TUNEL Assay for DNA Fragmentation: A Comprehensive Guide to Detecting Late-Stage Apoptosis
This article provides a comprehensive resource for researchers and drug development professionals on the TUNEL assay, a cornerstone technique for detecting DNA fragmentation during late-stage apoptosis.
TUNEL Assay for DNA Fragmentation: A Comprehensive Guide to Detecting Late-Stage Apoptosis
Abstract
This article provides a comprehensive resource for researchers and drug development professionals on the TUNEL assay, a cornerstone technique for detecting DNA fragmentation during late-stage apoptosis. It covers the foundational principles of apoptosis and the assay's biochemical basis, details step-by-step methodologies and advanced applications across various sample types, offers extensive troubleshooting guidance for common pitfalls, and presents a critical validation against alternative DNA damage assays. By synthesizing current research and protocol innovations, this guide aims to empower scientists to implement robust, reproducible, and insightful TUNEL assays in their experimental workflows.
The Biology of Apoptosis and the Principle of TUNEL Staining
Apoptosis, or programmed cell death, is a fundamental biological process essential for the normal development and maintenance of multicellular organisms [1]. This regulated cellular suicide mechanism eliminates potentially harmful, damaged, or unnecessary cells through a controlled process characterized by distinct morphological changes including cell shrinkage, membrane blebbing, and nuclear fragmentation [2] [3]. Unlike necrosis, which results from external damage and triggers inflammatory responses, apoptosis is an actively executed process that is a regular component of cellular metabolism [1].
A key hallmark of the late stages of apoptosis is the systematic fragmentation of nuclear DNA by endonucleases [4]. The TUNEL (Terminal deoxynucleotidyl transferase dUTP Nick End Labeling) assay, first described in 1992, has become a gold-standard technique for detecting this DNA fragmentation in situ [4] [5] [1]. This method provides researchers with a powerful tool to visualize and quantify apoptotic cells within tissue sections or cultured cell samples, making it invaluable for research in developmental biology, oncology, neuroscience, and toxicology [4] [5].
Core Principles of the TUNEL Assay
Biochemical Basis
During the execution phase of apoptosis, endogenous endonucleases—such as Caspase-Activated DNase (CAD)—cleave the cell's genomic DNA between nucleosomes, generating millions of DNA fragments with exposed 3'-hydroxyl (3'-OH) ends [4]. The TUNEL assay capitalizes on this biochemical signature by utilizing the enzyme Terminal deoxynucleotidyl transferase (TdT), a unique DNA polymerase that catalyzes the template-independent addition of labeled deoxynucleotides (dUTPs) to these 3'-OH termini [4] [6] [7].
The TdT enzyme sequentially adds labeled nucleotides to the free 3'-OH ends, creating a polymer that can be detected through various methods [4]. The extensive DNA fragmentation that occurs during apoptosis results in a high density of these labeled nucleotides incorporated at the damage sites, generating a strong signal that distinguishes apoptotic cells from healthy counterparts [7].
Detection Methodologies
The TUNEL assay offers flexibility in detection strategies to accommodate different experimental needs and platforms:
Fluorescent Detection: This widely used approach employs directly fluorescent-dUTP (e.g., FITC-dUTP) or indirect labeling methods using hapten-labeled dUTP (e.g., Br-dUTP, EdUTP) followed by detection with a fluorescent antibody or click chemistry reaction [4] [8]. Fluorescent TUNEL signals are typically visualized by fluorescence microscopy, confocal microscopy, or quantified via flow cytometry [4] [3].
Colorimetric Detection: For bright-field microscopy applications, biotin-labeled dUTP can be incorporated, followed by sequential incubation with streptavidin-horseradish peroxidase (HRP) and a chromogenic substrate like 3,3'-diaminobenzidine (DAB), which produces a dark brown precipitate at the site of DNA fragmentation [4] [8].
Table 1: Comparison of TUNEL Assay Detection Methodologies
| Detection Method | dUTP Label | Detection Reagent | Readout | Applications |
|---|---|---|---|---|
| Direct Fluorescence | Fluorescein-dUTP | None | Green fluorescence (495/519 nm) | Fluorescence microscopy, flow cytometry |
| Indirect Fluorescence | BrdUTP | Anti-BrdU-Alexa Fluor antibody | Fluorophore-dependent | High-sensitivity imaging |
| Click Chemistry | EdUTP | Fluorescent azide | Fluorophore-dependent | Multiplexing with fluorescent proteins |
| Colorimetric | Biotin-dUTP | Streptavidin-HRP + DAB | Brown precipitate | Bright-field microscopy, histology |
Comprehensive TUNEL Assay Protocol
Sample Preparation
Proper sample preparation is critical for successful TUNEL staining and accurate results:
Cell Culture Samples: Wash adherent cells with phosphate-buffered saline (PBS) and fix with 1-4% paraformaldehyde (PFA) in PBS for 15-30 minutes at room temperature [4]. Over-fixation should be avoided as it can cross-link DNA ends and reduce enzyme accessibility.
Tissue Sections: For formalin-fixed, paraffin-embedded (FFPE) tissues, deparaffinize sections and rehydrate through a graded ethanol series [4]. Antigen retrieval using citrate buffer steam treatment may improve signal [4]. Frozen tissue sections should be fixed with 4% PFA for 15-30 minutes [4].
Plant Tissues: Plant materials present unique challenges due to cellulosic cell walls and phenolic compounds that can inhibit TdT activity [9]. An optimized protocol includes extended fixation and specialized permeabilization using citric acid buffer [9].
Permeabilization and Controls
Permeabilization: The large size of the TdT enzyme (∼60 kDa) necessitates effective permeabilization to allow nuclear access. For cultured cells, incubate with 0.1-0.5% Triton X-100 in PBS for 5-15 minutes on ice [4]. Tissue sections often require harsher permeabilization using 20 μg/mL Proteinase K for 10-20 minutes at room temperature [4].
Essential Controls:
- Positive Control: Treat a sample with DNase I (1 μg/mL for 15-30 minutes) before the labeling step to artificially create DNA breaks; all nuclei should stain positive [4] [7].
- Negative Control: Omit the TdT enzyme from the reaction mix; this sample should show no specific signal and reveals non-specific background [4] [7].
Labeling Reaction and Detection
The following workflow diagram illustrates the key steps in the TUNEL assay procedure:
Equilibration: Incubate samples with equilibration buffer for 10 minutes to prepare the DNA for enzymatic labeling [4].
TdT Reaction: Prepare the TdT reaction mix according to kit specifications (typically containing TdT enzyme, labeled dUTP, and reaction buffer). Apply to samples and incubate for 60 minutes at 37°C in a humidified chamber to prevent evaporation [4].
Reaction Termination: Stop the enzymatic reaction by incubating with stop/wash buffer for 10 minutes, followed by 2-3 PBS rinses [4].
Signal Detection:
Counterstaining and Mounting: Incubate with a nuclear counterstain (DAPI for fluorescence, Methyl Green/Eosin for colorimetric) to visualize all nuclei [4]. Mount coverslips with appropriate antifade mounting medium [4].
Research Reagent Solutions
Table 2: Essential Reagents for TUNEL Assay Implementation
| Reagent Category | Specific Examples | Function | Optimization Notes |
|---|---|---|---|
| Fixatives | 1-4% Paraformaldehyde (PFA) | Preserves cellular architecture and cross-links fragmented DNA | Over-fixation can mask DNA ends; 15-30 min optimal |
| Permeabilization Agents | 0.1-0.5% Triton X-100, 20 μg/mL Proteinase K | Enables TdT enzyme access to nuclear DNA | Concentration and time must be optimized for each sample type |
| Enzymes | Terminal Deoxynucleotidyl Transferase (TdT) | Catalyzes addition of labeled dUTPs to 3'-OH DNA ends | Recombinant TdT ensures consistent activity |
| Labeled Nucleotides | BrdUTP, FITC-dUTP, EdUTP, Biotin-dUTP | Provides detectable tag at DNA break sites | EdUTP enables flexible click chemistry detection |
| Detection Reagents | Anti-BrdU antibodies, Fluorescent azides, Streptavidin-HRP | Visualizes incorporated nucleotides | Antibody-based methods offer signal amplification |
| Buffers | Equilibration buffer, Reaction buffer, SSC stop solution | Maintains optimal enzymatic activity and terminates reactions | Cacodylate-free buffers reduce toxicity [1] |
Critical Optimization and Troubleshooting
Addressing Specificity Challenges
The TUNEL assay, while powerful, is notoriously prone to artifacts that must be carefully controlled:
False Positives: TdT will label any free 3'-OH DNA ends, not just those generated during apoptosis. False positives can arise from necrotic cell death, DNA repair processes, autolysis, or harsh sample treatment [4] [5]. Over-fixation or over-permeabilization can artificially create DNA breaks or increase non-specific background [4].
False Negatives: Insufficient permeabilization may prevent TdT from accessing nuclear DNA, while excessive cross-linking from over-fixation can block the 3'-OH ends, both resulting in reduced signal [4].
The Anastasis Consideration: Recent research indicates that cells can be TUNEL-positive and still recover from the apoptotic process through a phenomenon called "anastasis," meaning a positive signal does not always equate to irreversible cell death [4].
Methodological Validation
To ensure accurate interpretation of TUNEL results, researchers should:
- Always include appropriate positive and negative controls [4] [7]
- Combine TUNEL with complementary apoptosis markers such as cleaved caspase-3 immunostaining (for early apoptosis) or Annexin V staining (for membrane changes) [4]
- Correlate TUNEL positivity with morphological assessment of apoptotic features (nuclear condensation, cell shrinkage) [2]
Advanced Applications and Methodological Integration
Versatile Research Applications
The TUNEL assay has been successfully implemented across diverse research contexts:
Kidney Injury Assessment: The high activity of DNase I in kidney tissue makes TUNEL particularly valuable for evaluating toxic or hypoxic injury in this organ [5]. The assay has detected cell death in various kidney injury models including ischemia-reperfusion, toxic compound exposure, and progressive renal diseases [5].
Plant Programmed Cell Death: Optimized TUNEL protocols enable the study of DNA fragmentation in plant root cells subjected to various stress conditions, despite technical challenges posed by cell walls and phenolic compounds [9].
Cancer Research and Drug Development: TUNEL assays are widely used to evaluate the efficacy of chemotherapeutic agents by quantifying apoptosis induction in tumor cells [2] [10].
Neurodegenerative Disease Research: The assay helps characterize neuronal cell death in models of Alzheimer's disease, Parkinson's disease, and other neurological disorders [10].
Comparative Analysis with Other Apoptosis Detection Methods
Table 3: Comparison of TUNEL with Other Apoptosis Detection Methods
| Method | Target | Sensitivity | Specificity | Advantages | Limitations |
|---|---|---|---|---|---|
| TUNEL Assay | DNA fragmentation (3'-OH ends) | High | Moderate to High | In situ detection, Broad applicability, Quantitative potential | Not apoptosis-specific, Requires careful controls |
| DNA Laddering | Oligonucleosomal DNA fragments | Moderate | High for apoptosis | Characteristic apoptotic pattern, Semi-quantitative | Requires many cells, No spatial information, Late apoptosis only |
| Annexin V Staining | Phosphatidylserine externalization | High | High for early apoptosis | Early detection, Live cell capability | Requires intact membranes, Cannot use with fixed tissues |
| Caspase Activity Assays | Caspase enzyme activity | High | High | Early apoptosis detection, Mechanistic insight | Does not confirm cell death commitment |
The TUNEL assay remains an indispensable technique in the cell death researcher's toolkit, providing sensitive in situ detection of DNA fragmentation—a hallmark of late-stage apoptosis. When properly optimized and validated with appropriate controls, this method generates reliable, publication-quality data across diverse biological systems from mammalian tissues to plant specimens. As research into programmed cell death continues to evolve, with growing recognition of non-apoptotic cell death pathways and the discovery of reversal mechanisms like anastasis, the TUNEL assay maintains its relevance through adaptability to new technological developments such as click chemistry detection and compatibility with multiplexed analytical approaches. By understanding both the power and limitations of this technique, researchers can effectively employ the TUNEL assay to advance our comprehension of programmed cell death in development, homeostasis, and disease pathogenesis.
DNA Fragmentation: The Biochemical Hallmark of Late Apoptosis is a definitive event in the programmed cell death cascade, representing the point of no return for a cell destined to die. This process is characterized by the systematic cleavage of nuclear DNA into oligonucleosomal fragments, typically in multiples of 180-200 base pairs, producing a characteristic "DNA ladder" pattern when separated by gel electrophoresis [2]. The execution of this biochemical signature is primarily mediated by the activation of specific endonucleases, most notably the Caspase-Activated DNase (CAD), which is activated upon cleavage by caspases during apoptosis [11].
The detection of this specific DNA fragmentation pattern has become a cornerstone methodology in cell biology research, particularly for distinguishing apoptotic cell death from other forms of cell death such as necrosis. While necrosis typically displays a random DNA fragmentation pattern appearing as a "smear" on gels, the organized, internucleosomal cleavage of apoptosis creates a distinctive laddering pattern that serves as a biochemical fingerprint [2]. Among the various techniques developed to detect this phenomenon, the TUNEL (Terminal deoxynucleotidyl transferase dUTP Nick-End Labeling) assay has emerged as the gold standard for in situ detection, allowing researchers to identify apoptotic cells within tissue sections and cell cultures while preserving spatial context [11].
This application note provides a comprehensive technical resource for researchers investigating apoptotic processes, detailing the underlying principles of DNA fragmentation, presenting optimized protocols for its detection, summarizing current methodological approaches in an accessible format, and highlighting recent technical advancements that enhance the utility of apoptosis detection in complex research scenarios.
Biochemical Principles of Apoptotic DNA Fragmentation
The systematic degradation of nuclear DNA during apoptosis results from the activation of an evolutionarily conserved biochemical pathway specifically designed to dismantle the cellular genome. Understanding this process requires examining the key enzymes, molecular triggers, and sequential events that characterize this form of programmed cell death.
The Apoptotic Pathway and DNA Cleavage
The cleavage of DNA during apoptosis occurs as part of the execution phase of programmed cell death, typically initiated after the commitment to apoptosis has been made through either the intrinsic (mitochondrial) or extrinsic (death receptor) pathways. Both pathways converge on the activation of caspase enzymes, which serve as the primary molecular executioners of apoptosis [2]. Of particular importance is the activation of caspase-3, which subsequently cleaves and activates the inhibitor of CAD (ICAD), thereby releasing active CAD to enter the nucleus and initiate DNA fragmentation [11].
CAD specifically targets the linker regions between nucleosomes, the fundamental repeating units of chromatin structure. Each nucleosome consists of approximately 146 base pairs of DNA wrapped around a histone core, with linker DNA spanning between these structures. The endonucleolytic cleavage at these linker regions results in DNA fragments whose sizes are multiples of the nucleosome unit, creating the characteristic ladder pattern observed in agarose gel electrophoresis [2]. This systematic fragmentation contrasts sharply with the random DNA degradation observed in necrotic cell death, where uncontrolled enzyme release and activity produce a continuous smear of DNA fragments without discrete banding patterns.
Molecular Mechanism of the TUNEL Assay
The TUNEL assay capitalizes on the biochemical signature created by CAD activity. During apoptosis, the endonucleolytic cleavage generates countless DNA fragments with exposed 3'-hydroxyl (3'-OH) termini [11]. The TUNEL assay utilizes the enzyme terminal deoxynucleotidyl transferase (TdT), a unique DNA polymerase that catalyzes the template-independent addition of deoxynucleotides to these 3'-OH ends [11] [12].
TdT incorporates labeled deoxynucleotides (most commonly modified dUTPs) to the free 3'-OH ends of fragmented DNA. These labels can include:
- Directly fluorescent tags (e.g., FITC-dUTP, CF-Dye 488-dUTP)
- Haptens for indirect detection (e.g., biotin-dUTP, Br-dUTP, EdUTP)
- Alkyne groups for subsequent click chemistry conjugation [11] [13] [12]
The incorporated labels are then visualized through various detection methods appropriate to the specific application, including fluorescence microscopy, flow cytometry, or bright-field microscopy for colorimetric detection [11] [12].
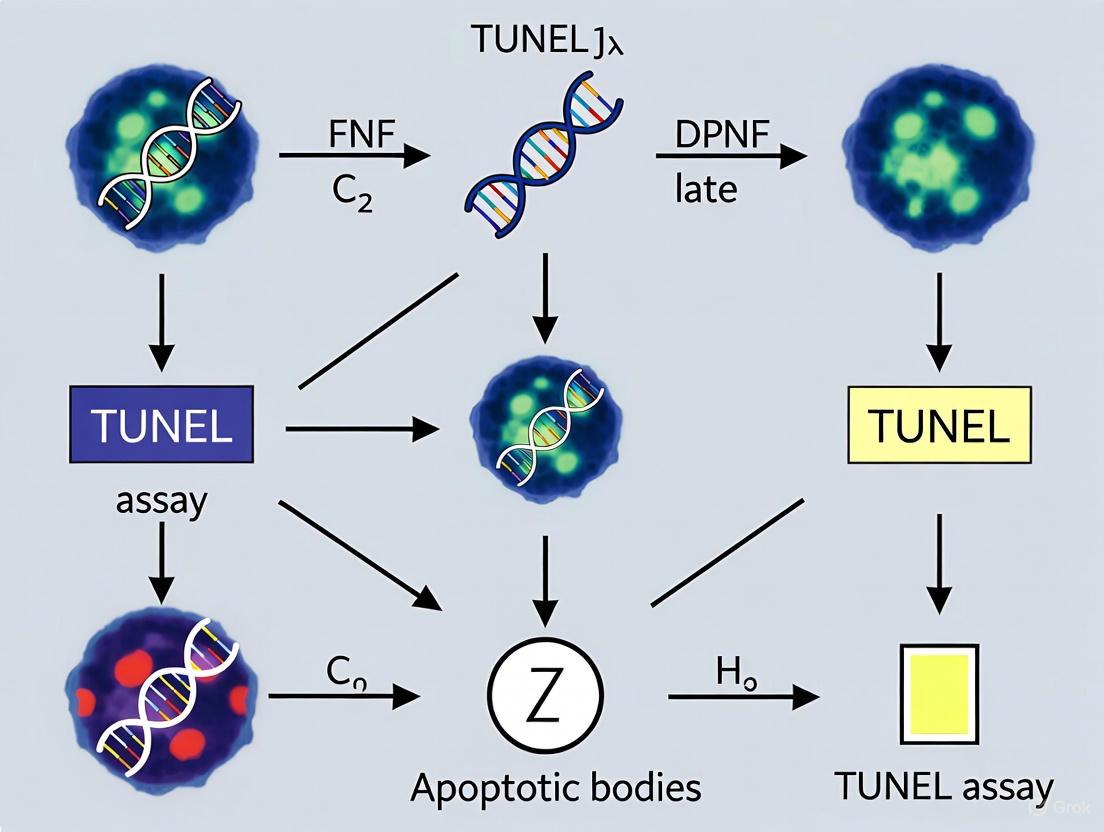
Figure 1: Biochemical Pathway of DNA Fragmentation and TUNEL Detection. This diagram illustrates the sequential process from apoptotic stimulus to DNA fragmentation and subsequent detection via the TUNEL assay. The yellow boxes represent key steps in the apoptotic pathway, blue boxes show the TUNEL labeling mechanism, and the green box indicates the final detection step.
Comprehensive TUNEL Assay Protocols
The successful implementation of the TUNEL assay requires careful attention to sample preparation, labeling conditions, and detection methodologies. Below, we present detailed protocols optimized for different sample types and research applications.
Standard TUNEL Protocol for Cultured Cells and Tissue Sections
This generalized protocol synthesizes best practices from multiple technical sources and commercial kits, providing a robust foundation for detecting DNA fragmentation in most sample types [11] [13].
Materials Required
- Fixative: 4% Paraformaldehyde (PFA) in PBS
- Permeabilization Solution: 0.1-0.5% Triton X-100 in PBS or 20 µg/mL Proteinase K
- TdT Reaction Buffer (commercial kits)
- Terminal deoxynucleotidyl transferase (TdT) enzyme
- Labeled dUTP (e.g., FITC-dUTP, Br-dUTP, EdUTP)
- Stop/Wash Buffer: Saline-sodium citrate (SSC) buffer
- Detection Reagents (if using indirect method): Anti-BrdU antibody, Streptavidin-HRP, or Click-iT reaction cocktail
- Counterstains: DAPI (fluorescence) or Methyl Green (colorimetric)
- Mounting Medium: Antifade mounting medium
Step-by-Step Procedure
Sample Preparation and Fixation
- Adherent Cells: Wash cells with PBS, then fix with 4% PFA for 15-30 minutes at room temperature [11].
- Tissue Sections (FFPE): Deparaffinize and rehydrate through graded ethanol series. Perform antigen retrieval using steam heating with citrate buffer [11].
- Frozen Tissue Sections: Fix with 4% PFA for 15-30 minutes at room temperature [11].
Permeabilization
- Cultured Cells: Incubate with 0.1-0.5% Triton X-100 in PBS for 5-15 minutes on ice [11].
- Tissue Sections: Treat with 20 µg/mL Proteinase K for 10-20 minutes at room temperature or 0.5-1% Triton X-100 [11].
- Critical Note: Permeabilization must be optimized for each cell type. Under-permeabilization prevents TdT access to nuclear DNA, while over-permeabilization can damage nuclear integrity.
Experimental Controls
TdT Labeling Reaction
- Equilibrate samples with TdT reaction buffer for 10 minutes [11].
- Prepare TdT Reaction Mix containing TdT enzyme and labeled dUTP in reaction buffer.
- Remove equilibration buffer and apply TdT Reaction Mix to samples.
- Incubate for 60 minutes at 37°C in a humidified chamber to prevent evaporation [11].
Reaction Termination and Detection
- Stop the reaction with Stop/Wash Buffer for 10 minutes [11].
- Rinse samples 2-3 times with PBS.
- For direct detection (fluorescently-tagged dUTP): Proceed to counterstaining.
- For indirect detection:
Counterstaining and Mounting
- Incubate with nuclear counterstain (DAPI for fluorescence) for 5-10 minutes [11].
- Perform final PBS rinse.
- Mount coverslips with antifade mounting medium.
Analysis and Interpretation
Advanced Protocol: TUNEL Integration with Multiplexed Immunofluorescence
Recent advancements have enabled the harmonization of TUNEL with spatial proteomic methods, allowing rich contextualization of cell death within tissue microenvironments. This protocol modification replaces proteinase K with pressure cooker antigen retrieval to preserve protein antigenicity for multiplexed imaging [15] [16].
Key Modifications for Multiplex Compatibility
Antigen Retrieval
- Replace Proteinase K treatment with pressure cooker-mediated antigen retrieval using citrate buffer [16].
- This preservation of protein epitopes enables subsequent iterative antibody staining.
TUNEL Staining Integration
- Perform TUNEL assay as described above following pressure cooker retrieval.
- For antibody-based TUNEL detection systems, apply TUNEL in the first staining cycle [16].
Erasure and Iterative Staining (MILAN Protocol)
- After imaging, erase antibodies by incubating in 2-mercaptoethanol/SDS (2-ME/SDS) at 66°C [16].
- Confirm TUNEL signal erasure by re-imaging.
- Proceed with subsequent rounds of antibody staining for spatial proteomics.
Validation
Figure 2: Workflow for TUNEL Integration with Multiplexed Iterative Immunofluorescence. This diagram illustrates the advanced protocol that enables TUNEL detection followed by multiple rounds of antibody staining for spatial proteomics, preserving precious tissue samples while generating rich multidimensional data.
Research Reagent Solutions
Selecting appropriate detection methodologies and reagents is critical for successful TUNEL assay implementation. The table below summarizes the primary detection approaches with their respective advantages and applications.
Table 1: Comparison of TUNEL Detection Methodologies
| Detection Method | Principle | Advantages | Limitations | Popularity* | Best Applications |
|---|---|---|---|---|---|
| Direct Fluorescence (e.g., FITC-dUTP) [12] | Fluorescently-tagged dUTP directly incorporated | Fast protocol (fewer steps); Reduced background | Potentially lower signal intensity | 50% | Routine apoptosis detection; Flow cytometry |
| Biotin-Streptavidin [12] | Biotin-dUTP + Streptavidin-HRP + chromogen | Signal amplification; Compatible with bright-field microscopy | Endogenous biotin may cause background; Additional blocking needed | 15% | Histology sections; Colorimetric detection |
| Antibody-Based (e.g., BrdUTP) [11] [12] | BrdUTP + Anti-BrdU antibody conjugated to fluorophore | Bright signal; BrdU easily incorporated by TdT | More staining steps; Potential antibody variability | 8% | Sensitive detection; Low-copy DNA fragmentation |
| Click Chemistry (e.g., EdUTP) [13] | EdUTP + Copper-catalyzed azide-alkyne cycloaddition | Small label size improves penetration; Efficient incorporation | Copper catalyst may affect some fluorophores | Emerging | Multiplexed assays; Difficult-to-penetrate tissues |
Based on survey of 50 research papers published in 2017 containing "TUNEL Assay" or "TUNEL Staining" [12].
Methodological Considerations and Troubleshooting
Despite its widespread use, the TUNEL assay presents several technical challenges that researchers must address to ensure accurate interpretation of results.
Specificity Challenges and Optimization
The TUNEL assay's principal limitation is its potential lack of absolute specificity for apoptosis. The TdT enzyme will label any DNA fragment with exposed 3'-OH ends, regardless of origin [11]. This necessitates careful experimental design and interpretation to avoid false positives.
Common Sources of False Positives:
- Necrotic Cell Death: Random DNA degradation during necrosis generates 3'-OH ends detectable by TUNEL [11].
- DNA Repair Intermediates: Cells actively repairing DNA damage may incorporate labeled nucleotides [11].
- Sample Processing Artifacts: Over-fixation or harsh permeabilization can artificially create DNA breaks [11].
- The Anastasis Phenomenon: Cells can be TUNEL-positive yet recover from the apoptotic process, demonstrating that DNA fragmentation is not always terminal [11].
Strategies for Specificity Enhancement:
- Morphological Correlation: Always correlate TUNEL staining with nuclear morphology (chromatin condensation, nuclear fragmentation) [11].
- Multiparametric Analysis: Combine TUNEL with other apoptosis markers:
- Appropriate Controls: Always include both positive (DNase-treated) and negative (no TdT) controls in every experiment [11].
Technical Troubleshooting Guide
Table 2: TUNEL Assay Troubleshooting Guide
| Problem | Potential Causes | Solutions |
|---|---|---|
| Weak or No Signal | Under-permeabilization; Over-fixation; Inadequate enzyme activity; Impromed reaction conditions | Optimize permeabilization duration/concentration; Reduce fixation time; Verify enzyme activity with positive control; Ensure proper pH and cation concentration in reaction buffer |
| High Background | Over-permeabilization; Non-specific antibody binding; Endogenous enzyme activity; Inadequate blocking | Titrate permeabilization reagent; Include appropriate blocking steps; Use antibody dilutions with carrier proteins; Quench endogenous peroxidases (HRP detection) |
| Inconsistent Staining | Uneven reagent application; Sample drying; Variable fixation | Use humidified chamber during incubations; Ensure complete coverage of samples; Standardize fixation protocols across samples |
| Positive Control Failure | DNase I degradation; Improper DNase application; Compromised reagents | Aliquot and store DNase properly; Verify DNase concentration and incubation time; Use fresh reagents |
| Tissue Detachment | Excessive washing; Over-permeabilization; Adhesive coating issues | Use gentle washing techniques; Optimize permeabilization; Use charged or coated slides for tissue sections |
Applications and Advanced Techniques
The utility of DNA fragmentation detection extends across multiple research domains, with continuous methodological advancements expanding its applications.
Research Applications
Male Infertility Assessment
- Sperm DNA fragmentation (SDF) measured by TUNEL is a crucial parameter in male fertility evaluation [14].
- Flow cytometric TUNEL analysis provides standardized assessment of sperm DNA integrity, correlating with fertilization potential and embryonic development [14].
Cancer Research and Therapeutic Development
- Evaluation of chemotherapeutic efficacy through apoptosis induction in tumor cells [2].
- Assessment of treatment response and resistance mechanisms [2].
Neurodegenerative Disease Research
- Detection of apoptotic cells in neurological tissues to quantify disease-associated cell loss [17].
- Investigation of gliotoxic factors in multiple sclerosis through astrocyte apoptosis models [17].
Developmental Biology
- Mapping programmed cell death during embryonic development [11].
- Studying tissue remodeling and homeostasis maintenance [11].
Emerging Technologies and Innovations
Artificial Intelligence-Enhanced Analysis
- Recent developments apply AI tools for automated TUNEL signal quantification [18].
- Phase contrast microscopy combined with ensemble AI models can predict DNA fragmentation, enabling non-destructive sperm selection for assisted reproduction [18].
Spatial Proteomics Integration
- The harmonization of TUNEL with multiplexed iterative immunofluorescence (MILAN, CycIF) enables comprehensive cellular phenotyping of dying cells within tissue architecture [15] [16].
- Replacement of proteinase K with pressure cooker retrieval preserves protein antigenicity while maintaining TUNEL sensitivity [16].
Advanced Detection Chemistries
- Click-iT TUNEL assays utilizing EdUTP and copper-catalyzed azide-alkyne cycloaddition offer improved penetration and labeling efficiency compared to traditional methods [13].
- These advanced chemistries enable more rapid protocols (completed within 2 hours) with enhanced sensitivity [13].
DNA fragmentation remains a definitive biochemical hallmark of late apoptosis, and its detection via the TUNEL assay continues to be an indispensable methodology in cell death research. While the fundamental principles of the assay remain consistent, ongoing technical advancements have substantially expanded its applications and improved its reliability. The development of multiplex-compatible protocols, AI-enhanced analysis platforms, and novel detection chemistries represents significant progress in the field.
Successful implementation of DNA fragmentation analysis requires careful consideration of methodological options, appropriate control strategies, and awareness of technical limitations. The protocols and guidelines presented in this application note provide researchers with a comprehensive resource for designing, executing, and interpreting experiments focused on apoptotic DNA fragmentation. As research continues to reveal the complexities of cell death mechanisms, the TUNEL assay remains a cornerstone technique for investigating apoptotic processes in health and disease.
The understanding of apoptosis, or programmed cell death, represents a fascinating journey in biological sciences, evolving from initial morphological observations to a sophisticated field of molecular research. This conceptual evolution is crucial for contemporary applications, such as the TUNEL assay, which provides a direct method for detecting DNA fragmentation during late apoptosis. The journey began with Carl Vogt's foundational observations in 1842 while studying the metamorphosis of tadpoles of the common midwife toad, where he documented the phenomenon of cells self-destructing during development [1] [19]. However, this discovery lay dormant for more than a century, awaiting the tools and scientific context for its full appreciation. The field remained largely unexplored until the mid-20th century when biologist Alfred Glücksmann revived the concept in his 1951 paper "Cell Deaths in Normal Vertebrate Ontogeny," which described apoptosis as it occurs during embryonic development [20].
The term "apoptosis" (from an ancient Greek word meaning "falling off") was formally coined in a landmark 1972 paper by John F.R. Kerr, Andrew Wyllie, and Alastair R. Currie [20] [1] [19]. This paper refined the concept of programmed cell death by asserting that cell death occurring during embryonic development was the same process as cellular suicide triggered by hormones or toxins [20]. The study of apoptosis increased dramatically after the 1988 discovery of Bcl-2, a protein that plays a key role in regulating programmed cell death, leading to the definitive conclusion in 1992 that apoptosis and programmed cell death were identical processes [20]. This historical progression from observation to mechanistic understanding laid the essential groundwork for developing sophisticated detection methods like the TUNEL assay, which now plays a vital role in both basic research and drug development.
The Birth of a Discovery: Key Historical Milestones
The conceptualization of apoptosis required scientists to first recognize and describe the phenomenon of programmed cell death, then develop the terminology to discuss it systematically, and finally create the tools to study it experimentally. The following timeline visualizes the key historical milestones in the understanding of apoptosis before the development of modern detection methods:
Distinguishing Apoptosis from Necrosis
A critical advancement in apoptosis research was the clear distinction between apoptosis and necrosis, another form of cell death. While both processes result in cell death, their mechanisms and consequences differ fundamentally. Apoptosis is an orderly, naturally occurring, and genetically controlled process that typically occurs in single cells without causing inflammation or damage to surrounding tissues [20]. In contrast, necrosis results from external factors like bodily injury, poisoning, or disrupted blood supply, leading to uncontrolled cell death where cells swell and burst, causing inflammation and potential damage to neighboring cells [20].
This distinction is particularly relevant for the TUNEL assay, as it specifically detects the organized DNA fragmentation characteristic of apoptosis rather than the random DNA degradation found in necrosis. During apoptosis, cells undergo a series of orchestrated steps including cell shrinkage, DNA fragmentation, and the formation of membrane-bound apoptotic bodies that are efficiently cleared by other cells [20]. The biochemical hallmark of this process is internucleosomal DNA cleavage, generating characteristic fragments of approximately 180-200 base pairs in length [1], which serves as the fundamental basis for TUNEL assay detection.
Modern Applications: The TUNEL Assay in Contemporary Research
The historical understanding of apoptosis has directly enabled the development of powerful research tools like the TUNEL assay, which now plays a crucial role across diverse fields of biomedical research. This assay has become the most widely used in situ test for apoptosis study since its introduction in 1992 [13], capitalizing on the unique DNA fragmentation pattern that characterizes apoptotic cell death.
Principles and Market Significance of Apoptosis Detection
The TUNEL (Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling) assay operates on the principle that during late apoptosis, DNA is fragmented by endonucleases that cleave chromatin into nucleosomal units [21]. The assay detects this fragmentation by employing the enzyme terminal deoxynucleotidyl transferase (TdT), which attaches deoxynucleotides to the 3'-hydroxyl terminus of DNA breaks [21] [12]. These nucleotides are tagged with either a direct fluorescent label or a chemical label that can be indirectly linked to a detection system [12].
The significance of apoptosis detection in general, and TUNEL assay in particular, is reflected in the substantial and growing market for apoptosis assays. The global apoptosis assay market continues to expand rapidly, demonstrating the technique's importance in both basic research and applied drug development:
Table 1: Apoptosis Assay Market Growth and Segmentation (2024-2034)
| Market Segment | 2024 Value (USD Billion) | Projected 2034 Value (USD Billion) | CAGR (%) |
|---|---|---|---|
| Total Market | 6.5 | 14.6 | 8.5 |
| Consumables | 3.6 | 8.2 | 8.9 |
| Instruments | 2.9 | 6.4 | 8.4 |
Source: [22]
This market growth is fueled by several factors, including the rising incidence of chronic diseases such as cancer and neurodegenerative disorders, increasing demand for personalized medicine, and technological advancements in detection platforms like flow cytometry [22]. The consumables segment dominates the market, reflecting the recurring need for reagents and assay kits in routine laboratory workflows [22].
Key Research Applications of the TUNEL Assay
The TUNEL assay serves as a critical tool across multiple research domains, providing sensitive detection of apoptotic cells in various experimental contexts:
- Sperm DNA Fragmentation Analysis: The TUNEL assay has become a standardized method for assessing sperm DNA fragmentation, which is crucial for understanding male infertility. It directly measures both single- and double-DNA strand breaks in sperm cells, with flow cytometry-based TUNEL assays growing in popularity for this application [14].
- Cancer and Drug Development Research: In cancer studies, TUNEL assays help evaluate the efficacy of chemotherapeutic agents by measuring their ability to induce apoptosis in tumor cells [22] [2]. This application is particularly valuable in personalized medicine approaches that aim to tailor therapies based on individual cellular responses [22].
- Neurodegenerative Disease Research: Apoptosis plays a significant role in neurodegenerative conditions such as Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis (ALS), where excessive apoptosis may contribute to disease progression [20]. TUNEL assays enable researchers to quantify and characterize apoptotic cells in neurological tissues.
- Post-COVID Immune Dysregulation Studies: Recent research has employed TUNEL assays to investigate persistent apoptotic signatures in peripheral blood mononuclear cells (PBMCs) of elderly individuals following COVID-19 recovery. These studies reveal prolonged immune dysregulation with significantly elevated proportions of apoptotic PBMCs, particularly within CD4+ and CD8+ T-cell subsets [23].
- Developmental Biology: The TUNEL assay helps researchers study programmed cell death during embryonic development, including processes like the elimination of webbing between fingers and toes in fetuses and the removal of unnecessary neural cells during brain formation [20].
Methodological Guide: TUNEL Assay Protocols and Reagents
The practical application of TUNEL assays requires careful attention to methodology and reagent selection. Below, we present detailed protocols and essential reagent information to facilitate successful experimental implementation.
Experimental Workflow for TUNEL Assay
The following diagram illustrates the generalized workflow for conducting a TUNEL assay, from sample preparation through final analysis:
Detailed Protocol for Cells Grown on Coverslips
The following step-by-step protocol is adapted from established TUNEL assay methods and optimized for adherent cells grown on coverslips [13]:
Cell Fixation and Permeabilization
- Remove media and wash coverslips once with PBS. Note: If cells may be lost during washing, proceed directly to fixation without this wash step.
- Add sufficient volume of fixative (4% paraformaldehyde) to completely cover the coverslips.
- Incubate samples for 15 minutes at room temperature.
- Remove fixative.
- Add sufficient volume of permeabilization reagent (0.25% Triton X-100 in PBS) to completely cover the coverslips.
- Incubate samples for 20 minutes at room temperature.
- Wash twice with deionized water.
Preparing a Positive Control (Optional)
- Wash coverslips with deionized or molecular biology grade water.
- Prepare DNase I solution according to manufacturer's instructions. Note: Do not vortex the DNase I solution as vigorous mixing can denature the enzyme.
- Add 100 µL of the DNase I solution to each coverslip and incubate for 30 minutes at room temperature.
- Wash coverslips once with deionized water before proceeding to the TUNEL reaction.
TUNEL Reaction
- Prepare the TUNEL reaction mixture according to kit specifications, typically containing TdT reaction buffer, TdT enzyme, and modified nucleotides.
- Apply the TUNEL reaction mixture to the samples.
- Incubate in a humidified chamber for 60 minutes at 37°C.
- Terminate the reaction by washing with the recommended buffer.
- For indirect detection methods, apply the appropriate detection reagent (e.g., streptavidin-HRP or antibody conjugates) and incubate as specified.
- Apply counterstain if required (e.g., DAPI or Hoechst 33342 for nuclear staining).
- Mount samples and proceed with appropriate detection method (microscopy, flow cytometry, etc.).
Research Reagent Solutions for TUNEL Assay
Successful implementation of TUNEL assays requires specific reagents, each serving distinct functions in the detection process. The table below outlines essential materials and their applications:
Table 2: Essential Research Reagents for TUNEL Assays
| Reagent | Function | Examples & Specifications |
|---|---|---|
| Terminal Deoxynucleotidyl Transferase (TdT) | Enzymatically incorporates modified nucleotides at 3'-OH ends of fragmented DNA | Recombinant enzyme; 15 U/μL concentration; requires cobalt cofactor in buffer [21] [13] |
| Modified Nucleotides (dUTP) | Serves as label incorporated at DNA break sites | Directly labeled (FITC-dUTP) or indirectly labeled (BrdUTP, biotin-dUTP); 50X solution [13] [12] |
| TdT Reaction Buffer | Provides optimal enzymatic reaction conditions | Contains potassium cacodylate and cobalt chloride; 1X solution [13] |
| Fixative | Preserves cellular structure and antigen integrity | 4% paraformaldehyde in PBS; 15-minute incubation at room temperature [13] |
| Permeabilization Reagent | Enables reagent access to nuclear DNA | 0.25% Triton X-100 in PBS; 20-minute incubation at room temperature [13] |
| Detection Reagents | Visualizes incorporated nucleotides | Varies by method: streptavidin-HRP with DAB substrate, antibody conjugates, or direct fluorescence [12] |
| DNase I | Generates DNA strand breaks for positive controls | Validates assay performance; requires careful handling without vortexing [13] |
Comparison of TUNEL Detection Methods
Researchers can choose from several detection methodologies for TUNEL assays, each with distinct advantages and limitations:
Table 3: Comparison of TUNEL Assay Detection Methods
| Detection Method | Principle | Advantages | Disadvantages | Popularity |
|---|---|---|---|---|
| Direct Fluorescence | dUTP directly conjugated to fluorescent dye (e.g., FITC) | Faster protocol; fewer steps; reduced background | Potentially lower signal intensity | 50% of published studies [12] |
| Biotin-Streptavidin | Biotin-dUTP detected with streptavidin-HRP and chromogenic substrate | Signal amplification; high sensitivity | Requires endogenous biotin blocking; additional steps | 15% of published studies [12] |
| BrdU-Based | BrdUTP detected with anti-BrdU antibody conjugates | Brighter signal; easier TdT incorporation | More expensive; additional incubation steps | 8% of published studies [12] |
| Click Chemistry | Alkyne-modified dUTP detected via copper-catalyzed click reaction with azide dyes | Small label size improves penetration; mild fixation sufficient; highly sensitive | Copper catalyst may affect some fluorophores | Increasing adoption [13] |
Technical Advancements and Methodological Considerations
Evolution of TUNEL Assay Technology
The original TUNEL assay methodology has undergone significant refinements since its introduction in 1992 [13]. Modern iterations offer improved sensitivity, safety, and compatibility with various detection platforms. Notable advancements include:
- Elimination of Toxic Reagents: Earlier TUNEL protocols utilized potassium or sodium cacodylate in reaction buffers, a carcinogenic arsenic derivative that could itself induce apoptosis and cause background signals [1]. Contemporary kits from manufacturers like AAT Bioquest have eliminated this toxic component, resulting in safer handling and reduced false-positive rates [1].
- Click Chemistry Integration: The introduction of click chemistry-based TUNEL assays, such as Thermo Fisher's Click-iT TUNEL platform, represents a significant technological advancement [13]. These assays utilize a dUTP modified with a small alkyne group that is more readily incorporated by TdT than bulkier modified nucleotides. Detection occurs through a copper-catalyzed reaction between the alkyne and an azide dye, offering superior penetration with only mild fixation and permeabilization required [13].
- Enhanced Sensitivity: Comparative studies demonstrate that click chemistry-based TUNEL assays can detect a higher percentage of apoptotic cells under identical conditions compared to traditional methods using fluorescein-dUTP [13]. This improved sensitivity is particularly valuable for detecting early or low levels of apoptosis.
- Multiplexing Capabilities: Modern TUNEL assays allow researchers to simultaneously detect apoptosis and other biomarkers through multiplexing approaches [13]. However, compatibility considerations remain important, as the copper catalyst used in click chemistry may destabilize certain molecules like fluorescent proteins or phalloidin [13].
Comparison with Alternative Apoptosis Detection Methods
While the TUNEL assay represents a gold standard for detecting DNA fragmentation during apoptosis, researchers should consider its relative advantages and limitations compared to alternative methods:
- DNA Laddering Assay: This traditional approach detects the characteristic internucleosomal DNA cleavage pattern (approximately 180-200 base pairs) through agarose gel electrophoresis [2]. While straightforward and cost-effective, it is less sensitive than TUNEL, semi-quantitative, requires substantial cell numbers, and cannot identify individual apoptotic cells within heterogeneous samples [2].
- Annexin V Staining: This method detects the externalization of phosphatidylserine to the outer leaflet of the plasma membrane during early apoptosis [2]. While excellent for identifying early apoptotic events, it requires live cells and specialized equipment like flow cytometers, and cannot detect late apoptotic stages where DNA fragmentation has already occurred [2].
- Caspase Activity Assays: These assays measure the activation of caspase enzymes that orchestrate the apoptotic process [2]. While providing mechanistic insights into apoptosis signaling, they may not correlate perfectly with actual cell death outcomes, as caspase activation can sometimes be reversible [2].
The TUNEL assay's unique ability to specifically detect and localize DNA fragmentation within individual cells makes it particularly valuable for situ apoptosis analysis, though optimal experimental design often combines multiple complementary approaches for comprehensive apoptosis assessment.
The journey from Carl Vogt's initial observations in 1842 to the sophisticated TUNEL assays of today represents a remarkable evolution in our understanding of programmed cell death. The formal conceptualization of apoptosis by Kerr, Wyllie, and Currie in 1972 provided the critical foundation for developing targeted detection methods that capitalize on the biochemical hallmarks of this process. The TUNEL assay stands as a direct technological beneficiary of this historical progression, enabling researchers to visualize and quantify DNA fragmentation—a definitive characteristic of late apoptosis—with exceptional specificity and sensitivity.
In contemporary research, TUNEL assays continue to evolve with advancements in detection chemistry, reagent safety, and compatibility with high-throughput platforms. These improvements have expanded applications across diverse fields including cancer research, neurodegenerative disease studies, male infertility assessment, and emerging areas such as post-COVID immune dysregulation. The substantial market growth for apoptosis assays—projected to reach USD 14.6 billion by 2034—testifies to their enduring importance in both basic research and drug development contexts [22].
As the field advances, TUNEL methodology continues to refine with integration of novel technologies like click chemistry, automated imaging systems, and artificial intelligence-assisted analysis. These innovations build upon the historical foundations of apoptosis research while addressing contemporary needs for precision, throughput, and quantitative accuracy. Through this ongoing synthesis of historical insight and technological progress, TUNEL assays remain indispensable tools for unraveling the complexities of programmed cell death in health and disease.
The TUNEL (Terminal deoxynucleotidyl transferase dUTP Nick End Labeling) assay is a cornerstone technique for the specific detection of DNA fragmentation, a hallmark event of late-stage apoptosis [1] [24]. During apoptosis, endogenous endonucleases (such as Caspase-Activated DNase) are activated and cleave the cell's genomic DNA between nucleosomes, generating a multitude of DNA fragments with exposed 3'-hydroxyl (3'-OH) ends [25] [1]. The TUNEL assay harnesses this specific biochemical event for detection.
The key enzyme in this process, Terminal Deoxynucleotidyl Transferase (TdT), is a unique DNA polymerase that catalyzes the template-independent addition of deoxynucleotide triphosphates to the 3'-hydroxyl ends of DNA molecules [25] [12]. In the TUNEL assay, TdT is used to add labeled deoxyuridine triphosphate (dUTP) nucleotides to these exposed 3'-OH termini of fragmented DNA [26] [1]. The resulting labeled DNA strands can then be visualized using various detection methods, allowing for the precise identification and localization of apoptotic cells within a sample [25].
Diagram 1: Core principle of the TUNEL assay.
Key Methodological Approaches
The fundamental TUNEL principle can be implemented through several technical approaches, primarily categorized into direct and indirect detection methods, each with distinct advantages [12].
Direct TUNEL Assays utilize dUTP nucleotides that are directly conjugated to a fluorophore (e.g., FITC-dUTP, Tunnelyte Green-dUTP, or Tunnelyte Red-dUTP) [12] [24]. After TdT-mediated incorporation, the labeled DNA can be immediately visualized via fluorescence microscopy or quantified by flow cytometry without additional steps [12]. This method is faster and involves fewer procedural steps, reducing the potential for non-specific background [12].
Indirect TUNEL Assays employ hapten-labeled dUTPs, such as biotin-dUTP, BrdUTP, digoxigenin-dUTP, or EdUTP (an alkyne-modified dUTP) [25] [12] [27]. The detection requires a subsequent step:
- For biotin-dUTP, streptavidin conjugated to horseradish peroxidase (HRP) or a fluorophore is used [26] [12].
- For BrdUTP or digoxigenin-dUTP, a specific antibody (anti-BrdU or anti-digoxigenin) conjugated to a reporter molecule is applied [12].
- For EdUTP, a copper-catalyzed "click" chemistry reaction links an azide-bearing fluorophore or biotin to the alkyne group on the incorporated nucleotide [27].
Indirect methods can offer signal amplification, which is particularly beneficial for samples with low levels of DNA fragmentation [12]. A survey of recent literature indicates that direct methods using FITC-dUTP are the most prevalent, accounting for approximately 50% of published TUNEL assays, while indirect methods collectively cover the remaining applications [12].
Table 1: Comparison of TUNEL Assay Detection Methods
| Method | Label Used | Detection System | Key Features | Reported Usage |
|---|---|---|---|---|
| Direct | Fluorescein-dUTP (FITC-dUTP) | Fluorescence microscopy/flow cytometry [12] | Fastest protocol; fewer steps [12] | ~50% of papers [12] |
| Indirect (Biotin-Streptavidin) | Biotin-dUTP | Streptavidin-HRP + DAB (colorimetric) or fluorescent streptavidin [26] [12] | Signal amplification; requires endogenous biotin blocking [12] | ~15% of papers [12] |
| Indirect (Antibody-based) | BrdUTP, digoxigenin-dUTP | Anti-BrdU or anti-digoxigenin antibody conjugated to fluorophore or HRP [12] | Bright signal; more incubation steps [12] | ~20-35% of papers [12] |
| Click Chemistry | EdUTP | Azide-containing fluorophore or biotin via copper-catalyzed reaction [27] | Highly specific; flexible detection; compatible with multiplexing [27] | - |
Detailed Experimental Protocol
The following protocol is a generalized procedure for performing a TUNEL assay on cultured cells or tissue sections. Always refer to the specific instructions provided with your commercial kit for optimal results.
Sample Preparation and Fixation
The initial step aims to preserve cellular morphology and stabilize the fragmented DNA.
- Adherent Cells: Wash cells gently with phosphate-buffered saline (PBS). Fix with 4% paraformaldehyde (PFA) in PBS for 15–30 minutes at room temperature [25].
- Tissue Sections (FFPE): Deparaffinize and rehydrate sections through a graded series of xylenes and ethanol (e.g., 100% xylene, 96% ethanol, 90% ethanol, 80% ethanol, 70% ethanol, 50% ethanol) ending in distilled water [26]. Antigen retrieval using a pressure cooker is recommended for compatibility with subsequent multiplexed protein detection, as it preserves protein antigenicity better than proteinase K [16].
- Frozen Tissue Sections: Fix with 4% PFA for 15-30 minutes [25].
Permeabilization
This critical step creates pores in the cell membrane and nuclear envelope, allowing the large TdT enzyme and labeled nucleotides to access the nuclear DNA. Optimization is essential to avoid under-permeabilization (leading to false negatives) or over-permeabilization (causing artificial DNA breaks and false positives) [25].
- Cultured Cells: Incubate in 0.1%–0.5% Triton X-100 in PBS for 5–15 minutes on ice [25].
- Tissue Sections: A harsher permeabilization is often required. Use 20 µg/mL Proteinase K for 10–20 minutes at room temperature or 0.5-1% Triton X-100 [25]. Note that Proteinase K can degrade protein antigens, so if subsequent immunofluorescence is planned, pressure cooker retrieval is a superior alternative [16].
TdT Labeling Reaction
This is the core reaction where DNA breaks are labeled.
- Equilibration (Optional): Incubate the sample with the kit-specific equilibration buffer for ~10 minutes to prepare the DNA for the enzymatic reaction [25].
- Reaction Mix Incubation: Carefully remove the buffer and apply the prepared TdT Reaction Mix (containing TdT enzyme, labeled dUTP, and reaction buffer). Use a parafilm coverslip to ensure even distribution and prevent evaporation [26] [25].
- Incubate the samples in a humidified chamber at 37°C for 30-60 minutes [26] [25]. The TdT reaction buffer typically requires cobalt ions as a cofactor for optimal activity [1].
Reaction Stop and Detection
- Stop Reaction: Terminate the TdT reaction by incubating the samples with a stop/wash buffer (often provided in kits) for 10 minutes [25].
- Wash: Rinse the samples 2-3 times with PBS to remove unincorporated nucleotides [25].
- Detection (for indirect methods):
- BrdUTP/Digoxigenin-dUTP: Apply a fluorophore- or enzyme-conjugated anti-BrdU or anti-digoxigenin antibody for 30-60 minutes [25] [12].
- Click Chemistry (EdUTP): Perform the copper-catalyzed "click" reaction with an azide-containing reporter molecule (e.g., Alexa Fluor azides) according to the kit protocol [27].
- Final Washes: Wash the samples 2-3 times in PBS to remove excess detection reagents [25].
Counterstaining, Mounting, and Analysis
- Counterstaining: Incubate with a nuclear counterstain to visualize all cells.
- Wash and Mount: Perform a final rinse with water and mount coverslips using an appropriate antifade mounting medium [25].
- Analysis:
- Fluorescence: Use a fluorescence microscope with appropriate filter sets. Apoptotic cells display bright nuclear fluorescence, while non-apoptotic cells show only the counterstain [25].
- Colorimetric: View under a bright-field microscope. Apoptotic nuclei are stained a dark brown by the DAB precipitate [26] [12].
Diagram 2: TUNEL assay workflow.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents for TUNEL Assay
| Reagent / Solution | Function / Purpose | Examples / Notes |
|---|---|---|
| Terminal Deoxynucleotidyl Transferase (TdT) | Core enzyme that catalyzes the addition of labeled dUTPs to 3'-OH DNA ends [25] [1]. | Supplied in commercial kits. Critical to include a negative control without TdT [25]. |
| Labeled dUTP | The nucleotide that is incorporated into DNA breaks and provides the detectable signal [25]. | FITC-dUTP (direct), Biotin-dUTP, BrdUTP, EdUTP (indirect) [12] [27]. |
| Fixative | Preserves cellular architecture and cross-links fragmented DNA in place [25]. | 4% Paraformaldehyde (PFA) is standard [26] [25]. |
| Permeabilization Agent | Creates pores for reagent access to the nucleus [25]. | Triton X-100 (for cells) [25]; Proteinase K or pressure cooker (for tissues) [25] [16]. |
| Detection Reagents | Visualizes the incorporated label (for indirect methods). | Streptavidin-HRP, anti-BrdU antibodies, or click chemistry reagents [26] [12] [27]. |
| Chromogenic Substrate | Produces an insoluble colored precipitate for bright-field microscopy. | DAB (3,3'-Diaminobenzidine) produces a brown stain [26] [12]. |
| Counterstain | Provides contrast and visualizes overall tissue or cell structure. | DAPI (fluorescent) [25]; Methyl Green or Hematoxylin (colorimetric) [26] [12]. |
Critical Controls and Troubleshooting
Essential Experimental Controls
Including proper controls is non-negotiable for validating TUNEL assay results.
- Positive Control: Treat a sample with DNase I (1 µg/mL for 15-30 minutes) after permeabilization. This intentionally fragments all nuclear DNA, and should result in ~100% TUNEL-positive nuclei, confirming the assay is working [25].
- Negative Control: Process a sample where the TdT enzyme is omitted from the reaction mix. This sample should show no specific signal and helps identify non-specific binding of detection antibodies or dyes [25].
Addressing Common Artifacts and Limitations
The TUNEL assay is powerful but requires careful interpretation due to potential artifacts.
- False Positives: TdT will label any exposed 3'-OH end, which can occur in necrosis (random DNA degradation), cells undergoing DNA repair, or due to over-fixation/harsh permeabilization that artificially creates DNA breaks [25].
- False Negatives: Can result from under-permeabilization (preventing TdT access), over-fixation (cross-linking and blocking 3'-OH ends), or incomplete apoptosis where DNA fragmentation has not yet occurred [25].
- The Anastasis Problem: Research indicates that cells can be TUNEL-positive and still recover, a process called anastasis. Therefore, a TUNEL signal does not always equate to irreversible cell death [25].
To enhance specificity, it is highly recommended to combine TUNEL with another apoptosis marker, such as an immunofluorescence assay for cleaved Caspase-3 (an earlier apoptotic event) or an Annexin V assay for phosphatidylserine externalization [25]. A recent study also highlights that replacing Proteinase K with pressure cooker-based antigen retrieval preserves both TUNEL signal and protein antigenicity, enabling robust multiplexing with spatial proteomics methods [16]. Furthermore, some vendors offer advanced kits that eliminate toxic sodium cacodylate from the reaction buffer, improving safety and reducing background apoptosis induction [1] [24].
The TUNEL (Terminal deoxynucleotidyl transferase dUTP nick end labeling) assay serves as a cornerstone technique in apoptosis research, enabling specific detection of DNA fragmentation—a hallmark of programmed cell death. Its effectiveness hinges on the precise biochemical interplay between the terminal deoxynucleotidyl transferase (TdT) enzyme, modified deoxyuridine triphosphate (dUTP) nucleotides, and essential cationic cofactors. This application note details the roles of these core components within the context of late apoptosis research, providing validated protocols and analytical frameworks for researchers and drug development professionals. We summarize critical quantitative data, outline step-by-step methodologies, and visualize core reaction pathways to standardize and enhance experimental accuracy in both basic and translational research.
In late-stage apoptosis, the activation of caspase-activated DNase (CAD) cleaves chromosomal DNA into oligonucleosomal fragments, generating an abundance of double-stranded DNA breaks with exposed 3'-hydroxyl (3'-OH) termini [28] [29]. The TUNEL assay is specifically designed to tag these 3'-OH ends, providing a powerful tool for in situ identification and quantification of apoptotic cells within tissue sections or cultured cell samples [5] [30]. The specificity and sensitivity of this assay are fundamentally dependent on three key reaction components: the TdT enzyme, which catalyzes the reaction; labeled dUTP, which serves as the detection moiety; and essential cofactors, which optimize enzymatic activity. For researchers in drug development, understanding these components is crucial for accurately assessing the efficacy of therapeutic agents designed to either induce apoptosis in cancer cells or inhibit it in neurodegenerative conditions [28] [31]. This note delineates the function of each component and provides optimized protocols for robust, reproducible results.
Core Biochemical Components and Their Functions
The TUNEL reaction is a carefully orchestrated biochemical process. The table below summarizes the roles and key characteristics of its fundamental components.
Table 1: Key Components of the TUNEL Assay Reaction
| Component | Primary Function | Key Characteristics & Variants | Optimization Notes |
|---|---|---|---|
| Terminal Deoxynucleotidyl Transferase (TdT) | Template-independent DNA polymerase that catalyzes the addition of deoxynucleotides to the 3'-OH ends of single- and double-stranded DNA fragments [5] [30]. | • Highly purified forms are used for reduced background [32]. | • Enzyme activity is cation-dependent [28].• Omission serves as a critical negative control [31]. |
| Modified dUTP | The nucleotide substrate incorporated into the DNA breaks; the modification (e.g., fluorophore, hapten) enables detection [8]. | • Fluorescein-dUTP: Direct detection [30].• BrdUTP: Indirect detection via anti-BrdU antibodies; offers high sensitivity [8] [33].• EdUTP: Detection via click chemistry, offering flexibility and high specificity [8]. | • The dUTP:TdT molar ratio is critical (e.g., 5:1) [31]. |
| Cofactors (Cations) | Essential for activating the TdT enzyme and maximizing its catalytic efficiency [28]. | • Cobalt (Co²⁺): A common cofactor included in the reaction buffer [1] [28]. | • The concentration in the labeling buffer must be optimized to prevent inhibition or high background. |
The following diagram illustrates the coordinated interaction of these components in the core TUNEL reaction mechanism.
Figure 1: TUNEL Assay Core Reaction Mechanism. The TdT enzyme, activated by a cationic cofactor, catalyzes the template-independent addition of modified dUTP nucleotides to the 3'-OH termini of fragmented DNA.
Quantitative Data and Reagent Specifications
For robust experimental design, understanding the quantitative aspects of reagent use and expected outcomes is essential. The following table compiles key quantitative data from established protocols and validation studies.
Table 2: Quantitative Assay Parameters and Reagent Specifications
| Parameter | Typical Range / Value | Application Context & Significance |
|---|---|---|
| dUTP:TdT Molar Ratio | 5:1 [31] | Optimized for efficient labeling while minimizing non-specific background. |
| Reaction Incubation | 1–3 hours at 37°C [28] | Ensures sufficient nucleotide incorporation; varies with DNA break density. |
| Positive Signal Threshold | 5–10x background fluorescence [31] | Distinguishes specific apoptosis-associated fragmentation from random DNA damage. |
| Apoptotic DNA Fragment Size | ~180-200 base pairs [1] | Characteristic nucleosomal ladder pattern, a key biochemical hallmark. |
| Sperm DNA Fragmentation (Clinical Cutoff) | 16.8% (TUNEL-positive) [34] | Reference value for male infertility assessment (specificity: 91.6%). |
| Permeabilization Condition | 0.1% Triton X-100 for 8 minutes [31] | Balances membrane permeability with preservation of cellular morphology. |
Research Reagent Solutions
A successful TUNEL assay relies on a suite of essential materials. The table below lists key reagents and their functions.
Table 3: Essential Research Reagents for TUNEL Assay
| Reagent / Material | Function | Key Considerations |
|---|---|---|
| Terminal Deoxynucleotidyl Transferase (TdT) | Catalyzes the addition of labeled nucleotides to DNA breaks. | Use highly purified enzyme to reduce background noise [32]. |
| Labeled dUTP (e.g., FITC-dUTP, EdUTP) | Forms the basis for detection of incorporated nucleotides. | EdUTP with click chemistry offers high specificity and low background [8]. |
| Reaction Buffer with Cobalt | Provides optimal pH and ionic conditions; Co²⁺ acts as a critical cofactor. | Essential for maximal TdT enzyme activity [1] [28]. |
| Paraformaldehyde (4%) | Fixes cells/tissues, preserving structural integrity and preventing autolysis. | Prolonged fixation can mask antigenic sites and should be avoided [31]. |
| Permeabilization Agent (e.g., Proteinase K, Triton X-100) | Creates pores in the cell membrane, allowing TUNEL reagents to enter the nucleus. | Concentration and time must be optimized to prevent over-digestion [28] [31]. |
| DNase I | Used to intentionally fragment DNA in positive control samples. | Validates the assay procedure and helps set detection thresholds [31]. |
Standardized TUNEL Assay Protocol
This protocol is optimized for the detection of apoptotic cells in formalin-fixed, paraffin-embedded (FFPE) tissue sections and can be adapted for cultured cells with minor modifications [8] [28] [31].
The complete experimental workflow, from sample preparation to analysis, is visualized below.
Figure 2: Standard TUNEL Assay Workflow. The process involves sample preparation, the key labeling reaction, and subsequent detection steps.
Step-by-Step Procedure
Sample Preparation and Fixation
- Tissue: Fix tissue samples in 4% paraformaldehyde (PFA) for 4–24 hours at 4°C. For FFPE tissues, deparaffinize and rehydrate sections using standard xylene and ethanol series [28] [31].
- Cultured Cells: Grow cells on chamber slides or coverslips. Fix with 4% PFA for 20 minutes at room temperature. Avoid prolonged fixation to prevent epitope masking [8] [31].
- Critical Control: Include a positive control (e.g., treated with DNase I, 1 µg/mL for 10 minutes) to validate the assay [31].
Permeabilization
- Treat samples with a permeabilization solution. For FFPE tissues, proteinase K (20 µg/mL for 15–25 minutes at room temperature) is often used. For cultured cells or frozen sections, 0.1% Triton X-100 for 8 minutes is sufficient [28] [31].
- Rinse slides thoroughly with phosphate-buffered saline (PBS) to terminate permeabilization.
TUNEL Reaction Mixture Preparation
- Prepare the TUNEL reaction mixture on ice according to the table below. The exact volumes may vary by commercial kit; follow manufacturer instructions for optimal performance [34] [8].
- Table 4: TUNEL Reaction Mixture Composition
Component Volume/Final Concentration Reaction Buffer (with Co²⁺) 1X Modified dUTP (e.g., EdUTP) As per kit (e.g., ~50 µM) TdT Enzyme As per kit Deionized Water To final volume
Incubation and Reaction Termination
- Apply the TUNEL reaction mixture to the samples, ensuring complete coverage. Incubate in a humidified chamber protected from light for 1–3 hours at 37°C [28].
- Terminate the reaction by immersing the slides in a stop/wash buffer (provided in many kits) for 15 minutes. Rinse several times with PBS.
Detection and Counterstaining
- For direct detection (e.g., FITC-dUTP): Proceed to counterstaining and mounting.
- For indirect detection (e.g., BrdUTP, EdUTP): Perform the required detection step. For EdUTP, this involves a click reaction with a fluorescent azide. For BrdUTP, incubate with an Alexa Fluor-conjugated anti-BrdU antibody [8].
- Apply an appropriate nuclear counterstain (e.g., Hoechst 33342 or Propidium Iodide) to visualize all nuclei [29]. Mount slides with an anti-fade mounting medium.
Analysis and Quantification
- Visualize using fluorescence microscopy, high-content analysis systems, or flow cytometry.
- For imaging: Acquire images from 5–10 random fields (≥200 cells total). TUNEL-positive cells will exhibit bright nuclear staining, typically 5–10 times brighter than background [31].
- Quantification: Calculate the apoptotic index as (Number of TUNEL-positive cells / Total number of cells) × 100%. Use image analysis software (e.g., ImageJ) for automated, unbiased counting in large datasets [31].
Troubleshooting and Assay Limitations
A clear understanding of the assay's limitations is vital for accurate data interpretation.
- Specificity Considerations: While a hallmark of apoptosis, DNA fragmentation detectable by TUNEL can also occur in other cell death modes (e.g., necrosis, pyroptosis) and during DNA repair [5] [33]. Therefore, TUNEL positivity alone is not absolute proof of apoptosis. Correlation with morphological features (chromatin condensation, nuclear blebbing, apoptotic bodies) is crucial [31]. For definitive confirmation, perform multiplex assays combining TUNEL with caspase-3 activation markers or Annexin V staining [33] [31].
- Common Pitfalls:
- False Positives: Can arise from sample over-digestion during permeabilization, endogenous biotin, or DNA damage from sample processing [31].
- False Negatives: May occur in early apoptotic stages prior to extensive DNA fragmentation, or due to suboptimal fixation/permeabilization preventing reagent access [31].
- Solution: Always include and validate against positive and negative controls (DNase-treated and no-TdT enzyme, respectively) in every experiment [31].
The TUNEL assay remains a powerful and versatile method for detecting DNA fragmentation in apoptosis research. Its reliable application, however, is fundamentally dependent on a thorough understanding of its core components—the TdT enzyme, modified dUTP, and essential cofactors. By adhering to the detailed protocols, quantitative guidelines, and troubleshooting advice outlined in this document, researchers can design and execute robust, reproducible experiments. The ongoing development of novel detection chemistries, such as click chemistry, and integration with high-throughput platforms continue to enhance the utility of this gold-standard technique in both basic research and drug discovery pipelines.
Step-by-Step TUNEL Protocol and Advanced Applications
Proper sample preparation is the foundational step upon which reliable TUNEL (Terminal deoxynucleotidyl transferase dUTP Nick-End Labeling) assay results are built. The accuracy of detecting DNA fragmentation—a hallmark of late-stage apoptosis—is profoundly influenced by the fixation and permeabilization steps [35]. These processes preserve cellular architecture while enabling enzyme access to nuclear DNA, and their optimization is critical for distinguishing true apoptotic events from artifacts [35] [16]. This application note provides detailed methodologies and best practices for preparing cell and tissue samples for TUNEL assays, framed within the broader context of apoptosis research for scientists and drug development professionals.
Principles of Fixation and Permeabilization in TUNEL Assays
The primary objective of fixation in TUNEL assays is to cross-link proteins and preserve the structural integrity of cells and tissues at the moment of harvesting, thereby immobilizing the fragmented DNA characteristic of apoptosis [35] [36]. Paraformaldehyde (PFA) is the preferred fixative as it creates reversible cross-links that maintain morphology while still allowing enzymatic access to DNA after appropriate permeabilization [35].
Permeabilization follows fixation and involves disrupting lipid membranes to grant the large TdT enzyme (Terminal deoxynucleotidyl transferase) and labeled nucleotides access to the nuclear compartment [35]. The key challenge lies in achieving sufficient permeability without destroying antigenicity for multiplexed experiments or creating excessive DNA breaks that lead to false-positive signals [35] [16]. The optimal balance must be determined empirically for different sample types, as under-permeabilization results in false negatives, while over-permeabilization can cause artifactual DNA damage [35].
Table 1: Fixation and Permeabilization Conditions for Different Sample Types
| Sample Type | Recommended Fixative | Fixation Duration | Recommended Permeabilization Agent | Permeabilization Duration |
|---|---|---|---|---|
| Adherent Cells | 1%–4% PFA in PBS [35] | 15–30 minutes at room temperature [35] | 0.1%–0.5% Triton X-100 in PBS [35] | 5–15 minutes on ice [35] |
| Suspension Cells | 4% PFA in PBS [13] | 15 minutes at room temperature [13] | 0.25% Triton X-100 in PBS [13] | 20 minutes at room temperature [13] |
| FFPE Tissue Sections | Formalinfixed, paraffin-embedded [35] [16] | N/A (post-processing required) [35] | 20 µg/mL Proteinase K or 0.5-1% Triton X-100 [35] | 10–20 minutes at room temperature [35] |
| Frozen Tissue Sections | 4% PFA in PBS [35] | 15-30 minutes [35] | Proteinase K or pressure cooker retrieval [16] | Tissue-specific optimization required [16] |
Comprehensive Protocols
Protocol for Adherent Cells
Materials Required:
- Phosphate-buffered saline (PBS), pH 7.4
- Fixative: 4% Paraformaldehyde (PFA) in PBS [35]
- Permeabilization solution: 0.1%–0.5% Triton X-100 in PBS [35]
- Blocking solution: 3% Bovine serum albumin (BSA) in PBS (optional for reducing background) [13]
Procedure:
- Culture Preparation: Grow cells on appropriately sized coverslips until they reach 60-80% confluence.
- Washing: Gently wash cells with PBS to remove culture media and debris. If cells are particularly delicate, proceed directly to fixation to prevent cell loss [13].
- Fixation: Add sufficient 4% PFA to completely cover cells and incubate for 15-30 minutes at room temperature [35].
- PFA Removal and Washing: Remove fixative and wash cells twice with deionized water or PBS [13].
- Permeabilization: Apply 0.1%-0.5% Triton X-100 in PBS and incubate for 5-15 minutes on ice [35]. The optimal concentration and duration should be determined empirically for each cell type.
- Washing: Wash samples twice with PBS to remove permeabilization agent before proceeding to TUNEL reaction [35].
Protocol for Formalin-Fixed Paraffin-Embedded (FFPE) Tissues
Materials Required:
- Xylene or xylene substitute
- Ethanol series (100%, 95%, 70%, 50%)
- Antigen retrieval solution (e.g., citrate buffer, pH 6.0)
- Proteinase K (20 µg/mL) or permeabilization buffer (0.5-1% Triton X-100) [35]
Procedure:
- Deparaffinization: Immerse slides in xylene (2 changes, 5 minutes each) to remove paraffin.
- Rehydration: Transfer slides through a graded ethanol series: 100% ethanol (2 changes, 3 minutes each), 95% ethanol (2 minutes), 70% ethanol (2 minutes), and 50% ethanol (2 minutes).
- Rinsing: Rinse slides in PBS or deionized water.
- Antigen Retrieval: Heat-induced epitope retrieval using pressure cooker or steam in citrate buffer (pH 6.0) can significantly enhance both TUNEL signal and protein antigenicity for multiplexing [16].
- Permeabilization: Apply either:
- Washing: Rinse thoroughly with PBS to stop permeabilization.
Advanced Consideration: Compatibility with Multiplexed Spatial Proteomics
For researchers integrating TUNEL with advanced spatial proteomics methods like Multiple Iterative Labeling by Antibody Neodeposition (MILAN) or Cyclic Immunofluorescence (CycIF), recent evidence indicates that proteinase K treatment—common in many TUNEL protocols—consistently reduces or abrogates protein antigenicity [16]. As an alternative, pressure cooker-based antigen retrieval quantitatively preserves TUNEL signal without compromising protein antigenicity, enabling comprehensive multiplexed analysis of cell death in complex tissues [16].
Critical Controls and Optimization
Including appropriate controls is essential for validating TUNEL assay results and troubleshooting potential issues.
Table 2: Essential Controls for TUNEL Assay Validation
| Control Type | Purpose | Preparation Method | Expected Result |
|---|---|---|---|
| Positive Control | Verify assay functionality and accessibility of DNA ends | Treat sample with 1 µg/mL DNase I for 15-30 minutes before labeling step [35] | All nuclei should stain positive [35] |
| Negative Control | Identify non-specific background signal | Omit TdT enzyme from reaction mix [35] or omit labeled nucleotide [13] | No specific nuclear staining [35] |
| Biological Control | Confirm apoptosis induction method | Include known apoptotic and healthy cell populations | Differential staining between populations |
| Technical Control | Assess sample processing effects | Include healthy cells processed identically to test samples | Minimal background staining |
Troubleshooting Common Issues
Excessive Background Staining:
- Cause: Over-permeabilization or excessive fixation
- Solution: Titrate permeabilization agent concentration and duration; reduce fixation time [35]
Weak or No Signal:
- Cause: Under-permeabilization, over-fixation, or incomplete deparaffinization
- Solution: Increase permeabilization agent concentration/duration; optimize antigen retrieval method [35] [16]
Tissue Detachment:
- Cause: Over-permeabilization or harsh handling
- Solution: Use charged slides; monitor permeabilization time; handle slides gently during washing
Incompatibility with Protein Co-detection:
- Cause: Proteinase K-mediated degradation of protein epitopes
- Solution: Replace proteinase K with pressure cooker antigen retrieval [16]
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents for TUNEL Sample Preparation
| Reagent | Function | Example Formulations | Considerations |
|---|---|---|---|
| Paraformaldehyde (PFA) | Cross-linking fixative that preserves cellular structure and immobilizes fragmented DNA | 1%-4% in PBS [35] | Concentration and fixation time must be optimized; over-fixation can mask DNA ends |
| Triton X-100 | Non-ionic detergent that disrupts lipid membranes to enable reagent access | 0.1%-0.5% for cells; 0.5%-1% for tissues [35] | Concentration critical; too low prevents enzyme access, too high causes artifacts |
| Proteinase K | Proteolytic enzyme that digests proteins and enhances access to DNA | 20 µg/mL for tissue permeabilization [35] | Degrades protein epitopes; avoid if multiplexing with protein detection [16] |
| Ethanol Series | Dehydrates and rehydrates tissue sections for processing | 50%, 70%, 95%, 100% concentrations [35] | Essential for FFPE tissue processing |
| BSA (Bovine Serum Albumin) | Blocking agent that reduces non-specific binding | 3% BSA in PBS [13] | Minimizes background in indirect detection methods |
| Antigen Retrieval Buffers | Reverses formaldehyde cross-links to expose epitopes | Citrate buffer (pH 6.0) [16] | Pressure cooker method compatible with multiplexed spatial proteomics [16] |
Workflow Visualization
The following diagram illustrates the complete sample preparation workflow for different sample types in TUNEL assays:
TUNEL Sample Preparation Workflow
Proper fixation and permeabilization are critical determinants of success in TUNEL assays for apoptosis detection. The optimal protocol must balance preservation of cellular morphology with accessibility of fragmented DNA to the TdT enzyme, while considering the specific requirements of different sample types. Recent advances, particularly the replacement of proteinase K with pressure cooker antigen retrieval for multiplexed spatial proteomics applications, demonstrate the ongoing evolution of these fundamental techniques [16]. By adhering to these best practices and implementing appropriate controls, researchers can ensure reliable, reproducible detection of DNA fragmentation that accurately reflects apoptotic activity in their experimental systems.
The Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay stands as a cornerstone technique for detecting DNA fragmentation, a definitive biochemical hallmark of late-stage apoptosis [37] [24]. Since its introduction in 1992, it has become the most widely used in situ test for apoptosis study, prized for its sensitivity and capacity to provide quantitative measurements over several orders of magnitude [13] [24]. The core principle of the TUNEL assay relies on the enzyme Terminal Deoxynucleotidyl Transferase (TdT), which catalyzes the attachment of labeled deoxynucleotides to the 3'-hydroxyl termini of DNA double-strand breaks [24] [12]. The critical choice facing researchers lies in the detection method for these incorporated nucleotides—fluorescence or colorimetry—each with distinct advantages and limitations that impact experimental outcomes, sensitivity, and applicability across different research scenarios.
This application note provides a systematic comparison between fluorescence and colorimetric detection methods for TUNEL assays, framed within the context of DNA fragmentation analysis in late apoptosis research. We present quantitative performance data, detailed protocols, and strategic guidance to enable researchers, scientists, and drug development professionals to make informed methodological selections based on their specific experimental requirements, instrumentation capabilities, and application needs.
Fundamental Principles and Comparative Analysis
Mechanism of Action and Technical Foundations
The TUNEL assay detects the DNA fragmentation that occurs in the final phase of apoptosis, where caspase-activated DNase (CAD) cleaves DNA at internucleosomal linker sites [2]. The enzyme Terminal Deoxynucleotidyl Transferase (TdT) adds deoxynucleotides to the 3'-hydroxyl ends of these DNA breaks [12]. The detection strategy for these incorporated nucleotides forms the basis for the distinction between fluorescence and colorimetric methods, as shown in the experimental workflow below:
Fluorometric detection employs nucleotides directly conjugated to fluorescent dyes (e.g., FITC, Alexa Fluor dyes) [12]. The signal is generated when these fluorophores are excited by light at a specific wavelength and emit light at a longer wavelength, with intensity proportional to the amount of DNA fragmentation [38]. In contrast, colorimetric detection typically uses biotin-tagged nucleotides that are subsequently bound by streptavidin-HRP (horseradish peroxidase) complexes, then detected using chromogenic substrates like DAB (3,3'-diaminobenzidine) to generate a brown-colored precipitate [12].
Performance Comparison and Method Selection
The choice between fluorescence and colorimetric detection involves trade-offs between sensitivity, equipment requirements, throughput, and practicality. The table below summarizes the key characteristics of each method based on current literature and technical data:
Table 1: Quantitative Comparison of Fluorescence vs. Colorimetric TUNEL Assay Methods
| Parameter | Fluorometric Detection | Colorimetric Detection |
|---|---|---|
| Sensitivity | High (nanomolar to picomolar levels) [38] | Moderate (micromolar to millimolar levels) [38] |
| Detection Limit | Can detect higher percentage of apoptotic cells under identical conditions [13] | Limited by chromogen precipitation and visual detection |
| Equipment Needs | Fluorescence microscope, plate reader, or flow cytometer [12] [38] | Standard brightfield microscope [12] |
| Assay Time | Faster (∼2 hours post-fixation for direct methods) [13] | Longer due to additional incubation and amplification steps [12] |
| Multiplexing Potential | High - compatible with other fluorescent markers [13] | Limited to single-plex detection |
| Throughput | Excellent for flow cytometry and high-content screening [24] | Well-suited for individual sample analysis |
| Quantification | Highly quantitative via flow cytometry or plate readers [24] | Semi-quantitative via image analysis [12] |
| Sample Type | Cells, tissue sections (requires fluorescence-capable instrumentation) [12] | Primarily tissue sections for histology [12] |
| Cost Factors | Higher instrumentation costs; some kit options provide cost savings [24] [38] | Lower instrumentation costs; may require additional reagents for signal amplification [38] |
Survey data from recent scientific publications indicates that fluorescent methods, particularly those using dUTP directly conjugated to FITC, dominate current research applications, accounting for approximately 50% of TUNEL assays published in 2017 [12]. Biotin-dUTP with streptavidin-HRP represented about 15% of methods, while another 15% used FITC-dUTP with an anti-FITC antibody conjugated to HRP [12]. This distribution reflects the broader trend toward fluorescence-based detection in modern apoptosis research, particularly for applications requiring quantification and high sensitivity.
Experimental Protocols
Fluorometric TUNEL Assay Protocol for Adherent Cells
The following protocol is adapted from the Click-iT TUNEL Alexa Fluor Imaging Assay and optimized for adherent cells grown on coverslips or in 96-well plates [13]. This method utilizes a click chemistry approach that incorporates an alkyne-modified dUTP, followed by detection with a fluorescent azide, offering superior penetration and sensitivity compared to traditional antibody-based detection.
Materials and Reagents
Table 2: Essential Reagents for Fluorometric TUNEL Assay
| Reagent/Equipment | Function/Application | Notes/Specifications |
|---|---|---|
| Terminal deoxynucleotidyl transferase (TdT) | Catalyzes addition of modified nucleotides to 3'-OH DNA ends | Recombinant enzyme, supplied at 15 U/μL [13] |
| TdT Reaction Buffer | Provides optimal enzymatic reaction conditions | Contains cacodylate buffer and cobalt chloride; handle with appropriate precautions [13] |
| EdUTP Nucleotide Mixture | Alkyne-modified nucleotide for incorporation into DNA breaks | 50X solution; more readily incorporated by TdT than larger modifications [13] |
| Click-iT Reaction Buffer with Alexa Fluor Azide | Copper-catalyzed cycloaddition for fluorescence detection | Contains Alexa Fluor 488, 594, or 647 azide; choose based on available filter sets [13] |
| Hoechst 33342 | Nuclear counterstain | 10 mg/mL solution in water; known mutagen, use with appropriate precautions [13] |
| Fixative (4% Paraformaldehyde in PBS) | Preserves cellular morphology and crosslinks biomolecules | Prepare fresh or use commercially available solutions |
| Permeabilization Reagent (0.25% Triton X-100 in PBS) | Enables reagent access to nuclear DNA | Concentration and time may require optimization for different cell types |
| DNase I (Optional) | Generates DNA strand breaks for positive controls | Do not vortex to prevent denaturation [13] |
| Fluorescence Microscope or Plate Reader | Signal detection and quantification | Equipped with appropriate filter sets for chosen fluorophore [13] |
Step-by-Step Procedure
Day 1: Cell Preparation and Fixation
Cell Culture and Treatment: Plate adherent cells (e.g., HeLa, A549) on coverslips or in 96-well plates and allow to adhere overnight. Treat with apoptosis-inducing agents (e.g., 0.5-1 μM staurosporine for 4-18 hours) [13] [24]. Include untreated negative controls.
Fixation:
- Remove media and wash once with PBS. Note: If cells are prone to detachment, proceed directly to fixation without washing.
- Add sufficient 4% paraformaldehyde in PBS to completely cover cells.
- Incubate for 15 minutes at room temperature.
- Remove fixative and wash twice with deionized water.
Permeabilization:
- Add sufficient 0.25% Triton X-100 in PBS to cover cells.
- Incubate for 20 minutes at room temperature.
- Wash twice with deionized water.
Day 1: Positive Control Preparation (Optional)
- DNase I Treatment:
- Prepare DNase I solution by diluting Component G 1:100 in the provided DNase I buffer (Component H). Do not vortex.
- Add 100 μL of DNase I solution to designated positive control samples.
- Incubate for 30 minutes at room temperature.
- Wash once with deionized water.
Day 1: TdT Reaction
Reaction Mixture Preparation:
- Prepare TdT reaction buffer by combining (per sample): 98 μL TdT reaction buffer (Component A), 2 μL EdUTP nucleotide mixture (Component B), and 2 μL TdT enzyme (Component C).
Labeling:
- Add 102 μL of reaction mixture to each sample.
- Incubate in a humidified chamber for 60 minutes at 37°C.
- Wash twice with deionized water.
Day 1: Click Reaction
Click-iT Reaction Mixture Preparation:
- Prepare Click-iT reaction cocktail by combining (per sample): 103 μL Click-iT reaction buffer (Component D) and 2 μL Click-iT reaction buffer additive (Component E). Mix until additive is completely dissolved.
Detection:
- Add 105 μL of Click-iT reaction cocktail to each sample.
- Incubate for 30 minutes at room temperature, protected from light.
- Wash twice with deionized water.
Day 1: Counterstaining and Mounting
Nuclear Staining:
- Prepare a 1:2000 dilution of Hoechst 33342 (Component F) in PBS.
- Add to samples and incubate for 15 minutes at room temperature, protected from light.
- Wash twice with deionized water.
Mounting and Visualization:
- For coverslips, mount on slides using an appropriate antifade mounting medium.
- For 96-well plates, add 100 μL PBS to prevent drying.
- Visualize using a fluorescence microscope with appropriate filter sets or analyze by flow cytometry.
Data Analysis and Interpretation
For qualitative analysis, apoptotic cells display bright nuclear fluorescence corresponding to the fluorophore used (green for Alexa Fluor 488, red for Alexa Fluor 594, etc.), while non-apoptotic cells show only the Hoechst nuclear counterstain. For quantitative analysis using flow cytometry or high-content imaging, calculate the percentage of TUNEL-positive cells by gating on the population showing fluorescence intensity above the negative control threshold. Typical results show a dose-dependent increase in TUNEL-positive cells with increasing concentrations of apoptosis-inducing agents, as demonstrated in Figure 6 where HeLa cells treated with staurosporine for 18 hours showed increasing TUNEL positivity across concentrations ranging from 1 nM to 1 μM [13].
Colorimetric TUNEL Assay Protocol for Tissue Sections
This protocol outlines the colorimetric detection of apoptotic cells in tissue sections using biotin-dUTP and streptavidin-HRP with DAB chromogen, producing a brown precipitate that can be visualized by brightfield microscopy [12].
Materials and Reagents
- Biotin-dUTP: Nucleotide tag for incorporation by TdT
- Streptavidin-HRP: Enzyme conjugate for signal detection
- DAB Substrate: Chromogen producing brown precipitate
- Hydrogen Peroxide: Enzyme substrate for HRP
- Proteinase K: For antigen retrieval in tissue sections
- Endogenous Peroxidase Blocking Solution: (e.g., 3% H₂O₂ in methanol)
- Normal Serum: For blocking non-specific binding
- Methyl Green or Hematoxylin: Nuclear counterstain
Step-by-Step Procedure
Tissue Preparation:
- Deparaffinize and rehydrate formalin-fixed, paraffin-embedded tissue sections using standard histology protocols.
- Perform antigen retrieval with proteinase K (10-20 μg/mL in PBS) for 15-30 minutes at room temperature.
Endogenous Peroxidase Blocking:
- Incubate sections with 3% H₂O₂ in methanol for 10 minutes to quench endogenous peroxidase activity.
- Rinse with PBS.
Permeabilization:
- Treat sections with 0.1-0.5% Triton X-100 in PBS for 15 minutes.
- Rinse with PBS.
TdT Reaction:
- Prepare TdT reaction buffer: 100 μL TdT reaction buffer, 1 μL biotin-dUTP, and 1 μL TdT enzyme per sample.
- Apply to tissue sections and incubate in a humidified chamber for 60 minutes at 37°C.
- Rinse with PBS.
Signal Detection:
- Block with 2% normal serum for 30 minutes.
- Apply streptavidin-HRP (diluted according to manufacturer's instructions) for 30 minutes.
- Rinse with PBS.
- Prepare DAB solution according to manufacturer's instructions and apply to sections.
- Monitor development until brown color appears (typically 2-10 minutes).
- Rinse with distilled water to stop reaction.
Counterstaining and Mounting:
- Counterstain with Methyl Green or Hematoxylin.
- Dehydrate, clear, and mount with permanent mounting medium.
Visualization:
- Analyze by brightfield microscopy. Apoptotic cells display brown nuclear staining.
Technical Considerations and Troubleshooting
Method Selection Guidelines
The decision between fluorescence and colorimetric detection should be guided by specific experimental requirements. Fluorescence detection is preferable for: (1) applications requiring high sensitivity and quantification; (2) multiparametric analysis combining multiple markers; (3) flow cytometry or high-content screening applications; and (4) situations where background autofluorescence is manageable [13] [38]. Colorimetric detection may be more appropriate for: (1) single-parameter histological analysis; (2) laboratories without fluorescence capability; (3) archival purposes where fluorophores may fade over time; and (4) when a permanent record is needed [12].
Recent innovations in TUNEL methodology include the development of safer assay formulations that eliminate toxic components like sodium cacodylate, a carcinogenic arsenic derivative present in some traditional TUNEL reaction buffers that can itself induce apoptosis and create background noise [24]. Additionally, click chemistry-based approaches offer improved penetration and sensitivity due to the smaller size of the detection moieties compared to antibody-based methods [13].
Troubleshooting Common Issues
Table 3: Troubleshooting Guide for TUNEL Assay Performance Issues
| Problem | Potential Causes | Solutions |
|---|---|---|
| High Background | Inadequate blocking, over-fixation, endogenous enzyme activity, excessive TdT concentration | Optimize fixation time, include appropriate blocking steps, titrate TdT concentration, use recommended controls |
| Weak or No Signal | Insufficient permeabilization, low apoptosis incidence, enzyme inactivation, suboptimal substrate development | Validate with positive control (DNase-treated samples), optimize permeabilization, check reagent activity and storage conditions |
| Non-Specific Staining | Cell necrosis, inappropriate fixative, endogenous biotin (colorimetric) | Distinguish apoptosis from necrosis using morphological criteria, use alternative fixatives, block endogenous biotin |
| Cell Loss | Excessive washing, inadequate fixation, harsh handling | Use poly-lysine coated slides, optimize fixation protocol, minimize harsh washing steps |
Applications in Research and Drug Development
The TUNEL assay serves as a critical tool across multiple research domains. In cancer research and drug development, it is extensively used to evaluate the efficacy of chemotherapeutic agents by quantifying apoptosis induction in tumor cells [2]. In neurodegenerative disease research, the assay helps document apoptotic activity in neurons, which contributes to conditions like Alzheimer's and Parkinson's disease [24]. Developmental biologists employ TUNEL staining to identify areas of programmed cell death during embryogenesis and tissue remodeling [37]. Toxicological studies utilize the assay to assess cellular responses to environmental stressors and pharmaceutical compounds [2]. The choice between fluorescence and colorimetric detection in these applications depends on the specific requirements for throughput, quantification, and sample type.
The selection between fluorescence and colorimetric detection for TUNEL assays represents a critical methodological decision that directly impacts data quality, interpretation, and application scope. Fluorometric methods offer superior sensitivity, quantification capability, and multiplexing potential, making them ideal for most contemporary research applications, particularly drug screening and mechanistic studies where precise quantification is essential [38]. Colorimetric methods provide an accessible alternative for histological applications where permanent staining and brightfield microscopy are preferred [12].
Researchers should base their selection on experimental priorities, considering factors such as required sensitivity, available instrumentation, sample type, and desired throughput. Regardless of the chosen method, proper controls and validation are essential for accurate interpretation. As apoptosis research continues to evolve, particularly in the context of targeted therapies and personalized medicine, the TUNEL assay remains an indispensable tool for understanding cell death mechanisms and evaluating therapeutic efficacy.
The TUNEL (Terminal deoxynucleotidyl transferase dUTP Nick End Labeling) assay, first described in 1992, remains the most widely used in situ test for apoptosis research [13] [39] [12]. This technique specifically detects DNA fragmentation, a biochemical hallmark and ultimate determinate of late-stage apoptosis, where cells undergo controlled self-destruction [13] [1] [40]. The assay operates on the principle that the enzyme terminal deoxynucleotidyl transferase (TdT) catalyzes the template-independent addition of modified deoxynucleotides to the 3'-hydroxyl termini of DNA strand breaks [13] [1] [12]. These incorporated nucleotides are then detected directly via fluorescence or indirectly through antibody-based methods, enabling precise identification and quantification of apoptotic cells within tissue sections, adherent cells, or whole-mount specimens [13] [41] [39].
The critical importance of the TUNEL assay extends across multiple research domains, particularly in cancer development and autoimmune disease research, where understanding apoptotic mechanisms is fundamental [1] [40]. Its reliability stems from targeting a late-stage, committed event in the apoptotic cascade—internucleosomal DNA cleavage into characteristic 180-200 base pair fragments—making it a definitive marker for programmed cell death [1] [40]. Unlike necrosis, which represents pathological cell death with inflammatory consequences, apoptosis is a physiologically silent process essential for tissue homeostasis, development, and aging [1] [40]. The TUNEL assay's versatility allows adaptation to various experimental needs, from high-throughput drug screening to detailed morphological analysis in complex tissues [41] [42].
Principles and Mechanism
Biochemical Basis
The TUNEL assay capitalizes on the fundamental biochemical hallmark of apoptosis: systematic DNA cleavage by endonucleases activated during the cell death program [40]. In healthy cells, genomic DNA remains intact, but during apoptosis, activation of CAD (caspase-activated DNase) leads to targeted DNA cleavage between nucleosomes, generating abundant DNA fragments with exposed 3'-hydroxyl ends [1] [40]. The TUNEL assay specifically recognizes these exposed ends through the enzymatic activity of terminal deoxynucleotidyl transferase (TdT), which adds labeled nucleotides to the 3'-OH termini without requiring a template [13] [12].
The reaction requires cobalt ions as a cofactor in the buffer solution to facilitate efficient nucleotide incorporation [13] [1]. Detection strategies vary, with some protocols utilizing nucleotides directly conjugated to fluorophores (e.g., FITC-dUTP), while others employ hapten-labeled nucleotides (biotin-dUTP, DIG-dUTP, or EdUTP) that require subsequent detection with affinity reagents [13] [12]. The introduction of click chemistry-based approaches, such as the Click-iT TUNEL assay, has further enhanced detection efficiency by utilizing a small alkyne-modified dUTP that reacts with fluorescent azides in a copper-catalyzed cycloaddition, offering superior penetration and reduced background compared to antibody-based methods [13].
Key Steps and Their Purposes
The following diagram illustrates the core biochemical mechanism of the TUNEL assay, from DNA fragmentation in apoptotic cells to the visualization of labeled DNA ends.
Essential Reagents and Materials
The Scientist's Toolkit
A successful TUNEL experiment requires careful preparation of specific reagents and materials. The following table catalogs essential components referenced across established protocols, along with their functions and specifications.
| Reagent/Material | Function/Purpose | Specifications/Storage |
|---|---|---|
| Terminal Deoxynucleotidyl Transferase (TdT) | Catalyzes addition of modified nucleotides to 3'-OH ends of fragmented DNA [13] [12] | Recombinant enzyme; 15 U/µL in glycerol; store at ≤ -20°C [13] |
| Modified Nucleotide (dUTP) | Labeling substrate incorporated at DNA break sites [13] [12] | Alkyne-, Br-, DIG-, or biotin-modified; 50X solution; store at ≤ -20°C, desiccated, protected from light [13] |
| TdT Reaction Buffer | Provides optimal ionic environment for TdT activity [13] [1] | 1X solution; contains cobalt chloride; store at ≤ -20°C [13] |
| Fixative | Preserves cellular morphology and antigenicity [13] [41] | 4% paraformaldehyde in PBS; prepare fresh [13] [41] |
| Permeabilization Reagent | Creates pores in membrane for reagent entry [13] [41] | 0.25% Triton X-100 in PBS; can be stored at 4°C for up to a month [13] [41] |
| Blocking Solution | Reduces non-specific background binding [41] | 3% BSA in PBS or 10% Normal Donkey Serum in PBST; store in aliquots at -20°C [41] |
| Click-iT Reaction Buffer | Catalyzes fluorophore attachment via click chemistry [13] | 1X solution; contains Alexa Fluor azide; store protected from light [13] |
| DNase I | Generates DNA strand breaks for positive controls [13] | N/A; do not vortex to prevent denaturation [13] |
| Counterstain | Labels all nuclei for spatial context [13] [41] | Hoechst 33342 (10 mg/mL); known mutagen, handle with precautions [13] |
Detection Method Comparison
Different detection strategies offer distinct advantages depending on experimental requirements. The table below summarizes the primary approaches based on survey data from recent scientific publications.
| Detection Method | Principle | Relative Popularity | Key Advantages | Key Disadvantages |
|---|---|---|---|---|
| Direct Fluorescence | dUTP directly conjugated to fluorophore (e.g., FITC) [12] | 50% of publications [12] | Faster protocol (fewer steps) [12] | Potentially lower signal intensity |
| Biotin-Streptavidin | Biotin-dUTP detected with streptavidin-HRP and chromogenic substrate (e.g., DAB) [12] | 15% of publications [12] | Signal amplification via streptavidin-biotin complex [12] | Requires endogenous biotin blocking; more steps |
| Antibody-Based (FITC) | FITC-dUTP detected with anti-FITC antibody conjugated to HRP [12] | 15% of publications [12] | Bright signal | Requires additional antibody incubation |
| Antibody-Based (DIG) | DIG-dUTP detected with anti-digoxigenin antibody [12] | 12% of publications [12] | High specificity | Requires additional antibody incubation |
| BrdU-Based | BrdUTP detected with anti-BrdU antibody [12] | 8% of publications [12] | Brighter signal (easier TdT incorporation) [12] | Requires additional antibody incubation |
Standard Protocol Workflow
Comprehensive Workflow Diagram
The following flowchart provides a complete overview of the TUNEL assay procedure, integrating both core and optional steps to guide researchers from sample preparation through final analysis.
Step-by-Step Experimental Procedure
Day 1: Sample Preparation and Fixation
Sample Collection and Dissection
- For cell cultures: Grow cells on coverslips or in 96-well plates to approximately 60-80% confluence [13].
- For tissue samples: Dissect tissue promptly in cold 1X PBS. For Drosophila imaginal discs, dissection should not exceed 20 minutes at room temperature to prevent tissue disintegration [41].
- For whole-mount specimens (e.g., planarians): Transfer animals to 5% N-acetylcysteine (NAC) for 10 minutes at room temperature to remove mucus and kill humanely before fixation [39].
Fixation
Permeabilization
- Remove fixative and add permeabilization reagent (0.25% Triton X-100 in PBS).
- Incubate for 20 minutes at room temperature [13].
- Wash twice with deionized water or PBS.
Positive Control Preparation (Optional but Recommended)
- Prepare DNase I solution: Dilute Component G (DNase I) in the provided DNase I buffer according to kit specifications. Do not vortex, as DNase I denatures with vigorous mixing [13].
- Apply 100 µL of DNase I solution to designated positive control samples and incubate for 30 minutes at room temperature.
- Wash once with deionized water before proceeding [13].
Day 2: TdT Reaction and Detection
TdT Reaction Mix Preparation
- Prepare the TdT reaction mixture according to the chosen method:
TdT Reaction Incubation
Detection
- Direct Fluorescence Method: If using directly labeled nucleotides, wash samples thoroughly and proceed to counterstaining [12].
- Click Chemistry Method: Prepare Click-iT reaction cocktail according to manufacturer instructions. Apply to samples and incubate for 30-60 minutes at room temperature, protected from light. Wash thoroughly [13].
- Indirect Methods:
Counterstaining and Mounting
- Prepare nuclear counterstain (e.g., Hoechst 33342 or DAPI) at recommended dilution in PBS.
- Incubate samples for 10-15 minutes at room temperature, protected from light [13] [41].
- Wash twice with PBS or PBST.
- Mount samples using appropriate fluorescence mounting medium.
- For whole-mount specimens, use microscope slides and coverslips with mounting media designed for fluorescence microscopy [39].
Critical Experimental Considerations
Optimization and Troubleshooting
Successful TUNEL assay implementation requires attention to several critical variables that significantly impact results. Fixation time must be carefully controlled—under-fixation compromises morphology, while over-fixation (beyond 20 minutes in paraformaldehyde) can mask DNA breaks or create artificial ones, leading to false positives or reduced signal [41] [39]. Permeabilization optimization is equally crucial; insufficient permeabilization prevents TdT access to nuclear DNA, while excessive treatment damages cellular structure. The standard 20-minute incubation in 0.25% Triton X-100 works for most cell types, but may require adjustment for dense tissues or whole-mount specimens [13] [39].
Appropriate controls are essential for data interpretation. The positive control (DNase I treatment) should yield strong, uniform labeling across all nuclei, verifying assay functionality [13]. The negative control (omission of TdT enzyme) should show minimal signal, confirming specificity of labeling [12]. For antibody-based detection, additional controls excluding primary antibody help identify non-specific binding. Multiplexing considerations include careful fluorophore selection to avoid spectral overlap—for example, when using TMR red-based TUNEL detection, avoid Cy3-conjugated secondary antibodies for immunostaining [41]. Click chemistry-based TUNEL is incompatible with phalloidin staining but works well with antibody-based cytoskeletal markers [13].
Technical Advantages and Limitations
The TUNEL assay offers several significant advantages over alternative apoptosis detection methods. Its high sensitivity enables detection of DNA fragmentation at the single-cell level, providing spatial information within tissue architecture that bulk methods like gel electrophoresis cannot offer [1] [12]. The technique's versatility allows adaptation to various platforms including fluorescence microscopy, flow cytometry, and microplate readers, facilitating both qualitative and quantitative analysis [12]. Modern iterations like the Click-iT TUNEL assay demonstrate superior performance compared to traditional methods, detecting a higher percentage of apoptotic cells under identical conditions with faster processing times (complete within 2 hours) [13].
However, researchers must acknowledge important limitations. The potential for false positives exists, as the assay detects DNA breaks regardless of origin—including those from necrotic cell death, autolysis, or DNA repair processes [39] [40]. False negatives may occur in early apoptosis before significant DNA fragmentation, or with suboptimal fixation/permeabilization that prevents TdT access [39]. Recent innovations addressing these limitations include cacodylate-free reaction buffers that eliminate this carcinogenic component, reducing background signals and potential artifacts [1]. Electrochemical detection approaches using carbon nanotube-modified electrodes offer emerging alternatives for label-free, high-throughput screening of DNA fragmentation in apoptotic cells [42].
The TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) assay is a cornerstone method for detecting DNA fragmentation, a definitive hallmark of the late stages of programmed cell death or apoptosis [8] [2]. For decades, its utility was constrained by technical limitations of traditional detection methods. The integration of Click-iT chemistry with EdUTP incorporation represents a significant technological leap, offering researchers a superior tool for precise and multiplexed spatial analysis of apoptosis within complex biological systems [8] [13]. This application note details the principles, protocols, and recent advancements of this innovative approach, framing it within the context of modern apoptosis research and drug development.
Principles of Click-iT TUNEL Assay
The core innovation of the Click-iT TUNEL assay lies in its two-step detection strategy, which replaces bulky antibody-based or directly labeled nucleotide detection methods.
- Traditional TUNEL Limitations: Conventional assays use nucleotides modified with haptens like BrdU (detected with antibodies) or directly conjugated to fluorophores like fluorescein-dUTP. The former involves a multi-step, indirect detection process that can be time-consuming, while the latter uses a large modification that can hinder the enzyme's ability to incorporate the nucleotide efficiently [8] [13].
- Click-iT Advantage: The Click-iT approach utilizes a much smaller, alkyne-modified nucleotide called EdUTP (5-ethynyl-2’-deoxyuridine) [8]. The enzyme Terminal deoxynucleotidyl transferase (TdT) efficiently incorporates EdUTP at the 3'-OH ends of fragmented DNA. Detection is achieved via a copper-catalyzed "click" reaction—a bioorthogonal cycloaddition between the alkyne group on the incorporated EdUTP and an azide group conjugated to a bright, photostable Alexa Fluor dye [43] [8]. This method is highly specific, rapid, and, because of the small size of the EdUTP and azide, provides better access to DNA breaks, resulting in higher sensitivity and a greater percentage of apoptotic cells detected under identical conditions [13].
The following diagram illustrates the logical workflow and key chemical reaction of the Click-iT TUNEL assay.
Quantitative Performance and Compatibility
The superiority of the Click-iT TUNEL assay is demonstrated by direct comparisons with traditional methods. As shown in Table 1, the assay offers significant advantages in the percentage of apoptotic cells detected and the time required to complete the assay.
Table 1: Performance Comparison of TUNEL Assay Methods
| Assay Method | Modified Nucleotide | Detection Mechanism | % Apoptotic Cells Detected* | Total Assay Time | Key Advantages |
|---|---|---|---|---|---|
| Click-iT TUNEL | EdUTP (alkyne) | Click chemistry with Alexa Fluor azide | ~25% | ~2 hours | High sensitivity, mild conditions, efficient penetration [13] |
| BrdU TUNEL | BrdUTP | Antibody-based (anti-BrdU) | ~16% | >3 hours | Established protocol [8] |
| Fluorescein-dUTP | Fluorescein-dUTP | Direct fluorescence | ~10% | >3 hours | Direct detection, no antibody needed [13] |
| Fluorescein-dUTP (2) | Fluorescein-dUTP | Direct fluorescence | ~5% | >3 hours | Direct detection, no antibody needed [13] |
Data obtained from HeLa cells treated with 0.5 μM staurosporine for 4 hours [13].
Furthermore, the choice of detection azide allows for flexibility in experimental design, enabling multiplexing with other fluorescent markers. Table 2 summarizes the key fluorescent azides available.
Table 2: Click-iT TUNEL Alexa Fluor Azide Options
| Alexa Fluor Azide | Excitation/Emission Maxima (nm) | Compatible Standard Filter Set |
|---|---|---|
| Alexa Fluor 488 | 495/519 | FITC |
| Alexa Fluor 594 | 590/615 | Texas Red |
| Alexa Fluor 647 | 650/670 | Cy5 |
Detailed Experimental Protocol for Imaging in Cultured Cells
This protocol is adapted for adherent cells grown on coverslips and is based on the manufacturer's instructions [13]. The entire process, from fixation to mounting, can be completed in approximately 3.5 hours.
Cell Fixation and Permeabilization
- Fixation: Gently wash cells with PBS. Add enough 4% paraformaldehyde in PBS to completely cover the coverslips. Incubate for 15 minutes at room temperature. Remove the fixative.
- Permeabilization: Add sufficient 0.25% Triton X-100 in PBS to cover the coverslips. Incubate for 20 minutes at room temperature. Wash the coverslips twice with deionized water.
TdT-Mediated Incorporation of EdUTP
- Prepare the TdT reaction mixture by combining the following for each sample:
- 98 µL of TdT reaction buffer (Component A)
- 2 µL of EdUTP nucleotide mixture (Component B)
- 5 µL of TdT enzyme (Component C). Mix gently; do not vortex.
- Apply 100 µL of the TdT reaction mixture to each coverslip.
- Incubate for 60 minutes at 37°C in a humidified chamber.
Click-iT Reaction for Fluorescent Detection
- Prepare the Click-iT reaction cocktail for each sample as follows. Add components in the specified order:
- 430 µL of Click-iT reaction buffer (Component D, contains the Alexa Fluor azide)
- 20 µL of CuSO₄ (from a separate vial, not included in the initial table but standard in the reaction [43])
- 50 µL of 1X Click-iT reaction buffer additive (prepared by diluting Component E)
- Mix well. The total volume of 500 µL is sufficient for one coverslip.
- Apply 500 µL of the reaction cocktail to each coverslip.
- Incubate for 30 minutes at room temperature, protected from light.
- Remove the cocktail and wash the coverslips once with 1 mL of 3% BSA in PBS.
Counterstaining and Mounting
- To visualize all nuclei, stain the cells with 1 mL of a 1:2,000 dilution of Hoechst 33342 in PBS for 30 minutes at room temperature, protected from light [43].
- Wash the coverslips twice with PBS.
- Mount the coverslips onto slides using an appropriate anti-fade mounting media. The samples are now ready for imaging using fluorescence microscopy with the appropriate filter sets.
Advanced Application: Harmonizing TUNEL with Spatial Proteomics
A recent groundbreaking innovation is the successful integration of the Click-iT TUNEL assay with multiplexed spatial proteomic methods, such as Multiple Iterative Labeling by Antibody Neodeposition (MILAN) [16]. This allows for the rich spatial contextualization of cell death within tissues while simultaneously profiling dozens of protein markers.
The key challenge was the traditional use of proteinase K (ProK) for antigen retrieval in TUNEL, which was found to consistently reduce or abrogate protein antigenicity, making subsequent iterative antibody staining impossible [16]. The solution was to replace proteinase K treatment with heat-induced antigen retrieval using a pressure cooker. This modification:
- Preserved TUNEL Signal: The TUNEL signal was reliably produced independent of the antigen retrieval method, with pressure cooker treatment showing tissue-specific minor differences in signal-to-noise compared to ProK [16].
- Maintained Protein Antigenicity: Pressure cooker treatment enhanced protein antigenicity for the targets tested, enabling full compatibility with MILAN and cyclic immunofluorescence (CycIF) [16] [44].
- Enabled Erasable Staining: The antibody-based readout of the TUNEL signal was shown to be erasable using the 2-ME/SDS treatment of the MILAN protocol, allowing the same tissue section to be reused for multiple rounds of staining [16].
The following workflow diagram integrates this advanced, harmonized protocol for tissue sections.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents for Click-iT TUNEL and Related Assays
| Reagent / Kit | Primary Function | Research Application |
|---|---|---|
| Click-iT TUNEL Alexa Fluor Imaging Assay | Provides all components (TdT, EdUTP, Alexa Fluor azide) for fluorescent detection of apoptosis in cultured cells [13]. | Apoptosis detection via microscopy or high-content screening. |
| Click-iT Plus TUNEL Assay | Optimized with lower copper concentrations to preserve fluorescence of fluorescent proteins (e.g., GFP) and compatibility with phalloidin staining [8]. | Multiplexed apoptosis detection in samples expressing fluorescent proteins or requiring actin cytoskeleton labeling. |
| EdUTP (Component B) | Alkyne-modified nucleotide incorporated by TdT enzyme into sites of DNA fragmentation; the core of the click chemistry detection [8]. | Essential substrate for the Click-iT TUNEL reaction. |
| Alexa Fluor Azides | Bright, photostable dyes conjugated to an azide group that "clicks" with the incorporated EdUTP [43] [13]. | Flexible fluorescent detection with choice of color channels. |
| Terminal Deoxynucleotidyl Transferase (TdT) | Enzyme that catalyzes the template-independent addition of deoxynucleotides to the 3'-OH ends of DNA fragments [8] [13]. | Key enzyme for labeling DNA breaks in the TUNEL assay. |
| DNase I | Enzyme used to intentionally create DNA strand breaks in control samples, generating a positive TUNEL signal [13]. | Assay validation and protocol optimization. |
The integration of Click-iT chemistry with EdUTP incorporation has fundamentally enhanced the TUNEL assay, transforming it from a simple apoptosis detection tool into a powerful, sensitive, and versatile platform for cell death research. The provided detailed protocols and performance data empower researchers to robustly apply this method. Furthermore, its recent harmonization with spatial proteomics through antigen retrieval optimization opens new frontiers, allowing scientists to precisely map the tissue microenvironment of apoptotic cells. These innovative approaches are poised to accelerate discovery in fundamental biology, toxicology, and the development of novel therapeutics.
The TUNEL (Terminal deoxynucleotidyl transferase dUTP Nick-End Labeling) assay is a cornerstone method for detecting DNA fragmentation, a hallmark of late-stage apoptosis, in situ [1] [45] [8]. While it excels at spatially localizing cell death, its traditional implementation is limited to visualizing only a few additional protein targets via immunofluorescence (IF). This restriction hinders a comprehensive understanding of the complex cellular mechanisms and tissue microenvironments associated with programmed cell death [16].
Spatial proteomic technologies have emerged as powerful tools for profiling dozens of protein markers within a preserved tissue context. Among these, Multiple Iterative Labeling by Antibody Neodeposition (MILAN) and Cyclic Immunofluorescence (CyCIF) enable highly multiplexed protein imaging from a single specimen [46] [16]. However, the integration of TUNEL with these methods has been challenging due to incompatible tissue processing steps, particularly the use of proteinase K (ProK) for antigen retrieval in standard TUNEL protocols, which degrades protein epitopes and abrogates subsequent antibody binding [16].
This application note details a harmonized protocol that replaces ProK with heat-induced antigen retrieval, enabling the robust integration of TUNEL with both MILAN and CyCIF. This advancement allows researchers to richly contextualize cell death within the complex spatial architecture of tissues, providing unprecedented insights for disease research and drug development [16] [44].
The Core Challenge: Proteinase K Incompatibility
The key obstacle to combining TUNEL with iterative immunofluorescence methods is the fundamental incompatibility of proteinase K (ProK) with the preservation of protein antigenicity.
- ProK Treatment Severely Diminishes Protein Antigenicity: In comparative studies, ProK digestion, a standard step in most commercial TUNEL kits, consistently reduced or entirely abrogated signal for subsequent protein targets in MILAN and CycIF cycles. This irreversible damage prevents meaningful multiplexing [16].
- Pressure Cooker Retrieval Preserves Antigenicity: Heat-induced antigen retrieval using a pressure cooker was found to be an effective alternative. This method not only maintained TUNEL sensitivity but also enhanced antigenicity for the protein targets tested, making it ideal for multiplexed workflows [16] [44].
Table 1: Impact of Antigen Retrieval Method on TUNEL and Protein Antigenicity
| Antigen Retrieval Method | TUNEL Signal Quality | Preservation of Protein Antigenicity | Compatibility with MILAN/CycIF |
|---|---|---|---|
| Proteinase K (ProK) | Reliable | Severely diminished or abrogated | No |
| Pressure Cooker | Reliable (tissue-specific minor differences in S/N) | Enhanced | Yes |
Harmonized Protocol for TUNEL with MILAN
The following protocol is optimized for the seamless integration of an antibody-based TUNEL assay into the MILAN spatial proteomics workflow.
Key Reagent Solutions
Table 2: Essential Research Reagents for the Harmonized Workflow
| Reagent / Kit | Function in the Protocol |
|---|---|
| Click-iT Plus TUNEL Assay (or in-house BrdU-based TUNEL) | Detects DNA fragmentation via TdT-mediated incorporation of EdUTP/BrdUTP [16] [8]. |
| Pressure Cooker | For heat-induced epitope retrieval (HIER); preserves protein antigens [16]. |
| 2-Mercaptoethanol/SDS (2-ME/SDS) Solution | Antibody erasure solution for MILAN; removes primary and secondary antibodies between cycles [16]. |
| Primary Antibodies | For multiplexed detection of protein targets in MILAN cycles. |
| Fluorophore-conjugated Secondary Antibodies | For visualization of protein targets. |
Detailed Experimental Workflow
The integrated process involves performing TUNEL first, erasing the detection antibodies, and then proceeding with iterative rounds of multiplexed immunofluorescence.
Step-by-Step Methodology
Tissue Preparation:
- Use formalin-fixed, paraffin-embedded (FFPE) tissue sections (4-5 µm thick) mounted on glass slides.
- Deparaffinize and rehydrate sections using standard xylene and ethanol series.
Antigen Retrieval (Critical Step):
- Perform heat-induced epitope retrieval (HIER) using a pressure cooker in an appropriate buffer (e.g., citrate or EDTA buffer). Do not use proteinase K. [16]
TUNEL Assay:
- Reaction: Incubate sections with the TUNEL reaction mixture containing terminal deoxynucleotidyl transferase (TdT) and a modified nucleotide (EdUTP for Click-iT kits or BrdUTP for antibody-based detection) [16] [8].
- Detection:
- For Click-iT-based kits, perform the copper-catalyzed "click" reaction with a fluorescent azide.
- For antibody-based TUNEL, incubate with an anti-BrdU primary antibody followed by a fluorophore-conjugated secondary antibody [16].
Initial Imaging (Round 1):
- Acquire the TUNEL signal using a fluorescence microscope. This image will be registered with subsequent cycles.
Antibody Erasure:
- Remove coverslips and incubate slides in 2-ME/SDS erasure buffer at 66°C. This step completely removes the TUNEL detection antibodies (primary and secondary) while leaving the incorporated nucleotides and protein antigens intact [16].
MILAN Cyclic Immunofluorescence:
- Begin iterative cycles of multiplexed protein staining: a. Incubate with a panel of primary antibodies (typically 3-5 per cycle). b. Incubate with fluorophore-conjugated secondary antibodies. c. Image the slide to capture the signal for these protein targets. d. Erase antibodies using the 2-ME/SDS buffer at 66°C.
- Repeat steps a-d for the desired number of cycles to build a highly multiplexed protein profile [16].
Data Analysis:
- Align all image cycles using rigid registration.
- Segment cells and analyze multiplexed protein expression in conjunction with TUNEL positivity to spatially map cell death within specific cellular phenotypes and tissue neighborhoods [16].
Harmonized Protocol for TUNEL with CycIF
The protocol for CycIF follows a similar logic but is adapted to the specific staining and erasure conditions of CycIF, which often relies on harsher denaturants or high-pH buffers for fluorophore inactivation [46] [16].
Key Workflow Modifications
- Erasure Method: Instead of 2-ME/SDS, CycIF typically uses chemical erasure with buffers containing SDS or similar detergents at high pH, or mild acid treatment to inactivate fluorophores [46] [47].
- Integration Order: The TUNEL assay can be performed as the first cycle of the CycIF process. Following TUNEL imaging and erasure, standard CycIF cycles for protein markers proceed [16].
Experimental Validation and Data Output
The harmonized protocol was validated in defined murine models of cell death [16]:
- Necrosis: Acetaminophen (APAP)-induced hepatocyte necrosis in the liver.
- Apoptosis: Dexamethasone-induced adrenocortical apoptosis.
Table 3: Quantitative Performance of Harmonized vs. Standard TUNEL
| Performance Metric | Standard TUNEL (with ProK) | Harmonized TUNEL (with Pressure Cooker) |
|---|---|---|
| TUNEL Signal-to-Noise | Reliable in positive controls [16] | Reliable, with tissue-specific minor differences [16] |
| Preservation of Protein Antigenicity | Severely compromised | Fully preserved |
| Compatibility with Iterative Staining | No | Yes (validated for ≥4 subsequent MILAN/CycIF cycles) [16] |
| Spatial Contextualization | Limited (2-3 markers) | High (20+ markers) |
The data output is a multi-layered, spatially resolved image set where TUNEL-positive cells can be precisely phenotyped based on the expression of dozens of proteins, allowing for deep analysis of cell death in its tissue context.
The harmonization of the classic TUNEL assay with modern spatial proteomics platforms like MILAN and CycIF resolves a significant technical limitation in cell death research. By substituting proteinase K with pressure cooker antigen retrieval, researchers can now perform highly multiplexed spatial analysis on the same tissue section where apoptosis is detected. This protocol enables the precise correlation of DNA fragmentation with specific cell states, signaling pathways, and cellular interactions, greatly enriching our ability to investigate the role of programmed cell death in cancer, autoimmune diseases, and drug development.
Apoptosis, or programmed cell death, is a highly regulated process essential for maintaining tissue homeostasis in multicellular organisms. It is characterized by distinct morphological and biochemical markers, including cell shrinkage, chromatin condensation, and nuclear DNA fragmentation [24]. The inappropriate regulation of apoptosis is a hallmark of various diseases; excessive apoptosis is implicated in neurodegenerative disorders such as Alzheimer's and Parkinson's disease, while insufficient apoptosis can lead to cancerous growths [24]. Two primary pathways initiate apoptosis: the extrinsic pathway, activated by extracellular death receptor ligands, and the intrinsic pathway, triggered by intracellular stressors leading to mitochondrial membrane permeabilization [24]. Both pathways converge on the activation of caspases, which ultimately leads to the digestion of nuclear DNA by caspase-activated nucleases [24].
The TUNEL assay is a pivotal technique for detecting apoptotic cell death. The assay identifies DNA fragmentation—a key late-stage event in apoptosis—by leveraging the enzyme Terminal deoxynucleotidyl Transferase (TdT). This enzyme catalyzes the addition of fluorescently-labeled or modified deoxyuridine triphosphate (dUTP) to the 3'-hydroxyl termini of DNA double-strand breaks [24] [13]. Since its introduction in 1992, the TUNEL assay has become a widely accepted in situ method for identifying apoptotic cells due to its high sensitivity and capacity to provide quantitative measurements over a broad range [24] [13]. Its utility spans fundamental cancer research, drug discovery, and clinical diagnostics, particularly in the assessment of male infertility through sperm DNA fragmentation analysis [48] [49] [50].
Application Spectrum of the TUNEL Assay
The TUNEL assay serves as a versatile tool across diverse fields of biomedical research and clinical diagnostics. The table below summarizes its primary applications, key objectives, and specific research contexts.
Table 1: Spectrum of TUNEL Assay Applications
| Field of Application | Primary Objective | Specific Research or Clinical Context |
|---|---|---|
| Cancer Research | Study drug-induced apoptosis; evaluate efficacy of chemotherapeutic agents [24] [13] | HeLa, A549, and CHO K1 cells treated with apoptosis inducers like staurosporine [24] [13]. |
| Neurodegenerative Disease Research | Investigate excessive apoptotic activity contributing to neuronal loss [24] | Studies on Alzheimer's and Parkinson's disease models [24]. |
| Male Infertility Assessment | Evaluate sperm DNA integrity as a diagnostic and prognostic marker [48] [49] [50] | Unexplained infertility, recurrent pregnancy loss, varicocele, and prior to assisted reproductive techniques (IUI, IVF, ICSI) [48]. |
| Develop Biology | Quantify cell death during tissue remodeling and organogenesis [51] | Drosophila melanogaster eye-antennal imaginal discs [51]. |
| Drug Discovery & Therapy Monitoring | Screen compounds for pro- or anti-apoptotic activity; monitor treatment response [52] | Real-time apoptosis surveillance in complex biological settings for data-driven drug discovery [52]. |
In-Depth Focus: Sperm DNA Fragmentation Analysis
Sperm DNA fragmentation (SDF) is a critical factor in male infertility, with implications for fertilization success, embryo quality, and pregnancy outcomes [48]. The TUNEL assay is one of the most reliable and sensitive methods for directly assessing SDF [49] [50]. Infertile men frequently exhibit higher levels of SDF compared to fertile men, and conditions such as varicocele, infections, advanced paternal age, and lifestyle factors can contribute to elevated SDF [48].
Clinical Validity and Thresholds: The TUNEL assay has been validated for clinical use in andrology laboratories. A recognized cutoff value for TUNEL in sperm analysis is 26%, with reported sensitivity and specificity of 85% and 89%, respectively, for discriminating between fertile and infertile men [49]. Another meta-analysis established a threshold of 20% as optimal for this discrimination [48]. These thresholds help clinicians identify patients for whom high SDF may be a contributing factor to infertility.
Correlation with Standard Semen Parameters: While SDF is considered an independent marker of sperm quality, studies show a significant negative correlation between SDF levels and traditional parameters such as sperm motility, morphology, and vitality, particularly in sperm samples prepared through techniques like swim-up [49] [50]. This suggests that sperm with damaged DNA are often functionally deficient in other aspects.
Choosing the Right Sample for Testing: A key consideration is whether to perform the TUNEL assay on raw (neat) semen or on processed samples (e.g., after swim-up). Research indicates that while median SDF values may not differ significantly between pre- and post-swim-up samples, a substantial proportion (e.g., 39.1% in one study) can show a marked difference [49]. Processed samples often show a stronger correlation with motility and morphology [49]. The choice of sample should therefore be guided by the clinical context: a raw ejaculate may reflect the natural state, while a processed sample may better represent the sperm selected for use in assisted reproductive technologies [49].
Detailed Experimental Protocols
Protocol 1: TUNEL Assay for Adherent Cells (e.g., Cancer Cell Lines)
This protocol is optimized for adherent cells grown on coverslips or in 96-well plates, using the Click-iT TUNEL methodology for high sensitivity [13].
Key Materials:
- TdT (Terminal deoxynucleotidyl transferase): Enzyme that incorporates modified nucleotides into DNA breaks [13].
- Modified Nucleotide (e.g., EdUTP): A dUTP modified with an alkyne for bio-orthogonal click chemistry [13].
- Click-iT Reaction Buffer: Contains an Alexa Fluor azide for fluorescent detection [13].
- Fixative: 4% paraformaldehyde in PBS.
- Permeabilization Reagent: 0.25% Triton X-100 in PBS.
- DNase I (Optional): For generating positive control samples [13].
Table 2: Key Reagent Solutions for TUNEL Assay
| Reagent | Function | Key Considerations |
|---|---|---|
| TdT Enzyme | Catalyzes the addition of labeled nucleotides to 3'-OH ends of fragmented DNA. | Sensitive to storage conditions; requires -20°C storage [13]. |
| Modified dUTP (e.g., Tunnelyte, EdUTP) | The labeled substrate incorporated into DNA breaks. | Directly fluorescent tags or small alkyne groups allow for more efficient incorporation and simpler protocols [24] [13]. |
| Click-iT Reaction Buffer | Facilitates the copper-catalyzed "click" reaction between azide and alkyne for detection. | Contains Alexa Fluor azides; small size improves penetration [13]. |
| TdT Reaction Buffer | Provides optimal biochemical conditions for TdT enzyme activity. | May contain potassium cacodylate, a toxic arsenic derivative; handle with care [13]. Safer, cacodylate-free buffers are available [24]. |
Workflow Steps:
Cell Fixation and Permeabilization:
- Remove culture media and wash cells once with PBS.
- Fix cells by adding 4% paraformaldehyde in PBS for 15 minutes at room temperature.
- Remove the fixative and permeabilize cells by adding 0.25% Triton X-100 in PBS for 20 minutes at room temperature.
- Wash the cells twice with deionized water [13].
Preparing a Positive Control (Optional):
- On a separate coverslip, treat fixed and permeabilized cells with a DNase I solution (e.g., 1 U/mL in DNase I buffer) for 30 minutes at room temperature to intentionally introduce DNA strand breaks.
- Wash once with deionized water before proceeding [13].
TdT Reaction (Labeling DNA Breaks):
Click Reaction (Detection):
- Prepare the Click-iT reaction mixture from the provided components (Click-iT reaction buffer and additive).
- Apply this mixture to the cells and incubate for 30 minutes at room temperature, protected from light.
- Wash the cells with a buffer like 3% BSA in PBS [13].
Counterstaining and Visualization:
- Counterstain nuclei using a dye like Hoechst 33342 (a known mutagen; handle with care) or DAPI.
- Mount the coverslips and visualize using a fluorescence microscope with appropriate filter sets for the fluorophores used [13].
Protocol 2: TUNEL Assay for SDNA Fragmentation Analysis via Flow Cytometry
This protocol is designed for the quantitative assessment of sperm DNA fragmentation using flow cytometry, which allows for high-throughput analysis of thousands of cells [49] [50].
Key Materials:
- Semen Sample: Collected after 3-5 days of sexual abstinence [49].
- Propidium Iodide (PI): A DNA stain used to differentiate sperm and assess viability [49].
- TdT Enzyme, Modified dUTP, and Reaction Buffers.
Workflow Steps:
Sample Preparation and Basic Analysis:
Sperm Processing (Optional - Swim-Up):
Fixation and Permeabilization:
- Wash an aliquot of the sperm sample (e.g., 100 μL) in PBS and centrifuge.
- Fix and permeabilize the sperm cells to allow reagent entry. Methods can include a methanol:acetic acid fixative or a detergent-based buffer [50].
TUNEL Reaction:
Staining and Flow Cytometry:
- After the TUNEL reaction, stain the sperm with Propidium Iodide (PI) to label all sperm nuclei [49].
- Analyze the sample using a flow cytometer. The instrument will detect sperm with fragmented DNA (TUNEL-positive, fluorescein signal) and total sperm (PI-positive) [49] [50].
- The DNA Fragmentation Index (DFI) is calculated as the percentage of TUNEL-positive sperm out of the total sperm analyzed [50].
Apoptosis Signaling Pathways and Detection Logic
The TUNEL assay detects the end product of a cascade of biochemical events. Understanding the upstream pathways that lead to DNA fragmentation is crucial for interpreting TUNEL results.
Intrinsic Pathway (Mitochondrial): This pathway is activated by internal stressors like DNA damage, oxidative stress, or growth factor withdrawal. It leads to mitochondrial outer membrane permeabilization (MOMP), resulting in the release of pro-apoptotic factors such as cytochrome c into the cytoplasm. Cytochrome c, along with Apaf-1, forms the "apoptosome," which activates caspase-9. Caspase-9 then initiates the caspase cascade, culminating in the activation of effector caspases like caspase-3 [24] [52].
Extrinsic Pathway (Death Receptor): This pathway is triggered by the binding of extracellular death ligands (e.g., FasL, TNF-α) to their corresponding cell surface death receptors. This binding induces the formation of the Death-Inducing Signaling Complex (DISC), which directly activates caspase-8. Caspase-8 can then directly activate effector caspases [24].
Execution Phase and DNA Fragmentation: Both pathways converge on the activation of effector caspases (e.g., caspase-3). These caspases cleave and activate specific substrates, including the ICAD/DFF45 protein, which in turn releases the CAD nuclease. CAD is the ultimate executor that cleaves nuclear DNA into the characteristic oligonucleosomal fragments, creating the 3'-OH ends that are detected by the TUNEL assay [24].
The Scientist's Toolkit: Essential Reagents and Materials
Successful execution of the TUNEL assay requires a set of core reagents. The following table details these essential components and their functions.
Table 3: Essential Research Reagent Solutions for the TUNEL Assay
| Reagent/Material | Function in the Assay | Technical Notes & Safety |
|---|---|---|
| Terminal Deoxynucleotidyl Transferase (TdT) | The core enzyme that catalyzes the template-independent addition of labeled dUTP to 3'-OH ends of DNA breaks. | Recombinant enzymes offer high activity. Store at -20°C [13]. |
| Labeled dUTP | The substrate incorporated into DNA breaks. Can be directly fluorescent (e.g., Fluorescein-dUTP) or modified (e.g., EdUTP, Biotin-dUTP). | Direct labels simplify protocols; haptenated labels (Biotin, EdU) allow signal amplification [24] [13]. |
| TdT Reaction Buffer | Provides optimal pH and ionic strength (often containing cacodylate buffer) and cofactors (e.g., Co²⁺) for TdT activity. | Caution: Traditional buffers may contain potassium cacodylate, an arsenic compound that is highly toxic. Safer, cacodylate-free buffers are available [24] [13]. |
| Click-iT Chemistry Kit (for EdUTP) | Enables highly sensitive detection of the alkyne-modified EdUTP via a copper-catalyzed reaction with a fluorescent azide. | Offers superior penetration and sensitivity compared to antibody-based detection. The copper catalyst can interfere with some fluorophores [13]. |
| Paraformaldehyde (PFA) | Cross-linking fixative that preserves cellular morphology while immobilizing antigens and nucleic acids. | Typically used at 4% in PBS. Handle in a fume hood. |
| Permeabilization Agent (e.g., Triton X-100) | A detergent that creates pores in the cell membrane, allowing large molecules like TdT to enter the cell and access the nucleus. | Concentration and time must be optimized to balance access with preservation of structure [13]. |
| DNase I (Deoxyribonuclease I) | Enzyme used to intentionally introduce DNA strand breaks in control samples, ensuring the TUNEL reaction is working correctly. | Do not vortex the DNase I solution, as vigorous mixing can denature the enzyme [13]. |
| Propidium Iodide (PI) / DAPI | DNA counterstains used to label all nuclei, allowing for the calculation of the percentage of TUNEL-positive cells. | Caution: PI and DAPI are known mutagens. Handle with appropriate personal protective equipment [13] [49]. |
Solving Common TUNEL Assay Problems and Enhancing Performance
Addressing Weak or Absent Fluorescence Signals
In the context of a broader thesis on utilizing the TUNEL assay for DNA fragmentation research in late apoptosis, obtaining a clear and robust fluorescence signal is paramount. Weak or absent signals can compromise data integrity, leading to false negatives and an underestimation of apoptotic events. This application note provides a structured, evidence-based guide to diagnose and rectify the common issues of weak or absent fluorescence in TUNEL assays, ensuring reliable and reproducible results for researchers, scientists, and drug development professionals.
The TUNEL (Terminal deoxynucleotidyl transferase dUTP Nick-End Labeling) assay is a cornerstone method for detecting the extensive DNA fragmentation that characterizes the late stages of apoptosis [53]. The core principle involves using the enzyme Terminal deoxynucleotidyl transferase (TdT) to add fluorescently-labeled nucleotides to the 3'-hydroxyl ends of fragmented DNA, which are then visualized microscopically or via flow cytometry [8] [12]. When this signal is weak or absent, a systematic investigation of the protocol is required, focusing on sample preparation, enzyme activity, and detection efficiency.
Troubleshooting Weak TUNEL Signals: A Systematic Approach
A methodical approach is crucial for diagnosing the root cause of signal failure. The following workflow outlines the key decision points and corrective actions. The diagram below maps the logical pathway for troubleshooting weak or absent TUNEL signals, from initial control checks to specific investigative actions.
The first critical step is to run controls. A positive control, typically a sample treated with DNase I to artificially fragment all DNA, should show strong nuclear fluorescence [53]. If this signal is also weak, the problem lies with the assay reagents or detection system. If the positive control is strong, the issue is specific to your sample preparation or the apoptotic process in your experimental samples. Simultaneously, a negative control (omitting the TdT enzyme) should show no signal; high background here indicates non-specific binding [53].
Primary Causes and Corrective Actions
Based on the troubleshooting logic, the table below summarizes the most common causes of weak signals and their evidence-based solutions.
Table 1: Common Causes and Solutions for Weak TUNEL Fluorescence
| Category | Specific Cause | Proposed Solution | Rationale & Evidence |
|---|---|---|---|
| Sample Preparation | Insufficient Permeabilization [53] | Optimize concentration (0.1-1% Triton X-100) and incubation time (5-15 mins on ice) for cell type. | The large TdT enzyme cannot access nuclear DNA without adequate membrane disruption. |
| Over-fixation [53] | Limit fixation with 1-4% PFA to 15-30 minutes at room temperature. | Excessive cross-linking can mask DNA breaks, blocking the 3'-OH ends. | |
| Suboptimal Antigen Retrieval (FFPE tissue) [16] | Replace proteinase K with pressure cooker-based epitope retrieval. | Proteinase K drastically reduces protein antigenicity, while heat-induced retrieval preserves TUNEL signal and enables multiplexing [16]. | |
| Reaction & Detection | Compromised TdT Enzyme | Aliquot and avoid freeze-thaw cycles; include a positive control to confirm activity. | TdT is a critical, sensitive reagent. A failed positive control often points to enzyme inactivation. |
| Inefficient Nucleotide Incorporation | Consider BrdUTP-based methods or Click-iT Plus kits with EdUTP for brighter, more efficient labeling [8] [12]. | BrdUTP is more easily incorporated by TdT, and optimized "click" chemistry can enhance sensitivity [8] [12]. | |
| Fluorophore Degradation | Protect all fluorescent reagents from light; confirm the integrity of secondary antibodies and azide dyes. | Photobleaching during storage or handling can quench the signal before imaging. | |
| General Pitfalls | Inadequate Controls | Always run DNase I (positive) and No TdT (negative) controls in parallel. | Controls are non-negotiable for diagnosing whether the issue is technical or biological [53]. |
Experimental Protocols for Optimization
Protocol 1: Standardized TUNEL Assay with Enhanced Permeabilization
This protocol provides a robust baseline, emphasizing critical steps for signal optimization in cultured cells [53].
Materials:
- Fixation Solution: 4% Paraformaldehyde (PFA) in PBS
- Permeabilization Solution: 0.1-0.5% Triton X-100 in PBS (pre-chilled)
- TUNEL Reaction Kit (e.g., Click-iT Plus TUNEL Assay or equivalent [8])
- Blocking Buffer (e.g., 1-3% BSA in PBS)
- DAPI or other nuclear counterstain
- Antifade Mounting Medium
Step-by-Step Procedure:
- Sample Preparation and Fixation:
- Wash adherent cells gently with PBS.
- Fix cells with 4% PFA for 15 minutes at room temperature. Avoid prolonged fixation.
- Wash thoroughly with PBS 2-3 times.
Permeabilization (Critical Optimization Step):
- Incubate cells with 0.3% Triton X-100 in PBS for 10 minutes on ice.
- The concentration and time may require titration for different cell lines. Harsher conditions (e.g., 0.5-1% Triton X-100) may be needed for tissues [53].
Positive Control Preparation:
- On a separate coverslip, treat fixed and permeabilized cells with DNase I (1 µg/mL in PBS) for 15-30 minutes at room temperature to introduce DNA breaks. Rinse with PBS before proceeding.
TUNEL Reaction:
- Prepare the TUNEL reaction mix according to the manufacturer's instructions.
- For the negative control, prepare a reaction mix omitting the TdT enzyme.
- Apply the reaction mixes to the samples and incubate for 60 minutes at 37°C in a humidified dark chamber.
Detection and Washing:
- Terminate the reaction as per kit instructions (often with a wash buffer).
- Wash samples 2-3 times with PBS containing 0.1% Tween-20 (PBST) to reduce background.
Counterstaining and Mounting:
- Incubate with DAPI (1 µg/mL) for 5-10 minutes to label all nuclei.
- Wash with PBS.
- Mount coverslips with an antifade mounting medium and seal.
Protocol 2: Proteinase K-Free TUNEL for Multiplexing with Spatial Proteomics
Recent research demonstrates that proteinase K, a common TUNEL reagent, severely degrades protein antigenicity, preventing multiplexing with other protein biomarkers [16]. This advanced protocol replaces proteinase K with heat-induced antigen retrieval.
Materials:
- Formalin-fixed, paraffin-embedded (FFPE) tissue sections.
- Citrate or EDTA-based antigen retrieval buffer.
- Pressure cooker or commercial decloaking chamber.
- Standard TUNEL reagents (excluding proteinase K).
Step-by-Step Procedure:
- Dewax and Rehydrate: Follow standard procedures for FFPE sections.
- Heat-Induced Antigen Retrieval:
- Place slides in a cop jar filled with antigen retrieval buffer.
- Perform heat-induced retrieval in a pressure cooker for 10-15 minutes as per standard IHC protocols for your target antigens [16].
- Allow slides to cool slowly to room temperature in the buffer.
- Permeabilization:
- Optional: For some tissues, a brief, mild permeabilization with 0.1% Triton X-100 may be applied after cooling, though the heat retrieval is often sufficient.
- TUNEL Reaction and Detection:
- Proceed with the standard TUNEL protocol from Step 4 (TUNEL Reaction) as described in Protocol 1.
This harmonized protocol preserves tissue architecture and protein epitopes, enabling the rich contextualization of cell death within complex tissues through iterative staining methods like MILAN (Multiple Iterative Labeling by Antibody Neodeposition) [16].
The Scientist's Toolkit: Essential Reagents for Reliable TUNEL
The following table details key reagent solutions and their critical functions in achieving a successful TUNEL assay.
Table 2: Key Research Reagent Solutions for TUNEL Assays
| Reagent / Kit | Core Function | Key Considerations for Signal Strength |
|---|---|---|
| Terminal Deoxynucleotidyl Transferase (TdT) | Catalyzes the template-independent addition of labeled nucleotides to 3'-OH DNA ends. | Enzyme activity is paramount. Aliquot to avoid freeze-thaw cycles. A failed positive control often indicates inactive TdT. |
| Labeled Nucleotides (e.g., BrdUTP, EdUTP, FITC-dUTP) | Serves as the detectable label incorporated at DNA break sites. | BrdUTP/EdUTP often provide brighter signals than FITC-dUTP due to more efficient incorporation or detection amplification [12] [8]. |
| Click-iT Plus TUNEL Assay Kits [8] | Provides a complete system using EdUTP and optimized "click" chemistry for detection. | The "Plus" kits use lower copper concentrations, preserving signal from fluorescent proteins (e.g., GFP) and phalloidin binding during multiplexing. |
| Permeabilization Agent (Triton X-100, Proteinase K) | Creates pores in the cell membrane to allow TdT enzyme access to the nucleus. | Concentration and time are critical. Under-permeabilization causes false negatives; over-permeabilization can damage nuclear structure. |
| DNase I (Recombinant) | Creates universal DNA breaks for the positive control. | Essential for validating the entire workflow. Confirms that weak signal is not due to reagent failure. |
| Pressure Cooker & Retrieval Buffer [16] | Replaces proteinase K for antigen retrieval in FFPE tissues. | Crucial for multiplexing. Preserves protein antigenicity for downstream antibody staining while enabling effective TUNEL labeling. |
Addressing weak or absent fluorescence in TUNEL assays requires a disciplined, step-wise approach centered on rigorous controls and systematic optimization of sample preparation and detection. Key strategies include validating the assay with a DNase I positive control, carefully titrating permeabilization conditions, and considering modern kit-based solutions that offer enhanced sensitivity and compatibility. Furthermore, adopting proteinase K-free protocols opens the door to powerful multiplexed analyses, allowing apoptosis to be contextualized within a rich spatial proteomic landscape. By applying these detailed protocols and troubleshooting guidelines, researchers can overcome common pitfalls and generate robust, high-quality data on DNA fragmentation in their late apoptosis research.
Reducing High Background and Non-Specific Staining in TUNEL Assays
The TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) assay is a cornerstone technique for detecting DNA fragmentation, a hallmark biochemical event of late-stage apoptosis [37] [12]. Since its introduction in 1992, it has become the most widely used in situ method for apoptosis research due to its exceptional sensitivity in identifying DNA double-strand breaks [13] [24]. The assay operates on the principle that the enzyme terminal deoxynucleotidyl transferase (TdT) catalyzes the addition of labeled deoxynucleotides to the 3'-hydroxyl termini of fragmented DNA [12] [6]. These labels, typically fluorophores or haptens, enable visualization and quantification of apoptotic cells via microscopy or flow cytometry.
Despite its widespread adoption and utility, researchers frequently encounter the technical challenge of high background and non-specific staining, which can compromise data interpretation and experimental validity [13]. This background noise may arise from multiple sources, including inadequate fixation, suboptimal permeabilization, endogenous enzyme activities, or non-specific binding of detection reagents. The complexity is further amplified when working with diverse sample types, such as tissue sections versus cultured cells, each requiring tailored optimization approaches. Within the broader context of DNA fragmentation research in late apoptosis, distinguishing true apoptotic signals from artifacts is paramount for generating reliable data, particularly in pharmaceutical development where accurate apoptosis quantification directly impacts therapeutic agent evaluation.
The fundamental principle underlying TUNEL assay background stems from the specific biochemical reaction it exploits. During apoptosis, endonucleases such as CAD (caspase-activated DNase) cleave DNA at internucleosomal linker sites, generating fragments of approximately 180-200 base pairs [24] [2]. TdT recognizes the 3'-OH ends created by this fragmentation and incorporates modified nucleotides. However, DNA breaks can also occur through non-apoptotic mechanisms, including necrosis, pyroptosis, mechanical shearing during sample preparation, or fixed-tissue artifacts, all of which can contribute to false-positive signals if not properly controlled [37] [54].
The choice of nucleotide label and detection system significantly influences background levels. Survey data from 50 recently published studies reveals that approximately 50% of TUNEL assays use dUTP directly conjugated to FITC, while other methods employ biotin-dUTP with streptavidin-HRP (15%), FITC-dUTP with anti-FITC-HRP (15%), digoxygenin-dUTP with anti-digoxygenin antibodies (12%), or Br-dUTP with anti-BrdU antibodies (8%) [12]. Each detection methodology presents distinct advantages and potential background sources. For instance, assays utilizing biotin-tagged nucleotides require additional blocking steps to neutralize endogenous biotin, whereas BrdU-based methods often provide brighter signals due to more efficient TdT incorporation but may introduce antibody-related background [12].
Table 1: Common Sources of Background Staining in TUNEL Assays
| Background Source | Manifestation | Underlying Cause |
|---|---|---|
| Incomplete Fixation | Diffuse nuclear staining | Poor preservation of cellular architecture allowing non-specific probe access |
| Over-Permeabilization | Excessive staining intensity | Unmasking of non-apoptotic DNA breaks and increased non-specific binding |
| Endogenous Biotin | False-positive signals (HRP systems) | Interference with streptavidin-biotin detection systems |
| Free Aldehyde Groups | High background fluorescence | Inadequate removal of fixative leading to non-specific conjugate binding |
| Endogenous Nucleases | Nuclear staining in negative controls | Enzyme activity persisting after fixation |
| TdT Concentration | Non-specific labeling | Enzyme excess leading to non-template nucleotide addition |
Critically, the chemical composition of reaction buffers can directly impact background staining. Traditional TUNEL assays often incorporate sodium or potassium cacodylate, a carcinogenic arsenic derivative that is highly toxic by ingestion, inhalation, or skin contact [24]. Beyond safety concerns, this compound can itself induce apoptosis at certain concentrations, inevitably generating background noise during measurement. Novel TUNEL formulations that eliminate cacodylate buffers demonstrate enhanced sensitivity and specificity while providing safer handling characteristics [24].
Comparative Analysis of TUNEL Methodologies and Background Profiles
Different TUNEL detection methodologies exhibit characteristic performance profiles regarding sensitivity, specificity, and background propensity. Direct methods utilizing nucleotides pre-conjugated to fluorophores (e.g., FITC-dUTP) require fewer incubation steps, potentially reducing technical variability and background [12]. However, they may offer less signal amplification compared to indirect methods. Conversely, indirect systems employing hapten-labeled nucleotides (biotin, BrdU, or digoxigenin) with secondary detection reagents provide signal amplification but introduce additional potential background sources through the extra binding steps [13] [12].
Advanced detection chemistries have been developed to address these limitations. The Click-iT TUNEL assay utilizes a dUTP modified with a small alkyne group, which is more readily incorporated by TdT than larger fluorescent nucleotide conjugates [13]. Detection occurs via a copper-catalyzed "click" reaction between the alkyne and an Alexa Fluor azide, creating a highly specific conjugation that significantly reduces background. This system requires only mild fixation and permeabilization, better preserving cellular morphology while minimizing the exposure of non-apoptotic DNA breaks. Comparative studies demonstrate that the Click-iT technology detects a higher percentage of apoptotic cells under identical conditions compared to conventional TUNEL methods using fluorescein-dUTP [13].
Table 2: Performance Comparison of TUNEL Detection Methodologies
| Methodology | Relative Background | Signal Amplification | Protocol Steps | Optimal Application |
|---|---|---|---|---|
| Direct FITC-dUTP | Moderate | Low | Minimal (fastest) | Flow cytometry, basic imaging |
| Biotin-Streptavidin-HRP | High (without blocking) | High | Multiple (includes blocking) | Chromogenic IHC, low antigen expression |
| BrdU with Antibody | Moderate | High | Multiple | High-resolution imaging, multiplexing |
| Click-iT Chemistry | Low | Moderate | Minimal | Sensitive quantification, complex samples |
The physical size of detection reagents profoundly impacts background characteristics. The molecular weight of Alexa Fluor azides (~1,000 Da) in Click-iT systems is substantially smaller than antibodies (~150,000 Da) used in indirect detection methods [13]. This size difference enhances reagent penetration into complex samples like tissue sections, enabling more homogeneous labeling while requiring less detergent permeabilization—a common contributor to background staining when over-applied.
Optimized Protocols for Background Reduction
Standardized TUNEL Protocol for Low Background
This protocol has been optimized to minimize background staining across various sample types, incorporating critical control points for reliable apoptosis detection.
Materials and Reagents
- TUNEL assay kit (preferentially selecting kits without cacodylate buffers [24])
- 4% paraformaldehyde in PBS (freshly prepared or freshly thawed aliquots)
- Permeabilization reagent (0.25% Triton X-100 in PBS)
- Blocking solution (3% BSA in PBS for cells; 10% normal serum from detection antibody host for tissues)
- DNase I (for positive control)
- Phosphate-buffered saline (PBS), pH 7.4
- Optional: Sodium citrate buffer (100 mM) for antigen retrieval in tissue samples
Procedure for Adherent Cells
- Cell Fixation: Remove culture media and gently wash cells with room temperature PBS. Avoid vigorous pipetting directly onto cells to prevent mechanical induction of DNA breaks. Add sufficient 4% paraformaldehyde to completely cover cells and incubate for 15 minutes at room temperature [13] [41]. Critical step: Do not over-fixate, as extended aldehyde exposure can mask epitopes and increase autofluorescence.
Permeabilization: Remove fixative and wash twice with PBS. Apply 0.25% Triton X-100 in PBS for 20 minutes at room temperature [13]. Critical step: Titrate permeabilization concentration (0.1-0.5%) and duration for specific cell types, as over-permeabilization exposes non-apoptotic DNA breaks.
Blocking: Incubate samples with appropriate blocking solution for 30 minutes at room temperature. For systems utilizing biotin-streptavidin detection, include an endogenous biotin blocking step according to manufacturer recommendations [12].
TUNEL Reaction Preparation: Prepare TUNEL reaction mixture according to manufacturer instructions, ensuring optimal TdT concentration. Critical step: Include negative controls (omitting TdT enzyme) and positive controls (treating samples with DNase I after permeabilization) with every experiment [13] [6].
Labeling: Apply TUNEL reaction mixture to samples and incubate in a humidified chamber at 37°C for 60 minutes. Avoid prolonged incubation times which increase non-specific labeling.
Washing: Remove reaction mixture and wash samples three times with PBS containing 0.1% Tween-20 (5 minutes per wash). Thorough washing is critical for reducing unincorporated label.
Counterstaining and Mounting: Apply appropriate nuclear counterstain (e.g., Hoechst 33342, DAPI) if required and mount for microscopy [13].
Tissue Section Modifications For tissue sections, incorporate an additional antigen retrieval step after permeabilization: incubate slides in 100 mM sodium citrate buffer with 0.1% Triton X-100 for 30 minutes at 65°C [41]. This step enhances DNA accessibility while reducing variability in staining intensity.
Troubleshooting High Background Staining
The following troubleshooting guide addresses the most common causes of excessive background in TUNEL assays:
Diffuse Nuclear Staining in All Samples Including Negative Controls: This pattern suggests excessive TdT enzyme concentration or incomplete blocking. Reduce TdT concentration by 50% and ensure proper blocking with serum proteins. Verify that negative controls (without TdT) are included to identify this issue [12].
Cytoplasmic or Non-Nuclear Staining: Typically indicates inadequate washing or non-specific antibody binding (in indirect methods). Increase wash stringency (higher detergent concentration, extended wash times) and optimize blocking conditions. For indirect methods, titrate secondary antibody concentration and include relevant species-specific IgG controls [12].
High Background in Positive Controls Only: Suggests DNase I over-treatment or excessive permeabilization. Optimize DNase I concentration and incubation time (typically 1-10 µg/mL for 10-30 minutes). Reduce Triton X-100 concentration or permeabilization duration [13].
Variable Staining Between Replicates: Often results from inconsistent fixation or permeabilization. Standardize fixation time precisely across all samples and ensure consistent permeabilization conditions. Prepare fresh fixative and permeabilization solutions for each experiment [41].
Elevated Background in Tissue Sections Compared to Cell Cultures: Frequently caused by incomplete reagent penetration followed by excessive washing attempts. Incorporate gentle agitation during washes and consider extending incubation times rather than increasing reagent concentrations. Implement graded ethanol dehydration steps after fixation to better preserve tissue architecture [41].
Experimental Workflow for Background Optimization
The following workflow diagram systematically outlines the critical decision points for minimizing background in TUNEL assays, integrating both preventive measures and troubleshooting interventions:
Diagram 1: Systematic workflow for identifying and addressing common sources of background staining in TUNEL assays at critical experimental stages.
The Scientist's Toolkit: Essential Reagents for Optimal TUNEL Staining
Successful TUNEL staining with minimal background requires careful selection and preparation of key reagents. The following table catalogues essential solutions and their optimized formulations for background reduction:
Table 3: Essential Research Reagents for Low-Background TUNEL Assays
| Reagent | Optimal Formulation | Function | Background Consideration |
|---|---|---|---|
| Fixative | 4% paraformaldehyde in PBS, pH 7.4 | Preserves cellular architecture and prevents degradation | Must be freshly prepared; over-fixation increases autofluorescence |
| Permeabilization Solution | 0.1-0.5% Triton X-100 in PBS | Creates membrane pores for reagent access | Concentration must be titrated; excess exposes non-apoptotic DNA breaks |
| Blocking Solution | 3% BSA or 10% normal serum in PBS | Prevents non-specific binding of detection reagents | Serum should match host species of secondary antibodies |
| TUNEL Reaction Buffer | Cacodylate-free formulations preferred [24] | Provides optimal enzymatic environment for TdT | Traditional cacodylate buffers can induce apoptosis |
| Wash Buffer | PBS with 0.1% Tween-20 | Removes unbound reagents and reduces background | Increased detergent concentration improves stringency |
| Nuclear Counterstain | Hoechst 33342 (1-5 µg/mL) or DAPI | Identifies all nuclei in sample | Use minimal effective concentration to avoid signal bleed-through |
Additional specialized reagents include DNase I (for positive controls; typically used at 1-10 µg/mL for 10-30 minutes) [13] and sodium citrate buffer (for antigen retrieval in tissue sections; 100 mM, pH 6.0) [41]. For researchers using chromogenic detection, endogenous peroxidase blocking solutions (3% H₂O₂ in methanol) are essential when working with HRP-based systems.
Minimizing background and non-specific staining in TUNEL assays requires a systematic approach addressing fixation, permeabilization, reagent selection, and detection methodologies. The implementation of cacodylate-free reaction buffers, optimized TdT concentrations, and appropriate control samples significantly enhances assay specificity. Advanced detection chemistries such as the Click-iT system offer particular advantages for challenging applications by combining high sensitivity with low background characteristics. Through careful attention to protocol details and methodical troubleshooting, researchers can achieve the reliable, high-quality data essential for accurate assessment of DNA fragmentation in late-stage apoptosis, thereby supporting robust conclusions in both basic research and drug development contexts.
Within the context of DNA fragmentation research using the TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) assay, effective permeabilization of cell samples is a critical preparatory step. This process enables detection reagents to access the nucleus and label fragmented DNA, a hallmark of late-stage apoptosis. While proteinase K is a common permeabilization agent, its use requires precise optimization and consideration of alternatives to prevent the degradation of protein antigens, which is crucial for concurrent multiplexed analyses. Recent advancements highlight that alternative methods, such as heat-mediated antigen retrieval, can effectively resolve the incompatibility between TUNEL and modern spatial proteomic techniques like MILAN (multiple iterative labeling by antibody neodeposition) and CycIF (cyclic immunofluorescence) [16]. This Application Note provides detailed protocols and data to guide researchers in selecting and optimizing permeabilization strategies for robust and reliable TUNEL assay outcomes.
The Permeabilization Landscape in TUNEL Assays
Permeabilization is essential for facilitating the entry of the terminal deoxynucleotidyl transferase (TdT) enzyme and labeled nucleotides into the cell nucleus during a TUNEL assay. The choice of permeabilization agent and conditions directly impacts assay sensitivity, specificity, and compatibility with downstream applications.
- Role of Proteinase K: Proteinase K (ProK) is a broad-spectrum serine protease that digests proteins and enhances tissue permeability by breaking down cross-linked proteins, particularly in formalin-fixed paraffin-embedded (FFPE) samples. However, a key limitation is that Proteinase K treatment consistently reduced or even abrogated protein antigenicity, making it unsuitable for experiments that combine TUNEL with iterative immunofluorescence staining for multiple protein targets [16].
- Heat-Induced Epitope Retrieval (HIER): As an alternative to enzymatic digestion, heat-mediated methods using a pressure cooker or microwave with a retrieval buffer can effectively reverse formaldehyde cross-links. This approach enhanced protein antigenicity for the targets tested and enabled seamless integration of TUNEL with multiplexed spatial proteomic methods [16].
The following diagram illustrates the decision-making workflow for selecting an appropriate permeabilization method based on experimental goals.
Quantitative Comparison of Permeabilization Methods
Selecting the optimal permeabilization strategy requires balancing the efficiency of DNA labeling with the preservation of other cellular components. The table below summarizes the key characteristics of Proteinase K and heat-based alternatives.
Table 1: Comparative Analysis of Permeabilization Methods for TUNEL Assays
| Method | Typical Working Concentration/Conditions | Key Advantages | Key Limitations & Considerations |
|---|---|---|---|
| Proteinase K | 40 U/mL (for fixed cells in RNA-seq) [55]; Requires titration for specific tissues [56]. | Effective for dense tissues and FFPE samples; improves antibody penetration for some targets [56]. | Degrades protein antigenicity, hindering multiplexed protein imaging [16]. Over-digestion can damage morphology and reduce RNA yield [55]. |
| Pressure Cooker (HIER) | Not applicable. Relies on heat and buffer conditions (e.g., citrate buffer, pH 6.0). | Preserves full protein antigenicity; fully compatible with MILAN and CycIF [16]. No enzymatic titration needed. | May not be sufficient for some heavily cross-linked or dense tissue types; requires specialized equipment. |
| Combined Approaches | Sequential application of mild HIER followed by low-concentration ProK. | Can balance DNA access and antigen preservation in challenging samples. | Protocol is more complex; requires extensive optimization to avoid the pitfalls of both methods. |
Detailed Experimental Protocols
Protocol 1: Proteinase K Titration for Embryonic Tissues
This protocol, adapted from aphid embryo research, provides a framework for optimizing Proteinase K concentration based on tissue thickness and fragility [56].
A. Materials
- Fixed tissue samples (e.g., aphid embryos, murine tissues)
- Proteinase K (e.g., 20 mg/mL stock solution)
- Phosphate-Buffered Saline (PBS)
- Triton X-100 (optional, for enhanced permeabilization)
B. Step-by-Step Procedure
- Sample Preparation: Dissect and fix tissues according to standard protocols for your specimen. Preserve tissue integrity during dissection.
- Prepare Proteinase K Dilutions: Create a dilution series of Proteinase K in PBS. A suggested starting range is:
- Early/Thin Embryos: 1-5 µg/mL
- Mid-stage Embryos: 5-10 µg/mL
- Late-stage/Thick Embryos: 10-20 µg/mL
- Apply Treatment: Incubate samples in the different Proteinase K solutions for a fixed time (e.g., 10-30 minutes) at room temperature.
- Terminate Reaction: Gently wash samples 3-5 times with PBS to completely remove Proteinase K.
- Proceed with TUNEL Assay: Continue with the standard TUNEL staining protocol, incorporating appropriate positive and negative controls.
C. Troubleshooting and Optimization
- Weak TUNEL Signal: This may indicate under-digestion. Gradually increase the Proteinase K concentration or incubation time.
- Poor Morphology/High Background: This suggests over-digestion. Reduce the Proteinase K concentration or incubation time. Consider using a different permeabilization agent.
Protocol 2: Pressure Cooker-Based Antigen Retrieval for Multiplexed TUNEL
This protocol leverages heat-induced epitope retrieval to make TUNEL compatible with subsequent multiplexed protein imaging [16].
A. Materials
- FFPE tissue sections mounted on slides
- Antigen retrieval buffer (e.g., citrate-based, pH 6.0)
- Pressure cooker
- Staining dishes and coplin jars
B. Step-by-Step Procedure
- Dewax and Rehydrate: Process FFPE slides through xylene and a graded ethanol series to water.
- Antigen Retrieval: Place slides in a container filled with antigen retrieval buffer. Heat in a pressure cooker according to the standard protocol for your specific buffer (e.g., 15-20 minutes at full pressure).
- Cool Down: Allow the pressure cooker to cool down naturally or under running cool water.
- Rinse: Gently rinse slides with distilled water.
- Perform TUNEL Assay: Conduct the TUNEL assay using an antibody-based detection method (e.g., BrdU-incorporated).
- Proceed to Multiplexed Imaging: After TUNEL, the slides can be directly used for iterative immunofluorescence cycles (e.g., MILAN) without loss of protein antigenicity [16].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for TUNEL Assay Permeabilization and Detection
| Reagent | Function in the Protocol | Key Considerations |
|---|---|---|
| Proteinase K | Enzymatic permeabilization of fixed tissues; digests proteins to expose nucleic acids. | Concentration and time are critical; must be titrated for each tissue type to avoid over-digestion [56]. |
| Terminal Deoxynucleotidyl Transferase (TdT) | Core enzyme in TUNEL; catalyzes the addition of labeled dUTPs to 3'-OH ends of fragmented DNA. | Requires cobalt cofactor in buffer solution for optimal activity [1]. |
| Labeled dUTP (e.g., FITC, BrdU) | Provides the detectable label incorporated at DNA break sites. | BrdU-based methods can produce a brighter signal and are compatible with antibody-based erasure in iterative staining [16] [12]. |
| Click-iT Chemistry Kits | Allows for fluorescent labeling of EdU (a thymidine analog) via a click reaction; an alternative to TUNEL. | Offers a faster, non-enzymatic detection method but still requires optimized permeabilization [16]. |
| Antigen Retrieval Buffers | Used in heat-induced methods to reverse cross-links and expose epitopes. | pH and buffer composition (e.g., citrate, EDTA) can significantly impact retrieval efficiency. |
| 2-Mercaptoethanol/SDS (2-ME/SDS) | Erasure buffer for MILAN; removes antibodies between staining cycles. | Enables iterative multiplexing after TUNEL when pressure cooker retrieval is used [16]. |
Optimizing permeabilization is a decisive factor in the success of TUNEL assays, especially in complex experimental workflows. While Proteinase K is a powerful tool, its tendency to degrade protein targets limits its utility in multiplexed studies. The adoption of heat-mediated antigen retrieval methods, such as pressure cooking, presents a robust alternative that harmonizes TUNEL with advanced spatial proteomics by fully preserving protein antigenicity. The protocols and data provided herein empower researchers to make informed decisions, ensuring precise detection of apoptotic cells while maintaining the integrity of the cellular proteome for comprehensive analysis.
Preventing False Positives from Necrosis and Tissue Autolysis
A fundamental challenge in cell death research, particularly within the context of a broader thesis on the TUNEL assay for DNA fragmentation in late apoptosis, is the accurate differentiation of apoptosis from necrosis and tissue autolysis. The TUNEL (Terminal deoxynucleotidyl transferase dUTP Nick End Labeling) assay is a cornerstone technique for detecting the DNA fragmentation that is a hallmark of late-stage apoptosis [2] [8]. However, its widespread application is complicated by a significant limitation: the potential for false-positive signals. DNA degradation occurring during unprogrammed cell death, such as necrosis, or as a post-mortem artifact during tissue autolysis, can also be labeled by the TUNEL reaction, leading to misinterpretation of results and flawed scientific conclusions [2]. For researchers, scientists, and drug development professionals, this lack of specificity can compromise data integrity in critical areas like cancer research, toxicology, and the evaluation of chemotherapeutic agents [2]. This application note details strategic methodologies and refined protocols designed to enhance the specificity of apoptosis detection and mitigate the risk of false positives originating from necrotic processes and tissue autolysis.
Comparative Analysis of Apoptosis Detection Methods
A clear understanding of the capabilities and limitations of available assays is the first step in designing a robust strategy to prevent false positives. No single method is perfect, and the choice often depends on the required specificity, throughput, and available equipment. The table below summarizes key apoptosis detection methods, highlighting their propensity for false positives from necrosis.
Table 1: Comparison of Apoptosis Detection Methods and False Positive Risk
| Assay | Detects | Risk of False Positives from Necrosis | Key Advantages | Key Limitations |
|---|---|---|---|---|
| TUNEL Assay [2] [8] | DNA strand breaks | High; necrotic cells can also display DNA fragmentation. | High sensitivity; applicable to tissue sections and cells. | Cannot easily distinguish between apoptotic and necrotic DNA fragmentation. |
| DNA Laddering [2] | Internucleosomal DNA cleavage | Low; necrosis typically produces a "smear" pattern, not a ladder. | Direct visual evidence; cost-effective. | Semi-quantitative; not suitable for low cell numbers; requires careful handling. |
| Annexin V/PI Staining [2] | Phosphatidylserine externalization (early apoptosis) & membrane integrity. | Low for early apoptosis; necrotic cells are PI+/Annexin V+. | Distinguishes early apoptosis (Annexin V+/PI-) from late apoptosis/necrosis (Annexin V+/PI+). | Requires live cells; specialized equipment (flow cytometry). |
| Caspase Activity [2] | Activation of executioner caspases (e.g., Caspase-3/7). | Very Low; caspase activation is a specific feature of apoptosis. | High specificity for apoptosis; available in 96-well format. | Misses late apoptotic stages where caspase activity may decline. |
| Morphological Staining (EB/AO) [57] [2] | Chromatin condensation & membrane integrity. | Low; allows visual differentiation based on nuclear morphology. | Quantifies live, apoptotic, and necrotic cells simultaneously; simple and rapid. | Requires experience for morphological discrimination; semi-quantitative. |
Key Methodologies for Distinguishing Apoptosis from Necrosis
A Modified Ethidium Bromide & Acridine Orange (EB/AO) Staining Assay in a 96-Well Plate
This improved protocol leverages nuclear morphology to differentiate cell death states and is particularly advantageous for minimizing cell loss and handling artifacts that can confound results [57].
Principle: Acridine orange (AO) permeates all cells, staining nuclei green. Ethidium bromide (EB) is only taken up by cells with lost membrane integrity, staining nuclei red and dominating over AO. Crucially, apoptotic cells display condensed or fragmented chromatin (bright green or orange), while necrotic cells have a structurally normal orange nucleus [57].
Detailed Protocol:
- Cell Seeding and Treatment: Seed adherent or suspension cells in a 96-well plate and apply experimental treatments.
- Staining Solution Preparation: Prepare a working solution of EB/AO (e.g., 100 µg/mL acridine orange and 100 µg/mL ethidium bromide in PBS).
- Staining: Add the EB/AO solution directly to the wells containing the cell culture medium.
- Centrifugation: Centrifuge the 96-well plate to sediment all cells, including floaters (which may include late apoptotic and necrotic cells), to the bottom of the well [57].
- Incubation: Incubate the plate for a short period (e.g., 10-20 minutes) at room temperature, protected from light.
- Imaging and Quantification: Immediately visualize the cells using a fluorescence microscope with FITC and TRITC filters. A minimum of 200 cells per condition should be counted and classified [57]:
- Viable Cells: Normal green nucleus.
- Early Apoptotic Cells: Bright green nucleus with condensed or fragmented chromatin.
- Late Apoptotic Cells: Condensed and fragmented orange chromatin.
- Necrotic Cells: Structurally normal orange nucleus.
Advantages for False Positive Prevention: This method drastically reduces the possibility of losing floating cells (a significant population in cell death studies) and minimizes damage to adherent cells by eliminating detaching and washing steps, leading to a more accurate representation of the cell population [57].
The Click-iT Plus TUNEL Assay with Multiplexing for Verification
This advanced TUNEL protocol incorporates a copper-optimized click chemistry reaction, which improves sensitivity and allows for multiplexing with other markers to verify the apoptotic nature of TUNEL-positive cells [8].
Principle: The assay incorporates EdUTP (an alkyne-modified dUTP) into DNA strand breaks using Terminal deoxynucleotidyl transferase (TdT). The incorporated EdUTP is then detected using a fluorescent azide dye via a copper-catalyzed "click" reaction. The "Plus" version uses optimized copper concentrations to preserve the signal of fluorescent proteins, enabling multiplexing [8].
Detailed Protocol for Cultured Cells:
- Cell Fixation and Permeabilization:
- Wash cells (grown on coverslips or in a 96-well plate) with PBS.
- Fix with 4% paraformaldehyde for 15 minutes at room temperature.
- Remove fixative and permeabilize cells with 0.25% Triton X-100 in PBS for 20 minutes at room temperature.
- Wash twice with deionized water [13].
- TdT Reaction (Labeling DNA Breaks):
- Prepare the TdT reaction buffer according to the manufacturer's instructions (e.g., from Component A).
- Add the EdUTP nucleotide mixture (Component B) and the recombinant TdT enzyme (Component C) to the buffer.
- Apply the reaction mixture to the fixed and permeabilized cells and incubate for 60-90 minutes at 37°C [13] [8].
- Click-iT Reaction (Detection):
- Prepare the Click-iT reaction cocktail by mixing the Click-iT reaction buffer (Component D, which contains the Alexa Fluor azide) with the reaction buffer additive (Component E).
- Apply the cocktail to the cells and incubate for 30 minutes at room temperature, protected from light.
- Wash with a buffered solution, e.g., 3% BSA in PBS [13].
- Multiplexing with a Necrosis Marker or Morphological Stain:
- To confirm apoptosis, counterstain with a marker for a specific apoptotic protein (if using a transgenic cell line with a fluorescent protein tag) or with a morphological stain like Hoechst 33342 (Component F) to visualize condensed chromatin [8].
- Critical Note: The high copper concentration in the standard Click-iT (non-Plus) assay can quench fluorescent proteins and prevent phalloidin binding. The Click-iT Plus TUNEL assay is specifically designed to overcome this limitation, making it the preferred choice for multiplexing verification assays [8].
Advantages for False Positive Prevention: The ability to simultaneously detect TUNEL signal and another specific marker of apoptosis (e.g., caspase-cleaved proteins) or distinct nuclear morphology in the same cell provides a powerful internal validation, dramatically reducing the chance of falsely attributing a TUNEL signal to apoptosis when it originates from necrosis.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents for Apoptosis and Necrosis Discrimination
| Reagent / Kit | Function / Target | Utility in False Positive Prevention |
|---|---|---|
| Click-iT Plus TUNEL Assay Kits (e.g., Alexa Fluor variants) [8] | Fluorescent detection of DNA strand breaks via click chemistry. | Copper-optimized chemistry allows multiplexing with fluorescent proteins and phalloidin to confirm apoptosis. |
| Ethidium Bromide (EB) & Acridine Orange (AO) [57] [2] | Differential fluorescent nucleic acid stains for assessing membrane integrity and nuclear morphology. | Enables visual discrimination of apoptotic chromatin condensation from necrotic nuclear structure. |
| Annexin V Binding Assays (e.g., Alexa Fluor conjugates) [2] | Detects phosphatidylserine (PS) exposure on the outer leaflet of the cell membrane. | Used with a viability dye (e.g., PI) to identify early apoptotic (Annexin V+/PI-) and necrotic (Annexin V+/PI+) populations. |
| Hoechst 33342 [13] | Cell-permeable blue fluorescent DNA stain. | Allows visualization of nuclear morphology (condensation, fragmentation) to contextualize TUNEL positivity. |
| Anti-Caspase-3 (Active) Antibodies | Detect cleaved/activated executioner caspases. | Provides a highly specific biochemical marker of apoptosis to corroborate TUNEL results. |
| Propidium Iodide (PI) [2] | red fluorescent viability dye that is excluded by intact membranes. | Distinguishes cells with compromised membranes (necrotic, late apoptotic) from intact cells (viable, early apoptotic). |
| DNase I ( recombinant) [13] | Enzyme that introduces DNA strand breaks in a controlled manner. | Serves as an essential positive control for TUNEL assays to ensure technical success. |
Experimental Workflow for Specific Apoptosis Detection
The following workflow diagram integrates multiple methods to create a confirmatory strategy for accurately identifying apoptotic cells.
Diagram 1: Confirmatory workflow combining TUNEL assay with multiplex staining.
Decision Pathway for Cell Death Characterization
When a TUNEL-positive signal is detected, the following logical pathway should be followed to characterize the type of cell death.
Diagram 2: Decision pathway for characterizing TUNEL-positive cells.
Terminal deoxynucleotidyl transferase (TdT) is a unique template-independent DNA polymerase that catalyzes the addition of deoxynucleotides to the 3'-hydroxyl terminus of DNA molecules, without requiring a template strand [58]. This specialized function makes it invaluable for detecting DNA fragmentation, a hallmark event of late-stage apoptosis [32]. In the TUNEL (Terminal deoxynucleotidyl transferase dUTP Nick End Labeling) assay, TdT enzymatically incorporates labeled nucleotides at the 3'-ends of DNA fragments, enabling the precise identification and quantification of apoptotic cells within tissue samples [32].
The accuracy and sensitivity of TUNEL assays are fundamentally dependent on the catalytic activity of TdT, which in turn is heavily influenced by reagent stability and handling practices. Maintaining full enzymatic function requires strict adherence to specific storage conditions, buffer compositions, and handling protocols. This application note provides detailed methodologies and data-driven guidelines to preserve TdT enzyme activity, ensuring reliable and reproducible results in apoptosis research.
TdT Biochemical Characteristics and Stability Profile
Key Molecular Properties
TdT possesses distinct biochemical characteristics that dictate its handling requirements. The enzyme is a 58 kDa protein typically purified from calf thymus and expressed in recombinant E. coli systems for commercial production [59]. Unlike other DNA polymerases, TdT functions without exonuclease activity (both 5'→3' and 3'→5'), making it exclusively synthetic in function [59]. A critical feature of its catalytic mechanism is the dependence on cobalt ions (Co²⁺), which are included in the reaction buffer to stimulate the addition of nucleotides to the 3'-ends of DNA fragments [59].
The enzyme demonstrates versatility in substrate recognition, efficiently acting on protruding, recessed, or blunt-ended double or single-stranded DNA molecules [59]. This broad substrate specificity makes it particularly suitable for labeling the diverse DNA ends generated during apoptotic fragmentation.
Stability and Storage Conditions
Proper storage is essential for maintaining TdT activity over time. The enzyme should be stored at -20°C in a storage buffer typically consisting of 100 mM potassium acetate (pH 6.8), 2 mM 2-mercaptoethanol, 0.01% (v/v) Triton X-100, and 50% (v/v) glycerol [59]. The high glycerol concentration prevents ice crystal formation, while the slightly acidic pH and reducing environment preserve enzymatic structure and function.
Table 1: TdT Enzyme Storage Specifications and Stability Indicators
| Parameter | Specification | Stability Impact |
|---|---|---|
| Storage Temperature | -20°C | Prevents thermal denaturation |
| Specific Activity | 42,000 units/mg | Benchmark for quality assessment |
| Unit Definition | Incorporation of 1 nmol dTTP in 1 hour at 37°C | Standardized activity measurement |
| Purity Level | >99% (SDS-PAGE) | Reduces protease contamination risk |
| Optimal pH Range | 6.8 (storage); 7.9 (reaction) | Maintains structural integrity |
Factors Affecting TdT Activity and Stability
Reaction Buffer Composition
The 10X reaction buffer for TdT typically contains 500 mM potassium acetate, 200 mM Tris-acetate, 100 mM magnesium acetate (pH 7.9 at 25°C), and 25 mM CoCl₂ [59]. The cobalt chloride component is particularly crucial as it serves as a cofactor that dramatically enhances the catalytic efficiency of the enzyme. However, this required component presents a significant challenge for downstream applications, as Co²⁺ can interfere with subsequent analytical procedures. It is therefore necessary to remove CoCl₂ from the reaction mixture after the labeling step using spin column purification or phenol/chloroform extraction followed by ethanol precipitation [59].
Known Inhibitors and Inactivation Methods
Several chemical compounds profoundly inhibit TdT activity and must be avoided in experimental workflows. Key inhibitors include:
- Metal chelators (EDTA, EGTA) that sequester essential cofactor ions
- Ammonium, chloride, iodide, and phosphate ions at elevated concentrations
- Heparin and certain types of chemically modified DNA [59]
For intentional termination of TdT activity, the enzyme can be effectively inactivated by either heating at 70°C for 10 minutes or by the addition of EDTA to chelate the essential metal cofactors [59]. These inactivation methods are particularly useful for controlling reaction times and preventing non-specific labeling in TUNEL assays.
Comprehensive TUNEL Assay Protocol Using TdT
Sample Preparation Considerations
The integrity of starting material significantly impacts TUNEL assay outcomes. When working with formalin-fixed, paraffin-embedded (FFPE) tissues—common specimens in apoptosis research—proper processing is essential. Studies indicate that deparaffinization with xylene generally increases DNA yield from FFPE samples [60]. However, certain staining procedures prior to microdissection, such as methyl green staining, may cause additional DNA fragmentation [60]. The DNA extraction method also influences results, with column-based methods (e.g., QIAamp DNA FFPE Tissue Kit) generally producing less fragmented DNA with higher amplifiable yields compared to phenol-chloroform extraction and ethanol precipitation [60].
Step-by-Step TUNEL Assay Procedure
Sample Fixation and Sectioning
- Fix tissues in neutral-buffered formalin for optimal preservation
- Embed in paraffin and section at 4-5μm thickness
- Mount on positively charged glass slides to prevent detachment
Deparaffinization and Rehydration
- Bake slides at 60°C for 1 hour to melt paraffin
- Deparaffinize with xylene (3 changes, 10 minutes each)
- Rehydrate through graded ethanol series (100%, 95%, 70%) to water
Proteinase Digestion and Permeabilization
- Treat with Proteinase K (20-30μg/mL) for 15-30 minutes at room temperature
- Optimize digestion time to preserve tissue architecture while allowing TdT access
TdT Reaction Mixture Preparation
- Prepare reaction mixture fresh before use:
- Gently mix by pipetting; avoid vortexing to preserve enzyme activity
Enzymatic Labeling
- Apply 50μL of reaction mixture to each tissue section
- Incubate in a humidified chamber at 37°C for 60-90 minutes
- Include negative controls without TdT enzyme to assess non-specific labeling
Reaction Termination and Washing
- Stop the reaction by washing with 2× SSC buffer for 15 minutes
- Alternatively, incubate with EDTA-containing buffer to chelate Co²⁺
Detection and Visualization
- For colorimetric detection:
- Block endogenous peroxidases (if using HRP-based detection)
- Incubate with Streptavidin-Horseradish Peroxidase conjugate
- Develop with DAB or TACS Blue Label substrate [32]
- For fluorescence detection:
- Counterstain with DAPI or propidium iodide
- Apply anti-fade mounting medium
- Visualize by fluorescence microscopy
- For colorimetric detection:
Analysis and Interpretation
- Quantify apoptotic cells using image analysis software
- Express results as apoptotic index (percentage of TUNEL-positive cells)
- Correlate findings with morphological features of apoptosis
Table 2: Research Reagent Solutions for TUNEL Assay
| Reagent | Function | Application Notes |
|---|---|---|
| TdT Enzyme | Catalyzes nucleotide addition to DNA 3' ends | Source: recombinant calf thymus; Specific activity: 42,000 units/mg [59] |
| Labeled Nucleotides | Detection of DNA fragments | Biotin-dUTP, DIG-dUTP, or fluorescent conjugates (FITC-dUTP) |
| Cobalt Chloride | Essential reaction cofactor | 25 mM in 10X reaction buffer; must be removed post-labeling [59] |
| Proteinase K | Tissue permeabilization | Enables TdT access to nuclear DNA; concentration requires optimization |
| Streptavidin-HRP | Signal generation for colorimetric detection | Used with biotinylated nucleotides; develop with DAB substrate [32] |
Quality Control and Troubleshooting
Assessing TdT Enzyme Activity
Regular verification of TdT activity is crucial for assay consistency. The standard unit definition specifies that one unit incorporates 1 nmol of dTTP into acid-insoluble material in 1 hour at 37°C using d(A)₁₈ as a primer [59]. Researchers should periodically test enzyme performance using control reactions with standardized DNA substrates to detect any loss of activity before proceeding with valuable samples.
Troubleshooting Common Issues
- High Background Signal: Reduce TdT enzyme concentration; shorten incubation time; optimize Proteinase K treatment duration; include additional washing steps
- Weak or No Staining: Verify enzyme activity and storage conditions; check reaction buffer composition, particularly Co²⁺ concentration; ensure proper tissue permeabilization; confirm detection reagent functionality
- Tissue Damage: Reduce Proteinase K concentration or treatment time; avoid excessive heating during procedures
- Inconsistent Results Between Experiments: Standardize fixation times; prepare fresh reaction mixtures; control for room temperature fluctuations; use consistent lot numbers of critical reagents
Apoptosis Signaling Pathway and DNA Fragmentation
The following diagram illustrates the key apoptotic signaling events that lead to DNA fragmentation, which is detected by the TUNEL assay utilizing TdT enzyme activity.
TUNEL Assay Workflow
This diagram outlines the complete experimental workflow for the TUNEL assay, highlighting critical steps where TdT stability and handling are paramount for success.
Within the broader study of DNA fragmentation in late apoptosis using the TUNEL (Terminal deoxynucleotidyl transferase dUTP nick-end labeling) assay, a critical technical challenge has been the method of antigen retrieval. The TUNEL assay is a cornerstone technique for identifying programmed cell death in situ, as it leverages terminal deoxynucleotidyl transferase (TdT) to label the 3'-hydroxyl termini of fragmented DNA, a hallmark of late-stage apoptosis [24]. Traditionally, this process has relied on enzymatic retrieval using proteinase K (ProK) to unmask these DNA ends [16]. However, recent investigations reveal a significant incompatibility: ProK treatment consistently reduces or abrogates the antigenicity of protein epitopes, thereby preventing the multiplexed spatial contextualization of cell death with protein biomarkers using modern spatial proteomic methods [16]. This application note details a validated advanced optimization—replacing proteinase K with heat-induced epitope retrieval (HIER) using a pressure cooker. This harmonized protocol preserves superior TUNEL signal sensitivity while maintaining full protein antigenicity for multiplexed iterative immunofluorescence, thus enabling richer spatial analysis of apoptotic processes in complex tissues [16].
The Critical Limitation of Proteinase K in Multiplexed Workflows
The primary motivation for optimizing antigen retrieval in TUNEL assays stems from the critical limitations of enzymatic retrieval with proteinase K. While effective at digesting proteins and unmasking DNA termini for TdT enzyme access, Proteinase K's action is non-specific and irreversible [16]. Research demonstrates that Proteinase K treatment consistently reduces or even abrogates protein antigenicity [16]. This massive degradation of protein epitopes makes it impossible to perform high-plex protein co-detection on the same sample after the TUNEL assay.
This is a major impediment for researchers aiming to use spatial proteomic methods like Multiple Iterative Labeling by Antibody Neodeposition (MILAN) or Cyclic Immunofluorescence (CycIF). These techniques allow for the staining of 20-80 protein targets on a single tissue specimen, providing deep spatial context [16]. The incompatibility with Proteinase K means that elucidating the cell-type-specific context of apoptosis and the complex mechanistic relationships between cell death and its tissue microenvironment has been severely limited. Replacing this step with a non-destructive retrieval method is, therefore, essential for advanced apoptosis research.
Optimized Pressure Cooker Antigen Retrieval Protocol for TUNEL
The following section provides a detailed, step-by-step protocol for implementing heat-induced epitope retrieval (HIER) using a pressure cooker as a direct replacement for Proteinase K in TUNEL assays.
Materials and Reagent Setup
- Tissue Sections: Formalin-fixed, paraffin-embedded (FFPE) tissue sections mounted on slides.
- Pressure Cooker: A domestic stainless-steel pressure cooker is suitable [61].
- Hotplate: A capable hotplate for bringing the pressure cooker to pressure.
- Slide Rack: A metal rack suitable for holding microscopy slides.
- Antigen Retrieval Buffers: Two common buffers are recommended; selection may require empirical testing [61] [62].
- Citrate Buffer (10 mM, pH 6.0): 2.94 g of tri-sodium citrate (dihydrate) dissolved in 1 L distilled water. Adjust pH to 6.0 with HCl, then add 0.5 mL Tween 20 [61] [62]. Ideal for many targets and yields low background.
- EDTA Buffer (1 mM, pH 8.0): 0.37 g of EDTA dissolved in 1 L distilled water. Adjust pH to 8.0 with NaOH, then add 0.5 mL Tween 20 [61] [62]. Can be more effective for some targets but may yield higher background.
Step-by-Step Workflow
- Deparaffinization and Rehydration: Prior to antigen retrieval, standard deparaffinization and rehydration of FFPE sections must be performed.
- Buffer Preparation: Add the selected antigen retrieval buffer (e.g., Citrate pH 6.0 or EDTA pH 8.0) to the pressure cooker. Place the open pressure cooker on a hotplate set to full power and bring the buffer to a boil [61] [62].
- Slide Loading: Once the buffer is boiling, carefully transfer the deparaffinized and rehydrated slides into the pressure cooker using forceps. Secure the lid of the pressure cooker as per the manufacturer's instructions [61] [62].
- Pressurized Retrieval: Once the cooker reaches full pressure (approximately 121°C), start the timer. Maintain full pressure for 3 minutes [62]. Other protocols suggest a slightly different regime (125°C for 30 seconds followed by 90°C for 10 seconds), but the 3-minute duration is a robust starting point [63].
- Cooling: After 3 minutes, turn off the hotplate and move the pressure cooker to an empty sink. Activate the pressure release valve and run cold water over the cooker to depressurize and cool it quickly [61] [62]. Once depressurized, open the lid and run cold water into the cooker for an additional 10 minutes to cool the slides further [61].
- Protocol Continuation: After cooling, the slides are ready for the subsequent steps of the TUNEL assay protocol and any iterative immunofluorescence staining.
The following workflow diagram illustrates the optimized TUNEL assay procedure with the pressure cooker integration.
Comparative Data and Validation
The optimized protocol's efficacy is confirmed through direct comparison with the traditional Proteinase K method. The table below summarizes the key performance characteristics of both antigen retrieval methods in the context of a harmonized TUNEL and multiplexed immunofluorescence workflow.
Table 1: Quantitative Comparison of Antigen Retrieval Methods for TUNEL and Multiplexed Protein Detection
| Parameter | Proteinase K (Traditional) | Pressure Cooker (Optimized) |
|---|---|---|
| TUNEL Signal Quality | Reliable signal production [16] | Reliable signal production; tissue-specific minor differences in signal-to-noise [16] |
| Protein Antigenicity | Consistently reduced or abrogated [16] | Preserved or enhanced for targets tested [16] |
| Compatibility with MILAN/CycIF | Incompatible [16] | Fully compatible [16] |
| Erasability with 2-ME/SDS | Not determined | Fully erasable, allowing antibody removal and restaining [16] |
| Spatial Proteomic Context | Limited to 2-3 protein targets | Enables rich spatial contextualization with dozens of protein targets [16] |
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful implementation of this optimized protocol requires key reagents and materials. The following table lists essential solutions and their specific functions in the workflow.
Table 2: Key Research Reagent Solutions for Pressure Cooker TUNEL Assays
| Item | Function/Description | Application Note |
|---|---|---|
| Citrate Buffer (pH 6.0) | A common HIER buffer that breaks protein cross-links formed during fixation. | Provides intense TUNEL staining with very low background; a versatile first choice [62]. |
| EDTA Buffer (pH 8.0) | A higher-pHI HIER buffer that works via calcium ion extraction. | Can be highly effective for unmasking difficult epitopes; may require optimization to manage background [61] [62]. |
| Terminal Deoxynucleotidyl Transferase (TdT) | The core enzyme of the TUNEL assay; catalyzes the addition of labeled dUTP to DNA breaks. | Critical for labeling fragmented DNA; used in both traditional and optimized protocols [24]. |
| Labeled dUTP (e.g., Fluorescent) | The modified nucleotide incorporated by TdT, enabling detection of DNA fragmentation. | Fluorogenic detection (e.g., Tunnelyte Green/Red) allows for direct, safe, and sensitive detection [24]. |
| 2-Mercaptoethanol/SDS (2-ME/SDS) Buffer | A stripping buffer used in MILAN for antibody erasure between staining cycles. | Enables iterative staining on the same sample; compatible with the pressure-cooker-based TUNEL protocol [16]. |
Integrated Signaling Pathway and Experimental Logic
The pressure cooker optimization addresses a key step in a broader experimental workflow for studying apoptosis. The following diagram contextualizes the role of DNA fragmentation within the apoptotic signaling pathways and illustrates the logical flow of the integrated experimental approach, from cell death initiation to multiplexed spatial analysis.
Replacing Proteinase K with pressure cooker-based HIER resolves a fundamental incompatibility between the robust TUNEL assay and modern spatial biology techniques. This advanced optimization protocol provides a reliable method for detecting DNA fragmentation in late apoptosis while simultaneously preserving the antigenicity of protein biomarkers. By adopting this method, researchers and drug development professionals can achieve a rich, high-plex spatial contextualization of cell death, enabling deeper insights into disease mechanisms, drug efficacy, and tissue homeostasis in complex biological systems.
Validating TUNEL Results and Comparing DNA Damage Assays
Within the framework of a broader thesis on DNA fragmentation in late apoptosis research, the Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay stands as a cornerstone methodology. However, its accuracy is notoriously dependent on the implementation of stringent experimental controls. This application note details the essential control groups—positive, negative, and experimental—required to generate reliable, interpretable, and publication-quality TUNEL data. We provide validated protocols, quantitative benchmarks, and advanced visualization tools to empower researchers in the fields of cell biology, oncology, and drug development to confidently distinguish specific apoptosis from technical artifacts.
The TUNEL assay identifies a key hallmark of late-stage apoptosis: extensive DNA fragmentation. The assay utilizes the enzyme Terminal deoxynucleotidyl transferase (TdT) to incorporate labeled nucleotides at the 3'-hydroxyl (3'-OH) ends of fragmented DNA [64]. Despite its widespread use, the TUNEL assay is prone to significant pitfalls, including false-positive results from non-apoptotic DNA breaks generated during necrosis, DNA repair, or harsh sample processing [64] [65]. Consequently, establishing rigorous controls is not merely a recommendation but an absolute necessity to confirm that the observed signal genuinely reflects apoptotic DNA fragmentation.
This document outlines a comprehensive strategy for control groups, ensuring that your TUNEL assay data accurately reflects the underlying biology of apoptosis.
Core Control Groups: Definitions and Purpose
A properly controlled TUNEL experiment must include three fundamental groups, each serving a distinct and critical function in data interpretation and validation. The relationship and purpose of these groups are summarized in the following workflow.
Positive Control
- Purpose: To verify that every step of the assay protocol—from fixation and permeabilization to the TdT enzymatic reaction and detection—was performed correctly. A successful positive control demonstrates that the assay is technically capable of detecting DNA fragmentation.
- Expected Outcome: Nearly 100% of nuclei should show a strong TUNEL-positive signal. Failure to achieve this indicates a technical problem with the assay procedure [64] [13].
Negative Control
- Purpose: To identify non-specific background staining and false-positive signals. This control accounts for fluorescence or colorimetric signals not generated by the specific activity of the TdT enzyme on fragmented DNA.
- Expected Outcome: Minimal to no TUNEL signal should be present. A clear signal in the negative control indicates high background or non-specific staining, compromising the validity of the experimental results [64].
Experimental Group
- Purpose: To test the specific biological hypothesis (e.g., the effect of a chemotherapeutic agent on inducing apoptosis in a cancer cell line). The data from this group can only be trusted if the positive and negative controls have performed as expected.
- Expected Outcome: A variable percentage of TUNEL-positive cells, interpreted within the context of the established control baselines.
Detailed Experimental Protocols
Positive Control Generation Using DNase I
This protocol induces DNA strand breaks enzymatically, creating the substrate for the TdT enzyme and serving as a robust positive control [64] [13].
Materials
- DNase I (e.g., Component G from Click-iT TUNEL Kit [13])
- DNase I Buffer (e.g., 10X solution, Component H [13])
- Fixed and permeabilized sample (cell smear, coverslip, or tissue section)
Step-by-Step Procedure
- Prepare the sample: Fix and permeabilize cells or tissue sections according to your standard TUNEL protocol. Common fixation uses 4% paraformaldehyde for 15-30 minutes, followed by permeabilization with 0.1-0.5% Triton X-100 for 5-15 minutes on ice [64].
- Prepare DNase I Solution: For the Click-iT TUNEL kit, dilute the 10X DNase I buffer to 1X in molecular biology grade water. Add DNase I to a final concentration of ~1 µg/mL. Do not vortex, as vigorous mixing can denature the enzyme [13].
- Treat the sample: Apply 100 µL of the DNase I solution to the fixed/permeabilized sample and incubate for 30 minutes at room temperature [13].
- Terminate the reaction: Wash the sample once thoroughly with deionized water.
- Proceed with TUNEL labeling: Continue to the standard TUNEL assay protocol (Step 4: TdT Labeling Reaction) alongside your experimental and negative control samples.
Negative Control Generation (Enzyme Omission)
This is the most critical control for establishing assay specificity.
Procedure
- Prepare the TdT Reaction Mix without TdT Enzyme: For the sample designated as the negative control, prepare the labeling solution exactly as for the experimental group, but omit the Terminal deoxynucleotidyl transferase (TdT) enzyme [64]. All other components (reaction buffer, labeled dUTP) should be identical.
- Incubate and process: Apply this "-TdT" reaction mix to the negative control sample and incubate alongside the experimental samples. All subsequent wash, detection, and mounting steps should be identical.
Quantitative Analysis and Reference Values
For quantitative analyses, especially in clinical or translational research, establishing reference values is crucial. The following table summarizes key quantitative findings from sperm DNA fragmentation research, illustrating how controls enable the definition of diagnostically relevant thresholds.
Table 1: Quantitative Reference Values for TUNEL Assay in Sperm DNA Fragmentation Analysis [34]
| Parameter | Value | Context and Interpretation |
|---|---|---|
| Reference Value (Cutoff) | 16.8% | Sperm samples with DNA fragmentation below this value are considered normal. |
| Specificity | 91.6% | The probability that the test correctly identifies non-infertile men (low false-positive rate). |
| Sensitivity | 32.6% | The probability that the test correctly identifies infertile men with elevated SDF. |
| Positive Predictive Value (PPV) | 91.4% | The probability that a man with a positive test (SDF > 16.8%) is truly infertile due to SDF. |
| Upper Limit of Damage (Controls) | 19.6% | The maximum level of SDF observed in the control group with proven/unproven fertility. |
| Upper Limit of Damage (Infertile Patients) | 68.9% | The maximum level of SDF observed in the infertile patient group. |
The Scientist's Toolkit: Essential Reagents and Materials
A successful TUNEL assay relies on a suite of specialized reagents. The following table details the essential components and their functions.
Table 2: Key Research Reagent Solutions for TUNEL Assay [64] [13] [8]
| Reagent / Component | Function | Key Considerations & Variants |
|---|---|---|
| Terminal Deoxynucleotidyl Transferase (TdT) | Core enzyme that catalyzes the template-independent addition of labeled nucleotides to 3'-OH DNA ends. | Recombinant enzymes offer high consistency. The enzyme is sensitive to improper storage and handling. |
| Labeled dUTP | The detectable nucleotide incorporated into fragmented DNA. | BrdUTP (detected with antibody); Fluorescein-dUTP (direct detection); EdUTP (detected via click chemistry, offers small size and efficient incorporation [13] [8]). |
| Fixation Buffer (e.g., 4% PFA) | Cross-links cellular components, preserving morphology and locking fragmented DNA in place. | Over-fixation can mask DNA breaks and lead to false negatives [64]. |
| Permeabilization Buffer (e.g., Triton X-100, Proteinase K) | Creates pores in the cell and nuclear membranes, allowing the TdT enzyme to access the nuclear DNA. | Concentration and time must be optimized; under-permeabilization causes false negatives, while over-permeabilization can damage nuclei [64]. |
| Detection Reagents | Visualizes the incorporated nucleotide. | Varies by dUTP type: anti-BrdU antibodies, streptavidin-HRP/DAB for colorimetric detection, or copper-catalyzed "click" chemistry reagents for EdUTP [64] [8]. |
| Nuclear Counterstain (e.g., DAPI, Hoechst 33342, PI) | Labels all cell nuclei, enabling total cell counting and verification of nuclear localization of TUNEL signal. | Critical for accurate quantification and for distinguishing specific nuclear staining from artifacts [65]. |
| DNase I | Used to generate intentional DNA strand breaks for the positive control. | Essential for validating the entire assay workflow. |
Advanced Considerations and Multiplexing Strategies
To further enhance the rigor of apoptosis detection, the TUNEL assay should be integrated with other complementary methods.
Combating False Positives with Multiplexing: A positive TUNEL signal is not exclusively specific to apoptosis. It can also arise from DNA breaks during necrosis or active DNA repair processes [64]. To confirm apoptosis, multiplex the TUNEL assay with an antibody against an early apoptotic marker, such as cleaved Caspase-3 [64]. Cells positive for both cleaved Caspase-3 and TUNEL provide strong evidence of bona fide apoptosis. Furthermore, research has shown that cells can be TUNEL-positive and still recover, a process termed "anastasis" [64], further underscoring the need for complementary assays.
Advanced Quantification Techniques: Simple thresholding of TUNEL signal can lead to over-counting. Advanced image analysis methods, such as Multichannel Thresholding (MCT), are now recommended. MCT uses the nuclear counterstain (e.g., DAPI) to confirm that the TUNEL signal is co-localized with a cell nucleus, thereby avoiding counting non-nuclear artifacts and improving quantification accuracy [65].
Within the framework of research utilizing the TUNEL assay for detecting DNA fragmentation in late apoptosis, it is crucial to understand how this method correlates with other established techniques for assessing DNA integrity. The Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay detects DNA strand breaks by labeling the 3'-OH termini with modified nucleotides using the enzyme terminal deoxynucleotidyl transferase (TdT) [5] [8]. However, no single assay provides a complete picture, necessitating a comparative approach. This application note details the correlation of the TUNEL assay with three other major techniques: the Comet assay, Sperm Chromatin Structure Assay (SCSA), and Acridine Orange Test (AOT). We provide a structured quantitative comparison, detailed experimental protocols, and strategic guidance for researchers and drug development professionals to select and implement the most appropriate methodologies for their specific research contexts, particularly in the study of late-stage apoptotic DNA fragmentation.
Comparative Analysis of DNA Fragmentation Assays
The four major assays—TUNEL, Comet, SCSA, and AOT—each employ distinct principles to measure DNA strand breaks, offering complementary information [66] [67]. The following table summarizes their core characteristics, enabling researchers to make informed selections based on their specific experimental needs.
Table 1: Quantitative Comparison of Major DNA Fragmentation Assays
| Assay Name | Principle of Detection | Detection Output | Sample Type | Key Metric | Advantages | Limitations |
|---|---|---|---|---|---|---|
| TUNEL [5] [8] | Enzymatic labeling of 3'-OH ends with TdT and modified dUTP (e.g., EdU, BrdU) | Fluorescence or colorimetric signal at DNA break sites | Fixed cells/tissues (FFPE, frozen), cultured cells | Percentage of TUNEL-positive cells | High sensitivity, in situ detection, compatible with IHC/IF | May detect non-apoptotic DNA breaks; requires careful optimization [68] [5] |
| Comet Assay [66] [67] | Electrophoresis of lysed cells in agarose; DNA fragments migrate out of nucleus | Fluorescence microscopy; "comet" tail moment (tail length × intensity) | Fresh, suspended cells | Tail Moment, % DNA in Tail | Sensitive to low levels of damage, detects single/double-strand breaks | Limited to single-cell suspensions, complex image analysis |
| SCSA [66] [67] | Acid-induced DNA denaturation at break sites, staining with Acridine Orange (AO) | Flow cytometry; red (denatured) vs. green (native) fluorescence | Sperm cells, suspended cells | DNA Fragmentation Index (DFI), % HDS | High-throughput, objective, statistical reliability | Primarily validated for sperm, requires flow cytometer |
| Acridine Orange Test (AOT) [66] [67] | Metachromatic staining of native (green) vs. denatured (red) DNA | Fluorescence microscopy; green/red fluorescence ratio | Sperm cells, fixed cells | Ratio of red-to-green fluorescence | Simple, low-cost, light microscope-based | Semi-quantitative, more susceptible to subjective interpretation |
Figure 1: DNA Assay Selection Workflow. This diagram guides researchers in selecting the most appropriate DNA fragmentation assay based on their primary experimental requirement, such as the need for spatial context or high-throughput data.
Detailed Experimental Protocols
TUNEL Assay Protocol for Cultured Cells
The following protocol is adapted for the Click-iT TUNEL Alexa Fluor Imaging Assay and is typically completed within two hours [8] [13].
Key Research Reagent Solutions:
- Click-iT TUNEL Alexa Fluor Imaging Assay Kit: Contains TdT reaction buffer, EdUTP nucleotide mixture, recombinant TdT enzyme, Click-iT reaction buffer with Alexa Fluor azide, and Hoechst 33342 for nuclear counterstaining [13].
- 4% Paraformaldehyde (PFA) in PBS: A cross-linking fixative that preserves cellular morphology.
- 0.25% Triton X-100 in PBS: A detergent-based permeabilization reagent that creates pores in the cell membrane, allowing assay components to enter.
- DNase I (Component in Kit): An enzyme used to intentionally fragment DNA, serving as a essential positive control to validate the assay is working correctly [13].
Procedure:
- Cell Fixation: Grow cells on coverslips or in a 96-well microplate. Remove the culture media and wash once with 1X PBS. Add a sufficient volume of 4% PFA to completely cover the cells and incubate for 15 minutes at room temperature.
- Cell Permeabilization: Remove the fixative and add the permeabilization reagent (0.25% Triton X-100 in PBS). Incubate for 20 minutes at room temperature. Wash twice with deionized water.
- Positive Control Preparation (Optional but Recommended): Prepare a DNase I solution according to the kit instructions. Apply 100 µL to the designated control coverslip/well and incubate for 30 minutes at room temperature. Wash once with deionized water [13].
- TdT Reaction (Labeling): Prepare the TdT reaction cocktail by mixing TdT reaction buffer, EdUTP nucleotide mixture, and TdT enzyme. Add the cocktail to each sample and incubate in a humidified chamber for 60 minutes at 37°C.
- Click-iT Reaction (Detection): Prepare the Click-iT reaction mixture. Remove the TdT reaction cocktail from the samples and immediately add the Click-iT reaction mixture. Incubate for 30 minutes at room temperature, protected from light.
- Counterstaining and Imaging: Wash the samples with PBS. Add Hoechst 33342 or another nuclear stain diluted in PBS for 15 minutes. Wash and mount the coverslips for microscopy imaging. Analyze using standard fluorescence filters for the respective Alexa Fluor dye (e.g., FITC for Alexa Fluor 488) [13].
Comet Assay Protocol
The Comet assay, or single-cell gel electrophoresis, is a sensitive technique for detecting DNA damage at the level of individual cells [66] [67].
Procedure:
- Sample Preparation: Suspend cells in PBS at a density of approximately 1x10^5 cells/mL.
- Embedding in Agarose: Mix cells with melted low-melting-point agarose (e.g., 1% in PBS) at a ratio of 1:10 (v/v). Immediately pipet the mixture onto a comet slide or a pre-coated glass slide and place a coverslip on top. Allow the agarose to solidify at 4°C for 10-15 minutes.
- Lysis: Carefully remove the coverslip and immerse the slides in a chilled, neutral lysis buffer (e.g., containing 2.5 M NaCl, 100 mM EDTA, 10 mM Tris, 1% Triton X-100, pH 10) for at least 1 hour at 4°C, protected from light.
- Electrophoresis: After lysis, place the slides in a horizontal electrophoresis tank filled with neutral electrophoresis buffer (e.g., TBE). Run electrophoresis at a low voltage (e.g., 1 V/cm) for 20-30 minutes.
- Neutralization and Staining: Neutralize the slides by immersing in a neutral buffer (e.g., 0.4 M Tris, pH 7.5) for 5 minutes. Stain with a fluorescent DNA-binding dye such as SYBR Gold or propidium iodide (20 µg/mL) for 20 minutes.
- Analysis: Visualize using a fluorescence microscope. Analyze the "comet" images with specialized software to determine metrics like tail moment and % DNA in tail.
Sperm Chromatin Structure Assay (SCSA) Protocol
The SCSA is a flow cytometry-based technique that measures the susceptibility of sperm DNA to acid-induced denaturation [66] [67].
Procedure:
- Sample Preparation: Dilute a fresh or thawed sperm sample to a concentration of 1-2 x 10^6 cells/mL in TNE buffer (0.15 M NaCl, 0.01 M Tris-HCl, 1 mM EDTA, pH 7.4).
- Acid Denaturation: Mix 100 µL of sperm suspension with 200 µL of a low-pH detergent solution (0.1% Triton X-100, 0.15 M NaCl, 0.08 N HCl, pH 1.2). Incubate exactly for 30 seconds.
- Staining: Add 600 µL of acridine orange (AO) staining solution (6 µg/mL AO, 0.1 M Citric Acid, 0.2 M Na2HPO4, 1 mM EDTA, 0.15 M NaCl, pH 6.0). Incubate for 3 minutes.
- Flow Cytometric Analysis: Within 3-10 minutes of staining, analyze the sample using a flow cytometer. Acquire a minimum of 5,000 events per sample. AO intercalated into double-stranded DNA fluoresces green (530 nm, FL1), while AO associated with single-stranded DNA fluoresces red (>630 nm, FL3).
- Data Interpretation: The DNA Fragmentation Index (DFI) is calculated as the ratio of red to total (red + green) fluorescence. A DFI threshold of 27-30% is clinically significant for predicting reduced fertility outcomes [66] [67].
Acridine Orange Test (AOT) Protocol
The AOT is a simpler, microscopy-based version of the SCSA principle [66] [67].
Procedure:
- Slide Preparation: Prepare a sperm smear on a glass slide and allow it to air-dry.
- Fixation: Fix the smear in Carnoy's solution (3:1 methanol:glacial acetic acid) for at least 2 hours.
- Staining: Air-dry the fixed slide and stain with a fresh AO solution (0.19 mg/mL in citrate phosphate buffer, pH 2.5) for 5 minutes.
- Washing and Mounting: Rinse the slide gently with deionized water, mount with a coverslip, and blot away excess water.
- Analysis: Examine the slide immediately under a fluorescence microscope. A minimum of 200 cells should be counted and categorized based on fluorescence: green (normal double-stranded DNA) or red/orange (denatured single-stranded DNA).
Integration and Correlation in Apoptosis Research
In the context of late apoptosis research, these assays are not mutually exclusive but are often used to validate and complement each other's findings. The TUNEL assay is highly specific for in situ detection of DNA strand breaks, a hallmark of late apoptosis [5] [8]. However, it is critical to note that TUNEL can also label DNA breaks from other cell death mechanisms, such as necrosis, or even during DNA repair [68] [5]. Therefore, correlating TUNEL data with morphological analysis or other assays is essential for accurate interpretation.
The Comet assay offers superior sensitivity for detecting early, low-level DNA damage and can differentiate between single and double-strand breaks, providing a more granular view of the DNA damage process that precedes full-blown apoptosis [66]. The SCSA and AOT, while extensively used in andrology, are based on the principle of chromatin susceptibility, which reflects both DNA strand breaks and underlying chromatin packaging quality [66] [67]. This makes them excellent for assessing the overall health and maturity of cells, which can be a predisposing factor for apoptosis.
Figure 2: Assay Correlation in Apoptosis. This diagram illustrates how different DNA fragmentation assays provide complementary information when studying late apoptosis, from direct break detection to overall chromatin integrity.
The TUNEL, Comet, SCSA, and Acridine Orange tests each provide unique and valuable insights into DNA fragmentation. The choice of assay should be guided by the specific research question, sample type, and required throughput. For late apoptosis research focused on in situ confirmation within a tissue context, TUNEL is unparalleled. For highly sensitive detection of early damage or for samples requiring high-throughput analysis, the Comet assay or SCSA are more appropriate. The AOT remains a valuable, cost-effective tool for initial screening. A multi-assay approach, leveraging the strengths of each technique, often provides the most robust and comprehensive analysis of DNA fragmentation in complex biological systems and drug development pipelines.
Sperm DNA integrity is a critical parameter of male fertility, influencing not only fertilization success but also early embryonic development and offspring health. While DNA fragmentation is a well-established biomarker, the epigenetic implications of sperm DNA damage are increasingly recognized as equally vital. This application note provides a detailed comparative analysis of two predominant techniques for assessing sperm DNA damage: the TUNEL assay and the Comet assay. Framed within broader research on DNA fragmentation in late apoptosis, this document underscores the Comet assay's superior utility in predicting sperm epigenetic health, a crucial consideration for researchers and drug development professionals aiming to comprehensively evaluate male germline quality.
Quantitative Comparison of TUNEL and Comet Assays
Extensive clinical studies have systematically compared the diagnostic and predictive performance of these two assays in male infertility contexts. The following table consolidates key performance metrics from recent research.
Table 1: Comparative Performance Metrics of TUNEL and Comet Assays in Male Infertility
| Assay Parameter | TUNEL Assay | Alkaline Comet Assay | Clinical Context & Notes |
|---|---|---|---|
| Predictive Power for Infertility | Good | Best | Alkaline Comet was the best predictor of male infertility, followed by TUNEL [69] [70]. |
| Typical Cut-off Value for Infertility | 19.25% - 22.08% [70] [71] | 45.37% - 48.47% [69] [70] | Cut-off values are technique-dependent and should be established per laboratory [69]. |
| Correlation with Semen Parameters | Significant negative correlation with sperm count, motility, and morphology [72] | Significant negative correlation with standard semen parameters [69] | Both assays reflect conventional semen quality. |
| Association with DNA Methylation | Minimal (23 differentially methylated sites) [73] [74] | Strong (3,387 differentially methylated sites) [73] [74] | Comet assay is a significantly better indicator of sperm epigenetic health. |
| Detection Principle | Detects single and double-strand breaks via dUTP labeling [73] [71] | Detects single/double-strand breaks & alkali-labile sites via electrophoresis [75] [76] | Comet detects a broader spectrum of primary DNA lesions. |
Experimental Insights and Epigenetic Correlation
Predictive Value in Clinical Infertility
A comprehensive study comparing five Sperm DNA Fragmentation (SDF) assays found the alkaline Comet assay to be the most effective at distinguishing fertile donors from infertile patients, with TUNEL also showing significant diagnostic power [69] [70]. This establishes both as valuable tools for diagnosing male factor infertility.
Correlation with Assisted Reproductive Technology (ART) Outcomes
SDF levels measured by TUNEL have significant prognostic value for ART. One study found that patients with low-quality embryos had significantly higher SDF levels (30.02 ± 12.52%) compared to those with high-quality embryos (23.16 ± 8.41%) [72]. Furthermore, TUNEL values above 19.25% were shown to differentiate infertile men with 100% specificity [71].
Critical Differentiation for Epigenetic Health
The most compelling differentiator between these assays lies in their correlation with sperm DNA methylation patterns. A 2025 retrospective study of 1,470 men revealed a stark contrast:
- The Comet assay identified 3,387 significantly differentially methylated sites associated with high DNA damage.
- The TUNEL assay identified only 23 such sites [73] [74].
Gene ontology analysis further showed that DNA damage identified by the Comet assay was linked to biological pathways involved in germline development, while TUNEL produced no relevant pathways [74]. This evidence strongly positions the Comet assay as a superior tool for assessing epigenetic disruptions in sperm.
Detailed Experimental Protocols
Protocol for TUNEL Assay
The TUNEL assay detects DNA strand breaks by enzymatically labeling free 3'-OH termini with modified nucleotides.
Table 2: Key Research Reagent Solutions for TUNEL Assay
| Reagent/Material | Function / Specification |
|---|---|
| In situ Cell Death Detection Kit | Contains TdT enzyme and fluorescently-labeled dUTP. |
| Paraformaldehyde (3.9%) | Sample fixation. |
| Triton X-100 (2%) | Cell permeabilization to allow reagent entry. |
| Phosphate-Buffered Saline (PBS) | Washing and dilution buffer. |
| DAPI (4',6-diamidino-2-phenylindole) | Counterstain for total sperm head visualization. |
| Fluorescence Microscope or Flow Cytometer | Detection and quantification of labeled sperm. |
Step-by-Step Procedure:
- Smear and Fixation: Create air-dried smears of washed sperm. Fix slides in 3.9% paraformaldehyde at room temperature for 30 seconds [70].
- Permeabilization: Wash slides in PBS and permeabilize with 2% Triton X-100 for 30 seconds to create pores in the cell membrane [70] [71].
- Labeling: Apply the TUNEL reaction mixture (containing TdT enzyme and fluorescent-dUTP) to the slides. Incubate in a humidified chamber at 37°C for 58 minutes in the dark [70].
- Washing and Counterstaining: Wash slides thoroughly to remove unincorporated nucleotides. Counterstain with DAPI (8 mg/mL) to visualize all sperm nuclei [70].
- Analysis: Examine using fluorescence microscopy. TUNEL-positive sperm (green fluorescence) are counted against the total number of DAPI-stained sperm (blue fluorescence). A minimum of 300 sperm per sample should be scored [70]. For flow cytometry, analyze a minimum of 1,000 spermatozoa per examination [71].
Protocol for Alkaline Comet Assay
The Comet assay measures DNA strand breaks through single-cell gel electrophoresis under alkaline conditions, which also reveals alkali-labile sites.
Table 3: Key Research Reagent Solutions for Alkaline Comet Assay
| Reagent/Material | Function / Specification |
|---|---|
| Low-Melting Point Agarose (LMPA) | Forms a gel matrix that embeds cells for electrophoresis. |
| Normal-Melting Point Agarose (NMA) | Pre-coating slides to improve gel adhesion. |
| Lysis Solution (pH 10) | Contains NaCl (2.5M), Na₂EDTA (100mM), Tris base (10mM), DMSO (10%), and Triton X-100 (1%). Removes membranes and proteins. |
| Alkaline Electrophoresis Buffer (pH >13) | Contains NaOH and Na₂EDTA; enables DNA unwinding and expression of alkali-labile sites. |
| Neutralization Buffer (0.4M Tris, pH 7.5) | Neutralizes alkaline buffer post-electrophoresis. |
| Fluorescent DNA Stain (e.g., SYBR Green I) | Stains DNA for visualization and quantification. |
| Epifluorescence Microscope with Analysis Software | Captures and analyzes comet images (e.g., % tail DNA). |
Step-by-Step Procedure:
- Slide Preparation: Pre-coat microscopy slides with a layer of 1% Normal-Melting Point Agarose [76].
- Cell Embedding: Mix a 25 μL aliquot of sperm suspension (approx. 10x10⁶ sperm/mL) with 50 μL of 1% Low-Melting Point Agarose. Immediately pipet 10 μL of this mixture onto a pre-coated slide, cover with a coverslip, and allow the gel to solidify on a cold plate [70].
- Lysis: Carefully remove the coverslip and immerse slides in freshly prepared, cold lysis solution for a minimum of 1 hour at 4°C. For highly compacted sperm chromatin, lysis duration may be extended up to 24 hours [76].
- DNA Unwinding: Place slides in a horizontal electrophoresis tank filled with fresh, cold alkaline electrophoresis buffer (pH >13). Incubate for 20-40 minutes at 4°C to allow DNA unwinding and expression of alkali-labile sites [76].
- Electrophoresis: Perform electrophoresis under low voltage (e.g., 0.6-0.8 V/cm) for 20 minutes. The exact conditions (voltage, time) must be calibrated and kept constant for reproducibility [76].
- Neutralization and Staining: Gently neutralize slides by washing in neutralization buffer (e.g., 0.4M Tris, pH 7.5) for 5 minutes, repeated three times [76]. Stain with a fluorescent DNA stain such as SYBR Green I.
- Analysis: Visualize comets using an epifluorescence microscope. Analyze 100-150 comets per sample manually or using specialized software. The percentage of DNA in the comet tail (% tail DNA) is the most recommended and intuitive descriptor for DNA damage [77].
Workflow and Decision Pathways
The following diagram illustrates the key procedural steps and decision points for both the TUNEL and Comet assays, highlighting their parallel yet distinct methodologies.
Both the TUNEL and Comet assays are robust and validated methods for detecting sperm DNA fragmentation, offering significant diagnostic and prognostic value in male infertility and ART. The TUNEL assay provides a straightforward, specific measure of DNA strand breaks suitable for clinical flow cytometry. However, for research focused on the epigenetic consequences of sperm DNA damage, the alkaline Comet assay is the unequivocal tool of choice. Its superior sensitivity to a wider array of DNA lesions and its strong correlation with significant disruptions in sperm DNA methylation patterns make it indispensable for a comprehensive assessment of male germline epigenetic health. The choice of assay should therefore be guided by the specific research or clinical question: TUNEL for efficient quantification of strand breaks in a clinical setting, and the Comet assay for in-depth mechanistic studies linking DNA damage to epigenetic dysregulation.
The TUNEL (Terminal deoxynucleotidyl transferase dUTP nick end labeling) assay stands as a cornerstone technique in apoptosis research, specifically detecting the extensive DNA fragmentation that characterizes late-stage programmed cell death [1] [8]. Since its introduction in 1992, this method has become the most widely used in situ test for apoptosis study, enabling researchers to identify and quantify apoptotic cells within their physiological context [13] [8]. The fundamental principle relies on the enzyme Terminal deoxynucleotidyl transferase (TdT) catalyzing the incorporation of modified nucleotides at the 3'-OH ends of fragmented DNA, which are then detected through various strategies [1] [13] [8].
The selection of an appropriate quantification platform is crucial for generating accurate, reproducible data in DNA fragmentation studies. Each major platform—microscopy, flow cytometry, and microplate readers—offers distinct advantages and limitations regarding spatial information, statistical power, throughput, and multiplexing capabilities [17] [78] [79]. This application note provides detailed methodologies and comparative analysis to guide researchers in selecting and implementing the optimal quantification approach for their specific experimental needs in basic research and drug development.
Comparative Analysis of Quantification Platforms
The choice between microscopy, flow cytometry, and microplate reading depends heavily on the experimental goals, sample type, and required data output. The table below summarizes the key characteristics of each platform for TUNEL assay quantification.
Table 1: Comparison of TUNEL Assay Quantification Platforms
| Platform | Spatial Information | Throughput | Sensitivity | Primary Applications | Key Advantages |
|---|---|---|---|---|---|
| Microscopy | High (cellular/subcellular) | Low to moderate | High | Tissue sections, adherent cells, subcellular localization | Visual confirmation of morphology, spatial context within tissues |
| Flow Cytometry | None | High | High | Suspension cells, dissociated tissues, cell cycle analysis | Multiparametric analysis, objective quantification of large cell populations |
| Microplate Readers | None | Very high | Moderate | High-throughput compound screening, kinetic studies | Compatibility with 96/384-well formats, excellent for dose-response studies |
Microscopy-based quantification, including both fluorescence and brightfield imaging, provides unparalleled spatial context, allowing researchers to correlate TUNEL positivity with specific tissue architectures or subcellular compartments [8] [80]. This is particularly valuable in complex tissues where the spatial distribution of apoptosis is biologically meaningful. Flow cytometry excels in rapid, multiparametric analysis of individual cells in suspension, generating statistically robust data from thousands of events per sample [79]. This platform enables simultaneous assessment of DNA fragmentation alongside other parameters such as cell cycle phase or surface markers [79]. Microplate fluorescence assays offer the highest throughput capability, making them ideal for drug discovery applications requiring screening of multiple compounds or conditions with replicate sampling [1] [81].
When quantifying TUNEL assays, the modified nucleotide and detection strategy significantly impact sensitivity and specificity. BrdUTP detection with antibody conjugation typically offers the highest sensitivity for DNA break detection [79], while click chemistry-based approaches utilizing EdUTP provide efficient incorporation and flexible detection options [13] [8]. Studies comparing these methodologies under identical conditions have demonstrated that click chemistry-based TUNEL assays can detect a higher percentage of apoptotic cells than traditional methods using fluorescein-dUTP or BrdUTP [13] [8].
Detailed Experimental Protocols
TUNEL Assay for Fluorescence Microscopy
Sample Preparation (Adherent Cells):
- Culture cells on sterile glass coverslips or in chambered slides until 60-80% confluent.
- Apply apoptotic stimulus alongside appropriate negative (vehicle-treated) and positive controls (e.g., 0.5-1 μM staurosporine for 4 hours for HeLa cells) [13].
- Carefully remove media and wash cells once with phosphate-buffered saline (PBS), pH 7.4 [13].
Fixation and Permeabilization:
- Fix cells with 4% paraformaldehyde in PBS for 15 minutes at room temperature [13] [80].
- Remove fixative and wash twice with PBS.
- Permeabilize cells with 0.1-0.25% Triton X-100 in PBS for 20 minutes at room temperature [13] [80].
- Wash twice with PBS or deionized water to stop permeabilization.
TUNEL Reaction:
- Prepare TUNEL reaction mixture according to kit specifications. For example:
- Apply 20-100 μL of reaction mixture to completely cover the sample.
- Incubate for 1-4 hours at 37°C in a humidified, dark chamber [13] [39].
- Terminate reaction by washing twice with PBS containing 0.1% Triton X-100.
Detection and Visualization:
- For Click-iT chemistry: Prepare Click-iT reaction cocktail containing Alexa Fluor azide, reaction buffer, and additive. Incubate for 30 minutes at room temperature, protected from light [13].
- For antibody-based detection: Incubate with fluorochrome-conjugated anti-BrdU antibody (1:50-1:100 dilution) or anti-digoxigenin antibody in blocking buffer for 1-4 hours at room temperature [79].
- Wash thoroughly with PBS containing 0.1% Triton X-100.
- Counterstain nuclei with Hoechst 33342 (1-2 μg/mL) or DAPI for 15 minutes [13] [8].
- Mount coverslips with antifade mounting medium.
- Image using a fluorescence microscope with appropriate filter sets. For Alexa Fluor 488, use FITC filters (Ex/Em: 495/519 nm); for Alexa Fluor 594, use Texas Red filters (Ex/Em: 590/615 nm) [8].
Figure 1: TUNEL Microscopy Workflow
TUNEL Assay for Flow Cytometry
Sample Preparation:
- Harvest adherent cells using mild dissociation methods (PBS-EDTA with low trypsin concentration) to preserve cell integrity [17].
- For suspension cells, proceed directly to fixation.
- Wash cells once with PBS and resuspend at 1-2 × 10⁶ cells/mL in PBS.
Fixation and Permeabilization:
- Fix cells by adding them to ice-cold 1% formaldehyde (methanol-free) in PBS for 15 minutes on ice [79].
- Centrifuge at 300×g for 5 minutes, resuspend in PBS, and centrifuge again.
- Permeabilize cells by resuspending in ice-cold 70% ethanol and storing at -20°C for at least 2 hours (or up to several weeks).
TUNEL Labeling:
- Centrifuge ethanol-suspended cells at 200×g for 3 minutes and remove ethanol.
- Wash cells once with PBS and centrifuge at 300×g for 5 minutes.
- Prepare TUNEL reaction mixture (50 μL per sample):
- 10 μL TdT 5X reaction buffer
- 2.0 μL Br-dUTP stock solution (2 mM)
- 0.5 μL (12.5 units) TdT enzyme
- 5 μL CoCl₂ solution (10 mM)
- 32.5 μL distilled H₂O
- Resuspend cell pellet in TUNEL reaction mixture.
- Incubate for 40 minutes at 37°C [79].
Immunodetection:
- Add 2 mL of rinsing buffer (0.1% Triton X-100 + 0.5% BSA in PBS) and centrifuge.
- Resuspend in 100 μL of FITC-conjugated anti-BrdU monoclonal antibody (0.3 μg in PBS with 0.3% Triton X-100 and 1% BSA).
- Incubate for 1 hour at room temperature, protected from light.
- Add 2 mL of rinsing buffer, centrifuge, and resuspend in PI staining solution (5 μg/mL PI + 100 μg/mL RNase A in PBS).
- Incubate for 15-30 minutes at room temperature, protected from light.
- Analyze by flow cytometry using standard FITC and PI channels.
Table 2: Key Research Reagent Solutions for TUNEL Assays
| Reagent | Function | Example Formulations | Considerations |
|---|---|---|---|
| Fixative | Preserves cellular architecture and crosslinks DNA fragments | 4% paraformaldehyde in PBS, 1% formaldehyde (methanol-free) | Methanol-free formaldehyde prevents DNA denaturation; fixation time affects permeability |
| Permeabilization Agent | Enables reagent access to nuclear DNA | 0.1-0.25% Triton X-100, 70% ethanol, 0.1% sodium citrate with Triton X-100 | Concentration and duration must be optimized for each cell type to balance access and morphology |
| TdT Enzyme | Catalyzes nucleotide addition to 3'-OH DNA ends | 15 U/μL in glycerol storage buffer | Critical to maintain enzyme activity through proper storage and handling |
| Modified Nucleotides | Substrates for incorporation at DNA breaks | BrdUTP, EdUTP, FITC-dUTP, DIG-dUTP | BrdUTP/EdUTP offer higher sensitivity than direct FITC-dUTP; EdUTP enables click chemistry |
| Reaction Buffer | Provides optimal TdT enzyme activity | Cacodylate buffer (1M) with CoCl₂ | Cacodylate is toxic; some kits eliminate it for safety [1] |
| Detection Reagents | Visualize incorporated nucleotides | Fluorochrome-conjugated antibodies, Alexa Fluor azides, peroxidase substrates | Choice depends on detection method and desired multiplexing capabilities |
TUNEL Assay for Microplate Readers
Sample Preparation:
- Seed cells in black-walled, clear-bottom 96-well or 384-well microplates at optimal density.
- Apply experimental treatments alongside standards and controls.
- Include blank wells (no cells) for background subtraction.
Fixation and Permeabilization:
- Remove media and carefully wash once with PBS using multichannel pipette.
- Fix cells with 4% paraformaldehyde in PBS (50-100 μL/well) for 15 minutes at room temperature.
- Remove fixative and permeabilize with 0.1-0.25% Triton X-100 in PBS (50-100 μL/well) for 20 minutes.
- Wash twice with PBS.
TUNEL Reaction:
- Prepare TUNEL reaction mixture according to manufacturer's instructions.
- Add reaction mixture to each well (30-50 μL for 96-well plates, 10-15 μL for 384-well plates).
- Incubate for 1-2 hours at 37°C in the dark.
- Wash twice with PBS containing 0.1% Triton X-100.
Detection and Quantification:
- For fluorescence detection, add PBS or appropriate buffer to each well.
- Measure fluorescence using microplate reader with appropriate filters:
- FITC/Green fluorescence: Ex/Em ~490/520 nm [81]
- Red fluorescence: Ex/Em ~570-590/610-620 nm
- Normalize data to cell number using DNA-binding dyes (e.g., Hoechst 33342, Ex/Em ~350/461 nm) [13] [8].
Figure 2: Microplate TUNEL Workflow
Troubleshooting and Technical Considerations
Optimization and Validation:
- Always include appropriate controls: untreated cells (negative control), DNase I-treated cells (positive control for DNA break detection), and label-free samples (background control) [13] [39].
- Titrate TdT enzyme concentration and incubation time to maximize signal-to-noise ratio.
- For tissue sections, optimize proteinase K concentration and digestion time (typically 15-30 minutes) or consider alternative antigen retrieval methods like pressure cooking, which better preserves protein antigenicity for multiplexing [16].
- Validate TUNEL results with complementary apoptosis assays when possible, such as annexin V staining or caspase activation [17].
Troubleshooting Common Issues:
- High background signal: Reduce TdT enzyme concentration, shorten incubation times, increase wash stringency (higher detergent concentration or additional washes), or optimize fixation conditions.
- Low signal: Confirm TdT enzyme activity, ensure proper permeabilization, increase incubation time with TdT reaction mixture, or check reagent freshness.
- Cell loss (especially adherent cells): Use poly-lysine coated surfaces, avoid excessive pipetting directly on cells, and consider alternative fixation/permeabilization methods [17].
- Inconsistent results between technical replicates: Ensure consistent cell numbers per sample, uniform reagent distribution, and standardized washing procedures.
Multiplexing Capabilities: Modern TUNEL assays can be combined with other staining protocols to provide additional biological context:
- Click-iT Plus TUNEL assays allow multiplexing with fluorescent proteins and phalloidin staining by optimizing copper concentration in the detection reaction [8].
- Immunofluorescence staining for cell-type-specific markers or proteins of interest can be performed either before or after the TUNEL reaction, depending on the epitope sensitivity to TUNEL reagents [8] [39].
- For cell cycle analysis, combine TUNEL with DNA content staining using propidium iodide or DAPI [79].
The selection of an appropriate quantification platform for TUNEL assays fundamentally shapes the type and quality of data generated in apoptosis research. Microscopy provides invaluable spatial context at the cost of throughput, flow cytometry offers robust statistical power for population-level analyses, and microplate readers deliver maximum efficiency for screening applications. Recent methodological advancements, particularly the development of click chemistry-based detection and improved multiplexing capabilities, have expanded the applications of TUNEL across all platforms [13] [8] [16].
By following the detailed protocols outlined in this application note and understanding the comparative advantages of each platform, researchers can implement TUNEL quantification methods that yield precise, reproducible data tailored to their specific research questions in drug development and basic apoptosis mechanisms.
The Terminal deoxynucleotidyl transferase dUTP Nick-End Labeling (TUNEL) assay stands as a cornerstone method for detecting DNA fragmentation, a hallmark of apoptotic cell death. The assay operates on the principle of utilizing the enzyme terminal deoxynucleotidyl transferase (TdT) to catalyze the attachment of modified deoxynucleotides (dUTP) to the 3'-hydroxyl termini of DNA double-strand breaks [30]. These incorporated nucleotides are tagged with fluorochromes or haptens, enabling the visualization and quantification of cells containing fragmented DNA [82] [83]. Despite its widespread adoption and utility in apoptosis research since its development in 1992, a significant and persistent challenge surrounds the TUNEL assay: its lack of absolute specificity for apoptosis [68] [82]. This application note critically examines the limitations of the TUNEL assay, details the sources of false-positive and false-negative results, and provides validated protocols and strategies to enhance specificity, thereby ensuring more accurate interpretation in the context of DNA fragmentation research and drug development.
Core Limitations and Specificity Concerns of the TUNEL Assay
The TUNEL assay's fundamental limitation stems from its detection target—DNA strand breaks. While extensive DNA fragmentation is a definitive feature of the late stages of apoptosis, DNA breaks can arise from numerous other biological processes. Interpreting a positive TUNEL signal as synonymous with apoptotic cell death is a common misconception that can lead to erroneous conclusions in research and preclinical studies [68] [82].
Several pathological and physiological conditions distinct from apoptosis can generate DNA strand breaks detectable by the TUNEL assay. The primary sources of false-positive signals are summarized in the table below.
Table 1: Common Sources of False-Positive TUNEL Staining and Their Characteristics
| Source | Underlying Mechanism | Morphological Context |
|---|---|---|
| Necrosis | Unregulated DNA breakdown due to cellular energy failure and release of lysosomal nucleases [82]. | Loss of membrane integrity, cell swelling, and inflammatory response [17]. |
| Autolysis | Post-mortem DNA degradation occurring in tissues due to prolonged ischemia or inadequate fixation [82]. | Generalized tissue degradation without specific apoptotic morphology. |
| Active DNA Repair | DNA nicks generated during base excision repair or other repair pathways [82]. | Cells may appear normal or show non-apoptotic stress responses. |
| Cellular Senescence | Associated with persistent DNA damage signals and genomic instability [68]. | Enlarged, flattened cell morphology, and senescence-associated beta-galactosidase activity. |
| Proliferating Cells | DNA nicks generated during DNA replication and repair in highly proliferative tissues [82]. | Mitotic figures and evidence of active cell cycling. |
The Critical Distinction: Anastasis vs. Apoptosis
A profound challenge to the paradigm that TUNEL-positive cells are fated to die is the phenomenon of anastasis (Greek for "rising to life"). Anastasis is the process by which cells recover from the brink of apoptotic death, even after exhibiting canonical markers of apoptosis, including caspase activation, phosphatidylserine externalization, and genomic DNA breakage detected by TUNEL [68]. Compelling evidence from various biological systems demonstrates that cells can reverse early and even late stages of apoptosis upon removal of the apoptotic stimulus. This recovery has been observed in cancer cell lines expressing wild-type p53, mutant p53, and even in p53-null cells [68]. The implication for cancer therapy is critical: the administration of a chemotherapeutic agent may induce TUNEL-positive DNA fragmentation in tumor cells, but if the stimulus is not lethal or is removed, some cells may undergo anastasis, potentially leading to tumor repopulation and conferring therapy resistance [68]. This underscores the necessity of correlating TUNEL data with clonogenic survival assays or other viability tests in therapeutic contexts.
Technical and Interpretive Limitations
Beyond biological false positives, several technical factors influence the accuracy and reproducibility of the TUNEL assay:
- Variable Antigen Retrieval: The most common antigen retrieval method for TUNEL, proteinase K digestion, has been shown to consistently reduce or abrogate protein antigenicity, preventing robust multiplexing with immunofluorescence [16] [15]. This limits the ability to co-stain for other cell death or lineage markers, which is crucial for contextualization.
- Accessibility of DNA Breaks: The sensitivity of TUNEL is dependent on the accessibility of TdT to DNA breaks, which can be hindered by nuclear protein environment and the extent of fixative-induced cross-linking [82]. In sperm cells, the highly compact chromatin structure can prevent TdT from accessing DNA strand breaks, potentially leading to false negatives unless reducing agents like dithiothreitol are used [83].
- Lack of Standardization: The TUNEL assay lacks a universally standardized protocol, leading to variability in results between laboratories. Differences in fixation, permeabilization, and detection methods contribute to inconsistent thresholds for positivity and unreliable quantitative data [83].
Table 2: Advantages and Disadvantages of Common TUNEL Detection Methods
| Detection Method | Key Advantage | Key Disadvantage |
|---|---|---|
| Direct Fluorophore-dUTP (e.g., FITC-dUTP) | Faster protocol with fewer steps [12]. | Potential for lower signal intensity; susceptible to photo-bleaching. |
| Biotin-dUTP + Streptavidin-HRP | Signal amplification through streptavidin-biotin complex [12]. | Requires blocking of endogenous biotin; additional staining steps. |
| BrdU-dUTP + Anti-BrdU Antibody | Brighter signal due to efficient TdT incorporation [83] [12]. | Requires an antibody detection step; more complex protocol. |
| Click-iT (Alkyne-dUTP) | Superior penetration of small azide dyes; high sensitivity and compatibility with multiplexing [13]. | Copper catalyst can interfere with some fluorescent proteins or phalloidin [13]. |
Methodological Improvements and Enhanced Protocols
To overcome the limitations of traditional TUNEL, researchers have developed improved protocols that enhance specificity, sensitivity, and compatibility with modern spatial biology techniques.
Harmonized TUNEL with Spatial Proteomics
A significant recent advancement is the harmonization of TUNEL with multiplexed iterative immunofluorescence, such as Multiple Iterative Labeling by Antibody Neodeposition (MILAN). The key incompatibility was identified as proteinase K treatment, which massively degrades protein antigenicity. Replacing proteinase K with heat-mediated antigen retrieval using a pressure cooker quantitatively preserves the TUNEL signal without compromising the antigenicity of protein targets [16] [15]. This allows for the rich spatial contextualization of cell death within complex tissues, enabling researchers to simultaneously identify the cell type, activation state, and death status of individual cells.
Diagram 1: Workflow for TUNEL and Spatial Proteomics Integration
Optimized TUNEL Protocol for Enhanced Specificity
The following protocol is adapted for high specificity and compatibility with pressure cooker antigen retrieval, suitable for formalin-fixed paraffin-embedded (FFPE) tissues.
Materials & Reagents:
- TUNEL Assay Kit: e.g., Click-iT TUNEL Alexa Fluor Imaging Assay (Thermo Fisher) or equivalent.
- Fixative: 4% Paraformaldehyde (PFA) in PBS.
- Permeabilization Reagent: 0.25% Triton X-100 in PBS.
- Blocking Solution: 3% Bovine Serum Albumin (BSA) in PBS.
- Antigen Retrieval Buffer: Sodium citrate buffer (10mM, pH 6.0) or Tris-EDTA buffer (pH 9.0).
- Primary and Secondary Antibodies for multiplex immunofluorescence.
Experimental Procedure:
- Sample Preparation and Fixation:
- Fix cells or tissue sections in 4% PFA for 15 minutes at room temperature.
- Wash twice with PBS.
- Permeabilization:
- Permeabilize cells with 0.25% Triton X-100 in PBS for 20 minutes at room temperature.
- Wash twice with PBS.
- Heat-Mediated Antigen Retrieval (Critical for FFPE):
- For FFPE sections, perform deparaffinization and rehydration through xylene and a graded ethanol series.
- Place slides in a container with preheated antigen retrieval buffer.
- Perform heat-induced epitope retrieval using a pressure cooker or steamer according to standard protocols for your tissue type (e.g., 15-20 minutes at full pressure).
- Cool slides to room temperature and wash with distilled water, followed by PBS.
- TUNEL Reaction:
- Prepare the TUNEL reaction mixture according to the manufacturer's instructions. For the Click-iT TUNEL assay, this includes TdT reaction buffer, EdUTP, and TdT enzyme.
- Apply the reaction mixture to the samples and incubate in a dark, humidified chamber for 60 minutes at 37°C.
- Terminate the reaction by washing the samples with the provided buffer or PBS.
- Click-iT Detection (if applicable):
- Prepare the Click-iT reaction cocktail containing the fluorescent azide.
- Apply the cocktail to the samples and incubate for 30 minutes at room temperature, protected from light.
- Wash thoroughly with PBS.
- Multiplex Immunofluorescence:
- Block samples with 3% BSA for 30 minutes.
- Incubate with primary antibodies against target proteins (e.g., activated Caspase-3, cell-type-specific markers) overnight at 4°C.
- Wash and incubate with fluorophore-conjugated secondary antibodies for 1 hour at room temperature.
- Counterstain nuclei with Hoechst 33342 or DAPI.
- Mount and image.
Essential Research Reagent Solutions
Table 3: Key Reagents for a Modern TUNEL Workflow
| Reagent / Kit | Function | Specific Example |
|---|---|---|
| Click-iT TUNEL Kits | Provides a sensitive, click chemistry-based detection of DNA breaks with superior multiplexing capability [13]. | Click-iT Plus TUNEL Assay with Alexa Fluor dyes (Thermo Fisher) |
| Pressure Cooker / Steamer | Enables heat-mediated antigen retrieval, preserving protein epitomes for multiplexing while allowing TUNEL staining [16]. | Standard histology pressure cooker |
| DNase I (Recombinant) | Essential positive control to generate DNA strand breaks in situ, confirming assay functionality [13]. | Included in most commercial TUNEL kits |
| Caspase-3 (Active) Antibody | Key validation antibody to confirm the apoptotic pathway alongside DNA fragmentation [68]. | Anti-Cleaved Caspase-3 (Asp175) antibody |
| MILAN/CycIF Reagents | Enables iterative staining and erasure for high-plex protein marker analysis on the same section as TUNEL [16]. | 2-Mercaptoethanol/SDS erasure buffers, validated antibody panels |
A Strategic Framework for Accurate TUNEL Assay Interpretation
Given the documented limitations, relying solely on a positive TUNEL stain as definitive proof of apoptosis is not scientifically rigorous. The following strategic framework is recommended to ensure accurate data interpretation.
Mandatory Morphological and Multiplexed Validation
The gold standard for confirming apoptosis remains correlative morphological assessment. TUNEL-positive cells should be examined for classic apoptotic features such as cell shrinkage, chromatin condensation, nuclear fragmentation, and formation of apoptotic bodies [17]. This is best achieved by counterstaining with nuclear dyes like DAPI or Hoechst and using high-resolution microscopy.
Furthermore, the TUNEL assay must be combined with other markers of apoptosis in a multiplexed approach. Co-staining for executioner caspases (e.g., activated Caspase-3) provides independent biochemical confirmation that the apoptotic pathway has been triggered [68]. This multi-parametric analysis helps distinguish true apoptosis from other causes of DNA strand breaks.
Diagram 2: Decision Tree for Interpreting TUNEL-Positive Results
Utilizing Appropriate Controls and Complementary Assays
Rigorous experimental design requires the inclusion of comprehensive controls in every TUNEL experiment:
- Positive Control: Treat sample with DNase I to introduce DNA strand breaks and confirm the assay is working correctly [13] [12].
- Negative Control: Omit the TdT enzyme from the reaction mixture. This controls for non-specific labeling or background fluorescence.
- Technical Replicates: Ensure reproducibility by performing the assay on multiple biological and technical replicates.
For functional follow-up, especially in cancer therapy studies, TUNEL data should be correlated with clonogenic survival assays [68]. A cell may be TUNEL-positive but still retain the capacity to proliferate, a phenomenon central to anastasis. The clonogenic assay directly measures long-term reproductive viability, providing a functional counterpoint to the molecular snapshot provided by TUNEL.
The TUNEL assay remains a powerful and sensitive technique for detecting DNA fragmentation. However, its value is entirely dependent on the researcher's understanding of its significant limitations and a rigorous approach to experimental design and interpretation. The specificity of the TUNEL assay for apoptosis is not inherent but must be built through correlative morphological analysis, multiplexing with biochemical markers like activated caspases, and the use of functional viability assays. The adoption of modern protocols, particularly the replacement of proteinase K with heat-induced antigen retrieval, opens new doors for the rich spatial contextualization of cell death within complex tissue environments. By moving beyond the oversimplified equation of "TUNEL-positive equals dead," researchers can leverage this classic assay to generate more accurate, reliable, and biologically insightful data, ultimately advancing our understanding of cell death in development, disease, and therapeutic intervention.
The accurate assessment of sperm quality is a critical determinant of success in assisted reproductive technologies (ART). Traditional analysis, reliant on manual evaluation, is inherently subjective and prone to inter-operator variability. This is particularly consequential in the context of sperm DNA fragmentation (SDF), a key parameter reflecting male fertility potential. The TUNEL (Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling) assay stands as a gold-standard method for detecting DNA fragmentation, a hallmark of late-stage apoptosis [84] [85]. However, its manual interpretation can be time-consuming and subjective. The emergence of Artificial Intelligence (AI) and Computer-Aided Sperm Analysis (CASA) systems presents a transformative solution, enabling the automated, objective, and high-throughput analysis of sperm motility, morphology, and crucially, DNA integrity [86]. This document details the application of these AI-driven tools, framing their validation and protocols within the essential research context of apoptosis and DNA damage studies.
The Role of AI in Sperm Analysis and DNA Fragmentation
AI-powered CASA systems leverage advanced machine learning (ML) and deep learning (DL) techniques to overcome the limitations of manual analysis. These systems provide automated, objective, and high-throughput evaluation of key sperm parameters [86]. This is paramount for DNA fragmentation analysis, where AI can standardize the interpretation of assays like TUNEL, reducing human bias and enhancing reproducibility.
The TUNEL assay operates on the principle of detecting the extensive DNA fragmentation that occurs during the late stages of apoptosis. The key enzyme, Terminal deoxynucleotidyl transferase (TdT), catalyzes the addition of labeled nucleotides (e.g., fluorescein-dUTP) to the free 3'-hydroxyl termini of fragmented DNA [84]. AI integrates with this process by automating the identification and quantification of TUNEL-positive sperm cells from digital microscopy images or flow cytometry data, ensuring consistent and precise measurement of SDF levels.
Table 1: Core Analytical Parameters in AI-Based Sperm Analysis
| Parameter Category | Specific Measured Parameters | Clinical/Research Significance |
|---|---|---|
| Motility Analysis | Progressive motility, velocity, linearity | Assesses sperm movement efficiency and ability to reach the oocyte [86]. |
| Morphology Analysis | Head size/shape, midpiece and tail defects | Evaluates structural normality, crucial for successful fertilization [86]. |
| DNA Integrity Analysis | DNA Fragmentation Index (DFI) via TUNEL | Detects apoptotic sperm with damaged DNA, linked to embryo development failure [87] [86]. |
Quantitative Performance of AI-Based Sperm Analysis Tools
Validation studies demonstrate that AI-assisted systems perform on par with or exceed the capabilities of experienced embryologists. A 2025 observational study on an AI-based sperm selection system (Sperm ID) showed that its use led to biological outcomes, such as fertilization and blastocyst development rates, that were generally similar to those achieved by senior embryologists. Notably, in cycles with autologous oocytes, the use of top-quality sperm selected by AI was associated with a significantly higher blastocyst formation rate [87].
In flow cytometry, another area ripe for automation, an AI-assisted workflow (DeepFlow) reduced the analysis time for immunologic disorder cases from 10-20 minutes to under 5 minutes per case. When compared to manual analysis as the gold standard, the AI model exhibited a strong correlation (r > 0.9) across a wide range of lymphocyte subsets, confirming its diagnostic accuracy [88].
Table 2: Performance Metrics of Featured AI Systems in Validation Studies
| AI System / Software | Application Context | Key Performance Outcome | Comparison to Manual Standard |
|---|---|---|---|
| Sperm ID (v.1.0) [87] | Sperm selection for ICSI | Significantly higher blastocyst rate with "Best" sperm in autologous oocyte cycles. | Fertilization and embryo development rates were generally similar to senior embryologists. |
| DeepFlow (v.2.1.1) [88] | Flow cytometry for immunologic disorders | Reduced average analysis time to <5 minutes per case. | Strong correlation (r > 0.9) for cell subset enumeration across 379 clinical cases. |
| AI-CASA Systems [86] | General sperm quality assessment (motility, morphology, DNA integrity) | Automated, objective, and high-throughput evaluation. | Overcomes limitations of subjective, labor-intensive manual analysis. |
Integrated Experimental Protocol: AI-Assisted Sperm Analysis with TUNEL Assay
This protocol outlines the steps for assessing sperm DNA fragmentation using the TUNEL assay, with integrated AI-based analysis for objective quantification.
Stage 1: Sample Preparation and TUNEL Staining
Materials:
- Sperm Sample: Washed spermatozoa prepared via density gradient centrifugation or swim-up.
- Fixative: 4% Paraformaldehyde (PFA) in PBS.
- Permeabilization Solution: 0.1% Triton X-100 in PBS.
- TUNEL Assay Kit: Comprising TdT enzyme, labeled nucleotide (e.g., fluorescein-dUTP), and reaction buffer [85].
- Positive Control: DNase I (1 µg/mL) to induce DNA fragmentation.
- Negative Control: TUNEL reaction mix omitting the TdT enzyme.
Procedure:
- Fixation: Resuspend the sperm pellet in 4% PFA and incubate for 30-60 minutes at room temperature to cross-link and preserve cellular structures [84] [2].
- Permeabilization: Pellet the cells, wash with PBS, and permeabilize with 0.1% Triton X-100 for 5-15 minutes on ice. This creates pores in the cell membrane, allowing the TdT enzyme to access the nucleus [84].
- Control Setup:
- Positive Control: Treat a separate sample aliquot with DNase I for 15-30 minutes to create artificial DNA breaks.
- Negative Control: Designate a sample for the TUNEL reaction without the TdT enzyme.
- TUNEL Labeling:
- Prepare the TUNEL reaction mix according to the manufacturer's instructions.
- Incubate the fixed and permeabilized sperm cells (and controls) with the reaction mix for 60 minutes at 37°C in a humidified chamber to prevent evaporation [84].
- Stopping the Reaction: Wash the cells 2-3 times with PBS or a kit-specific stop/wash buffer.
- Counterstaining and Mounting: Counterstain nuclei with DAPI to visualize all cells. Mount the samples on slides with an antifade mounting medium [84].
Stage 2: AI-Assisted Image Acquisition and Analysis
Materials:
- Fluorescence Microscope: Equipped with high-resolution camera and automated stage.
- AI-CASA Software: Configured for TUNEL signal detection and sperm morphology analysis.
Procedure:
- Automated Image Acquisition: Use the fluorescence microscope to capture multiple, non-overlapping fields of view automatically. Acquire images using the appropriate filter sets for DAPI (all nuclei) and the fluorophore attached to the dUTP (e.g., FITC/Green for TUNEL-positive cells).
- AI-Based Quantification:
- Import the image set into the AI-CASA software.
- The AI algorithm, typically a deep learning model, will automatically:
- Identify and segment individual sperm heads based on the DAPI signal.
- Detect and quantify the fluorescence intensity of the TUNEL signal within each identified sperm nucleus.
- Classify each sperm cell as TUNEL-positive or TUNEL-negative based on a predefined intensity threshold.
- Data Output: The software generates a comprehensive report, including the DNA Fragmentation Index (DFI), which is the percentage of TUNEL-positive sperm in the total counted population.
Workflow Visualization
The following diagram illustrates the integrated experimental and computational pipeline for AI-assisted TUNEL analysis.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagent Solutions for AI-Assisted TUNEL Assay
| Item | Function / Role in the Protocol |
|---|---|
| Terminal Deoxynucleotidyl Transferase (TdT) | The core enzyme that catalyzes the addition of labeled dUTP to the 3'-OH ends of fragmented DNA [84] [85]. |
| Fluorophore-conjugated dUTP (e.g., FITC-dUTP) | The labeled nucleotide incorporated into damaged DNA, serving as the detectable marker for apoptosis [84]. |
| Paraformaldehyde (PFA) | A cross-linking fixative that preserves cellular morphology and locks fragmented DNA in place [84] [2]. |
| Triton X-100 | A detergent used for permeabilization, allowing the TdT enzyme to access the nuclear DNA [84]. |
| DNase I | Used to artificially fragment DNA in the positive control sample, validating the assay's functionality [84]. |
| DAPI (4',6-diamidino-2-phenylindole) | A nuclear counterstain that labels all cell nuclei, enabling the AI software to identify and count total sperm cells [84]. |
| AI-CASA Software | The computational tool that automates sperm identification, TUNEL signal quantification, and classification of sperm based on DNA integrity [86]. |
Conclusion
The TUNEL assay remains an indispensable, highly sensitive tool for the specific detection of late-stage apoptotic DNA fragmentation. Its utility is greatly enhanced by a thorough understanding of its biochemical principles, meticulous protocol execution informed by robust troubleshooting, and critical interpretation of results within the context of its specific limitations compared to other DNA damage assays. Future directions point toward deeper integration with multiplexed spatial biology techniques, the adoption of safer and more efficient chemistry like Click-iT, and the increasing use of AI for objective quantification. These advancements will further solidify the TUNEL assay's role in elucidating disease mechanisms, screening novel therapeutics, and advancing diagnostic applications in biomedical and clinical research.
References
- 1. Visualizing Apoptosis with AAT Bioquest's TUNEL Assays [https://resources.biomol.com/biomol-blog/cellular-self-destruction-visualizing-apoptosis-with-aat-bioquests-tunel-assays]
- 2. Apoptosis DNA fragmentation analysis protocol [https://www.abcam.com/en-us/technical-resources/protocols/apoptosis-dna-fragmentation]
- 3. TUNEL Assay Kit (Fluorescence, 594 nm) #48513 [https://www.cellsignal.com/products/cellular-assay-kits/tunel-assay-kit-fluorescence-594-nm/48513?srsltid=AfmBOopXc0lQIqZpVZs7oR_OgERXIlKG95Yq4-dLkQRgjCzAMT2GJSjE]
- 4. Mastering Apoptosis Detection: A Step-by-Step TUNEL Assay ... [https://www.clyte.tech/post/mastering-apoptosis-detection-a-step-by-step-tunel-assay-protocol?srsltid=AfmBOoqv18agpdiBklwBVMT6Vu-3dGQgMlZ_K2-X4G8j66w6NkXZi6F-]
- 5. TUNEL Assay: A Powerful Tool for Kidney Injury Evaluation [https://pmc.ncbi.nlm.nih.gov/articles/PMC7795088/]
- 6. TUNEL assay – Encyclopedia of Biological Methods [https://rwu.pressbooks.pub/encyclopediaofbiologicalmethods/chapter/tunel-assay/]
- 7. 32: TUNEL Assay [https://bio.libretexts.org/Bookshelves/Biotechnology/Encyclopedia_of_Biological_Methods_(Mattaini)/32%3A_TUNEL_Assay]
- 8. TUNEL Assays | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/cell-analysis/cell-viability-and-regulation/apoptosis/tunel-assays.html]
- 9. TUNEL Assay to Assess Extent of DNA Fragmentation and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8413497/]
- 10. Tunel Cell Apoptosis Assay Kit Market Size By Type [https://www.linkedin.com/pulse/tunel-cell-apoptosis-assay-kit-market-rgtyf/]
- 11. Mastering Apoptosis Detection: A Step-by-Step TUNEL Assay ... [https://www.clyte.tech/post/mastering-apoptosis-detection-a-step-by-step-tunel-assay-protocol?srsltid=AfmBOoouGAwbJnnfXF5je3RH4mFtDpbik7zcUq_2-5qsa3lI4RR6fFNz]
- 12. TUNEL staining [https://www.abcam.com/en-us/technical-resources/guides/cell-health-guide/tunel-staining-or-tunel-assay]
- 13. Click-iT TUNEL Alexa Fluor Imaging Assay Protocol [https://www.thermofisher.com/us/en/home/references/protocols/cell-and-tissue-analysis/apoptosis-protocol/tunel-protocols/click-it-tunel-alexa-fluor-imaging-assay-protocol.html]
- 14. TUNEL assay-Standardized method for testing sperm DNA ... [https://pubmed.ncbi.nlm.nih.gov/32706440/]
- 15. Harmonizing TUNEL with multiplexed iterative ... [https://www.sciencedirect.com/science/article/pii/S2667237525000839]
- 16. Harmonizing TUNEL with multiplexed iterative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12146639/]
- 17. Comparison of several techniques for the detection ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6495494/]
- 18. Validation of an Artificial Intelligence Tool for the Detection ... [https://arxiv.org/abs/2510.11142]
- 19. Advances in apoptosis research [https://pubmed.ncbi.nlm.nih.gov/9398063/]
- 20. Apoptosis | Research Starters [https://www.ebsco.com/research-starters/science/apoptosis]
- 21. TUNEL Assay Kit (Fluorescence, 594 nm) #48513 [https://www.cellsignal.com/products/cellular-assay-kits/tunel-assay-kit-fluorescence-594-nm/48513?srsltid=AfmBOopuTjx4mwnRCxbZJTe8DIMgMBhV2uq615mxk9hkckaVTAb0Nl2H]
- 22. Apoptosis Assay Market Size Report, 2025 – 2034 [https://www.gminsights.com/industry-analysis/apoptosis-assay-market]
- 23. Investigating apoptosis in peripheral blood mononuclear cells ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12577001/]
- 24. A Superior Direct Method for the Quantitative Analysis ... [https://www.aatbio.com/resources/assaywise/2018-7-1/a-superior-direct-method-for-the-quantitative-analysis-of-dna-fragments-in-late-stage-apoptosis]
- 25. Mastering Apoptosis Detection: A Step-by-Step TUNEL Assay ... [https://www.clyte.tech/post/mastering-apoptosis-detection-a-step-by-step-tunel-assay-protocol?srsltid=AfmBOoqX8iG9-WNfmiLFBgr4EmYEwgy6IApdkyq_gOa2FkccG-n5DYiD]
- 26. tunel染色检测凋亡细胞技术原理与步骤介绍 [https://cn.gempharmatech.com/resources/organism/4935353.html]
- 27. 染色质缩合和TUNEL试剂盒| Thermo Fisher Scientific - 赛默飞 [https://www.thermofisher.cn/cn/zh/home/life-science/cell-analysis/cellular-imaging/fluorescence-microscopy-and-immunofluorescence-if/imaging-apoptosis-assays/tunel-assays-dna-fragmentation-chromatin-condensation.html]
- 28. Video: The TUNEL Assay [https://www.jove.com/v/5651/the-tunel-assay]
- 29. Detection of DNA Fragmentation in Apoptotic Cells by TUNEL [https://pubmed.ncbi.nlm.nih.gov/27698233/]
- 30. TUNEL assay [https://en.wikipedia.org/wiki/TUNEL_assay]
- 31. TUNEL Assay: The Gold Standard Technique for Apoptosis ... [https://www.antbioinc.com/blogs/technical-articles/tunel-assay-the-gold-standard-technique-for-apoptosis-detection]
- 32. TUNEL Assay Principle - TACS TdT [https://www.rndsystems.com/resources/technical/tunel-assay-principle-tacs-tdt]
- 33. DNA Fragmentation in Apoptosis | Detection and TUNEL ... [https://www.bio-techne.com/research-areas/apoptosis/dna-fragmentation]
- 34. Terminal deoxynucleotidyl transferase dUTP nick end labeling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4758999/]
- 35. Mastering Apoptosis Detection: A Step-by-Step TUNEL Assay ... [https://www.clyte.tech/post/mastering-apoptosis-detection-a-step-by-step-tunel-assay-protocol?srsltid=AfmBOoojUfEfSb-YUkXjwMRxXRKTfTyQA5MP-7pnm7pJm_u9AAFKGdb_]
- 36. TUNEL Assay for Analyzing Apoptosis & Cell Death [https://opentrons.com/applications/flow-cytometry/tunel-assay?srsltid=AfmBOor7Rz7f7px4TUTFy3YoqgUqQfmpuQ8EkI_Mnu5OXm8sBjf3pPU1]
- 37. Programmed cell death detection methods - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC9308588/]
- 38. Colorimetric vs Fluorometric assay: Which is better for ... [https://eureka.patsnap.com/article/colorimetric-vs-fluorometric-assay-which-is-better-for-sensitivity]
- 39. Detection of Apoptotic Cells in Planarians by Whole-Mount ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6042512/]
- 40. Programmed cell death detection methods: a systematic ... [https://link.springer.com/article/10.1007/s10495-022-01735-y]
- 41. Protocol to study cell death using TUNEL assay in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8808282/]
- 42. Electrochemical approach for the analysis of DNA ... [https://www.sciencedirect.com/science/article/pii/S2405844024016335]
- 43. Click-iT EdU Imaging Cell Proliferation Protocol [https://www.thermofisher.com/us/en/home/references/protocols/cell-and-tissue-analysis/protocols/click-it-edu-imaging-protocol.html]
- 44. Harmonizing TUNEL with multiplexed iterative ... [https://hscrb.harvard.edu/publication/harmonizing-tunel-with-multiplexed-iterative-immunofluorescence-enriches-spatial-contextualization-of-cell-death-cell-rep-methods-2025-may-19-55101047/]
- 45. Apoptosis: A Review of Programmed Cell Death - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2117903/]
- 46. Highly multiplexed 3D profiling of cell states and immune ... [https://www.nature.com/articles/s41592-025-02824-x]
- 47. EACR 2025 - Lunaphore Technologies [https://lunaphore.com/our-events/eacr25/]
- 48. Sperm DNA fragmentation testing: Summary evidence and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7988559/]
- 49. Fragmentation of sperm DNA using the TUNEL method [https://www.sciencedirect.com/science/article/abs/pii/S2173578614001371]
- 50. TUNEL via Fluorescence Microscopy, and Flow Cytometry [https://www.mdpi.com/1648-9144/59/7/1313]
- 51. Protocol to study cell death using TUNEL assay in ... [https://www.sciencedirect.com/science/article/pii/S266616672200020X]
- 52. Emerging Electrochemical Approaches for the Early Detection ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12355238/]
- 53. Mastering Apoptosis Detection: A Step-by-Step TUNEL Assay ... [https://www.clyte.tech/post/mastering-apoptosis-detection-a-step-by-step-tunel-assay-protocol?srsltid=AfmBOooiHn4sNJYvqzPVnnocVXBkkJ7rLL9G7UMaD0sJDI6G1TTAHoud]
- 54. Guidelines for Regulated Cell Death Assays - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC7970050/]
- 55. High-throughput RNA sequencing of paraformaldehyde ... [https://www.nature.com/articles/s41467-021-25871-2]
- 56. A guided protocol for tissue dissection and proteinase K ... [https://www.sciencedirect.com/science/article/pii/S2215016124004333]
- 57. A simple technique for quantifying apoptosis in 96-well plates [https://pmc.ncbi.nlm.nih.gov/articles/PMC1142306/]
- 58. Terminal deoxynucleotidyl transferase: Properties and applications - ScienceDirect [https://www.sciencedirect.com/science/article/pii/S2667370324000419]
- 59. Terminal deoxynucleotidyl Transferase (TdT) [https://mclab.com/mc/terminal-deoxynucleotidyl-transferase-tdt/]
- 60. Using ddPCR to assess the DNA yield of FFPE samples [https://pmc.ncbi.nlm.nih.gov/articles/PMC6287546/]
- 61. IHC antigen retrieval protocol [https://www.abcam.com/en-us/technical-resources/protocols/ihc-antigen-retrieval]
- 62. Tips for Antigen retrieval [https://www.biossusa.com/blogs/news/tips-for-antigen-retrieval?srsltid=AfmBOoqTJOuCyen3BGtx3eRMv3mifblX8NzyIFn7RqQJASqyFQUkUNEk]
- 63. What is your recommended procedure for antigen retrieval ... [https://www.cellsignal.com/learn-and-support/technical-support/what-is-your-recommended-procedure-for-antigen-retrieval-with-a-pressure-cooker/000001093?srsltid=AfmBOorV6jJO2yyOBtx8rY4x5EpIBhWdR5elZxVHhYNp4Eo83jULd0ld]
- 64. Mastering Apoptosis Detection: A Step-by-Step TUNEL Assay ... [https://www.clyte.tech/post/mastering-apoptosis-detection-a-step-by-step-tunel-assay-protocol?srsltid=AfmBOorxlRN8K2VKPfdDSv5b_ju5-j7Bz31fOJUQXcU-LJP3_jzUGai9]
- 65. A new multichannel method quantitating TUNEL in ... [https://www.sciencedirect.com/science/article/abs/pii/S0014483518303786]
- 66. Clinical aspects of sperm DNA fragmentation detection and male... [https://pubmed.ncbi.nlm.nih.gov/16242181/]
- 67. (PDF) Clinical aspects of sperm DNA fragmentation detection and male... [https://www.academia.edu/24893557/Clinical_aspects_of_sperm_DNA_fragmentation_detection_and_male_infertility]
- 68. Do TUNEL and Other Apoptosis Assays Detect Cell Death ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7730366/]
- 69. TUNEL assay, SCSA, SCD test and alkaline and neutral ... [https://pubmed.ncbi.nlm.nih.gov/23843251/]
- 70. Evaluation of sperm DNA fragmentation using multiple ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6436467/]
- 71. TUNEL as a Test for Sperm DNA Damage in the Evaluation ... [https://www.sciencedirect.com/science/article/abs/pii/S0090429510005972]
- 72. The Utility of Sperm DNA Fragmentation as a Diagnostic ... [https://www.mdpi.com/1422-0067/26/13/6314]
- 73. The correlation between sperm DNA methylation and ... [https://www.frontiersin.org/journals/reproductive-health/articles/10.3389/frph.2025.1523386/full]
- 74. The correlation between sperm DNA methylation and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11882583/]
- 75. A Comprehensive Review on Clinical Applications of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4413081/]
- 76. Comet Assay - an overview [https://www.sciencedirect.com/topics/immunology-and-microbiology/comet-assay]
- 77. An Overview of Comet Assay Application for Detecting ... [https://www.mdpi.com/2077-0472/13/3/623]
- 78. Comparing flow cytometry and fluorescence microscopy for ... [https://pubmed.ncbi.nlm.nih.gov/18636473/]
- 79. Analysis of apoptosis by cytometry using TUNEL assay [https://pmc.ncbi.nlm.nih.gov/articles/PMC2295206/]
- 80. TUNEL Assay - an overview | ScienceDirect Topics [https://www.sciencedirect.com/topics/neuroscience/tunel-assay]
- 81. One-step TUNEL Apoptosis Assay Kit (Green Fluorescence ... [https://www.antibodies.com/catalog/assay-kits/one-step-tunel-apoptosis-assay-kit-green-fluorescence-a319767]
- 82. TUNEL Assay - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/tunel-assay]
- 83. TUNEL Assay - an overview [https://www.sciencedirect.com/topics/medicine-and-dentistry/tunel-assay]
- 84. Mastering Apoptosis Detection: A Step-by-Step TUNEL Assay ... [https://www.clyte.tech/post/mastering-apoptosis-detection-a-step-by-step-tunel-assay-protocol?srsltid=AfmBOopro6xqUB4XNO7N4BNyIZiPv3byr4uN5QEZhMS43VwrFSiOmD2z]
- 85. TUNEL Assay Kit (Fluorescence, 594 nm) #48513 [https://www.cellsignal.com/products/cellular-assay-kits/tunel-assay-kit-fluorescence-594-nm/48513?srsltid=AfmBOor-f8kCUd0wkv12trxBb7-Z3SB6A59D39OvCNRGYjBKYQOvTBZ7]
- 86. Revolutionizing Sperm Analysis with AI: A Review of ... [https://www.mdpi.com/2079-3197/13/6/132]
- 87. Automated AI for real-time sperm selection in ICSI [https://pubmed.ncbi.nlm.nih.gov/41299699/]
- 88. Validation of Artificial Intelligence (AI)-Assisted Flow ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10888253/]