Validating Apoptosis: A Comprehensive Guide to Caspase-3 Activation and PARP Cleavage by Western Blot
This article provides a complete resource for researchers and drug development professionals on the concurrent detection of caspase-3 activation and PARP cleavage, a gold-standard method for apoptosis validation.
Validating Apoptosis: A Comprehensive Guide to Caspase-3 Activation and PARP Cleavage by Western Blot
Abstract
This article provides a complete resource for researchers and drug development professionals on the concurrent detection of caspase-3 activation and PARP cleavage, a gold-standard method for apoptosis validation. It covers the foundational biology of these key apoptotic markers, detailed methodological protocols including optimized antibody usage and sample preparation, extensive troubleshooting for common issues like weak signals and non-specific bands, and strategies for experimental validation. By integrating the latest technical insights and troubleshooting guides, this guide aims to ensure the acquisition of reliable, publication-quality data in studies of cell death mechanisms, cancer biology, and therapeutic efficacy.
The Apoptotic Executioners: Understanding Caspase-3 and PARP in Cell Death Pathways
Caspase-3 is a critical executioner protease that serves as the central mediator of apoptotic cell death, responsible for the systematic dismantling of cellular structures through the proteolytic cleavage of key protein substrates. This cysteine-aspartic protease exists as an inactive 32 kDa zymogen (pro-caspase-3) that undergoes proteolytic processing into activated fragments of 17 kDa and 19 kDa (p17/p19) upon apoptotic stimulation [1] [2]. The activation of caspase-3 represents a point of convergence in apoptosis signaling, as it can be triggered by both the extrinsic (death receptor) and intrinsic (mitochondrial) pathways [3] [4]. Once activated, caspase-3 executes the terminal phase of apoptosis by cleaving over 500 cellular targets, including the well-characterized nuclear enzyme poly(ADP-ribose) polymerase (PARP), whose cleavage serves as a definitive biochemical marker of apoptosis [1] [5]. This review provides a comprehensive comparison of caspase-3 detection methodologies, experimental approaches for validating its activation, and essential reagent solutions for researchers studying apoptotic mechanisms in both physiological and pathological contexts.
Caspase-3 Activation Pathways and Detection Strategies
The transition of caspase-3 from inactive zymogen to active protease involves precise proteolytic cleavage at specific aspartic residue sites. Initiator caspases, particularly caspase-8 in the extrinsic pathway and caspase-9 in the intrinsic pathway, catalyze the proteolytic processing of pro-caspase-3 [4]. The activation mechanism involves cleavage at Asp175-Ser176, generating the large (p17/p19) and small (p12) subunits that form the active heterotetrameric enzyme [2]. This active caspase-3 then recognizes the tetra-peptide motif DE(V/T)D in target proteins, cleaving them to bring about the characteristic morphological and biochemical changes of apoptosis [6] [7].
Figure 1: Caspase-3 Activation Pathways. Caspase-3 serves as a convergence point for extrinsic (death receptor) and intrinsic (mitochondrial) apoptotic pathways, undergoing proteolytic activation that leads to PARP cleavage and execution of apoptosis.
Detection of caspase-3 activation relies primarily on immunological methods that distinguish between the inactive zymogen and the active cleavage fragments. Antibodies specific to the cleaved forms of caspase-3 (particularly those recognizing the Asp175 cleavage site) provide the most reliable indication of activation, as they avoid cross-reactivity with the abundant pro-caspase-3 pool [2]. The active p17/p19 fragments can be detected in various applications including western blotting, immunohistochemistry, immunofluorescence, and flow cytometry, with optimized protocols for each method ensuring specific and sensitive detection [1] [2].
Comparative Analysis of Caspase-3 Antibody Performance
Key Antibody Specifications and Applications
Table 1: Comparison of Commercial Caspase-3 Antibodies
| Product Name | Host Species | Reactivity | Applications | Specificity | Recommended Dilutions |
|---|---|---|---|---|---|
| Caspase 3/P17/P19 Polyclonal Antibody (19677-1-AP) [1] | Rabbit | Human, Mouse, Rat, and 8 more species | WB, IHC, IF/ICC, IP, ELISA | Recognizes p17, p19, and p32 (full-length) forms | WB: 1:500-1:2000IHC: 1:50-1:500IF/ICC: 1:50-1:500 |
| Cleaved Caspase-3 (Asp175) Antibody (#9661) [2] | Rabbit | Human, Mouse, Rat, Monkey | WB, IHC, IF, FC, IP | Specific for large fragment (17/19 kDa) of activated caspase-3; does not recognize full-length | WB: 1:1000IHC: 1:400IF: 1:400FC: 1:800 |
The Proteintech Caspase 3/P17/P19 antibody (19677-1-AP) stands out as the most cited caspase-3 antibody in the market with over 2,543 documented citations, reflecting its extensive validation across numerous research applications [1]. This antibody recognizes multiple forms of caspase-3 including the full-length zymogen (32-35 kDa) and the active cleavage fragments (p17 and p19), providing researchers with a comprehensive view of both expression and activation status. In contrast, Cell Signaling Technology's Cleaved Caspase-3 (Asp175) Antibody (#9661) offers exceptional specificity for the activated form only, making it particularly valuable for specifically detecting apoptosis without background from the inactive precursor [2].
Experimental Validation Data
Table 2: Experimental Performance Data of Caspase-3 Detection Methods
| Experimental Context | Sample Type | Key Findings | Validation Approach |
|---|---|---|---|
| USP48 Cleavage in AML [7] | U937, NB4, OCI-AML2 leukemia cells | Activated caspase-3 cleaves USP48 at DEQD611-614 motif during drug-induced apoptosis | Western blot with Proteintech USP48 antibody (12076-1-AP) and CST Cleaved Caspase-3 (#9661) |
| Real-time Caspase-3/7 Dynamics [6] | 2D/3D cell cultures, patient-derived organoids | ZipGFP reporter enabled live imaging of DEVD cleavage activity; verified by western blot | Concurrent western blot with cleaved caspase-3 and PARP antibodies |
| PARP-1 Activation in Bovine Mastitis [8] | Bovine milk leukocytes | Significant PAR content increase in infected samples correlated with active caspase-3 | Flow cytometry with PE-conjugated anti-active Caspase-3 (BD Biosciences) |
| Neuronal Apoptosis [9] | Neurally differentiated NT2 cells | Caspase-3 activation precedes neurodegeneration in APP-overexpressing neurons | Western blot and immunocytochemistry with anti-p20/17 antibodies |
The integration of caspase-3 activation detection with downstream substrate cleavage analysis, particularly PARP processing, provides a robust validation framework for apoptosis research. Studies consistently demonstrate that caspase-3 activation directly correlates with PARP cleavage, establishing this paired analysis as a gold standard for confirming apoptotic events [1] [5]. Furthermore, the development of advanced reporter systems incorporating the DEVD cleavage motif (the canonical caspase-3 recognition sequence) enables real-time monitoring of caspase-3 activity in live cells, organoids, and complex 3D culture systems [6].
Essential Protocols for Caspase-3 Activation Analysis
Western Blot Analysis of Caspase-3 Processing and PARP Cleavage
Sample Preparation: Lysate cells using RIPA buffer supplemented with protease and phosphatase inhibitors. For tissue samples, homogenize using a Dounce homogenizer or similar mechanical disruption method. Protein concentration should be determined using BCA or Bradford assay, with 20-50 µg of total protein typically loaded per lane for SDS-PAGE [1] [7].
Electrophoresis and Transfer: Separate proteins using 12-15% SDS-PAGE gels to optimally resolve the caspase-3 fragments (p17/p19) and PARP cleavage products (89 kDa fragment). Transfer to nitrocellulose or PVDF membranes using standard wet or semi-dry transfer systems [7].
Antibody Incubation: Block membranes with 5% non-fat dry milk or BSA in TBST for 1 hour at room temperature. Incubate with primary antibodies against caspase-3 (1:500-1:2000 for Proteintech 19677-1-AP) and/or cleaved caspase-3 (1:1000 for CST #9661) diluted in blocking buffer overnight at 4°C [1] [2]. For PARP cleavage analysis, use anti-PARP antibody (e.g., CST #9542) at 1:1000 dilution [7]. Follow with appropriate HRP-conjugated secondary antibodies and detect using enhanced chemiluminescence.
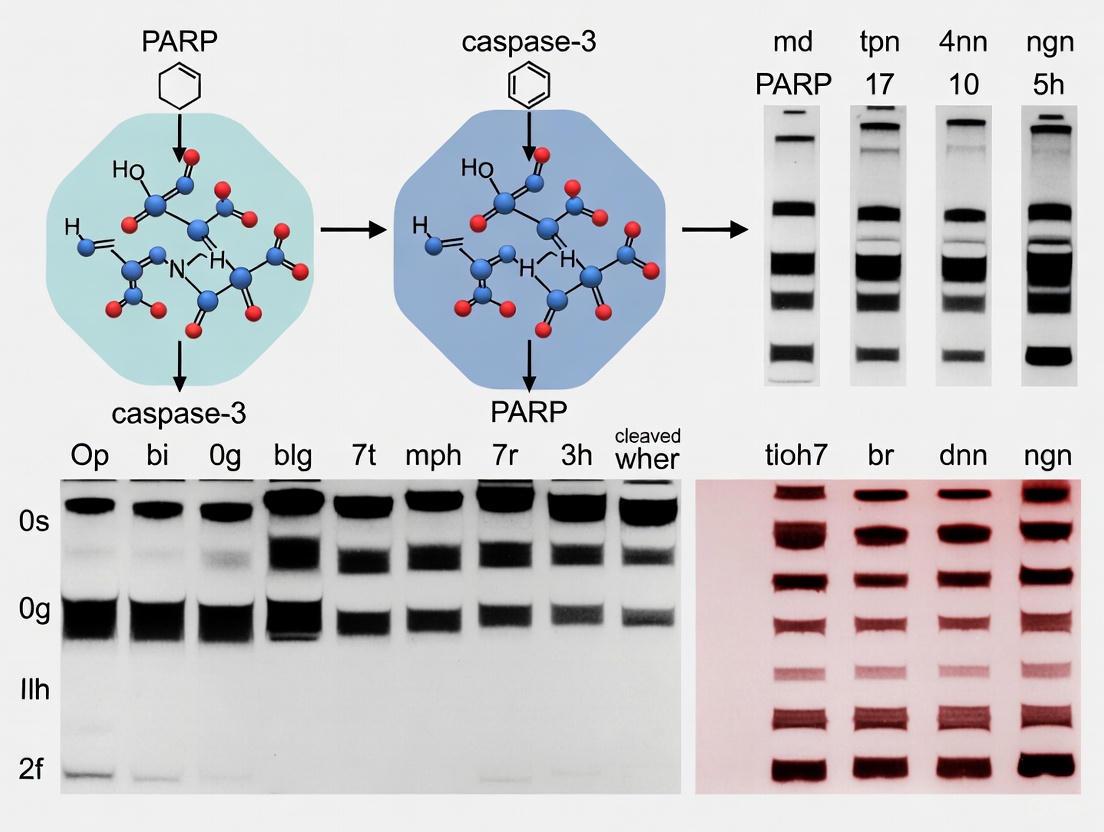
Interpretation: Activated caspase-3 is indicated by the appearance of p17/p19 bands, while PARP cleavage is demonstrated by the presence of the 89 kDa fragment alongside the diminution of the full-length 116 kDa protein [1] [7] [5].
Immunohistochemistry and Immunofluorescence Detection
Tissue Preparation: For paraffin-embedded sections, perform antigen retrieval using TE buffer (pH 9.0) or citrate buffer (pH 6.0) [1]. Alternatively, frozen tissue sections can be fixed with 4% paraformaldehyde for 10-15 minutes [9].
Staining Protocol: Block endogenous peroxidase activity with 3% H2O2 in methanol for IHC, or use appropriate serum blocking for IF. Apply primary antibodies at optimized dilutions (typically 1:50-1:500 for Proteintech 19677-1-AP in IHC; 1:400 for CST #9661 in IF) [1] [2]. For IHC, use HRP-based detection systems with DAB chromogen. For IF, employ fluorophore-conjugated secondary antibodies (e.g., Alexa Fluor series) and counterstain with DAPI or Hoechst for nuclear visualization [9].
Validation Controls: Include positive controls (e.g., apoptotic Jurkat cells treated with staurosporine) and negative controls (omission of primary antibody) in each experiment. For cleaved caspase-3 specificity, pre-absorption with the immunizing peptide can confirm signal specificity [2].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Caspase-3 and Apoptosis Research
| Reagent/Category | Specific Examples | Research Application | Function in Experimental Workflow |
|---|---|---|---|
| Caspase-3 Antibodies | Proteintech 19677-1-AP [1]; CST Cleaved Caspase-3 #9661 [2] | WB, IHC, IF, FC, IP | Detection of caspase-3 expression and activation status |
| PARP Antibodies | CST #9542 [7]; Enzo ALX-804-220 [8] | WB, IF, FC | Verification of apoptosis through substrate cleavage |
| Caspase Inhibitors | zVAD-FMK (pan-caspase) [6]; Z-DEVD-FMK (caspase-3 specific) [7] | Functional studies | Inhibition of caspase activity to establish mechanistic role |
| Apoptosis Inducers | Staurosporine [1]; Carfilzomib [6]; Etoposide (VP-16) [5] | Experimental apoptosis models | Induction of controlled apoptotic stimulation |
| Detection Kits | Annexin V/PI apoptosis detection [6]; TUNEL assay kits [3] | Apoptosis quantification | Complementary validation of cell death |
| Live-Cell Reporters | ZipGFP DEVD-based biosensors [6] | Real-time apoptosis imaging | Dynamic monitoring of caspase-3/7 activity in live cells |
Advanced Research Applications and Methodologies
Real-Time Caspase Activity Monitoring
Recent technological advances have enabled real-time visualization of caspase-3 dynamics using engineered fluorescent reporters. The ZipGFP-based caspase-3/7 reporter system incorporates a DEVD cleavage motif within a split-GFP architecture, where caspase-3 mediated cleavage permits GFP reconstitution and fluorescence emission [6]. This system allows for longitudinal tracking of apoptotic events at single-cell resolution in both 2D and 3D culture systems, including patient-derived organoids that better recapitulate in vivo physiology. When combined with constitutive mCherry expression for normalization, this approach provides quantitative metrics of caspase activation kinetics while accounting for variations in cell viability and reporter expression levels [6].
Flow Cytometric Analysis of Caspase Activation
Multiparametric flow cytometry enables simultaneous assessment of caspase-3 activation alongside other apoptotic markers in heterogeneous cell populations. The protocol involves cell fixation and permeabilization using solutions such as Cytofix/Cytoperm, followed by intracellular staining with fluorochrome-conjugated anti-active caspase-3 antibodies (e.g., PE-conjugated clone C92-605) [8]. This approach can be combined with surface marker staining to evaluate cell-type specific apoptosis and with other intracellular markers such as poly(ADP-ribose) (PAR) to correlate caspase-3 activation with PARP-1 activity [8]. This methodology is particularly valuable for analyzing rare cell populations and for pharmacodynamic studies in drug development.
The critical role of caspase-3 as the key executioner protease in apoptotic pathways necessitates rigorous methodological approaches for its detection and quantification. The comprehensive comparison presented herein demonstrates that antibody-based detection methods, particularly those specific to the activated p17/p19 fragments, provide reliable and reproducible results across multiple experimental platforms. The integration of caspase-3 activation assessment with downstream substrate cleavage analysis, especially PARP processing, establishes a robust framework for validating apoptotic events in both research and drug discovery contexts. As technological advances continue to enhance our ability to monitor caspase dynamics in real-time within physiologically relevant model systems, researchers are better equipped than ever to decipher the complex regulatory mechanisms governing cell death and survival decisions.
The cleavage of poly (ADP-ribose) polymerase 1 (PARP1) is a well-established biochemical hallmark of apoptosis, serving as a critical marker for researchers validating caspase activation in cell death studies. The generation of the specific 89 kDa fragment, resulting from caspase-mediated proteolysis, not only signifies apoptotic induction but also participates in active signaling processes that regulate cell fate. This guide provides a comprehensive comparison of the 89 kDa fragment's roles, detection methodologies, and functional significance within the broader landscape of apoptotic substrates, offering researchers in drug development a foundation for experimental design and data interpretation in caspase activation studies.
PARP1 is a 116 kDa nuclear enzyme that plays a central role in the cellular response to DNA damage, participating in DNA repair mechanisms through its poly(ADP-ribosyl)ation activity [10] [11]. During apoptosis, PARP1 becomes a primary substrate for executioner caspases (particularly caspase-3 and -7), which cleave the protein at a specific aspartic acid residue (Asp214) to generate two characteristic fragments: a 24 kDa DNA-binding fragment and an 89 kDa catalytic domain fragment [12] [13]. This proteolytic event represents more than merely an inactivation mechanism for DNA repair; it constitutes a definitive biochemical signature of caspase activation that researchers routinely monitor through Western blot analysis. The detection of the 89 kDa fragment has become a gold standard in apoptosis assessment across diverse fields, from cancer drug development to neurodegeneration research, providing a reliable indicator of caspase-3 activation in experimental models [14] [11].
Comparative Analysis of PARP1 Cleavage Fragments
Molecular Characteristics and Functions
PARP1 cleavage produces fragments with distinct molecular properties and cellular functions, which researchers must recognize for accurate experimental interpretation.
Table 1: Characteristics of PARP1 and Its Major Cleavage Fragment
| Parameter | Full-Length PARP1 (116 kDa) | 89 kDa Cleavage Fragment |
|---|---|---|
| Domains Contained | DNA-binding domain (two zinc fingers), automodification domain, catalytic domain | Automodification domain, catalytic domain |
| Cellular Localization | Nuclear | Translocates to cytoplasm after cleavage |
| Primary Function | DNA damage repair via poly(ADP-ribosyl)ation | Serves as PAR carrier; induces AIF release from mitochondria |
| Detection Methods | Western blot with antibodies against N-terminal or full-length epitopes | Western blot with cleavage-specific antibodies (e.g., anti-cleaved PARP Asp214) |
| Role in Cell Death | Protects genome integrity; overactivation leads to parthanatos | Facilitates crosstalk between apoptosis and parthanatos |
The 89 kDa fragment encompasses the automodification and catalytic domains of PARP1 but loses the nuclear localization signal located near the DNA-binding domain [10] [15]. This alteration in domain architecture explains its distinct subcellular redistribution following cleavage. Unlike the 24 kDa fragment that remains tightly associated with DNA lesions in the nucleus, the 89 kDa fragment translocates to the cytoplasm, where it executes novel functions in apoptosis signaling [10] [16].
Comparative Signaling Pathways in Programmed Cell Death
PARP1 cleavage patterns and the subsequent fate of the 89 kDa fragment differ significantly between apoptotic pathways, providing researchers with contextual clues for interpreting cell death mechanisms.
Table 2: PARP1 Cleavage in Different Cell Death Pathways
| Pathway | Protease Involved | Cleavage Fragments | Functional Outcome | Key Signaling Molecules |
|---|---|---|---|---|
| Caspase-Dependent Apoptosis | Caspase-3 and -7 | 24 kDa + 89 kDa | Inactivation of DNA repair; facilitation of AIF release | Caspase-3, caspase-7, AIF |
| Parthanatos | Not applicable (PARP1 overactivation) | No specific cleavage | PAR translocation to cytoplasm; energy depletion | PAR polymer, AIF, hexokinase |
| Hybrid Apoptosis-Parthanatos | Caspase-3 | Poly(ADP-ribosyl)ated 89 kDa + 24 kDa | 89 kDa fragment serves as cytoplasmic PAR carrier | Caspase-3, PAR polymer, AIF |
The 89 kDa fragment plays a particularly intriguing role in scenarios where caspase activation occurs alongside PARP1 hyperactivation. Research by Mashimo et al. (2021) demonstrated that under staurosporine and actinomycin D treatment, caspase activation induces both PARP1 autopoly(ADP-ribosyl)ation and fragmentation, generating poly(ADP-ribosyl)ated 89 kDa fragments that translocate to the cytoplasm [10] [16]. In this capacity, the fragment acts as a specific carrier of PAR polymers to the cytoplasm, where it facilitates apoptosis-inducing factor (AIF) release from mitochondria—a mechanism traditionally associated with the caspase-independent parthanatos pathway [15] [16]. This crosstalk between apoptosis and parthanatos represents a significant expansion of the 89 kDa fragment's biological significance beyond a mere caspase substrate.
Figure 1: Signaling Pathway of PARP1 Cleavage in Apoptosis. This diagram illustrates the sequence of events from initial DNA damage to apoptotic cell death, highlighting the distinct nuclear and cytoplasmic roles of the 24 kDa and 89 kDa PARP1 fragments following caspase-mediated cleavage.
Experimental Detection and Validation
Western Blot Methodology for 89 kDa Fragment Detection
The detection of the 89 kDa PARP1 fragment via Western blotting remains the most widely accepted method for validating caspase-3 activation in apoptosis research. The following protocol outlines the key steps for reliable detection and interpretation:
Sample Preparation and Electrophoresis:
- Collect cells by scraping in RIPA lysis buffer [14].
- Determine protein concentration and prepare 10-30 μg of protein per sample for SDS-PAGE separation [14].
- Use precast gradient gels (4-20%) or standard Tris-Glycine gels for optimal separation of the 116 kDa full-length PARP1 and the 89 kDa fragment.
Antibody Selection and Detection:
- Select appropriate antibodies based on experimental goals: For total PARP1 (full-length + 89 kDa), use antibodies like PARP Antibody #9542 (Cell Signaling Technology) that recognize both forms [12]. For specific detection of the cleaved fragment, use cleavage-specific antibodies such as Cleaved PARP (Asp214) (7C9) Mouse Monoclonal Antibody #9548 that specifically target the 89 kDa fragment without cross-reacting with full-length PARP1 [13].
- Incubate with appropriate secondary antibodies and detect using chemiluminescence systems.
- Reprobe blots with loading controls (e.g., anti-GAPDH) to ensure equal protein loading [14].
Troubleshooting Considerations:
- Incomplete cleavage may yield intermediate bands; optimize apoptosis induction time courses.
- Cell-type specific variations in PARP1 expression levels may require antibody titration.
- Always include positive controls (e.g., staurosporine-treated cells) to validate antibody performance.
Research Reagent Solutions
Table 3: Essential Reagents for PARP Cleavage Detection
| Reagent/Category | Specific Examples | Research Application | Technical Considerations |
|---|---|---|---|
| PARP Antibodies | PARP Antibody #9542 (Cell Signaling) | Detects both full-length (116 kDa) and cleaved (89 kDa) PARP1 | Suitable for most applications; works in human, mouse, rat, monkey [12] |
| Cleavage-Specific Antibodies | Cleaved PARP (Asp214) (7C9) Mouse mAb #9548 | Specifically detects the 89 kDa fragment resulting from caspase cleavage | Ideal for specific apoptosis confirmation; mouse-specific [13] |
| Apoptosis Inducers | Staurosporine, Actinomycin D | Positive controls for caspase activation and PARP cleavage | Concentration and time course must be optimized for each cell type [10] [16] |
| Caspase Inhibitors | zVAD-fmk | Negative control to confirm caspase-dependent cleavage | Can be used to distinguish caspase-dependent vs independent death [10] [17] |
| PARP Inhibitors | PJ34, ABT-888 | Tools to investigate PARP1-dependent cell death mechanisms | Helpful in dissecting parthanatos contributions [10] |
Functional Consequences of PARP1 Cleavage
DNA Repair Inactivation and Apoptotic Progression
The traditional understanding of PARP1 cleavage centers on its role in shutting down DNA repair processes to facilitate apoptotic progression. Cleavage separates the 24 kDa DNA-binding domain from the 89 kDa catalytic domain, effectively eliminating PARP1's ability to respond to DNA damage [17] [11]. The 24 kDa fragment remains bound to DNA breaks, acting as a trans-dominant inhibitor of DNA repair by blocking access to damage sites [10]. This irreversible binding prevents DNA repair enzymes from accessing lesions, thereby conserving cellular ATP pools that would otherwise be depleted by PARP1 overactivation [17] [18]. The 89 kDa fragment, while retaining the catalytic domain, cannot localize to DNA damage sites due to the loss of its DNA-binding domain, rendering it unable to participate in DNA repair [10] [11]. This coordinated inactivation represents a point of no return in the commitment to apoptotic cell death.
Novel Signaling Functions of the 89 kDa Fragment
Beyond its passive role as an indicator of caspase activity, emerging research reveals that the 89 kDa fragment actively participates in signaling pathways that regulate cell death:
Cytoplasmic PAR Carrier Function: The 89 kDa fragment serves as a specific vehicle for transporting poly(ADP-ribose) (PAR) polymers to the cytoplasm during apoptosis [10] [16]. When PARP1 undergoes auto-poly(ADP-ribosyl)ation prior to caspase cleavage, the covalently attached PAR polymers remain associated with the 89 kDa fragment after proteolysis. This PAR-bound fragment then translocates to the cytoplasm, where the PAR polymers interact with apoptosis-inducing factor (AIF) anchored to mitochondrial membranes [15] [16]. This interaction facilitates AIF release and subsequent translocation to the nucleus, where it contributes to large-scale DNA fragmentation—a hallmark of apoptotic execution [10].
RNA Polymerase III Interaction: Recent research has revealed that the 89 kDa fragment (also referred to as tPARP1) interacts with the RNA polymerase III (Pol III) complex in the cytoplasm during apoptosis [19]. Through its BRCT domain, tPARP1 recognizes Pol III subunits and mediates ADP-ribosylation of the complex, which facilitates IFN-β production and enhances apoptosis in response to cytosolic DNA stimuli [19]. This function appears particularly relevant in pathogen defense mechanisms, where tPARP1 promotes innate immune responses through Pol III activation.
Cross-talk Between Cell Death Pathways: The 89 kDa fragment represents a molecular bridge between caspase-dependent apoptosis and caspase-independent cell death mechanisms. By transporting PAR to the cytoplasm and facilitating AIF release, the fragment creates amplification loops that ensure cell death commitment even when caspase activity is suboptimal [10] [16]. This hybrid pathway demonstrates the sophisticated integration of different cell death mechanisms and positions the 89 kDa fragment as an active contributor rather than a passive bystander in cell fate decisions.
Discussion and Research Implications
The detection of the 89 kDa PARP1 fragment remains a cornerstone methodology for apoptosis assessment in basic research and drug development. However, the emerging roles of this fragment in active signaling pathways necessitate a more nuanced interpretation of its appearance in experimental systems. Researchers should consider that the 89 kDa fragment may participate in both the initiation and amplification of cell death signals beyond its traditional role as a caspase substrate.
From a therapeutic perspective, the multiple functions of the 89 kDa fragment offer potential opportunities for intervention in pathological conditions. In neurodegenerative diseases where parthanatos contributes to neuronal loss, understanding the precise role of the 89 kDa fragment in amplifying death signals could inform combination therapies targeting both caspase-dependent and independent pathways [11]. Similarly, in cancer therapeutics, the fragment's role in cell death execution may influence the efficacy of PARP inhibitors and other genotoxic agents.
Future research directions should focus on quantifying the differential signaling outcomes based on the concentration of the 89 kDa fragment, identifying potential post-translational modifications that regulate its functions, and exploring tissue-specific variations in its activities. The development of more sophisticated detection methods that can distinguish between the unmodified and poly(ADP-ribosyl)ated forms of the fragment would provide deeper insights into its functional status in different experimental and pathological contexts.
The 89 kDa PARP1 cleavage fragment represents far more than a simple proteolytic relic of caspase activation; it serves as an active participant in cell death signaling with distinct functions that extend beyond the nuclear compartment. Its detection via Western blotting provides researchers with a robust tool for validating caspase-3 activation, but the interpretation of results should consider the complex biological activities associated with this fragment. As research continues to elucidate the multifaceted roles of the 89 kDa fragment in coordinating cell death pathways, its significance as a biomarker and potential therapeutic target will undoubtedly expand, offering new opportunities for intervention in diseases characterized by dysregulated apoptosis.
When and Why to Monitor this Apoptotic Axis
Apoptosis, or programmed cell death, is a controlled cellular process essential for maintaining tissue homeostasis, eliminating damaged cells, and enabling proper embryonic development [20]. At the heart of the apoptotic execution phase lies a critical proteolytic axis comprising caspase-3 and its substrate, Poly (ADP-ribose) Polymerase (PARP). Caspase-3 serves as a major executioner caspase that becomes activated through proteolytic processing during apoptosis, while PARP is a nuclear DNA repair enzyme that undergoes specific cleavage by active caspase-3 [20] [21]. This caspase-3/PARP axis represents a fundamental biochemical pathway that researchers monitor to objectively confirm and quantify the induction of programmed cell death in experimental models. The detection of caspase-3 activation concurrent with PARP cleavage provides compelling evidence that cells are undergoing apoptosis through a canonical pathway, making this axis a cornerstone for apoptosis validation in research contexts ranging from cancer biology to neurodegenerative diseases [20] [17]. The biological significance of this axis extends beyond mere correlation, as PARP cleavage is thought to inactivate DNA repair processes during cell death, thereby facilitating the apoptotic process [17].
Key Markers and Experimental Detection
Molecular Markers of the Apoptotic Axis
Western blot analysis allows for the specific detection of both inactive precursors and activated components of the caspase-3/PARP axis. The key molecular markers include:
- Procaspase-3: The inactive zymogen form of caspase-3 with a molecular weight of approximately 32 kDa [22]. Its presence indicates the potential for apoptosis initiation, while its decrease suggests conversion to active forms.
- Cleaved Caspase-3: The activated form resulting from proteolytic processing at Asp175, producing p17 and p12 fragments [21] [22]. The p17 fragment (17-19 kDa) is most commonly detected and serves as a direct marker of caspase activation.
- Full-length PARP: The intact DNA repair enzyme with a molecular weight of 116 kDa [20] [17]. Its presence indicates inactive apoptotic pathways.
- Cleaved PARP: The apoptosis-specific fragment of 89 kDa generated by caspase-mediated cleavage at the DEVD site [20] [22]. This fragment represents the proteolytic consequence of caspase-3 activity.
Table 1: Key Molecular Markers in the Caspase-3/PARP Apoptotic Axis
| Marker | Molecular Weight | Biological Significance | Detection Antibody |
|---|---|---|---|
| Procaspase-3 | 32 kDa | Inactive precursor form | Caspase-3 Antibody [22] |
| Cleaved Caspase-3 | 17 kDa (p17 fragment) | Activated executioner caspase | Cleaved Caspase-3 (Asp175) Antibody [21] |
| Full-length PARP | 116 kDa | DNA repair enzyme | PARP Antibody [20] |
| Cleaved PARP | 89 kDa | Apoptosis-specific cleavage product | Cleaved PARP Antibody [22] |
Experimental Design and Data Interpretation
Monitoring the caspase-3/PARP axis requires careful experimental design with appropriate controls and normalization strategies. Proper interpretation of western blot data involves analyzing specific band patterns that indicate apoptotic progression:
- Apoptosis Induction: Activated caspase-3 (p17/p19 fragments) accompanied by the appearance of the 89 kDa cleaved PARP fragment, with corresponding decreases in procaspase-3 (32 kDa) and full-length PARP (116 kDa) [22].
- Basal State: Presence of procaspase-3 (32 kDa) and full-length PARP (116 kDa) without detectable cleaved fragments [22].
- Quantitative Analysis: Using densitometry software (e.g., ImageJ) to calculate the ratio of cleaved to full-length proteins, normalized to loading controls [20].
Table 2: Experimental Controls for Apoptosis Detection
| Control Type | Purpose | Example | Expected Result |
|---|---|---|---|
| Negative Control | Baseline apoptosis | Untreated Jurkat cells [23] | Procaspase-3 present; no cleaved PARP |
| Positive Control | Induced apoptosis | Anti-FAS treated Jurkat cells [23] [22] | Cleaved caspase-3 and PARP fragments |
| Loading Control | Normalization | β-actin, GAPDH, or muscle actin [20] [22] | Consistent expression across samples |
Analytical Protocols for Axis Validation
Western Blot Protocol for Apoptosis Detection
The following protocol provides a standardized approach for detecting caspase-3 activation and PARP cleavage in cell culture models:
Sample Preparation: Harvest cells and lyse in appropriate buffer (e.g., CHAPS cell extract buffer or RIPA buffer) containing protease inhibitors [24]. For tissue samples, homogenize using a Dounce homogenizer in lysis buffer [24].
Protein Quantification: Determine protein concentration using a standardized assay (e.g., BCA Protein Assay) to ensure equal loading across samples [20] [24].
Gel Electrophoresis: Separate proteins (20-50 μg per lane) by SDS-PAGE using appropriate percentage gels (e.g., 10-15% acrylamide) [24] [22]. Include molecular weight markers and positive/negative controls.
Protein Transfer: Transfer proteins to PVDF or nitrocellulose membranes using standard wet or semi-dry transfer systems [24].
Blocking: Incubate membrane with 5% non-fat milk or BSA in TBST or PBST for 1 hour at room temperature to prevent non-specific antibody binding [20] [24].
Primary Antibody Incubation: Incubate with specific primary antibodies diluted in blocking buffer:
Membrane Washing: Wash membrane 3-5 times for 5 minutes each with TBST or PBST [24].
Secondary Antibody Incubation: Incubate with appropriate HRP-conjugated secondary antibodies (e.g., 1:1000-1:5000 dilution) for 1 hour at room temperature [24] [22].
Detection: Develop blots using enhanced chemiluminescence (ECL) reagents and image with a digital imaging system [24].
Alternative Detection Methods
While western blotting is a cornerstone technique, several complementary methods can validate caspase-3/PARP axis activation:
- Caspase Activity Assays: Fluorometric or colorimetric assays using synthetic tetrapeptide substrates (DEVD-AMC/AFC for caspase-3/7) to measure enzymatic activity in cell lysates [24].
- Immunohistochemistry: Detection of cleaved caspase-3 and PARP fragments in tissue sections using specific antibodies (e.g., 1:400 dilution for cleaved caspase-3) [24] [21].
- Flow Cytometry: Analysis of caspase activation in individual cells using fluorochrome-labeled inhibitors (FLICA) or specific antibodies [21].
Signaling Pathways and Molecular Interactions
The caspase-3/PARP axis functions as a critical execution point within the broader apoptotic signaling network. The following diagram illustrates the key pathways regulating this axis and the molecular interactions that define its activity:
The caspase-3/PARP axis integrates signals from both extrinsic (death receptor) and intrinsic (mitochondrial) apoptotic pathways [20]. Initiator caspases (caspase-8 and -9) activate executioner caspase-3 through proteolytic cleavage, converting the 32 kDa pro-caspase-3 to active fragments of 17 and 19 kDa [21]. Active caspase-3 then cleaves PARP at the DEVD216–G217 motif, separating the N-terminal DNA-binding domain from the C-terminal catalytic domain [17]. This cleavage event inactivates PARP's DNA repair function and is considered a hallmark of apoptotic commitment, as it prevents futile DNA repair efforts during cell death execution [17]. The detection of both active caspase-3 and cleaved PARP fragments provides complementary verification of this proteolytic cascade, serving as a definitive indicator of apoptotic progression.
Research Reagent Solutions
Selecting appropriate reagents is crucial for accurate detection of the caspase-3/PARP axis. The following table outlines essential reagents and their applications in apoptosis research:
Table 3: Essential Research Reagents for Caspase-3/PARP Axis Detection
| Reagent Category | Specific Examples | Application Notes | References |
|---|---|---|---|
| Cleaved Caspase-3 Antibodies | Cleaved Caspase-3 (Asp175) Antibody #9661 | Detects endogenous 17/19 kDa fragments; suitable for WB, IHC, IF, FC; 1:1000 dilution for WB | [21] |
| PARP Antibodies | Cleaved PARP Antibodies | Detect 89 kDa apoptosis-specific fragment; some antibodies detect both full-length and cleaved forms | [20] [22] |
| Apoptosis Antibody Cocktails | Apoptosis WB Cocktail (ab136812) | Contains antibodies for pro/p17-caspase-3, cleaved PARP1, and muscle actin loading control | [22] |
| Positive Controls | Caspase-3 Control Cell Extracts #9663 | Cytochrome c-treated Jurkat cell extracts provide positive control for caspase activation | [23] |
| Caspase Substrates | DEVD-AMC, DEVD-AFC | Fluorogenic/colorimetric substrates for caspase-3/7 activity assays | [24] |
| Caspase Inhibitors | zVAD-fmk | Broad-spectrum caspase inhibitor used to confirm caspase-dependent apoptosis | [17] [25] |
Applications in Disease Research and Drug Development
Monitoring the caspase-3/PARP axis provides critical insights across multiple research domains:
- Cancer Research: Evaluating the efficacy of chemotherapeutic agents by assessing their ability to activate the caspase-3/PARP axis in tumor cells [20] [26]. For example, studies in B-cell lymphomas have demonstrated that resistant cells show impaired caspase activation and PARP cleavage following Fas stimulation [26].
- Neurodegenerative Disease Research: Investigating excessive apoptosis in Alzheimer's and Parkinson's diseases by detecting aberrant activation of the caspase-3/PARP pathway [20].
- Viral Pathogenesis Studies: Examining viral modulation of host cell apoptosis, as demonstrated in Crimean-Congo hemorrhagic fever virus infection where caspase-3 activation and subsequent cleavage of viral nucleocapsid protein occurs [27].
- Drug Screening and Development: Validating pro-apoptotic compounds by monitoring caspase-3 activation and PARP cleavage as biomarkers of therapeutic efficacy [20].
- Radiation Biology: Studying cell death mechanisms following radiation exposure, where alternative proteases like calpain may activate the apoptotic pathway upstream of caspases [25].
The caspase-3/PARP axis represents a critical biochemical pathway whose monitoring provides researchers with a definitive method for apoptosis validation across diverse experimental contexts. Through western blot analysis and complementary techniques, scientists can obtain quantifiable data on the activation status of this key apoptotic pathway, enabling robust assessment of cell death mechanisms in basic research and drug development. The concurrent detection of caspase-3 activation and PARP cleavage serves as a gold standard for confirming apoptotic induction, offering higher specificity than single-marker approaches. As research continues to elucidate the complex regulation of programmed cell death, the caspase-3/PARP axis remains an essential checkpoint for distinguishing apoptotic mechanisms from other forms of cell death, making its monitoring indispensable for studies of cellular homeostasis, disease pathogenesis, and therapeutic development.
For decades, apoptosis was considered the primary form of programmed cell death, characterized by caspase activation, DNA fragmentation, and controlled cellular dismantling that avoided inflammatory responses. However, the discovery of pyroptosis has fundamentally expanded our understanding of regulated cell death mechanisms and their biological significance. Unlike apoptosis, pyroptosis represents a lytic, inflammatory form of programmed cell death characterized by cellular swelling, membrane rupture, and release of pro-inflammatory intracellular contents [28]. This form of cell death has emerged as a critical component in host defense, inflammation, and disease pathogenesis, with distinct molecular regulators and cellular consequences.
The paradigm shift in cell death classification began with the identification of gasdermin proteins as the executioners of pyroptosis, providing a molecular distinction from apoptotic pathways [28]. Initially observed in the 1990s during bacterial infection studies, pyroptosis was formally distinguished from apoptosis in 2001 and linked to inflammatory caspase activation [28]. The landmark discovery in 2015 that identified gasdermin D (GSDMD) as the key substrate of inflammatory caspases solidified pyroptosis as a unique cell death pathway with profound implications for understanding immune responses and disease mechanisms [28]. This guide provides a comprehensive comparison of pyroptosis against other cell death modalities, with specific focus on experimental validation through caspase-3 activation and PARP cleavage analysis.
Molecular Mechanisms of Pyroptosis
Gasdermin Proteins: The Executioners of Pyroptosis
The gasdermin protein family represents the fundamental executioners of pyroptosis, with six identified members in humans (GSDMA, GSDMB, GSDMC, GSDMD, GSDME, and PJVK) [28] [29]. These proteins share a characteristic two-domain structure: an N-terminal cytotoxic domain capable of forming plasma membrane pores, and a C-terminal inhibitory domain that maintains autoinhibition through intramolecular binding [28]. Proteolytic cleavage within the flexible linker region releases the N-terminal domain, enabling it to target various cellular membranes including the plasma membrane, nuclear envelope, and mitochondrial membranes [28].
GSDMD remains the most extensively characterized gasdermin family member, serving as the primary effector for inflammasome-activated pyroptosis. Upon activation, the GSDMD N-terminal domain (GSDMD-NT) associates with acidic phospholipids in the plasma membrane's inner leaflet, forming large transmembrane pores approximately 10-14 nanometers in diameter [28] [29]. These pores disrupt electrochemical gradients, leading to water influx, cellular swelling, and eventual osmotic lysis [28]. The membrane protein ninjurin-1 (NINJ1) subsequently oligomerizes to drive complete plasma membrane rupture, facilitating the release of large danger-associated molecular patterns (DAMPs) [28].
Table 1: Gasdermin Family Proteins and Their Characteristics
| Protein | Primary Activators | Key Functions | Disease Associations |
|---|---|---|---|
| GSDMA | Streptococcal SpeB protease | Keratinocyte pyroptosis, antimicrobial defense | Asthma, alopecia, systemic sclerosis [29] |
| GSDMB | Granzyme A from cytotoxic lymphocytes | Immune-mediated tumor clearance | Inflammatory bowel disease, asthma [29] |
| GSDMC | Caspase-8 (under hypoxic conditions) | Tumor progression, apoptosis-to-pyroptosis switch | Metastatic melanoma, colorectal cancer [29] |
| GSDMD | Caspase-1, -4, -5, -11 | Inflammasome-mediated pyroptosis, host defense | Cardiovascular diseases, inflammatory disorders [28] [30] |
| GSDME | Caspase-3 | Apoptosis-to-pyroptosis switch, hearing loss | Cancer, drug-induced tissue damage [29] |
| PJVK | Not well characterized | Hearing function, neuronal development | Hearing loss [28] |
Signaling Pathways Activating Pyroptosis
Pyroptosis can be initiated through multiple signaling pathways that converge on gasdermin activation:
Canonical Inflammasome Pathway: Cytosolic pattern recognition receptors (PRRs) detect pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs), leading to inflammasome assembly and caspase-1 activation. Active caspase-1 cleaves GSDMD and pro-IL-1β/pro-IL-18, resulting in pore formation and cytokine maturation [28] [30].
Non-Canonical Inflammasome Pathway: Direct sensing of intracellular lipopolysaccharide (LPS) by caspase-4/5 (human) or caspase-11 (mouse) triggers GSDMD cleavage and pyroptosis independently of inflammasome complexes [28] [30].
Caspase-3-Mediated Pathway: In certain contexts, apoptotic caspase-3 activation can cleave GSDME (DFNA5), converting apoptotic signals into pyroptotic outcomes [29]. This pathway represents a crucial molecular switch between apoptosis and pyroptosis.
Granzyme-Mediated Pathway: Cytotoxic lymphocytes release granzymes that directly cleave gasdermins, particularly GSDMB via granzyme A, bypassing caspase requirements to induce pyroptosis in target cells [29].
The following diagram illustrates the key signaling pathways activating pyroptosis:
Comparative Analysis of Cell Death Modalities
Pyroptosis vs. Apoptosis: Key Distinctions
While both pyroptosis and apoptosis represent forms of programmed cell death, they differ fundamentally in mechanism, morphology, and immunological consequences. Apoptosis is characterized by caspase-3/7 activation, DNA fragmentation, cell shrinkage, membrane blebbing, and formation of apoptotic bodies that are phagocytosed without triggering inflammation [28]. In contrast, pyroptosis features inflammatory caspase activation (caspase-1/4/5/11), gasdermin-mediated pore formation, cellular swelling, membrane rupture, and release of pro-inflammatory cytokines and DAMPs that amplify immune responses [28] [30].
The caspase-3/GSDME axis represents a critical molecular switch between these pathways. When caspase-3 cleaves GSDME, it converts apoptotic signals into pyroptotic outcomes, demonstrating the plasticity between these cell death modalities [29]. This switch has significant implications for cancer therapy, as certain chemotherapeutic agents induce caspase-3 activation that subsequently triggers GSDME-mediated pyroptosis rather than apoptosis.
Table 2: Comparative Features of Cell Death Modalities
| Characteristic | Pyroptosis | Apoptosis | Necroptosis | Ferroptosis |
|---|---|---|---|---|
| Key Regulators | Gasdermins, inflammatory caspases | Caspase-3/6/7, Bcl-2 family | RIPK1, RIPK3, MLKL | GPX4, lipid peroxidation [31] |
| Morphology | Cell swelling, membrane pore formation, lysis | Cell shrinkage, nuclear condensation, apoptotic bodies | Organelle swelling, membrane rupture | Mitochondrial shrinkage, membrane rupture [31] |
| Inflammatory Response | Strongly pro-inflammatory | Anti-inflammatory | Pro-inflammatory | Variable context-dependent inflammation |
| Membrane Integrity | Disrupted by pores | Maintained in early stages | Disrupted | Disrupted |
| Biomarkers | GSDMD cleavage, IL-1β/IL-18 release | PARP cleavage, caspase-3 activation | p-MLKL, RIPK1/RIPK3 activation | Lipid peroxides, GPX4 inactivation [31] |
PANoptosis: Integrated Cell Death Communication
Emerging evidence reveals significant crosstalk between pyroptosis, apoptosis, and necroptosis through a concept termed PANoptosis - a coordinated inflammatory cell death pathway incorporating components from all three mechanisms [32]. PANoptosis is regulated by multiprotein complexes called PANoptosomes that simultaneously activate key effectors from multiple cell death pathways [32].
In inflammatory bone diseases, TNF-α-driven PANoptosis inhibits osteogenic differentiation through coordinated activation of pyroptotic, apoptotic, and necroptotic pathways [32]. Inhibition of NLRP3 in this context rescues cells from PANoptosis and restores osteogenic differentiation, highlighting the therapeutic potential of targeting PANoptosis regulators [32].
Experimental Validation: Caspase-3 Activation and PARP Cleavage
Methodologies for Detecting Caspase-3-Mediated Pyroptosis
The intersection between caspase-3 activation and pyroptosis requires specific experimental approaches to distinguish these pathways:
Western Blot Analysis for PARP Cleavage and Gasdermin Activation:
- Cell Lysis: Use RIPA buffer supplemented with protease and phosphatase inhibitors on ice for 30 minutes, followed by centrifugation at 12,000 rpm and protein quantification via BCA assay [32].
- Electrophoresis and Transfer: Load 30μg total protein per lane for SDS-PAGE at 160V, then transfer to PVDF membrane at 400mA [32].
- Antibody Detection: Probe membranes with anti-PARP1 (cleaved 89kDa fragment), anti-caspase-3 (active p17 subunit), and anti-GSDME (N-terminal fragment) antibodies [32] [31].
- Validation: PARP cleavage confirms caspase-3 activation, while GSDME cleavage indicates subsequent pyroptosis induction.
Functional Assays for Cell Death Characterization:
- LDH Release Assay: Quantitate lactate dehydrogenase release to measure plasma membrane integrity and cell lysis [32].
- Annexin V/PI Staining: Differentiate apoptotic (Annexin V+/PI-) from pyroptotic (Annexin V+/PI+) cells using flow cytometry [31].
- Cytokine Measurement: ELISA for IL-1β and IL-18 release confirms inflammatory component of pyroptosis [30].
The following workflow diagram outlines the key experimental steps for validating caspase-3-mediated pyroptosis:
Research Reagent Solutions for Pyroptosis Studies
Table 3: Essential Research Reagents for Pyroptosis and Cell Death Analysis
| Reagent/Category | Specific Examples | Research Application | Experimental Considerations |
|---|---|---|---|
| Caspase Inhibitors | Z-VAD-FMK (pan-caspase), VX-765 (caspase-1), DEVD-CHO (caspase-3) | Pathway dissection, mechanism studies | Use concentration ranges (10-100μM); assess effects on both PARP and gasdermin cleavage [30] [31] |
| Gasdermin Inhibitors | Necrosulfonamide, disulfiram | Specific pyroptosis inhibition | Evaluate pore formation prevention via LDH release assays [30] |
| Cytokine Analysis | IL-1β, IL-18 ELISA kits | Pyroptosis inflammatory readout | Correlate with cell death markers; measure in supernatant [30] |
| Cell Death Detection | LDH assay kits, Annexin V/PI staining, propidium iodide uptake | Quantification of lytic cell death | Combine multiple methods for comprehensive assessment [32] [31] |
| Western Blot Antibodies | Anti-GSDMD (NT), anti-GSDME, anti-cleaved PARP, anti-caspase-3 | Molecular pathway activation | Validate specificity with knockout controls; detect both full-length and cleaved forms [32] [31] |
| Inflammasome Activators | Nigericin, ATP, nigericin | NLRP3 inflammasome studies | Use positive controls for canonical pyroptosis induction [30] |
Pathophysiological Roles and Therapeutic Targeting
Cardiovascular Diseases
Pyroptosis significantly contributes to cardiovascular pathogenesis through multiple mechanisms. In myocardial infarction, ischemia-reperfusion injury releases DAMPs that activate NLRP3 inflammasomes in cardiomyocytes and immune cells, triggering GSDMD-mediated pyroptosis that amplifies myocardial injury [30]. Genetic ablation of GSDMD in mouse models reduces infarct sizes by approximately 40-50%, improves cardiac function, and decreases inflammatory cell infiltration [30]. Clinically, acute MI patients exhibit significantly elevated plasma GSDMD levels that correlate with infarct biomarkers and inflammatory cytokines [30].
The cGAS-STING pathway has emerged as a key contributor to pyroptosis in atrial fibrillation, where mitochondrial DNA release activates this pathway, promoting inflammasome assembly and gasdermin activation that drives structural and electrical remodeling [30]. Therapeutic targeting of pyroptosis with NLRP3 inhibitors (MCC950) or caspase-1 inhibitors (VX-765) demonstrates cardioprotective effects in preclinical models, reducing infarct size and improving ventricular function [30].
Pulmonary and Metabolic Diseases
In pulmonary fibrosis, pyroptosis drives persistent inflammation and tissue remodeling through gasdermin activation in alveolar epithelial cells [29]. SARS-CoV-2 infection can trigger pyroptosis in severe COVID-19 cases, contributing to both acute lung injury and long-term fibrotic sequelae [29]. The dual targeting of pyroptosis and fibrotic pathways represents a promising therapeutic approach for these conditions.
Diabetic foot ulcers exhibit enhanced pyroptosis signaling that impairs wound healing through sustained inflammation [33]. Bioinformatic analyses identify six key pyroptosis-related genes (FSTL1, PINK1, HDAC3, ULK1, CPTP, and NOD2) as potential diagnostic biomarkers and therapeutic targets, with diagnostic models showing exceptional accuracy (AUC=1.000) [33].
Cancer and Therapy Resistance
Pyroptosis induction represents a promising strategy for overcoming apoptosis resistance in cancer therapy. The RSL3 ferroptosis inducer promotes PARP1 apoptotic functions through distinct mechanisms, demonstrating therapeutic potential against PARP inhibitor-resistant malignancies [31]. RSL3 triggers caspase-dependent PARP1 cleavage while simultaneously reducing full-length PARP1 through inhibition of METTL3-mediated m6A modification [31].
Cytotoxic lymphocytes induce tumor cell pyroptosis through granzyme-mediated gasdermin cleavage, particularly targeting GSDMB in cancer cells, thereby enhancing immune checkpoint blockade efficacy [29]. This mechanism provides a complementary approach to T-cell mediated killing that may overcome immunosuppressive tumor microenvironments.
The expanding understanding of pyroptosis has fundamentally transformed our perspective on regulated cell death, revealing sophisticated molecular networks that integrate inflammatory signaling with cell fate decisions. The experimental framework centered on caspase-3 activation and PARP cleavage provides a critical methodology for distinguishing pyroptosis from other cell death modalities, particularly through detection of gasdermin cleavage fragments. As research continues to elucidate the complex interactions within PANoptosis networks and context-specific gasdermin functions, targeting these pathways holds significant promise for therapeutic intervention across cardiovascular, inflammatory, neoplastic, and metabolic diseases. The integration of pyroptosis modulation with established treatment paradigms represents the next frontier in combating cell death-driven pathologies.
In the molecular orchestration of programmed cell death, specific proteolytic events serve as critical markers and regulatory mechanisms. The cleavage of Caspase-3 at Asp175 and PARP1 at Asp216 represents one such pivotal pairing, where one activated protease systematically inactivates a key DNA repair enzyme to facilitate apoptotic progression. This proteolytic relationship has become a cornerstone in biological research for validating caspase-3 activation, particularly in therapeutic development for cancer and neurodegenerative diseases. This guide objectively examines the specificity, experimental validation, and functional consequences of this key molecular event, providing researchers with comprehensive methodological and analytical frameworks for its investigation.
Molecular Specificity and Cleavage Fragment Analysis
Caspase-3, a critical executioner protease of apoptosis, recognizes specific aspartate residues within its substrate proteins. For PARP1, a nuclear enzyme involved in DNA repair, caspase-3 cleaves at the Asp216-Gly217 bond within the DEVD214G sequence, separating the 116-kDa full-length protein into distinct fragments [17] [11].
Table 1: Characteristics of Caspase-3 and PARP1 Cleavage
| Feature | Caspase-3 | PARP1 |
|---|---|---|
| Cleavage Site | Asp175 | Asp216 |
| Recognition Motif | DEVD | DEVD |
| Full-length Size | 32-35 kDa (pro-caspase-3) | 116 kDa |
| Cleavage Fragments | p17 and p12 subunits | 24 kDa and 89 kDa fragments |
| Primary Function | Executioner protease in apoptosis | DNA repair enzyme |
| Functional Consequence of Cleavage | Activation of protease activity | Inactivation of DNA repair function |
The cleavage of PARP1 by caspase-3 produces two primary fragments with distinct cellular fates: a 24-kDa DNA-binding domain fragment that remains nuclear and acts as a trans-dominant inhibitor of DNA repair, and an 89-kDa fragment containing the automodification and catalytic domains [17] [11] [10]. Recent research indicates that the 89-kDa fragment, when modified with poly(ADP-ribose) (PAR) polymers, can translocate to the cytoplasm during apoptosis and facilitate AIF (apoptosis-inducing factor) release from mitochondria, creating a bridge between caspase-dependent apoptosis and parthanatos [10].
Experimental Validation: Methodologies and Protocols
Western Blot Analysis for Cleavage Detection
Western blotting remains the gold standard technique for detecting PARP1 cleavage and caspase-3 activation. The protocol below outlines a optimized workflow for simultaneous assessment of both proteins.
Table 2: Key Antibodies for Detecting Caspase-3 and PARP1 Cleavage
| Target | Antibody Specificity | Expected Bands | Application |
|---|---|---|---|
| Caspase-3 | Anti-cleaved caspase-3 | p17 subunit (activated form) | Western Blot, IHC |
| PARP1 | Anti-PARP1 (full-length) | 116 kDa (full-length) | Western Blot |
| PARP1 | Anti-cleaved PARP1 | 89 kDa fragment | Western Blot |
| Caspase-3 | Anti-caspase-3 | 32-35 kDa (pro-form) and p17 | Western Blot |
Sample Preparation and Electrophoresis
Begin with protein extraction using RIPA buffer (25 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) supplemented with protease inhibitors [34]. For tissue samples, manual maceration followed by homogenization in extraction buffer at approximately 1:10 w/v (tissue weight/buffer volume) is recommended. Centrifuge homogenates at 20,000 × g for 20 minutes at 4°C and collect the supernatant for protein determination using BCA or Bradford assays with R-squared values ≥0.99 for standard curves [34]. Load 15-30 μg of protein per lane onto 4-12% Bis-Tris gradient gels for optimal separation across molecular weights. Use MES running buffer for proteins between 3.5-160 kDa or MOPS buffer for higher molecular weight proteins above 200 kDa [34].
Transfer and Blocking
Electrophoretically transfer proteins to nitrocellulose or PVDF membranes. For far-western analysis requiring protein-protein interaction studies, eliminate SDS during transfer to enhance protein renaturation [35]. Block membranes with 5% BSA in TBST for 1 hour at room temperature, as BSA generally provides superior signal-to-noise ratios compared to milk-based blockers [35] [34].
Antibody Incubation and Detection
Incubate membranes with primary antibodies specific for target proteins. For comprehensive cell death assessment, simultaneously probe for multiple caspases (caspase-1, -3, -7, -8, -9) and PARP1 from the same cellular population [36]. Use fluorescently-labeled secondary antibodies for quantitative fluorescence-based detection, which provides a linear detection profile superior to chemiluminescence for accurate quantification [34]. Image using systems such as the LI-COR Odyssey with appropriate channels (700 nm and 800 nm) for multiplex detection [34].
Caspase-3 Activity Assay
The Caspase-3 Activity Assay Kit utilizes a fluorogenic substrate (Ac-DEVD-AMC) that emits fluorescence at 420-460 nm when cleaved between DEVD and AMC by activated caspase-3 [37]. The assay requires 0.5-2×10⁵ cells/well or 100 μg/well of total lysate protein, though concentration titrations are recommended for optimal results [37]. Note that this substrate may also detect caspase-7 activity due to shared substrate specificity [37].
Research Reagent Solutions
Table 3: Essential Research Reagents for Caspase-3/PARP1 Studies
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Caspase-3 Activity Assays | Fluorogenic substrates (Ac-DEVD-AMC) | Detection of caspase-3 enzymatic activity in cell lysates |
| PARP1 Cleavage Detection | Anti-PARP1 antibodies (full-length and cleaved) | Western blot detection of PARP1 and its 89 kDa fragment |
| Caspase-3 Detection | Anti-caspase-3 antibodies (pro-form and cleaved) | Western blot detection of caspase-3 activation |
| Apoptosis Inducers | Staurosporine, Actinomycin D, 5-FU | Positive controls for inducing caspase-3-dependent apoptosis |
| Caspase Inhibitors | zVAD-fmk (pan-caspase inhibitor) | Negative control for caspase-dependent processes |
| Fluorescent Reporters | DEVD-inserted GFP mutants | Real-time detection of caspase-3 activation in live cells |
Data Interpretation and Technical Considerations
Quantification and Normalization
For accurate quantification, identify the linear range where signal intensity correlates linearly with protein concentration [38]. Avoid saturated signals in the "shoulder region" of intensity curves, which distort accurate quantification. Dilute protein lysates, reduce antibody concentration, or decrease exposure time to address saturation [38]. Normalize target protein bands to housekeeping proteins (e.g., actin, beta-tubulin, Hsp70) to account for loading variations, though researchers should validate the consistency of these controls under experimental conditions [38].
Specificity Controls and Validation
Include both positive controls (recombinant proteins or apoptosis-induced samples) and negative controls (caspase inhibitor-treated samples) in experimental designs. For caspase-3 activity assays, specificity can be confirmed using caspase inhibitors. For apoptosis induction in gastric cancer models, 5-fluorouracil treatment provides a relevant physiological context for observing PARP1 cleavage [39].
Functional Significance in Research and Therapeutics
The caspase-3/PARP1 cleavage axis provides critical insights into cell fate decisions, particularly in cancer therapy and neurodegenerative diseases. In cancer research, the detection of PARP1 cleavage serves as a marker for effective chemotherapy-induced apoptosis, with CAD (a pyrimidine synthesis enzyme) also being cleaved by caspase-3 at Asp1371 in sensitive gastric and colorectal cancer cells [39]. In neurodegenerative contexts, PARP1 cleavage fragments generated by various proteases (caspases, calpains, cathepsins, granzymes) serve as signature biomarkers for specific protease activities in unique cell death programs [11].
The interplay between these cleavage events extends beyond simple apoptosis regulation. Recent evidence indicates that the 89-kDa PARP1 fragment generated by caspase-3 cleavage can function as a PAR carrier to the cytoplasm, facilitating AIF release and creating a bridge between caspase-dependent apoptosis and caspase-independent parthanatos [10]. This demonstrates the complex regulatory networks governed by specific proteolytic events and their relevance to therapeutic development across the disease spectrum.
From Theory to Bench: A Step-by-Step Western Blot Protocol for Apoptosis Detection
Apoptosis, or programmed cell death, is a fundamental biological process crucial for embryonic development, tissue homeostasis, and the elimination of damaged cells. The caspase family of cysteine proteases serves as central executioners of apoptosis, with caspase-3 being a key effector that is activated in response to various apoptotic stimuli. Caspase-3 is synthesized as an inactive pro-enzyme (pro-caspase-3) that undergoes proteolytic cleavage at specific aspartic acid residues to generate its active form, comprised of 17 kDa and 12 kDa subunits. This activation process is a pivotal point in the apoptotic cascade, as caspase-3 is responsible for the proteolytic cleavage of numerous cellular substrates, including the nuclear enzyme PARP (poly-ADP-ribose polymerase). Cleavage of PARP (from 116 kDa to an 85 kDa fragment) serves as a well-established biochemical marker of apoptosis, as it inactivates the enzyme's DNA repair function and facilitates cellular dismantling. Within the context of drug development and basic research, validating caspase-3 activation through the detection of both cleaved caspase-3 and cleaved PARP provides compelling evidence of apoptosis engagement, making the selection of specific antibodies for these targets a critical consideration for researchers.
The temporal dynamics of caspase-3 activation reveal the remarkable speed of apoptotic commitment. Research utilizing fluorescence resonance energy transfer (FRET) biosensors has demonstrated that once initiated, caspase-3 activation completes within 5 minutes or less in individual cells, occurring almost simultaneously with mitochondrial membrane depolarization and just prior to characteristic morphological changes associated with apoptosis [40]. This rapid activation underscores the importance of sensitive and specific detection reagents capable of capturing these transitional molecular events.
Antibody Characterization and Comparison
Pro-Caspase-3 vs. Cleaved Caspase-3 Antibodies
The distinction between antibodies recognizing pro-caspase-3 versus cleaved caspase-3 is fundamental to experimental interpretation. Antibodies targeting pro-caspase-3 identify the inactive zymogen form (approximately 32-35 kDa), providing information about total protein expression levels but not activity. In contrast, cleaved caspase-3 antibodies are specifically designed to recognize neo-epitopes exposed only after proteolytic activation, offering a direct readout of enzymatic activation.
Pro-Caspase-3 Antibody (Clone 31A893): This mouse monoclonal antibody is generated against recombinant full-length human caspase-3 protein and recognizes the inactive precursor [41]. It is suitable for Western blot (WB), immunohistochemistry (IHC), and immunocytochemistry (ICC) applications. When using this antibody, researchers detect a single band at approximately 32-35 kDa in non-apoptotic cells, with decreased signal upon apoptosis induction due to conversion to the cleaved form.
Cleaved Caspase-3 (Asp175) Antibody (#9661): This rabbit polyclonal antibody is raised against a synthetic peptide corresponding to amino-terminal residues adjacent to Asp175 in human caspase-3 [42]. It is highly specific for the large fragment (17/19 kDa) of activated caspase-3 and does not recognize full-length caspase-3 or other cleaved caspases. This antibody has extensive validation across multiple applications, including WB, IHC, IF, and flow cytometry.
Table 1: Comparison of Key Antibodies for Caspase-3 Detection
| Parameter | Pro-Caspase-3 (MA1-41163) | Cleaved Caspase-3 (#9661) | Cleaved PARP (#9541) |
|---|---|---|---|
| Target | Inactive zymogen (32-35 kDa) | Activated fragments (17/19 kDa) | Cleaved fragment (89 kDa) |
| Host Species | Mouse | Rabbit | Rabbit |
| Clonality | Monoclonal | Polyclonal | Polyclonal |
| Applications | WB, IHC, ICC | WB, IHC, IF, FC, IP | WB, IHC, IF |
| Recommended Dilution (WB) | 2 µg/mL | 1:1000 | 1:1000 |
| Specificity | Full-length caspase-3 | Caspase-3 cleaved at Asp175 | PARP cleaved at Asp214 |
| Species Reactivity | Human, Mouse | Human, Mouse, Rat, Monkey | Human, Mouse, Rat, Monkey |
Cleaved PARP Antibodies
The detection of PARP cleavage represents a downstream verification of caspase-3 activity. Antibodies specific for cleaved PARP provide complementary evidence of apoptosis execution.
- Cleaved PARP (Asp214) Antibody: While not detailed in the search results, this antibody would specifically recognize the 89 kDa cleavage fragment of PARP generated by caspase-3 cleavage at Asp214, serving as a crucial secondary validation in apoptosis assays.
Neo-Epitope Antibodies for Caspase-Cleaved Proteins
Beyond target-specific antibodies, innovative approaches have emerged for broader detection of caspase-cleaved proteins. Research has demonstrated that immunization with C-terminal tetrapeptide sequences (DXXD motifs) exposed after caspase cleavage can generate neo-epitope antibodies (NEAs) that recognize multiple caspase substrates [43]. These antibodies exhibit structure-based specificity rather than pure sequence specificity, enabling detection of various caspase-cleaved proteins, including those with cleavage sites not directly used in immunization (e.g., DALD in cytokeratin-18) [43]. This approach offers potential for discovering novel caspase substrates and pathway-specific cleavage events.
Experimental Design and Protocols
Western Blot Methodology for Detecting Caspase Activation
Western blotting remains the gold standard technique for quantifying apoptosis-related protein cleavage events. The following protocol outlines a standardized approach for detecting pro-caspase-3, cleaved caspase-3, and cleaved PARP:
Sample Preparation: Harvest cells at appropriate time points after apoptosis induction. Use RIPA buffer supplemented with protease and phosphatase inhibitors. For cleaved caspase-3 detection, process samples quickly to prevent post-lysis artifactual activation. Protein concentration should be determined via BCA or Bradford assay, with 20-50 µg total protein typically loaded per lane.
Gel Electrophoresis and Transfer: Separate proteins using 4-20% gradient SDS-PAGE gels to resolve both high (pro-caspase-3, PARP) and low (cleaved caspase-3, cleaved PARP) molecular weight targets. Transfer to PVDF membrane using wet or semi-dry transfer systems. PVDF is preferred for its superior binding capacity for low abundance proteins.
Blocking and Antibody Incubation: Block membranes with 5% non-fat dry milk or BSA in TBST for 1 hour at room temperature. Incubate with primary antibodies diluted in blocking buffer overnight at 4°C with gentle agitation. Use the recommended dilutions specified in Table 1 as starting points. Include loading controls (e.g., GAPDH, β-actin) to ensure equal protein loading.
Detection and Visualization: After secondary antibody incubation (HRP-conjugated anti-mouse or anti-rabbit), use enhanced chemiluminescence (ECL) substrates for detection. For low-abundance targets like cleaved caspase-3, consider using high-sensitivity ECL reagents. Multiple exposure times may be necessary to capture both strong (pro-caspase-3) and weak (cleaved caspase-3) signals on the same blot.
Addressing Common Experimental Challenges
High Background Signal: For antibodies with non-specific binding, pre-adsorption of the antiserum with cell lysates from protein knockout cells or related species can deplete background-recognizing antibodies [44]. Alternatively, use antigen-affinity purified antibodies when working with low-abundance targets in complex cellular compartments.
Multiple Band Detection: Some caspase-3 antibodies may detect non-specific bands. Ensure proper antibody validation using positive controls (e.g., staurosporine-treated HeLa or HL-60 cells) and negative controls (caspase inhibitor pre-treatment) [41] [42].
Optimization for Different Sample Types: Tissue extracts may require different preparation conditions than cell culture samples. For formaldehyde-fixed paraffin-embedded tissues, antigen retrieval methods may be necessary for optimal IHC detection of cleaved caspase-3.
Caspase-3 Activation Pathway
Diagram 1: Caspase-3 activation and PARP cleavage pathway during apoptosis. Caspase-3 is activated by cleavage at Asp175, then proteolytically cleaves PARP at Asp214, generating signature fragments detectable by specific antibodies.
Research Reagent Solutions
Table 2: Essential Research Reagents for Caspase-3 and PARP Detection
| Reagent | Specification | Research Function |
|---|---|---|
| Pro-Caspase-3 Antibody (MA1-41163) | Mouse monoclonal, 1.0 mg/mL | Detects inactive caspase-3 precursor (32-35 kDa) to assess total protein levels |
| Cleaved Caspase-3 Antibody (#9661) | Rabbit polyclonal, specific for Asp175 | Identifies activated caspase-3 fragments (17/19 kDa) to confirm apoptosis initiation |
| Cleaved PARP Antibody | Rabbit polyclonal, specific for Asp214 | Recognizes PARP cleavage fragment (89 kDa) to verify caspase-3 activity |
| Apoptosis Inducers (Staurosporine) | 0.1-1 µM for 3-6 hours | Positive control for caspase-3 activation and PARP cleavage |
| Caspase Inhibitors (QVD-OPH, zVAD-fmk) | 10-50 µM, pre-treatment 1-2 hours | Negative control to confirm caspase-dependent cleavage events |
| Positive Control Cell Lysates | Staurosporine-treated HeLa or HL-60 cells | Verification of antibody specificity and functionality |
Data Interpretation Guidelines
Proper interpretation of experimental data requires understanding the temporal relationship and relative abundance of these apoptotic markers. During apoptosis induction, researchers should observe a progressive decrease in pro-caspase-3 levels accompanied by a corresponding increase in cleaved caspase-3 fragments (17/19 kDa). Subsequently, detection of the cleaved PARP fragment (89 kDa) should follow caspase-3 activation, serving as confirmation of downstream apoptotic signaling.
The ratio of cleaved to full-length proteins provides valuable quantitative information about the extent of apoptosis in a population. Densitometric analysis of Western blot bands can calculate this ratio, allowing for comparative assessment of apoptotic response across experimental conditions. For cleaved caspase-3 detection, the 19 kDa fragment often appears initially, followed by further processing to the 17 kDa form, with both fragments indicating activation.
When utilizing neo-epitope antibodies that recognize multiple DXXD-containing caspase substrates, interpretation should consider that these reagents detect a broader spectrum of cleavage events beyond specific targets, potentially revealing novel caspase substrates or cell-type specific patterns of apoptosis [43]. This approach is particularly valuable for discovery-based research where comprehensive profiling of caspase activity is desired.
Western Blot Experimental Workflow
Diagram 2: Western blot workflow for detecting caspase-3 activation and PARP cleavage, highlighting critical experimental controls necessary for data validation.
The strategic selection of antibodies for detecting pro-caspase-3, cleaved caspase-3, and cleaved PARP is fundamental to accurate apoptosis assessment. Antibodies specific for cleaved forms provide the highest specificity for detecting active apoptotic signaling, while pro-form antibodies establish baseline expression. The complementary use of both cleavage-specific and neo-epitope antibodies offers researchers flexible approaches for either targeted pathway analysis or discovery-based apoptosis research. As caspase-3 activation represents a rapid, commitment point in programmed cell death, the reagents and methodologies outlined in this guide enable precise capture of this critical biological transition, supporting advancements in both basic research and drug development pipelines.
In research focused on validating caspase-3 activation through PARP cleavage, the reliability of your Western blot data is paramount. This process hinges on the very first step: sample preparation. Inefficient or improper protein extraction can lead to the degradation or modification of key biomarkers, rendering subsequent data on caspase-3 activity and PARP cleavage inconclusive. This guide objectively compares common protein extraction methodologies and provides supporting experimental data to help researchers select the optimal protocol for their apoptosis studies, ensuring the integrity of proteins like caspase-3 and its substrate, PARP.
The Critical Role of Caspase-3 and PARP Cleavage in Apoptosis Research
Caspase-3 is a central executioner protease in the apoptotic pathway, responsible for the cleavage of numerous key cellular proteins. One of its most well-characterized substrates is Poly(ADP-ribose) polymerase-1 (PARP-1). During apoptosis, caspase-3 cleaves the 116-kDa PARP-1 protein into a characteristic 24-kDa and an 85-kDa fragment, separating its DNA-binding domain from its catalytic domain [17]. This cleavage event is considered a hallmark of apoptosis and is widely used as a biochemical marker to confirm caspase-3 activation [17]. Therefore, in experiments designed to study apoptosis, preserving the integrity of both the full-length and cleaved forms of PARP is essential for accurate interpretation.
The diagram below illustrates this key signaling relationship and the consequence of its detection in a Western blot.
Core Principles of Sample Preparation for Western Blotting
The primary goals of sample preparation for Western blotting are to efficiently extract the target proteins, maintain their native state as required for detection, and prevent post-lytic modifications. Two key challenges threaten these goals:
- Proteolytic Degradation: Upon cell lysis, intracellular proteases are released, which can rapidly digest proteins of interest. This is particularly problematic when studying proteolytic events like PARP cleavage, as it can create misleading bands or destroy epitopes recognized by antibodies [45].
- Protein Modification: Phosphatases and other enzymes can alter the post-translational modification state of proteins after the cell is lysed, which may affect antibody binding or protein mobility [45].
To mitigate these risks, a standard practice is to perform all pre-lytic steps on ice and use ice-cold buffers. Furthermore, the addition of protease and phosphatase inhibitor cocktails to the lysis buffer is essential to arrest all enzymatic activity and "freeze" the cellular state at the moment of lysis [46] [45].
Comparison of Lysis Buffer Compositions and Their Impact on Protein Integrity
The choice of lysis buffer determines the efficiency of protein extraction and the solubility of different protein classes. It can also affect the antigenicity of the target protein. The table below compares common lysis buffers used in apoptosis research.
Table 1: Comparison of Common Lysis Buffers for Western Blotting
| Lysis Buffer Type | Key Detergents & Components | Mechanism of Action | Best For | Impact on PARP/Caspase-3 Detection |
|---|---|---|---|---|
| RIPA Buffer | Ionic detergents (SDS, deoxycholate), Non-ionic (Triton X-100) [47] | Powerful solubilization; disrupts membranes and protein-protein interactions [47]. | Total protein lysates for denaturing SDS-PAGE; efficient for nuclear proteins like PARP. | Excellent for solubilizing PARP; may disrupt some protein complexes. |
| NP-40/Triton X-100 Buffer | Non-ionic detergents (NP-40, Triton X-100) [46] | Milder disruption; solubilizes cytoplasmic and membrane proteins without denaturing. | Preserving protein complexes for co-IP; cytoplasmic extracts. | May be less efficient for nuclear proteins; preserves native structures. |
| SDS Lysis Buffer | Strong ionic detergent (SDS) [48] | Denatures proteins, masks intrinsic charge, and imparts uniform negative charge. | Difficult-to-solubilize proteins; ensures complete denaturation. | Highly effective for total protein recovery; requires boiling for gel loading. |
Essential Research Reagent Solutions
A successful experiment relies on a suite of specialized reagents. The following toolkit outlines essential materials for preparing samples for caspase-3 and PARP Western blotting.
Table 2: Research Reagent Toolkit for Sample Preparation
| Research Reagent | Function & Role in Experiment |
|---|---|
| Protease Inhibitor Cocktail | Inhibits serine, cysteine, and aspartic proteases, and aminopeptidases to prevent protein degradation post-lysis [45]. |
| Phosphatase Inhibitor Cocktail | Preserves the phosphorylation state of proteins by inhibiting serine/threonine and tyrosine phosphatases [45]. |
| RIPA Lysis Buffer | A versatile, denaturing buffer for efficient extraction of total cellular proteins, including nuclear factors like PARP [47]. |
| DTT or β-Mercaptoethanol | Reducing agents that break disulfide bonds to ensure proteins are fully unfolded and linearized for SDS-PAGE [48]. |
| Caspase-3 Inhibitor (e.g., Ac-DEVD-fmk) | A specific, competitive inhibitor used as an experimental control to confirm caspase-3-dependent cleavage events [49] [50]. |
Detailed Experimental Protocol for Validating Caspase-3 Activation via PARP Cleavage
The following step-by-step protocol is adapted from established methods and is designed to reliably detect PARP cleavage as a marker of caspase-3 activation [49] [45].
Materials and Reagents
- Ice-cold PBS
- Pre-chilled RIPA Lysis Buffer
- Commercially available protease inhibitor cocktail (add fresh to lysis buffer)
- Phosphatase inhibitor cocktail (add fresh to lysis buffer)
- Cell scraper (for adherent cells)
- Microcentrifuge tubes (pre-cooled)
- Dry ice or liquid nitrogen
- 2X SDS-PAGE Sample Loading Buffer
- Heating block or water bath
Step-by-Step Method
- Treatment and Collection: Induce apoptosis in your cell culture model. Quickly place the culture dish on ice and wash cells twice with ice-cold PBS to remove serum and dead cells.
- Lysis: Aspirate the PBS completely. Add an appropriate volume of freshly prepared, ice-cold RIPA lysis buffer containing protease and phosphatase inhibitors directly to the culture dish (e.g., 1 mL per 10⁷ cells) [45].
- Scraping and Transfer: Use a cell scraper to dislodge the cells from the plate and transfer the viscous lysate to a pre-cooled microcentrifuge tube.
- Incubation: Keep the lysate on ice for 15-30 minutes with occasional vortexing to ensure complete lysis.
- Clarification: Centrifuge the lysate at maximum speed (≥13,000 rpm) for 15 minutes at 4°C to pellet insoluble cellular debris, including genomic DNA and large organelles.
- Sample Preparation: Carefully transfer the clarified supernatant to a new tube. Add an equal volume of 2X SDS-PAGE sample loading buffer containing a reducing agent like β-mercaptoethanol.
- Denaturation: Boil the samples for 5-10 minutes at 95-100°C to fully denature the proteins [48].
- Storage or Analysis: The samples can be stored at -80°C or loaded directly onto an SDS-PAGE gel for Western blotting.
The entire workflow, from cell culture to analysis, is summarized below.
Supporting Experimental Data and Validation
The critical importance of proper sample preparation and the use of caspase-3 inhibitors for validation is demonstrated in a bead-based cleavage assay study. As shown in Figure 2A of the associated research, the treatment of cell lysate with active caspase-3 led to the cleavage of both the positive control PARP-1 and a novel substrate, Nek9. Crucially, this degradation was completely rescued when the caspase-3 inhibitors Ac-DEVD-fmk or Ac-VAD-fmk were included in the reaction, confirming that the cleavage was specifically mediated by caspase-3 [49]. This type of control is essential for unequivocally attributing observed PARP cleavage to caspase-3 activity in an experimental setting.
The path to a clean, interpretable Western blot showing PARP cleavage begins long before the gel is run. Robust and reproducible data requires a meticulous approach to sample preparation, starting with the immediate inhibition of proteases and phosphatases upon cell lysis. The choice of lysis buffer must be tailored to the experimental goals—with powerful denaturing buffers like RIPA being a strong choice for total PARP extraction. By adhering to these best practices and employing specific caspase-3 inhibitors as experimental controls, researchers in drug development and basic science can generate highly reliable data to validate caspase-3 activation, a key event in the regulation of programmed cell death.
Optimized Electrophoresis and Transfer for Clear Resolution of 17-19 kDa and 89 kDa Bands
Validating caspase-3 activation through detection of its cleaved fragments (17-19 kDa) and the subsequent cleavage of PARP (89 kDa fragment) is a cornerstone assay in apoptosis research. Achieving clear, simultaneous resolution of these low and high molecular weight targets on a single blot is technically challenging due to their differing electrophoretic and transfer characteristics. This guide objectively compares optimized electrophoresis and transfer methods against standard protocols, providing supporting experimental data to help researchers in drug development select the most reliable strategies for their caspase activation studies.
Electrophoresis System Comparison
Optimal protein separation requires different gel compositions and buffer systems based on protein size. The table below compares system parameters for resolving the 17-19 kDa caspase-3 fragments and 89 kDa PARP cleavage product.
Table 1: Electrophoresis System Optimization for Caspase-3 and PARP Detection
| Parameter | Standard Protocol | Optimized for 17-19 kDa | Optimized for 89 kDa |
|---|---|---|---|
| Recommended Gel | 10-15% Tris-Glycine [51] | 12-20% Tris-Glycine [51] | 4-12% Bis-Tris (MOPS buffer) [51] or 8% Tris-Glycine [51] |
| Gel Chemistry | Fixed % Tris-Glycine | High-percentage acrylamide (12-20%) [51] | Low-percentage acrylamide (4-8%) [51] |
| Running Buffer | Tris-Glycine [51] | Tris-Glycine [51] | MOPS (for Bis-Tris gels) or Tris-Glycine [51] |
| Separation Principle | Size-based through dense polymer | Restricts migration for better band sharpness | Larger pores allow easier migration of big proteins [52] |
For simultaneous detection, a gradient gel (4-12% or 4-20%) is highly recommended as it provides a wide range of pore sizes, effectively separating both low and high molecular weight targets on the same gel [51].
Protein Transfer Method Comparison
Efficient transfer of proteins from gel to membrane is critical for detection, with specific considerations for different protein sizes.
Table 2: Transfer Method Optimization for Multi-Size Protein Detection
| Parameter | Standard Wet Transfer | Optimized for 17-19 kDa | Optimized for 89 kDa |
|---|---|---|---|
| Transfer Type | Wet transfer, 100V, 1h [51] | Wet transfer, 100V, 45-60 min | Wet transfer, 500 mA, 1h, 4°C [52] |
| Membrane | Nitrocellulose or PVDF | PVDF (better retention of small proteins) | Methanol-activated PVDF [52] |
| Key Considerations | Standard buffer, room temp | Risk of over-transfer; monitor time | Pre-chilled buffer, extended equilibration [52] |
Critical Consideration for Simultaneous Transfer: The conditions ideal for transferring high molecular weight proteins (89 kDa PARP) – specifically, longer transfer times or higher power – can sometimes lead to the complete loss of low molecular weight proteins like the 17-19 kDa caspase-3 fragments through the membrane [52]. Careful optimization and validation are essential.
Experimental Protocol for Simultaneous Detection
This detailed protocol ensures clear resolution of both caspase-3 fragments and PARP on the same blot.
Sample Preparation
- Lysis: Use RIPA buffer supplemented with fresh protease and phosphatase inhibitors to prevent degradation [51] [53].
- Protein Quantification: Perform BCA or Bradford assay to ensure equal loading [51].
- Denaturation: Dilute lysates in Laemmli buffer containing DTT or β-mercaptoethanol to reduce disulfide bonds [51] [53]. Boil samples at 100°C for 10 minutes [51].
- Loading: Load 10-40 µg of total protein per lane [51]. Include a pre-stained molecular weight marker.
Gel Electrophoresis
- Gel Selection: Use a 4-20% gradient polyacrylamide gel.
- Apparatus Setup: Place gel in the running chamber and fill with 1X running buffer (e.g., Tris-Glycine-SDS) [51].
- Running Conditions: Apply constant voltage (150-200V) until the dye front reaches the bottom. Use ice packs or a cooling unit if running for extended periods to prevent overheating and band smearing [51] [52].
Western Blot Transfer
- Gel Equilibration: After electrophoresis, immerse the gel in 1X transfer buffer for 15-20 minutes [52].
- Membrane Activation: For PVDF membranes, activate in 99.5% methanol for 15 seconds, then equilibrate in transfer buffer [52].
- Cassette Assembly: Build the transfer stack (sponge, filter paper, gel, membrane, filter paper, sponge) ensuring no air bubbles are trapped.
- Transfer Conditions: Perform wet transfer at 4°C. For a balanced transfer of both 19 kDa and 89 kDa proteins, a setting of 100V (constant voltage) for 1 hour is a good starting point. Monitor to ensure smaller proteins are not lost.
Immunodetection
- Blocking: Incubate membrane in 5% non-fat dry milk (NFDM) or BSA in TBST for 1 hour at room temperature or overnight at 4°C [51] [52].
- Primary Antibody Incubation: Dilute primary antibodies (anti-caspase-3 and anti-PARP) in blocking buffer. Incubate membrane for 1 hour at room temperature or overnight at 4°C with gentle shaking [52].
- Washing: Wash membrane three times for 10 minutes each with TBST (Tris-Buffered Saline with 0.1% Tween-20) with agitation [54].
- Secondary Antibody Incubation: Incubate with HRP-conjugated secondary antibodies diluted in blocking buffer for 1 hour at room temperature [52].
- Washing: Repeat washing step as above [52] [54].
- Detection: Use a high-sensitivity chemiluminescent substrate and image with a system capable of capturing a wide dynamic range.
Quantitative Normalization and Publication Standards
Accurate quantification is essential for validating caspase-3 activation.
- Housekeeping Protein (HKP) Normalization: Traditional method using proteins like GAPDH or β-actin. However, HKP expression can vary with experimental conditions, potentially leading to misinterpretation [55].
- Total Protein Normalization (TPN): The gold standard for quantitative western blotting. This method normalizes the target protein signal to the total amount of protein in each lane, is not affected by experimental manipulations, and provides a larger dynamic range [55]. TPN can be achieved with fluorescent total protein stains or labels [55].
Table 3: Key Reagents for Apoptosis Signaling Research
| Reagent / Solution | Function / Rationale | Example |
|---|---|---|
| RIPA Lysis Buffer | Efficiently extracts proteins from various subcellular compartments, including nuclear fractions containing PARP [53]. | ab156034 [51] |
| Protease Inhibitor Cocktail | Prevents proteolytic degradation of protein targets, especially critical for detecting cleavage fragments [51] [53]. | ab65621 [51] |
| Phosphatase Inhibitor Cocktail | Preserves post-translational modifications like phosphorylation, which can regulate caspase activity and PARP function [51]. | ab201112 [51] |
| DTT (Dithiothreitol) | A reducing agent that breaks disulfide bonds, ensuring proteins are linearized for accurate molecular weight separation [51]. | ab141390 [51] |
| Methanol-Activated PVDF Membrane | PVDF has high protein binding capacity and is robust for stripping/reprobing; methanol activation is essential for its function [52] [56]. | - |
| No-Stain Protein Labeling Reagent | A fluorogenic label for highly sensitive, wash-free total protein normalization, superior to traditional Coomassie staining [55]. | - |
Successful simultaneous detection of 17-19 kDa caspase-3 and 89 kDa PARP fragments hinges on strategic compromises during electrophoresis and transfer. Employing a 4-20% gradient gel with a MOPS buffer system facilitates separation across molecular weights. A cooled, standard-duration wet transfer onto PVDF membrane provides a balanced approach for efficient transfer of both targets without significant loss of smaller fragments. For conclusive quantification in drug development studies, total protein normalization is strongly recommended over traditional housekeeping proteins. By systematically optimizing these parameters, researchers can generate highly reproducible and reliable data for validating caspase-3 activation.
Validating caspase-3 activation through the detection of its canonical substrate, poly (ADP-ribose) polymerase (PARP) cleavage, is a fundamental assay in apoptosis research and drug development. The Western blot, despite being a cornerstone technique, presents significant challenges in reproducibility and reagent consumption, particularly with costly antibodies. The pursuit of efficient probing strategies is not merely a technical exercise but a critical step toward generating robust, quantitative data. This guide objectively compares conventional antibody incubation methods with a novel, resource-conscious technique—the Sheet Protector (SP) strategy—providing researchers with experimental data and protocols to optimize their caspase-3 activation studies. Framed within the broader thesis of apoptosis validation, this comparison highlights how methodological innovations can enhance data reliability while conserving precious reagents.
Conventional Western Blot Probing: Established Practices and Limitations
The conventional (CV) method for antibody incubation in Western blotting is characterized by the use of a large volume of antibody solution to ensure even coverage and binding.
Standard Protocol and Typical Data
In the CV method, a nitrocellulose (NC) membrane is typically placed in a container with a large volume of primary antibody working solution—often around 10 mL—to ensure complete submersion [57]. The container is then placed on an orbital shaker or rocker at low speed (e.g., 60 RPM) to promote constant agitation and even distribution of the antibody. Incubation is commonly performed at 4°C for an extended period, frequently overnight (approximately 18 hours) [57]. Following primary incubation, the membrane is washed and then incubated with a secondary antibody, usually for about 1 hour at room temperature with agitation. This method, while widely used, consumes substantial quantities of antibody, making it costly and inefficient for laboratories using rare or expensive antibodies.
The Sheet Protector Method: A Novel Approach for Antibody Conservation
The Sheet Protector (SP) strategy represents a paradigm shift in Western blot probing, moving from a bulk liquid incubation to a minimal-volume, surface-tension-based system.
Principle and Workflow
The core innovation of the SP strategy lies in its use of a common stationery item—a transparent sheet protector—to create a uniform, thin layer of antibody solution over the membrane [57]. The method involves blotting the blocked membrane to remove excess moisture, placing it on a leaflet of a cropped sheet protector, and applying a small, calculated volume of primary antibody working solution directly onto the membrane. Another leaflet of the sheet protector is then gently overlaid, allowing the antibody solution to disperse across the entire membrane surface via surface tension, forming a SP unit. This unit can be incubated under various conditions without agitation, and for some targets, incubation can be completed in minutes at room temperature [57].
Table 1: Key Parameters of the Sheet Protector Method
| Parameter | Specification | Experimental Basis |
|---|---|---|
| Antibody Volume | 20 - 150 µL for a mini-gel membrane | Adjustable based on membrane size and number of lanes [57] |
| Agitation Requirement | No agitation needed | Incubation occurs within the sealed SP unit [57] |
| Incubation Temperature | Room temperature or 4°C | Effective at both temperatures; RT enables rapid protocols [57] |
| Incubation Time | 15 minutes to several hours | Demonstrated for apoptosis time-series samples with 15-min incubation [57] |
| Key Advantage | Drastic antibody conservation, faster turnaround | Uses ~1/50th to 1/100th of the volume of the CV method [57] |
Experimental Protocol for the SP Strategy
- Post-Block Membrane Preparation: After blocking with a solution like 5% skim milk, briefly immerse the membrane in wash buffer (e.g., TBST) to remove excess blocking agent. Thoroughly blot the membrane with a paper towel to absorb residual moisture, achieving a "semi-dried" state [57].
- Assembly of the SP Unit: Place the prepared membrane on a cut-to-size sheet protector leaflet. Apply the pre-calculated volume of primary antibody solution (e.g., 20 µL for a small membrane) directly onto the membrane. Gently lower a second sheet protector leaflet on top, allowing the solution to spread evenly across the membrane by surface tension, avoiding bubble formation [57].
- Incubation: Leave the assembled SP unit flat on the benchtop for room temperature incubation or in a sealed bag with a damp paper towel to prevent evaporation for longer incubations (e.g., >2 hours). Agitation is not required [57].
- Post-Incubation Processing: After incubation, carefully open the SP unit and proceed with standard wash, secondary antibody incubation, and detection steps.
Head-to-Head Comparison: Sheet Protector vs. Conventional Method
A direct comparison reveals that the SP strategy can achieve sensitivity and specificity comparable to the CV method while offering significant advantages in efficiency and resource utilization.
Table 2: Performance Comparison of Conventional vs. Sheet Protector Method
| Feature | Conventional Method | Sheet Protector Method | Experimental Evidence |
|---|---|---|---|
| Antibody Consumption | ~10,000 µL | 20 - 150 µL | Volume reduction of >98% for a mini-gel [57] |
| Typical Incubation Time | ~18 hours (overnight) | 15 minutes - 2 hours (for many targets) | Apoptosis time-series validated with 15-min protocol [57] |
| Agitation | Required | Not required | SP unit enables passive, even distribution [57] |
| Incubation Temperature | Typically 4°C | Room temperature or 4°C | Effective performance demonstrated at RT [57] |
| Sensitivity & Specificity | Standard | Comparable to conventional | Validated using housekeeping proteins (GAPDH, β-actin) [57] |
| Required Equipment | Laboratory rocker, cold room | Common stationery (sheet protector) | No specialized equipment needed [57] |
Determining Antibody Concentration for the SP Strategy
A critical consideration for the SP method is the potential for local antibody depletion due to the minimal volume. Research indicates that to achieve a signal intensity similar to the CV method with a 0.1 µg/mL antibody concentration, the SP method may require a slightly higher antibody concentration, such as 0.2 µg/mL [57]. This adjustment ensures that the binding reaction proceeds efficiently despite the lack of a large reservoir, and it still results in substantial overall antibody savings.
The Biological Context: Caspase-3 Activation and PARP Cleavage
Efficient Western blot protocols are crucial for accurately studying key apoptotic events. The executioner caspase-3 is proteolytically activated by cleavage at aspartic acid 175, generating ~17 kDa and ~12 kDa active fragments [58]. A primary and well-characterized substrate of active caspase-3 is PARP-1. During apoptosis, caspase-3 cleaves the 116 kDa PARP-1 protein at the DEVD216↓G217 motif, generating a characteristic 89 kDa fragment (containing the catalytic domain) and a 24 kDa DNA-binding fragment [11]. This cleavage event inactivates PARP-1's role in DNA repair, preventing futile energy consumption and facilitating cellular dismantling [59]. The 89 kDa fragment is a definitive biochemical signature of caspase-mediated apoptosis.
Diagram 1: Caspase-3 and PARP Cleavage Pathway.
The Scientist's Toolkit: Essential Reagents for Apoptosis Detection by Western Blot
Table 3: Key Research Reagent Solutions for Caspase-3/PARP Blotting
| Reagent / Material | Function / Role | Example & Notes |
|---|---|---|
| Cleaved Caspase-3 (Asp175) Antibody | Detects activated caspase-3 (p17/p19 fragments); does not recognize full-length protein [58]. | Cell Signaling Technology #9661; specific for the large fragment resulting from cleavage adjacent to Asp175 [58]. |
| PARP-1 Antibody | Detects both full-length PARP-1 (116 kDa) and its caspase-derived cleavage fragment (89 kDa). | The 89 kDa fragment is a hallmark of apoptosis; specific antibodies can distinguish the cleaved form [11]. |
| HRP-Conjugated Secondary Antibody | Binds to primary antibody; enables chemiluminescent detection. | Species-specific (e.g., anti-rabbit IgG); critical for signal generation [57]. |
| Chemiluminescent Substrate | Enzyme substrate for HRP; produces light signal for imaging. | WesternBright Quantum; signal intensity depends on membrane wetness [57] [60]. |
| Sheet Protector | Creates a sealed, uniform layer for minimal-volume antibody incubation. | Common stationery item; enables the SP strategy by distributing antibody via surface tension [57]. |
| Nitrocellulose Membrane | Matrix for protein transfer and immobilization. | Amersham Protran 0.2 µm NC; pore size affects protein binding capacity [57]. |
This objective comparison establishes that the Sheet Protector method is a universally accessible and efficient alternative to conventional Western blot probing. By slashing antibody consumption by over 98% and reducing incubation times from hours to minutes for many applications, the SP strategy directly addresses the pressing needs of life sciences research for cost-effectiveness and speed without compromising data quality [57]. For researchers dedicated to validating caspase-3 activation and PARP cleavage, integrating this method streamlines workflow and enhances the sustainability of long-term apoptosis studies. As the field moves toward more quantitative and reproducible biochemical assays, adopting resource-efficient techniques like the SP method will be paramount in advancing both basic science and drug discovery.
In cell biology and death signaling research, accurately distinguishing apoptotic cell death from other forms is a fundamental requirement. Western blot analysis of specific protein cleavage events serves as a cornerstone technique for this purpose, with caspase-3 activation and PARP cleavage representing two of the most validated and widely utilized apoptotic markers. This guide provides a comprehensive comparison of the expected band patterns in apoptotic versus non-apoptotic samples, delivering essential reference data for researchers conducting experiments in cancer research, neurodegenerative diseases, and drug development.
Core Signaling Pathway and Proteolytic Events in Apoptosis
The intrinsic and extrinsic apoptotic pathways converge on the activation of executioner caspases, primarily caspase-3, which then systematically cleave key cellular substrates including PARP-1. The diagram below illustrates this central signaling axis and the resulting proteolytic fragments that serve as detection targets in Western blotting.
Expected Band Patterns: Apoptotic vs. Non-Apoptotic Samples
The table below summarizes the definitive band patterns that distinguish apoptotic from non-apoptotic samples in Western blot analyses, focusing on the core apoptotic markers caspase-3 and PARP-1.
| Target Protein | Sample Condition | Expected Band Patterns | Molecular Weights | Biological Significance |
|---|---|---|---|---|
| Caspase-3 | Non-Apoptotic | Single band: pro-caspase-3 | ~32 kDa | Inactive zymogen form [61] [62] |
| Apoptotic | Disappearance of pro-caspase-3 band;Appearance of cleaved fragments | ~17/19 kDa (large subunit) | Active executioner caspase [61] [20] | |
| PARP-1 | Non-Apoptotic | Single band: full-length PARP-1 | ~113-116 kDa | DNA repair enzyme in intact form [63] [11] |
| Apoptotic | Reduction of full-length band;Appearance of 89 kDa fragment | ~89 kDa (catalytic fragment) | Hallmark of caspase-mediated apoptosis [63] [22] [11] |
Differential PARP-1 Cleavage Across Cell Death Modalities
PARP-1 is cleaved by different proteases in various cell death processes, producing signature fragments that can help distinguish the death modality. The following table compares these patterns beyond classical apoptosis.
| Cell Death Mode | Primary Proteases | PARP-1 Cleavage Fragments | Key Inhibitors | Functional Consequences |
|---|---|---|---|---|
| Apoptosis | Caspase-3 and -7 [11] [5] | 24 kDa (DBD) + 89 kDa (catalytic) [63] [11] | zVAD-fmk [17] | Inactivation of DNA repair; Energy conservation [17] [11] |
| Necrosis | Lysosomal proteases (e.g., cathepsins) [64] | ~50 kDa major fragment [64] | Not inhibited by zVAD-fmk [64] | Cellular catabolism; Inflammatory response [64] |
Detailed Experimental Methodology
Cell Culture and Apoptosis Induction
- Cell Lines: Commonly used models include SH-SY5Y human neuroblastoma cells, Jurkat T-cells, HL-60 promyelocytic leukemia cells, and MCF-7 breast cancer cells (caspase-3 deficient) [63] [19] [5].
- Apoptosis Inducers: Staurosporine (0.1-1 μM for 2-6 hours), etoposide (VP-16) (10-100 μM for 4-24 hours), anti-FAS antibody (for death receptor activation), or poly(dA-dT) transfection (for cytosolic DNA-induced apoptosis) [22] [19] [5].
- Validation: Include caspase inhibitors (zVAD-fmk, 20-50 μM) as negative controls to confirm caspase-dependent events [17].
Protein Extraction and Western Blot Protocol
- Lysis: Use RIPA or similar lysis buffer supplemented with protease inhibitors to prevent post-lysis protein degradation [62].
- Protein Quantification: Perform with BCA or Bradford assay to ensure equal loading across samples (typically 20-50 μg protein per lane) [22] [62].
- Electrophoresis: Separate proteins using 10-12% SDS-PAGE gels to optimally resolve target proteins in the 15-120 kDa range [62].
- Transfer: Use nitrocellulose or PVDF membranes for protein transfer [62].
- Blocking: Incubate membrane in 5% non-fat dry milk in TBST for 1 hour at room temperature to prevent non-specific antibody binding [62].
- Antibody Incubation:
- Detection: Apply ECL reagent and visualize using chemiluminescence imaging systems [62].
Essential Antibody Specifications
The table below details critical antibodies required for these experiments and their specific characteristics.
| Target | Antibody Specificity | Recommended Dilution | Clonality / Host | Key Characteristics |
|---|---|---|---|---|
| Caspase-3 | Cleaved Caspase-3 (Asp175) [61] | 1:1000 (WB) [61] | Polyclonal Rabbit [61] | Detects 17/19 kDa fragments; Does not recognize full-length [61] |
| PARP-1 | Cleaved PARP-1 (89 kDa fragment) [22] | 1:250 (WB cocktail) [22] | Monoclonal Mouse [22] | Apoptosis-specific; No cross-reactivity with full-length [22] |
| Loading Control | β-Actin, GAPDH, or Muscle Actin [22] [20] | 1:1000-1:5000 | Variable | Sample normalization for quantitative comparisons [20] |
Advanced Interpretation Guidelines
Temporal Dynamics of Proteolytic Events
In many apoptotic models, caspase-3 activation precedes PARP-1 cleavage. When using time-course experiments, researchers may observe the appearance of the 17/19 kDa caspase-3 fragments before detectable levels of the 89 kDa PARP-1 fragment. In caspase-3-deficient MCF-7 cells, caspase-7 primarily cleaves PARP-1, demonstrating compensatory mechanisms [5].
Alternative Cleavage Patterns in Non-Apoptotic Cell Death
During necrotic cell death, PARP-1 undergoes distinct cleavage patterns mediated by lysosomal proteases such as cathepsins B and G, producing a characteristic 50 kDa fragment instead of the apoptotic 89 kDa fragment [64]. This cleavage is not inhibited by zVAD-fmk, providing a key diagnostic feature [64].
Functional Consequences of PARP-1 Cleavage
The 24 kDa PARP-1 fragment containing the DNA-binding domain remains nuclear and acts as a trans-dominant inhibitor of DNA repair by occupying DNA strand breaks, while the 89 kDa catalytic fragment translocates to the cytoplasm where it may engage in non-canonical functions, including modulation of RNA polymerase III activity during innate immune responses [11] [19].
Research Reagent Solutions
The table below provides a consolidated overview of essential materials and reagents for conducting these apoptosis detection experiments.
| Reagent Category | Specific Examples | Research Applications |
|---|---|---|
| Apoptosis Inducers | Staurosporine, Etoposide (VP-16), Anti-FAS antibody [22] [62] [5] | Positive controls for caspase activation |
| Caspase Inhibitors | zVAD-fmk (pan-caspase inhibitor) [17] | Specificity controls for caspase-dependent processes |
| Antibody Cocktails | Apoptosis Western Blot Cocktail (e.g., ab136812) [22] | Simultaneous detection of multiple apoptotic markers |
| Detection Substrates | Ac-DEVD-AFC (caspase-3/7 substrate) [62] | Fluorometric caspase activity assays |
| Cell Death Modulators | 3-Aminobenzamide (PARP inhibitor) [17] | Investigating PARP activity in cell death |
The simultaneous detection of caspase-3 activation and PARP-1 cleavage provides a highly reliable methodological approach for identifying apoptotic cells and distinguishing apoptosis from other cell death modalities. The characteristic band patterns of ~17/19 kDa for activated caspase-3 and ~89 kDa for cleaved PARP-1 serve as definitive biomarkers when properly validated with appropriate controls. Researchers should remain alert to alternative cleavage patterns that may indicate non-apoptotic cell death processes or cell-type specific variations in protease activity.
Solving the Puzzle: Troubleshooting Weak Signals, High Background, and Unexpected Bands
For researchers validating caspase-3 activation, observing no signal or a weak signal on a Western blot for cleaved caspase-3 or its substrate, PARP, can bring critical experiments to a halt. This guide systematically troubleshoots this common problem, from initial protein transfer verification to antibody-specific issues, providing a clear framework for diagnosis and resolution. Within the context of apoptosis research, demonstrating caspase-3 activation via detection of its cleaved form and the subsequent cleavage of PARP is a cornerstone for confirming cell death mechanisms [7] [65]. A failure in this detection can stem from numerous factors in the Western blot workflow, which this article will compare and contrast through an objective lens, supported by experimental data and protocols.
The Core Signaling Pathway: Caspase-3 Activation and PARP Cleavage
A clear understanding of the biological relationship between caspase-3 and PARP is fundamental to troubleshooting. During apoptosis, executioner caspases like caspase-3 are activated by cleavage. Once active, caspase-3 cleaves specific cellular substrates, including PARP, to dismantle the cell. Detecting cleaved caspase-3 and the 89 kDa cleaved fragment of PARP are definitive markers for this process [65].
The diagram below illustrates this key signaling relationship and the points of detection in a Western blot assay.
Troubleshooting Workflow: A Step-by-Step Diagnostic Guide
A systematic approach is the most efficient way to identify the source of a weak or absent signal. The following workflow guides you from the most common to the more specific causes.
Comparative Analysis of Caspase-3 Antibodies
Antibody selection is one of the most critical factors influencing signal strength. Different antibodies, even those targeting the same protein, can exhibit vastly different performance characteristics. The table below provides an objective comparison of commonly used caspase-3 antibodies, highlighting key differences in reactivity and recommended applications [66].
Table 1: Caspase-3 Antibody Comparison for Western Blotting
| Antibody (Clone / Catalog) | Reactivity | Western Blot (WB) | Immunohistochemistry (IHC) | Key Features and Notes |
|---|---|---|---|---|
| Cleaved Caspase-3 (Asp175) (D3E9) Rabbit mAb #9579 | H, (M, R, Mk, B, Pg) | N/A | ++++ | Cleavage-specific; ideal for IHC/IF/Flow. Not recommended for WB [66]. |
| Cleaved Caspase-3 (Asp175) (5A1E) Rabbit mAb #9664 | H, M, R, Mk, (Dg) | ++++ | +++ | Highly recommended for WB; also works well for IP [66]. |
| Cleaved Caspase-3 (Asp175) Antibody #9661 | H, M, R, Mk, (B, Dg, Pg) | ++++ | ++++ | Strong performance in both WB and IHC; broad species reactivity [66]. |
| Caspase-3 (3G2) Mouse mAb #9668 | H | +++ | - | Detects full-length and cleaved caspase-3; human-specific [66]. |
| Caspase-3 Antibody #9662 | H, M, R, Mk | +++ | ++ | Detects full-length caspase-3; less specific for cleavage alone [66]. |
Application Key: (++++)=Very Highly Recommended, (+++)=Highly Recommended, (++)=Recommended, (-)=Not Recommended, N/A=Not Applicable. Reactivity Key: H=Human, M=Mouse, R=Rat, Mk=Monkey, B=Bovine, Dg=Dog, Pg=Pig. Species in parentheses are predicted based on 100% sequence homology [66].
Supporting Data: Antibody Performance in Recent Research
Recent studies on caspase-3 substrates consistently rely on specific, high-performance antibodies. For instance, research identifying USP48 as a novel caspase-3 substrate utilized the Cleaved Caspase-3 (Asp175) Antibody #9661 from Cell Signaling Technology for Western blotting, validating its effectiveness in detecting caspase-3 activation in drug-induced apoptosis models [7]. Similarly, a 2025 study investigating the cleavage of CAD during chemotherapy used antibodies against cleaved caspase-3 to establish the functional link between caspase-3 activation and pyrimidine synthesis shutdown [39].
For PARP cleavage detection, the Cleaved PARP (Asp214) (D64E10) Rabbit Monoclonal Antibody #5625 is widely used. This antibody is highly specific for the 89 kDa cleavage fragment generated by caspases and does not recognize full-length PARP, making it an excellent companion for caspase-3 antibodies in apoptosis validation [65].
Key Experimental Protocols for Troubleshooting
Protocol: Validating Protein Transfer with Ponceau S Staining
A failed or inefficient transfer of proteins from the gel to the membrane is a common point of failure.
- Procedure: After transfer, immerse the nitrocellulose or PVDF membrane in Ponceau S staining solution (e.g., Merck, P7170) for 5 minutes with gentle agitation [57].
- Analysis: Observe the membrane for uniform red staining, which indicates the presence of transferred proteins. The molecular weight ladder lanes should be clearly visible.
- Destaining: Rinse the membrane with distilled water or TBST until the red stain fades, revealing a pink background where proteins are bound. The membrane can then proceed to blocking. A lack of strong, uniform staining indicates a transfer problem.
Protocol: The Sheet Protector (SP) Strategy for Antibody Conservation and Rapid Testing
When dealing with weak signals or precious antibody stocks, optimizing the incubation protocol can yield significant improvements. The Sheet Protector (SP) strategy is a recently developed method that drastically reduces antibody consumption and incubation time while maintaining sensitivity and specificity [57].
- Preparation: After blocking, briefly immerse the membrane in TBST and thoroughly blot it with a paper towel to absorb residual moisture. The membrane should be semi-dry.
- Forming the SP Unit: Place the membrane on a leaflet of a cropped sheet protector. Apply a small volume of the primary antibody working solution directly onto the membrane (empirically, 20–150 µL for a mini-gel membrane). Gently place the upper leaflet of the sheet protector over the membrane. The antibody solution will disperse as a thin layer by surface tension [57].
- Incubation: Incubate the SP unit at room temperature. For cleaved caspase-3 antibody #9661, a 15-minute to 2-hour incubation can be sufficient for detection. For longer incubations, place the SP unit on a wet paper towel and seal it inside a zipper bag to prevent evaporation [57].
- Comparison to Conventional Method: This SP strategy uses 20-150 µL of antibody versus the conventional 10 mL, allows for room temperature incubation without agitation, and enables detection on the order of minutes to a few hours, vastly improving efficiency [57].
Protocol: Using a Positive Control for Apoptosis Induction
The most specific troubleshooting step is to confirm that your experimental system is indeed undergoing apoptosis.
- Control Lysate Preparation: Treat a well-characterized cell line (e.g., HeLa, U937, or 293T) with a known apoptosis inducer. Staurosporine (1 µM for 4-6 hours) or anti-Fas antibody (for sensitive cells) are robust inducers. Camptothecin (10 µM for 4-6 hours) is another excellent option as a DNA-damaging agent [7] [39].
- Validation: Harvest cells and prepare lysates using RIPA buffer. Confirm apoptosis by analyzing the lysates for cleaved caspase-3 and cleaved PARP alongside your experimental samples. The clear signal in the positive control lane confirms that all reagents and the blotting process are functional, localizing the problem to your sample preparation or apoptosis induction if your samples remain negative.
The Scientist's Toolkit: Key Research Reagents
Table 2: Essential Reagents for Validating Caspase-3 Activation
| Reagent / Kit | Function / Application | Example Product / Citation |
|---|---|---|
| Cleaved Caspase-3 (Asp175) Antibody | Detects activated caspase-3 in WB; critical for specificity. | Cell Signaling Technology #9661, #9664 [66] [7] |
| Cleaved PARP (Asp214) Antibody | Detects caspase-derived 89 kDa fragment; confirms downstream activity. | Cell Signaling Technology #5625 [65] |
| Apoptosis Inducers (Positive Control) | Provides a reliable positive control for assay validation. | Staurosporine, Camptothecin, Anti-Fas Antibody |
| Caspase-3 Inhibitor | Confirms caspase-3-specific cleavage; used as a negative control. | Z-DEVD-FMK (MedChemExpress HY-12466) [7] |
| DUB Inhibitor | Induces caspase-3-mediated apoptosis in research models. | PR-619 (MedChemExpress HY-13814) [7] |
| Chemiluminescent Substrate | For signal detection in Western blotting. | WesternBright Quantum (Advansta) [57] |
| Cell Viability/Proliferation Assay | Correlates molecular apoptosis markers with cellular health. | Cell Counting Kit-8 (CCK-8) [7] |
| Annexin V / PI Apoptosis Kit | Quantifies apoptosis by flow cytometry for orthogonal validation. | Annexin-V-FITC/PtdIns Kit (Bestbio) [7] |
Eradicating High Background and Non-Specific Bands Through Blocking and Wash Optimization
In the context of research focused on validating caspase-3 activation via PARP cleavage, the clarity and specificity of a Western blot are not merely aesthetic concerns—they are fundamental to data integrity. The characteristic cleavage of PARP from its full-length 116-kDa form to an 89-kDa fragment is a definitive biochemical hallmark of apoptosis and serves as a key metric for caspase-3 activity [11]. However, high background noise and non-specific bands can obscure these critical results, leading to misinterpretation and unreliable quantification. This guide objectively compares the performance of various blocking and wash optimization strategies, providing a structured framework for researchers to achieve publication-quality data with high signal-to-noise ratios. The principles outlined are particularly pertinent for studies in neurodegeneration and drug development, where precise measurement of apoptotic markers is paramount.
Blocking Buffer Comparison: Selecting the Primary Defense Against Noise
The blocking step is the first and most crucial line of defense against high background. It works by saturating the "sticky" protein-binding sites on the PVDF or nitrocellulose membrane with an inert agent, preventing antibodies from binding non-specifically [67]. The choice of blocking agent, however, is not one-size-fits-all and must be tailored to the specific primary antibody and detection system.
Table 1: Comparison of Common Blocking Buffers for Western Blotting
| Blocking Agent | Best For/Advantages | Limitations and Potential Issues | Performance in PARP Cleavage Studies |
|---|---|---|---|
| Non-Fat Dry Milk | General purpose use; inexpensive and readily available [67]. | Contains phosphoproteins and endogenous biotin; can interfere with phospho-specific and biotin-streptavidin detection systems [67] [68]. | Not recommended for phospho-specific targets; casein may cause cross-reactivity [69]. |
| Bovine Serum Albumin (BSA) | Phosphoprotein detection; biotin-streptavidin systems; ideal when milk causes high background [67] [68]. | More expensive than milk; may not be effective for all antibodies. | Preferred for detecting cleaved PARP and other proteins where phosphate groups are of interest [68]. |
| Specialized Commercial Buffers | Often optimized for low background with specific detection methods (e.g., fluorescence); may contain proprietary additives [67]. | Higher cost; performance can vary by manufacturer and application. | Can provide superior signal-to-noise ratios for multiplex fluorescent detection of full-length and cleaved PARP [67]. |
| Normal Serum | Can be used when other blockers fail. | Risk of cross-reactivity with secondary antibodies; can be variable and expensive. | Use with caution, ensuring serum host species does not interfere with antibody reagents. |
| Non-Protein Blockers (e.g., PVP-40) | Situations where cross-reactivity with protein-based blockers is a persistent issue [67]. | Less common; may not provide adequate blocking for all membranes and targets. | A troubleshooting option if protein-based blockers yield consistently high background. |
As shown in Table 1, for apoptosis research involving PARP cleavage, BSA-based buffers are often the superior choice over milk. This is because milk contains active phosphatases and phosphoproteins like casein, which can cross-react with antibodies and generate a high background, particularly when investigating phosphorylation events that frequently coincide with signaling pathways upstream of caspase activation [67] [68]. Furthermore, the dilution of primary and secondary antibodies in a buffer containing a small percentage of your blocking agent (e.g., 1% BSA) can further reduce non-specific binding throughout the incubation steps [69].
Experimental Protocol: Blocking Buffer Titration
To empirically determine the optimal blocking conditions for your specific experimental setup, a blocking gradient is recommended.
- Membrane Preparation: After transfer, confirm the transfer efficiency with a reversible stain like Ponceau S [70]. Cut the membrane into vertical strips, each containing a full set of lanes (e.g., ladder, positive control, experimental samples).
- Blocking Incubation: Block each strip with a different blocking buffer (e.g., 5% BSA, 5% non-fat dry milk, a commercial buffer) or different concentrations of the same buffer (e.g., 1%, 3%, and 5% BSA). Blocking should be performed for 30-60 minutes at room temperature with mild agitation [71]. Do not exceed 2 hours at room temperature, as this can lead to the displacement of proteins from the membrane [67].
- Standardized Probing: Follow identical protocols for primary antibody incubation, washing, secondary antibody incubation, and detection for all membrane strips.
- Analysis: Compare the strips for specific signal intensity versus background noise. The condition that yields the strongest target band (e.g., the 89-kDa PARP fragment) with the cleanest background should be selected for future experiments.
Wash Optimization: The Strategic Removal of Non-Specific Binding
If blocking is the primary defense, then rigorous washing is the active cleanup process that removes weakly bound antibodies. Inadequate washing is a primary contributor to high background noise [69] [72].
Wash Buffer Composition and Technique
A standard wash buffer consists of Tris-Buffered Saline (TBS) or Phosphate-Buffered Saline (PBS) with a low concentration (0.1%) of a mild detergent like Tween 20 (TBST or PBST). The detergent helps to solubilize and displace antibodies that are bound non-specifically. For persistent background, consider the following adjustments:
- Increased Detergent Concentration: Temporarily increasing Tween 20 to 0.2% or, for severe cases, adding a small amount of SDS (0.1%) can help strip non-specifically bound antibodies [68]. However, this must be done cautiously as it can also weaken the specific signal.
- Wash Frequency and Volume: Opt for a higher frequency of washes with sufficient buffer volume. For example, five or six washes of 5 minutes each are often more effective than three washes of 10 minutes [72]. Ensure the membrane is fully submerged and agitated during washes.
- Temperature Optimization: Performing all washing steps at 4°C has been shown to maximize the signal-to-noise ratio by reducing nonspecific interactions, which are often weaker at lower temperatures [73].
Table 2: Troubleshooting Guide for High Background and Non-Specific Bands
| Problem | Potential Cause | Recommended Solution |
|---|---|---|
| High Uniform Background | Insufficient blocking [69]. | Increase blocking time or concentration of blocking agent; switch blocking buffer (e.g., milk to BSA) [67] [72]. |
| Insufficient washing [69]. | Increase wash frequency, duration, and/or detergent concentration [68] [72]. | |
| Primary antibody concentration too high [69]. | Titrate the primary antibody to find the optimal dilution. | |
| Secondary antibody concentration too high or non-specific binding [69] [72]. | Titrate secondary antibody; test secondary alone on a control membrane. | |
| Membrane dried out during processing [69]. | Ensure the membrane remains wet from transfer until final detection. | |
| Non-Specific Bands | Antibody cross-reactivity with other proteins or blocking agent [72]. | Change blocking buffer; use a more specific antibody (e.g., monoclonal vs. polyclonal). |
| Protein degradation or aggregation [70]. | Use fresh protease inhibitors during lysis; optimize lysis temperature to prevent aggregation. | |
| Overloaded gel [69]. | Titrate down the amount of total protein loaded per lane. | |
| Smeared Bands | Gel ran too hot or at high voltage [70]. | Run gel at lower voltage (e.g., 100V) or in a cold room/with ice packs. |
The PARP Cleavage Context: A Case Study in Specificity
The PARP cleavage assay is an excellent model for applying these optimization principles. During apoptosis, caspase-3 cleaves PARP-1 at the DEVD motif, generating signature 89 kDa (catalytic domain) and 24 kDa (DNA-binding domain) fragments [11]. Detecting this shift requires a clean blot to distinguish the full-length protein from its cleaved products unequivocally. Non-specific bands or high background can easily be mistaken for alternative cleavage fragments or obscure the cleavage event entirely. The following diagram illustrates the key steps in the apoptosis pathway where caspase-3 activation leads to PARP cleavage, and how optimal blocking and washing are critical for its clear detection.
Integrated Workflow for a Perfect Blot
Achieving a publication-ready blot for PARP cleavage validation requires a systematic approach, integrating the optimal blocking and washing strategies into a cohesive workflow. The following diagram outlines this step-by-step process.
The Scientist's Toolkit: Essential Reagents for Apoptosis Signaling Research
Table 3: Key Research Reagent Solutions for PARP Cleavage Studies
| Reagent / Material | Function / Application | Key Considerations |
|---|---|---|
| Caspase-3 Resistant PARP Mutant | Control for confirming caspase-specific cleavage; demonstrates that cell death acceleration is due to failure of PARP cleavage [59]. | Generated via site-directed mutagenesis of the DEVD cleavage site [59]. |
| PVDF Membrane (0.2 µm & 0.45 µm) | Protein immobilization post-transfer. | Use 0.2 µm for small proteins (<15 kDa); 0.45 µm for larger proteins like PARP [70]. |
| BSA (IgG-Free, Protease-Free) | Blocking agent for phosphoprotein detection and biotin-streptavidin systems; reduces background vs. milk [67] [68]. | Ensures no antibody cross-reactivity with contaminants. |
| Protease Inhibitor Cocktail | Prevents protein degradation during sample preparation, preserving the integrity of full-length PARP and its cleavage fragments [70]. | Essential for accurate assessment of cleavage ratios. |
| Phosphatase Inhibitors | Preserves phosphorylation states of signaling proteins upstream of caspase activation [71]. | Critical for studies of signaling pathways regulating apoptosis. |
| Anti-PARP Primary Antibody | Binds specifically to PARP protein to detect both full-length (116 kDa) and cleaved (89 kDa) forms. | Must be validated for Western blot; cleavage site-specific antibodies are available. |
| Fluorescent or HRP-conjugated Secondary Antibodies | Enables detection of the primary antibody. | Fluorescent secondaries allow for multiplexing; HRP requires chemiluminescent substrates [71]. |
In the rigorous field of apoptosis research, particularly in validating caspase-3 activation through PARP cleavage, the reliability of your data is directly contingent on the quality of your Western blots. High background and non-specific bands are not minor inconveniences; they are significant sources of error that can compromise experimental conclusions. By systematically comparing and optimizing blocking buffers—favoring BSA for phosphoprotein-related work—and implementing stringent, cold-temperature washing protocols, researchers can consistently achieve high-fidelity results. This guide provides a direct performance comparison and a detailed roadmap to eradicate noise, ensuring that the critical 89-kDa PARP cleavage fragment stands out with clarity, thereby strengthening the validation of caspase-3 activity and contributing to robust, reproducible scientific findings.
In the study of programmed cell death, the activation of caspase-3 and subsequent cleavage of key substrates like poly(ADP-ribose) polymerase (PARP) serves as a fundamental biomarker for apoptosis. However, Western blot analysis often reveals unexpected molecular weights that can complicate interpretation and validation. These anomalies primarily arise from three interconnected biological phenomena: proteolytic degradation, post-translational modifications (PTMs), and the expression of alternative protein isoforms. For researchers in drug development and basic research, accurately distinguishing between these possibilities is not merely a technical exercise but a critical component of data validation. This guide provides a structured approach to address these challenges, with a specific focus on validating caspase-3 activation through PARP cleavage, offering comparative experimental methodologies to identify artifacts and confirm genuine biological signals.
The following diagram illustrates the core decision pathway for troubleshooting unexpected molecular weights in apoptosis research:
Key Causes of Unexpected Molecular Weights
Proteolytic Degradation by Caspases and Other Enzymes
During apoptosis, caspase-3 activation triggers the systematic cleavage of numerous cellular proteins, producing characteristic fragment sizes that often appear as unexpected bands on Western blots.
PARP Cleavage: The classic apoptosis substrate PARP (113-116 kDa) is cleaved by caspase-3 at the DEVD²¹⁴│G²¹⁵ site, generating signature 89 kDa and 24 kDa fragments [17] [59]. This cleavage separates PARP's DNA-binding domain from its catalytic domain, effectively inactivating the enzyme. The 89 kDa fragment is typically detected on Western blots using antibodies against the N-terminal region, while the 24 kDa fragment requires C-terminal specific antibodies.
Additional Caspase Substrates: Beyond PARP, caspase-3 cleaves multiple essential proteins, each producing distinctive fragments:
- Eukaryotic Initiation Factor 2α (eIF2α): Cleaved by caspase-3, altering its function and contributing to translation inhibition during apoptosis [74].
- Polypyrimidine Tract-Binding Protein (PTB): Caspase-3 cleaves PTB isoforms at three non-canonical sites (Asp⁷, Asp¹³⁹, and Asp¹⁷²), generating fragments that relocate from the nucleus to the cytoplasm and inhibit IRES-mediated translation [75].
- Actin: Limited cleavage by caspase-3 produces a characteristic 14 kDa fragment that facilitates subsequent degradation by the ubiquitin-proteasome system in catabolic muscle conditions [76].
Necrotic Cleavage Patterns: Unlike apoptosis, necrosis induces PARP cleavage through lysosomal proteases like cathepsins B and G, producing a distinct 50 kDa fragment instead of the classic 89 kDa apoptotic fragment [64]. This differential cleavage provides a valuable diagnostic tool for distinguishing cell death mechanisms.
Alternative Splicing and Protein Isoforms
Alternative splicing generates multiple protein isoforms from a single gene, often yielding products with different molecular weights and potentially distinct functions.
Ebp1 Isoforms: The ErbB3-binding protein Ebp1 expresses as p48 and p42 isoforms through alternative splicing. The p48 isoform localizes to both cytoplasm and nucleolus, suppresses apoptosis, and promotes cell proliferation. In contrast, the p42 isoform resides exclusively in the cytoplasm, promotes differentiation, and inhibits cell growth [77]. These isoforms display different subcellular localizations and opposing biological functions despite originating from the same gene.
PTB Isoform Diversity: Multiple PTB isoforms (1, 2, and 4) are differentially cleaved by caspase-3 during apoptosis, generating distinct fragment patterns that complicate Western blot interpretation [75].
Table 1: Characteristic Cleavage Fragments of Apoptosis-Related Proteins
| Protein | Full-length (kDa) | Cleavage Fragment(s) | Protease Responsible | Biological Consequence |
|---|---|---|---|---|
| PARP-1 | 113-116 | 89 kDa + 24 kDa | Caspase-3 | Inactivation of DNA repair [17] |
| PARP-1 (Necrosis) | 113-116 | 50 kDa | Cathepsins B/G | Distinct necrosis pathway [64] |
| Actin | 42 | 14 kDa | Caspase-3 | Facilitates ubiquitin-proteasome degradation [76] |
| eIF2α | 36 | ~30 kDa (C-terminal) | Caspase-3, 6, 8, 10 | Inhibition/alteration of protein synthesis [74] |
| PTB | 57-60 | Multiple (N & C-terminal) | Caspase-3 | Altered IRES-mediated translation [75] |
Post-Translational Modifications
PTMs significantly alter protein migration without changing the amino acid sequence, potentially leading to misinterpretation of Western blot results.
Phosphorylation Impact: The Ebp1 protein demonstrates how phosphorylation controls subcellular localization. PKC-mediated phosphorylation at Ser-360 causes Ebp1 translocation from the nucleolus to the nucleoplasm, which could potentially alter its apparent molecular weight due to conformational changes or interactions [77].
ADP-Ribosylation: PARP-1 undergoes extensive automodification with poly(ADP-ribose) polymers, dramatically increasing its apparent molecular weight and creating smeared bands on Western blots [17]. This modification occurs rapidly in response to DNA damage and can obscure cleavage events if not properly accounted for.
Experimental Approaches for Identification and Validation
Protease Inhibition Assays
Protease inhibition provides the most direct method to identify caspase-mediated cleavage.
Protocol: Caspase Inhibition Assay
- Cell Treatment: Divide cells into three treatments: (1) untreated control, (2) apoptosis inducer (e.g., 40 ng/mL TNF-α with 1 μg/mL actinomycin D), and (3) apoptosis inducer + 50 μM pan-caspase inhibitor Z-VAD-fmk [59].
- Incubation: Treat cells for 4-12 hours based on apoptosis kinetics.
- Protein Extraction: Lyse cells in RIPA buffer (25 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) supplemented with protease inhibitor cocktail [34].
- Western Blot Analysis: Separate 15-30 μg protein on 4-12% Bis-Tris gradient gels using MES running buffer for optimal resolution of 3.5-160 kDa proteins [34].
- Interpretation: disappearance of cleavage fragments in Z-VAD-fmk treated samples confirms caspase-dependent processing.
Application Example: In L929 cells, Z-VAD-fmk prevents CD95-mediated apoptosis but potentiates TNF-induced necrosis, demonstrating stimulus-specific caspase involvement [17].
Subcellular Fractionation and Localization Studies
Compartment-specific localization helps distinguish isoforms and cleavage products.
Protocol: Cellular Fractionation
- Cell Harvesting: Wash cells with ice-cold PBS and scrape gently.
- Hypotonic Lysis: Resuspend cell pellet in buffer A (20 mM HEPES pH 7.5, 10 mM MgCl₂, 1 mM EDTA, 1 mM EGTA, 1 mM DTT) and incubate on ice for 30 minutes [76].
- Homogenization: Use 20 strokes of a Dounce homogenizer on ice.
- Centrifugation: Spin at 1,600 × g for 10 minutes to separate nuclear (pellet) and cytoplasmic (supernatant) fractions [76].
- Analysis: Detect proteins by Western blotting using compartment-specific markers for validation.
Application Example: Ebp1 isoform localization - p48 appears in both nuclear and cytoplasmic fractions, while p42 resides exclusively in the cytoplasm [77].
Advanced Western Blot Normalization and Quantitation
Proper normalization ensures accurate quantification of cleavage events and isoform expression.
Total Protein Normalization (TPN) Protocol:
- Protein Labeling: Use No-Stain Protein Labeling Reagent or similar fluorescent total protein stains according to manufacturer instructions [55].
- Gel Imaging: Capture total protein signal before immunodetection using appropriate fluorescence imaging systems.
- Immunoblotting: Proceed with standard blocking, primary antibody incubation, and fluorescent secondary antibody detection.
- Quantitation: Calculate target protein signal normalized to total protein in each lane using imaging software [34].
Advantages Over Housekeeping Proteins: TPN avoids issues with variable expression of traditional loading controls (GAPDH, β-actin) under different experimental conditions and provides a broader dynamic range for accurate quantitation [55].
Table 2: Troubleshooting Unexpected Bands in Apoptosis Western Blots
| Unexpected Band | Possible Causes | Confirmatory Experiments | Interpretation |
|---|---|---|---|
| ~89 kDa (PARP blot) | Caspase-3-mediated cleavage | Z-VAD-fmk inhibition; DEVD-ase activity assay | Apoptosis marker [17] |
| ~50 kDa (PARP blot) | Lysosomal protease cleavage | Cathepsin inhibition; zVAD-fmk non-responsiveness | Necrosis indicator [64] |
| ~14 kDa (Actin blot) | Caspase-3 cleavage in muscle | Caspase-3 inhibitor in vivo; insulin/PI3K pathway modulation | Muscle atrophy indicator [76] |
| Doublet at ~48/42 kDa | Alternative Ebp1 isoforms | Subcellular fractionation; RT-PCR for splice variants | Differential regulation of survival vs. differentiation [77] |
| Multiple PTB fragments | Caspase-3 cleavage at multiple sites | Caspase-3 inhibitor; mapping with cleavage-site mutants | Apoptosis-dependent translation regulation [75] |
Research Reagent Solutions
The following table outlines essential reagents for studying caspase-mediated cleavage and addressing molecular weight anomalies:
Table 3: Essential Research Reagents for Apoptosis Signaling Studies
| Reagent Category | Specific Examples | Application & Function | Considerations |
|---|---|---|---|
| Caspase Inhibitors | Z-VAD-fmk (broad-spectrum), Ac-DEVD-CHO (caspase-3 specific) | Inhibit caspase activity to confirm caspase-dependent cleavage [17] [59] | Cell-permeable Z-VAD-fmk for live cells; Ac-DEVD-CHO for in vitro assays |
| Apoptosis Inducers | TNF-α + actinomycin D, anti-CD95/Fas antibody | Activate death receptor pathways to induce caspase activation [59] | Concentration and timing vary by cell type; confirm sensitivity |
| Phosphorylation Modulators PKC inhibitors/activators | Modulate phosphorylation-dependent localization and function [77] | Assess potential PTM effects on protein mobility | |
| Protein Extraction Buffers | RIPA buffer (whole cell), NP-40 buffer (membrane proteins), Tris-Triton (cytoskeletal) | Optimal extraction based on protein localization and solubility [34] | Detergent compatibility with downstream protein assays |
| Antibodies | PARP (N-terminal for 89 kDa fragment), cleaved caspase-3, isoform-specific antibodies | Detect specific cleavage fragments and isoforms [77] [17] | Validate specificity with appropriate controls |
| Normalization Reagents | No-Stain Protein Labeling Reagents, fluorescent secondary antibodies | Accurate total protein normalization for quantitative Westerns [55] [34] | Compatible with fluorescence imaging systems |
Integrated Experimental Workflow
The following diagram outlines a comprehensive experimental strategy for validating unexpected bands in apoptosis research:
Addressing unexpected molecular weights in apoptosis research requires a systematic approach that discriminates between proteolytic degradation, alternative isoforms, and post-translational modifications. Through targeted experimental designs incorporating specific protease inhibitors, subcellular fractionation, and appropriate normalization techniques, researchers can accurately validate caspase-3 activation and PARP cleavage while avoiding misinterpretation of Western blot results. The integrated workflow and troubleshooting guidelines presented here provide a robust framework for confirming apoptosis signaling events, ultimately strengthening research validity in both basic science and drug development contexts. As methodological standards evolve toward quantitative fluorescent Western blotting and total protein normalization, the accuracy and reproducibility of apoptosis detection will continue to improve, facilitating more reliable translation of research findings into therapeutic applications.
In caspase-3 activation research, robust experimental controls are fundamental for generating reliable and interpretable data. The cleavage of poly(ADP-ribose) polymerase (PARP) serves as a well-established biochemical hallmark of apoptosis, making it a critical readout for validating caspase-3 activity. However, without proper control strategies, researchers risk misinterpretation due to technical artifacts, inefficient apoptosis induction, or suboptimal sample preparation. This guide objectively compares control methodologies and reagent solutions essential for rigorous PARP cleavage Western blot analysis, providing scientists with a framework for experimental validation.
The Caspase-3/PARP Cleavage Signaling Pathway
The extrinsic and intrinsic apoptosis pathways converge on the activation of executioner caspases, primarily caspase-3. This protease then cleaves specific cellular substrates, with PARP being one of the most prominent targets. Cleavage of the 116 kDa PARP protein between Asp214 and Gly215 generates an 89 kDa fragment, irreversibly inactivating its DNA repair function and committing the cell to death. This specific cleavage event provides a definitive biochemical marker for detecting caspase-3 activity in apoptotic cells [78] [59].
Comparative Analysis of Control Strategies
Effective experimental design for studying caspase-3 activation requires implementing multiple control types, each serving a distinct purpose in data validation and interpretation.
Table 1: Control Types for Caspase-3 Activation and PARP Cleavage Research
| Control Type | Purpose | Common Implementation | Interpretation of Expected Results |
|---|---|---|---|
| Positive Control | Verify apoptosis induction and antibody functionality | Pre-treated control cell extracts (e.g., etoposide or cytochrome c-treated Jurkat cells) [79] | Clear detection of cleaved caspase-3 and 89 kDa PARP fragment confirms experimental system works |
| Negative Control | Confirm specificity of observed cleavage events | Untreated cell extracts or caspase inhibitor-treated cells (e.g., Q-VD-OPh) [43] | Absence of cleaved bands indicates apoptosis-specific signaling; presence suggests non-specific cleavage |
| Loading Control | Normalize for protein loading and transfer variations | Housekeeping proteins (e.g., tubulin, GAPDH) in total cell lysates [80] | Ensures equal protein loading across all lanes, enabling accurate quantification of cleavage events |
| Caspase Inhibition Control | Specific verification of caspase-dependent cleavage | Pan-caspase inhibitors (e.g., QVD-OPH, z-DEVD.fmk) [43] [59] | Attenuation or elimination of PARP cleavage confirms caspase-dependent processing |
Essential Research Reagent Solutions
The following reagents and tools are critical for implementing comprehensive control strategies in apoptosis research.
Table 2: Key Research Reagents for Apoptosis Western Blotting
| Reagent / Resource | Function / Application | Example Specifications |
|---|---|---|
| Control Cell Extracts | Pre-validated positive controls for apoptosis markers | Jurkat Apoptosis Cell Extracts (etoposide-treated) [79] |
| Caspase-3 Control Extracts | Cytochrome c-induced caspase activation control | Caspase-3 Control Cell Extracts (cytoplasmic fraction) [79] |
| Cleaved PARP Antibodies | Specific detection of caspase-cleaved PARP fragment | Cleaved PARP (Asp214) Antibody #9545; detects 89 kDa fragment [78] |
| Caspase Inhibitors | Negative controls to confirm caspase dependence | z-DEVD.fmk (caspase-3 inhibitor), QVD-OPH (pan-caspase inhibitor) [43] |
| Apoptosis Inducers | Chemical induction of apoptosis for positive controls | Etoposide (25 μM, 5 hours), cytochrome c + dATP [79] |
Experimental Protocols for Control Implementation
Protocol 1: Generating Positive Controls with Etoposide Treatment
- Cell Culture: Maintain Jurkat cells in appropriate medium supplemented with 10% fetal bovine serum [79].
- Treatment: Apply 25 μM etoposide to cells for 5 hours to induce apoptosis [79].
- Harvesting: Collect cells, wash with ice-cold PBS, and lyse in modified RIPA buffer containing protease inhibitors [59].
- Analysis: Subject 50 μg of protein to SDS-PAGE and Western blotting with cleaved PARP and caspase-3 antibodies [59].
Protocol 2: Caspase Inhibition Negative Control
- Pre-treatment: Incubate cells with 20 μM caspase-3 inhibitor (z-DEVD.fmk) or pan-caspase inhibitor (QVD-OPH) for 30-60 minutes prior to apoptosis induction [43].
- Apoptosis Induction: Apply apoptotic stimulus (e.g., TNF-α/actinomycin D or etoposide) while maintaining inhibitor presence [59].
- Sample Processing: Prepare cell lysates as described in Protocol 1.
- Analysis: Compare PARP cleavage in inhibitor-treated versus non-inhibited samples to confirm caspase dependence [43].
Protocol 3: Western Blot Analysis for PARP Cleavage
- Gel Electrophoresis: Separate 50-100 μg of total protein on 10% SDS-polyacrylamide gels [59].
- Transfer: Transfer proteins to nitrocellulose membranes at 100V for 1 hour using Tris-glycine buffer with 20% methanol [27].
- Blocking: Incubate membranes with 5% non-fat dried milk overnight at 4°C [27].
- Antibody Probing: Incubate with primary antibodies against cleaved PARP (Asp214) at 1:1000 dilution [78], followed by HRP-conjugated secondary antibodies.
- Detection: Develop blots using ECL Plus Western blotting detection reagents [27].
Experimental Workflow for Apoptosis Control Validation
A comprehensive approach to validating caspase-3 activation through PARP cleavage requires systematic implementation of controls throughout the experimental workflow.
Discussion: Optimizing Control Strategies
The implementation of comprehensive control strategies significantly enhances the reliability of caspase-3 activation studies. Pre-validated control cell extracts provide critical benchmarks for experimental apoptosis induction and antibody performance [79]. Meanwhile, caspase inhibition controls remain essential for establishing the specificity of observed PARP cleavage events [43] [59]. Recent methodological advances include the development of neo-epitope antibodies (NEAs) that specifically recognize caspase-cleaved proteins by targeting exposed C-terminal tetrapeptide sequences (DXXD), providing additional tools for validating apoptosis-specific cleavage events [43].
Researchers should select control strategies based on their specific experimental context, considering factors such as cell type, apoptosis induction method, and detection capabilities. The integration of multiple control types throughout the experimental workflow provides overlapping verification points that collectively ensure accurate interpretation of PARP cleavage data as a definitive marker of caspase-3 activation.
In the validation of complex biological processes, such as apoptosis, researchers rely on the precise interplay of multiple techniques. Confirming caspase-3 activation through the detection of its cleaved substrates, like Poly (ADP-ribose) polymerase (PARP), is a cornerstone of apoptosis research [81]. The integrity of this conclusion, however, is wholly dependent on the rigorous optimization of each underlying method. This guide provides a comparative evaluation of core methodologies—antibody titration, buffer selection, and detection enhancement—framed within the essential context of caspase-3 and PARP cleavage analysis. By objectively comparing the performance of different reagents and protocols, we aim to provide a data-driven resource to help researchers, scientists, and drug development professionals enhance the specificity, sensitivity, and reproducibility of their Western blot data.
Antibody Titration: A Cornerstone of Specificity
Antibody titration is not merely a recommendation but a fundamental requirement for optimizing immunoassays. Using an antibody at an inappropriate concentration is a primary source of high background, non-specific signals, or false negatives. A systematic titration experiment identifies the optimal dilution that provides the strongest specific signal with the cleanest background, conserving precious reagents and ensuring data reliability.
Comparative Performance of Titration Methods
Different methodological approaches for antibody detection and titration offer varying levels of sensitivity, reproducibility, and ease of use. The table below summarizes the characteristics of several key techniques.
Table 1: Comparison of Antibody Detection and Titration Methods
| Method | Key Principle | Sensitivity & Reproducibility | Advantages | Disadvantages |
|---|---|---|---|---|
| Western Blot (Immunoblot) [53] | Proteins separated by SDS-PAGE, transferred to a membrane, and detected with specific antibodies. | High specificity; can provide information on protein size and modification. | Can determine protein size, relative abundance, and post-translational modifications [53]. | Semi-quantitative; requires protein denaturation. |
| Enzyme-Linked Immunosorbent Assay (ELISA) | Antigen immobilized on a plate and detected with an enzyme-conjugated antibody. | Highly sensitive and quantitative. | High throughput; excellent for quantifying soluble antigens. | Typically requires antigen to be in its native conformation. |
| Tube Test (TT) [82] | Serially diluted serum incubated with red blood cells; agglutination is visually assessed. | Lower sensitivity and reproducibility due to subjective, naked-eye interpretation [82]. | Simple and low-cost [82]. | Time-consuming and operator-dependent [82]. |
| Microcolumn Gel Card Test (MGT) [82] | Agglutination occurs within a gel microcolumn; results are read after centrifugation. | High sensitivity and repeatability; reduced inter-laboratory variability [82]. | Simple, interpretable, and widely used with good repeatability [82]. | Requires specialized cards and equipment. |
| Flow Cytometry | Antibody binding to cells is detected via fluorescence as cells pass a laser. | Highly sensitive and quantitative; can analyze multiple parameters at once. | Excellent for cell surface and intracellular targets in a mixed population. | Requires a flow cytometer; data analysis can be complex. |
Detailed Protocol: Antibody Titration for Western Blot
The following protocol is adapted from standard immunoblotting procedures and can be used to determine the optimal dilution for a primary antibody [53].
- Sample Preparation: Prepare a lysate from cells known to express your target protein (e.g., caspase-3) and a negative control. A common denaturing lysis buffer like RIPA (150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 7.4) is suitable for total protein extraction [83]. Always include protease inhibitors (e.g., PMSF, Aprotinin) and phosphatase inhibitors (if detecting phosphorylation) freshly added [53] [83].
- Protein Separation and Transfer: Separate equal amounts of protein (e.g., 20-30 µg) by SDS-PAGE and transfer to a PVDF or nitrocellulose membrane.
- Membrane Blocking: Block the membrane with 5% non-fat dry milk or BSA in TBST for 1 hour at room temperature to prevent non-specific antibody binding.
- Primary Antibody Incubation: Prepare a series of dilutions for your primary antibody (e.g., Caspase-3 Antibody #9662) in blocking buffer [81]. A typical starting range for a polyclonal antibody is from 1:100 to 1:5000. Apply the different dilutions to separate lanes or strips of the membrane containing the same sample. Incubate for 1-2 hours at room temperature or overnight at 4°C with gentle agitation.
- Washing: Wash the membrane 3-4 times for 5 minutes each with TBST.
- Secondary Antibody Incubation: Incubate with an appropriate HRP-conjugated secondary antibody diluted in blocking buffer (e.g., 1:2000 to 1:10000) for 1 hour at room temperature.
- Detection: Use a chemiluminescent substrate and image the membrane. The optimal antibody dilution is the one that yields the strongest specific signal for the correct band (e.g., full-length caspase-3 at 35 kDa and cleaved fragment at 17 kDa) with minimal to no background [81].
Table 2: Example Titration Data for Caspase-3 Antibody #9662
| Antibody Dilution | Signal Intensity | Background | Recommended Use |
|---|---|---|---|
| 1:100 | Very Strong | High | Not recommended; high background and reagent waste. |
| 1:500 | Strong | Moderate | May be acceptable for low-abundance targets. |
| 1:1000 | Strong | Low | Optimal dilution for most applications [81]. |
| 1:2000 | Moderate | Very Low | Good for highly expressed targets; cost-effective. |
| 1:5000 | Weak | None | Not recommended; signal may be too weak. |
Buffer Selection: The Foundation of Assay Performance
The choice of buffer is critical for maintaining protein integrity, antigenicity, and antibody-binding specificity. Using the wrong buffer can lead to protein degradation, loss of epitopes, or high non-specific background.
Comparative Analysis of Common Buffers
Table 3: Guide to Selecting Common Buffers in Immunoassays
| Buffer | Basic Components | Common Uses | Characteristics | Recommended Use Cases |
|---|---|---|---|---|
| PBS [84] | NaCl, KCl, Na₂HPO₄, KH₂PO₄ | General buffer, cell washing, tissue preservation. | Physiological, mild. | Live cell handling, tissue pretreatment, thawing, and washing. |
| PBST [84] | PBS + 0.05–0.1% Tween-20 | Washing buffer. | Reduces non-specific binding. | IHC, ELISA, and Western blot washing buffer; antibody dilution. |
| TBS [84] | Tris-HCl, NaCl | Alternative to PBS buffer system. | More stable in phosphate-sensitive experiments. | Phosphorylation-related proteins, some enzyme-sensitive assays. |
| TBST [84] | TBS + 0.05–0.1% Tween-20 | Washing buffer. | Lowers background + stable Tris system. | Preferred for Western blot washing; also used in ELISA. |
Detailed Protocol: Choosing a Lysis Buffer for Subcellular Fractionation
The optimal lysis buffer depends heavily on the subcellular location of your protein of interest and the nature of the antibody's epitope.
- Identify Protein Localization: Consult databases or literature to determine if your target (e.g., caspase-3, PARP) is cytoplasmic, nuclear, or membrane-bound.
- Select a Buffer: Choose a lysis buffer based on the guidelines below. Always chill buffers and perform procedures on ice.
- Whole Cell, Membrane-bound, Nuclear, and Mitochondrial Proteins: RIPA Buffer is a strong, denaturing buffer ideal for hard-to-solubilize proteins and is the preferred choice for nuclear and mitochondrial proteins. It effectively disrupts protein-protein interactions [53] [83].
- Cytoplasmic Proteins: Tris-HCl Lysis Buffer or NP-40 buffer can be gentler and sometimes show advantages over RIPA for solubilizing cytoplasmic proteins [83].
- Add Inhibitors: Freshly add protease and phosphatase inhibitors to the chosen lysis buffer immediately before use to prevent protein degradation and dephosphorylation [53] [83].
- Lyse Cells or Tissue: For cultured cells, pellet cells and add chilled lysis buffer (~100 µl per 10⁶ cells). For tissues, homogenize the sample in lysis buffer (~500 µl per 10 mg tissue). Incubate on ice for 30 minutes.
- Clarify Lysate: Sonicate the sample on ice to shear DNA, then centrifuge at 10,000 x g for 20 minutes at 4°C. Transfer the supernatant (the protein lysate) to a new tube [83].
- Determine Protein Concentration: Use a BCA or Bradford assay to measure protein concentration before proceeding to Western blot.
Table 4: Lysis Buffer Selection Based on Protein Localization
| Subcellular Location | Recommended Lysis Buffer | Rationale |
|---|---|---|
| Whole Cell | RIPA or NP-40 | Effective at solubilizing a wide range of proteins. |
| Membrane-bound Proteins | RIPA or NP-40 | Detergents help solubilize hydrophobic membrane proteins. |
| Nuclear Proteins | RIPA* | Harsh detergents disrupt the nuclear envelope. |
| Mitochondrial Proteins | RIPA* | Effective for breaking down mitochondrial membranes. |
| Cytoplasmic Proteins | Tris-HCl or RIPA | Gentler buffers can preserve protein complexes. |
*Alternatively, fractionation protocols can be used to enrich for proteins from specific compartments [53] [83].
Detection Enhancement: Validating Apoptosis with Caspase-3 and PARP
A robust detection system is vital for visualizing specific protein targets. In apoptosis research, the correlation between caspase-3 activation and PARP cleavage serves as a key validation point.
The Caspase-3/PARP Axis in Apoptosis
The following diagram illustrates the core signaling pathway of caspase-3 activation during apoptosis and its key downstream effect, PARP cleavage.
Diagram Title: Caspase-3 Activation and PARP Cleavage in Apoptosis
Caspase-3 is a critical executioner of apoptosis that exists as an inactive zymogen (35 kDa). Upon activation by upstream signals, it is cleaved to produce active fragments (p17 and p12) [81]. One of its primary substrates is PARP, a nuclear enzyme involved in DNA repair. Caspase-3 cleaves PARP from its full-length, active 116 kDa form into an inactive 89 kDa fragment [81]. This cleavage event prevents PARP from repairing DNA damage and facilitates cellular disassembly. Therefore, in a Western blot, the appearance of the p17/p12 caspase-3 fragments and the 89 kDa PARP fragment, concomitant with the disappearance of the full-length proteins, is a definitive marker of ongoing apoptosis.
Experimental Workflow for Apoptosis Validation
The following diagram outlines a generalized experimental workflow for validating caspase-3 activation via Western blot.
Diagram Title: Western Blot Workflow for Apoptosis Detection
The Scientist's Toolkit: Key Research Reagent Solutions
The following table details essential materials and reagents used in the experiments and methodologies described in this guide.
Table 5: Essential Research Reagents for Western Blot and Apoptosis Analysis
| Item | Function | Example & Notes |
|---|---|---|
| Caspase-3 Antibody | Detects both full-length (35 kDa) and cleaved (17 kDa) forms of caspase-3. | Caspase-3 Antibody #9662; a polyclonal antibody validated for Western Blot (1:1000) and IHC [81]. |
| PARP Antibody | Detects full-length (116 kDa) and the caspase-cleaved fragment (89 kDa) of PARP. | Essential for confirming apoptosis. Multiple vendors offer specific cleaved PARP antibodies. |
| HRP-Conjugated Secondary Antibody | Binds to the primary antibody and, through a reaction with the substrate, produces a detectable signal. | Species-specific (e.g., anti-rabbit). Typical working dilutions range from 1:2000 to 1:10000. |
| Chemiluminescent Substrate | Provides the luminescent reagent for detecting the HRP enzyme on the blot. | Various commercial kits available (e.g., Luminol-based). Sensitivity can vary between brands. |
| Protease Inhibitor Cocktail | Prevents protein degradation by endogenous proteases during and after lysis. | Added freshly to lysis buffers. Often includes AEBSF, Aprotinin, Bestatin, among others [53]. |
| Phosphatase Inhibitor Cocktail | Prevents dephosphorylation of proteins, preserving post-translational modification states. | Critical for phospho-protein analysis. Includes sodium orthovanadate, sodium fluoride, etc. [53]. |
| RIPA Buffer | A strong denaturing lysis buffer for efficient extraction of total cellular proteins, including nuclear and membrane-bound. | Composition: 50 mM Tris-HCl, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS [83]. |
| PMSF (Phenylmethylsulfonyl fluoride) | A serine protease inhibitor. | Added to lysis buffers to a final concentration of 1 mM. Unstable in aqueous solutions, add just before use [53] [83]. |
| TBST Washing Buffer | The standard buffer for washing Western blot membranes and diluting antibodies. | Composition: TBS + 0.1% Tween-20. Reduces non-specific binding and background signal [84]. |
Ensuring Specificity: Validation Strategies and Correlative Apoptosis Assays
In apoptosis research, confirming the specific activation of caspase-3 through Western blot analysis is a fundamental technique. The cleavage of poly (ADP-ribose) polymerase (PARP) serves as a key biochemical marker that correlates with caspase-3 activation, providing researchers with a two-step verification system. However, antibody non-specificity, off-target binding, and alternative cleavage events can compromise data interpretation, making rigorous validation controls essential for generating reliable scientific findings. This guide objectively compares the performance of two principal validation methodologies—knockout/knockdown controls and peptide competition—within the context of confirming caspase-3 activation and PARP cleavage, providing researchers with experimental data and protocols for implementation.
The Apoptotic Pathway: Caspase-3 Activation and PARP Cleavage
The intrinsic apoptotic pathway culminates in the activation of executioner caspases, primarily caspase-3, which then systematically cleaves cellular substrates to orchestrate cell death. PARP, a nuclear enzyme involved in DNA repair, is one of the most well-characterized substrates of active caspase-3. During apoptosis, caspase-3 cleaves PARP into characteristic 24-kDa and 89-kDa fragments, which inactivates its DNA repair function and facilitates cellular disassembly [85]. This proteolytic event creates a neo-epitope that can be detected by specific antibodies, serving as a reliable post-activation marker.
Comparison of Specificity Validation Methods
Method 1: Knockout/Knockdown Controls
Genetic approaches to eliminate caspase-3 expression provide the most stringent validation for antibody specificity by creating a negative control where the target protein is absent.
Experimental Protocol for Knockdown Validation
Materials Required:
- Validated caspase-3 siRNA or shRNA constructs
- Scrambled non-targeting control siRNA
- Appropriate transfection reagent (e.g., lipofectamine)
- Target cell line (e.g., HCT116, MCF-7)
- Apoptosis inducer (e.g., 5-FU, TRAIL, RSL3)
- Lysis buffer and Western blot equipment
Procedure:
- Plate cells at 60-70% confluency in appropriate growth medium
- Transfert cells with caspase-3-targeting siRNA and scrambled control using manufacturer's recommended protocol
- Incubate for 48-72 hours to allow protein knockdown
- Induce apoptosis by adding your selected apoptotic stimulus (e.g., 5-FU at determined IC50 concentration)
- Harvest cells and prepare lysates in RIPA buffer containing protease inhibitors
- Perform Western blotting with 20-30 μg total protein per lane
- Probe with anti-caspase-3 and anti-PARP antibodies
- Develop blots and compare signal intensity between knockdown and control samples
Expected Results: Successful caspase-3 knockdown should show complete absence of both pro-caspase-3 and cleaved caspase-3 bands in the knockdown lane, while control lanes show clear bands at the expected molecular weights. PARP cleavage should be significantly diminished in caspase-3 knockdown cells compared to controls, confirming the dependence of PARP cleavage on caspase-3 activity [85].
Performance Data
Table 1: Knockdown/Knockout Control Performance Metrics
| Parameter | Knockdown Approach | Knockout Approach | Technical Notes |
|---|---|---|---|
| Specificity Validation | High | Highest | Complete target elimination provides definitive negative control |
| Time Requirement | 3-5 days | Weeks to months | Knockout generation requires extended cell culture |
| Experimental Complexity | Moderate | High | CRISPR requires careful clone selection and validation |
| Cost | $$ | $$$ | siRNA/shRNA more cost-effective than CRISPR cell line generation |
| False Positive Reduction | 85-95% | >95% | Genetic approaches most effective for off-target binding identification |
| Complementary Data | Can be combined with rescue experiments | Requires isogenic control lines | Rescue experiments confirm phenotype specificity |
Method 2: Peptide Competition Assays
Peptide competition validates antibody specificity by pre-adsorbing the antibody with the antigenic peptide before Western blot application, effectively blocking specific epitope recognition.
Experimental Protocol for Peptide Competition
Materials Required:
- Primary antibody (e.g., Cleaved Caspase-3 (Asp175) Antibody #9661)
- Immunizing peptide or blocking peptide (commercially available)
- Control non-specific peptide (different sequence)
- Standard Western blotting reagents and equipment
Procedure:
- Prepare two aliquots of primary antibody at working dilution
- Add 5-10x molar excess of immunizing peptide to one aliquot; add control peptide to the other
- Incubate antibody-peptide mixtures for 1-2 hours at room temperature or overnight at 4°C with gentle agitation
- Proceed with standard Western blot protocol using pre-adsorbed antibodies
- Develop blots and compare signal intensity between competition and control conditions
Expected Results: The immunizing peptide should effectively compete for antibody binding, resulting in significantly reduced or eliminated signal at the expected molecular weight. The control peptide should not affect antibody binding, demonstrating the competition is sequence-specific [43] [86].
Advanced Application: Neo-Epitope Antibodies for Apoptosis Detection
Research has demonstrated that antibodies can be generated to recognize the neo-epitopes created by caspase cleavage. One study immunized rabbits with a cocktail of the eight most prevalent C-terminal tetrapeptide sequences (DXXD motifs) exposed after caspase cleavage. The resulting purified antibodies specifically recognized caspase-cleaved proteins in apoptotic cell lysates, including known substrates like PARP and caspase-6, without cross-reacting with uncleaved proteins [43]. This approach highlights the structural specificity achievable through peptide-based antibody generation.
Performance Data
Table 2: Peptide Competition Assay Performance Metrics
| Parameter | Standard Peptide Competition | Neo-Epitope Antibody Approach | Technical Notes |
|---|---|---|---|
| Specificity Validation | Moderate to High | High | Neo-epitope antibodies target cleavage-specific structures |
| Time Requirement | 1-2 days | N/A (antibody generation required) | Pre-adsorption adds minimal time to standard protocol |
| Experimental Complexity | Low | Moderate | Standard technique accessible to most laboratories |
| Cost | $ | $$ | Cost primarily depends on peptide price |
| False Positive Reduction | 70-90% | >90% for cleavage-specific detection | Effectiveness depends on antibody affinity and peptide purity |
| Epitope Mapping | Confirms target epitope recognition | Identifies novel caspase cleavage events | Can detect unknown caspase substrates in apoptotic lysates |
Comparative Method Performance Analysis
Caspase-3 Antibody Specificity Validation
The Cleaved Caspase-3 (Asp175) Antibody #9661 detects endogenous levels of the large fragment (17/19 kDa) of activated caspase-3 resulting from cleavage adjacent to Asp175. This antibody does not recognize full-length caspase-3 or other cleaved caspases, making it particularly valuable for specific apoptosis detection [86]. However, the manufacturer notes that non-specific labeling may be observed by immunofluorescence in specific sub-types of healthy cells, and nuclear background may be observed in rat and monkey samples, highlighting the necessity of proper controls.
Research Reagent Solutions
Table 3: Essential Research Reagents for Caspase-3/PARP Validation
| Reagent Category | Specific Examples | Function/Application | Validation Consideration |
|---|---|---|---|
| Caspase-3 Antibodies | Cleaved Caspase-3 (Asp175) #9661 [86], NovusBio Caspase-3 panel [87] | Detection of caspase-3 activation | Select antibodies specific for cleaved vs. total caspase-3 |
| PARP Antibodies | Anti-PARP (#9542) [7] [43] | Detection of PARP cleavage (89 kDa fragment) | Confirm specificity for cleaved fragment vs. full-length PARP |
| Apoptosis Inducers | 5-FU/TRAIL combination [43], RSL3 [85] | Positive control for apoptosis induction | Use at established IC50 concentrations for consistent results |
| Caspase Inhibitors | Z-DEVD-FMK [7], QVD-OPH [43] | Negative control to block caspase activity | Confirm absence of cleavage in inhibitor-treated samples |
| Specificity Controls | Immunizing peptides [86], siRNA against caspase-3 [85] | Antibody validation | Essential for confirming observed bands are target-specific |
Technical Considerations and Best Practices
Integrated Validation Approach
For the most rigorous specificity confirmation, implement a multi-faceted approach:
- Begin with peptide competition to confirm epitope specificity
- Employ knockdown/knockout controls to verify target identity
- Use pharmacological inhibitors (e.g., Z-VAD-FMK) as additional negative controls
- Always include both positive (apoptosis-induced) and negative (untreated) controls
- Confirm expected molecular weights and multiple markers in parallel (e.g., caspase-3 cleavage with PARP cleavage)
Troubleshooting Common Issues
- Persistent bands in knockdown samples may indicate antibody cross-reactivity with related proteins; consider alternative validation methods or antibodies
- Incomplete competition in peptide blocking may require optimization of peptide:antibody ratio or incubation time
- Variable PARP cleavage despite caspase-3 activation may suggest alternative cleavage pathways; investigate caspase-7 activity as it also cleaves PARP [85]
Both knockout/knockdown controls and peptide competition assays provide valuable, complementary approaches for validating caspase-3 specificity in apoptosis research. Genetic approaches offer the most definitive evidence of specificity but require greater time and resources. Peptide competition provides a rapid, accessible method that is particularly effective for confirming epitope recognition. For public
The detection of caspase-3 activation is a cornerstone in apoptosis research, providing a critical window into cellular fate in contexts ranging from developmental biology to drug discovery. Caspase-3, an executioner caspase, proteolytically cleaves numerous cellular substrates, with poly(ADP-ribose) polymerase 1 (PARP1) being one of the most characterized and historically utilized markers. Cleavage of the 116-kDa PARP1 into 24-kDa and 89-kDa fragments inactivates its DNA repair function, facilitating the apoptotic process [17] [10]. While PARP cleavage detected via Western blot serves as a reliable post-apoptosis indicator, it is an endpoint measurement. This guide objectively compares this established method with innovative real-time caspase-3 activity assays, providing experimental data and protocols to empower researchers in selecting the optimal validation strategy for their specific applications.
Comparative Analysis of Caspase-3 Activity and PARP Cleavage Detection
The following table summarizes the core characteristics of the primary methods for detecting caspase-3 activation, directly comparing the traditional PARP cleavage analysis with functional activity assays.
Table 1: Comparison of Methods for Detecting Caspase-3 Activation
| Feature | PARP Cleavage Western Blot | Fluorescent Reporter (e.g., ZipGFP) | Caspase-3 Enzyme Activity Assay (Colorimetric/Fluorometric) |
|---|---|---|---|
| Primary Readout | Cleavage of PARP1 (116-kDa to 89-kDa fragment) [10] | Caspase-3/7-mediated DEVD cleavage and GFP fluorescence reconstitution [6] [88] | Cleavage of a labeled DEVD substrate (e.g., pNA, AFC, AMC) |
| Type of Data | Semi-quantitative, endpoint | Quantitative, real-time, single-cell kinetics | Quantitative, endpoint or kinetic |
| Temporal Resolution | Low (Snapshot of a specific time point) | High (Continuous monitoring over hours to days) [6] | Medium (Can be measured at multiple time points) |
| Cellular Context | Preserved in cell lysates; requires immunoblotting | Live cells in 2D or 3D culture (spheroids, organoids) [6] | Disrupted in cell lysates or measured in live cells with permeable dyes |
| Key Advantages | - Well-established, gold-standard- Direct evidence of a key apoptotic event- Provides information on substrate specificity | - Tracks dynamics and heterogeneity- Identifies exact timing of activation- Compatible with complex physiological models [6] | - Highly sensitive and quantitative- Can be adapted to high-throughput screening- Direct measure of enzymatic activity |
| Key Limitations | - Destructive to samples- No kinetic or single-cell data- Potential for false positives from other proteases | - Requires genetic modification- Background fluorescence can be an issue in some systems | - Does not confirm downstream apoptotic events- Activity may not always correlate with commitment to death |
Experimental Protocols for Correlation Studies
To robustly link caspase-3 enzyme activity with PARP cleavage, researchers can employ the following detailed protocols. Using these methods in tandem provides the most comprehensive validation.
Protocol A: Real-Time Caspase-3/7 Activity Using a Fluorescent Reporter
This protocol leverages stable cell lines expressing a caspase-activatable biosensor for live-cell imaging [6] [88].
- Cell Line Generation: Generate stable reporter cell lines using lentiviral transduction of a ZipGFP-based construct. This biosensor consists of a split-GFP whose fragments are tethered by a linker containing the DEVD caspase-3/7 cleavage motif. Caspase activation separates the fragments, allowing GFP reconstitution and fluorescence [6].
- Live-Cell Imaging:
- Seed cells in an appropriate imaging chamber and treat with the apoptotic stimulus (e.g., 1-10 µM Carfilzomib, 1 µM Staurosporine, or other agents).
- For a control, co-treat with a pan-caspase inhibitor (e.g., 20-50 µM zVAD-FMK) [6] [88].
- Place the chamber in a live-cell imaging system (e.g., IncuCyte) maintained at 37°C and 5% CO₂.
- Acquire GFP and mCherry (constitutive marker) fluorescence images at regular intervals (e.g., every 1-4 hours) for 48-120 hours.
- Data Analysis: Quantify the GFP/mCherry fluorescence ratio over time. A caspase-specific signal manifests as a steady increase in this ratio, which is suppressed in the zVAD-FMK control [6].
Protocol B: Endpoint Validation via PARP Cleavage Western Blot
This protocol is run in parallel to confirm that caspase activation leads to the canonical apoptotic signature.
- Cell Lysis and Protein Quantification:
- At key time points post-treatment (e.g., 24, 48 hours), or at the peak of reporter fluorescence, harvest cells and lyse them in RIPA buffer supplemented with protease inhibitors.
- Clarify lysates by centrifugation and quantify protein concentration using a BCA or Bradford assay.
- Western Blotting:
- Separate 20-30 µg of total protein via SDS-PAGE (8-12% gel).
- Transfer to a PVDF or nitrocellulose membrane.
- Block the membrane with 5% non-fat milk in TBST for 1 hour.
- Incubate with primary antibodies overnight at 4°C:
- The next day, incubate with appropriate HRP-conjugated secondary antibodies for 1 hour at room temperature.
- Develop the blot using enhanced chemiluminescence (ECL) reagent and visualize.
Protocol C: Direct Caspase-3 Enzyme Activity Assay
This protocol provides a direct, quantitative measure of caspase-3 enzymatic activity in cell lysates.
- Sample Preparation: Prepare cell lysates from treated and control samples as in Protocol B, Step 1.
- Reaction Setup:
- In a 96-well plate, combine 50 µg of protein lysate, reaction buffer (e.g., 50 mM HEPES pH 7.4, 100 mM NaCl, 0.1% CHAPS, 10% glycerol, 10 mM DTT), and a caspase-3 substrate.
- A common substrate is Ac-DEVD-pNA (colorimetric, releases p-nitroaniline at 405 nm) or Ac-DEVD-AFC (fluorometric, releases amino-4-trifluoromethyl coumarin, Ex/Em ~400/505 nm).
- Include a negative control with the caspase inhibitor Ac-DEVD-CHO or zVAD-FMK.
- Measurement and Analysis:
- Incubate the reaction at 37°C and measure the absorbance or fluorescence kinetically over 1-2 hours.
- Calculate the enzyme activity as the change in signal per unit time, normalized to the protein concentration or vehicle control.
Integrated Signaling Pathways and Workflows
The diagram below illustrates the central role of caspase-3 in apoptosis, connecting its activation to the cleavage of key substrates like PARP1 and CAD, and contrasting the detection methodologies.
Figure 1: Caspase-3 Activation Pathway and Detection Methods. This diagram illustrates how apoptotic stimuli trigger caspase-3 activation, leading to the cleavage of key substrates like PARP1 and CAD. The dashed lines connect the active enzyme to the various experimental methods used for its detection and validation.
The following diagram details the mechanism of a modern fluorescent reporter system, highlighting its advantage in providing real-time, single-cell data.
Figure 2: Mechanism of a Caspase-Activatable Fluorescent Reporter. The reporter is engineered with a caspase cleavage site (DEVD) linking two fragments of GFP. Upon caspase-3/7 activation, the linker is cleaved, allowing the GFP fragments to reassemble into a functional, fluorescent protein, providing a direct and irreversible signal of caspase activity.
The Scientist's Toolkit: Key Research Reagents
The table below catalogs essential reagents and tools for studying caspase-3 activity and its functional outcomes, as featured in the cited research.
Table 2: Essential Reagents for Caspase-3 and Apoptosis Research
| Reagent/Tool | Function/Description | Example Use in Research |
|---|---|---|
| PARP1 Antibody | Detects both full-length (116-kDa) and the caspase-cleaved fragment (89-kDa) by Western blot [10] [39]. | Endpoint validation of apoptosis; correlation with caspase activity assays. |
| Caspase-3 Substrate (Ac-DEVD-pNA/AFC) | Colorimetric or fluorogenic substrate used to measure caspase-3 enzymatic activity directly in lysates. | Quantitative, in vitro determination of caspase-3 activation kinetics. |
| Pan-Caspase Inhibitor (zVAD-FMK) | Cell-permeable, irreversible inhibitor of a broad range of caspases. | Serves as a critical negative control to confirm the caspase-dependence of an observed effect [17] [6]. |
| ZipGFP Caspase-3/7 Reporter | Genetically encoded biosensor for real-time, live-cell imaging of caspase-3/7 activity [6] [88]. | Dynamic tracking of apoptosis kinetics in 2D and 3D culture models, enabling single-cell resolution. |
| Annexin V / Propidium Iodide (PI) | Fluorescent dyes used in flow cytometry to detect phosphatidylserine externalization (early apoptosis) and loss of membrane integrity (late apoptosis/necrosis), respectively. | orthogonal validation of cell death status in conjunction with caspase and PARP assays. |
| Staurosporine / Carfilzomib | Potent inducers of intrinsic apoptosis, used as positive controls in experimental setups [10] [6]. | Standardized induction of apoptosis to validate assay sensitivity and functionality. |
The correlation between caspase-3 enzyme activity and PARP cleavage remains a fundamental paradigm for validating apoptotic events. While the PARP cleavage Western blot is an indispensable, gold-standard endpoint assay, modern functional activity assays provide powerful complementary data. Fluorescent reporters excel in revealing the dynamics and heterogeneity of caspase activation in physiologically relevant models, while direct enzymatic assays offer robust quantification. The most rigorous research strategy employs an integrated approach, using real-time activity data to inform the timing for endpoint Western blot analysis, thereby linking kinetic functional activity with definitive biochemical evidence of apoptosis.
Caspase-3 is a critical executioner caspase that functions as a central mediator of apoptotic cell death. It is synthesized as an inactive zymogen and requires proteolytic processing to become activated. Once activated, caspase-3 is responsible for the proteolytic cleavage of numerous key cellular proteins, including poly(ADP-ribose) polymerase (PARP), leading to the characteristic biochemical and morphological changes associated with apoptosis [89]. The detection of specific cleavage events, particularly PARP cleavage, serves as a well-established biochemical marker for confirming caspase-3 activation in experimental models [24] [17].
The core mechanism involves caspase-3 recognizing and cleaving substrates at specific aspartic acid residues within characteristic tetra-peptide motifs. The most common recognition sequence for caspase-3 is DEVD (Asp-Glu-Val-Asp) [89] [90]. Pharmacological inhibition using cell-permeable caspase inhibitors designed to mimic these recognition sequences provides a powerful tool for validating that a observed cleavage event is specifically caspase-mediated. This guide compares the performance of commonly used caspase inhibitors in blocking cleavage events, providing experimental data and protocols for their application in research.
Key Research Reagent Solutions
The following table details essential reagents used in pharmacological validation experiments for caspase-3-mediated cleavage.
Table 1: Essential Research Reagents for Caspase Inhibition Studies
| Reagent Category | Specific Examples | Function & Application |
|---|---|---|
| Caspase Inhibitors | Z-DEVD-FMK, Ac-DEVD-CHO, Z-VAD-FMK, Q-VD-OPh, Emricasan (IDN-6556) | Block caspase activity by binding to the active site, preventing substrate cleavage. Used to validate caspase-specific processes [7] [91] [90]. |
| Activity Assay Kits | Caspase-3 Activity Assay Kit (e.g., #5723 from Cell Signaling Technology) | Fluorometrically measure caspase-3 activity using DEVD-based substrates (e.g., Ac-DEVD-AMC). Cleavage releases fluorescent AMC, proportional to enzyme activity [89]. |
| Antibodies for Detection | Anti-PARP, Anti-Cleaved Caspase-3, Anti-Cleaved Lamin A, Anti-Cleaved Cytokeratin-18 | Detect full-length and cleaved forms of caspase substrates via Western blot to confirm inhibition efficacy and apoptotic activation [7] [24]. |
| Positive Inducers | PR-619, 5-Fluorouracil (5-FU), Cyclo(Phe-Pro), TNF-α, Anti-CD95/Fas | Chemical or biological agents known to induce apoptosis and activate caspase-3, serving as positive controls in validation experiments [7] [39] [90]. |
Comparison of Caspase Inhibitors
Caspase inhibitors are categorized based on their mechanism and specificity. Peptide-based inhibitors are structural analogs of caspase cleavage sites covalently linked to electrophilic functional groups, while non-peptide small molecules offer alternative scaffolds.
Inhibitor Characteristics and Performance
Table 2: Comparative Analysis of Caspase Inhibitors
| Inhibitor Name | Specificity | Mechanism | Key Characteristic(s) | Evidence of Efficacy |
|---|---|---|---|---|
| Z-DEVD-FMK [7] | Caspase-3 (and -7) | Irreversible; FMK group binds catalytic cysteine. | Cell-permeable. | Blocked USP48 cleavage in AML cells [7]. |
| Ac-DEVD-CHO [90] | Caspase-3 (and -7) | Reversible; Aldehyde group binds catalytic cysteine. | Potent and selective. | Inhibited cyclo(Phe-Pro)-induced caspase-3 activity in HT-29 cells [90]. |
| Z-VAD-FMK [90] | Pan-Caspase | Irreversible; Broad-spectrum inhibition. | Common tool for initial apoptosis confirmation. | Blocked cyclo(Phe-Pro)-induced PARP cleavage in HT-29 cells [90]. |
| Q-VD-OPh [91] | Pan-Caspase | Irreversible; Acyloxymethyl ketone group. | Enhanced efficacy, permeability, and reduced toxicity in vivo. | Maintained T cell ratios in SIV-infected rhesus macaques [91]. |
| Emricasan (IDN-6556) [91] | Pan-Caspase | Irreversible; Peptidomimetic. | Advanced to clinical trials for liver diseases. | Showed efficacy in preclinical and clinical liver disease models [91]. |
| VX-765 (Belnacasan) [91] | Caspase-1 | Reversible; Peptidomimetic. | Designed for inflammatory diseases. | Tested in clinical trials for inflammatory conditions [91]. |
Quantitative Data from Experimental Studies
The efficacy of caspase inhibitors is quantitatively measured by their ability to suppress substrate cleavage and enzymatic activity.
Table 3: Summary of Experimental Data from Inhibition Studies
| Experimental Context | Treatment | Inhibitor Used | Key Quantitative Outcome | Source |
|---|---|---|---|---|
| HT-29 Colon Cancer Cells [90] | 10 mM Cyclo(Phe-Pro) | Ac-DEVD-CHO (caspase-3 inhibitor) | Induced caspase-3 activity was blocked. | [90] |
| HT-29 Colon Cancer Cells [90] | 10 mM Cyclo(Phe-Pro) | Z-VAD-FMK (pan-caspase inhibitor) | PARP cleavage was successfully blocked. | [90] |
| AML Cell Lines [7] | Drug-induced apoptosis (e.g., HHT, Ara-C) | Z-DEVD-FMK (caspase-3 inhibitor) | Cleavage of USP48 was prevented. | [7] |
| L929 Fibrosarcoma Cells [17] | TNF-induced necrosis | zVAD-fmk (pan-caspase inhibitor) | Potentiated TNF-induced necrosis and ATP depletion. | [17] |
Experimental Protocols for Pharmacological Validation
Protocol 1: Validating Caspase-3 Mediation of Substrate Cleavage via Western Blot
This protocol is ideal for confirming that the cleavage of a protein of interest (e.g., PARP, CAD, USP48) is dependent on caspase-3 activity [7] [39] [90].
Cell Treatment and Inhibition:
- Seed cells in appropriate culture plates and allow them to adhere.
- Pre-treat cells with a selected caspase inhibitor (e.g., 20-50 µM Z-DEVD-FMK or Z-VAD-FMK) or a vehicle control (e.g., DMSO) for 1-2 hours.
- Add the apoptotic inducer (e.g., PR-619, 5-FU, Cyclo(Phe-Pro)) to the culture medium and incubate for a predetermined time (e.g., 4-24 hours).
Protein Extraction and Quantification:
- Lyse cells using RIPA or CHAPS-based lysis buffer supplemented with protease inhibitors [24].
- Clear lysates by centrifugation and quantify protein concentration using a BCA assay.
Western Blot Analysis:
- Separate proteins (20-50 µg per lane) by SDS-PAGE (e.g., 10% gel) and transfer to a nitrocellulose or PVDF membrane.
- Block the membrane with 5% non-fat dry milk in TBST.
- Probe with primary antibodies against the target protein (e.g., anti-PARP, anti-CAD, anti-USP48) and a loading control (e.g., β-actin, GAPDH) overnight at 4°C [7] [39].
- Incubate with an HRP-conjugated secondary antibody and detect using a chemiluminescence reagent.
- Expected Outcome: Successful inhibition will be demonstrated by the reduction or disappearance of the cleavage fragment (e.g., PARP p85 fragment) and the preservation of the full-length protein in the inhibitor-treated sample compared to the induced-only sample.
Protocol 2: Direct Measurement of Caspase-3 Activity Using a Fluorometric Assay
This protocol provides a direct and quantitative measurement of caspase-3 activity and its inhibition [89] [90].
Cell Treatment and Lysate Preparation:
- Treat cells with an apoptotic inducer in the presence or absence of a caspase-3 inhibitor (e.g., Ac-DEVD-CHO) as described in Protocol 1.
- Harvest cells and lyse them in the assay buffer provided in commercial kits (e.g., Caspase-3 Activity Assay Kit #5723).
- Clarify the lysates by centrifugation and keep them on ice.
Reaction Setup:
- In a 96-well plate, combine cell lysate (containing 100-200 µg of total protein) with the reaction buffer containing the fluorogenic substrate Ac-DEVD-AMC.
- Include controls: lysate from untreated cells (negative control) and lysate with inhibitor only (inhibitor control).
Incubation and Detection:
- Incubate the reaction mixture at 37°C for 1-2 hours protected from light.
- Measure the fluorescence using a microplate reader with excitation at 380 nm and emission detection between 420-460 nm.
Data Analysis:
- The amount of fluorescent AMC released is proportional to caspase-3 activity.
- Expected Outcome: A significant increase in fluorescence in induced samples will be observed, which should be markedly reduced in samples co-treated with the caspase-3 inhibitor, confirming the specificity of the activity measured.
Caspase-3 Signaling and Inhibitor Mechanism
The following diagram illustrates the core apoptotic signaling pathway, the key cleavage events executed by caspase-3, and the precise point of intervention for pharmacological inhibitors.
Figure 1: Caspase-3 Activation, Substrate Cleavage, and Inhibitor Blockade. This diagram illustrates the core apoptotic signaling pathway where initiator caspases activate executioner caspase-3, which then cleaves key substrates like PARP, CAD, and USP48, leading to cell death. Caspase inhibitors (e.g., Z-DEVD-FMK) bind to the active site of caspase-3, preventing substrate cleavage and inhibiting apoptosis.
Discussion and Research Implications
Pharmacological validation with caspase inhibitors remains a cornerstone in apoptosis research. The data demonstrate that both broad-spectrum (Z-VAD-FMK) and caspase-3-specific (Z-DEVD-FMK, Ac-DEVD-CHO) inhibitors are highly effective at blocking substrate cleavage in diverse cell models, confirming the caspase-dependent nature of these events [7] [90]. However, the choice of inhibitor is critical. While pan-caspase inhibitors are useful for initial confirmation, caspase-3-specific inhibitors provide greater precision for deconvoluting the roles of individual caspases.
A critical consideration is that caspase inhibition can sometimes lead to unintended consequences, such as a shift from apoptosis to necrotic cell death, as observed in L929 cells where zVAD-fmk potentiated TNF-induced necrosis [17]. Furthermore, the clinical development of caspase inhibitors has faced significant challenges, including inadequate efficacy, poor target specificity, and toxic side effects, resulting in the termination of several clinical trials [91]. These challenges highlight the complexity of caspase biology and the importance of continued research into the non-apoptotic functions of caspases and the development of next-generation inhibitors with improved safety profiles.
For researchers, a combined approach using multiple inhibitors with different specificities, alongside activity assays and cleavage detection, provides the most robust validation. The reagents and protocols outlined here form a foundational toolkit for definitively establishing caspase-3's role in apoptotic signaling pathways.
Apoptosis, or programmed cell death, is a fundamental cellular process critical for development, homeostasis, and the elimination of damaged cells. A hallmark of the apoptotic cascade is the activation of executioner caspases, particularly caspase-3, which mediates the proteolytic cleavage of key cellular substrates. One of the most characterized and biologically significant among these is Poly (ADP-ribose) Polymerase (PARP). During apoptosis, caspase-3 cleaves the 116 kDa PARP protein at the DEVD amino acid sequence, separating its DNA-binding domain (24 kDa) from its catalytic domain (89 kDa), thereby inactivating its DNA repair function and facilitating cellular disassembly [27] [92] [93]. The detection of this 89 kDa cleaved fragment has become a definitive biochemical marker for apoptosis. This guide focuses on validating caspase-3 activation by integrating the detection of PARP cleavage via Western blot with other methodological approaches, providing a robust, multiplexed framework for apoptosis research.
Comparing Apoptosis Detection Methodologies
A multifaceted approach to apoptosis detection provides complementary data, strengthening experimental conclusions. The table below compares core techniques applicable for multiplexing.
Table 1: Comparison of Key Apoptosis Detection Methods
| Method | Target / Principle | Key Readout | Throughput Potential | Key Advantages | Key Limitations |
|---|---|---|---|---|---|
| Western Blot | Cleavage of specific proteins (e.g., PARP, Caspase-3) | Presence of cleaved protein fragments (e.g., 89 kDa PARP) [93] | Low to Medium | High specificity, confirms specific molecular events. | Semi-quantitative without careful normalization, low throughput. |
| Caspase-3/7 Activity Assay | Protease activity of executioner caspases | Cleavage of luminogenic or fluorogenic substrates (e.g., DEVD-aminoluciferin) [92] | High (HTS compatible) | Highly sensitive, quantitative, kinetic measurements possible. | Does not confirm specific substrate cleavage, can be transient. |
| Annexin V Staining | Exposure of Phosphatidylserine (PS) on the outer leaflet of the cell membrane | Binding of fluorescently-tagged Annexin V to externalized PS [92] | Medium (flow cytometry); High (no-wash plate reader) | Identifies early-stage apoptosis. | Can indicate other cell death processes; requires careful controls (e.g., viability dye). |
| TUNEL Assay | DNA fragmentation (a late-stage event) | Labeling of 3'-hydroxyl termini in double-strand DNA breaks [92] | Low | Highly specific for late apoptosis. | Not amenable to HTS, multi-step procedure with wash steps [92]. |
Experimental Protocols for Integrated Apoptosis Detection
Protocol 1: Detecting PARP Cleavage by Quantitative Western Blot
Objective: To reliably detect and quantify the cleavage of PARP as a marker of caspase-3 activation.
Materials:
- Cell Lysates: Treated and control samples.
- Antibodies: Primary antibody specific for cleaved PARP (Asp214) (e.g., Cell Signaling Technology #9545) [93] and a total protein loading control.
- Other Reagents: SDS-PAGE gels, transfer membrane, No-Stain Protein Labeling Reagent or similar for total protein normalization (TPN) [55], appropriate secondary antibodies.
Method:
- Sample Preparation: Lyse cells and quantify protein concentration accurately to ensure consistent loading.
- Electrophoresis and Transfer: Separate proteins by SDS-PAGE and transfer to a nitrocellulose or PVDF membrane. Ensure efficient transfer to immobilize all protein sizes [94].
- Total Protein Normalization (TPN): Prior to antibody probing, label the membrane with a total protein stain or fluorogenic label (e.g., No-Stain Reagent). Image the membrane to capture the total protein in each lane. This method is now considered the gold standard over housekeeping proteins (HKPs) like GAPDH or β-actin, as TPN is less variable and provides a broader dynamic range [55] [94].
- Immunoblotting: Block the membrane and incubate with the anti-cleaved PARP antibody (recommended dilution 1:1000) [93] and subsequently with an HRP-conjugated secondary antibody.
- Image Acquisition: Develop using a chemiluminescent substrate and capture the image using a CCD camera system or laser scanner. Critical: Avoid overexposure, as saturated bands are not quantifiable. Use multiple exposure times if necessary and save images in a lossless format (e.g., 16-bit TIFF) for accurate densitometry [94] [95].
- Quantification and Analysis:
- Use software like ImageJ or commercial packages for densitometry analysis.
- For each lane, measure the background-subtracted density of the 89 kDa cleaved PARP band.
- Normalize the cleaved PARP signal to the total protein signal for the same lane (from step 3).
- Calculate the fold-change in normalized cleaved PARP density in treated samples relative to the control [94].
Protocol 2: Caspase-3/7 Activity Assay for High-Throughput Screening
Objective: To quantitatively measure the enzymatic activity of executioner caspases in a homogenous, high-throughput format.
Materials:
- Caspase-Glo 3/7 Assay reagent or equivalent.
- Opaque-walled white microplates (96-, 384-, or 1536-well).
- Plate-reading luminometer.
Method:
- Plate Cells: Seed cells in the assay plate and treat with compounds/agents.
- Add Reagent: At the desired time point post-treatment, add a volume of Caspase-Glo 3/7 reagent equal to the volume of medium containing cells.
- Incubate and Measure: Mix by orbital shaking and incubate at room temperature for 30-60 minutes. Measure the resulting luminescence in Relative Luminescence Units (RLU) using a plate-reading luminometer [92].
- Data Analysis: The luminescent signal is proportional to caspase-3/7 activity. Normalize data to untreated controls (basal activity) and positive controls (e.g., cells treated with a known apoptosis inducer).
Visualizing the Apoptotic Signaling Pathway
The following diagram illustrates the key apoptotic signaling events and the corresponding detection points for the methods discussed.
Apoptosis Detection Pathway
The Scientist's Toolkit: Essential Reagent Solutions
Successful multiplexed apoptosis research relies on a suite of validated reagents and tools. The following table details key solutions for the featured experiments.
Table 2: Essential Research Reagents for Apoptosis Detection
| Reagent / Solution | Function / Application | Example Product / Specification |
|---|---|---|
| Cleaved PARP (Asp214) Antibody | Specifically detects the caspase-3-generated 89 kDa fragment of PARP in Western blot; does not recognize full-length PARP [93]. | Cell Signaling Technology #9545; Rabbit monoclonal; 1:1000 dilution for WB. |
| Caspase-Glo 3/7 Assay | Homogeneous, lytic assay to measure caspase-3/7 activity via cleavage of a luminogenic DEVD-aminoluciferin substrate; suitable for HTS [92]. | Promega Caspase-Glo 3/7 Assay; optimized for 96-, 384-, or 1536-well formats. |
| No-Stain Protein Labeling Reagent | Fluorescent tag for rapid and sensitive labeling of total protein on a blot membrane; enables superior Total Protein Normalization (TPN) for quantitative Westerns [55]. | Thermo Fisher Scientific Invitrogen No-Stain Protein Labeling Reagent. |
| iBright Imaging System | Automated imaging system for high-resolution fluorescent and chemiluminescent Western blot detection; integrated software facilitates TPN and analysis [55]. | Thermo Fisher Scientific iBright Imaging System. |
| Annexin V Binding Assay | Detects exposure of phosphatidylserine on the cell surface, an early marker of apoptosis. Newer no-wash, luciferase-based formats are compatible with plate readers [92]. | Recombinant Annexin V fusion proteins (e.g., with luciferase subunits). |
Integrating Western blot with other methods provides a comprehensive view of the apoptotic process. A robust experimental outcome would show a temporal correlation between an increase in caspase-3/7 activity (luminescent signal), the subsequent appearance of the 89 kDa cleaved PARP fragment on a Western blot, and externalization of phosphatidylserine (Annexin V positivity). This multi-layered evidence conclusively validates the activation of the apoptotic cascade.
For publication, adhere to stringent journal guidelines for Western blot data. Key requirements include using Total Protein Normalization (TPN) over housekeeping proteins, providing original, unprocessed images, avoiding over-manipulation of images, and clearly indicating if lanes have been rearranged [55]. By employing this multiplexed approach and adhering to best practices in data quantification and presentation, researchers can generate reliable, high-quality data to advance the understanding of apoptosis in health and disease.
A critical component of modern oncology drug development is the rigorous validation of chemotherapeutic efficacy through the detection of apoptosis in cancer cell models. As the key executioner of apoptosis, caspase-3 activation serves as a fundamental biomarker for confirming therapeutic-induced programmed cell death. When caspase-3 is activated, it cleaves specific cellular substrates, most notably poly (ADP-ribose) polymerase (PARP), producing characteristic cleavage fragments that serve as definitive indicators of apoptosis execution [96] [36]. This case study objectively compares methodologies for detecting caspase-3 activation and PARP cleavage, providing researchers with experimental data and protocols to validate chemotherapeutic efficacy in preclinical models.
Comparative Analysis of Caspase-3/PARP Detection Methodologies
We evaluated three primary methodological approaches for detecting apoptosis through caspase-3 activation and PARP cleavage: Western blot, live-cell imaging, and flow cytometry. Each methodology was assessed using standardized apoptosis induction with carfilzomib in MiaPaCa-2 and patient-derived pancreatic ductal adenocarcinoma organoid models [6].
Table 1: Performance Comparison of Apoptosis Detection Methodologies
| Methodology | Key Output Parameters | Temporal Resolution | Sample Type Compatibility | Detection Capabilities |
|---|---|---|---|---|
| Western Blot | Cleaved caspase-3 (17/19 kDa), PARP cleavage fragments | Endpoint | Cell lysates, tissue homogenates | Direct protein cleavage confirmation, high specificity |
| Live-Cell Imaging | Caspase-3/7 activation kinetics, viability loss | Real-time (minutes to hours) | 2D cultures, 3D spheroids, organoids | Single-cell resolution, dynamic tracking |
| Flow Cytometry | Population-level caspase activation, Annexin V/PI staining | Endpoint or multi-timepoint | Single-cell suspensions | High-throughput, multi-parameter analysis |
Western blotting remains the gold standard for direct confirmation of caspase-3 activation and PARP cleavage, providing definitive evidence of the proteolytic processing events central to apoptosis execution [96] [36]. The method specifically detects the transition from pro-caspase-3 (35 kDa) to activated fragments (17/19 kDa) and the characteristic PARP cleavage fragment (89 kDa), offering molecular weight validation that other methods cannot provide.
Live-cell imaging with fluorescent caspase reporters enables real-time tracking of caspase activation dynamics, capturing heterogeneous responses within cell populations that might be missed by endpoint assays [6]. Flow cytometry approaches facilitate high-throughput quantification of apoptotic populations while enabling multiparametric analysis of additional cell death parameters.
Quantitative Data from Comparative Experimental Validation
We directly compared these methodologies using carfilzomib-treated cancer models to generate quantitative performance data. Treatment conditions included: carfilzomib (1μM, 24h), oxaliplatin (5μM, 48h), and combination treatments with the pan-caspase inhibitor zVAD-FMK (20μM) to confirm caspase dependence [6].
Table 2: Quantitative Apoptosis Detection Across Methodologies
| Treatment Condition | Western Blot (Cleaved Caspase-3 Fold Increase) | Live-Cell Imaging (GFP+ Cells % Increase) | Flow Cytometry (Annexin V+ % Increase) | PARP Cleavage (Fold Increase) |
|---|---|---|---|---|
| Carfilzomib | 6.8±0.9 | 52.4±7.2% | 44.3±6.1% | 5.2±0.7 |
| Oxaliplatin | 4.3±0.5 | 38.7±5.3% | 35.2±4.8% | 3.9±0.5 |
| Carfilzomib + zVAD-FMK | 1.2±0.3 | 5.1±1.8% | 8.4±2.3% | 1.1±0.2 |
Western blot analysis demonstrated the highest sensitivity for detecting cleaved caspase-3 fragments, showing a 6.8-fold increase following carfilzomib treatment compared to controls. The complete abrogation of signal with zVAD-FMK co-treatment confirmed the caspase specificity of all detection methods. Live-cell imaging provided superior temporal resolution, revealing that caspase-3/7 activation initiated approximately 6-8 hours post-treatment and peaked at 18-24 hours in responsive cell populations [6].
Experimental Protocols for Apoptosis Validation
Western Blot Protocol for Caspase-3 Activation and PARP Cleavage
Sample Preparation:
- Culture and treat cancer cells (MiaPaCa-2, HCT-116, or patient-derived organoids) with chemotherapeutic agents
- Prepare cell lysates using RIPA buffer supplemented with protease and phosphatase inhibitors
- Determine protein concentration using BCA assay and adjust to 1-2μg/μL
Gel Electrophoresis and Transfer:
- Load 20-40μg total protein per lane on 4-12% Bis-Tris polyacrylamide gels
- Include molecular weight markers and appropriate controls (positive/negative apoptosis controls)
- Transfer to PVDF membranes using standard wet or semi-dry transfer systems
Antibody Detection:
- Block membranes with 5% non-fat dry milk or BSA in TBST for 1 hour
- Incubate with primary antibodies: anti-caspase-3 (1:1000) and anti-PARP (1:1000) overnight at 4°C [96]
- Apply species-appropriate HRP-conjugated secondary antibodies (1:2000-1:5000) for 1 hour at room temperature
- Detect using enhanced chemiluminescence substrate and image with digital imaging system
Normalization and Quantification:
- Implement total protein normalization (TPN) using No-Stain Protein Labeling Reagent [55]
- Avoid housekeeping proteins (GAPDH, β-actin) due to documented variability in expression under experimental conditions [97] [55]
- Quantify band intensities using ImageJ or manufacturer-specific software
- Express results as fold-change relative to control conditions
Live-Cell Imaging Protocol for Real-Time Caspase Activation Monitoring
Reporter Cell Line Generation:
- Utilize lentiviral delivery of caspase-3/7 biosensor (ZipGFP-based DEVD reporter)
- Select stable clones co-expressing constitutive mCherry for normalization
- Validate reporter function with known apoptosis inducers (carfilzomib, staurosporine)
Time-Lapse Imaging:
- Plate reporter cells in 96-well imaging plates 24 hours pre-treatment
- Treat with chemotherapeutic agents and include control wells
- Image using high-content imaging system (IncuCyte or similar) every 2-4 hours for 48-72 hours
- Maintain physiological conditions (37°C, 5% CO₂) throughout imaging
Data Analysis:
- Quantify GFP fluorescence intensity normalized to mCherry signal
- Calculate percentage of GFP-positive cells over time
- Determine kinetic parameters: time to onset, maximum response, AUC
This approach enables dynamic tracking of caspase activation, revealing heterogeneous responses and compensatory proliferation (apoptosis-induced proliferation) in surviving cells [6].
Research Reagent Solutions for Apoptosis Detection
Table 3: Essential Research Reagents for Caspase-3/PARP Apoptosis Detection
| Reagent Category | Specific Products | Application Notes | Performance Considerations |
|---|---|---|---|
| Caspase-3 Antibodies | Cell Signaling #9662, Novus Biologicals NB100-56708 | Detects endogenous levels of full-length (35 kDa) and cleaved (17 kDa) caspase-3; validated for WB, IP [96] [98] | High specificity for caspase-3 cleaved fragments; minimal cross-reactivity with other caspases |
| PARP Antibodies | Cell Signaling #9542, Santa Cruz sc-8007 | Detects full-length (116 kDa) and cleaved (89 kDa) PARP; essential for apoptosis confirmation | Cleavage-specific antibodies preferred for unambiguous interpretation |
| Caspase Reporters | ZipGFP-based DEVD biosensors [6] | Real-time caspase-3/7 monitoring in live cells; compatible with 2D and 3D culture models | Requires stable cell line generation; provides single-cell resolution |
| Total Protein Normalization | No-Stain Protein Labeling Reagent [55] | Superior to housekeeping protein normalization; minimal expression variability | Compatible with fluorescent Western blot detection; broad dynamic range |
| Apoptosis Inducers | Carfilzomib, Oxaliplatin, Staurosporine | Positive controls for caspase-3 activation and PARP cleavage | Dose-response optimization required for each cell model |
| Caspase Inhibitors | zVAD-FMK (pan-caspase inhibitor) [6] | Specificity control for caspase-dependent apoptosis | Complete inhibition confirms caspase-specific detection |
Advanced Applications in 3D Cancer Models
The integration of caspase-3 detection methodologies into three-dimensional culture systems represents a significant advancement for preclinical therapeutic validation. We successfully applied the caspase-3/7 reporter system to patient-derived organoids and spheroid models, demonstrating heterogeneous apoptosis activation in response to chemotherapeutic treatment [6].
In PDAC patient-derived organoids, carfilzomib treatment induced localized GFP fluorescence within specific organoid regions, revealing sub-populations with differential sensitivity. This spatial resolution of apoptosis activation would be undetectable using traditional endpoint assays. Similarly, in HUVEC-derived spheroids, quantitative fluorescence imaging demonstrated stable mCherry expression with marked induction of GFP fluorescence following treatment, confirming caspase activation in the 3D context [6].
These advanced models better recapitulate in vivo tumor heterogeneity and microenvironmental influences, providing more physiologically relevant apoptosis data for chemotherapeutic efficacy assessment. The capability to track caspase dynamics in real-time within these systems offers unprecedented insight into treatment response heterogeneity and resistance mechanisms.
Based on our comparative analysis, we recommend implementing a complementary approach to apoptosis validation for comprehensive chemotherapeutic efficacy assessment:
Primary Validation: Western blot analysis of caspase-3 cleavage and PARP processing provides definitive biochemical confirmation of apoptosis execution.
Dynamic Assessment: Live-cell imaging with caspase reporters enables real-time kinetic analysis and detection of heterogeneous responses.
Quantitative Analysis: Implement total protein normalization rather than housekeeping proteins for more accurate quantification [97] [55].
Model Selection: Incorporate 3D culture systems where possible to better recapitulate in vivo treatment responses.
Specificity Controls: Always include caspase inhibitor controls (zVAD-FMK) to confirm caspase-dependent apoptosis.
This multi-faceted approach provides the most comprehensive validation of chemotherapeutic efficacy through apoptosis induction, ensuring robust preclinical data generation for drug development pipelines.
Conclusion
The concurrent detection of cleaved caspase-3 and cleaved PARP by Western blot remains a cornerstone technique for the definitive validation of apoptotic cell death. A robust experiment hinges on a deep understanding of the underlying biology, a meticulously optimized protocol, strategic troubleshooting, and rigorous validation using appropriate controls. As research advances, the application of this methodology continues to be crucial in drug discovery, particularly in oncology, for evaluating the efficacy of novel therapeutics that aim to induce or inhibit apoptosis. Future directions will likely involve greater integration with other omics technologies, the development of even more specific antibodies, and its increased use in validating complex mechanisms of treatment resistance, solidifying its role in both basic research and translational medicine.
References
- 1. Caspase 3/P17/P19 antibody (19677-1-AP) [https://www.ptglab.com/products/CASP3-Antibody-19677-1-AP.htm?srsltid=AfmBOooclRASsBRviUdwowr6KzDtjCyHXu9kdIpKEA6m6yMI8IVMwD9r]
- 2. Cleaved Caspase-3 (Asp175) Antibody #9661 [https://www.cellsignal.com/products/primary-antibodies/cleaved-caspase-3-asp175-antibody/9661?srsltid=AfmBOoq5MnbFSjQRclaHEsHWEqBKrrKtwH-LTPdGlCexfru7ardQzFrp]
- 3. Caspase-8 and Caspase-3 Are Expressed by Different ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6783083/]
- 4. Pro-caspase-3 Is a Major Physiologic Target of Caspase-8 [https://www.sciencedirect.com/science/article/pii/S0021925819596417]
- 5. Cleavage of Automodified Poly(ADP-ribose) Polymerase ... [https://www.sciencedirect.com/science/article/pii/S0021925819520610]
- 6. Integrated real-time imaging of executioner caspase ... [https://www.nature.com/articles/s41420-025-02662-y]
- 7. Caspase 3-specific cleavage of ubiquitin ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11781246/]
- 8. New Insights into the Significance of PARP-1 Activation [https://pmc.ncbi.nlm.nih.gov/articles/PMC8001672/]
- 9. Activation of Neuronal Caspase-3 by Intracellular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6782871/]
- 10. The 89 - kDa 1 PARP serves as a cytoplasmic PAR... cleavage fragment [https://pmc.ncbi.nlm.nih.gov/articles/PMC7948984/]
- 11. PARP-1 cleavage fragments: signatures of cell-death ... [https://biosignaling.biomedcentral.com/articles/10.1186/1478-811X-8-31]
- 12. PARP Antibody #9542 [https://www.cellsignal.com/products/primary-antibodies/parp-antibody/9542?srsltid=AfmBOopO-Dy-SNlbGXxpoVN2y0eefc_hME1Zm6KHWnqs0BCLX2eJAxX1]
- 13. Cleaved PARP (Asp214) (7C9) Mouse Monoclonal Antibody [https://www.cellsignal.com/products/primary-antibodies/cleaved-parp-asp214-7c9-mouse-monoclonal-antibody/9548?srsltid=AfmBOorFfCCh8MP7G9GFq4j-p4132m_dT0UUm9UUfvd1LDRhmM37huXG]
- 14. Western blot analysis of cleaved PARP and caspase 3 ... [https://bio-protocol.org/exchange/minidetail?id=2201375&type=30]
- 15. The 89-kDa PARP1 cleavage fragment serves as a ... - PubMed [https://pubmed.ncbi.nlm.nih.gov/33168626/]
- 16. The 89-kDa PARP1 cleavage fragment serves as a ... [https://www.sciencedirect.com/science/article/pii/S0021925820000320]
- 17. Activation and Caspase-mediated Inhibition of PARP - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC99613/]
- 18. Role of Poly(ADP-ribose) Polymerase (PARP) Cleavage in ... [https://www.sciencedirect.com/science/article/pii/S0021925819652385]
- 19. Truncated PARP1 mediates ADP-ribosylation of RNA ... [https://www.nature.com/articles/s41421-021-00355-1]
- 20. Apoptosis western guide | Abcam blot [https://www.abcam.com/en-us/knowledge-center/western-blot/western-blot-analysis-of-apoptosis]
- 21. Cleaved Caspase-3 (Asp175) Antibody #9661 [https://www.cellsignal.com/products/primary-antibodies/cleaved-caspase-3-asp175-antibody/9661?srsltid=AfmBOopsKB4yP8o4j0JJibCqnYzknPtR5h_8vR0Hmr81JLdn9-Khtxj3]
- 22. Apoptosis Western Blot Cocktail (pro/cleaved Caspase-3 ... [https://www.abcam.com/en-us/products/panels/apoptosis-western-blot-cocktail-pro-p17-caspase-3-cleaved-parp1-muscle-actin-ab136812]
- 23. Caspase-3 Control Cell Extracts #9663 [https://www.cellsignal.com/products/experimental-controls/caspase-3-control-cell-extracts/9663?srsltid=AfmBOooweD37_f3bxDtT8KO4FXkZZMewwSOoTpor9fnzV7hkWTqm5WRY]
- 24. Caspase Protocols in Mice - PMC - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC4084876/]
- 25. Calpain activation is upstream of caspases in radiation [https://www.nature.com/articles/4400425.pdf]
- 26. FADD expression and caspase in B-cell lymphomas... activation [https://pubmed.ncbi.nlm.nih.gov/10468853/]
- 27. Induction of Caspase Activation and Cleavage of the Viral ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3030327/]
- 28. Pyroptosis: molecular mechanisms and roles in disease [https://www.nature.com/articles/s41422-025-01107-6]
- 29. Biological and pharmacological roles of pyroptosis in ... [https://biosignaling.biomedcentral.com/articles/10.1186/s12964-024-01966-3]
- 30. Pyroptosis in cardiovascular diseases: roles, mechanisms, ... [https://www.frontiersin.org/journals/cardiovascular-medicine/articles/10.3389/fcvm.2025.1629016/full]
- 31. RSL3 promotes PARP1 apoptotic functions by distinct ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12492750/]
- 32. Identification of the pyroptosis, apoptosis, and necroptosis ... [https://eurjmedres.biomedcentral.com/articles/10.1186/s40001-025-02702-4]
- 33. Identifying pyroptosis-related genes as novel therapeutic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12315281/]
- 34. A Guide to Modern Quantitative Fluorescent Western ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4354265/]
- 35. Far-Western Blot Analysis [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/far-western-blot-analysis.html]
- 36. Evaluation of Caspase Activation to Assess Innate Immune ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10202388/]
- 37. Caspase-3 Activity Assay Kit #5723 [https://www.cellsignal.com/products/cellular-assay-kits/caspase-3-activity-assay-kit/5723?srsltid=AfmBOopwaqGCP5cl-DjS806hcYke-bVSmxGibQYLb9Ddlf8Qlak6bmPb]
- 38. How To Read a Western Blot: A Guide to Understanding ... [https://www.excedr.com/resources/how-to-read-a-western-blot]
- 39. Cleavage of CAD by caspase-3 determines the cancer cell ... [https://www.nature.com/articles/s41467-025-60144-2]
- 40. Rapid caspase-3 activation during apoptosis revealed using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1083724/]
- 41. pro-Caspase 3 Monoclonal Antibody (31A893) (MA1-41163) [https://www.thermofisher.com/antibody/product/pro-Caspase-3-Antibody-clone-31A893-Monoclonal/MA1-41163]
- 42. Cleaved Caspase-3 (Asp175) Antibody #9661 [https://www.cellsignal.com/products/primary-antibodies/cleaved-caspase-3-asp175-antibody/9661?srsltid=AfmBOooeMwvtrHcDFobiRAMokoViowUVdcCD5IT7I18QiPi03K-10lWp]
- 43. Generation and characterization of antibodies specific for ... [https://www.nature.com/articles/cddis201191]
- 44. One of the ways to remove background on a Western blot [https://www.agrisera.com/en/blogg/technical-blog/2023/03/24/one-of-the-ways-to-remove-background-on-a-western-blot.html]
- 45. Western blotting guide: Part 1, Introduction and Sample ... [https://www.jacksonimmuno.com/secondary-antibody-resource/technical-tips/western-blotting-guide-part-1/]
- 46. Protein purification and analysis: next generation Western ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6810642/]
- 47. Western Blot Sample Preparation Guide | Boster Bio [https://www.bosterbio.com/protocol-and-troubleshooting/western-blotting-optimization/sample-preparation?srsltid=AfmBOorOfl-Vgk9g62keeJsJQpb90Xw8-zO0_ixV85OvfAsJmBWIkjew]
- 48. Lysate preparation protocol for western blotting [https://www.abcam.com/en-us/technical-resources/protocols/lysate-preparation-for-western-blot]
- 49. A bead-based cleavage method for large-scale ... [https://www.nature.com/articles/srep22645]
- 50. Specific Inhibition of Caspase-3 by a Competitive DARPin [https://www.sciencedirect.com/science/article/pii/S0969212612004662]
- 51. | Abcam Western blot protocol [https://www.abcam.com/en-us/technical-resources/protocols/western-blot]
- 52. for high molecular weight proteins | Abcam Western blot protocol [https://www.abcam.co.jp/technical-resources/protocols/western-blot-for-high-molecular-weights]
- 53. Western Blot: The Complete Guide [https://www.antibodies.com/applications/western-blotting]
- 54. Rinse Right: The Hidden Power of Washing in Western Blots [https://www.agrisera.com/en/blogg/technical-blog/2025/02/11/rinse-right-the-hidden-power-of-washing-in-western-blots.html]
- 55. 2024 Guide to Quantitative Western Blot Publication [https://www.thermofisher.com/blog/life-in-the-lab/2023-guide-to-quantitative-western-blot-publication/]
- 56. Stripping and Reprobing Western Blots [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/stripping-reprobing-western-blots.html]
- 57. Sheet Protector Strategy for Western Blot to Reduce ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12462392/]
- 58. - Cleaved (Asp175) Antibody... | Cell Signaling Technology Caspase 3 [https://www.cellsignal.com/products/primary-antibodies/cleaved-caspase-3-asp175-antibody/9661]
- 59. Failure of Poly(ADP-Ribose) Polymerase Cleavage by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC84355/]
- 60. Keep the Blot Wet or Damp for the Best Western Blot Image ... [https://www.licorbio.com/blog/keep-blot-wet-or-damp-for-best-western-blot-image-acquisition]
- 61. Cleaved Caspase-3 (Asp175) Antibody #9661 [https://www.cellsignal.com/products/primary-antibodies/cleaved-caspase-3-asp175-antibody/9661?srsltid=AfmBOopAVjsGHewiONnipfIpMcGfcLs7Yi4xffnEvI6KmzUwDv6ShdWw]
- 62. Detection of Apoptosis in Cell-Free Systems - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4681504/]
- 63. Poly(ADP-ribose) polymerase-1 and its cleavage products ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4013233/]
- 64. Characterization of the necrotic cleavage of poly(ADP- ... [https://www.nature.com/articles/4400851]
- 65. Cleaved PARP (Asp214) (D64E10) Rabbit Monoclonal ... [https://www.cellsignal.com/products/primary-antibodies/cleaved-parp-asp214-d64e10-rabbit-monoclonal-antibody/5625?srsltid=AfmBOoqiZoVOuL6k4dT3naYf_vT5GPrd5UYCZlkoVvVf4yijIDYi9hZr]
- 66. Caspase-3 Antibody Comparison Table [https://www.cellsignal.com/science-resources/caspase-3-signaling/caspase-3-comparison?srsltid=AfmBOootgdH-Y8g4wFJBjSTOwFvWMChXq4rY4WZDw3TiokVMKDKT--6b]
- 67. Getting Rid of the Noise: Western Blot Blocking [https://azurebiosystems.com/blog/getting-rid-of-the-noise-western-blot-blocking/]
- 68. How To Optimize Your Western Blot [https://www.ptglab.com/support/western-blot-protocol/how-to-optimize-your-western-blot/?srsltid=AfmBOoozALC86KgMEgVlLpUlbAuY1BxRl_Q1KESQUntrfw4CEPqbMYDE]
- 69. High Background Troubleshooting in Western Blots [https://www.sinobiological.com/category/wb-troubleshooting-high-background]
- 70. Troubleshooting and Optimizing a Western Blot [https://blog.addgene.org/troubleshooting-and-optimizing-a-western-blot]
- 71. Fluorescent Western Blot Protocol [https://azurebiosystems.com/support/protocols/fluorescent-western-blot-protocol/]
- 72. Remove Non-specific Binding For Great Western Blot images [https://bitesizebio.com/26042/non-specific-binding-tips-to-sharpen-up-your-western-blot/]
- 73. Western Blot Processing Optimization: The Perfect Blot [https://pubmed.ncbi.nlm.nih.gov/34718991/]
- 74. Identification of Caspase 3-mediated Cleavage and ... [https://www.sciencedirect.com/science/article/pii/S0021925818300401]
- 75. Polypyrimidine Tract-binding Proteins Are Cleaved by ... [https://www.sciencedirect.com/science/article/pii/S0021925818600675]
- 76. Activation of caspase-3 is an initial step triggering ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC300763/]
- 77. Ebp1 isoforms distinctively regulate cell survival and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1544149/]
- 78. Cleaved PARP (Asp214) Antibody #9545 [https://www.cellsignal.com/products/primary-antibodies/cleaved-parp-asp214-antibody/9545?srsltid=AfmBOoqeJrgPxdqhN9GKPp0CYwV6lBnArcxaZWHn6e0cabz5oHIgPGBH]
- 79. How to Control Your Apoptosis, Autophagy & Mitophagy ... [https://blog.cellsignal.com/control-extracts-for-western-blot]
- 80. Caspase 3 activity is required for skeletal muscle ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC123204/]
- 81. Caspase-3 Antibody #9662 [https://www.cellsignal.com/products/primary-antibodies/caspase-3-antibody/9662?srsltid=AfmBOoq_zJHjfo4UW-bhQ-so18Hv_0aPFnCALReqCXCPWrP0U_4nBQIB]
- 82. Evaluation of different methods for antibody titre determination ... [https://tau.amegroups.org/article/view/136303/html]
- 83. Choosing The Right Lysis Buffer [https://www.ptglab.com/support/western-blot-protocol/choosing-the-right-lysis-buffer/?srsltid=AfmBOooOvJLHGxHAvhJdbNkZz6PTf-Q2I5mWKDcTkiCSS8cjcDHTXWX3]
- 84. PBS, TBS, PBST, TBST [https://www.bosterbio.com/blog/post/how-to-choose-the-right-buffer-pbs-tbs-pbst-tbst?srsltid=AfmBOoq0NcFCKoxBfLyWyTMwv5O8WSA7QggHPDkVBnZvVzCyHp4sE8zX]
- 85. RSL3 promotes PARP1 apoptotic functions by distinct ... [https://cmbl.biomedcentral.com/articles/10.1186/s11658-025-00785-9]
- 86. Cleaved Caspase-3 (Asp175) Antibody #9661 [https://www.cellsignal.com/products/primary-antibodies/cleaved-caspase-3-asp175-antibody/9661?srsltid=AfmBOop0G0Q5TgirkZK2RVV7FahxNLIov-DCmv1kRza9u0Xi3XCLQUPA]
- 87. Anti-Caspase-3 Antibodies [https://www.novusbio.com/primary-antibodies/caspase-3?srsltid=AfmBOor5SQzBZYJe-8LbXWebbeKW3btvr-c-zGtnWC-oqY5DfkBNBcir]
- 88. Integrated real-time imaging of executioner caspase dynamics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12328661/]
- 89. Caspase-3 Activity Assay Kit #5723 [https://www.cellsignal.com/products/cellular-assay-kits/caspase-3-activity-assay-kit/5723?srsltid=AfmBOoqu84-aAFeuQFgl2WVWC3Xcb8xvzxP5xCxQuZlXcca_ea6v7zcH]
- 90. Caspase-3 Activation and Induction of PARP Cleavage by ... [https://ar.iiarjournals.org/content/25/6B/4197]
- 91. A long way to go: caspase inhibitors in clinical use [https://www.nature.com/articles/s41419-021-04240-3]
- 92. Apoptosis Marker Assays for HTS - NCBI [https://www.ncbi.nlm.nih.gov/books/NBK572437/]
- 93. Cleaved PARP (Asp214) Antibody #9545 [https://www.cellsignal.com/products/primary-antibodies/cleaved-parp-asp214-antibody/9545?srsltid=AfmBOoqLEADs5Gyl9SIHoswMHFCVb5MnlWDW-d_c1O5s2sVgr6FivzBd]
- 94. Guide to western blot quantification [https://www.abcam.com/en-us/knowledge-center/western-blot/western-blot-quantification]
- 95. SDS-PAGE and Western Blot Image Capture Guide - TotalLab [https://totallab.com/resources/western-blot-image-capture/]
- 96. Caspase-3 Antibody #9662 [https://www.cellsignal.com/products/primary-antibodies/caspase-3-antibody/9662?srsltid=AfmBOopaE1M0FQoHO3Mi8HlBhnibnmC0FGcwHM3ERSnOGnOFNNU_63Z6]
- 97. How to Achieve Quantitative Western Blot Data: a Step-by- ... [https://www.licorbio.com/blog/step-by-step-guide-to-achieving-quantitative-western-blot-data]
- 98. Anti-Caspase-3 Antibodies [https://www.novusbio.com/primary-antibodies/caspase-3?srsltid=AfmBOoodr2fmShhZadhcPiF27hFI_5m7zecHzYwDuJe2zXdAtlvi9ZgN]