Validating Apoptotic Signaling: Advanced Analysis of PARP-1 and BCL-2 Family Protein Interplay
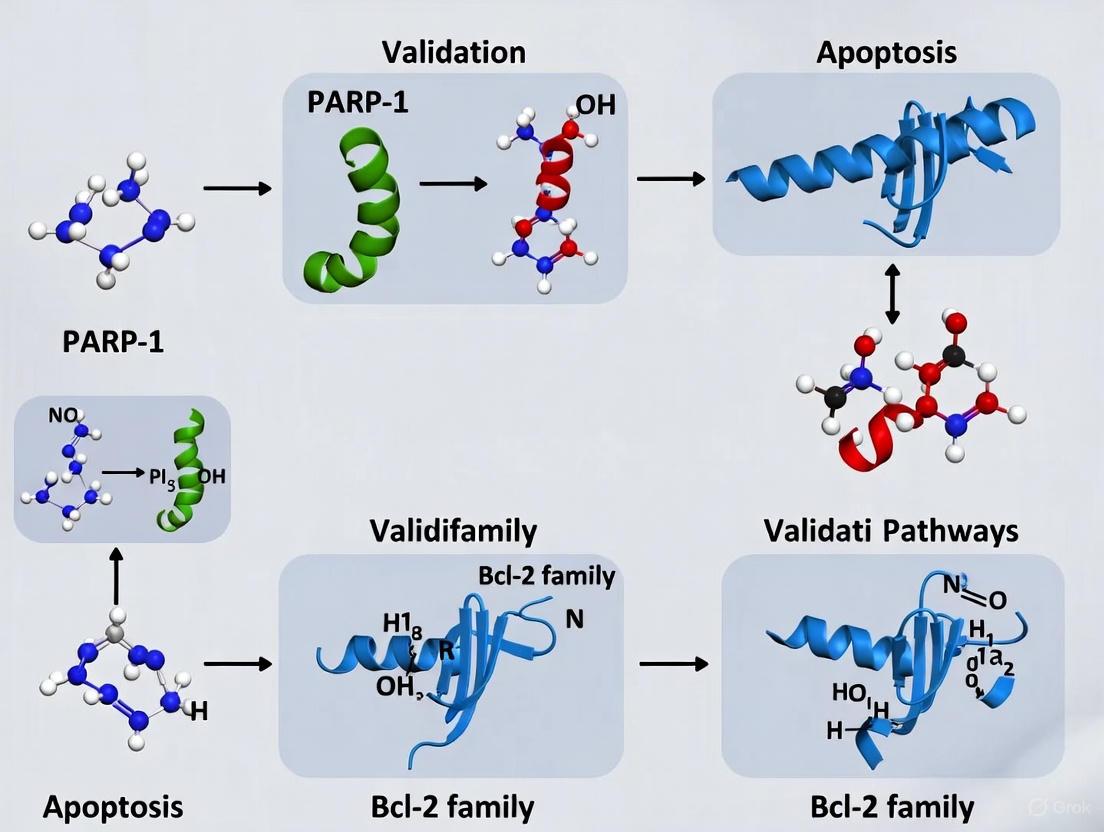
This article provides a comprehensive resource for researchers and drug development professionals on validating core apoptotic pathways.
Validating Apoptotic Signaling: Advanced Analysis of PARP-1 and BCL-2 Family Protein Interplay
Abstract
This article provides a comprehensive resource for researchers and drug development professionals on validating core apoptotic pathways. It covers the foundational biology of PARP-1 and BCL-2 family proteins, details established and emerging methodological approaches for their analysis, and offers troubleshooting strategies for common experimental challenges. A central focus is the validation and comparative analysis of crosstalk between apoptotic, ferroptotic, and other cell death pathways, with emphasis on translating these mechanisms into therapeutic strategies, particularly for overcoming treatment resistance in cancer.
Core Apoptotic Machinery: Defining the Roles of PARP-1 and BCL-2 Family Proteins
Apoptosis, a form of programmed cell death, is a highly regulated process essential for development, homeostasis, and the elimination of damaged or harmful cells [1]. It is characterized by distinct morphological changes, including cell shrinkage, chromatin condensation, membrane blebbing, and DNA fragmentation, culminating in the formation of apoptotic bodies that are phagocytosed by immune cells without triggering inflammation [1] [2]. The proper functioning of this process is critical, and its dysregulation is a hallmark of many diseases, particularly cancer [3]. Two principal signaling pathways—the intrinsic and extrinsic pathways—orchestrate apoptosis, both converging on the activation of a family of cysteine proteases known as caspases that execute the cell death program [4].
This article provides a comparative overview of these two pillars of apoptosis, framed within the context of validating these pathways through research on Bcl-2 family proteins and PARP-1. Analysis of these key regulators provides critical insights into cellular life-and-death decisions and mechanisms of treatment resistance in cancer.
Pathway Mechanisms: A Comparative Analysis
The intrinsic and extrinsic pathways of apoptosis are initiated by distinct stimuli and involve unique molecular components, yet they exhibit significant crosstalk and ultimately converge on a common execution phase.
The Intrinsic Apoptotic Pathway
The intrinsic pathway, also known as the mitochondrial pathway, is activated by internal cellular disturbances such as DNA damage, oxidative stress, hypoxia, survival factor deprivation, and oncogene activation [4] [1]. The central regulators of this pathway are the B-cell lymphoma 2 (Bcl-2) family of proteins, which govern mitochondrial outer membrane permeabilization (MOMP) [3]. Following cellular stress, the tumor suppressor protein p53 becomes activated and transcriptionally upregulates pro-apoptotic Bcl-2 family members like PUMA, Noxa, and Bax [4]. This disrupts the balance between pro-apoptotic and anti-apoptotic signals.
The subsequent permeabilization of the mitochondrial membrane leads to the release of several pro-apoptotic proteins into the cytosol, including cytochrome c, SMAC (Second Mitochondria-derived Activator of Caspases), and AIF (Apoptosis-Inducing Factor) [4]. Cytochrome c, together with apoptotic protease-activating factor-1 (APAF-1) and dATP, forms a complex called the apoptosome. The apoptosome then recruits and activates initiator caspase-9, which in turn cleaves and activates effector caspases-3, -6, and -7, leading to the systematic dismantling of the cell [1] [2].
The Extrinsic Apoptotic Pathway
The extrinsic pathway, or death receptor pathway, is initiated outside the cell through the engagement of death receptors (DRs) on the plasma membrane by specific extracellular ligands [3] [4]. Key death receptors include Fas (CD95), TNFR1 (Tumor Necrosis Factor Receptor 1), and TRAIL receptors (DR4/DR5) [4] [5]. These receptors belong to the tumor necrosis factor (TNF) receptor superfamily and are characterized by a conserved intracellular death domain (DD) [5].
Upon ligand binding (e.g., FasL binding to Fas), the receptors trimerize and recruit adapter proteins such as FADD (Fas-Associated protein with Death Domain) via death domain interactions. FADD then recruits initiator procaspase-8 via death effector domain (DED) interactions, forming the Death-Inducing Signaling Complex (DISC) [4] [1]. Within the DISC, caspase-8 undergoes auto-proteolytic activation. Active caspase-8 then directly cleaves and activates effector caspases-3 and -7, committing the cell to apoptosis [2].
Critical Crosstalk and Integration
A critical point of integration between the two pathways is the Bcl-2 family protein Bid. In some cell types (designated Type II cells), the amount of caspase-8 activated at the DISC is insufficient to fully activate effector caspases. In this scenario, caspase-8 cleaves Bid into its active truncated form (tBid), which translocates to the mitochondria and amplifies the apoptotic signal by engaging the intrinsic pathway, leading to cytochrome c release and apoptosome formation [4] [1].
Table 1: Comparative Overview of Intrinsic and Extrinsic Apoptotic Pathways
| Feature | Intrinsic Pathway | Extrinsic Pathway |
|---|---|---|
| Also Known As | Mitochondrial Pathway | Death Receptor Pathway |
| Initiating Stimulus | Internal stress (DNA damage, oxidative stress, hypoxia) [4] | External ligand binding (FasL, TRAIL, TNF-α) [3] [1] |
| Key Initiators | p53, Bcl-2 protein family, mitochondrial stress [1] | Death Receptors (Fas, DR4/DR5, TNFR1) [5] |
| Key Signaling Complex | Apoptosome (Cytochrome c, APAF-1, Caspase-9) [1] | DISC (Death Receptor, FADD, Caspase-8) [1] |
| Initiator Caspase | Caspase-9 [2] | Caspase-8, Caspase-10 [2] |
| Key Regulatory Proteins | Bcl-2, Bcl-xL (anti-apoptotic); Bax, Bak, Bid, BIM (pro-apoptotic) [3] [1] | c-FLIP (inhibits DISC), FADD (promotes DISC) [3] |
| Mitochondrial Involvement | Central (MOMP required) [3] | Variable (Occurs in Type II cells via Bid cleavage) [4] |
Research Focus: Validating Pathways via Bcl-2 and PARP-1
A deeper understanding of apoptosis is achieved by investigating key regulatory proteins and their complex interactions. The Bcl-2 family and the enzyme PARP-1 serve as critical focal points for experimental validation of apoptotic signaling.
Bcl-2 Family Proteins as Central Gatekeepers
The Bcl-2 protein family is the definitive regulator of the intrinsic apoptotic pathway. These proteins are categorized into three groups based on their function and Bcl-2 homology (BH) domains:
- Anti-apoptotic proteins (e.g., Bcl-2, Bcl-xL, Mcl-1): They possess multiple BH domains and preserve mitochondrial integrity by binding and neutralizing pro-apoptotic family members [3] [1].
- Pro-apoptotic effectors (e.g., Bax, Bak): These proteins, upon activation, oligomerize to form pores in the mitochondrial outer membrane, leading to MOMP and the release of cytochrome c [3].
- BH3-only proteins (e.g., Bid, BIM, Bad, PUMA): These are sensors of cellular stress and initiate apoptosis by either directly activating Bax/Bak or by neutralizing anti-apoptotic Bcl-2 proteins [3] [1].
The development of BH3 mimetics, such as venetoclax (ABT-199), represents a successful translational application of this knowledge. Venetoclax is a small molecule that specifically binds to Bcl-2, displacing pro-apoptotic proteins like BIM and thereby triggering apoptosis in cancer cells dependent on Bcl-2 for survival [3].
PARP-1 in DNA Damage Response and Cell Death
PARP-1 is a nuclear enzyme that is rapidly activated in response to DNA single-strand breaks, playing a key role in the DNA damage response and repair [6]. However, upon excessive DNA damage, PARP-1 overactivation can lead to a caspase-independent form of cell death by depleting cellular NAD+ and ATP levels [6].
Research has revealed a novel, non-canonical interaction between Bcl-2 and PARP-1. Bcl-2, which can localize to the nucleus in certain cancer cells, was found to bind to PARP-1 and suppress its enzymatic activity, thereby inhibiting DNA repair [6]. When the BH3 mimetic ABT-737 is introduced, it displaces PARP-1 from Bcl-2, restoring PARP-1 activity and DNA repair function, and can promote non-apoptotic cell death. This mechanism persists even in cells resistant to the apoptotic effects of ABT-737, suggesting a therapeutically exploitable vulnerability [6].
Table 2: Key Research Reagent Solutions for Apoptosis Research
| Research Reagent / Tool | Primary Function / Target | Research Application |
|---|---|---|
| Venetoclax (ABT-199) | BCL-2 specific BH3 mimetic [3] | Induces intrinsic apoptosis in BCL-2 dependent cancers (e.g., CLL, AML) [3] |
| ABT-737 | BH3 mimetic (Bcl-2, Bcl-xL, Bcl-w inhibitor) [6] | Displaces PARP1 from BCL2; used to study novel non-apoptotic cell death [6] |
| Recombinant Human TRAIL (rhTRAIL) | Agonist for DR4/DR5 Death Receptors [3] | Activates the extrinsic apoptosis pathway selectively in cancer cells [3] |
| TLY012 | PEGylated recombinant human TRAIL [3] | Second-generation TRAIL with prolonged half-life; used to overcome limitations of first-gen TRAIL [3] |
| ONC201 | TRAIL and DR5-inducing compound [3] | Used in combination with TLY012 to overcome TRAIL resistance in models like pancreatic cancer [3] |
| Caspase Inhibitors (e.g., Z-VAD-FMK) | Pan-caspase inhibitor | Determines caspase-dependency of cell death in experimental models [2] |
| PARP Inhibitors (e.g., ABT-888, Olaparib) | Inhibits PARP1 enzymatic activity [6] | Studies synthetic lethality in BRCA-deficient cancers and explores non-apoptotic cell death [6] |
Experimental Protocols for Pathway Validation
Protocol 1: Assessing BCL-2 Dependence with BH3 Profiling
Objective: To determine the dependence of cancer cells on anti-apoptotic Bcl-2 family proteins for survival, a technique known as BH3 profiling is employed [6]. Methodology:
- Cell Preparation: Permeabilize isolated tumor cells or primary cancer cells to allow intracellular access to synthetic BH3 peptides.
- BH3 Peptide Exposure: Expose the permeabilized cells to a panel of synthetic peptides derived from the BH3 domains of different BH3-only proteins (e.g., BIM, BAD, PUMA). A negative control peptide is also used.
- MOMP Measurement: Measure the loss of mitochondrial membrane potential (ΔΨm) using a fluorescent dye like JC-1 or Tetramethylrhodamine (TMRM). Alternatively, cytochrome c release can be quantified by immunofluorescence.
- Data Analysis: Cells dependent on Bcl-2 will show significant MOMP in response to the BAD BH3 peptide, as BAD selectively antagonizes Bcl-2 and Bcl-xL. A response to BIM peptide indicates a high level of overall "priming" for death, meaning the cells are close to the apoptotic threshold.
Protocol 2: Evaluating the BCL-2/PARP-1 Interaction
Objective: To investigate the novel interaction between BCL-2 and PARP-1 and its functional consequences, as described in [6]. Methodology:
- Co-Immunoprecipitation (Co-IP):
- Lyse cells (e.g., DLBCL cell lines known to overexpress BCL-2).
- Incubate the lysate with an antibody against BCL-2 or a control IgG.
- Precipitate the antibody-protein complex using Protein A/G beads.
- Analyze the immunoprecipitate by Western blotting using an anti-PARP1 antibody to confirm the physical interaction.
- Functional PARP1 Activity Assay:
- Treat cells with a DNA-damaging agent like N-Methyl-N′-nitro-N-nitrosoguanidine (MNNG).
- In a fraction of the treated cells, displace BCL-2 from PARP1 using ABT-737.
- Use a PARP ELISA kit that measures the incorporation of ADP-ribose units onto histone proteins to quantify PARP1 enzymatic activity in the different treatment groups.
- Assessment of DNA Repair:
- Use the Alkaline Comet Assay to assess DNA strand breaks.
- Treat cells with MNNG and then with ABT-737 or a PARP inhibitor (ABT-888).
- After a repair period, embed cells in agarose, lyse, and subject to electrophoresis. DNA damage appears as a "comet tail"; longer tails indicate more unrepaired DNA damage.
Signaling Pathway Diagrams
Diagram 1: The Intrinsic Apoptotic Pathway. Internal cellular stress triggers a signaling cascade that converges on the mitochondria, leading to caspase activation.
Diagram 2: The Extrinsic Apoptotic Pathway and Crosstalk. Ligation of death receptors initiates a caspase cascade. In some cells, signal amplification occurs via cleavage of Bid (tBid), which engages the mitochondrial pathway.
Diagram 3: BCL-2 and PARP-1 Interaction Model. Nuclear BCL-2 can bind to and inhibit PARP-1. BH3 mimetics can disrupt this interaction, restoring PARP-1 activity and leading to an alternative cell death pathway.
Poly(ADP-ribose) polymerase-1 (PARP-1) is a ubiquitous nuclear enzyme that serves as a critical molecular switch governing cellular fate in response to stress signals. As the most abundant member of the PARP family, PARP-1 processes diverse stress signals and directs cells toward specific outcomes—DNA repair versus cell death—based on the type and intensity of the stress stimulus [7]. This multifaceted protein exhibits a dual nature in cellular homeostasis: it maintains genomic integrity through its canonical DNA repair function while simultaneously acting as a central executioner in multiple cell death pathways when damage proves irreparable. PARP-1's functions are intimately tied to nuclear NAD+ metabolism and the broader metabolic profile of the cell, positioning it as a key regulator of cellular energy management during stress responses [7]. The enzyme's activity is particularly crucial in pathological conditions including cancer, neurodegenerative diseases, and metabolic dysregulation, making it both a biomarker and therapeutic target of considerable interest. This review comprehensively examines PARP-1's structural domains, its canonical DNA repair functions, and the molecular consequences of its caspase-mediated cleavage, with particular emphasis on how these processes inform current understanding of apoptotic pathway validation in the context of Bcl-2 family protein research.
Structural Organization and DNA Repair Functions of PARP-1
Domain Architecture and Activation Mechanism
PARP-1 is a 116-kDa protein consisting of three principal functional domains that dictate its cellular functions [8] [7]. The DNA-binding domain (DBD) located at the N-terminus contains three zinc-finger motifs (Zn1, Zn2, and Zn3) that recognize and bind to various DNA lesions, including single-strand breaks, double-strand breaks, and cruciform structures [7] [9]. This domain also harbors a nuclear localization signal (NLS) that ensures PARP-1's nuclear retention. The central automodification domain (AMD) contains a BRCT (BRCA1 C-terminal) phosphopeptide-binding motif that facilitates protein-protein interactions and serves as the primary acceptor site for auto-poly(ADP-ribosyl)ation [7]. The C-terminal catalytic domain houses a conserved "PARP signature" sequence essential for synthesizing poly(ADP-ribose) (PAR) polymers using NAD+ as a substrate [7]. Within this domain, a tryptophan-, glycine-, and arginine-rich (WGR) region mediates DNA-dependent allosteric activation [7].
PARP-1 activation follows an elegant molecular mechanism: upon detecting DNA damage through its zinc finger domains, the enzyme undergoes conformational changes that expose its catalytic active site, enabling the covalent attachment of ADP-ribose units from NAD+ to target proteins, including PARP-1 itself [8] [7]. This auto-poly(ADP-ribosyl)ation creates a negatively charged PAR meshwork that serves as a docking platform for DNA repair proteins such as XRCC1, thereby facilitating the recruitment of DNA repair machinery to damage sites [8] [9]. The PAR polymers synthesized by PARP-1 are rapidly turned over in the cell, with a half-life on the order of minutes, ensuring dynamic regulation of this signaling pathway [7].
PARP-1 in DNA Damage Response Pathways
PARP-1 plays a central role in multiple DNA repair mechanisms, with its most established function in base excision repair (BER)—the primary pathway for correcting single-strand breaks [9]. PARP-1-deficient cells demonstrate impaired BER activity, highlighting its essential role in this process [9]. Beyond BER, PARP-1 contributes to nucleotide excision repair, single-strand break repair mediated by DNA ligase III and XRCC1, and double-strand break repair through an alternate non-homologous end-joining pathway with DNA ligase III [9]. Interestingly, PARP-1's enzymatic activity is not always required for its repair functions, as overexpression of its DNA-binding domain alone can influence double-strand break repair efficiency [9].
The following diagram illustrates PARP-1's domain structure and its transition from DNA repair to apoptosis:
Table 1: PARP-1 Fragments Generated by Caspase-Mediated Cleavage
| Fragment | Domains Contained | Cellular Localization | Primary Functions | Impact on Cell Fate |
|---|---|---|---|---|
| 24-kDa | DNA-binding domain (Zn1, Zn2), NLS | Nuclear | Irreversibly binds DNA breaks; acts as trans-dominant inhibitor of DNA repair [8] [9] | Prevents DNA repair, conserves ATP, facilitates apoptotic dismantling |
| 89-kDa | Third zinc finger, BRCT domain, WGR domain, catalytic domain | Cytoplasmic (after translocation) | Serves as PAR carrier; binds AIF; can ADP-ribosylate cytoplasmic targets [8] [10] | Amplifies death signals via parthanatos; activates innate immune response |
Caspase-Mediated Cleavage of PARP-1: A Hallmark of Apoptosis
Proteolytic Processing and Fragment Generation
During caspase-dependent apoptosis, PARP-1 undergoes specific proteolytic cleavage by executioner caspases-3 and -7, which recognize a conserved DEVD motif (Asp-Glu-Val-Asp) located between the DNA-binding domain and the automodification domain [8] [9]. This cleavage event generates two prominent fragments: a 24-kDa N-terminal fragment containing the DNA-binding domain and nuclear localization signal, and an 89-kDa C-terminal fragment encompassing the automodification and catalytic domains [8]. The 24-kDa fragment remains tightly associated with DNA damage sites in the nucleus, where it acts as a trans-dominant inhibitor of DNA repair by blocking access to DNA breaks [8] [9]. Meanwhile, the 89-kDa fragment translocates to the cytoplasm, where it can engage in non-canonical functions that amplify cell death signaling [8].
This cleavage event serves as a biochemical hallmark of apoptosis and represents a crucial point of commitment to cell death. By dismantling PARP-1's DNA repair capacity, the cell ensures that damaged DNA is not redundantly repaired while apoptotic processes are underway, thereby preventing potentially mutagenic survival. The 89-kDa fragment's translocation to the cytoplasm represents a significant shift in PARP-1's functional paradigm, transforming it from a nuclear DNA guardian to a cytoplasmic death amplifier.
Non-Canonical Functions of the 89-kDa PARP-1 Fragment
Recent research has revealed that the 89-kDa PARP-1 fragment (tPARP1) possesses unique biological activities beyond its role as a caspase substrate. Once in the cytoplasm, tPARP1 can serve as a carrier for poly(ADP-ribose) (PAR) polymers, facilitating their translocation from the nucleus [8]. These PAR polymers bound to tPARP1 can interact with apoptosis-inducing factor (AIF), leading to AIF's release from mitochondria and subsequent translocation to the nucleus, where it contributes to caspase-independent DNA fragmentation—a pathway known as parthanatos [8].
Strikingly, tPARP1 also interacts with the RNA polymerase III (Pol III) complex in the cytoplasm, which it can mono-ADP-ribosylate during poly(dA-dT)-stimulated apoptosis [10]. This modification enhances Pol III-mediated detection of cytoplasmic DNA, promoting IFN-β production and amplifying apoptotic signaling in response to pathogenic insults [10]. This function appears to depend on the BRCT domain of tPARP1, which mediates protein-protein interactions with Pol III subunits [10]. These findings reveal an elegant evolutionary conservation, as PARP-1 orthologs in lower organisms naturally lack the N-terminal zinc fingers cleaved by caspases in higher organisms, suggesting that tPARP1 represents a functional ancestral form of the enzyme [10].
Experimental Approaches for PARP-1 Function Analysis
Methodologies for Detecting PARP-1 Cleavage and Localization
The analysis of PARP-1 cleavage and function employs a multifaceted experimental approach combining molecular, biochemical, and cell biological techniques. Western blot analysis remains the gold standard for detecting PARP-1 cleavage fragments, using antibodies that recognize either the full-length protein (116-kDa) or specific fragments (89-kDa and 24-kDa) [8]. For localization studies, subcellular fractionation followed by immunoblotting provides quantitative assessment of PARP-1 and its fragments distribution, while immunofluorescence and confocal microscopy offer spatial resolution of these localization changes in fixed cells [8] [6].
Functional PARP-1 activity can be measured using enzyme-linked immunosorbent assays (ELISAs) that quantify PAR synthesis on immobilized histone substrates in response to DNA damage [6]. Viability assays under PARP inhibition (using pharmacological inhibitors like PJ34, ABT-888, or AG14361) or PARP-1 knockdown (via shRNA) help delineate PARP-1's contribution to cell survival versus death pathways [8] [11]. For DNA repair assessment, the alkaline comet assay quantitatively measures DNA strand breaks under different experimental conditions, revealing PARP-1's role in DNA damage resolution [6].
The following workflow outlines a comprehensive experimental approach for analyzing PARP-1 cleavage and function:
Table 2: Key Experimental Reagents for PARP-1 and Apoptosis Research
| Reagent Category | Specific Examples | Primary Research Application | Mechanistic Insight |
|---|---|---|---|
| PARP Inhibitors | PJ34, ABT-888, AG14361 | Distinguish PARP-1-dependent cell death; chemopotentiation [8] [11] | Suppress PAR formation; block parthanatos; conserve cellular energy |
| Caspase Inhibitors | zVAD-fmk | Validate caspase-dependent apoptosis; block PARP-1 cleavage [8] | Prevent 89-kDa fragment generation; distinguish apoptosis from other death forms |
| Apoptosis Inducers | Staurosporine, Actinomycin D, Etoposide | Activate intrinsic apoptotic pathway; trigger caspase cascade [8] | Initiate mitochondrial outer membrane permeabilization; caspase activation |
| BH3 Mimetics | ABT-737, Venetoclax | Disrupt BCL2-PARP1 interaction; target anti-apoptotic BCL2 proteins [6] [12] | Release PARP1 from BCL2 inhibition; promote non-apoptotic death |
| DNA Damage Agents | MNNG, H₂O₂, Ionizing Radiation | Activate PARP-1 DNA repair function; induce parthanatos at high doses [8] [6] | Generate DNA strand breaks; activate PARP1 catalytic activity |
Quantitative Assessment of PARP-1-Mediated Cell Death
The functional consequences of PARP-1 activation and cleavage can be quantified through multiple complementary approaches. Cell viability assays (MTT, CellTiter-Glo) measure metabolic activity and ATP levels, reflecting cellular energy status during PARP-1-mediated cell death [6]. Flow cytometry with Annexin V/propidium iodide staining distinguishes early apoptotic, late apoptotic, and necrotic populations, allowing correlation with PARP-1 cleavage status [8] [13]. NAD+ and ATP quantification provides direct assessment of metabolic collapse associated with excessive PARP-1 activation [6].
For DNA damage assessment, the alkaline comet assay quantitatively measures DNA strand breaks, while γH2AX immunostaining detects double-strand break formation [6] [13]. PARP-1 enzymatic activity can be directly measured using ELISA-based methods that quantify PAR synthesis on histone substrates, with or without BCL2 co-incubation to study this regulatory interaction [6]. Advanced techniques like BH3 profiling can assess mitochondrial priming and apoptotic propensity in cells with different PARP-1 status, connecting PARP-1 function to BCL2 family regulation [6].
Interplay Between PARP-1 and BCL2 Family Proteins
BCL2-PARP1 Interaction: A Novel Regulatory Axis
Emerging research has revealed a direct molecular interaction between PARP-1 and the anti-apoptotic protein BCL2, creating a novel regulatory axis that influences cellular fate decisions [6] [14]. This interaction occurs within the nucleus of various tumor cell lines, including diffuse large B-cell lymphoma (DLBCL) cells harboring t(14;18) translocation [6]. BCL2 binding suppresses PARP-1 enzymatic activity and inhibits PARP-1-dependent DNA repair, creating a functional link between the key regulators of apoptosis and DNA damage response [6].
The BH3 mimetic ABT-737 can disrupt the BCL2-PARP1 interaction in a dose-dependent manner, restoring PARP1 activity and DNA repair capacity while promoting non-apoptotic cell death [6]. This form of cell death remains effective even in apoptosis-resistant cells that have upregulated other anti-apoptotic BCL2 family members, suggesting therapeutic potential for targeting this interaction in treatment-resistant malignancies [6]. Conversely, ectopic BCL2 expression kills PARP inhibitor-sensitive breast and lung cancer cells, effects reversible by ABT-737 treatment, highlighting the complex interplay between these pathways [6].
Therapeutic Implications and Future Directions
The molecular interplay between PARP-1 and BCL2 family proteins presents promising therapeutic opportunities, particularly for malignancies resistant to conventional apoptosis-based treatments. The development of BH3 mimetics like venetoclax (ABT-199) has transformed treatment for hematologic malignancies, showing remarkable efficacy in chronic lymphocytic leukemia and other BCL2-dependent cancers [12]. These agents target the hydrophobic groove of anti-apoptotic BCL2 proteins, displacing pro-apoptotic partners and, as recently discovered, potentially releasing PARP1 to execute alternative cell death programs [6] [12].
Combination approaches exploiting both PARP inhibition and BCL2 targeting hold particular promise. In PARP inhibitor-resistant settings, agents like RSL3 (a ferroptosis inducer) can promote caspase-dependent PARP1 cleavage and simultaneously reduce full-length PARP1 through inhibition of METTL3-mediated m6A modification, effectively bypassing resistance mechanisms [13]. Meanwhile, novel targeting strategies including proteolysis targeting chimeras (PROTACs) and antibody-drug conjugates (ADCs) offer potential for more selective inhibition of specific BCL2 family members with reduced toxicity [12].
The following table summarizes key experimental findings linking PARP-1 and BCL2 family proteins:
Table 3: Experimental Evidence for PARP-1 and BCL2 Family Interplay
| Experimental System | Key Intervention | Observed Outcome | Interpretation |
|---|---|---|---|
| DLBCL cell lines [6] | ABT-737 treatment | Displacement of PARP1 from BCL2; restored PARP1 activity; non-apoptotic death | BCL2 directly suppresses PARP1 function; BH3 mimetics release this inhibition |
| PARP inhibitor-sensitive cancer cells [6] | Ectopic BCL2 expression | 90-100% reduction in survival; reversible by ABT-737 | BCL2 overexpression creates synthetic lethality with PARP inhibition |
| Stroma-associated CLL cells [6] | MNNG + ABT-737/ABT-888 | Overcame stroma-mediated resistance to apoptosis | PARP1-dependent non-apoptotic death bypasses conventional resistance mechanisms |
| PARPi-resistant tumor models [13] | RSL3 treatment | Caspase-dependent PARP1 cleavage; reduced full-length PARP1; apoptosis restoration | Dual-pathway targeting overcomes PARPi resistance via epitranscriptomic regulation |
PARP-1 stands as a critical molecular decision-maker at the intersection of DNA repair and cell death pathways. Its canonical function in DNA damage response ensures genomic integrity, while its caspase-mediated cleavage represents a commitment to apoptotic execution. The generation of distinct PARP-1 fragments with unique functions—the nuclear 24-kDa dominant-negative inhibitor and cytoplasmic 89-kDPE1 death amplifier—illustrates the biochemical sophistication of cell fate regulation. The newly discovered interplay between PARP-1 and BCL2 family proteins expands our understanding of apoptotic regulation and reveals novel therapeutic opportunities for targeting non-apoptotic cell death in treatment-resistant malignancies. As research continues to unravel the complexities of PARP-1 functions in different cellular contexts, the integration of PARP-targeting strategies with BCL2 family inhibition represents a promising frontier in cancer therapeutics, particularly for malignancies that have evolved resistance to conventional apoptosis-based treatments.
The B-cell lymphoma 2 (BCL-2) family of proteins represents a crucial class of evolutionarily conserved regulators that determine cellular life-or-death decisions through control of the intrinsic apoptosis pathway [15] [12]. These proteins functionally comprise both inhibitors and inducers of programmed cell death, working in concert to regulate the process by which mitochondria contribute to cell death [15]. The discovery of BCL-2 in 1984 as the gene involved in the t(14;18) chromosomal translocation in follicular lymphoma marked a pivotal moment in cancer biology, representing the first example of an oncogene that promotes cancer by blocking cell death rather than stimulating proliferation [15] [12]. This foundational finding established the critical importance of regulated cell death in maintaining tissue homeostasis and preventing malignancy.
The BCL-2 family members are structurally characterized by the presence of BCL-2 homology (BH) domains, numbered BH1-BH4, which mediate complex interactions between family members [16] [17]. These proteins primarily localize to the outer mitochondrial membrane (OMM) where they control mitochondrial outer membrane permeabilization (MOMP), the key step in intrinsic apoptosis that leads to cytochrome c release and subsequent caspase activation [12] [16]. Beyond their canonical role in apoptosis regulation, emerging research has revealed non-apoptotic functions for BCL-2 family proteins in processes including neuronal activity, autophagy, calcium handling, and mitochondrial dynamics [15]. The delicate balance between pro- and anti-apoptotic BCL-2 family members serves as a critical rheostat for cellular survival, with dysregulation contributing to various pathologies including cancer, neurodegenerative diseases, and autoimmune disorders [12].
Structural and Functional Classification of BCL-2 Family Members
Hierarchical Organization Based on Structure and Function
The BCL-2 family is organized into three principal functional subgroups based on their structural characteristics and biological effects on apoptosis. This hierarchical classification system provides a framework for understanding the complex interactions that govern cellular fate decisions.
Table 1: Functional Classification of BCL-2 Family Proteins
| Functional Group | Representative Members | BH Domains | Primary Function |
|---|---|---|---|
| Anti-apoptotic | BCL-2, BCL-xL, MCL-1, BCL-w | BH1-BH4 | Inhibit MOMP by sequestering pro-apoptotic members |
| Multi-domain Pro-apoptotic | BAX, BAK, BOK | BH1-BH3 | Direct mediators of MOMP |
| BH3-only Pro-apoptotic | BIM, BID, BAD, PUMA, NOXA | BH3 only | Initiators/sensitizers that antagonize anti-apoptotic members |
Anti-apoptotic Proteins
The anti-apoptotic members, including BCL-2, BCL-xL, MCL-1, BCL-w, BFL-1/A1, and BCL-B, characteristically contain four BH domains (BH1-BH4) and serve as guardians of mitochondrial integrity [12] [16]. These proteins display a conserved three-dimensional structure featuring a hydrophobic groove formed by their BH1, BH2, and BH3 domains that serves as the primary interaction site for binding the BH3 domains of pro-apoptotic family members [17]. Structurally, they consist of two central hydrophobic α-helices surrounded by six or seven amphipathic α-helices, adopting a fold remarkably similar to the pore-forming domains of bacterial toxins [17]. This structural similarity enables their ability to form ion channels in artificial membranes, though the physiological relevance of this activity remains under investigation [17]. Their localization to the OMM, facilitated by a C-terminal transmembrane domain, positions them strategically to prevent MOMP and cytochrome c release [12] [16].
Pro-apoptotic Effectors
The multi-domain pro-apoptotic effectors, primarily BAX and BAK, contain BH1-BH3 domains and serve as the direct executioners of MOMP [18] [12]. In healthy cells, BAX predominantly resides in the cytoplasm in an inactive conformation, while BAK is integrated into the OMM [12]. Upon activation by BH3-only proteins, both undergo conformational changes that lead to their oligomerization and formation of pores in the OMM, permitting the release of cytochrome c and other apoptogenic factors into the cytosol [15] [12]. The three-dimensional structures of BAX and BID reveal a similar fold to their anti-apoptotic counterparts, though the hydrophobic groove of BID is neither as long nor as deep as that found in BCL-xL or BCL-2 [17]. The functional activity of these effectors is tightly controlled through interactions with anti-apoptotic proteins, providing a critical regulatory checkpoint in apoptosis initiation.
BH3-only Proteins
The BH3-only proteins, including BIM, BID, BAD, PUMA, NOXA, BIK, BMF, and HRK, represent the sentinels of cellular stress that initiate the apoptotic cascade [15] [12]. These proteins share only the BH3 domain, which is both necessary and sufficient for their pro-apoptotic function [16]. They can be further subdivided into "activators" (such as BIM and tBID) that directly engage and activate BAX/BAK, and "sensitizers" (such as BAD and NOXA) that indirectly promote apoptosis by neutralizing specific anti-apoptotic proteins [15]. Most BH3-only proteins are intrinsically disordered, which may facilitate their dynamic functions and rapid activation in response to diverse death signals [15]. The BH3 domain forms an amphipathic α-helix that binds into the hydrophobic groove of anti-apoptotic BCL-2 proteins, with binding specificity determined by complementary interactions within discrete hydrophobic pockets [12].
Quantitative Expression Profiles Across Tissues and Cancers
The expression patterns of BCL-2 family members vary significantly across different tissue types and cancer subtypes, contributing to tissue-specific apoptotic sensitivity and therapeutic responses.
Table 2: Expression Patterns and Cancer Associations of Key BCL-2 Family Members
| Protein | Expression in Normal Tissues | Cancer Associations | Therapeutic Targeting |
|---|---|---|---|
| BCL-2 | Lymphoid tissues, neuronal stem cells | Follicular lymphoma, CLL, ALL | Venetoclax (FDA-approved) |
| BCL-xL | Wide distribution, hematopoietic cells | Solid tumors, platelet survival | Navitoclax (thrombocytopenia limitation) |
| MCL-1 | Wide distribution, essential for development | Myeloma, AML, solid tumors | Clinical development (cardiac toxicity concerns) |
| BAX | Wide distribution | Microsatellite instability cancers | - |
| BIM | Wide distribution | TKI resistance in CML | - |
| BID | Liver, hematopoietic cells | Chemosensitivity marker | - |
The differential expression of these proteins across tissues explains the varying apoptotic thresholds and contributes to the therapeutic index of BH3-mimetic drugs. For instance, BCL-2 dependence in chronic lymphocytic leukemia (CLL) has been identified as a favorable predictive biomarker for response to therapy, with greater BCL-2 dependence associated with prognostically favorable genetic biomarkers and treatment sensitivity [19]. This functional dependence can be measured using BH3-profiling, which assesses cytochrome c release as an indicator of survival dependence on specific anti-apoptotic proteins, providing a predictive biomarker beyond genetic markers alone [19].
Molecular Mechanisms of Apoptotic Regulation
The BCL-2 Family Interactome: Balancing Life and Death Decisions
The regulation of mitochondrial apoptosis occurs through an intricate network of protein-protein interactions between pro- and anti-apoptotic BCL-2 family members. The prevailing "direct activation" model proposes that in response to cellular stress, activator BH3-only proteins (such as BIM and tBID) directly engage and conformationally activate BAX and BAK, leading to their oligomerization and MOMP [15] [12]. Anti-apoptotic proteins preserve mitochondrial integrity by sequestering these activator BH3-only proteins and directly inhibiting BAX/BAK activation [18]. Sensitizer BH3-only proteins (such as BAD and NOXA) promote apoptosis by displacing activators from their anti-apoptotic binding partners, thereby tilting the balance toward cell death [15].
This complex interactome functions as a tunable rheostat rather than a simple binary switch, with cellular fate determined by the relative ratios and binding affinities between competing family members. The system exhibits significant redundancy and compensatory mechanisms, as evidenced by the embryonic lethality of combined BAX/BAK deficiency but relatively mild phenotypes in single knockout mice [12]. Additional regulatory layers include post-translational modifications that modulate protein function, subcellular localization dynamics, and interactions with non-BCL-2 proteins that influence apoptotic sensitivity.
Integration with PARP1 and DNA Damage Response
Emerging research has revealed critical integration between BCL-2 family regulation and PARP1-mediated DNA damage response pathways. PARP1 [poly(ADP-ribose) polymerase 1] serves as a molecular sensor for DNA damage and plays a decisive role in determining cell fate in response to genomic insults [13]. Upon detecting DNA damage, PARP1 becomes activated and facilitates DNA repair through multiple mechanisms, including recruitment of repair complexes to damage sites [20]. However, excessive DNA damage leads to PARP1 hyperactivation and caspase-dependent cleavage, generating 24-kDa and 89-kDa fragments with distinct pro-apoptotic functions [13]. The 24-kDa fragment irreversibly binds DNA breaks, preventing repair and enhancing apoptosis, while the 89-kDa fragment translocates to the cytoplasm and promotes caspase-mediated DNA fragmentation [13].
Recent investigations have demonstrated that the ferroptosis inducer RSL3 triggers apoptosis through PARP1-mediated mechanisms, illustrating the cross-talk between different cell death pathways [13]. RSL3 activates two parallel apoptotic pathways: caspase-dependent PARP1 cleavage and DNA damage-dependent apoptosis resulting from reduced full-length PARP1 levels via inhibition of METTL3-mediated m6A modification [13]. This PARP1-centered mechanism operates in PARP inhibitor-resistant cells, suggesting therapeutic potential for overcoming treatment resistance in malignancies [13].
Figure 1: Integrated PARP1 and BCL-2 Family Apoptotic Signaling Pathway. This diagram illustrates the cross-talk between DNA damage response mediated by PARP1 and the mitochondrial apoptotic pathway regulated by BCL-2 family proteins. Excessive DNA damage leads to PARP1 cleavage and activation of BCL-2 family-mediated apoptosis through mitochondrial outer membrane permeabilization (MOMP).
Experimental Methodologies for Analyzing BCL-2 Family Functions
BH3 Profiling: Assessing Apoptotic Priming and Dependencies
BH3 profiling has emerged as a powerful functional technique for interrogating BCL-2 family interactions and measuring cellular proximity to the apoptotic threshold. This methodology involves permeabilizing cells and exposing them to synthetic peptides derived from the BH3 domains of various pro-apoptotic proteins, followed by measurement of cytochrome c release from mitochondria [19]. The pattern of cytochrome c release in response to different BH3 peptides reveals which anti-apoptotic proteins a cell depends on for survival, a concept known as "apoptotic priming" [19].
The experimental workflow consists of several key steps: (1) isolation of viable primary cells or cell lines with appropriate viability (>60%) and purity criteria (>85% for primary cells); (2) permeabilization with digitonin to allow BH3 peptide access to mitochondria; (3) incubation with a panel of BH3 peptides including MS1 (MCL-1-specific), HRK (BCL-xL-specific), BAD (BCL-2/BCL-xL-specific), and BIM (pan-BCL-2 family binder); (4) quantification of cytochrome c release by flow cytometry or ELISA; and (5) data analysis to determine differential dependence on anti-apoptotic proteins [19]. This technique has proven particularly valuable in predicting response to targeted therapies in hematologic malignancies like CLL, where BCL-2 dependence strongly correlates with treatment sensitivity independent of genetic background [19].
Molecular and Biochemical Assays
Multiple complementary approaches provide comprehensive analysis of BCL-2 family functions in experimental systems:
Protein-Protein Interaction Studies: Co-immunoprecipitation and proximity ligation assays quantify interactions between anti-apoptotic and pro-apoptotic family members, while surface plasmon resonance and isothermal titration calorimetry provide quantitative binding affinity measurements [18] [17].
Structural Biology Techniques: X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy have revealed the three-dimensional structures of multiple BCL-2 family members, illuminating the molecular details of the hydrophobic groove and BH3 domain interactions [17]. These structural insights have been instrumental in guiding the rational design of BH3-mimetic drugs [12].
Gene Expression Analysis: RNA sequencing and real-time quantitative PCR (RT-qPCR) measure transcript levels of BCL-2 family members, while Western blotting and flow cytometry assess protein expression and post-translational modifications [13] [18]. Epitranscriptomic regulation through mechanisms such as METTL3-mediated m6A modification can influence BCL-2 family protein expression and apoptotic sensitivity [13].
Functional Viability Assays: Luciferase-based killing assays and MTT viability tests evaluate the functional consequences of BCL-2 family manipulations in various cellular contexts, including cancer cells and engineered immune cells [18].
Table 3: Key Research Reagents for BCL-2 Family Studies
| Reagent Category | Specific Examples | Research Applications |
|---|---|---|
| BH3 Mimetics | Venetoclax (ABT-199), Navitoclax (ABT-263), AZD5991 | Selective pharmacological inhibition of anti-apoptotic BCL-2 proteins |
| BH3 Peptides | BIM, BID, BAD, HRK, MS1 peptides | BH3 profiling to assess apoptotic dependencies |
| Antibodies | Anti-BCL-2, Anti-BCL-xL, Anti-BAX, Anti-cleaved caspase-3 | Protein detection, localization, and quantification |
| Cell Lines | Jeko-1, Nalm6, Kuramochi, HCC1395 | In vitro models for functional studies |
| Expression Vectors | BCL-2, BCL-xL, MCL-1 with P2A/T2A self-cleaving peptides | Engineered protein overexpression in cellular models |
| Apoptosis Detection Kits | Annexin V, JC-1, caspase activation assays | Quantification of apoptotic parameters |
Therapeutic Targeting of BCL-2 Family Proteins
BH3-Mimetics: From Bench to Bedside
The structural characterization of the hydrophobic groove on anti-apoptotic BCL-2 proteins enabled the rational design of BH3-mimetic compounds that competitively inhibit protein-protein interactions within the BCL-2 family [12]. Venetoclax (ABT-199), a first-in-class selective BCL-2 inhibitor, received FDA approval in 2016 and has transformed treatment paradigms for several hematologic malignancies, including chronic lymphocytic leukemia and acute myeloid leukemia [12]. Its development followed earlier compounds such as ABT-737 and navitoclax (ABT-263), which targeted multiple anti-apoptotic proteins but faced limitations due to on-target thrombocytopenia from BCL-xL inhibition [12].
The clinical success of venetoclax has spurred development of next-generation BH3-mimetics with improved properties. Novel compounds such as sonrotoclax and lisaftoclax are currently under clinical evaluation, both as monotherapies and in rational combinations [12]. Additionally, indolyl-triazole derivatives have shown promise as BCL-2 inhibitors in preclinical studies, with compound R23 demonstrating potent anticancer activity through induction of apoptosis and cell cycle arrest [21]. These advances highlight the ongoing optimization of therapeutic targeting strategies against BCL-2 family proteins.
Combination Strategies and Resistance Mechanisms
The therapeutic efficacy of BH3-mimetics is enhanced through rational combination strategies that address compensatory survival pathways and resistance mechanisms. In CLL, the combination of venetoclax with anti-CD20 monoclonal antibodies has demonstrated improved outcomes, while in AML, combinations with hypomethylating agents have become standard of care [12]. Emerging approaches include combining BH3-mimetics with CAR T-cell therapy, where BCL-xL overexpression in CAR T cells protects them from BH3-mimetic-induced apoptosis while sensitizing tumor cells to treatment [18].
Resistance to BH3-mimetics can occur through multiple mechanisms, including upregulation of alternative anti-apoptotic proteins (particularly MCL-1 or BCL-xL), mutations in BCL-2 that reduce drug binding (such as the G101V mutation), and changes in the expression or activation of pro-apoptotic proteins [18] [12]. Functional BH3 profiling can identify these adaptive dependencies and guide subsequent treatment selection, illustrating the clinical utility of understanding the hierarchical relationships within the BCL-2 family [19].
Novel Targeting Approaches
Innovative strategies beyond traditional small-molecule inhibitors are emerging to expand the therapeutic targeting of BCL-2 family proteins:
PROTACs (Proteolysis Targeting Chimeras): These bifunctional molecules simultaneously bind to target proteins and E3 ubiquitin ligases, directing specific protein degradation. BCL-2-targeting PROTACs offer potential advantages in overcoming resistance mutations and achieving more complete target inhibition [12].
Antibody-Drug Conjugates (ADCs): ADCs enable selective delivery of cytotoxic payloads to malignant cells expressing specific surface markers, bypassing systemic toxicity associated with conventional chemotherapy [12].
BH4 Domain Targeting: The N-terminal BH4 domain is critical for the anti-apoptotic function of BCL-2 and BCL-xL. Compounds targeting this domain represent an alternative approach to inhibiting anti-apoptotic function [12].
Natural Product-Derived Inhibitors: Natural products provide novel structural scaffolds for inhibitor development. Coixol derivatives, for instance, have demonstrated PARP1 inhibitory activity with anticancer effects, offering new structural frameworks for drug discovery [20].
The BCL-2 family hierarchy represents a sophisticated regulatory network that balances cellular survival and death decisions through complex interactions between anti-apoptotic and pro-apoptotic members. The structural characterization of these proteins and their binding interfaces has enabled the development of targeted therapies that have transformed treatment for specific hematologic malignancies. Current research continues to elucidate the non-canonical functions of BCL-2 family proteins, their integration with other cell death pathways including those mediated by PARP1, and innovative approaches to overcome therapeutic resistance.
Future directions in the field include the development of more selective inhibitors, biomarker-driven combination strategies, and novel therapeutic modalities that exploit the intricate hierarchy of BCL-2 family interactions. As our understanding of this protein family continues to evolve, so too will our ability to therapeutically modulate apoptosis in cancer and other diseases characterized by dysregulated cell death.
The B-cell lymphoma 2 (BCL-2) protein and poly(ADP-ribose) polymerase 1 (PARP1) represent critical regulators of cellular survival and death, operating through distinct yet interconnected biochemical pathways. BCL-2, the founding member of the BCL-2 protein family, functions as a key anti-apoptotic regulator that maintains mitochondrial integrity and prevents cytochrome c release [12] [16]. PARP1, a nuclear enzyme, responds to DNA damage by catalyzing the transfer of ADP-ribose units to target proteins, playing essential roles in DNA repair and cellular stress response [22] [9]. While these proteins have been extensively studied within their respective domains, emerging evidence reveals a direct molecular interaction between BCL-2 and PARP1 that transcends their canonical functions, representing a significant convergence point in cell death regulation [6]. This interaction not only expands our understanding of apoptotic and non-apoptotic cell death mechanisms but also carries profound implications for therapeutic interventions in cancer and other pathologies.
The investigation of this direct interaction provides a framework for validating apoptotic pathways through PARP-1 and BCL-2 family protein analysis research. As resistance to conventional apoptosis-inducing therapies remains a significant clinical challenge, particularly in hematological malignancies and solid tumors, understanding non-canonical death mechanisms suppressed by BCL-2 becomes increasingly important [6] [12]. This review systematically examines the experimental evidence for the BCL-2-PARP1 interaction, details the methodological approaches for its validation, analyzes the functional consequences of this interplay, and explores the therapeutic implications for drug development.
Molecular Interaction Between BCL-2 and PARP1
Direct Binding and Structural Considerations
The direct interaction between BCL-2 and PARP1 was definitively established through a series of biochemical experiments demonstrating physical association between these proteins. Research has shown that BCL-2, traditionally considered a mitochondrial protein, also localizes to the nucleus in various tumor cell types, including diffuse large B-cell lymphoma (DLBCL) cells harboring the t(14;18) translocation [6]. Immunoblotting of nuclear and cytoplasmic fractions from multiple DLBCL cell lines (OCI-LY1, OCI-LY8, and Toledo) confirmed BCL-2 presence in the nuclear compartment regardless of DNA damage induction [6]. This nuclear localization enables spatial proximity for interaction with PARP1, a predominantly nuclear protein.
The binding mechanism involves the enzymatic suppression of PARP1 by BCL-2. Enzyme-linked immunosorbent assays utilizing purified GST-BCL-2 and fractionated lysates from HT cells demonstrated that BCL-2 directly inhibits PARP1 enzymatic activity in a dose-dependent manner [6]. This suppression occurs through BCL-2's interaction with PARP1, which blocks PARP1's catalytic function without necessarily displacing it from DNA damage sites. The interaction is functionally significant, as BCL-2 overexpression in PARP inhibitor-sensitive breast and lung cancer cells reduced their survival by 90-100%, effects that were reversible with the BCL-2 inhibitor ABT-737 [6].
Table 1: Key Experimental Evidence for BCL-2-PARP1 Interaction
| Experimental Approach | Key Findings | Cellular Context | Citation |
|---|---|---|---|
| Nuclear/Cytoplasmic Fractionation | BCL-2 detected in nuclear fractions | DLBCL cell lines with t(14;18) translocation | [6] |
| ELISA with Purified Proteins | BCL-2 suppresses PARP1 enzymatic activity dose-dependently | GST-BCL-2 with lysates from HT cells | [6] |
| Co-immunoprecipitation | Direct physical interaction confirmed | 293T cells with ectopic expression | [6] |
| ABT-737 Treatment | Displaces PARP1 from BCL-2 | DLBCL and CLL primary cells | [6] |
| Colony Formation Assay | BCL-2 expression kills PARP inhibitor-sensitive cells | Breast and lung cancer cells | [6] |
BCL-2 Domains and PARP1 Cleavage
The structural determinants of the BCL-2-PARP1 interaction involve specific domains within both proteins. BCL-2 contains four BCL-2 homology (BH) domains that facilitate its interactions with other proteins [16]. The hydrophobic groove formed by BH1-3 domains typically engages pro-apoptotic proteins through their BH3 domains, but evidence suggests this region may also participate in PARP1 binding [6] [16]. The BH3 mimetic ABT-737, designed to occupy this groove, displaces PARP1 from BCL-2 in a dose-dependent manner, indicating potential overlap in binding sites [6].
PARP1 contains three major functional domains: an N-terminal DNA-binding domain (DBD) with two zinc finger motifs, a central automodification domain (AMD), and a C-terminal catalytic domain (CAT) [9] [23]. The DBD facilitates PARP1's recognition of DNA strand breaks, while the CAT domain catalyzes poly(ADP-ribosyl)ation using NAD+ as substrate [23]. The specific PARP1 domains involved in BCL-2 interaction remain to be fully elucidated, though the disruption of PARP1 enzymatic function suggests BCL-2 may interact with or sterically hinder the catalytic domain.
Historical context for this interaction dates to 1997 when researchers demonstrated that BCL-2 acts upstream of PARP cleavage by proteases now known as caspases, preventing PARP1 activation during apoptosis [24]. This established an early functional connection between BCL-2 and PARP1 regulation, preceding the discovery of their direct physical interaction.
Diagram 1: Molecular Interplay Between BCL-2 and PARP1. BCL-2 binds to and suppresses PARP1 activity in the nucleus, preventing DNA repair and promoting non-apoptotic cell death. The BH3 mimetic ABT-737 disrupts this interaction, restoring PARP1 function and DNA repair capacity.
Experimental Methodologies for Validation
Protein Interaction Assays
The direct interaction between BCL-2 and PARP1 has been validated using multiple complementary biochemical approaches. Co-immunoprecipitation (Co-IP) experiments provide critical evidence for physical association, wherein antibodies targeting BCL-2 successfully co-precipitate PARP1 from cell lysates, and vice versa [6]. This methodology typically involves preparing whole-cell or nuclear extracts from relevant cell lines (e.g., DLBCL lines like OCI-LY8 or Toledo), incubating with specific antibodies, and capturing immune complexes with protein A/G beads. Following extensive washing to remove non-specifically bound proteins, the complexes are resolved by SDS-PAGE and probed with antibodies against both proteins to confirm interaction.
Enzyme-linked immunosorbent assays (ELISA) with purified components have further characterized this interaction. Experiments adding increasing concentrations of purified GST-BCL-2 to fractionated lysates from HT cells demonstrated dose-dependent suppression of PARP1 enzymatic activity [6]. Similarly, titration of ABT-737 into lysates from OCI-LY1-10R cells displaced PARP1 from BCL-2, confirming competitive binding at the BH3-binding groove [6]. These in vitro reconstitution assays provide quantitative data on binding affinity and functional consequences.
Cellular localization studies through subcellular fractionation and immunofluorescence establish the spatial context for this interaction. Immunoblotting of nuclear and cytoplasmic fractions from multiple B-cell lymphoma lines confirmed BCL-2 presence in the nucleus, while immunofluorescence microscopy visualized BCL-2 and PARP1 co-localization in nuclear compartments [6]. Chromatin fractionation experiments further revealed that DNA damage promotes BCL-2 recruitment to chromatin, potentially facilitating PARP1 interaction at DNA damage sites [6].
Functional Consequences Assessment
Determining the functional outcomes of the BCL-2-PARP1 interaction requires methodologies that assess PARP1 enzymatic activity, DNA repair capacity, and cell death modalities. PARP1 enzymatic activity is routinely measured using ELISA-based approaches with immobilized histones as substrates, detecting poly(ADP-ribose) formation through specific antibodies [6]. This allows quantification of how BCL-2 binding influences PARP1 catalytic function.
DNA repair capacity is frequently evaluated through comet assays (single-cell gel electrophoresis), which measure DNA strand breaks at the individual cell level [6]. In studies of the BCL-2-PARP1 interaction, cells treated with DNA-damaging agents like N-Methyl-N′-nitro-N-nitrosoguanidine (MNNG) show impaired DNA repair when BCL-2 is overexpressed, while ABT-737 treatment restores repair function by displacing PARP1 from BCL-2 [6].
Cell death modalities are distinguished using flow cytometry with Annexin V/propidium iodide (PI) staining to identify apoptotic cells, complemented by measurements of metabolic markers like NAD+ and ATP levels [6]. PARP1 overactivation depletes cellular NAD+ and ATP pools, promoting a non-apoptotic cell death pathway distinct from classical apoptosis [6] [22]. These multiparameter approaches confirm that BCL-2 suppression of PARP1 shifts cell fate toward survival, while BH3 mimetics like ABT-737 restore PARP1-mediated cell death even in apoptosis-resistant contexts.
Table 2: Key Methodologies for Studying BCL-2-PARP1 Interaction
| Methodology | Application | Key Outcomes | Technical Considerations |
|---|---|---|---|
| Co-immunoprecipitation | Protein-protein interaction | Confirms physical association between BCL-2 and PARP1 | Requires validation with multiple antibodies; controls for non-specific binding |
| Cellular Fractionation | Subcellular localization | Identifies nuclear BCL-2 population | Must ensure fraction purity; nuclear integrity critical |
| PARP Activity ELISA | Enzymatic function | Quantifies PARP1 suppression by BCL-2 | Uses immobilized histones as substrates; antibody detection |
| Alkaline Comet Assay | DNA repair capacity | Measures strand break repair efficiency | Single-cell approach; sensitive to DNA damage conditions |
| Annexin V/PI FACS | Cell death modality | Distinguishes apoptotic vs. non-apoptotic death | Requires careful timing after treatments |
| NAD+/ATP Assays | Metabolic status | Indicators of PARP1 overactivation | Luminescence-based assays; normalized to cell number |
Diagram 2: Experimental Workflow for Validating BCL-2-PARP1 Interaction. Comprehensive methodology combining cellular treatments with biochemical and functional assays to characterize the interaction and its consequences.
Signaling Pathways and Molecular Consequences
Apoptotic and Non-Apoptotic Cross-Regulation
The BCL-2-PARP1 interaction creates a sophisticated cross-regulatory node between apoptotic and non-apoptotic cell death pathways. Canonically, BCL-2 family proteins govern mitochondrial outer membrane permeabilization (MOMP), the pivotal event in intrinsic apoptosis that leads to cytochrome c release and caspase activation [12] [16]. Anti-apoptotic members like BCL-2 sequester pro-apoptotic proteins BAX and BAK or their activators (BH3-only proteins), preventing MOMP and maintaining cell survival [12].
PARP1 activation initiates an alternative cell death pathway through excessive poly(ADP-ribosyl)ation. Following severe DNA damage, PARP1 hyperactivation consumes cellular NAD+ pools, leading to ATP depletion and necrotic cell death [22] [23]. This process involves the nuclear-to-mitochondrial translocation of apoptosis-inducing factor (AIF), which triggers caspase-independent chromatin condensation and large-scale DNA fragmentation [22]. The BCL-2-PARP1 interaction represents a mechanistic bridge between these pathways, with BCL-2 suppressing the PARP1-mediated death program independently of its anti-apoptotic function [6].
This cross-regulation has particular significance in apoptosis-resistant contexts. Cells with upregulated anti-apoptotic BCL-2 family members, BAX/BAK deficiencies, or stromal protection remain susceptible to PARP1-mediated death when the BCL-2-PARP1 interaction is disrupted [6]. This explains why BH3 mimetics like ABT-737 can effectively kill apoptosis-resistant lymphoma cells through PARP1 reactivation rather than solely through canonical apoptosis induction.
DNA Repair Impairment and Genomic Instability
BCL-2-mediated suppression of PARP1 enzymatic activity directly compromises the base excision repair (BER) pathway, the primary mechanism for repairing single-strand DNA breaks [6] [22]. PARP1 serves as a DNA damage sensor in BER, detecting strand breaks and recruiting repair machinery through poly(ADP-ribosyl)ation of itself and other repair proteins [22] [25]. When BCL-2 binds and inhibits PARP1, this initial damage response is disrupted, leaving DNA lesions unrepaired.
The functional consequence of this repair impairment is demonstrated through comet assays showing persistent DNA strand breaks in BCL-2-overexpressing cells after damage induction [6]. While control cells efficiently repair DNA damage within hours, cells with active BCL-2-PARP1 interaction maintain elevated DNA break levels, indicating defective repair. This genomic instability potentially contributes to tumorigenesis in early stages and therapeutic vulnerability in established malignancies.
The repair deficiency creates a conditional synthetic lethality relationship. BCL-2 overexpression kills PARP inhibitor-sensitive cells [6], suggesting that tumors dependent on alternative repair pathways become vulnerable when BCL-2 suppresses PARP1-mediated BER. This synthetic lethal interaction expands the therapeutic applications of BCL-2 inhibition beyond apoptotic restoration to include DNA repair reactivation.
Therapeutic Implications and Drug Development
BH3 Mimetics and PARP Inhibitors
The BCL-2-PARP1 interaction presents novel therapeutic opportunities, particularly for leveraging BH3 mimetics in PARP1-dependent contexts. BH3 mimetics like ABT-737 and its clinical derivative venetoclax (ABT-199) occupy the hydrophobic groove of BCL-2, displacing both pro-apoptotic partners and PARP1 [6] [12]. This dual displacement has distinct functional consequences: releasing pro-apoptotic proteins restores apoptotic competence, while releasing PARP1 reactivates its enzymatic function and promotes DNA repair.
In apoptosis-competent cells, BH3 mimetics primarily induce classical apoptosis through mitochondrial pathway activation. However, in apoptosis-resistant settings (e.g., upregulated MCL1, BFL1, or stromal protection), the PARP1 reactivation component becomes predominant, triggering non-apoptotic death [6]. This explains why BH3 mimetics retain efficacy against apoptosis-resistant malignancies and suggests that PARP1 functional status may predict BH3 mimetic responsiveness.
Conversely, PARP inhibitors (PARPi) like olaparib, rucaparib, and talazoparib exploit synthetic lethality in homologous recombination-deficient cancers, particularly those with BRCA mutations [25]. The BCL-2-PARP1 interaction suggests additional considerations for PARPi application, as BCL-2 overexpression may mimic PARP inhibition by suppressing PARP1 function. This could create unexpected synthetic lethal relationships or alternatively contribute to PARPi resistance mechanisms.
Combination Therapy Strategies
Strategic combinations targeting both BCL-2 and PARP1 show promise for overcoming therapeutic resistance. Simultaneous BCL-2 inhibition (releasing PARP1) and PARP inhibition (blocking BER) might create dual vulnerability in DNA repair-deficient tumors [6] [25]. Preclinical models demonstrate that BCL-2 expression selectively kills PARP inhibitor-sensitive cells, while ABT-737 reverses this effect [6], suggesting careful sequencing would be essential in such combinations.
The temporal dynamics of these interactions significantly influence treatment efficacy. Simultaneously administering BH3 mimetics with PARP inhibitors might be counterproductive, as BH3 mimetics reactivate PARP1 while PARP inhibitors block its function. Sequential approaches—inducing PARP1 dependence through BH3 mimetics followed by PARP inhibition, or vice versa—may produce more favorable interactions, though this requires further investigation.
Emerging resistance mechanisms to both BH3 mimetics and PARP inhibitors highlight the need for such sophisticated combinations. BCL-2-positive lymphomas develop resistance to ABT-737 through upregulation of other anti-apoptotic family members like MCL1 and BFL1 [6], while PARPi resistance emerges through restoration of homologous recombination or drug efflux [25]. Simultaneously targeting both pathways may circumvent these resistance mechanisms by engaging multiple death programs.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Studying BCL-2-PARP1 Interactions
| Reagent/Category | Specific Examples | Research Application | Key Functions |
|---|---|---|---|
| BH3 Mimetics | ABT-737, ABT-263 (navitoclax), ABT-199 (venetoclax) | Disrupt BCL-2-PARP1 interaction; restore apoptosis | Competitively bind BCL-2 hydrophobic groove; displace binding partners |
| PARP Inhibitors | ABT-888 (veliparib), olaparib, rucaparib, talazoparib | Inhibit PARP1 enzymatic activity; study synthetic lethality | Block PARP1 catalytic site; prevent PAR formation; trap PARP1 on DNA |
| DNA Damaging Agents | MNNG, etoposide, hydrogen peroxide, ionizing radiation | Induce PARP1 activation; create DNA repair demand | Generate specific DNA lesions (strand breaks, base damage) |
| Apoptosis Assays | Annexin V/PI staining, caspase-3/7 activity assays, cytochrome c release | Distinguish cell death modalities | Detect phosphatidylserine exposure, caspase activation, mitochondrial events |
| DNA Repair Assays | Alkaline comet assay, γH2AX staining, PAR formation detection | Measure DNA damage and repair capacity | Quantify strand breaks, repair foci, PARP1 activity |
| Protein Interaction Tools | Co-immunoprecipitation kits, recombinant BCL-2/PARP1 proteins, proximity ligation assays | Validate direct interaction and complex formation | Capture protein complexes; measure binding affinity; visualize proximity |
| Cell Line Models | DLBCL lines (OCI-LY1, OCI-LY8, Toledo), primary CLL cells, engineered overexpression lines | Provide physiological and pathophysiological contexts | Endogenous BCL-2 expression; nuclear localization; therapeutic response |
The direct molecular interaction between BCL-2 and PARP1 represents a significant expansion of our understanding of cell death regulation, revealing a sophisticated interplay between apoptotic and non-apoptotic pathways. The experimental evidence demonstrates that BCL-2 localizes to the nucleus, physically associates with PARP1, and suppresses its enzymatic activity, thereby inhibiting DNA repair and modulating cell fate decisions. This interaction provides a mechanistic explanation for how BCL-2 contributes to tumorigenesis beyond its canonical anti-apoptotic function and suggests why BH3 mimetics retain efficacy against apoptosis-resistant malignancies.
From a therapeutic perspective, the BCL-2-PARP1 interface offers novel approaches for cancer treatment, particularly in leveraging non-apoptotic cell death programs when classical apoptosis is compromised. The ability of BH3 mimetics to disrupt this interaction and reactivate PARP1 function presents an underappreciated mechanism of action that complements their pro-apoptotic effects. Furthermore, the conditional synthetic lethality relationships emerging from this interplay suggest new biomarker strategies and combination therapies worth exploring in clinical settings.
As drug development continues to target both BCL-2 and PARP1 in various malignancies, acknowledging their direct molecular interaction becomes increasingly important for predicting efficacy, understanding resistance mechanisms, and designing optimal therapeutic sequences. Future research should focus on elucidating the structural determinants of this interaction, its prevalence across tumor types, and its contribution to therapeutic responses in clinical settings.
Abstract Regulated cell death (RCD) is a fundamental process in maintaining cellular homeostasis, and its dysregulation is a hallmark of cancer. While apoptosis has been extensively studied and targeted therapeutically, resistance to apoptosis-inducing therapies is a significant clinical challenge. This has spurred interest in alternative RCD modalities, such as ferroptosis, and the complex crosstalk between these pathways. This guide provides an objective comparison of key non-apoptotic RCD pathways—ferroptosis, pyroptosis, and necroptosis—against the benchmark of apoptosis. We summarize their molecular mechanisms, morphological features, and experimental data, with a specific focus on their interactions with core apoptotic components, including Bcl-2 family proteins and PARP1. The content is framed within the context of validating apoptotic pathways, offering researchers a structured overview of the expanding RCD landscape and its therapeutic implications.
Molecular Mechanisms and Morphological Hallmarks of RCD Pathways
The characterization of distinct RCD pathways is based on unique molecular triggers, executioners, and morphological features. The table below provides a comparative summary of apoptosis, ferroptosis, pyroptosis, and necroptosis.
Table 1: Comparative Analysis of Key Regulated Cell Death Pathways
| Feature | Apoptosis | Ferroptosis | Pyroptosis | Necroptosis |
|---|---|---|---|---|
| Primary Triggers | DNA damage, growth factor withdrawal, ER stress [26] [27] | Iron overload, lipid peroxidation, GPX4 inhibition [28] [29] | Pathogen-associated molecular patterns (PAMPs), danger signals, inflammasome activation [26] [27] | TNF receptor activation, TLR signaling, caspase-8 inhibition [26] [27] |
| Key Executioners | Caspase-3/7 [26] [27] | Lipid peroxides (no single known executioner protein) [26] | Gasdermin D (GSDMD) pore formation [26] [27] | MLKL pore formation [26] [27] |
| Morphological Features | Cell shrinkage, membrane blebbing, chromatin condensation, apoptotic bodies [28] [26] | Small mitochondria with ruptured outer membrane, intact plasma membrane, no chromatin condensation [28] | Cell swelling, plasma membrane pore formation, release of inflammatory cytokines [26] | Organelle swelling, plasma membrane rupture, release of DAMPs [26] |
| Immunogenicity | Immunologically silent [26] | Inflammatory [26] | Highly inflammatory [26] | Highly inflammatory [26] |
| Key Regulators | Bcl-2 family, caspases [26] [27] | GPX4, System Xc-, ACSL4 [28] | Inflammatory caspases, GSDMD [26] [27] | RIPK1, RIPK3, MLKL [26] [27] |
Experimental Data and Pathway Crosstalk
The boundaries between RCD pathways are not rigid, and extensive crosstalk exists, which can be exploited therapeutically. Key experimental findings and quantitative data are summarized below.
Table 2: Experimental Evidence of RCD Crosstalk and Therapeutic Modulation
| Experimental Context | Key Finding | Implication for RCD Crosstalk | Supporting Data |
|---|---|---|---|
| BCL2-PARP1 Interaction in Lymphoma [6] | BCL2 binds to and inhibits PARP1 activity, suppressing a non-apoptotic cell death pathway. | BCL2, a key anti-apoptotic protein, can directly regulate non-apoptotic (potentially parthanatos) death. | ABT-737 displaced PARP1 from BCL2, restoring PARP1 activity and promoting cell death in apoptosis-resistant cells. |
| Pan-Bcl-2 Inhibition & ER Stress in Glioblastoma [30] | Obatoclax (Mcl-1/Bcl-xL inhibitor) synergizes with tunicamycin (ER stress inducer) to trigger apoptosis. | Inhibition of anti-apoptotic proteins can lower the threshold for death from other stress pathways. | Combinatorial knockdown of Mcl-1 and Bcl-xL significantly increased cleaved PARP and apoptosis under ER stress. |
| Transcriptomic Landscape of Apoptosis vs. Ferroptosis [31] | Apoptosis and ferroptosis share common upstream regulators (e.g., p53) but have distinct transcriptomic profiles. | Stress signals can activate multiple death pathways simultaneously or sequentially; shared nodes exist. | p53 can transcriptionally repress SLC7A11 (promoting ferroptosis) while activating PUMA (promoting apoptosis). |
| PANoptosis in Infection & Cancer [26] [27] | Combined loss of pyroptosis, apoptosis, and necroptosis pathways, but not individual loss, prevents cell death triggered by specific stimuli. | Establishes a unified, innate immune death pathway with components of all three RCDs, highlighting profound crosstalk. | Activation of biochemical markers from all three RCD pathways observed in response to viral and bacterial infections. |
Diagram 1: RCD Pathway Crosstalk and Key Molecular Nodes. This diagram illustrates the triggers, key executioner proteins, and documented crosstalk between different RCD modalities. Solid lines represent direct activation or inhibition, while dashed lines represent transcriptional regulation or the convergence of pathways into complex phenomena like PANoptosis. Key regulatory nodes like BCL2, p53, and PARP1 are highlighted, showing their connectivity across multiple pathways.
Essential Reagents and Experimental Methodologies
To investigate RCD crosstalk, a specific toolkit of reagents and validated experimental protocols is required. The table below details key research solutions.
Table 3: The Scientist's Toolkit: Key Reagents for RCD Research
| Reagent / Assay | Primary Function | Application in RCD Research |
|---|---|---|
| BH3 Mimetics(e.g., ABT-737, Venetoclax, Obatoclax) | Small molecule inhibitors that bind to and antagonize anti-apoptotic Bcl-2 family proteins (Bcl-2, Bcl-xL, Mcl-1) [6] [30] [32]. | To probe dependence on anti-apoptotic proteins, sensitize cells to other death inducers, and investigate non-apoptotic functions of Bcl-2 proteins (e.g., PARP1 inhibition) [6] [30]. |
| Ferroptosis Inducers(e.g., Erastin, RSL3) | Erastin inhibits system Xc-, depleting glutathione; RSL3 directly inhibits GPX4. Both lead to lipid peroxidation [28]. | To selectively induce ferroptosis and study its unique biochemical hallmarks (iron dependence, lipid ROS) and its intersection with other stress pathways [28] [31]. |
| Caspase Inhibitors(e.g., z-VAD-fmk) | Pan-caspase inhibitor that blocks apoptotic signaling [32]. | To confirm caspase-dependent apoptosis or to block apoptosis to uncover/study alternative death pathways like necroptosis or ferroptosis [26]. |
| Annexin V / Propidium Iodide (PI) Staining | Flow cytometry assay to detect phosphatidylserine externalization (Annexin V, early apoptosis) and loss of membrane integrity (PI, late apoptosis/necrosis) [6]. | A foundational assay to quantify and distinguish early and late-stage cell death. Used in combination with pathway-specific inhibitors to characterize the mode of death [6]. |
| Western Blot for Cleaved PARP | Detects the caspase-mediated cleavage of PARP, a hallmark of apoptosis execution [30]. | A standard biochemical method to confirm the activation of the apoptotic cascade in experimental settings [30]. |
| GPX4 Activity Assay / Lipid Peroxidation Probes | Measures the activity of GPX4 or directly quantifies levels of lipid-reactive oxygen species (e.g., with BODIPY 581/591 C11) [28]. | Essential for the biochemical validation of ferroptosis induction and execution. |
Detailed Experimental Protocol: Validating BCL2-PARP1 Interaction & Non-Apoptotic Death
The following methodology, adapted from a study in diffuse large B-cell lymphoma, provides a framework for investigating non-apoptotic death pathways regulated by apoptotic proteins [6].
- 1. Cell Line Model: Use BCL2-overexpressing lymphoma cell lines (e.g., OCI-LY1, OCI-LY8) with established apoptosis resistance (e.g., upregulation of MCL1 or BFL1).
- 2. Treatment Conditions:
- Control: Vehicle (DMSO)
- DNA Damage: 50-500 µM MNNG (N-Methyl-N'-nitro-N-nitrosoguanidine) for 15 minutes.
- BCL2 Inhibition: 100 nM ABT-737 for 16 hours.
- Combination: MNNG + ABT-737.
- 3. Cell Death Measurement:
- After treatment, harvest cells and stain with Annexin V-FITC and Propidium Iodide (PI).
- Analyze by flow cytometry. Death is quantified as the percentage of Annexin V+/PI+ (late apoptotic/necrotic) and Annexin V+/PI- (early apoptotic) cells.
- 4. PARP1 Activity Assay:
- Prepare fractionated cell lysates from treated and control cells.
- Use a commercial PARP ELISA kit with immobilized histones to measure PARP1 enzymatic activity. Increased activity is indicated by higher poly(ADP-ribose) formation.
- 5. Disruption of BCL2-PARP1 Complex:
- Perform co-immunoprecipitation (Co-IP) using an anti-BCL2 antibody on nuclear fractions of cells.
- Probe the immunoprecipitate with an anti-PARP1 antibody via Western Blot. A reduction in co-precipitated PARP1 after ABT-737 treatment indicates displacement.
- 6. Functional Rescue:
- To confirm PARP1's role, repeat the cell death assay in the presence of a PARP inhibitor (e.g., ABT-888). A reduction in ABT-737-induced death upon PARP inhibition confirms the functional significance of the BCL2-PARP1 interaction in this death pathway.
The move beyond a purely apoptosis-centered view of cell death is crucial for advancing cancer research and therapy. The experimental evidence clearly shows that RCD pathways operate not in isolation but as an interconnected network. Key apoptotic regulators like BCL2 can modulate non-apoptotic death, as seen in its inhibition of PARP1, while master regulators like p53 can influence both apoptosis and ferroptosis. This intricate crosstalk, exemplified by the PANoptosis concept, presents both a challenge and an opportunity. Future therapeutic strategies should aim to systematically map these interactions within specific cancer types, leveraging combination therapies that simultaneously target multiple RCD vulnerabilities. This approach holds significant promise for overcoming the drug resistance that often plagues treatments focused on a single death pathway.
Techniques for Apoptosis Detection: From Western Blot to Functional Assays
The validation of apoptotic pathways is a cornerstone of cancer research, pivotal for understanding drug resistance mechanisms and developing novel therapeutic strategies. At the heart of this process lies the precise detection and analysis of key regulatory proteins, primarily PARP-1 and members of the BCL-2 family. PARP-1 (poly(ADP-ribose) polymerase 1) functions as a critical DNA damage sensor and facilitator of DNA repair mechanisms. Its cleavage by caspases serves as a well-established biochemical marker of apoptosis execution [33] [24]. Conversely, the BCL-2 protein family constitutes a complex network of regulators that govern mitochondrial outer membrane permeabilization, the decisive step in intrinsic apoptosis. This family includes pro-survival members (e.g., BCL-2, BCL-xL, MCL-1) and pro-apoptotic members [34] [35]. Research has revealed a direct interaction between BCL-2 and PARP-1, through which BCL-2 suppresses PARP1 enzymatic activity and DNA repair function, highlighting the interconnected nature of these pathways [6]. This guide provides a comparative analysis of key reagents and antibodies essential for researchers validating these critical apoptotic components.
Key Research Reagent Solutions
Successful investigation of apoptotic pathways requires a toolkit of highly specific reagents. The table below outlines essential materials for studying PARP-1 and BCL-2 family members, along with their primary applications in research.
Table 1: Essential Research Reagents for Apoptosis Studies
| Reagent Type | Specific Examples | Research Application |
|---|---|---|
| PARP Antibodies | PARP Antibody #9542 (Cell Signaling Technology) [33] | Detects endogenous full-length PARP1 (116 kDa) and its large caspase-cleaved fragment (89 kDa) via Western blot. |
| BCL-2 Family Antibody Kits | Pro-Survival Bcl-2 Family Antibody Sampler Kit II #17229 [35] | Provides multiple antibodies (e.g., against BCL-2, BCL-xL, MCL-1) for simultaneous screening of pro-survival protein levels by Western blot. |
| BH3 Mimetics | ABT-737, Venetoclax (ABT-199) [6] [36] [34] | Small molecule inhibitors that disrupt interactions between anti-apoptotic BCL-2 family proteins and their pro-apoptotic BH3-only partners. |
| PARP Inhibitors | ABT-888 (Veliparib), Olaparib, Niraparib [6] [37] [38] | Inhibit PARP enzymatic activity, used to induce synthetic lethality in HR-deficient cells or to study PARP function. |
| Caspase Inhibitors | QVD-OPh, zVAD-fmk [36] | Pan-caspase inhibitors used to confirm the caspase-dependent nature of cell death or PARP cleavage. |
PARP-1 Specificity: Reagent Comparison and Validation
Antibody-Based Detection
A critical step in apoptosis research is the specific detection of PARP-1 and its cleavage status. The widely used PARP Antibody #9542 from Cell Signaling Technology serves as a prime example of a well-validated reagent. It is a rabbit polyclonal antibody produced by immunizing animals with a synthetic peptide corresponding to the caspase cleavage site in human PARP, which contributes to its high specificity. This antibody detects endogenous levels of full-length PARP1 (116 kDa) and the 89 kDa fragment resulting from caspase cleavage, but does not cross-react with related proteins or other PARP isoforms [33]. This specificity is crucial for accurately interpreting apoptosis assays.
Pharmacological Inhibition
PARP inhibitors are another class of essential reagents, useful both as chemical probes and therapeutic agents. They function by blocking PARP enzymatic activity, which is central to DNA repair. When PARP is inhibited, unrepaired single-strand breaks can lead to double-strand breaks during replication, resulting in genomic instability and cell death, particularly in homologous recombination-deficient cancers (synthetic lethality) [37]. Non-selective PARP-1/-2 inhibitors like olaparib have demonstrated clinical success but are associated with hematologic toxicity, driving the development of more selective PARP-1 inhibitors to improve safety profiles [38]. The specificity of these compounds is determined by their structural interaction with the NAD+ binding site in the PARP catalytic domain.
Table 2: Comparison of PARP-Targeting Reagents
| Reagent Name | Specificity / Type | Key Feature / Application | Experimental Validation |
|---|---|---|---|
| PARP Antibody #9542 [33] | Rabbit Polyclonal; detects cleaved and full-length PARP1. | Targets caspase cleavage site; ideal for apoptosis confirmation in WB. | Validated for WB; recognizes human, mouse, rat, monkey. |
| Anti-PARP1 Antibody (HPA045168) [39] | Rabbit Polyclonal; for human PARP1. | Validated for IHC, ICC-IF, and WB. | Enhanced validation protocols for application-specific use. |
| ABT-888 (Veliparib) [6] | PARP-1/-2 inhibitor (non-selective). | Used in research combinations with BH3 mimetics like ABT-737. | Shows synergy in inducing non-apoptotic cell death in lymphoma models [6]. |
| PARP-1 Selective Inhibitors [38] | PARP-1 selective inhibitor. | Designed to overcome hematologic toxicity of pan-PARP inhibitors. | Emerging class; analysis of structure-activity relationships (SAR) is ongoing. |
BCL-2 Family Specificity: Reagent Comparison and Validation
Antibody Sampler Kits
Given the functional redundancy and complex interactions among BCL-2 family proteins, researchers often need to screen multiple members simultaneously. Antibody sampler kits provide an economical solution. For instance, the Pro-Survival Bcl-2 Family Antibody Sampler Kit II includes antibodies against key pro-survival members like BCL-2, BCL-xL, and MCL-1 [35]. This allows for a coordinated analysis of how these proteins are regulated in response to apoptotic stimuli, which is vital as resistance to BH3 mimetics can occur through upregulation of alternative anti-apoptotic family members like MCL1 or BFL1 [6].
BH3 Mimetics
BH3 mimetics are a breakthrough class of small molecules that mimic the function of pro-apoptotic BH3-only proteins. They bind to the hydrophobic groove of anti-apoptotic BCL-2 family proteins, displacing sequestered pro-apoptotic effectors like BIM and BAK to initiate apoptosis [6] [34]. The specificity and efficacy of these drugs vary significantly.
- Venetoclax (ABT-199) is a highly selective BCL-2 inhibitor approved for clinical use [36] [34].
- ABT-737 is a predecessor that inhibits BCL-2, BCL-xL, and BCL-W. Its use in research has been instrumental in revealing non-apoptotic functions of BCL-2, such as its inhibition of PARP1 activity. Studies show ABT-737 can displace PARP1 from BCL-2, re-establishing PARP1 activity and promoting a non-apoptotic cell death, even in some apoptosis-resistant contexts [6].
Advanced techniques like quantitative fast fluorescence lifetime imaging microscopy (qF3) are now being used to compare the efficacy and unexpected selectivity of 15 different inhibitors against four anti-apoptotic proteins in live cells, highlighting the complex selectivity landscape of these compounds [34].
Table 3: Comparison of BCL-2 Family-Targeting Reagents
| Reagent Name | Specificity / Type | Key Feature / Application | Experimental Validation |
|---|---|---|---|
| Pro-Survival BCL-2 Family Ab Sampler Kit II [35] | Antibody Kit (multiple targets). | Economical parallel screening of BCL-2, BCL-xL, MCL-1, more. | Confirmed for Western Blot; monitors phosphorylation status. |
| Venetoclax (ABT-199) [36] [34] | BCL-2 selective inhibitor. | FDA-approved for leukemias; used to overcome adaptive resistance in UVM [36]. | Synergistic with MEK+FAK inhibition in uveal melanoma models [36]. |
| ABT-737 [6] [34] | BCL-2/BCL-xL/BCL-W inhibitor. | Research tool to study BCL-2 biology and PARP1 interaction. | Displaces PARP1 from BCL-2, restoring PARP1 activity and DNA repair [6]. |
| Navitoclax (ABT-263) [34] | BCL-2/BCL-xL/BCL-W inhibitor. | In-trial anticancer drug; limited by on-target thrombocytopenia. | Live-cell FLIM-FRET assays reveal unexpected efficacy and specificity profiles [34]. |
Experimental Protocols for Pathway Validation
Detecting Apoptosis via PARP Cleavage by Western Blot
Purpose: To confirm the induction of apoptosis by detecting the cleavage of endogenous PARP1. Reagents: PARP Antibody #9542 [33], cell lysis buffer, SDS-PAGE gels, transfer apparatus. Protocol:
- Treatment and Lysate Preparation: Treat cells with the apoptotic stimulus (e.g., etoposide, ABT-737, etc.) for a predetermined time. Harvest cells and lyse them using RIPA buffer supplemented with protease inhibitors.
- Protein Quantification and Separation: Determine protein concentration using a BCA assay. Separate equal amounts of protein (e.g., 20-30 µg) by SDS-PAGE on a 4-12% gradient gel.
- Western Blotting: Transfer proteins to a PVDF membrane. Block the membrane with 5% non-fat milk in TBST.
- Antibody Incubation: Incubate the membrane with a 1:1000 dilution of PARP Antibody #9542 overnight at 4°C [33]. This antibody will detect both the full-length (116 kDa) and the characteristic caspase-cleaved fragment (89 kDa).
- Detection: After washing, incubate with an HRP-conjugated secondary antibody and develop using enhanced chemiluminescence (ECL). Data Interpretation: The presence of the 89 kDa band, often with a concomitant decrease in the 116 kDa band, is a definitive marker of caspase-3 activation and apoptosis.
Assessing BCL-2-PARP1 Interaction via Immunoprecipitation
Purpose: To validate the physical interaction between BCL-2 and PARP1. Reagents: BCL-2 antibody for immunoprecipitation, PARP antibody for detection, protein A/G beads. Protocol:
- Cell Fractionation: Prepare nuclear fractions from B-cell lymphoma cells (e.g., OCI-LY8, Toledo) known to harbor the BCL2-PARP1 interaction [6].
- Immunoprecipitation (IP): Incubate clarified nuclear lysates with a BCL-2-specific antibody conjugated to protein A/G beads overnight at 4°C.
- Washing and Elution: Wash the beads extensively with lysis buffer to remove non-specifically bound proteins. Elute the immunoprecipitated complexes by boiling in SDS sample buffer.
- Analysis: Analyze the eluates by Western blotting using the PARP Antibody #9542 to probe for the presence of co-precipitated PARP1. Intervention: To demonstrate the specificity of the interaction, repeat the IP in cells treated with the BH3 mimetic ABT-737, which disrupts the BCL2-PARP1 complex, leading to a loss of the PARP1 signal in the BCL-2 IP [6].
Functional PARP1 Activity ELISA
Purpose: To measure the functional consequence of BCL-2 binding on PARP1 enzymatic activity. Reagents: PARP ELISA kit (e.g., from Trevigen), purified GST-BCL2 protein, ABT-737. Protocol:
- Sample Preparation: Use fractionated lysates from lymphoma cells or incubate purified PARP1 with increasing concentrations of purified GST-BCL2 protein [6].
- Activity Assay: Add the samples to an ELISA plate coated with histones. Follow the manufacturer's instructions to perform the PARP reaction in the presence of NAD+.
- Detection and Quantification: Detect the incorporated poly(ADP-ribose) using a specific detector antibody in a colorimetric or chemiluminescent readout.
- Inhibition Studies: To reactivate PARP1, include the BH3 mimetic ABT-737 in the reaction, which will displace PARP1 from BCL-2 and restore enzymatic activity [6].
Signaling Pathway and Experimental Workflow Diagrams
Diagram 1: BCL-2-PARP1 interaction in cell fate.
Diagram 2: Experimental workflow for pathway validation.
The rigorous validation of apoptotic pathways depends on the specific and reproducible performance of research reagents. Antibodies targeting PARP cleavage sites and BCL-2 family members, coupled with highly specific chemical probes like BH3 mimetics and PARP inhibitors, form the foundation of this research. The experimental data and comparative tables presented in this guide underscore that reagent choice must be guided by a clear understanding of their documented specificities, strengths, and limitations. As research unveils more complex interactions, such as the direct suppression of PARP1 by BCL-2, the demand for even more selective tools, such as PARP-1-specific inhibitors, will continue to grow. By carefully selecting and validating these key reagents, researchers can confidently decipher the intricate signaling networks that control cell survival and death, accelerating the development of novel cancer therapeutics.
Poly(ADP-ribose) polymerase-1 (PARP-1) is a 116 kDa nuclear enzyme that plays a critical role in DNA repair and maintenance of genomic integrity [9] [8]. During the execution phase of apoptosis, PARP-1 becomes one of the primary cleavage targets of activated caspase-3 and caspase-7, generating characteristic 24 kDa and 89 kDa fragments [9] [40]. This proteolytic cleavage separates the DNA-binding domain (24 kDa) from the catalytic domain (89 kDa), effectively inactivating the enzyme's DNA repair capacity and facilitating cellular disassembly [8] [40]. The detection of the 89 kDa cleaved PARP-1 fragment by Western blot has become a gold standard method for identifying apoptotic cells in experimental systems, particularly in cancer research and therapeutic development where validating apoptotic pathways is essential [36] [41].
The significance of PARP-1 cleavage extends beyond merely serving as an apoptotic marker. The 24 kDa fragment retains the DNA-binding capacity and can act as a trans-dominant inhibitor of intact PARP-1, thereby preventing DNA repair and promoting apoptotic progression [9]. Meanwhile, recent evidence suggests that the 89 kDa fragment may function as a carrier for poly(ADP-ribose) (PAR) polymers, facilitating their translocation to the cytoplasm where they contribute to apoptosis-inducing factor (AIF)-mediated DNA fragmentation [8]. This dual role of PARP-1 fragments underscores the importance of accurate detection and interpretation of PARP-1 cleavage patterns in apoptotic research, particularly in the context of Bcl-2 family protein interactions and mitochondrial apoptotic pathway activation [36] [41].
PARP-1 Cleavage Fragment Characteristics
The cleavage of PARP-1 by caspases occurs at a specific aspartic acid residue (Asp214 in human PARP-1), located between the DNA-binding domain and the automodification domain [40]. This proteolytic event produces two major fragments with distinct molecular weights and biological functions, which serve as specific signatures of caspase activation in apoptotic cells.
Table 1: Characteristics of PARP-1 Cleavage Fragments
| Parameter | 24 kDa Fragment | 89 kDa Fragment |
|---|---|---|
| Domain Composition | DNA-binding domain (DBD) with two zinc finger motifs | Automodification domain (AMD) and catalytic domain (CD) |
| Cellular Localization | Remains tightly bound to DNA breaks in nucleus | Translocates from nucleus to cytoplasm during apoptosis |
| Biological Function | Acts as trans-dominant inhibitor of PARP-1; blocks DNA repair | Serves as PAR carrier; may facilitate AIF-mediated DNA fragmentation |
| Detection Specificity | Requires antibodies targeting N-terminal epitopes; less commonly detected | Detected by antibodies specific to C-terminal epitopes surrounding Asp214; widely used as apoptosis marker |
| Protease Specificity | Caspase-3 and caspase-7 (primary); other caspases in vitro | Caspase-3 and caspase-7 (primary); other caspases in vitro |
The differential behavior of these fragments following cleavage provides important insights into the apoptotic process. The 24 kDa fragment's irreversible binding to DNA strand breaks effectively halts DNA repair processes, thereby conserving cellular energy for the apoptotic program [9]. Meanwhile, the translocation of the 89 kDa fragment to the cytoplasm represents a critical step in certain forms of apoptosis, particularly in pathways involving parthanatos, a caspase-independent programmed cell death [8]. This fragment-specific behavior underscores the importance of detecting both fragments for a comprehensive understanding of apoptotic signaling in experimental systems.
Comparative Analysis of PARP-1 Cleavage Detection Methods
Western Blot Protocol for PARP-1 Cleavage Detection
The detection of PARP-1 cleavage by Western blot remains the most widely utilized method for apoptosis assessment in research settings. The following protocol, adapted from established methodologies, provides reliable detection of both full-length and cleaved PARP-1 fragments [42]:
Nuclear Extraction and Sample Preparation:
- Harvest cells and detach with trypsin-EDTA
- Incubate cells on ice for 10 minutes in hypotonic buffer (10 mM HEPES, pH 8.0, 10 mM KCl, 1.5 mM MgCl₂, 0.5 mM DTT) with complete EDTA-free protease inhibitor cocktail
- Lyse cells by adding 0.1% NP-40 and centrifuge at 1,500 ×g for 10 minutes at 4°C
- Resuspend nuclear pellet in RIPA buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) with protease inhibitors
- Incubate on ice for 30 minutes with occasional vortexing
- Centrifuge at 1,500 ×g for 30 minutes at 4°C
- Determine protein concentration in supernatant using Bradford assay
Gel Electrophoresis and Western Blotting:
- Separate 30 μg of nuclear protein extracts by 10% SDS-PAGE
- Transfer proteins to PVDF or nitrocellulose membranes
- Block membranes with 5% BSA in TBS-T (Tris-buffered saline with 0.1% Tween 20)
- Incubate with primary antibodies:
- Use B23 monoclonal antibody (1:2000 dilution) or other nuclear protein as loading control
- Incubate with HRP-conjugated secondary antibody (e.g., goat anti-mouse IgG)
- Detect using chemiluminescent method and appropriate imaging system
This protocol consistently detects the characteristic 89 kDa PARP-1 fragment in apoptotic cells, while the full-length 116 kDa PARP-1 decreases correspondingly. The inclusion of a nuclear protein loading control ensures equal loading and accurate interpretation of results.
Comparison of Detection Antibodies and Reagents
The selection of appropriate antibodies is critical for specific detection of PARP-1 cleavage fragments. Commercial antibodies vary in their specificity, sensitivity, and applications, necessitating careful selection based on experimental requirements.
Table 2: Comparison of Antibodies for PARP-1 Cleavage Detection
| Antibody Specificity | Clone/Product | Dilution | Specificity Profile | Applications |
|---|---|---|---|---|
| Cleaved PARP (Asp214) | #9541 (CST) | 1:1000 | Detects only 89 kDa fragment; does not recognize full-length PARP-1 | Specific apoptosis detection; ideal for quantifying caspase activity |
| Total PARP-1 | C2-10 (Santa Cruz) | 1:2000 | Recognizes both full-length (116 kDa) and cleaved (89 kDa) PARP-1 | Apoptosis progression monitoring; ratio of cleaved to full-length PARP |
| PARP-1 p85 Fragment | N/A (Promega) | Varies | Detects 89 kDa fragment in human and rodent samples | Cross-species apoptosis studies; caspase-3 activation assays |
The Cleaved PARP (Asp214) Antibody (#9541) offers superior specificity for apoptosis detection as it exclusively recognizes the caspase-generated 89 kDa fragment and does not cross-react with full-length PARP-1 [40]. This characteristic makes it particularly valuable for quantifying apoptotic induction without interference from the intact protein. In contrast, the C2-10 antibody provides a comprehensive view of PARP-1 status by detecting both full-length and cleaved forms, enabling calculation of cleavage ratios that correlate with apoptotic progression [42].
PARP-1 in Apoptotic Signaling Pathways
PARP-1 cleavage occupies a critical position in the execution phase of apoptosis, integrating signals from both intrinsic and extrinsic apoptotic pathways. The relationship between PARP-1 cleavage and other apoptotic events, particularly Bcl-2 family protein dynamics, provides a comprehensive framework for validating apoptotic pathways in experimental systems.
Figure 1: PARP-1 Cleavage in Apoptotic Signaling Pathway
The integration of PARP-1 cleavage with Bcl-2 family protein analysis creates a powerful approach for comprehensive apoptosis validation. As illustrated in Figure 1, apoptotic stimuli trigger mitochondrial pathway activation, leading to Bcl-2 family-mediated cytochrome c release and subsequent caspase activation [36] [43]. Caspase-3 then cleaves PARP-1, generating the characteristic 89 kDa fragment that serves as a definitive marker of apoptotic execution [40]. This pathway highlights the functional relationship between Bcl-2 family proteins and PARP-1 cleavage, enabling researchers to map apoptotic progression from initiation to execution.
Experimental Evidence: PARP-1 Cleavage in Therapeutic Contexts
Recent studies in cancer therapy development have demonstrated the utility of PARP-1 cleavage detection for validating apoptotic pathway activation. In uveal melanoma cells treated with combined FAK and MEK inhibitors, Western blot analysis revealed increased PARP-1 cleavage coinciding with upregulation of pro-apoptotic BIM and PUMA, confirming apoptosis induction through the intrinsic pathway [36]. Similarly, research on the ferroptosis inducer RSL3 demonstrated concurrent PARP-1 cleavage and caspase-3 activation, revealing crosstalk between ferroptotic and apoptotic cell death mechanisms [41].
The adaptive upregulation of anti-apoptotic BCL2 family proteins observed in treatment-resistant cells further underscores the importance of monitoring PARP-1 cleavage in therapeutic contexts [36]. Cells with elevated BCL2 levels showed reduced PARP-1 cleavage in response to FAK and MEK inhibition, indicating apoptosis suppression. However, combination therapies incorporating BCL2 inhibitors restored PARP-1 cleavage and apoptotic response, demonstrating how PARP-1 cleavage analysis can guide effective therapeutic combinations [36].
Research Reagent Solutions Toolkit
Table 3: Essential Reagents for PARP-1 Cleavage Analysis
| Reagent Category | Specific Products | Application Purpose | Technical Notes |
|---|---|---|---|
| PARP-1 Cleavage Antibodies | Cleaved PARP (Asp214) #9541 (CST); C2-10 (Santa Cruz) | Specific detection of 89 kDa fragment; total PARP-1 monitoring | Validate specificity with caspase inhibitor controls; optimize dilution for each cell type |
| Caspase Inhibitors | zVAD-fmk (pan-caspase); DEVD-fmk (caspase-3) | Apoptosis inhibition controls; pathway validation | Use concentration range 20-50 μM; pre-incubate 1-2 hours before apoptotic stimuli |
| Apoptosis Inducers | Staurosporine; Actinomycin D; Etoposide | Positive controls for PARP-1 cleavage | Titrate for cell type-specific response; establish time course (typically 4-24 hours) |
| Loading Controls | B23/nucleophosmin; Lamin B1; Histone H3 | Nuclear protein normalization | Essential for quantitative comparison; avoid cytoplasmic proteins (GAPDH, actin) for nuclear extracts |
| Detection Systems | HRP-conjugated secondary antibodies; chemiluminescent substrates | Signal detection and visualization | Optimize exposure time to avoid saturation; use linear range for quantification |
This curated toolkit provides the essential components for robust PARP-1 cleavage analysis. The antibody selection covers both specific detection of the apoptotic fragment (Cleaved PARP Asp214) and comprehensive monitoring of PARP-1 status (C2-10). Caspase inhibitors serve as critical experimental controls to confirm the caspase-dependence of observed cleavage events, while appropriate apoptosis inducers generate positive controls for protocol validation [36] [8]. Nuclear-specific loading controls are particularly important given PARP-1's nuclear localization, ensuring accurate normalization in subcellular fractionation experiments.
Interpretation Guidelines and Technical Considerations
Specificity Controls and Validation
Proper interpretation of PARP-1 cleavage data requires implementation of rigorous controls to ensure specificity and biological relevance:
Caspase Dependence Validation: Include caspase inhibitor controls (zVAD-fmk, 20-50 μM) to confirm that observed PARP-1 cleavage is caspase-mediated. The absence of the 89 kDa fragment in inhibitor-treated samples confirms caspase-specific processing [36] [8].
Time Course Analysis: Perform time course experiments (typically 2-24 hours post-treatment) to track cleavage progression. Early appearance of the 89 kDa fragment indicates rapid apoptotic commitment, while delayed cleavage may suggest alternative cell death pathways [41].
Fragment Specificity: Verify antibody specificity using cells with genetic PARP-1 knockdown or knockout. The Cleaved PARP (Asp214) antibody (#9541) should detect only the 89 kDa fragment and not cross-react with other proteins [40].
Alternative Protease Exclusion: Distinguish apoptotic cleavage from necrotic processing. Apoptosis generates 24 kDa and 89 kDa fragments, while necrosis produces a characteristic 50 kDa fragment through lysosomal protease (cathepsin) activity [44].
Integration with Bcl-2 Family Protein Analysis
Integrating PARP-1 cleavage data with Bcl-2 family protein analysis provides a comprehensive view of apoptotic signaling:
Pro-apoptotic Protein Correlation: Monitor concurrent upregulation of pro-apoptotic proteins (BIM, PUMA, Bax) with PARP-1 cleavage to confirm intrinsic pathway activation [36].
Anti-apoptotic Protein Dynamics: Assess compensatory upregulation of anti-apoptotic proteins (BCL2, BCL-xL, MCL1) that may limit PARP-1 cleavage despite apoptotic initiation [36].
Therapeutic Response Assessment: Evaluate PARP-1 cleavage restoration following targeted inhibition of anti-apoptotic BCL2 family proteins (e.g., venetoclax) as an indicator of resensitization to apoptotic stimuli [36].
This integrated approach enables researchers to distinguish between complete apoptotic execution (evidenced by robust PARP-1 cleavage) and abortive apoptotic signaling (characterized by initiator caspase activation without significant PARP-1 cleavage), providing deeper insights into cellular response mechanisms.
The detection of PARP-1 cleavage by Western blot remains an essential methodology for validating apoptotic execution in diverse research contexts, from basic mechanism studies to therapeutic development. The characteristic 89 kDa fragment provides a specific and reliable marker of caspase-mediated apoptotic commitment, with well-established protocols and commercial reagents ensuring reproducible detection across experimental systems. When integrated with Bcl-2 family protein analysis, PARP-1 cleavage monitoring offers a comprehensive framework for mapping apoptotic pathway activation and identifying potential resistance mechanisms. As research continues to reveal novel functions of PARP-1 fragments beyond their traditional role as apoptotic markers, this methodology will maintain its position as a cornerstone of cell death research and therapeutic validation.
Mitochondrial Outer Membrane Permeabilization (MOMP) is a decisive event in the intrinsic apoptotic pathway, serving as a point of no return for programmed cell death. [45] [46] This process is precisely regulated by the Bcl-2 protein family and results in the release of intermembrane space proteins, including cytochrome c, which activates caspase cascades and leads to apoptotic destruction. [47] [45] Assessing MOMP and cytochrome c release is therefore fundamental for validating apoptotic pathways in research, particularly in studies investigating Bcl-2 family proteins and their interactions with other key regulators like PARP1. [6] This guide provides a objective comparison of established methodologies for probing these critical events, offering experimental data and protocols to support research and drug discovery efforts.
Key Assays and Methodologies
Researchers employ several techniques to measure MOMP and cytochrome c release, each with distinct advantages and limitations. The choice of assay depends on the required readout (qualitative vs. quantitative, single-cell vs. population), available equipment, and the specific research context.
Mitochondrial Functional Assay with BH3 Profiling
This cell-free system utilizes isolated mitochondria to decipher the mode of action of BH3 peptides derived from BH3-only proteins, providing insights into mitochondrial priming and dependence on specific anti-apoptotic Bcl-2 family proteins. [47]
- Principle: Mitochondria isolated from cancer cells are incubated with BH3 peptides that mimic the activity of pro-apoptotic proteins. The release of cytochrome c and Smac/Diablo into the supernatant is then measured, typically via Western blotting, to determine the susceptibility of mitochondria to permeabilization. [47]
- Key Optimization: The use of a high ionic strength (HIS) buffer during the MOMP initiation is required for optimal cytochrome c release. This method allows for the functional assessment of dependencies on Bcl-2, Bcl-xL, and Mcl-1. [47]
- Application: It serves as a reliable screening tool for identifying BH3 mimetics with selective toxicity against cancer cell mitochondria. For instance, this assay demonstrated that Bad and Noxa BH3 peptides synergize in inducing MOMP in mitochondria dually protected by Bcl-2/Bcl-xL and Mcl-1. [47]
Immunodetection-Based Cytochrome c Release Assays
These assays detect the translocation of cytochrome c from the mitochondrial intermembrane space to the cytoplasm using antibodies.
Cellular Fractionation with Western Blotting
This traditional method involves physically separating cytoplasmic and mitochondrial fractions from cell populations.
- Workflow: After inducing apoptosis, cells are lysed with a digitonin-based buffer that selectively permeabilizes the plasma membrane but leaves mitochondrial membranes intact. Centrifugation separates the cytosolic fraction (supernatant) from the organellar fraction containing mitochondria (pellet). Cytochrome c levels in each fraction are then analyzed by Western blot. [48]
- Advantages: Provides semi-quantitative data on cytochrome c release from a cell population and is a widely established technique. [47] [48]
- Disadvantages: It is labor-intensive, requires a large number of cells, and lacks single-cell resolution. It also risks cross-contamination between fractions and cannot determine the heterogeneity of the response within a population. [48]
Immunofluorescence Microscopy
This method visualizes cytochrome c release at the single-cell level while preserving spatial information.
- Workflow: Cells are fixed, permeabilized, and stained with an antibody against cytochrome c along with a fluorescent secondary antibody. Its subcellular localization is then examined using a fluorescence microscope. A punctate, mitochondrial pattern indicates retention, while a diffuse, cytoplasmic pattern indicates release. [48]
- Advantages: Offers single-cell resolution and can be combined with other markers, such as cleaved caspase-3, to correlate events.
- Disadvantages: It is difficult to accurately quantitate the number of cells with released cytochrome c, as the diffuse signal can be faint and background fluorescence may interfere. The method is also low-throughput. [48]
Flow Cytometry-Based Assays
Flow cytometry offers a quantitative, high-throughput approach to measure mitochondrial events in single cells.
Selective Permeabilization Assay for Cytochrome c
This method, an adaptation for flow cytometry, uses controlled digitonin permeabilization to label intracellular cytochrome c.
- Principle: Apoptotic and non-apoptotic cells are treated with a low concentration of digitonin, which creates pores in the plasma membrane but leaves the mitochondrial membrane of healthy cells intact. Cells are then fixed and stained with an anti-cytochrome c antibody. In healthy cells, the antibody cannot access cytochrome c retained within mitochondria, resulting in low fluorescence. In apoptotic cells, where cytochrome c has been released into the cytoplasm, the antibody can bind, resulting in high fluorescence intensity, which is measured by the flow cytometer. [49]
- Advantages: Quantitative, allows for single-cell analysis, and can be combined with DNA-binding dyes to correlate cytochrome c release with cell cycle phase. [49]
- Limitations: The assay can underestimate the true extent of apoptosis due to the selective loss of some fragile, digitonin-treated apoptotic cells during processing. [49]
Assessment of Complementary Mitochondrial Parameters
Flow cytometry can also measure other aspects of mitochondrial health that are closely linked to MOMP.
- Loss of Mitochondrial Membrane Potential (ΔΨm): This is measured using potentiometric dyes like Tetramethylrhodamine Ethyl Ester (TMRE). A decrease in TMRE fluorescence indicates mitochondrial membrane depolarization, an event often associated with MOMP. [49]
- Loss of Mitochondrial Cardiolipin: The dye 10-N-Nonyl Acridine Orange (NAO) binds to cardiolipin, a phospholipid in the inner mitochondrial membrane. A decrease in NAO binding, detected by flow cytometry, can indicate mitochondrial damage. [49]
Table 1: Comparison of Key Assays for MOMP and Cytochrome c Release
| Assay Method | Readout | Throughput | Single-Cell Resolution | Key Advantages | Key Limitations |
|---|---|---|---|---|---|
| Mitochondrial BH3 Profiling [47] | Cytochrome c release (Western blot) | Medium | No | Functional insight into Bcl-2 family dependencies; useful for drug screening. | Uses isolated organelles, not whole cells; semi-quantitative. |
| Cellular Fractionation + WB [48] | Cytochrome c localization (Western blot) | Low | No | Widely accepted; no specialized equipment beyond a centrifuge. | Population-average only; risk of fraction cross-contamination. |
| Immunofluorescence [48] | Cytochrome c localization (microscopy) | Low | Yes | Visual confirmation; spatial context preserved. | Subjective and difficult to quantitate accurately; low-throughput. |
| Flow Cytometry (Cytochrome c) [49] | Cytochrome c release (fluorescence intensity) | High | Yes | Quantitative; high-throughput; can be multiplexed with other dyes. | May underestimate apoptosis due to cell loss; requires flow cytometer. |
| Flow Cytometry (ΔΨm/Cardiolipin) [49] | TMRE/NAO fluorescence intensity | High | Yes | Provides complementary data on mitochondrial health. | Does not directly measure cytochrome c release. |
Table 2: Comparative Performance of Flow Cytometry Assays in a Model Study [49] This table summarizes data from a study comparing three flow cytometry methods during staurosporine-induced apoptosis in Jurkat cells.
| Assay Method | Parameter Measured | Correlation with Apoptosis (Cleaved Caspase-3) | Notes on Accuracy |
|---|---|---|---|
| Cytochrome c Release | Cytosolic cytochrome c | Underestimated apoptosis | Selective loss of apoptotic cells during digitonin treatment led to underestimation. |
| TMRE Staining | Mitochondrial membrane potential | Strong correlation | Provided a reliable measure of mitochondrial damage. |
| NAO Staining | Mitochondrial cardiolipin content | Strong correlation | Another reliable indicator of mitochondrial integrity loss. |
Experimental Protocols
This protocol outlines the steps for a functional MOMP assay using mitochondria isolated from cultured cells.
Key Reagent Solutions:
- HIS Buffer: High ionic strength buffer required for optimal cytochrome c release.
- BH3 Peptides: Synthetic peptides corresponding to the BH3 domains of proteins like Bim, Bad, and Noxa.
- Recombinant Proteins: Purified anti-apoptotic Bcl-2 family proteins (e.g., Bcl-2, Bcl-xL, Mcl-1) for protection studies.
Methodology:
- Mitochondria Isolation: Harvest cultured cells (e.g., breast cancer cell lines) and homogenize in isotonic buffer. Isolate mitochondria via differential centrifugation.
- Incubation: Incubate isolated mitochondria in HIS buffer with various BH3 peptides (e.g., Bim, Bad, Noxa) alone or in combination. To test protection, pre-incubate mitochondria with recombinant anti-apoptotic proteins (Bcl-2, Bcl-xL, Mcl-1) before adding a challenging BH3 peptide like Bim.
- Centrifugation: Pellet the mitochondria by high-speed centrifugation.
- Analysis: Collect the supernatant (released content) and dissolve the mitochondrial pellet. Analyze both fractions for cytochrome c and Smac/Diablo by Western blotting.
This protocol details a method for quantifying cytochrome c release in single cells using selective permeabilization and immunofluorescence.
Key Reagent Solutions:
- Digitonin Buffer: Phosphate-buffered saline (PBS) containing a low, optimized concentration of digitonin (e.g., 0.005%) to permeabilize the plasma membrane.
- Fixative: 4% Paraformaldehyde (PFA) in PBS.
- Antibodies: Primary antibody against cytochrome c and a fluorescently-labeled secondary antibody.
Methodology:
- Induce Apoptosis: Treat cells (e.g., Jurkat cells) with an apoptotic agent like staurosporine.
- Selective Permeabilization: Harvest cells and treat them with digitonin buffer for a few minutes at a controlled temperature to create pores in the plasma membrane.
- Fixation: Immediately add PFA to fix the cells, preserving the subcellular localization of cytochrome c.
- Immunostaining: Incubate fixed cells with anti-cytochrome c primary antibody, followed by a fluorescent secondary antibody.
- Flow Cytometry: Analyze the cells using a flow cytometer. Cells that have released cytochrome c will show high fluorescence intensity, while cells with retained cytochrome c will show low fluorescence.
Integration with Broader Apoptosis Research
Understanding MOMP is critical within the broader context of apoptotic pathway validation, especially concerning key proteins like PARP1 and the Bcl-2 family.
- Connection to Bcl-2 Family Proteins: MOMP is directly controlled by the balance between pro- and anti-apoptotic Bcl-2 family members. [46] Research shows that inhibiting anti-apoptotic proteins like Bcl-2, Bcl-xL, and Mcl-1 can overcome resistance to other therapies by promoting MOMP. For example, in uveal melanoma, resistance to combined FAK and MEK inhibition was driven by adaptive BCL2 upregulation, which was overcome by adding the BCL2 inhibitor venetoclax. [36] Similarly, in glioblastoma, simultaneous inhibition of Mcl-1 and Bcl-xL synergized with ER stress inducers to trigger apoptosis. [30]
- Novel Interaction with PARP1: Beyond its established role in DNA repair, PARP1 has been found to interact with BCL2. This interaction suppresses PARP1 enzymatic activity. The BH3 mimetic ABT-737 can displace PARP1 from BCL2, restoring PARP1 activity and promoting a non-apoptotic cell death pathway, even in cells resistant to apoptotic stimuli. [6] This reveals a complex cross-talk where BCL2 can influence cell survival through mechanisms beyond its classic anti-apoptotic function.
Research Reagent Solutions
Table 3: Essential Research Reagents for MOMP and Apoptosis Studies
| Reagent / Tool | Function in Research | Example Use Case |
|---|---|---|
| BH3 Peptides [47] | Mimic pro-apoptotic proteins to probe mitochondrial priming and dependence on anti-apoptotic family members. | Classifying tumors as "primed" or "unprimed" for death; testing efficacy of BH3-mimetic drugs. |
| BH3 Mimetics (e.g., ABT-737, Venetoclax, Obatoclax) [36] [6] [30] | Small molecule inhibitors that bind to and antagonize anti-apoptotic Bcl-2 family proteins to induce MOMP. | Venetoclax for hematological malignancies; Obatoclax to target Mcl-1 in glioblastoma. [30] |
| Selective Permeabilization Agents (Digitonin) [49] | Creates pores in the plasma membrane while leaving mitochondrial membranes intact, enabling analysis of cytochrome c localization. | Used in flow cytometry and fractionation protocols to access the cytosol without damaging mitochondria. |
| Mitochondrial Dyes (TMRE, NAO) [49] | Measure loss of mitochondrial membrane potential (TMRE) or cardiolipin content (NAO), which are downstream consequences of MOMP. | Multiplexing with cytochrome c antibodies in flow cytometry to provide a more comprehensive view of mitochondrial health. |
| Apoptosis Markers (Antibodies vs. Cytochrome c, cleaved PARP, activated Caspases) [47] [36] [6] | Serve as key readouts for confirming apoptosis activation via Western blot, flow cytometry, or immunofluorescence. | Cleaved PARP is a widely used marker for late-stage apoptosis. [36] [6] |
Signaling Pathways and Experimental Workflows
The following diagrams illustrate the core apoptotic pathway and a key experimental workflow discussed in this guide.
Caspases, a family of cysteine-dependent proteases, are crucial mediators of programmed cell death, or apoptosis. These enzymes function as central regulators in maintaining cellular homeostasis, and their dysregulation is implicated in a range of diseases, including cancer and neurodegenerative disorders [50]. Caspases are synthesized as inactive zymogens and undergo proteolytic activation at specific aspartic acid residues in response to apoptotic signals [50]. The caspase family is categorized into initiator caspases (e.g., caspases-2, -8, -9, -10), executioner caspases (e.g., caspases-3, -6, -7), and inflammatory caspases (e.g., caspases-1, -4, -5, -11) [50]. A critical event in the apoptotic cascade is the cleavage of key cellular substrates, such as Poly (ADP-ribose) polymerase (PARP-1), which serves as a marker of apoptosis execution [50] [6]. The Bcl-2 family of proteins acts as a key regulator upstream of caspase activation, modulating mitochondrial outer membrane permeabilization and preventing the activation of caspases and subsequent PARP-1 cleavage [6] [24]. This guide provides a comparative analysis of the primary methods for profiling caspase activation, focusing on activity assays and cleavage-specific antibodies, to support research on apoptotic pathway validation.
Key Methodologies for Detecting Caspase Activation
Activity Assays: Measuring Proteolytic Function
Activity assays measure the catalytic function of caspases using substrates that mimic the natural cleavage site of caspase targets. The core principle involves the use of synthetic peptides containing a caspase-specific cleavage motif (e.g., DEVD for caspase-3/7) linked to a chromophore, fluorophore, or luminescent molecule [51] [52]. Upon substrate cleavage by active caspases in cell lysates or live cells, the signal is generated and can be quantified [51].
A significant challenge in using these assays is the overlapping substrate specificity among caspases. For example, the DEVD sequence, often considered specific for caspase-3, can also be efficiently cleaved by caspase-7 and, to a lesser extent, by other caspases such as caspase-2 [51] [53]. Therefore, data from a single substrate-based assay should not be used in isolation to identify a specific caspase [51]. Best practice involves using these assays in combination with other methods, such as western blotting, to confirm the identity of the active caspase [51].
Cleavage-Specific Antibodies: Detecting Proteolytic Fragments
Cleavage-specific (neo-epitope) antibodies are immunological tools that recognize the new amino or carboxy terminus of a caspase or a caspase substrate only after it has been cleaved [54] [55]. Unlike activity assays, these antibodies do not measure enzymatic activity but instead provide a snapshot of proteolysis by detecting the resulting fragments [55]. A prominent example is the detection of cleaved PARP-1, a well-established hallmark of apoptosis execution [50] [6].
These antibodies are highly specific because they recognize neo-epitopes that are exposed only after caspase cleavage and are absent in the full-length protein [55]. This makes them excellent tools for specifically identifying apoptotic cells in complex mixtures, such as tissue sections or heterogeneous cell populations, via techniques like western blotting, immunohistochemistry (IHC), and flow cytometry [54] [55]. For instance, antibodies specific to the cleaved form of caspase-3 at Asp175 are widely used to mark apoptotic cells in IHC experiments [54].
Comparative Analysis of Methodologies
The table below summarizes the core characteristics, strengths, and limitations of caspase activity assays and cleavage-specific antibodies.
Table 1: Direct Comparison of Caspase Profiling Methodologies
| Feature | Activity Assays | Cleavage-Specific Antibodies |
|---|---|---|
| What is Detected | Enzymatic (proteolytic) activity [51] | Presence of specific cleavage fragments (neo-epitopes) [55] |
| Key Applications | - Kinetic studies in live cells [52]- High-throughput screening [51]- Measuring activity in cell lysates [51] | - Western blotting [54]- Immunohistochemistry (IHC) [54]- Immunofluorescence (IF) [54]- Flow Cytometry [54] |
| Key Advantages | - Provides kinetic data- Amenable to live-cell imaging- Suitable for automation & HTS [52] | - High specificity for apoptosis- Spatial context in tissues (IHC)- Identifies specific cleaved protein [55] |
| Major Limitations | - Overlapping substrate specificity can lead to cross-reactivity [51] [53]- Does not distinguish between different caspases with similar motifs [53] | - Does not measure enzyme activity directly- Typically an endpoint measurement- Dependent on antibody quality and specificity [50] |
| Integration with PARP-1/Bcl-2 Research | Ideal for screening compounds that induce caspase activation upstream of PARP-1 cleavage. | Directly confirms PARP-1 cleavage; Bcl-2 overexpression can be correlated with suppression of cleavage [6] [24]. |
Experimental Workflows and Protocols
Protocol: Caspase-3/7 Activity Assay in Live Cells Using a Fluorescent Reporter
This protocol utilizes a stable fluorescent reporter system for real-time, single-cell analysis of executioner caspase dynamics [52].
- Stable Cell Line Generation: Transduce cells of interest with a lentiviral construct encoding a caspase-3/7 biosensor (e.g., ZipGFP with a DEVD cleavage motif) and a constitutive fluorescent marker (e.g., mCherry) for normalization [52].
- Experimental Treatment and Imaging:
- Plate the reporter cells in an appropriate imaging chamber.
- Treat cells with apoptosis-inducing agents (e.g., carfilzomib, oxaliplatin) in the presence or absence of a pan-caspase inhibitor (e.g., zVAD-FMK, 20 µM) as a control [52].
- Perform time-lapse live-cell imaging over 24-120 hours using a fluorescence microscope equipped with environmental control (37°C, 5% CO₂) [52].
- Acquire images in both GFP (reporter activation) and mCherry (cell presence) channels at regular intervals (e.g., every 2-4 hours).
- Data Analysis: Quantify the GFP/mCherry fluorescence ratio over time. An increase in the ratio indicates caspase-3/7 activation. Use automated image analysis software to track single-cell fates and generate kinetic curves [52].
Protocol: Detecting Cleaved Caspase-3 and PARP by Western Blotting
This is a standard endpoint protocol to confirm caspase activation and downstream substrate cleavage.
- Sample Preparation:
- Treat and lyse cells in RIPA buffer supplemented with protease inhibitors.
- Centrifuge lysates and quantify protein concentration.
- Gel Electrophoresis and Transfer:
- Separate equal amounts of protein (20-30 µg) by SDS-PAGE (4-20% gradient gel).
- Transfer proteins to a PVDF or nitrocellulose membrane.
- Immunoblotting:
- Block the membrane with 5% non-fat milk in TBST for 1 hour.
- Incubate with primary antibodies overnight at 4°C.
- Cleaved Caspase-3 (Asp175) antibody (e.g., Cell Signaling #9664) [54].
- Cleaved PARP (Asp214) antibody.
- Loading Control (e.g., GAPDH or β-Actin).
- Wash the membrane and incubate with an HRP-conjugated secondary antibody for 1 hour at room temperature.
- Develop the blot using enhanced chemiluminescence (ECL) substrate and visualize.
- Expected Results: Apoptotic samples will show bands for cleaved caspase-3 (~17/19 kDa) and cleaved PARP (~89 kDa), while untreated controls should not [6].
Visualizing the Experimental Workflow for Apoptosis Validation
The following diagram illustrates a logical workflow that integrates these methods to validate apoptotic signaling, placing the Bcl-2/PARP-1 axis in context.
Research Reagent Solutions
The table below lists essential reagents for profiling caspase activation, with specific examples from the literature.
Table 2: Key Research Reagents for Caspase and Apoptosis Analysis
| Reagent / Tool | Specific Example (where provided) | Function and Application |
|---|---|---|
| Cleavage-Specific Caspase-3 Antibody | Asp175 (e.g., Cell Signaling #9664, #9579) [54] | Detects activated caspase-3 fragment by WB, IHC, IF, and Flow Cytometry. Essential for confirming caspase-3 processing. |
| Caspase Activity Assay Kit | Fluorometric or Colorimetric DEVD-based kits (e.g., Abcam ab39401) [51] | Measures caspase-3/7 activity in cell lysates in a plate-reader format. Suitable for higher-throughput screening. |
| Live-Cell Caspase Reporter | ZipGFP-DEVD-mCherry construct [52] | Enables real-time, kinetic imaging of caspase-3/7 activation at single-cell resolution in 2D and 3D cultures. |
| Pan-Caspase Inhibitor | zVAD-FMK [52] | A cell-permeable broad-spectrum caspase inhibitor. Serves as a critical control to confirm the caspase-dependent nature of an observed phenotype. |
| Neo-Epitope Antibodies (Pan) | Anti-DXXD C-terminal antibodies [55] | A pool of antibodies that recognize a common "DXXD" motif exposed after caspase cleavage. Can be used to immunoprecipitate and discover novel caspase substrates. |
| PARP-1 Cleavage Antibody | - | Detects the signature 89 kDa fragment of PARP-1 generated by executioner caspases, serving as a gold-standard marker for apoptosis execution. |
Profiling caspase activation is fundamental to apoptosis research. Activity assays and cleavage-specific antibodies offer distinct yet complementary insights: the former provides dynamic, functional data on protease activity, while the latter delivers precise, spatial information on proteolytic events. Acknowledging the limitation of substrate cross-reactivity in activity assays is critical for accurate interpretation [51] [53]. The most robust strategy for validating apoptotic pathways involves an integrated approach, combining real-time activity kinetics with endpoint immunoblotting for key markers like cleaved caspase-3 and PARP-1. This multi-faceted methodology, framed within the context of regulatory proteins like Bcl-2, provides a comprehensive and convincing analysis of cell death mechanisms in both basic research and drug discovery.
The functional validation of combination therapies targeting apoptotic pathways represents a critical frontier in overcoming treatment resistance in oncology. This guide objectively compares the performance of combined Bcl-2 family inhibition and PARP suppression across diverse model systems. The Bcl-2 protein family, comprising both anti-apoptotic (e.g., BCL2, BCL-XL, MCL1) and pro-apoptotic members, constitutes the primary regulatory checkpoint for mitochondrial apoptosis [12]. Simultaneously, PARP1, a key DNA damage sensor, coordinates repair of single-strand breaks through poly(ADP-ribosyl)ation [56]. Mounting evidence reveals direct crosstalk between these pathways, including a novel protein interaction between BCL2 and PARP1 that suppresses PARP1 enzymatic activity [6]. This scientific foundation provides the mechanistic rationale for combining BH3 mimetics—small molecules that inhibit anti-apoptotic BCL2 family proteins—with PARP inhibitors to overcome therapeutic resistance. The following sections present comparative experimental data, detailed methodologies, and essential research tools for validating this combination strategy across hematologic and solid tumor models.
Comparative Performance Data: Combination Therapy Across Model Systems
Table 1: Efficacy of BH3 Mimetics and PARP Inhibitors in Hematologic Malignancy Models
| Cell Line/Model | BH3 Mimetic | PARP Inhibitor | Combination Effect | Key Metrics | Proposed Mechanism |
|---|---|---|---|---|---|
| Diffuse Large B-Cell Lymphoma (DLBCL) [6] | ABT-737 | ABT-888 | Re-established PARP1 activity & DNA repair; Promoted non-apoptotic death | • Dose-dependent PARP1 displacement from BCL2• Increased PARP1 enzymatic activity• Enhanced DNA repair (Comet assay) | Disruption of BCL2-PARP1 complex |
| Primary CLL Cells (with stromal protection) [6] | ABT-737 | ABT-888 | Overcame stroma-mediated resistance to MNNG | • Increased Annexin V/PI positivity• Reduced NAD/ATP levels | BCL2 inhibition overcoming microenvironmental protection |
| Acute Myeloid Leukemia (AML) - M4/M5 subtypes [57] | (Not combined) | (Frontline Chemo) | PARP1-mediated parthanatos associated with superior clinical outcomes | • 3-fold improvement in survival (HR=0.28-0.37)• Characteristic nuclear "ring" morphology | Caspase-independent parthanatos cell death pathway |
Table 2: Efficacy of BH3 Mimetics and PARP Inhibitors in Solid Tumor Models
| Cancer Type/Cell Line | BH3 Mimetic | PARP Inhibitor | Combination Effect | Synergy Score/Effect Size | Proposed Mechanism |
|---|---|---|---|---|---|
| Ovarian Cancer Models [58] | IS21 (pan-BH3 mimetic) | Olaparib | Sensitized ovarian cancer cells to PARP inhibition | • Additive to synergistic Loewe scores• Reduced colony formation | Potentiation of apoptotic pathway via Mcl-1 targeting |
| Melanoma Models [58] | IS21, ABT-199, S63845 | MAPK inhibitors | Sensitized melanoma to MAPK pathway inhibition | • Additive to synergistic Loewe scores• Reduced cell viability (MTT assay) | Bcl-xL and Mcl-1 protein levels predicted sensitivity |
| T-cell Acute Lymphoblastic Leukemia (T-ALL) [58] | IS21, ABT-199 | Chemotherapy (Doxorubicin, Vincristine) | Sensitized leukemia cells to chemotherapy | • Reduced IC50 values• Enhanced apoptosis (caspase activation) | BAX/BAK-dependent apoptosis |
Experimental Protocols for Combination Therapy Validation
Cell Viability and Synergy Assessment
Protocol 1: Cell Viability and Combination Synergy Screening
- Objective: Quantify dose-response relationships and synergistic effects of BH3 mimetics and PARP inhibitors.
- Key Reagents: BH3 mimetics (e.g., ABT-199, IS21, ABT-737), PARP inhibitors (e.g., Olaparib, ABT-888), cell culture media, viability assay reagents (MTT, MTS, CellTiter-Glo).
- Procedure:
- Cell Plating: Plate cells in 96-well plates at a density ensuring logarithmic growth throughout the experiment.
- Compound Treatment: After 24 hours, treat cells with a matrix of BH3 mimetic and PARP inhibitor concentrations (e.g., 8x8 dosing matrix). Include single-agent and vehicle (DMSO) controls.
- Incubation: Incubate for 72-96 hours, depending on cell doubling time.
- Viability Measurement:
- Data Analysis:
- Calculate IC50 values for single agents using GraphPad Prism or equivalent software.
- Analyze combination data with SynergyFinder 2.0 to calculate Loewe synergy scores. Scores <0 indicate antagonism, 0-5 additivity, and ≥5 synergy [58].
Apoptosis and Cell Death Mechanism Analysis
Protocol 2: Distinguishing Apoptosis from Parthanatos
- Objective: Determine the mode of cell death (caspase-dependent apoptosis vs. caspase-independent parthanatos) induced by combination treatment.
- Key Reagents: Annexin V-FITC/PI staining kit, pan-caspase inhibitor (zVAD-fmk), PARP inhibitor, DAPI, antibodies for PAR polymer and AIF.
- Procedure:
- Treatment Groups: Establish treatment groups: Vehicle, BH3 mimetic, PARP inhibitor, Combination, and Combination + zVAD-fmk.
- Phosphatidylserine Exposure: Harvest cells after treatment (e.g., 24h) and stain with Annexin V-FITC and Propidium Iodide (PI). Analyze by flow cytometry. Annexin V+/PI- indicates early apoptosis [6] [57].
- Caspase Dependency: Pre-treat cells with zVAD-fmk (20-50 µM) for 1 hour before adding combination therapy. Persistent cell death in the presence of zVAD suggests a non-apoptotic, caspase-independent pathway like parthanatos [57].
- Parthanatos Markers:
- PAR Accumulation: Fix cells and immunostain for PAR polymers. Detect increased nuclear PAR signal by fluorescence microscopy or flow cytometry [57].
- AIF Translocation: Perform subcellular fractionation or immunofluorescence to monitor the translocation of Apoptosis-Inducing Factor (AIF) from mitochondria to the nucleus, a hallmark of parthanatos [57].
- Nuclear Morphology: Stain nuclei with DAPI and examine for morphology. Apoptosis typically shows condensed, fragmented chromatin, while parthanatos can exhibit distinctive peripheral "ring-shaped" chromatin patterns [57].
Protein Interaction and Signaling Pathway Mapping
Protocol 3: Validating BCL2-PARP1 Interaction and Disruption
- Objective: Confirm the physical interaction between BCL2 and PARP1 and assess its disruption by BH3 mimetics.
- Key Reagents: Antibodies for BCL2, PARP1, and relevant BCL2 family proteins (Bim, Bax), RIPA lysis buffer, Protein A/G beads.
- Procedure:
- Cell Lysis and Fractionation: Lyse treated cells in RIPA buffer with protease/phosphatase inhibitors. For subcellular localization studies, perform nuclear and cytoplasmic fractionation [6].
- Immunoprecipitation (IP): Incubate cell lysates with an antibody against BCL2 or a control IgG. Capture immune complexes with Protein A/G beads.
- Western Blot Analysis:
- Resolve IP eluates and total cell lysates by SDS-PAGE.
- Transfer to PVDF membrane and probe with antibodies against PARP1 and BCL2. Co-precipitation of PARP1 with BCL2 confirms interaction [6].
- To assess functional consequences, probe lysates for markers of DNA damage (e.g., γH2AX) and apoptosis (cleaved caspases, PARP1 cleavage).
- Functional Enzyme Assay: Use a PARP enzymatic activity ELISA to measure the effect of BCL2 and BH3 mimetics on PARP1 function. Purified GST-BCL2 suppresses PARP1 activity, which is restored by adding ABT-737 [6].
Signaling Pathways and Experimental Workflows
Pathway Diagram: Mechanistic Basis for BH3 Mimetic and PARP Inhibitor Combination
Diagram 1: Mechanism of BH3 Mimetic and PARP Inhibitor Combination. BH3 mimetics (red) promote apoptosis by inhibiting anti-apoptotic BCL2 proteins, freeing pro-apoptotic proteins to trigger MOMP. They also disrupt the BCL2-PARP1 complex, restoring PARP1 activity. Combined with PARP inhibitors (which block SSBR and can trap PARP1), this drives dual death pathways: apoptosis and parthanatos [58] [6] [57].
Workflow Diagram: Experimental Strategy for Functional Validation
Diagram 2: Experimental Workflow for Validating Combination Therapy. A sequential approach begins with model characterization, progresses through quantitative synergy screening, and culminates in mechanistic dissection of cell death pathways and functional genetic validation [58] [6] [57].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Investigating BH3 Mimetic and PARP Inhibitor Combinations
| Reagent Category | Specific Examples | Key Function/Application | Experimental Notes |
|---|---|---|---|
| BH3 Mimetics | ABT-199 (Venetoclax), ABT-737, ABT-263 (Navitoclax), IS21, S63845 | Inhibit anti-apoptotic BCL2 family proteins; ABT-199 is BCL2-selective; IS21 is a pan-inhibitor; S63845 targets MCL1 [58] [12] [59]. | Check selectivity profile; BCL-XL inhibitors can cause thrombocytopenia [12]. |
| PARP Inhibitors | Olaparib, ABT-888 (Veliparib), Rucaparib, Niraparib | Inhibit PARP enzymatic activity; induce PARP "trapping" on DNA with varying potency [60] [56]. | Different inhibitors have varying trapping potency; consider catalytic inhibition vs. trapping in experimental design [60]. |
| Viability/Proliferation Assays | MTT, MTS, CellTiter-Glo, Trypan Blue Exclusion | Quantify cell viability and proliferation in response to treatment [58]. | Choose assay based on model: CellTiter-Glo for suspension cells, MTT/MTS for adherent lines [58]. |
| Apoptosis Assays | Annexin V/PI staining, Caspase-3/7 activity assays, zVAD-fmk | Detect phosphatidylserine exposure, caspase activation, and confirm caspase-dependent apoptosis [6] [57]. | zVAD is a pan-caspase inhibitor used to rule out caspase-dependent death [57]. |
| Parthanatos Detection | Anti-PAR antibody, Anti-AIF antibody, DAPI staining | Detect PAR polymer accumulation, AIF nuclear translocation, and characteristic nuclear morphology [57]. | "Ring-shaped" nuclear DAPI staining is indicative of parthanatos [57]. |
| Protein Interaction Tools | Antibodies (BCL2, PARP1, Bim, Bax), IP kits, Western Blot reagents | Validate protein-protein interactions and downstream signaling events [58] [6]. | Nuclear fractionation may be required to study BCL2-PARP1 interactions [6]. |
| Genetic Tools | siRNA/shRNA (BAK, BAX, PARP1) | Validate mechanistic dependency on specific proteins [58]. | Combined BAX/BAK knockout confers strong resistance to intrinsic apoptosis [58] [61]. |
Resolving Experimental Challenges in Apoptosis Pathway Analysis
Addressing Off-Target Effects in Pharmacological Apoptosis Induction
The therapeutic induction of apoptosis in cancer cells represents a cornerstone of modern oncology. However, the efficacy of pro-apoptotic drugs is often limited by off-target effects that can compromise therapeutic outcomes and patient safety. This guide provides a comparative analysis of experimental approaches for validating apoptotic pathways, with a specific focus on the interplay between PARP1 and BCL2 family proteins. As research in this field advances, understanding and addressing off-target effects has become crucial for developing more precise and effective cancer therapies. The complex interactions between key apoptotic regulators require sophisticated experimental methodologies to unravel potential secondary effects that may influence treatment efficacy and toxicity profiles. This review synthesizes current methodologies and findings to guide researchers in designing robust experiments that account for these complexities.
Comparative Analysis of Apoptosis-Targeting Agents
Table 1: Comparative profiles of pharmacological apoptosis inducers and their documented off-target effects
| Agent | Primary Target | Documented Off-Target Effects | Experimental Evidence | Therapeutic Implications |
|---|---|---|---|---|
| ABT-737 | BCL2, BCL-XL, BCL-w | PARP1 activation; Enhanced DNA repair [6] | Displaced PARP1 from BCL2, restoring PARP1 activity and promoting non-apoptotic cell death in lymphoma models [6] | May counteract intrinsic efficacy through PARP1-mediated survival pathways |
| Obatoclax | Pan-BCL2 (MCL-1, BCL-XL, BCL-2) | Autophagy disruption; Enhanced ER stress [30] | Disrupted autolysosome formation, potentiating tunicamycin-induced ER stress in glioblastoma [30] | Synergistic with ER stress inducers; potential for combination therapies |
| Venetoclax | BCL2 (selective) | Limited off-target effects reported | Selective inhibition reduces thrombocytopenia compared to navitoclax [12] | Improved safety profile for hematological malignancies |
| Navitoclax | BCL2, BCL-XL, BCL-w | Dose-limiting thrombocytopenia [12] | BCL-XL inhibition impairs platelet survival [12] | Clinical use limited by on-target, off-tissue toxicity |
| PARP inhibitors (Olaparib, Niraparib) | PARP1/2 | Variable efficacy in cisplatin-resistant models [62] | Complex response profiles in ovarian cancer cell lines; collagen I-mediated resistance [62] | Resistance mechanisms may limit efficacy beyond BRCA-mutant contexts |
Experimental Approaches for Pathway Validation
Assessing BCL2-PARP1 Interaction Dynamics
The interface between BCL2 and PARP1 represents a critical node for potential off-target effects that requires rigorous experimental validation. A seminal study demonstrated that BCL2 directly binds to and suppresses PARP1 enzymatic activity, thereby inhibiting PARP1-dependent DNA repair in diffuse large B-cell lymphoma cells. Treatment with the BH3 mimetic ABT-737 displaced PARP1 from BCL2 in a dose-dependent manner, re-establishing PARP1 activity and DNA repair while promoting non-apoptotic cell death [6]. This unexpected effect reveals a compensatory survival mechanism that may undermine therapeutic efficacy.
Protocol for Co-Immunoprecipitation of BCL2-PARP1 Complex:
- Prepare fractionated lysates from target cells (e.g., HT, Toledo, or OCI-LY8 DLBCL lines)
- Incubate lysates with anti-BCL2 antibody conjugated to beads overnight at 4°C
- Include controls with normal IgG and BCL2-overexpressing cells
- Wash beads extensively with lysis buffer
- Elute bound proteins and subject to Western blotting with anti-PARP1 antibody
- To test drug effects, pre-treat cells with ABT-737 (100nM) for 6 hours before lysis [6]
Table 2: Research reagent solutions for apoptosis pathway analysis
| Research Tool | Specific Application | Key Utility in Off-Target Assessment |
|---|---|---|
| ABT-737 | BH3-mimetic targeting BCL2/BCL-XL/BCL-w | Displaces PARP1 from BCL2; reveals non-apoptotic death pathways [6] |
| Comet Assay (Alkaline) | DNA strand break detection | Quantifies ABT-737-induced PARP1 activity through DNA repair assessment [6] |
| Obatoclax | Pan-BCL2 inhibitor (including MCL-1) | Uncovers autophagy disruption as off-target effect enhancing ER stress [30] |
| Annexin V/PI Staining | Apoptosis quantification | Differentiates apoptotic vs. non-apoptotic cell death mechanisms [6] |
| PARP Activity ELISA | Direct PARP1 function measurement | Quantifies BCL2-mediated suppression of PARP1 enzymatic activity [6] |
| JC-1 Mitochon-drial Membrane Potential Dye | Apoptosis early detection | Measures BCL2 family functional activity at mitochondria [30] |
Evaluating DNA Repair Pathway Modulation
Beyond its canonical apoptotic functions, BCL2 influences DNA repair pathways through direct protein interactions, creating another dimension of potential off-target effects. Research has demonstrated that BCL2 overexpression diminishes non-homologous end joining (NHEJ) efficiency by sequestering KU80 protein outside the nucleus. This effect is associated with a repair switch to error-prone PARP1-dependent end-joining (PARP1-EJ) [63]. This mechanism provides a rationale for combining PARP inhibitors with radiation therapy in BCL2-overexpressing prostate cancer cells.
Protocol for Gamma-H2AX Foci Analysis for DSB Repair:
- Culture BCL2-overexpressing and control cells on coverslips
- Treat cells with ionizing radiation (2-4 Gy)
- At specific timepoints post-irradiation (0.5, 2, 6, 24h), fix cells with 4% PFA
- Permeabilize with 0.25% Triton X-100 and block with 5% BSA
- Incubate with anti-phospho-histone H2AX (Ser139) antibody overnight at 4°C
- Apply fluorescent secondary antibody and counterstain with DAPI
- Quantify foci per nucleus using fluorescence microscopy [63]
Analyzing Cell Death Modalities
Comprehensive assessment of cell death modalities is essential for identifying off-target effects of apoptotic agents. Researchers should employ multiple complementary approaches to distinguish between apoptotic and non-apoptotic death mechanisms.
Protocol for Differentiating Apoptotic vs. Non-Apoptotic Death:
- Treat cells with apoptotic inducers (e.g., ABT-737, obatoclax) with/without modulators
- Assess cell death using Annexin V/PI staining with flow cytometry
- Simultaneously measure caspase-3/7 activity using luminescent substrates
- Evaluate PARP cleavage by Western blotting (89 kDa cleavage fragment)
- Determine metabolic markers (NAD+/ATP levels) to identify necrotic components
- For autophagy assessment, monitor LC3-I to LC3-II conversion and p62 degradation [6] [30]
Integrated Signaling Pathways in Apoptosis Modulation
Figure 1: Integrated apoptotic signaling network showing primary targets and documented off-target effects. Solid lines represent canonical pathways; dashed lines indicate off-target or alternative pathways. BCL2 overexpression sequesters KU80, impairing NHEJ and promoting PARP1-EJ. BH3 mimetics like ABT-737 disrupt BCL2-PARP1 interaction, activating PARP1 as an off-target effect. Obatoclax disrupts autophagy, enhancing ER stress-induced death.
Discussion and Research Recommendations
The experimental evidence compiled in this guide demonstrates that off-target effects in apoptosis induction can significantly influence therapeutic outcomes. The unexpected activation of PARP1 by ABT-737 illustrates how targeted agents may engage compensatory mechanisms that potentially limit their efficacy [6]. Similarly, the disruption of autophagy by obatoclax reveals how apparently synergistic combinations may work through unanticipated mechanisms [30]. These findings highlight the critical importance of comprehensive pathway validation in apoptosis research.
To effectively address off-target effects in future research, we recommend:
- Implementing multiple complementary cell death assessment methods to distinguish apoptotic from non-apoptotic mechanisms
- Systematically evaluating DNA repair pathway engagement following BCL2 inhibition
- Testing apoptosis inducers in relevant co-culture systems that mimic tumor microenvironment protection
- Assessing both short-term and long-term adaptive responses to apoptotic stress
- Considering the balance between on-target efficacy and off-target consequences in combination therapy design
As the field advances toward more selective agents like PARP1-specific inhibitors [64] and next-generation BCL2 antagonists, rigorous assessment of off-target effects will remain essential for developing safe and effective apoptosis-based therapies. The experimental frameworks presented here provide a foundation for such comprehensive evaluations.
In cell biology, distinguishing between different forms of regulated cell death (RCD) is crucial for accurate experimental interpretation, especially in therapeutic contexts like cancer research and drug development. Apoptosis, necroptosis, and ferroptosis represent distinct RCD pathways with unique molecular mechanisms and functional consequences. While apoptosis is typically immunologically silent, both necroptosis and ferroptosis are lytic, inflammatory forms of cell death that release damage-associated molecular patterns (DAMPs) and potent inflammatory mediators [27]. Understanding these differences is fundamental to research focusing on apoptotic pathway validation through PARP-1 and Bcl-2 family protein analysis.
The molecular pathways of these cell death modalities employ different executioners: apoptosis is executed by caspase-3/7, necroptosis by phosphorylated MLKL, pyroptosis by gasdermin D pores, and ferroptosis through iron-dependent lipid peroxidation without a known specific executioner protein [65]. This review provides a comprehensive comparison of detection methodologies, experimental protocols, and key reagents to enhance assay specificity when investigating these critical cellular processes.
Comparative Analysis of Cell Death Pathways
Key Characteristics and Detection Methods
Table 1: Comparative characteristics of apoptosis, necroptosis, and ferroptosis.
| Feature | Apoptosis | Necroptosis | Ferroptosis |
|---|---|---|---|
| Morphology | Cell shrinkage, chromatin condensation, apoptotic bodies, preserved membrane integrity [27] [66] | Cell swelling, plasma membrane rupture, loss of cellular integrity [67] | Plasma membrane rupture driven by lipid peroxidation [68] |
| Key Regulators | Caspases (CASP3/7, CASP8/9), Bcl-2 family, PARP-1 [27] [24] | RIPK1, RIPK3, pMLKL [27] [67] | GPX4, SLC7A11, ACSL4, iron metabolism [69] [68] |
| Inflammatory Response | Non-inflammatory [27] | Strongly inflammatory [67] | Immunogenic (releases DAMPs) [68] |
| Primary Biochemical Markers | DNA fragmentation (laddering), caspase activation, PARP cleavage [66] [24] | Phospho-MLKL oligomerization, membrane disruption [67] | Lipid ROS accumulation, iron dependency, GSH depletion [69] |
| Inhibitors | Z-VAD-FMK (pan-caspase), Bcl-2 overexpression [24] | Necrostatin-1 (RIPK1), GSK'872 (RIPK3) [67] | Ferrostatin-1, Liproxstatin-1, iron chelators [69] |
Table 2: Detection assays for differentiating cell death pathways.
| Detection Method | Apoptosis | Necroptosis | Ferroptosis | Key Considerations |
|---|---|---|---|---|
| Microscopy | Membrane blebbing, chromatin condensation (Hoechst/DAPI) [66] | Cellular swelling, membrane rupture [67] | Membrane rupture without nuclear condensation [69] | Electron microscopy reveals ultrastructural differences [66] |
| Viability Assays | Metabolic activity decrease (MTT, resazurin) [70] | Metabolic activity decrease with lytic features [67] | Metabolic activity decrease with iron dependence [69] | Use matched DMSO controls; avoid evaporation [70] |
| Membrane Integrity | Annexin V+/PI- (early), Annexin V+/PI+ (late) [6] | Annexin V+/PI+ (primary necrosis) [67] | PI+ with lipid peroxidation [69] | Timing is critical for interpretation |
| Biochemical Assays | Caspase activity, DNA laddering, TUNEL [66] | p-MLKL detection (Western blot, immunofluorescence) [67] | C11-BODIPY for lipid ROS, intracellular iron detection [69] | Multiparametric approach recommended [69] |
| Pathway-Specific | PARP cleavage detection [24] | Necrosome formation (RIPK1/RIPK3) [67] | GPX4 degradation, GSH depletion [69] | Confirm with genetic or pharmacological inhibition |
Molecular Pathways and Crosstalk
The following diagram illustrates the core molecular components and critical crosstalk points between apoptosis, necroptosis, and ferroptosis pathways:
The intricate relationship between PARP-1 and Bcl-2 represents a critical node in cell death regulation. Bcl-2 not only suppresses apoptosis by binding pro-apoptotic BH3-domain factors but also localizes to the nucleus where it directly interacts with PARP-1, suppressing its enzymatic activity and inhibiting PARP-1-dependent DNA repair [6]. This interaction creates a functional link between anti-apoptotic signaling and DNA repair mechanisms, which can be therapeutically exploited. The BH3 mimetic ABT-737 can displace PARP-1 from Bcl-2, re-establishing PARP-1 activity and promoting non-apoptotic cell death even in apoptosis-resistant contexts [6].
Experimental Protocols for Pathway Discrimination
Multiparametric Assessment Strategy
The following workflow diagram outlines an integrated experimental approach for distinguishing cell death mechanisms:
Specific Methodological Details
Annexin V/PI Staining for Flow Cytometry:
- Protocol: Harvest cells and wash with cold PBS. Resuspend in binding buffer containing Annexin V-FITC and propidium iodide (PI) [6]. Incubate for 15 minutes in the dark before analysis by flow cytometry.
- Interpretation: Annexin V+/PI- indicates early apoptosis; Annexin V+/PI+ suggests late apoptosis or secondary necrosis; Annexin V-/PI+ indicates primary necrosis/necroptosis [6] [66].
- Considerations: Always include unstained, single-stained, and compensation controls. Analyze within 1 hour of staining.
Caspase Activity Assay:
- Protocol: Use fluorogenic caspase substrates (e.g., DEVD-AFC for caspase-3/7, IETD-AFC for caspase-8). Lyse cells and incubate with substrate at 37°C. Measure fluorescence release over time [66].
- Interpretation: Increased caspase activity confirms apoptotic pathway engagement. Lack of activity in dying cells suggests alternative death mechanisms.
Lipid Peroxidation Measurement:
- Protocol: Load cells with C11-BODIPY 581/591 (2 µM) for 30 minutes at 37°C. After stimulation, analyze by flow cytometry monitoring shift from red to green fluorescence [69].
- Interpretation: Increased green/red fluorescence ratio indicates lipid peroxidation characteristic of ferroptosis.
Western Blot Analysis for Key Markers:
- Target Proteins: PARP-1 cleavage (89 kDa fragment), phosphorylated MLKL, GPX4 degradation, caspase-3 cleavage [24] [67] [69].
- Protocol: Standard SDS-PAGE followed by transfer. Use specific antibodies and normalize to loading controls.
- Critical Controls: Include positive controls (e.g., cells treated with known inducers) and specificity controls (e.g., inhibitor pretreatment).
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key research reagents for distinguishing cell death pathways.
| Reagent | Function/Application | Specificity | Example Usage |
|---|---|---|---|
| Z-VAD-FMK | Irreversible pan-caspase inhibitor | Apoptosis inhibitor | Confirm caspase-dependent death (10-20 µM) [66] |
| Necrostatin-1 | RIPK1 inhibitor | Necroptosis inhibitor | Differentiate necroptosis (10-30 µM) [67] |
| Ferrostatin-1 | Lipid antioxidant | Ferroptosis inhibitor | Confirm ferroptosis (1-10 µM) [69] |
| Annexin V Conjugates | Phosphatidylserine exposure detection | Early apoptosis marker | Flow cytometry or microscopy [6] |
| C11-BODIPY 581/591 | Lipid peroxidation sensor | Ferroptosis detection | Live-cell imaging or flow cytometry [69] |
| Anti-pMLKL Antibody | Detects phosphorylated MLKL | Necroptosis confirmation | Western blot, immunofluorescence [67] |
| Anti-cleaved PARP-1 Antibody | Detects PARP-1 cleavage (89 kDa) | Apoptosis marker | Western blot analysis [24] |
| ABT-737 | BH3 mimetic, Bcl-2 inhibitor | Disrupts Bcl-2/PARP-1 interaction | Research Bcl-2 function (100 nM) [6] |
Accurate discrimination between apoptosis, necroptosis, and ferroptosis requires a multiparametric approach that integrates morphological assessment, biochemical assays, and molecular confirmation. The interplay between PARP-1 and Bcl-2 family proteins represents a critical regulatory node that influences cell fate decisions following death stimuli. Researchers should employ pharmacological inhibitors alongside pathway-specific molecular markers to unambiguously identify the dominant cell death mechanism in their experimental systems. This comprehensive approach enables more accurate interpretation of experimental results, particularly in therapeutic contexts where targeting specific cell death pathways holds promise for cancer treatment and other diseases.
Strategies for Overcoming Intrinsic and Acquired Resistance to Apoptosis Inducers
A cornerstone of successful cancer therapy is the elimination of malignant cells through the induction of programmed cell death, or apoptosis. However, the development of resistance to apoptosis inducers represents a critical barrier to treatment efficacy, leading to disease progression and poor clinical outcomes. This resistance can be intrinsic, present before treatment begins, or acquired, developing after exposure to therapeutic agents [71] [72]. Research has increasingly demonstrated that key proteins within cancer cells, particularly those in the Bcl-2 family and the DNA repair protein PARP-1, play pivotal roles in mediating this resistance. The interaction between Bcl-2 and PARP-1 presents a compelling axis for understanding and overcoming treatment failure. This guide provides a comparative analysis of strategies designed to counteract these resistance mechanisms, offering researchers and drug development professionals a data-driven overview of the most promising therapeutic avenues.
Core Resistance Mechanisms: PARP-1 and Bcl-2 Family Proteins
The PARP-1 and Bcl-2 Interaction Axis
A critical, non-canonical function of the anti-apoptotic protein Bcl-2 is its direct interaction with and suppression of PARP-1 activity. PARP-1 is a nuclear enzyme essential for the detection and repair of DNA single-strand breaks. When active, it synthesizes poly(ADP-ribose) chains on itself and other nuclear proteins, which serves as a signal for the DNA repair machinery.
- Mechanism of Suppression: Bcl-2, which can localize to the nucleus, binds directly to PARP-1, thereby inhibiting its enzymatic activity. This interaction suppresses PARP-1-dependent DNA repair, leaving DNA damage unresolved [6] [73].
- Therapeutic Exploitation: The BH3 mimetic ABT-737 can displace PARP-1 from Bcl-2 in a dose-dependent manner. This displacement re-establishes PARP-1 activity and DNA repair capacity. Paradoxically, in the context of DNA damage, this re-activation can push cells toward a non-apoptotic cell death pathway, offering a strategy to kill cells resistant to classic apoptosis [6].
- Clinical Correlation: In B-lymphoblastic leukemia/lymphoma (B-ALL), a positive correlation between PARP-1 and Bcl-2 protein overexpression has been observed. This co-expression is furthermore associated with a complex karyotype, indicating greater genomic instability [73].
Dysregulation of the Apoptotic Machinery
The intrinsic (mitochondrial) apoptotic pathway is frequently disabled in cancer cells, primarily through an imbalance in the Bcl-2 family of proteins.
- Anti-apoptotic Protein Overexpression: Malignant cells often overexpress anti-apoptotic proteins like BCL-2, BCL-xL, and MCL-1. These proteins sequester pro-apoptotic activators (like BIM) and prevent the activation of the executioner proteins BAX and BAK. Without active BAX/BAK, the mitochondrial outer membrane remains intact, and cytochrome c cannot be released to initiate caspase activation [72] [74].
- Pro-apoptotic Protein Downregulation: Resistance is further reinforced by the decreased expression or functional inactivation of pro-apoptotic proteins such as BAX, BAK, and BIM [72].
- Efflux Pumps and Metabolic Adaptations: Beyond the core apoptosis machinery, resistance is mediated by the overexpression of drug efflux pumps (e.g., MDR1/ABCB1) that reduce intracellular drug concentrations, and metabolic shifts like the Warburg effect, which promotes cell survival [71] [72].
Table 1: Core Mechanisms of Resistance to Apoptosis Inducers
| Resistance Mechanism | Key Proteins/Pathways Involved | Functional Consequence |
|---|---|---|
| Direct Protein Interaction | Bcl-2 binding to PARP-1 [6] [73] | Suppression of PARP-1 enzymatic activity & inhibition of DNA repair |
| Anti-apoptotic Protein Overexpression | BCL-2, BCL-xL, MCL-1 [75] [72] | Sequestration of pro-apoptotic factors; blockade of MOMP |
| Pro-apoptotic Protein Deficiency | BAX, BAK, BIM, p53 mutation [75] [72] | Failure to initiate intrinsic apoptosis signaling |
| Drug Efflux | MDR1/ABCB1, MRP1 [71] [72] | Reduced intracellular concentration of chemotherapeutic drugs |
| Altered Glucose Metabolism | HK2, PI3K/AKT pathway [71] | Increased ATP production and anti-apoptotic signaling |
| Enhanced Antioxidant Defenses | NRF2, Glutathione (GSH) [71] | Scavenging of therapy-induced ROS, reducing DNA damage |
Comparative Analysis of Strategic Interventions
Several targeted strategies have been developed to overcome the aforementioned resistance mechanisms. The following table provides a comparative overview of these approaches.
Table 2: Strategic Comparison of Apoptosis-Resistance Interventions
| Therapeutic Strategy | Molecular Target | Mechanism of Action | Representative Agents | Overcomes Resistance to |
|---|---|---|---|---|
| BH3 Mimetics | BCL-2, BCL-xL, MCL-1 [75] [76] | Displace pro-apoptotic proteins from anti-apoptotic partners, triggering MOMP | Venetoclax (ABT-199), ABT-737 [6] [75] | Intrinsic resistance due to BCL-2 overexpression |
| PARP Inhibitors | PARP-1/2 enzymes [73] | Trap PARP on DNA; induce synthetic lethality in HR-deficient cells | Olaparib, ABT-888 [6] | Acquired resistance from enhanced DNA repair |
| SMAC Mimetics | IAP proteins (XIAP, survivin) [75] | Antagonize IAPs to relieve caspase inhibition and promote cell death | LCL161, BV6 [75] | Resistance from IAP-mediated caspase suppression |
| Death Receptor Agonists | Extrinsic Pathway (DR4/DR5) [75] [72] | Activate caspase-8 directly, bypassing intrinsic pathway blocks | TRAIL, DR5 agonists [75] | Intrinsic resistance from BCL-2 overexpression |
| Natural Product Sensitizers | Multiple (e.g., ROS, BCL-2) [74] [76] | Modulate signaling pathways; downregulate anti-apoptotic proteins | Curcumin, Quercetin [75] [76] | Broad-spectrum resistance; can chemosensitize |
| Combination: BH3 Mimetic + PARP Inhibitor | BCL-2 & PARP-1 [6] | Bcl-2 inhibition releases PARP-1; PARPi blocks repair → synergistic cell death | ABT-737 + ABT-888 [6] | Acquired, multi-factorial resistance |
Essential Experimental Protocols for Validating Efficacy
To evaluate the success of strategies aimed at overcoming apoptosis resistance, several key experimental protocols are routinely employed in both basic and translational research. The methodologies below are critical for assessing drug efficacy and mechanism of action.
Protocol 1: BH3 Profiling to Measure Apoptotic Priming
Purpose: To dynamically measure how close a cell is to the apoptotic threshold ("primed"), which predicts sensitivity to BH3 mimetics and other apoptosis inducers [72].
Workflow:
- Cell Preparation: Obtain single-cell suspensions from cell cultures or patient-derived samples (e.g., bone marrow, tumor biopsies).
- Permeabilization: Treat cells with digitonin to create pores in the plasma membrane while keeping mitochondrial membranes intact.
- BH3 Peptide Exposure: Incubate permeabilized cells with synthetic peptides corresponding to the BH3 domains of various pro-apoptotic proteins (e.g., BIM, BAD, NOXA). Each peptide has a specific binding profile to anti-apoptotic proteins.
- Mitochondrial Output Measurement: Measure the release of cytochrome c from the mitochondria, typically via immunofluorescence or flow cytometry.
- Data Interpretation: A high degree of cytochrome c release after exposure to a specific BH3 peptide indicates dependence on the corresponding anti-apoptotic protein for survival and predicts sensitivity to its inhibitor.
Protocol 2: PARP Activity ELISA
Purpose: To directly quantify the enzymatic activity of PARP-1 in cell extracts, useful for confirming target engagement by inhibitors or detecting functional changes due to Bcl-2 interaction [6].
Workflow:
- Sample Preparation: Lyse cells and fractionate to obtain nuclear extracts. Determine protein concentration.
- Reaction Setup: Add cell extracts to a 96-well plate coated with histones (a PARP substrate), along with a reaction buffer containing NAD+ (the substrate for poly(ADP-ribosylation)).
- Inhibition/Stimulation (Optional): To test the effect of a drug, pre-incubate extracts with a PARP inhibitor (e.g., ABT-888) or a Bcl-2 inhibitor (e.g., ABT-737).
- Detection: After the reaction, use an antibody specific for poly(ADP-ribose) (PAR) conjugated to a detection enzyme. Add a chemiluminescent or colorimetric substrate to generate a signal proportional to PARP activity.
Protocol 3: Alkaline Comet Assay for DNA Repair Capacity
Purpose: To measure the ability of a cell to repair DNA strand breaks, a key factor in resistance to DNA-damaging agents [6].
Workflow:
- Treatment and Embedding: Treat cells with a DNA-damaging agent (e.g., MNNG, etoposide). Subsequently, embed single cells in low-melting-point agarose on a microscope slide.
- Lysis and Unwinding: Lyse cells in situ to remove membranes and proteins, leaving "nucleoids" (supercoiled DNA attached to a nuclear matrix). Incubate in an alkaline solution (pH >13) to unwind DNA and reveal strand breaks.
- Electrophoresis: Subject the slides to electrophoresis under alkaline conditions. DNA fragments migrate away from the nucleus towards the anode, forming a "comet tail."
- Staining and Analysis: Stain DNA with a fluorescent dye (e.g., SYBR Gold). The percentage of DNA in the tail and the tail moment are quantitative measures of DNA strand breaks. Less repair is indicated by more residual damage (larger tails) at later time points post-damage.
Signaling Pathway and Experimental Workflow Visualizations
Apoptosis Resistance and Therapeutic Intervention Pathways
The following diagram illustrates the core mechanisms of intrinsic and acquired resistance to apoptosis inducers, highlighting the key nodes where therapeutic interventions act.
Diagram 1: Key nodes in apoptosis resistance and therapeutic intervention. Dashed red lines indicate inhibitory actions of resistance mechanisms. Solid green lines and arrows show the inhibitory or activating actions of therapeutic strategies. The core intrinsic apoptosis pathway is shown in blue.
Bcl-2 and PARP-1 Interaction Experimental Workflow
This diagram outlines a key experimental workflow used to validate the functional interaction between Bcl-2 and PARP-1 and to test strategies that disrupt it.
Diagram 2: Experimental workflow for validating Bcl-2 and PARP-1 interaction and therapeutic disruption.
The Scientist's Toolkit: Essential Research Reagents
The following table details key reagents and assays essential for researching apoptosis resistance mechanisms and screening potential therapeutic compounds.
Table 3: Essential Research Reagents for Apoptosis Resistance Studies
| Reagent / Assay Kit | Primary Function | Specific Application in Resistance Research |
|---|---|---|
| BH3 Mimetics (Venetoclax, ABT-737) | Inhibit anti-apoptotic BCL-2 family proteins [6] [75] | Tool compounds to test "addiction" to BCL-2/BCL-xL; used in BH3 profiling. |
| PARP Inhibitors (Olaparib, ABT-888) | Inhibit PARP-1/2 enzymatic activity [6] | Induce synthetic lethality; test synergy with Bcl-2 inhibitors; assess DNA repair dependency. |
| PARP Activity ELISA Kit | Quantifies PARP enzyme activity in cell extracts [6] | Directly measure the functional consequence of Bcl-2 binding or PARP inhibitor efficacy. |
| Comet Assay Kit | Measures DNA single/double-strand breaks [6] | Evaluate the DNA repair capacity of cells and the impact of its inhibition. |
| Annexin V / Propidium Iodide (PI) | Flow cytometry-based apoptosis detection [6] | Quantify early/late apoptosis and necrosis in response to treatment combinations. |
| siRNA/shRNA Libraries | Knockdown specific gene expression (e.g., BCL2, PARP1) [77] | Validate the functional role of a specific protein in mediating resistance. |
| Selective MCL-1 Inhibitors | Target MCL-1 protein for degradation or inhibition | Overcome resistance conferred by MCL-1 upregulation, often used in combination with BCL-2 inhibitors. |
Interpreting Inconclusive Results in PARP-1 Cleavage and BCL-2 Interaction Studies
The interplay between Poly(ADP-ribose) polymerase-1 (PARP-1) and B-cell lymphoma 2 (BCL-2) family proteins represents a critical interface in cell death regulation. While PARP-1 cleavage is a established apoptotic biomarker, its functional consequences and relationship with anti-apoptotic BCL-2 family members continue to yield complex, sometimes contradictory findings. This guide systematically compares experimental approaches for investigating PARP-1/BCL-2 interactions, synthesizes conflicting data from key studies, and provides standardized methodologies to resolve mechanistic ambiguities. We present quantitative analyses of PARP-1 cleavage patterns, BCL-2 interaction interfaces, and functional outcomes across different cellular contexts, offering researchers a framework for interpreting inconclusive results in apoptosis pathway validation.
Apoptosis regulation hinges on precise interactions between protein families that control cell survival decisions. The BCL-2 protein family, comprising both pro-apoptotic and anti-apoptotic members, constitutes a critical regulatory node for mitochondrial outer membrane permeabilization and intrinsic apoptosis execution [12]. PARP-1, a nuclear enzyme involved in DNA repair, becomes proteolytically cleaved during cell death, generating signature fragments that serve as apoptosis biomarkers [9]. While these pathways have been extensively studied independently, emerging research reveals direct molecular interactions between BCL-2 and PARP-1 that extend beyond canonical apoptosis regulation [6] [14].
The investigative landscape is complicated by several factors: the dual roles of PARP-1 fragments in promoting either survival or death, the cell-type specific expression of BCL-2 family members, and the diverse death stimuli that engage different proteolytic systems. This guide objectively compares experimental approaches, reconciles contradictory findings, and provides standardized methodologies for validating apoptotic pathways through PARP-1 and BCL-2 family protein analysis.
PARP-1 Cleavage Fragments: Signatures of Cell Death Proteases
PARP-1 serves as a substrate for multiple proteases activated during cell death, with each protease generating specific cleavage fragments that serve as signatures of particular death pathways.
Protease-Specific PARP-1 Cleavage Patterns
Table 1: PARP-1 Cleavage Fragments by Different Proteases
| Protease | Cleavage Fragments | Cell Death Context | Molecular Functions of Fragments |
|---|---|---|---|
| Caspase-3/7 | 24 kDa (DBD) + 89 kDa (CD+AMD) | Apoptosis | 24 kDa: DNA binding, inhibits repair; 89 kDa: reduced DNA binding [9] |
| Calpain | 55 kDa + 62 kDa | Necrosis, excitotoxicity | Not well characterized [9] |
| Granzyme A | 50 kDa + 62 kDa | Immune-mediated killing | Potential role in necrosis-like death [9] |
| MMPs | 35 kDa + 52 kDa | Inflammation-associated death | Potential role in necroptosis [9] |
Functional Consequences of PARP-1 Cleavage
The functional outcomes of PARP-1 cleavage are fragment-dependent and context-specific:
- Caspase-generated 24 kDa fragment: Contains the DNA-binding domain and acts as a trans-dominant inhibitor of intact PARP-1, potentially conserving cellular energy during apoptosis [9].
- Caspase-generated 89 kDa fragment: Comprises the automodification and catalytic domains with reduced DNA binding capacity, potentially enabling specific signaling functions [9].
- Uncleavable PARP-1 mutants: Studies demonstrate that expression of uncleavable PARP-1 (PARP-1UNCL) or the 24 kDa fragment (PARP-124) confers protection from oxygen/glucose deprivation damage, while the 89 kDa fragment (PARP-189) is cytotoxic [78].
- Regulation of inflammatory response: PARP-1 cleavage fragments differentially modulate NF-κB activity, with PARP-189 increasing NF-κB and iNOS promoter activity while PARP-1UNCL and PARP-124 decrease inflammatory markers [78].
BCL-2 Family Proteins: Regulators of Mitochondrial Apoptosis
The BCL-2 protein family constitutes a critical regulatory system for mitochondrial apoptosis, with members categorized by their structural and functional characteristics.
BCL-2 Family Classification and Functions
Table 2: BCL-2 Protein Family Members and Functions
| Protein | Class | BH Domains | Primary Function | Cancer Relevance |
|---|---|---|---|---|
| BCL-2 | Anti-apoptotic | BH1-4 | Inhibits MOMP, binds pro-apoptotic members | Overexpressed in follicular lymphoma, CLL [12] |
| BCL-XL | Anti-apoptotic | BH1-4 | Inhibits MOMP, high expression in solid tumors | Thrombocytopenia with inhibitors [12] |
| MCL-1 | Anti-apoptotic | BH1-4 | Inhibits MOPP, regulates development | Cardiac toxicity with inhibitors [12] |
| BAX | Pro-apoptotic | BH1-3 | Mediates MOMP, cytosolic in healthy cells | Frequently mutated in cancer [79] |
| BAK | Pro-apoptotic | BH1-3 | Mediates MOMP, mitochondrial in healthy cells | Frequently mutated in cancer [79] |
| BIM | BH3-only | BH3 | Activator, binds and activates BAX/BAK | Essential for apoptosis initiation [36] [79] |
| PUMA | BH3-only | BH3 | Activator/sensitizer, p53 target | Dysregulated in cancer [36] |
BCL-2 Interaction Networks
BCL-2 family proteins establish a complex interaction network that determines cell fate:
- Hydrophobic groove binding: Anti-apoptotic BCL-2 proteins contain a surface groove formed by BH1-3 domains that engages the BH3 domains of pro-apoptotic partners [12].
- BH3-only proteins as sensors: Diverse cellular stresses activate specific BH3-only proteins (e.g., BIM, PUMA) that either directly activate BAX/BAK or neutralize anti-apoptotic members [79].
- Complex interaction patterns: Bimolecular Fluorescence Complementation (BiFC) studies reveal that BCL-2 family members establish a complex interaction network in living cells, with differential binding affinities between specific pairs [79].
Experimental Approaches to PARP-1 and BCL-2 Analysis
Methodologies for Detecting Protein Interactions
- Co-immunoprecipitation and Western Blotting: Dutta et al. demonstrated direct BCL-2-PARP1 interaction through co-immunoprecipitation in diffuse large B-cell lymphoma cells, showing that this interaction suppresses PARP1 enzymatic activity [6] [14].
- Bimolecular Fluorescence Complementation (BiFC): This technique enables visualization of weak and transient interactions between BCL-2 family members in living cells, revealing that BH3-only proteins Bim, Puma, and Noxa can directly interact with Bax and Bak with different affinities [79].
- Enzyme-linked Immunosorbent Assay (ELISA): Quantitative assessment of PARP1 enzymatic activity showed that purified GST-BCL2 protein inhibits PARP1 activity on immobilized histones in a dose-dependent manner [6].
- Comet Assay: Used to evaluate DNA repair capacity, this method demonstrated that BCL2-PARP1 interaction inhibits PARP1-dependent DNA repair, which can be restored by BH3 mimetic ABT-737 [6].
Modulation of BCL-2 Family Interactions
- BH3-mimetic compounds: Small molecules like ABT-737, navitoclax (ABT-263), and venetoclax (ABT-199) bind the hydrophobic groove of anti-apoptotic BCL-2 proteins, displacing pro-apoptotic partners and promoting apoptosis [12].
- Genetic approaches: siRNA-mediated knockdown of specific BCL-2 family members identifies essential survival proteins in different cancer types, with combinatorial knockdown of MCL-1 and BCL-XL showing synergistic induction of apoptosis in glioblastoma [30].
- Pharmacological combinations: BH3-mimetics show enhanced efficacy when combined with other agents, such as ER stress inducers, through disruption of adaptive survival mechanisms [30].
Interpreting Inconclusive Results: Key Considerations
Contextual Factors Influencing Experimental Outcomes
- Cell type specificity: BCL-2 family expression patterns vary significantly between cell types, influencing dependence on specific anti-apoptotic members and responses to BH3-mimetics [30].
- Death stimulus differences: The same death stimulus can activate different proteases and signaling pathways depending on intensity and duration, generating distinct PARP-1 fragments [9].
- Subcellular localization: BCL-2 localizes to both mitochondrial and nuclear compartments, with nuclear BCL-2 potentially engaging PARP-1 in DNA damage response [6].
- Compensatory mechanisms: Inhibition of one anti-apoptotic BCL-2 family member often leads to upregulation of others, creating resistance to single-agent therapy [36].
Resolution of Contradictory Findings
Several approaches can help resolve contradictory findings in PARP-1/BCL-2 studies:
- Multiple protease inhibition: Using specific inhibitors for different protease classes (caspases, calpains, cathepsins, granzymes) can identify which pathways are active in specific death contexts [9].
- BH3 profiling: This functional assay measures mitochondrial priming to determine dependence on specific anti-apoptotic BCL-2 family members, predicting sensitivity to BH3-mimetics [6].
- Time-course experiments: PARP-1 cleavage occurs at different timepoints depending on the death stimulus and cellular context, requiring detailed kinetic analysis [9].
- Interaction mapping: Techniques like BiFC can detect weak or transient interactions that might be missed by traditional co-immunoprecipitation [79].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for PARP-1 and BCL-2 Studies
| Reagent | Type | Primary Function | Example Applications |
|---|---|---|---|
| ABT-737 | BH3-mimetic | Inhibits BCL-2, BCL-XL, BCL-w | Displaces PARP1 from BCL-2, restores PARP1 activity [6] |
| Venetoclax (ABT-199) | BH3-mimetic | Selective BCL-2 inhibitor | Clinical use in CLL, AML; research on BCL-2 dependence [12] |
| Obatoclax | Pan-BCL-2 inhibitor | Inhibits MCL-1, BCL-XL, BCL-2 | Synergizes with ER stress inducers in glioblastoma [30] |
| Q-VD-OPh | Caspase inhibitor | Broad-spectrum caspase inhibitor | Distinguishes caspase-dependent and independent death [36] |
| Tunicamycin | ER stress inducer | Inhibits N-linked glycosylation | Combined with BCL-2 inhibitors to disrupt autophagy [30] |
| PARP inhibitor (ABT-888) | PARP inhibitor | Inhibits PARP enzymatic activity | Investigates synthetic lethality with BCL-2 inhibition [6] |
Signaling Pathway Integration
The following diagram illustrates the complex interactions between BCL-2 family proteins and PARP-1 cleavage events in apoptosis regulation:
The investigation of PARP-1 cleavage and BCL-2 family interactions requires integrated experimental approaches that account for cellular context, dynamic protein networks, and complementary death pathways. While PARP-1 cleavage remains a valuable apoptosis biomarker, its functional interpretation must consider the specific proteases involved and the cellular context. Similarly, BCL-2 family interactions represent a complex regulatory network where functional outcomes depend on the balance between pro- and anti-apoptotic members. The emerging direct interaction between BCL-2 and PARP-1 reveals additional complexity in apoptosis regulation, suggesting non-canonical roles for these proteins in cell death control. By applying the standardized methodologies and interpretive frameworks presented in this guide, researchers can more effectively resolve contradictory findings and advance our understanding of apoptotic signaling networks in health and disease.
The selection of appropriate experimental models is a cornerstone of biomedical research, particularly in the complex study of apoptotic pathways such as those involving PARP-1 and Bcl-2 family proteins. These models serve as critical intermediaries between molecular discoveries and clinical applications, providing insights into fundamental biological processes and therapeutic potential. The validation of apoptotic pathways requires sophisticated model systems that can accurately recapitulate the intricate balance of cell survival and death mechanisms, especially in the context of cancer research where dysregulation of these pathways is a hallmark of disease progression. Model selection directly influences data interpretation, reproducibility, and ultimately, the translational potential of research findings. As such, researchers must navigate a complex landscape of experimental systems, each with distinct advantages, limitations, and appropriate applications. This guide provides a comprehensive comparison of prevalent model systems—from traditional two-dimensional cell cultures to advanced co-culture platforms and in vivo validation—focusing on their utility for investigating PARP-1 and Bcl-2 mediated apoptotic pathways.
Comparative Analysis of Experimental Model Systems
Defining the Model Spectrum: From Simple to Complex Systems
Experimental models for apoptosis research exist along a spectrum of biological complexity, each offering distinct advantages for specific research questions. Two-dimensional (2D) cell cultures represent the most simplified system, where cells grow as monolayers on plastic surfaces, allowing for straightforward experimental manipulation and high-throughput screening. However, these models lack the physiological context of three-dimensional tissue organization [80]. Three-dimensional (3D) cultures, including spheroids and organoids, bridge this gap by enabling cell-cell and cell-matrix interactions that more closely mimic in vivo conditions, resulting in more physiologically relevant cell signaling, differentiation, and drug response patterns [80] [81]. Co-culture systems further enhance complexity by incorporating multiple cell types to model the tumor microenvironment (TME), allowing researchers to study the paracrine relationships between cancer cells and stromal components such as fibroblasts, endothelial cells, and immune cells [82]. At the highest end of the complexity spectrum, in vivo models provide the complete biological context, including systemic effects, intact tissue architecture, and functional readouts, but present challenges in mechanistic analysis and scalability [82].
Quantitative Comparison of Model Characteristics
Table 1: Comprehensive Comparison of Experimental Model Systems for Apoptosis Research
| Model Type | Physiological Relevance | Throughput | Cost | Technical Complexity | Key Applications in Apoptosis Research |
|---|---|---|---|---|---|
| 2D Monoculture | Low: Altered cell morphology, polarity, and signaling due to artificial substrate [80] | High: Suitable for large-scale drug screening [82] | Low: Simple media requirements, minimal specialized equipment [80] | Low: Established protocols, easy maintenance [80] | Initial drug screening, mechanistic studies of isolated pathways, genetic manipulation |
| 3D Models (Spheroids/Organoids) | Medium-High: 3D architecture, cell-cell interactions, gradient formation [80] [81] | Medium: More complex than 2D but adaptable to screening formats [82] | Medium: Require specialized matrices and culture conditions [80] | Medium: Specialized culture techniques, characterization challenges [82] | Therapy response studies, microenvironmental effects on apoptosis, spatial organization of cell death |
| Co-culture Systems | Medium-High: Cell-cell signaling, stromal interactions, TME modeling [82] [83] | Medium-Low: Increased complexity reduces throughput [82] | Medium-High: Multiple cell sources, potential specialized equipment [83] | Medium-High: Requires optimization of multiple cell type ratios and interactions [82] [83] | Stroma-mediated therapeutic resistance, bidirectional signaling in apoptosis, tumor-stroma dynamics |
| In Vivo Models | High: Intact physiology, systemic effects, immune context [82] | Low: Time-intensive, ethical considerations limit scale [82] | High: Animal maintenance, specialized facilities | High: Surgical procedures, monitoring, regulatory compliance [84] | Validation of in vitro findings, systemic toxicity assessment, therapeutic efficacy in complex organisms |
Table 2: Model-Specific Insights into PARP-1 and Bcl-2 Apoptosis Pathways
| Model Type | PARP-1 Function Assessment | Bcl-2 Family Interactions | Limitations for Apoptosis Studies |
|---|---|---|---|
| 2D Monoculture | Direct pharmacological inhibition studies; DNA damage response assessment [6] | BH3-mimetic testing; overexpression/silencing studies [6] | Lack of microenvironment-mediated survival signals; exaggerated drug sensitivity [80] |
| 3D Models | Spatial assessment of PARP-1 activation in response to gradient-forming agents | Evaluation of penetration and efficacy of Bcl-2 inhibitors in tissue-like structures [81] | Limited stromal component may overlook critical survival signals |
| Stroma-Co-culture | Study of stromal protection from PARP inhibitor-induced apoptosis [82] | Investigation of stromal influence on anti-apoptotic protein expression [82] [6] | Complex data interpretation due to multiple cell populations; potential masking of cell-specific effects |
| In Vivo Models | Integrated assessment of PARP-1 function in physiological DNA repair and cell death contexts [84] | Evaluation of Bcl-2 dependency in intact tissue context with complete survival signaling [6] | Difficulty in real-time monitoring of pathway activation; challenges in mechanistic dissection |
Experimental Protocols for Model Establishment
Establishing a Stroma-Cancer Cell Co-culture System
Co-culture models provide invaluable platforms for studying apoptotic signaling in physiologically relevant contexts, particularly for investigating how stromal elements influence cancer cell survival and treatment resistance. The following protocol outlines the establishment of a fibroblast-cancer cell co-culture system for apoptosis studies:
Materials Required:
- Cancer cell line of interest (e.g., diffuse large B-cell lymphoma lines for Bcl-2 studies [6])
- Stromal cells (cancer-associated fibroblasts, bone marrow-derived stromal cells, or commercially available fibroblast lines)
- Appropriate culture medium (DMEM or RPMI-1640 supplemented with 10% FBS and 1% penicillin/streptomycin)
- Tissue culture-treated plates (standard or low-attachment for 3D co-cultures)
- Extracellular matrix components (Matrigel, collagen I) for 3D systems [82] [85]
- Small molecule inhibitors (PARP inhibitors, BH3 mimetics) for pathway modulation [6]
Methodology:
- Stromal Layer Preparation: Plate stromal cells at appropriate density (e.g., 5×10^4 cells/well in 48-well plates) and allow adherence for 24 hours [6]. For 3D systems, embed stromal cells in ECM hydrogels at concentrations optimized for the specific matrix (typically 1-5 mg/mL collagen I or diluted Matrigel) [82] [85].
Cancer Cell Introduction: Seed cancer cells at predetermined ratios relative to stromal cells. Optimal ratios vary by system but typically range from 1:1 to 20:1 (cancer:stromal cells) [6] [83]. In the established blood-brain barrier/glioblastoma co-culture model, a sequential seeding approach is used where stromal components are established before introducing cancer cells [83].
Culture Maintenance: Maintain co-cultures in media formulations that support both cell types, typically using a 1:1 mixture of condition-specific media or selecting a compromise medium. Monitor cultures regularly for morphological changes and cell viability.
Experimental Treatment: Administer apoptotic stimuli (e.g., PARP inhibitors, BH3 mimetics, chemotherapeutic agents) once co-cultures are established (typically 24-72 hours after cancer cell introduction). Include monoculture controls for both cell types to distinguish cell-autonomous from microenvironment-mediated effects.
Endpoint Analysis: Utilize appropriate assays to quantify apoptosis (annexin V/PI staining, caspase activation) with cell-type-specific markers to distinguish responses in different compartments [6]. For advanced models, live-cell imaging can track real-time apoptosis dynamics.
This co-culture approach has demonstrated significant utility in apoptosis research, exemplified by studies showing that stromal cells can protect lymphoma cells from PARP inhibitor-induced death through Bcl-2-mediated mechanisms [6].
Validation Methodologies for Apoptotic Pathway Analysis
Rigorous validation of apoptotic pathway activation is essential across all model systems. The following methodologies provide comprehensive assessment of PARP-1 and Bcl-2 function:
PARP-1 Activity Assessment:
- Enzymatic Activity Assays: Measure PARP-1 activity using ELISA-based formats with immobilized histones as substrates. Monitor the impact of Bcl-2 expression on PARP-1 function, as nuclear Bcl-2 has been shown to suppress PARP-1 enzymatic activity [6].
- PARP-1 Cleavage Analysis: Detect caspase-mediated PARP-1 cleavage (89 kDa fragment) by western blotting as a hallmark of apoptotic commitment. Note that certain cell death stimuli can activate PARP-1 independently of caspase cleavage [6].
- Functional DNA Repair Assessment: Utilize comet assays under alkaline conditions to evaluate single-strand break repair capacity, which is PARP-1 dependent. Studies have demonstrated that Bcl-2 overexpression can suppress PARP-1-dependent DNA repair, measurable through increased DNA damage in comet assays [6].
Bcl-2 Family Protein Analysis:
- Subcellular Localization: Perform fractionation studies followed by immunoblotting to determine Bcl-2 localization. Importantly, Bcl-2 has been documented in nuclear fractions of lymphoid tumor cells, where it interacts with PARP-1 [6].
- Inhibition Studies: Employ BH3 mimetics (e.g., ABT-737) to disrupt Bcl-2-PARP-1 interactions and monitor reactivation of PARP-1 activity and DNA repair capacity [6].
- Flow Cytometric Analysis: Use cell surface and intracellular staining to evaluate Bcl-2 family protein expression in specific cellular compartments within co-culture systems, employing cell-type-specific markers for multiplexed analysis [86].
Integrated Cell Death Assessment:
- Multiparametric Flow Cytometry: Combine annexin V/propidium iodide staining with cell lineage markers to quantify apoptosis in specific cell populations within co-cultures [6] [86].
- Metabolic Profiling: Monitor NAD+ and ATP levels following PARP activation, as PARP-1 hyperactivation consumes NAD+, leading to energetic crisis and non-apoptotic cell death [6].
- Clonogenic Survival: Perform colony formation assays following transient drug exposure to evaluate long-term reproductive cell death, particularly relevant for DNA damage response studies involving PARP-1 function [6].
Visualizing Experimental Workflows and Signaling Pathways
Apoptosis Signaling Pathway Involving PARP-1 and Bcl-2
Integrated Model Selection Workflow
Essential Research Reagent Solutions
Table 3: Key Reagents for Apoptosis Pathway Analysis Across Model Systems
| Reagent Category | Specific Examples | Research Applications | Model System Compatibility |
|---|---|---|---|
| PARP Inhibitors | ABT-888 (Veliparir), Olaparib, Talazoparib | Selective targeting of PARP-1 enzymatic activity; combination studies with other agents [6] | 2D, 3D, Co-culture, In Vivo |
| BH3 Mimetics | ABT-737, ABT-199 (Venetoclax), Navitoclax | Disruption of Bcl-2 interactions with pro-apoptotic partners and PARP-1 [6] | 2D, 3D, Co-culture, In Vivo |
| Extracellular Matrices | Matrigel, Collagen I, Fibrin, Synthetic PEG Hydrogels | 3D culture establishment; stromal co-culture support; migration/invasion studies [82] [81] [85] | 3D, Co-culture |
| Cell Death Assays | Annexin V/Propidium Iodide, Caspase Activity Assays, TUNEL, CellTiter-Glo | Quantification of apoptotic versus non-apoptotic cell death; temporal analysis of cell death [6] [81] | 2D, 3D, Co-culture |
| DNA Damage Assessment | Comet Assay, γH2AX Staining, Alkaline Unwinding Assay | Evaluation of DNA strand break formation and repair capacity [6] | 2D, 3D, Co-culture |
| Stromal Cell Markers | Anti-FAP, Anti-αSMA, Anti-Vimentin, Anti-CD90 | Identification and validation of stromal components in co-culture systems [82] [85] | Co-culture, In Vivo |
The selection of appropriate experimental models for studying PARP-1 and Bcl-2 mediated apoptosis requires careful consideration of research objectives, methodological constraints, and translational aspirations. Simple 2D systems offer unparalleled utility for reductionist mechanistic studies and high-throughput compound screening, while 3D models and co-culture systems provide essential physiological context that modulates apoptotic responses. The emerging recognition of non-canonical Bcl-2 functions, including its nuclear localization and interaction with PARP-1, underscores the importance of model systems that preserve appropriate subcellular compartmentalization [6]. Similarly, the influence of stromal elements on therapeutic response highlights the value of co-culture platforms that recapitulate tumor microenvironment interactions [82] [83]. Ultimately, a strategic approach that leverages complementary models across the complexity spectrum—from reductionist systems to in vivo validation—provides the most robust framework for advancing our understanding of apoptotic pathways and developing novel therapeutic interventions. This integrated methodology ensures that mechanistic insights gained from simplified systems are validated in increasingly physiological contexts, enhancing the translational potential of research findings in apoptosis and cancer biology.
Cross-Pathway Validation and Therapeutic Translation
Apoptosis, or programmed cell death, is a fundamental biological process crucial for maintaining tissue homeostasis, eliminating damaged or unnecessary cells, and ensuring proper embryonic development [87] [88]. Its dysregulation is a hallmark of numerous diseases, most notably cancer, making the accurate detection and quantification of apoptosis a critical task in basic research and drug discovery [89]. The process is characterized by a cascade of biochemical and morphological events, including caspase activation, phosphatidylserine externalization, DNA fragmentation, and changes in mitochondrial membrane potential [87] [88].
Given the complexity and rapid progression of apoptosis, no single assay captures the entire spectrum of these changes. Instead, researchers must choose from a plethora of available techniques, each with unique advantages, limitations, and windows of detection within the apoptotic timeline [90] [89]. The choice of assay is further complicated when research is focused on specific molecular pathways. This guide provides an objective, data-driven comparison of common apoptosis assays, with a particular emphasis on their utility in research centered on validating apoptotic pathways through the analysis of key biomarkers like PARP-1 and the Bcl-2 protein family.
Core Apoptotic Signaling Pathways
The Intrinsic and Extrinsic Pathways of Apoptosis
A clear understanding of the molecular pathways is essential for selecting the appropriate detection assay. The following diagram illustrates the core apoptotic signaling cascades.
The intrinsic pathway (mitochondrial) is initiated by internal cellular stressors like DNA damage, leading to the disruption of the Bcl-2 family protein balance and subsequent mitochondrial outer membrane permeabilization (MOMP). This results in the release of cytochrome c, which forms the apoptosome and activates caspase-9 [87]. In contrast, the extrinsic pathway (death receptor) begins with the binding of extracellular ligands to cell surface death receptors, forming a death-inducing signaling complex (DISC) that activates caspase-8 [87] [88]. Both pathways converge on the activation of executioner caspases-3 and -7, which orchestrate the hallmark morphological and biochemical changes of apoptosis, such as the cleavage of PARP-1 and DNA fragmentation [87].
Comparative Analysis of Apoptosis Assay Performance
The following tables provide a comparative summary of common apoptosis assays based on their fundamental characteristics and performance metrics, synthesizing data from multiple methodological studies [91] [90] [92].
Table 1: Characteristics and Applications of Key Apoptosis Assays
| Assay Category | Specific Target/Principle | Phase of Apoptosis Detected | Key Advantages | Key Limitations |
|---|---|---|---|---|
| Phosphatidylserine Exposure (Annexin V) | Externalized PS on outer leaflet | Early | Detects early apoptosis; can distinguish early apoptotic (Annexin V+/PI-) from late apoptotic/necrotic (Annexin V+/PI+) cells [91]. | Susceptible to false positives from mechanical damage; requires live cell handling [91]. |
| Caspase Activity | Activated caspase-3/7, -8, or -9 | Early/Mid | High specificity for apoptosis; indicates commitment to death pathway; various formats (fluorescent, luminescent) available [88]. | Transient activation window may be missed in endpoint assays; does not confirm cell death has occurred [89]. |
| Mitochondrial Potential (e.g., JC-1, TMRM) | ΔΨm collapse | Early/Mid | Can detect apoptosis before caspase activation; useful for intrinsic pathway studies [88]. | Loss of ΔΨm is not exclusively apoptotic; can occur in necrosis [89]. |
| DNA Fragmentation (TUNEL) | DNA strand breaks | Late | Highly specific for late apoptosis; can be used on fixed tissues [87]. | Misses early apoptotic cells; can label necrotic cells [90]. |
| Cell Permeability Dyes (YO-PRO-1) | Uptake by apoptotic cells | Early | YO-PRO-1 is more sensitive than Annexin V for early apoptosis, selectively entering apoptotic cells [91]. | Requires flow cytometry or fluorescent microscopy for detection [91]. |
| Bodipy-FL-Cystine (BFC) Uptake | xCT antiporter / GSH redox stress | Early | Novel method detecting early oxidative stress; correlates well with live/dead assays; can distinguish apoptosis stages [92]. | Newer method with less established protocols; uptake can be inhibited (e.g., by sulfasalazine) [92]. |
Table 2: Quantitative Performance Metrics of Apoptosis Assays
| Assay Method | Reported Sensitivity | Correlation with Reference Methods | Temporal Resolution | Remarks from Comparative Studies |
|---|---|---|---|---|
| Annexin V / PI | Lower maximum apoptosis detection compared to morphological and DNA fragmentation assays [90]. | Detects apoptosis 4-5 hours earlier than morphology, 8 hours earlier than DNA fragmentation [90]. | Endpoint (with kinetic potential) | Maximum apoptotic response is often the lowest among compared methods [90]. |
| YO-PRO-1 / 7-AAD | The most sensitive stain for apoptosis when combined with 7-AAD [91]. | Provides accurate simultaneous measure of apoptosis and mortality [91]. | Endpoint | Proposed as a low-cost, highly sensitive alternative [91]. |
| Caspase-3 Activation | Varies with timing, as activation is transient. | N/A | Endpoint | Serves as a specific biochemical hallmark but does not confirm cell death [87] [89]. |
| DNA Fragmentation | High maximum apoptosis detection [90]. | Detects apoptosis later than other methods (e.g., 8 hours after Annexin V) [90]. | Late Endpoint | Often yields the highest maximum extent of apoptosis but is a late-stage marker [90]. |
| BFC Uptake (Flow Cytometry) | High; can distinguish early, intermediate, and late apoptotic peaks [92]. | Strong correlation (R² = 0.7–0.9) with cell viability and distinct from PI staining [92]. | Endpoint (indicative of early event) | Accurate distinction between live and apoptotic cells, independent of drug mechanism [92]. |
| Microculture Kinetic (MiCK) Assay | High for kinetic measurement. | Correlates well with time-lapse video microscopy of membrane blebbing [90]. | Real-time Kinetic | Provides continuous, real-time data on the kinetics of apoptotic response [90]. |
Methodological Deep Dive: PARP-1 and Bcl-2 Analysis
Research focused on the Bcl-2 family and PARP-1 requires assays that can probe the intrinsic apoptotic pathway and its execution.
Bcl-2 Family Protein Analysis
The Bcl-2 family is comprised of both anti-apoptotic (e.g., Bcl-2, Bcl-xL, Mcl-1) and pro-apoptotic (e.g., Bax, Bak, BIM, PUMA) members that regulate MOMP [87] [36]. Investigating this system often involves:
- Western Blotting: To measure protein expression levels of Bcl-2 family members (e.g., Mcl-1, Bcl-xL) and downstream markers like cleaved PARP in response to treatments [36] [30].
- BH3 Profiling: A functional assay that measures mitochondrial sensitivity to pro-apoptotic BH3 peptides, predicting dependency on specific anti-apoptotic proteins for survival [6].
- Genetic Manipulation: Using siRNA or CRISPR to knock down specific anti-apoptotic members like Mcl-1 and Bcl-xL, then assessing apoptosis via cleaved PARP and viability assays to confirm their functional role [30].
- Pharmacological Inhibition: Using small-molecule inhibitors (e.g., Venetoclax for Bcl-2, Obatoclax for Mcl-1/Bcl-2/Bcl-xL) to disrupt protein-protein interactions and induce apoptosis, often in combination with other agents [36] [6] [30].
PARP-1 Analysis
PARP-1 is a nuclear enzyme involved in DNA repair. Its cleavage by executioner caspases is a definitive marker of apoptosis [36] [6].
- Cleaved PARP (c-PARP) Detection: Western blotting using antibodies specific for the cleaved fragment of PARP is a gold-standard method to confirm apoptosis has been initiated [36] [30]. Its appearance is closely linked to caspase-3/7 activation.
- PARP Activity Assays: ELISA-based methods can measure PARP enzymatic activity. Research has shown that BCL2 can directly bind to and suppress PARP1 activity. Displacing this interaction with BH3 mimetics like ABT-737 can restore PARP1 function, a mechanism relevant for non-apoptotic cell death [6].
Experimental Workflow for Pathway Validation
A typical integrated workflow to investigate these pathways is outlined below.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for Apoptosis and Pathway Analysis
| Reagent / Assay | Function / Target | Application Example |
|---|---|---|
| Annexin V Conjugates | Binds externalized phosphatidylserine (PS) | Flow cytometry or microscopy to identify early apoptotic cell populations when combined with a viability dye like PI or 7-AAD [91] [88]. |
| Caspase-Glo 3/7 Assay | Luciferase-based detection of caspase-3/7 activity | High-throughput, homogenous assay to quantify executioner caspase activity as a key apoptotic milestone [88]. |
| Bodipy-FL-Cystine (BFC) | Monitors xCT antiporter activity & oxidative stress | Flow cytometry assay to detect early apoptosis initiation via cellular stress response, validated with inhibitors like sulfasalazine [92]. |
| Anti-Cleaved PARP Antibody | Detects caspase-cleaved fragment of PARP1 | Western blotting to provide definitive biochemical evidence of ongoing apoptosis [36] [30]. |
| BCL-2 Family Antibodies | Detect specific pro- and anti-apoptotic proteins | Western blotting to measure expression changes in proteins like Bcl-2, Bcl-xL, Mcl-1, BIM, and PUMA in response to stimuli [36] [30] [88]. |
| BH3 Mimetics (e.g., ABT-737, Venetoclax, Obatoclax) | Small-molecule inhibitors of anti-apoptotic BCL-2 proteins | Used to experimentally induce intrinsic apoptosis or overcome treatment resistance by disrupting protein interactions [36] [6] [30]. |
| Cell Titer Blue | Measures metabolic activity via resazurin reduction | Spectroscopic viability assay often used in combination with specific apoptosis assays to comprehensively assess drug effects [92]. |
The optimal selection of apoptosis assays is contingent upon the specific research question, the apoptotic pathway of interest, and the required temporal resolution. For a comprehensive analysis, a combination of assays is strongly recommended.
- For early, high-throughput screening of treatment effects, a combination of a metabolic viability assay (like Cell Titer Blue) and a sensitive early apoptosis assay (like BFC flow cytometry or YO-PRO-1) has been shown to provide accurate and independent assessment of live and apoptotic cells [92].
- For definitive confirmation of apoptosis and mechanistic studies, Western blot analysis for cleaved PARP is a critical endpoint [36].
- For research focused on the intrinsic pathway and drug resistance, incorporating Bcl-2 family protein analysis and functional assays like BH3 profiling is essential to understand the underlying regulatory dynamics [36] [30].
No single assay is infallible. The inherent asynchrony of apoptosis in cell populations means that different methods, each targeting a distinct temporal event, will yield different quantitative results from the same culture [90]. Therefore, the most reliable strategy for validating apoptotic pathways, particularly through PARP-1 and Bcl-2 family analysis, is to employ a multi-parametric approach that leverages the strengths of complementary techniques.
The ferroptosis inducer RSL3, traditionally known for inhibiting glutathione peroxidase 4 (GPX4) to trigger iron-dependent lipid peroxidation, demonstrates a surprising capacity to activate apoptotic signaling through convergence on PARP1. This crosstalk provides a promising therapeutic strategy, particularly for overcoming resistance in malignancies. This guide compares the established apoptotic pathways with the emergent RSL3-mediated mechanisms, providing a side-by-side analysis of molecular players, experimental data, and functional outcomes to inform future research and drug development.
Cell death pathways have long been categorized into distinct types, with apoptosis and the more recently characterized ferroptosis representing mechanistically different processes. Apoptosis is an orchestrated proteolytic cascade mediated by caspases, while ferroptosis is driven by iron-catalyzed phospholipid peroxidation. However, emerging evidence reveals significant molecular crosstalk between these pathways, challenging this strict classification.
Central to this discussion is PARP1, a DNA repair enzyme that plays a decisive role in cell fate. During apoptosis, PARP1 is cleaved by executioner caspases into pro-apoptotic fragments. Simultaneously, the BCL-2 family of proteins governs the mitochondrial pathway of apoptosis, with anti-apoptotic members like BCL-2 itself suppressing cell death. Recent groundbreaking research establishes that the ferroptosis activator RSL3 directly engages these core apoptotic components, orchestrating a convergent death signaling network with significant implications for cancer therapy, especially in PARP inhibitor (PARPi)-resistant contexts.
Comparative Analysis of Cell Death Pathway Activation
The following table summarizes the core characteristics of the established apoptotic pathway in contrast to the RSL3-induced crosstalk mechanism, highlighting key molecular events and functional outcomes.
Table 1: Comparative Analysis of Apoptotic and RSL3-Induced PARP1 Activation Pathways
| Feature | Classical Apoptosis | RSL3-Induced PARP1 Convergence |
|---|---|---|
| Primary Inducer | DNA damage, cytotoxic drugs | RSL3 (GPX4 inhibitor) |
| Key Initiators | Caspase-8 (extrinsic), Caspase-9 (intrinsic) | Reactive Oxygen Species (ROS) from lipid peroxidation |
| BCL-2 Role | Directly inhibits MOMP and caspase activation [24] | Interaction with PARP1 suppressed; role in crosstalk context |
| PARP1 Fate | Cleaved by caspase-3 into 24-kDa and 89-kDa pro-apoptotic fragments | 1. Caspase-dependent cleavage & 2. Full-length depletion via translational suppression |
| Downstream Effect | DNA fragmentation, apoptotic body formation | Enhanced DNA damage, apoptosis via dual PARP1 mechanisms |
| Therapeutic Implication | Basis for many chemotherapies | Overcomes PARPi resistance in malignancies [93] |
Detailed Mechanisms of RSL3-PARP1 Crosstalk
The convergence of RSL3-induced ferroptosis on PARP1 occurs through two parallel, synergistic mechanisms that are triggered by a surge in reactive oxygen species (ROS).
Mechanism 1: Caspase-Dependent PARP1 Cleavage
The first mechanism follows a more traditional apoptotic route. RSL3 treatment leads to ROS accumulation, which acts as a signal that ultimately triggers the activation of caspase-3. This key executioner caspase then cleaves full-length PARP1 (116-kDa) into characteristic 89-kDa and 24-kDa fragments [93] [41]. These fragments are not merely inactive byproducts; the 89-kDa fragment translocates from the nucleus to the cytoplasm where it actively promotes caspase-mediated DNA fragmentation, thereby amplifying the apoptotic signal [41].
Mechanism 2: METTL3-mediated m6A Modification and PARP1 Translation Suppression
The second mechanism is more novel and highlights the intersection of ferroptosis and epitranscriptomic regulation. RSL3-induced ROS inhibits METTL3-mediated N6-methyladenosine (m6A) modification of PARP1 mRNA [93] [41]. This m6A mark is crucial for the efficient translation of PARP1 mRNA. Its loss leads to a significant reduction in full-length PARP1 protein levels. The depletion of this key DNA repair enzyme results in the accumulation of unresolved DNA damage, marked by increased γH2AX foci, which in turn drives DNA damage-dependent apoptosis [41].
The following diagram illustrates the logical flow of these two parallel pathways.
Key Experimental Data and Validation
The proposed model of RSL3-PARP1 crosstalk is supported by robust in vitro and in vivo data. The table below quantifies key experimental findings that validate this pathway convergence.
Table 2: Summary of Key Experimental Findings Supporting RSL3-PARP1 Crosstalk
| Experimental Model | Treatment | Key Observation | Outcome/Implication |
|---|---|---|---|
| Multiple Cancer Cell Lines [93] [41] | RSL3 | Increased caspase-3 activity & PARP1 cleavage fragments | Confirms activation of apoptotic machinery |
| Multiple Cancer Cell Lines [93] [41] | RSL3 | ↓ PARP1 mRNA m6A modification; ↓ Full-length PARP1 protein | Confirms epitranscriptomic regulation |
| PARPi-Resistant Cells [93] | RSL3 | Induction of apoptosis & cytotoxicity | Overcomes PARP inhibitor resistance |
| Mouse Xenograft Model [93] | RSL3 | Inhibition of PARPi-resistant tumor growth | Confirms therapeutic potential in vivo |
| Cell-Free System / Lymphoma Cells [6] [24] | ABT-737 (BCL-2 inhibitor) | Displacement of PARP1 from BCL-2; restored PARP1 activity | Validates BCL-2-PARP1 interaction and its functional impact |
Essential Experimental Protocols
To validate RSL3-induced pathway crosstalk in a research setting, the following key methodologies are essential:
Assessing Cell Death and PARP1 Cleavage:
- Western Blot Analysis: Use antibodies against full-length PARP1 and its 89-kDa cleavage product to simultaneously detect both convergent mechanisms. Monitor caspase-3 cleavage and lipid peroxidation markers like GPX4 [93] [41].
- Flow Cytometry: Employ Annexin V/PI staining to quantify apoptosis and use ROS-sensitive dyes (e.g., DCFH-DA) to measure reactive oxygen species generation [93].
Evaluating the Epitranscriptomic Mechanism:
- m6A RNA Immunoprecipitation (MeRIP)-qPCR: Immunoprecipitate m6A-modified RNA with a specific antibody and use qPCR with primers for PARP1 mRNA to quantify its m6A modification levels [93] [41].
- RNA Immunoprecipitation (RIP)-qPCR: Identify specific reader proteins (e.g., YTHDF1) that bind to the m6A-modified site on PARP1 mRNA [41].
Measuring DNA Damage Response:
The workflow for a comprehensive experimental validation is outlined below.
The Researcher's Toolkit: Essential Reagents and Assays
Successfully investigating the RSL3-PARP1 crosstalk requires a specific set of reagents and tools. The following table catalogs essential solutions for this field of study.
Table 3: Key Research Reagent Solutions for Pathway Crosstalk Investigation
| Reagent / Assay | Function / Target | Specific Examples & Utility |
|---|---|---|
| Ferroptosis Inducers | Inhibit GPX4 to initiate ferroptosis | RSL3: The canonical inducer used to study PARP1 crosstalk [93] [94] |
| Pathway Inhibitors | Block specific pathways to validate mechanisms | Ferrostatin-1 (Fer-1): Inhibits ferroptosis; confirms ROS role [93].Z-VAD-FMK: Pan-caspase inhibitor; tests caspase-dependence [93]. |
| Antibodies for Detection | Detect key proteins and modifications | Anti-PARP1: For full-length and cleaved fragments [93] [41].Anti-γH2AX: For DNA damage quantification [41].Anti-Cleaved Caspase-3: For apoptosis confirmation [41]. |
| m6A Detection Kits | Study epitranscriptomic regulation | MeRIP-qPCR Kits: Measure m6A modification on specific mRNAs like PARP1 [93] [41]. |
| Viability & Apoptosis Assays | Quantify cell death | Annexin V Apoptosis Kits: Distinguish apoptotic cells [93] [6].CellTiter-Glo Assay: Measure ATP levels as a viability proxy [6] [94]. |
Therapeutic Implications and Future Directions
The convergence of RSL3-induced ferroptosis on PARP1 is not merely a mechanistic curiosity; it holds direct and promising therapeutic implications. The most significant finding is that RSL3 retains its pro-apoptotic function in PARP inhibitor (PARPi)-resistant cells and can effectively inhibit the growth of PARPi-resistant tumors in mouse xenograft models [93]. This suggests that leveraging this crosstalk could be a viable strategy to treat malignancies that have developed resistance to PARP-targeted therapies.
Furthermore, the interaction between BCL-2 and PARP1 adds another layer of complexity. BCL-2 can directly bind to and suppress PARP1 enzymatic activity, and this suppression can be reversed by the BH3-mimetic drug ABT-737, leading to non-apoptotic cell death [6]. This indicates that the BCL-2-PARP1 axis is a druggable target, and that combining BCL-2 inhibitors with ferroptosis inducers or other agents could be a potent synthetic lethal strategy, especially in apoptosis-resistant cancers.
Future research should focus on:
- Elucidating the precise sensor that links RSL3-induced lipid peroxidation to the inhibition of METTL3 activity.
- Exploring the therapeutic efficacy and potential toxicities of combining RSL3 (or other ferroptosis inducers) with BH3-mimetics in different cancer types.
- Developing more stable and potent ferroptosis inducers suitable for clinical application.
The efficacy of many cancer therapeutics hinges on their ability to trigger mitochondrial apoptosis, a precisely regulated form of programmed cell death. The foundation of this pathway is the BCL-2 protein family, which integrates diverse cellular stress signals to determine cellular fate. This family encompasses anti-apoptotic guardians (BCL-2, BCL-xL, MCL-1), pro-apoptotic executioner proteins (BAX, BAK), and pro-apoptotic sensitizer proteins (known as BH3-only proteins) [95]. A cell's position within this delicate equilibrium—its proximity to the apoptotic threshold—is termed mitochondrial apoptotic priming. Highly primed cells are on the verge of death and are more susceptible to chemotherapeutic agents, whereas poorly primed cells can withstand significant insult [96] [95]. BH3 profiling has emerged as a powerful functional assay that directly measures this priming, moving beyond static protein level measurements to predict cell fate in response to treatment [96].
This guide objectively compares BH3 profiling methodologies and their application in predicting responses to conventional chemotherapeutics and novel targeted agents like BH3 mimetics. Furthermore, we frame this discussion within the broader thesis of validating apoptotic pathways, highlighting a novel, non-apoptotic interaction between BCL-2 and PARP1, a key DNA repair enzyme. This interaction reveals that BCL-2's role extends beyond regulating apoptosis to directly suppressing PARP1 activity, and that BH3 mimetics can reverse this suppression to induce a non-apoptotic cell death—a critical mechanism for overcoming apoptosis resistance [6].
Comparative Analysis of BH3 Profiling Platforms
BH3 profiling functionally measures the readiness of a cell to undergo mitochondrial apoptosis by exposing mitochondria to synthetic peptides that mimic the killing domains of native BH3-only proteins. The subsequent release of cytochrome c is quantified, serving as a surrogate for mitochondrial outer membrane permeabilization (MOMP), the point of no return in intrinsic apoptosis [96] [95]. The following table compares the core BH3 profiling platforms.
Table 1: Comparison of Fundamental BH3 Profiling Methodologies
| Feature | Standard BH3 Profiling | Dynamic BH3 Profiling (DBP) |
|---|---|---|
| Primary Measurement | Basal mitochondrial priming, dependence on specific anti-apoptotic proteins [96] [95] | Drug-induced changes in mitochondrial priming [97] |
| Typical Application | Predicting inherent sensitivity to chemotherapy or BH3 mimetics; identifying dependencies on BCL-2, MCL-1, etc. [98] [95] | Identifying effective single agents and combination therapies by measuring early death signaling after ex vivo drug treatment [97] |
| Key Readout | Cytochrome c release after BH3 peptide exposure in untreated cells [95] | Delta priming (%) - the difference in cytochrome c release between drug-treated and control cells [97] |
| Throughput | Lower, often manual or semi-automated | Higher, adaptable to high-throughput (HTDBP) 384-well formats [97] |
| Therapeutic Prediction | Predicts response to agents like ABT-737 (navitoclax) based on basal protein dependencies [98] [95] | Identifies drug combinations that synergize to prime tumors, validated in PDX models [97] |
The predictive power of BH3 profiling is well-established. In a panel of 18 lymphoma cell lines, the assay successfully stratified cells into distinct classes of apoptotic blocks and predicted sensitivity to the BCL-2 antagonist ABT-737. Strikingly, it also forecasted responses to conventional chemotherapeutic agents like etoposide, vincristine, and adriamycin [98]. The binding specificity of different BH3 peptides forms the basis for identifying dependencies on specific anti-apoptotic proteins. For instance, sensitivity to the BAD BH3 peptide indicates BCL-2 dependence, while sensitivity to NOXA BH3 indicates MCL-1 dependence [95].
Connecting Apoptotic Function to PARP1 and Broader Cell Death Research
The BCL-2 and PARP1 Axis: A Non-Apoptotic Pathway
Research has uncovered that the anti-apoptotic protein BCL-2 localizes to the nucleus in certain lymphomas and can directly interact with PARP1, an enzyme critical for DNA repair. This interaction suppresses PARP1 enzymatic activity, thereby inhibiting DNA repair. The BH3 mimetic ABT-737 can displace PARP1 from BCL-2, restoring its activity [6]. This pathway represents a therapeutically exploitable, non-apoptotic function of BCL-2. In cells resistant to apoptotic death, targeting this interaction with BH3 mimetics can re-establish PARP1 function and promote a non-apoptotic cell death, bypassing classic resistance mechanisms [6].
Apoptosis in the Context of Other Cell Death Modalities
It is crucial to distinguish apoptosis from other regulated cell death (RCD) pathways. Apoptosis is characterized by cell shrinkage, membrane blebbing, and caspase activation, culminating in non-inflammatory phagocytosis. In contrast, necroptosis and pyroptosis are lytic and pro-inflammatory, while ferroptosis is an iron-dependent, lipid peroxidation-driven process [99] [100]. Accurate detection requires specific markers: caspase activation and DNA fragmentation (TUNEL) for apoptosis, versus MLKL oligomerization for necroptosis or gasdermin cleavage for pyroptosis [99].
Diagram 1: BCL-2 regulates apoptosis and PARP1. BCL-2 proteins integrate stress signals to control mitochondrial apoptosis. Nuclear BCL-2 also binds and inhibits PARP1. BH3 mimetics can trigger apoptosis and, by displacing PARP1, induce non-apoptotic death.
Essential Protocols for Functional Apoptosis Assessment
Core Protocol: BH3 Profiling to Measure Apoptotic Priming
The following protocol is adapted from established methodologies for performing BH3 profiling on primary cells or cell lines [96] [97].
- Step 1: Mitochondrial Isolation or Permeabilization. Cells are harvested and permeabilized with a digitonin-containing buffer to allow BH3 peptides access to the mitochondria while retaining cytochrome c within the intermembrane space.
- Step 2: BH3 Peptide Incubation. Permeabilized cells are incubated with a panel of synthetic BH3 peptides (e.g., BIM, BAD, NOXA, HRK). Each peptide is used at a defined concentration to probe dependence on specific anti-apoptotic proteins. A negative control (DMSO) and positive control (e.g., Alamethicin or BIM peptide) are always included.
- Step 3: Cytochrome c Release Quantification. After incubation, samples are fixed, and cytochrome c release is measured. This can be done via immunofluorescence staining and high-content microscopy (for HTDBP) [97], ELISA, or flow cytometry. The percentage of cells that have lost cytochrome c is calculated for each peptide.
- Step 4: Data Interpretation. A cell is considered primed if its mitochondria release cytochrome c in response to sensitizer BH3 peptides like BAD or NOXA. The pattern of response across the peptide panel reveals which anti-apoptotic proteins (BCL-2, BCL-xL, MCL-1) the cell is dependent on for survival.
Supporting Protocol: Differentiating Apoptosis from Other RCD
To validate apoptosis as the mechanism of death, especially in the context of BH3 mimetic treatment, a combination of assays is recommended [99].
- Morphology: Use light or electron microscopy to identify classic apoptotic features: cell shrinkage, chromatin condensation (pyknosis), and nuclear fragmentation (karyorrhexis) [99] [100].
- Biochemical Markers:
- Flow Cytometry: Stain cells with Annexin V (for phosphatidylserine exposure) and propidium iodide (PI). Early apoptotic cells are Annexin V+/PI-; late apoptotic/necrotic cells are Annexin V+/PI+ [99].
- Western Blot: Detect cleavage of executioner caspases (Caspase-3, -7) and their substrates, such as PARP1. Cleaved PARP1 is a classic apoptosis marker [101] [6].
- Functional Assays: BH3 profiling itself is a functional assay. To rule out other RCDs, use specific pharmacological inhibitors (e.g., necrostatin-1 for necroptosis, ferrostatin-1 for ferroptosis) [99].
Diagram 2: BH3 profiling workflow. Cells are permeabilized, exposed to a panel of BH3 peptides, and assessed for cytochrome c release to determine apoptotic priming.
The Scientist's Toolkit: Key Reagents for Apoptosis Research
Table 2: Essential Research Reagents for Apoptosis and BH3 Profiling Studies
| Reagent / Tool | Core Function | Specific Application Example |
|---|---|---|
| Synthetic BH3 Peptides (BIM, BAD, NOXA, HRK, MS1) | Functionally probe dependencies on specific anti-apoptotic proteins by mimicking native BH3-only proteins [96] [95]. | NOXA BH3 peptide identifies MCL-1 dependent cells; BAD BH3 identifies BCL-2/BCL-xL dependent cells [95]. |
| BH3 Mimetics (Venetoclax, Navitoclax, S63845) | Small molecule inhibitors that bind the BH3-binding cleft of anti-apoptotic proteins, displacing pro-apoptotic partners to induce apoptosis [98] [97]. | Navitoclax (BCL-2/BCL-xL inhibitor) combined with mTOR inhibitor AZD8055 showed efficacy in mesothelioma models by increasing BIM and decreasing MCL-1 [97]. |
| PARP Inhibitors (Olaparib, ABT-888) | Inhibit PARP1 enzymatic activity, inducing synthetic lethality in BRCA-deficient cancers and trapping PARP1 on DNA [6]. | Used to study the novel BCL-2-PARP1 interaction; ectopic BCL2 expression kills PARP inhibitor-sensitive cells [6]. |
| Caspase Activity Assays | Quantify the activation of executioner caspases (3/7) via fluorogenic or luminogenic substrates, confirming apoptotic engagement. | Validates that cell death induced by a BH3 mimetic occurs through the canonical apoptotic pathway. |
| Annexin V / Propidium Iodide | Flow cytometry-based kit to distinguish early apoptosis (Annexin V+/PI-) from late apoptosis/necrosis (Annexin V+/PI+) [6] [99]. | A standard method for quantifying the percentage of cells undergoing apoptosis after drug treatment. |
BH3 profiling stands as a preeminent functional metric for directly quantifying a cell's apoptotic competence, offering superior predictive power for therapy response compared to static genomic or proteomic measurements. Its utility spans from identifying dependencies on specific anti-apoptotic proteins to guiding the rational design of combination therapies via Dynamic BH3 Profiling. The integration of this assay into a broader apoptotic validation framework, which includes the recognition of non-apoptotic regulatory mechanisms like the BCL-2-PARP1 axis, provides a more comprehensive understanding of cell fate decisions. For researchers and drug developers, mastering BH3 profiling and its associated toolkit is indispensable for advancing the next generation of cancer therapeutics that effectively target the core regulators of cell survival and death.
The quest to overcome apoptosis resistance represents a central challenge in modern oncology. Within this landscape, the strategic combination of BH3 mimetics and PARP inhibitors (PARPi) has emerged as a promising therapeutic approach grounded in the molecular validation of apoptotic pathways. BH3 mimetics target the Bcl-2 family of anti-apoptotic proteins, thereby directly promoting mitochondrial outer membrane permeabilization (MOMP) and the initiation of the intrinsic apoptosis pathway [102]. PARP inhibitors, initially developed to exploit synthetic lethality in homologous recombination-deficient cancers, also engage cell death pathways through multiple mechanisms, including the induction of parthanatos—a caspase-independent form of programmed cell death driven by PARP1 overactivation [103]. The synergistic potential of these drug classes arises from their complementary actions on interconnected cell death networks, simultaneously disrupting cancer cell survival mechanisms while activating multiple lethal pathways. This review systematically compares the performance of prominent BH3 mimetics and PARP inhibitor combinations across various cancer models, providing supporting experimental data and methodological frameworks for researchers investigating apoptotic pathway validation.
Mechanistic Foundations: Pathway Interactions and Synergistic Potential
Molecular Mechanisms of BH3 Mimetics
BH3 mimetics function as small-molecule inhibitors that structurally mimic the BH3 domain of pro-apoptotic proteins. By binding to the hydrophobic groove of anti-apoptotic Bcl-2 family members (including Bcl-2, Bcl-xL, Mcl-1, Bcl-w, and Bfl-1), they prevent these guardians from sequestering pro-apoptotic effectors like Bax and Bak [102]. This displacement initiates mitochondrial outer membrane permeabilization (MOMP), leading to cytochrome c release, caspase activation, and apoptotic cell death. The specificity profile of various BH3 mimetics determines their therapeutic application: venetoclax (ABT-199) selectively targets Bcl-2; navitoclax (ABT-263) inhibits Bcl-2, Bcl-xL, and Bcl-w; A1331852 selectively targets Bcl-xL; S63845 specifically inhibits Mcl-1; while obatoclax (GX15-070) acts as a pan-Bcl-2 inhibitor with particular potency against Mcl-1 [104] [30] [105]. The functional dependence of cancer cells on specific anti-apoptotic proteins can be mapped using BH3 profiling, a technique that measures mitochondrial susceptibility to synthetic BH3 peptides to determine "mitochondrial priming" and identify the specific anti-apoptotic proteins maintaining cell survival [106].
PARP Inhibition and Engagement of Multiple Cell Death Pathways
PARP inhibitors engage multiple cell death pathways, with their effects extending beyond synthetic lethality in HR-deficient cells. While PARP inhibition alone can induce apoptosis through DNA damage accumulation, particularly in BRCA-mutated backgrounds, it can also trigger parthanatos—a caspase-independent programmed necrosis pathway initiated by PARP1 overactivation [103]. During parthanatos, severe DNA damage causes PARP1 hyperactivation, resulting in the accumulation of poly(ADP-ribose) (PAR) polymers, mitochondrial dysfunction, and apoptosis-inducing factor (AIF) nuclear translocation, culminating in large-scale DNA fragmentation [103]. The specific PARP inhibitor used influences the mechanistic emphasis; BMN 673 (talazoparib) exhibits particularly potent DNA-PARP trapping activity compared to other PARP inhibitors, creating a stronger death signal [107].
Theoretical Framework for Synergy
The synergistic interaction between BH3 mimetics and PARP inhibitors emerges from their engagement of complementary cell death pathways that converge on mitochondrial integrity. PARP inhibition creates cellular stress through DNA damage accumulation and energy depletion (via NAD+ and ATP consumption), which increases dependence on anti-apoptotic Bcl-2 family members for survival [107]. This dependency creates a vulnerable state susceptible to BH3 mimetics, which directly target these anti-apoptotic proteins. Additionally, evidence suggests that PARP inhibition can modulate the expression or activity of Bcl-2 family members, potentially enhancing the efficacy of BH3 mimetics through direct effects on the apoptotic machinery [104]. The resulting synergistic cell death represents the outcome of simultaneous induction of mitochondrial stress (via PARP inhibition) and lowering of the apoptotic threshold (via BH3 mimetics).
The following diagram illustrates the key molecular interactions and synergistic mechanisms between these drug classes:
Diagram Title: Molecular Pathways of BH3 Mimetic and PARP Inhibitor Synergy
Comparative Performance Analysis of Key Drug Combinations
Quantitative Comparison of Combination Efficacy
The therapeutic potential of BH3 mimetic and PARP inhibitor combinations has been evaluated across diverse cancer types, with particular emphasis on high-grade serous ovarian cancer (HGSOC), glioblastoma (GBM), and acute myeloid leukemia (AML). The following table summarizes key findings from pivotal preclinical studies, providing comparative efficacy data and synergy metrics.
Table 1: Comparative Performance of BH3 Mimetic and PARP Inhibitor Combinations in Preclinical Models
| Cancer Type | BH3 Mimetic | PARP Inhibitor | Synergy Metric | Experimental Model | Key Findings | Citation |
|---|---|---|---|---|---|---|
| HGSOC | ABT-263 (Navitoclax) | BMN 673 (Talazoparib) | CI <0.9; Synergy volume >100 μM²% | BRCA-wt cell lines (OVCAR3, OVCAR8, OV90) | Significant increase in sub-G1 population and Annexin V-positive cells; Enhanced caspase-3/7 activity and PARP cleavage | [107] |
| Ovarian Cancer | WEHI-539 (Bcl-xL inhibitor) | Carboplatin | Strong synergy in cell growth assays | Ovarian cancer cell lines | Augmented carboplatin-induced caspase 3/7 activity, PARP cleavage and annexin V labelling | [108] |
| Ovarian Cancer | GX15-070 (Obatoclax) | Niraparib | Significant synergy in CDX/PDX models | Ovarian cancer cell lines and xenografts | Disrupted Mcl-1/Ku70 interaction; Shifted DNA repair from HR to NHEJ; Synergy independent of BRCA status | [104] |
| Glioblastoma | GX15-070 (Obatoclax) | - | Enhanced ER stress-induced death | GBM cell lines (Onda7, DK-MG, YH-13) | Combinatorial knockdown of Mcl-1 and Bcl-xL enhanced apoptosis under ER stress; Disrupted autophagy cargo degradation | [30] |
| AML | S55746 (Bcl-2 inhibitor) + S63845 (Mcl-1 inhibitor) | - | High synergy scores | AML cell lines and PDX models | Co-targeting BCL-2 and MCL1 more effective against leukemic vs. normal hematopoietic progenitors | [105] |
| Various Cancers | A1331852 (Bcl-xL inhibitor) | - | Universal senolytic response | Therapy-induced senescent (TIS) cancer cells | BCL-xL identified as conserved anti-apoptotic effector across TIS phenotypes; Effective senolytic in "one-two punch" strategy | [106] |
Analysis of Combination Specificity and Context Dependencies
The efficacy of specific BH3 mimetic and PARP inhibitor combinations demonstrates significant dependence on cellular context, particularly the expression patterns of anti-apoptotic Bcl-2 family members. In ovarian cancer models, Bcl-xL inhibition appears particularly crucial, as WEHI-539 (Bcl-xL selective inhibitor) and ABT-263 (which inhibits Bcl-2, Bcl-xL, and Bcl-w) both synergize with DNA-damaging agents, while the Bcl-2 selective inhibitor ABT-199 (venetoclax) shows limited activity [108]. This aligns with tissue expression patterns, as Bcl-xL is more frequently deregulated in ovarian cancer than Bcl-2 [108]. Similarly, in glioblastoma, the potency of the pan-Bcl-2 inhibitor obatoclax (which effectively targets Mcl-1) over navitoclax (which has poor Mcl-1 affinity) corresponds with dominant Mcl-1 and Bcl-xL expression in GBM cell lines [30]. These findings highlight the importance of profiling anti-apoptotic protein dependencies to guide rational combination therapies.
Experimental Methodologies for Combination Therapy Assessment
Standardized Protocols for Synergy Quantification
Robust assessment of BH3 mimetic and PARP inhibitor combinations requires standardized methodologies for quantifying synergy. The following experimental approaches represent best practices derived from the cited literature:
Cell Viability and Synergy Assays: Cells are seeded in 96-well plates at optimized densities (e.g., 2,000-5,000 cells/well for ovarian cancer lines) and treated with serial dilutions of single agents or combinations after 24 hours. Cell viability is typically assessed after 72 hours of drug exposure using metabolic assays (XTT, MTT, or Cell Counting Kit-8) or luminescence-based assays (CellTiter-Glo) [108] [107]. For combination studies, drugs are typically administered simultaneously using a checkerboard design with multiple concentration permutations. Synergy is quantified using established models: the Chou-Talalay method (Calculating Combination Index values where CI <0.9 indicates synergy, 0.9-1.1 additive effect, and >1.1 antagonism) and/or the Prichard-Shipman method (MacSynergy II software calculating synergy volumes at 95% confidence, with values >25 μM²% indicating significant synergy) [107].
Apoptosis-Specific Detection Methods: Beyond viability assays, specific apoptosis measurement is crucial for validating the mechanistic basis of combinations. Flow cytometric analysis of Annexin V/propidium iodide staining provides quantitative assessment of phosphatidylserine externalization and membrane integrity [108] [107]. Caspase activation is measured using luminescent caspase-Glo 3/7 assays or Western blot detection of cleaved caspase-3 and its substrate poly(ADP-ribose) polymerase (PARP) [108] [107]. Additional apoptosis confirmation includes DNA fragmentation analysis (TUNEL assay) and cell cycle analysis to quantify the sub-G1 population representing apoptotic cells with fragmented DNA [104] [107].
Mechanistic Investigation Techniques
DNA Damage Response Assessment: Immunofluorescence staining for γ-H2AX foci (phosphorylated histone H2AX) serves as a sensitive marker for DNA double-strand breaks. Cells are typically fixed at various timepoints after drug exposure (e.g., 24 hours), stained with anti-γ-H2AX antibodies, and visualized by confocal microscopy, with foci quantified per nucleus [107]. Additional DNA damage markers include Western blot analysis of other phosphorylation events in the DNA damage response (e.g., ATM, ATR, CHK1, CHK2).
Protein Interaction and Pathway Mapping: Co-immunoprecipitation experiments elucidate changes in protein-protein interactions following combination treatment. For example, the interaction between Mcl-1 and Ku70 can be assessed by immunoprecipitating Mcl-1 and immunoblotting for Ku70 [104]. Mitochondrial priming and dependencies on specific anti-apoptotic proteins are determined using BH3 profiling, which measures mitochondrial membrane depolarization in response to specific BH3 domain peptides [106]. This functional assay identifies which anti-apoptotic proteins (Bcl-2, Bcl-xL, Mcl-1) are primarily maintaining survival in specific cancer contexts.
Table 2: Essential Research Reagents and Experimental Tools
| Reagent/Category | Specific Examples | Research Application | Key Function |
|---|---|---|---|
| BH3 Mimetics | ABT-263 (Navitoclax), ABT-199 (Venetoclax), WEHI-539, S63845, GX15-070 (Obatoclax) | Target validation, combination studies | Inhibit anti-apoptotic Bcl-2 family proteins |
| PARP Inhibitors | BMN 673 (Talazoparib), Olaparib, Niraparib | DNA damage response studies, combination therapies | Induce DNA damage accumulation and parthanatos |
| Viability Assays | XTT, MTT, CellTiter-Glo, CCK-8 | High-throughput screening, synergy quantification | Measure metabolic activity/cell number |
| Apoptosis Detection | Annexin V/Propidium iodide, Caspase-Glo 3/7, TUNEL, PARP cleavage (Western) | Mechanistic studies, cell death validation | Specific detection of apoptotic pathways |
| DNA Damage Markers | γ-H2AX immunofluorescence, COMET assay | DNA repair efficiency assessment | Quantify DNA double-strand breaks |
| Protein Analysis | Co-immunoprecipitation, Western blotting | Pathway mechanism studies | Detect protein interactions/expression |
| BH3 Profiling | Synthetic BH3 peptides (BAD, HRK, NOXA, etc.) | Functional dependency mapping | Identify anti-apoptotic protein dependencies |
| In Vivo Models | Cell line-derived xenografts (CDX), Patient-derived xenografts (PDX) | Preclinical efficacy validation | Assess therapeutic efficacy in physiological context |
The systematic comparison of BH3 mimetic and PARP inhibitor combinations reveals a compelling therapeutic strategy with robust synergistic potential across diverse cancer models. The experimental data consistently demonstrates that simultaneous targeting of anti-apoptotic proteins and DNA repair pathways can overcome traditional resistance mechanisms, with efficacy influenced by the specific anti-apoptotic dependencies of each cancer type. The methodological framework presented provides researchers with standardized approaches for quantifying synergy and investigating mechanistic foundations. Future research directions should prioritize biomarker development for patient stratification, particularly through functional BH3 profiling and DNA repair deficiency assessment. Additionally, optimizing dosing schedules to maximize therapeutic windows while minimizing toxicity—especially thrombocytopenia associated with Bcl-xL inhibition—represents a critical translational challenge. As these targeted combinations progress through clinical development, they offer the potential to validate apoptotic pathway manipulation as a cornerstone of precision oncology, particularly for apoptosis-resistant malignancies.
The validation of apoptotic pathways represents a cornerstone of modern cancer research, offering critical insights for therapeutic development. Within this framework, the status of two key proteins, Poly(ADP-ribose) polymerase 1 (PARP-1) and B-cell lymphoma 2 (BCL-2), has emerged as a critical determinant of treatment efficacy and patient outcomes. PARP-1 plays a central role in the DNA damage response (DDR), functioning as a molecular sensor for DNA strand breaks and facilitating repair processes [37]. Conversely, BCL-2 family proteins constitute the essential regulatory network governing mitochondrial apoptosis, thereby determining a cell's commitment to survival or death [109] [110]. The intricate interplay between these DNA damage and survival pathways creates a complex biological interface that influences responses to diverse anticancer therapies. This guide systematically compares the biomarker potential of PARP-1 and BCL-2, providing a structured analysis of their functions, detection methodologies, and validated correlations with therapeutic outcomes, thereby offering a resource for researchers and drug development professionals focused on apoptotic pathway validation.
Biomarker Comparison: PARP-1 and BCL-2 at a Glance
Table 1: Core Characteristics and Biomarker Functions of PARP-1 and BCL-2
| Feature | PARP-1 | BCL-2 |
|---|---|---|
| Primary Function | DNA damage sensor and repair enzyme [37] | Guardian of mitochondrial integrity; inhibits apoptosis [109] [110] |
| Role in Cancer | Facilitates DNA repair in cancer cells; target for synthetic lethality in HR-deficient cancers [13] [37] | Promotes tumor cell survival by blocking intrinsic apoptosis; overexpression confers resistance [111] [110] |
| Key Biomarker Readouts | Protein levels, cleavage fragments (89 kDa, 24 kDa), enzymatic (PAR) activity [13] [112] | Protein expression levels, localization, interaction with pro-apoptotic partners (e.g., BIM, BAX) [112] [110] |
| Therapeutic Correlation | PARP inhibitor sensitivity; parthanatos induction with chemotherapy [13] [113] | Sensitivity to BH3-mimetics (e.g., venetoclax); chemoresistance predictor [109] [110] |
| Detection Platforms | Western blot (cleavage), IHC, PAR activity assays [13] [112] | IHC, Western blot, flow cytometry, BH3 profiling [112] [110] |
Table 2: Correlative Biomarker Status and Associated Therapeutic Outcomes
| Biomarker Status | Experimental/Therapeutic Context | Correlated Outcome | Supporting Evidence |
|---|---|---|---|
| PARP-1 Cleavage (89 kDa fragment) | Caspase-3 activation during RSL3-induced ferroptosis-apoptosis crosstalk [13] | Execution of apoptosis; positive correlation with cell death | In vitro cell line models [13] |
| High Nuclear BCL-2 Protein | Diffuse Large B-Cell Lymphoma (DLBCL) [6] | Suppressed PARP1 activity; inhibition of DNA repair and non-apoptotic death | Cell line fractionation and immunofluorescence [6] |
| Functional PARP-1 Mediated Parthanatos | Frontline chemotherapy (Cytarabine/Idarubicin) in Acute Myeloid Leukemia (AML) [113] | 3-fold improved survival (HR=0.28-0.37); favorable risk factor | Analysis of primary patient samples (n=39) [113] |
| Combined Low Mcl-1 & Bcl-xL | Glioblastoma (GBM) cells under ER stress [30] | Synergistic apoptosis induction with obatoclax (pan-BCL-2 inhibitor) | Combinatorial siRNA knockdown in GBM cell lines [30] |
| BCL-2 Overexpression | Chronic Lymphocytic Leukemia (CLL) and Follicular Lymphoma [111] [109] | Sensitivity to Venetoclax (BCL-2 selective inhibitor) | Clinical trial data leading to FDA approval [109] |
Experimental Protocols for Biomarker Analysis
Protocol 1: Assessing PARP-1 Cleavage via Western Blotting
The detection of PARP-1 cleavage by caspases serves as a definitive biochemical hallmark of apoptotic commitment [13] [112]. This protocol allows for the discrimination of full-length PARP-1 (113 kDa) from its characteristic apoptotic fragments (89 kDa and 24 kDa).
Detailed Methodology:
- Cell Lysis: Harvest treated and control cells. Lyse cells using RIPA buffer supplemented with protease and phosphatase inhibitors. Incubate on ice for 30 minutes, followed by centrifugation at 14,000 x g for 15 minutes at 4°C to collect the supernatant.
- Protein Quantification: Determine protein concentration using the Pierce BCA Protein Assay Kit or an equivalent method [13].
- Gel Electrophoresis: Load 20-30 μg of total protein per lane onto a 4-12% Bis-Tris polyacrylamide gel. Perform electrophoresis at constant voltage (120-150V) until the dye front reaches the bottom of the gel.
- Membrane Transfer: Transfer proteins from the gel to a PVDF or nitrocellulose membrane using a wet or semi-dry transfer system.
- Immunoblotting: Block the membrane with 5% non-fat milk in TBST for 1 hour. Incubate with primary antibodies against PARP-1 (specific for the 89 kDa fragment or full-length protein) overnight at 4°C [13]. Following TBST washes, incubate with an appropriate horseradish peroxidase (HRP)-conjugated secondary antibody for 1 hour at room temperature.
- Detection: Visualize protein bands using enhanced chemiluminescence (ECL) substrate and imaging system. The cleavage is indicated by the appearance of the 89 kDa fragment and the concomitant decrease in the 113 kDa full-length band.
Protocol 2: Evaluating BCL-2 Family Protein Interactions via Co-Immunoprecipitation
Understanding the functional status of BCL-2 involves assessing its interactions with pro-apoptotic binding partners like BIM or BAX. Co-immunoprecipitation (Co-IP) is a key technique for validating these critical protein-protein interactions [6] [110].
Detailed Methodology:
- Preparation of Cell Lysates: Lyse cells in a mild, non-denaturing lysis buffer (e.g., containing 1% CHAPS or Triton X-100) to preserve protein interactions. Maintain samples at 4°C throughout the process.
- Pre-Clearing: Incubate the cell lysate with Protein A/G Agarose beads for 30-60 minutes to remove non-specifically binding proteins. Pellet the beads and collect the supernatant.
- Immunoprecipitation: Incubate the pre-cleared lysate with an antibody specific for your target protein (e.g., anti-BCL-2) overnight at 4°C with gentle agitation. Then, add Protein A/G Agarose beads and incubate for an additional 2-4 hours to capture the antibody-protein complex.
- Washing and Elution: Pellet the beads and wash them thoroughly with lysis buffer 3-5 times to remove unbound proteins. Elute the bound complexes by boiling the beads in 2X Laemmli sample buffer.
- Analysis: Analyze the eluted proteins by Western blotting as described in Protocol 3.1. Probe the membrane for the immunoprecipitated protein (e.g., BCL-2) and its suspected binding partners (e.g., BIM, BAX) to confirm interaction.
Visualization of Signaling Pathways and Experimental Workflows
PARP-1 and BCL-2 in Apoptotic Signaling Crosstalk
Diagram 1: Apoptotic Signaling Crosstalk. This pathway illustrates the interplay between DNA damage-induced PARP-1 activation and the BCL-2 regulated mitochondrial apoptotic pathway. Key biomarkers (PARP-1 cleavage, MOMP) and therapeutic intervention points (BH3-mimetics) are highlighted.
Experimental Workflow for Combined Biomarker Profiling
Diagram 2: Biomarker Profiling Workflow. A logical flow for the simultaneous experimental assessment of PARP-1 and BCL-2 status, integrating specific molecular readouts into a cohesive correlative analysis.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents and Kits for Apoptotic Biomarker Discovery
| Reagent / Kit | Primary Function | Specific Application |
|---|---|---|
| Anti-PARP-1 Antibody (cleavage-specific) | Detect apoptotic PARP-1 fragments (89 kDa) by Western Blot or IHC [13] | Discriminate apoptosis from other cell death modes; measure caspase-3 activity. |
| Anti-BCL-2 Family Antibodies | Measure protein levels of BCL-2, BCL-xL, MCL-1, BIM, BAX via Western Blot, IHC, or Flow Cytometry [30] [110] | Profile pro- and anti-apoptotic protein expression; stratify patients for targeted therapy. |
| Caspase-3/7 Activity Assay Kits | Quantify effector caspase activity using fluorometric or luminescent substrates [112] [30] | Functional confirmation of apoptosis engagement in high-throughput formats. |
| BH3 Profiling Peptides | Synthetic BH3 domain peptides to measure mitochondrial priming and dependence on anti-apoptotic proteins [109] | Functional assay to predict sensitivity to BH3-mimetics; assess apoptotic readiness. |
| PARP Activity Assay Kits | Quantify PARP enzymatic activity by measuring poly(ADP-ribose) (PAR) polymer formation [37] | Assess PARP-1 functional status independent of protein levels; monitor inhibitor efficacy. |
| Annexin V Apoptosis Detection Kits | Detect phosphatidylserine externalization on the plasma membrane via flow cytometry [112] [99] | Identify early-stage apoptotic cells in a heterogeneous population. |
The correlative analysis of PARP-1 and BCL-2 status provides a powerful, multi-faceted framework for predicting therapeutic outcomes. PARP-1 biomarkers, particularly its cleavage and role in parthanatos, offer a direct readout of cell death execution, while the expression and interaction profiles of BCL-2 family proteins reveal the cell's underlying survival threshold. As evidenced by the clinical success of PARP inhibitors and BH3-mimetics, integrating these biomarkers into research and diagnostic workflows is no longer optional but essential for advancing targeted cancer therapy. The experimental data and methodologies outlined in this guide provide a foundational toolkit for researchers to validate these critical apoptotic pathways, ultimately contributing to more precise and effective cancer treatments. Future directions will likely focus on standardized multiplex assays that simultaneously capture the dynamic state of both pathways, further refining our ability to predict and monitor patient responses.
Conclusion
The rigorous validation of apoptotic pathways through the integrated analysis of PARP-1 and BCL-2 family proteins is paramount for advancing our understanding of cell fate decisions. The foundational principles and methodological frameworks outlined provide a roadmap for dissecting the complex crosstalk between different regulated cell death mechanisms. The ability to troubleshoot experimental challenges and perform robust comparative analyses is critical for translating these insights into actionable therapeutic strategies. Future directions should focus on exploiting this crosstalk to overcome drug resistance in oncology, developing novel BH3 mimetics and PARP inhibitors with improved efficacy, and establishing standardized, functional biomarkers to predict patient response in clinical settings.
References
- 1. Insights on the crosstalk among different cell death ... [https://www.nature.com/articles/s41420-025-02328-9]
- 2. Regulation of Apoptosis [https://www.cellsignal.com/pathways/regulation-of-apoptosis-pathway?srsltid=AfmBOoovjotnhiq4bfyIA471fFQ1gdDRUAmvlBn8YEGtT-cbJ_dpSb5P]
- 3. Targeting apoptotic pathways for cancer therapy - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11245162/]
- 4. Intrinsic and Extrinsic Pathways of Apoptosis [https://www.thermofisher.com/us/en/home/life-science/antibodies/antibodies-learning-center/antibodies-resource-library/cell-signaling-pathways/cellular-apoptosis-pathway.html]
- 5. Apoptosis signaling pathways and lymphocyte ... [https://www.nature.com/articles/cr200752]
- 6. BCL2 suppresses PARP1 function and non-apoptotic cell death [https://pmc.ncbi.nlm.nih.gov/articles/PMC4075432/]
- 7. On PAR with PARP: cellular stress signaling through poly ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3305980/]
- 8. The 89-kDa PARP1 cleavage fragment serves as a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7948984/]
- 9. PARP-1 cleavage fragments: signatures of cell-death ... [https://biosignaling.biomedcentral.com/articles/10.1186/1478-811X-8-31]
- 10. Truncated PARP1 mediates ADP-ribosylation of RNA ... [https://www.nature.com/articles/s41421-021-00355-1]
- 11. PARP-1 inhibition influences the oxidative stress response ... [https://www.sciencedirect.com/science/article/pii/S2213231716300222]
- 12. The BCL2 family: from apoptosis mechanisms to new ... [https://www.nature.com/articles/s41392-025-02176-0]
- 13. RSL3 promotes PARP1 apoptotic functions by distinct ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12492750/]
- 14. BCL2 suppresses PARP1 function and nonapoptotic cell death [https://pubmed.ncbi.nlm.nih.gov/22689920/]
- 15. Multiple Functions of BCL-2 Family Proteins - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3552500/]
- 16. Bcl-2 family [https://en.wikipedia.org/wiki/Bcl-2_family]
- 17. Review Structural biology of the Bcl-2 family of proteins [https://www.sciencedirect.com/science/article/pii/S0167488903001757]
- 18. Comprehensive analysis of Bcl-2 family protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11737343/]
- 19. BCL-2 dependence is a favorable predictive marker of ... [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-025-02260-7]
- 20. Coixol derivatives induce DNA damage by targeting PARP1 [https://www.sciencedirect.com/science/article/abs/pii/S0223523425010724]
- 21. Next-Generation Bcl-2 Inhibitors: Design and Evaluation of ... [https://pubmed.ncbi.nlm.nih.gov/40963496/]
- 22. PARP and the Release of Apoptosis-Inducing Factor ... - NCBI [https://www.ncbi.nlm.nih.gov/books/NBK6179/]
- 23. Biological function, mediate cell death pathway and their ... [https://www.frontiersin.org/journals/nutrition/articles/10.3389/fnut.2022.1093939/full]
- 24. Bcl-2 acts upstream of the PARP protease and prevents its ... [https://www.nature.com/articles/4400200.pdf]
- 25. Current Evidence and Future Perspectives about the Role ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10362869/]
- 26. Regulated cell death pathways and their roles in homeostasis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10474065/]
- 27. Regulated cell death pathways and their roles in ... [https://www.nature.com/articles/s12276-023-01069-y]
- 28. Ferroptosis: Emerging mechanisms, biological function ... [https://www.nature.com/articles/s41420-024-01825-7]
- 29. Molecular mechanisms and potential implications of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12394052/]
- 30. Inhibition of anti-apoptotic Bcl-2 family members promotes ... [https://www.nature.com/articles/s41420-025-02632-4]
- 31. A snapshot into the transcriptomic landscape of apoptosis ... [https://www.nature.com/articles/s41419-024-06766-8]
- 32. Targeting the anti-apoptotic Bcl-2 family proteins [https://www.thno.org/v12p2427.htm]
- 33. PARP Antibody #9542 [https://www.cellsignal.com/products/primary-antibodies/parp-antibody/9542?srsltid=AfmBOor7KC2kSH1TRKs8Qml4AxXkql9hwirLPpAG8whQsBGFzPIWWsAc]
- 34. Efficacy and specificity of inhibitors of BCL-2 family protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9020777/]
- 35. Pro-Survival Bcl-2 Family Antibody Sampler Kit II #17229 [https://www.cellsignal.com/products/primary-antibodies/pro-survival-bcl-2-family-antibody-sampler-kit-ii/17229?srsltid=AfmBOoqliq7GpOn8xFXC1VzsW7yURpgPQOVLTPFwI3eJa0-jDERVE3jK]
- 36. Inhibition of anti-apoptotic BCL2 overcomes adaptive ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12557573/]
- 37. PARP inhibitors as radiosensitizers: a comprehensive ... [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2025.1702121/full]
- 38. Design, development, and therapeutic applications of ... [https://pubmed.ncbi.nlm.nih.gov/40948328/]
- 39. Anti-PARP1 Antibody [https://www.atlasantibodies.com/products/primary-antibodies/triple-a-polyclonals/anti-parp1-antibody-hpa045168/]
- 40. Cleaved PARP (Asp214) Antibody #9541 [https://www.cellsignal.com/products/primary-antibodies/cleaved-parp-asp214-antibody/9541?srsltid=AfmBOoo_OZRhRE0o26DauDjz7hKHUhErmdBuw6Ht8abPEsdn_AJYn_C5]
- 41. RSL3 promotes PARP1 apoptotic functions by distinct ... [https://cmbl.biomedcentral.com/articles/10.1186/s11658-025-00785-9]
- 42. 2.10. PARP-1 Cleavage [https://bio-protocol.org/exchange/minidetail?id=622538&type=30]
- 43. Comprehensive landscape of cell death mechanisms [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2025.1611055/full]
- 44. Characterization of the necrotic cleavage of poly(ADP- ... [https://www.nature.com/articles/4400851]
- 45. Apoptosis: A Comprehensive Overview of Signaling Pathways ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11592877/]
- 46. permeabilization - Wikipedia Mitochondrial outer membrane [https://en.wikipedia.org/wiki/Mitochondrial_outer_membrane_permeabilization]
- 47. Optimization and validation of mitochondria -based functional assay ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3717276/]
- 48. A new quantitative assay for cytochrome c release in ... [https://www.nature.com/articles/4401263]
- 49. A Comparison of Three Flow Cytometry Methods for ... [https://pubmed.ncbi.nlm.nih.gov/17654655/]
- 50. A Comprehensive Exploration of Caspase Detection Methods [https://pmc.ncbi.nlm.nih.gov/articles/PMC11121653/]
- 51. Caspase assay selection guide [https://www.abcam.com/en-us/products/selection-guides/caspase-assay-selection-guide]
- 52. Integrated real-time imaging of executioner caspase ... [https://www.nature.com/articles/s41420-025-02662-y]
- 53. Overlapping cleavage motif selectivity of caspases [https://www.nature.com/articles/4402260]
- 54. Caspase-3 Antibody Comparison Table [https://www.cellsignal.com/science-resources/caspase-3-signaling/caspase-3-comparison?srsltid=AfmBOoogtU-5z0TL3aad1rRluTZPpzUdsuVGeuw42VZ8PKKRpo16BbTo]
- 55. Generation and characterization of antibodies specific for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3186909/]
- 56. PARP Inhibitors: Clinical Relevance, Mechanisms of Action ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2020.564601/full]
- 57. PARP-1 improves leukemia outcomes by inducing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10518631/]
- 58. Bcl-2 family inhibitors sensitize human cancer models to therapy | Cell Death & Disease [https://www.nature.com/articles/s41419-023-05963-1]
- 59. Targeting BCL-2 family proteins using BH3 mimetic drugs ... [https://www.sciencedirect.com/science/article/pii/S2590098624000241]
- 60. Expanding the Perspective on PARP1 and Its Inhibitors in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11275100/]
- 61. Targeting the Bcl-2-regulated apoptosis pathway by BH3 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4563920/]
- 62. resistant ovarian cancer cells mediated via ATR and ATM ... [https://www.nature.com/articles/s41420-025-02740-1]
- 63. BCL2-overexpressing prostate cancer cells rely on PARP1 ... [https://www.sciencedirect.com/science/article/abs/pii/S0304383518301964]
- 64. Targeting PARP1: A Promising Approach for Next- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12162764/]
- 65. Review The biochemical pathways of apoptotic, necroptotic ... [https://www.sciencedirect.com/science/article/pii/S1097276523010171]
- 66. Apoptosis detection: a purpose-dependent approach ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8208110/]
- 67. Targeting regulated cell death: Apoptosis, necroptosis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11921819/]
- 68. Targeting ferroptosis, pyroptosis and necroptosis for cancer ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12605190/]
- 69. MiniResource Brief guide to detecting ferroptosis [https://www.sciencedirect.com/science/article/pii/S1016847825001001]
- 70. Optimization of cell viability assays to improve replicability ... [https://www.nature.com/articles/s41598-020-62848-5]
- 71. Strategies to Overcome Intrinsic and Acquired Resistance ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11719474/]
- 72. Targeting Apoptosis to Overcome Chemotherapy Resistance [https://www.ncbi.nlm.nih.gov/books/NBK580869/]
- 73. overexpression of nuclear DNA repair protein PARP-1 ... [https://www.sciencedirect.com/science/article/abs/pii/S0046817714001488]
- 74. Overcoming cancer drug resistance: insights into Apoptotic ... [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2025.1677380/full]
- 75. Therapeutic Targeting of Apoptosis, Autophagic Cell Death ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12293011/]
- 76. Targeting anti-apoptotic mechanisms in tumour cells [https://www.sciencedirect.com/science/article/abs/pii/S0045206825002688]
- 77. Apoptosis-independent inhibition of DNA repair by Bcl-2 ... [https://www.sciencedirect.com/science/article/abs/pii/S1383574212000415]
- 78. Poly(ADP-ribose) polymerase-1 and its cleavage products ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4013233/]
- 79. Study of the Bcl-2 Interactome by BiFC Reveals Differences ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10000386/]
- 80. 2D and 3D cell cultures – a comparison of different types of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6040128/]
- 81. A comparative analysis of 2D and 3D experimental data for ... [https://www.nature.com/articles/s41598-023-42486-3]
- 82. Key aspects for conception and construction of co-culture ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10119418/]
- 83. Establishment and Validation of a New Co-Culture for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10222603/]
- 84. Data alchemy, from lab to insight: Transforming in vivo ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11361667/]
- 85. Validation of a Novel EUS-FNB-Derived Organoid Co ... [https://www.mdpi.com/2072-6694/15/14/3677]
- 86. Establishment and validation of an in vitro co-culture model ... [https://www.nature.com/articles/s41598-020-73941-0]
- 87. Programmed cell death detection methods: a systematic review and a categorical comparison - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9308588/]
- 88. Apoptosis Assays | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/cell-analysis/cell-viability-and-regulation/apoptosis.html]
- 89. Assay technologies for apoptosis and autophagy [https://www.sciencedirect.com/science/article/pii/S259009862100021X]
- 90. Comparative analysis of different methodological approaches to the in vitro study of drug-induced apoptosis - PubMed [https://pubmed.ncbi.nlm.nih.gov/10514415/]
- 91. Comparison of apoptosis and mortality measurements in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6496915/]
- 92. Comparison of cell-based assays to quantify treatment ... [https://www.nature.com/articles/s41598-018-34696-x]
- 93. RSL3 promotes PARP1 apoptotic functions by distinct ... [https://pubmed.ncbi.nlm.nih.gov/41039223/]
- 94. Potent ferroptosis agent RSL3 induces cleavage of ... [https://www.nature.com/articles/s41598-025-11368-1]
- 95. BH3 profiling – measuring integrated function of the mitochondrial apoptotic pathway to predict cell fate decisions - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3770266/]
- 96. BH3 Profiling: A Functional Assay to Measure Apoptotic Priming and Dependencies - PubMed [https://pubmed.ncbi.nlm.nih.gov/30535998/]
- 97. Dynamic BH3 profiling identifies pro-apoptotic drug ... [https://www.nature.com/articles/s41467-023-38552-z]
- 98. BH3 Profiling Identifies Three Distinct Classes of Apoptotic ... [https://www.sciencedirect.com/science/article/pii/S1535610807002000]
- 99. Guidelines for Regulated Cell Death Assays: A Systematic ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2021.634690/full]
- 100. Targeting Cancer Cell Fate: Apoptosis, Autophagy, and ... [https://www.mdpi.com/1467-3045/47/6/460]
- 101. Comparison of Apoptosis Detection Markers Combined ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2716774/]
- 102. BH3-mimetics: recent developments in cancer therapy [https://jeccr.biomedcentral.com/articles/10.1186/s13046-021-02157-5]
- 103. Parthanatos and apoptosis: unraveling their roles in cancer ... [https://www.excli.de/vol24/2025-8251/2025-8251.htm]
- 104. GX15-070 enhances niraparib efficacy in ovarian cancer by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12607189/]
- 105. Combining BH3-mimetics to target both BCL-2 and MCL1 ... [https://www.nature.com/articles/s41375-018-0261-3]
- 106. Mitochondrial priming and response to BH3 mimetics in “ ... [https://www.nature.com/articles/s41420-025-02379-y]
- 107. International Journal of Oncology [https://www.spandidos-publications.com/10.3892/ijo.2017.3914]
- 108. Antagonism of Bcl-XL is necessary for synergy between ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4832520/]
- 109. The BCL2 family: from apoptosis mechanisms to new ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11926181/]
- 110. The role of BCL-2 family proteins in regulating apoptosis ... [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2022.985363/full]
- 111. The BCL-2 protein family: from discovery to drug ... [https://www.nature.com/articles/s41418-025-01481-z]
- 112. Biomarkers of apoptosis - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2538762/]
- 113. Article PARP-1 improves leukemia outcomes by inducing ... [https://www.sciencedirect.com/science/article/pii/S2666379123003580]