather than inducing direct allosteric activation. This refined understanding opens new therapeutic avenues for manipulating apoptotic thresholds in cancer, neurodegenerative disorders, and ischemic conditions. Future research must focus on developing isoform-specific modulators, understanding non-apoptotic functions in cellular physiology, and translating mechanistic insights into clinical applications that can selectively enhance or suppress apoptosome activity based on pathological context. The integration of apoptosome targeting with other cell death pathways presents particularly promising ground for combinatorial therapeutic strategies.
References
- 1. Regulation of the Apaf-1–caspase-9 apoptosome - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2939798/]
- 2. Apoptosome [https://en.wikipedia.org/wiki/Apoptosome]
- 3. Caspases as master regulators of programmed cell death [https://pmc.ncbi.nlm.nih.gov/articles/PMC12229594/]
- 4. The Apoptosome Activates Caspase-9 by Dimerization [https://www.sciencedirect.com/science/article/pii/S1097276506001730]
- 5. Atomic structure of the apoptosome: mechanism of cytochrome c [https://pmc.ncbi.nlm.nih.gov/articles/PMC4691890/]
- 6. 3JBT: Atomic structure of the Apaf-1 apoptosome [https://www.rcsb.org/structure/3JBT]
- 7. New insights into apoptosome structure and function - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6030056/]
- 8. Caspase-9: structure, mechanisms and clinical application [https://pmc.ncbi.nlm.nih.gov/articles/PMC5410359/]
- 9. Caspases as master regulators of programmed cell death [https://www.nature.com/articles/s12276-025-01470-9]
- 10. the cellular engine for the activation of caspase-9 [https://pubmed.ncbi.nlm.nih.gov/12005427/]
- 11. A Systems Biology Analysis of Apoptosome Formation and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4176236/]
- 12. A histidine switch regulates pH-dependent filament formation by the... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12157618/]
- 13. - Caspase - Wikipedia 9 [https://en.wikipedia.org/wiki/Caspase-9]
- 14. - Caspase : 9 , mechanisms and clinical application | Oncotarget structure [https://www.oncotarget.com/article/15098/text/]
- 15. Caspase-9 activates β-catenin signaling to promote ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-025-07020-1]
- 16. Apoptosis: A Comprehensive Overview of Signaling Pathways ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11592877/]
- 17. Mechanisms of cytochrome c release from mitochondria [https://www.nature.com/articles/4401950]
- 18. Formation of apoptosome is initiated by cytochrome c-induced ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1266161/]
- 19. structural and molecular mechanisms and functions in cell ... [https://www.nature.com/articles/s41421-025-00791-3]
- 20. Cytochrome c Promotes Caspase-9 Activation by Inducing ... [https://www.sciencedirect.com/science/article/pii/S0021925820893958]
- 21. Quantitative Analysis of Pathways Controlling Extrinsic ... [https://www.sciencedirect.com/science/article/pii/S1097276508001317]
- 22. Proteases for Cell Suicide: Functions and Regulation of Caspases ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC99015/]
- 23. (PDF) Caspase - 9 , Bcl-XL, and Apaf-1 Form a Ternary Complex [https://www.academia.edu/15496519/Caspase_9_Bcl_XL_and_Apaf_1_Form_a_Ternary_Complex]
- 24. The Caenorhabditis elegans CED- 9 protein does not directly inhibit the... [https://www.nature.com/articles/4401501?error=cookies_not_supported]
- 25. Apoptosome: a platform for the activation of initiator caspases [https://www.nature.com/articles/4402028]
- 26. Caspase-9: A Multimodal Therapeutic Target With Diverse ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2021.701301/full]
- 27. Caspase-9 Activation of Procaspase-3 but not ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8489496/]
- 28. Alternative splicing and cell survival: from tissue ... [https://www.nature.com/articles/cdd201691]
- 29. Apoptosis and autophagy: Regulation of caspase-9 by ... [https://pubmed.ncbi.nlm.nih.gov/19788417/]
- 30. Caspase-9 activates β-catenin signaling to promote ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12406446/]
- 31. Structural biology of the intrinsic cell death pathway [https://pmc.ncbi.nlm.nih.gov/articles/PMC3962096/]
- 32. The Apaf-1 apoptosome induces formation of caspase-9 ... [https://www.nature.com/articles/ncomms13565]
- 33. Caspase-9 and APAF-1 form an active holoenzyme [https://genesdev.cshlp.org/content/13/24/3179.short]
- 34. Activation of caspase-9 on the apoptosome as studied by ... [https://pubmed.ncbi.nlm.nih.gov/38085782/]
- 35. Activation of caspase-9 on the apoptosome as studied by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10743466/]
- 36. The Cellular Engine for the Activation of Caspase-9 [https://www.sciencedirect.com/science/article/pii/S0969212602007323]
- 37. A Comprehensive Exploration of Caspase Detection Methods [https://www.mdpi.com/1422-0067/25/10/5460]
- 38. Cell-to-cell variability in inducible Caspase9-mediated cell ... [https://www.nature.com/articles/s41419-021-04468-z]
- 39. Caspase-9 driven murine model of selective cell apoptosis ... [https://www.nature.com/articles/s41419-023-05594-6]
- 40. The Apoptosome: Emerging Insights and New Potential ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2279152/]
- 41. The Apoptosome: Physiological, Developmental, and ... [https://www.sciencedirect.com/science/article/pii/S1534580706001699]
- 42. Bio-mimetic strategies to re-activate apoptotic cell death for ... [https://www.explorationpub.com/Journals/eds/Article/100874]
- 43. Apaf-1 is an evolutionarily conserved DNA sensor that ... [https://www.nature.com/articles/s41421-024-00750-4]
- 44. Smac mimetic increases chemotherapy response and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2848888/]
- 45. Structure of an apoptosome-procaspase-9 CARD complex [https://pmc.ncbi.nlm.nih.gov/articles/PMC2874686/]
- 46. Novel Small Molecules Relieve Prothymosin α-Mediated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2852139/]
- 47. High-Content Screening (HCS) and High-Throughput ... [https://biobide.com/difference-between-hcs-and-hts]
- 48. High-Throughput Screening - Drug Discovery [https://www.technologynetworks.com/drug-discovery/articles/high-throughput-screening-principles-applications-and-advancements-405121]
- 49. Accessing the High Throughput Screening Data Landscape [https://pmc.ncbi.nlm.nih.gov/articles/PMC5613246/]
- 50. Proximity- or Conformation-Induced Caspase Activation? [https://pmc.ncbi.nlm.nih.gov/articles/PMC1088975/]
- 51. Molecule of the Month: Apoptosomes - PDB-101 [https://pdb101.rcsb.org/motm/177]
- 52. A degron-mimicking molecular glue drives CRBN homo-dimerization and degradation | Nature Communications [https://www.nature.com/articles/s41467-025-65094-3]
- 53. Dimerization and substrate recognition of human taurine transporter | Nature Communications [https://www.nature.com/articles/s41467-025-60967-z]
- 54. Caspase-9 swings both ways in the apoptosome - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5383358/]
- 55. Surface hydrophobics mediate functional dimerization of CYP121A1 of Mycobacterium tuberculosis | Scientific Reports [https://www.nature.com/articles/s41598-020-79545-y]
- 56. Allosteric Mechanisms Triggering Substrate and Cofactor ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12529757/]
- 57. Recent Advances in the Use of the Dimerization Strategy as a Means to Increase the Biological Potential of Natural or Synthetic Molecules [https://www.mdpi.com/1420-3049/26/8/2340]
- 58. Caspase-9: A Multimodal Therapeutic Target With Diverse ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8299054/]
- 59. The anti-apoptotic activity of XIAP is retained upon ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2173256/]
- 60. The apoptosome molecular timer synergises with XIAP to ... [https://www.nature.com/articles/s41418-020-0545-9]
- 61. Identification of an endogenous dominant-negative short ... [https://pubmed.ncbi.nlm.nih.gov/10070954/]
- 62. SRSF1 (SRp30a) regulates the alternative splicing of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3140550/]
- 63. SRp30a (ASF/SF2) regulates the alternative splicing of ... [https://www.sciencedirect.com/science/article/pii/S0022227520332375]
- 64. Regulation and Mechanistic Functions of Caspase-9 RNA ... [https://scholarscompass.vcu.edu/etd/3578/]
- 65. Targeting apoptotic pathways for cancer therapy - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11245162/]
- 66. Apoptosis defects and chemotherapy resistance: molecular ... [https://pubmed.ncbi.nlm.nih.gov/15077155/]
- 67. Targeting Apoptosis to Overcome Chemotherapy Resistance [https://www.ncbi.nlm.nih.gov/books/NBK580869/]
- 68. The BCL2 family: from apoptosis mechanisms to new ... [https://www.nature.com/articles/s41392-025-02176-0]
- 69. Anti-apoptotic PROTACs in Cancer Therapy: A Review [https://esmed.org/anti-apoptotic-protacs-in-cancer-therapy-a-review/]
- 70. Apoptosome - an overview | ScienceDirect Topics [https://www.sciencedirect.com/topics/neuroscience/apoptosome]
- 71. Apoptosome dysfunction in human cancer | Apoptosis [https://link.springer.com/article/10.1023/B:APPT.0000045786.98031.1d]
- 72. Apoptosome dysfunction in human cancer [https://pubmed.ncbi.nlm.nih.gov/15505412/]
- 73. Large transient assemblies of Apaf1 constitute the ... [https://www.nature.com/articles/s41467-025-64478-9]
- 74. Molecular mechanisms of cell death in neurological diseases [https://www.nature.com/articles/s41418-021-00814-y]
- 75. Mechanisms of Caspase-Independent Neuronal Death [https://pmc.ncbi.nlm.nih.gov/articles/PMC6741034/]
- 76. Cross-Talk in Cell Death Signaling - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2195865/]
- 77. Insights on the crosstalk among different cell death ... [https://www.nature.com/articles/s41420-025-02328-9]
- 78. BID is cleaved by caspase-8 within a native complex on the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3132005/]
- 79. Caught in the act between death receptors and mitochondria [https://www.sciencedirect.com/science/article/pii/S0167488911000346]
- 80. Caspase-8 Induces Lysosome-Associated Cell Death in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7132614/]
- 81. Control of Cell Differentiation by Mitochondria, Typically ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4030964/]
- 82. Mitochondrial dynamics coordinate cell differentiation [https://www.sciencedirect.com/science/article/abs/pii/S0006291X17312251]
- 83. Mitochondria as Regulators of Nonapoptotic Cell Death in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12284444/]
- 84. Mitochondrial dynamics and apoptosis - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC2732420/]
- 85. Mitochondrial dynamics in health and disease [https://www.nature.com/articles/s41392-023-01547-9]
- 86. Non-apoptotic functions of BCL-2 family proteins [https://www.nature.com/articles/cdd201722]
- 87. Mechanistic insights from inflammasome structures [https://www.nature.com/articles/s41577-024-00995-w]
- 88. Inflammatory caspases and inflammasomes [https://www.nature.com/articles/4402038]
- 89. A death effector domain chain DISC model reveals ... [https://pubmed.ncbi.nlm.nih.gov/22683266/]
- 90. The Fas/FADD death domain complex structure unravels ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2661029/]
- 91. Inflammasome: structure, biological functions, and therapeutic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10560975/]
- 92. The hierarchical structural architecture of inflammasomes ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4476925/]